CADTH Reimbursement Review
Durvalumab (Imfinzi)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Biliary tract cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
5-FU
5-fluorouracil
AE
adverse event
AoV
ampulla of Vater
BICR
blinded independent central review
BOR
best overall response
BTC
biliary tract cancer
CA 19-9
carbohydrate antigen 19-9
CCRAN
Colorectal Cancer Resource and Action Network
CCSN
Canadian Cancer Survivor Network
CGEON
Canadian Gastrointestinal Oncology Evidence Network
CI
confidence interval
CR
complete response
DAC
Drug Advisory Committee
DCO
data cut-off
DCR
disease control rate
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EHCC
extrahepatic cholangiocarcinoma
EORTC QLQ-BIL21
European Organisation for Research and Treatment of Cancer 21-Item Cholangiocarcinoma and Gallbladder Cancer Quality of Life Questionnaire
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EQ-5D-5L
5-Level EQ-5D
ES-SCLC
extensive-stage small cell lung cancer
FAS
full analysis set
FH
Fleming-Harrington
FOLFIRI
5-fluorouracil, leucovorin, and irinotecan
FOLFOX
5-fluorouracil, leucovorin, and oxaliplatin
GBC
gallbladder cancer
GHS
global health status
GI
gastrointestinal
HR
hazard ratio
HRQoL
health-related quality of life
IA-1
first interim analysis
IA-2
second interim analysis
IHCC
intrahepatic cholangiocarcinoma
imAE
immune-mediated adverse event
IRR
infusion-related reactions
IVRS
Interactive Voice Response System
IWRS
Interactive Web Response System
MID
minimally important difference
MSI
microsatellite instability
NMA
network meta-analysis
NOC
Notice of Compliance
NSCLC
non–small cell lung cancer
OH-CCO
Ontario Health Cancer Care Ontario
OR
odds ratio
ORR
objective response rate
OS
overall survival
PD
progressive disease
PD-1
programmed cell death 1 protein
PD-L1
programmed cell death 1 ligand 1
PFS
progression-free survival
PGI-S
Patient Global Impression of Severity
PR
partial response
PRO
patient-reported outcome
PRO-CTCAE
Patient Reported Outcomes version of the Common Terminology Criteria for Adverse Events
q.3.w.
every 3 weeks
q.4.w.
every 4 weeks
QoL
quality of life
RCT
randomized controlled trial
SAE
serious adverse event
SAP
statistical analysis plan
SLR
systematic literature review
SOC
standard of care
TEAE
treatment-emergent adverse event
VAS
visual analogue scale
WHO
World Health Organization
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Durvalumab (50 mg/mL) concentrate for solution for IV infusion, 1,500 mg in combination with chemotherapy q.3.w., followed by 1,500 mg q.4.w. as monotherapy until disease progression or unacceptable toxicity |
Indication | In combination with gemcitabine-based chemotherapy, for the treatment of patients with locally advanced or metastatic BTC |
Reimbursement request | Per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review (Project Orbis) |
NOC date | September 28, 2022 |
Sponsor | AstraZeneca Canada Inc. |
BTC = biliary tract cancer; NOC = Notice of Compliance; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks.
Introduction
Biliary tract cancer (BTC) refers to a heterogeneous group of gastrointestinal (GI) adenocarcinomas in the liver, gallbladder, and bile ducts. There are 4 subtypes of BTC: intrahepatic cholangiocarcinoma (IHCC), extrahepatic cholangiocarcinoma (EHCC), gallbladder cancer (GBC), and ampulla of Vater (AoV) cancer.1-3 Although BTCs comprise less than 1% of all cancers, they account for 10% to 15% of primary liver cancers,4,5 which are and 12th and 18th most common cancer in males and females in Canada, respectively, in 2021.6 The most common subtype of BTC is GBC.7,8 IHCC makes up approximately 10% to 20% of BTC cases, whereas EHCC makes up in 30% to 40% of BTC cases.5,9 The incidence of BTCs varies globally, depending on various risk factors,4 with an incidence of cholangiocarcinoma and GBC of 0.3 to 3.5 per 100,000 and 1.6 to 2.0 per 100,000, respectively, in Europe, the US, and Australasia2,5 although incidence rates are said to be increasing.1,10 Although there are few estimates for BTC in Canada, 1 study estimated the average national incidence rate of GBC and extrahepatic BTCs to be 30.92 cases per 1,000,000 individuals per year (approximately 3 per 100,000),11 which increased between 1994 and 2012.12 In Canada and the US, it has been estimated that there are approximately 400 and 5,000 new cases of cholangiocarcinoma diagnosed each year, respectively.4,13 although these numbers are nearly 20 years old and may not be reflective of current incidence rates.
Symptoms of BTCs are often nonspecific and include nausea, emesis, anorexia, weight loss, abdominal pain, and jaundice. As such, the majority of patients present with locally advanced or metastatic BTC and up to 90% of BTC cases are inoperable at the time of diagnosis.14,15 Symptoms often reflect the location of the cancer; patients with IHCC often present with nonspecific symptoms that include fever, weight loss, and/or abdominal pain, and patients with EHCC present with jaundice due to biliary obstruction.16,17
For patients with locally advanced or metastatic BTC, platinum-based chemotherapy, most commonly the combination of gemcitabine (1,000 mg/m2) plus cisplatin (25 mg/m2), has remained, for more than 10 years, the preferred first-line standard of care (SOC) regimen for patients with advanced BTC.9 There is currently no standard second-line treatment option for patients with locally advanced or metastatic BTC who experience disease progression after first-line treatment. Palliative treatment options include 5-fluorouracil (5-FU) or capecitabine monotherapy, a 5-FU, leucovorin, and irinotecan (FOLFIRI) combination, or a 5-FU, folinic acid (leucovorin), and oxaliplatin (FOLFOX) combination. In addition, pemigatinib has Health Canada market authorization with conditions for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma that has a fibroblast growth factor receptor 2 fusion or other rearrangement,18 although it recently received a do not reimburse CADTH recommendation and is not funded in Canada.19
Durvalumab (Imfinzi) is a fully humanized immunoglobulin G1 kappa monoclonal antibody that selectively blocks the interaction of programmed cell death 1 ligand 1 (PD-L1) with programmed cell death 1 protein (PD-1) and CD80. Durvalumab has received a Notice of Compliance (NOC) from Health Canada for the treatment of patients with locally advanced or metastatic BTC in combination with gemcitabine-based chemotherapy. Durvalumab is also indicated for the treatment of locally advanced, unresectable, stage III, non–small cell lung cancer (NSCLC) and for extensive-stage small cell lung cancer (ES-SCLC). In addition, durvalumab has received marketing authorization with conditions for the treatment of locally advanced or metastatic urothelial carcinoma, pending the results of clinical trials.20 Durvalumab has been previously reviewed by CADTH for the treatment of patients with locally advanced, unresectable NSCLC who have received curative-intent, platinum-based chemoradiation therapy, for a maximum of 12 months, as well as for the first-line treatment of adults with ES-SCLC in combination with etoposide and either carboplatin or cisplatin.21,22
The objective of the current report is to review the beneficial and harmful effects of durvalumab (50 mg/mL) 1,500 mg in combination with chemotherapy for the treatment of locally advanced or metastatic BTC.
Stakeholder Perspectives
The information in this section is a summary of input provided by patient groups that responded to CADTH’s call for patient input, by clinician groups, and by clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Patient group input for the review of durvalumab was provided in a submission by the Canadian Cancer Survivor Network (CCSN), with participation from the Canadian Liver Foundation, Canadian Organization for Rare Disorders, Cholangiocarcinoma Foundation, Colorectal Cancer Resource and Action Network (CCRAN), Gastrointestinal Society, and Regroupement québécois des maladies orphelines. The CCSN is a national network of patients, families, survivors, friends, community partners, funders, and sponsors who have come together to promote the best SOC for patients with cancer.
The CCSN and participating patient groups conducted an online survey between July 18 and August 2, 2022, to collect quantitative data on durvalumab. Of the 58 individuals who responded to the survey, 12 had experience with durvalumab, 25 did not have experience with durvalumab, and 17 identified as caregivers. Of the 58 survey respondents, 21 were living in Canada, 35 were living in the US, 1 was living in the UK, and 1 was living in Spain. Additionally, the CCRAN conducted 7 interviews between July 7 and July 22, 2022, with 4 patients and 3 caregivers in Canada and the US who had experience with durvalumab.
In the CCSN survey, 29 respondents disclosed their disease stage, including 18 with late-stage or metastatic, 4 with middle-stage, 3 with early-stage, and 4 with unknown disease stage. Respondents to the CCSN survey reported symptoms related to BTC that had an impact on their quality of life (QoL) and day-to-day life, which included abdominal pain, loss of appetite and weight loss, nausea and vomiting, itching, dark urine, fever, jaundice, and light coloured, greasy stools. Caregivers indicated that caring for a loved 1 with BTC had an impact on their lifestyle, and reported feeling emotionally drained, experiencing challenges in managing medications and medical appointments, and being unable to plan ahead. The most commonly used treatments reported by respondents to the CCSN survey included gemcitabine plus cisplatin, immunotherapy, radiation, surgical therapy, targeted therapy, and FOLFOX. Most CCSN respondents reported tiredness, difficulty sleeping, hair loss, nausea and vomiting, muscle weakness, numbness and tingling of the arms and legs, and diarrhea as adverse effects associated with treatment. Although most respondents indicated no issues with accessing treatments, they did cite travel costs, limited availability in the community, and financial hardship due to cost as challenges.
Most survey respondents with durvalumab experience indicated that there was little to no difference in symptom management, side effects, ease of use, or disease progression between durvalumab and other therapies they had received for BTC. Respondents reported fatigue, constipation, decreases in white blood cell and platelet counts, and anemia as side effects of durvalumab. In the CCRAN interviews, 4 patients and 3 caregivers described their experience with durvalumab for BTC. Patients accessed durvalumab through compassionate access programs, clinical trials, private insurance coverage, and by paying out of pocket. Most CCRAN interviewees described few to no side effects associated with durvalumab and reported that their cancer had regressed with treatment. Further, CCRAN respondents indicated that durvalumab was easier to use, with a shorter duration of infusion, compared to other treatment options. There was a consensus among patients that durvalumab should be made available to eligible patients.
Survey respondents reported that new treatments should maintain QoL, prolong life, provide a cure, reduce side effects from current treatments, delay the onset of symptoms, and be easy to use. When asked to describe how much of an improvement would be needed from a new treatment to make it better than current treatments, the consensus was that prolonged life with similar or reduced side effects was most important, and that QoL and ease of access should be maintained as much as possible.
Clinician Input
Input From Clinical Experts Consulted by CADTH
First-line therapy for advanced BTC has remained gemcitabine and cisplatin since publication of the ABC-02 study in 2010; however, median overall survival (OS) is less than 12 months, so the experts considered prolonged survival an important unmet need for patients with locally advanced or metastatic BTC. The clinical experts highlighted that surgery is currently the only curative treatment for BTC, but because the disease is often detected at an advanced stage, it is usually inoperable at diagnosis. Aside from the current SOC of gemcitabine and cisplatin, patients with a relatively poor performance status often receive gemcitabine alone. The experts noted that patients commonly progress after first-line treatment and there is currently no standard second-line treatment available. There are no predictive biomarkers for locally advanced or metastatic BTC. Therefore, according to the clinical experts, patients most suitable for durvalumab plus gemcitabine and cisplatin are those with preserved organ function and good performance status, regardless of the presence of cancer-related symptoms. Conversely, patients least suitable for durvalumab plus gemcitabine and cisplatin are those with a contraindication to immunotherapy. The experts also felt that it would not be appropriate to recommend other treatments before initiating treatment with durvalumab.
The experts noted that, in clinical practice, patients would be assessed every 3 weeks during routine follow-up for changes in symptoms such as fatigue and pain, and clinical and functional status, and response to treatment would be assessed every 9 to 12 weeks with imaging. The experts also noted that tumour marker assessments of carbohydrate antigen 19 to 9 (CA19 to 9) are often evaluated and followed for those with adequate biliary decompression and elevated CA 19 to 9 levels. Per the experts, durvalumab would be discontinued at clinical or radiologic progression, a confirmed worsening of symptoms, or unacceptable immune-related toxicity. The clinical experts stated that durvalumab should only be prescribed by medical oncologists and administered by qualified nurses under the supervision of a medical oncologist in a systemic treatment unit.
Clinician Group Input
Two clinician groups provided input for the review of durvalumab: the Canadian Gastrointestinal Oncology Evidence Network (CGOEN), represented by 7 clinicians, and the Ontario Health Cancer Care Ontario (OH-CCO) Gastrointestinal Cancer Drug Advisory Committee (DAC), represented by 5 clinicians. The CGOEN is a virtual network of Canadian GI oncology clinicians who contribute knowledge about GI cancer and its treatments. The OH-CCO DAC provides evidence-based clinical and health system guidance on drug-related issues.
Both clinician groups noted that current treatment goals for patients with unresectable, metastatic BTC include extending patients’ lives, delaying disease progression, and maintaining QoL. The CGEON indicated that cisplatin plus gemcitabine is the only currently available treatment option for patients with unresectable BTC, although the OH-CCO DAC also indicated that carboplatin and gemcitabine may be used in the first-line setting. The CGEON clinicians noted that the majority of BTC patients do not reach second-line treatment as the disease progresses rapidly, and that second-line treatment with 5FU and oxaliplatin provides a modest survival benefit but is poorly tolerated. The clinicians highlighted the fact that none of the molecularly targeted drugs for BTC are funded in Canada. Thus, both clinician groups emphasized that the limited number of treatment options and the moderate survival benefit provided by gemcitabine and cisplatin treatment constitute a significant unmet need for more effective treatments in this setting.
Given the lack of available options, both clinician groups indicated that there was no rationale for patients to try other treatments before initiating durvalumab plus gemcitabine and cisplatin, and that the addition of durvalumab to the current SOC would not affect the sequencing of subsequent therapy. The CGEON and OH-CCO DAC inputs indicated that patients with unresectable BTC who align with the clinical trial criteria would be most suited for treatment with durvalumab. Patients least suitable for treatment were identified as those with contraindications to immunotherapy, inadequate liver or renal function, or an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 3 or more. The CGEON and OH-CCO DAC both indicated that in clinical practice, clinical condition and/or radiologic progression are used to determine response to treatment. The CGEON indicated that a clinically meaningful response to treatment would be maintenance or improvement in QoL and prolongation of survival, and that disease progression or intolerance would be factors for discontinuing treatment. The groups agreed that durvalumab could be administered in any setting where standard chemotherapy is delivered, under the supervision of a specialist. The OH-CCO DAC agreed with the weight-based dosing method with a cap, which is consistent with other disease-site regimens, and noted that flat dosing can result in overtreatment.
Drug Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, considerations for initiation of therapy, considerations for continuation or renewal of therapy, considerations for discontinuation of therapy, considerations for prescribing of therapy, generalizability, and system and economic issues. Refer to Table 3 for more details.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Study
TOPAZ-1 was a double-blind, placebo-controlled, international, randomized, phase III study to evaluate the efficacy and safety of adding durvalumab to the established chemotherapy regimen of gemcitabine and cisplatin in patients with previously untreated, unresectable, locally advanced, or metastatic BTC. Patients were randomized in a 1:1 ratio to either durvalumab 1,500 mg (n = 341) or placebo (n = 344) delivered by IV infusion (on day 1 and every 3 weeks [q.3.w.] thereafter) in combination with cisplatin 25 mg/m2 and gemcitabine 1,000 mg/m2 (each administered on days 1 and 8, and q.3.w. thereafter) for up to 8 cycles, followed by 1,500 mg durvalumab or placebo delivered by IV infusion every 4 weeks (q.4.w.) until clinical progression (or RECIST 1.1-defined radiological progressive disease [PD]), unacceptable toxicity, withdrawal of consent, or another discontinuation criterion. The primary end point of the TOPAZ-1 trial was OS, with secondary end points of progression-free survival (PFS), objective response rate (ORR), duration of response (DOR), disease control rate (DCR), and health-related quality of life (HRQoL), and treatment tolerability.23,24
Baseline characteristics of the TOPAZ-1 trial were well balanced between treatment groups; however, according to the clinical experts, the trial may have enrolled a healthier group of patients with a lower ECOG PS than would be seen in the population in Canada. In the TOPAZ-1 trial, patients were mostly Asian (56.4%), with an even balance of males (50.4%) and females (49.6%) and a median age of 64 years, and most patients had initially unresectable disease (█████████). Most patients had IHCC (█████████), followed by GBC (█████████) and EHCC (█████████). There were 3 planned data cut-offs (DCOs); 2 interim analyses and 1 final analysis, although the second interim analysis (IA-2) was considered the final analysis. As of the most recent DCO (February 25, 2022), the median follow-up in the TOPAZ-1 trial was ██████ ███████ ███████████ ███ months and █████████████ ███████████ ███ months for the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups for OS.23,24
Efficacy Results
Results of the efficacy analyses of the TOPAZ-1 trial are summarized in Table 2. As of the final efficacy analysis of the TOPAZ-1 trial (IA-2, DCO of August 11, 2021), the median OS was 12.8 months (95% confidence interval [CI], 11.1 to 14.0 months) in the durvalumab plus gemcitabine and cisplatin group, and 11.5 months (95% CI, 10.1 to 12.5 months) in the placebo plus gemcitabine and cisplatin group. Durvalumab plus gemcitabine and cisplatin was associated with a prolonged OS compared to placebo plus gemcitabine and cisplatin (hazard ratio [HR], 0.80; 95% CI, 0.66 to 0.97; P = 0.021).23 With an additional 6.5 months of follow-up (DCO of February 25, 2022), the median OS was 12.9 months (95% CI, 11.6 to 14.1 months) for durvalumab plus gemcitabine and cisplatin, and 11.3 months (95% CI, 10.1 to 12.5 months) for placebo plus gemcitabine and cisplatin, and durvalumab plus gemcitabine and cisplatin remained favoured over placebo plus gemcitabine and cisplatin (HR, 0.76; 95% CI, 0.64 to 0.91). Results for OS rate were consistent at IA-2 and the 6.5-month follow-up, with landmark OS rates at 12 months, 18 months, and 24 months of 54.3% versus 47.1%, 34.8% versus 24.1%, and 23.6% versus 11.5%, respectively.24
At IA-2, the key secondary end point of PFS was in line with the primary end point. Durvalumab plus gemcitabine and cisplatin was associated with a prolonged PFS compared to placebo plus gemcitabine and cisplatin (HR, 0.75; 95% CI, 0.63 to 0.89; P = 0.001), with a median PFS of 7.2 months (95% CI, 6.7 to 7.4 months) for durvalumab plus gemcitabine and cisplatin, and 5.7 months (95% CI, 5.6 to 6.7 months) for placebo plus gemcitabine and cisplatin.23 Results for PFS were not available at the 6.5-month additional follow-up DCO.
