CADTH Reimbursement Review
Lutetium (177Lu) Vipivotide Tetraxetan (Pluvicto)
Sponsor: Advanced Accelerator Applications Canada, Inc.
Therapeutic area: Metastatic castration-resistant prostate cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Stakeholder Input
Clinical Review
Abbreviations
177Lu
lutetium-177
68Ga
gallium-68
AE
adverse event
ARPI
androgen receptor pathway inhibitor
BPI-SF
Brief Pain Inventory-Short Form
BSC
best supportive care
BSoC
best standard of care
CCS
Canadian Cancer Society
CCSN
Canadian Cancer Survivor Network
CI
confidence interval
CMII
Canadian Medical Imaging Inventory
CR
complete response
CrCl
creatinine clearance
CRPC
castration-resistant prostate cancer
CUA
Canadian Urological Association
DCR
disease control rate
DOR
duration of response
EC
ethics committee
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EOT
end of treatment
EQ-5D-5L
5-Level EQ-5D
EQ-VAS
EQ visual analogue scale
FACT-G
Functional Assessment of Cancer Therapy – General
FACT-P
Functional Assessment of Cancer Therapy – Prostate
FAPSI-8
8-item Functional Assessment of Cancer Therapy Advanced Prostate Symptoms Index
FAS
full analysis set
FDG
18F-fluorodeoxyglucose
HR
hazard ratio
HRQoL
health-related quality of life
IRB
institutional review board
ITC
indirect treatment comparison
ITT
intention to treat
LDH
lactase dehydrogenase
MCID
minimum clinically important difference
mCSPC
metastatic castration-sensitive prostate cancer
NAAD
novel androgen axis drug
NMA
network meta-analysis
OR
odds ratio
ORR
overall response rate
OS
overall survival
PCWG3
Prostate Cancer Working Group 3
PFS
progression-free survival
PPI
present pain intensity
PR
partial response
PSA
prostate-specific antigen
PSMA
prostate-specific membrane antigen
QoL
quality of life
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours Version 1.1
rPFS
radiographic progression-free survival
SAE
serious adverse event
SD
standard deviation
SSE
symptomatic skeletal event
SUVmax
maximum standardized uptake value
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | 177Lu vipivotide tetraxetan (Pluvicto), solution for injection, 1 GBq/mL (27 mCi/mL) at calibration |
Indication | The treatment of adults with PSMA-positive mCRPC who have received at least 1 ARPI and taxane-based chemotherapy |
Reimbursement request | Per indication |
Health Canada approval status | Approved (NOC) |
Health Canada review pathway | Priority review |
NOC date | August 25, 2022 |
Sponsor | Advanced Accelerator Applications |
177Lu = lutetium-177; ARPI = androgen receptor pathway inhibitor; mCRPC = metastatic castration-resistant prostate cancer; NOC = Notice of Compliance; PSMA = prostate-specific membrane antigen.
Introduction
Prostate cancer is the most common cancer among men in Canada (excluding nonmelanoma skin cancers), affecting 1 in 9 men during their lifetime.1 Prostate cancer represents approximately 20% of all new cancers diagnosed in men in Canada and 10% of cancer deaths in men.2 In 2022, it was estimated that 24,600 men in Canada would be diagnosed with prostate cancer and 4,600 men would die from prostate cancer.2 Patients who die from prostate have typically progressed to the metastatic castration-resistant prostate cancer (mCRPC) stage, which has a 5-year survival rate of approximately 30%.3 Castration-resistant prostate cancer (CRPC) is defined as disease progression despite castrate levels of testosterone that may present as a continuous rise in serum prostate-specific antigen (PSA) levels, the progression of preexisting disease, and/or the appearance of new metastases.4
Prostate-specific membrane antigen (PSMA) is a transmembrane glycoprotein that is highly expressed in prostate cancer cells. Lutetium-177 (177Lu) vipivotide tetraxetan (Pluvicto) contains the radionuclide lutetium-177, which is linked to a targeting moiety that binds to PSMA. Upon the binding of 177Lu vipivotide tetraxetan to PSMA-expressing cancer cells, the beta-minus emission from lutetium-177 delivers therapeutic radiation to the targeted cell, as well as to surrounding cells, and induces DNA damage that can lead to cell death.5
177Lu vipivotide tetraxetan injection is indicated for the treatment of adults with PSMA-positive mCRPC who have received at least 1 androgen receptor pathway inhibitor (ARPI) and taxane-based chemotherapy.5 The sponsor has requested that 177Lu vipivotide tetraxetan be reimbursed in accordance with the Health Canada–approved indication.6 Based on the approved indication for 177Lu vipivotide tetraxetan, there are 3 relevant subpopulations for consideration in this review:
patients previously treated with docetaxel who are considered eligible to receive cabazitaxel
patients previously treated with docetaxel who are considered ineligible to receive cabazitaxel
patients previously treated with both docetaxel and cabazitaxel.
177Lu vipivotide tetraxetan is administered intravenously, and the recommended dose is 7.4 GBq every 6 weeks (± 1 week), for a total of 6 doses.5 It is available as a 1,000 MBq/mL solution for injection in single-dose vials that contain a total amount of radioactivity of 7.4 GBq (± 10%) at the time of administration.5
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, the Canadian Cancer Society (CCS) and the Canadian Cancer Survivor Network (CCSN), provided input on the treatment of adults with PSMA-positive mCRPC who have been treated with an ARPI and taxane-based chemotherapy or who are not medically suitable for taxanes. Patient input was gathered from surveys and interviews with patients with mCRPC and their caregivers across Canada in August 2022. Of the 27 survey respondents, 19 were from the CCS and 8 were from the CCSN. Of the 7 patients included in the submissions who had experience with the treatment under review, 4 were from the CCS and 3 were from the CCSN.
Patients noted that mCRPC has a substantial negative impact on their quality of life (QoL) and their ability to perform the activities of daily living, including the ability to engage in sexual activity, travel and exercise, fulfill family obligations, maintain mental health, work, perform household chores, concentrate, spend time with family and friends, and fulfill practical needs (e.g., preparing meals, dressing, bathing). Patients can suffer from frequent urination, erectile dysfunction, bone and/or skeletal pain, hot flashes, weight gain, memory loss, and cognitive problems. Patient groups noted that patients are seeking new treatment options that will prolong life, maintain QoL, delay the onset of symptoms, and improve sexual function. The groups noted that existing treatment options can be associated with negative side effects and highlighted the need for effective and more tolerable treatment options.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH noted that there are limited effective treatments for patients with mCRPC who have progressed after treatment with an ARPI and docetaxel. Overall survival (OS) is poor for patients with disease that has been demonstrated to be refractory to multiple treatment options and for whom the symptoms of cancer progression pose a considerable burden. Other standard of care treatments, such as cabazitaxel, are associated with significant toxic effects for patients. The clinical experts noted that there is a need for therapies that improve OS and QoL for this patient population and that are better tolerated and more convenient (e.g., less need for supportive medications, less frequent administration) than current standard of care options.
The clinical experts noted that 177Lu vipivotide tetraxetan could be considered for patients whose disease progressed after treatment with both an ARPI and docetaxel. The experts noted that there is uncertainty regarding the place of 177Lu vipivotide tetraxetan in therapy relative to cabazitaxel for patients who are considered appropriate candidates for treatment with a second chemotherapy regimen. The clinical experts consulted by CADTH also identified the requirement for suitable PSMA-targeted PET expression, per the inclusion criteria of the pivotal VISION trial, to be a candidate for therapy. The clinical experts noted that 177Lu vipivotide tetraxetan should be discontinued in patients who experience disease progression characterized by at least 2 specific adverse occurrences (i.e., sustained PSA rise, clinical progression [sustained, nonanalgesic, responsive pain; performance status decline], and radiographic progression); significant toxicity related to the treatment; or worsening of performance status (i.e., Eastern Cooperative Oncology Group Performance Status [ECOG PS] ≥ 3).
Clinician Group Input
Clinician group input (coordinated by the CCS) was received from prostate-treating clinicians in Canada with a special interest in the care of patients with metastatic prostate cancer. The clinician group noted that there are unmet needs for patients with mCRPC and a need for additional lines of therapy that can preserve QoL and provide meaningful survival benefits for patients with progressive metastatic prostate cancer. According to the clinician group, a new treatment would be most suited to patients with progressive (symptomatic, imaging, or biochemical) mCRPC, PSMA-expressing metastases identified on a diagnostic PSMA-targeted PET scan, and with adequate performance status (ECOG PS of 0 to 2) and organ function (liver and bone marrow). The clinician group also pointed out that the most meaningful clinical response to treatment for this disease would be the prevention of progression, reflected in stability or improvement in biochemical and imaging biomarkers such as serum PSA, bone scan, and CT. The clinician group emphasized that appropriate facilities, certifications, and licensed personnel for the safe delivery of unsealed radiopharmaceutical treatments would be needed for the treatment under review, as would access to diagnostic PSMA-targeted PET for proper patient selection.
Drug Program Input
The drug programs that participated in the CADTH reimbursement review process identified possible implementation issues related to the following: potential comparators for 177Lu vipivotide tetraxetan; the application of PSMA testing in Canada as a diagnostic modality for identification of the target patient population and as a potential evaluator of response to therapy; the criteria used in practice to identify patients who would not be suitable for treatment with cabazitaxel; and the potential use of 177Lu vipivotide tetraxetan in combination with other systemic anticancer therapies (most notably ARPIs).
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
The evidence for the review of 177Lu vipivotide tetraxetan for the treatment of adults with PSMA-positive mCRPC who have received at least 1 ARPI and taxane-based chemotherapy was derived from a systematic literature review of pivotal and phase III studies, supplemented with additional information to address important gaps in the randomized controlled trial (RCT) evidence. One RCT met the eligibility criteria for the systematic review. VISION (N = 831) is a phase III, open-label, RCT conducted to evaluate the efficacy and safety of 177Lu vipivotide tetraxetan administered to patients with progressive PSMA-positive mCRPC in addition to best supportive care (BSC) or best standard of care (BSoC) (the 177Lu vipivotide tetraxetan + BSC/BSoC group) relative to BSC or BSoC alone (the BSC/BSoC alone group).7 Patients were randomized in a 2:1 ratio to 177Lu vipivotide tetraxetan + BSC/BSoC or BSC/BSoC alone, with allocation stratified by lactase dehydrogenase (LDH) (≤ 260 IU/L versus > 260 IU/L), presence of liver metastases (yes versus no), ECOG PS (0 or 1 versus 2), and inclusion of a novel androgen axis drug (NAAD) in BSC/BSoC (yes versus no).
The VISION trial had considerable early withdrawal of consent and a disproportionate dropout in the BSC/BSoC alone group (patients typically cited disappointment that they would not receive 177Lu vipivotide tetraxetan). This was a major limitation of the study and required the sponsor to introduce the following protocol amendments: the overall target sample size was increased; educational measures were introduced to try to bolster the retention of patients in the comparator group; and, most important from a critical appraisal perspective, the definition of a new analysis set that would be limited to patients enrolled after the protocol amendments were introduced (i.e., the progression-free survival [PFS] full analysis set [FAS]). This new analysis set was used for the primary evaluation of all end points, with the exception of OS (FAS) and the overall response rate (ORR) and disease control rate (DCR), which evaluated an even smaller subset of patients (i.e., those in the PFS-FAS who had Response Evaluation Criteria in Solid Tumours Version 1.1 (RECIST)-evaluable disease).
Efficacy Results
Table 2 summarizes results for the efficacy end points from the VISION trial. The primary and secondary end points of the VISON trial were aligned with those recommended by the Prostate Cancer Working Group 3 (PCWG3) (i.e., OS, radiographic progression-free survival [rPFS], time to first symptomatic skeletal event [SSE], health-related quality of life [HRQoL], PFS, and biochemical response [e.g., PSA]). As noted previously, only the analysis of OS was conducted using the FAS dataset.
OS: There was a statistically significant improvement in OS for patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group, compared with those in the BSC/BSoC alone group (hazard ratio [HR] = 0.62; 95% confidence interval [CI], 0.52 to 0.74; P < 0.001). Median OS was 15.3 months (95% CI, 14.2 to 16.9 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 11.3 months (95% CI, 9.8 to 13.5 months) in the BSC/BSoC alone group. Subgroup analyses based on the number of prior taxane regimens favoured 177Lu vipivotide tetraxetan + BSC/BSoC over BSC/BSoC alone for patients who received a single prior taxane regimen (HR = 0.59; 95% CI, 0.46 to 0.75) and those who received 2 or more prior taxane regimens (HR = 0.73; 95% CI, 0.53 to 0.99).
rPFS: There was a statistically significant improvement in rPFS for patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group, compared with those in the BSC/BSoC alone group (HR = 0.40; 99.2% CI, 0.29 to, 0.57; P < 0.001). Events of radiographic progression or death were reported for 66.0% of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group (171 radiographic progression events and 83 deaths) and 47.4% of patients in the BSC/BSoC alone group (59 radiographic progression events and 34 deaths). Median rPFS was 8.7 months (95% CI, 7.9 to 10.8 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 3.4 months (95% CI, 2.4 to 4.0 months) in the BSC/BSoC alone group. The sponsor reported that median follow-up time for rPFS was longer in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC group (16.4 months and 3.9 months, respectively).
ORR: The ORR was statistically significantly greater in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC group (29.8% versus 1.7%), with an odds ratio (OR) of 24.99 (95% CI, 6.05 to 103.24).
DOR: Median duration of response (DOR) in patients who demonstrated a response to treatment (i.e., complete response [CR] or partial response [PR]) was 9.8 months (95% CI, 9.1 to 11.7 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC group. Only 2 patients in the BSC/BSoC only group demonstrated a response to treatment, and only 1 of those met the criteria for RECIST radiographic progression or death; therefore, the sponsor reported that median DOR could not be reliably estimated for the BSC/BSoC alone group.
DCR: The DCR was statistically significantly greater in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (89.0% versus 66.7%), with an OR of 5.79 (95% CI, 3.18 to 10.55; P < 0.001).
Time to first SSE: There were 256 SSEs in the 177Lu vipivotide tetraxetan + BSC/BSoC group (66.5%; 60 events and 196 deaths) and 137 SSEs (69.9%; 34 events and 103 deaths) in the BSC/BSoC alone group. 177Lu vipivotide tetraxetan + BSC/BSoC was associated with a statistically significant reduction in the risk of SSE (or death), compared to BSC/BSoC alone (HR = 0.5; 95% CI, 0.40 to 0.62).
PFS: Progression events or death were reported for █████ of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group (█████ radiographic progression, █████ clinical progression, █████ PSA progression, ████ death) and █████ of patients in the BSC/BSoC group (█████ radiographic progression, █████ clinical progression; █████ PSA progression; ████ death). 177Lu vipivotide tetraxetan + BSC/BSoC was associated with a statistically significant reduction in the risk of radiographic disease progression, clinical progression, PSA progression, or death, compared to BSC/BSoC alone (███ █████ ███ ███ ████ ██ █████ ██ █████). Median PFS was ███ ██████ (███ ███ ███ ██ ███) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and ███ ██████ ████ ███ ███ ██ ████ in the BSC/BSoC alone group.
PSA levels: The sponsor reported a large disparity in the proportion of patients who could be evaluated for PSA doubling time between the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (█████ and ██████ respectively). For the subset of patients who could be evaluated, mean PSA doubling time was ████ ██████ (███ ███ ████ ██ ████) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and ████ ██████ (███ ███ ███ ██ ████) in the BSC/BSoC alone group.
BPI-SF: Worsening in pain intensity was defined as an increase from baseline of at least 30% or an increase from baseline of at least 2 points on the Brief Pain Inventory-Short Form (BPI-SF) scale at any time up to the end-of-treatment (EOT) visit, clinical disease progression, or death. Time to worsening pain was longer in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (HR = 0.52; 95% CI, 0.43 to 0.63; P < 0.001). Median time to deterioration was 5.9 months (95% CI, 4.8 to 6.9 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 2.2 months (95% CI, 1.8 to 2.8 months) in the BSC/BSoC alone group.
FACT-P: Time to worsening in Functional Assessment of Cancer Therapy-Prostate (FACT-P) scores was defined as the time from randomization to the first occurrence of a decrease of at least 10 points in total score from baseline, clinical disease progression, or death. Total events were similar in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (87.0% and 85.7%, respectively). Median time to worsening of the FACT-P score was shorter in the 177Lu vipivotide tetraxetan + BSC/BSoC group (5.7 months; 95% CI, 4.8 to 6.6 months) than in the BSC/BSoC alone group (2.2 months; 95% CI, 1.8 to 2.8 months), with an HR of 0.54 (95% CI, 0.45 to 0.66; P < 0.001).
FACT-G: Time to worsening in Functional Assessment of Cancer Therapy-General (FACT-G) scores was defined as the time from randomization to the first occurrence of a decrease of at least 10 points in total score from baseline, clinical disease progression, or death. Median time to worsening of the FACT-G score was lower in the 177Lu vipivotide tetraxetan + BSC/BSoC group (███ ███████ ███ ███ ███ ██ ███) than in the BSC/BSoC alone group (███ ███████ ███ ███ ███ ██ ███) (HR = █████ ███ ███ ████ ██ █████ ██ █████).
FAPSI-8: Time to worsening in 8-item Functional Assessment of Cancer Therapy Advanced Prostate Symptoms Index (FAPSI-8) scores was defined as the time from randomization to the first occurrence of a decrease of at least 10 points in total score from baseline, clinical disease progression, or death. Total events were nearly identical in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (█████ ███ ██████ respectively). Median time to worsening of the FAPSI-8 score was lower in the 177Lu vipivotide tetraxetan + BSC/BSoC group (███ ███████ ███ ███ ███ ██ ███) than in the BSC/BSoC alone group (███ ███████ ███ ███ ███ ██ ███) (HR = █████ ███ ███ ████ ██ █████ ██ █████).
Harms Results
Table 2 summarizes key adverse event (AE) data from the VISION trial. The sponsor reported that the following events were reported more commonly with the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (i.e., a difference of ≥ 10.0% between the groups): fatigue (43.1% versus 22.9%), dry mouth (38.8% versus 0.5%), nausea (35.3% versus 16.6%), anemia (31.8% versus 13.2%), diarrhea (18.9% versus 2.9%), vomiting (18.9% versus 6.3%), thrombocytopenia (17.2% versus 4.4%), lymphopenia (14.2% versus 3.9%), leucopenia (12.5% versus 2.0%), and urinary tract infection (11.0% versus 1.0%).7
A greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group reported at least 1 AE of grade 3 AE or higher than in the BSC/BSoC alone group (52.7% versus 38.0%). Events of grade 3 or higher that were more commonly reported in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group included anemia (12.9% versus 4.9%), thrombocytopenia (7.9% versus1.0%), lymphopenia (7.8% versus 0.5%), and fatigue (5.9% versus 1.5%). Spinal cord compression was reported more commonly in the BSC/BSoC alone group than in the 177Lu vipivotide tetraxetan + BSC/BSoC group (5.4% versus 1.3%).7 At least 1 serious adverse event (SAE) was reported for a greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (36.3% versus 27.8%).
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies
Result | 177Lu vipivotide tetraxetan + BSC/BSoC | BSC/BSoC alone |
|---|---|---|
OS (FAS) | n = 551 | n = 280 |
Deaths, n (%) | 343 (62.3) | 187 (66.8) |
Censored, n (%) | 208 (37.7) | 93 (33.2) |
Median OS (95% CI), months | 15.3 (14.2 to 16.9) | 11.3 (9.8 to 13.5) |
HR (95% CI); P value | 0.62 (0.52 to 0.74); P < 0.001 | |
rPFS (PFS-FAS) | n = 385 | n = 196 |
Events (progression or death), n (%) | 254 (66.0) | 93 (47.4) |
Radiographic progression, n (%) | 171 (44.4) | 59 (30.1) |
Deaths | 83 (21.6) | 34 (17.3) |
Censored | 131 (34.0) | 103 (52.6) |
Median rPFS (99.2% CI) | 8.7 (7.9 to 10.8) | 3.4 (2.4 to 4.0) |
HR (95% CI); P value | 0.40 (0.29 to 0.57); P < 0.001 | |
ORR (PFS-FAS) | n = 319 | n = 120 |
Patients with response (CR + PR), n (%) | 95 (29.8) | 2 (1.7) |
OR (95% CI); P value | 24.99 (6.05 to 103.24); P < 0.001 | |
DOR (PFS-FAS) | n = 319 | n = 120 |
Median DOR (95% CI) | 9.8 (9.1 to 11.7) | 10.6 (NE to NE) |
EDOR (months) | 3.7 | 0.2 |
Ratio of EDOR (95% CI); P value | 21.05 (5.27 to 84.05); P < 0.001 | |
DCR (PFS-FAS) | n = 319 | n = 120 |
Patients with event (CR, PR, stable disease ≥ 6 months), n (%) | 284 (89.0) | 80 (66.7) |
OR (95% CI); P value | 5.79 (3.18 to 10.55); P < 0.001 | |
Time to first SSE (PFS-FAS) | n = 385 | n = 196 |
Events (SSE or death), n (%) | 256 (66.5) | 137 (69.9) |
Median time to first SSE (95% CI) | 11.5 (10.3 to 13.2) | 6.8 (5.2 to 8.5) |
HR (95% CI);a,b P value | 0.50 (0.40 to 0.62); P < 0.001 | |
PFS (PFS-FAS) | n = 385 | n = 196 |
Events (progression or death), n (%) | ███ ██████ | ███ ██████ |
Median PFS (95% CI) | ███ ████ ██ ████ | ███ ████ ██ ████ |
HR (95% CI);b,c P value | ████ █████ ██ ██████ ██ █████ | |
Time to worsening in BPI-SF (PFS-FAS) | n = 385 | n = 196 |
Events (worsening, progression, or death), n (%) | 328 (85.2) | 166 (84.7) |
Median time to worsening (95% CI) | 5.9 (4.8 to 6.9) | 2.2 (1.8 to 2.8) |
HR (95% CI); P value | 0.52 (0.43 to 0.63); P < 0.001 | |
AEs (PFS-FAS) | n = 529 | n = 205 |
All AEs, n (%) | 519 (98.1) | 170 (82.9) |
SAEs, n (%) | 192 (36.3) | 57 (27.8) |
Grade 3, 4, or 5 AEs, n (%) | 279 (52.7) | 78 (38.0) |
Fatal AEs, n (%) | 19 (3.6) | 6 (2.9) |
177Lu = lutetium-177; AE = adverse event; BPI-SF = Brief Pain Inventory-Short Form; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; CR = complete response; DCR = disease control rate; DOR = duration of response; EDOR = expected duration of response; FAS = full analysis set; HR = hazard ratio; NE = not evaluable; OR = odds ratio; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; rPFS = radiographic progression-free survival; SAE = serious adverse event; SSE = symptomatic skeletal event.
Source: Clinical Study Report.7
Critical Appraisal
Internal Validity
Randomization was stratified by important prognostic factors, and baseline and demographic characteristics were generally well balanced in the 177Lu vipivotide tetraxetan + BSC/BSoC and the BSC/BSoC alone groups (including receipt of prior systemic anticancer therapy). The sponsor reported that the open-label design of the VISION study was used because blinding would not be practical, owing to the specialized precautions required for administration of a radiopharmaceutical and the toxicities related to targeted radioligand therapy and because it would not be appropriate to subject patients who did not receive a radiopharmaceutical to posttreatment radiation protection protocols (e.g., maintaining physical distancing from family members). Radiographic images were evaluated using blinded independent central review, and those results were used in the primary evaluations of rPFS and ORR (local assessments were used for patient management and in sensitivity analyses).
The open-label study design contributed to the high rate of early withdrawal for those who were randomized to the BSC/BSoC alone group (i.e., patients were disappointed at not receiving 177Lu vipivotide tetraxetan, leading to a lack of willingness to comply with the study protocol and/or interest in receiving therapies that were prohibited in the study protocol). The sponsor established corrective actions with a protocol amendment that included site calls to discuss the management of patients in the BSC/BSoC alone, or control, arm; investigator letters clarifying aspects of the study; and prescreening updates to improve patient education about the trial. After implementation of these measures, the sponsor noted that withdrawal of consent decreased.8 However, withdrawal rates in the BSC/BSoC alone group were █████ and █████ before and after the protocol amendment, respectively, compared with ████ and ████ in the 177Lu vipivotide tetraxetan + BSC/BSoC group (i.e., although the rate of discontinuation from the BSC/BSoC alone group improved after the protocol amendment, it remained considerably higher than the rate observed in the 177Lu vipivotide tetraxetan + BSC/BSoC group). As a result of the high dropout rate in the BSC/BSoC alone group, the sponsor also amended the protocol so that all end points, with the exception of OS, were analyzed using a newly established PFS-FAS dataset, which was composed of patients enrolled after the educational protocol amendments were introduced.9 The approach used is a way to handle early withdrawals; however, the analyses based on the PFS-FAS would not likely have followed the intention-to-treat (ITT) principle, which would affect many of the assumptions made about the comparisons. This approach was acceptable to the FDA and Health Canada;8,9 however, both regulatory agencies stated that the interpretation of the magnitude of the rPFS effect was limited because of the high degree of censoring from early drop-outs in the control arm (neither the approved US label nor the Canadian product monograph include the effect size for rPFS from the VISION trial).5,10
The high and disproportionate number of patients who withdrew from the control group could bias the study results in favour 177Lu vipivotide tetraxetan + BSC/BSoC, as those who remained in the study may have had poorer prognoses than those who withdrew and subsequently received treatment with regimens that were not permitted in the VISION protocol. Similarly, patients who remained in the trial may have had fewer therapeutic options (e.g., more advanced disease) and may have lacked the resources to obtain access to alternative regimens outside of the clinical trial setting (e.g., socioeconomic factors).11
External Validity
The clinical experts consulted by CADTH noted that the baseline and demographic characteristics in the VISION trial are a reasonable reflection of the target patient population in Canada. The clinical experts consulted by CADTH noted that the duration of survival in the control group (i.e., 11.3 months) exceeds what would be anticipated for the target population in Canadian practice. The experts estimated that survival is typically in the range of 6 to 9 months for patients with progressive mCRPC who have demonstrated disease progression after prior treated with ARPI(s) and taxane regimen(s). It was noted that this is commonly observed in clinical trials of prostate cancer in which patients are often healthier and have fewer comorbidities than the overall patient population encountered in routine Canadian clinical practice.
All of the patients included in the VISION trial had prior exposure to at least 1 taxane regimen. At the time of screening, █████ of patients had received 2 taxane regimens and ████ had received more than 2 taxane regimens. At the time of enrolment in the VISION trial, █████ of the total study population had been treated with a single taxane and, therefore, should not have been medically suitable for another taxane regimen, per the study protocol. The clinical experts consulted by CADTH noted that this number is greater than would be anticipated in Canadian practice for the target population, in which approximately ███ ██ ███ of patients would be considered not medically suitable for cabazitaxel. An important limitation of the external validity of the VISION trial was the large proportion of patients who received cabazitaxel in the poststudy treatment setting (i.e., the VISION trial enrolment criteria stated that patients who had received a single taxane regimen would be medically unsuitable for an additional taxane regimen). The clinical experts consulted by CADTH noted that this would not be reflective of Canadian practice, in which a patient with mCRPC who is considered ineligible for a further taxane regimen is unlikely to become eligible at a later point in time, as this disease is progressive and improvements in functional status or physiologic reserve are not anticipated. Other than these issues, the clinical experts noted that subsequent therapies could be reflective of routine care for patients for whom there are no other therapies that have been shown to increase OS.
177Lu vipivotide tetraxetan was administered as an add-on therapy in the VISION trial, which included concomitant administration of other systemic cancer therapies. There are no Canadian clinical practice guidelines that address the use of 177Lu vipivotide tetraxetan, and the clinical experts consulted by CADTH noted that it is unclear whether the use of 177Lu vipivotide tetraxetan in combination with other systemic anticancer therapies would be adopted in practice because of uncertainty regarding additional clinical benefit and harms for patients.
Several potential comparators for 177Lu vipivotide tetraxetan were not permitted in the acceptable BSoC treatment regimes. These included cytotoxic chemotherapy (e.g., cabazitaxel), immunotherapies, and other systemic radioisotopes (e.g., radium-223 dichloride [Xofigo], or hemi-body radiotherapy). The rationale provided by the sponsor was that these therapies could confound the analysis of results and that systemic anticancer options in the comparator group were limited to hormone therapies, including ARPIs (e.g., abiraterone and enzalutamide). All of the patients enrolled in the trial had exposure to novel ARPIs before enrolment. This approach may have biased the treatment effects in favour of 177Lu vipivotide tetraxetan, as the majority those in the BSC/BSoC alone group had already been treated with and demonstrated disease progression on the only systemic therapies that were permitted in the trial.11
177Lu vipivotide tetraxetan could be administered for up to 6 cycles in the VISION trial, which is consistent with recommendations in the Canadian product monograph.5,7 The VISION trial protocol also included an additional step, in which the patient was to be evaluated after 4 cycles by the investigator for evidence of treatment response (specified as radiological response, PSA response, or clinical benefit in the opinion of the investigator), signs of residual disease on CT with contrast and/ or MRI or bone scan, and good tolerance of the treatment. Patients meeting all those criteria could receive up to 2 additional cycles at the discretion of the treating physician.5,7 The clinical experts consulted by CADTH noted that evaluation of response to treatment in the target patient population (i.e., those with progressive mCRPC) is multifactorial and would be based on clinical response, radiographic imaging, biochemical measures, and need for medications to manage pain. It was noted that a formal assessment of response after 4 cycles (as performed in the VISION trial) is unlikely to be standardized in Canadian clinical practice and could be a challenge to implement if it was included as renewal criteria for 177Lu vipivotide tetraxetan. Overall, the clinical experts consulted by CADTH noted that the distribution of doses in the VISION is likely an accurate reflection of what would occur with patients in Canada, as the treatment is generally well tolerated and relatively few AEs lead to dose reductions, interruptions, or discontinuations.
Indirect Comparisons
Description of Studies
The sponsor-submitted indirect treatment comparison (ITC) included a systematic review that used a Bayesian network meta-analysis (NMA) to evaluate the efficacy of 177Lu vipivotide tetraxetan relative to other comparators(including radium-223 plus BSC, cabazitaxel plus prednisone, olaparib, mitoxantrone or placebo plus prednisone, and ARPI) for the treatment of patients with pretreated, progressive mCRPC. The NMA was based on a systematic review of the literature, and data from 8 studies were used to inform the analyses. The efficacy outcomes of interest were rPFS and OS.
Efficacy Results
The sponsor-submitted ITC reported that the results for OS favoured 177Lu vipivotide tetraxetan over radium-223 plus BSC (HR = █████ ███ ████ ████ ██ ████) and over ARPI (███ █████ ███ ████ ████ ██ ████). The sponsor-submitted ITC reported that the results for rPFS favoured 177Lu vipivotide tetraxetan over cabazitaxel plus prednisone (███ █████ ███ ████ ████ ██ ████), mitoxantrone or placebo plus prednisone (███ █████ ███ ████ ████ ██ ████) and over ARPI (███ █████ ███ ████ ████ ██ ████). HRs for OS and rPFS were reported as comparator versus 177Lu vipivotide tetraxetan.
Critical Appraisal
The clinical heterogeneity in the analysis was related to variation in patient characteristics across the included trials. In the absence of statistical adjustment, sensitivity analyses, or subgroup analyses, the potential impact of the between-study heterogeneity cannot be evaluated. The clinical experts consulted by CADTH noted that there was heterogeneity in clinically important patient characteristics (i.e., prior receipt of chemotherapy, disease severity, and treatment indication); therefore, the ITC analysis may be subject to bias. Of particular concern was the fact that patients included in the 177Lu vipivotide tetraxetan trial (i.e., VISION) had more severe disease at baseline, as indicated by a higher prior treatment count, and the fact that at least 40% of patients had received cabazitaxel before enrolment. Inconsistency in the network was not reported, likely because of the limited ability to do so, given that the network only had 1 closed loop.
Summary
The sponsor-submitted ITC had several limitations, including the lack of reporting of certain items that would better inform the certainty of the indirect evidence. Despite the heterogeneity of many patient and study characteristics, the ITC authors did not adequately conduct sensitivity or subgroup analyses to investigate the root of heterogeneity or conduct a meta-regression to adjust for effect modifiers that could influence the results. Consequentially, there is substantial uncertainty around the ITC results, and firm conclusions cannot be drawn about the efficacy of 177Lu vipivotide tetraxetan relative to relevant comparators.
Other Relevant Evidence
Inclusion criteria in VISION trial specified that patients who were previously treated with docetaxel and considered eligible to receive cabazitaxel were to be excluded from the study. As this population is included in the Health Canada–approved indication, CADTH considered this to be an important gap in the evidence and, therefore, summarized the phase II TheraP trial, which enrolled patients with prior exposure to docetaxel for whom cabazitaxel was considered the appropriate treatment option.
Description of Study
TheraP was a multicentre, open-label, phase II RCT comparing the activity and safety of 177Lu vipivotide tetraxetan with cabazitaxel in patients with mCRPC. The study was conducted by the Australian and New Zealand Urogenital and Prostate Cancer Trials Group. As in the VISION trial, the study enrolled patients with PSMA-positive mCRPC, but the TheraP trial used a more rigorous 2-stage screening process to determine PSMA status.
Gallium-68 (68Ga)-PSMA PET-CT: patients were eligible if they demonstrated a maximum standardized uptake value (SUVmax) of at least 20 at a site of disease, and an SUVmax of more than 10 at sites of measurable disease of at least10 mm.
18F-fluorodeoxyglucose (FDG) PET-CT: patients were ineligible if they demonstrated FDG-positive lesions with minimal PSMA expression, defined as an FDG intensity greater than 68Ga-PSMA activity or a 68Ga-PSMA SUVmax of less than 10 (i.e., discordant imaging).
Eligible patients were randomized in a 1:1 ratio to receive either 177Lu vipivotide tetraxetan or cabazitaxel. Randomization was stratified by disease burden (> 20 sites versus ≤ 20 sites, assessed by PSMA PET-CT), previous treatment with enzalutamide or abiraterone, and study site.
Patients randomized to the 177Lu vipivotide tetraxetan group received IV infusions once every 6 weeks for a maximum of 6 cycles. The starting dose was 8.5 GBq, which decreased by 0.5 GBq each subsequent cycle (i.e., not the dosage recommended in the Canadian product monograph, which is 7.4 GBq). Patients in the cabazitaxel group received IV infusions of 20 mg/m2 once every 3 weeks for a maximum of 10 cycles. Patients enrolled in the TheraP trial continued to receive supportive cancer therapies (e.g., zoledronic acid, denosumab, or palliative radiotherapy). An important difference between the TheraP trial and the VISION trial is that patients were prohibited from using other systemic anticancer therapies in the TheraP trial (i.e., the study investigated use as monotherapy, which is more reflective of how 177Lu vipivotide tetraxetan would be administered in Canadian clinical practice). Patients could receive any treatment after completion or discontinuation of the study drugs at the discretion of the treating clinician(s).
A total of 291 patients were screened for eligibility and 200 patients were randomized. As in the VISION trial, a greater proportion of patients in the comparator group (in this case, cabazitaxel) withdrew before receiving any doses of the study medications (16 of101 patients [15.8%] in cabazitaxel group versus 1 of 99 patients [1.0%] in the 177Lu vipivotide tetraxetan group).
Efficacy Results
After 3 years of follow-up, there was no statistically significant difference in OS between 177Lu vipivotide tetraxetan and cabazitaxel (HR = 0.97; 95% CI, 0.70 to 1.4; P = 0.99). 177Lu vipivotide tetraxetan was statistically superior to cabazitaxel for the primary end point of PSA response (i.e., reduction of ≥ 50% from baseline) (risk difference = 29%; 95% CI, 16% to 42%); PFS (HR = 0.63; 95% CI, 0.46 to 0.86); rPFS (HR = 0.64; 95% CI, 0.46 to 0.88); ORR (relative risk = 2.12; 95% CI, 1.10 to 4.08); PSA PFS (HR = 0.60; 95% CI, 0.44 to 0.83); and pain (measured on the present pain intensity [PPI] index) PFS (HR = 0.72; 95% CI, 0.53 to 0.97).
Harms Results
Grade 1 or 2 AEs were more commonly reported in the 177Lu vipivotide tetraxetan group than in the cabazitaxel group (54% versus 40%), and grade 3 or 4 AEs were more commonly reported in the cabazitaxel group than in the 177Lu vipivotide tetraxetan group (53% versus 33%).
Critical Appraisal
Internal Validity
Randomization was stratified by a different set of baseline parameters than in the VISION trial (i.e., disease burden based on metastatic sites [> 20 sites versus ≤ 20], whether or not the patient had received previous treatment with enzalutamide or abiraterone, and study site). Overall, baseline and demographic characteristics were well balanced in the 177Lu vipivotide tetraxetan and cabazitaxel groups in the TheraP trial. As in the VISION trial, the study drugs in the TheraP trial were administered in an open-label manner (for the reasons outlined for the VISION trial). Radiographic images in the TheraP trial were evaluated centrally, but not in a manner that was blinded to the evaluator.
As in the VISION trial, the internal validity of the TheraP trial was limited by the high and disproportionate dropout rate in the comparator group before any doses of the study medications were administered (15.8% in cabazitaxel group versus 1.0% in the 177Lu vipivotide tetraxetan group). The rationale provided was similar to that for the VISION trial (i.e., patient disappointment at not having access to 177Lu vipivotide tetraxetan). As in the VISION trial, the high and disproportionate number of patients who withdrew from the control group could bias the study results in favour 177Lu vipivotide tetraxetan, as those who remained in the study may have had poorer prognoses in than those who withdrew (although the direction and magnitude of any potential bias is uncertain).
The phase II TheraP study was not designed or powered to evaluate differences between 177Lu vipivotide tetraxetan and cabazitaxel for the primary end points that are recommended by the PCWG3 (e.g., OS). The investigators reported an OS analysis after 3 years of follow-up and noted no statistically significant differences between the 2 treatment groups; however, this analysis may be confounded by crossover and other potential differences in the subsequent therapy setting.
External Validity
Unlike in the VISION trial, 177Lu vipivotide tetraxetan was administered as monotherapy in the TheraP trial (no other systemic anticancer drugs were permitted as part of the study protocol in the TheraP trial). This is likely more generalizable to the Canadian setting, as the clinical experts consulted by CADTH noted that 177Lu vipivotide tetraxetan is likely to be used as monotherapy, there is a lack of evidence with which to evaluate the potential benefits of combination regimens; there is a potential for increased drug-related AEs; and there is a likelihood that reimbursement status would be limited to monotherapy.
The comparator in the TheraP trial (cabazitaxel) is highly relevant in the Canadian context for patients who have previously been treated with docetaxel and an ARPI. Unlike in the VISION trial, the TheraP study did not include the eligibility criterion that patients must be considered medically unsuitable to receive further treatment with a taxane regimen. The maximum number of cycles used in the TheraP trial (i.e., 6 cycles) was consistent with the VISION trial and the Canadian product monograph; however, the dosage strength was not consistent with recommendations in the product monograph. Patients in the TheraP trial received an initial dose of 8.5 GBq, which was decreased by 0.5 GBq each subsequent cycle; this is not reflective of the standardized dose of 7.4 GBq recommended in the product monograph.
PSMA status in the TheraP trial was determined using a 2-stage screening process, in which patients were initially screened using 68Ga-PSMA PET-CT and then subsequently evaluated using FDG PET-CT. Patients with discordant imaging between 68GA-PSMA PET-CT and FDG PET-CT (e.g., FDG intensity levels were greater than those observed with the 68Ga-PSMA PET-CT) were excluded from the trial. The clinical experts consulted by CADTH noted that the more rigorous criteria applied in the TheraP trial could help identify patients who may be most likely to response to 177Lu vipivotide tetraxetan; however, the need for 2 diagnostic PET-CT scans to determine PSMA status would likely pose implementation challenges in clinical practice for clinicians and the health system in Canada.
Conclusions
177Lu vipivotide tetraxetan injection is indicated for the treatment of adults with PSMA-positive mCRPC who have received at least 1 ARPI and taxane-based chemotherapy. Effective treatments for patients with mCRPC who have progressed after treatment with an ARPI and docetaxel are currently limited, and all stakeholders identified important unmet medical needs, particularly for patients who are be ineligible to receive additional therapy with taxane regimens.
The CADTH review included 1 phase III RCT (VISION; N = 831) that compared the use of 177Lu vipivotide tetraxetan as an add-on therapy to BSC/BSoC with BSC/BSoC alone. The VISION trial suggested that 177Lu vipivotide tetraxetan was superior to BSC/BSoC alone on a series of outcomes that are considered important in the evaluation of prostate cancer therapies (i.e., OS, rPFS, time to first SSE, HRQoL, and PFS). However, there is uncertainty regarding the internal validity of the results because of several important limitations, most notably the considerable early withdrawal of consent and the disproportionate dropout rate in the BSC/BSoC alone group. The extent of the early withdrawal was substantial enough to require amendments to the VISION protocol, which included the use of a new analysis set that was limited to patients enrolled after the amendments. This new analysis set was used for the primary evaluation of all end points, with the exception of OS (as the sponsor was able to obtain mortality data for those who withdrew from the study) and ORR (which was based on a smaller subset of patients). Regulatory authorities and the clinical experts consulted by CADTH considered the results for OS to be clinically important, given that the patients enrolled in the VISION trial were heavily pretreated. The choice of BSC/BSoC as the comparator in the VISION trial and the limitations of the sponsor’s ITC preclude the drawing of any conclusions regarding the efficacy of 177Lu vipivotide tetraxetan relative to other relevant comparators for OS.
The inclusion criteria in the VISION trial specified that patients who were previously treated with docetaxel and considered eligible to receive cabazitaxel were to be excluded from the study (despite this, approximately 18% of patients received cabazitaxel in the poststudy treatment setting). To address this important gap in the evidence, CADTH summarized results from the TheraP trial (N = 200), which enrolled patients with prior exposure to docetaxel for whom cabazitaxel was considered the appropriate treatment option. TheraP was a phase II study that was not designed or powered to evaluate potential differences in OS, and there was no statistically significant difference between 177Lu vipivotide tetraxetan and cabazitaxel for OS. Treatment with 177Lu vipivotide tetraxetan was statistically superior to cabazitaxel for the primary end point of PSA response, PFS, rPFS, ORR, PSA PFS, and pain PFS.
177Lu vipivotide tetraxetan is the first drug specifically indicated for use in patients with PSMA-positive prostate cancer. The evaluation of PSMA status requires PET-CT imaging with a PSMA-targeted radioligand; at the time of CADTH’s review, this process was not routinely performed in Canadian clinical practice. The potential health system implications and imaging resource requirements may pose implementation challenges that could affect the integration and uptake of 177Lu vipivotide tetraxetan in Canadian practice.
Introduction
Disease Background
Prostate cancer is the most common cancer among Canadian men (excluding nonmelanoma skin cancers), affecting 1 in 9 men during their lifetime.1 Prostate cancer represents approximately 20% of all new cancers diagnosed in men in Canada and 10% of cancer deaths in men.2 In 2022, it was estimated that 24,600 men in Canada would be diagnosed with prostate cancer and that 4,600 men would die from prostate cancer, according to the CCS.2 Patients who die from prostate cancer typically progress to the mCRPC stage, and the 5-year survival rate is approximately 30%.3 The Canadian Urological Association (CUA) defines CRPC as disease progression despite castrate levels of testosterone that can present as a continuous rise in serum PSA levels, the progression of preexisting disease, and/or the appearance of new metastases.4
PSMA is a transmembrane glycoprotein that is highly expressed in prostate cancer cells. The CUA notes that the expression of PSMA has been observed to increase with the emergence of androgen independence in prostate cancers.4
Figure 1: Progression of Prostate Cancer
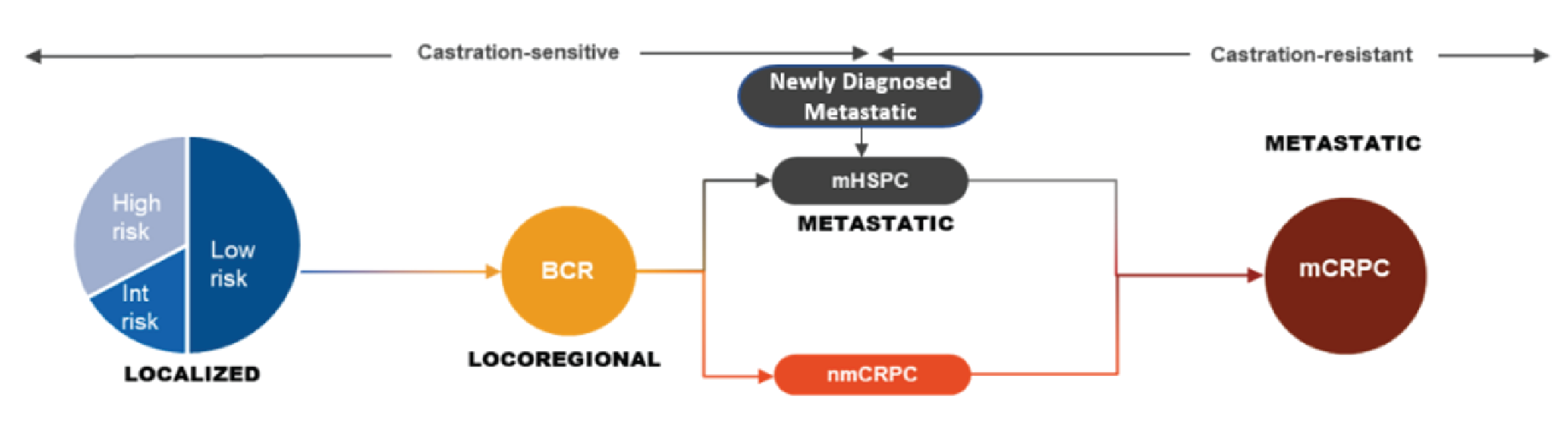
BCR = biochemical relapse; Int = intermediate; mCRPC = metastatic castration-resistant prostate cancer; mHSPC = metastatic hormone-sensitive prostate cancer; nmCRPC = nonmetastatic castration-resistant prostate cancer.
Source: Sponsor’s clinical summary.6
Standards of Therapy
Systemic Cancer Therapies
Figure 2 provides a summary of the CUA guidelines for the management of CRPC.12 The CUA states that the optimal sequence of the available therapeutic options remains uncertain, but that changing the therapeutic mechanism of action with each line of therapy may lead to better and longer-lasting responses in patients. Cytotoxic chemotherapy with docetaxel has been the standard of care for patients with mCRPC who progress on first- or second-line androgen-deprivation therapy. The clinical experts consulted by CADTH noted that patients with mCRPC who experience disease progression after treatment with docetaxel have a poor prognosis and that there are limited effective treatment options available for these patients. For patients who are considered suitable for further chemotherapy, cabazitaxel is currently considered the standard of care and is recommended by the CUA, based on a phase III study that demonstrated that cabazitaxel had a statistically significant survival advantage over mitoxantrone for patients previously treated with docetaxel.4
Figure 2: CUA Guidelines for the Management of CRPC (Redacted)

CRPC = castration-resistant prostate cancer; CUA = Canadian Urological Association; mCRPC = metastatic castration-resistant prostate cancer; PSADT = prostate-specific antigen doubling time.
Figure redacted to respect copyright terms.
Source: Saad et al. (2022).12
CUA guidelines were updated in November 2022 to address the use of 177Lu vipivotide tetraxetan.12 The CUA notes that 177Lu vipivotide tetraxetan for up to 6 cycles is recommended for patients with mCRPC and PSMA-expressing metastatic lesions who have progressed on at least 1 previous taxane chemotherapy and an ARPI (Level 1, strong recommendation). In support of the recommendation, the CUA cites the results of the TheraP trial (improvement in PSA response and fewer grade 3 or 4 AEs than cabazitaxel) and the VISION trial (improvements in all primary and secondary outcomes).
The CUA guidelines state that the following Health Canada–approved drugs have shown improvements in survival for patients with mCRPC: abiraterone acetate plus prednisone, enzalutamide, docetaxel plus prednisone or prednisolone, cabazitaxel plus prednisone or prednisolone, radium-223, olaparib, and 177Lu vipivotide tetraxetan.5,13-18 Among these treatment options, the following were considered relevant comparators for the CADTH review of 177Lu vipivotide tetraxetan:
Cabazitaxel in combination with prednisone or prednisolone, which is indicated for the treatment of patients with mCRPC previously treated with a docetaxel-containing regimen.16
Olaparib, which is indicated as monotherapy for the treatment of adults with deleterious or suspected deleterious germline and/or somatic BRCA- or ATM-mutated mCRPC who have progressed after treatment with a new hormonal drug.14 Olaparib was previously reviewed by CADTH for use in the indicated population and received a recommendation in favour of reimbursement from the CADTH pan-Canadian Oncology Drug Review Expert Review Committee.19
Radium-223, which is indicated for the treatment of patients with mCRPC, symptomatic bone metastases, and no known visceral metastatic disease.13 The clinical experts consulted by CADTH estimated that approximately 60% of patients with mCRPC who have progressed after treatment with a taxane would have bone-only metastases, and noted that the current reimbursement status of radium-223 varies across Canada.
Another ARPI (e.g., enzalutamide if the patient was previously treated with abiraterone plus prednisone) can be administered for some patients but is considered to have limited activity.4
The European Society for Medical Oncology and the European Association of Urology prostate cancer guidelines were also updated to address 177Lu vipivotide tetraxetan. The European Association of Urology guidelines state that 177Lu vipivotide tetraxetan be offered to pretreated patients with mCRPC with 1 or more metastatic lesions and high-level expression of PSMA (exceeding the uptake in the liver) on the diagnostic radiolabelled PSMA PET-CT scan.20 The European Society for Medical Oncology guidelines state that 177Lu vipivotide tetraxetan should be considered (when available) for patients who have progressed on an APRI and a taxane regimen.21 The National Comprehensive Cancer Network treatment guidelines for mCRPC state that 177Lu vipivotide tetraxetan can be a useful treatment option for patients with PSMA-positive prostate cancer who were previously treated with docetaxel and an ARPI.22
Patients with mCRPC can receive supportive therapy with systemic corticosteroids (e.g., low-dose prednisone or dexamethasone), which can lead to improvements in palliative outcomes; palliative radiation for bone metastases; and treatment with bisphosphonates or denosumab to reduce the risk of skeletal-related events (e.g., pathological fractures, spinal cord compression, or the need for surgery or radiation therapy to bone).4
PSMA Testing in Canada
Figure 3 provides a summary of the CUA recommendations for the application of PSMA testing in Canadian practice. These recommendations predated the Canadian regulatory approval of PSMA-targeted pharmacotherapy and do not specifically address the use of PSMA testing to identify candidates for treatment with 177Lu vipivotide tetraxetan. The CUA statement recommends that PSMA-targeted PET should not be routinely offered outside a clinical trial for patients with mCRPC (i.e., the target population of 177Lu vipivotide tetraxetan).
Figure 3: CUA Recommendations for PSMA Testing (Redacted)

CRPC = castration-resistant prostate cancer; CUA = Canadian Urological Association; mCRPC = metastatic castration-resistant prostate cancer; PSADT = prostate-specific antigen doubling time; PSMA = prostate-specific membrane antigen.
Figure redacted to respect copyright terms.
Source: Shaygan et al. (2021).27
There is currently 1 68Ga-labelled PSMA-targeted radiopharmaceutical approved for use in Canada (68Ga-gozetotide [Illucix]).23 Two others are currently listed as being under review by Health Canada, including 1 manufactured by the sponsor of 177Lu vipivotide tetraxetan (Advanced Accelerator Applications US, Inc.).24 An additional PSMA diagnostic radiopharmaceutical has been approved by the FDA (piflufolastat F 18 [Pylarify]),25 but it has not been approved and is not listed as being under consideration by Health Canada at the time of this review. At least 1 additional PSMA diagnostic radiopharmaceutical is currently under development (i.e., 18F-PSMA-1007).26
Drug Under Review
177Lu vipivotide tetraxetan injection is indicated for the treatment of adults with PSMA-positive mCRPC who have received at least 1 ARPI and taxane-based chemotherapy.5 The sponsor has requested that 177Lu vipivotide tetraxetan be reimbursed in accordance with the Health Canada–approved indication.6 Based on the approved indication for 177Lu vipivotide tetraxetan, there are 3 relevant subpopulations for consideration in this review:
patients previously treated with docetaxel who are considered eligible to receive cabazitaxel
patients previously treated with docetaxel who are considered ineligible to receive cabazitaxel
patients previously treated with both docetaxel and cabazitaxel.
Mechanism of Action
The product monograph states that the active moiety of 177Lu vipivotide tetraxetan is the radionuclide lutetium-177, which is linked to a targeting moiety that binds to PSMA, a transmembrane protein that is highly expressed in prostate cancer. Upon the binding of 177Lu vipivotide tetraxetan to PSMA-expressing cancer cells, the beta-minus emission from lutetium-177 delivers therapeutic radiation to the targeted cell, as well as to surrounding cells, and induces DNA damage, which can lead to cell death.5
Figure 4: Sponsor’s Proposed Place in Therapy for 177Lu Vipivotide Tetraxetan
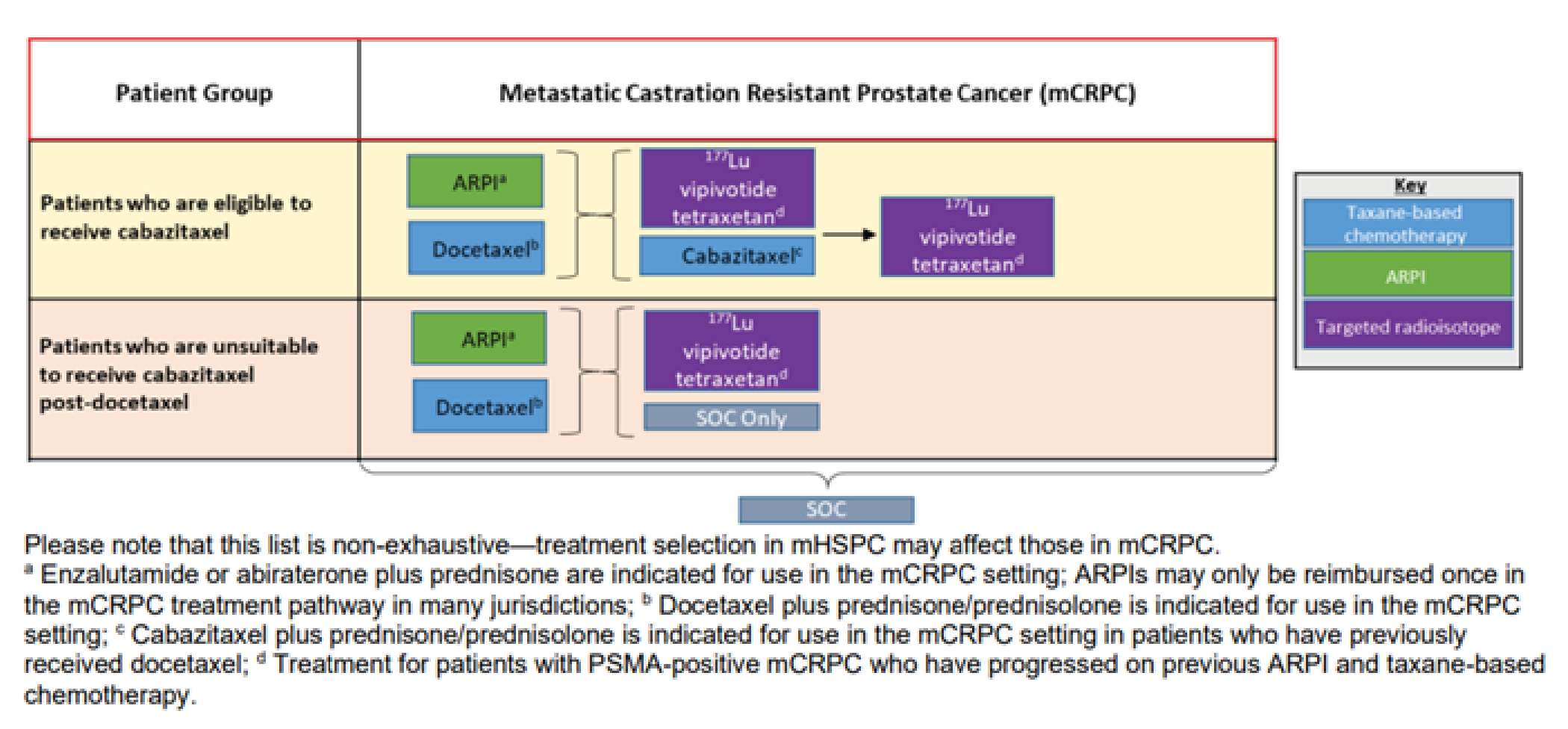
177Lu = lutetium-177; ARPI = androgen receptor pathway inhibitor; mHSPC = metastatic hormone-sensitive prostate cancer; mCRPC = metastatic castration-resistant prostate cancer; PSMA = prostate-specific membrane antigen; SCO = standard of care.
Source: Sponsor’s application.6
Dosage and Administration
177Lu vipivotide tetraxetan is administered intravenously. The recommended dose of 177Lu vipivotide tetraxetan is 7.4 GBq (7,400 MBq or200 mCi) every 6 weeks (± 1 week) for a total of 6 doses.5 Table 3 provides recommended dose modifications for the management of adverse drug reactions. The management of severe or intolerable adverse drug reactions may require temporary dose interruption (extending the dosing interval from every 6 weeks to every 10 weeks), dose reduction, or permanent discontinuation of treatment with 177Lu vipivotide tetraxetan.5 No dose adjustment is recommended for patients with hepatic impairment or for patients with mild (baseline creatinine clearance [CrCl] of 60 mL/min to 89 mL/min, according to the Cockcroft-Gault equation) to moderate (CrCl of 30 mL/min to 59 mL/min) renal impairment. The pharmacokinetic profile and safety of 177Lu vipivotide tetraxetan has not been studied in patients with severe (CrCl of 15 mL/min to 29 mL/min) renal impairment or end-stage renal disease.5
177Lu vipivotide tetraxetan is available as a 1,000 MBq/mL solution for injection in single-dose vials that contain a total amount of radioactivity of 7.4 GBq (7,400 MBq or 200 mCi) (± 10%) at the time of administration.5
Table 3: Recommended Dose Modifications for Adverse Reactions
Adverse drug reaction | Severitya | Dose modification |
|---|---|---|
Dry mouth |
|
|
Gastrointestinal toxicity |
|
|
Anemia, thrombocytopenia, leukopenia, neutropenia, pancytopenia |
|
|
Renal toxicity |
|
|
|
| |
|
| |
Spinal cord compression | Any |
|
Fracture in weight-bearing bones | Any |
|
AST or ALT > 5 times ULN in the absence of liver metastases | — |
|
ALT = alanine aminotransferase; AST = aspartate aminotransferase; CrCl = creatinine clearance; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ULN = upper limit of normal.
aThe same thresholds are also applicable to baseline values at the time of treatment initiation with 177Lu vipivotide tetraxetan.
Source: Product monograph.5
Precautions for Post-treatment Contamination
As with other radiopharmaceuticals, the product monograph states that 177Lu vipivotide tetraxetan should be used only by health professionals who are appropriately qualified in the use of radioactive prescribed substances in or on humans.5 Before the patient is released, the health care provider should explain the necessary radioprotection precautions that the patient should follow to minimize radiation exposure to others. After the administration of 177Lu vipivotide tetraxetan, patients should be advised to limit close contact (less than 1 m) with household contacts for 2 days and with children and pregnant women for 7 days. After the administration of 177Lu vipivotide tetraxetan, patients should be advised to sleep in a bedroom separate from household contacts for 3 days, from children for 7 days, or from pregnant women for 15 days.5
Table 4: Key Characteristics of 177Lu Vipivotide Tetraxetan, Cabazitaxel, Olaparib, and Radium-223
Characteristic | 177Lu vipivotide tetraxetan | Cabazitaxel | Olaparib | Radium-223 |
|---|---|---|---|---|
Mechanism of action | PSMA-targeted radioligand therapy composed of vipivotide tetraxetan, which is the targeting component recognizing PSMA, and the antitumour radionuclide lutetium-177 | Cytotoxic chemotherapeutic | PARP inhibitor | Alpha particle-emitting pharmaceutical with targeted antitumour effect on bone metastases |
Indicationa | The treatment of adults with PSMA-positive mCRPC who have received at least 1 ARPI and taxane-based chemotherapy | In combination with prednisone or prednisolone, is indicated for the treatment of patients with mCRPC previously treated with a docetaxel-containing regimen | As monotherapy for patients with deleterious or suspected deleterious germline and/or somatic BRCA- or ATM-mutated mCRPC who have progressed after treatment with a new hormonal drug | CRPC with symptomatic bone metastases and no known visceral metastatic disease |
Route of administration | IV | IV | Oral | IV |
Recommended dose | 7.4 GBq every 6 weeks (± 1 week) for up to 6 doses | 20 mg/m2 administered every 3 weeks in combination with oral prednisone (or prednisolone) 10 mg, administered daily during cabazitaxel treatment | 600 mg per day (two 150 mg tablets twice daily) | 55 kBq per kg body weight, given at 4-week intervals for a total of 6 injections |
Diagnostic testing or imaging requirements | PSMA-positive imaging with PET-CT | N/A | BRCA or ATM mutations must be confirmed | Bone metastases with no known visceral metastases |
Serious warnings and precautions | Black-box warnings regarding the risk of myelosuppression and renal toxicity | Black-box warnings regarding the risk of neutropenia and gastrointestinal hemorrhage and perforation | Black-box warnings regarding the risk of myelodysplastic syndrome or acute myeloid leukemia and pneumonitis | Black-box warnings regarding the risk of bone marrow suppression |
177Lu = lutetium-177; ARPI = androgen receptor pathway inhibitor; CRPC = castration-resistant prostate cancer; mCRPC = metastatic castration-resistant prostate cancer; N/A = not applicable; PARP = poly (ADP-ribose) polymerase; PSMA = prostate-specific membrane antigen.
aHealth Canada–approved indications.
Sources: Product monographs.5,13,14,16
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient inputs received by CADTH have been included in the stakeholder section at the end of this report.
Two patient groups, the CCS and the CCSN, provided input for the treatment of adults with PSMA-positive mCRPC who have been treated with ARPI and taxane-based chemotherapy or who are not medically suitable for taxanes. Patient input was gathered from surveys of and interview with patients with mCRPC and their caregivers in Canada in August 2022. Of the 27 survey respondents, 19 were from the CCS and 8 were from the CCSN. Of the 7 patients included in the submissions who had experience with the treatment under review, 4 were from the CCS and 3 were from the CCSN. Of these 7 patients, 2 participated in an in-depth interview.
Patients surveyed by the CCS reported that their disease had a negative effect on their QoL and their ability to perform certain routine functions, such as sexual activity, travel and exercise, family obligations, maintaining mental health, work, household chores, concentrating, spending time with family and friends, and practical needs (e.g., preparing meals, dressing, bathing). Patients surveyed by the CCSN reported frequent urination, erectile dysfunction, bone and/or skeletal pain, loss of QoL, hot flashes, weight gain, and slight memory loss related to current day-to-day experiences as a result of their disease. These patients also reported AEs related to their current treatments, such as incontinence, fatigue, diarrhea, weight gain, erectile dysfunction, urinary issues, infection, hot flashes, loss of muscle, hair loss, breast enlargement, and loss of libido. Patients surveyed by the CCS reported that changes in libido and sexual function had the most significant impact on their day-to-day lives.
Respondents to the CCSN survey reported no issues accessing therapies, but 11 of 19 respondents to the CCS survey reported experiencing 1 or more barriers to treatment. The most common barrier reported was the cost of transportation to appointments, followed by costs associated with complementary medicines recommended by their health care team (e.g., vitamins and supplements.), loss of income due to absence from work, costs associated with medical tests and procedures, and lack of familiarity with the health care system.
Patients who responded to the CCSN survey reported some outcomes they expect from a new treatment, including maintaining QoL, delaying the onset of symptoms, prolonging life, providing a cure, reducing side effects from current medications or treatments, and the return of libido, along with ease of use and easy access. When describing experiences with 177Lu vipivotide tetraxetan, patients reported some side effects, including dry mouth, weakness, fatigue, low blood platelet counts, low red blood cell counts and/or anemia, nausea and/or vomiting, loss of appetite, constipation, diarrhea, abdominal pain, shortness of breath, and brain fog. All 4 patients using the treatment under review who responded to the CCS survey strongly agreed that they would recommend it to others with mCRPC, and 3 patients stated that they would choose to continue the treatment despite side effects.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). A panel of 3 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations for which there are gaps in the evidence that could be addressed with the collection of additional data, promote the early identification of potential implementation challenges, gain insight into the clinical management of patients living with mCRPC, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion is presented here.
Unmet Needs
The clinical experts consulted by CADTH noted that there are limited effective treatments for patients with mCRPC who have progressed after treatment with docetaxel. OS is poor for those with disease that is refractory to multiple treatment options, and the symptoms of cancer progression pose a considerable burden for patients. Standard of care treatments, such as cytotoxic chemotherapy with cabazitaxel, are associated with significant toxicities (e.g., neuropathy and febrile neutropenia). The experts noted that many patients are often unsuitable to receive treatment with cabazitaxel because of comorbidities and/or a poor performance status. As with most advanced malignancies, there are limited treatments that can meaningfully reverse the course mCRPC for a prolonged amount of time. As in the input from the patient groups, the clinical experts noted that there is a need for therapies that improve OS and QoL and that are better tolerated and more convenient (e.g., less need for supportive medications, less frequent administration) than the current standard of care for this patient population.
Place in Therapy
The clinical experts consulted by CADTH noted that 177Lu vipivotide tetraxetan could be considered a treatment option for patients whose disease has progressed after both an androgen receptor axis-targeted therapy and docetaxel. The experts noted that there is uncertainty regarding the place of 177Lu vipivotide tetraxetan in therapy relative to cabazitaxel for patients who are considered appropriate candidates for a second chemotherapy regimen. They noted that the eligibility criteria for the phase III VISION trial specifically excluded such patients (although 14.9% and 18.9% of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups, respectively, received cabazitaxel in the subsequent therapy setting), but that the phase II TheraP trial provided some evidence of comparative efficacy for 177Lu vipivotide tetraxetan and cabazitaxel.
Patient Population
This treatment would be most suited for patients with the following characteristics:
Progressive mCRPC: symptomatic, imaging, or biochemical.
PSMA: Evidence of PSMA-expressing metastases based on a diagnostic PSMA-targeted PET scan. The criteria used in the VISION trial were considered acceptable for the identification of patients based on input from the clinical experts consulted by CADTH and input from stakeholders. It was noted that the criteria used in the phase II TheraP trial were more restrictive and could be used as alternative criteria; however, the application of those criteria would pose additional challenges for clinicians.
Adequate performance status: ECOG PS of 0 to 2.
Adequate organ function: liver and bone marrow.
Assessing Response to Treatment
The clinical experts noted that imaging for patients with mCRPC would typically be performed once every 12 weeks in Canadian clinical practice or earlier in response to changes in symptoms and/or clinical examination. The clinical experts consulted by CADTH noted that evaluating response to treatment in the target patient population (i.e., those with progressive mCRPC) is multifactorial and would be based on clinical response, radiographic imaging, biochemical measures, and analgesic requirements. It was noted that a formal assessment of response after 4 cycles (as conducted in the VISION trial) is unlikely to be standardized in Canadian clinical practice and could be a challenge to implement if included as renewal criteria for 177Lu vipivotide tetraxetan.
Discontinuing Treatment
The clinical experts consulted by CADTH noted that 177Lu vipivotide tetraxetan should be discontinued in patients who experience disease progression characterized by at least 2 specific adverse occurrences (i.e., sustained PSA rise, clinical progression [sustained, analgesic, nonresponsive pain; performance status decline], and radiographic progression); significant toxicity related to the treatment; or worsening of performance status (i.e., ECOG PS ≥ 3).
Prescribing Conditions
The product monograph states that 177Lu vipivotide tetraxetan should be administered under the supervision of a health professional who is experienced in the use of radiopharmaceuticals. Appropriate management of therapy and complications is only possible when adequate diagnostic and treatment facilities are readily available. The clinical experts consulted by CADTH noted that these requirements, although necessary, would likely pose challenges to patient access, based on the number and/or locations of currently available facilities (e.g., regional variation, existing capacity constraints). Overall, there was consensus that 177Lu vipivotide tetraxetan is ideally delivered in the setting of multidisciplinary care, with collaboration between specialists experienced in treating prostate cancer (i.e., those specializing in urologic oncology, medical oncology, nuclear medicine, radiation oncology, and diagnostic radiology).
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input received by CADTH have been included in the stakeholder section at the end of this report.
Clinician group input was received from prostate-treating clinicians in Canada with a special interest in the care of those with metastatic prostate cancer. The CCS coordinated the submission.
The clinician group agreed that there have been some unmet needs for additional lines of therapy and a scarcity of treatments that induce disease modification to preserve QoL and provide meaningful survival benefits for patients with progressive metastatic prostate cancer. The clinician group indicated that radioligand therapies that target PSMA confer both disease-modifying and symptom-management benefits on this population, as suggested by the phase III VISION trial and the phase II TheraP trial. Because 177Lu vipivotide tetraxetan is a PSMA-targeted radioligand therapeutic, the clinicians expect similar benefits in patients who have PSMA-expressing prostate cancer identified on diagnostic PSMA-targeted PET. The clinicians pointed out that the drug under review can be considered well tolerated, based on evidence from the randomized trials, and a good additional line of therapy compared to third-line chemotherapy or supportive care only.
The clinician group noted that the treatment would be best suited to patients with progressive (symptomatic, imaging, or biochemical) mCRPC, PSMA-expressing metastases identified on a diagnostic PSMA-targeted PET scan, and with an adequate performance status (ECOG PS of 0 to 2) and organ function (liver and bone marrow). The clinician group also pointed out that the most meaningful clinical response to treatment for this disease would be the prevention of progression, reflected in stability or improvement in biochemical and imaging biomarkers such as serum PSA, bone scan, and CT. The clinician group emphasized that appropriate facilities, certifications, and personnel licensed to deliver unsealed radiopharmaceutical treatments would be needed for the safe delivery of 177Lu vipivotide tetraxetan, in addition to access to diagnostic PSMA-targeted PET for proper patient selection.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that could affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation issues | Clinical expert response |
|---|---|
Relevant comparators | |
BSC/BSoC in the VISION trial included abiraterone or enzalutamide, bone-directed therapies (e.g., denosumab, zoledronic acid), corticosteroids, and/or radiation. Cytotoxic chemotherapy, other radioisotopes (e.g., radium-223), immunotherapy, and investigational drugs (e.g., olaparib) were not permitted as comparators. Many of the therapies excluded in the VISION trial are relevant comparators to 177Lu vipivotide tetraxetan in practice. Funded relevant comparators depend upon drugs used in prior lines of therapy; comparators include taxane-based chemotherapy, alternate chemotherapy (e.g., carboplatin, mitoxantrone), and abiraterone or enzalutamide. For patients with bone-only metastases, radium-223 is a relevant comparator as well. Olaparib may be a relevant comparator in patients with a confirmed BRCA or ATM mutation. | The clinical experts consulted by CADTH agreed that the VISION trial excluded relevant comparators. However, the clinical experts noted that olaparib was an investigational drug for mCRPC when the VISION trial was initiated (i.e., first patient was enrolled in May 2018 and olaparib did not receive regulatory approval in any jurisdiction until May 2020),7,28 and this drug is indicated for only a small subset of patients with mCRPC (i.e., those with documented deleterious or suspected deleterious germline and/or somatic BRCA or ATM mutations). Therefore, the exclusion of this drug from the BSoC regimen is understandable and not considered to be a major limitation with respect to the generalizability of the study results. The clinical experts consulted by CADTH noted that radium-223 is indicated only for patients with bone metastases and is not available in all Canadian jurisdictions. |
Considerations for initiation of therapy | |
Eligible patients have had previous treatment with ARPIs and taxanes and must have CRPC to be eligible for 177Lu vipivotide tetraxetan. Are patients eligible for 177Lu vipivotide tetraxetan only if prior ARPIs and/or taxanes were given for mCRPC? Are patients who only received ARPIs and/or taxanes for castrate-sensitive disease eligible for 177Lu vipivotide tetraxetan? | In the absence of high-quality data regarding treatment sequencing, the clinical experts commented that patients that who received either ARPIs or taxanes in the castrate-sensitive prostate cancer disease state setting would be eligible for 177Lu vipivotide tetraxetan in the mCRPC setting. |
Patients required 68Ga-labelled PSMA-11 PET-CT scans to confirm PSMA-positive disease eligibility for 177Lu vipivotide tetraxetan. This requires access to and/or funding for 68Ga and 68Ga-labelled PET-CT, which is not currently available in all jurisdictions. | The clinical experts consulted by CADTH noted that PSMA testing with PET-CT is not widely available in routine practice in Canada and is typically only performed as part of clinical studies, accessed through private mechanisms, or in very rare cases in which there is the potential for another malignant diagnosis and the clinical team requires clarity on the histology of the disease. The experts noted that patients may encounter financial and logistical challenges (e.g., interprovincial travel to access PSMA testing). PSMA PET is considered to be a prerequisite diagnostic test to determine eligibility for 177Lu vipivotide tetraxetan. |
The VISION trial included patients with PSMA-positive mCRPC, defined as at least 1 PSMA-positive metastatic lesion and no PSMA-negative lesions that would be excluded because of protocol criteria. The VISION trial defined PSMA-positive lesions as those having a 68Ga uptake greater than that of liver parenchyma in 1 or more metastatic lesions of any size. PSMA-negative lesions were defined as those having a PSMA uptake equal to or lower than that of liver parenchyma in any lymph node with a short axis of at least 2.5 cm, in any metastatic solid-organ lesion with a short axis of at least 1.0 cm, or in any metastatic bone lesion with a soft-tissue component of at least 1.0 cm in the short axis. Patients with any PSMA-negative lesions were ineligible. In clinical practice, are the eligibility criteria and definitions of PSMA-positive and PSMA-negative lesions used in the VISION trial appropriate for identifying the eligible population? | The clinical experts consulted by CADTH noted that the criteria used in the VISION trial are acceptable for the identification of patients. It was noted that the criteria used in the phase II TheraP trial were more restrictive and could be used as alternative criteria; however, the sequential 68Ga-PSMA PET-CT scan followed by a FDG PET-CT scan to determine PSMA status would pose additional implementation challenges for clinicians and the health system (i.e., resource constraints currently limit existing access to PET-CT scans for prostate cancer; the need for 2 diagnostic scans to determine PSMA status poses a challenge). |
The VISION trial stated that patients who had been treated with just 1 taxane regimen could only be eligible if the physician deemed them unsuitable to receive a second taxane regimen. What is the definition of “not medically suitable for taxanes”? | The clinical experts consulted by CADTH noted that patients with the following characteristics would not be medically suited for taxane-based therapy:
|
Considerations for continuation or renewal of therapy | |
The VISION trial included imaging at baseline, then every 8 weeks for 24 weeks, then every 12 weeks until the end of treatment. Radiologic evaluations included CT or MRI and bone scans. Are imaging assessments in the VISION trial appropriate in clinical practice? | The clinical experts noted that the intensity of imaging used in the VISION trial is common in clinical trials for mCRPC but not in routine clinical practice. It is anticipated that imaging for patients with mCRPC would be performed once every 12 weeks in practice, or earlier in response to changes in symptoms and/or clinical examination. |
Is there a role for repeat 68Ga-labelled PET-CT to assess treatment response? | The clinical experts noted that the utility of evaluating response to treatment based on repeated 68Ga-labelled PSMA PET-CT assessments was not part of the phase III VISION trial, and that this approach has not been investigated in a prospective, adequately powered fashion. It was noted that the phase II TheraP trial included repeat PSMA PET-CT to establish 177Lu retention in target and off-target tissues, with suspension of therapy for patients who demonstration low or no PSMA uptake at sites of metastatic disease; however, no efficacy outcomes were reported based on these subgroups of patients. |
Considerations for discontinuation of therapy | |
The VISION trial required patients to have castrate testosterone levels throughout therapy. Is a castrate level of testosterone required for continuation of therapy in clinical practice? | The clinical experts consulted by CADTH noted that it is well established in clinical practice to require patients to have castrate levels of testosterone for continuation of systemic therapy. |
Should 177Lu vipivotide tetraxetan be discontinued if testosterone levels are no longer castrate level during therapy? | The clinical experts consulted by CADTH noted that treatment with 177Lu vipivotide tetraxetan should be discontinued if testosterone levels are no longer castrate level after initiation of therapy. It was suggested that testosterone levels should be decreased to castrate levels before resumption of therapy. |
Considerations for prescribing of therapy | |
177Lu vipivotide tetraxetan is administered by IV infusion at a dose of 7.4 GBq once every 6 weeks for 4 cycles. Up to 2 additional cycles can be administered at the discretion of the treating physician in patients with evidence of disease response. In clinical practice, in which scenarios would 2 additional cycles be indicated? | The clinical experts noted that the median number of cycles in the VISION trial was 5 (range, 1 to 6) and that 46.5% of patients received 6 cycles. The clinical experts consulted by CADTH noted that evaluating response to treatment for the target patient population (i.e., those with progressive mCRPC) is multifactorial and would be based on clinical response, radiographic imaging, biochemical measures, and the need for medications to manage pain. It was noted that a formal assessment of response after 4 cycles (as performed in the VISION trial) is unlikely to be standardized in Canadian clinical practice and could be a challenge to implement if included as renewal criteria for 177Lu vipivotide tetraxetan. |
Should 177Lu vipivotide tetraxetan be added to an existing systemic treatment for patients who meet trial criteria? | The clinical experts consulted by CADTH noted that combination use in Canada may be limited by reimbursement status. Public reimbursement for ARPIs after a patient has demonstrated disease progression on the therapy varies across jurisdictions, with some provinces mandating discontinuation of coverage and others permitting continuation of therapy. Overall, the experts noted that it is uncertain if 177Lu vipivotide tetraxetan used in combination with other systemic anticancer therapies offers additional clinical benefit for patients. |
177Lu = lutetium-177; 68Ga = gallium-68; ALT = alanine aminotransferase; ARPI = androgen receptor pathway inhibitor; AST = aspartate aminotransferase; BSC = best supportive care; BSoC = best standard of care; CRPC = castration-resistant prostate cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FDG = 18F-fluorodeoxyglucose; mCRPC = metastatic castration-resistant prostate cancer; PSMA = prostate-specific membrane antigen.
Clinical Evidence
The clinical evidence included in the review of 177Lu vipivotide tetraxetan is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of 177Lu vipivotide tetraxetan for the treatment of adults with PSMA-positive mCRPC who have received at least 1 ARPI and taxane-based chemotherapy.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.29
Table 6: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults with PSMA-positive mCRPC who have received at least 1 ARPI and taxane-based chemotherapy Subgroups:
|
Intervention | 177Lu vipivotide tetraxetan; IV at recommended dosages |
Comparators |
|
Outcomes | Efficacy outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
177Lu = lutetium-177; AE = adverse events; ARPI = androgen receptor pathway inhibitor; ECOG PS = Eastern Cooperative Oncology Group Performance Status; mCRPC = metastatic castration-resistant prostate cancer; NAAD = novel androgen axis drug; PSA = prostate-specific antigen; PSMA = prostate-specific membrane antigen; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aThe clinical experts consulted by CADTH noted that a small subset of patients could be candidates for re-treatment with docetaxel.
bRe-treatment with ARPI was not considered to be a relevant comparator by the clinical experts consulted by CADTH (as patients would have to have previously been treated with an ARPI to be considered a candidate for 177Lu vipivotide tetraxetan). This has been included as a comparator in the CADTH review protocol as it was included in the sponsor’s economic evaluation and was identified as a potential comparator by the participating drug programs.
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Pluvicto (177Lu vipivotide tetraxetan). The clinical trials registries searched were the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on September 7, 2022. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on January 11, 2023.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.30 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 1 study was identified from the literature for inclusion in the systematic review (Figure 5). The included study is summarized in Table 7. A list of excluded studies is presented in Appendix 2.
Table 7: Details of Included Studies
Detail | VISION |
|---|---|
Designs and populations | |
Study design | Open-label, phase III, RCT |
Locations | Belgium, Canada, Denmark, France, Netherlands, Sweden, UK, US |
Patient enrolment dates | Study initiation date: May 29, 2018 (first patient’s first visit) Data cut-off date: January 27, 2021 |
Randomized (N) | 831
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | 177Lu vipivotide tetraxetan was administered as a slow IV injection at a dose of 7.4 GBq (± 10%) once every 6 weeks (± 1 week) for a maximum of 6 cycles |
Comparator(s) | BSC/BSoC (physician’s discretion) |
Duration | |
Phase | |
Run-in | NA |
Open-label treatment phase | Treatment with 177Lu vipivotide tetraxetan initiated in the 28 days after randomization and continued for 6 cycles (6 weeks) |
Follow-up | Up to 24 months or until 508 deaths occurred |
Outcomes | |
Primary end point |
|
Secondary and exploratory end points |
|
Notes | |
Publications | Sartor et al. (2021)31 |
177Lu = lutetium-177; 68Ga = gallium-68; BSC = best supportive care; BSoC = best standard of care; CNS = central nervous system; FACT-G = Functional Assessment of Cancer Therapy-General; FACT-P = Functional Assessment of Cancer Therapy-Prostate; FAPSI-8 = 8-item Functional Assessment of Cancer Therapy Advanced Prostate Symptoms Index; mCRPC = metastatic castration-resistant prostate cancer; NA = not applicable; NYHA = New York Heart Association; PCWG3 = Prostate Cancer Working Group 3; PSA = prostate-specific antigen; PSMA = prostate-specific membrane antigen; RCT = randomized controlled trial; RLT = targeted radioligand therapy.
Note: The 5 additional reports included were the Clinical Study Report,7 FDA Multidiscipline Review,9 Health Canada Biologics Safety and Efficacy Assessment Report,8 Common Technical Document,6 and clinicaltrials.gov.32
Source: Clinical Study Report.7
Description of Studies
VISION is a phase III, open-label, RCT conducted to compare the efficacy and safety of 177Lu vipivotide tetraxetan + BSC/BSoC with BSC/BSoC alone in patients with progressive PSMA-positive mCRPC (Figure 6). The sponsor reported that the open-label design was used because blinding would not be practical, owing to the specialized precautions required for administration of a radiopharmaceutical and the toxicities related to targeted radioligand therapy. In addition, the sponsor noted that it would not be appropriate to subject patients who did not receive a radiopharmaceutical to posttreatment radiation protection protocols (e.g., maintaining physical distancing from family members).
Patients were randomized in a 2:1 ratio to receive either 177Lu vipivotide tetraxetan + BSC/BSoC or BSC/BSoC alone (a rationale for the 2:1 ratio was not stated in the protocol). Randomization was stratified by LDH (≤ 260 IU/L versus > 260 IU/L), presence of liver metastases (yes versus no), ECOG PS (0 or 1 versus 2), and inclusion of NAAD in BSC/BSoC (yes versus no).
Patients were enrolled in Belgium (n = 17); Canada (n = 49); Denmark (n = 24); France (n = 70); the Netherlands (n = 38); Sweden (n = 33); the UK (n = 47); and the US (n = 553).7
Figure 6: Design of the VISION Study
177Lu = lutetium-177; ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; mCRPC = metastatic castration-resistant prostate cancer; NAAD = novel androgen axis drug; OS = overall survival; PSA = prostate-specific antigen; PSMA = prostate-specific membrane antigen; RECIST = Response Evaluation Criteria in Solid Tumours Version 1.1; rPFS = radiographic progression-free survival.
Source: Clinical Study Report.7
Populations
Inclusion and Exclusion Criteria
Eligibility criteria are summarized in Table 7. The VISION trial enrolled patients with PSMA-positive, progressive, mCRPC (i.e., serum PSA progression, soft-tissue progression, or progression of bone disease) who had received prior treatment with at least 1 NAAD and at least 1 taxane regimen. Patients who had received treatment with only 1 taxane regimen were required to be medically unsuitable to receive a second taxane regimen. The trial was limited to those with an ECOG PS of 0 to 2.7
PSMA-positive patients were identified using 68Ga-PSMA-11 PET-CT scans that were evaluated centrally based on the following criteria:
At least 1 68Ga-PSMA-11-positive lesion. On PET-CT, a positive lesion was defined as having an uptake greater than normal liver parenchyma, whereas a negative lesion was defined as having an uptake less than or equal to liver uptake.
All lymph nodes that measured at least 2.5 cm on the short axis had to be 68Ga-PSMA-11 positive.
All bone metastases with a soft-tissue component of at least 1.0 cm on the short axis had to be 68Ga-PSMA-11-positive (patients with PSMA-negative osseous metastases without a soft-tissue component were not excluded).
All solid-organ metastases (e.g., lung, liver, adrenal glands.) of at least 1.0 cm on the short axis had to be 68Ga-PSMA-11-positive.
Only patients with at least 1 PSMA-positive lesion identified on PET (i.e., criterion 1) and no negative lesions (i.e., criteria 2 to 4) were enrolled in the study, provided all other inclusion and exclusion criteria were met.7
Baseline and Demographic Characteristics
Table 8 provides a summary of baseline and demographic characteristics for the FAS (i.e., all randomized patients) and PFS-FAS (i.e., all randomized patients on or after March 5, 2019) in the VISION trial. Baseline characteristics were generally similar in the 2 study populations and across the treatment groups.
Data are presented separately for the FAS (all randomized patients who received at least 1 dose of the study treatment) and the PFS-FAS (all patients randomized on or after March 5, 2019). The FAS was used for the evaluation of OS and the PFS-FAS was used for the evaluation of rPFS and all secondary end points (except ORR and DCR). Details regarding these populations are reported in the Statistical Analysis section.
Table 8: Summary of Baseline Characteristics
Characteristics | FASa | PFS-FASb | ||
|---|---|---|---|---|
177Lu vipivotide tetraxetan + BSC/BSOC (N = 551) | BSC/BSOC alone (N = 280) | 177Lu vipivotide tetraxetan + BSC/BSOC (N = 385) | BSC/BSOC alone (N = 196) | |
Age (years) | ||||
Mean (SD) | ████ █████ | ████ █████ | ████ ██████ | ████ ██████ |
Median (range) | 70.0 (48 to 94) | 71.5 (40 to 89) | 71.0 (52 to 94) | 72.0 (51 to 89) |
< 65 years | ███ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
≥ 65 years | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
≥ 65 to 84 years | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
≥ 85 years | █ █████ | █ █████ | █ █████ | █ █████ |
Race, n (%) | ||||
White | 486 (88.2) | 235 (83.9) | 336 (87.3) | 166 (84.7) |
Black/African American | 34 (6.2) | 21 (7.5) | 29 (7.5) | 14 (7.1) |
Asian | 9 (1.6) | 11 (3.9) | 6 (1.6) | 9 (4.6) |
Other | 2 (0.4) | 0 | 2 (0.5) | 0 |
Missing | 20 (3.6) | 13 (4.6) | 12 (3.1) | 7 (3.6) |
ECOG PS, n (%) | ||||
0 to 1 | 510 (92.6) | 258 (92.1) | 352 (91.4) | 179 (91.3) |
2 | 41 (7.4) | 22 (7.9) | 33 (8.6) | 17 (8.7) |
Time since initial cancer diagnosis (years) | ||||
Mean (SD) | 8.3 (5.5) | 8.9 (5.8) | 8.37 (5.683) | 8.80 (5.871) |
Median (range) | 7.4 (0.9 to 28.9) | 7.4 (0.7 to 26.2) | 7.26 (0.9 to 28.9) | 7.01 (0.7 to 26.2) |
Initial histopathological classification, n (%) | ||||
Adenocarcinoma | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
Neuroendocrine | █ █████ | █ | █ █████ | █ |
Unknown | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
Other | █ █████ | █ █████ | █ █████ | █ █████ |
Initial Gleason score, categorized, n (%) | ||||
2 to 3 | █ █████ | █ | █ █████ | █ |
4 to 7 | ███ ██████ | ██ ██████ | ███ ██████ | ██ ██████ |
8 to 10 | 324 (58.8) | 170 (60.7) | 226 (58.7) | 118 (60.2) |
Unknown | 42 (7.6) | 24 (8.6) | 28 (7.3) | 19 (9.7) |
Staging at initial diagnosis, n (%) | ||||
I | █ █████ | █ █████ | █ █████ | █ █████ |
IA | █ | █ █████ | █ | █ █████ |
IB | █ █████ | █ █████ | █ █████ | █ █████ |
II | ██ █████ | ██ █████ | ██ █████ | █ █████ |
IIA | ██ █████ | █ █████ | ██ █████ | █ █████ |
IIB | ██ █████ | ██ █████ | ██ █████ | █ █████ |
III | ██ █████ | ██ █████ | ██ █████ | █ █████ |
IIIA | ██ █████ | █ █████ | ██ █████ | █ █████ |
IIIB | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
IIIC | █ █████ | █ █████ | █ █████ | █ █████ |
IV | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
IVA | ██ █████ | █ █████ | █ █████ | █ █████ |
IVB | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
Unknown | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
Baseline target lesions, n (%) | ||||
Yes | 279 (50.6) | 140 (50.0) | 203 (52.7) | 100 (51.0) |
No | 272 (49.4) | 140 (50.0) | 182 (47.3) | 96 (49.0) |
Nontarget to target lesions, n (%) | ||||
Yes | 429 (77.9) | 212 (75.7) | 303 (78.7) | 144 (73.5) |
No | 122 (22.1) | 68 (24.3) | 82 (21.3) | 52 (26.5) |
Total sum of target lesion diameters (mm) | ||||
Mean (SD) | ████ ██████ | ████ ██████ | ████ ███████ | ████ ███████ |
Median (range) | 45.0 (10 to 351) | 46.2 (10 to 249) | 44.0 (10 to 351) | 43.5 (10 to 209) |
Disease site (target and/or nontarget to target lesions), n (%) | ||||
Lung | 49 (8.9) | 28 (10.0) | 35 (9.1) | 20 (10.2) |
Liver | 63 (11.4) | 38 (13.6) | 47 (12.2) | 26 (13.3) |
Lymph node | 274 (49.7) | 141 (50.4) | 193 (50.1) | 99 (50.5) |
Bone | 504 (91.5) | 256 (91.4) | 351 (91.2) | 179 (91.3) |
PSA doubling time (months) | ||||
Mean (SD) | 3.2 (5.3) | 4.3 (9.1) | 3.07 (3.474) | 5.19 (11.288) |
Median (range) | 2.4 (0.0 to 74.4) | 2.6 (0.0 to 93.1) | 2.40 (0.0 to 37.3) | 2.82 (0.0 to 93.1) |
PSA doubling time (categorized), n (%) | ||||
n | 269 | 131 | 182 | 82 |
Stable | █ █████ | █ █████ | █ █████ | █ █████ |
≤ 6 months | 245 (91.1) | 115 (87.8) | 167 (91.8) | 70 (85.4) |
> 6 months | ██ █████ | ██ █████ | ██ █████ | █ ██████ |
PSA (ng/mL) | ||||
Mean (SD) | █████ ███████ | █████ ███████ | █████ ████████ | █████ ████████ |
Median (range) | 77.5 (0 to 6988) | 74.6 (0 to 8995) | 93.2 (0 to 6988) | 90.7 (0 to 6600) |
Baseline ALP (IU/L) | ||||
Mean (SD) | █████ ███████ | █████ ███████ | █████ ████████ | █████ |
Median (range) | 105.0 (17 to 2524) | 94.5 (28 to 1355) | 108.0 (26 to 2524) | 96.0 (34 to 1355) |
Baseline LDH (IU/L) | ||||
Mean | █████ ███████ | █████ ███████ | █████ ████████ | █████ |
Median (range) | 221.0 (88 to 5387) | 224.0 (105 to 2693) | 230.5 (119 to 5387) | 232.0 (105 to 2693) |
177Lu = lutetium-177; ALP = alkaline phosphatase; BSC = best supportive care; BSOC = best standard of care; ECOG PS = Eastern Cooperative Oncology Group Performance Status; FAS = full analysis set; LDH = lactase dehydrogenase; PFS = progression-free survival; PSA = prostate-specific antigen; SD = standard deviation.
aThe FAS was used for the evaluation of OS.
bThe PFS-FAS was used for the evaluation of rPFS and all secondary end points (except ORR and DCR).
Source: Clinical Study Report.7
Prior Cancer Therapies
Table 9 provides a summary of prior surgery, radiotherapy, and systemic cancer therapy for the FAS and PFS-FAS populations in the VISION trial. Overall, exposure to all types of prior therapies was well balanced across the treatment groups in both the FAS and PFS-FAS. For prior cancer surgery, 43.2% and 39.9% of patients had received at least 1 prior therapeutic surgery for prostate cancer in the FAS and PFS-FAS populations, respectively. The majority of patients had received at least 1 prostate cancer-related radiotherapy (76.1% and 75.4% in the FAS and PFS-FAS, respectively). The most frequent site for radiotherapy was the prostate gland.7
The number and type of systemic therapies were well balanced between the 2 treatment groups in both the FAS and PFS-FAS populations. In the FAS, the mean number of prior systemic regimens was 5.3, and the majority of patients (79.1%) had received at least 4 different categories of treatment. All patients had received prior taxane treatment (1 regimen in 57.9% of patients and 2 regimens in 41.2%), and all patients had received prior therapy with a NAAD (1 regimen in 51.3% of patients, 2 regimens in 41.0%, and more than 2 regimens in 7.7%).7 As shown in Table 9, results were similar for patients in the PFS-FAS population.
Table 9: Summary of Prior Cancer Therapies
Prior cancer therapy | FASa | FAS-PFSb | ||
|---|---|---|---|---|
177Lu vipivotide tetraxetan + BSC/BSOC (N = 551) | BSC/BSOC alone (N = 280) | 177Lu vipivotide tetraxetan + BSC/BSOC (N = 385) | BSC/BSOC (N = 196) alone | |
Prior cancer surgery | ||||
≥ 1 PC-related surgery (including biopsies), n (%) | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
Prior number of surgeries and/or biopsies, mean (SD) | ███ █████ | ███ █████ | ███ █████ | ███ ██████ |
Reason for surgery, n (%) | ||||
Diagnostic and/or biopsy | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
Therapeutic | ███ ██████ | ███ ██████ | ███ ██████ | ██ ██████ |
Palliative | ██ █████ | ██ █████ | █ | █ █████ |
Other | █ █████ | █ | █ █████ | █ |
Prior radiotherapy | ||||
≥ 1 PC-related radiotherapy, n (%) | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
Number of radiotherapies, mean (SD) | ███ █████ | ███ █████ | ███ ██████ | ███ ██████ |
Prior systemic therapy | ||||
Prior number of regimens | ||||
Mean (SD) | ███ █████ | ███ █████ | ███ ██████ | ███ ██████ |
1, n (%) | 1 (0.2) | 0 | 1 (0.3) | 0 |
2, n (%) | 22 (4.0) | 15 (5.4) | 16 (4.2) | 13 (6.6) |
3, n (%) | 95 (17.2) | 41 (14.6) | 74 (19.2) | 33 (16.8) |
> 3, n (%) | 433 (78.6) | 224 (80.0) | 294 (76.4) | 150 (76.5) |
Number of taxane-containing regimens | ||||
Mean (SD) | ███ █████ | ███ █████ | ███ ██████ | ███ ██████ |
1, n (%) | 325 (59.0) | 156 (55.7) | 207 (53.8) | 102 (52.0) |
2, n (%) | 220 (39.9) | 122 (43.6) | 173 (44.9) | 92 (46.9) |
> 2, n (%) | 6 (1.1) | 2 (0.7) | 5 (1.3) | 2 (1.0) |
Prior number of NAAD-containing regimens | ||||
Mean (SD) | ███ █████ | ███ █████ | ███ ██████ | ███ ██████ |
1, n (%) | 298 (54.1) | 128 (45.7) | 213 (55.3) | 98 (50.0) |
2, n (%) | 213 (38.7) | 128 (45.7) | 150 (39.0) | 86 (43.9) |
> 2, n (%) | 40 (7.3) | 24 (8.6) | 22 (5.7) | 12 (6.1) |
Reason for therapy, n (%) | ||||
Therapeutic | ███ ██████ | ███ ██████ | ███ ██████ | ███ ██████ |
Adjuvant | ███ ██████ | ██ ██████ | ███ ██████ | ██ ██████ |
Unknown | ███ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
Neoadjuvant | ██ ██████ | ██ ██████ | ██ ██████ | ██ ██████ |
Maintenance | ██ █████ | ██ █████ | ██ █████ | ██ █████ |
Prophylaxis | ██ █████ | █ █████ | █ █████ | █ █████ |
Other | ██ █████ | █ █████ | █ █████ | █ █████ |
177Lu = lutetium-177; BSC = best supportive care; BSOC = best standard of care; FAS = full analysis set; NAAD = novel androgen axis drug; PC = prostate cancer; PFS = progression-free survival; SD = standard deviation.
aThe FAS was used for the evaluation of OS.
bThe PFS-FAS was used for the evaluation of rPFS and all secondary end points (except ORR and DCR).
Source: Clinical Study Report.7
Interventions
Intervention of Interest
Administration: 177Lu vipivotide tetraxetan was administered as a slow IV injection at a dose of 7.4 GBq (± 10%) once every 6 weeks (± 1 week) for a maximum of 6 cycles. An infusion with at least 10 mL of normal saline was administered to ensure patency of the IV line before 177Lu vipivotide tetraxetan administration. After 177Lu vipivotide tetraxetan administration, a saline infusion of 500 mL was recommended. Cooling of the salivary glands from 30 minutes before and up to 4 hours after the 177Lu vipivotide tetraxetan injection was optional to reduce the risk of salivary gland radiation injuries and depended on the centre’s standard practice. Additionally, allopurinol could be started from 7 days to 10 days after 177Lu vipivotide tetraxetan therapy in patients with a high tumour burden or gout, at the investigator’s discretion.7
Treatment cycles: Patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group began administration with the study drug no more than 28 days after randomization. These patients received 7.4 GBq (± 10%) of 177Lu vipivotide tetraxetan once every 6 weeks (± 1 week) for a maximum of 6 cycles while receiving BSC/BSoC. After the cycle 4 treatment and before the scheduled cycle 5 treatment, the investigator had to determine whether:
the patient showed evidence of response (reported as radiological, PSA, or clinical benefit)
the patient had signs of residual disease on CT with contrast, MRI, or bone scan
the patient had shown good tolerance to the 177Lu vipivotide tetraxetan treatment.
If the patient met all of these criteria and agreed to continue with 177Lu vipivotide tetraxetan treatment, the investigator could administer 2 additional cycles. A maximum of 6 cycles of 177Lu vipivotide tetraxetan was allowed. If the patient did not meet any of the criteria or did not agree to additional dosing, no further treatment with 177Lu vipivotide tetraxetan would be administered during the study period.
After the final cycle of 177Lu vipivotide tetraxetan, patients continued to be treated with BSC/BSoC as long as the investigator felt they were clinically benefiting (regardless of radiographic progressive disease, based on investigator's assessment, per PCWG3 criteria) or until they required a treatment regimen that was not permitted in the VISION study. For both treatment arms, the cycle duration for cycles 1 to 6 was 6 weeks, and for cycle 7 and beyond was 12 weeks. From cycle 7 onward, all patients from both treatment arms should only receive BSC/BSoC.7
Dose reductions and discontinuations: Only 1 dose reduction (by 20%) in administered activity was permitted. Once a dose was reduced, the 177Lu vipivotide tetraxetan dose could not be re-escalated. If a patient had further toxicity that required an additional reduction in administered activity, treatment with 177Lu vipivotide tetraxetan was discontinued. If a treatment delay due to an AE or toxicity management persisted for more than 4 weeks, treatment with 177Lu vipivotide tetraxetan was discontinued. If treatment with 177Lu vipivotide tetraxetan was discontinued due to an AE, abnormal laboratory value, or toxicity, patients continued to receive BSC/BSoC alone as long as the investigator felt they were clinically benefiting or until they required a treatment regimen not allowed in the VISION study.7
Comparator
The BSC/BSoC for each patient was selected by the patient’s physician before randomization, was administered per the physician’s orders before randomization, and was to continue until the patient completed the randomized treatment period and entered the long-term follow-up period. Patients were treated with BSC/BSoC as long as the investigator felt they were clinically benefiting (regardless of radiographic progressive disease, based on investigator's assessment, per PCWG3 criteria) or until they required a treatment regimen not allowed in the VISION study. Treatments for prostate cancer could include but were not limited to:
supportive measures (e.g., pain medications, hydration, transfusions)
ketoconazole
androgen-reducing drugs (including any corticosteroid and 5-alpha reductases)
abiraterone, enzalutamide, apalutamide, or any other NAAD
radiation in any external beam or seeded form (however, systemic radioisotopes [e.g., radium-223] and hemi-body radiotherapy treatment were not permitted in the study)
bone-targeted drugs, including zoledronic acid, denosumab, and any bisphosphonate
combinations of any or all of the aforementioned treatments (these could be modified over time as needed).7
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 10. These end points are summarized here. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Overall Survival
OS was defined as the time (in months) from the date of randomization to the date of death from any cause. If the patient was not known to have died, then OS was censored. The censoring date was the date of the last study visit, or contact, before the cut-off date. The cut-off date was not used as the last contact date unless the patient was seen or contacted on that date.7 In response to questions from Health Canada, the sponsor provided the following explanation for how survival status was obtained for patients who were reported as lost to follow-up or off study due to withdrawal of consent.
Preemptively
Where permitted by ethics committees (ECs) and/or institutional review boards (IRBs), informed consent included a clause that allowed survival follow-up through public registries, local physicians, or medical records in health systems or institutions.
Study sites were instructed to explain to patients who opted to withdraw consent from the study that if they were willing to allow survival data to continue to be collected, such limited follow-up consisted only of survival status data.
Retrospectively
Where allowed by ECs and/ or IRBs, patients who had withdrawn consent from the study were contacted and asked whether they would be willing to allow limited follow-up data collection that consisted of survival status only.
Recurrent listings were created through data management to identify patients who were lost to follow-up or withdrew from the study.
These patients were discussed between the sponsor and local clinical research associates, and clinical research associates discussed the patients with the respective sites.
All informed consents forms were checked to confirm whether survival data could be searched and retrieved from public health records, whether local physicians could be contacted for survival status, and whether medical records in health systems or institutions could be used to confirm survival status. Whichever form of survival data collection was allowed was pursued.
Where required, ECs and/or IRBs were informed and/or consulted and additional submissions were obtained, where needed, to gather survival data from the aforementioned options.
Where permitted, searches in public health records and survival data retrieval were performed by sites for eligible patients.8
Radiographic PFS
rPFS was defined as the time (in months) from the date of randomization to the date of radiographic disease progression, based on the central review assessment, per PCWG3 criteria, or death from any cause. Patients who were alive without radiographic progression at the analysis data cut-off were censored for rPFS at the time of their last evaluable radiographic assessment.
The date of a radiographic progressive disease event was the date of the first CT, MRI, or bone scan identifying progressive disease or of death from any cause (whichever occurred first) for patients with no more than 1 immediately prior assessment missing.
Progressive disease was based on overall response assessed using RECIST.
The date of progressive disease was the date of the first appearance of new lesion(s), if applicable.
Progressive disease identified on a bone scan had to be confirmed with the following 2 rules (which allow for a flare effect at the first posttreatment scan):
Rule 1 (progression at week 8, confirmed at week 16) — If there were at least 2 new lesions on the first posttreatment scan, they had to be confirmed with at least 2 additional lesions on the next scan (2 + 2 rule). The date of progression was the date of the first posttreatment scan.
Rule 2 (progression at week 16 or later confirmed at next scan) — For scans performed after the first posttreatment scan, there had to be at least 2 new lesions after the first posttreatment scan (treated as a new baseline) that were persistent (confirmed) on a subsequent scan. The date of progression was the date of the scan that first documented the second lesion relative to the first posttreatment scan.
The censoring date was the date when the last evaluable radiographic assessment (CT, MRI, or bone scan) determined a lack of progression. If there were no evaluable assessments, censoring occurred at the date of randomization. Patients who had 2 or more consecutive missed tumour assessments immediately before progressive disease or death were censored at the date of the last evaluable tumour assessment before the missing tumour assessments.7
Progression-Free Survival
PFS was defined as the time (in months) from the date of randomization to the date of first evidence of radiographic, clinical, or PSA progression or of death from any cause, whichever occurred first.
The date of radiographic progression was per central review assessment.
The date of clinical progression was the earliest date of assessment in which the investigator indicated clinical progression.
Date of PSA progression: Where a decline from baseline was documented, the date that an increase of at least 25% in PSA and an absolute increase of 2 ng/mL or more from the nadir (from all scheduled and unscheduled visits before the current visit being evaluated) was documented and confirmed by a second consecutive value obtained at least 3 weeks later. Rises in PSA within the first 12 weeks of the date of first dose of randomized treatment were ignored. Where there was no decline from baseline documented, PSA progression was defined as an increase of at least 25% increase from baseline along with an increase in absolute value of at least 2 ng/mL after 12 weeks from the first dose of the study treatment.7
Skeletal-Related Events
Time to first SSE was defined as the date of randomization to the date of first new symptomatic pathological bone fracture, spinal cord compression, tumour-related orthopedic surgical intervention, requirement for radiation therapy to relieve bone pain, or death from any cause (whichever occurred first).7
Brief Pain Inventory-Short Form
The BPI-SF is a generic pain assessment tool used in research and practice for pain assessment in musculoskeletal conditions. The higher the BPI-SF score, the worse the pain. The BPI-SF measures pain intensity (on a range from 0 [no pain] to 10 [ worst pain]), as well as the way pain interferes with daily activities (on a range from 0 [no interference] to 10 [completely interferes]).7 Worsening pain intensity was defined as an increase from baseline of at least 30% or an increase from baseline of at least 2 points on the BPI-SF scale at any time up to the EOT visit, clinical disease progression, or death. A minimally important difference of 2 or more points or a 30% change in pain intensity items from baseline has previously been used in studies of patients with mCRPC.33,34
Functional Assessment of Cancer Therapy-Prostate
FACT-P is a prostate cancer–specific HRQoL measure. The FACT-P total score (range, 0 to 156) consists of 5 subscales: physical well-being, social/family well-being, emotional well-being, functional well-being, and prostate cancer. Higher FACT-P scores reflect a better QoL.7 Improvement in FACT-P was defined as an increase from baseline in FACT-P total score of at least 10 points any time up to the EOT visit (i.e., an increase of ≥ 10 points at any single assessment would be considered an improvement event). Time to worsening of FACT-P total score was defined as the time (in months) from randomization to the first decrease from baseline greater than 10 points in FACT-P, clinical disease progression (excluding radiographic and PSA progression), or death. If no event has occurred, the censoring date is the time of the last QoL assessment.7 The minimally important difference for FACT-P has been estimated to be in the range of 6 to 10 points.35
Functional Assessment of Cancer Therapy-General
The FACT-G total score (range, 0 to 108) consist of 4 subscales: physical well-being, social/family well-being, emotional well-being, and functional well-being.7 Improvement in FACT-G was defined as an increase from baseline in FACT-G total score of at least 9 points any time up to the EOT visit (i.e., an increase of ≥ 9 points at any single assessment would be considered an improvement event). Time to worsening in FACT-G score was defined as the time from randomization to the first decrease from baseline of at least 10 points in FACT-G total score, clinical disease progression, or death.
Eight-Item Functional Assessment of Cancer Therapy Advanced Prostate Symptoms Index
FAPSI-8 (range, 0 to 32) covers 8 prostate cancer–specific symptoms: 3 related to pain, 1 related to fatigue, 1 related to weight loss, 2 related to urinary difficulties, and 1 related to concerns about the condition getting worse.7 Improvement in FAPSI-8 score was defined as an increase from baseline in FAPSI-8 total score of at least 3 points any time up to the EOT visit (i.e., an increase of ≥ 3 points at any single assessment would be considered an improvement event). Time to worsening in FAPSI-8 score was defined as the time from randomization to the first decrease from baseline of at least 10 points in FAPSI-8 total score, clinical disease progression, or death.
Five-Level EQ-5D
HRQoL was measured using the 5-Level EQ-5D (EQ-5D-5L). The EQ-5D-5L descriptive system has 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each is rated on 5 levels of perceived problems (level 1 = no problems, level 2 = slight problems, level 3 = moderate problems, level 4 = severe problems, and level 5 = extreme problems) measured on a single day. A Canadian-specific minimum clinically important difference (MCID) of 0.037 has been reported.36,37 An MCID of the EQ-5D-5L was not identified in patients with mCRPC. The instrument also includes a 20 cm EQ visual analogue scale (EQ-VAS) that has end points labelled 0 [worst imaginable health state] and 100 [best imaginable health state]. Respondents are asked to rate their health by drawing a line from an end point to the point on the EQ-VAS that best represents their health on that day. No MCID for the EQ-VAS in patients with mCRPC was identified.
Statistical Analysis
Power Calculation
The sample size was determined from the alternate primary end points of rPFS and OS. The term “alternate end points” is used because the study was designed so that statistical significance for either the rPFS or OS end point would be considered sufficient to have the study declared positive (i.e., it was not required that both rPFS and OS be statistically significant for the study to be declared positive).
A total of 814 patients randomized and followed on an ITT basis for a minimum of 13 months was expected to yield 508 deaths. This would provide at least 90% power to test the hypothesis that the HR for OS is 0.7306 or better with a 1-sided alpha level of at least 0.020. For rPFS, approximately 557 of 814 patients were expected to be randomized on or after March 5, 2019, with a minimum of approximately 6 months follow-up; these patients were expected to yield 364 rPFS events. This would provide 84% power to test the hypothesis that the HR of rPFS is 0.67 or better with a 1-sided alpha level of 0.004.7
Statistical Testing and Models
Table 10 summarizes the statistical testing and modelling approaches used for the primary, key secondary, and secondary end points in the VISION trial.
Table 10: Statistical Analysis of Efficacy End Points
End point | Analysis set | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|---|
Alternate primary end points | ||||
rPFS | PFS-FAS |
|
|
|
OS | FAS |
| Same as for rPFS |
|
Key secondary end points | ||||
ORR DCR | Response-evaluable analysis set |
| Same as rPFS | NA |
Time to first SSE | PFS-FAS |
| Same as rPFS | NA |
Secondary end points | ||||
PFS | PFS-FAS |
| Same as rPFS | NA |
PSA response | PFS-FAS | Same as for ORR | Same as rPFS | NA |
FACT-P FACT-G FAPSI-8 BPI-SF EQ-5D-5L | PFS-FAS | Time to worsening:
| Same as rPFS | NA |
BPI-SF = Brief Pain Inventory-Short Form; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; DCR = disease control rate; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EQ-5D-5L = 5-Level EQ-5D; FACT-G = Functional Assessment of Cancer Therapy-General; FACT-P = Functional Assessment of Cancer Therapy-Prostate; FAPSI-8 = 8-item Functional Assessment of Cancer Therapy Advanced Prostate Symptoms Index; FAS = full analysis set; HR = hazard ratio; LDH = lactate dehydrogenase; NA = not applicable; NAAD = novel androgen axis drug; OR = odds ratio; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PSA = prostate-specific antigen; rPFS = radiographic progression-free survival; SSE = symptomatic skeletal event.
aThe additional events included: events regardless of intervening missed assessments; bone progressive disease indicated by progression at week 8 and confirmed at week 16 or progression at week 16 or later without confirmation; and included all radiographic progressive disease and deaths captured in the study, including scans not centrally read that were captured on the long-term follow-up.
Source: Clinical Study Report.7
Statistical Testing Hierarchy
The VISION trial had 2 alternate primary efficacy end points (OS and rPFS) and used the following testing approach: a primary analysis of rPFS with an interim analysis of OS when 364 rPFS events had occurred; and a final analysis of OS when 508 deaths had occurred. The interim OS analysis was not completed by the sponsor as the targeted number of OS events for the final analysis (using the FAS population) were observed before the targeted number of rPFS events (using the PFS-FAS population).9
The key secondary end points were tested using a Hochberg closed-test procedure to control the overall type I error rate, whereas the key secondary efficacy end points were only statistically tested if OS was statistically significant.7
Table 11: Analysis of Key Secondary End Points at the Interim and Final Analyses
Scenario | Final rPFS 1-sided P value | Interim OS 1-sided P value | 2-sided alpha (CI) for key secondary end points | Final OS 1-sided P value | 2-sided alpha (CI) for key secondary end points |
|---|---|---|---|---|---|
Final rPFS or interim OS analysis and final OS analysis | |||||
A | P < 0.004 | P < 0.001 | 0.01 (99%) | Not re-testeda | Not re-testedb |
B | P < 0.004 | NS | Not testedc | P < 0.024 | 0.048 (95.2%) |
NS | Not testedd | ||||
C | NS | P < 0.001 | 0.002 (99.8%) | Not re-testeda | Not re-testedb |
D | NS | NS | Not testedc | P < 0.02 | 0.040 (96%) |
Final rPFS or final OS analysis | |||||
A | P < 0.004 | P < 0.025 | 0.05 (95%) | — | — |
B | P < 0.004 | NS | Not testedd | — | — |
C | NS | P < 0.021 | 0.042 (95.8%) | — | — |
D | NS | NS | Not testede | — | — |
CI = confidence interval; NS = not statistically significant at the prespecified alpha level; OS = overall survival; rPFS = radiographic progression-free survival.
aFinal OS will not be re-tested if the interim OS is met. The final OS results will be presented descriptively, including 95% CI and the nominal P value.
bThe key secondary end points will not be re-tested if the final OS is met; however, the results will be presented descriptively, including 95% CI and the nominal P value.
cThe key secondary end points will not be tested if the interim OS is not met; however, the results will be presented descriptively, including 95% CI and the nominal P value.
dIf the key secondary end points are tested when the final OS is not met using the successful rPFS 1-sided (2-sided) alpha level of 0.004 (0.008), there will be, at most, a type I error inflation of 0.004 because the rPFS alpha level has already been allocated to the final OS test. Therefore, if the final OS is not met, the key secondary end points will only be presented descriptively, including 95% CI and the nominal P value.
eThe key secondary end points will not be tested if the final OS is not met; however, the results will be presented descriptively, including 95% CI and the nominal P value.
Source: Clinical Study Report.7
Subgroup Analyses
Subgroups of interest reported in the VISION trial for rPFS and OS included the following: inclusion of NAADs as part of the assigned BSC/BSoC treatment at the start of the study (without NAAD versus with NAAD); ECOG PS at baseline (0 or 1 versus 2); and the presence of liver metastases at baseline (yes versus no). Additional subgroups reported were baseline LDH levels (≤ 260 IU/L versus > 260 IU/L); age (< 65 years versus ≥ 65 years); and race (white versus Black or African American versus Asian versus other).7 No formal statistical test of hypotheses was performed for the subgroups.
Analysis Populations
The analysis sets that were used to evaluate the safety and efficacy end points in the VISION trial are summarized in Table 12.
The sponsor noted that the open-label study design resulted in a high rate of early withdrawal. Patients were often disappointed at not receiving 177Lu vipivotide tetraxetan, leading to a lack of willingness to comply with the study protocol and/or an interest in receiving therapies that were prohibited in the study protocol (e.g., taxanes). As part of the plan to address the early withdrawal of consent in the BSC/BSoC alone group, the sponsor amended the trial protocol so that the primary analysis of rPFS would be conducted using patients prospectively randomized on or after March 5, 2019 (i.e., the PFS-FAS population) (refer to Figure 7).7
Analysis set | Description | Use |
|---|---|---|
FAS | All randomized patients. Patients were included in the treatment arm to which they were randomized regardless of actual treatment received. This is an ITT analysis set. | Analysis of OS |
PFS-FAS | All patients randomized on or after March 5, 2019. Patients were included in the treatment arm to which they were randomized regardless of actual treatment received. | Primary analyses of rPFS and all secondary end points except ORR and DCR |
Response-evaluable analysis set | The subset of patients in the PFS-FAS with evaluable disease by RECIST at baseline (i.e., at least 1 target and/or nontarget lesion per independent central review radiologist assessment was used as the final radiology assessment). Patients were included in the treatment arm to which they were randomized. Soft-tissue response, measured by RECIST, was assessed in this dataset. | Primary analyses of ORR and DCR |
FAS safety analysis set | The subset of patients in the FAS who received at least 1 dose of randomized treatment. Patients were included in the treatment arm corresponding to the actual treatment received. | Safety analyses |
DCR = disease control rate; FAS = full analysis set; ITT = intention to treat; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumours Version 1.1; rPFS = radiographic progression-free survival.
Source: Clinical Study Report.7
Results
Patient Disposition
Patient disposition for the VISION trial is summarized in Figure 8 and Table 13. A total of 1179 patients were screened for eligibility. Of the 176 patients, 141 were excluded from undergoing a 68Ga-PSMA-11 PET-CT scan because they failed to meet the eligibility criteria before imaging and 24 withdrew their consent. A total of 1,003 patients underwent a 68Ga-PSMA-11 PET-CT scan; of the 172 patients excluded, 123 were excluded because of a negative 68Ga-PSMA-11 PET-CT scan.7
A total of 831 patients were randomized to either the 177Lu vipivotide tetraxetan + BSC/BSoC group (N = 551, but for patients enrolled on or after March 5, 2019, N = 385) or the BSC/BSoC alone group (N = 280, but for patients enrolled on or after March 5, 2019, N = 196). Nearly all patients randomized to 177Lu vipivotide tetraxetan (96.7%) received at least 1 dose of the study drug (the primary reason for not receiving the treatment was cited as AEs occurring between randomized at the first treatment cycle).7 Of the 280 patients randomized to the BSC/BSoC alone group, 71.8% received at least 1 dose of treatment.
A substantial proportion of the patients in the BSC/BSoC alone group withdrew early from the VISION trial (28.3% before receiving any part of the BSC/BSoC alone regimen). As shown in Figure 7, 56.0% of patients in the BSC/BSoC alone group who were enrolled before protocol amendment 4 withdrew before receiving a study treatment or a radiographic assessment, compared with 1.2% in the 177Lu vipivotide tetraxetan + BSC/BSoC group. After the protocol amendment designed to address early withdrawal, the rate of withdrawal was reduced to 16.3% (which was still far greater than the 4.2% rate of early withdrawal in the 177Lu vipivotide tetraxetan group).8,11 Withdrawn consent was the primary reason for the high rate of early withdrawal from the BSC/BSoC alone group.
Figure 7: Protocol Amendments to Address Early Withdrawal
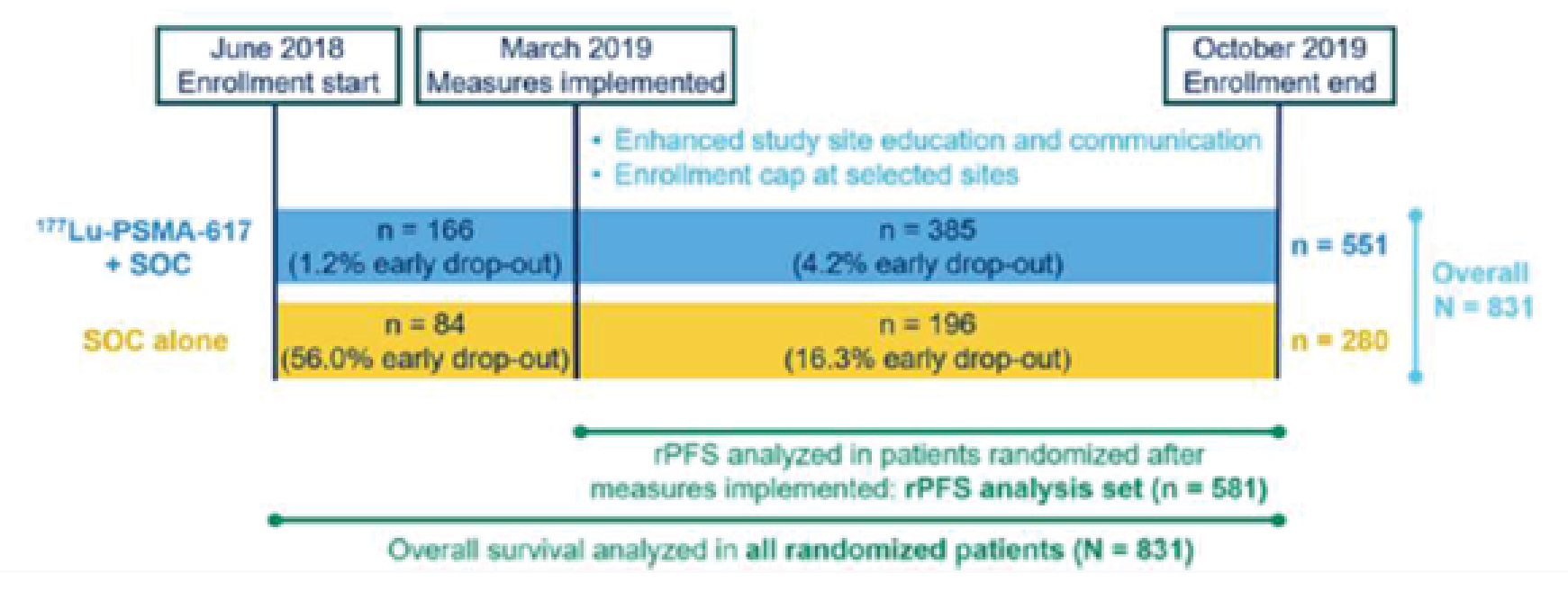
177Lu-PSMA-617 = lutetium-177 vipivotide tetraxetan; rPFS = radiographic progression-free survival; PMSA = prostate-specific membrane antigen; SOC = standard of care.
Source: Health Canada biologics safety and efficacy assessment report8
█████ patients in the 177Lu vipivotide tetraxetan +BSC/BSoC group discontinued the study treatment, primarily because of progressive disease (█████), AEs ███████, and lack of clinical benefit (████). As patients in both treatment groups received BSC/BSoC, the sponsor also reported discontinuations from the BSC/BSoC regimens. The primary reasons for discontinuing BSC/BSoC treatment were (177Lu vipivotide tetraxetan + BSC/BSoC versus BSC/BSoC alone) progressive disease (█████ ██████ █████), lack of clinical benefit (█████ ██████ ██████), and withdrawal of consent for treatment (████ ██████ █████). The sponsor reported that the relatively higher proportion of patients who discontinued due to progressive disease in the 177Lu vipivotide tetraxetan + BSC/BSoC arm may be related to the longer period of time in which patients in this group received study-related treatment (e.g., at the data cut-off, █████ of those in the 177Lu vipivotide tetraxetan + BSC/BSoC group and █████ in the BSC/BSoC alone group had discontinued the study).7
If a patient chose to discontinue randomized treatment in the VISION study for a reason other than radiographic progression, the patient was asked to confirm whether they consented to continue to be followed for long-term safety, rPFS, and OS. If the patient did not specifically withdraw consent for long-term follow-up evaluations, the patient remained in the long term follow-up.7
Figure 8: Patient Disposition in the VISION Trial
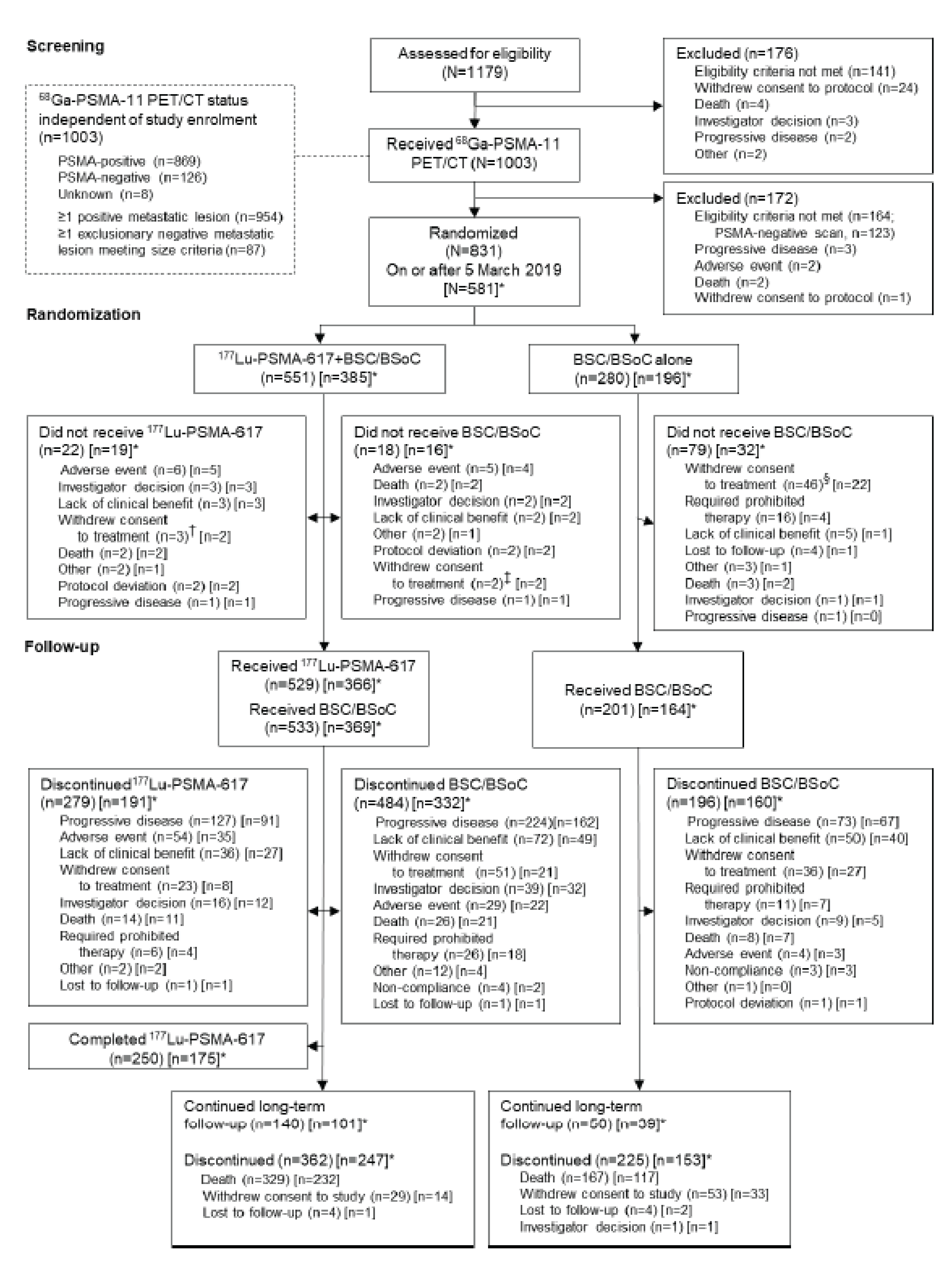
177Lu = lutetium-177; 68Ga = gallium-68; BSC = best supportive care; BSoC = best standard of care; PSMA = prostate-specific membrane antigen.