ORR was a secondary end point of the TOPAZ-1 study but was a primary outcome at the first interim analysis (IA-1). █████ █ ██ █████ █████ ███████ █████████ █████████ ████ █ ████ █████ ██ ████ ██ ██████ ██████████ █████ █ ██ ████ ██████ ███████ █████████ █████████ ██ ████████████ ████ ███ █████████. At IA-2, the ORR was 26.7% for durvalumab plus gemcitabine and cisplatin and 18.7% for placebo plus gemcitabine and cisplatin (odds ratio [OR], 1.60; 95% CI, 1.11 to 2.31; P = 0.011). The statistical test for this outcome was not adjusted for multiplicity, so there is an increased risk of type I error. Only 7 (2.1%) patients in the durvalumab plus gemcitabine and cisplatin group and 2 (0.6%) patients in the placebo plus gemcitabine and cisplatin group achieved a complete response (CR). No results for ORR were available at the 6.5-month update. The median DOR was 6.4 months ████ ███████████ ██████████████████ ███ in the durvalumab plus gemcitabine and cisplatin group and 6.2 months ██████ ███████ ███████████ ████████ ████ in the placebo plus gemcitabine and cisplatin group.23
Secondary end points for HRQoL consisted of the time to deterioration and improvement rates for the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and the EORTC 21-Item Cholangiocarcinoma and Gallbladder Cancer Quality of Life Questionnaire (EORTC QLQ-BIL21). For global health status (GHS)/QoL, the median time to deterioration was 7.4 months (95% CI, 5.6 to 8.9 months) for durvalumab plus gemcitabine and cisplatin compared to 6.7 months (95% CI, 5.6 to 7.9 months) for placebo plus gemcitabine and cisplatin. The median time to deterioration in functional groups ranged from 5.6 months to 10.1 months for durvalumab plus gemcitabine and cisplatin and 6.5 months to 10.0 months for placebo plus gemcitabine and cisplatin. The median time to deterioration in multiple and single symptom items ranged from 3.0 months for fatigue to 18.2 months for diarrhea in the durvalumab plus gemcitabine and cisplatin group and 3.5 months for fatigue to 11.0 months for diarrhea in the placebo plus gemcitabine and cisplatin group. █████ █ ██ ██████████ █ ██████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ███ ██ █████ ██ █████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████ ████ ████ ███ █████████ █████████ ████ ████ ██████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██ ████ ████ ██ █████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ █████ ███ ███ █ ██ █████ ███ ██ ████ ███ █████████ █████ ████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ███ █████ █ ██ █ █ ███ █████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ █ ████ ██████ █ ██ ██ ██ ██ ████ ███████ █████████ ██ ███████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ██ ███ ████ █ ██ █ ███ ██████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ██ ██████ ██ █████████ █ ██ █ ████ █████ ███████ █████████ █████████ ████ ████████ ██ ██ ████ ██ ██████ ██ ████████ █████ █ ██ ██ ████████ ███████ █████████ █████████ █████
The median time to symptom deterioration on the EORTC QLQ-BIL21 ranged from 3.5 months to 11.7 months for durvalumab plus gemcitabine and cisplatin, and 3.7 months to 14.2 months for placebo plus gemcitabine and cisplatin. The proportion of patients experiencing improvement in symptom domains ranged from ███ ███████ ████████ █████████ for durvalumab plus gemcitabine and cisplatin, and from ████ ██████████████ █████ ██ ██ for placebo plus gemcitabine and cisplatin.23
Harms Results
As of the final analysis (IA-2), the overall incidence of treatment-emergent adverse events (TEAEs) in the TOPAZ-1 study was comparable between durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin (336 [99.4%] versus 338 [98.8%]). The most frequent TEAEs for both durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin included anemia (163 [48.2%] versus ███ ██████ ██████ ███) and nausea █ ███ ██ ███████████ ██████████ ███ ████████ ███████, with differences of at least 5% only observed between the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups for nausea. Grade 3 or 4 adverse events (AEs) were reported at a similar frequency in the 2 treatment groups, with 250 patients (74.0%) and 257 patients (75.1%) in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group, respectively. The most common grade 3 or 4 AEs included anemia ███████ █████ ███████████ ████, decreased neutrophil count (21.0% versus 25.7%), and neutropenia (20.1% versus 21.1%). The incidence of serious adverse events (SAEs) was similar in the durvalumab plus gemcitabine group and the cisplatin and placebo plus gemcitabine and cisplatin group █████ █████████ ████ versus 151 [44.2%]), with the most common SAE being cholangitis in both groups (██ ███████ ████ █████ █████████ [5.0%]). The proportion of patients that discontinued treatment due to TEAEs was █ ████████ in the durvalumab plus gemcitabine and cisplatin group, and 15.2% in the placebo plus gemcitabine and cisplatin group, driven mainly by █████ █ ██ ██████████ ███████ ████ █████ ███ ██████ ██ ███ in the placebo group. Deaths due to AEs were reported in 13 (3.8%) patients in the durvalumab plus gemcitabine and cisplatin group, and 14 (4.1%) patients in the placebo plus gemcitabine and cisplatin group, with most deaths in the durvalumab group due to █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██ ███ █████ ██ ████ ██ ██████ ██████████ █████ █ ██ ██████████ ███████ █████████ █████████ ██ █ ███ ███ █ ███████ █████████ █████ █████████ ████████ in the placebo group.23,24
The incidence of notable harms, including immune-mediated AEs (imAEs), infusion-related reactions (IRRs), infections, and GI events was generally more frequent in the durvalumab plus gemcitabine and cisplatin group than in the placebo plus gemcitabine and cisplatin group. At the final analysis, imAEs were identified for 43 (12.7%) patients in the durvalumab plus gemcitabine and cisplatin group and 16 (4.7%) patients in the placebo plus gemcitabine and cisplatin group; of these, 8 (2.4%) and 5 (1.5%) patients, respectively, experienced grade 3 or 4 imAEs. IRRs were reported in ███ ████ ███ ████ ███████████ ████████ ███ patients in the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups, respectively. Infections and infestations occurred in ███████ ████ ███████ patients in the durvalumab plus gemcitabine and cisplatin group, and ████ █████████ ███ ██ patients in the placebo plus gemcitabine and cisplatin group. GI disorders occurred in █ ██ ██████ ███ ████ ██████ ███ ██████████ █████ █████ patients in the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups, respectively. Immune-mediated GI events occurred in 3 ███ ██████ ██████ ███ patients in the durvalumab plus gemcitabine and cisplatin group and in 1 (0.3%) patient in placebo plus gemcitabine and cisplatin group.23,24
Table 2: Summary of Key Results From TOPAZ-1
Outcome | IA-2 (DCO: August 11, 2021) | 6.5-month update (DCO: February 25, 2022) | ||
|---|---|---|---|---|
D + Gem and Cis | Placebo + Gem and Cis | D + Gem and Cis | Placebo + Gem and Cis | |
Efficacy outcomes (FAS) | ||||
N | 341 | 344 | 341 | 344 |
Median OS (95% CI) | 12.8 (11.1 to 14.0) | 11.5 (10.1 to 12.5) | 12.9 (11.6 to 14.1) | 11.3 (10.1 to 12.5) |
Event, n (%) | 198 (58.1) | 226 (65.7) | 248 (72.7) | 279 (81.1) |
Censored, n (%) | 143 (41.9) | 118 (34.3) | 93 (27.3) | 65 (18.9) |
HR (95% CI) | 0.80 (0.66 to 0.97) | 0.76 (0.64 to 0.91) | ||
P value | 0.021 | NR | ||
Median PFS (95% CI) | 7.2 (6.7 to 7.4) | 5.7 (5.6 to 6.7) | NR | NR |
Event, n (%) | 276 (80.9) | 297 (86.3) | NR | NR |
Censored, n (%) | 65 (19.1) | 47 (13.7) | NR | NR |
HR (95% CI) | 0.75 (0.63 to 0.89) | NR | ||
P value | 0.001 | NR | ||
ORR (CR plus PR), n (%) | 91 (26.7) | 64 (18.7) | NR | NR |
CR | 7 (2.1) | 2 (0.6) | NR | NR |
PR | 84 (24.6) | 62 (18.1) | NR | NR |
OR (95% CI) | 1.60 (1.11 to 2.31) | NR | ||
P valuea | 0.011 | NR | ||
Median DOR (95% CI) | 6.4 (5.9 to 8.1) | 6.2 (4.4 to 7.3) | NR | NR |
Harms, n (%) (Safety Analysis Set) | ||||
N | 338 | 342 | 338 | 342 |
AEs | 336 (99.4) | 338 (98.8) | 336 (99.4) | 338 (98.8) |
SAEs | 160 (47.3) | 149 (43.6) | █████████ | █████████ |
WDAE | 44 (13.0) | 52 (15.2) | 43 (12.7) | 52 (15.2) |
Deaths | 12 (3.6) | 14 (4.1) | 13 (3.8) | 14 (4.1) |
Notable Harms | ||||
imAEs, n (%) | 43 (12.7) | 16 (4.7) | █████████ | █████████ |
IRRs, n (%) | 13 (3.8) | 6 (1.8) | █████████ | █████████ |
Infections, n (%) | 120 (35.5) | 113 (33.0) | 127 (37.6) | 120 (35.1) |
GI events | 266 (78.7) | 238 (69.6) | 268 (79.3) | 239 (69.9) |
AE = adverse event; CI = confidence interval; Cis = cisplatin; CR = complete response; D = durvalumab; DCO = data cut-off; FAS = full analysis set; Gem = gemcitabine; GI = gastrointestinal; HR = hazard ratio; IA-2 = interim analysis 2; imAE = immune-mediated adverse event; IRR = infusion-related reaction; NR = not reported; OR = odds ratio; OS = overall survival; ORR = objective response rate; PFS = progression-free survival; PR = partial response; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aP value for ORR was not controlled for multiple testing.
Sources: TOPAZ-1 CSR (IA-2 and 6.5-month update).23,24
Critical Appraisal
TOPAZ-1 was an international, double-blind, phase III randomized controlled trial (RCT). Appropriate methods of randomization, treatment allocation, and stratification were employed using Interactive Web Response Systems (IWRS) and Interactive Voice Response Systems (IVRS). The methods for randomization were considered by the CADTH review team to result in a low risk of bias. Patients did not differ with regard to baseline demographic or disease characteristics, indicating that randomization was successful. There was potential for unblinding in the TOPAZ-1 study in the event of differences in rates of disease progression and known treatment-related toxicities associated with durvalumab, such as imAEs. Overall, withdrawals and discontinuations due to AEs were similar across treatment groups and, therefore, likely did not result in unblinding. Methods to maintain blinding were also appropriate, although the number of patients who were unblinded for various reasons was not reported. Two interim analyses and 1 final analysis were planned for the TOPAZ-1 trial. As of IA-2, the study met its primary end point ██ ██████ ██████ ████ ████ █████ and was considered the final analysis by the Independent Data Monitoring Committee. Trials that stop early for benefit may overestimate the relative benefits of treatment,25,26 although the presence and extent of overestimation is uncertain. The 6.5-month follow-up was performed at 76.9% maturity for OS, which demonstrated consistent benefit compared to IA-2, although comparisons between DCOs were not formally conducted. Acceptable methods to account for multiplicity were conducted in the TOPAZ-1 trial for the primary efficacy end point of OS (via family-wise error rate) and the selected key secondary end point, PFS, using a hierarchical testing procedure. Other secondary outcomes, including ORR, DOR, EORTC QLQ-C30 and EORTC QLQ-BIL21, and exploratory outcomes (such as Patient Global Impression of Severity [PGI-S], Patient Reported Outcomes version of the Common Terminology Criteria for Adverse Events [PRO-CTCAE], and the 5-Level EQ-5D [EQ-5D-5L]), were not controlled for multiplicity, so need to be interpreted with consideration of the increased risk of type I error. For all time points for all HRQoL and patient-reported outcome (PRO) end points, there was a notable amount of missing data; thus, there was a high risk of attrition bias for these outcomes.The inclusion and exclusion criteria for TOPAZ-1 trial were generally as expected for patients with locally advanced or metastatic BTC, although histologic diagnosis is not always confirmed, the clinical experts consulted by CADTH explained. It was considered that patients included in the study may be a less sick population for various reasons, including ECOG PS, where the clinical experts noted that patients with an ECOG PS of 2 would also be eligible to receive durvalumab plus gemcitabine and cisplatin, provided they have adequate organ function. The clinical experts consulted by CADTH highlighted some characteristics that were not reflective of clinical practice in Canada, primarily that there was an overrepresentation of Asian patients in the TOPAZ-1 trial (56.4%), which is not representative of what would be seen in the population in Canada. This was a reflection of where the trial was conducted, as no Canadian sites were included, and most patients were enrolled from Asian countries (n = 374 [54.6%]). Regarding BTC subtypes, the experts felt that the proportion of patients with IHCC was higher than seen in the real world, and the proportion with EHCC was lower than seen in the real world, which may have a subtle effect on prognosis. The clinical experts consulted by CADTH noted that the proportion of patients receiving subsequent anticancer therapy was higher than expected, although that likely reflected the location where patients were enrolled in the trial. It was hypothesized by the clinical experts that the elevated proportion of patients receiving subsequent anticancer therapy was due to the baseline ECOG PS, where patients likely remained fit enough for subsequent lines of therapy. This was not considered reflective of clinical practice in Canada by the experts, so may not be generalizable to the population in Canada. Outcomes of the TOPAZ-1 trial were clinically relevant and important to both clinicians and patients. Measures of HRQoL were prespecified in the protocol and suggested no detriment to QoL with durvalumab plus gemcitabine and cisplatin; however, no conclusions for HRQoL or treatment tolerability can be drawn, owing to the methodological limitations associated with these analyses.
Indirect Comparisons
No indirect evidence was included in the sponsor’s submission to CADTH. One systematic literature review (SLR) and network meta-analysis (NMA) was identified in the literature search that matched the inclusion and exclusion criteria of this review. Feng et al. (2022)27 conducted an NMA comparing the OS and PFS of different first-line treatment regimens for patients with advanced BTC using methods of random-effects. A total of 19 different treatment regimens were included in the NMA, although only comparisons involving treatments identified in the CADTH review protocol (Table 4) were of interest to this review. Data provided for comparisons of interest were derived from the TOPAZ-1 trial. There was insufficient evidence to draw conclusions for the outcome of OS (HR, 0.27; 95% CI, 0.06 to 1.29), whereas for PFS, the results produced by the NMA for comparisons of interest were in favour of durvalumab plus gemcitabine and cisplatin (HR, 0.22; 95% CI, 0.08 to 0.62).27 There was notable heterogeneity in the populations from the studies included in the NMA, including differences in age, ECOG PS, and primary tumour location, with different proportions of patients with IHCC, EHCC and GBC, and some trials including patients with AoV cancer. This heterogeneity likely resulted in the notable imprecision, given the wide 95% CIs, and uncertainty in the comparative efficacy estimates.
Other Relevant Evidence
No long-term extension studies or other relevant studies were included in the sponsor’s submission to CADTH.
Conclusions
Patients and clinicians highlighted the need for new effective treatments that prolong life, maintain QoL, and reduce side effects compared to current treatments. In the randomized, phase III TOPAZ-1 trial, durvalumab plus gemcitabine and cisplatin demonstrated a statistically significant, albeit modest, improvement in median OS compared with placebo plus gemcitabine and cisplatin, which is the current SOC in Canada for patients with locally advanced or metastatic BTC. However, the clinical importance of the modest survival gain afforded by treatment with durvalumab compared with SOC is subjective and may vary from patient to patient, although the OS rates may also suggest that patients achieve longer OS benefit. Analyses of secondary outcomes, including PFS, and ORR supported the efficacy of adding durvalumab to the SOC. Because the findings for survival and response outcomes were based on an interim analysis, the benefits of durvalumab relative to SOC may be overestimated, although the presence and extent of any overestimation is uncertain. No definitive conclusions could be drawn about the impact of treatment with durvalumab compared with SOC on HRQoL or time to deterioration, owing to limitations of the analyses, including a lack of statistical testing and risk of attrition bias. The safety of durvalumab is well known, considering its use in other indications; however, aside from the AEs specific to durvalumab, the frequency of AEs and SAEs was similar in the 2 treatment groups. Overall, although there are limitations associated with stopping the trial at the second interim analysis, and also surrounding the statistical analysis for secondary end points, the TOPAZ-1 study demonstrated improvement in OS with the addition of durvalumab to gemcitabine and cisplatin. However, given the modest gain in OS observed in the trial, the clinical benefit is likely to be incremental, and there remains uncertainty about the magnitude of clinical benefit in the locally advanced or metastatic BTC population in Canada.
Introduction
Disease Background
BTC refers to a heterogeneous group of GI adenocarcinomas in the liver, gallbladder, and bile ducts. There are 4 subtypes of BTC: IHCC, which occurs in the small bile ducts in the periphery of the liver; EHCC, which occurs in the main ducts of the hilum or distal region of the bile duct; GBC; and AoV.1-3
Although BTCs comprise less than 1% of all cancers, they account for 10% to 15% of primary liver cancers,4,5 which are the fifth most common cancers worldwide,28,29 and the 12th and 18th most common cancers in males and females in Canada, respectively, in 2021.6 The other 85% to 90% of primary liver cancers are hepatocellular carcinomas.28,29 The most common subtype of BTC is GBC.7,8 IHCC makes up approximately 10% to 20% of BTC cases, whereas EHCC makes up 30% to 40% of BTC cases.5,9 The incidence of BTC varies globally, depending on various risk factors,4 with an incidence of cholangiocarcinoma and GBC of 0.3 to 3.5 per 100,000 and 1.6 to 2.0 per 100,000, respectively,2,5 in Europe, the US, and Australasia, although incidence rates are said to be increasing.1,10 Rates of BTC tend to be higher in Asian cultures, with China, Thailand, South Korea, Japan, and Taiwan having some of the highest rates of incidence and mortality;30 the highest incidence, of 90 per 100,000, is seen in Northeast Thailand.31 Although there are few estimates of BTC in Canada, 1 study estimated the average national incidence rate of GBC and extrahepatic BTC at 30.92 cases per 1,000,000 individuals per year (approximately 3 per 100,000),11 which increased between 1994 and 2012.12 It has been estimated that there are approximately 400 and 5,000 new cases of cholangiocarcinoma diagnosed each year in Canada and the US,, respectively,4,13 although these numbers are nearly 20 years old and may not reflect current incidence rates.