* Number in square brackets indicate patients randomized on or after Mach 5, 2019
† Reasons for withdrawal of consent to treatment: none given (n = 2), travel or procedure “fatigue” (n = 1)
ǂ Reasons for withdrawal of consent to treatment: none given (n = 1), travel or procedure “fatigue” (n = 1)
§ Reasons for withdrawal of consent to treatment: receiving BSC/BSoC without 177Lu (n = 31), none given (n = 7), decided to pursue off-study treatment (n = 5), travel or procedure “fatigue” (n = 2), perceived lack of benefit (n = 1)
Source: Clinical Study Report.7
Disposition | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 551) | BSC/BSoC alone (N = 280) |
|---|---|---|
Randomized (total) | 551 | 280 |
Randomized (after March 5, 2019) | 385 | 196 |
Patients treated | 533 (96.7) | 201 (71.8) |
Patients not treated | 18 (3.3) | 79 (28.2) |
Patients still on treatment | 49 (8.9) | 5 (1.8) |
Patients who discontinued all study treatments | 484 (87.8) | 196 (70.0) |
Treated with 177Lu vipivotide tetraxetan | 529 (96.0) | NA |
Not treated with 177Lu vipivotide tetraxetan | 22 (4.0) | NA |
Adverse event | 6 (1.1) | NA |
Investigator decision | 3 (0.5) | NA |
No longer clinically beneficial | 3 (0.5) | NA |
Withdrew consent (treatment) | 3 (0.5) | NA |
Death | 2 (0.4) | NA |
Other | 2 (0.4) | NA |
Protocol deviation | 2 (0.4) | NA |
Progressive disease | 1 (0.2) | NA |
Patients who completed 177Lu vipivotide tetraxetan | 250 (45.4) | NA |
Patients who discontinued 177Lu vipivotide tetraxetan | 279 (50.6) | NA |
Progressive disease | 127 (23.0) | NA |
Adverse event | 54 (9.8) | NA |
No longer clinically beneficial | 36 (6.5) | NA |
Withdrew consent (treatment) | 23 (4.2) | NA |
Investigator decision | 16 (2.9) | NA |
Death | 14 (2.5) | NA |
Patient requires care not allowed in study | 6 (1.1) | NA |
Other | 2 (0.4) | NA |
Patient lost to follow-up | 1 (0.2) | NA |
Patients treated with BSC/BSoC | 533 (96.7) | 201 (71.8) |
Patients not treated with BSC/BSoC | 18 (3.3) | 79 (28.2) |
Reason not treated with BSC/BSoC | ||
Withdrew consent (treatment) | 2 (0.4) | 46 (16.4) |
Requires care not allowed in study | 0 | 16 (5.7) |
No longer clinically beneficial | 2 (0.4) | 5 (1.8) |
Patient lost to follow-up | 0 | 4 (1.4) |
Death | 2 (0.4) | 3 (1.1) |
Other | 2 (0.4) | 3 (1.1) |
Progressive disease | 1 (0.2) | 1 (0.4) |
Investigator decision | 2 (0.4) | 1 (0.4) |
Adverse event | 5 (0.9) | 0 |
Protocol deviation | 2 (0.4) | 0 |
Reason for discontinuation from BSC/BSoC | ||
Progressive disease | 224 (40.7) | 73 (26.1) |
No longer clinically beneficial | 72 (13.1) | 50 (17.9) |
Withdrew consent (treatment) | 51 (9.3) | 36 (12.9) |
Investigator decision | 39 (7.1) | 9 (3.2) |
Adverse event | 29 (5.3) | 4 (1.4) |
Death | 26 (4.7) | 8 (2.9) |
Patient requires care not allowed in study | 26 (4.7) | 11 (3.9) |
Other | 12 (2.2) | 1 (0.4) |
Patient noncompliance | 4 (0.7) | 3 (1.1) |
Patient lost to follow-up | 1 (0.2) | 0 |
Protocol deviation | 0 | 1 (0.4) |
Continuing in long-term follow-up period | 140 (25.4) | 50 (17.9) |
Patients who discontinued study | 362 (65.7) | 225 (80.4) |
Death | 329 (59.7) | 167 (59.6) |
Withdrew consent (protocol) | 29 (5.3) | 53 (18.9) |
Patient lost to follow-up | 4 (0.7) | 4 (1.4) |
Investigator decision | 0 | 1 (0.4) |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; NA = not applicable.
Source: Clinical Study Report.7
Exposure to Study Treatments
Study Treatments
Table 14 provides a summary of exposure to the study treatments in the VISION trial. The median number of 177Lu vipivotide tetraxetan cycles received was 5 (range, 1 to 6), and 46.5% of patients received the full 6 cycles of the study treatment.
Concomitant Medications and Cointerventions
Table 15 summarizes the protocol-permitted use of standard of care therapies.
Subsequent Treatments
Subsequent treatments (i.e., those initiated after discontinuation of the study drug) used by the patients enrolled in the VISION trial are summarized in Table 16 for nonradiation therapies and in Table 17 for radiation therapies.
Table 14: Exposure to Study Treatments
Exposure | Parameter | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | BSC/BSoC alone (N = 205) |
|---|---|---|---|
Duration of exposure to 177Lu vipivotide tetraxetan + BSC/BSoC (months) | Mean (SD) | ███ █████ | ██ |
Median (range) | ███ ████ ██ █████ | ██ | |
Duration of exposure to BSC/BSoC (months) | Mean (SD) | ███ █████ | ███ █████ |
Median (range) | ███ ████ ██ █████ | ███ ████ ██ | |
Duration of exposure to 177Lu vipivotide tetraxetan (months) | Mean (SD) | ███ █████ | ██ |
Median (range) | ███ ████ ██ █████ | ██ | |
Number of cycles started by patient | Mean (SD) | ███ █████ | ██ |
Median (range) | ███ ██ ██ ██ | ██ | |
1 cycle, n (%) | ██ █████ | ██ | |
2 cycles, n (%) | ██ ██████ | ██ | |
3 cycles, n (%) | ██ ██████ | ██ | |
4 cycles, n (%) | ██ ██████ | ██ | |
5 cycles, n (%) | ██ █████ | ██ | |
6 cycles, n (%) | ███ ██████ | ██ | |
Average duration of treatment cycles (months) | Mean (SD) | ███ █████ | ██ |
Median (range) | ███ ████ ██ ████ | ██ | |
Number of cycles delayed | ≥ 1 cycle delayed, n (%) | ██ | ██ |
Mean (SD) | ███ █████ | ██ | |
Median (range) | ███ ██ ██ ██ | ██ | |
Reason for delay of cycle(s), n (%) | Delayed due to scheduling | ██ ██████ | ██ |
Delayed due to AE | ██ █████ | ██ | |
Cumulative dose (GBq) | Mean (SD) | ████ ██████ | ██ |
Median (range) | ████ ████ ██ | ██ | |
Dose intensity (GBq/month) | Mean (SD) | ███ █████ | ██ |
Median (range) | ███ ████ ██ █████ | ██ | |
Relative dose intensity (%) | Mean (SD) | █████ ██████ | ██ |
Median (range) | █████ █████ ██ | ██ |
177Lu = lutetium-177; AE = adverse event; BSC = best supportive care; BSoC = best standard of care; SD = standard deviation.
Source: Clinical Study Report.7
Table 15: Protocol-Permitted Use of Standard of Care Therapies (SAS)
Concomitant medication and interventions, n (%) | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | BSC/BSoC alone (N = 205) |
|---|---|---|
██████████ | ███ ███████ | ███ ███████ |
████████████ | ██ ██████ | ██ ██████ |
█████ █████████████ | ██ █████ | █ █████ |
███ ███████████ ████████ ██ ███ ██ ████████ | ||
██████████████████████ ███████ █████████ | ███ ██████ | ███ ██████ |
███████████████ | ███ ██████ | ███ ██████ |
████ | ███ ██████ | ███ ██████ |
████████████ | ███ ██████ | ██ ██████ |
███████████ | ███ ██████ | ██ ██████ |
███████████ | ██ █████ | █ █████ |
████████████ | █ █████ | █ █████ |
█████████ | ███ ██████ | ██ ██████ |
███████████████ | ██ █████ | ██ ██████ |
████████████ ██ █████████ ██████████ | ██ █████ | ██ █████ |
█████████ ███████ | ██ █████ | █ █████ |
█████████ | █ █████ | █ █████ |
█████████ | ██ █████ | █ █████ |
177Lu = lutetium-177; ARPI = androgen receptor pathway inhibitor; BSC = best supportive care; BSoC = best standard of care; SAS = safety analysis set; SoC = standard of care.
Source: Sponsor Clinical Summary.6
Table 16: Subsequent Exposure to Nonradiation Therapies
Nonradiation therapy | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 551) | BSC/BSoC alone (N = 280) |
|---|---|---|
██ █████████████ ██████ ███████ ███████ | ███ ██████ | ██ ██████ |
██████████████ ███████████ | █ █████ | █ █████ |
████████████ | █ █████ | █ |
████████████ █████████ | █ █████ | █ █████ |
██████████████ ███ ███████ ██████████ | █ █████ | █ █████ |
████████████ | █ █████ | █ █████ |
██████████████ | ██ █████ | ██ █████ |
████████████ | ██ █████ | █ █████ |
████████████ | █ █████ | █ █████ |
███████████ | █ █████ | █ █████ |
█████████████ | █ █████ | █ █████ |
████████████ | █ █████ | █ █████ |
██████████ | █ █████ | █ |
█████████ | █ █████ | █ |
████████████ ██ ██████████████ ██████ | █ █████ | █ █████ |
████████████ ███████████ | █ █████ | █ |
████████████ █████████ | █ █████ | █ █████ |
█████████ ██████████ | █ █████ | █ |
███████ | █ █████ | █ |
█████████ | █ | █ █████ |
█████████ | █ | █ █████ |
███████████████ | █ █████ | █ |
██████████ | █ █████ | █ |
█████████████ | █ █████ | █ |
████████████ █████████ ███████ █████████ | █ | █ █████ |
█████████ | █ | █ █████ |
███████████ | █ | █ █████ |
███ ███ █████████ ██████████ | █ █████ | █ |
███████████ | █ █████ | █ |
████████████ | █ | █ █████ |
████████ ████ | █ | █ █████ |
██████████ ██████████ | ██ █████ | ██ █████ |
█████████ | █ █████ | █ █████ |
█████████████ | █ █████ | ██ █████ |
███████████ | █ █████ | █ █████ |
██████████ | █ █████ | █ █████ |
████████████ | █ █████ | █ █████ |
██████████ | █ █████ | █ |
███ ████████ | █ █████ | █ |
████████████ | █ █████ | █ |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; CAR-t = chimeric antigen receptor T-cells; CI = confidence interval; HR = hazard ratio; NOS = not otherwise specified; OS = overall survival.
Some redacted cells have been deleted.
Source: Clinical Study Report.7
Table 17: Subsequent Exposure to Posttreatment Radiotherapy
Posttreatment radiotherapy | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 551) | BSC/BSoC alone (N = 280) |
|---|---|---|
██ ██████████████ ███████████████ ████ | ██ █████ | ██ ██████ |
██████ ██ ██████████████ | ██ | ██ |
████ ██ █████████████ ████ | ||
████ | ██ █████ | ██ █████ |
█████ | ██ █████ | █ █████ |
███ | ██ █████ | █ █████ |
█████████ ██████ | █ █████ | █ █████ |
█████ | █ █████ | █ █████ |
███ | █ █████ | █ |
█████ | █ █████ | █ █████ |
██████ ████ | █ █████ | █ █████ |
██████ | █ █████ | █ █████ |
███████ | █ █████ | █ |
████ | █ █████ | █ █████ |
███████ ███████ ██████ | █ █████ | █ |
████ ███ ████ | █ █████ | █ |
█████ | █ █████ | █ █████ |
█████ ████ | █ █████ | █ █████ |
████ | █ █████ | █ |
████████ █████ | █ █████ | █ |
█████ | █ █████ | █ █████ |
█████ █████████ ████ | █ █████ | █ |
███████ | █ | █ █████ |
███████████████ █████ ████ | █ | █ █████ |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care.
Source: Clinical Study Report.7
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 3 for detailed efficacy data.
Overall Survival
Table 18 summarizes the results for the primary analysis of OS. There was a statistically significant improvement in OS for patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group compared with those in the BSC/BSoC alone group (HR = 0.62; 95% CI, 0.52 to 0.74; P < 0.001). Deaths were reported for 62.3% of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group and for 66.8% in the BSC/BSoC alone group. Median OS was 15.3 months (95% CI, 14.2 to 16.9 months) in 177Lu vipivotide tetraxetan + BSC/BSoC group and 11.3 months (95% CI, 9.8 to 13.5 months) in the BSC/BSoC alone group. The sponsor reported that median follow-up times for OS were similar in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (20.3 months and 19.8 months, respectively).7 Kaplan-Meier curves for OS are provided in Figure 9.
Table 18: Overall Survival (FAS)
Survival | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 551) | BSC/BSoC alone (N = 280) |
|---|---|---|
OS, n (%) | ||
Deaths | 343 (62.3) | 187 (66.8) |
Censored | 208 (37.7) | 93 (33.2) |
Alivea | 189 (34.3) | 55 (19.6) |
Lost to follow-upb | 4 (0.7) | 5 (1.8) |
Withdrew consentc | 15 (2.7) | 33 (11.8) |
Kaplan-Meier estimates (months) | ||
25th percentile (95% CI) | 9.0 (7.9 to 9.7) | 5.1 (4.2 to 6.3) |
Median OS (95% CI) | 15.3 (14.2 to 16.9) | 11.3 (9.8 to 13.5) |
75th percentile (95% CI) | 26.8 (23.9 to NE) | 19.8 (17.3 to 23.0) |
OS rates (%) | ||
6 months (95% CI) | 86.6 (83.5 to 89.2) | 71.5 (65.5 to 76.7) |
12 months (95% CI) | 61.7 (57.5 to 65.6) | 49.0 (42.6 to 55.1) |
18 months (95% CI) | 43.0 (38.7 to 47.2) | 28.8 (23.1 to 34.7) |
HR (95% CI)d,e | 0.62 (0.52 to 0.74) | |
Stratified log-rank 1-sided P valuee | < 0.001 | |
Follow-up time (months)f | ||
Median (95% CI) | 20.3 (19.8 to 21.0) | 19.8 (18.3 to 20.8) |
Minimum to maximum | 0.0 to 31.5 | 0.0 to 27.1 |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NE = not evaluable; OS = overall survival.
aPatients without event and still on study at data cut-off date.
bPatients who discontinued the study for reasons other than withdrawn consent.
cPatients who withdrew consent from the study.
dHR for 177Lu vipivotide tetraxetan + BSC/BSoC vs. BSC/BSoC alone.
eBoth the Cox proportional hazards model and log-rank test are stratified for LDH (≤ 260 IU/L vs. > 260 IU/L); the presence of liver metastases (yes vs. no); ECOG PS (0 or 1 vs. 2); and the inclusion of NAAD in BSC/BSoC at the time of randomization (yes vs. no).
fFollow-up time = (date of event or censoring – randomization date + 1)/30.4375 (months) censoring for deaths.
Source: Clinical Study Report.7
Sensitivity Analyses of OS
Results of the sensitivity analysis using the PFS-FAS population were similar to those of the primary analysis (HR = 0.63; 95% CI, 0.51 to 0.79), with a median OS of 14.6 months (95% CI, 13.2 to 16.0 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 10.4 months (95% CI, 8.5 to 13.6 months) in the BSC/BSoC alone group.7
The FDA reported numerous sensitivity analyses for OS. In addition to those reported in Figure 10, the FDA reported that they conducted an additional OS sensitivity analysis by excluding patients who withdrew consent. The HR was 0.62 (95% CI, 0.52 to 0.75), which was also consistent with the primary analysis of OS. The FDA concluded that although the disproportionate dropout rate in the BSC/BSoC arm was a concern, ascertainment of many of the OS events from withdrawn patients, as well as multiple sensitivity analyses that considered extreme cases, and the possibility of informative censoring continued to support the statistical significance of the primary analysis. The FDA report highlighted the following sensitivity analyses.
Extreme case: All drop-outs in the 177Lu vipivotide tetraxetan + BSC/BSoC group were considered to be deaths (i.e., assumed a shortened survival time in the 177Lu vipivotide tetraxetan + BSC/BSoC group) (HR = 0.66; 95% CI, 0.55 to 0.79).
Best-case analysis: Elongated survival was assumed in the BSC/BSoC alone group by imputing data for drop-outs in the control arm based on the HR in the 20% of patients with the longest survival, either overall or in the BSC/BSoC alone arm (HR = 0.8; 95% CI, 0.67 to 0.96).
Tipping-point analyses: The increase or decrease in the risk of death in patients who withdrew from either treatment group that would make the primary analysis of OS lose statistical significance was quantified. In tipping-point analysis 1, the hazard of survival would need to decrease by 99% in the BSC/BSoC group for OS to become nonstatistically significant, which is considered extreme and very unlikely to occur.
Figure 9: Kaplan-Meier Plot of OS (FAS)

BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; FAS = full analysis set; Lu = lutetium-177; n/N = number of events/number of patients in treatment arm; OS = overall survival; PSMA = prostate-specific membrane antigen.
Source: Clinical Study Report.7
Figure 10: Sensitivity Analyses of OS Reported by the FDA (FAS)
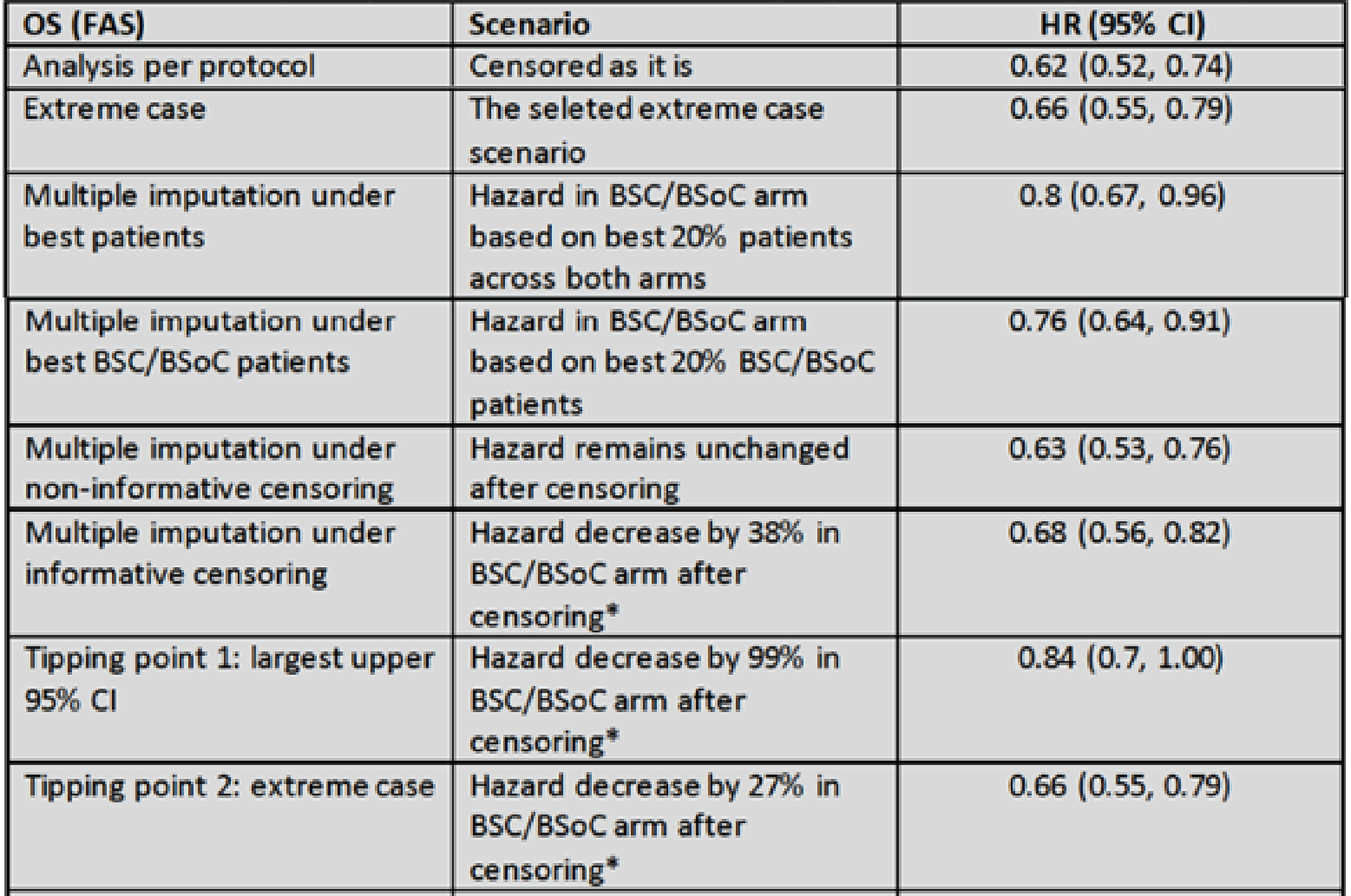
BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; OS = overall survival.
* Risk of event remains unchanged after censoring in the investigational arm.
Source: FDA Multidiscipline review.9
Subgroup Analyses of OS
Subgroup analyses for OS are summarized in Figure 11. Subgroup analyses based on the number of prior taxane regimens favoured 177Lu vipivotide tetraxetan over BSC/BSoC alone for patients who received a single prior taxane regimen (HR = 0.59; 95% CI, 0.46 to 0.75) and for those who received 2 or more prior taxane regimens (HR = 0.73; 95% CI, 0.53 to 0.99). Because of the small number of patients, there is considerable uncertainty about the estimate of effect for the subgroup analysis of patients with a ECOG PS of 2.7
Figure 11: Forest Plots With Subgroup Analyses for OS (FAS)
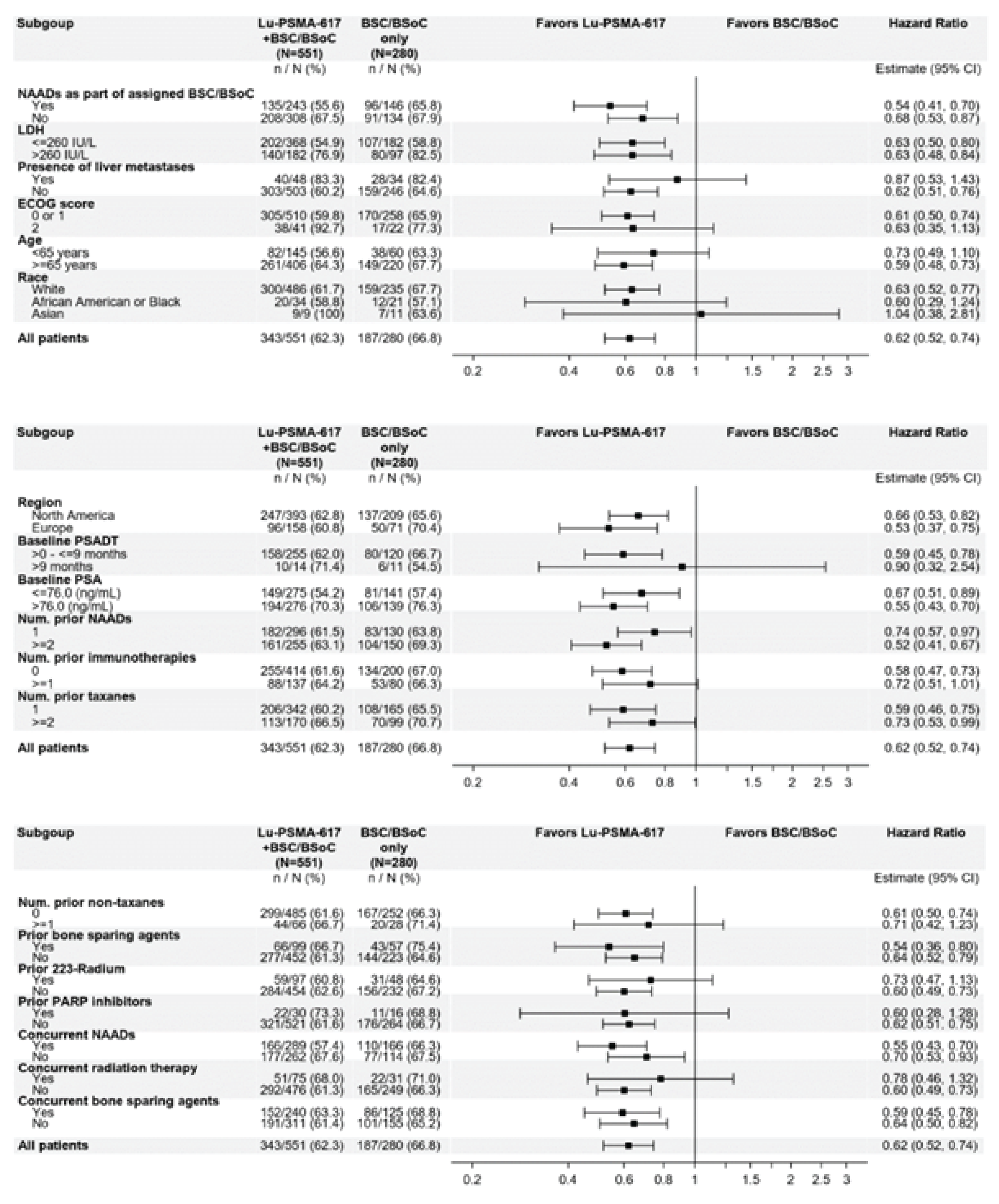
BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; LDH = lactase dehydrogenase; Lu = lutetium-177; n/N = number of events/number of patients in treatment group; NAAD = novel androgen axis drug; Num. = number; OS = overall survival; PARP = poly (ADP-ribose) polymerase; PSA = prostate-specific antigen; PSADT = prostate-specific antigen doubling time; PSMA = prostate-specific membrane antigen.
Source: FDA Multidisciplinary Review9
Radiographic PFS
Table 19 summarizes the results of the primary analysis for rPFS. There was a statistically significant improvement in rPFS for patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group compared with those in the BSC/BSoC alone group (HR = 0.40; 99.2% CI, 0.29 to, 0.57; P < 0.001). Events of radiographic progression or death were reported for 66.0% of patients in the 177Lu vipivotide tetraxetan group (171 radiographic progression events and 83 deaths) and for 47.4% in the BSC/BSoC alone group (59 radiographic progression events and 34 deaths). Median rPFS was 8.7 months (95% CI, 7.9 to 10.8 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 3.4 months (95% CI, 2.4 to 4.0 months) in the BSC/BSoC alone group. The sponsor reported that median follow-up time for rPFS was longer in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC group (16.4 months versus 3.9 months).7 Kaplan-Meier curves for rPFS are provided in Figure 12.
As shown in Figure 13, results in the prespecified sensitivity analyses were similar to those of the primary analysis. Subgroup analyses for rPFS are summarized in Figure 13 and generally suggested similar results for 177Lu vipivotide tetraxetan + BSC/BSoC group compared with BSC/BSoC alone group as the primary rPFS analysis.
Table 19: rPFS Analysis (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC (N = 196) alone |
|---|---|---|
rPFS, n (%) | ||
Events (progression or death) | 254 (66.0) | 93 (47.4) |
Radiographic progressions | 171 (44.4) | 59 (30.1) |
Deaths | 83 (21.6) | 34 (17.3) |
Censored | 131 (34.0) | 103 (52.6) |
Ongoing without event | 90 (23.4) | 24 (12.2) |
Event documented after ≥ 2 missed tumour assessments | 36 (9.4) | 44 (22.4) |
Adequate assessment not availablea | 5 (1.3) | 35 (17.9) |
Kaplan-Meier estimates (months) | ||
25th percentile (99.2% CI) | 4.1 (2.6 to 4.9) | 2.1 (2.0 to 2.3) |
Median rPFS (99.2% CI) | 8.7 (7.9 to 10.8) | 3.4 (2.4 to 4.0) |
75th percentile (99.2% CI) | 16.2 (12.9 to NE) | 7.0 (4.2 to NE) |
rPFS rates (%) | ||
3 months (99.2% CI) | 79.8 (73.6 to 84.7) | 54.3 (42.0 to 65.1) |
6 months (99.2% CI) | 64.6 (57.5 to 70.9) | 27.8 (16.7 to 40.1) |
12 months (99.2% CI) | 33.2 (26.2 to 40.3) | 19.1 (9.0 to 32.1) |
HR (99.2% CI)b,c | 0.40 (0.29 to 0.57) | |
Stratified log-rank 1-sided P value | < 0.001 | |
Follow-up time (months)d | ||
Median (95% CI) | 16.4 (14.3 to 17.0) | 3.9 (2.4 to 5.4) |
Minimum to maximum | 0.0 to 22.6 | 0.0 to 19.8 |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; NE = not evaluable; PFS = progression-free survival; rPFS = radiographic progression-free survival.
aPatients censored without adequate postbaseline evaluations or adequate baseline assessment.
bHR for 177Lu vipivotide tetraxetan + BSC/BSoC vs. BSC/BSoC alone.
cBoth the Cox proportional hazards model and log-rank test are stratified for LDH (≤ 260 IU/L vs. > 260 IU/L); the presence of liver metastases (yes vs. no); ECOG PS (0 or 1 vs. 2); and the inclusion of NAAD in BSC/BSoC at the time of randomization (yes vs. no).
dFollow-up time = (date of event or censoring – randomization date + 1)/30.4375 (months) censoring for death or radiographic progression.
Source: Clinical Study Report.7
Figure 12: Kaplan-Meier Plot of rPFS, per Independent Central Review (PFS-FAS)
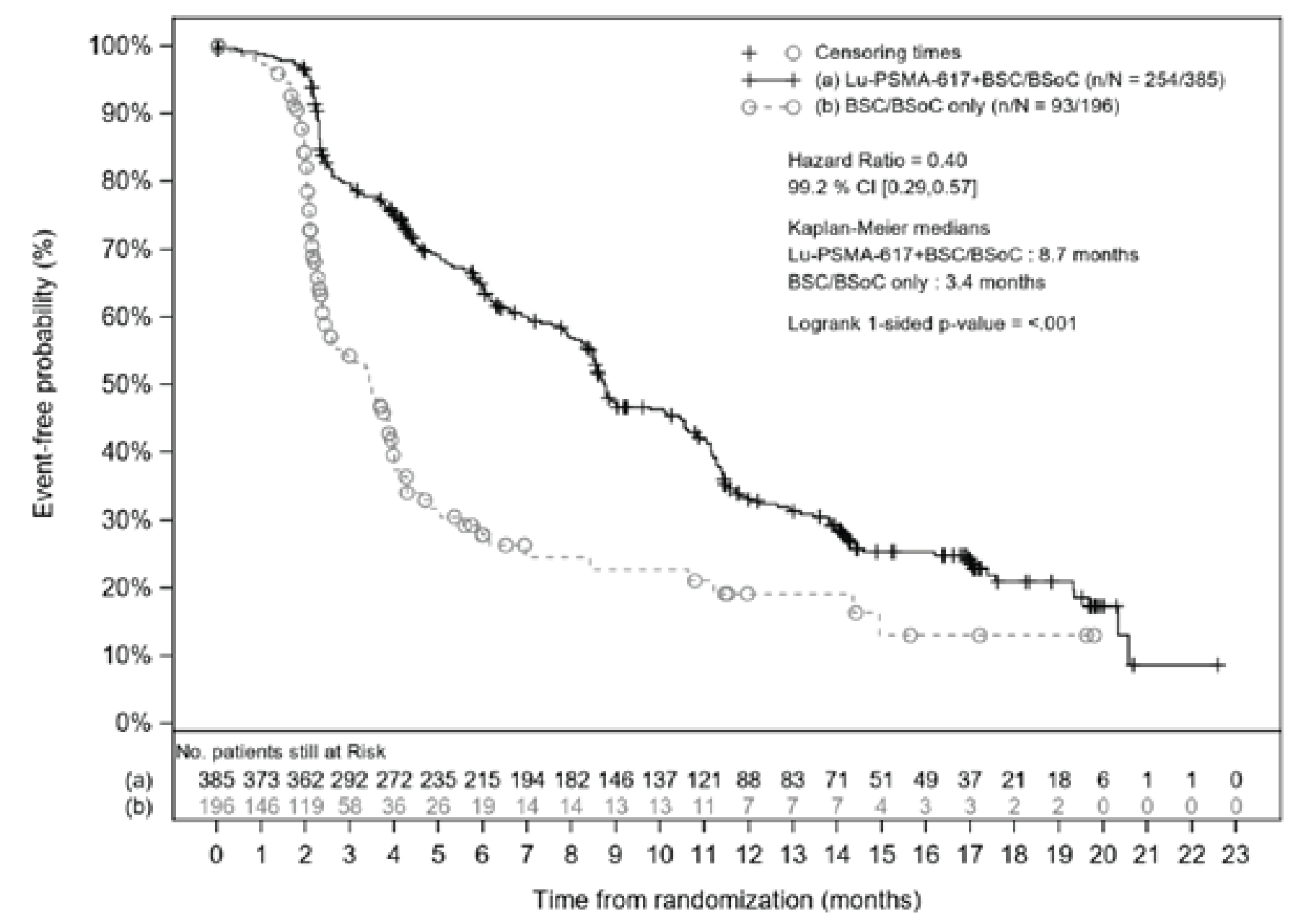
BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; FAS = full analysis set; Lu = lutetium-177; n/N = number of events/number of patients in treatment arm; PSMA = prostate-specific membrane antigen; rPFS = radiographic progression-free survival.
Note: Stratified log-rank test and stratified Cox model using strata, per Interactive Response Technology, defined by LDH level, the presence of liver metastases, ECOG PS, and the inclusion of NAAD in BSC/BSoC at the time of randomization.
Source: Clinical Study Report.7
Figure 13: Forest Plots With Sensitivity Analyses for rPFS (PFS-FAS)
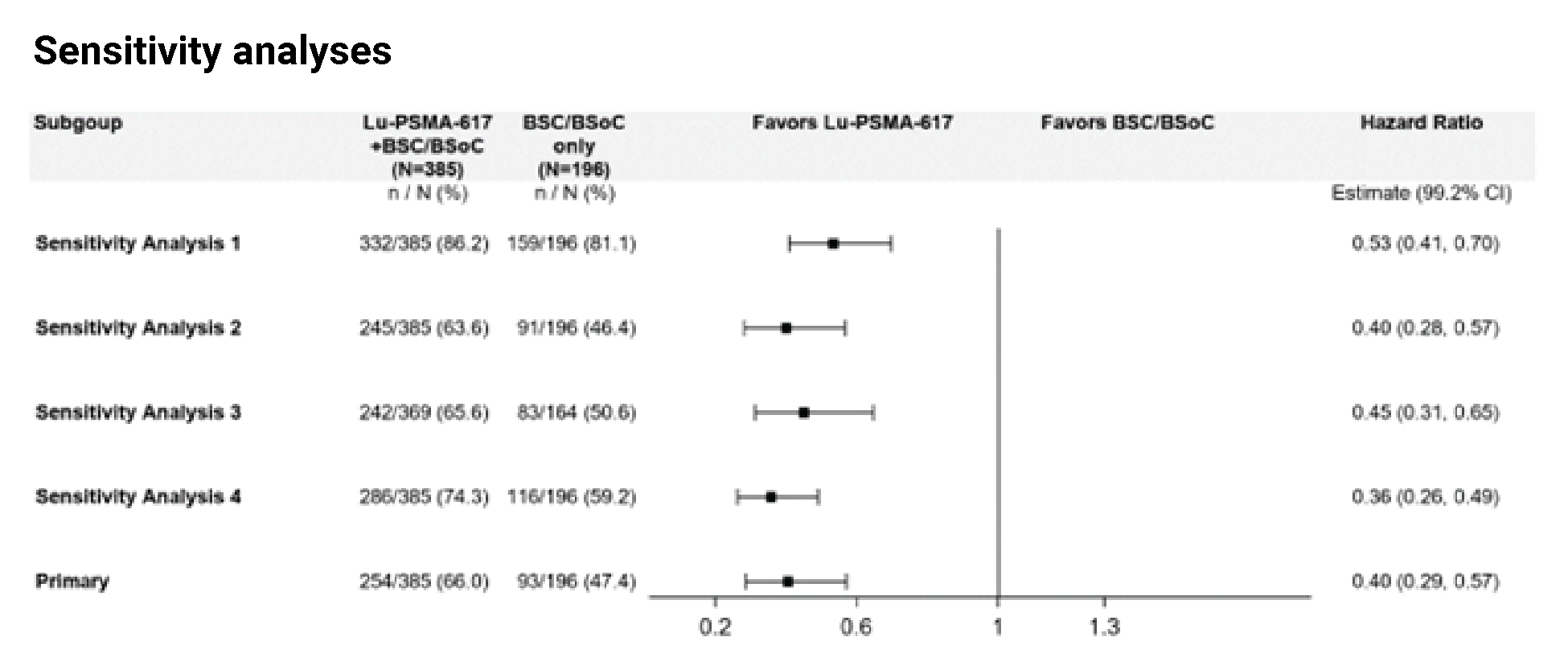
BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; FAS = full analysis set; Lu = lutetium-177; n / N = number of events/number of patients in treatment arm; PFS = progression-free survival; PSMA = prostate-specific membrane antigen; rPFS = radiographic progression-free survival.
Notes: Sensitivity analysis 1 includes events regardless of intervening missed assessments and includes all radiographic progressive disease and deaths captured in the study, including scans not centrally read; bone progressive diseases were indicated, per PCWG3 guidelines, with modified rules for confirmation after week 16.
Sensitivity analysis 2 includes deaths occurring after the start of a new anticancer therapy were censored at the start date of the new therapy.
Sensitivity analysis 3 assesses rPFS from the date of the first dose of randomized treatment.
Sensitivity analysis 4 used local investigator assessments instead of central reading.
Source: Clinical Study Report.7
Progression-Free Survival
Results for PFS are summarized in Table 20. 177Lu vipivotide tetraxetan + BSC/BSoC was associated with a statistically significant reduction in events of radiographic disease progression, clinical progression, PSA progression or death, compared to BSC/BSoC alone (HR = 0.30; 95% CI, 0.24 to 0.38; P < 0.001). Median PFS was 5.9 months (95% CI, 5.2 to 6.6 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 2.4 months (95% CI, 2.2 to 3.0 months) in the BSC/BSoC alone group.7 Kaplan-Meier curves are presented in Figure 14.
Table 20: Progression-Free Survival (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC alone (N = 196) |
|---|---|---|
██████ ████████████ ██ ██████ | ███ ██████ | ███ ██████ |
████████████ ████████████ | ███ ██████ | ██ ██████ |
████████ ████████████ | ██ ██████ | ██ ██████ |
██ ████ ████████ ██ ███ ███████ ██ ███████████ | ██ █████ | ██ █████ |
██████ █████████████ ██ ████ ██ ██ ██████ █ | █ █████ | █ █████ |
█████████ ████ ███ ███ ██████████ █████████ | █ █████ | █ █████ |
██████ ██████████ ██ ██████████████ ████ | █ █████ | █ █████ |
███ ████████████ | ███ ██████ | ██ ██████ |
██████ | ██ █████ | ██ █████ |
████████ | ██ ██████ | ██ ██████ |
███████ ███████ █████ | ██ ██████ | ██ █████ |
█████ ██████████ █████ ██ ██████ ██████ | ██ █████ | ██ ██████ |
████████ ██████████ ███ █████████ █ | █ █████ | ██ ██████ |
████████████ █████████ ████████ | ||
████ ██████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
██████ ███ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
████ ██████████ ████ ███ | ████ ████ ██ | ███ ████ ██ ████ |
███ █████ ███ | ||
█ ██████ ████ ███ | ████ █████ ██ | ████ █████ ██ |
█ ██████ ████ ███ | ████ █████ ██ | ████ ████ ██ |
██ ██████ ████ ███ | ████ █████ ██ | ███ ████ ██ █████ |
██ ████ ███ ███ | ████ █████ ██ █████ | |
█████████ ███████ | █ █████ | |
█████████ ████ ████████ █ | ||
██████ ████ ███ | ████ █████ ██ | ███ ████ ██ ████ |
███████████████ | ███ ██ ████ | ███ ██ ████ |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; FAS = full set analysis; HR = hazard ratio; PFS = progression-free survival; PSA = prostate-specific antigen.
aPatients censored without adequate postbaseline evaluations or adequate baseline assessment.
bHR for 177Lu vipivotide tetraxetan + BSC/BSoC vs. BSC/BSoC alone.
cThe Cox proportional hazards model is stratified for LDH (≤ 260 IU/L vs. > 260 IU/L); the presence of liver metastases (yes vs. no); ECOG PS (0 or 1 vs. 2); and the inclusion of NAAD in BSC/BSoC at the time of randomization (yes vs. no). Interactive Response Technology data for stratification are used.
dFollow-up time = (date of event or censoring – randomization date + 1)/30.4375, censoring for death, radiographic progression, clinical progression, or PSA progression.
Source: Clinical Study Report.7
Figure 14: Kaplan-Meier Plot of PFS (PFS-FAS) (Redacted)

BSC/BSoC = best supportive care/best standard of care; CI = confidence interval; FAS = full analysis set; Lu-PSMA-617 = lutetium-177 vipivotide tetraxetan; n/N = number of events/number of patients in treatment arm; PFS = progression-free survival.
Confidential figure removed at the sponsor’s request.
Source: Clinical Study Report.7
ORR and DOR
Table 21 summarizes the results for ORR and DOR in the VISION trial. The ORR was statistically significantly greater in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (29.8% versus 1.7%), with an OR of 24.99 (95% CI, 6.05 to 103.24). The median DOR in patients who demonstrated a response to treatment (i.e., CR or PR) was 9.8 months (95% CI, 9.1 to 11.7 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC group. Only 2 patients in the BSC/BSoC alone group demonstrated a response to treatment, and only 1 of those met the RECIST criteria for radiographic progression or death; therefore, the sponsor reported that the median DOR could not be reliably estimated for the BSC/BSoC alone group.7
Disease Control Rate
Results for the DCR are summarized in Table 22. The DCR was statistically significantly greater in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (89.0% versus 66.7%), with an OR of 5.79 (95% CI, 3.18 to 10.55; P < 0.001).7
Table 21: ORR and DOR (Response-Evaluable Analysis Set)
Response | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 319) | BSC/BSoC alone (N = 120) |
|---|---|---|
Best overall response, n (%) | ||
CR | 18 (5.6) | 0 |
PR | 77 (24.1) | 2 (1.7) |
Stable disease | 68 (21.3) | 30 (25.0) |
Non-CR and/or non-PD | 121 (37.9) | 48 (40.0) |
PD | 33 (10.3) | 35 (29.2) |
Unknown | 2 (0.6) | 5 (4.2) |
ORR | ||
Patients with response (CR + PR), n (%) | 95 (29.8) | 2 (1.7) |
Odds ratio (95% CI)a | 24.99 (6.05 to 103.24) | |
Two-sided P valuea | < 0.001 | |
DOR (months) | ||
Patients with response, n | ██ | █ |
Events (progression or death), n (%) | ██ ██████ | █ ██████ |
Radiographic progressions, n (%) | ██ ██████ | █ ██████ |
Deaths, n (%) | ██ ██████ | █ |
Censored, n (%) | ██ ██████ | █ ██████ |
Ongoing without event, n (%) | ██ ██████ | █ ██████ |
Event documented after ≥ 2 missed tumour assessments, n (%) | ██ ██████ | █ |
Kaplan-Meier estimates (months) | ||
25th percentile (95% CI) | ███ ████ ██ ████ | ████ ███ █ |
Median DOR (95% CI) | ███ ████ ██ █████ | ████ ███ █ |
75th percentile (95% CI) | ████ █████ ██ █████ | ████ ███ █ |
DOR (months), mean (SD)b | ████ ███████ | ████ ████ |
EDOR (months)b | ███ | ███ |
Ratio of EDOR (95% CI)b | █████ █████ ██ ██████ | |
Two-sided P valueb | █ █████ | |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; CR = complete response; DOR = duration of response; EDOR = expected duration of response (mean DOR × ORR); ORR = overall response rate; PD = progressive disease; PR = partial response; SD = standard deviation.
aOR of 177Lu vipivotide tetraxetan + BSC/BSoC vs. BSC/BSoC alone based on logistic regression model stratified for the following randomization factors: LDH (≤ 260 IU/L vs. > 260 IU/L); presence of liver metastases (yes vs. no); ECOG PS (0 or 1 vs. 2); and inclusion of NAAD in BSC/BSoC at the time of randomization (yes vs. no). P value based on Wald chi-square distribution.
bAnalyzed using mixture distribution methodology.
cOnly 2 patients in the BSC/BSoC alone group demonstrated a response to treatment, and only 1 of those met the RECIST for radiographic progression or death; therefore, the sponsor reported that the median DOR could not be reliably estimated for the BSC/BSoC alone group.
Source: Clinical Study Report.7
Figure 15: Forest Plots With Subgroup Analyses for rPFS (PFS-FAS)
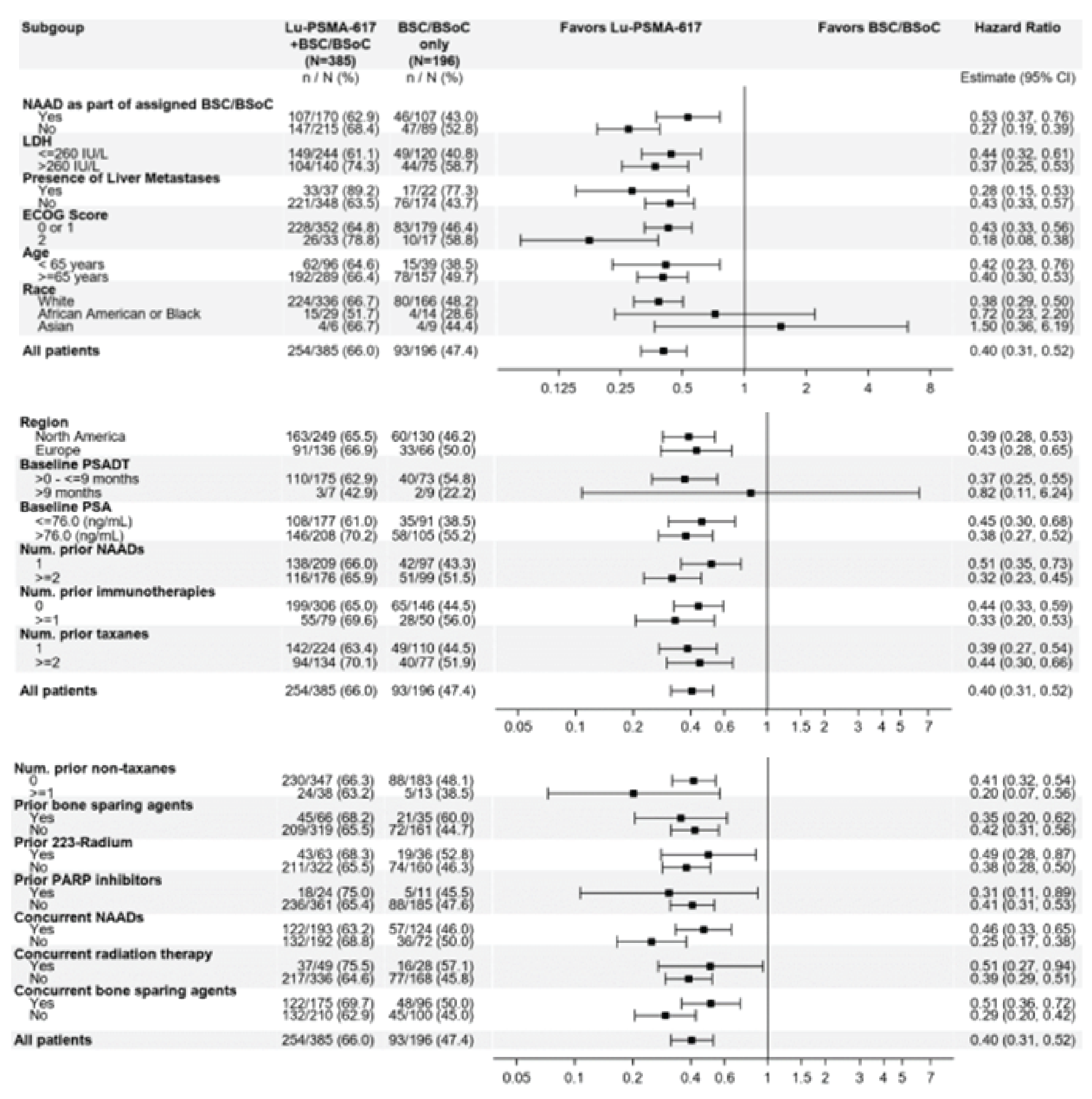
BSC = best supportive care; BSoC = best standard of care; CI = confidence interval ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; LDH = lactase dehydrogenase; Lu = lutetium-177; n / N = number of events/number of patients in treatment group; NAAD = novel androgen axis drug; PARP = poly (ADP-ribose) polymerase; PFS = progression-free survival; PSA = prostate-specific antigen; PSADT = prostate-specific antigen doubling time; PSMA = prostate-specific membrane antigen; rPFS = radiographic progression-free survival.
Source: FDA Multidiscipline Review9
Time to First SSE
Results for time to first SSE are summarized in Table 23. There were 256 events in the 177Lu vipivotide tetraxetan + BSC/BSoC group (66.5% of patients; 60 SSE events and 196 deaths) and 137 events (69.9% of patients; 34 SSE events and 103 deaths) in the BSC/BSoC alone group. 177Lu vipivotide tetraxetan + BSC/BSoC was associated with a statistically significant reduction in SSE (or death), compared to BSC/BSoC alone (HR = 0.5; 95% CI, 0.40 to 0.62). The median time to first SSE was 11.5 months (95% CI, 10.3 to 13.2 months) in the 177Lu vipivotide tetraxetan vipivotide tetraxetan + BSC/BSoC group and 6.8 months (95% CI, 5.2 to 8.5 months) in the BSC/BSoC alone group.7 Kaplan-Meier curves are presented in Figure 16.
Table 22: Disease Control Rate
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 319) | BSC/BSoC alone (N = 120) |
|---|---|---|
Patients with event (CR, PR, stable disease ≥ 6 months), n (%) | 284 (89.0) | 80 (66.7) |
Odds ratio (95% CI)a | 5.79 (3.18 to 10.55) | |
Two-sided P valuea | < 0.001 | |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; CR = complete response; PR = partial response; SD = standard deviation.
aOR for 177Lu vipivotide tetraxetan + BSC/BSoC vs. BSC/BSoC alone based on a logistic regression model that stratifying for the following randomization factors: LDH (≤ 260 IU/L vs. > 260 IU/L); presence of liver metastases (yes vs. no); ECOG PS (0 or 1 vs. 2); and inclusion of NAAD in BSC/BSoC at time of randomization (yes vs. no). P value based on Wald chi-square distribution.
Source: Clinical Study Report7
Table 23: Analysis of Time to First SSE (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC alone (N = 196) |
|---|---|---|
Events (SSE or death), n (%) | 256 (66.5) | 137 (69.9) |
SSEs | ██ ██████ | ██ ██████ |
Deaths | ███ ██████ | ███ ██████ |
Censored | ███ ██████ | ██ ██████ |
Kaplan-Meier estimates (months) | ||
25th percentile (95% CI) | ███ ████ ██ ████ | ███ ████ ██ ████ |
Median time to first SSE (95% CI) | ████ █████ ██ █████ | ███ ████ ██ ████ |
75th percentile (95% CI) | ████ █████ ██ █████ | ████ █████ ██ ███ |
First SSE rates (%) | ||
3 months (95% CI) | ████ █████ ██ █████ | ████ █████ ██ ███ |
6 months (95% CI) | ████ █████ ██ █████ | ████ █████ ██ ███ |
12 months (95% CI) | ████ █████ ██ █████ | ████ █████ ██ ███ |
HR (95% CI)a,b | 0.50 (0.40 to 0.62) | |
Stratified log-rank 2-sided P value | < 0.001 | |
Follow-up time (months)c | ||
Median (95% CI) | ████ █████ ██ █████ | ████ █████ ██ ███ |
Minimum to maximum | ███ ██ ████ | ███ ██ ████ |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; FAS = full analysis set; HR = hazard ratio; PFS = progression-free survival; SSE = symptomatic skeletal event.
aHR for 177Lu vipivotide tetraxetan + BSC/BSoC vs. BSC/BSoC.
bCox proportional hazards model is stratified for LDH (≤ 260 IU/L vs. > 260 IU/L); the presence of liver metastases (yes vs. no); ECOG PS (0 or 1 vs. 2); and inclusion of NAAD in BSC/BSoC at the time of randomization (yes vs. no).
cFollow-up time = (date of event or censoring – randomization date + 1)/30.4375, censoring for death or SSE.
Source: Clinical Study Report7
Figure 16: Kaplan-Meier Plot of Time to First SSE (PFS-FAS) (Redacted)

BSC/BSoC = best supportive care/best standard of care; CI = confidence interval; Lu-PSMA-617 = lutetium-177 vipivotide tetraxetan; n/N = number of events/number of patients in treatment arm; PFS-FAS = progression-free survival – full analysis set; SSE = symptomatic skeletal event
Note: Stratified log-rank test and stratified Cox model stratified by LDH level, presence of liver metastases, ECOG PS, and the inclusion of NAAD in BSC/BSoC at the time of randomization.
Confidential figure removed at the sponsor’s request.
Source: Clinical Study Report.7
PSA Levels
Table 24 provides results for PSA doubling time, proportion of patients with PSA response (i.e., ≥ 50% decrease from baseline), proportion of patients with a decrease from baseline of at least 80%, and duration of PSA response.
The sponsor reported a large disparity between the 2 treatment groups in the proportion of patients who could be evaluated for PSA doubling time (██████ and ██████, respectively). For the subset of patients who could be evaluated, mean PSA doubling time was ████ ██████ ████ ███ ████ ██ █████ for the 177Lu vipivotide tetraxetan + BSC/BSoC group and (████ ██████ ████ ███ ███ ██ ████) for the BSC/BSoC alone group.
Compared with the BSC/BSoC alone group, a greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group had a decrease from baseline of at least 50% than in the BSC/BSoC alone group ██████ ██████ █████ ███ ██████ ███ ███ ███ ██ █████ ██ ██████ and a decrease from baseline of at least ███ █████ ██████ █████ ███ █████ ███ ███ ███ ██ █████ ██ ███████7
Table 24: PSA Doubling Time and PSA Response (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC alone (N = 196) |
|---|---|---|
███ ████████ ████ ████████ █ | ||
████████ ██████████ ██ ████ | ███ ████ ██████ | ██ ████ ██████ |
███████ ██████████████ ██ | ███ ██████ | ██ ██████ |
██████████ | ███ ██████ | ██ ██████ |
████ ████ ███ | ████ █████ ██ █████ | ████ ████ ██ █████ |
██████ ███████ | ███ ████ ██ ██████ | ███ ████ ██ █████ |
███ ████████ █ | ||
████████ ████ ███ █████████ █ | ███ ██████ | ██ █████ |
████ █████ ████ ███ | █████ ████ ██ █████ | |
█████████ ███████ | █ █████ | |
███ ████ ████████ █ | ||
████████ ████ ███ ████████ █ | ███ ██████ | █ █████ |
████ █████ ████ ███ █ | ████ ████ ██ █████ | |
█████████ ███████ | █ █████ | |
████████ ██ ███ ████████ | ||
█ | ███ | ██ |
███████ ████ | ██ ██████ | ██ ██████ |
█████████ ████ | ██ ██████ | █ ██████ |
████████████ █████████ | ||
████ ██████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
██████ ████████ ██ ███ | ███ ████ ██ █████ | ███ ████ ██ █████ |
████ ██████████ ████ ███ | ████ █████ ██ ███ | ████ ████ ██ ███ |
███ █████████ ████ █████ | ████ ███████ | ███ ███████ |
████ ████████ | ███ | ███ |
█████ ██ ████ ████ ███ | ████ ████ ██ █████ | |
█████████ ███████ | █ █████ | |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; PFS = progression = free survival; PSA = prostate-specific antigen.
██████ ████ ████████ ██ ███ ███████ ███████ ████████ ██ ███ ████ ██████████████ █████████ ████ ████████ ████ █████████ █████ ███ ██ █████ ███ ███████████ █████████████ █████ ██████████ ███ ███████████████ ███ ████████ ████ ███ ███████ ███ ████ ███████ ██ ███ ███████ ███ ████████ ██ ███ ███ ██ ███ █████ ███ ██████ ██████ ██ ███ ██████ ███████████ ██████ █████ ███████ ███████ ███ ██ ███ ███ ████ ██ ███ ███████████ ███ ████████ ████████ ████ ████████ ███ ██ █████ ████████████ █████████████ ███ ██████ ████ ████████ ██ ███ ████████ ███ ████████ ███ ███████ ██ ███ ██████████ ██ ████████ ███ ███ ██ ███ ████████ ██ ███ ████ ████████ █████████ ██ ████ ███████████ ██ ████████
Source: Clinical Study Report.7
Brief Pain Inventory-Short Form
Time to worsening on the BPI-SF pain intensity scale is reported in Table 25. Time to worsening pain was delayed in the 177Lu vipivotide tetraxetan + BSC/BSoC group compared with the BSC/BSoC alone group (HR = 0.52; 95% CI, 0.43 to 0.63; P < 0.001). The median time to deterioration was 5.9 months (95% CI, 4.8 to 6.9 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC arm and 2.2 months (95% CI, 1.8 to 2.8 months) in the BSC/BSoC alone group.7 Kaplan-Meier curves are presented in Figure 17.
Table 25: Time to Worsening on BPI-SF Pain Intensity Scale (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC alone (N = 196) |
|---|---|---|
████ ██ █████████ ██ ███████ ████ █ | ||
██████ | ███ ██████ | ███ ██████ |
█████████ ████████ ██ ████████ | ███ ██████ | ██ ██████ |
████████ ████████████ | ███ ██████ | ██ ██████ |
██████ | ██ ██████ | ██ ██████ |
████████ | ██ ██████ | ██ ██████ |
████████████ █████████ ████████ | ||
████ ██████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
██████ ████ ██ █████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
████ ██████████ ████ ███ | ████ █████ ██ █████ | ███ ████ ██ ████ |
████ ██ █████████ █████ ███ | ||
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
██ ██████ ████ ███ | ████ █████ ██ █████ | ███ ████ ██ █████ |
██ ████ ██████ | ████ █████ ██ █████ | |
█████████ ███████ | ██████ | |
177Lu = lutetium-177; BPI-SF = Brief Pain Inventory-Short Form; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; PFS = progression = free survival.
██████ ██████ ████ █████████ ██████ █████ ████████ █████ ██████████ █████████ ████████ ██ █████ ██ ███████████ █████████ ██ ██████ ██████ ███████ █████████████████ ████████ █████ ████████ ███ ██ ████ ██ █████████ ██ ███████ ██ ████ ████ █████████████ ██ ███ █████ █████████ ██ ██ ████████ ██ █████████ █████████ █████ ██ ████████ ██ ████████ █████████ ██ ███ ████ ██ ███████ ███ █████ ████ ████ ██ █████████ ████████ ███████ ████████████ ██ ████████ ██████ █████ ██ ████████ ██ ███ ███ ██ ██████████ ██████████ ██████████ █████████ ██████ ███████████ ███ ██ █████ ██ ███ ███████ ███ ███ ██ ███ ████ ██████ ████ ██████ ████████ ██ █████ ██████████ ████ ██████ ████ ████ ███ ██ ██ ██ ███████ ███ ███ █████████ ██ ████ ██ ████ ███████████████████ ██ ████ ██ ████ ██ █████ ████████ ████ ██████ ████
Source: Clinical Study Report.7
Functional Assessment of Cancer Therapy-Prostate
Table 26 summarizes improvement, worsening, and time to worsening in FACT-P in the VISION trial. Improvement in at least 1 assessment was reported for a greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (45.7% versus 15.3%). The proportion of patients with FACT-P scores that worsened from baseline at any time was similar in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (47.5% versus 44.9%).7
Total events for time to worsening in FACT-P scores were similar in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (87.0% and 85.7%, respectively). Median time to worsening was shorter in those who received 177Lu vipivotide tetraxetan + BSC/BSoC (5.7 months; 95% CI, 4.8 to 6.6 months) than in those who received BSC/BSoC alone (2.2 months; 95% CI, 1.8 to 2.8 months), with a HR of 0.54 (95% CI, 0.45 to 0.66; P < 0.001).7
Figure 17: Kaplan-Meier Plot of Time to Worsening on BPI-SF Pain Intensity Scale (PFS-FAS)
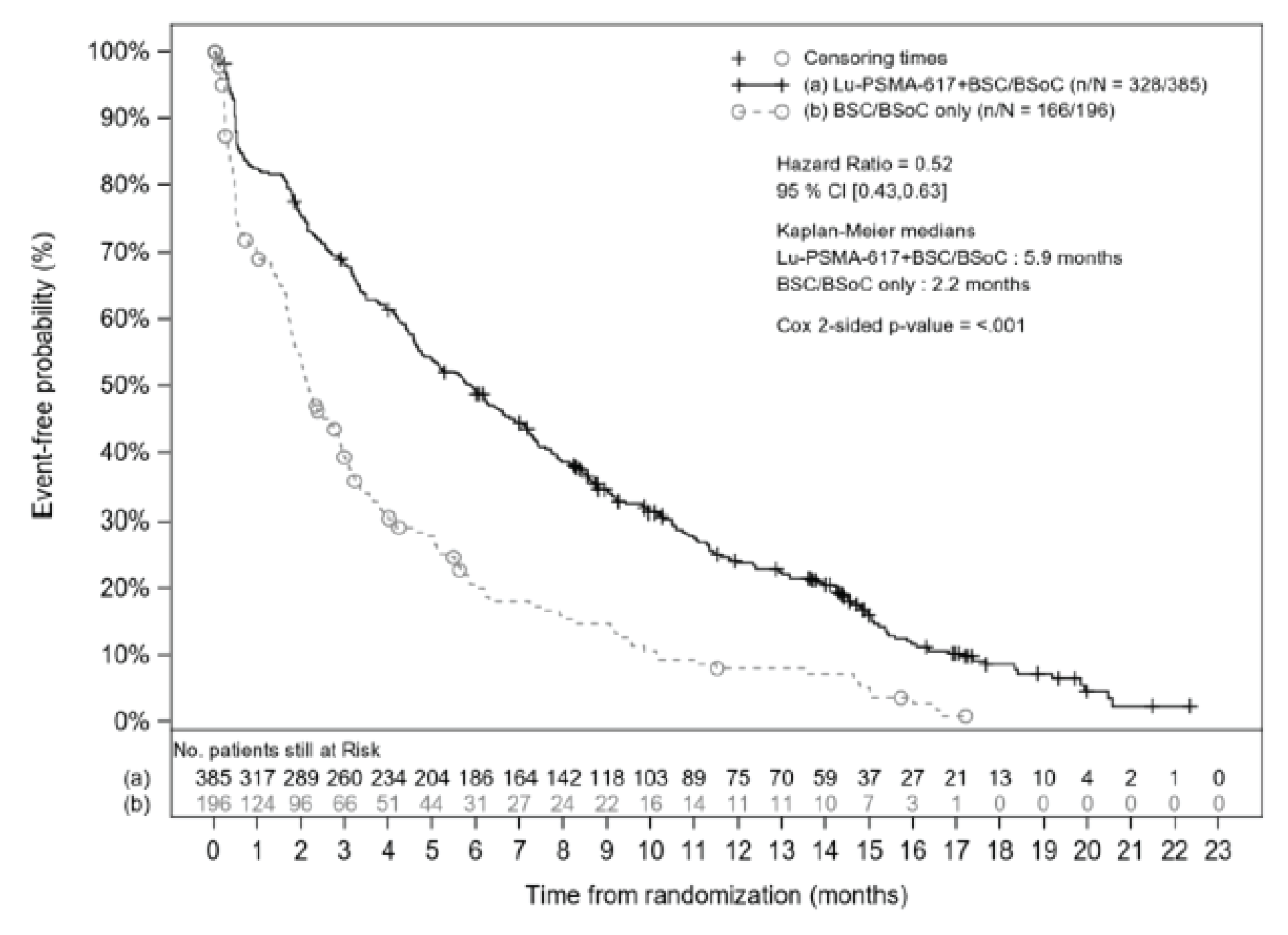
BPI-SF = Brief Pain Inventory-Short Form; BSC = best supportive care; BSoC = best standard of care; CI = confidence interval; FAS = full analysis set; Lu-PSMA-617 = lutetium-177 vipivotide tetraxetan; n/N = number of events/number of patients in treatment arm; PFS = progression-free survival.
Note: Log-rank test and Cox model stratified by LDH level, presence of liver metastases, ECOG PS, and inclusion of NAAD in BSC/BSoC at the time of randomization.
Source: Clinical Study Report.7
Table 26: Improvement and Worsening From Baseline and Time to Worsening in FACT-P Total Score (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC alone (N = 196) |
|---|---|---|
███████████ ████████ ██ █████████ █████ | ███ ██████ | ██ ██████ |
█████████ ████████ ██ █████████ █████ | ███ ██████ | ██ ██████ |
████ ██ ██████████ ████ █ | ||
██████ | ███ ██████ | ███ ██████ |
█████████ ████████ ██ ████████ | ███ ██████ | ██ ██████ |
████████ ████████████ | ███ ██████ | ██ ██████ |
██████ | ██ ██████ | ██ ██████ |
████████ | ██ ██████ | ██ ██████ |
████████████ █████████ ████████ | ||
████ ██████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
██████ ████ ██ █████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
████ ██████████ ████ ███ | ████ █████ ██ █████ | ███ ████ ██ ████ |
████ ██ █████████ █████ ███ | ||
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
██ ██████ ████ ███ | ████ █████ ██ █████ | ████ ████ ██ █████ |
██ ████ ██████ | ████ █████ ██ █████ | |
█████████ ███████ | ██████ | |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; FACT-P = Functional Assessment of Cancer Therapy-Prostate FAS = full analysis set; PFS = progression = free survival.
████████ █████ ██████████ █████████ ████████ ██ █████ ██ ███████████ █████████ ██████ █ ██████████ ██████████ ██ ██████ ███████ ██████████ ██ ███████ ██████ ██ ██████ ██ ████████ ████ ██████ ███████ █████████████████ ████████ █████ ████████ █████ ███████████ ██ ███████ ██ ██ ████████ ████████ ██ ████████ ██ ██████ █████ █████ ██ ███ ███ ████ ██ ███████ ███ ████████ █████████ ██ ███████ ██ █████████ ████████ ██ ████████ ██ ██████ █████ █████ ██ ███ ███ ████ ██ ███████ ███ ████████ ████ ██ █████████ ██ ███████ ██ ████ ████ █████████████ ██ ███ █████ █████████ ██ ████ █████ ████████ ██ ██████ █████ █████ ████████ ██ █████████ ████████ ███████ ████████████ ██ ████████ ██████ █████ ██ █████ ██████████ ██████████ █████████ ███ ███████████ ███ ██ █████ ██ ██████████ ██ ███ ██████ ████████ ██ █████ ███████████ ████ █████ ███ █████████ ██ ████ ██ ████████ ██ ████ ██ ██████████████
Source: Clinical Study Report.7
Functional Assessment of Cancer Therapy-General
Table 26 summarizes improvement, worsening, and time to worsening in FACT-G in the VISION trial. Among those assessed with FACT-G, at least 1 assessment of improvement or worsening was reported for a greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (39.2% versus 14.8%). The proportion of patients with FACT-G scores that worsened from baseline were was in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (43.1% versus 48.0%).7 Total events for time to worsening in FACT-G scores were similar in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (85.5% and 85.2%, respectively). Median time to worsening was shorter in those who received 177Lu vipivotide tetraxetan + BSC/BSoC (6.6 months; 95% CI, 5.5 to 7.3 months) than in those who received BSC/BSoC alone (2.4 months; 95% CI, 2.0 to 3.1 months), with a HR of 0.53 (95% CI, 0.44 to 0.65; P < 0.001).7
Table 27: Improvement and Worsening From Baseline and Time to Worsening in FACT-G Total Score (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC alone (N = 196) |
|---|---|---|
███████████ ████████ ██ | ███ ██████ | ██ ██████ |
█████████ ████████ ██ | ███ ██████ | ██ ██████ |
████ ██ ██████████ ████ █ | ||
██████ | ███ ██████ | ███ ██████ |
█████████ ████████ ██ | ███ ██████ | ██ ██████ |
████████ ████████████ | ███ ██████ | ██ ██████ |
██████ | ██ ██████ | ██ ██████ |
████████ | ██ ██████ | ██ ██████ |
████████████ █████████ ████████ | ||
████ ██████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
██████ ████ ██ █████████ | ███ ████ ██ ████ | ███ ████ ██ ████ |
████ ██████████ ████ ███ | ████ █████ ██ █████ | ███ ████ ██ ████ |
████ ██ █████████ █████ ███ | ||
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
██ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
██ ████ ██████ | ████ █████ ██ █████ | |
█████████ ███████ | ██████ | |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; FACT-G = Functional Assessment of Cancer Therapy-General; FAS = full analysis set; PFS = progression = free survival.
████████ █████ ██████████ █████████ ████████ ██ █████ ██ ███████████ █████████ ██████ ██████████ ██████████ ██ ██████ ███████ █████████ ██ ███████ ██████ ██ ██████ ██ ████████ ████ █████████████ ██ ███████ ██ ██ ████████ ████████ ██ ████████ ██ ██████ █████ █████ ██ ██ ███ ████ ██ ███████ ███ ████████ █████████ ██ ███████ ██ █████████ ████████ ██ ████████ ██ ██████ █████ █████ ██ ██ ███ ████ ██ ███████ ███ ████████ ████ ██ █████████ ██ ███████ ██ ████ ████ █████████████ ██ ███ █████ █████████ ██ ███ █████ ████████ ██ ██████ █████ █████ ████████ ██ █████████ ████████ ███████ ████████████ ██ ████████ ██████ █████ ██ █████ ██████████ ██████████ █████████ ██████ ███████████ ███ ██ █████ ██ ██████████ ██ ███ ██████ ████████ ██ █████ ███████████ ████ █████ ███ █████████ ██ ████ ██ ████████ ██ ████ ██ ██████████████
Source: Clinical Study Report7
Eight-Item Functional Assessment of Cancer Therapy Advanced Prostate Symptoms Index
Table 26 summarizes improvement, worsening, and time to worsening on FAPSI-8 in the VISION trial. Improvement in at least 1 assessment was reported for a greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC group (50.6% versus 26.0%). Worsening on FAPSI-8 was defined as a decrease from baseline of at least 3 points at any time up to the EOT visit. The proportion of patients with FAPSI-8 scores that worsened from baseline was the same in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (42.3% for both).7
Total events for time to worsening were nearly identical in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (86.0% and 86.2%, respectively). Median time to worsening on FAPSI-8 was shorter in those who received 177Lu vipivotide tetraxetan + BSC/BSoC (5.9; 95% CI, 4.8 to 6.9 months) than in those who received BSC/BSoC alone group (2.0 months; 95% CI, 1.7 to 2.6 months), with an HR of 0.52 (95% CI, 0.43 to 0.64; P < 0.001).7
Table 28: Improvement and Worsening From Baseline and Time to Worsening in FAPSI-8 Score (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC alone (N = 196) |
|---|---|---|
███████████ ████████ ██ | ███ ██████ | ██ ██████ |
█████████ ████████ ██ █ | ███ ██████ | ██ ██████ |
████ ██ ██████████ ████ █ | ||
██████ | ███ ██████ | ███ ██████ |
█████████ ████████ | ███ ██████ | ██ ██████ |
████████ ████████████ | ███ ██████ | ██ ██████ |
██████ | ██ ██████ | ██ ██████ |
████████ | ██ ██████ | ██ ██████ |
████████████ █████████ ████████ | ||
████ ██████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
██████ ████ ██ ██████ | ███ ████ ██ ████ | ███ ████ ██ ████ |
████ ██████████ ███████ | ████ █████ ██ █████ | ███ ████ ██ ████ |
████ ██ █████████ █████ ███ | ||
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
██ ██████ ████ ███ | ████ █████ ██ █████ | ███ ████ ██ █████ |
██ ████ ██████ | ████ █████ ██ █████ | |
█████████ ███████ | ██████ | |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; FAPSI-8 = 8-item Functional Assessment of Cancer Therapy Advanced Prostate Symptoms Index; PFS = progression = free survival.
████████ █████ ██████████ █████████ ████████ ██ █████ ██ ███████████ █████████ █████████████████ ████████ ███████ ████████ ██ ███████ ██████ ██ ██████ ██ ████████ ████ ██████ ███████████████████ ████████ █████ ████████ █████ ███████████ ██ ███████ ██ ██ ███████████ ████████ ██ ████████ ██ ██ █████ ████████ ██ ███ ████ ██ ███████ ███ ████████ █████████ ██ ███████ ██ █████████ ████ ████████ ██ ██ █████ ████████ ██ ███ ████ ██ ███████ ███ ████████ ████ ██ █████████ ██
Source: Clinical Study Report.7
Five-Level EQ-5D
Table 29 provides a summary of improvement and worsening relative to baseline and time to worsening on the EQ-5D-5L utility score.
Harms
Only harms identified in the review protocol are reported here. Table 30 provides a summary of AEs reported in the VISION trial.
Table 29: Improvement and Worsening in EQ-5D-5L Utility Score (PFS-FAS)
Analysis | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 385) | BSC/BSoC alone (N = 196) |
|---|---|---|
███████████ ████████ ██ █████████ ████ | ███ █████ ██ █████ | ██ █████ ██ █████ |
█████████ ████████ ██ █████████ ████ | ███ █████ ██ █████ | ███ █████ ██ █████ |
████ ██ ██████████ ████ █ | ||
██████ | ███ ██████ | ███ ██████ |
█████████ ████████ ██ ████████ | ███ ██████ | ███ ██████ |
████████ ████████████ | ██ ██████ | ██ █████ |
██████ | ██ ██████ | ██ ██████ |
████████ | ██ █████ | ██ ██████ |
████████████ █████████ ████████ | ||
████ ██████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
██████ ████ ██ █████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
████ ██████████ ████ ███ | ███ ████ ██ ████ | ███ ████ ██ ████ |
████ ██ █████████ █████ ███ | ||
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ █████ ██ █████ |
█ ██████ ████ ███ | ████ █████ ██ █████ | ████ ████ ██ █████ |
██ ██████ ████ ███ | ████ ████ ██ █████ | ███ ████ ██ ████ |
██████ █████ ████ ██████ | ████ █████ ██ █████ | |
█████████ ███████ | ██████ | |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; EQ-ED-5L = 5 Level EQ-5D; FAS = full analysis set; PFS = progression = free survival.
████████ █████ ██████████ █████████ ████████ ██ █████ ██ ███████████ █████████ ████████ ████████ ███████████ ██████ ██ ███████ ██████ ██ ██████ ██ ████████ ████ ██████ ███████ ██ █████ █ ██ ██████ ████████ █████ ████████ █████ ███████████ ████████████ ████████ ██ ████████ ██ ██████ █ █████████ ██ ███ █████ ████████ ██ ███ ████ ██ ███████ ███ ██ █████████ ████████ █████████ ████████████████ ██ ████████ ██ ███████ █████ █████████ ██ ██ ██████ ██ ███ ████████ ██ ███ ████ ██ █████ ██ ███ ██ █████████ ████████ ████ ██ █████████ ███ ███████ █████ ██ ███████ ██ ████ ████ █████ ████████ ██ ███ █████ ██████████ ██ █████████ ██ ███████ █████ ████████ ██ ████████
Source: Clinical Study Report.7
Table 30: Summary of AEs (FAS-SAS)
Summary of AEs, n (%) | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | BSC/BSoC alone (N = 205) |
|---|---|---|
All AEs | 519 (98.1) | 170 (82.9) |
SAEs | 192 (36.3) | 57 (27.8) |
Grade 3, 4, or 5 AEs | 279 (52.7) | 78 (38.0) |
AEs leading to reduction of 177Lu vipivotide tetraxetan | 30 (5.7) | 0 |
AEs leading to reduction of BSC/BSoC | 17 (3.2) | 7 (3.4) |
AEs leading to interruption of 177Lu vipivotide tetraxetan | 85 (16.1) | 2 (1.0) |
AEs leading to interruption of BSC/BSoC | 50 (9.5) | 14 (6.8) |
AEs leading to discontinuation of 177Lu vipivotide tetraxetan | 63 (11.9) | 1 (0.5) |
AEs leading to discontinuation of BSC/BSoC | 45 (8.5) | 16 (7.8) |
Fatal AEs | 19 (3.6) | 6 (2.9) |
177Lu = lutetium-177; AE = adverse event; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; SAE = serious adverse event; SAS = safety analysis set.
Source: Clinical Study Report.7
Adverse Events
Table 31 provides a summary of AEs that were reported for at least 5% of the patients in either treatment group. At least 1 AE was reported for a greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (98.1% versus 82.9%). The sponsor reported that the following events were more common (i.e., a difference of ≥ 10.0% between groups) with 177Lu vipivotide tetraxetan + BSC/BSoC than with BSC/BSoC alone: fatigue (43.1% versus 22.9%), dry mouth (38.8% versus 0.5%), nausea (35.3% versus 16.6%), anemia (31.8% versus 13.2%), diarrhea (18.9% versus 2.9%), vomiting (18.9% versus 6.3%), thrombocytopenia (17.2% versus 4.4%), lymphopenia (14.2% versus 3.9%), leucopenia (12.5% versus 2.0%), and urinary tract infection (11.0% versus 1.0%).7
A greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group reported at least 1 AE of at least grade 3 than in the BSC/BSoC alone group (52.7% versus 38.0%). Events of at least grade 3 that were more commonly reported in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group included the following: anemia (12.9% versus 4.9%), thrombocytopenia (7.9% versus1.0%), lymphopenia (7.8% versus 0.5%), and fatigue (5.9% versus 1.5%). Spinal cord compression was reported more commonly in the BSC/BSoC alone group than in the 177Lu vipivotide tetraxetan + BSC/BSoC group (5.4% versus 1.3%).7
Serious Adverse Events
Table 32 provides a summary of SAEs that were reported for at least 3 patients in either treatment group. At least 1 SAE was reported for a greater proportion of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group (36.3% versus 27.8%). As previous noted, spinal cord compression was reported more commonly in the BSC/BSoC alone group than in the 177Lu vipivotide tetraxetan + BSC/BSoC alone group.7
Withdrawals, Interruptions, and Reductions Due to Adverse Events
Table 33 summarizes AEs that led to permanent discontinuation of 177Lu vipivotide tetraxetan or required a dosage interruption or reduction to manage.
Table 31: AEs Occurring in ≥ 5% of Patients in Either Arm (FAS-SAS)
Preferred term | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | BSC/BSoC alone (N = 205) | ||
|---|---|---|---|---|
All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | |
Patients with ≥ 1 event, n (%) | 519 (98.1) | 279 (52.7) | 170 (82.9) | 78 (38.0) |
Fatigue | 228 (43.1) | 31 (5.9) | 47 (22.9) | 3 (1.5) |
Dry mouth | 205 (38.8) | 0 | 1 (0.5) | 0 |
Nausea | 187 (35.3) | 7 (1.3) | 34 (16.6) | 1 (0.5) |
Anemia | 168 (31.8) | 68 (12.9) | 27 (13.2) | 10 (4.9) |
Back pain | 124 (23.4) | 17 (3.2) | 30 (14.6) | 7 (3.4) |
Arthralgia | 118 (22.3) | 6 (1.1) | 26 (12.7) | 1 (0.5) |
Decreased appetite | 112 (21.2) | 10 (1.9) | 30 (14.6) | 1 (0.5) |
Constipation | 107 (20.2) | 6 (1.1) | 23 (11.2) | 1 (0.5) |
Diarrhea | 100 (18.9) | 4 (0.8) | 6 (2.9) | 1 (0.5) |
Vomiting | 100 (18.9) | 5 (0.9) | 13 (6.3) | 1 (0.5) |
Thrombocytopenia | 91 (17.2) | 42 (7.9) | 9 (4.4) | 2 (1.0) |
Lymphopenia | 75 (14.2) | 41 (7.8) | 8 (3.9) | 1 (0.5) |
Leukopenia | 66 (12.5) | 13 (2.5) | 4 (2.0) | 1 (0.5) |
Bone pain | 59 (11.2) | 13 (2.5) | 17 (8.3) | 5 (2.4) |
Urinary tract infection | 58 (11.0) | 20 (3.8) | 2 (1.0) | 1 (0.5) |
Weight decreased | 57 (10.8) | 2 (0.4) | 18 (8.8) | 0 |
Dyspnea | 53 (10.0) | 7 (1.3) | 20 (9.8) | 3 (1.5) |
Peripheral edema | 51 (9.6) | 2 (0.4) | 13 (6.3) | 0 |
Hematuria | 45 (8.5) | 13 (2.5) | 9 (4.4) | 1 (0.5) |
Neutropenia | 45 (8.5) | 18 (3.4) | 3 (1.5) | 1 (0.5) |
Pain in extremity | 45 (8.5) | 3 (0.6) | 12 (5.9) | 0 |
Dizziness | 44 (8.3) | 5 (0.9) | 9 (4.4) | 0 |
Cough | 42 (7.9) | 0 | 13 (6.3) | 0 |
Hypokalemia | 40 (7.6) | 5 (0.9) | 8 (3.9) | 0 |
Fall | 38 (7.2) | 1 (0.2) | 12 (5.9) | 2 (1.0) |
Headache | 37 (7.0) | 4 (0.8) | 4 (2.0) | 0 |
Hypocalcemia | 36 (6.8) | 4 (0.8) | 7 (3.4) | 1 (0.5) |
Pyrexia | 36 (6.8) | 2 (0.4) | 7 (3.4) | 0 |
Asthenia | 34 (6.4) | 6 (1.1) | 16 (7.8) | 2 (1.0) |
Pain | 33 (6.2) | 7 (1.3) | 9 (4.4) | 1 (0.5) |
Abdominal pain | 32 (6.0) | 5 (0.9) | 7 (3.4) | 1 (0.5) |
Hypertension | 30 (5.7) | 17 (3.2) | 12 (5.9) | 3 (1.5) |
Blood creatinine increase | 28 (5.3) | 1 (0.2) | 5 (2.4) | 1 (0.5) |
Hypophosphatemia | 28 (5.3) | 5 (0.9) | 7 (3.4) | 1 (0.5) |
Insomnia | 28 (5.3) | 0 | 9 (4.4) | 0 |
Spinal cord compression | 7 (1.3) | 7 (1.3) | 11 (5.4) | 11 (5.4) |
177Lu = lutetium-177; AE = adverse event; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; SAS = safety analysis set.
Source: Clinical Study Report.7
Table 32: SAEs Occurring in ≥ 3 Patients in Either Arm (FAS-SAS)
Preferred term, n (%) | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | BSC/BSoC alone (N = 205) | ||
|---|---|---|---|---|
All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | |
████████ ████ ██ █████ | ███ ██████ | ███ ██████ | ██ ██████ | ██ ██████ |
███████ | ██ █████ | ██ █████ | █ █████ | █ |
███████ █████ █████████ | ██ █████ | ██ █████ | █ █████ | █ █████ |
██████████ | ██ █████ | ██ █████ | █ █████ | █ █████ |
██████ | ██ █████ | █ █████ | █ █████ | █ █████ |
█████ ██████ ██████ | █ █████ | █ █████ | █ █████ | █ █████ |
████ ████ | █ █████ | █ █████ | █ █████ | █ █████ |
█████████ | █ █████ | █ █████ | █ █████ | █ █████ |
███████ | █ █████ | █ █████ | █ | █ |
████ ████ | █ █████ | █ █████ | █ █████ | █ █████ |
████████████ | █ █████ | █ █████ | █ | █ |
█████████ ████████ | █ █████ | █ █████ | █ █████ | █ █████ |
██████ ████ ███████████ | █ █████ | █ █████ | ██ █████ | ██ █████ |
████████████ | █ █████ | █ █████ | █ █████ | █ █████ |
███████████ | █ █████ | █ █████ | █ █████ | █ █████ |
████████ | █ █████ | █ █████ | █ █████ | █ █████ |
████ | █ █████ | █ █████ | █ █████ | █ █████ |
███████ █████████ | █ █████ | █ █████ | █ █████ | █ █████ |
████████ | █ █████ | █ █████ | █ █████ | █ █████ |
█████████ ████ | █ █████ | █ █████ | █ █████ | █ █████ |
███████████ | █ █████ | █ █████ | █ | █ |
████████ █████████ | █ █████ | █ █████ | █ █████ | █ █████ |
███████ | █ █████ | █ █████ | █ | █ |
███████ █████ ███████████ | █ █████ | █ █████ | █ | █ |
████ ████ ██████████ | █ █████ | █ █████ | █ | █ |
█████████ | █ █████ | █ █████ | █ █████ | █ █████ |
█████████ ██████ | █ █████ | █ █████ | █ | █ |
██████ ██████ ███████ | █ █████ | █ █████ | █ | █ |
██████ | █ █████ | █ █████ | █ █████ | █ █████ |
████████████████ | █ █████ | █ █████ | █ | █ |
█████████ | █ █████ | █ █████ | █ | █ |
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; SAE = serious adverse event; SAS = safety analysis set.
Source: Clinical Study Report.7
Table 33: AEs Leading to Discontinuation, Dose Interruption, or Dose Reduction (FAS-SAS)
Preferred term, n (%) | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | |
|---|---|---|
All grades | Grade ≥ 3 | |
███████████ ███ ██ ███ | ||
████████ ████ ██ █████ | ██ ██████ | ██ █████ |
███████ | ██ █████ | █ █████ |
████████████████ | ██ █████ | ██ █████ |
██████████ | █ █████ | █ █████ |
███████████ | █ █████ | █ █████ |
████████████ | █ █████ | █ █████ |
███████ | █ █████ | █ █████ |
██████████ | █ █████ | █ █████ |
███████████ | █ █████ | █ █████ |
█████████ | █ █████ | █ █████ |
██████████ ████████████████ | █ █████ | █ █████ |
██████ █████████ | █ █████ | █ |
█████ ███████ ███████ | █ █████ | █ █████ |
██████████ | █ █████ | █ █████ |
███████ | █ █████ | █ |
█████ ██████████ █████████ | █ █████ | █ |
████ ████ | █ █████ | █ |
███████ ███████████ | █ █████ | █ █████ |
███ █████ | █ █████ | █ |
████████ | █ █████ | █ █████ |
███ ████████ | █ █████ | █ |
████ | █ █████ | █ |
█████████████████████████ | █ █████ | █ █████ |
████████ | █ █████ | █ |
██████████ ██ ███████ ███████ | █ █████ | █ █████ |
██████ ██████████ | █ █████ | █ |
██████ | █ █████ | █ █████ |
████ █████ | █ █████ | █ |
██████ ████ ███████████ | █ █████ | █ █████ |
████████ █████████ | █ █████ | █ █████ |
███████ █████ █████████ | █ █████ | █ █████ |
████████ | █ █████ | █ |
███ ███████ ██ ████████████ ██ █████ ██ ████████ | ||
████████ ████ ██ █████ | ██ ██████ | ██ █████ |
███████ | ██ █████ | █ █████ |
████████████████ | ██ █████ | █ █████ |
██████████ | █ █████ | █ █████ |
███████████ | █ █████ | █ █████ |
███ █████████ | █ █████ | █ █████ |
██████████ | █ █████ | █ █████ |
███ ███████ ██ █████████ ██ █████ ██ ████████ | ||
████████ ████ ██ █████ | ██ █████ | ██ █████ |
████████████████ | ██ █████ | █ █████ |
███████ | █ █████ | █ █████ |
███ █████ | █ █████ | █ |
██████████ | █ █████ | █ █████ |
███████████ | █ █████ | █ █████ |
177Lu = lutetium-177; AE = adverse event; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; SAS = safety analysis set.
███ ████████ ███████ ███ ██████████ █████████████████ ████████ █████ ██████████ █████████ ████████ ██ █████ ███████ █████ ████████ ███ ███████ ████████ ███
Source: Clinical Study Report7
Mortality on Study Treatment
Overall, 66 patients (12.5%) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 19 patients (9.3%) in the BSC/BSoC alone group died while on the study treatments. Disease progression was the most frequently cited cause of death in both the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (8.3% and 6.8%, respectively). The primary cause of death was cited as an AE related to the study treatments for 17 patients (3.2%) in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 4 patients (2.0%) in the BSC/BSoC alone group.7
Notable Harms
Table 34 provides a summary of the AEs of special interest for 177Lu vipivotide tetraxetan.
Table 34: Summary of AEs of Special Interest (FAS-SAS)
Preferred term, n (%) | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | BSC/BSoC alone (N = 205) | ||
|---|---|---|---|---|
All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | |
███████ | ███ ██████ | ██ █████ | ██ ██████ | █ █████ |
████████████████ | ███ ██████ | ███ ██████ | ██ ██████ | ██ █████ |
███ █████ | ███ ██████ | █ | █ █████ | █ |
██████ ███ ████████ | ███ ██████ | █ █████ | ██ ██████ | █ █████ |
██████████████ | ██ ██████ | ██ █████ | ██ █████ | █ █████ |
█████ ███████ | ██ █████ | ██ █████ | ██ █████ | █ █████ |
██ ████████████ | █ █████ | █ █████ | █ █████ | █ █████ |
177Lu = lutetium-177; AE = adverse event; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; SAS = safety analysis set.
Source: Clinical Study Report.7
Myelosuppression
The sponsor included myelosuppression as a prespecified AE of special interest for the VISION trial, and the results are reported in Table 35. The sponsor noted that AEs of special interest included multiple event terms covering all blood cell lines. Total events were more commonly reported with 177Lu vipivotide tetraxetan + BSC/BSoC than with BSC/BSoC alone (47.4% versus 17.6%).
The clinical experts consulted by CADTH identified the following events as being important for this review: anemia requiring transfusion with packed red blood cells; systemic infections requiring antimicrobial treatment; and clinically significant bleeds. The sponsor reported that 19.2% and 6.6% of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups, respectively, received transfusions with packed red blood cells during the VISION trial. AE data were not specifically reported for systemic infections requiring antimicrobial treatment or clinically significant bleeds.
Table 35: Summary of Myelosuppression AEs (FAS-SAS)
Myelosuppression AEs, n (%) | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | BSC/BSoC alone (N = 205) |
|---|---|---|
████████ ████ ██ █████ ███ █████ | ███ ██████ | ██ ██████ |
███████ | ███ ██████ | ██ ██████ |
████████████████ | ██ ██████ | █ █████ |
███████████ | ██ ██████ | █ █████ |
██████████ | ██ ██████ | █ █████ |
███████████ | ██ █████ | █ █████ |
████████████ | █ █████ | █ |
███████ ███████████ | █ █████ | █ |
███████████ | █ █████ | █ |
████ ██████ ███████ | █ █████ | █ |
██████████ ███████ | █ █████ | █ |
███████ █████ | ||
█████ ████ | ███ ██████ | ██ █████ |
█████ ████ | ██ █████ | █ |
█████ ████ | █ █████ | █ |
████ | ██ █████ | █ █████ |
177Lu = lutetium-177; AE = adverse event; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; SAS = safety analysis set.
Source: Clinical Study Report.7
Renal AEs
The sponsor included renal AEs as a prespecified AE of special interest for the VISION trial, and the results are reported in Table 36. Total renal AEs were reported for 8.7% and 5.9% of those in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups, respectively.
Table 36: Summary of Renal AEs (FAS-SAS)
Renal AEs, n (%) | 177Lu vipivotide tetraxetan + BSC/BSoC (N = 529) | BSC/BSoC alone (N = 205) |
|---|---|---|
████████ ████ ██ █████ ███ | ██ █████ | ██ █████ |
█████ ██████████ █████████ | ██ █████ | █ █████ |
█████ ██████ ██████ | ██ █████ | █ █████ |
█████ ████ █████████ | █ █████ | █ |
███████████ | █ █████ | █ |
█████ ███████ | █ █████ | █ |
█████ ██████ █████████ | █ █████ | █ █████ |
███████ █████ | ||
█████ ████ | ██ █████ | █ █████ |
█████ ████ | █ | █ |
█████ ████ | █ | █ |
████ | █ █████ | █ █████ |
177Lu = lutetium-177; AE = adverse event; BSC = best supportive care; BSoC = best standard of care; FAS = full analysis set; SAS = safety analysis set.
Source: Clinical Study Report.7
Critical Appraisal
Internal Validity
Allocation: Randomization in the VISION was performed using an appropriate methodology with adequate allocation concealment (i.e., Interactive Response Technology). Randomization was stratified by LDH level at screening, the presence of liver metastases, ECOG PS, and the inclusion of NAADs in BSC/BSoC. Health Canada and the clinical experts consulted by reviewers noted that the stratification variables are important prognostic factors and would increase the balance in the treatment groups. Overall, baseline and demographic characteristics were generally well balanced across the 177Lu vipivotide tetraxetan and BSC groups in the VISION trial.
The treatment groups were well balanced in the FAS and PFS-FAS populations with respect to prior cancer-related surgery, prior radiation therapy, and prior systemic anticancer therapy. All patients in the VISION trial had received at least 1 prior systemic anticancer therapy and all patients had received at least 1 prior ARPI and taxane-based chemotherapy. Health Canada noted that the proportion of patients who had received 2 prior NAAD therapies was slightly higher in the BSC/BSoC alone group than in the 177Lu vipivotide tetraxetan + BSC/BSoC group (45.7% versus 38.7%), as was the proportion that received 2 prior taxane therapies (43.6% versus 39.9%). CADTH reviewers agreed with the Health Canada assessment that these differences were not likely to affect interpretation of VISION trial results.
Blinding: The sponsor reported that the open-label design was used because blinding would not be practical because of the specialized precautions required for administration of a radiopharmaceutical and the toxicities related to targeted radioligand therapy.7 In addition, the trial protocol noted that it would not be appropriate to subject patients who did not receive a radiopharmaceutical to the posttreatment radiation protection protocols (e.g., maintaining physical distancing from family members). Radiographic images were evaluated using blinded independent central review, and those results were used in the primary evaluations of rPFS and ORR (local assessments were used for patient management and in sensitivity analyses).7 Regulatory authorities agreed with the sponsor that blinding would not be feasible or ethical in the VISION trial. In addition, it was noted that OS is an objective end point and would not be subject to bias in the open-label administration of the study drugs.8,9
The open-label study design contributed to the high rate of early withdrawal for those who were randomized to BSC/BSoC alone. The sponsor noted that patients were often disappointed at not receiving 177Lu vipivotide tetraxetan, leading to a lack of willingness to comply with the study protocol and/or interest in receiving therapies that were prohibited in the study protocol (e.g., taxanes).
Comparator: In the VISION trial, 177Lu vipivotide tetraxetan was administered with BSC/BSoC (which included systemic anticancer therapies) and the comparator group was BSC/BSoC alone.7 As shown in Table 15, systemic standard of care therapies were wide ranging and generally balanced in the 2 treatment groups. Health Canada reviewers commented on the wide range of BSC/BSoC therapies that were used in the VISION trial, which ranged from ARPIs to palliative care. They further noted that none of the standard of care options included n the permitted BSC/BSoC options have been shown to improve OS for patients with mCRPC with progressive disease after ARPI and 1 or 2 taxane regimens.8 Re-treatment with an ARPI was not considered to be a relevant comparator by the clinical experts consulted by CADTH (as patients would have previously been treated with an ARPI to be considered a candidate for 177Lu vipivotide tetraxetan, based on the approved indication). This is similarly reflected in the proposed place in therapy for 177Lu vipivotide tetraxetan that was submitted by the sponsor (Figure 4), which underscores the fact that many Canadian jurisdictions limit reimbursement to a single ARPI.
The lack of an active comparator group, particularly cabazitaxel, was noted as an important limitation of the VISION trial by regulatory authorities and the clinical experts consulted by CADTH. To try to address this gap in the evidence, CADTH summarized the phase II TheraP trial (refer to the Other Relevant Evidence section) and the sponsor submitted an indirect comparison (refer to the Indirect Evidence section).
Concomitant systemic anticancer therapies: To limit potential confounding, randomization was stratified by baseline use of an ARPI. During the VISION trial, 52.6% and 67.8% of patients received concomitant treatment with ARPIs in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups, respectively.7 The clinical experts consulted by CADTH noted that there is uncertainty regarding the potential additional benefit and harms of continuing therapy with ARPIs in the target patient population, including those who received the ARPI in combination with 177Lu vipivotide tetraxetan. As noted previously, concerns have been raised about the use of ARPIs in the BSC/BSoC comparator arm, as patients had already demonstrated disease progression on at least 1 ARPI (in a situation where the protocol restricted access to therapies that are approved for use in patients with mCRPC).11
Concomitant supportive therapies: Supportive cancer therapies were generally well balanced in the 2 the groups. The clinical experts consulted by CADTH noted that the BSoC therapies that were used as supportive cancer therapies to decrease the risk or severity of skeletal-related events (e.g., denosumab and bisphosphonates) have not been demonstrated to have a survival benefit for patients with mCRPC. Continuation of these therapies is consistent with routine clinical practice.
Subsequent anticancer therapies: The enrolment criteria for the VISION trial stated that patients who had received 1 taxane regimen must be deemed unsuitable to receive a second taxane regimen to be eligible for the study (on the basis of frailty assessed by geriatric or health status evaluation or expected intolerance, for example).7 Despite these criteria, 14.9% and 18.9% of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups, respectively, received cabazitaxel after discontinuing treatment with the study drug regimens. It is unclear why these patients were eligible for therapy with a taxane regimen after discontinuation of the study drugs but considered ineligible to receive a taxane at the time of screening. CADTH asked the sponsor to explain why these patients would be considered ineligible to receive cabazitaxel at the time of enrolment but eligible after withdrawing from or progressing on the study drug regimens. The sponsor reported that this should not be considered in conflict with the eligibility criteria of the study, as the initial determination was made by the study investigator at the time of screening and the subsequent decision to initiate cabazitaxel could have been made at the discretion of the patient’s local physician after withdrawal from the study treatments.38
CADTH noted that the 2021 CUA-Canadian Uro Oncology Group guidelines for the management of mCPRC recommend cabazitaxel as a treatment option after demonstrated disease progression following therapy with an initial taxane regimen (docetaxel). The clinical experts consulted by CADTH agreed with this approach, and emphasized the following key limitations of the VISION trial: the exclusion of cabazitaxel as 1 of the comparators in the BSoC therapies (i.e., the initiation criterion stated that patients with prior exposure to a single taxane regimen were not considered suitable for another taxane regimen); and the fact that approximately 18% of the trial population would go on to receive an additional taxane regimen in the poststudy treatment setting. In a critique of the VISION trial, Olivier et al. (2022)11 state that prostate cancer specialists have suggested that these patients should have received cabazitaxel before enrolment in the trial, as the permitted BSC/BSoC regimens represented suboptimal care in comparison.
In the subsequent therapy setting, a greater proportion of patients in the BSC/BSoC alone group than in the177Lu vipivotide tetraxetan + BSC/BSoC group received at least 1 radiation treatment (34.6% versus 28.1%), at least 1 monoclonal antibody (7.9% versus 2.9%; the majority in both groups received programmed cell death 1 ligand 1 (PD-L1) inhibitors, which are not approved for use in Canada), and at least 1 radiopharmaceutical (8.2% versus 2.9%).7 Overall, the clinical experts consulted by CADTH did not feel that the distribution of the subsequent therapies would likely bias the treatment effect in favour or against 177Lu vipivotide tetraxetan, as there is no evidence that any of the therapies have demonstrated meaningful improvement in OS for patients with mCRPC in the target population (i.e., those with disease progression after an ARPI, docetaxel, and cabazitaxel [or who would not be medically suitable for cabazitaxel]).
Once a patient entered the long-term follow-up portion of the study (i.e., after discontinuation of the study drug regimens), the sponsor noted that any treatment deemed necessary could be pursued by the patient and/or their treating physician. This included medications that were prohibited in the treatment period of the VISION trial.7 These new treatments were to be recorded and reported as part of the long-term follow-up (as shown in Table 16). An important limitation of the VISION trial is that the sponsor would not have been able to obtain a complete list of subsequent treatments received by patients who elected to withdraw completely from the trial.8 Although survival data could be obtained through public registries, it would not be possible to obtain information regarding the therapeutic regimens offered to those patients in the interval between withdrawal from the VISION trial and death. The sponsor has reported that the survival status of 34 patients was obtained through public registries and that the data were used in the analysis of OS (14 patients [2.5%] were in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 20 [7.1%] were in the BSC/BSoC alone group).38
Patient disposition: As shown in Table 13, there was considerable withdrawal of consent in the BSC/BSoC group in the VISION trial, particularly early in the course of the trial. The FDA noted that this was attributable to the nonblinded trial design, with patients withdrawing consent when they realized they were assigned to the control arm and were not going to receive 177Lu vipivotide tetraxetan (and leading, for example, to disappointment, the desire to try other therapeutic options [e.g., taxanes], and an unwillingness to comply with the study protocol without access to the experimental therapy).9 The clinical experts consulted by CADTH noted that it is understandable that patients would withdraw from the study after realizing they would not be receiving 177Lu vipivotide tetraxetan, as these patients are at the end of their life and may not be interested in complying with the study protocol. The clinical experts also noted that the ability to seek treatment options that were not permitted in the VISION trial could have been an additional consideration for these patients. The sponsor established corrective actions in February 2019 that included site calls to discuss the management of patients in the control arm, investigator letters clarifying study aspects, and updates to prescreening to improve patient education about the trial. After implementation of these measures, the sponsor noted that withdrawal of consent decreased.8 However, as shown in Figure 7, withdrawal rates in the BSC/BSoC alone group were 56.0% before and 16.3% after the protocol amendment, compared with 1.2% before and 4.2% after in the 177Lu vipivotide tetraxetan group (i.e., although the rate of discontinuation from the BSC/BSoC alone group improved after the protocol amendment, it remained considerable higher than the rate observed in the 177Lu vipivotide tetraxetan group).
As a result of the high dropout rate in the comparator group (BSC/BSoC alone), the sponsor amended the protocol and increased the total number of patients to 814 (from the original plan of 750), and all end points, with the exception of OS, were analyzed using the newly established PFS-FAS dataset that was composed of patients enrolled after the educational protocol amendments were introduced in February 2019.9 The approach used is a method to handle the early withdrawals; however, the analyses based on the PFS-FAS would not likely have followed the ITT principle, which would have had an impact on many of the assumptions about the comparisons. This approach was acceptable to the FDA and Health Canada;8,9 however, both regulatory agencies stated that the interpretation of the magnitude of the rPFS effect was limited because of the high degree of censoring from early dropout in the control arm (neither the approved US label nor the Canadian product monograph include the effect size for rPFS from the VISION trial).5,10
The high and disproportionate number of patients who withdrew from the BSC/BSoC group could bias the study results in favour 177Lu vipivotide tetraxetan, as those who remained in the study may have had a poorer prognosis than those who withdrew and subsequently received treatment with regimens that were not permitted by the VISION protocol (although the magnitude of any bias is uncertain). Similarly, Olivier et al. (2022)11 noted that the patients who remained in the trial may have had fewer therapeutic options (e.g., more advanced disease) and may have lacked resources to obtain alternative regimens outside of the clinical trial setting (e.g., due to socioeconomic factors). Hence, the VISION trial may not have satisfied the Kaplan-Meier method assumption that patients who were censored would have had the same probability of survival as those who remained in the study.
In the primary analysis for OS, the proportion of patients who were censored for withdrawn consent was different between the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (2.7% and 11.8%, respectively). Reviewers for the FDA noted that the sensitivity analyses performed by the sponsor and the FDA review team to investigate the differences in censoring due to withdrawal were supportive of the primary analysis.9 The FDA concluded that 177Lu vipivotide tetraxetan had a robust statistically significant effect on OS compared with BSC/BSoC alone. They noted that sensitivity analyses suggested that there could be some diminution of the OS magnitude of effect (i.e., potential for bias in favour in 177Lu vipivotide tetraxetan); however, they concluded that the precision of OS as an end point and its meaningfulness as a clinical outcome are such that even a slightly smaller magnitude in the delay of death than what is reported would still be clinically meaningful and support a favourable benefit-risk profile for the target population of mCRPC patients.
End points: The primary and secondary end points of the VISON trial were aligned with those recommended by the PCWG3 (i.e., OS, rPFS, time to first SSE, HRQoL, PFS, and biochemical response [e.g., PSA and LDH]). The analysis of OS was performed using the FAS dataset, which was acceptable to Health Canada.8 The evaluation of rPFS was based on a smaller subset of patients who were enrolled after the protocol amendment to address the proportion of patients who withdrew from the BSC/BSoC alone group after randomization (i.e., the PFS-FAS). This analysis may be subject to bias as it was not done in accordance with ITT principles, an appraisal that was noted by Health Canada reviewers.8
Subgroup analyses: No concrete conclusions should be drawn from the results for the subgroup analyses because no formal statistical test of hypotheses was performed for any of the subgroup analyses. Health Canada noted that the sample sizes for several subgroups were small, limiting the ability to interpret the results (e.g., those with an ECOG PS of 2 and those with liver metastases at baseline).8
External Validity
Patient population: The clinical experts consulted by CADTH noted that the baseline and demographic characteristics in the VISION trial are a reasonable reflection of the target patient population in Canada. Health Canada and FDA reviewers noted that patients identifying as Black or Asian were underrepresented in the VISION study (6.6% and 2.4%, respectively).8,9 The clinical experts noted that the results of the VISION would be generalizable to these patients.
Median survival: Median survival in the VISION trial was 15.3 months (95% CI, 14.2 to 16.9 months) and 11.3 months (95% CI, 9.8 to 13.5 months) in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups, respectively. The clinical experts consulted by CADTH noted that the duration of survival in the BSC/BSoC alone group (i.e., 11.3 months) exceeds what would be anticipated for the target population in Canadian practice. The experts estimated that survival is typically in the range of 6 to 9 months for patients with progressive mCRPC who have demonstrated disease progression after treatment with both ARPI and taxane regimens. It was noted that this is commonly observed in clinical trials in which patients with prostate cancer are often healthier with have fewer comorbidities than the overall patient population encountered in routine clinical practice in Canada.
Prior taxane regimens: All the patients in the VISION trial had prior exposure to at least 1 taxane regimen. At the time of screening, 41.2% of patients had received 2 taxane regimens and 1.0% had received more than 2 taxane regimens. At the time of enrolment, 57.9% of the total study population had been treated with a single taxane and, therefore, should not have been medically suitable to receive another taxane regimen, in accordance with the study protocol. The clinical experts consulted by CADTH noted that this is greater than the number of patients that would be anticipated in Canadian practice for the target population, approximately 30% to 40% of whom would be considered not medically suitable to receive cabazitaxel based on clinical experience and recently published Flatiron real-world data.39
Other therapies: Radium-223 is approved in Canada for use in the treatment of patients with mCRPC and bone metastases, but there is variability across jurisdictions with respect to reimbursement by the drug programs that participate in the CADTH reimbursement review process. In some Canadian jurisdictions, this therapy can be accessed by patients with mCRPC and bone-only metastases early in the treatment algorithm (e.g., before docetaxel), but it is not currently reimbursed in other jurisdictions. In the VISION trial, 17.4% of patients had received prior therapy with radium-223 at the time of enrolment, and 2.5% of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 5.4% in the BSC/BSoC alone group received this therapy in the subsequent therapy setting.
Concomitant supportive therapies: 177Lu vipivotide tetraxetan was administered in combination with other systemic therapies in the VISION trial, including systemic anticancer therapies used as supportive cancer therapies.7 The supportive cancer therapies that are used to reduce the risk or severity of skeletal-related events (e.g., denosumab and bisphosphonates) are consistent with Canadian practice for patients with CRPC.4 As approximately 91% of patients enrolled in the VISION trial had bone metastases at baseline, the clinical experts noted that the proportion of patients receiving concomitant treatment with denosumab (34.8% in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 39.0% in the BSC/BSoC group) or bisphosphonates (8.5% and 13.7%, respectively) is likely lower than would be expected in routine practice in Canada (denosumab is not available for the treatment of prostate cancer in all Canadian jurisdictions). The clinical experts noted that androgen-deprivation therapy (gonadotropin-releasing hormone analogues and/or degarelix) would be continued, as it is a foundational medicine in this context and continued through every line of therapy for mCRPC.
Concomitant systemic anticancer therapies: 177Lu vipivotide tetraxetan was administered as an add-on therapy in the VISION trial, which included concomitant administration with other systemic cancer therapies. The product monograph does not specify that 177Lu vipivotide tetraxetan should be administered as an add-on therapy. As of the drafting of this review report, there are no Canadian clinical practice guidelines that address the use of 177Lu vipivotide tetraxetan. The clinical experts consulted by CADTH noted that combination use in Canada can be limited by reimbursement status. Public reimbursement for ARPIs after a patient has demonstrated disease progression on the therapy varies across jurisdictions, with some provinces mandating discontinuation of coverage and others permitting the continuation of therapy.40 Overall, the experts noted that it is unclear if the use of 177Lu vipivotide tetraxetan in combination with other systemic anticancer therapies will be adopted in practice because of uncertainty about the additional clinical benefit for patients.
When evaluating the AE data from the VISION trial, Health Canada noted that the increase in overall treatment-emergent AEs with177Lu vipivotide tetraxetan + BSC/BSoC would be expected, given that the drug is being used as an add-on therapy. Given the uncertainty about whether the drug will be used in combination with other systemic anticancer therapies in Canadian practice, the overall AE profile when used as monotherapy may be different.
Subsequent cancer therapies: After the EOT visit, patients were allowed to take any type of anticancer treatment deemed by their local physician to be in their best interest (these medications include those were not permitted during the treatment phase of the VISION trial, and are summarized in Table 16).7 As noted, an important limitation related to the external validity of the VISION trial was the large proportion of patients who received cabazitaxel in the poststudy treatment setting (i.e., the VISION trial enrolment criteria stated that patients who had received a single taxane regimen must be medically unsuitable for an additional taxane regimen). The clinical experts consulted by CADTH noted that this would not be reflective of Canadian practice because a patient with mCRPC who is considered ineligible for a further taxane regimen is unlikely to become eligible at a later point in time, as this disease is progressive and improvements in functional status or physiologic reserve are not anticipated. Other than these issues, the clinical experts noted that subsequent therapies are reflective of routine care for patients for whom there are no other therapies shown to increase OS.
Comparator: Several potential comparators for 177Lu vipivotide tetraxetan were not permitted in the acceptable BSoC treatment regimes. These include cytotoxic chemotherapy (e.g., cabazitaxel), immunotherapies, and other systemic radioisotopes (e.g., radium-223 or hemi-body radiotherapy). The rationale provided by the sponsor was that these therapies could confound the analysis in the VISION trial. The VISION protocol limited systemic anticancer options in the comparator group to hormone therapies, including novel ARPIs (e.g., abiraterone and enzalutamide). All the patients enrolled in the trial had exposure to novel ARPIs before enrolment. As shown in Table 9, 45.7% of patients in the comparator group had prior exposure to 1 novel ARPI regimen and 45.7% had prior exposure to 2 regimens. This approach may have biased the treatment effects in favour of 177Lu vipivotide tetraxetan, as the majority those in the BSC/BSoC alone group had already been treated with and demonstrated disease progression on the only systemic therapies that were permitted.11
The clinical experts noted that olaparib was an investigational drug for mCRPC when the VISION trial was initiated (the first patient was enrolled in May 2018 and olaparib did not receive regulatory approval in any jurisdiction until May 2020),7,28 and is indicated for only a small subset of patients with mCRPC (i.e., those with documented deleterious or suspected deleterious germline and/or somatic BRCA or ATM mutations). Therefore, the exclusion of this drug from the BSoC regimen is understandable and not considered to be a major limitation with respect to the generalizability of the study results. In addition, the experts noted that radium-223 is indicated only for patients with bone metastases (without visceral metastases) and is not available in all Canadian jurisdictions.
Withdrawals: The clinical experts consulted by CADTH noted that the distributions and reasons for discontinuation from 177Lu vipivotide tetraxetan in the VISION trial are a reasonable reflection of what would likely be observed in routine clinical practice, although they noted that discontinuation due to AEs could occur more frequently in the real-world setting (e.g., patients may be less healthy than those enrolled in the trial and there would be no pressure to try to have patients complete the treatment regimen as specified in the trial protocol).
Dosing: 177Lu vipivotide tetraxetan could be administered for up to 6 cycles in the VISION trial, which is consistent with recommendations in the Canadian product monograph.5,7 The VISION trial protocol included an additional step in which the patient was to be evaluated by the investigator after 4 cycles for evidence of treatment response (specified as either radiological response, PSA response, or clinical benefit in the opinion of the investigator), signs of residual disease on CT with contrast and/or MRI or bone scan, and tolerance of the treatment. Patients meeting all those criteria could receive up to 2 additional cycles at the discretion of the treating physician.5,7 The clinical experts consulted by CADTH noted that the evaluation of response to treatment for the target patient population (i.e., those with progressive mCRPC) is multifactorial and would be based on clinical response, radiographic imaging, biochemical measures, and the need for medications to manage pain. It was noted that a formal assessment of response after 4 cycles (as performed in the VISION trial) is unlikely to be standardized in Canadian clinical practice and could be a challenge to implement if included as renewal criteria for 177Lu vipivotide tetraxetan. The FDA noted that there is insufficient evidence to evaluate the efficacy of 4 cycles of 177Lu vipivotide tetraxetan and, hence, the recommended dosage regimen is for “up to 6 cycles, disease progression, or unacceptable toxicity.”9 Overall, the clinical experts consulted by CADTH noted that the distribution of doses observed in the VISION trial is likely an accurate reflection of what would occur with patients in Canada, as the treatment is generally well tolerated and relatively few AEs lead to dose reductions, interruptions, or discontinuations.
Treatment setting: The product monograph states that 177Lu vipivotide tetraxetan should be administered under the supervision of a health professional who is experienced in the use of radiopharmaceuticals. Appropriate management of therapy and complications is only possible when adequate diagnostic and treatment facilities are readily available.5 This is consistent with administration in the study centres involved in the VISION trial and consistent with the way this drug would be administered in Canada.5
Study locations: The VISION trial was multinational; all sites were in the US (66.0% of patients), Europe (27.6% of patients), and Canada (6.0% of patients).8 The clinical experts consulted by CADTH noted that the study locations (primarily in the US) would not be expected to limit generalizability of the study results to the Canadian setting, but noted that the use of subsequent therapies can be different in the US than in Canada and Europe, where public drug programs may have more strict criteria for reimbursement.
PSMA screening: Routine PSMA screening in patients with mCRPC is not currently recommended by the CUA, and the clinical experts consulted by CADTH noted that PSMA testing with PET-CT is not widely available in routine practice in Canada. The VISION trial included patients with PSMA-positive mCRPC, defined as at least 1 PSMA-positive metastatic lesion and no PSMA-negative lesions (i.e., patients with any PSMA-negative lesions were ineligible).7 The clinical experts anticipated that patients would show a mix of PSMA-positive and PSMA-negative lesions in clinical practice, but noted that the criteria used in the VISION trial are acceptable for the identification of patients who could be candidates for 177Lu vipivotide tetraxetan.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
One sponsor-submitted ITC was summarized and critically appraised. The sponsor-submitted ITC was used to inform the pharmacoeconomic model because of a lack of head-to-head RCTs. CADTH conducted a literature search to identify other potentially relevant ITCs of patients with mCRPC. The Ovid MEDLINE database was searched using a combination of MeSH (Medical Patient Headings) and keywords. The main search concept was adults with mCRPC. An NMA filter was applied to limit study type to NMAs. Retrieval was not limited by publication date or by language. Titles, abstracts, and full-text articles were screened for inclusion by 1 reviewer based on the population, intervention, comparator, and outcome criteria outlined in the CADTH systematic review protocol (Table 6).
Description of Indirect Comparison
The sponsor-submitted ITC, which is an NMA, aimed to evaluate the efficacy of 177Lu vipivotide tetraxetan relative to other regimens for adults with advanced mCRPC who have received ARPI(s) and taxanes-based chemotherapy or are not medically suitable for taxanes. The sponsor performed a systematic review to identify relevant studies for inclusion in the ITC. Outcomes that were included in the review were efficacy and safety outcomes. The population, interventions, comparators, outcomes, and design of studies included in the sponsor’s ITC are provided in in Table 37 and subsequently described in detail.
Table 37: Study Selection Criteria and Methods for ITCs
Characteristic | Sponsor-submitted ITC |
|---|---|
Population | █████ █████ ████ █████ ████ ████ ████████████ ███████████ ██ |
Intervention | ██ ███████████ ██ █████ ██ ████████████ |
Comparator | ██ ███████████ ██ █████ ██ ██████████ |
Outcome | ████████ ███████████████████ ████████ █████████████████████ ████████████ ██ ███ ████████████████ ██ ██████ ████████████████ ██ ███████████ ████████ ██████████ ████████████████ ███████ █████████████ ████ ███████████ ████████ ███████████████ ████ ██████ ██ ███ ████████████████ ██ █████ █████████████ ██ █████████████████████ ████████████ ██ █████████ ██████████████ ████████████ ██ ████ ███████████████████ █████████████████ ██████ ██████ ███ ███ ████████████████████████ ███ ██ ███ █ |
Study design | ████ ██████ ████ |
Publication characteristics | ███████ ████████ |
Exclusion criteria | ███████ ████ ████████ ████████ ████████ ████████ ███ ███████████ ███████████████ █████ ████████ █████ ███ ███ ██████ ███ ██ ███ ████████ ██ ██████ ████████ ██ ████████ █████ ████ █████████ ████████ ███████████ ███████████ ████████ ██████ █████ ███████ ████ ██████ ██ ████████ ██████ ██ ██ █████ ████████ ██████████ ███████████ █████ ████████ ██████████ ███████ ███████ ████████ ████ ██████ ███████████ █████████████ ███████ █████ ████ ███████████ ████████████ |
Databases searched | ████████ ███████ ████████ ████████ ███████ ██████ ████████ |
Selection process | ████████ ████████ █████████████ ██ ███ ███████████ |
Data extraction process | ████ ██████████ ███ █████████ ██ █████ ██ █████████ ███ ████████ ███ █████████████ ██ ██████ ████████ |
Quality assessment | █████ █████████ |
ITC = indirect treatment comparison; mCRPC = metastatic castration-resistant prostate cancer; PICOS = Population, Intervention, Comparison, Outcomes and Study; RCT = randomized controlled trial.
Source: Sponsor-submitted ITC.
Methods of Sponsor-Submitted ITC
Study Selection Methods
The systematic review and NMA were conducted in accordance with the principles set out in the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses).
███████ ████████ ████████ ████ ████████ ██ ███████ ██████████ ███████ ███████████████ ███████████ ███████████ ███████ ███ ████████ ███████ ██████ ████████████████████████ █████ ████████ █████████ ██ ████ █████ ██████ █████ ████████ ███ ███ ███ ███ █████████ █████████ ███ █████ ██████████ ███ ██████ ██████████ █████████ ██ █████ █████ ████████ ████ ████ ████████████ ███████████ █████ ██ ███ ██████████ █████ ██ ███████ ██████████ ██████ ███ ███ ████████ ███ █████████ ██ ███████ █████ ██ ███ ██████████ ██████████████ █████ ███ ███████████████ █████████████ █████████ ████████████████ ████ █████████
█████████ ████ █████ ████████ █████ ██ █████ ███ █████████ ███ ██████ █████ ███████████ ███ █████████ █████ █████████ ███████ ████ █████ ███ ██████ ████ █████████████████████ ██████████ ██ █████ ████████ ████ █████ ████ █████████ ███ ███████████ ███████████████ ███ █████████ ████ █████████████ ██████████ ██ ██████ ███████████ █████████ █████████ ███ ████ ██ ████ ██████████ █████████ ██████████ ████ █████████ ██ ███ ███████████████ ███ ███ █████████████ ████ ██████████ ██ █████ ████████ ██ █████ ███████████ █████████ ████████ ███████ ███ █████ ████████ ██ ████████ █████████ █████████████ ████████████ ███ █████ ██ ███ ████████ ██ █████ ███
ITC Analysis Methods
███ ███████ ██ ███ █████████ ███ ████ █████████ ███ █████████ ████ ████████████ ███ █████████████ ██████ ████ ███████████ ███ ██ ███████ ██████ ██ ████ ████████ ██ ███ ████ ███ ███ ████████ █████████████ ███████ ██ █████ ██ ██████ ███ ███████████████ ██ ██████████ ██ ████ ████████████ ██████████ ███ █████████ ███ █████████ ██ █████████ ███ ███████████ ██ ████████ ██████████ ████████ ██ █████████ ███ ████ ███████████ ███████████ ██ ████████ ███ ████ ██ █████████ █████████████ ███████ ██████ ███████ ███ █████ ████████████████ ██ █████████ ████████████ ██ ███████ ███████████ ██████ █████ ███ ██ █████████ ████████████ ██ ███████ ███████████████ ██████ ███ █████
A comprehensive set of statistical analyses were performed that comprised:
Base-case NMA: ████ ████████ ███ ███████ ███ ███████ ███ █████████ ███ ██ ████ ███ ███ █████ ███████████ █████████████████████████ █████████████ ████████████
Sensitivity analysis: █████ █████████ ███████ █████████ ██ ███ ████ █████████ ███ ███████ ████████████ ██ ████████████
The analyses were performed for the ITT populations from the included studies.
███ ███ ████████ ███ ██ █████ ██ ████ ███ ██ █████ ███ ██████████ ██ ████████████ ███████ ███████ ██████████ ███ █████████ █████ ███████████ █████ ████ ███████ ██████████ ███ ███ ███ ██ ████ █████████████ ████████ ██████ ██ ████ █████ ███ ███████████ ██ ███ ████████ █████ █████ █████████████ ███ ███ ██████████ ████ █████
███ ███████ ██ ███ ███ ███ ████ ███ ██ ████ █████████ ████ █████████ ███ █████████ ███████ ██ ████ ████████████ ████████ ██ ███ █████████ █████ ██████████ ██████████ █████████ ████████████ ███ █████████ █████████████ ██ ████████ █████████ ███████ ████ ██████████ ██ ███ ██████ ███ ███ ████████ █████████ ███████ ███ ███████ ██ ███ ███ ████ █████████ ██ ███ ████ ██ ████████████ ████ ████████ █████████ ██████ █████████ █████ ███████ ███ █████████████ ██ ████████ █████ ████ ███ ███ ███ ███ █████████
Table 38: ITC Analysis Methods
Method | ITC |
|---|---|
ITC methods | █████ ███████ ███████ █████████████ █████ █ |
Priors | █████ ██████ |
Assessment of model fit | ████████ ███████████ █████████ █████ |
Assessment of consistency | ███ ████████ |
Assessment of convergence | ██████ ██████████ ██ █████ █████ |
Outcomes | █████ ██ |
Follow-up time points | ███ ████████ |
Construction of nodes | ███ ████████ |
Sensitivity analyses | █████████ ███████ █████████ ██ ███ ████ |
Subgroup analysis | ███ █████████ |
Methods for pairwise meta-analysis | ███ █████████ |
ITC = indirect treatment comparison; OS = overall survival; rPFS = radiographic progression-free survival.
Source: Sponsor-submitted ITC.
Results of Sponsor-Submitted ITC
Summary of Included Studies
A systematic literature review was conducted and █████ citations were identified. Of these, █ citations pertaining to █████ unique trials met the inclusion criteria for the NMA. The sponsor included the VISION trial so that there would be a total of █████ unique trials in the NMA.
Figure 18: Network of Trials Included in the NMA (Redacted)

NMA = network meta-analysis.
Confidential figure removed at the sponsor’s request.
██████████████ █████████ ███████ ██████████ ███████████ ████ █████████ ████████ ███████ ██████████ ███ █████ ██████████ █████ ███ ███████████ ██████████ █████████████ █████████████████ █████
Study Characteristics
Of the █ included RCTs, █ were phase III and █ was phase IV. Regarding the blinding status, █ RCTs were open-label and █ were double-blinded. █ trials assessed OS as the primary outcome, rPFS was the primary outcome in █ trials, and time to tumour progression was assessed as the primary outcome in █ trial.
Treatment Characteristics
████ was the most frequently used intervention, and was included in █ of the █ RCTs. ███████████ was included in █ of the RCTs. █████████████ ███████████ █████████ ███ █████ ██████████ ██████████ ████ ████ ████████ ██ ███ ████████ ███. The sponsor did not report specific dosages for these treatment regimens from their RCT.