Symptoms of BTCs are often nonspecific and include nausea, emesis, anorexia, weight loss, abdominal pain, and jaundice. As such, the majority of patients present with locally advanced or metastatic BTC, and up to 90% of BTC cases are inoperable at the time of diagnosis.14,15 Symptoms often reflect the location of the cancer; patients with IHCC often present with nonspecific symptoms, including fever, weight loss, and/or abdominal pain, whereas patients with EHCC present with jaundice due to biliary obstruction.16,17 Generally, a diagnosis of BTC is made with MRI, and magnetic resonance cholangiopancreatography, ultrasound, or CT scan are used to identify the disease, locate the level of biliary obstruction, and identify any regional lymphadenopathy or metastatic disease. In general, a pathologic diagnosis with endoscopic retrograde cholangiopancreatography, percutaneous transhepatic cholangiography, or magnetic resonance cholangiopancreatography should be obtained before the initiation of nonsurgical treatments.5,9 In cases of advanced disease, serum CA19 to 9 has some added value as a tumour marker, but it is not considered diagnostic.32
Risk factors for BTC vary by subtype and by region. Well-established risk factors in the Western world include primary sclerosing cholangitis, chronic inflammation or infection, age, hepatitis C infection, cirrhosis, and exposure to various toxins, such as dioxins and vinyl chlorides. In regions where liver flukes are common, patients with liver fluke infections are at an increased risk of BTC.1,4,5,9 The incidence and risk of GBC is closely associated with cholelithiasis (gall stones), gallbladder polyps, primary sclerosing cholangitis, chronic infection (e.g., Salmonella typhi), congenital malformations, obesity, and other risk factors for gallbladder disease.1,5,33 Globally, GBC is more common in females, whereas cholangiocarcinoma, particularly EHCC, is more common in males.1,5,34
BTCs are typically staged using the American Joint Committee on Cancer tumour, node, metastases staging system. The prognosis for patients with BTCs is poor, with estimated 5-year survival rates in the US of 30%, 24%, and 2% for local, regional, and distant metastatic EHCC, respectively, and of 15%, 6%, and 2%, respectively, for IHCC. Five-year survival rates for GBC are similar, at 8% and 7% for patients with stages IIIA and IIIB disease, respectively, and at 4% and 2% for patients with stages IVA and IVB disease, respectively.14
Standards of Therapy
In patients with resectable, early-stage disease, surgery is the only potentially curative option. However, few patients are diagnosed with resectable disease, and nearly 50% relapse after surgery.14,15,35
For patients with locally advanced or metastatic disease, treatment is often palliative. Platinum-based chemotherapy, most commonly the combination of gemcitabine (1,000 mg/m2) plus cisplatin (25 mg/m2), has remained, for more than 10 years, the preferred first-line SOC regimen for suitable patients with advanced BTC.9 The combination of gemcitabine plus cisplatin was established after results of the ABC-02 study in patients with locally advanced or metastatic BTC demonstrated better OS (11.7 months versus 8.1 months; HR, 0.64; 95% CI, 0.52 to 0.80) and better PFS (8.0 months versus 5.0 months; HR, 0.63; 95% CI, 0.51 to 0.77) than gemcitabine monotherapy.36
Although not the focus of this review, there is currently no standard second-line treatment option for patients with locally advanced or metastatic BTC who experience disease progression after first-line treatment. Palliative treatment options include 5-FU monotherapy, FOLFIRI, or FOLFOX. Pemigatinib also has Health Canada market authorization with conditions for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma that has a fibroblast growth factor receptor 2 fusion or other rearrangement,18 although it recently received a do not reimburse CADTH recommendation and is not publicly funded in Canada.19
Drug
Durvalumab is a fully immunoglobulin G1 kappa monoclonal antibody that selectively blocks the interaction of PD-L1 with PD-1 and CD80. Expression of PD-L1 helps tumours evade detection and elimination by the immune system. When PD-L1 interacts with PD-1 and CD80, the PD-L1/ PD-1 pathway decreases cytotoxic T-cell activity, proliferation, and cytokine production. By selectively blocking the interaction of PD-L1 with PD-1 and CD80, durvalumab inhibits the immunosuppressive effects of the PD-L1/PD-1 pathway, enhancing antitumour immune responses.20
Durvalumab is provided as a single-use vial of 120 mg durvalumab/2.4 mL (nominal concentration of 50 mg/mL) and 500 mg durvalumab/10 mL (nominal concentration of 50 mg/mL) and is administered as an IV infusion over 60 minutes. The recommended dose of durvalumab is 1,500 mg in combination with chemotherapy every 3 weeks, followed by 1,500 mg every 4 weeks as monotherapy until disease progression or unacceptable toxicity. Patients with a body weight of 30 kg or less must receive weight-based dosing of durvalumab at 20 mg/kg in combination with chemotherapy dosed every 3 weeks, followed by monotherapy at 20 mg/kg every 4 weeks until weight increases to greater than 30 kg. When given on the same day, durvalumab should be administered before chemotherapy.20
Durvalumab has received an NOC from Health Canada for the treatment of patients with locally advanced or metastatic BTC in combination with gemcitabine-based chemotherapy.20 Durvalumab is also indicated for the treatment of locally advanced, unresectable, stage III NSCLC and for ES-SCLC. In addition, durvalumab has received marketing authorization with conditions for the treatment of locally advanced or metastatic urothelial carcinoma, pending the results of clinical trials.20 Durvalumab has been previously reviewed by CADTH for the treatment of patients with locally advanced, unresectable NSCLC following curative-intent, platinum-based chemoradiation therapy, for up to a maximum of 12 months, as well as for first-line treatment of adult patients with ES-SCLC in combination with etoposide and either carboplatin or cisplatin.21,22
Stakeholder Perspectives
Patient Group Input
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient group input for the review of durvalumab was provided as a submission by the CCSN, with participation from the Canadian Liver Foundation, Canadian Organization for Rare Disorders, Cholangiocarcinoma Foundation, CCRAN, Gastrointestinal Society, and Regroupement québécois des maladies orphelines. The CCSN is a national network of patients, families, survivors, friends, community partners, funders, and sponsors who have come together to promote the best SOC for patients with cancer.
The CCSN and participating patient groups conducted an online survey between July 18 and August 2, 2022, to collect quantitative data on durvalumab. Of the 58 individuals who responded to the survey, 12 had experience with durvalumab, 25 did not have experience with durvalumab, and 17 identified as caregivers. Of the 58 survey respondents, 21 were living in Canada, 35 were living in the US, 1 was living in the UK, and 1 was living in Spain. Additionally, the CCRAN conducted 7 interviews between July 7 and July 22, 2022, with 4 patients and 3 caregivers in Canada and the US who had experience with durvalumab.
In the CCSN survey, 29 respondents disclosed their disease stage, including 18 with late-stage or metastatic, 4 with middle-stage, 3 with early-stage, and 4 with unknown disease stage. Respondents to the CCSN survey reported BTC symptoms that had an impact on their QoL and day-to-day life, which included abdominal pain, loss of appetite and weight loss, nausea and vomiting, itching, dark urine, fever, jaundice, and light coloured, greasy stools. Caregivers indicated that caring for a loved 1 with BTC had an impact on their lifestyle, and reported feeling emotionally drained, experiencing challenges in managing medications and medical appointments, and being unable to plan ahead. The most commonly used treatments reported by respondents to the CCSN survey included gemcitabine plus cisplatin, immunotherapy, radiation, surgical therapy, targeted therapy, and FOLFOX. Most CCSN respondents reported tiredness, difficulty sleeping, hair loss, nausea and vomiting, muscle weakness, numbness and tingling of the arms and legs, and diarrhea as adverse effects associated with treatment. Although most respondents indicated no issues with accessing treatments, they did cite travel costs, limited availability in the community, and financial hardship due to cost as challenges.
Most survey respondents with durvalumab experience indicated that there was little to no difference in symptom management, side effects, ease of use, or disease progression between durvalumab and other therapies they had received for BTC. Respondents reported fatigue, constipation, decreases in white blood cell and platelet counts, and anemia as side effects of durvalumab. In the CCRAN interviews, 4 patients and 3 caregivers described their experience with durvalumab for BTC. Patients accessed durvalumab through compassionate access programs, clinical trials, private insurance coverage, and by paying out of pocket. Most CCRAN interviewees described few to no side effects associated with durvalumab and reported that their cancer had regressed with treatment. Further, CCRAN respondents indicated that durvalumab was easier to use, with a shorter duration of infusion compared to other treatment options. There was a consensus among patients that durvalumab should be made available to eligible patients.
Survey respondents reported that new treatments should maintain QoL, prolong life, provide a cure, reduce side effects from current treatments, delay the onset of symptoms, and be easy to use. When asked to describe how much of an improvement would be needed from a new treatment to make it better than current treatments, the consensus was that prolonged life with similar or reduced side effects was most important, and that QoL and ease of access should be maintained as much as possible.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of BTC.
Unmet Needs
First-line therapy for advanced BTC has not changed in more than a decade and remains cytotoxic chemotherapy in the form of gemcitabine and cisplatin. The median OS with current SOC is just less than 12 months. As such, the experts considered prolonged survival an important unmet need for patients with locally advanced or metastatic BTC because less than 15% of patients remain alive 24 months after diagnosis. Additionally, improvement in disease-related symptoms was considered important.
Place in Therapy
According to the clinical experts consulted by CADTH, surgery is currently the only curative treatment for BTC; however, the disease is often detected at an advanced stage and is usually inoperable at diagnosis. The standard first-line treatment for locally advanced or metastatic BTC has been the combination of gemcitabine and cisplatin since publication of the ABC-02 study in 2010, which demonstrated better OS and PFS than gemcitabine alone.36 For patients with a relatively poor performance status, gemcitabine alone is often used. One expert noted that for patients with biliary obstruction, interventions such as ERCP or percutaneous transhepatic cholangiography often have to be considered to relieve the obstruction before treatment.
The experts emphasized that there is currently no standard second-line treatment for locally advanced or metastatic BTC, and patients commonly progress after first-line treatment. Several clinical trials have aimed to improve survival outcomes with the addition of more targeted therapies, the clinical experts noted, but the results of these studies have largely been negative. However, a recent RCT comparing FOLFOX to active symptom control showed survival benefits, the experts reported.37
Given that the mechanism of action of durvalumab differs from that of cytotoxic chemotherapy, the experts explained that its complementary use with the current gemcitabine and cisplatin SOC (up to 8 cycles) is expected to shift the first-line treatment paradigm and is expected to provide incremental improvement in outcomes.
Patient Population
There is currently no companion diagnostic required, or there are no predictive biomarkers for locally advanced or metastatic BTC. Microsatellite instability (MSI) is rare in this population, the experts noted, although many Canadian centres have reflex testing for MSI. And not all patients will have had a pathologic diagnosis, they added, as some patients with extrahepatic disease are not amenable to a biopsy.
According to the clinical experts and given that the TOPAZ study23 did not specify a biomarker, such as PD-L1 expression, patients who are candidates for gemcitabine and cisplatin and who have preserved organ function and a good performance status are most suitable for durvalumab plus gemcitabine and cisplatin, regardless of the presence of cancer-related symptoms.
Conversely, the experts added, durvalumab plus gemcitabine and cisplatin would only be suitable for patients who do not have a contraindication to immunotherapy. The experts also felt that it would not be appropriate to recommend other treatments before initiating treatment with durvalumab.
Assessing Response to Treatment
The clinical experts agreed that patients would be assessed clinically during routine follow-up (i.e., q.3.w., given the cycle length) for changes in symptoms, such as fatigue and pain, and for clinical and functional status. Response to treatment would be assessed every 9 to 12 weeks with cross sectional imaging, most often a CT scan, although MRI or ultrasound may be used to evaluate tumour response or stability.
The experts also emphasized that tumour marker assessments of CA 19 to 9 are often evaluated and followed in patients with adequate biliary decompression and elevated CA 19 to 9 levels.
Discontinuing Treatment
The clinical experts stated that durvalumab would be discontinued at objective, clinical, or radiologic disease progression, confirmed worsening of symptoms, or unacceptable immune-related toxicity.
Prescribing Conditions
The clinical experts stated that durvalumab should only be prescribed by medical oncologists. Durvalumab should be administered in a systemic chemotherapy unit by qualified nurses under the supervision of a medical oncologist. The experts also noted that, at this time, all centres would likely be familiar with immune checkpoint inhibitors and many would have had experience with durvalumab administration.
Clinician Group Input
Two clinician groups provided input for the review of durvalumab: the CGOEN, represented by 7 clinicians; and the OH-CCO DAC, represented by 5 clinicians. The CGOEN is a virtual network of Canadian GI oncology clinicians who contribute knowledge about GI cancer and its treatments. The OH-CCO DAC provides evidence-based clinical and health system guidance on drug-related issues.
Both clinician groups noted that current treatment goals for patients with unresectable, metastatic BTC include extending lives, delaying disease progression, and maintaining QoL. The CGEON indicated that cisplatin plus gemcitabine is the only currently available treatment option for patients with unresectable BTC, although the OH-CCO DAC also indicated that carboplatin and gemcitabine may be used in the first-line setting. The CGEON clinicians noted that the majority of BTC patients do not reach second-line treatment as the disease progresses rapidly, and that second-line treatment with 5FU and oxaliplatin provides a modest survival benefit but is poorly tolerated. The clinicians noted that no molecularly targeted drugs for BTC are funded in Canada. Thus, both clinician groups emphasized that the limited number of treatment options and the moderate survival benefit provided by gemcitabine and cisplatin treatment constitute a significant unmet need for more effective treatments in this setting.
Given the lack of available options, both clinician groups indicated that there was no rationale for patients to try other treatments before initiating durvalumab plus gemcitabine and cisplatin, and that the addition of durvalumab to the current SOC would not affect the sequencing of subsequent therapy. The CGEON and OH-CCO DAC inputs indicated that patients with unresectable BTC who align with the clinical trial criteria would be most suited for treatment with durvalumab. Patients least suitable for treatment were identified as those with contraindications to immunotherapy, inadequate liver or renal function, or an ECOG PS of 3 or more. The CGEON and OH-CCO DAC both indicated that, in clinical practice, clinical condition and/or radiologic progression are used to determine response to treatment. The CGEON indicated that a clinically meaningful response to treatment would be maintenance or improvement in QoL and prolongation of survival, whereas disease progression or intolerance would be factors for discontinuing treatment. The groups agreed that durvalumab could be administered in any setting where standard chemotherapy is delivered, under the supervision of a specialist. The OH-CCO DAC agreed with the weight-based dosing method with a cap, which is consistent with other disease-site regimens, and noted that flat dosing can result in overtreatment.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The comparator arm of the TOPAZ-1 trial was gemcitabine plus cisplatin. This is a funded therapy and is considered SOC for the first-line treatment of patients with locally advanced or metastatic BTC. If there are concerns about a patient’s renal function, carboplatin or oxaliplatin may be substituted for cisplatin. For patients with a poor performance status, gemcitabine monotherapy may be used as a first-line treatment. If a patient is not able to tolerate cisplatin-based chemotherapy, is it reasonable to combine durvalumab with alternate chemotherapy? | Decisions on alternative chemotherapy options must be considered on a case-by-case basis, based on age, renal function, and other factors. Substitution with carboplatin may be an option, although the experts believe this would be uncommon. The experts noted that they might consider using durvalumab in combination with gemcitabine monotherapy for select patients who are not receiving the platinum doublet. |
Considerations for initiation of therapy | |
Is histologic diagnosis of BTC required for patients to be eligible for durvalumab? Is a diagnosis of BTC ever made without histologic confirmation? | Yes, histologic confirmation of BTC is required for durvalumab eligibility. The experts noted that the diagnosis of BTC is often made without histologic confirmation, and the current treatment paradigm of platinum-based chemotherapy would be given regardless of histologic confirmation. |
Can durvalumab be restarted if treatment was stopped for reasons other than disease progression? | The clinical experts noted that as long as durvalumab was not stopped for immune-related toxicities, then restarting durvalumab is reasonable. |
Considerations for continuation or renewal of therapy | |
What is the recommended type and frequency of follow-up for patients on durvalumab maintenance? | Patients should be assessed clinically every 3 weeks, with imaging conducted every 2 to 3 months. |
Considerations for discontinuation of therapy | |
In the trial, treatment could be continued beyond disease progression at the discretion of the investigator if there was continued clinical benefit. What are the criteria for discontinuing durvalumab? | Durvalumab should be discontinued when there is objective evidence of disease progression or severe immune-related toxicity. Given the modest improvement in OS, the experts stated that it is unlikely that clinical benefit would be observed in the presence of progression. The experts also noted that if durvalumab was discontinued, they would also consider discontinuing treatment with gemcitabine and cisplatin (if during chemotherapy phase), unless toxicities were specific to individual treatments. |
If there is progression during a drug holiday, can treatment be resumed? If re-treatment with durvalumab is permitted in this scenario, would therapy consist of durvalumab monotherapy or durvalumab plus chemotherapy? Is there a minimum number of cycles of chemotherapy that must be given with durvalumab (e.g., what if the patient must discontinue the chemotherapy portion after 1 cycle)? | The experts stated that re-treatment with durvalumab after progression during a drug holiday would be reasonable; however, durvalumab would only be administered in combination with chemotherapy and not given as monotherapy. They also noted that this would only be considered in patients who had evidence of progression but were still well enough to receive treatment with chemotherapy. The experts considered that a minimum of 4 cycles of chemotherapy and durvalumab must be received to inform clinical decisions about re-treatment. |
Considerations for prescribing of therapy | |
In the trial, patients received durvalumab at a flat dose of 1,500 mg q.3.w. in combination with gemcitabine and cisplatin for up to 8 cycles, followed by 1,500 mg q.4.w. as a single drug until disease progression or unacceptable toxicity. If a patient’s weight fell to ≤ 30 kg, they received a weight-based dose equivalent to 20 mg/kg of durvalumab q.3.w. in combination with chemotherapy, followed by 20 mg/kg q.4.w. as a single drug. Jurisdictions use weight-based dosing up to a cap. Can weight-based dosing up to a cap be used in place of flat dosing for patients weighing more than 30 kg? If weight-based dosing up to a cap can be used, what mg/kg dose(s) of durvalumab should be used when it is given in combination with chemotherapy q.3.w. and then as a single drug q.4.w.? Should the weight-based dosing be 15 mg/kg up to 1,500 mg q.3.w. in combination with chemotherapy, followed by 20 mg/kg up to 1,500 mg q.4.w. as a single drug, rather than 20 mg/kg q.3.w. in combination with chemotherapy and then q.4.w. as a single drug? | The clinical experts emphasized that flat-based dosing is preferred from a clinical standpoint and reflects how durvalumab was administered in the clinical trial. The experts also noted that very few patients weigh less than 30 kg, and it is unlikely that they would be treated with chemotherapy at this weight. |
Generalizability | |
Should durvalumab be considered in patients with an ECOG PS of 2 or greater, or in patients with AoV cancer, as these patients were excluded from the trial? | Patients with an ECOG PS of 2 or greater were not eligible for the TOPAZ-1 trial. The experts stated that patients with an ECOG PS of 2 should be treated with durvalumab at the discretion of the treating physician if they are fit enough for treatment. However, they noted that patients with an ECOG PS greater than 2 would not be eligible for treatment with durvalumab. The clinical experts emphasized that AoV cancers are treated differently than BTC, so such patients should not be considered for treatment with durvalumab. |
Should durvalumab be added to patients currently on, or who have just completed, a first-line chemotherapy regimen? | In patients who are currently receiving first-line chemotherapy with no evidence of disease progression, the clinical experts felt that durvalumab may be initiated in these patients. However, if patients have already completed their first-line chemotherapy regimen, durvalumab should not be added. |
System and economic issues | |
PAG has potential concerns regarding the feasibility of adoption. Are these concerns valid? | No response required. For pERC consideration. |
AoV = ampulla of Vater; BTC = biliary tract cancer; ECOG PS = Eastern Cooperative Oncology Group Performance Status; OS = overall survival; PAG = Provincial Advisory Group; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; SOC = standard of care.
Clinical Evidence
The clinical evidence included in the review of durvalumab is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence selected from the literature that met the selection criteria specified in the review. No indirect evidence was provided by the sponsor. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the Systematic Review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of durvalumab (50 mg/mL) 1,500 mg in combination with chemotherapy for the treatment of locally advanced or metastatic BTC.
Methods
Studies selected for inclusion in the Systematic Review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 4. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Of note, the systematic review protocol presented below was established before the granting of an NOC from Health Canada.