Baseline Characteristics
Various baseline patient characteristics were examined to evaluate the clinical heterogeneity between the trials included in the NMA. These characteristics included ████ ███████ ██████ ███ ███████ █████ █████████ ███████ ███ ████ ███████████ ██████ ███████ █████ ████████ ███████████████ ████ ██████████ ███████ ███████ ██████ ███ ██████ ███ ███ ████ ██ ███ ██ ████████ ██████ ███ ██████ ██ ██████ ██ ███████████ ████ █████ ██ ████████ ███ ████ ████ ████████ ██ ███ ██████ ██████ ██████ ████████ ██ ███ ██████ ███ ███████ ██ █████████ ███ ███████████ █████ ███ ████ ██ ████████ ████████ ███ ████████ ██ ████████ ███ ████ ████████ █████████ ██ █████ ███████ ████ ███████ ███████ ████ █████ ████████ ██ ███ ███████████ ███████ █████ ██ ██████ ████ █████ ██ ██████ ██████ ███ ███████ ██████ ████████ ██████ ██████████ ██ ████████ ████ ███████ █████ ██ ██████ ██████ ██████ ████████ ██ █████████ ██████ ████ ████████ ██ ██████ ████████ ███ ███████ ██████.
Reported Outcomes
██████ ███ ███ ████ █████████ ███ ██████████ ██ █████ ██ ███████████ ███ ████████ ██ ██ ███ ████████ ███████ ██ ███ ████ ████ █████████████ ██ ███████ ███████████ ██ ██████ ██ █████████ ██ ███████████ ████ ████████ ███████ ███ ███ ████ █████████ ███ ██ ███████████ ████ ███ ████ ████ █████████████ ██ █████ ████ ███ ██████
Results
███ ███████ ██ ███ ███████████ ██████████ █████████ ████ ████████ █████████ ███████ ███████████ ██ ███ ███ ██ ██ █████████████ ██ ███ ██████ ██████ █████ ██████ ███ ███████ █████████ ██████ ███████████ ██ ███ ███ ██ ████ █████████████ ██ ██████ ██████ ██████ ██████ █████ ███████ ████████ ███ ██ ████████ ██ ██████████ ████████ ██ ███ ███ ██████████ █████████ ████████ █████████████ ███ ███████ ██ ███ ████████ ███ ██ ███████ ███████ ███████████████ ██████ ███████
Overall Survival
OS was reported in █ RCTs pertaining to █ unique treatments. The NMA showed that 177Lu vipivotide tetraxetan suggested improvements in OS compared to ██████████ ███████ █████ ███ ████ ████ ██ ████████████████ ███ ████████ ████ █████ ███ ████ ████ ██ ████████████████████████ ████████████ █████ ███ ████ ████ ██ ████ and ████ ████ █████ ███ ████ ████ ██ ████). HRs were reported as the comparator versus 177Lu vipivotide tetraxetan. The NMA results showed favourable OS outcome for 177Lu vipivotide tetraxetan when compared to ████████; however, statistical significance was not reached. The base-case NMA results are presented in Table 39.
Figure 19: Network of Trials Included in the NMA for OS (Redacted)

NMA = network meta-analysis; OS = overall survival.
Confidential figure removed at the sponsor’s request.
██████████████ █████████ ███████ ██████████ ███████████ ████ █████████ ████████ ███████ ██████████ ███ █████ ██████████ █████ ███ ███████████ ██████████ ████ █████████ █████████████████ █████
Table 39: Fixed-Effects ITC Results
████████ | ██████████████ | ████████████ | ████████ | ███████████████████ | ████ |
|---|---|---|---|---|---|
███ ██████████████ █ ███ | |||||
██ ███ █ | ████ █████ ██ | ████ █████ | ████ █ | ████ █████ ██ █████ | ████ |
██████ | ██ | ████ █████ ██ | █████ | ████ █████ ██ █████ | ████ █ |
CrI = credible interval; HR = hazard ratio; ITC = indirect treatment comparison; NA = not applicable; OS = overall survival; rPFS = radiographic progression-free survival; SoC = standard of care.
Source: sponsor-submitted ITC.41
Table 40: Fixed-Effects ITC Results — Sensitivity Analysis
████████ | █████████ | ███████████ | ████████ | ██████████ | ████ |
|---|---|---|---|---|---|
███ █████ ██████████ ██████████ ████ | |||||
██ ███ ███ | ████ █████ | ████ █████ | ████ ███ | ████ █████ █ | ████ ███ |
███████ | ██ | ████ █████ | ████ ████ | ████ █████ ██ | ████ ████ |
CrI = credible interval; HR = hazard ratio; ITC = indirect treatment comparison; NA = not applicable; OS = overall survival; rPFS = radiographic progression-free survival; SoC = standard of care.
Source: sponsor-submitted ITC.41
Sensitivity analysis: ██ ███ ████████ ██ █████ ██████████ ██ ███████ ███████████ ███ ███ ██ █ ███ █████ ██████████ ██████████ ████████████ ███████████ ██ ████ ████████ ██ ████ ████ █████ ███ ████ ████ ██ ██████ ███ ████ ████████ ██ ██████████ ██████ █████ ██ ███ █████ ███████████ ███ ███ ███████ ██████ ██████████ ██ ███████ ███ █████ ████ ██ ██ ██ ███ ██ ██ ███ ████ ████████ ██ ██████████ █████ ███████████ ████████████ ███ ██ ██ ███ ██████████ ████████████ ███ █████████ ████████ ███████████ ████████████ ███ ███ ████████ ███ ███████████ ████████ ███ ███████ ███ █████████ ██ █████ ███
Radiographic Progression-Free Survival
rPFS was reported in █ RCTs pertaining to █ unique treatments. The NMA showed that 177Lu vipivotide tetraxetan demonstrated significant rPFS gains compared to ███████████ ███████████ (███ █████ ███ ████ ████ ██ ████), ████████████████████ ███████████ (███ █████ ███ ████ ████ ██ ████), and ████ (███ █████ ███ ████ ████ ██ ████). HRs were reported as the comparator versus 177Lu vipivotide tetraxetan. The NMA results showed a favourable rPFS outcome for 177Lu vipivotide tetraxetan compared to ████████; however, statistical significance was not reached. The base-case NMA results are presented in Table 39.
Sensitivity analysis: rPFS was reported in █ RCTs pertaining to █ unique treatments. The NMA showed that 177Lu vipivotide tetraxetan demonstrated significant rPFS gains compared to ████████████████████ ███████████ (███ █████ ███ ████ ████ ██ ████) and APRI (███ █████ ███ ████ ████ ██ ████). HRs were reported as the comparator versus 177Lu vipivotide tetraxetan. The NMA results showed a favourable OS outcome for 177Lu vipivotide tetraxetan compared to ███████████ ███████████ ███ ████████; however, statistical significance was not reached. The sensitivity analysis NMA results are presented in Table 40.
Figure 20: Network of Trials Included in the NMA for rPFS (Redacted)

NMA = network meta-analysis; rPFS = radiographic progression-free survival.
Confidential figure removed at the sponsor’s request.
██████████████ █████████ ███████ ██████████ ███████████ ████ █████████ ████████ ██ █████ ██████████ ███ █████ ██████████ █████ ███ ████████ ██████████████ ███ ███ ████████ ██████████ ██████ ████ █████████████ ████████████████ ████████████████ ██ ███████████████ █████
Critical Appraisal of Sponsor-Submitted ITC
The sponsor’s rationale for conducting the ITC (i.e., absence of head-to-head studies that compare 177Lu vipivotide tetraxetan with other treatments in adults with mCRPC who have received ARPI and at least 1 taxane-based chemotherapy). From the ITC, ███████████ ██████████ █████ ███████████ ████████████ █████████ ███ ████ were considered relevant to the Canadian clinical context, whereas mitoxantrone or placebo plus prednisone was considered less relevant. A comprehensive systematic review was performed to identify relevant clinical studies. The efficacy outcomes of interest were rPFS and OS. Several relevant outcomes, including AEs, HRQoL, and ORR, were not assessed in the sponsor-submitted ITC (no rationale for their exclusion was provided by the sponsor).
Clinical heterogeneity was present in the analysis owing to variation in patient characteristics across the included trials. In the absence of statistical adjustment, sensitivity analyses, or subgroup analyses, the potential impact of the between-study heterogeneity cannot be evaluated. The clinical experts consulted by CADTH noted that there was heterogeneity in clinically important patient characteristics (i.e., historical use of chemotherapy, disease severity, and treatment indication); therefore, the ITC analysis may be subject to bias. Of particular concern was the fact that patients who received 177Lu vipivotide tetraxetan in the VISION trial had more severe disease at baseline, as indicated by the higher prior treatment count (at least 40% of patients had received cabazitaxel before enrolment). ████ was selected as the reference comparator for all trials and, although there was no statistical heterogeneity between the reference arms, different definitions of ████ were considered. The base-case analysis results were sensitive to the various definitions, as shown by the sensitivity analysis results. In addition, inconsistency of the network was not reported, likely because of the limited ability to do so, given that the network only had 1 closed loop. As a result, the 2 key measurable assumptions of an NMA, between-trial homogeneity and network consistency, could not be confirmed if these were met.
Overall, the sponsor-submitted ITC has important limitations that preclude the drawing of conclusions regarding the efficacy of 177Lu vipivotide tetraxetan relative to relevant comparators for the target patient population.
Summary
The sponsor-submitted ITC was based on a systematic review of relevant clinical studies and used a Bayesian NMA approach to evaluate the efficacy of 177Lu vipivotide tetraxetan relative to other comparators, including ██████████ █████ ███████████ ████████████ █████████ ████████████████████ ████████████ ███ ████, for the treatment of patients with pretreated, progressive mCRPC. The NMA was based on a systematic review of the literature, and data from █ studies were used to inform the analyses. The efficacy outcomes of interest were rPFS and OS.
The sponsor-submitted ITC reported that the results for OS favoured 177Lu vipivotide tetraxetan versus ██████████ ██ (HR = █████ ███ ████ ████ ██ ████) and versus ████ (HR = █████ ███ ████ ████ ██ ████). The sponsor-submitted ITC reported that the results for rPFS favoured 177Lu vipivotide tetraxetan versus ███████████ ███████████ (HR = █████ ███ ████ ████ ██ ████), ████████████████████ ███████████ (HR = █████ ███ ████ ████ ██ ████), and ████ ████ █████ ███ ████ ████ ██ ██████ HRs for OS and rPFS were reported as the comparator versus 177Lu vipivotide tetraxetan.
The sponsor-submitted ITC had several limitations, including the lack of reporting on certain items that would help to inform the certainty of the indirect evidence. Despite the heterogeneity of many patient and study characteristics, the ITC authors did not adequately conduct sensitivity and subgroup analyses to investigate the root of the heterogeneity or conduct a meta-regression that would adjust for effect modifiers that could influence the results. Consequentially, there is substantial uncertainty around the ITC results, and firm conclusions cannot be drawn about the efficacy of 177Lu vipivotide tetraxetan relative to relevant comparators.
Other Relevant Evidence
Comparison of 177Lu Vipivotide Tetraxetan Versus Cabazitaxel
The inclusion criteria for the VISION trial specified that patients who were previously treated with docetaxel and considered eligible to receive cabazitaxel were to be excluded from the study. As this population is included in the Health Canada–approved indication, CADTH considered this to be an important gap in the evidence and, therefore, summarized the phase II TheraP trial, which enrolled patients with prior exposure to docetaxel and for whom cabazitaxel was considered the appropriate treatment option.42-44
Description of the TheraP Study
Figure 21 provides a summary of the study design of TheraP, a multicentre, open-label, phase II, RCT comparing the activity and safety of 177Lu vipivotide tetraxetan with cabazitaxel in patients with mCRPC. The study was conducted by the Australian and New Zealand Urogenital and Prostate Cancer Trials Group. As in the VISION trial, the study enrolled patients with PSMA-positive mCRPC, but the TheraP trial used a 2-stage screening process to determine PSMA status.
68Ga-PSMA PET-CT: patients were eligible if they demonstrated an SUVmax of at least 20 at a site of disease, and an SUVmax of more than 10 at sites of measurable disease at least 10 mm.
FDG PET-CT: patients were ineligible if they were FDG-positive with minimal PSMA expression, defined as an FDG intensity of more than 68Ga-PSMA activity or a 68Ga-PSMA SUVmax of less than 10 (i.e., discordant imaging).44
Eligible patients were randomized (1:1) to receive either 177Lu vipivotide tetraxetan or cabazitaxel. Randomization was stratified by disease burden (> 20 sites versus ≤ 20 sites, as assessed by PSMA PET-CT), previous treatment with enzalutamide or abiraterone, and study site.44
Used with permission of John Wiley and Sons - Books, from Hofman MS, Emmett L, Violet J, et al. TheraP: a randomized phase II trial of 177Lu-PSMA-617 theranostic treatment vs cabazitaxel in progressive metastatic castration-resistant prostate cancer (Clinical Trial Protocol ANZUP 1603). BJU Int. 2019;124 Suppl 1:5 to 13. Copyright: 2019; permission conveyed through Copyright Clearance Center, Inc.
Interventions
Study Treatments
Patients who were randomized to receive 177Lu vipivotide tetraxetan received IV infusions once every 6 weeks for a maximum of 6 cycles. The starting dose was 8.5 GBq, which was decreased by 0.5 GBq each subsequent cycle (i.e., not administered at the dosages recommended in the Canadian product monograph, which is 7.4 GBq). As in the VISION trial, the dosage of 177Lu vipivotide tetraxetan in the protocol for the TheraP trial included dose reduction and interruption scenarios for the management of AEs (e.g., 20% dose reduction if the patient demonstrated a nadir platelet count < 100 × 109/L, a nadir neutrophil count < 1.0 × 109/L, dry mouth or dry eyes of grade ≥ 2, or other significant drug-related toxicities). Patients in the cabazitaxel group received IV infusions of 20 mg/m2 once every 3 weeks for a maximum of 10 cycles. In the event of drug-related toxicity, the investigator was permitted to reduce the dosage of cabazitaxel to 15 mg/m2 and 10 mg/m2.
Figure 21: The Design of the TheraP Trial
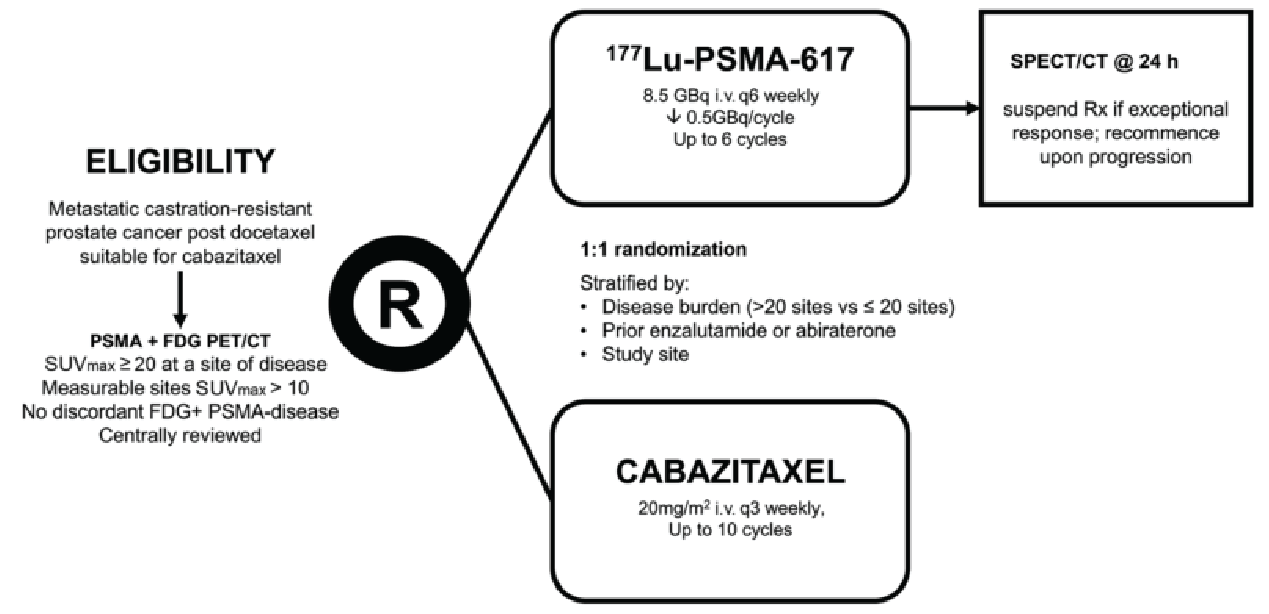
177Lu-PSMA-617 = lutetium-177 vipivotide tetraxetan; FDG = 18F-fluorodeoxyglucose; PSMA = prostate-specific membrane antigen; R = randomization; Rx = prescription; SPECT = single-photon emission CT; SUVmax = maximum standardized uptake value.
Source: Hofman et al. (2021).44
Concomitant Medications
Patients enrolled in the TheraP trial continued to receive supportive cancer therapies (e.g., zoledronic acid, denosumab, or palliative radiotherapy). An important difference between the TheraP and VISION trials is that patients were prohibited from using other systemic anticancer therapy in the TheraP trial (i.e., the study investigated 177Lu vipivotide tetraxetan as monotherapy).
Subsequent Treatments
Patients could receive any treatment after completion or discontinuation of the study drugs at the discretion of the treating clinician(s).
Outcomes
The primary end point was PSA response rate (defined as the proportion of patients with a PSA reduction of ≥ 50% from baseline). Secondary end points included PFS, rPFS, objective response rate (based on RECIST); pain response (defined as a reduction from baseline of ≥ 2 points) for patients with a PPI score at baseline of at least 2.
Table 41: Details of Other Relevant Study — the TheraP Trial
Details | TheraP |
|---|---|
Designs and populations | |
Study design | Open-label, randomized, stratified, 2-arm, phase II trial |
Locations | 11 centres in Australia |
Patient enrolment dates | Study initiation date: January 29, 2018 (first patient enrolled) Data cut-off date: July 20, 2020 |
Randomized (N) | 200
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | 177Lu vipivotide tetraxetan was administered as a slow IV injection at a dose of 8.5 GBq, decreased by 0.5 GBq per cycle, once every 6 weeks for a maximum of 6 cycles |
Comparator(s) | cabazitaxel IV at a dose of 20 mg/m2 once every 3 weeks for a maximum of 10 cycles |
Duration | |
Phase | |
Run-in | NA |
Open-label treatment phase | Treatment with 177Lu vipivotide tetraxetan continued for 6 cycles (once every 6 weeks) and treatment with cabazitaxel continued for 10 cycles (once every 3 weeks) Safety assessment was done at 30 days and 12 weeks after the last dose of study treatment |
Follow-up | Continued every 12 weeks after safety assessment |
Outcomes | |
Primary end point | PSA response ratec |
Secondary and exploratory end points |
|
Notes | |
Publications | Hofman et al. (2021)45 Hofman et al. (2019)44 NCT0339242842 |
177Lu = lutetium-177; 68Ga = gallium-68; CRPC = castration-resistant prostate cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; FDG = 18F-fluorodeoxyglucose; ITT = intention to treat; LHRH = luteinizing hormone-releasing hormone; NA = not applicable; PCWG3 = Prostate Cancer Working Group 3; PFS = progression-free survival; PPI = present pain intensity; PSA = prostate-specific antigen; PSMA = prostate-specific membrane antigen; RECIST = Response Evaluation Criteria in Solid Tumours Version 1.1.
aDefined as an SUVmax ≥ 20 at a site of disease, and an SUVmax > 10 at sites of measurable disease ≥ 10 mm.
bDefined as an FDG intensity > 68Ga-PSMA activity or a 68Ga-PSMA SUVmax < 10.
cDefined as the proportion of participants in each group with a PSA reduction of ≥ 50% from baseline.
dDefined as the interval from randomization to first evidence of PSA progression defined by an increase of at least 25% and at least 2 ng/mL after 12 weeks (per PCWG3), radiographic progression using locally reported CT and bone scanning (RECIST and PCWG3 criteria for bone lesions), commencement of nonprotocol anticancer treatment, or death from any cause.
Sources: Hofman et al. (2021)45 and NCT03392428.42
Secondary end points included:
PFS
PPI-PFS (interval from randomization to the first increase of at least 1 point from the nadir PPI score, commencement of nonprotocol anticancer treatment, or death)
The McGill Pain Questionnaire
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30)
The Patient Disease and Treatment Assessment Form
Patient Disposition
Figure 22 summarizes patient disposition in the TheraP trial. A total of 291 patients were screened for eligibility and 200 patients were randomized. PSMA screening in the TheraP 2 trial was more rigorous than in the VISION trial. As in the VISION trial, a greater proportion of patients in the comparator group (in this case, cabazitaxel) than in the 177Lu vipivotide tetraxetan group withdrew before receiving any doses of the study medications (16/101 [15.8%] versus 1/99 [1.0%]).
Used with permission of Lancet Publishing Group, from Hofman MS, Emmett L, Sandhu S, et al. [177Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomized, open-label, phase II trial. Lancet. 2021;397(10276):797 to 804. Copyright: 2021; permission conveyed through Copyright Clearance Center, Inc.
Patient Characteristics
The baseline characteristics for the ITT population in the TheraP trial are summarized in Figure 23. Overall, baseline characteristics were similar in the 177Lu vipivotide tetraxetan and cabazitaxel groups. A greater proportion of patients in the cabazitaxel group than in the 177Lu vipivotide tetraxetan group had a Gleason score no higher than 7 (35% versus 25%), and a greater proportion of patients in the 177Lu vipivotide tetraxetan group than in the cabazitaxel group had received prior therapy with both enzalutamide and abiraterone (21% versus 9%).
Figure 22: Patient Disposition in the TheraP Trial
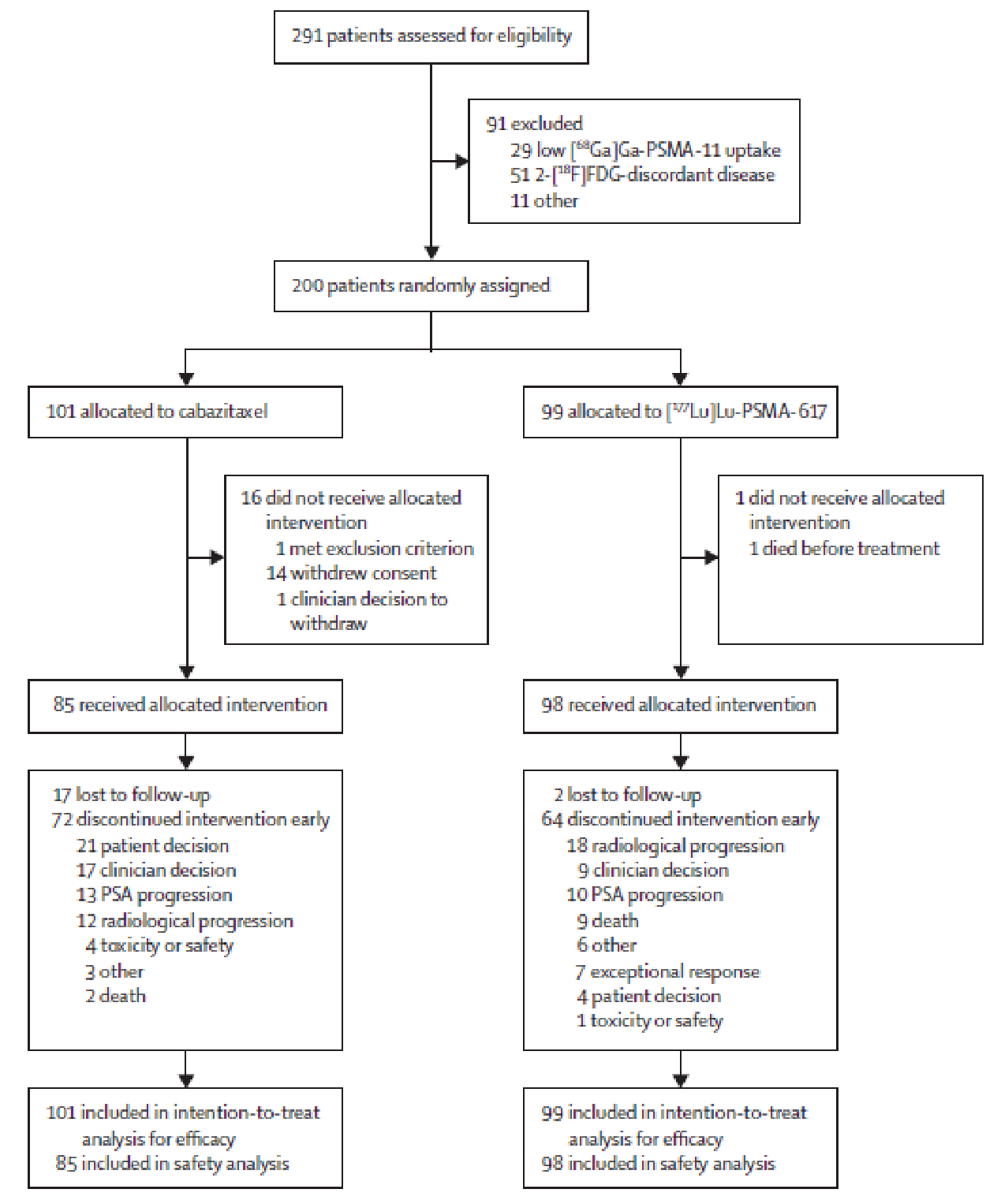
[177Lu]Lu-PSMA-617 = lutetium-177 vipivotide tetraxetan; [18F]FDG = 18F-fluorodeoxyglucose; 68Ga = gallium-68; PSA = prostate-specific antigen.
Source: Hofman et al. (2021).45
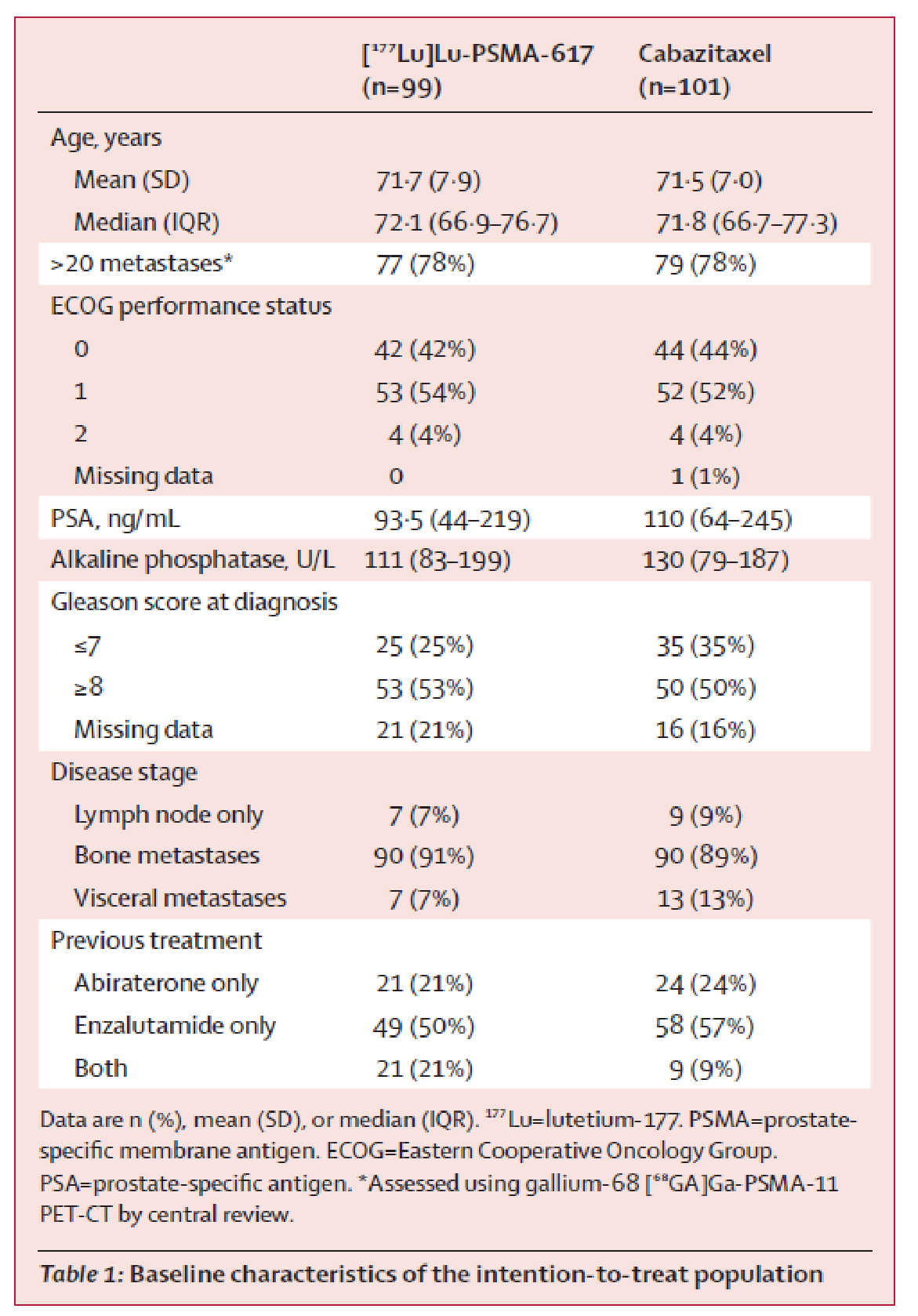
Used with permission of Lancet Publishing Group, from Hofman MS, Emmett L, Sandhu S, et al. [177Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomized, open-label, phase II trial. Lancet. 2021;397(10276):797 to 804. Copyright: 2021; permission conveyed through Copyright Clearance Center, Inc.
Efficacy Results
Efficacy results for the TheraP trial are summarized in Table 42. After 3 years of follow-up, there was no statistically significant difference between 177Lu vipivotide tetraxetan and cabazitaxel for OS (HR = 0.97; 95% CI, 0.70 to 1.4; P = 0.99). 177Lu vipivotide tetraxetan was better than cabazitaxel for the primary end point of PSA response and for PFS (HR = 0.63; 95% CI, 0.46 to 0.86), rPFS (HR = 0.64; 95% CI, 0.46 to 0.88), ORR (relative risk = 2.12; 95% CI, 1.10 to 4.08), PSA PFS (HR = 0.60; 95% CI, 0.44 to 0.83), and pain (PPI) PFS (HR = 0.72; 95% CI, 0.53 to 0.97).
Harms Results
Figure 24 provides a summary of AEs reported in the TheraP trial for patients who received at least 1 dose of the study drugs (n = 98 for 177Lu vipivotide tetraxetan and n = 85 for cabazitaxel). Grade 1 or 2 AEs were more commonly reported in the 177Lu vipivotide tetraxetan group than in the cabazitaxel group (54% versus 40%), and grade 3 or 4 AEs were more commonly reported in the cabazitaxel group than in the 177Lu vipivotide tetraxetan group (53% versus 33%).
Table 42: Summary of Key Results From Pivotal and Protocol-Selected Studies
Key results | 177Lu vipivotide tetraxetan (N = 99) | Cabazitaxel (N = 101) | |
|---|---|---|---|
OS | |||
Deaths, n (%) | 77 (77.8) | 70 (69.3) | |
Censored, n (%) | NR | NR | |
Restricted mean survival time (95% CI) | 19.1 (16.9 to 21.4) | 19.6 (17.4 to 21.8) | |
HR (95% CI); P value | 0.97 (0.70 to 1.4); P = 0.99 | ||
PFS | |||
HR (95% CI); P value | 0.63 (0.46 to 0.86); P = 0.0028 | ||
rPFS | |||
HR (95% CI); P value | 0.64 (0.46 to 0.88); P = 0.0070 | ||
ORR | |||
Relative risk (95% CI); P value | 2.12 (1.10 to 4.08); P = 0.019 | ||
PSA PFS | |||
HR (95% CI); P value | 0.60 (0.44 to 0.83); P = 0.0017 | ||
PSA reduction of ≥ 50% | |||
Events, n (%) | 65 (65.7%) | 37 (36.6) | |
Risk Difference (95% CI) | 29% (95% CI, 16 to 42); P < 0.0001 | ||
Pain (PPI) PFS | |||
HR (95% CI); P value | 0.72 (0.53 to 0.97); P = 0.0328 | ||
177Lu = lutetium-177; CI = confidence interval; HR = hazard ratio; NR = not reported; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PPI = present pain intensity; PSA = prostate-specific antigen; rPFS = radiographic progression-free survival.
Sources: Hofman et al. (2021)45 and Hofman et al. (2022).43
Used with permission of Lancet Publishing Group, from Hofman MS, Emmett L, Sandhu S, et al. [177Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomized, open-label, phase II trial. Lancet. 2021;397(10276):797 to 804. Copyright: 2021; permission conveyed through Copyright Clearance Center, Inc.
Critical Appraisal
Internal Validity
Allocation: Randomization in the TheraP was performed using an appropriate methodology with adequate allocation concealment (i.e., centralized web-based system). Randomization was stratified by a different set of baseline parameters than in the VISION trial (i.e., disease burden based on metastatic sites [> 20 sites versus ≤ 20 sites], whether or not the patient had received previous treatment with enzalutamide or abiraterone, and the study site). Overall, baseline and demographic characteristics were well balanced in the 177Lu vipivotide tetraxetan and cabazitaxel groups in the TheraP trial.
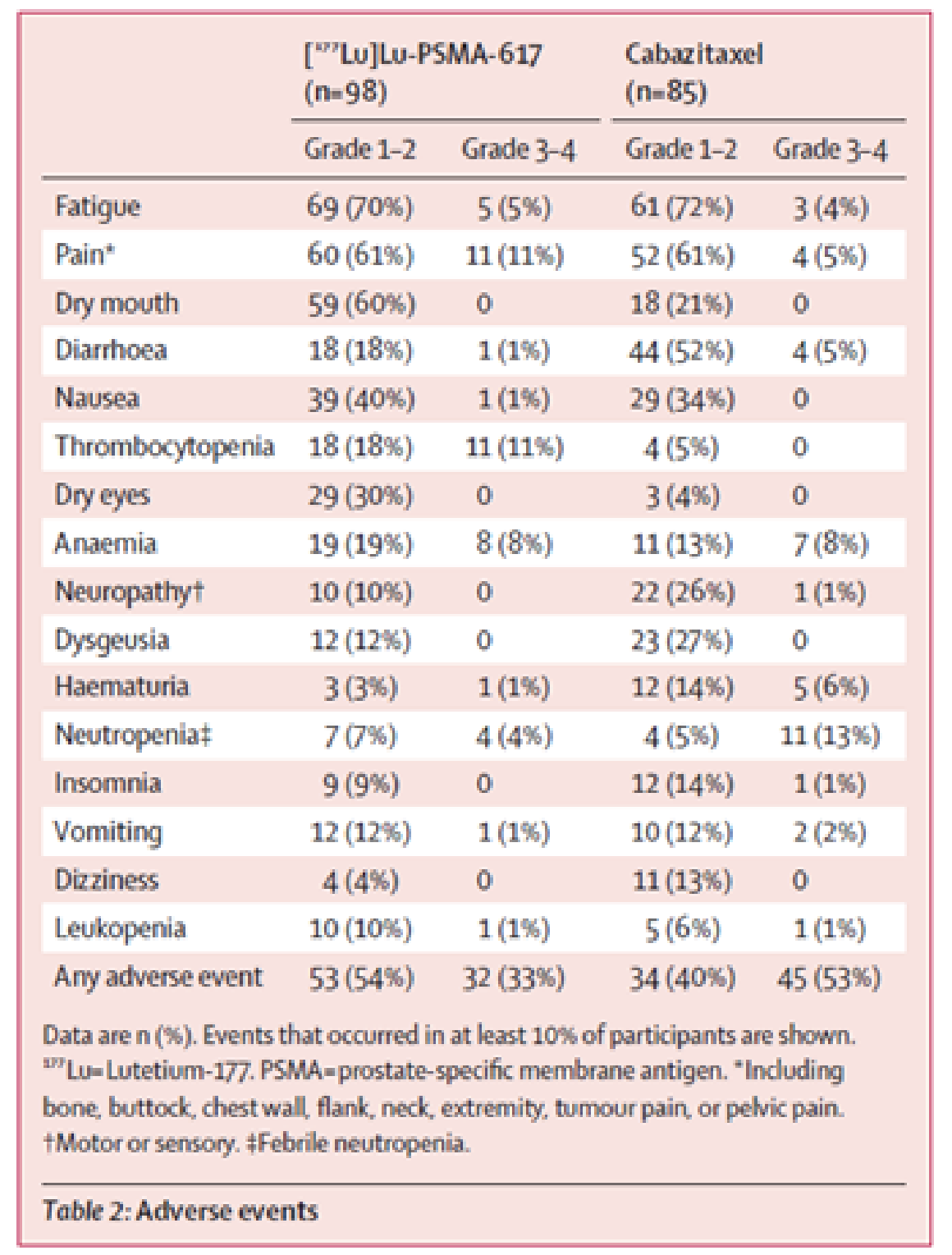
Blinding: As in the VISION trial, the study drugs in the TheraP trial were administered in an open-label manner (the rationale for open-label administration has been described previously). Radiographic images in the TheraP trial were evaluated centrally, but not in a manner that was blinded to the evaluator.
Concomitant and subsequent medications: A detailed description of concomitant supportive cancer therapies or subsequent anticancer therapies was not reported for the TheraP trial.
Patient disposition: As in the VISION trial, the internal validity of the TheraP trial was limited by the high and disproportionate early dropout rate in the comparator group (15.8% of patients in the cabazitaxel group compared with 1.0% in the 177Lu vipivotide tetraxetan group withdrew before receiving any doses of the study medications). The rationale provided was similar to that in the VISION trial (i.e., patient disappointment at not having access to 177Lu vipivotide tetraxetan). As in the VISION trial, the high and disproportionate number of patients who withdrew from the control group could bias the study results in favour 177Lu vipivotide tetraxetan, as those who remained in the study may have had a poorer prognosis than those who withdrew (although the magnitude of the potential bias is uncertain). The investigators performed a sensitivity analysis for the primary outcome using only patients who received the study treatments and noted that the results were supportive of the primary analysis.
End points: TheraP was a phase II study that was not designed or powered to evaluate differences between 177Lu vipivotide tetraxetan and cabazitaxel for the primary end points that are recommended by the PCWG3 (e.g., OS). The investigators reported an OS analysis after 3 years of follow-up, which noted no statistically significant difference between the 2 treatment groups; however, that analysis may be confounded by crossover or other potential differences in the subsequent therapy setting.
External Validity
Patient population: The clinical experts consulted by CADTH noted that the baseline and demographic characteristics for the TheraP trial are a reasonable reflection of the target patient population in Canada of those considered to be candidates for treatment with cabazitaxel.
Combination with systemic anticancer therapies: Unlike in the VISION trial, 177Lu vipivotide tetraxetan was administered as monotherapy (no other systemic anticancer drugs were permitted in the TheraP trial). Results of this study are likely more generalizable to the Canadian setting than those of the VISION trial, the clinical experts consulted by CADTH noted, because 177Lu vipivotide tetraxetan is expected to be used as monotherapy and evidence of the potential benefits of combination use, which could lead to increased AEs, is uncertain.
Comparator: The comparator in the TheraP trial (cabazitaxel) was relevant in the Canadian context for patients who have previously been treated with docetaxel and an ARPI. Unlike in the VISION trial, the TheraP study did not exclude patients who were medically unsuitable to receive further treatment with taxane regimen. The dosage of cabazitaxel (20 mg/m2 IV once every 3 weeks for a maximum of 10 cycles) is consistent with recommendations in the Canadian product monograph.16
Dosing: The maximum number of cycles used in the TheraP trial (i.e., 6 cycles) was consistent with the VISION trial and the Canadian product monograph; however, the dosage strength was not consistent with recommendations in the product monograph. Patients in the TheraP trial received an initial dose of 8.5 GBq, which was then decreased by 0.5 GBq each subsequent cycle; this is not reflective of the standardized dose of 7.4 GBq that is recommended in the Canadian product monograph.5
Treatment setting: The TheraP trial was conducted exclusively in Australia, which was not expected to limit generalizability of the results to the Canadian setting.
PSMA screening: PSMA status in the TheraP trial was determined using a 2-stage screening process: patients were initially screened using 68Ga-PSMA PET-CT, and subsequently evaluated using FDG PET-CT. Those who demonstrated discordant imaging between 68GA-PSMA PET-CT and FDG PET-CT (e.g., FDG intensity levels were greater than those observed with 68Ga-PSMA PET-CT) were excluded from the trial. The clinical experts consulted by CADTH noted that the more rigorous criteria applied in the TheraP could help identify patients who would be most likely to respond to 177Lu vipivotide tetraxetan; however, the need for 2 diagnostic PET-CT scans to determine PSMA status would likely pose implementation challenges in clinical practice for clinicians and the health system.
Discussion
Summary of Available Evidence
One RCT met the eligibility criteria for the Systematic Review. VISION (N = 831) was a phase III, open-label, RCT conducted to compare the efficacy and safety of 177Lu vipivotide tetraxetan administered in addition to BSC/BSoC with BSC/BSoC alone in patients with progressive PSMA-positive mCRPC. Patients were randomized in a 2:1 ratio to either 177Lu vipivotide tetraxetan + BSC/BSoC or BSC/BSoC alone, with allocation stratified by LDH (≤ 260 IU/L versus > 260 IU/L), the presence of liver metastases (yes versus no), ECOG PS (0 or 1 versus 2), and the inclusion of NAADs in BSC/BSoC (yes versus no). The primary and secondary end points of the VISON trial were aligned with those recommended by the PCWG3 (i.e., OS, rPFS, time to first SSE, HRQoL, PFS, and biochemical response [e.g., PSA]).
The VISION trial had considerable early withdrawal of consent and a disproportionate dropout rate in the BSC/BSoC alone group (patients typically cited disappointment that they would not receive 177Lu vipivotide tetraxetan). Withdrawal rates in the BSC/BSoC alone group were 56.0% before and 16.3% after the protocol amendment, compared with 1.2% before and 4.2% after in the 177Lu vipivotide tetraxetan group. This was a major limitation of the study and required the sponsor to introduce protocol amendments that included an increase in the overall target sample size, the introduction of educational measures to try to bolster the retention patients in the comparator group, and, most important from a critical appraisal perspective, the establishment of a new analysis set that would be limited to those enrolled after the protocol amendments were introduced (i.e., the PFS-FAS set). This new analysis set was used for the primary evaluation of all end points, with the exception of OS (FAS) and of ORR and DCR, which were evaluating using an even smaller subset of patients (i.e., those in the PFS-FAS who had RECIST-evaluable disease).
Evidence from the VISION trial was supplemented with data from a phase II trial comparing 177Lu vipivotide tetraxetan with cabazitaxel and an indirect comparison submitted by the sponsor.41-44 In addition, stakeholder input was received from 2 patient groups (the CCS and the CCSN) and 1 clinician group.
Based on the approved indication for 177Lu vipivotide tetraxetan, there are 3 relevant subpopulations for consideration in this review: patients previously treated with docetaxel who are considered eligible to receive cabazitaxel; patients previously treated with docetaxel who are considered ineligible to receive cabazitaxel; and patients previously treated with both docetaxel and cabazitaxel. The evidence for each of these populations is summarized and discussed in the next section.
Interpretation of Results
Efficacy
The results of the VISION trial suggest that treatment with 177Lu vipivotide tetraxetan + BSC/BSoC is associated with improvement in OS compared with BSC/BSoC alone, with a median survival of 15.3 months (95% CI, 14.2 to 16.9 months) versus 11.3 months (95% CI, 9.8 to 13.5 months), and an HR of 0.62 (95% CI, 0.52 to 0.74). Unlike all other efficacy end points in the VISION trial, the primary analysis of OS included all randomized patients (i.e., the FAS dataset). There was still a high and disproportionate number of discontinuations in the BSC/BSoC alone group, however, the VISION protocol allowed patients to continue to be followed for long-term safety, rPFS, and OS follow-up. Health Canada noted that this aspect of the protocol mitigated concerns about the differential dropout rate with respect to the OS analysis, as it allowed the sponsor to ascertain survival (e.g., missing data were obtained for 34 patients through public registries).8 As such, the results for OS were considered to be internally valid and clinically meaningful by Health Canada and the FDA;8,9 these results are the only data presented in the Canadian product monograph.5 The FDA examined numerous sensitivity analyses conducted for the OS end point and concluded that 177Lu vipivotide tetraxetan + BSC/BSoC had a robust statistically significant effect on OS compared with BSC/BSoC alone. They noted that sensitivity analyses suggested that there could be some diminution of the OS magnitude of effect (i.e., potential for bias in favour in 177Lu vipivotide tetraxetan); however, they concluded that the precision of OS as an end point and its meaningfulness as a clinical outcome are such that even a slightly smaller magnitude in the delay of death than what is reported would still be clinically meaningful and support a favourable benefit-risk profile for the target population of patients with mCRPC. The clinical experts consulted by CADTH also noted that the effect size would surpass thresholds for clinical relevance for patients with mCRPC who were heavily pretreated before the study.
Although the sponsor was able to obtain mortality data for patients who withdrew from the study, it is unclear if these patients were receiving the same level of optimized care that would be provided in the clinical trial setting (e.g., there are missing data regarding the supportive medications and anticancer therapies administered outside the trial protocol). In addition to the issues posed by the high rate of early discontinuation from the VISION trial and the subsequent missing information, the choice of comparator in the VISION trial poses some external validity challenges, as the protocol restricted access to some therapies. Canadian clinical practice guidelines for mCRPC recommend the use of a drug with a different mechanism of action in progressive lines of therapy, and the clinical experts consulted by CADTH noted that re-treatment with an ARPI was not considered to be a relevant comparator, given that the approved indication for 177Lu vipivotide tetraxetan requires prior treatment with and progression on an ARPI.4 This is also reflected in the proposed place in therapy for 177Lu vipivotide tetraxetan that was submitted by the sponsor, which underscores the fact that reimbursement in many Canadian jurisdictions is limited to a single ARPI for patients with mCRPC.6 Acknowledging these limitations, the clinical experts consulted by CADTH agreed with the perspective of regulatory authorities that the OS benefit reported with 177Lu vipivotide tetraxetan is clinically meaningful for a patient population with no effective treatment alternatives.
Patients Previously Treated With Docetaxel Who Are Considered Ineligible to Receive Cabazitaxel
The inclusion criteria for the phase III VISION trial limited enrolment to patients who had received prior therapy with at least 1 taxane regimen, and those with exposure to only a single taxane regimen (57.9% of study participants) must have been deemed unsuitable to receive a second taxane regimen (e.g., because of frailty assessed with a geriatric or health status evaluation or intolerance). The subgroup analysis based on the number of prior taxane regimens favoured 177Lu vipivotide tetraxetan + BSC/BSoC over BSC/BSoC alone for those who had received a single prior taxane regimen (HR = 0.59; 95% CI, 0.46 to 0.75).
Despite the eligibility criteria for the VISION trial stating that patients must be ineligible to receive a taxane regimen, 14.9% and 18.9% of patients in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups, respectively, received cabazitaxel after discontinuing the study drug regimens. It is unclear why these patients were eligible for therapy with a taxane regimen after discontinuation of the study drugs but were considered ineligible to receive a taxane at the time of screening. In a critique of the VISION trial, Olivier et al. (2022)11 state that prostate cancer specialists have suggested that these patients should have received cabazitaxel before enrolment in the trial, as the permitted BSC/BSoC regimens represented suboptimal care in comparison. The clinical experts consulted by CADTH also noted that those patients should likely have been treated with cabazitaxel before enrolment in the VISION trial.
The clinical experts consulted by CADTH provided guidance on the patient characteristics that would be used in Canadian clinical practice to identify patients who are not be medically suited for taxane-based therapy (these are described in Table 5 and include ECOG PS, laboratory measures, and cognitive function and/or social support to monitor and report toxicities). Such criteria were not specifically stated in the protocol for the VISION trial and the decision was based on investigator judgment.7
Patients Previously Treated With Both Docetaxel and Cabazitaxel
Clinician group input identified a significant unmet medical need for patients who have demonstrated disease progression after therapy with 2 taxane regimens and a lack of therapies with evidence supporting an improvement in survival. In the VISION trial, 41.2% of the trial population had received 2 prior taxane-containing regimens at the time of enrolment.7 The subgroup analysis of OS based on the number of prior taxane regimens favoured 177Lu vipivotide tetraxetan + BSC/BSoC over BSC/BSoC alone for those who had received 2 or more prior taxane regimens (HR = 0.73; 95% CI, 0.53 to 0.99).7 The clinical experts consulted by CADTH noted that this is a patient population with a high-level of unmet needs, as there are no standard therapies that have been shown to increase OS. The clinical experts also noted that, in this context, the availability of an active drug shown to improve OS would fill an unmet need in this population, given the clinically meaningful magnitude of benefit in this subgroup of patients.
Previously Treated With Docetaxel Who Are Considered Eligible to Receive Cabazitaxel
As mentioned, the inclusion criteria in the VISION trial specified that patients previously treated with docetaxel and considered eligible to receive cabazitaxel were to be excluded from the study.7 As this population is included in the Health Canada–approved indication,5 CADTH considered this to be an important gap in the evidence and, therefore, summarized the phase II TheraP trial, which enrolled patients with prior exposure to docetaxel and for whom cabazitaxel was considered the appropriate treatment option.42-44 Although the TheraP trial met its primary objective (more patients in the 177Lu vipivotide tetraxetan group than in the cabazitaxel group had a PSA reduction of at least 50%), as a phase II trial, TheraP was not designed to evaluate potential differences in PFS or OS between 177Lu vipivotide tetraxetan and cabazitaxel. Three-year follow-up data demonstrated no statistically significant difference between the 2 treatments for OS. The internal validity of the study is limited by the open-label design, the high and differential withdrawal rate in the control group (much like in the VISION trial), and crossover and other potential differences in the subsequent therapy setting.
Additional End Points
Because of the postwithdrawal follow-up that occurred for the OS end point, it was not possible to determine rPFS, as it would require information about radiographic disease progression that could not be readily obtained by the sponsor outside of the clinical trial setting. The impacts on the integrity of the randomization that the early and disproportionate withdrawals in the control group had and the limited information related to subsequent treatments and outcomes means that concrete conclusions should not be drawn from the PFS results. Regulatory authorities likewise noted that there is the potential for bias and uncertainty in the analysis of rPFS for the following reasons: the high proportion of patients excluded from the PFS-FAS (i.e., those randomized before the March 5, 2019, amendment); the imbalance in censoring (34% in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 52.6% in the BSC/BSoC alone group), which was identified as potentially biasing the results in favour of 177Lu vipivotide tetraxetan + BSC/BSoC; the high number of protocol deviations and the imbalance of those deviations across treatment groups (50.6% in the 177Lu vipivotide tetraxetan + BSC/BSoC group and 38.6% in the BSC/BSoC alone group); and the higher proportion of patients in the BSC/BSoC alone group than in the 177Lu vipivotide tetraxetan + BSC/BSoC group with no adequate assessment of rPFS (17.9% vs 1.3%).8 The sponsor conducted sensitivity analyses to examine the robustness of the rPFS results; however, Health Canada reviewers concluded that these did not eliminate the uncertainty and did not feel that the results of the sensitivity analyses were consistent with or supportive of the primary rPFS analysis (citing the heterogenous results, with an HR that ranged from 0.4 to 0.77 and a 99.2% CI that ranged from 0.29 to 1.07). Overall, the CADTH reviewers agreed with the Health Canada conclusion that the rPFS results were not sufficiently robust or reliable.8
As with rPFS, ORR and DCR were only evaluated in the subset of patients enrolled after the March 5, 2019 amendment.7 In addition, these end points were only evaluated for patients who met the criteria for evaluable disease at baseline (i.e., the response-evaluable analysis set), which was compromised of an even smaller set of randomized patients (42.9% and 57.9% in the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone arms, respectively). Although 177Lu vipivotide tetraxetan + BSC/BSoC, compared with BSC/BSoC alone, was associated with an improvement in ORR (29.8% versus 1.7%) and DCR (89.0% versus 66.7%),7 Health Canada concluded that there are important limitations with these analyses and that the results cannot be included in the Canadian product monograph.8 Specific concerns included the exclusion of such a large proportion of the overall trial population from the response-evaluable analysis set, imbalance in the proportion of patients excluded in the 2 treatment groups, and the very high proportion of patients excluded from the BSC/BSoC alone group (i.e., 57.1%). Health Canada reviewers questioned the reliability of the ORR evaluation, which found that only 2 patients in the BSC/BSoC alone group demonstrated a PR to the BSoC regimen (there were no CRs), and the sponsor acknowledged that the estimated OR and 95% CI in the PFS-FAS could be attributed to the observed low response rate in the BSC/BSoC alone arm. Overall, the estimated relative benefit of ORR may be overestimated.7
177Lu vipivotide tetraxetan + BSC/BSoC was associated with a reduction in SSEs (or death) compared to BSC/BSoC alone in the VISION trial (HR = 0.5; 95% CI, 0.40 to 0.62). This analysis is associated with the same statistical and interpretation issues that were previously described for the PFS-FAS dataset. As well, the PCWG3 advises that the reporting of SSEs in prostate cancer clinical trials should be limited to events that are symptomatic because of their clinical significance. Health Canada noted that this recommendation was not followed in the VISION trial and that most of the SSEs were death driven (i.e., 103 of the 196 events in the 177Lu vipivotide tetraxetan + BSC/BSoC group were deaths).8 Health Canada further noted that the censoring of events that occurred at the EOT visit creates further uncertainty and limits the ability to interpret the results, and stated that SSEs should not be reflected in the Canadian product monograph.8
The PCWG3 recommends that prostate cancer trials include measures of biochemical response, and the VISION trial included PSA, LDH, and alkaline phosphatase levels. There were no statistical comparisons for change from baseline in LDH or alkaline phosphatase levels; therefore, the results are not discussed further. There were also no statistical comparisons provided for PSA doubling time, and the sponsor noted that the proportion of patients who could be evaluated for this end point was not balanced between the 177Lu vipivotide tetraxetan + BSC/BSoC and BSC/BSoC alone groups (73.8% and 37.8%, respectively). For change from baseline in PSA levels, 177Lu vipivotide tetraxetan + BSC/BSoC was associated with statistically significant improvements in PSA response and duration of PSA response. These analyses are limited by the issues previously described for the PFS-FAS dataset, the high and disproportionate amount of missing data, and the lack of control for type I error. Health Canada concluded that these end points should be interpretated with caution, and the results are not reflected in the Canadian product monograph.8
177Lu vipivotide tetraxetan + BSC/BSoC demonstrated improvements from baseline in time to worsening measured with FACT-P, FACT-G, FAPSI-8, and EQ-5D-5L, compared with BSC/BSoC alone. HRQoL end points can be subject to bias in trials with the open-label administration of study drugs. This may be particularly challenging in populations such as that enrolled in the VISION trial, as evidenced by the high early withdrawal of patients randomized to the BSC/BSoC alone group (e.g., many of those who withdrew cited disappointment with their allocated group as a reason). Overall, these analyses were not considered reliable by CADTH or by regulatory authorities because of the probability of bias.
In the absence of head-to-head studies comparing 177Lu vipivotide tetraxetan with other treatments in adults with mCRPC who have received ARPI and at least 1 taxane-based chemotherapy, the sponsor submitted a Bayesian NMA approach that evaluated the efficacy of 177Lu vipivotide tetraxetan relative to relevant comparators (i.e., radium-223 plus BSC, cabazitaxel plus prednisone, olaparib, mitoxantrone or placebo plus prednisone, and ARPI). The sponsor-submitted ITC reported that 177Lu vipivotide tetraxetan was superior to radium-223 plus BSC and to ARPI for OS. The clinical experts consulted by CADTH noted that there was heterogeneity in clinically important patient characteristics (i.e., historical use of chemotherapy, disease severity, biomarker status for BRCA1, BRCA2, and ATM mutations, and treatment indication); therefore, the ITC analysis may be subject to bias. The clinical experts did not feel that the comparisons in the sponsor’s NMA could be applied to reasonably reflect either a potential purported clinical trial or real-world comparisons of effectiveness. Of particular concern was the fact that the patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group in the VISION trial had more severe disease at baseline than those in the BSC/BSoC alone group, as indicated by a higher prior treatment count, and that at least 40% of patients had received cabazitaxel before enrolment. In the absence of statistical adjustment, sensitivity analyses, or subgroup analyses, the potential impact of the between-study heterogeneity cannot be evaluated. Overall, the sponsor-submitted ITC has important limitations that preclude the drawing of conclusions regarding the efficacy of 177Lu vipivotide tetraxetan relative to relevant comparators in the target patient population.
Harms
The product monograph for 177Lu vipivotide tetraxetan provides detailed recommendations for the management of AEs that require temporary interruption, dose reduction, or discontinuation of treatment (Table 3).5 The clinical experts consulted by CADTH indicated that this is a reasonable reflection of the way patients would be managed in clinical practice.
The product monograph for 177Lu vipivotide tetraxetan contains black-box warnings regarding the risk of myelosuppression and renal toxicity, so the following tests are recommended before and during treatment: hematology (hemoglobin, white blood cell count, absolute neutrophil count, platelet count); kidney function (serum creatinine, calculated creatinine clearance); and liver function (alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, blood serum albumin, total blood bilirubin).5 The clinical experts consulted by CADTH noted that these are routine pretreatment laboratory measures that are performed before the administration of any systemic anticancer therapy and would be performed before the administration of each treatment cycle with 177Lu vipivotide tetraxetan. The experts noted that these requirements would not lead to an increase in laboratory testing for patients receiving treatment with 177Lu vipivotide tetraxetan rather than with alternative treatments.
More patients in the 177Lu vipivotide tetraxetan + BSC/BSoC group than in the BSC/BSoC alone group experienced at least 1 SAE (36.3% versus 27.8%), a grade 3, 4, and 5 AE (52.7% versus 38.0%), and withdrawal due to adverse event (11.9% versus 0.5%). The increase in AEs overall is expected, given that the VISION trial examined the use of 177Lu vipivotide tetraxetan as an add-on therapy and both groups received BSC/BSoC.8 As expected, the high rate of withdrawal from the BSC/BSoC alone group resulted in less overall exposure to the study treatments and less time for AEs to accrue during the trial. Nonetheless, the higher frequency of withdrawal due to adverse event and cytopenias secondary to myelosuppression reported in the VISION trial was notable. In the TheraP trial, grade 3 or 4 AEs were more commonly reported in the cabazitaxel group than in the 177Lu vipivotide tetraxetan group (53% versus 33%).
Other Considerations
Access to PSMA Testing
The product monograph for 177Lu vipivotide tetraxetan states that tumour PSMA expression should be verified before the initiation of treatment. There is no specific companion diagnostic test for 177Lu vipivotide tetraxetan (there are several PSMA-targeted radiopharmaceuticals approved, undergoing regulatory review, or in clinical development);23,24,26 however, the product monograph notes that PSMA expression was detected with PET diagnostic imaging using 68Ga-PSMA-11 in the VISION trial.5 The CUA recommendations regarding the application of PSMA testing in Canadian practice predate the regulatory approval of 177Lu vipivotide tetraxetan and do not specifically address the use of PSMA testing in the context of identifying candidates for treatment with the drug under review.27 The CUA statement recommends that PSMA-targeted PET should not be routinely offered outside a clinical trial for patients with mCRPC (i.e., the target population for 177Lu vipivotide tetraxetan), and the clinical experts consulted by CADTH noted that PSMA testing with PET-CT is not widely available in Canadian routine practice and typically only performed as part of clinical studies, through private mechanisms, or in very rare cases for which there is the potential for another malignant diagnosis and the clinical team requires clarity on the histology of the disease. The experts noted that patients may encounter financial and logistical challenges (e.g., interprovincial travel to access PSMA testing).
Through its Canadian Medical Imaging Inventory (CMII), CADTH tracks and analyzes imaging capacity, exam volume, hours of operation, and types of use for medical imaging.46,47 CADTH recently used the CMII to conduct a review of PSMA PET-CT imaging for the staging of prostate cancer in Canada48 to examine health system infrastructural readiness for the widespread adoption of PSMA PET-CT imaging. The CADTH analysis estimated that the diagnostic use of PSMA PET-CT would require a minimum increase of 23% in the existing PET-CT exam volume. The estimated increase was based solely on changes to accommodate PSMA PET-CT and did not consider other competing health system demands for this imaging modality. With 57 units in Canada, patient access to PET-CT is limited and, as such, it is unlikely that all eligible patients with prostate cancer will be able to receive a PSMA PET-CT exam in a reasonable time frame.48
Access to Radiopharmaceutical Facilities
The product monograph states that 177Lu vipivotide tetraxetan should be administered under the supervision of a health professional experienced in the use of radiopharmaceuticals and that appropriate management of therapy and complications is only possible when adequate diagnostic and treatment facilities are readily available.5 The clinical experts consulted by CADTH noted that these requirements, although necessary, would likely pose a challenge to patient access based on the number and/or location of currently available facilities (e.g., regional variation, existing capacity constraints). The experts noted that the use of radiopharmaceuticals in the treatment of prostate cancer varies across Canada because of differences in the reimbursement status of radium-223 across jurisdictions. They noted that, even in large academic treatment centres in Canada, there are likely few medical oncologists who have practical experience with radioligand therapy for prostate cancer.
CADTH’s CMII report on PSMA PET-CT imaging noted that an increase in imaging capacity to accommodate PSMA testing would likely require investment in the health care professionals required to perform the diagnostic test in the specialized radiopharmaceutical facilities.48 As PSMA testing is not currently performed in routine clinical practice,27 health care professionals who interpret PSMA test results may require additional education and training.47 An additional CADTH CMII report on the future of PET-CT in Canada47 noted that shortages in nuclear medicine technologists, particularly those with experience in PET-CT, are common in provinces with existing PET-CT capacity and, in some jurisdictions, shortages of medical physicists and radiochemists have also been reported.
Combination Use of 177Lu Vipivotide Tetraxetan With Other Systemic Anticancer Therapies
177Lu vipivotide tetraxetan was administered as an add-on therapy in the VISION trial, which included the concomitant administration of other systemic cancer therapies. It is currently unclear whether clinical specialists in Canada would be interested in using 177Lu vipivotide tetraxetan in combination with other systemic anticancer therapies. The clinical experts consulted by CADTH noted that combination use in Canada may be limited by the reimbursement status of other systemic anticancer therapies used to treat patients with mCRPC. Public reimbursement of ARPIs after a patient has demonstrated disease progression on the therapy varies across jurisdictions, with some provinces mandating discontinuation of coverage and others permitting the continuation of therapy.40 Overall, the experts noted that it is uncertain if the use of 177Lu vipivotide tetraxetan in combination with other systemic anticancer therapies offers additional clinical benefit for patients. An ongoing phase II study (Enza-P) is specifically investigating the safety and efficacy of 177Lu vipivotide tetraxetan used in combination with enzalutamide compared with enzalutamide alone in patients with mCRPC who are at high risk of early progression.49
The clinical experts noted that a previous phase III clinical trial (ERA 223) involving the combination of radium-223 and an ARPI regimen (abiraterone plus prednisone) in 806 patients demonstrated no increase in OS and increased toxicity.50,51 It is uncertain whether use of 177Lu vipivotide tetraxetan in combination with an ARPI would increase harms relative to those who received 177Lu vipivotide tetraxetan alone or those who received the ARPI as part of the BSC/BSoC regimen.
Potential Expanded Indications for 177Lu Vipivotide Tetraxetan
The sponsor is currently conducting 2 phase III studies to examine the efficacy of 177Lu vipivotide tetraxetan: the PSMAddition trial,52 with a planned enrolment of 1126 patients with metastatic hormone-sensitive prostate cancer; and the PSMAfore trial,53 with a planned enrolment of 450 patients with mCRPC who have progressed on an ARPI but who are naive to taxane therapy.
Conclusions
177Lu vipivotide tetraxetan injection is indicated for the treatment of adults with PSMA-positive mCRPC who have received at least 1 ARPI and taxane-based chemotherapy. There are currently few effective treatments for patients with mCRPC who have progressed after treatment with an ARPI and docetaxel, and all stakeholders identified important unmet medical needs, particularly for patients who may be ineligible to receive additional therapy with taxane regimens.
The CADTH review included 1 phase III RCT (VISION; N = 831) that compared the use of 177Lu vipivotide tetraxetan as an add-on therapy to BSC/BSoC with BSC/BSoC alone. The VISION trial suggested that 177Lu vipivotide tetraxetan + BSC/BSoC was superior to BSC/BSoC alone for a series of outcomes considered important in the evaluation of prostate cancer therapies (i.e., OS, rPFS, time to first SSE, HRQoL, and PFS). However, there is uncertainty regarding the internal validity of the results because of several important limitations, most notably the considerable early withdrawal of consent and disproportionate dropout rate in the BSC/BSoC alone group. The extent of early withdrawal was substantial enough to require amendments to the VISION protocol, including the establishment of a new analysis set that was limited to those enrolled after the amendments. This new analysis set was used for the primary evaluation of all end points, with the exception of OS (as the sponsor was able to obtain mortality data for those who withdrew from the study) and ORR (which was based on a smaller subset of patients). Regulatory authorities and the clinical experts consulted by CADTH considered the results for OS to be clinically important, given that the patients enrolled in the VISION trial were heavily pretreated. The choice of BSC/BSoC as the comparator in the VISION trial and the limitations of sponsor’s ITC preclude the drawing of any conclusions regarding the efficacy of 177Lu vipivotide tetraxetan relative toother relevant comparators for OS.
The inclusion criteria in the VISION trial specified that patients who were previously treated with docetaxel and considered eligible to receive cabazitaxel were to be excluded from the study (despite this, approximately 18% of patients received cabazitaxel in the poststudy treatment setting). To address this important gap in the evidence, CADTH summarized results from the TheraP trial (N = 200), which enrolled patients with prior exposure to docetaxel and for whom cabazitaxel was considered the appropriate treatment option. The phase II TheraP trial was not designed or powered to evaluate potential differences in OS, and there was no statistically significant difference between 177Lu vipivotide tetraxetan and cabazitaxel for OS. Treatment with 177Lu vipivotide tetraxetan was statistically superior to cabazitaxel for the primary end point of PSA response, PFS, rPFS, ORR, PSA PFS, and pain PFS.
177Lu vipivotide tetraxetan is the first drug specifically indicated for use in patients with PSMA-positive prostate cancer. The evaluation of PSMA status requires PET-CT imaging with a PSMA-targeted radioligand, a process not routinely performed in Canadian clinical practice at the time of CADTH’s review. The potential health system implications and imaging resource requirements may pose important implementation challenges that could affect the integration and uptake of 177Lu vipivotide tetraxetan in Canadian practice.
References
1.Public Health Agency of Canada. Prostate cancer in Canada. 2021: https://www.canada.ca/en/public-health/services/publications/diseases-conditions/prostate-cancer.html. Accessed 2022 Oct 22.
2.Prostate cancer statistics. Toronto (ON): Canadian Cancer Society; 2022: https://www.cancer.ca/en/cancerinformation/cancer-type/prostate/statistics/?region=on. Accessed 2022 Oct 27.
3.American Cancer Society. Survival Rates for Prostate Cancer. 2021: https://www.cancer.org/cancer/prostatecancer/detection-diagnosis-staging/survival-rates.html. Accessed 2022 Oct 27.
4.Saad F, Aprikian A, Finelli A, et al. 2021 Canadian Urological Association (CUA)-Canadian Uro Oncology Group (CUOG) guideline: Management of castration-resistant prostate cancer (CRPC). Can Urol Assoc J. 2021;15(2):E81-e90. PubMed
5.Pluvicto: lutetium (177 Lu) vipivotide tetraxetan injection, solution for injection/infusion 1 000 MBq/mL at calibration; Therapeutic Radiopharmaceutical [product monograph]. Millburn (NJ): Advanced Accelerator Applications USA, Inc.; 2022 Jun 29.
6.Drug Reimbursement Review sponsor submission: lutetium (177Lu) vipivotide tetraxetan [internal sponsor's package]. Mississauga (ON): Advanced Accelerator Applications Canada Inc.; 2022 Jul 21.
7.Clinical Study Report: PSMA-617-01. VISION: An international, prospective, open label, multicenter, randomized phase 3 study of 177Lu-PSMA-617 in the treatment of patients with progressive PSMA-positive metastatic castration-resistant prostate cancer (mCRPC) [internal sponsor's report]. Millburn (NJ): Advanced Accelerator Applications; 2021 Jun 28.
8.Health Canada reviewer's report: Pluvicto (lutetium (177Lu) vipivotide tetraxetan injection) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lutetium (177Lu) vipivotide tetraxetan injection. Ottawa (ON): Sponsor name; 2022.
9.Center for Drug Evaluation Research. Multidiscipline review(s). Pluvicto (lutetium lu-177 vipivotide tetraxetan injection solution. Company: Advanced Accelerator Applications USA, Inc., A Novartis Company. Application No.:215833. Approval date: 03/23/2022 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2022 Mar 23: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/215833Orig1s000MultidisciplineR.pdf. Accessed 2022 Jul 28.
10.Pluvicto (lutetium Lu 177 vipivotide tetraxetan) injection, for intravenous use [package insert]. Millburn, NJ: Advanced Accelerator Applications USA, Inc.; 2022: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215833s000lbl.pdf. Accessed 2022 Oct 2.
11.Olivier T, Powell K, Prasad V. Lutetium-177-PSMA-617 in Metastatic Castration-resistant Prostate Cancer: Limitations of the VISION Trial. Eur Urol. 2022 Sep 9;S0302-2838(22)02613-6.
12.Saad F, Aprikian A, Finelli A, et al. 2022 Canadian Urological Association (CUA)-Canadian Uro Oncology Group (CUOG) guideline: Management of castration-resistant prostate cancer (CRPC). Canadian Urological Association Journal. 2022;16(11):E506-515. PubMed
13.Xofigo; radium Ra 223 dichloride solution for injection [product monograph]. Mississauga (ON): Bayer Inc.; 2019 Aug 1: https://pdf.hres.ca/dpd_pm/00052465.PDF. Accessed 2022 Oct 2.
14.Lynparza; olaparib 100 mg and 150 mg tablets [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; August 5, 2022: https://pdf.hres.ca/dpd_pm/00066912.PDF. Accessed 2022 Oct 22.
15.Xtandi, enzalutamide capsules 40 mg [product monograph]. Markham (ON): Astellas Pharma Canada, Inc.; 2022 Jan 24: https://pdf.hres.ca/dpd_pm/00064474.PDF. Accessed 2022 Oct 2.
16.Cabazitaxel For Injection; Concentrated Solution 10 mg/mL (45 mg/4.5 mL & 60 mg/6 mL) [product monograph]. Boucherville (QC): Sandoz Canada Inc.; July 18, 2022: https://pdf.hres.ca/dpd_pm/00066736.PDF. Accessed 2022 Oct 2.
17.Docetaxel, 10 mg/mL for intravenous infusion [product monograph] Kirkland (QC): Pfizer Canada ULC; March 26, 2021: https://pdf.hres.ca/dpd_pm/00060616.PDF. Accessed 2022 Oct 2.
18.Zytiga, abiraterone acetate, 250 mg uncoated tablets, 500 mg film-coated tablets, oral [product monograph]. Toronto (ON): Janssen Inc.; 2021 Nov 15: https://pdf.hres.ca/dpd_pm/00063577.PDF. Accessed 2022 Oct 2.
19.CADTH Drug Reimbursement Expert Review Committee final recommendation: Olaparib (Lynparza) (AstraZeneca Canada Inc.). Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/pcodr/Reviews2021/10223OlaparibmCRPC_fnRec_REDACT_EC21Apr2021_final.pdf. Accessed 2022 Oct 2.
20.EAU - EANM - ESTRO - ESUR - ISUP - SIOG Guidelines on Prostate Cancer. Arnhem, The Netherlands: EAU Guidelines Office; 2022: https://d56bochluxqnz.cloudfront.net/documents/full-guideline/EAU-EANM-ESTRO-ESUR-ISUP_SIOG-Guidelines-on-Prostate-Cancer-2022_2022-04-25-063938_yfos.pdf. Accessed 2022 Oct 22.
21.European Society for Medical Oncology. Genitourinary Cancers Pocket Guideline. 2022: https://interactiveguidelines.esmo.org/esmo-web-app/toc/index.php?subjectAreaId=14&pdfId=4. Accessed 2022 Oct 22.
22.NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Prostate Cancer Version 4.0. National Comprehensive Cancer Network; 2022: https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf. Accessed 2022 Aug 16.
23.Illucix, gallium (68Ga) PSMA-11, 25 microgram PSMA-11 per vial, Kit for the preparation for intravenous injection [product monograph]. Fishers (IN): Telix Pharmaceuticals (US), Inc.; October 13, 2022: https://pdf.hres.ca/dpd_pm/00067755.PDF. Accessed 2022 Oct 22.
24.Health Canada. Drug and Health Product Submissions Under Review (SUR): New drug submissions under review. 2022; https://www.canada.ca/en/health-canada/services/drug-health-product-review-approval/submissions-under-review/new-drug-submissions-under-review.html. Accessed 2022 Oct 22.
25.Pylarify (piflufolastat F 18) injection, for intravenous use [package insert]. Progenics Pharmaceuticals, Inc.; 2022: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214793s000lbl.pdf. Accessed 2022 Oct 2.
26.Centre for Probe Development and Commercialization. NCT04644822: Safety and Efficacy of [18F]PSMA-1007 Injection in Suspected Persistent or Recurrent Prostate Cancer. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT04644822. Accessed 2022 Oct 2.
27.Shaygan B, Zukotynski K, Bénard F, et al. Canadian Urological Association best practice report: Prostate-specific membrane antigen positron emission tomography/computed tomography (PSMA PET/CT) and PET/magnetic resonance (MR) in prostate cancer. Can Urol Assoc J. 2021;15(6):162-172. PubMed
28.U.S. Food and Drug Administration. FDA approves olaparib for HRR gene-mutated metastatic castration-resistant prostate cancer. 2020; https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer. Accessed 2022 Oct 22.
29.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
30.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Oct 6.
31.Sartor O, de Bono J, Chi KN, et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2021;385(12):1091-1103. PubMed
32.Endocyte. NCT03511664: Study of 177Lu-PSMA-617 In Metastatic Castrate-Resistant Prostate Cancer (VISION). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT03511664. Accessed 2022 Oct 2.
33.Logothetis CJ, Basch E, Molina A, et al. Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: exploratory analysis of data from the COU-AA-301 randomised trial. Lancet Oncol. 2012;13(12):1210-1217. PubMed
34.Kuppen MCP, Westgeest HM, van den Eertwegh AJM, et al. Health-related quality of life and pain in a real-world castration-resistant prostate cancer population: results from the PRO-CAPRI study in the Netherlands. Clin Genitourin Cancer. 2020;18(3):e233-e253. PubMed
35.Cella D, Nichol MB, Eton D, Nelson JB, Mulani P. Estimating Clinically Meaningful Changes for the Functional Assessment of Cancer Therapy—Prostate: Results from a Clinical Trial of Patients with Metastatic Hormone-Refractory Prostate Cancer. Value Health. 2009;12(1):124-129. PubMed
36.Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727-1736. PubMed
37.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-Defined Estimates of the Minimally Important Difference for EQ-5D-5L Index Scores. Value Health. 2017;20(4):644-650. PubMed
38.Advanced Accelerator Applications response to request for additional information regarding Pluvicto review [internal additional sponsor's information]. Advanced Accelerator Applications; 2022.
39.George DJ, Sartor O, Miller K, et al. Treatment Patterns and Outcomes in Patients With Metastatic Castration-resistant Prostate Cancer in a Real-world Clinical Practice Setting in the United States. Clin Genitourin Cancer. 2020;18(4):284-294. PubMed
40.Exceptional Access Program Reimbursement Criteria for Frequently Requested Drugs. Ontario Ministry of Health; September 29, 2022: https://health.gov.on.ca/en/pro/programs/drugs/docs/frequently_requested_drugs.pdf. Accessed 2022 Oct 22.
41.Network meta-analysis in progressive, PSMA–positive metastatic castration-resistant prostate cancer [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lutetium (177Lu) vipivotide tetraxetan. Mississauga (ON): Advanced Accelerator Applications Canada Inc. 2022 Jul 21.
42.Australian and New Zealand Urogenital and Prostate Cancer Trials Group. NCT03392428: A Trial of 177Lu-PSMA617 Theranostic Versus Cabazitaxel in Progressive Metastatic Castration Resistant Prostate Cancer (TheraP). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT03392428. Accessed 2022 Oct 2.
43.Hofman MS, Emmett L, Sandhu S, et al. TheraP: 177Lu-PSMA-617 (LuPSMA) versus cabazitaxel in metastatic castration-resistant prostate cancer (mCRPC) progressing after docetaxel—Overall survival after median follow-up of 3 years (ANZUP 1603). J Clin Oncol. 2022;40(16_suppl):5000-5000.
44.Hofman MS, Emmett L, Violet J, et al. TheraP: a randomized phase 2 trial of 177 Lu-PSMA-617 theranostic treatment vs cabazitaxel in progressive metastatic castration-resistant prostate cancer (Clinical Trial Protocol ANZUP 1603). BJU Int. 2019;124 Suppl 1:5-13. PubMed
45.Hofman MS, Emmett L, Sandhu S, et al. [<sup>177</sup>Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomised, open-label, phase 2 trial. Lancet. 2021;397(10276):797-804. PubMed
46.Chao YS, Sinclair A, Morrison A, Hafizi D, Pyke L. The Canadian Medical Imaging Inventory 2019-2020. (CADTH health technology review). Can J Health Technol. 2021;1(1):1-215. https://cadth.ca/sites/default/files/ou-tr/op0546-cmii3-final-report.pdf. Accessed 2022 Jan 10.
47.Morrison A. The future of PET-CT in Canada. Canadian Medical Imaging Inventory Report. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/attachments/2021-10/cmii_service_future_of_PET_CT_0.pdf. Accessed 2021 Oct 8.
48.Prostate-Specific Membrane Antigen PET-CT Imaging for the Staging of Prostate Cancer in Canada. Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/attachments/2022-11/CM0001-PSMA-PET3.0-KH-AM-KH-KH2%20-%20with%20updates%20-%20QC-meta.pdf. Accessed 2022 Nov 15.
49.Emmett L, Subramaniam S, Joshua AM, et al. ENZA-p trial protocol: a randomized phase II trial using prostate-specific membrane antigen as a therapeutic target and prognostic indicator in men with metastatic castration-resistant prostate cancer treated with enzalutamide (ANZUP 1901). BJU Int. 2021;128(5):642-651. PubMed
50.Smith M, Parker C, Saad F, et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(3):408-419. PubMed
51.Bayer. NCT02043678: Radium-223 Dichloride and Abiraterone Acetate Compared to Placebo and Abiraterone Acetate for Men With Cancer of the Prostate When Medical or Surgical Castration Does Not Work and When the Cancer Has Spread to the Bone, Has Not Been Treated With Chemotherapy and is Causing no or Only Mild Symptoms (ERA 223). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT02043678. Accessed 2022 Oct 2.
52.Novartis Pharmaceuticals. NCT04720157: An International Prospective Open-label, Randomized, Phase III Study Comparing 177Lu-PSMA-617 in Combination With SoC, Versus SoC Alone, in Adult Male Patients With mHSPC (PSMAddition). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT04720157. Accessed 2022 Oct 2.
53.Novartis Pharmaceuticals. NCT04689828:177Lu-PSMA-617 vs. Androgen Receptor-directed Therapy in the Treatment of Progressive Metastatic Castrate Resistant Prostate Cancer (PSMAfore) ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT04689828. Accessed 2022 Oct 2.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: September 7, 2022
Alerts: Bi-weekly search updates until project completion.
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq = # | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
cctr | Ovid database code; Cochrane Central Register of Controlled Trials |
Multidatabase Strategy
(pluvicto* or ((177lu or 177 lu or lu 177 or lu177 or lutetium 177 or 177 lutetium or 177lutetium) adj3 (psma-617 or psma617 or vipivotide tetraxetan or dota vipivotide or vipivotide dota)) or G6UF363ECX).ti,ab,kf,ot,hw,rn,nm.
177Lu-PSMA.ti,ab,kf.
1 or 2
3 use medall
(*vipivotide tetraxetan lutetium lu 177/ or *vipivotide tetraxetan/) and *lutetium 177/
(pluvicto* or ((177lu or 177 lu or lu 177 or lu177 or lutetium 177 or 177 lutetium or 177lutetium) adj3 (psma-617 or psma617 or vipivotide tetraxetan or dota vipivotide or vipivotide dota))).ti,ab,kf,dq.
177Lu-PSMA.ti,ab,kf.
or/5-7
8 use oemezd
9 not (conference abstract or conference review).pt.
4 or 10
remove duplicates from 11
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search terms: Pluvicto (177lu vipivotide tetraxetan) AND metastatic castration-resistant prostate cancer (mCRPC)]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms: Pluvicto (177lu vipivotide tetraxetan) AND metastatic castration-resistant prostate cancer (mCRPC)]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms: Pluvicto (177lu vipivotide tetraxetan) AND metastatic castration-resistant prostate cancer (mCRPC)]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms: Pluvicto (177lu vipivotide tetraxetan) AND metastatic castration-resistant prostate cancer (mCRPC)]
Grey Literature
Search dates: August 25 to 31, 2022
Keywords: Pluvicto (177lu vipivotide tetraxetan) AND metastatic castration-resistant prostate cancer (mCRPC)
Limits: No publication limits
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for Exclusion |
|---|---|
Hofman MS, Emmett L, Sandhu S, et al. [177Lu]Lu-PSMA-617 vs. cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomized, open-label, phase 2 trial. Lancet. 2021 02 27;397(10276):797 to 804. PubMed: PM33581798 Hofman MS, Emmett L, Violet J, et al. TheraP: a randomized phase 2 trial of 177 Lu-PSMA-617 theranostic treatment vs cabazitaxel in progressive metastatic castration-resistant prostate cancer (Clinical Trial Protocol ANZUP 1603). BJU Int. 2019 11;124 Suppl 1:5 to 13. | Does not meet the eligibility criteria for the CADTH review as TheraP is a phase 2 trial that did not use the doses recommended in the Canadian product monograph; however, this study is summarized within the report as it provides evidence for the following important gaps:
|
177Lu-PSMA-617 = lutetium (177Lu) vipivotide tetraxetan; mCRPC = metastatic castration-resistant prostate cancer; PSMA = prostate-specific membrane antigen.
Pharmacoeconomic Review
Abbreviations
177Lu
lutetium-177
AE
adverse event
APRI
androgen receptor pathway inhibitor
BIA
budget impact analysis
G-CSF
granulocyte colony-stimulating factor
HR
hazard ratio
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LHRH
luteinizing hormone-releasing hormone
mCRPC
metastatic castration-resistant prostate cancer
NMA
network meta-analysis
OS
overall survival
PFS
progression-free survival
PSMA
prostate-specific membrane antigen
QALY
quality-adjusted life-year
rPFS
radiographic progression-free survival
SOC
standard of care
SSE
symptomatic skeletal event
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | 177Lu vipivotide tetraxetan (Pluvicto), solution |
Submitted price | 177Lu vipivotide tetraxetan, 1,000 MBq/mL, vial of solution for IV injection = $27,000 |
Indication | Treatment of adults with PSMA-positive mCRPC who have been treated with an ARPI and taxane-based chemotherapy |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | August 25, 2022 |
Reimbursement request | Per indication |
Sponsor | Advanced Accelerator Applications Canada, Inc. |
Submission history | Not previously reviewed |
177Lu = lutetium-177; ARPI = androgen receptor pathway inhibitor; mCRPC = metastatic castration-resistant prostate cancer; NOC = Notice of Compliance; PSMA = prostate-specific membrane antigen.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population(s) | Patients with PSMA PET-CT scan positive mCRPC who have received an ARPI and taxane-based chemotherapy. Aligns with reimbursement request. |
Treatment | 177Lu vipivotide tetraxetan |
Comparator(s) | BSC/BSoC (hereafter referred to as SOC), per the VISION triala Cabazitaxel 60 mg |
Perspective | Canadian publicly funded health care payer |
Outcome(s) | QALYs, LYs |
Time horizon | 10 years |
Key data source | The VISION trial: efficacy and safety of 177Lu vipivotide tetraxetan vs. BSC/BSoC, health utility values for 177Lu vipivotide tetraxetan and SOC1 Sponsor-commissioned NMA: efficacy of 177Lu vipivotide tetraxetan vs. cabazitaxel2 NICE TA391: health utility values for cabazitaxel3 |
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
177Lu = lutetium-177; BSC = best supportive care; BSoC = best standard of care; ICER = incremental cost-effectiveness ratio; LY = life-year; mCRPC = metastatic castration-resistant prostate cancer; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; OS = overall survival; PET-CT = PET–CT; PSMA = prostate-specific membrane antigen; QALY = quality-adjusted life-year; rPFS = radiographic progression-free survival; SOC = standard of care; SSE = symptomatic skeletal event; WTP = willingness to pay.
aSOC — referred to as best supportive care or best standard of care in the VISION trial Clinical Study Report — is per investigator or physician choice from the VISION trial. In line with the Clinical Study Report, this included ketoconazole, androgen-reducing drugs (including any corticosteroid and 5-alpha reductases), abiraterone, enzalutamide, apalutamide or any other novel androgen axis drug, radiation in any external beam or seeded form, and bone-targeting drugs (including zoledronic acid, denosumab, and any bisphosphonates).
Conclusions
Evidence from the VISION trial suggests that adding lutetium (177Lu) vipivotide tetraxetan (Pluvicto) to best supportive care or best standard of care (hereafter referred to as standard of care [SOC]) was statistically superior to SOC alone for outcomes that are considered important in the evaluation of prostate cancer therapies (i.e., overall survival [OS], progression-free survival [PFS], time to first symptomatic skeletal event [SSE], health-related quality of life, and radiographic progression-free survival [rPFS]). The CADTH Clinical Review Report highlighted several concerns with the trial’s internal and external validity, which may bias these results; however, clinical expert feedback noted that the median OS for 177Lu vipivotide tetraxetan was longer than what has previously been observed in heavily pretreated patients with metastatic castration-resistant prostate cancer (mCRPC).
The VISION trial only represents a portion of the patients eligible for 177Lu vipivotide tetraxetan, as it excluded patients who had received prior docetaxel treatment but were otherwise eligible for cabazitaxel. To address the gap in evidence, the phase II TheraP trial of patients with prior exposure to docetaxel and for whom cabazitaxel was considered the appropriate treatment option was summarized. Although this study was not designed or powered to evaluate potential differences in OS, there was no statistically significant difference between 177Lu vipivotide tetraxetan and cabazitaxel for OS. No stratified economic analysis was provided to CADTH that assessed the relative impact of 177Lu vipivotide tetraxetan on patients based on cabazitaxel eligibility. The sponsor submitted an indirect treatment comparison (ITC), but CADTH noted that the ITC is associated with uncertainty, owing to the heterogeneity of patient characteristics in the included studies. The choice of comparator for the VISION trial and the limitations with sponsor’s ITC preclude the drawing of any conclusions regarding the efficacy of 177Lu vipivotide tetraxetan relative to other relevant comparators, including radium-223 and cabazitaxel, for all important outcomes. As the clinical evidence parameterized the sponsor’s economic analysis, the limitations noted about the clinical evidence were also considered limitations of the sponsor’s economic analysis.
In addition, CADTH identified several other key limitations in the sponsor's economic analysis, including uncertainty regarding the long-term survival benefit assumption associated with 177Lu vipivotide tetraxetan, the omission of radium-223 as a relevant comparator, and the use of treatment-specific health state utilities. In CADTH's base case, the following revisions were made: comparable efficacy of 177Lu vipivotide tetraxetan and cabazitaxel was assumed, alternative survival models to predict long-term rPFS and OS were used, state-specific utility values were applied, correct publicly listed prices were used, the treatments comprising SOC were revised, and the duration of granulocyte colony-stimulating factor (G-CSF) was revised to align with Canadian practice. The CADTH base-case results align with the available clinical evidence, which suggests that there may be a survival benefit for 177Lu vipivotide tetraxetan over SOC. In a sequential analysis with equivalent efficacy assumed between 177Lu vipivotide tetraxetan and cabazitaxel, 177Lu vipivotide tetraxetan generated the same number of QALYs but was more expensive than cabazitaxel; 177Lu vipivotide tetraxetan was therefore dominated by cabazitaxel. The probability that 177Lu vipivotide tetraxetan is cost-effective compared to cabazitaxel or SOC was 0% at a willingness-to-pay (WTP) threshold of $50,000 per QALY. This finding was robust to changes in the model’s assumptions and input parameters. Based on publicly available list prices, a price reduction of 92% is required to make 177Lu vipivotide tetraxetan cost-effective at a WTP threshold of $50,000 per QALY and a price reduction of approximately 82% is required for 177Lu vipivotide tetraxetan to achieve cost parity with cabazitaxel. Scenario analyses highlighted the fact that the comparative efficacy of 177Lu vipivotide tetraxetan and cabazitaxel was a key model driver. The sponsor’s economic model did not allow for consideration of the cost-effectiveness of 177Lu vipivotide tetraxetan in 2 key distinct subgroups: patients eligible for cabazitaxel, and patients ineligible for cabazitaxel. As such, the cost-effectiveness of 177Lu vipivotide tetraxetan in these populations is unknown.
Ultimately, as CADTH could not make firm conclusions regarding the efficacy of 177Lu vipivotide tetraxetan relative to other relevant treatment options, the cost-effectiveness estimates provided are uncertain, and in some cases (e.g., relative to radium-223 and olaparib) are unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from the Canadian Cancer Society, which conducted surveys and interviews of 19 patients with mCRPC. All the survey respondents resided in Canada, with the majority residing in Ontario (42%) and Alberta (37%). Patients reported previous experience with antiandrogen drugs, luteinizing hormone-releasing hormone (LHRH) agonists, external beam radiation, surgery, steroids, LHRH antagonists, and injection radiotherapy. The most common side effects of currently available therapies that had a significant impact on daily life were changes in libido and sexual function, fatigue, loss of muscle mass, incontinence, hot flushes, and weight changes. Four of the patients surveyed had current experience with 177Lu vipivotide tetraxetan, and 1 was interviewed in detail. Side effects of 177Lu vipivotide tetraxetan included shortness of breath, abdominal pain, nausea or vomiting, fatigue, feelings of weakness, and dry mouth. Of the 12 side effects selected, 67% were rated as tolerable or very tolerable by patients; only 1 patient rated low red blood cell count as intolerable. Most patients who had experience with 177Lu vipivotide tetraxetan reported that they would choose to take the treatment even with the side effects they experienced, and all patients would recommend the treatment to others.
CADTH received 1 registered clinician input submission for this review from prostate-treating clinicians in Canada with a special interest in the care of those with metastatic prostate cancer, which was coordinated by the Canadian Cancer Society. The current pathway of care for patients with mCRPC is dependent on the sequencing of therapies during the earlier stages of the disease. At the diagnosis of castration-resistant prostate cancer, the pathway includes treatment with an LHRH agonist and either a taxane or an androgen receptor pathway inhibitor (ARPI); clinicians may consider radium-223 if patients have isolated bone metastases. If patients progress to mCRPC, the LHRH agonist would be continued and a systemic drug, such as cabazitaxel, may be added; poly (ADP-ribose) polymerase inhibitors or other treatments being studied may be considered, as needed and available. Supportive measures such as focal radiotherapy, pain management, and bone-protecting drugs should be used throughout the treatment pathway and continued after cessation of systemic therapies. The clinicians suggested 177Lu vipivotide tetraxetan would be used as a treatment for patients who have prostate-specific membrane antigen (PSMA)-positive mCRPC and who have been treated with ARPIs, and would be integrated with other prostate cancer therapies. The goals of treatment are to delay the clinical progression of disease, improve OS, maintain quality of life, improve pain or constitutional symptoms, and delay events that compromise function.
Drug plan input for this review indicated that many therapies excluded from the VISION trial were relevant comparators to 177Lu vipivotide tetraxetan in practice, including radium-223, poly (ADP-ribose) polymerase inhibitors, and alternate chemotherapy. The public drug plans also sought input on how clinicians would determine whether to use radium-223 or 177Lu vipivotide tetraxetan in eligible patients. The drug plans requested clarity regarding the eligibility criteria and definitions of PSMA-positive lesions; specifically, whether the criteria used in the VISION trial were generalizable to Canadian clinical practice. Drug plan feedback noted that up to 6 cycles of 177Lu vipivotide tetraxetan could be administered at the discretion of the physician and wondered how the duration of 177Lu would be determined. The drug plans noted that the costs of 177Lu vipivotide tetraxetan and the diagnostic test were high, and may have a large budget impact; they also observed that market share assumptions may be underestimated. Questions regarding capacity were also raised.
Several of these concerns were addressed in the sponsor’s model:
Most common side effects of 177Lu vipivotide tetraxetan, cabazitaxel, and best SOC were considered.
The sponsor’s model accounted for the impact of 177Lu vipivotide tetraxetan on disease progression, survival, and quality of life.
In addition, CADTH addressed some of these concerns, as follows:
The drug plan’s question about the number of cycles of 177Lu vipivotide tetraxetan is noted. CADTH assessed the impact of the number of cycles in a scenario analysis.
CADTH was unable to address the following concerns that arose from stakeholder input:
Input from clinicians and drug plans questioned the exclusion of radium-223 as a relevant a comparator.
Economic Review
The current review is for 177Lu vipivotide tetraxetan for patients with PSMA PET-CT scan positive mCRPC who have received an ARPI and taxane-based chemotherapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing costs and outcomes for 177Lu vipivotide tetraxetan, SOC as determined in the VISION trial, and cabazitaxel for the treatment of mCRPC.4 In the VISION trial, SOC comprised investigator’s and/or physician’s choice and included the following: palliative care, ketoconazole, androgen-reducing drugs (including any corticosteroid and 5-alpha reductases), abiraterone, enzalutamide, apalutamide or any other novel androgen axis drug, radiation in any external beam or seeded form, and bone-targeting drugs (including zoledronic acid, denosumab, and any bisphosphonates).1 The modelled population was in line with the reimbursement request and Health Canada–approved indication.
177Lu vipivotide tetraxetan is available in a vial that contains 1,000 MBq/mL (27 mCi/mL) of 177Lu vipivotide tetraxetan. Each vial contains a volume of solution that can range from 7.5 mL to 12.5 mL, corresponding to a radioactivity of 7.4 GBq (200 mCi) (± 10%) in a single-dose vial. According to the product monograph, the recommended dosage is 7.4 GBq (7,400 MBq, 200 mCi; i.e., 1 vial) intravenously every 6 weeks for up to 6 doses, or until disease progression or unacceptable toxicity.5 The cost of 177Lu vipivotide tetraxetan is $27,000 per vial (1 treatment cycle), and the sponsor assumed a mean cycle of 4.54 cycles per patient, as in the VISION trial, so the total treatment cost was $122,489 per patient. The submitted economic model included the cost of PSMA testing for patients receiving 177Lu vipivotide tetraxetan. The sponsor assumed the cost of a PSMA PET-CT scan was $1,200, and using a number needed to test of 1.15 tests per diagnosis (derived from published literature), this resulted in an estimated total cost of $1,386 per eligible patient. Drug costs associated with SOC were estimated based on the cost of concomitant drugs used in the SOC arm of the VISION trial. The cost of cabazitaxel was calculated by multiplying the cost per treatment cycle ($2,134) with the mean number of doses, derived from in the CARD trial (7.33 doses), equating to $15,647 per patient.6
Model Structure
The sponsor used a partitioned survival model with 3 health states: PFS, progressed disease, and death (Figure 1 in Appendix 3). The proportion of patients who were progression-free, who experienced progressed disease, or who were dead at any time over the model horizon was derived from nonmutually exclusive survival curves. All patients entered in the PFS state and were assumed to receive either 177Lu vipivotide tetraxetan, SOC, or cabazitaxel. Patients could discontinue treatment but remain in the progression-free health state until disease progression. At the end of each weekly cycle, the proportion of patients with progressed disease or death was derived from the area under the survival curves. Specifically, OS was partitioned to estimate the proportion of patients in the death state, whereas rPFS was used to estimate the proportion of patients in the PFS health state. The definition of rPFS was per the VISION trial.1 The difference between the OS curve and rPFS curve was partitioned at each time point to estimate the proportion of patients in the progressed disease health state. Disease progression was defined as radiographic disease progression determined by blinded independent central review.
Model Inputs
The modelled population reflected the baseline patient characteristics of the enrolled population in the VISION trial, an international, prospective, open-label, multicenter, randomized, phase III study of 177Lu vipivotide tetraxetan for the treatment of patients with progressive PSMA-positive mCRPC.1 Guided by the VISION trial, the sponsor's model used a median age of 70 years, a mean weight of 88 kg, and a mean body surface area of 2.1 m2.
For 177Lu vipivotide tetraxetan, the sponsor derived transitions from the PFS health state to the progressed disease health state from the rPFS curves reported in the VISION trial. The sponsor derived the relative treatment benefits of 177Lu vipivotide tetraxetan and cabazitaxel from a sponsor-commissioned ITC — in the form of a network meta-analysis (NMA) — which used the summary results reported in published studies and pooled the hazard ratios (HRs) of the time-to-event end points of OS and rPFS.2 The stratified flexible Weibull (2 knots) curve was used to predict long-term PFS and OS for 177Lu vipivotide tetraxetan and SOC. HRs from the NMA were applied to the extrapolated rPFS and OS data to represent long-term rPFS and OS for patients receiving cabazitaxel.
The model accounted for symptomatic skeletal events (SSEs), and a log-normal model was fitted to time to first SSE data from the VISION trial to extrapolate SSEs beyond the trial time horizon. The sponsor applied similar SSE rates for 177Lu vipivotide tetraxetan and cabazitaxel. The model also considered adverse events (AEs) of grade 3 or higher with an incidence of at least 2% for each comparator.
Health state–specific utility values were assumed to depend on the type of treatment. For 177Lu vipivotide tetraxetan, the sponsor derived a health state–specific value for each treatment option by fitting a generalized linear mixed model to EQ-5D data collected as part of the VISION trial. For cabazitaxel, preprogression health utility data were assumed to be the same as those for the health utility of patients receiving SOC in the VISION trial, whereas health utility for progressed disease was based on the National Institute for Health and Care Excellence (NICE) TA391 technology appraisal guidance,3 which evaluated cabazitaxel for hormone-relapsed metastatic prostate cancer treated with docetaxel. The sponsor did not account for health utility decrements related to SSEs or AEs to avoid double counting.
Costs included drugs (drug acquisition and drug administration), PSMA testing, concomitant medications, premedication, subsequent therapies, disease-management costs, and AE costs. The drug-acquisition cost for 177Lu vipivotide tetraxetan was provided by the sponsor, whereas the costs for comparators were sourced from the IQVIA DeltaPA database. The sponsor assumed that there were no concomitant treatments associated with 177Lu vipivotide tetraxetan or cabazitaxel, but it assigned premedication costs to patients receiving cabazitaxel, based on feedback from clinical experts in Canada. The costs of concomitant treatments were applied to SOC, and the proportion of patients receiving each therapy was informed by the VISION trial. The cost of subsequent systemic treatment was applied in the model as a 1-off cost at the time of disease progression. Subsequent treatments (when 2% or more of patients continued on a treatment) for the 177Lu vipivotide tetraxetan and SOC arms were based on the VISION trial. Subsequent treatments for cabazitaxel were informed by the CARD trial (where available).6 For subsequent therapies with no data from the CARD trial (i.e., carboplatin, olaparib, and radiotherapy), the model assumed a frequency equal to the overall proportion observed in the VISION trial. The model assumed a duration of 6 months for any subsequent drug therapy for all comparators. Disease-management costs were informed by a combination of available published literature, feedback from clinical experts in Canada, and product monographs. These included costs associated with appointments and diagnostic procedures. The unit costs for physician visits were obtained from the Ontario Schedule of Benefits for Physician Services, whereas the unit costs for laboratory tests were obtained from the Ontario Schedule of Benefits for Laboratory Services. A 1-off terminal-care cost of $5,050.49 was sourced from a retrospective, population-based, cohort study by de Oliveira et al. (2016)7 that reported phase-specific and lifetime costs of cancer in Ontario.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 2,000 iterations. Deterministic and probabilistic results were consistent. The probabilistic findings are presented here.
Base-Case Results
In the sponsor's base-case analysis over a 10-year time horizon, all treatments were on the cost-effectiveness frontiers. Cabazitaxel was associated with an incremental cost-effectiveness ratio (ICER) of $227,206 per QALY compared to SOC, and 177Lu vipivotide tetraxetan was associated with an ICER of $281,361 per QALY compared to cabazitaxel (Table 3 and Table 12). At a WTP threshold of $50,000 per QALY, the probability of 177Lu vipivotide tetraxetan being cost-effective was 0% compared to cabazitaxel.
At the end of the model time horizon (i.e., 10 years), all patients were dead. A breakdown of the sponsor-submitted results for the base-case population by trial duration (i.e., 2.26 years, based on the submitted Kaplan-Meier curve) and extrapolated period shows that approximately one-third of the expected QALY gains for 177Lu vipivotide tetraxetan (31% compared to SOC and 30% compared to cabazitaxel) come from the time beyond the VISION trial follow-up period.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
SOC | 35,808 | 0.73 | Reference |
Cabazitaxel | 55,443 | 0.81 | 227,206 |
177Lu vipivotide tetraxetan | 153,533 | 1.16 | 281,361 |
177Lu = lutetium; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Source: Sponsor’s Pharmacoeconomic Submission.4
Sensitivity and Scenario Analysis Results
The sponsor performed scenario analyses by removing the cost of PSMA testing, adding APRI as a part of SOC comparator, using alternate rates of AEs, varying the time horizon, changing discount rates, changing assumptions regarding duration of therapy, changing parametric survival models (for rPFS, SSE, and OS prediction), using alternate assumptions about disease-management costs and subsequent therapies, and changing assumptions regarding utility decrements related to SSEs and AEs. Key drivers of the cost-effectiveness of 177Lu vipivotide tetraxetan included the choice of survival extrapolation for OS and assumptions surrounding health state utility values.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations of the sponsor’s analysis that have notable implications on the economic analysis:
Comparative efficacy of 177Lu vipivotide tetraxetan and relevant comparators is uncertain. For the comparison of 177Lu vipivotide tetraxetan with SOC, the sponsor derived rPFS and OS data from the VISION trial. As indicated in the CADTH Clinical Review Report, based on clinical expert feedback and CADTH methodologists, concerns were identified regarding the trial’s internal and external validity. In particular, the proportion of patients who had been treated with a single taxane at the time of enrolment in the VISION trial was higher than the estimated proportion of patients not medically suitable to receive cabazitaxel in Canadian practice (57.9% in the VISION trial versus 30% to 40% estimated in Canadian practice). Additionally, 177Lu vipivotide tetraxetan was used as an add-on to SOC in the VISION trial; however, it is unclear whether 177Lu vipivotide tetraxetan would be used in this manner in routine Canadian practice. For example, a large proportion of patients received ARPIs as part of SOC, and whether these would be continued in Canadian practice in this population is highly uncertain. In a critique of the VISION trial, international prostate cancer specialists suggested that eligible patients should have received cabazitaxel before enrolment in the trial, as the permitted SOC regimens represented suboptimal care in comparison.8 However, clinical expert feedback obtained by CADTH considered the SOC used in the VISION trial to be sufficient. Moreover, use of 177Lu vipivotide tetraxetan in combination with other systemic anticancer therapies may be limited by reimbursement status. CADTH further noted that the sponsor’s economic report did not adequately describe whether 177Lu vipivotide tetraxetan was used as a monotherapy or an add-on therapy, but excluded the costs of concomitant treatments from 177Lu vipivotide tetraxetan and cabazitaxel arms. Exclusion of such costs would underestimate the total costs and ICERs of 177Lu vipivotide tetraxetan and cabazitaxel compared to SOC, given that SOC was inherently captured in the treatment effects, although concomitant systemic anticancer therapies are not expected to have a notable impact on incremental treatment effects. In addition, the CADTH Clinical Review Report highlighted the potential bias and uncertainty associated with rPFS and SSE outcomes, owing to imbalance in censoring and protocol deviations in the treatment groups observed from the VISION trial.
Another validity concern was related to subsequent cancer therapies. A significant proportion of patients enrolled in the VISION trial received cabazitaxel as a subsequent therapy (after both 177Lu vipivotide tetraxetan and SOC), although the trial’s inclusion criteria indicated that patients who had received a single taxane regimen must be medically unsuitable for an additional taxane regimen. Clinical expert feedback obtained by CADTH agreed that a patient with mCRPC who is considered ineligible for a further taxane regimen is unlikely to become eligible at a later point in time.
For the comparison with cabazitaxel, the sponsor submitted an ITC to inform relative rPFS and OS. CADTH identified several limitations of the ITC, including the lack of reporting on specific items that would better inform the certainty of the indirect evidence. Additionally, CADTH highlighted concerns regarding the heterogeneity and differences in key baseline characteristics (i.e., historical use of chemotherapy, disease severity, and treatment indication) of included studies, which were not adequately investigated or adjusted for. As a result, it was inconclusive if there would be an additional benefit associated with 177Lu vipivotide tetraxetan compared to cabazitaxel and other comparators.
CADTH was unable to address most limitations related to the validity of the VISION trial.
In the reanalysis, CADTH included some concomitant supportive therapies (antiemetics, bisphosphonates, denosumab, corticosteroids, G-CSF, and opioid analgesics) but excluded APRIs for all treatment arms. CADTH maintained the sponsor assumptions that there were rPFS and OS benefits for 177Lu vipivotide tetraxetan relative to SOC, despite concerns related to the validity of the trial results. CADTH attempted to account for the limitations of the submitted ITC in the reanalysis by assuming no difference in clinical efficacy between 177Lu vipivotide tetraxetan and cabazitaxel (i.e., setting HRs for rPFS and OS to 1).
Treatment benefits of 177Lu vipivotide tetraxetan in patients previously treated with docetaxel who are considered eligible for cabazitaxel is inconclusive. As noted in the CADTH Clinical Review Report, the VISION trial included a subset of patients eligible for 177Lu vipivotide tetraxetan according to the Health Canada indication. Patients who had received prior docetaxel treatment but were otherwise eligible for cabazitaxel were not allowed to participate in the trial, although this population is included in the Health Canada–approved indication. The TheraP trial reported the treatment benefits of 177Lu vipivotide tetraxetan compared to cabazitaxel in this population.9 However, although the TheraP trial indicated no statistical differences between 177Lu vipivotide tetraxetan and cabazitaxel, as a phase II trial, it was not intended to evaluate potential differences in rPFS or OS between these treatments.10 CADTH identified concerns regarding the study's validity related to its open-label design, high and differential withdrawal rates in the control group (similar to those in the VISION trial), crossover, and other possible differences in the subsequent therapy setting.
CADTH was unable to address this limitation because of the structure of the submitted model and important concerns regarding the validity of the TheraP trial, which enrolled patients who had been previously treated with docetaxel but for whom cabazitaxel could be an option.
Long-term rPFS and OS were overestimated. The sponsor’s use of a partitioned survival model introduces structural assumptions about the relationship between rPFS and OS (i.e., nonmutually exclusive curves) that could not be adjusted for in reanalysis. These assumptions may introduce a postprogression survival bias that favours 177Lu vipivotide tetraxetan.
The sponsor used survival models to extrapolate the trial data over the entire time horizon of the model using separate curves for 177Lu vipivotide tetraxetan and SOC, whereas rPFS and OS rates for of patients receiving cabazitaxel were calculated by applying HRs estimated from the NMA to 177Lu vipivotide tetraxetan curves. The sponsor indicated that survival model selection was based on statistical considerations (i.e., Akaike information criterion and Bayesian information criteria), graphical assessments of fit, and clinical plausibility. The sponsor selected the stratified flexible Weibull model with 2 knots as the best fit for both rPFS and OS data. Clinical expert feedback obtained by CADTH for this review indicated that predicted rPFS and OS rates obtained from the selected model were overestimated at year 5. Given the poor prognosis and frailty of this patient population, the expected 5-year rPFS and OS rates would likely be 0%, but the sponsor’s model predicted that 2.7% of patients receiving 177Lu vipivotide tetraxetan would survive at 5 years.
Given the structural uncertainty of the survival curves and aligned with feedback received from clinical experts consulted by CADTH, CADTH selected alternative curves and incorporated treatment waning in the base case to arrive at the most conservative and clinically plausible estimates for OS in patients with mCRPC in Canada. CADTH explored other survival curves in scenario analyses.
A relevant comparator was omitted: The sponsor included SOC and cabazitaxel as relevant comparators in the submitted economic model. As noted in the CADTH Clinical Review Report and stakeholder feedback section, clinical experts indicated that radium-223 is a relevant comparator. Radium-223 is approved in Canada for the treatment of patients with mCRPC and bone metastases, although there is variability across provinces regarding access and reimbursement. CADTH noted that radium-223 was included in the sponsor’s NMA, but because of limitations identified with the NMA, the effectiveness of 177Lu vipivotide tetraxetan relative to radium-223 is uncertain.
CADTH was unable to address this limitation; because of the structure of the submitted model, CADTH could not add radium-223 as a comparator. CADTH noted that the cost per cycle of radium-233 is cheaper than that of 177Lu vipivotide tetraxetan (Table 9).
Health utility benefits associated with 177Lu vipivotide tetraxetan are highly uncertain. The sponsor’s model assumed that health utility values would differ by the type of treatment received and the patient’s health state. Health utility values for 177Lu vipivotide tetraxetan and SOC were derived from the VISON trial, whereas those for cabazitaxel were obtained from the published National Institute for Health and Care Excellence TA391 technology appraisal guidance for cabazitaxel.3 Treatment-specific utilities are inappropriate and health state–specific utilities are preferred, per CADTH Guidelines for the Economic Evaluation of Health Technologies: Canada.11 No rationale was provided as to why 177Lu vipivotide tetraxetan is expected to result in a different quality of life than cabazitaxel for patients in the same health state. Any benefit associated with the treatment should be captured in the derivation of the health states or incorporation of disutilities (e.g., AEs). As noted in the CADTH Clinical Review, although the 5 Level EQ-5D data suggested a statistically significant benefit associated with 177Lu vipivotide tetraxetan compared to SOC, these analyses were not considered reliable, given inherent biases associated with the collection of health-related quality of life data in open-label trials. Furthermore, the CADTH Clinical Review Report indicates that 177Lu vipivotide tetraxetan is associated with an increase in serious AEs and SSEs (once deaths are removed) compared with SOC (refer to appraisal point related to SSEs) and, as a result, an increase in disutilities associated with 177Lu vipivotide tetraxetan are expected. This does not align with the sponsor’s health state utility values, which suggest higher utility values for 177Lu vipivotide tetraxetan than SOC, regardless of whether a patient has progressed or not. Furthermore, the sponsor’s use of trial-based values using data from a generalized linear mixed models of utility are highly uncertain, given the lack of information on how the sponsor handled dropout and missing data, which is critical, given the high rate of dropouts for patients receiving SOC in the VISION study.
CADTH used the average health utility values specific to a health state estimated from the VISION trial and applied these values to all treatment arms in the reanalysis. Although CADTH’s approach could not address the concern regarding the reliability of health utility values derived from the VISION trial, these values are considered the best available evidence and their inherent bias was unlikely to affect the results of CADTH’s reanalysis because of the assumption of equal health utility across treatment arms. CADTH retained the sponsor’s assumption regarding the exclusion of utility decrements associated with AEs and SSEs to avoid double counting.
Time to first SSE was driven by death events. In the clinical study report of the VISION trial submitted by the sponsor, time to first SSE was defined as the date of randomization to the date of first new symptomatic pathological bone fracture, spinal cord compression, tumour-related orthopedic surgical intervention, requirement for radiation therapy to relieve bone pain, or death from any cause (whichever occurred first). Inclusion of deaths in the SSE definition might have biased the predicted SSE in favour of 177Lu vipivotide tetraxetan because the risk of death was higher among patients receiving SOC. CADTH also noted the difference between time to SSE curves shown in the sponsor’s economic and clinical study reports. The sponsor did not specify which dataset (full analysis set or rPFS full analysis set) was used to inform SSE data in the submitted economic model.
CADTH was unable to assess the uncertainty associated with the time to first SSE data, but explored its impact using different modelling approaches in a scenario analysis.
Additional limitations were identified but were not considered to be key limitations:
Unit costs of some concomitant treatments did not reflect the public drug costs in Canada. CADTH corrected these costs using data from public sources. Moreover, the sponsor’s model used treatment duration (5.06 months) to calculate the total costs of premedications (chlorpheniramine, ranitidine, dexamethasone, and filgrastim) for cabazitaxel. The sponsor should have used the mean doses of cabazitaxel (7.33 doses), as these medications were used before each dose of cabazitaxel. CADTH corrected these estimates in the reanalysis.
Additionally, the key assumptions outlined in Table 4 were made by the sponsor and have been appraised by CADTH.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations of the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Patient characteristics (i.e., age, sex, weight, body surface area) were based on patients enrolled in the VISION trial. | CADTH obtained clinical expert feedback noting that this assumption was acceptable, but also that the use of subsequent therapies in other countries can be different from those used in Canada and Europe, where public drug programs may have stricter criteria for reimbursement. |
There were no concomitant treatments associated with 177Lu vipivotide tetraxetan or cabazitaxel. | Excluding all concomitant treatments would underestimate total costs. CADTH obtained clinical expert feedback that indicated that some concomitant drugs (including antiemetics, bisphosphonates or denosumab, corticosteroids, and opioid analgesics) should be continued in view of what represents current standard of care. CADTH included a list of concomitant drugs suggested by the clinical experts in the reanalysis. |
40% of patients who received cabazitaxel were assumed to have received filgrastim for 14 days of each 21-day cycle of cabazitaxel. | CADTH obtained clinical expert feedback suggesting that the proportion of patients receiving cabazitaxel who received G-CSF was reasonable, but that in Canada, filgrastim is prescribed for 7 days for each cycle of cabazitaxel. CADTH changed treatment duration for filgrastim to 7 days in the reanalysis. |
Patients who received cabazitaxel were assumed to receive 7.33 doses, per the CARD trial. | CADTH obtained clinical expert feedback advising that the mean doses of cabazitaxel were higher than what is used in Canadian practice. The expected mean dose is approximately 4. CADTH performed a scenario analysis by varying the mean doses of cabazitaxel. |
Utility decrements related to SSEs and AEs were excluded to avoid double counting, given the use of treatment-specific health state utilities in the sponsor’s approach. | Acceptable, given the sponsor’s approach. However, as noted previously, the sponsor’s approach was not appropriate. |
177Lu = lutetium-177; AE = adverse event; G-CSF = granulocyte macrophage colony-stimulating factor; SSE = symptomatic skeletal event.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by correcting unit costs for APRIs, zoledronic acid, denosumab, corticosteroids, morphine, and oxycodone using publicly listed prices and changing the number of doses of premedications for patients receiving cabazitaxel. In addition, CADTH addressed limitations of the sponsor’s submitted clinical evidence and methodological approaches, as previously noted, by removing APRIs and antifungal and erythropoietin-stimulating drugs from a list of concomitant treatments, reducing treatment duration for filgrastim to 7 days of each cycle of cabazitaxel, applying HRs of 1 to represent comparable effects of 177Lu vipivotide tetraxetan and cabazitaxel on rPFS and OS, applying the same health state–specific utility values for all treatment arms, and using alternate survival models and a treatment-waning assumption to bring predicted rPFS and OS curves closer to the patient outcomes observed in clinical practice. Table 5 details the change made to derive the CADTH base case, and the summary results of the CADTH's base case are presented in Table 7. Additional results are shown in Table 6 and Appendix 4 (Table 13).
Results from CADTH's revised base case show that only cabazitaxel and SOC were on a cost-effectiveness frontier, and that compared to SOC, cabazitaxel was associated with higher costs and increased QALYs, with an ICER of $91,220 per QALY. Compared to cabazitaxel, 177Lu vipivotide tetraxetan was more expensive but generated the same QALYs; it was therefore dominated by cabazitaxel. The cost-effectiveness of 177Lu vipivotide tetraxetan shown by CADTH was different than that in the sponsor's base case because of the assumptions regarding comparable treatment effects and health utility values between 177Lu vipivotide tetraxetan and cabazitaxel. The probability that 177Lu vipivotide tetraxetan is cost-effective compared to cabazitaxel or SOC was 0% at the WTP threshold of $50,000 per QALY.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
Unit costs (cost/pack) of some concomitant drugs were not reflective of costs seen in Canadian practice | Abiraterone $15.31 Enzalutamide $28.35 Zoledronic acid $134.61 Denosumab $629.24 Dexamethasone $0.30 Prednisolone $1.46 Morphine $0.62 Oxycodone $0.37 | Abiraterone $52.06 Enzalutamide $29.20 Zoledronic acid $415.46 Denosumab $648.11 Dexamethasone $0.61 Prednisolone $0.02 Morphine $0.99 Oxycodone $1.48 |
Changes to derive the CADTH base case | ||
1. Relative efficacy of 177Lu and cabazitaxel was uncertain | HR rPFS (vs. 177Lu): ████ HR OS (vs. 177Lu): ████ | HR rPFS (vs. 177Lu): 1.00 HR OS (vs. 177Lu): 1.00 |
2. Predicted rPFS and OS 1111curves were overestimated | Stratified flexible Weibull model with 2 knots without treatment waning was used to predict rPFS and OS curves | rPFS curve: stratified flexible Weibull model with 2 knots with treatment waning OS curve: Weibull model with treatment waning |
3. Quality of life benefits of 177Lu compared to SOC and cabazitaxel was uncertain | Health utility values specific to each treatment were used.
| Health utility values were assumed to be independent of the type of treatment.
|
4. Concomitant treatments were not reflective of routine practice in Canada | All concomitant treatments were excluded from the 177Lu and cabazitaxel arms | A selected list of concomitant treatments (including antiemetics, bisphosphonates, denosumab, corticosteroids, and opioid analgesics), was considered in all treatment arms |
5. Treatment duration of filgrastim for patients receiving cabazitaxel did not align with Canadian practice | 14 days for each cycle of cabazitaxel | 7 days for each cycle of cabazitaxel |
CADTH base case | — | 1 + 2 + 3 + 4 + 5 |
177Lu = lutetium-177 vipivotide tetraxetan; HR = hazard ratio; OS = overall survival; rPFS = radiographic progression-free survival; SOC = standard of care.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Treatment | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | SOC | 35,808 | 0.726 | Reference |
Cabazitaxel | 55,443 | 0.813 | 227,206 | |
177Lu | 153,533 | 1.161 | 281,361 | |
Sponsor’s corrected base case | SOC | 37,455 | 0.726 | Reference |
Cabazitaxel | 56,828 | 0.813 | 224,158 | |
177Lu | 153,790 | 1.161 | 278,128 | |
CADTH reanalysis 1 | SOC | 37,455 | 0.726 | Reference |
Cabazitaxel | 57,486 | 1.055 | 60,992 | |
177Lu | 153,790 | 1.161 | 903,178 | |
CADTH reanalysis 2 | SOC | 37,545 | 0.729 | Reference |
Cabazitaxel | 56,731 | 0.775 | Extendedly dominated | |
177Lu | 153,586 | 1.074 | 335,878 | |
CADTH reanalysis 3 | SOC | 37,455 | 0.769 | Reference |
Cabazitaxel | 56,828 | 0.873 | 186,626 | |
177Lu | 153,790 | 1.136 | 368,358 | |
CADTH reanalysis 4 | SOC | 37,455 | 0.726 | Reference |
Cabazitaxel | 66,879 | 0.813 | Extendedly dominated | |
177Lu | 163,638 | 1.161 | 290,043 | |
CADTH reanalysis 5 | SOC | 37,455 | 0.726 | Reference |
Cabazitaxel | 52,478 | 0.813 | 173,832 | |
177Lu | 153,790 | 1.161 | 290,604 | |
CADTH base case | SOC | 37,545 | 0.771 | Reference |
Cabazitaxel | 62,985 | 1.050 | 91,220 | |
177Lu | 163,434 | 1.050 | Dominated |
177Lu = lutetium-177 vipivotide tetraxetan; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: All results are probabilistic.
Table 7: Summary of the CADTH Reanalysis Results
Treatment | Total costs | Total QALYs | ICER vs. SOC | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor-corrected base case | ||||
SOC | 37,455 | 0.726 | Reference | Reference |
Cabazitaxel | 56,828 | 0.813 | 224,158 | 224,158 |
177Lu | 153,790 | 1.161 | 267,407 | 278,128 |
CADTH base case | ||||
SOC | 37,545 | 0.771 | Reference | Reference |
Cabazitaxel | 62,985 | 1.050 | 91,220 | 91,220 |
177Lu | 163,434 | 1.050 | 451,407 | Dominated |
177Lu = lutetium-177 vipivotide tetraxetan; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Scenario Analysis Results
A series of scenario analyses was conducted based on CADTH base case. These analyses explored the impact of the following model parameters and assumptions: the number of doses of cabazitaxel; the proportion of patients receiving cabazitaxel as a subsequent therapy in the cabazitaxel arm; survival models for rPFS and OS predictions; cost of PSMA testing; proportion and type of G-CSF; approaches to estimate SSEs; and assumptions about health utility values.
Results from scenario analyses (Table 13 in Appendix 4) demonstrated that the assumption about the relative efficacy of 177Lu vipivotide tetraxetan and cabazitaxel had the largest impact on the cost-effectiveness results. Cost-effectiveness results ranged from 177Lu vipivotide tetraxetan being dominated to an ICER of $451,043 per QALY compared to cabazitaxel (scenario 2 [using the upper bound of the HRs for rPFS and OS] for 177Lu versus cabazitaxel). The ICERs for 177Lu vipivotide tetraxetan were found to align robustly to the assumptions and to changes in the risk of SSE, health utility values, and the cost of PSMA testing.
A price-reduction analysis was conducted using the sponsor-corrected and CADTH base case (Table 8). Based on the sponsor’s corrected base case, cabazitaxel is extendedly dominated after the price of 177Lu vipivotide tetraxetan is reduced by 20% or more. A price reduction of up to 78% is required for 177Lu vipivotide tetraxetan to be considered cost-effective compared to SOC at a WTP threshold of $50,000 per QALY. A price-reduction analysis using the CADTH base case suggested that 177Lu vipivotide tetraxetan would dominate cabazitaxel if its price was decreased by 82.1%. Compared to SOC, a price reduction of 92% is required for 177Lu vipivotide tetraxetan to be cost-effective at a WTP threshold of $50,000 per QALY.
Table 8: CADTH Price-Reduction Analyses
Analysis | ICERs for 177Lu vs. cabazitaxel vs. SOC | |
|---|---|---|
Price reduction | Sponsor-corrected base case | CADTH reanalysis |
No price reduction | If WTP < $224,158/QALY, SOC is optimal If WTP ≥ $224,158/QALY but < $278,128/QALY, cabazitaxel is optimal If WTP ≥ $278,128/QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $100,449) If WTP < $451,407/QALY, SOC is optimal If WTP ≥ $451,407/QALY cabazitaxel is optimal |
10% | If WTP < $224,158/QALY, SOC is optimal If WTP ≥ $224,158/QALY but < $243,004/QALY, cabazitaxel is optimal If WTP ≥ $243,004 /QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $88,204) If WTP < $407,499/QALY, SOC is optimal If WTP ≥ $407,499/QALY cabazitaxel is optimal |
20% | Cabazitaxel is extendedly dominate If WTP < $211,114/QALY, SOC is optimal If WTP ≥ $211,114/QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $75,959) If WTP < $363,592/QALY, SOC is optimal If WTP ≥ $363,592/QALY, cabazitaxel is optimal |
30% | Cabazitaxel is extendedly dominated If WTP < $182,968/QALY, SOC is optimal If WTP ≥ $182,968/QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $63,714) If WTP < $319,684/QALY, SOC is optimal If WTP ≥ $319,684/QALY, cabazitaxel is optimal |
40% | Cabazitaxel is extendedly dominated If WTP < $154,821/QALY, SOC is optimal If WTP ≥ $154,821/QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $51,469) If WTP < $275,776/QALY, SOC is optimal If WTP ≥ $275,776/QALY, cabazitaxel is optimal |
50% | Cabazitaxel is extendedly dominated If WTP < $126,675/QALY, SOC is optimal If WTP ≥ $126,675/QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $39,224) If WTP < $231,869/QALY, SOC is optimal If WTP ≥ $231,869/QALY cabazitaxel is optimal |
60% | Cabazitaxel is extendedly dominated. If WTP < $98,528/QALY, SOC is optimal If WTP ≥ $98,528/QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $26,979) If WTP < $187,961/QALY, SOC is optimal If WTP ≥ $187,961/QALY, cabazitaxel is optimal |
70% | Cabazitaxel is extendedly dominated. If WTP < $70,382/QALY, SOC is optimal If WTP ≥ $70,382/QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $14,734) If WTP < $144,053/QALY, SOC is optimal If WTP ≥ $144,053/QALY, cabazitaxel is optimal |
78% | Cabazitaxel is extendedly dominated. If WTP < $47,865/QALY, SOC is optimal If WTP ≥ $47,865/QALY, 177Lu is optimal | 177Lu is dominated by cabazitaxel (Δcost = $4,938) If WTP < $108,927/QALY, SOC is optimal If WTP ≥ $108,927/QALY, cabazitaxel is optimal |
80% | NR | 177Lu is dominated by cabazitaxel (Δcost = $2,489) If WTP < $100,146/QALY, SOC is optimal If WTP ≥ $100,146/QALY, cabazitaxel is optimal |
90% | NR | Cabazitaxel is dominated by 177Lu (Δcost = –$9,756). If WTP < $56,238/QALY, SOC is optimal If WTP ≥ $56,238/QALY, 177Lu is optimal |
92% | NR | Cabazitaxel is dominated by 177Lu (Δcost = –$12,205) If WTP < $47,456/QALY, SOC is optimal If WTP ≥ $47,456/QALY, 177Lu is optimal |
177Lu = lutetium-177 vipivotide tetraxetan; ICER = incremental cost-effectiveness ratio; NR = not reported; SOC = standard of care; WTP = willingness to pay.