Table 4: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients with locally advanced or metastatic BTC Subgroups:
|
Intervention | Durvalumab 1,500 mg IV (plus chemotherapy) q.3.w. followed by 1,500 mg IV q.4.w. as monotherapy |
Comparator | Gemcitabine alone or in combination with cisplatin or carboplatin |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse events; BTC = biliary tract cancer; DCR = disease control rate; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EHCC = extrahepatic cholangiocarcinoma; GBC = gall bladder cancer; GI = gastrointestinal; HRQoL = health-related quality of life; IHCC = intrahepatic cholangiocarcinoma; ORR = objective response rate; OS = overall survival; PD-L1 = programmed cell death 1 ligand 1; PFS = progression-free survival; PGI-S = Patient Global Impression of Severity; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SAE = serious adverse event; TTD = time to treatment discontinuation; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.38
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Imfinzi or durvalumab and biliary cancer. The clinical trials registries searched were the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on August 10, 2022. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on December 7, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.39 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 1 study was identified from the literature for inclusion in the Systematic Review (Figure 1). That study is summarized in Table 5.
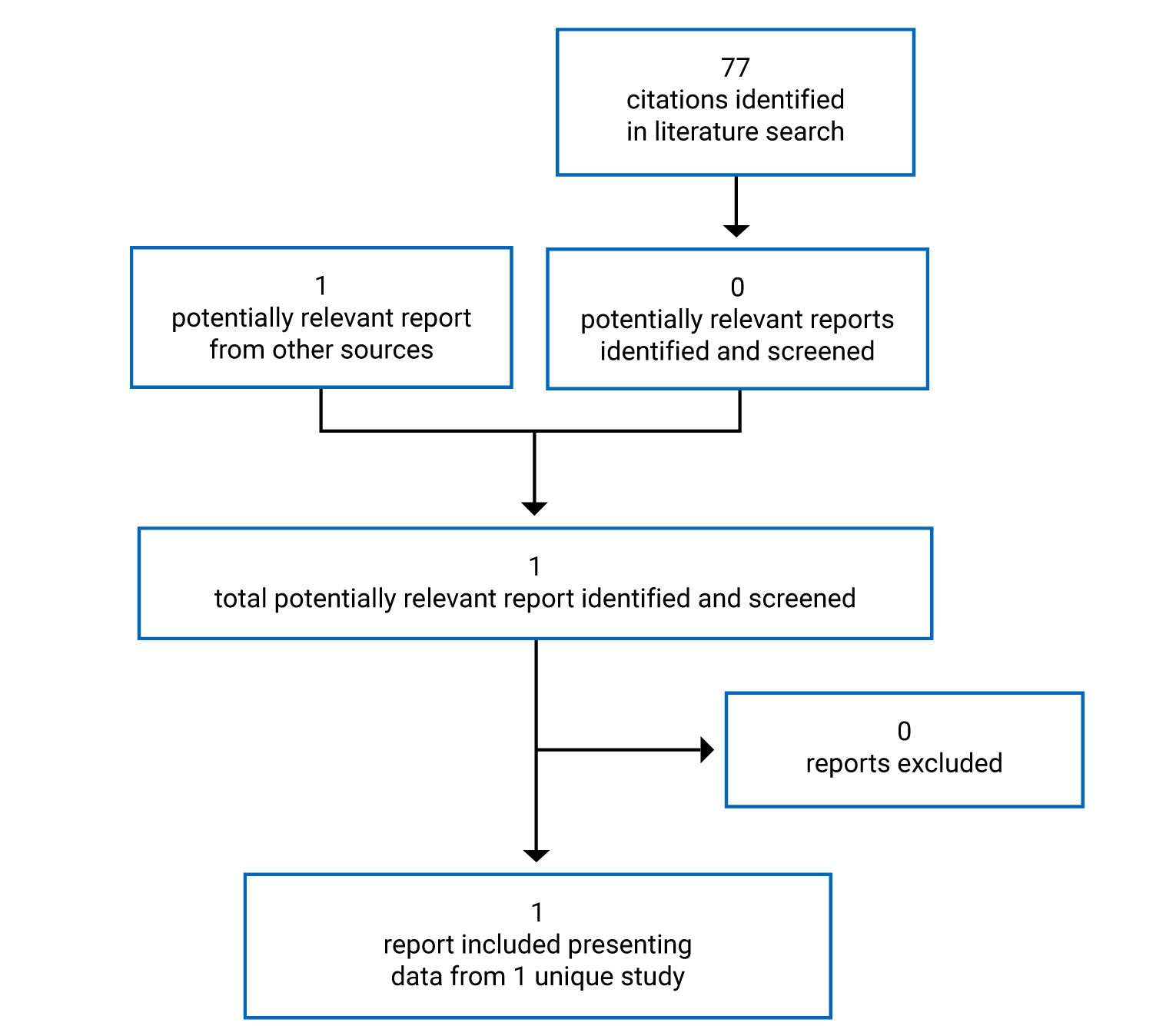
Table 5: Details of Included Studies
Detail | TOPAZ-1 Trial |
|---|---|
Designs and populations | |
Study design | Phase III, placebo-controlled, international, double-blind RCT. |
Locations | Patients enrolled in 17 countries from Asia, Europe, North America (all US), and South America. |
Patient enrolment dates | First patient enrolled: April 16, 2019 Last patient enrolled: December 11, 2020 |
Randomized (N) | 685 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Durvalumab plus gemcitabine and cisplatin: 1,500 mg durvalumab via IV infusion q.3.w., starting on cycle 1 in combination with 25 mg/m2 cisplatin, followed by 1,000 mg/m2 gemcitabine (each administered on days 1 and 8 q.3.w.) up to 8 cycles, followed by durvalumab 1,500 mg as monotherapy q.4.w. |
Comparator(s) | Placebo plus gemcitabine and cisplatin: Placebo via IV infusion q.3.w., starting on cycle 1 in combination with 25 mg/m2 cisplatin, followed by 1,000 mg/m2 gemcitabine (each administered on days 1 and 8 q.3.w.) up to 8 cycles, followed by placebo monotherapy q.4.w. |
Duration | |
Phase | |
Screening | 28 days |
Treatment | Up to eight 21-day cycles during the combination chemotherapy period, followed by 28-day cycles of durvalumab monotherapy thereafter. |
Follow-up | Until clinical progression (or RECIST 1.1-defined radiological PD), unacceptable toxicity, withdrawal of consent, or another discontinuation criterion was met. |
Outcomes | |
Primary end point | OS, defined as the time from date of randomization to date of death by any cause. |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publications | Oh et al. (2022)40 |
ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; CHF = congestive heart failure; CrCl = creatinine clearance; CSP = clinical study protocol; ctDNA = circulating tumour DNA; CTLA-4 = cytotoxic T-lymphocyte-associated antigen-4; DCR = disease control rate; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-BIL21 = European Organisation for Research and Treatment of Cancer 21-Item Cholangiocarcinoma and Gallbladder Cancer Quality of Life Questionnaire; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GI = gastrointestinal; HBc = hepatitis B core; HBV = hepatitis B virus; HCV = hepatitis C virus; HRQoL = health-related quality of life; IBD = inflammatory bowel disease; ILD = interstitial lung disease; IP = investigational product; MSI = microsatellite instability; NCI-CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; ORR = objective response rate; OS = overall survival; PD = progressive disease; PD-1 = programmed cell death 1 protein; PD-L1 = programmed cell death 1 ligand 1; PD-L2 = programmed cell death 1 ligand 2; PFS = progression-free survival; PGI-S = Patient Global Impression of Severity; PRO-CTCAE = Patient Reported Outcomes version of the Common Terminology Criteria for Adverse Events; q.3.w. = every 3 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; RECIST = Response Evaluation Criteria in Solid Tumours; SLE = systemic lupus erythematosus; TMB = tumour mutation burden; TL = target lesions; ULN = upper limit of normal; WHO = WHO.
Source: TOPAZ-1 CSR (IA-2).23
Description of Studies
One study, TOPAZ-1, was included in this review. The TOPAZ-1 trial was a double-blind, placebo-controlled, international, randomized, phase III study designed to evaluate the efficacy and safety of adding durvalumab to the established chemotherapy regimen of gemcitabine and cisplatin in patients with previously untreated, unresectable, locally advanced or metastatic BTC. The primary objective of the TOPAZ-1 trial was to confirm superiority of the combination of durvalumab plus gemcitabine and cisplatin over gemcitabine and cisplatin alone in terms of OS in patients with first-line advanced BTC.23
A total of 685 patients were centrally randomized 1:1 ratio by interactive response technology to either cisplatin 25 mg/m2 and gemcitabine 1,000 mg/m2 (each administered on days 1 and 8 q.3.w.) in combination with durvalumab 1,500 mg (n = 341) or placebo (n = 344) delivered by IV infusion (on day 1 q.3.w.); starting on cycle 1, for up to 8 cycles. After completion of the chemotherapy treatment period, patients received 1,500 mg durvalumab or placebo delivered by IV infusion every 4 weeks until clinical progression (or RECIST 1.1-defined radiological PD), unacceptable toxicity, withdrawal of consent, or another discontinuation criterion. Randomization was stratified by disease status (initially unresectable versus recurrent) and primary tumour site (IHCC versus EHCC versus GBC). The TOPAZ-1 trial enrolled patients from 17 countries,23 but no Canadian investigative sites were included.
The TOPAZ-1 trial planned to include 3 DCOs consisting of 2 interim analyses and 1 final analysis. The DCO for IA-1 was December 18, 2020. The DCO for IA-2 was August 11, 2021 (database lock: September 13, 2021). The TOPAZ-1 trial met its primary end point at IA-2, so this was considered the final formal analysis.23 A 6.5-month update of IA-2 was conducted based on a DCO of February 25, 2022 (database lock: March 22, 2022).24
Populations
Inclusion and Exclusion Criteria
The inclusion and exclusion criteria for the TOPAZ-1 trial are summarized in Table 5. Briefly, eligible patients were required to have histologically confirmed, unresectable advanced or metastatic adenocarcinoma of the biliary tract, including IHCC, EHCC, or GBC, as well as a WHO/ECOG PS of 0 or 1. Additionally, if unresectable or metastatic at diagnosis, patients were required to have untreated disease; however, patients who had received prior adjuvant therapy were permitted, provided that it was completed more than 6 months ahead of this study. Given the different genetic profile ampullary cancer, such patients were excluded to minimize diversity. Patients with active or prior autoimmune disorders were excluded because of the mechanism of action of durvalumab and the known AEs associated with immunotherapy.23
Baseline Characteristics
Baseline characteristics of patients in the TOPAZ-1 trial are summarized in Table 6. Baseline characteristics were well balanced between the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group. Patients were mostly Asian in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group (54.3% and 58.4%, respectively), with an even split between male (49.6% and 51.2%, respectively) and female (50.4% and 48.8%, respectively) patients. The median age was 64 years (range, 20 to 85 years), with most patients younger than 65 years (53.1% and 53.5%, respectively, in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group). The majority of patients had metastatic disease at baseline (88.9% and 83.1%, respectively, in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group). Across groups, the proportion of patients with a WHO/ECOG PS of 0 (50.7% and 49.3%, respectively, in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group) or 1 (47.4% and 52.6%, respectively) was well balanced. Patients were stratified by disease status (initially unresectable [80.4% and 80.2%, respectively, in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group] versus recurrent [19.6% versus 19.8%, respectively]) and by primary tumour location (IHCC [56.9% and 57.3% respectively, in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group] versus EHCC [18.2% and 18.3%, respectively] versus GBC [24.9% and 24.4%, respectively]).23
Table 6: Summary of Baseline Characteristics (IA-2; DCO of August 11, 2021)
Characteristic | TOPAZ-1 (FAS) | |
|---|---|---|
D + Gem and Cis (N = 341) | Placebo + Gem and Cis (N = 344) | |
Demographic characteristics | ||
Age (years) | ||
█████████ | █████████ | █████████ |
Median (range) | 64 (20 to 84) | 64 (31 to 85) |
█████████ | █████████ | █████████ |
█████████ | █████████ | █████████ |
█████████ | █████████ | █████████ |
█████████ | █████████ | █████████ |
Sex, n (%) | ||
Male | 169 (49.6) | 176 (51.2) |
Female | 172 (50.4) | 168 (48.8) |
Race, n (%) | ||
Asian | 185 (54.3) | 201 (58.4) |
█████████ | █████████ | █████████ |
█████████ | █████████ | █████████ |
█████████ | █████████ | █████████ |
Region, n (%) | ||
Asia | 178 (52.2) | 196 (57.0) |
Rest of the world | 163 (47.8) | 148 (43.0) |
BMI (kg/m2) | ||
█████████ | █████████ | █████████ |
Disease and clinical characteristics | ||
WHO/ECOG PS, n (%) | ||
(0) Normal activity | 173 (50.7) | 163 (47.4) |
(1) Restricted activity | 168 (49.3) | 181 (52.6) |
Overall disease classification, n (%) | ||
Locally advanced | 38 (11.1) | 57 (16.6) |
Metastatic | 303 (88.9) | 286 (83.1) |
Disease status per IVRS, n (%) | ||
Initially unresectable | 274 (80.4) | 276 (80.2) |
Recurrent | 67 (19.6) | 68 (19.8) |
Primary tumour location per IVRS, n (%) | ||
IHCC | █████████ | █████████ |
EHCC | █████████ | █████████ |
GBC | █████████ | █████████ |
PD-L1 expression, n (%) | ||
High (TIP ≥ 1%) | 197 (57.8) | 205 (59.6) |
Low/negative (TIP < 1%) | 103 (30.2) | 103 (29.9) |
Missing | 41 (12.0) | 36 (10.5) |
MSI status, n (%) | ||
High | 3 (0.9) | 2 (0.6) |
Stable | 160 (46.9) | 168 (48.8) |
Missinga | 178 (52.2) | 174 (50.6) |
BMI = body mass index; Cis = cisplatin; D = durvalumab; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EHCC = extrahepatic cholangiocarcinoma; FAS = full analysis set; GBC = gallbladder cancer; Gem = gemcitabine; IA-2 = second interim analysis; IHCC = intrahepatic cholangiocarcinoma; IVRS = Interactive Voice Response System; MSI = microsatellite instability; PD-L1 = programmed cell death 1 ligand 1; TIP = tumour and/or immune cell positivity; WHO = WHO.
aOverall, 5 of 333 (1.5%) patients with an MSI result were MSI high. MSI status missing includes MSI-unknown and not tested.
Source: TOPAZ-1 CSR (IA-2).23
Interventions
The dosing schedule of durvalumab or placebo plus gemcitabine and cisplatin is displayed in Figure 2. In the durvalumab plus gemcitabine and cisplatin arm, patients received 25 mg/m2 cisplatin and 1,000 mg/m2 gemcitabine administered on days 1 and 8 every 3 weeks plus 1,500 mg durvalumab starting on cycle 1, for a maximum of 8 cycles, delivered by IV infusion every 3 weeks. Patients randomized to the comparator arm received 25 mg/m2 cisplatin and 1,000 mg/m2 gemcitabine administered on days 1 and 8 every 3 weeks plus placebo delivered by IV infusion every 3 weeks starting on cycle 1, for a maximum of 8 cycles. During day 1 visits of each cycle, administration of durvalumab or placebo occurred first, followed by chemotherapy. Given the potential disadvantages, including cumulative toxicities, continuing chemotherapy after 8 cycles was not allowed in the TOPAZ-1 trial. After completing the gemcitabine and cisplatin treatment period (first 8 cycles), patients received 1,500 mg durvalumab or placebo as monotherapy delivered by IV infusion every 4 weeks until clinical progression (or RECIST 1.1-defined radiological PD), unacceptable toxicity, withdrawal of consent, or any other discontinuation criterion was met.23
Patients, investigators, and study centre staff were blinded to treatment allocation; IV solutions of durvalumab and placebo were blinded using a translucent-coloured or opaque sleeve, fastened with tamper-evident tape to maintain the double-blind conditions. An unblinded pharmacist prepared the durvalumab and placebo for patients as specified by the randomization scheme and IVRS. Investigators were unblinded to treatment allocation only in cases of medical emergencies or, at progression of disease, patients may have been unblinded at the request of the investigator to inform decisions regarding subsequent anticancer treatment. After the final analysis, open-label durvalumab was provided up to the time that patients discontinued treatment.23
Figure 2: Durvalumab or Placebo Plus Gemcitabine and Cisplatin Dosing Schedule

d/c = discontinuation; PD = progressive disease; q3w = every 3 weeks; q4w = every 4 weeks; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: TOPAZ-1 CSR (IA-2).23
If a patient’s weight fell to 30 kg or below, the patient received weight-based dosing equivalent to 20 mg/kg of durvalumab (or placebo) every 3 weeks (i.e., with chemotherapy) or every 4 weeks (i.e., during monotherapy) after consultation between the investigator and the study physician, until the patient’s weight increased to more than 30 kg, at which point the patient was to start receiving the fixed dosing of durvalumab 1,500 mg (or placebo) every 3 weeks or every 4 weeks.23
Dose Modifications, Reductions, or Interruptions
Local standard clinical practice regarding dose modifications were followed for the combination of gemcitabine and cisplatin. Gemcitabine and cisplatin may have been delayed and subsequently resumed, per local standard clinical practice. If dosing was delayed for reasons other than causally related toxicity, dosing occurred as soon as possible. Dose reductions were permitted for gemcitabine and cisplatin. For patients who discontinued gemcitabine and cisplatin before completion of cycle 8 due to toxicity, treatment could continue with durvalumab or placebo monotherapy (q.4.w.) at the investigator’s discretion once toxicity resolved to grade 2 or less.23
Dose reductions were not permitted for durvalumab (or placebo). Guidelines for the management of imAEs, IRRs, and nonimmune-mediated reactions for durvalumab were provided in the dosing modification and toxicity management guidelines. During the first 8 cycles of durvalumab or placebo in combination with gemcitabine and cisplatin, if durvalumab or placebo was discontinued due to toxicity related to either drug, the patient was discontinued from study treatment, completed the treatment discontinuation visit, and entered the follow-up period. Subsequent anticancer treatment was to be held until the toxicity resolved to grade 2 or less. At the discretion of the investigator, gemcitabine and cisplatin could be continued as the postdiscontinuation anticancer treatment.23,41
Treatment Discontinuation and Withdrawal From Study
Patients were discontinued from study treatment for the following reasons23,41:
Withdrawal of consent from further treatment. Patients were free to discontinue treatment or withdraw from the study at any time. Patients who discontinued treatment were to continue to participate in the study for safety and survival follow-up, unless consent was specifically withdrawn for all further participation in any study procedures and assessments.
An AE that, in the opinion of the investigator or the sponsor, contraindicated further dosing.
Any AE that met the criteria for discontinuation as defined in the dosing modification and toxicity management guidelines or as defined in the local prescribing information for gemcitabine and cisplatin.
Pregnancy or intent to become pregnant.
Noncompliance with the study protocol that, in the opinion of the investigator or sponsor, warranted withdrawal from treatment (e.g., refusal to adhere to scheduled visits).
Initiation of alternative anticancer therapy, including another investigational drug.
Clinical progression or radiological progression and investigator determination that the patient is no longer benefiting from treatment.