Issues for Consideration
In Canada, treatment options for patients with mCRPC who have previously received an ARPI and taxane-based chemotherapy may include olaparib. However, as olaparib is only recommended for a small subgroup of adults with deleterious or suspected deleterious germline and/or somatic BRCA-mutated or ATM-mutated mCRPC, accounting for less than 5% of the population, this was not deemed to be a relevant comparator, based on the clinical expert feedback.
According to the product monograph, 177Lu vipivotide tetraxetan should only be used under the guidance of a medical professional with training in the use of radiopharmaceuticals in designated clinical settings. Based on the quantity and/or locations of currently available facilities, clinical expert feedback obtained by CADTH noted that these requirements would limit patient access (e.g., based on regional variation or existing capacity constraints). The use of radiopharmaceuticals in the treatment of prostate cancer varies across Canada; as previously noted, access to the other radiopharmaceutical available for a subgroup of patients with mCRPC varies across jurisdictions. It was also noted that there may be a limited number of medical oncologists in Canada with experience using radiopharmaceuticals for prostate cancer, even in major academic treatment facilities.
Feedback from public drug plans and clinical experts highlighted the challenges associated with the need for PSMA testing with PET-CT to meet the eligibility for 177Lu vipivotide tetraxetan. This test is not widely available in routine practice in Canada and is typically only performed as part of clinical studies or accessed through private mechanisms. Limitations associated with access to PSMA PET-CT, challenges with capacity for testing patients, and information on cost considerations associated with public funding for PSMA PET-CT imaging in Canada are also highlighted in a recent publication by CADTH on PSMA PET-CT imaging in Canada.12 These considerations have not been incorporated in the economic evaluation of 177Lu vipivotide tetraxetan.
There will be a need for patients and potentially their caregivers to miss work and travel out of province for PSMA testing and 177Lu vipivotide tetraxetan treatment. The diagnostic imaging product used in the VISION trial for all patients was still under review by Health Canada at the time of this review. It is unclear whether this imaging product will be funded by Canadian public drug plans or whether an alternate diagnostic imaging product will be used with 177Lu vipivotide tetraxetan.
The sensitivity and specificity of PSMA PET-CT testing was not incorporated in the sponsor’s economic model. As a result, the costs and outcomes for patients who are misdiagnosed were not captured in the economic analysis. This is likely to lead to an increase in the estimated ICER of 177Lu vipivotide tetraxetan, although the magnitude of the impact is uncertain.
In line with the CADTH economic evaluation guidelines, base case analyses by the sponsor and by CADTH did not account for time missed from work or travel expenses (e.g., travel, lodging, booking). If these costs were considered in the economic evaluation, it would likely increase the expected costs of 177Lu vipivotide tetraxetan, which may have an added impact on the quality of life of caregivers. Disparities in testing and treatment access may vary, depending on the province or territory, and the requirement for access to a tertiary care centre for the administration of 177Lu vipivotide tetraxetan may have equity implications that were not considered in the economic submission.
Overall Conclusions
Evidence from the VISION trial suggests that adding 177Lu vipivotide tetraxetan to SOC is statistically superior to SOC alone for outcomes that are considered important in the evaluation of prostate cancer therapies (i.e., OS, PFS, time to first SSE, health-related quality of life, and rPFS). However, the CADTH Clinical Review Report highlighted several concerns with the trial’s internal and external validity, which may bias these results. Health Canada considered the results for OS to be clinically important, given that the patients enrolled in the VISION trial were heavily pretreated, and CADTH obtained clinical expert feedback that noted that the median OS for 177Lu vipivotide tetraxetan was longer than what has previously been observed for heavily pretreated patients with mCRPC. Although clinical expert feedback noted that SOC in Canada is different than that used in the VISION trial, this was not expected to have an impact on the generalizability of the results. However, CADTH noted that published literature has identified concerns about whether patients receiving SOC in the VISION trial were optimally treated.
The VISION trial only represents a portion of the patients eligible for 177Lu vipivotide tetraxetan, as it excluded patients who had received prior docetaxel treatment but were otherwise eligible for cabazitaxel. To address the gap in evidence, the phase II TheraP trial of patients with prior exposure to docetaxel and for whom cabazitaxel was considered the appropriate treatment option was summarized. Although this study was not designed or powered to evaluate potential differences in OS, there was no statistically significant difference between 177Lu vipivotide tetraxetan and cabazitaxel for OS. No stratified economic analysis was provided to CADTH that assessed the relative impact of 177Lu vipivotide tetraxetan on patients based on cabazitaxel eligibility. The sponsor submitted an ITC, but it was associated with uncertainty, owing to the heterogeneity of patient characteristics in the included studies.
The choice of comparators in the VISION trial and the limitations of sponsor’s ITC preclude the drawing of any conclusions regarding the efficacy of 177Lu vipivotide tetraxetan relative to other relevant comparators for all important outcomes. As the clinical evidence parameterized the sponsor’s economic analysis, the limitations noted in the clinical evidence were further noted as limitations in the sponsor’s economic analysis. In addition, CADTH identified several other key limitations in the sponsor's economic analysis, including uncertainty regarding the long-term survival benefit assumption associated with 177Lu vipivotide tetraxetan, the omission of radium-223 as a relevant comparator, and the use of treatment-specific health state utilities.
CADTH made minor corrections to cost calculations and revised the sponsor's base case to derive the CADTH base case. CADTH used publicly listed prices for APRIs, zoledronic acid, denosumab, corticosteroids, and opioid analgesics, and corrected the number of doses of premedications for patients receiving cabazitaxel. CADTH also removed APRIs and antifungal and erythropoietin-stimulating drugs from a list of concomitant treatments, reduced the treatment duration for filgrastim to 7 days for each cycle of cabazitaxel, applied HRs of 1 to represent comparable effects of 177Lu vipivotide tetraxetan and cabazitaxel on rPFS and OS, applied the same health state–specific utility values for all treatment arms, and used alternate survival models and a treatment-waning assumption to bring predicted rPFS and OS curves closer to the patient outcomes observed in clinical practice.
CADTH's base case reported that 177Lu vipivotide tetraxetan was more expensive than but generated the same QALYs as cabazitaxel; therefore, 177Lu vipivotide tetraxetan was dominated by cabazitaxel, but not on the cost-effectiveness frontier. The probability that 177Lu vipivotide tetraxetan is cost-effective compared to cabazitaxel or SOC was 0% at a WTP threshold of $50,000 per QALY. Scenario analyses highlighted that the relative efficacy of 177Lu vipivotide tetraxetan was a key driver. 177Lu vipivotide tetraxetan was dominated by cabazitaxel if its efficacy was assumed to be comparable to cabazitaxel. This finding was aligned robustly to changes in the model’s assumptions and input parameters. A price reduction of 92% of more is required to make 177Lu vipivotide tetraxetan cost-effective at the WTP threshold of $50,000 per QALY compared to SOC; whereas a price reduction of approximately 82% was required for 177Lu vipivotide tetraxetan to achieve cost parity with cabazitaxel.
CADTH conducted a series of scenario analyses to explore the impact of alternate relative efficacy of 177Lu vipivotide tetraxetan and cabazitaxel; different survival approaches for rPFS, SSEs, and OS; assumptions regarding the number of doses of cabazitaxel; and health utility values. The results of these scenarios were generally aligned with the CADTH base case.
The economic results align with the available clinical evidence suggesting that there is a survival benefit for 177Lu vipivotide tetraxetan compared with SOC. However, CADTH was unable to comment on the cost-effectiveness of 177Lu vipivotide tetraxetan for patients previously treated with docetaxel and considered eligible for cabazitaxel because of inconclusive efficacy evidence. Further, CADTH could not make firm conclusions regarding the relative efficacy and cost-effectiveness of 177Lu vipivotide tetraxetan and cabazitaxel, given substantial limitations of the comparative evidence. Furthermore, the cost-effectiveness of 177Lu vipivotide tetraxetan and other potentially relevant treatment options, including radium-223 and olaparib, is unknown.
References
1.Clinical Study Report: PSMA-617-01. VISION: An international, prospective, open label, multicenter, randomized phase 3 study of 177Lu-PSMA-617 in the treatment of patients with progressive PSMA-positive metastatic castration-resistant prostate cancer (mCRPC) [internal sponsor's report]. Millburn (NJ): Advanced Accelerator Applications; 2021 Jun 28.
2.Network meta-analysis in progressive, PSMA–positive metastatic castration-resistant prostate cancer [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lutetium (177Lu) vipivotide tetraxetan. Mississauga (ON): Advanced Accelerator Applications Canada Inc. 2022 Jul 21.
3.Cabazitaxel for hormone-relapsed metastatic prostate cancer treated with docetaxel. Technology appraisal guidance [TA391]. 2016; https://www.nice.org.uk/guidance/ta391. Accessed 2022 Nov 2.
4.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lutetium (177Lu) vipivotide tetraxetan. Mississauga (ON): Advanced Accelerator Applications Canada Inc. 2022 Jul 21.
5.Pluvicto: lutetium (177Lu) vipivotide tetraxetan injection, solution for injection/infusion 1 000 MBq/mL at calibration; Therapeutic Radiopharmaceutical [product monograph]. Millburn (NJ): Advanced Accelerator Applications USA, Inc.; 2022 Jun 29.
6.de Wit R, de Bono J, Sternberg CN, et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. N Engl J Med. 2019;381(26):2506-2518. PubMed
7.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):1-12. PubMed
8.Olivier T, Powell K, Prasad V. Lutetium-177-PSMA-617 in Metastatic Castration-resistant Prostate Cancer: Limitations of the VISION Trial. Eur Urol. 2022:S0302-2838 (0322) 02613.
9.Hofman MS, Emmett L, Sandhu SK, et al. 177Lu-PSMA-617 (LuPSMA) versus cabazitaxel in metastatic castration-resistant prostate cancer (mCRPC) progressing after docetaxel: Updated results including progression-free survival (PFS) and patient-reported outcomes (PROs)(TheraP ANZUP 1603). Journal of Clinical Oncology 39, no. 6_suppl (February 20, 2021) 6-6. 2021. https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.6_suppl.6. Accessed 2022 Dec 12.
10.Hofman MS, Emmett L, Violet J, et al. TheraP: a randomized phase 2 trial of (177) Lu-PSMA-617 theranostic treatment vs cabazitaxel in progressive metastatic castration-resistant prostate cancer (Clinical Trial Protocol ANZUP 1603). BJU Int. 2019;124 Suppl 1:5-13. PubMed
11.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2022 Nov 2.
12.Prostate-Specific Membrane Antigen PET-CT Imaging for the Staging of Prostate Cancer in Canada. Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/attachments/2022-11/CM0001-PSMA-PET3.0-KH-AM-KH-KH2%20-%20with%20updates%20-%20QC-meta.pdf. Accessed 2022 Nov 15.
13.Cabazitaxel (45mg/4.5mL & 60mg/6mL) for injection. [product monograph]. Sandoz Canada Inc; 2022 Jul 18: https://pdf.hres.ca/dpd_pm/00066736.PDF. Accessed 2022 Nov 2.
14.DeltaPA. [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/.
15.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/.
16.Docetaxel (20mg/1mL,80mg/4mL, 160mg/8mL) for intravenous infusion. [product monograph]. St. Catharines, ON: Biolyse Pharma Corportion; 2016 Sep 23: https://pdf.hres.ca/dpd_pm/00036571.PDF. Accessed 2022 Nov 2.
17.Apo-abiraterone (250mg & 500mg) film coated tablets. [product monograph]. Toronto, ON: Apotex; 2021 Apr 1: https://pdf.hres.ca/dpd_pm/00060444.PDF. Accessed 2022 Nov 2.
18.Xtandi (Enzalutamide) 40mg capsules. [product monograph]. Markham, ON: Astellas Pharma Canada, Inc; 2022 Jan 24: https://pdf.hres.ca/dpd_pm/00064474.PDF. Accessed 2022 Nov 2.
19.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx.
20.Woon DT, Chandrasekar T, Aaron L, et al. Disparity in public funding of therapies for metastatic castrate-resistant prostate cancer across Canadian provinces. Canadian Urological Association Journal. 2018;12(10):328. PubMed
21.Firmagon (Degarelix) 120mg & 80mg for injection. [product monograph]. North York, ON: Ferring Pharmaceuticals; 2016 Mar 18: https://pdf.hres.ca/dpd_pm/00034229.PDF. Accessed 2022 Nov 2.
22.Lupron (Leurprolide acetate for injection) 3.75mg, 7.5mg, 11.25mg, 22.5mg & 30mg. [product monograph]. St. Laurent, QC: AbbVie Corporation; 2021 Nov 18: https://pdf.hres.ca/dpd_pm/00063626.PDF. Accessed 2022 Nov 2.
23.Morphine sufate (10mg, 15mg, 30mg, 60mg, 100mg, 200mg extended release capsules). [product monograph]. Montreal, QC: Ethypharm Inc.; 2019 Jun 11: https://pdf.hres.ca/dpd_pm/00051786.PDF. Accessed 2022 Nov 2.
24.OxyNEO (oxycodone hydrochlride controlled release tablets) 10mg, 15mg, 20mg, 30mg, 40mg, 60mg, & 80mg [product monograph]. Toronto, ON: Purdue Pharma; 2022 Jul 12: https://pdf.hres.ca/dpd_pm/00066651.PDF. Accessed 2022 Nov 2.
25.Trelstar (Triptorelin for injection) 3.75mg, 11.25mg & 22.5mg. [product monograph]. Montreal, QC: Knight Therapeutics Inc.; 2022 Sep 8: https://pdf.hres.ca/dpd_pm/00067340.PDF. Accessed 2022 Nov 2.
26.Xgeva (denosumab injection) 120mg/1.7mL single use vial. [product monograph]. Mississauaga, ON: Amgen Canada, Inc; 2011 May 10: https://pdf.hres.ca/dpd_pm/00059342.PDF. Accessed 2022 Nov 2.
27.Zoladex (Goserelin) 3.6mg depot. [product monograph]. Mississauga, ON: AstraZeneca; 2017 Dec 21: https://pdf.hres.ca/dpd_pm/00042728.PDF. Accessed 2022 Nov 2.
28.Zoledronic Acid for inejction. 4mg/5mL. [product monograph]. Boucherville, QC: JAMP Pharma Corporation; 2018 Nov 7: https://pdf.hres.ca/dpd_pm/00048158.PDF. Accessed 2022 Nov 2.
29.Zytram XL (tramadol hydrochloride controlled release tablets) 75, 100, 150, 200, 300 & 400mg [product monograph]. Toronto, ON: Purdue Pharma; 2022 Mar 14: https://pdf.hres.ca/dpd_pm/00065233.PDF. Accessed 2022 Nov 2.
30.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: lutetium (177Lu) vipivotide tetraxetan. Mississauga (ON): Advanced Accelerator Applications Canada Inc. 2021 Jul 21.
31.Canadian Cancer Society. Canadian Cancer Statistics. https://cancer.ca/en/research/cancer-statistics/canadian-cancer-statistics. Accessed 2022 Jun.
32.pCODR Expert Review Committee (pERC) Final Recommendation: Darolutamide (Nubeqa). 2020; https://www.cadth.ca/sites/default/files/pcodr/Reviews2020/10196DarolutamidenmCRPC_fnRec_REDACT_EC_22Apr2020_final.pdf. Accessed 2022 Jun.
33.Sartor O, De Bono J, Chi KN, et al. Lutetium-177–PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med. 2021;385(12):1091-1103. PubMed
34.Shayegan B, Wallis CJD, Malone S, et al. Real-world use of systemic therapies in men with metastatic castration resistant prostate cancer (mCRPC) in Canada. Urol Oncol. 2022;40(5):192 e191-192 e199.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 9: CADTH Cost Comparison Table for the Treatment of PSMA-Positive mCRPC
Treatment | Strength / concentration | Form | Price | Recommended dosage | 28-Day cost ($) | Total treatment Duration cost ($) |
|---|---|---|---|---|---|---|
lutetium (177Lu) vipivotide tetraxetan | 7.40 GBq | Vial | 27,000.0000a | 7.40 GBq every 6 weeks for a maximum of 6 cycles | 18,000 | 122,489 |
Antineoplastic drug | ||||||
Cabazitaxel (Generic)b | 40 mg / 1 mL 45 mg / 4.5 mL 60 mg / 6 mL | Vial | 2,465.0000 to 4,930.0002c | 20 mg/m2 every 3 weeksd | 3,285 to 6,570 | 18,069 to 36,137 |
Docetaxel (Generic) | 20 mg/1.0 mL 80 mg/4.0 mL 160 mg/8.0 mL | 1 mL Vial 4 mL Vial 8 mL Vial | 249.0000c 497.0000c 990.0000c | 75 mg/m2 as a 1-hour fusion every 3 weeks for 6 cyclesd,e | 1,320 | 5,940 |
Androgen synthesis inhibitor | ||||||
Abiraterone Acetate (Generic) | 250 mg 500 mg | Tab | 26.0313f 52.0625f | 1,000 mg daily | 2,916 | 12,630 |
Androgen receptor antagonist | ||||||
Enzalutamide (Xtandi) | 40 mg | Tab | 29.1954g | 160 mg daily | 3,270 | 14,166 |
Radiopharmaceutical | ||||||
Radium-223 (Xofigo) | 1,100 kBq/mL | Vial | 5,640.0000h | 55 kBq per kg, given at 4-week intervals for a total of 6 injections | 5,640 | 33,840 |
Note: as olaparib is only recommended for a small subset of the mCRPC population, it was excluded as a comparator and not considered in the cost table.
aSponsor-submitted price.
bCabazitaxel pre-treatment regimen includes prednisone, dexamethasone, filgrastim/pegfilgrastim and ranitidine.13 Patients receiving all treatments are pretreated with prednisone. The costs associated with pre-treatment regimens are not considered in the cost table.
cWholesale price reported by IQVIA DeltaPA. October 2022.14
dBased on an assumed body surface area of 2.05 m2. According to the product monograph, a dose of 25 mg/m2 may be used in select patients at the discretion of the treating health care provider. A median of 7.33 cycles is given to patients based on methods from the CARD trial.6
eA maximum of 6 cycles is given to patients based on the methods from the CHAARTED trial.
fOntario Benefit Formulary (accessed October 2022).15 Recommended dosages are from the respective monographs.5,13,16-18
gOntario Exceptional Access Program (accessed October 2022).19
hSponsor-submitted price sourced from Woon et al. 2018.20
Table 10: CADTH Cost Comparison Table for Standard Care of PSMA-Positive mCRPC
Treatment | Strength / concentration | Form | Price | Recommended dosage | 28-Day cost ($) | Average Treatment Duration cost ($) |
|---|---|---|---|---|---|---|
Androgen deprivation therapy | ||||||
Degarelix (Firmagon) | 80 mg | Inj | 274.1760a | Initial dose: 240mg given as 2 SC injections of 120mg Maintenance: monthly administration of 80mg as 1 SC injection | Year 1: 742 Subsequent: 252 | Year 1: 3,758 Subsequent: 3,290 |
120 mg | Inj | 370.9440a | ||||
Goserelin (Zoladex) | 3.6 mg 10.8 mg | Inj | 422.6778a 1,204.7322a | 3.6mg SC injection every 28 days 10.8mg SC injection every 13 weeks | 423 370 | 5,510 4,819 |
Leuprorelin / Leuprolide (Lupron) | 3.75 mg 7.5 mg 11.25 mg 22.5 mg 30 mg | Inj | 370.6000a 387.9700a 1,104.3000a 1,071.0000a 1,428.0000a | 7.5 mg monthly; 22.5 mg every 3 months; 30 mg every 4 months | 329 to 388 | 4,284 to 4,656 |
Triptorelin (Trelstar) | 3.75 mg 11.25 mg 22.5 mg | Inj | 346.3100a 1,038.9700a 1,659.9000a | 3.75 mg monthly; 11.25 every 3 months; 22.5 mg every 6 months | 255 to 346 | 3,320 to 4,156 |
Bone-targeting drugs | ||||||
Denosumab (Xgeva) | 120 mg/1.7 mL | Inj | 648.1100b | 120 mg every 3 monthsc | 199 | 2,592 |
Zoledronic Acid (Generic) | 4 mg/5 mL | Inj | 415.5600a | 4 mg every 3 monthsc | 127 | 1,662 |
Corticosteroids | ||||||
Dexamethasone (Generic) | 0.5 mg 4 mg | Tab | 0.1564a 0.3046a | 8 mg dailyd or 8 mg, 12 hours, 3 hours and 1 hour before infusione | 0.17e or 1.22d | 22e or 188d |
Prednisone (Generic) | 5 mg 50 mg | Tab | 0.0220a 0.1735a | 10 mg dailyd,e | 0.04d,e | 6e or 7d |
Opioid analgesics | ||||||
Morphine Sulphate (Generic) | 10 mg 15 mg 30 mg 60 mg 100 mg | Cap | 0.3250a 0.3750a 0.5590a 0.9900a 2.1460a | 100 mg twice daily | 110 to 182 | 1,139 to 1,892 |
Oxycodone Hydrochloride (OxyNEO) | 10 mg 15 mg 20 mg 30 mg 40 mg 80 mg | Tab | 0.9850b 1.1905b 1.4770b 1.9555b 2.5515b 4.7330b | 10 mg twice daily | 41 to 71 | 430 to 573 |
Tramadol Hydrochloride (Zytram) | 75 mg 100 mg 150 mg 200 mg 300 mg 400 mg | Tab | 1.2050g 1.5615g 2.2930g 3.0135g 4.3510g 5.7390g | 75 mg twice daily | 64 to 67 | 667 to 701 |
Cap = capsule; Inj = injection; SC = subcutaneous; Tab = tablet.
aOntario Benefit Formulary (accessed October 2022).15 Recommended dosages are from the respective monographs.21-29
bOntario Exceptional Access Program (accessed October 2022).19
cCADTH obtained clinical expert feedback that the recommended time between treatments with bone-targeting drugs is typically every 3 months.
dCabazitaxel regimen.13
eDocetaxel regimen.16
fAbiraterone Acetate regimen.17
gWholesale price reported by IQVIA DeltaPA. October 2022.14
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CADTH appraisal section. |
Model has been adequately programmed and has sufficient face validity | Yes | CADTH identified minor errors in the calculation of premedication costs for cabazitaxel and G-CSF costs. |
Model structure is adequate for decision problem | Yes | No Comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CADTH appraisal section. Imbalance in censoring and outcome assessment between treatment arms in the VISION trial make the validity of rPFS and SSE outcomes questionable. There is also a question about the difference in rPFS curve shown in the economic report and the clinical study report submitted by the sponsor. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The report did not clearly specify whether 177Lu vipivotide tetraxetan were used as a combination therapy with SOC or a monotherapy. In addition, the sponsor did not justify why radium-223 was not considered as a relevant comparator despite being widely used in some jurisdictions. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
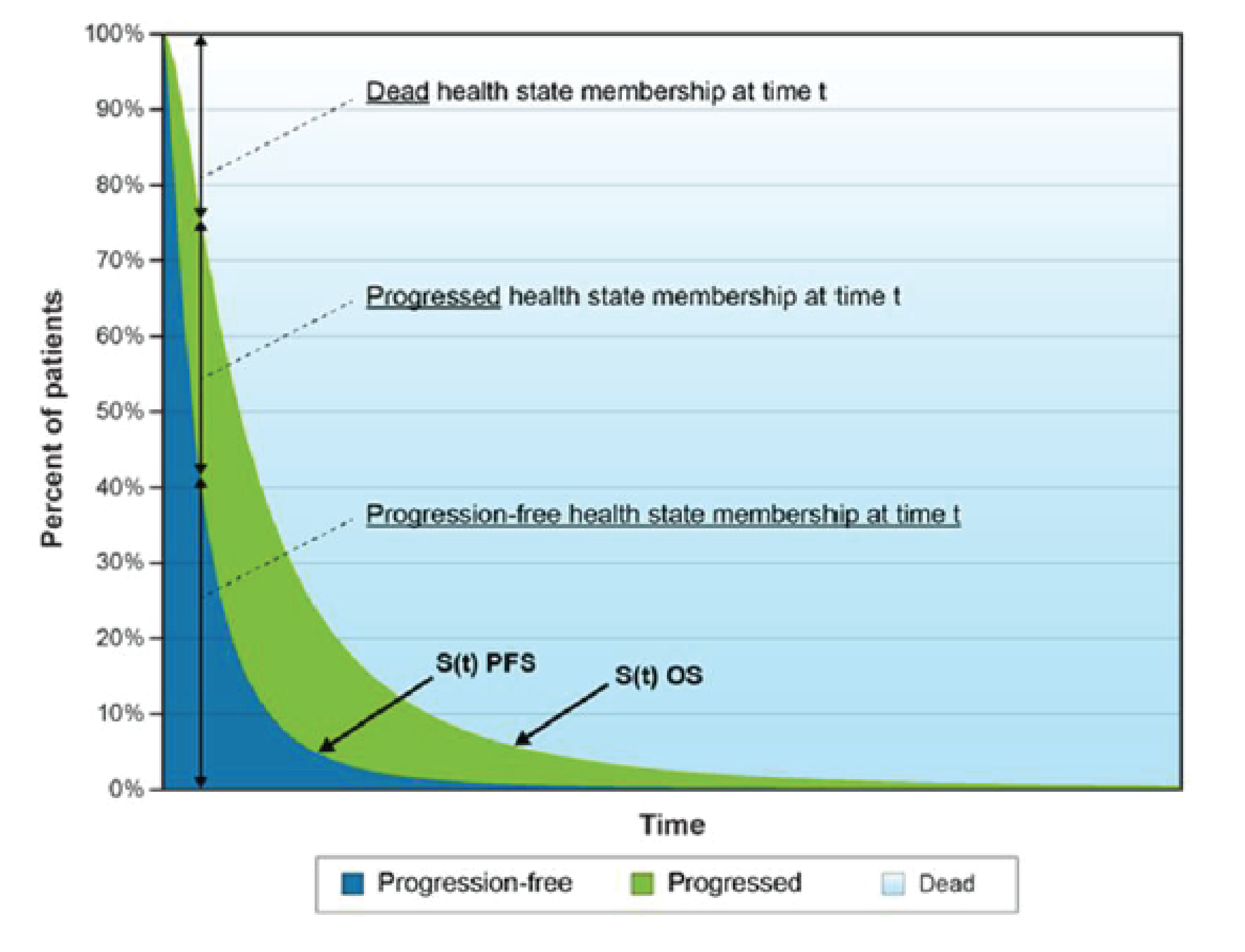
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 12: Disaggregated Summary of the CADTH Economic Evaluation Results
Treatment | Component | Value | Incremental (vs. standard of care) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
Standard of care | Progression-free | 0.59 | NA | NA |
Progressed | 0.53 | NA | NA | |
Total | 1.13 | NA | NA | |
Cabazitaxel | Progression-free | 1.02 | 0.43 | NA |
Progressed | 0.49 | −0.04 | NA | |
Total | 1.51 | 0.39 | NA | |
177Lu vipivotide tetraxetan | Progression-free | 1.02 | 0.43 | 0.00 |
Progressed | 0.49 | −0.04 | 0.00 | |
Total | 1.51 | 0.39 | 0.00 | |
Discounted QALYs | ||||
Standard of care | Progression-free | 0.422 | NA | NA |
Progressed | 0.350 | NA | NA | |
Total | 0.771 | NA | NA | |
Cabazitaxel | Progression-free | 0.726 | 0.304 | NA |
Progressed | 0.324 | −0.025 | NA | |
Total | 1.050 | 0.279 | NA | |
177Lu vipivotide tetraxetan | Progression-free | 0.726 | 0.304 | 0.00 |
Progressed | 0.324 | −0.025 | 0.00 | |
Total | 1.050 | 0.279 | 0.00 | |
Discounted costs ($) | ||||
Standard of care | Acquisition | 0 | NA | NA |
Administration | 0 | NA | NA | |
Concomitant treatments | 13,167 | NA | NA | |
Subsequent treatments | 8,121 | NA | NA | |
Health state costs | 7,591 | NA | NA | |
SSEs | 6,854 | NA | NA | |
AEs | 1,813 | NA | NA | |
Total | 24,378 | NA | NA | |
Cabazitaxel | Acquisition | 15,645 | 15,645 | NA |
Administration | 549 | 549 | NA | |
Concomitant treatments | 14,420 | 1,253 | NA | |
Subsequent treatments | 8,464 | 343 | NA | |
Health state costs | 8,372 | 781 | NA | |
SSEs | 6,079 | −774 | NA | |
AEs | 9,457 | 7,644 | NA | |
Total | 48,565 | 24,187 | NA | |
177Lu vipivotide tetraxetan | Acquisition | 123,835 | 123,835 | 108,191 |
Administration | 712 | 712 | 163 | |
Concomitant treatments | 9,848 | −3,320 | −4,572 | |
Subsequent treatments | 6,306 | −1,814 | −2,157 | |
Health state costs | 8,372 | 781 | 0 | |
SSEs | 8,265 | 1,412 | 2,186 | |
AEs | 6,096 | 4,283 | −3,361 | |
Total | 153,586 | 129,209 | 105,022 | |
Treatment | ICER vs. standard of care ($) | Sequential ICER ($) | ||
Standard of care | Ref. | Ref. | ||
Cabazitaxel | 91,220 | 91,220 | ||
177Lu vipivotide tetraxetan | 451,407 | Dominated | ||
177Lu = lutetium; AE = adverse event; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; Ref. = reference; SSE = symptomatic skeletal events; vs. = versus.
Detailed Results of CADTH Base Case
Scenario Analyses
Table 13: Summary of the CADTH Scenario Analyses
Drug | Total costs ($) | Total QALYs | Sequential ICER |
|---|---|---|---|
Sponsor's corrected base case | |||
SOC | $37,455 | 0.726 | Reference |
Cabazitaxel | $56,828 | 0.813 | $224,158 |
177Lu | $153,790 | 1.161 | $278,128 |
CADTH's base case | |||
SOC | $37,545 | 0.771 | Reference |
Cabazitaxel | $62,985 | 1.050 | $91,220 |
177Lu | $163,434 | 1.050 | Dominated |
CADTH's scenario analysis 1: Using the lower bound of HRs rPFS and OS of 177Lu vs. cabazitaxel | |||
SOC | 37,563 | 0.771 | Reference |
Cabazitaxel | 62,819 | 0.984 | 118,552 |
177Lu | 163,541 | 1.051 | 1,519,930 |
CADTH's scenario analysis 2: Using the upper bound of HRs rPFS and OS of 177Lu vs. cabazitaxel | |||
SOC | 37,563 | 0.771 | Reference |
Cabazitaxel | 62,052 | 0.691 | Dominated |
177Lu | 163,541 | 1.051 | 451,043 |
CADTH's scenario analysis 3: Using stratified flexible Weibull (1 knot) model to predict rPFS | |||
SOC | 37,562 | 0.773 | Reference |
Cabazitaxel | 62,990 | 1.050 | 91,657 |
177Lu | 163,549 | 1.050 | Dominated |
CADTH's scenario analysis 4: Using flexible Weibull (3 knot) model to predict rPFS | |||
SOC | 37,605 | 0.767 | Reference |
Cabazitaxel | 62,982 | 1.050 | 89,699 |
177Lu | 163,541 | 1.050 | Dominated |
CADTH's scenario analysis 5: Using Gamma model to predict OS | |||
SOC | 37,857 | 0.781 | Reference |
Cabazitaxel | 63,066 | 1.085 | 83,044 |
177Lu | 163,625 | 1.085 | Dominated |
CADTH's scenario analysis 6: Using stratified Gamma model to predict OS | |||
SOC | 37,917 | 0.791 | Reference |
Cabazitaxel | 63,036 | 1.072 | 89,462 |
177Lu | 163,595 | 1.072 | Dominated |
CADTH's scenario analysis 7: Using stratified log-logistic model to predict SSE | |||
SOC | 37,771 | 0.771 | Reference |
Cabazitaxel | 60,318 | 1.051 | 80,724 |
177Lu | 159,915 | 1.051 | Dominated |
CADTH's scenario analysis 8: Using stratified exponential model to predict SSE | |||
SOC | 37,740 | 0.771 | Reference |
Cabazitaxel | 61,326 | 1.051 | 84,445 |
177Lu | 161,293 | 1.051 | Dominated |
CADTH's scenario analysis 9: Using alternate health state utility values | |||
SOC | 37,563 | 0.749 | Reference |
Cabazitaxel | 62,983 | 1.024 | 92,186 |
177Lu | 163,541 | 1.024 | Dominated |
CADTH's scenario analysis 10: Using alternate health state utility values and applying utility decrement due to AEs/SSEs | |||
SOC | 37,563 | 0.817 | Reference |
Cabazitaxel | 62,983 | 1.075 | 98,740 |
177Lu | 163,541 | 1.063 | Dominated |
CADTH's scenario analysis 11: Changing the proportion of G-CSF to 100% | |||
SOC | 37,563 | 0.771 | Reference |
Cabazitaxel | 69,498 | 1.051 | 114,339 |
177Lu | 163,541 | 1.051 | Dominated |
CADTH's scenario analysis 12: Replacing filgrastim with peg-filgrastim | |||
SOC | 37,563 | 0.771 | Reference |
Cabazitaxel | 62,678 | 1.051 | 89,921 |
177Lu | 163,541 | 1.051 | Dominated |
CADTH's scenario analysis 13: Excluding PSMA testing cost | |||
SOC | 37,563 | 0.771 | Reference |
Cabazitaxel | 62,983 | 1.051 | 91,011 |
177Lu | 162,155 | 1.051 | Dominated |
CADTH's scenario analysis 14: Reducing the number of doses of cabazitaxel to 4 | |||
SOC | 37,563 | 0.771 | Reference |
Cabazitaxel | 58,647 | 1.051 | 75,487 |
177Lu | 163,498 | 1.051 | Dominated |
CADTH's scenario analysis 15: Removing subsequent cabazitaxel for those receiving cabazitaxel | |||
SOC | 37,565 | 0.771 | Reference |
Cabazitaxel | 62,545 | 1.051 | 89,437 |
177Lu | 163,534 | 1.051 | Dominated |
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 14: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted budget impact analysis (BIA) assessed expected budgetary impact resulting from reimbursing 177Lu vipivotide tetraxetan for the treatment Prostate-specific Membrane Antigen (PSMA)-positive Metastatic Castration Prostate Cancer (mCRPC).30 The BIA was conducted from the perspective of the Canadian public drug plans over a 3-year time horizon using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec). The sponsor assumed the costs of the therapy would be paid by the province in which the treating hospital was located for the Non-Insured Health Benefit (NIHB) population. Therefore, this BIA did not evaluate the impact on the NIHB population.30 The analysis was performed using jurisdiction-specific values by summing up individual provincial results to obtain consolidated results. Key inputs to the BIA are documented in Table 15.
The following key assumptions were made by the sponsor:
The sponsor used Ontario project cancer incidence as example to estimate the eligible Canadian population.
The sponsor assumed ██% of the eligible patient population would be registered in clinical trials.
The sponsor assumed 60% were suitable and 40% unsuitable for a second taxane among the population who already received ARPI and prior taxane therapy.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Prostate Cancer Incidence Progress to mCRPC PSMA-positive disease Tested for PSMA positivity Received ARPI Received 2 or more therapies in mCRPC Received ARPI and Docetaxel Eligible for Cabazitaxel Eligible for 177Lu vipivotide tetraxetan Unsuitable for Cabazitaxel Eligible for 177Lu vipivotide tetraxetan | 19,89031 28%32 86.6%33 90%a 100%34 28.5%34 71.8%a 60%b 100%b 40%b 80%b |
Number of patients eligible for drug under review (pan-Canadian) | 827 / 837 / 847 |
Market uptake (3 years) | |
Uptake (reference scenario) Cabazitaxel SoC Clinical Trials | ███% / ██% / ██% ██% / ██% / ██% ██% / ██% / ██% |
Uptake (new drug scenario) 177Lu vipivotide tetraxetan Cabazitaxel SoC Clinical Trials | ███% / ██% / ██% ██% / ██% / ██% ██% / ██% / ██% ██% / ██% / ██% |
Cost of treatment (per patient) | |
Cost of treatment over regimen 177Lu vipivotide tetraxetan Cabazitaxel SOC | $122,489 $18,077 $5,486 |
ARPI = androgen receptor pathway inhibition; mCRPC = metastatic castration-resistant prostate cancer; PSMA = prostate-specific membrane antigen; SOC = standard of care.
aAdvanced Accelerator Application on sponsor file.
bClinical Expert feedback obtained from sponsor.
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of 177Lu vipivotide tetraxetan for the treatment of PSMA-positive mCRPC would lead to an incremental budget impact of $13,131,824 in Year 1, $22,146,704 in Year 2, and $31,374,741 in Year 3. The total 3-year incremental cost was $66,653,269. Scenario analyses were completed to (i) account for administration costs, (ii) include upcharges, dispensing fees, and co-payments, (iii) include costs for subsequent therapies and (iv) include vial sharing. These scenario analyses impacted the 3-year incremental budget impact from + 1.6% to −1.6% in terms of % change from base case. The sponsor also explored the impact of alternative assumptions through sensitivity analyses, which suggested the 3-year total incremental budget impact may vary from $35,707,108 to $79,983,922.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Relevant comparator excluded: The sponsor’s submitted budget impact detailed 2 main competitors of 177Lu vipivotide tetraxetan: cabazitaxel and SOC. Clinical expert feedback obtained by CADTH highlighted that in Canadian clinical practice, clinicians may prescribe radium-223 and that it is a relevant comparator to 177Lu vipivotide tetraxetan. While this treatment was listed as a subsequent therapy and is not reimbursed in all jurisdictions, clinical expert feedback suggested that radium-223 would be a comparator in jurisdictions in which it is accessible.
CADTH could not undertake reanalysis to address this limitation due to inflexibility of the budget impact model and lack of information on market shares.
Underestimated market capture of comparators: The sponsor’s submitted budget impact analysis indicated that clinical trials would result in a yearly ██% market share from baseline to Year 3. The clinical experts consulted by CADTH did not agree with this market share assumption by the sponsor and instead indicated that the sponsor likely underestimated SOC and cabazitaxel uptake.
To address this limitation, CADTH undertook a reanalysis by removing the market share for clinical trials and distributing the share proportionally among the other comparators.
Uncertainty associated with PSMA-positive disease testing costs not included: The sponsor’s submitted budget impact analysis did not include PSMA positivity testing costs. In the economic evaluation, the sponsor assumed a unit cost of $1,200 applied per patient to determine the costs associated with diagnostic testing of the patient. Additionally, the sponsor assumed 1.15 tests would be required to diagnose 1 patient, resulting in the costs associated with diagnostic testing per treated patient to total to $1,386. By not including these costs, the sponsor’s submitted BIA may have underestimated the costs to the public health system associated with the reimbursement of 177Lu vipivotide tetraxetan. The sponsor assumed that 90% of patients are tested for PSMA positivity. The clinical experts consulted by CADTH suggested this assumption may be higher than what is observed in practice. Furthermore, the clinical experts noted that there are limited facilities that have the appropriate equipment to test PSMA-positive patients and the patients that fall within this category are sick and may not travel to further facilities for the testing. This associated with public funding of PSMA PET-CT imaging is highlighted in a recent CADTH Technology Report on this topic.12
To address this limitation, CADTH undertook exploratory analyses to include PSMA PET-CT testing costs using the sponsor’s assumed cost of $1,200 per test (1 test per person). CADTH also undertook a scenario analysis assuming only 50% of the eligible population is tested for PSMA positivity, to reflect clinical expert feedback obtained by CADTH regarding capacity limitations currently associated with PSMA PET-CT testing.
Sponsor-submitted concomitant drugs in SOC arm do not reflect those used in clinical practice: The sponsor’s submitted budget impact detailed several concomitant drugs in the SOC treatment arm that were deemed unnecessary by clinical expert feedback obtained by CADTH. The expert’s found the therapies to be symptom controlling rather than anticancer therapy. Additionally, the clinical experts highlighted that the use of ARPIs (Abiraterone and Enzalutamide) are not likely to be considered a concomitant therapy or SOC for PSMA-positive mCRPC patients, given that to be eligible for 177Lu vipivotide tetraxetan, the patients must have previously received an ARPI. Therefore, the inclusion of these ARPIs may have led to an overestimation in the SOC cost arm as well as a concomitant therapy.
To address this limitation, CADTH removed the irrelevant concomitant therapies for an updated base case. Additionally, CADTH undertook a scenario analysis including ARPIs abiraterone and enzalutamide.
The sponsor’s submitted model for the budget impact analysis is not user-friendly and unnecessarily complicated: Several of the model inputs and assumptions in the sponsor’s submitted budget impact analysis were difficult to test or modify with alternate inputs and assumptions due to the unnecessary complexity (e.g., illogical formulas and disconnected worksheets) and structural constraints of the submitted budget impact model.
CADTH was unable to address this limitation, resulting in several exploratory analyses
Additional limitations were identified but were not considered to be key limitations. These limitations include that unit costs of concomitant treatments did not reflect publicly available drug costs in Canada. CADTH corrected these costs using data identified from public sources. The sponsor also noted that the dosage for cabazitaxel was 25 mg/m2 per cycle, while CADTH identified a dose of 20 mg/m2 per cycle from the product monograph. Lastly, the sponsor submission did not include olaparib as a comparator but was listed as a subsequent treatment. As olaparib is only recommended for a small subset of this population, and 177Lu vipivotide tetraxetan is not expected to take market share from olaparib in this population, its exclusion as a comparator was considered reasonable.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18. Based on the CADTH base case, the budget impact associated with 177Lu vipivotide tetraxetan’s reimbursement in the indicated target population is expected to be $13,670,690 in Year 1, $23,120,229 in Year 2, and $32,793,211 in Year 3, with a 3-year total of $69,584,130.
CADTH Reanalyses of the BIA
Table 16: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Concomitant treatment costs | 28-day cost: Abiraterone: $858 Enzalutamide: $3,175 Zoledronic Acid: $135 Denosumab: $629 Dexamethasone: $26 Morphine: $58 Oxycodone: $10 | 28-day cost: Abiraterone: $2,916 Enzalutamide: $3,270 Zoledronic Acid: $127 Denosumab: $199 Dexamethasone: $34 Morphine: $110 Oxycodone: $41 |
2. Treatment dosage | 25 mg/m2 per cycle of cabazitaxel 12 mg per day of dexamethasone 4 mg / 3 weeks of zoledronic acid 120 mg / 4 weeks of denosumab 5mcg/kg daily for 14 days of filgrastim | 20 mg/m2 per cycle of cabazitaxel 8 mg per day of dexamethasone 4 mg / 3 months of zoledronic acid 120 mg / 3 months of denosumab 5mcg/kg daily for 7 days of filgrastim |
Corrected sponsor’s base case | 1 + 2 | |
Changes to derive the CADTH base case | ||
1. Market share | Reference Scenario: Cabazitaxel: ██% / ██% / ██% SoC: ██ % / ██% / ██% Clinical Trials: ██% / ██% / ██% New Drug Scenario: 177Lu -PSMA-617: ██% / ██% / ██% Cabazitaxel: ██% / ██% / ██% SoC: ██% / ██% / ██% Clinical Trials: ██% / ██% / ██% | Reference Scenario: Cabazitaxel: 64% / 64% / 64% SoC: 36% / 36% / 36% Clinical Trials: 0% / 0% / 0% New Drug Scenario: 177Lu -PSMA-617: ██% / ██% / ██% Cabazitaxel: ██% / ██% / ██% SoC: ██% / ██% / ██% Clinical Trials: 0% / 0% / 0% |
2. Updated SoC arm to reflect concomitant drugs used in clinical practice | ARPIs: Abiraterone Enzalutamide Antiemetics: Prochlorperazine Antifungals: Ketoconazole Bone-Targeting Drugs Zoledronic Acid Denosumab Corticosteroids: Dexamethasone Erythropoietin-Stimulating Drugs: Darbepoetin Alpha Opioid Analgesics: Morphine Oxycodone Tramadol | Bone-Targeting Drugs: Zoledronic Acid Denosumab Corticosteroids: Prednisone Dexamethasone Opioid Analgesics: Morphine Oxycodone Tramadol ADTs: Goserelin Degarelix Leuprolide Triptorelin |
CADTH base case | Reanalysis 1 + 2 | |
ARPI = androgen receptor pathway inhibition; SoC = standard of care.
Table 17: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $66,653,269 |
Corrected base case | $68,607,064 |
CADTH reanalysis 1: Market shares | $68,367,820 |
CADTH reanalysis 2: SoC update | $69,932,572 |
CADTH base case (reanalysis 1 + 2) | $69,584,130 |
BIA = budget impact analysis; SoC = standard of care.
CADTH conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 18. The scenario analyses conducted included exploring a 92% price reduction, consistent with that required for the CADTH base case to be considered cost-effective at a WTP threshold of $50,000 per QALY. In this scenario analysis the budget impact led to cost savings. This was driven by the fact that CADTH uses publicly available list prices. Additionally, scenario analyses were conducted to assess the impact of: substituting filgrastim for pegfilgrastim in the preregimen of cabazitaxel, 50% PSMA positivity testing, and having 4 cycles of cabazitaxel highlighted by clinical experts in comparison to 7.33.
Due to the limitations on the transparency and usability of the sponsor’s submitted BIA, exploratory analyses were conducted to explore the cost of PSMA PET-CT testing costs based on the annual incidence of prostate cancer and for the pan-Canadian estimate of patients that progress to mCRPC; these costs were applied to the new drug scenario as they are not part of the reference scenario (current practice). Additional exploratory analyses were conducted to review the impact of the populations eligible and ineligible for cabazitaxel, as the inflexibility of the BIA model did not allow for a scenario analysis on these key subgroup populations. The results of these exploratory analyses, particularly the population specific analyses, are provide as guidance, as CADTH could not address the relevant comparators for each group appropriately, given the limitations with the sponsor-submitted budget impact model.
Table 18: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $13,646,416 | $13,808,489 | $13,972,736 | $14,139,189 | $41,920,414 |
New drug | $13,646,416 | $26,940,313 | $36,119,440 | $45,313,930 | $108,573,683 | |
Budget impact | $0 | $13,131,824 | $22,146,704 | $31,374,741 | $66,653,269 | |
Corrected base case | Reference | $11,175,257 | $11,307,981 | $11,442,486 | $11,578,796 | $34,329,263 |
New drug | $11,175,257 | $24,824,736 | $34,238,372 | $43,873,219 | $102,936,327 | |
Budget impact | $0 | $13,516,755 | $22,795,886 | $32,294,423 | $68,607,064 | |
CADTH base case | Reference | $13,177,326 | $13,337,452 | $13,499,784 | $13,664,354 | $40,501,590 |
New drug | $13,177,326 | $27,008,143 | $36,620,013 | $46,457,565 | $110,085,721 | |
Budget impact | $0 | $13,670,690 | $23,120,229 | $32,793,211 | $69,584,130 | |
CADTH scenario analysis 1: 92% price reduction | Reference | $13,177,326 | $13,337,452 | $13,499,784 | $13,664,354 | $40,501,590 |
New drug | $13,177,326 | $13,031,117 | $13,047,884 | $13,063,455 | $39,142,456 | |
Budget impact | $0 | -$306,335 | -$451,900 | -$600,899 | -$1,359,134 | |
CADTH scenario analysis 2: pegfilgrastim substitution | Reference | $12,711,268 | $12,865,860 | $13,022,582 | $13,181,467 | $39,069,909 |
New drug | $12,711,268 | $26,595,499 | $36,239,743 | $46,110,490 | $108,945,732 | |
Budget impact | $0 | $13,729,640 | $23,217,161 | $32,929,023 | $69,875,823 | |
CADTH scenario analysis 3: Four cabazitaxel cycles | Reference | $8,880,121 | $8,989,212 | $9,099,823 | $9,211,978 | $27,301,013 |
New drug | $8,880,121 | $23,203,432 | $33,113,794 | $43,257,419 | $99,574,646 | |
Budget impact | $0 | $14,214,220 | $24,013,971 | $34,045,442 | $72,273,633 | |
CADTH scenario analysis 4: PSMA positivity testing 50% | Reference | $7,320,736 | $7,409,696 | $7,499,880 | $7,591,308 | $22,500,883 |
New drug | $7,320,736 | $15,004,524 | $20,344,452 | $25,809,758 | $61,158,734 | |
Budget impact | $0 | $7,594,828 | $12,844,572 | $18,218,451 | $38,657,850 | |
CADTH exploratory analysis 1: cabazitaxel-eligible population | Reference | $13,177,326 | $8,669,344 | $8,774,860 | $8,881,830 | $26,326,034 |
New drug | $13,177,326 | $17,555,293 | $23,803,008 | $30,197,417 | $71,555,719 | |
Budget impact | $0 | $8,885,949 | $15,028,149 | $21,315,587 | $45,229,685 | |
CADTH exploratory analysis 2: cabazitaxel ineligible population | Reference | $13,177,326 | $4,668,108 | $4,724,924 | $4,782,524 | $14,175,557 |
New drug | $13,177,326 | $9,452,850 | $12,817,005 | $16,260,148 | $38,530,002 | |
Budget impact | $0 | $4,784,742 | $8,092,080 | $11,477,624 | $24,354,446 | |
CADTH exploratory analysis 3: PSMA positivity testing costs on % who progress to mCRPC | Reference | $13,177,326 | $13,337,452 | $13,499,784 | $13,664,354 | $40,501,590 |
New drug | $13,177,326 | $33,771,484 | $43,464,745 | $53,384,795 | $130,621,024 | |
Budget impact | $0 | $20,434,031 | $29,964,961 | $39,720,441 | $90,119,433 | |
CADTH exploratory analysis 4: PSMA positivity testing costs on annual incidence of prostate cancer | Reference | $13,177,326 | $13,337,452 | $13,499,784 | $13,664,354 | $40,501,590 |
New drug | $13,177,326 | $51,162,931 | $61,065,485 | $71,197,672 | $183,426,088 | |
Budget impact | $0 | $37,825,478 | $47,565,701 | $57,533,319 | $142,924,498 |
ARPI = androgen receptor pathway inhibition; BIA = budget impact analysis; PSMA = prostate-specific membrane antigen.
Ethics Review
Abbreviations
177Lu
lutetium-177
ARPI
androgen receptor pathway inhibitor
BSC
best supportive care
BSoC
best standard of care
CMII
Canadian Medical Imaging Inventory
CRPC
castration-resistant prostate cancer
CUA
Canadian Urological Association
CUOG
Canadian Uro Oncology Group
GAHT
gender-affirming hormone therapy
GAS
gender-affirming surgery
mCRPC
metastatic Castration-Resistant Prostate Cancer
OS
overall survival
PSMA
prostate-specific membrane antigen
rPFS
radiographic progression-free survival
Summary
Input provided by patient groups, clinician groups, and provincial drug programs, as well as direct engagement with clinical experts and relevant literature, was reviewed to identify ethical considerations relevant to the use of lutetium-177 (177Lu) vipivotide tetraxetan (Pluvicto) for the treatment of adults with metastatic castration-resistant prostate cancer (mCRPC).
Ethical considerations arising in the context of mCRPC highlight the impact on patients, as well as disparities in the incidence, treatment, and outcomes of mCRPC, especially as they affect racialized, transgender, and gender nonbinary people. The treatment space of mCRPC is complex, and although there may be general guidance on the types of interventions that could be useful at different stages, there is currently no optimal treatment sequence. This implies a heavy reliance on clinical expertise and a provider’s ability to involve patients in a process of shared decision-making to choose treatment options that are reflective of the ideals discussed during the process. This is particularly important in the context of mCRPC as it is incurable, but it can also be a challenge as providers may not always be well trained in the process of shared decision-making.
Ethical considerations arising in the evidence used to evaluate 177Lu vipivotide tetraxetan highlight limitations related to the definition of “standard of care” used in the VISION trial, whether the inclusion and exclusion criteria were adequately applied, and the high withdrawal rate from the control arm. It was also indicated that participants in the VISION trial may not be reflective of those seen in clinical practice, even if clinical experts felt the trial data would be generalizable to patients with mCRPC.
As a radiopharmaceutical that requires extensive health system resourcing, the context of 177Lu vipivotide tetraxetan raises several ethical considerations related to its access and use. The need to confirm prostate-specific membrane antigen (PSMA) status is a prerequisite for being considered a candidate for 177Lu vipivotide tetraxetan, yet access to PET-CT, and more specifically PSMA PET-CT, is very limited in Canada. The logistics associated with the supply and delivery of 177Lu vipivotide tetraxetan also raise ethical considerations related to equitable access. Variable access to both PSMA PET-CT and 177Lu vipivotide tetraxetan may pose a challenge to clinicians deciding when or how to discuss 177Lu vipivotide tetraxetan as a treatment option for patients who might be strong candidates.
The already limited availability of PET-CT broadly is further narrowed in the context of 177Lu vipivotide tetraxetan, which requires the onsite or regional production of radiotracers that can specifically target PSMA-positive tumours. Funding the development of further PSMA PET-CT capacity will likely be an extensive financial and logistical burden on the health care system.
Objectives
To identify and describe ethical considerations associated with the use of 177Lu vipivotide tetraxetan for the treatment of adults with PSMA-positive mCRPC, including those related to the broader context of mCRPC, the evidentiary basis and use of 177Lu vipivotide tetraxetan as a targeted radiopharmaceutical, and other considerations relevant to health systems.
Research Questions
This report addresses the following research questions:
What ethical considerations arise in the context of mCRPC?
What ethical considerations arise related to the evidence used (e.g., clinical and economic data) to evaluate 177Lu vipivotide tetraxetan?
What ethical considerations arise from the use of 177Lu vipivotide tetraxetan for clinicians, patients, and their caregivers?
What ethical considerations for health systems are involved in the context of 177Lu vipivotide tetraxetan and its complementary diagnostic, PSMA PET-CT?
Methods
Overview
To identify ethical considerations relevant to the use of 177Lu vipivotide tetraxetan in the treatment of PSMA-positive mCRPC, this ethics report was driven by relevant questions identified in the EUnetHTA Core Model 3.0, Ethics Analysis Domain,1 and supplemented by relevant questions from the Equity Checklist for Health Technology Assessment.2 These guiding questions were organized to respond to the 4 research questions posed. In response to each of these 4 questions, this report investigated ethical considerations for 4 domains of interest:
The treatment of patients living with mCRPC and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies).
The evidence used to demonstrate the benefits, harms, and value of 177Lu vipivotide tetraxetan (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, appropriateness of analytical methods and models to all population groups; ethical considerations related to the data or assumptions in the economic evaluation).
The use of 177Lu vipivotide tetraxetan, including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, and society, and considerations related to access to these therapies.
The uptake of 177Lu vipivotide tetraxetan in health systems, including considerations related to the distribution of health care resources (e.g., access to its complementary diagnostic, PSMA PET-CT) and the presence of 177Lu vipivotide tetraxetan as a radiopharmaceutical that requires special handling and disposal.
These were explored through a review and synthesis of project inputs and relevant literature to highlight ethical considerations in each of the domains.
Data Collection Approach: Review of Project Inputs and Literature
Data to inform this ethics report were drawn from the identification of ethical considerations (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis) raised from input provided by patient and clinician groups, clinical experts, and drug programs collected during this review, as well as a complementary review of the published literature. Ongoing collaboration and communication with the CADTH review team assisted in the clarification and identification of the ethical considerations raised.
Review of Project Inputs
Over the course of this CADTH review, a single reviewer collected and considered input from various sources to inform this ethics report. All input was reviewed for content related to ethical considerations (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues on an evidentiary basis) relevant to the research questions driving this report. This report considered the following sources:
the sponsor’s submission
clinician group input that was coordinated by the Canadian Cancer Society
patient group input that was gathered by both the Canadian Cancer Society and the Canadian Cancer Survivor Network
drug program input that was submitted to CADTH
written input and discussion with 2 clinical experts directly engaged by CADTH
CADTH clinical and health economics reviewers who were engaged to identify domains of ethical interest arising from their reviews and to identify further questions or sources to pursue.
Literature Search Methods
A literature search was conducted by an information specialist on key resources, including MEDLINE All (1946–) via Ovid, Philosopher’s Index via Ovid, CINAHL, and Scopus. Duplicates were removed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Pluvicto (177Lu vipivotide tetraxetan), metastatic castration-resistant prostate cancer (mCRPC), and prostate-specific membrane antigen PET-CT (PSMA PET-CT).
CADTH-developed search filters were applied to limit retrieval to citations related to empirical and normative ethical considerations. The searches for Pluvicto and mCRPC were completed on September 15, 2022, and the search for PSMA PET-CT was completed on September 27, 2022. Search results were limited to English-language documents.
Grey literature (literature that is not commercially published) was identified by searching sources listed in relevant sections of the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.3 The grey literature search for ethical considerations was conducted on September 29, 2022. The main search concepts were Pluvicto (177Lu vipivotide tetraxetan) and metastatic castration-resistant prostate cancer (mCRPC), and prostate-specific membrane antigen PET-CT (PSMA PET-CT). The search was limited to English-language documents. Google was used to search for additional internet-based materials. These searches were supplemented by reviewing bibliographies of key papers and through contacts with experts and CADTH reviewers, as appropriate.
Literature Screening and Selection
Literature was screened for relevance to ethical considerations in the context of 177Lu vipivotide tetraxetan for the treatment of PSMA-positive mCRPC, driven by the 4 research questions driving this report. Both empirical and nonempirical publications were included in the review. The selection of relevant literature proceeded in 2 stages. In the first stage, the titles and abstracts of citations were screened for relevance by a single reviewer. Publications were marked as either “retrieve” or “unsure” if they identified normative or empirical ethical considerations related to:
evidence on, use of, or implications of 177Lu vipivotide tetraxetan for the treatment of adults with PSMA-positive mCRPC
screening for PSMA-positive mCRPC using PET-CT technology
living with or treating mCRPC.
Citations marked retrieve were ordered for full-text review. For citations marked unsure, a second reviewer was consulted to determine whether full texts should be retrieved. Citations the 2 reviewers agreed on were then retrieved for full-text review. In the second stage, the primary reviewer read and assessed the eligibility of the full-text publications. Publications meeting the aforementioned criteria were included in the ethics report and those that did not were excluded.
As a parallel process, grey literature and other sources drawn from relevant bibliographies or in consultation with experts or other CADTH reviewers were retrieved and reviewed in accordance with the aforementioned selection criteria.
Data Analysis
The data analysis for this ethics report included the collection, coding, and thematic analysis of data drawn from the literature and project inputs, driven by the 4 research questions guiding this report. The reviewer conducted 2 cycles of coding to abstract, identify, and synthesize relevant ethical considerations in the literature and from relevant project inputs.
In the initial coding phase, publications and input sources were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using methods of qualitative description.4 Initial descriptive coding of the reports focused broadly on categories concerning the ethical considerations that were described. In the second coding phase, major themes and subcodes were identified through repeated readings of the data4 and summarized into the thematic categories within each domain or research question. When ethical content emerged that did not fit into the categories or domains outlined in the research questions, this was noted.
Data collected and analyzed from these sources were thematically organized and described according to the 4 research questions and domains driving this report. The results of this analysis and its limitations and conclusions are described here.
Results
Description of Included Sources
Data for this ethics report were drawn from a review of the input provided by patients, clinician groups, and drug programs, and in consultation with clinical experts engaged for this review. A description and summary of these sources are included in the clinical report of this review.
A total of 471 citations were identified in the search of published literature. After title and abstract screening, 454 citations were excluded and 17 potentially relevant publications from the electronic search were retrieved for full-text review. Of the potentially relevant publications, 6 were excluded, as they did not discuss ethical issues arising in the context of 177Lu vipivotide tetraxetan for the treatment of mCRPC. The 11 publications that met the inclusion criteria were included in this report. In addition, 5 relevant publications were retrieved from other sources.
Of the 16 publications used to inform this report, 14 examined ethical considerations related to diagnosis, treatment, and outcomes for people with mCRPC; 1 examined ethical considerations related to the evidence used to evaluate 177Lu vipivotide tetraxetan; and 1 examined ethical considerations related to the production and use of 177Lu vipivotide tetraxetan’s complementary diagnostic, PSMA PET-CT. Details regarding the characteristics of included publications are reported in Table 1.
Key Ethical Considerations
The context of mCRPC and determination of the role that 177Lu vipivotide tetraxetan might play in the care of people with mCRPC are complex and multifaceted. The range of ethical considerations related to the fact that mCRPC is incurable is an important overarching consideration. 177Lu vipivotide tetraxetan is being considered for adults who have mCRPC refractory to multiple therapies, meaning that providers and their patients who engage with 177Lu vipivotide tetraxetan are operating in the context of incremental survival benefits in a way that is supportive of an improved quality of life at the end of life.
Ethical Considerations Related to the Experiences and Treatment of mCRPC
Patient Experiences With mCRPC
Published literature and patient input received by CADTH identified the physical and psychosocial aspects of mCRPC and their impact on people living with the disease. Some of the more prominent concerns raised were the way mCRPC can affect libido and a person’s ability to engage in sexual activity, travel, exercise, maintain a sense of mental wellbeing, and fulfill perceived family obligations.5,6 Patient input described challenges related to access to care and treatment experienced by patients with mCRPC, including costs associated with transportation to appointments, tests, procedures not publicly funded or covered by private insurance, and complementary medicines recommended by health care teams (e.g., vitamins and supplements), and loss of income.
Some of the improvements people with mCRPC are seeking from future treatments include effective therapies with fewer side effects, more affordable and accessible drugs, and treatments that can prolong life or cure mCRPC altogether. As mCRPC is currently considered incurable, supportive care (e.g., mental health programming, nursing staff, and end-of-life planning services) that is well resourced could help alleviate some of the psychosocial pressures experienced by people living with mCRPC or dealing with the side effects of treatment.5,6
Disparities in Identification, Treatment, and Outcomes of Prostate Cancer and mCRPC
The published literature has identified several populations that experience significant disparities in the identification, treatment, and outcomes of prostate cancer and, more specifically, mCRPC. In the US, for example, Black patients have a significantly greater chance of dying from prostate cancer — around 2.5 times higher — than white patients.7 As well, discriminatory and exclusionary health care spaces may prevent transgender women and people who area gender nonbinary with prostate cancer from engaging with the health care system, which can lead to delays in accessing diagnostic and specialist services.8,9 Older patients may also face barriers to treatment, and despite the fact that improved therapeutic options for mCRPC are helping people live longer, there are concerns that ageist assumptions around mental acuity and desired outcomes may foster paternalistic approaches to care for older patients with mCRPC.10
Disparities in access to and participation in clinical trials for all novel castration-resistant prostate cancer (CRPC) or mCRPC therapeutics were identified in 1 study as leading to further disparities in treatment and outcomes for Black patients in the US.7 The authors of that study noted that Black patients are consistently underrepresented in trials of novel mCRPC therapies, by upwards of 80%.7 Although there is no clear evidence indicating that prostate cancer in Black patients is any different biologically than in other populations, the authors expressed concern that the limited data from randomized controlled trials on Black patients using these therapies makes it a challenge to understand the efficacy and determine optimal care strategies in this population.7 Other authors suggest, however, that it is not enough to think about the different outcomes for Black patients with mCRPC primarily along biologic lines.11 Instead, based on an analysis conducted using data from across Canada, those authors report that when adjusted for nonbiological differences (e.g., access to health care and socioeconomic status), the difference in the risk of death from prostate cancer between Black and white patients was nearly eliminated.11 In addition to developing clinical evidence of responses to various mCRPC therapeutics in Black patients, this points to the importance of attending to social determinants and access challenges when considering disparities in the incidence, treatment, and outcomes for all mCRPC therapies.