Patients who permanently discontinued treatment for reasons other than RECIST 1.1-defined radiological PD were to continue to have RECIST scans performed every 6 weeks (± 1 week) for the first 24 weeks (relative to the date of randomization) and then every 8 weeks (± 1 week) thereafter (relative to the date of randomization) until PD plus at least 1 additional follow-up scan or death (whichever came first).23,41
Treatment Through Progression
During the treatment period, patients who were clinically stable at an initial RECIST 1.1-defined radiological PD could continue to receive study treatment at the discretion of the investigator (with input from the patient) if they were deemed to be receiving clinical benefit. For these patients, imaging for tumour assessments were to be continued at a maximum scan interval of 8 weeks for the duration of treatment unless there was unacceptable toxicity, withdrawal of consent, or another discontinuation criterion was met.23,41
Patients with RECIST 1.1-defined radiological PD who continued to receive their assigned treatment at the discretion of the investigator (with input from the patient) could receive treatment until it no longer provided clinical benefit, and imaging for tumour assessments were continued at a maximum scan interval of 8 weeks for the duration of treatment unless there was unacceptable toxicity, withdrawal of consent, or another discontinuation criterion is met. For patients who discontinued treatment due to radiological PD, 1 additional follow-up scan was performed at the next scheduled imaging visit and no less than 4 weeks after the prior assessment of PD. All patients were followed for survival until the end of the study.23,41
Patients with rapid tumour progression or with symptomatic progression that required urgent medical intervention (e.g., central nervous system metastasis, respiratory failure due to tumour compression, or spinal cord compression) were not eligible to continue durvalumab (or placebo). For all patients who were treated through progression, the investigator was to ensure that the patients did not have any significant, unacceptable, or irreversible toxicities that indicated that continued treatment would not further benefit them.23,41
Crossover from the placebo plus gemcitabine and cisplatin arm to the durvalumab plus gemcitabine and cisplatin arm was not permitted.23,41
Concomitant and Rescue Therapy
Concomitant medications or treatments included those that were deemed necessary to provide adequate prophylactic or supportive care (e.g., acetaminophen or diphenhydramine); best supportive care (including antibiotics, nutritional support, correction of metabolic disorders, optimal symptom control, and pain management) and inactivated viruses (i.e., the influenza vaccine) were permitted for use, when necessary, for all patients.23,41
Restricted, prohibited, and permitted concomitant medications included:23,41
any investigational anticancer therapy, monoclonal antibodies against CTLA-4, PD-1, or PD-L1 other than those under investigation in this study were not to be given while the patient was on study treatment.
any concurrent chemotherapy, radiotherapy, immunotherapy, or biologic or hormonal therapy for cancer treatment other than those under investigation in this study. Concurrent use of hormones for noncancer-related conditions (e.g., insulin for diabetes and hormone replacement therapy) was acceptable. Local treatment of isolated lesions, excluding target lesions, for palliative intent was acceptable (e.g., radiotherapy for bone metastasis).
live attenuated vaccines, which should not be administered until 30 days after the last dose of investigational product.
immunosuppressive medications, including but not limited to systemic corticosteroids at doses exceeding 10 mg/day of prednisone or equivalent, methotrexate, azathioprine, and tumour necrosis factor-alpha blockers, which should not be given concomitantly or used for premedication before the intraosseous infusions.
epidermal growth factor receptor tyrosine kinase inhibitors, which should not be given concomitantly and should be used with caution in the 90 days after the last dose of durvalumab, owing to the potential for pneumonitis and transaminase increases.
herbal and natural remedies, which may have immune-modulating effects and should not be given concomitantly.
Rescue medications (steroids and other immunosuppressants) were available to manage imAEs experienced by patients receiving durvalumab. The permitted immunosuppressants were infliximab (e.g., for colitis) and mycophenolate (e.g., for hepatitis). Blinded and unblinded access and notifications were to be controlled using the IVRS or IWRS.23
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 7 and is summarized in the subsequent text. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 3.
Table 7: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | TOPAZ-1 |
|---|---|
OS | Primary |
PFS | Secondary |
ORR | Secondary |
BOR | Secondary |
DOR | Secondary |
DCR | Secondary |
HRQoL (EORTC QLQ-BIL21 and EORTC QLQ-C30) | Secondary |
HRQoL (EQ-5D-5L) | Exploratory |
Symptom severity (PGI-S) | Exploratory |
Treatment tolerability (PRO-CTCAE and EORTC QLQ-BIL21 item 49) | Exploratory |
Harms | Secondary |
BOR = best overall response; DCR = disease control rate; DOR = duration of response; EORTC QLQ-BIL21 = European Organisation for Research and Treatment of Cancer 21-Item Cholangiocarcinoma and Gallbladder Cancer Quality of Life Questionnaire; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level ED-EQ; HRQoL = health-related quality of life; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PGI-S = Patient Global Impression of Severity; PRO-CTCAE = PRO-CTCAE = Patient Reported Outcomes version of the Common Terminology Criteria for Adverse Events.
Efficacy Outcomes
In line with the primary objective of the TOPAZ-1 study, the primary end point was OS. PFS was a key secondary end point of the TOPAZ-1 trial. Other secondary end points in the TOPAZ-1 trial consisted of ORR, DOR, and DCR, safety, and HRQoL as measured by EORTC QLQ-C30 and EORTC QLQ-BIL21. Exploratory outcomes included symptom severity as measured by the PGI-S, treatment tolerability as measured by the PRO-CTCAE, and HRQoL as measured by the EQ-5D-5L.23
The analysis of secondary end points of PFS, ORR, DOR, and DCR were conducted by investigator RECIST 1.1 assessments. For IA-1 to support early registration, a blinded independent central review (BICR) was performed for the end points of ORR and DOR for the first 200 randomized patients for at least 32 weeks. After IA-1, all images were collected and stored for potential BICR if considered necessary by the sponsor.23
Overall Survival
The primary outcome of the TOPAZ-1 trial, OS, was defined as the time from the date of randomization until death from any cause. Any patient not known to have died at the time of analysis was censored on the last recorded date on which the patient was known to be alive.23
Progression-Free Survival
PFS was a key secondary end point of the TOPAZ-1 trial and was defined as the time from the date of randomization until the date of RECIST 1.1-defined radiological PD or death, regardless of whether the patient withdrew from therapy or received another anticancer therapy before progression. Patients who had not progressed or died at the time of analysis were censored at the time of the most recent date of assessment after their last evaluable RECIST 1.1 assessment. However, if the patient progressed or died after 2 or more missed visits, the patient was censored at the time of the most recent evaluable RECIST 1.1 assessment before the 2 missed visits.23
Objective Response Rate
The secondary end point of ORR, defined as the percentage of patients with confirmed CR or partial response (PR), per investigator, according to RECIST 1.1 criteria. The ORR was analyzed using all scans, regardless of whether they were scheduled or not. Data obtained up to progression, or the most recent evaluable assessment in the absence of progression, were included in the assessment of ORR. Patients who discontinued treatment without progression, received a subsequent anticancer therapy, and then responded were not considered responders in the ORR. The ORR was also defined using the BICR data (at IA-1 only) to establish a visit response of CR or PR, with the denominator being a subset of all randomized patients with measurable disease at baseline, per BICR.23
Duration of Response
The DOR, per investigator assessment, was a secondary end point of the TOPAZ-1 trial and was defined as the time from the date of first documented response to the date of documented progression or death in the absence of disease progression. For patients with a measurable disease at baseline, DOR was summarized by treatment group. At IA-1, DOR was also defined using BICR data.23
Disease Control Rate
The DCR, per investigator assessment, was defined as the rate of best objective response of no evidence of disease, CR, PR, or stable disease, according to RECIST 1.1. The DCR was summarized by treatment for the overall treatment duration at 24 weeks, 32 weeks, and 48 weeks. For IA-1, the DCR was also assessed by BICR.23
HRQoL and Patient-Reported Outcomes
PRO assessments were completed using electronic case report forms at the study site. Questionnaires were completed before treatment dosing and before any other study procedures in the following order: EORTC QLQ-C30, EORTC QLQ-BIL21, PGI-S, EQ-5D-5L, and PRO-CTCAE.23
EORTC QLQ-C30 and EORTC QLQ-BIL21
The EORTC QLQ-C30 assesses the impact of treatment on disease-related symptoms, impact of the disease, and HRQoL. It includes 30 items and assesses HRQoL and health status through 5 functional scales (physical, role, cognitive, emotional, and social), 3 symptom scales (fatigue, pain, and nausea and vomiting), and 1 GHS/QoL scale. It also includes 6 single-item symptom and impact measures: dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties. A high score on a functional or GHS/QoL scale indicates a high level of functioning or global HRQoL, whereas a high score on a symptom scale or item indicates a high level of symptom burden.23,42
The EORTC QLQ-BIL21 was developed to measure QoL specifically for patients with BTC. It consists of 21 questions: 3 single-item assessments related to treatment side effects, difficulties with drainage bags or tubes, and concerns regarding weight loss; and 18 items grouped into 5 symptom scales (pain [4 items], tiredness [3 items], jaundice [3 items], anxiety [4 items], and eating [4 items]). The EORTC QLQ-BIL21 has demonstrated content validity in BTC patients. It was developed based on a literature review, in-depth patient interviews, and health care provider interviews specific to BTC populations.43 Its measurement properties have also been tested in BTC populations and include evidence of acceptable internal consistency reliability, test-retest reliability (reproducibility), and construct validity.23,44
A clinically meaningful change or difference on the EORTC QLQ-C30 and EORTC QLQ-BIL21, based on patients with breast or lung cancer, was defined in the TOPAZ-1 trial as an absolute change in score from baseline of at least 10 points;23,45 however, no minimally important difference (MID) for the EORTC QLQ-C30 or EORTC QLQ-BIL21 for patients with BTC was identified in the literature by the CADTH review team (Appendix 3).
Five-Level EQ-5D
The EQ-5D-5L is a generic questionnaire that provides a simple descriptive profile of health and a single index value for health status. The EQ-5D-5L questionnaire comprises 5 questions that cover 5 dimensions of health: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Respondents also assess their health today using the EQ-visual analogue scale (VAS), which ranges from 0 (worst imaginable health) to 100 (best imaginable health).23 No evidence of the validity, reliability, or responsiveness of the EQ-5D-5L, or of an MID among patients with BTC was identified in the literature by the CADTH review team (Appendix 3).
Symptom Severity
Patient Global Impression of Severity
The PGI-S item is included to assess how patients perceive the overall severity of their cancer symptoms over the previous 7 days. It is a single-item questionnaire, in which patients choose response options on a 6-point scale that range from no symptoms to very severe.23 No evidence of the validity, reliability, or responsiveness of this single item, or of an MID among patients with BTC was identified in the literature by the CADTH review team (Appendix 3).
Treatment Tolerability
PRO-CTCAE and EORTC QLQ-BIL21 Item 49
Patient-reported treatment tolerability was explored using the PRO-CTCAE and EORTC QLQ-BIL21 item 49. The PRO-CTCAE is a tool developed by the National Cancer Institute to address tolerability from the patients’ perspective. The PRO-CTCAE is an item-bank of symptoms. Six different symptomatic AEs were selected for the PRO-CTCAE, based on available safety reports of durvalumab and gemcitabine and cisplatin. Generally, symptomatic AEs that were not included in the EORTC were prioritized for the PRO-CTCAE to reduce patient burden, per published recommendations.23,46 No evidence of the validity, reliability, or responsiveness of the PRO-CTCAE, nor of an MID among patients with BTC was identified in the literature by the CADTH review team (Appendix 3). The psychometric properties of the PRO-CTCAE were evaluated in 940 adults with various cancer types.47
Item 49 of the EORTC QLQ-BIL21 assesses a patient’s global impression of treatment tolerability with the question, “To what extent have you been troubled with side effects from your treatment?” Item 49 was included to complement the findings of the PRO-CTCAE.23,41
Harms Outcomes
Safety and tolerability were assessed in terms of AEs, SAEs, deaths, physical examinations, laboratory findings, WHO/ECOG PS, vital signs, electrocardiograms, and exposure, which were collected for all patients. AEs were defined as an undesirable medical condition (other than the progression of the malignancy under evaluation) or the deterioration of a preexisting medical condition after or during exposure to a pharmaceutical product, whether or not it was considered causally related to the product. The severity of AEs was defined according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0.23,48
Statistical Analysis
Sample Size and Power Calculation
Approximately 672 patients were to be randomized in a 1:1 ratio to either durvalumab plus gemcitabine and cisplatin or placebo plus gemcitabine and cisplatin in the global cohort. Approximately 130 patients from China were to be randomized. Once the global cohort was closed, sites in China continued recruitment until 130 patients were randomized.23,41
The TOPAZ-1 trial was powered to demonstrate the superiority in OS of durvalumab plus gemcitabine and cisplatin over placebo plus gemcitabine and cisplatin in patients previously untreated for unresectable, locally advanced or metastatic BTC.23,41
The hypothesis of improved OS was to be tested when:23,41
approximately 397 OS events had occurred across both groups (59% maturity) (IA-2), and
approximately 496 OS events had occurred across both groups (74% maturity) (final analysis).
The primary analysis of OS was based on a log-rank test for the interim analysis and a Fleming-Harrington (FH) (0,1) test for the final analysis. The log-rank test was also performed at the final analysis as a sensitivity analysis.23,41
Sample size calculations were performed under the assumption that the true average HR for OS is 0.745, corresponding to an approximate 4-month improvement in median OS over the control group of 11.7 months. Using a 2-sided log-rank test for the final analysis, approximately 496 OS events were estimated to provide 90% power to demonstrate statistical significance at the 2-sided level of 4.20%. The 4.9% (2-sided) alpha allocation for the OS analysis was controlled at IA-2 and final analysis using the Lan-DeMets spending function that approximates the O’Brien-Fleming approach, in which the significance level applied depends on the information fraction. The smallest treatment difference that could be statistically significant at the final analysis was an HR of 0.833 using log-rank test, or a 2.3-month improvement in median OS. With a planned 21-month nonlinear recruitment period, with accrual weight equal to 1.5 and a minimum follow-up period of 19 months assumed, it was anticipated that the final analysis will be performed approximately 40 months after the first patient is randomized. With a log-rank test at IA-2 and a FH(0,1) test at the final analysis, the overall power was at least 86%, based on an assumed average HR of 0.745 under the assumption of proportional hazards or up to a 6-month delayed effect (i.e., delayed separation of the OS curves by up to 6 months).23,41
Interim and Final Analyses
Two interim analyses and 1 final analysis were planned for the evaluation of efficacy.23,41
IA-1: The objective of IA-1 was to assess clinical activity with ORR and DOR to support early registration of durvalumab plus gemcitabine and cisplatin using both investigator assessments and BICR assessments according to RECIST 1.1. The minimum efficacy criterion for IA-1 was that the lower bound of the 2-sided exact 95% CI for ORR in the durvalumab plus gemcitabine and cisplatin arm was higher than the ORR point estimate in the placebo plus gemcitabine and cisplatin arm. The planned DCO for IA-1 was to occur when at least 200 patients (i.e., approximately 100 per arm) completed at least 32 weeks of follow-up. No formal comparison or statistical testing between arms was performed, although a nominal significance level of 0.001 was allocated to IA-1. Descriptive summaries of ORR, including a 2-sided exact Clopper-Pearson 95% CI, were presented by treatment group. Further descriptive summaries of ORR were produced by subgroups based on confirmed BICR assessments.23,41
IA-1 was conducted in March 2021 and included 369 randomized patients who had had an opportunity to be followed up for at least 32 weeks. At the time of IA-1, the Independent Data Monitoring Committee determined that an acceptable safety profile was observed and the prespecified efficacy criteria for ORR had been met. The study blinding was maintained for all study personnel until full unblinding, at the time of IA-2.23,41
IA-2: IA-2 tested for early superiority of durvalumab plus gemcitabine and cisplatin relative to placebo plus gemcitabine and cisplatin. This analysis was performed when approximately 80% of the final number of OS events was expected to be reached (approximately 397 of 496 OS events). The 2-sided alpha level allocated to OS was 0.049 and was controlled at the interim and final analyses, accounting for the correlation structure between the test statistics at IA-2 and the final analysis. The significance level for the primary confirmatory OS analysis at the final analysis was determined based on the actual alpha spending at IA-2 and the correlation structure between the IA-2 log-rank statistic and the final analysis FH(0,1) statistic. As a secondary evaluation, PFS was also analyzed at the time of OS for IA-2 and the final analysis, but only if superiority is confirmed with OS. The alpha level for PFS was controlled at the interim and final analysis using the Lan-DeMets spending function that approximates a Pocock approach. The significance levels for the PFS analyses using the log-rank test were calculated by specifying the information fraction for each analysis. The information fraction was calculated as the number of PFS events at the analysis time point divided by the total number of events at the final analysis time point.23,41
Results from IA-2, a 6.5-month update for additional follow-up on OS, and safety outcomes are included in this report.
Statistical and Analytical Plans
Primary Efficacy End Point Analyses
For the primary end point of OS, the stratified log-rank test (adjusted for disease status and primary tumour location as entered into the IVRS or IWRS at randomization) was used for statistical inference, the stratified Cox proportional hazards model was used for quantification of the treatment effect under the assumption of proportional hazards with Efron ties, and the CI was calculated using the profile likelihood approach. Kaplan-Meier plots of OS were presented by treatment, and median OS and estimated OS rates at 12, 18, and 24 months were presented.23
Because of the mechanism of action of immunotherapy, delayed separation of survival curves may be observed between groups in clinical trials, violating the proportional hazards assumption. The standard log-rank test, although optimal under proportional hazards, suffers substantial power loss in handling survival data with delayed separation of survival curves. At IA-2, the proportional hazards assumption was assessed by examining plots of complementary log-log (event times) versus log (time) and, if concerning, by fitting a time-dependent covariate (adding a treatment-by-time or treatment-by-ln[time] interaction term) to assess the extent to which this represents random variation. As a lack of proportionality was evident, the variation in treatment effect was described by presenting piecewise HR (Cox modelling) calculated over distinct time periods.23
Sensitivity Analyses: No adjustment to the significance level for testing of the sensitivity analyses was made. Sensitivity analysis was conducted by examining censoring patterns to rule out attrition bias using a Kaplan-Meier plot of time to censoring, in which the censoring indicator of OS was reversed. The number of patients prematurely censored (i.e., the last known alive date is prior to DCO) was summarized by treatment group. Additionally, median durations of follow-up were provided for censored patients alive at DCO (i.e., time from randomization to date of censoring [i.e., date last known to be alive]) for each group.23
Additional sensitivity analysis using Cox proportional hazards modelling, which contained treatment, the stratification factors, and the covariates of age, sex, race, ECOG PS, and BTC stage (locally advanced versus metastatic), were used to assess the effect of covariates on the HR estimate interactions between treatments, and stratification factors were also tested to rule out any qualitative interaction using the approach of Gail and Simon (1985).23,49
Subgroup Analyses: Comparisons of efficacy between durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin were performed for the following prespecified subgroups of interest in the full analysis set (FAS):23
sex (male versus female)
age at randomization (< 65 years versus ≥ 65 years)
PD-L1 expression
disease status (initially unresectable versus recurrent)
primary tumour location (IHCC versus EHCC versus GBC)
race (Asian versus non-Asian)
region (Asia versus rest of the world)
WHO/ECOG PS (0 versus 1 at screening)
locally advanced versus metastatic BTC.
For these subgroup analyses, any patient with missing values was excluded from that particular subgroup. No adjustment to the significance level for testing of the subgroups was conducted, and all analyses were considered supportive of the analysis of OS and PFS. For each subgroup, the HR and 95% CI were calculated from a Cox proportional hazards model that only contained a term for treatment.23
To assess the consistency of the treatment effect between subgroups for plausible subgroups, the presence of quantitative interactions between treatment and stratification factors was checked using an overall global interaction test (at the 2-sided 10% significance level). Where statistically significant, an attempt to determine the cause and type of interaction was made using stepwise backward selection on the saturated model until a final model was reached in which all included interactions were significant and all excluded interactions were nonsignificant. All main effects were included in the model, regardless of whether the corresponding interaction term was still present to identify factors that independently alter the treatment effect and prevent identification of multiple correlated interactions. Quantitative interactions identified using this procedure were then tested to rule out any qualitative interaction using the approach of Gail and Simon (1985).23,49
Secondary Efficacy End Point Analyses
The analysis of the secondary efficacy end points of PFS, ORR, DOR, and DCR were based on investigator assessments using RECIST 1.1.23
Progression-Free Survival: The analysis of the key secondary end point of PFS was based on RECIST 1.1, per investigator assessments, using a methodology similar to that described for OS, including estimated PFS rates at 6, 9, 12, and 24 months, using the Kaplan-Meier curves. The assumption of proportional hazards was examined, and other sensitivity analyses were performed analogous to OS. An additional sensitivity analysis of PFS was performed to assess possible evaluation-time bias that could be introduced if scans were not performed at the protocol-scheduled time points. The midpoint between the time of progression and the previous evaluable RECIST 1.1 assessment was analyzed using a stratified log-rank test, per the main PFS analysis. For patients whose death was treated as a PFS event, the date of death was used to derive the PFS time used in the analysis.23
Attrition bias was assessed by repeating the PFS analysis, except that the actual PFS event times, rather than the censored times, of patients who progressed or died in the absence of progression immediately after 2 or more nonevaluable tumour assessments were included. In addition, patients receiving subsequent therapy before progression or death were censored at their last evaluable assessment before receiving the subsequent therapy; this was further supported by a Kaplan-Meier plot of the time to censoring, in which the censoring indicator of the PFS analysis was reversed. Cox proportional hazards modelling (conducted analogous to OS) was used to assess the effect of covariates on the HR estimate. Subgroup analyses comparing PFS (per RECIST 1.1 using investigator assessments) were conducted analogous to OS.23
Objective Response Rate: ORR was the primary end point for IA-1. The ORR, using the BICR data (at IA-1 only) to define a visit response of CR or PR, was analyzed for the FAS at 32 weeks. A comparison of confirmed best overall response (BOR) for BICR versus investigator assessment was provided.23
At IA-2, the ORR was compared between treatment groups using a stratified Cochran-Mantel Haenszel test as the primary test. That test used the same stratification factors as for OS, and results were presented in terms of OR and P value.23
As a sensitivity analysis, ORR was analyzed using logistic regression models (adjusted for the same stratification factors as for OS) and presented in terms of OR (OR greater than 1 favouring treatment with durvalumab plus gemcitabine and cisplatin) together with its associated profile likelihood 95% CI and P value. The ORR analysis was performed in the subset of patients in the FAS who had measurable disease at baseline, with a sensitivity analysis based on the complete FAS. Additionally, for each treatment group, BOR was summarized by n (%) for each category (CR, PR, stable disease, PD, no evidence of disease, and not evaluable).23
Subgroup analyses for ORR were performed using the subgroups specified for OS.23
Duration of Response: Kaplan-Meier plots of DOR (per investigator) were presented, along with median DOR. In addition, the number of censored responders (split by lost to follow-up and still in response), number (%) of patients with a DOR of at least 3 months, at least 6 months, at least 9 months, and at least 12 months, and the time to onset of response from randomization were summarized.23
Further descriptive summaries of DOR were produced by subgroups (disease status, primary tumour location, and region). Swimmer plots that show the profile of each responder were also produced. Subgroup analyses were performed based on geographic region, primary tumour location, and disease status.23
Health-Related Quality of Life: The PRO analyses for EORTC QLQ-C30 and EORTC QLQ-BIL21 included time to deterioration (using a stratified log-rank test that provided a P value and a stratified Cox proportional hazard model that provided HR and 95% CI), adjusted mean change from baseline score (using a mixed-effect model for repeated measures that provided an estimate of the treatment difference, 95% CI, and P value), and BOR (using a logistic regression model that provided OR, 95% CI, and P value). Visit responses (improvement, no change, and deterioration) and visit-specific and overall scores (across all visits) were summarized descriptively. Deterioration and improvement were defined as an increase or decrease of at least 10 points, respectively, on symptom scales and items. Changes of less than 10 points were considered to be no change.23
For the EQ-5D-5L, descriptive statistics (e.g., n, mean, median, standard deviation, minimum, and maximum) were reported for the EQ-5D index score and the EQ-VAS score, as were the change from baseline for the EQ-5D index score and the EQ-VAS score.23
Symptom Severity (PGI-S): Data for the PGI-S were summarized descriptively as the number of patients and corresponding percentages for each category in the questionnaire at each visit by treatment group.23
Treatment Tolerability (PRO-CTCAE, QLQ-BIL21 Item 49): Data for the PRO-CTCAE and EORTC QLQ-BIL21 were summarized descriptively.23
Safety Analyses
Safety data were not formally analyzed but were summarized descriptively according to the treatment received.23
Multiple Testing
The objective of IA-1 was to evaluate the efficacy of durvalumab plus gemcitabine and cisplatin in terms of clinical activity based on all patients in the global cohort randomized at least 32 weeks before the IA-1 DCO (December 18, 2020). A small alpha expenditure of 0.001 was allocated to IA-1 for ORR.23,41
Multiplicity adjustment was incorporated in IA-2 and the final analysis of the primary end point (OS) and the selected key secondary end point (PFS). For the planned OS analysis, control of the family-wise error rate at the remaining 4.9% level (2-sided) was achieved with a combined approach of alpha allocation to the OS analyses (IA-2 and the FA) using the O’Brien-Fleming alpha spending function and a hierarchical testing procedure; that is, PFS was to be tested only if OS met statistical significance at IA-2 or the final analysis.23,41
The IA-2 OS analysis was conducted when 424 of the 496 expected OS events at the final analysis (61.9% overall maturity for OS) had occurred, using the Lan-DeMets spending function, approximating O’Brien-Fleming boundaries, to ensure strong control of the type I error (with a 2-sided interim P value of < 0.0300) for the treatment comparison. As OS met statistical significance at IA-2, significance levels for PFS at IA-2 for the log-rank test were derived based on the Lan-DeMets alpha spending function, approximating Pocock boundaries, which strongly controls the type I error at the 0.049 level (2-sided).23,41
Analysis Populations
The following analysis populations of interest were defined in the TOPAZ-1 trial:23
FAS, which consisted of all randomized patients and was analyzed on an intention-to-treat basis. The FAS was used for all efficacy analyses.
Safety analysis set, which consisted of all patients who received at least 1 dose of the study treatment. Patients were analyzed according to the treatment they received.
PRO analysis set, which was defined as all patients from the FAS except those with no questionnaire translation available or those who did not complete the questionnaire because of other physical or language reasons (included with statistical analysis plan (SAP) amendment on June 7, 2021).
Protocol Amendments, Deviations, and Changes to Planned Analyses
Protocol Amendments
The original clinical study protocol was finalized on December 19, 2018. There were subsequently 6 global protocol amendments, for a total of 7 versions. Changes to the study protocol after the initiation of patient recruitment are summarized in Table 8. It was unclear how many patients were enrolled at each protocol amendment; therefore, the impact of potential biases due to protocol amendments remains unknown. According to the investigators, none of the protocol amendments affected the conduct or integrity of the study, nor did it affect the patients’ safety.23,41
Table 8: Important Amendments to the Original Study Protocol After the Start of Patient Recruitment
Amendment number (date) | Key changes |
|---|---|
Version 2 (March 21, 2019) |
|
Version 3 (September 18, 2019) | Objective and end point:
Inclusion criteria:
Exclusion criteria:
Patient enrolment and randomization:
Removal of the requirement that the sponsor must be notified before the blind is broken. Addition of exploratory genetic testing |
Version 4 (April 17, 2020) | Sample size determination:
Interim analyses:
|
Version 5 (October 7, 2020) | Version 5 was not implemented because of text formatting errors found after document finalization; Therefore, version 6 follows version 4 |
Version 6 (October 28, 2020) | Update of the information fraction for OS at IA-2 to 80% and the related numbers (number of events, maturity, significance levels, critical HR and timing of IA-2), accordingly |
Version 7 (March 1, 2021) | Statistical considerations:
Addition of subsection detail for mitigation in the case of study disruption due to civil crisis, natural disaster, or public health crisis (i.e., COVID-19 pandemic) Addition of appendix details for mitigation in the case of study disruption due to civil crisis, natural disaster, or public health crisis (i.e., COVID-19) (not applicable for IA-1 CSR, as it was implemented after the DCO) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; CMH = Cochran-Mantel Haenszel; CSR = Clinical Study Report; ctDNA = circulating tumour DNA; DCO = data cut-off; FA = final analysis; FH = Fleming-Harrington; HR = hazard ratio; IA-1 = first interim analysis; IA-2 = second interim analysis; MSI = microsatellite instability; ORR = objective response rate; OS = overall survival; PD = progressive disease; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; TMB = tumour mutation burden.
Sources: TOPAZ-1 Protocol and CSR (IA-2).23,41
Protocol Deviations
The number of patients with important protocol deviations in each treatment group are summarized in Table 9. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ████████ ███ █ ██ ███ ███████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ████████ ██ ███ ███ █████ █ ██ █████ █████ ███████ ██████ ███ █████████ ████ ██████████ ██ ████ ██ ██████ ██████ ████ ████ ███ ██ ██ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ███ ██ ████ █ ██ ██████ ████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ████ ██████ ██ █ ████ ████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █████ █████ ██ █ ███ █████ ██████ █████ ███████ █████████ ████████
Table 9: Important Protocol Deviations (FAS)
Protocol deviations | D + Gem and Cis (N = 341) | PBO + Gem and Cis (N = 344) |
|---|---|---|
██████████████████████████████████ | ██████████████ | ██████████████ |
██████████████████████████████████ | ██████████████ | ██████████████ |
██████████████████████████████████ | ██████████████ | ██████████████ |
██████████████████████████████████ | ██████████████ | ██████████████ |
██████████████████████████████████ | ██████████████ | ██████████████ |
██████████████████████████████████ | ██████████████ | ██████████████ |
██████████████████████████████████ | ██████████████ | ██████████████ |
Cis = cisplatin; D = durvalumab; FAS = full analysis set; Gem = gemcitabine; PBO = placebo; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
█████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ █████ ██ ████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ██ ██████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ███ █ ██ ██████████ ███████ █████████ █████████ ████
Source: TOPAZ-1 CSR (IA-2).23
Changes to Planned Analyses
Changes to the SAP between August 2020 to October 2021 (SAP Amendments version 2 to version 6) were implemented to align with the clinical study protocol. Deviations from the planned analysis in the protocol included the following:23
all PRO analyses were performed on the PRO analysis set and not the FAS, as stated in protocol
no subgroup analysis was performed for DCR
no sensitivity analysis was carried out for ORR or DOR for patients with 32 weeks of follow-up as assessed by BICR, as BICR data were only collected until IA-1.
After unblinding of the study data, the following posthoc analyses were conducted as supplemental analyses.23,41
For OS:
The plot of kernel-smoothed estimates of the hazard functions versus time to show the departure from proportional hazards.
OS by confirmed ORR (RECIST 1.1, per investigator).
A forest plot of HR by additional PD-L1 tumour and/or immune cell positivity cut-offs (5% and 10%) was produced.
Forest plot of HR for ORR by subgroup to visually present this prespecified subgroup analysis.
A waterfall plot for DOR.
Demographic and disease characteristics (including stratification factors) presented by region (Asia versus rest of the world).
Time to treatment discontinuation for patients with any causally related AE leading to discontinuation of any study treatment.
The potential for hepatitis reactivation was evaluated by shifts in viral load for patients with hepatitis B or C.
Additional by-patient data to provide clarity around certain cases.
Results
Patient Disposition
TOPAZ-1 was a randomized, phase III clinical trial. Table 10 summarizes the disposition of enrolled patients at IA-2 (DCO of August 11, 2021) and as of the 6.5-month update (DCO of February 25, 2022). A total of 914 patients were screened for eligibility and 685 were randomized to receive durvalumab plus gemcitabine and cisplatin (n = 341) or placebo plus gemcitabine and cisplatin (n = 344).23
With 6.5 months of additional follow-up, the proportion of patients who discontinued treatment was slightly higher than at IA-2. At the 6.5-month update (DCO of February 25, 2022), ██████████████ patients discontinued treatment in the durvalumab plus gemcitabine and cisplatin group, as did ██████████████ patients in the placebo plus gemcitabine and cisplatin group. █████ █ ██ ██████████ ███████ ██ ███████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ████████ ████ ███████ █ ██ █ █████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ████ ████ ██████ █████ █ ██ ███ ███████ ███████ █████████ ████ █████ ████ ██████████ ██ ████ ██ ██████ ██████████ █████ █ ██ ██████████ ███████ █████████ █████████ ███████ ██████ ███████████24
Disposition | TOPAZ-1 (IA-2) | TOPAZ-1 (6.5-month update) | ||
|---|---|---|---|---|
D + Gem and Cis | PBO + Gem and Cis | D + Gem and Cis | PBO + Gem and Cis | |
Screened, n | 914 | N/A | ||
████████████████████ | ███████████████ | ███████████████ | ||
Randomized, n (%) | 341 (100.0) | 344 (100.0) | 341 (100.0) | 344 (100.0) |
Treated | 338 (99.1) | 342 (99.1) | 338 (99.1) | 342 (99.4) |
Discontinued from treatment, n (%) | 275 (81.4) | 322 (94.2) | 306 (90.5) | 335 (98.0) |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
Discontinued study, N (%) | ||||
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
████████████████████ | ███████████ | ███████████ | ███████████ | ███████████ |
Cis = cisplatin; D = durvalumab; DCO = data cut-off; FAS = full analysis set; Gem = gemcitabine; IA-2 = second interim analysis; N/A = not applicable; PBO = placebo; RECIST = Response Evaluation Criteria in Solid Tumours; SAS = safety analysis set.
Sources: TOPAZ-1 CSR (IA-2 and 6.5-month update).23,24
Exposure to Study Treatments
Exposure to study treatments at IA-2 and the 6.5-month update is summarized in Table 11. At the 6.5-month update, the median duration of treatment was ████████ █████████████ ██████████████ █████ █████ with durvalumab plus gemcitabine and cisplatin and ████████████ ██████ ████ ███ ██████████ ██████████ with placebo plus gemcitabine and cisplatin. The difference in duration of treatment was due to the monotherapy period for durvalumab after 8 cycles of chemotherapy.24
Table 11: Duration of Durvalumab or Placebo Exposure (Safety Analysis Set)
Duration of exposure | IA-2 (DCO: August 11, 2021) | 6.5-month update (DCO: February 25, 2022) | ||
|---|---|---|---|---|
D + Gem and Cis (N = 338) | PBO + Gem and Cis (N = 342) | D + Gem and Cis (N = 338) | PBO + Gem and Cis (N = 342) | |
Duration of exposure (all treatments) | ||||
Total (intended) treatment duration (months)a | ||||
Mean (SD) | ████████████ | ████████████ | ███████████ | █████████████ |
Median (range) | ████████████ | ████████████ | ███████████ | █████████████ |
Total treatment years | ████████████ | ████████████ | ███████████ | █████████████ |
Actual treatment duration (months)b | ||||
Mean (SD) | ████████████ | ████████████ | ███████████ | █████████████ |
Median (range) | 7.33 (0.1 to 24.5) | 5.77 (0.2 to 21.5) | ████████████ | █████████████ |
Total treatment years | ████████████ | ████████████ | ████████████ | █████████████ |
Duration of durvalumab or placebo exposure during chemotherapy period | ||||
Total treatment duration (months)c | ||||
Mean (SD) | ████████████ | ████████████ | ███████████ | █████████████ |
Median (range) | ████████████ | ████████████ | ███████████ | █████████████ |
Total treatment years | ████████████ | ████████████ | ███████████ | █████████████ |
Actual treatment duration (months)d | ||||
Mean (SD) | ████████████ | ████████████ | ███████████ | █████████████ |
Median (range) | ████████████ | ████████████ | ███████████ | █████████████ |
Total treatment years | ████████████ | ████████████ | ███████████ | █████████████ |
Duration of durvalumab or placebo exposure during monotherapy period | ||||
Total treatment duration (months)e | ||||
Mean (SD) | ████████████ | ████████████ | ███████████ | █████████████ |
Median (range) | ████████████ | ████████████ | ███████████ | █████████████ |
Total treatment years | ████████████ | ████████████ | ███████████ | █████████████ |
Actual treatment duration (months)d | ||||
Mean (SD) | ████████████ | ████████████ | ███████████ | █████████████ |
Median (range) | ████████████ | ████████████ | ███████████ | █████████████ |
Total treatment years | ████████████ | ████████████ | ███████████ | █████████████ |
Cis = cisplatin; D = durvalumab; DCO = data cut-off; Gem = gemcitabine; PBO = placebo.
aTotal treatment duration = (min [last dose date where dose > 0 plus {20 if last dose in period 1 (combination) or 27 if last dose in period 2 (maintenance)}, date of death, date of DCO] – first dose date plus 1)/(365.25/12).
bActual treatment duration = total treatment duration minus the total duration of delays.
cTotal treatment duration = (min [last dose date period 1 where dose > 0 plus 20, date of death, date of DCO] – first dose date plus 1)/(365.25/12).
dActual treatment duration = total treatment duration minus the total duration of delays.
eTotal treatment duration = (min [last dose date period 2 where dose > 0 plus 27, date of death, date of DCO] – first dose date plus 1)/(365.25/12).
Sources: TOPAZ-1 CSR (IA-2 and 6.5-month update).23,24
The median number of treatment cycles for each treatment is summarized in Table 12. The median number of cycles for durvalumab was 10 (range, 1 to 29) and the median number of cycles for placebo was 8 (range, 1 to 26). A total of 60.1% and 59.2% of patients in the durvalumab plus gemcitabine and cisplatin group received at least 8 cycles of gemcitabine and cisplatin, respectively. Conversely, 52.0% and 50.0% of patients in the placebo plus gemcitabine and cisplatin group received at least 8 cycles of gemcitabine and cisplatin, respectively. For durvalumab, 30.8% of patients received 12 or more treatment cycles, whereas 19.6% of patients received 12 or more treatment cycles of placebo.23
The median dose intensity for both treatment groups were identical. The median dose intensity for durvalumab and placebo was 100%, whereas the median dose intensity for both gemcitabine and cisplatin was 93.8%.23
Table 12: Number of Treatment Cycles Received and Relative Dose Intensity (IA-2, Safety Analysis Set)
Variable | Durvalumab + gemcitabine and cisplatin (N = 338) | Placebo + gemcitabine and cisplatin (N = 342) | ||||
|---|---|---|---|---|---|---|
Durvalumab | Gemcitabine | Cisplatin | Placebo | Gemcitabine | Cisplatin | |
Number of cycles | ||||||
Mean | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
Median (range) | 10.0 (1 to 29) | 8.0 (0 to 8) | 8.0 (0 to 8) | 8.0 (1 to 26) | 8.0 (1 to 8) | 7.5 (1 to 8) |
Patients who initiated treatment for at least, n (%) | ||||||
0 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 1 cycle | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 2 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 3 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 4 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 5 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 6 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 7 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 8 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 9 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 10 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 11 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 12 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 15 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
≥ 20 cycles | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
Dose Intensity, % | ||||||
Mean (SD) | ████████ | ████████ | ████████ | ████████ | ████████ | ████████ |
Median (range) | 100.0 (25 to 109) | 93.8 (36 to 150) | 93.8 (36 to 150) | 100.0 (50 to 100) | 93.8 (33 to 133) | 93.8 (33 to 133) |
IA-2 = second interim analysis; N/A = not available; SD = standard deviation.