Published literature indicated that there is a notable absence of guidance on how to provide appropriate screening, diagnostics, treatment, and care for transgender women and people who are gender nonbinary with prostate cancer.8,9 The absence of guidance may further entrench existing inequities in access to gender-affirming health care and seriously affect outcomes for this population. In particular, even when transgender women on gender-affirming hormone therapy (GAHT) or who have undergone gender-affirming surgery (GAS) are able to access diagnostic and specialist services, the absence of any guidance on what qualifies as normal or abnormal results in common risk stratification tools (e.g., prostate-specific antigen levels) may pose a challenge to providers trying to appropriately identify the presence or stage of prostate cancer in this population.8 Cancer staging is an important element for all people presenting with suspected or actual prostate cancer, but the challenges around staging are particularly problematic for transgender women on long-term GAHT, who seem more likely to present with advanced CRPC, potentially mCRPC, because GAHT is focused on decreasing testosterone to castrate levels.8,9 Presentation at an advanced stage can affect the treatment options available and negatively affect the types of outcome that can be achieved.
Although CRPC and mCRPC treatments can help people live longer after diagnosis, published studies have identified challenges related to the organization and provision of care for older patients.10,12 Assumptions about older patients may lead to paternalistic approaches to care and neglect the importance of shared decision-making, the authors of those studies report.10,12 Rather than focus on age as the determining factor in what to offer and how to engage, the authors have argued that providers should elevate abilities and lifestyle over age when making care decisions.10,12
Challenges of Ensuring Equitable Access to Existing mCRPC Therapies
The disparities in access to mCRPC care faced by racialized, transgender, and people who are gender nonbinary are particularly problematic, but the clinical experts engaged in this review suggested that some challenges related to access apply to all people living with mCRPC. Notably, ensuring equitable access to mCRPC therapies may be difficult, given the absence of clear guidelines on the optimal sequencing of therapies, the geographic differences in reimbursement, and the broad reliance on a clinician’s ability to engage patients in a process of shared decision-making.
Therapeutic options for mCRPC have expanded over the past decade and brought with them challenges related to clinical determinations of the appropriateness and optimal sequencing of these novel therapies.13-17 Although this expansion in options has an implied positive benefit, none have been curative and mCRPC remains incurable.14 Nonetheless, with the development and FDA approval of docetaxel nearly 2 decades ago, in 2004, life-prolonging therapies for mCRPC are available.
Following its establishment as the first therapeutic drug to have a proven overall survival (OS) benefit in the mCRPC setting, docetaxel remained the only option for improving OS for another 6 years.13 This longstanding role as the sole option for mCRPC established a treatment paradigm that was centred on docetaxel and provided some sequencing clarity for novel therapeutics in line to become the next line of treatment when docetaxel failed.13 However, with approval of the radiopharmaceutical radium-223 (for patients with symptomatic bone metastases and without visceral metastases) and the androgen receptor pathway inhibitors (ARPIs) enzalutamide and abiraterone in both the prechemotherapy and postchemotherapy spaces, by 2015 this clarity was beginning to blur.13,15
Current guidelines in Canada17 and provincial funding schemas18 are reflective of the ongoing challenges around the optimal sequencing of mCRPC therapies. Although the most recent Canadian Urological Association (CUA) and Canadian Uro Oncology Group (CUOG) joint guidelines for the management of CRPC provide general parameters for which therapies should be considered in the first, second, and third lines for mCRPC, they note that the optimal sequencing is currently unknown.17 Given the variability in funding regimens across provinces,18 there may be a concern that access to the most effective mCRPC therapies for individual patients may be inequitably distributed across Canada. This could be particularly challenging in the case of radiopharmaceuticals used in mCRPC settings, given the finding by Woon et al. (2018)18 that, at the time of publication, radium-223, the other radiopharmaceutical used in the mCRPC setting for patients with symptomatic bone metastases and without visceral metastases, was only publicly funded in 5 provinces.
This uncertainty around optimal sequencing and the resultant variability in access across settings may place added pressure on clinical decision-making. In a qualitative study conducted by Dutch urologists, oncologists, and oncology nurses, participants repeatedly detailed the importance of shared decision-making in the context of mCRPC.12 However, they indicated that 1 of the primary challenges with shared decision-making was that Dutch providers were often not skilled in ways to support this process and needed well-developed decision aids.12 Relatedly, patient input collected for this CADTH review suggested that there may be some confusion about the potential to be cured from mCRPC, and called for providers and support workers to provide clear and consistent communication about expectations.14 The context of clinical communication and decision-making may be further complicated by the language of “castration resistance.”15 The authors of 1 study claim that the language of “castration” is uncomfortable for patients, as it carries negative connotations associated with punishment or submission.15 As such, they noted that it can make conversations with patients difficult and alienate them from the decision-making process.15
Ethics of Evidence and Evaluation of 177Lu Vipivotide Tetraxetan
The primary clinical evidence used to evaluate 177Lu vipivotide tetraxetan for PSMA-positive mCRPC is drawn from the VISION trial. Clinical experts and published literature19 have pointed to 3 overarching concerns with the evidence collected in the VISION trial. These concerns pivot around the definition of “standard of care” used in the trial, whether the inclusion and exclusion criteria were adequately applied, and the high withdrawal rate from the control arm.19 Additionally, clinical experts and published literature indicated that participants in the VISION trial, and in mCRPC trials more broadly, may not have been reflective of those likely to be seen in clinical practice.7,9,10
Ethical Considerations of the VISION Trial
A foundational challenge to the evidence from the VISION trial has been whether the trial’s definition of standard of care (using the language of best supportive care [BSC] and best standard of care [BSoC]) appropriately reflected current clinical practice.19 As has already been described, there is no clear guidance on the optimal sequencing of therapies in the context of mCRPC, which can place considerable emphasis on clinical expertise and a provider’s capacity to engage patients in a process of shared decision-making. Clinical experts and published sources took issue with the definition used in the VISION trial, the use of BSC/BSoC, and the exclusion of systemic therapies (e.g., cabazitaxel) that they would have considered for patients with mCRPC refractory to multiple therapies in current clinical practice.19 Instead, the only systemic anticancer therapies allowed in the trial were hormone therapies like enzalutamide and abiraterone. When accounting for the treatment histories of patients in the control arm, the clinical experts and authors indicated that nearly 3-quarters of patients were unlikely to experience any response, owing to the overlap of treatments or previous failure on the therapeutics meeting trial definition of BSC/BSoC.19 Although the authors of a critique of the VISION trial did suggest that this definition may have been chosen because of uncertainty around the safety of using some drugs alongside 177Lu vipivotide tetraxetan in the active arm, it is unclear why this definition could not have been expanded for the control arm.19
Relatedly, clinical experts and those authors worried that inclusion and exclusion criteria may have been inadequately applied across trial sites.19 According to the VISION protocol, patients who were eligible for a second taxane-based chemotherapy (e.g., cabazitaxel) should have been excluded from the trial. However, the authors and clinical experts reported a discrepancy between this exclusion criteria and the postprotocol therapies trial participants went on to receive. With nearly 20% of participants in the trial receiving a postprotocol taxane regimen, and 18.9% of those in the control arm receiving cabazitaxel specifically, it is unclear how these participants were able to meet the inclusion criteria of the VISION trial.19 The authors and clinical experts alike felt that these participants should have received cabazitaxel before enrolment in the VISION trial.19 This is particularly concerning, given the possibility that cabazitaxel may have been a more effective option for some participants in the control arm and is available in clinical practice.19 By removing the only drug with a demonstrated improvement in OS in the phase III trial setting for patients with mCRPC refractory to multiple therapies (i.e., after ARPI and docetaxel), the clinical experts felt that providers with patients randomized to the control arm might have seen that their patients were unlikely to benefit and be concerned about their continued participation. This also signals that there may have been some concern that participation in the control arm might constitute undue harm to these patients because of a lack of access to established therapies.
This leads the third challenge described in the critique of the VISION trial19: the disproportionate rate of early withdrawal from the control arm and the resultant possibility that informative censoring biased the analysis in favour of 177Lu vipivotide tetraxetan. Informative censoring occurs when trial participants are disproportionately lost to follow-up in the trial arms owing to some fault of the trial itself. In the case of the VISION trial, 56% of potential participants randomized to the control arm dropped out during the first phase of enrolment, and another 16.2% withdrew after the second phase. Given that the corresponding dropout rates during enrolment for the experimental arm were 1.2% and 4.2% respectively, the authors suggested that the remaining control arm participants were likely living with more advanced disease and had fewer therapeutic options than those who dropped out to pursue alternative interventions.19 Importantly, it was suggested that this may have further disadvantaged patients with fewer resources and a limited support system to help them pursue other care options.19
All of this may call the reliability of some trial end point data into question.19 Although the clinical experts felt that the results for OS were internally valid, the sponsor acknowledged (and the clinical experts agreed) that interpretation of the magnitude of effect of the trial’s primary alternative end point, radiographic progression-free survival (rPFS), is limited because of this censoring. The authors of the critique agreed with this limitation and implied that pursuing the trial despite these high rates of early withdrawal may have been in contravention of the Helsinki Declaration’s statement that the goals of medical research should never outstrip the rights or interests of research subjects.19
Ethical Considerations of Trial Representativeness
Age
When considering the populations included in the VISION trial, the clinical experts and published literature suggested that the study population may not be reflective of regular clinical practice. Although the clinical experts did not think this would affect the generalizability of the trial data, they did suggest that their patients may have multiple comorbidities and would, on average, be older than those included in the trial. As most trial participants (92.4%) presented with an Eastern Cooperative Oncology Group Performance Status of 0 or 1, it seems likely that the VISION trial was not representative of those living with several comorbidities. Similarly, although the median age of trial participants was 70.0 years (range, 48 to 94 years) in the treatment arm and 71.5 years (range, 40 to 89 years) in the control arm, the oldest subgroup analysis provided is for participants 65 years and older. This can make it a challenge to discern the treatment effect and risk of adverse events in older populations. The authors of the critique suggested that RCTs of novel mCRPC therapeutics include subgroup analyses for much older populations, given the increasing likelihood that people with mCRPC will live well into their late 70s and 80s.10
Race
Similarly, the sponsor, in its submission, acknowledged that Black or African American patients (6.6% overall) and Asian patients (2.4%) were underrepresented in the VISION trial. As previously noted, this is unsurprising, given that Black patients are consistently underrepresented in trials of novel mCRPC therapies, by upwards of 80%.7 Of all the trial participants enrolled in 7 landmark phase III RCTs on novel CRPC therapies conducted from 2009 to 2015, only 3.3% (n = 240) were Black and only 2.1% (n = 150) were enrolled in an experimental arm.7 In the US context, the representation of Black patients with mCRPC would have to be closer to 24%.7 The sponsor clearly indicated that there is balanced underrepresentation in both the control and active treatment arms of the VISION trial, but that this underrepresentation may still raise concerns about equity, inclusion, and representativeness, given the greater likelihood that Black patients are more likely to die from their prostate cancer than white patients.7
Gender
As with Black patients, transgender women and people who are gender nonbinary are underrepresented in clinical trials of novel prostate cancer therapies.9 One reason for this may be that the eligibility criteria for these trials often require participants to be identified as male.9 This language may prevent transgender women or people who are gender nonbinary from participating in trials and can hamper efforts to understand how these populations will respond to novel treatment regimens.9 This is particularly concerning, given that people who are gender nonbinary (who were assigned male sex at birth) and transgender women who are not on GAHT or who have not undergone GAS have the same level of risk for developing prostate cancer as cisgender men.8,9 Given that there is no explicit mention of gender nonbinary or transgender women who have undergone GAS or who are on GAHT in trial data, it is possible that this population was excluded from the VISION trial.
Ethical Considerations in the Use of 177Lu Vipivotide Tetraxetan
Resource Challenges in Determining Candidates for 177Lu Vipivotide Tetraxetan
According to the sponsor’s submission, 177Lu vipivotide tetraxetan’s value lies in the novelty of its mechanism of action for the treatment of mCRPC. This novelty is 177Lu vipivotide tetraxetan’s ability to bond with PSMA-expressing tumour cells and deliver localized radiotherapy through the decay of an attached radionuclide, lutetium-177. Given this mechanism of action, determining eligibility and which patients will have a stronger chance of benefiting from 177Lu vipivotide tetraxetan means identifying whether a potential candidate has PSMA-positive mCRPC. To do this, patients are required to undergo PSMA-targeted PET-CT. Given current limitations in access to PSMA PET-CT in Canada, providing access to 177Lu vipivotide tetraxetan in a way that is equitable and attentive to considerations of fairness and distributive justice presents several challenges.
As PSMA-targeted PET-CT is currently the only way to identify PSMA-positive tumour cells, the clinical experts indicated that, in practice, PSMA PET-CT would be considered a companion, rather than complementary, diagnostic, given its necessity in determining the applicability of 177Lu vipivotide tetraxetan. There is currently limited availability of PSMA PET-CT across Canada,20 and this has been identified as a barrier to the use of 177Lu vipivotide tetraxetan by clinical experts and in input from patient groups, clinician groups, and drug programs. A report from CADTH’s Canadian Medical Imaging Inventory (CMII) initiative indicated that access to PSMA PET-CT through clinical trials may be available in 5 provinces in Canada,20 and the clinical experts noted that access is even more limited for patients not already enrolled in a clinical trial, as only 2 provinces, British Columbia and Quebec, currently allow off-trial access to PSMA PET-CT. For those who can access PSMA PET-CT off trial, they must cover their own costs.
The CMII initiative has indicated that Canada is currently ranked 21 out of 32 Organization for Economic Co-operation and Development (OECD) countries for PET-CT units per million population.20 Although limited access to PET-CT units, broadly, is a systemic challenge that has an impact on all current (or potential) radioligand therapies, it is unclear whether centres that already have access to a PET-CT unit are capable of producing the radiotracers necessary to conduct PSMA-targeted imaging, which presents a particular challenge to the use of 177Lu vipivotide tetraxetan.20
Input from patient groups and the clinical experts described how this limited availability implies that potential patients would first need to travel (possibly out of province) to undergo diagnostic imaging before being offered 177Lu vipivotide tetraxetan. Input from patient groups indicated that the need to travel and isolate in a hotel room would be too burdensome for some patients to follow through. The clinical experts added that the costs associated with travel and lodging would make this an untenable option for many of their patients. As such, the clinical experts worried that the need to travel may prevent some patients who would be eligible for 177Lu vipivotide tetraxetan from accessing the therapy.
Not only does the limited access to PSMA PET-CT in Canada make it challenging to identify candidates for 177Lu vipivotide tetraxetan, there is some concern that this test alone may not adequately identify the candidates who are most likely to benefit. Although suggesting that phase II data from the TheraP trial should not be used in the evaluation of 177Lu vipivotide tetraxetan, the clinical experts did point to the use of fluorodeoxyglucose PET-CT in this trial population as a potentially beneficial diagnostic test to add to PSMA PET-CT. This may help select people more likely to benefit from 177Lu vipivotide tetraxetan, but it would also increase demand on the already limited availability of PET-CT. Given the growth in expected uses of and demands for PET-CT across disease spaces (e.g., oncology, cardiology, neurology, and infectious disease), it is possible that the ability to access PSMA-targeted PET-CT could remain a challenge in the near future and that there may be a significant demand that could outstrip capacity and place limitations on patient access.20
Challenges of 177Lu Vipivotide Tetraxetan as a Radiopharmaceutical
Once a patient has been identified as an appropriate candidate for 177Lu vipivotide tetraxetan, ordering, manufacturing, transporting, and delivering 177Lu vipivotide tetraxetan represent the next stage of challenges. As a radiopharmaceutical, radiation is emitted by the decay of 177Lu vipivotide tetraxetan’s PSMA-labelled radionuclide, lutetium-177. With a half-life of around 6.65 days, lutetium-177 lasts long enough that it can be manufactured outside of the local or regional settings in which 177Lu vipivotide tetraxetan would be delivered. However, the clinical experts noted that the logistics associated with manufacturing, transporting, and administering 177Lu vipivotide tetraxetan safely and effectively could be a challenge. Any delay between manufacturing and administration could render the radioisotope ineffective and prevent patients from accessing 177Lu vipivotide tetraxetan altogether, as they are often near the end of their lives.
In its submission, the sponsor notes that 177Lu vipivotide tetraxetan would need to be delivered by, or under the supervision of, providers with training in how to handle radiopharmaceuticals and who are licensed to do so. Input provided by the drug programs similarly indicated the need for specialized staff and facilities. Although this may be standard for the provision of oncology therapeutics that use radionuclides, it is worth noting that the clinical experts consulted for this review had various levels of experience with radiotherapies, given the differential funding across Canada for the only other radiotherapy indicated for mCRPC: radium-223. With this in mind, those with limited access to radium-223 currently may face uncertainties regarding how, where, and by whom 177Lu vipivotide tetraxetan would be administered.
Similarly, there are some risks taken on by people receiving 177Lu vipivotide tetraxetan. Patient input noted that the side effects of 177Lu vipivotide tetraxetan were tolerable and would not affect their recommendation that other people living with mCRPC be offered this drug. However, given the radioactivity of patients after the administration of 177Lu vipivotide tetraxetan, it is important they can modify activities and proximity to other members of their household to limit radiation exposure. According to the sponsor’s submission, these modifications can last anywhere from 2 to 15 days, depending on the member of the household and the activity in question. For example, patients are asked to sleep in a bedroom separate from anyone for 3 days, from children for 7 days, and from a pregnant person for 15 days. General proximity to others, toileting, washing clothes, showering, waste disposal, and care provision for people with mobility challenges or comorbidities also need to be modified for 2 to 7 days. Although this may not be difficult for some (as noted by 1 interviewee in the patient input), it is possible that these adaptations may pose serious challenges for others (such as those who live in a 1-bedroom home or use a laundromat) and increase their risk of exposing others to radiation.
Clinical Judgment and Referral
As previously noted, the absence of defined guidelines for the optimal sequencing of therapies for mCRPC places extensive responsibility on care providers and their ability to navigate shared decision-making with patients. Deciding when to offer 177Lu vipivotide tetraxetan is no different. The sponsor has positioned 177Lu vipivotide tetraxetan as either a third-line option alongside cabazitaxel or, for those who are not suitable for a second taxane regimen, as a third-line option alongside current standard of care. To support this, the sponsor has indicated that 177Lu vipivotide tetraxetan’s focus on PSMA aligns with the joint CUA and CUOG guidelines, which suggest changing the mechanism of action at each line.17 The sponsor also notes that European Society for Medical Oncology guidelines21 suggest that 177Lu vipivotide tetraxetan should be used as a third-line therapy, where available, and that results from an informal survey22 at the 2021 Advanced Prostate Cancer Consensus Conference suggest that most (voting) attendees preferred 177Lu vipivotide tetraxetan to a second taxane (i.e., cabazitaxel) in this setting.
The clinical experts consulted for this review thought it was plausible that 177Lu vipivotide tetraxetan could be sequenced alongside cabazitaxel; however, they were concerned that there is insufficient evidence comparing the head-to-head effectiveness of cabazitaxel with 177Lu vipivotide tetraxetan for patients who are eligible for treatment with a second taxane regimen. As such, they suggested that, as of this review, placing the 2 on the same line would be inappropriate. Instead, they suggested that cabazitaxel should come first for those who are medically suitable for a second taxane and expressed some uncertainty about how and when 177Lu vipivotide tetraxetan would be used in practice. Although the joint CUA and CUOG guidelines have recommended that 177Lu vipivotide tetraxetan be considered as an option for this population,17 there remains an absence of clarity about which preforms better and for whom.
Health System and Funding Considerations
The use of 177Lu vipivotide tetraxetan for people with mCRPC raises several health system considerations related to resource allocation and trade-offs, infrastructural needs for screening programs, and the high costs associated with these infrastructural needs.
As noted previously, access to PSMA PET-CT is limited in Canada. Work done through the CADTH CMII initiative has estimated that there would need to be a 23% increase in current PET-CT capacity to meet demands for PSMA-targeted PET-CT diagnostic services.20 Not only are the PET-CT machines themselves largely absent in Canada, the radiotracers needed to conduct PSMA PET-CT present another challenge. To conduct PSMA-targeted PET-CT, departments of nuclear medicine need to be able to create radiotracers that can bond with and highlight PSMA-positive tumour cells. In nuclear medicine, radiotracers involve 2 basic components: carrier molecules; and a radioactive atom (i.e., radioisotope and/or radionuclide) that has been bonded to these carrier molecules. Depending on the half-life of the radionuclide being used, the radiotracers may need to be manufactured onsite using table-top generators or manufactured nearby using a cyclotron.20 Work done through the CADTH CMII initiative notes that only 1 radiotracer has received Health Canada regulatory approval to date: 68Ga-PSMA-11.20
At least 1 challenge to accessing the radiotracer 68Ga-PSMA-11 is that the radionuclide 68Ga has a short half-life, of just under 68 minutes, and requires onsite production.20 Typically, 68Ga is produced using a table-top generator, although it may also be produced onsite using a cyclotron.20 The CMII initiative notes that table-top generators can cost upwards of US$100,000, need to be replaced every 250 to 400 uses, and can only provide the necessary 68Ga for a maximum of 3 patients a day.20 Another radiotracer not yet approved by Health Canada for PSMA-targeted PET-CT, fluorine-18, has a longer half-life, of 110 minutes, but it can only be created using a cyclotron.20 Cyclotrons are able to produce higher yields in shorter turn-around times, but only one-third of PET-CT sites in Canada currently have access to a cyclotron.20 The CMII report suggested that existing cyclotrons may not have the capacity to meet the demands of newly installed PET-CT units in Canada and that building new cyclotrons can be expensive (i.e., between $2.5 million and $6.6 million per unit).20
Regardless of the method chosen, the need for onsite or regional capacity to produce radiotracers can be a major financial and logistical undertaking. As such, the clinical experts and provincial drug programs indicated that funding decisions for 177Lu vipivotide tetraxetan may be complicated by the fact that costs for diagnostic services and the radiopharmaceuticals needed to conduct these diagnostics would be borne by radiology and nuclear medicine departments. Additionally, the CMII report indicated that some settings have seen a monopolization of radiopharmaceuticals over the past decade.20 If this were to happen with the raw materials needed to created either 68Ga or fluorine-18 for PSMA PET-CT, or for the production of 177Lu vipivotide tetraxetan itself,23 disruptions to the supply chain could require the cancellation or rebooking of appointments.20 For a population at the end of their lives, this may not be possible and could limit a patient’s ability to access PSMA PET-CT and 177Lu vipivotide tetraxetan altogether.
Limitations
This review is limited by the paucity of published literature examining ethical issues directly related to the use of 177Lu vipivotide tetraxetan for the treatment of adults with mCRPC. Given the novelty of 177Lu vipivotide tetraxetan, this limitation is neither unexpected nor the same as suggesting there are no ethical issues related to the use of 177Lu vipivotide tetraxetan. As such, this report has relied on input from patient and clinician groups, provincial drug programs, the sponsor, and clinical experts to augment the limitations of the literature. Although this has helped to identify challenges related to the use of 177Lu vipivotide tetraxetan, limitations in opportunities to engage these groups in direct (and repeated) interviews may have diminished the breadth and depth of information available to inform this report. This report was also limited by the types of experts engaged. Additional engagement with policy-makers, patients, and citizens especially affected by the generation, use, and disposal of radiopharmaceuticals may have augmented the nature and types of ethical considerations explored in this report.
Conclusion
Input from patient groups, clinician groups, and provincial drug programs, as well as direct engagement with clinical experts and published literature, was reviewed for ethical considerations relevant to the use of 177Lu vipivotide tetraxetan for the treatment of adults with mCRPC. Ethical considerations in the context of mCRPC highlight the impact on patients; disparities in the incidence, treatment, and/or outcomes for racialized, transgender or nonbinary, and advanced-age groups; and challenges with decisions about the appropriate sequencing of mCRPC therapies. Clinical trial evidence used to evaluate 177Lu vipivotide tetraxetan was challenged by a suboptimal control arm, issues around participant inclusion, and very high rates of early withdrawal from the control arm. There were also concerns about the accessibility of PSMA PET-CT testing, which is a prerequisite to be considered a candidate for 177Lu vipivotide tetraxetan. The introduction of 177Lu vipivotide tetraxetan into the health care system in Canada would involve issues related to resource allocation and the high costs and logistical challenges associated with infrastructural needs to further develop the availability of PSMA PET-CT testing.
Table 1: Details of Included Publications
Publication | Publication type | Objective | Key ethical considerations | Funding source |
|---|---|---|---|---|
Bertoncelli Tanaka et al. (2022)8 | Literature review | To review existing literature, current guidelines, and standard of practice related to prostate cancer in transgender women | Although the overall incidence of prostate cancer in transgender women on GAHT is generally lower than in cisgender men, some evidence suggests that transgender women on GAHT present with more advanced prostate cancer than cisgender men. Marginalization and discrimination of transgender women in health care settings may prevent these women from sharing background information with their providers and could lead to longer wait times than cisgender men for prostate cancer–related diagnostic or specialist services. Although PSA levels and mpMRI images are commonly used as risk-stratification tools in prostate cancer, there is no guidance on what PSA levels should be considered normal for transgender women on GAHT or how to read mpMRI images for this population. The risk of prostate cancer in gender nonbinary and transgender women who are not on GAHT or have not had GAS is the same as it is in cisgender men. | None reported |
Morrison (2022)20 | Service report | To summarize information on the infrastructural readiness of Canada’s health care system for the adoption of PSMA PET-CT for the staging and restaging of prostate cancer | Access to PET-CT imaging may be limited If publicly funded, PSMA PET-CT will increase demand for PET-CT imaging by a minimum of 23% The possible monopolization of the radiopharmaceutical supply could cause major service disruptions if there are issues with the supply chain | CADTH receives support from Canada’s federal, provincial, and territorial governments, with the exception of Quebec |
Olivier et al. (2022)19 | Commentary | To outline 3 primary limitations of the VISION trial | The VISION trial limited the choice of standard of care, which “unfairly led to a suboptimal control arm, beneath best available care outside of the trial setting.” Inadequate inclusion and exclusion criteria that affected the quality of the control arm. High attrition rates in the control arm, which affected the randomization of the control arm. | None reported |
Saad et al. (2022)17 | Guidelines | To provide guidance on the management of both CRPC and mCRPC | Optimal sequencing of mCRPC treatments is unknown. | None reported |
Stern et al. (2021)11 | Commentary | To describe the underrepresentation of Black patients in RCTs for mCRPC therapeutics | Black patients have higher risk of dying from prostate cancer than white patients. When adjusted for social determinants such as socioeconomic status and access to health care, the increased risk of Black patients dying is nearly eliminated. | None reported |
Burbridge et al. (2020)6 | Interview study | To explore the symptomatic experience of patients recently told that their CRPC has metastasized, the emotional response to this diagnosis, and the emotional burden of monitoring metastatic status | mCRPC has an impact on functional, emotional, and mental wellbeing. | Janssen |
Catt et al. (2019)5 | Interview study | To explore the experiences of treatment decisions, information provision, perceived benefits and harms of treatment, and effects of these in the lives of people living with mCRPC and their partners | Supportive care (for both patients and their partners) is generally underresourced, leaving people uncertain about how to manage side effects or other issues arising from their treatment. | None reported |
de Angst et al. (2019)12 | Survey study | To investigate perspectives of Dutch urologists, oncologists, and oncology nurses on patient involvement in the decision-making process, to explore their views on the added value of decision aids, and to assess their treatment recommendations for different patients with mCRPC | Although patients should be involved in the decision-making process for mCRPC interventions, providers may not be adequately trained in shared decision-making. Wide provider variability in treatment recommendations in response to hypothetical cases demonstrates how sensitive decision-making can be to patient input. Assumptions around the capacity or interest of older patients with mCRPC to be involved in treatment decisions may lead to physician paternalism. The focus should be on individual health status and lived experience rather than age when considering treatment options | None reported |
Ingham et al. (201)89 | Review | To review the available literature and provide guidance on how best to diagnose, manage, and follow transgender women diagnosed with prostate cancer | Transgender women face significant barriers to care and are at an increased risk of suicide and poor health. Transgender women may present at a more advanced disease stage and may not respond to standard ADT as well as cisgender men. | None reported |
Woon et al. (2018)18 | Review | To describe the nuances of mCRPC therapy availability in each province to characterize interprovincial disparities to access, explore barriers and the potential consequences this disparity may introduce, and to contrast access with treatment preferences and perceived barriers as reported in the Canadian Genitourinary Research Consortium survey | Interprovincial differences in mCRPC funding could lead to inequitable access across Canada. The disconnect between the providers’ preferred sequencing of mCRPC therapies and actual sequencing available through the public payer. | Janssen |
Caffo et al. (2017)10 | Review | To describe available treatments for older patients with mCRPC, paying particular attention to the benefit:harm ratio in octogenarians | The increasing availability of life-prolonging therapeutic options for people with mCRPC means that people are now living longer and may be older when receiving mCRPC treatments. Clinical trials should now include a subgroup analysis of older patients with an age cut-off of 80 and older, rather than 75 and older, to be representative of practice. People being categorized as elderly (older patients) should not be prevented from accessing new treatments based on their age, but should be treated on the basis of careful geriatric and comorbidity evaluations. | None reported |
Pezaro et al. (2017)15 | Commentary | To argue for a shift in language in advanced prostate cancer away from castrate-resistance and toward a more descriptive nomenclature focused on which lines of therapy have failed | Categorizing advanced prostate cancer into clinical states that are reflective of cancer biology is becoming more complex as people are living longer with new therapeutics. Term “castration-resistant” carries negative connotations and should be replaced with language that is descriptive of the complexity of advanced prostate cancer. | None reported |
Lewis and Sartor (2015)13 | Review | To describe the current landscape of mCRPC therapeutic options and potential future directions | Absent or limited head-to-head trials comparing the effectiveness of novel mCRPC therapeutics makes optimal sequencing unclear and leaves decisions up to clinical gestalt. | None reported |
Spratt and Osborne (2015)7 | Commentary | To describe disparities in the representation of Black patients in CRPC clinical trials | In the US, Black patients are 2.5 times more likely than white patients to have lethal prostate cancer. Black patients are underrepresented in clinical trials for novel mCRPC therapies. Given the underrepresentation of Black patients in clinical trials for mCRPC, it is difficult to assess clinical benefit in this population. | None reported |
Flynn (2013)14 | Commentary | To examine the physical and psychosocial effects of mCRPC and its treatment, alongside QoL issues and service-delivery implications | Ensuring that palliative services are optimized and accessible is imperative in the mCRPC setting because it is incurable. Optimal sequencing for mCRPC treatment is unclear and should be tailored to the person living with mCRPC and include input from a multidisciplinary treatment team. The importance of balancing moves toward OS with improvements in QoL. When interpreting data from end-of-life studies, it is important to be aware of the possibility of responder bias because those who live longer have more time to develop a response than those whose QoL is deteriorating quickly. Providers should pay attention to the potential for distress in their patients with mCRPC and know how to support them or where to refer them. | None reported |
Armstrong et al. (2012)16 | Review | To discuss potential biomarkers in mCRPC and how they inform prognosis, aid in treatment selection, and relate to survival outcomes | “A major clinical and research dilemma in CRPC has been to define and standardize progression as an objective end point and therefore optimize duration of therapy of a given systemic agent.” | Duke Cancer Institute The Sidney Kimmel Center for Prostate and Urologic Cancers |
ADT = androgen-deprivation therapy; CRPC = castration-resistant prostate cancer; GAHT = gender-affirming hormone therapy; GAS = gender-affirming surgery; mCRPC = metastatic castration-resistant prostate cancer; mpMRI = multiparametric MRI; OS = overall survival; PSA = prostate-specific antigen; PSMA = prostate-specific membrane antigen; QoL = quality of life; RCT = randomized controlled trial.
References
1.EUnetHTA Joint Action 2 Work Package 8, HTA Core Model version 3.0. 2016: https://www.htacoremodel.info/BrowseModel.aspx. Accessed 2022 Apr 25.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37:e17. PubMed
3.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jun 28.
4.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
5.Catt S, Matthews L, May S, Payne H, Mason M, Jenkins V. Patients' and partners' views of care and treatment provided for metastatic castrate-resistant prostate cancer in the UK. European Journal of Cancer Care. 2019;28(6):e13140. PubMed
6.Burbridge C, Randall JA, Lawson J, et al. Understanding symptomatic experience, impact, and emotional response in recently diagnosed metastatic castration-resistant prostate cancer: a qualitative study. Support Care Cancer. 2020;28(7):3093-3101. PubMed
7.Spratt DE, Osborne JR. Disparities in castration-resistant prostate cancer trials. J Clin Oncol. 2015;33(10):1101-1103. PubMed
8.Bertoncelli Tanaka M, Sahota K, Burn J, et al. Prostate cancer in transgender women: what does a urologist need to know? BJU Int. 2022;129(1):113-122. PubMed
9.Ingham MD, Lee RJ, MacDermed D, Olumi AF. Prostate cancer in transgender women. Urologic Oncology: Seminars and Original Investigations. 2018;36(12):518-525. PubMed
10.Caffo O, Maines F, Rizzo M, Kinspergher S, Veccia A. Metastatic castration-resistant prostate cancer in very elderly patients: challenges and solutions. Clin Interv Aging. 2017;12:19-28. PubMed
11.Stern N, Ly TL, Welk B, et al. Association of Race and Ethnicity With Prostate Cancer–Specific Mortality in Canada. JAMA netw. 2021;4(12):e2136364-e2136364.
12.de Angst IB, Kil PJM, Bangma CH, Takkenberg JJM. Should we involve patients more actively? Perspectives of the multidisciplinary team on shared decision-making for older patients with metastatic castration-resistant prostate cancer. J Geriatr Oncol. 2019;10(4):653-658. PubMed
13.Lewis B, Sartor O. The Changing Landscape of Metastatic Prostate Cancer. Am J Hematol Oncol. 2015;11(8):11-20.
14.Flynn K. Metastatic castrate-resistant prostate cancer: A discussion of the physical and psychosocial effects. International Journal of Urological Nursing. 2013;7(2):98-105.
15.Pezaro CJ, Omlin A, Mastris K, et al. Precision, complexity and stigma in advanced prostate cancer terminology: it is time to move away from 'castration-resistant' prostate cancer. Ann Oncol. 2017;28(8):1692-1694. PubMed
16.Armstrong AJ, Eisenberger MA, Halabi S, et al. Biomarkers in the management and treatment of men with metastatic castration-resistant prostate cancer. Eur Urol. 2012;61(3):549-559. PubMed
17.Saad F, Aprikian A, Finelli A, et al. 2022 Canadian Urological Association (CUA)-Canadian Uro Oncology Group (CUOG) guideline: Management of castration-resistant prostate cancer (CRPC). Can Urol Assoc J. 2022;16(11):E506-e515. PubMed
18.Woon DTS, Chandrasekar T, Aaron L, et al. Disparity in public funding of therapies for metastatic castrate-resistant prostate cancer across Canadian provinces. Can Urol Assoc J. 2018;12(10):328-336. PubMed
19.Olivier T, Powell K, Prasad V. Lutetium-177-PSMA-617 in Metastatic Castration-resistant Prostate Cancer: Limitations of the VISION Trial. Eur Urol. 2022;09:09.
20.Prostate-Specific Membrane Antigen PET-CT Imaging for the Staging of Prostate Cancer in Canada. Canadian Journal of Health Technologies. 2022 Nov 2(11). https://canjhealthtechnol.ca/index.php/cjht/article/view/HC0024/HC0024. Accessed 2022 Dec 12.
21.European Society for Medical Oncology. Genitourinary Cancers: Pocket Guideline 2022. 2022: https://interactiveguidelines.esmo.org/esmo-web-app/toc/index.php?subjectAreaId=14&pdfId=4. Accessed 2022 Nov 1.
22.APCCC 2021: Summary of Panel Responses Regarding PSMA Diagnostics and Therapy. 2021; https://www.urotoday.com/conference-highlights/apccc-2021/133259-apccc-2021-summary-of-panel-responses-regarding-psma-in-diagnostics-and-therapy.html. Accessed 2022 Nov 9.
23.Novartis Pauses US Pluvicto Production Due to Possible Quality Issues. 2022 May 5; https://www.precisiononcologynews.com/cancer/novartis-pauses-us-pluvicto-production-due-possible-quality-issues#.Y3JL2vfMKUl. Accessed 2022 Nov 14.
Stakeholder Input
Patient Input
Canadian Cancer Survivor Network
About the Canadian Cancer Survivor Network
The Canadian Cancer Survivor Network (CCSN) is a national network of patients, families, survivors, friends, community partners, funders, and sponsors who have come together to take action to promote the very best standard of care, whether it be early diagnosis, timely treatment and follow-up care, support for cancer patients, or issues related to survivorship or quality of end-of-life care. https://survivornet.ca/
Information Gathering
The Canadian Cancer Survivor Network utilized SurveyMonkey to create and collect data for the survey on lutetium. CCSN then utilized their newsletter, social media, and email to disseminate the survey to collect the responses. The survey was open from August 10, 2022 – August 22, 2022, to obtain responses. All the respondents to the survey reside in Canada; three live in British Columbia, four live in Ontario, and one lives in Alberta. Out of the eight respondents, seven identify as male and one preferred not to say. Out of the eight respondents, three patients have or are taking lutetium, four have not taken lutetium, and one is a caregiver.
Disease Experience
The respondents were asked how their cancer was diagnosed; this resulted in eight people who provided responses:
Contrast-enhanced computed tomography (CT): 1
Magnetic resonance imaging (MRI): 3
Blood Tests: 7
Biopsy: 8
Other: 2 (1 Digital Exam and 1 Bone Scan)
When asked what stage of prostate cancer they have and experience with lutetium, the survey provided three people who were all diagnosed with late stage (4), metastatic prostate cancer.
Current treatments that were identified by the survey respondents include:
ADT: 2
Lutetium: 3
Clinical Trials: 1
Targeted Therapy: 2
Radiation: 2
Chemotherapy: 1
Surgical Therapy: 1
Experiences With Currently Available Treatments
When asked about their stage of prostate cancer, among those with no experience with lutetium, the survey provided four responses; two patients responded with being diagnosed at late stage (4), metastatic and two patients did not know what stage their cancer was at.
When asked about their current quality of life and day-to-day living experience, the following responses were provided:
Frequency in urination: 4
Erectile Dysfunction (ED): 2
Bone/Skeletal Pain: 2
Loss of quality of life: 2
Other: 1 (1 Hot flashes, weight gain, slight memory loss)
Survey respondents indicated these adverse effects due to their current treatment:
Incontinence: 2
Fatigue: 4
Diarrhea: 1
Weight Gain: 2
Erectile Dysfunction (ED): 2
Urinary Issues: 1
Infection: 1
Hot flashes: 1
Loss of muscle: 1
Hair loss: 1
Breast enlargement: 1
Other: 1 (total loss of libido)
Survey respondents were asked if they have had any issues accessing any therapies and all four respondents who gave a response indicated that they haven’t had any issues accessing therapy.
Improved Outcomes
When asked about the following issues that they would hope to see a new treatment address to manage their disease, the survey respondents replied:
Maintain quality of life: 2
Delay onset of symptoms: 3
Access to a new option for treatment: 2
Ease of use: 2
Prolong life: 4
Provide a cure: 4
Reduce side effects from current medications or treatments: 1
Other: 1 (return of libido)
Survey respondents were asked to describe how much of an improvement would be needed from a new treatment to make it better than the current treatment that they are undergoing. Of the two respondents who gave responses, they believe that there should be a treatment with less side effects. Some more detail is included in the following:
“Tiredness and memory but change of diet and better supplements have contributed to a better life.”
“Less severe side effects. Return of muscle mass.”
Experience With Drug Under Review
When asked to compare their experience with lutetium to other therapies in treating their prostate cancer, respondents rated their experience as follows:
Symptom management: Much Better: 3, Little or no difference, Much worse
Side effects: Much Better, Little or no difference: 1, Much worse: 2
Ease of use: Much Better, Little or no difference: 3, Much worse
Disease progression: Much Better: 3, Little or no difference, Much worse
Other: Much Better: 1
Survey respondents were asked what adverse effects were caused by taking lutetium, 3 indicated the following:
Fatigue: 3
Dry Mouth: 3
Anemia: 1
Decreased appetite: 1
Nausea: 1
Constipation: 1
Other: 2 (1 Low blood counts platelets and 1 Brain fog)
Survey respondents were asked to describe the positive and negative effects of lutetium:
“I’m a little tired, maybe a little weaker. My PSA results have greatly improved. I’ve only had 2 treatments.”
“Removed lesions on back and liver.”
“TBC (lutetium) has added to my life expectancy. I am currently cancer free. The side effects of TBC affected my quality of life including the symptoms I have listed.”
Survey respondents were asked if they would recommend that lutetium be made available to all patients who qualify for it. All respondents agreed that it seems to be working for them and that it should be made available to others as well:
“Yes. It seems to work.”
“Absolutely. Seems to work.”
“It seems to be working for me. I am not an oncologist.”
Survey respondents were asked to share about their own cancer journey:
“Why is it always drugs being put to us when certain foods and supplements make quite a big difference?”
“I was diagnosed 3 years ago with metastatic prostate cancer. I was 67 and got yearly check-ups. My GP should have been sending me for PSA tests much earlier. I didn’t even know what a PSA test was. Lu 177 seems to be my best bet for a prolonged life.”
“My oncologist addresses all my concerns and refers me to other specialists to deal with my issues.”
As a caregiver for someone with prostate cancer, the one respondent selected the following issues that they encounter:
Fatigue: 1
Emotional Drain: 1
Anxiety/Worrying: 1
Management of side effects: 1
Hours spent in medical appointments: 1
Monetary concerns (absence at work, driving expenses, etc.): 1
Lifestyle changes: 1
Inability to plan ahead: 1
Feelings of “doom” due to challenging prognosis: 1
Feelings of helplessness: 1
The caregiver described how caring for someone with prostate cancer has affected their daily routine or lifestyle:
“Daily routine is affected in terms of having less time for household chores due to time spent caregiving. No time for myself.”
The caregiver described the most challenging adverse effects related to their loved one and their current therapy or treatment:
“The treatment made them moody and emotional, plus the side effects are affecting quality of life.”
The caregiver also had this to share about their experience as caring for someone with prostate cancer:
“In general partners in a relationship have developed roles and since PC is common in older men those roles were established long ago. Traditionally the female partner does most of the household chores.”
Companion Diagnostic Test
Not applicable.
Anything Else?
The Canadian Cancer Survivor Network is aware of the limitations of this submission given the small number of respondents and that only three patients are on lutetium.
Conflict of Interest Declaration — Canadian Cancer Survivor Network
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for the Canadian Cancer Survivor Network
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novartis – 2021 | X | — | — | — |
Novartis – 2022 | — | — | X | — |
Clinician Input
Canadian Cancer Society
About Canadian Cancer Society
Our purpose: To unite and inspire all Canadians to take control of cancer. Our mission: In trusted partnership with donors and volunteers, we improve the lives of all those affected by cancer through world-class research, transformative advocacy and compassionate support. We set ourselves apart from other cancer charities by taking a comprehensive approach against cancer. We are also the only national charity that supports all Canadians living with all cancers across the country.
The Canadian Cancer Society sourced several Canadian prostate cancer expert clinicians with a special interest in the care of people with metastatic prostate cancer for this submission. https://cancer.ca/en
Information Gathering
The initial version of the submission was drafted by Dr Bauman and circulated to a select group of Canadian thought leaders in the care of advanced prostate cancer for input. The final version of the submission integrated all feedback before obtaining approval for submission by all participants.
Current Treatments
Describe the current treatment paradigm for the disease.
Most men with prostate cancer present with localized disease that may be managed through a variety of means including active surveillance, surgery, radiotherapy or combinations of treatments. A subset of men with localized disease may recur after primary therapy presenting with biochemical recurrence (rising prostate specific antigen (PSA) alone) or with overt relapse in bone, lymph nodes or viscera typically detected with imaging. Some may present with de novo metastatic disease at the time of initial diagnosis. When isolated localized relapse is detected in the prostate gland, prostate bed or pelvic lymph nodes after primary therapy loco-regional salvage may be curative and/or associated with prolonged disease control. Distant disease relapse is incurable, though the disease may be managed through a variety of systemic and localized treatments providing disease control and maintaining quality of life.
Prostate cancer is broadly divided into “hormone sensitive metastatic prostate cancer” and “castrate resistant prostate cancer”. The use of castration, whether surgical or medical (LHRH agonists or antagonists), that suppresses circulating testosterone to sub physiological levels has been the mainstay of treatment for years and even in the setting of overt metastatic disease may provide 1-2 years of disease control; disease control may be longer in the setting of biochemical failure without overt metastases where hormone therapy may provide biochemical control for 5 or more years.
In the absence of other life limiting events, men with hormone sensitive disease will invariably progress to castrate resistant prostate cancer (CRPC) where disease progression based on biochemical or imaging parameters occurs in the setting of castrate levels of testosterone. Such progression may be asymptomatic initially but eventually is accompanied by symptoms such as pain (due to bone involvement), constitutional symptoms or signs of organ failure (i.e. liver dysfunction due to liver metastases, renal failure due to ureteric obstruction from adenopathy or local pelvic recurrence in the prostate/prostate bed). Ultimately men with metastatic CRPC (mCRPC) die of prostate cancer due to general debilitation or other intercurrent medical events.
In the past, management options for men with mCRPC were limited with most interventions directed at managing symptoms form the progressive cancer rather than modifying the disease course. For example, low dose prednisone could address constitutional symptoms, focal radiotherapy could address focal bone pain and systemic radiopharmaceuticals were found to be effective for multifocal bone pain. The use of mitoxantrone was found to be superior to prednisone for the management of symptomatic mCRPC in randomized trials in the 1990s and subsequently taxane based chemotherapy was found to be beneficial for both symptom management as well as improving overall survival in mCRPC. Currently, taxanes remain a mainstay of treatment for mCRPC and may be of benefit in other settings such as men with hormone sensitive metastatic prostate cancer or high risk localized prostate cancer.
More recent research has led to the understanding that even in the setting of castrate resistance, men with mCRPC may still respond to agents directed at the androgen receptor pathway. Androgen synthesis inhibitors such as abiraterone and potent androgen receptor inhibitors such as enzalutamide have shown benefit in symptom and disease control in mCPRC and they too have found their way into the management of earlier stages of prostate cancer like the taxanes. Additionally newer taxanes such as cabazitaxel are of benefit in late stage mCRPC.
Other classes of drugs which have been shown to be of benefit in mCRPC include the bone targeted radionuclide Radium-223 which has been associated with both symptom and survival benefits. Other emerging/investigational agents for mCRPC include PARP inhibitors and immunotherapy with checkpoint inhibitors for specific subsets of men with specific molecular signatures of DNA pathway repair defects.
Another form of immunotherapy Sipuleucel-T is available in the United States but is not approved for use in Canada.
Thus, today, there are a number of options available to treat mCRPC, depending on the sequencing of therapies during earlier stages of the disease. The general paradigm in place is as follows:
Hormone sensitive metastatic prostate cancer: LHRH agonist with consideration of addition of either a taxane or androgen receptor pathway agent
On development of CRPC: continue LHRH agonist and add either a taxane or androgen receptor pathway agent; consider Radium-223 if isolated bone metastases
On progression of mCRPC: continue LHRH agonist and switch from taxane or androgen receptor pathway agent to the other; consider Radium-223 if isolated bone metastases
On progression of mCRPC: continue LHRH agonist and consider third line systemic agents like cabazitaxel, consider PARP inhibitor depending on molecular profile; consider clinical trials/investigational therapies such as immunotherapy where available
Supportive measures such as focal radiotherapy, pain control, bone protecting agents (i.e., zoledronic acid, denosumab) etc. are integral to optimal patient management through all stages of disease and after there are no more disease modifying systemic therapies available or patient declines disease modifying therapies.
In parallel with the development of the above therapeutic approaches for prostate cancer has been the development of theranostic approaches for prostate cancer that exploit the molecular target Prostate Specific Membrane Antigen (PSMA) for the imaging and therapy of advanced and recurrent prostate cancer. Prostate-specific membrane antigen (PSMA) is a transmembrane glutamate carboxypeptidase that is highly expressed on prostate cancer cells with high expression noted in high grade, metastatic and castrate resistant prostate cancer. PSMA targeted Positron Emission Tomography (PET) imaging agents use diagnostic radioisotopes such as 18F and 68Ga for the detection of prostate cancer and men with PSMA avid disease may be treated with PSMA targeting ligands conjugated to therapeutic radiopharmaceuticals such as 177Lu.
The agent currently under consideration, lutetium vipivotide tetraxetan is a PSMA targeting radiopharmaceutical for the treatment of patients with prostate-specific membrane antigen (PSMA)-positive mCRPC who have been treated with androgen receptor pathway inhibition and taxane-based chemotherapy or who are not medically suitable for taxanes. Thus, this therapy will integrate with other prostate cancer therapies: from point 3-5 if not suitable for taxanes after androgen receptor therapy and point 4-5 if progression after taxane and androgen receptor pathway agent occurs.
The main alternatives at these points of disease progression would be consideration of PARP inhibitors (for those 10 to 20% of men with molecular signatures of specific DNA pathway repair defects), Radium-223 for men with bone only metastatic disease, cabazitaxel or supportive measures only or a clinical trial if available.
Given the incurable nature of metastatic prostate cancer, when considering sequencing of treatments and decisions for treatment, the primary concerns are interventions that will maintain quality of life first and foremost and secondly modify the disease course to provide control of disease progression. In many cases the alternatives available are suitable only for subsets of men with mCRPC (i.e., Rn223 for men with bone only disease, PARP inhibitors for those with molecular evidence of DNA repair pathway defects)
Treatment Goals
What are the most important goals that an ideal treatment would address?
Understanding that complete eradication of metastatic solid malignancies like mCRPC (e.g. “cure”) is unlikely to be realized with current therapies, the important goals for therapies for mCRPC are therapies that are able to delay the clinical progression of disease and improve overall survival, maintain quality of life, improve symptoms like pain or constitutional symptoms and delay or avoid events that compromise function or increase symptoms (e.g. bone fracture, organ compromise) and are associated with an acceptable toxicity profile.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
For men with mCRPC who have progressed on taxanes and androgen receptor agents, there is a paucity of agents available for disease modification to preserve quality of life and reduce symptoms. For example, PARP inhibitors are applicable to only a small proportion of men (typically ,15%) with molecular signatures of specific DNA pathway repair defects. Radium-223 is only applicable to those men with predominantly bone metastatic disease, an increasingly small subset with increasing use of PSMA imaging, which is more sensitive at detecting sites of disease outside of bone (lymph nodes, viscera etc). Additionally, none of these agents provide long term disease control and inevitably men progress to therapy refractory states where supportive care only is the standard of care. Even then, supportive care measures may be inadequate to address the symptom burden from prostate cancer, such as pain due to progressive bone metastasis. In general, as men progress through lines of therapy, tolerance for these therapies decreases and for some therapies, such as chemotherapy-based regimens, bone marrow reserve may become a limiting factor. Thus, there is an unmet need for additional lines of therapy that provide meaningful survival and quality of life benefits for those men with progressive metastatic prostate cancer.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Most men with mCRPC die of progressive disease once they become refractory to the currently available lines of therapy. Approximately 4600 men die from prostate cancer in Canada annually, about 10% of all cancer deaths in men. Almost all of these men die from mCRPC refractory to current therapies. Available data suggests the majority (>80%) of men with mCRPC have cancers that express Prostate Specific Membrane Antigen (PSMA) based on diagnostic molecular imaging using PSMA targeted positron emission tomography (PET). As such, therapeutic agents directed at this target could be applicable to a large proportion of men with mCRPC after progression on other agents. Randomized trials such as the Phase III VISION trial (vs. supportive care) and the Phase II TheraP trial (vs. cabazitaxel) suggest PSMA targeting radioligand therapies confer both disease modifying and symptom management benefits among this population of men with progressive castrate resistant prostate cancer. The drug under review, lutetium vipivotide tetraxetan, is a PSMA targeting radioligand therapeutic and would be expected to confer similar benefits among men who have PSMA expressing prostate cancer on diagnostic PSMA targeted PET.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The drug under review lutetium vipivotide tetraxetan (also known as 177-Lu PSMA-617) is currently positioned as a disease modifying drug that also provides disease and symptom control for men with mCRPC who have progressed after taxane based chemotherapy and androgen receptor targeted therapy, and who are remaining on LHRH agonist or antagonist therapy. For example, in the TheraP trial, lutetium vipivotide tetraxetan compared favourably with cabazitaxel in men with mCRPC leading to a higher PSA response and fewer grade 3 or 4 adverse events. In the VISION trial, lutetium vipivotide tetraxetan, plus standard of care significantly prolonged life, as compared with standard care alone (median overall survival 15.3 vs. 11.3 months; hazard ratio for death, 0.62; 95% CI, 0.52 to 0.74; P<0.001); imaging-based progression-free survival was improved (median, 8.7 vs. 3.4 months; hazard ratio for progression or death, 0.40; 99.2% confidence interval [CI], 0.29 to 0.57; P<0.001). Further, secondary end points significantly favored lutetium vipivotide tetraxetan. While the incidence of adverse events of grade 3 or above was higher with lutetium vipivotide tetraxetan than without (52.7% vs. 38.0%), the quality of life was not adversely affected. Thus, PSMA targeting radioligand therapies, like lutetium vipivotide tetraxetan, can be considered well-tolerated, and a good additional line of therapy compared to third line chemotherapy or supportive care only. This is the current indication being sought for the agent. It is reasonable to expect that with further clinical trials, this drug could be found to be efficacious in earlier stages of disease (i.e. first or second line treatment of mCRPC or treatment of hormone sensitive metastatic prostate) and trials are ongoing to address these questions (e.g. NCT04663997: docetaxel vs. PSMA radioligand therapy in second line therapy of mCRPC; NCT04647526: PSMAradioligand therapy vs second line hormonal therapy in second line therapy of mCRPC; NCT04720157: PSMA radioligand therapy vs standard of care in hormone sensitive metastatic prostate cancer).
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
For men with rare variants of mCRPC, treatment with alternate therapies could be considered. For example, for men with neuroendocrine differentiation, treatment with chemotherapy may be appropriate (and such men typically have cancer that does not express PSMA). For men with molecular profiling and evidence of DNA pathway repair deficiencies, treatment with PARP inhibitors is a consideration. Clinical trials of other approaches such as immunotherapy may be available to men depending on clinical and molecular characteristics. Those men who have metastatic disease that does not exhibit PSMA avidity on diagnostic PSMA targeted PET should be offered therapies other than lutetium vipivotide tetraxetan as the response rate would be expected to be low.
How would this drug affect the sequencing of therapies for the target condition?
For the indication being sought for the drug under review, the sequencing of other therapies would not be affected as the drug approval is for men who have already progressed after first and second line therapies for mCRPC (taxanes and androgen receptor targeted therapy) or are ineligible for these therapies. For small subsets of men, sequencing issues may arise with other agents like PARP inhibitors or Radium-223 depending on the molecular profile of their tumor (where available) and/or pattern of metastases (bone only vs. bone plus nodal and/or visceral metastases)
Which patients would be best suited for treatment with the drug under review?
This treatment would be most suited for men with the following characteristics:
Progressive (symptomatic, imaging or biochemical) mCRPC
Evidence of PSMA expressing metastases based on a diagnostic PSMA targeted PET scan
Adequate performance status (ECOG 0-2) and organ function (liver and bone marrow)
How would patients best suited for treatment with the drug under review be identified?
Men with progression on current therapy would be identified using clinical, biochemical (PSA) and standard imaging (bone scan and CT scan) methods. Men with progression would require a diagnostic PSMA targeted PET scan to confirm that PSMA expressing metastatic disease is present.
Which patients would be least suitable for treatment with the drug under review?
Men with poor performance status and extensive visceral metastases or limited organ reserve (compromised liver function and/or cytopenias due to bone marrow compromise) would be least suitable for this therapy due to concerns about response rate and tolerance. Additionally, men without PSMA avid lesions on PSMA targeted PET would be least suitable for treatment as the response rate would be expected to be low. For example, in the VISION trial, to be elibile men had to have PSMA-positive metastatic castration-resistant prostate cancer, which was defined as at least one PSMA-positive metastatic lesion and no PSMA-negative lesions. PSMA-negative lesions were defined as those metastatic lesions visible on conventional imaging that had PSMA uptake equal to or lower than that of physiologic liver parenchymal PSMA uptake.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
In the VISION trial of PSMA radioligand therapy vs. standard of care treatment in men with progressive mCRPC after prior taxane and androgen receptor therapy, 1179 men were assessed for eligibility. Of these 1003 were deemed eligible and underwent PSMA targeted PET. Men had to have at least one measurable PSMA PET avid lesion and no PSMA non-avid lesions. The presence of PSMA-negative lesions was defined in the protocol as PSMA uptake equal to or lower than that of liver parenchyma in any lymph node with a short axis of at least 2.5 cm, in any metastatic solid-organ lesions with a short axis of at least 1.0 cm, or in any metastatic bone lesion with a soft-tissue component of at least 1.0 cm in the short axis. Ultimately 831 (~83%) met imaging criteria and underwent randomization. Similarly, in the Thera-P trial, men were required to have PSMA avid disease on PSMA targeted PET imaging. At a minimum PSMA PET targeting PET is needed to identify those men most likely to respond to therapy.
The combination of PSMA targeting PET and radioligand therapy is considered a theranostic pair (i.e. an agent that targets a molecule or pathway for both diagnostic and therapeutic purposes)
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
In the VISION trial, the alternate primary endpoints were imaging progression as defined by Prostate Cancer Working Group 3 criteria and overall survival. In clinical practice conventional imaging (bone scan and CT) and biochemical biomarkers (PSA) are routinely used to assess response and outcomes.
What would be considered a clinically meaningful response to treatment?
The most meaningful clinical endpoint would be avoiding progression. Generally, this endpoint would be reflected in stability or improvement in biochemical and imaging biomarkers such as serum PSA and bone scan and CT. This would also correspond to a stabilization or improvement in quality of life and function as assessed by patient reported symptom scales and avoidance of excess treatment related toxicity.
How often should treatment response be assessed?
Patient reported outcomes and treatment tolerance as measured by biochemical indices for hematologic, hepatic and renal function should be assessed prior to each cycle of treatment (i.e. every 6 weeks).
Radiographic response should be assessed after every 2-3 cycles of therapy (every 12-18 weeks).
What factors should be considered when deciding to discontinue treatment?
In the VISION trial patients continued to receive treatment until imaging-documented disease progression was detected, an unacceptable level of toxic effects occurred or a lack of clinical benefit was recognized. In practice, radiographic progression, a patient reported decline in quality of life or treatment related toxicity (i.e. compromised hematologic, renal or liver function) would be indications to discontinue treatment.
What settings are appropriate for treatment with the drug under review?
In order to deliver this treatment safely, sites would need appropriate facilities, certifications and licensed personnel for delivering unsealed radiopharmaceutical treatments. Additionally, access to diagnostic PSMA targeted PET is necessary for proper patient selection. As well, this therapy is ideally delivered in the setting of multidisciplinary care and management to these patients with collaboration between specialists experienced in treating prostate cancer (urologic oncology, medical oncology, nuclear medicine, radiation oncology, diagnostic radiology).
Additional Information
Is there any additional information you feel is pertinent to this review?
Published guidelines endorse the indications proposed for this agent: The European Association of Nuclear Medicine in its 2019 guidelines proposed that PSMA targeting radioligand therapy should be considered for men with mCRPC who have failed or are not eligible to standard of care managements and those with adequate uptake of a PSMA‐targeted radiotracer on a prior PET scan. National Comprehensive Cancer Network guidelines similarly identify PSMA targeting radioligand therapy as an option for men with PSMA-avid metastases with progression after prior taxane chemotherapy and androgen pathway agents. A recent systematic review, including the TheraP and VISION trials summarizes the benefits of this therapy in terms of PSA response and survival compared to standard of care control patients (Prostate. 2022 May;82(7):826-835. doi: 10.1002/pros.24325). Canadian centres have been participants in clinical trials like VISION and there is Canadian clinical experience using lutetium vipivotide tetraxetan through these trials and special access programs.
Conflict of Interest Declarations — Canadian Cancer Society
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation.
Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
The Canadian Cancer Society helped to coordinate this submission.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Not applicable.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input.
Declaration for Clinician 1
Name: Glenn Bauman
Position: Radiation Oncologist and Distinguished University Professor, Departments of Oncology and Medical Biophysics, London Health Sciences Cancer and Western University, London, Ontario, Canada
Date: August 8, 2022
Table 2: COI Declaration for Canadian Cancer Society — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Point Biopharm | X | — | — | — |
Siemens Healthineers | — | — | X | — |
Invicro | — | — | X | — |
Declaration for Clinician 2
Name: Dr Urban Emmenegger
Position: Staff Medical Oncologist, Odette Cancer Centre, Toronto, ON
Date: August 8, 2022
Table 3: COI Declaration for Canadian Cancer Society — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Amgen | — | X | — | — |
Astellas | — | — | X | — |
AstraZeneca | — | — | X | — |
Bayer | — | — | X | — |
Ferring | X | — | — | — |
Janssen | — | X | — | — |
Knight | X | — | — | — |
Merck | — | X | — | — |
Novartis (including Endocyte and Advanced Accelerator Applications) | — | X | — | — |
Pfizer | X | — | — | — |
Point Biopharma | X | — | — | — |
Roche-Genentech | X | — | — | — |
Declaration for Clinician 3
Name: Christopher Wallis
Position: Urologic Oncologist; Assistant Professor
Date: August 4, 2022
Table 4: COI Declaration for Canadian Cancer Society — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen Oncology | — | — | X | — |
SESEN Bio | — | — | X | — |
Precision Point Specialty LLC | — | X | — | — |
Bayer | X | — | — | — |
EMD Serono | X | — | — | — |
Haymarket Media | X | — | — | — |
Healing and Cancer Foundation | X | — | — | — |
Knight Theraputics | X | — | — | — |
TerSera Canada | X | — | — | — |
Tolmar Pharmaceuticals | X | — | — | — |
Declaration for Clinician 4
Name: Katherine Zukotynski
Position: Professor
Date: 2022-09-02
Table 5: COI Declaration for Canadian Cancer Society — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Point Biopharm | X | — | — | — |
Siemens Healthineers | X | — | — | — |
Invicro | — | — | X | — |
Declaration for Clinician 5
Name: Jean-Mathieu Beauregard
Position: Associate professor, Department of Radiology Faculty of Medicine, Université Laval
Date: August 30, 2022
Table 6: COI Declaration for Canadian Cancer Society — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novartis/AAA | X | — | — | — |
Declaration for Clinician 6
Name: Frédéric Pouliot
Position: Associate professor, Urologist oncologist, CHU de Québec – Laval University
Date: August 20, 2022
Table 7: COI Declaration for Canadian Cancer Society — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Point Biopharm | X | — | — | — |
Siemens Healthineers | X | — | — | — |
Invicro | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.