Note: Relative dose intensity is the percentage of the actual dose intensity delivered relative to the intended dose intensity through treatment discontinuation.
Source: TOPAZ-1 CSR (IA-2).23
Dose Modifications
Results for dose modifications, including dose delays, infusion interruptions, and dose reductions, as of IA-2 are summarized in Table 13. No results for dose modifications were available at the 6.5-month update █████ █ ██ ██████████ ███████ ██ ██ █████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ███████ ██ ██ █████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ███████ █ █ ███████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ █████ █ ██ ██████████ ███████ █ ███ █████ █████████ ██████████████. Dose reductions were not permitted for durvalumab or placebo. █████ █ ██ ███ ███ ████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ███ ██ ████ █ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ █████ ██████ █ ██ ██ █ ███ ████ ███████ █████████ █████ ████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██ ███ █████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ███ ███ ███ █ ██ ██████ ████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ██ ███████ █ ██ ████ ██████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ██ ██████ █ ██ █████ █████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ████ █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ██ ██████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ███ █████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ █ ██ ███ ██ █████ █ ██ ██████████ ███████ ████ █████ █████████ ███ ███████ ███████ ███████ ███████ █ █ ███ ████ █████
Table 13: TOPAZ-1 Dose Delays, Interruptions, Reductions (IA-2, Safety Analysis Set)
Dose modification | Durvalumab + gemcitabine and cisplatin (N = 338) | Placebo + gemcitabine and cisplatin (N = 342) | ||||
|---|---|---|---|---|---|---|
Durvalumab | Gemcitabine | Cisplatin | Placebo | Gemcitabine | Cisplatin | |
█████████████████████████████████████████████ | ||||||
███████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
███████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
█████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
█████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
█████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
█████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
█████████████████████████████████████████████ | ||||||
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
█████████████████████████████████████████████ | ||||||
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
██████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
█████████████████████████████████████████████ | ||||||
████████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
████████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
████████████ | █████████ | █████████ | █████████ | █████████ | █████████ | █████████ |
AE = adverse event; NA = not applicable; NR = not reported.
Source: TOPAZ-1 CSR (IA-2).23
Subsequent Anticancer Therapy
Results for subsequent anticancer therapy received at IA-2 and as of the 6.5-month follow-up (DCO of February 25, 2022) are summarized in Table 14. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██ ████ ██████████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ███ ███ █████ █████ ██████ █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ███████ ███ ████████ ██████ █████ ███ ██████ ████ █████████████24
Although not considered subsequent anticancer therapy, █████ █ ██ ██████████ ███████ █████████ █████████ ████ █ ██ ███████ ██ ████ ██ ██████ ██ ████████ ███████████ █ ██ ██████████ ███ ████ █████████ █████████ █ ███ ██ █████ ███ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ████ ██████ ███████ █████████ █████████ █ ███ ██ ████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ███████ █████████ █ ███ █████ █ ███ █ █████████ ██ ████ ██ ██████ ████ ██████ ███████████ █ ██ ███ ███████ ███████ █████████ ███ ██ ████ ████ █████ █████ ██ ████ ██ ██████ ██████████ █████████████████████████████████████████ patients in the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups, respectively.23
Table 14: Subsequent Anticancer Therapy (FAS)
Therapy | IA-2 (DCO: August 11, 2021) | 6.5-month update (DCO: February 25, 2022) | ||
|---|---|---|---|---|
D + Gem and Cis (N = 338) | PBO + Gem and Cis (N = 342) | D + Gem and Cis (N = 338) | PBO + Gem and Cis (N = 342) | |
Any subsequent anticancer therapy | 145 (42.5) | 170 (49.4) | ██████████ | ████████████ |
Cytotoxic chemotherapy | 137 (40.2) | 156 (45.3) | ██████████ | ████████████ |
Targeted therapy | 12 (3.5) | 16 (4.7) | ██████████ | ████████████ |
Immunotherapy | 3 (0.9) | 16 (4.7) | ██████████ | ████████████ |
Taxane chemotherapy | 5 (1.5) | 9 (2.6) | ██████████ | ████████████ |
Antiangiogenic therapy | 1 (0.3) | 1 (0.3) | ██████████ | ████████████ |
Other | 15 (4.4) | 28 (8.1) | ██████████ | ████████████ |
████████████ | ████████████ | ████████████ | ██████████ | ████████████ |
Cis = cisplatin; D = durvalumab; DCO = data cut-off; FAS = dull analysis set; Gem = gemcitabine; IA-2 = second interim analysis; PBO = placebo.
Note: Therapies post-discontinuation of study treatment were displayed. A patient could have more than 1 cancer therapy.
Sources: TOPAZ-1 CSR (IA-2 and 6.5-month update).23,24
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 3 for detailed efficacy data.
Overall Survival
OS was the primary end point of IA-2 of the TOPAZ-1 study. Results for OS at IA-2 and 6.5-month additional follow-up are summarized in Table 15 and by Kaplan-Meier plot in Figure 3.
At the preplanned IA-2 (DCO of August 11, 2021), a total of 424 death events had occurred (198 [58.1%] in the durvalumab plus gemcitabine and cisplatin group, and 226 [65.7%] in the placebo plus gemcitabine and cisplatin group), representing ████ ███ ███ ███ ███████ ██████ ██████████. The median duration of follow-up in censored patients was ███ ██████ ██████ ███████ ██ █████ ███████ in the durvalumab plus gemcitabine and cisplatin group, and ██ █████ ██████ ███ █████ ███ █████ █ █████ ███ █████ █████ in the placebo plus gemcitabine and cisplatin group. The HR for the durvalumab plus gemcitabine and cisplatin versus placebo plus gemcitabine and cisplatin was 0.80 (95% CI, 0.66 to 0.97; P = 0.021) in favour of durvalumab plus gemcitabine and cisplatin, with a median OS of 12.8 months (95% CI, 11.1 to 14.0 months) for durvalumab plus gemcitabine and cisplatin and 11.5 months (95% CI, 10.1 to 12.5 months) for placebo plus gemcitabine and cisplatin.
Table 15: Overall Survival (FAS)
Overall survival | IA-2 (DCO: August 11, 2021) | 6.5-month update (DCO: February 25, 2022) | ||
|---|---|---|---|---|
D + Gem and Cis (N = 341) | Placebo + Gem and Cis (N = 344) | D + Gem and Cis (N = 341) | Placebo + Gem and Cis (N = 344) | |
Death, n (%) | 198 (58.1) | 226 (65.7) | 248 (72.7) | 279 (81.1) |
████████████ | ██████████ | ████████████ | ████████████ | ████████████ |
████████████ | ██████████ | ████████████ | ████████████ | ████████████ |
████████████ | ██████████ | ████████████ | ████████████ | ████████████ |
████████████ | ██████████ | ████████████ | ████████████ | ████████████ |
████████████ | ██████████ | ████████████ | ████████████ | ████████████ |
Median OS, months (95% CI)d | 12.8 (11.1 to 14.0) | 11.5 (10.1 to 12.5) | 12.9 (11.6 to 14.1) | 11.3 (10.1 to 12.5) |
HR (95% CI)e | 0.80 (0.66 to 0.97) | 0.76 (0.64 to 0.91) | ||
P valuef | 0.021 | NR | ||
OS rate, % (95% CI)d | ||||
12 months | 54.1 (48.4 to 59.4) | 48.0 (42.4 to 53.4) | 54.3 (48.8 to 59.4) | 47.1 (41.7 to 52.3) |
18 months | 35.1 (29.1 to 41.2) | 25.6 (19.9 to 31.7) | 34.8 (29.6 to 40.0) | 24.1 (19.6 to 28.9) |
24 months | 24.9 (17.9 to 32.5) | 10.4 (4.7 to 18.8) | 23.6 (18.7 to 28.9) | 11.5 (7.6 to 16.2) |
CI = confidence interval; Cis = cisplatin; D = durvalumab; DCO = data cut-off; FAS = full analysis set; Gem = gemcitabine; HR = hazard ratio; IA-2 = second interim analysis 2; OS = overall survival.
aIncludes patients known to be alive at DCO.
bIncludes patients with unknown survival status, patients who were lost to follow-up, or patients with “other” recorded on the case report form.
cAt IA-2, the investigator made a transcription error that was clarified at the 6.5-month update analysis. At IA-2, the last known date that patient was known to be alive was erroneously reported as June, 29 2021. The last known date that the patient was known to be alive is July 24, 2020.
dCalculated using the Kaplan-Meier technique. CI for median OS derived using the Brookmeyer-Crowley method.
eThe analysis was performed using a stratified Cox proportional hazards model (ties = Efron), adjusted for disease status and primary tumour location. The CI was calculated using a profile likelihood approach. HRs < 1 favour durvalumab, and is associated with a longer OS than placebo.
fThe primary end point of OS was controlled for multiple testing using the Lan-DeMets spending function, approximating O’Brien-Fleming boundaries. The 2-sided significance level was < 0.0300.
Sources: TOPAZ-1 CSR (IA-2 and 6.5-month update).23,24
At the 6.5-month update (DCO of February 25, 2022), a total of ███████ death events had occurred (248 [72.7%] in the durvalumab plus gemcitabine and cisplatin group, and 279 [81.1%] in the placebo plus gemcitabine and cisplatin group), representing 76.9% overall maturitsy for OS. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ █ ███ █ █████ ██ ████ ██ ██████ ██████████ █████ █ ██ ██████████ ███████ █████████ █████████ ██ ██ ██ █ █ ████ █████ █████████████ ██████████. With 6.5 months of additional follow-up, the HR for the durvalumab plus gemcitabine and cisplatin versus placebo plus gemcitabine and cisplatin was 0.76 (95% CI, 0.64 to 0.91) in favour of durvalumab plus gemcitabine and cisplatin, with a median OS of 12.9 months (95% CI, 11.6 to 14.1 months) for durvalumab plus gemcitabine and cisplatin and 11.3 months (95% CI, 10.1 to 12.5 months) for placebo plus gemcitabine and cisplatin.
Results for the OS rate were consistent at IA-2 and at the 6.5-month additional follow-up. The OS rate at 18 months was 34.8% (95% CI, 29.6% to 40.0%) for durvalumab plus gemcitabine and cisplatin and 24.1% (95% CI, 19.6% to 28.9%) for placebo plus gemcitabine and cisplatin, and at 24 months was 23.6% (95% CI, 18.7% to 28.9%) and 11.5% (95% CI, 7.6% to 16.2%), respectively.23,24
Posthoc Analyses of the Primary End Point
A posthoc analysis was conducted confirming the nonproportionality of the hazards assumption, given that delayed separation of Kaplan-Meier curves is common in clinical trials of immunotherapy in combination with chemotherapy. Results of the posthoc analysis were consistent with the primary analysis, confirming nonproportional hazards, and the piecewise HR was █████ █ ██ ███ ███████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ █ ██████ ███ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ████ ██████ █████████ ████ █████ ██ ██████23,24 An additional posthoc analysis analyzing OS by response status was conducted, given that the OS benefit for patients receiving durvalumab plus gemcitabine and cisplatin was driven not only by responders but also by patients with a BOR of stable disease. Results for this posthoc analysis are summarized in Figure 4 of Appendix 2. █████ █ ██ ██████████ ███████ █████ ████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ████████ ██ ████ █ ██ ██████ ███ ███ ██████ ████ ██████████ ██ ████ ██ ██████ ███ ███████ ███████████ █ ██ ████████ ██ █ ███ ███ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ███ ███████ ██ █████ █████████ ███ ██████ ████ ████ ██████ ██ ████ ██ ██████ ██████████ █████ ██████ █ ██ ██ █████ ███ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████ ████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ █████ █ ██ ██████ ████ ███████ █████████ █████████ ███████████ █████████
Sensitivity Analyses
Results for the prespecified sensitivity analysis examining censoring patterns to identify potential attrition bias by reversing the censoring indicator of OS suggested that the HR for OS was ██████ █████████ ███████ ███████ ████████ █████████ ██. The sponsor considered this supportive of the primary analysis.23
An additional sensitivity analysis of OS HR estimates adjusted for the potential effect of covariates using Cox proportional hazards modelling was similar to that obtained without such adjustment █████ █ ██ ███████ ███ ███████ █████████ █████████ ██ ███████ █████.23
Results of the sensitivity analyses at the 6.5-month follow-up were consistent with the results at IA-2.24
Subgroup Analyses
Results for prespecified subgroups in the subgroups of interest to this review at IA-2 and after 6.5 months of additional follow-up are summarized in Table 16. The full subgroup analysis from the TOPAZ-1 trial is summarized in Figure 4 of Appendix 2. At IA-2, the OS was generally consistent across subgroups. Results of the subgroup analyses of interest after 6.5 months of additional follow-up were generally consistent with the IA-2 subgroups, as well as with the primary analysis.24
Progression-Free Survival
PFS was a key secondary end point of the TOPAZ-1 study, and results are summarized in Table 17 and by Kaplan-Meier plot for IA-2 in Figure 5. No results for PFS were available at the 6.5-month update. At the time of the IA-2 DCO (August 11, 2021), a total of ███ █████████ PFS events had occurred; ██ ██████ ████ in the durvalumab plus gemcitabine and cisplatin group, and ████ █████ █ ██ in the placebo plus gemcitabine and cisplatin group ██████ █████████ ████ ██████████ ██ █████. The HR for durvalumab plus gemcitabine and cisplatin versus placebo gemcitabine and cisplatin was 0.75 (95% CI, 0.63 to 0.89; P = 0.001) in favour of durvalumab. The median PFS was 7.2 months (95% CI, 6.7 to 7.4 months) in the durvalumab plus gemcitabine and cisplatin group, and 5.7 months (95% CI, 5.6 to 6.7 months) in the placebo plus gemcitabine and cisplatin group. The PFS rate at 6, 9, and 12 months was █████ █ ██ █████ █████ ███████ █████████ █████████ ███████████ █████████ ███ ███ for the durvalumab plus gemcitabine and cisplatin group, and █████ █ ██ ██████████ ███████ ████ █████ █████████ ██ ██ ████ ████████████ ███████ ███████ ████ in the placebo plus gemcitabine and cisplatin group, respectively.23
Table 16: OS by Subgroup of Interest (FAS)
Subgroup | IA-2 (DCO: August 11, 2021) | 6.5-month update (DCO: February 25, 2022) | ||||
|---|---|---|---|---|---|---|
D + Gem and Cis, Event/N | PBO + Gem and Cis, Event/N | HR (95% CI) | D + Gem and Cis, Event/N | PBO + Gem and Cis, Event/N | HR (95% CI) | |
All patients | ||||||
N | 198/341 | 226/344 | 0.80 (0.66 to 0.97) | 248/341 | 279/344 | 0.76 (0.64 to 0.91) |
Sex | ||||||
Male | 99/169 | 122/176 | 0.82 (0.62 to 1.08) | 126/169 | 148/176 | 0.81 (0.64 to 1.04) |
Female | 99/172 | 104/168 | 0.82 (0.62 to 1.08) | 122/172 | 131/168 | 0.81 (0.64 to 1.04) |
Disease status | ||||||
Initially unresectable | 176/274 | 194/279 | 0.84 (0.69 to 1.03) | 209/274 | 240/279 | 0.79 (0.65 to 0.95) |
Recurrent | 22/67 | 32/64 | 0.56 (0.32 to 0.96) | 39/67 | 39/64 | 0.76 (0.49 to 1.20) |
Locally advanced | 16/38 | 36/57 | 0.49 (0.26 to 0.88) | 22/38 | 45/57 | 0.54 (0.32 to 0.88) |
Metastatic | 182/303 | 190/286 | 0.83 (0.68 to 1.02) | 226/303 | 234/286 | 0.80 (0.76 to 0.97) |
Primary tumour location | ||||||
IHCC | 105/190 | 126/193 | 0.76 (0.58 to 0.98) | 136/190 | 153/193 | 0.78 (0.62 to 0.99) |
EHCC | 38/66 | 42/65 | 0.76 (0.49 to 1.19) | 45/66 | 55/65 | 0.61 (0.41 to 0.91) |
GBC | 55/85 | 58/86 | 0.94 (0.65, 1.37) | 67/85 | 71/86 | 0.90 (0.64 to 1.25) |
PD-L1 status (TIP 1%) | ||||||
High (≥ 1) | 120/197 | 138/205 | 0.79 (0.61, 1.00) | 149/199 | 172/207 | 0.75 (0.60 to 0.93) |
Low/negative (< 1%) | 57/103 | 66/103 | 0.86 (0.60, 1.23) | 71/103 | 81/103 | 0.79 (0.58 to 1.09) |
WHO/ECOG PS | ||||||
0 | 95/173 | 93/163 | 0.90 (0.68, 1.20) | 126/173 | 125/163 | 0.87 (0.68 to 1.12) |
1 | 103/168 | 133/181 | 0.72 (0.56, 0.94) | 122/168 | 154/181 | 0.70 (0.55 to 0.89) |
Region | ||||||
Asia | 103/178 | 137/196 | 0.72 (0.56, 0.94) | 130/178 | 170/196 | 0.68 (0.54 to 0.85) |
Rest of the world | 95/163 | 89/148 | 0.89 (0.66, 1.19) | 118/163 | 109/148 | 0.91 (0.70 to 1.18) |
CI = confidence interval; Cis = cisplatin; D = durvalumab; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EHCC = extrahepatic cholangiocarcinoma; FAS = full analysis set; GBC = gallbladder cancer; Gem = gemcitabine; HR = hazard ratio; IA-2 = second interim analysis; IHCC = intrahepatic cholangiocarcinoma; PBO = placebo; PD-L1 = programmed cell death 1 ligand 1; TIP = tumour and/or immune cell positivity; WHO = WHO.
Note: HRs < 1 favour durvalumab plus gemcitabine plus cisplatin.
Sources: TOPAZ-1 CSR (IA-2 and 6.5-month update).23,24
Table 17: PFS by Investigator Assessment According to RECIST 1.1 (FAS)
PFS | IA-2 (DCO: August 11, 2021) | |
|---|---|---|
D + Gem and Cis (N = 341) | PBO + Gem and Cis (N = 344) | |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
Median PFS, months (95% CI)e | 7.2 (6.7 to 7.4) | 5.7 (5.6 to 6.7) |
HR (95% CI)f | 0.75 (0.63 to 0.89) | |
95.19% CI for HRf | 0.63 to 0.89 | |
2-sided P valueg | 0.001 | |
PFS rate, % (95% CI)e | ||
6 months | 58.3 (52.8 to 63.4) | 47.2 (41.6 to 52.5) |
9 months | 34.8 (29.6 to 40.0) | 24.6 (20.0 to 29.5) |
12 months | 16.0 (12.0 to 20.6) | 6.6 (4.1 to 9.9) |
CI = confidence interval; Cis = cisplatin; D = durvalumab; DCO = data cut-off; FAS = full analysis set; Gem = gemcitabine; HR = hazard ratio; IA-2 = second interim analysis; PBO = placebo; PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumours.
aOnly includes progression events that occurred within 2 missed visits of the last evaluable assessment.
bTarget lesions, nontarget lesions, and new lesions are not necessarily mutually exclusive categories.
cRECIST 1.1 progression event occurred 2 or more consecutive missed visits after last evaluable RECIST 1.1 assessment.
dDeath occurred after 2 or more consecutive missed visits after last evaluable RECIST 1.1 assessment.
eCalculated using the Kaplan-Meier technique.
fThe HR and its CI were estimated using a stratified Cox proportional hazards model (ties = Efron) adjusted for disease status and primary tumour location.
gThe P value is based on a stratified log-rank test and tested at the 0.0481 significance level. HRs < 1 favour durvalumab plus gemcitabine plus cisplatin, and are associated with a longer PFS than placebo plus gemcitabine plus cisplatin. PFS was included in the multiple-testing procedure and was formally tested after a statistically significant result for the primary outcome, OS.
Source: TOPAZ-1 CSR (IA-2).23
Sensitivity Analyses
Sensitivity analyses of the key secondary end point of PFS to evaluate the potential for attrition bias and the impact of covariates on the treatment effect were consistent with the primary analysis. █████ █ ██ ████████ ██ ███████ █████████ █████ ████ █ ███ █████ █████ ██ ████ ██ ██████ ████ ██████ ███████████ █ ██ ██████████ ███████ ███ ██████ ██████ ███ ████ ███ ██ █████ ██ ████ ██ ██████ ████ ██████ █████ █ ██ ██████████ ███████ █████████ ███████ ██ ████.
██ ███ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ███ ██████ █ ██ ████████ ██ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ███ █████ █ ██ █████ █████ ███████ ██ ████ ███ █████████ ████ ███████ ███ ██ ████ ██ ██████ ██ ███ █████ ██████
Subgroup Analyses
Results of subgroup analyses of PFS from the TOPAZ-1 trial are summarized in Figure 6. For subgroups of interest to this review, results appeared consistent with the primary analysis, although no HRs were presented.23
Clinical Response
Objective Response Rate
ORR was the primary outcome at IA-1 and was a secondary outcome at IA-2. Results for ORR at both IA-1 and IA-2 are summarized in Table 18. No results for ORR were available at the 6.5-month update. Results at IA-1 were assessed by the investigator and by BICR. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████ ████ ██ ████ ██ ██ ██ ██ ██████ ████ ███████ ████ █ ██ ████ ██████ ███████ █████████ █████████ ████ ██████ ████ ██ ████ ██ ██ ████ ███ ██ █████ █████ █ ██ ██████████ ███████ █████████ █████████ ██████ ████████████ ██ ██████ ████ ███ ███████.
Table 18: ORR — Stratified CMH Test (FAS Patients With Measurable Disease at Baseline)
ORR | D + Gem and Cis | PBO + Gem and Cis |
|---|---|---|
IA-1 (DCO: December 18, 2020) | ||
████████████ | ████████████ | ████████████ |
████████████ | ████████████ | ████████████ |
IA- 2 (DCO: August 11, 2021, investigator assessed) | ||
Complete response, n/N (%) | 7/341 (2.1) | 2/343 (0.6) |
Partial response, n/N (%) | 84/341 (24.6) | 62/343 (18.1) |
ORRa, n/N (%) | 91/341 (26.7) | 64/343 (18.7) |
OR (95% CI)b | 1.60 (1.11 to 2.31) | |
Nominal 2-sided P valuec | 0.011 | |
DCR, n/N (%) | 291/341 (85.3) | 284/343 (82.6) |
CI = confidence interval; Cis = cisplatin; CMH = Cochran-Mantel Haenszel; D = durvalumab; DCO = data cut-off; DCR = disease control rate; FAS 32w = full analysis set at 32 weeks; Gem = gemcitabine; IA-2 = interim analysis 2; OR = odds ratio; ORR = objective response rate; PBO = placebo.
aResponses included confirmed complete or partial response, per investigator, according to RECIST 1.1. They do not include patients who discontinued randomized treatment without progression or received a subsequent anticancer therapy and then responded.
bORs > 1 favour durvalumab plus gemcitabine plus cisplatin.
cNominal 2-sided P value was not adjusted for multiple testing.
Source: TOPAZ-1 CSR (IA-2).23
At IA-2, per the investigator, 91 (26.7%) patients in the durvalumab plus gemcitabine and cisplatin group and 64 (18.7%) in the placebo plus gemcitabine and cisplatin group achieved an ORR. The OR of achieving a response with durvalumab plus gemcitabine and cisplatin versus placebo plus gemcitabine and cisplatin was 1.60 (95% CI, 1.11 to 2.31; P = 0.011) in favour of durvalumab plus gemcitabine and cisplatin. The median time to response was 1.6 months for durvalumab plus gemcitabine and cisplatin and 2.7 months for placebo plus gemcitabine and cisplatin.23
DCR (Table 18) was a secondary outcome of the TOPAZ-1 trial. At IA-2, the DCR in the FAS was 85.3% in the durvalumab plus gemcitabine and cisplatin group and 82.6% in the placebo plus gemcitabine and cisplatin group.23
Sensitivity Analysis: █████ █ ██ ██████████ ███████ ████ █████ █████████ ████ ██████████ ██ ████ ██ ███ ███ ████ ██████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ████ ██████ ██ ████ ██ ██████ ██ ████ ████ █████ █ ██ ██████████ ███████ █████████ █████████ ██████████████ ██████ ████████.
Subgroup Analysis: A post-hoc subgroup analysis of ORR at IA-2 was conducted. Results of the subgroup analyses are summarized in Figure 7. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██ █ ███ ██ ██ ██████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██ ████ █████ █████ █████ █ ██ ██████████ ███████ █████████ █████████ █████████ █████ ██████████████.
Duration of Response
DOR in responders was a secondary outcome of the TOPAZ-1 trial. Results for DOR at IA-2 are summarized in Table 19. No results for DOR were available at the 6.5-month update. More confirmed responses were observed in the durvalumab plus gemcitabine and cisplatin group (91 [26.7%] patients) than in the placebo plus gemcitabine and cisplatin group (64 [18.7%] patients). The median DOR was 6.4 months (95% CI, 5.9 to 8.1 months) with durvalumab plus gemcitabine and cisplatin and 6.2 months (95% CI, 4.4 to 7.3 months) with placebo plus gemcitabine and cisplatin. Most patients in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group subsequently progressed or died (65.9% versus 79.7%).23
The percent of patients remaining in response at 9 and 12 months was 32.6% and 26.1%, respectively, for durvalumab plus gemcitabine and cisplatin and 25.3% and 15.0%, respectively, for placebo plus gemcitabine and cisplatin.23
Table 19: Duration of Objective Response per Investigator (FAS; Patients With Objective Response and Measurable Disease at Baseline)
Duration of objective response | IA-2 (DCO: August 11, 2021) | |
|---|---|---|
D + Gem and Cis (N = 91) | Placebo + Gem and Cis (N = 64) | |
████████████████████████ | ████████████ | ████████████ |
████████████████████████████████████ | ████████████ | ████████████ |
████████████████████████ | ████████████ | ████████████ |
████████████████████████████████████ | ████████████ | ████████████ |
DOR from onset of response (months)d,e | ||
Median (95% CI) | 6.4 (5.9 to 8.1) | 6.2 (4.4 to 7.3) |
Percent remaining in responsee | ||
≥ 3 months | 88.9 | 89.0 |
≥ 6 months | 59.3 | 54.2 |
≥ 9 months | 32.6 | 25.3 |
≥ 12 months | 26.1 | 15.0 |
CI = confidence interval; Cis = cisplatin; D = durvalumab; DCO = data cut-off; DOR = duration of response; FAS = full analysis set; Gem = gemcitabine; IA-2 = second interim analysis.
aOne patient received surgical resection for BTC and remained on study treatment after surgery. The site continued scans (each resulting in RECIST not evaluable). Per the SAP, DOR was censored at patient’s last evaluable RECIST 1.1 assessment.
bFor 2 patients, the ongoing responses were apparently maintained with subsequent anticancer therapy. Both patients discontinued treatment due to subjective disease progression. Per PFS censoring methodology, DOR was censored at patient’s last evaluable RECIST 1.1 assessment for both cases.
cOne patient responded to treatment but then withdrew consent for study participation, without RECIST progressive disease, and later died. Due to withdrawal of consent, follow-up scans were not performed. In the absence of at least 2 or more missed visits, the patient was censored at the time of the latest evaluable RECIST 1.1 assessment.
dDOR is the time from the first documentation of CR or PR until the date of progression, death, or the last evaluable RECIST assessment for patients who do not progress. The DOR was calculated following the PFS methodology.
eCalculated using the Kaplan-Meier technique.
Source: TOPAZ-1 CSR (IA-2).23
Health-Related Quality of Life
The PRO analysis set was used for all HRQoL and PRO measures; however, the sample size varied by measure. HRQoL measured by the EORTC QLQ-C30 and EORTC QLQ-BIL21 were secondary end points in the TOPAZ-1 trial. The PGI-S, PRO-CTCAE, and EQ-5D measures were exploratory end points of the TOPAZ-1 trial.23
EORTC QLQ-C30
The PRO analysis set for the EORTC QLQ-C30 included 318 patients in the durvalumab plus gemcitabine and cisplatin group, and 328 patients in the placebo plus gemcitabine and cisplatin group. PRO analyses for EORTC QLQ-C30 at IA-2 included time to deterioration and improvement rates (Table 20). For GHS/QoL, 132 (49.1%) and 135 (48.8%) patients in the durvalumab plus gemcitabine and cisplatin and the placebo plus gemcitabine and cisplatin groups, respectively, experienced deterioration. The HR for durvalumab plus gemcitabine and cisplatin versus placebo plus gemcitabine and cisplatin for GHS/QoL was 0.87 (95% CI, 0.69 to 1.12). For GHS/QoL, the median time to deterioration was 7.4 months (95% CI, 5.6 to 8.9 months) for durvalumab plus gemcitabine and cisplatin and 6.7 months (95% CI, 5.6 to 7.9 months) for placebo plus gemcitabine and cisplatin. Results for the proportion of patients experiencing deterioration in functional domains ranged from ████████████ in the durvalumab plus gemcitabine and cisplatin group and ████████████████████████ in the placebo plus gemcitabine and cisplatin group. The median time to deterioration in functional domains ranged from █ ████ ██████ █████ ████████ for durvalumab plus gemcitabine and cisplatin and █████ █████ █████ ███████ ██ for placebo plus gemcitabine and cisplatin. In the multiple and single symptom domains, the proportion of patients experiencing deterioration ranged from 26.5% (diarrhea) to 52.4% (fatigue) in the durvalumab plus gemcitabine and cisplatin group, and from 26.8% (diarrhea) to 50.0% (fatigue) in the placebo plus gemcitabine and cisplatin group. The median time to deterioration in multiple and single symptom items ranged from ████ ███████ █████ █████ ██ █ █ █████████ ████████ ██████ for diarrhea with durvalumab plus gemcitabine and cisplatin and from █████ █ ███ ████ ███████ █ █ for fatigue to ██ ███ ██ █████ ██████ ██████ for diarrhea with placebo plus gemcitabine and cisplatin.23
Improvements in GHS/QoL or functional domains were defined by the investigators as 2 consecutive assessments at least 14 days apart that show an increase from baseline score greater than or equal to 10 points. For GHS/QoL at IA-2, █████ █████ ███ ██ ███████ ██ patients in the durvalumab plus gemcitabine and cisplatin group and █████ ██ ██ ███ patients in the placebo plus gemcitabine and cisplatin group showed improvement. Across functional domains, ██████ █████████ █████ ████ of patients in the durvalumab plus gemcitabine and cisplatin group and ██ █████ ███ ██ ██████████ ██ of patients in the placebo plus gemcitabine and cisplatin group showed improvement. Improvement in multiple and single symptom domains was defined as 2 consecutive assessments at least 14 days apart that show a decrease from baseline score greater than or equal to 10 points. In total, █ ██████ ████ ██ ████ ███████ of patients in the durvalumab plus gemcitabine and cisplatin group and ████ ██████████ ███████████ ███████ ████ of patients in the placebo plus gemcitabine and cisplatin group showed improvement in symptoms at IA-2.23
Table 20: Time to Deterioration and Improvement Based on BOR for EORTC QLQ-C30 (IA-2, PRO Analysis Set)
Variable | Time to deterioration | Improvement based on BOR | ||
|---|---|---|---|---|
D + Gem and Cis (N = 318) | PBO + Gem and Cis (N = 328) | D + Gem and Cis (N = 318) | PBO + Gem and Cis (N = 328) | |
GHS/QoL | ||||
Event/n (%) | 132/269 (49.1) | 135/279 (48.4) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 7.4 (5.6 to 8.9) | 6.7 (5.6 to 7.9) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.87 (0.69 to 1.12) | |||
Functional domains | ||||
Physical | ||||
Event/n (%) | 138/271 (50.9) | 125/280 (44.6) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 6.6 (4.9 to 8.6) | 7.7 (6.4 to 9.2) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.05 (0.83 to 1.35) | ████████████ | ||
Role | ||||
Event/n (%) | 144/264 (54.5) | 142/278 (51.1) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 5.6 (3.7 to 6.7) | 6.5 (4.7 to 7.8) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.08 (0.85 to 1.36) | |||
Cognitive | ||||
Event/n (%) | 137/271 (50.6) | 126/282 (44.7) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 6.0 (4.8 to 8.9) | 7.7 (6.6 to 9.0) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.09 (0.86 to 1.39) | ████████████ | ||
Emotional | ||||
Event/n (%) | 110/269 (40.9) | 95/280 (33.9) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 10.1 (7.9 to 12.2) | 10.0 (8.0 to NC) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.98 (0.75 to1.30) | ████████████ | ||
Social | ||||
Event/n (%) | 138/271 (50.9) | 130/281 (46.3) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 6.0 (4.0 to 9.6) | 6.8 (4.8 to 8.4) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.98 (0.77 to 1.25) | ████████████ | ||
Multiple symptoms | ||||
Fatigue | ||||
Event/n (%) | 163/311 (52.4) | 163/326 (50.0) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 3.0 (2.4 to 4.5) | 3.5 (2.8 to 4.4) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.97 (0.78 to 1.20) | ████████████ | ||
Pain | ||||
Event/n (%) | 137/316 (43.4) | 132/325 (40.6) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 6.5 (5.1 to 8.9) | 7.0 (5.7 to 8.3) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.98 (0.77 to 1.25) | ████████████ | ||
Nausea and/or vomiting | ||||
Event/n (%) | 137/318 (43.1) | 134/325 (41.2) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 6.6 (4.3 to 9.3) | 6.6 (4.2 to 8.0) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.95 (0.74 to 1.21) | ████████████ | ||
Single item | ||||
Dyspnea | ||||
Event/n (%) | 109/314 (34.7) | 110/322 (34.2) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 8.8 (7.2 to NC) | 8.1 (7.2 to 10.7) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.93 (0.71 to 1.22) | ████████████ | ||
Insomnia | ||||
Event/n (%) | 110/305 (36.1) | 117/320 (36.6) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 8.8 (6.9 to 14.1) | 7.2 (6.5 to 9.3) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.87 (0.67 to 1.14) | ████████████ | ||
Appetite loss | ||||
Event/n (%) | 138/307 (45.0) | 111/315 (35.2) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 6.0 (4.3 to 8.8) | 8.5 (6.8 to 10.2) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.24 (0.96 to 1.60) | ████████████ | ||
Constipation | ||||
Event/n (%) | 135/306 (44.1) | 127/318 (39.9) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 5.7 (3.5 to 7.9) | 7.2 (5.1 to 8.1) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.09 (0.86 to 1.39) | ████████████ | ||
Diarrhea | ||||
Event/n (%) | 84/317 (26.5) | 88/328 (26.8) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 18.2 (11.1 to NC) | 11.0 (9.0 to 12.7) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.86 (0.63 to 1.16) | ████████████ | ||
BOR = best overall response; CI = confidence interval Cis = cisplatin; D = durvalumab; Gem = gemcitabine; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GHS = global health status; HR = hazard ratio; IA-2 = second interim analysis; N/A = not applicable; NC = not calculable; NR = not reported; OR = odds ratio; PBO = placebo; PRO = patient-reported outcome; QoL = quality of life; TTD = time to deterioration.
aHRs are presented for time to deterioration. HRs < 1 favour durvalumab plus gemcitabine plus cisplatin. ORs are presented for improvement rates based on BOR. ORs > 1 favour durvalumab plus gemcitabine plus cisplatin.
Source: TOPAZ-1 CSR (IA-2).23
█████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████ ████ █ █ ██ ██ █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ███ ██ ████ █ ██ ██████████ ███████ █████████ ████ █████ ████ ██████████ ██ ████ ██ ██████ █████████ █ ███ ███ █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ███ ███ ████ █ ████ ██████ █ ██ ████ ██████ ███████ █████████ ███ ██████ ████ ██████████ ██ ████ ██ ██████ █ ██ ███████ ██ ████ █████████████ ███████
EORTC QLQ-BIL21
The PRO analysis set for the EORTC QLQ-BIL21 included 305 patients in the durvalumab plus gemcitabine and cisplatin group and 322 patients in the placebo plus gemcitabine and cisplatin group. PRO analyses for EORTC QLQ-BIL21 at IA-2 included time to deterioration and improvement rates (Table 21). Clinically meaningful symptom deterioration was defined by the investigators as an increase from baseline of greater than or equal to 10 points for symptom scales or items. The proportion of patients experiencing deterioration in symptoms ranged from 20.6% (jaundice) to 48.5% (tiredness) with durvalumab plus gemcitabine and cisplatin and from 19.9% (jaundice) to 47.9% (tiredness) with placebo plus gemcitabine and cisplatin. The median time to symptom deterioration ranged from █████ █ ██ ██████████ ███████ █████████ █████████ ██████████████ for durvalumab plus gemcitabine and cisplatin and from █████ █ ██ ██████████ ███████ █████████ █████████ ██████████████ with placebo plus gemcitabine and cisplatin.23
Improvement rates for EORTC QLQ-BIL21 at IA-2 are also summarized in Table 21. Improvement in symptoms was defined by the investigators as 2 consecutive assessments at least 14 days apart that show a decrease from baseline score greater than or equal to 10 points. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █████ █████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ██ ██████ █████ █ ██ ██████████ ███████ █████████ █████████ ████.
Table 21: Time to Symptom Deterioration and Improvement Rates for the EORTC QLQ-BIL21 (IA-2, PRO Analysis Set)
Variable | Time to deterioration | Improvement based on BOR | ||
|---|---|---|---|---|
D + Gem and Cis (N = 305) | PBO + Gem and Cis (N = 322) | D + Gem and Cis (N = 305) | PBO + Gem and Cis (N = 322) | |
Single Item | ||||
Abdominal pain | ||||
Event/n (%) | 93/297 (31.3) | 101/315 (32.1) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 11.1 (8.3 to NC) | 8.5 (7.0 to 11.5) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.92 (0.69 to 1.23) | ████████████ | ||
Pruritis | ||||
Event/n (%) | 97/301 (32.2) | 91/320 (28.4) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 9.8 (7.8 to 11.6) | 8.9 (8.0 to 11.2) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.00 (0.75, 1.33) | ████████████ | ||
Jaundice | ||||
Event/n (%) | 62/301 (20.6) | 64/321 (19.9) | ████████████ | ████████████ |
Median TTD, months (95% CI) | NC (14.7 to NC) | 14.2 (11.3 to NC) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.88 (0.62 to 1.25) | ████████████ | ||
Weight loss | ||||
Event/n (%) | 90/297 (30.3) | 83/316 (26.3) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 11.7 (9.2 to NC) | 11.5 (8.7 to NC) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.11 (0.82 to 1.50) | ████████████ | ||
Multiple symptoms | ||||
Eating | ||||
Event/n (%) | 118/305 (38.7) | 110/320 (34.4) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 7.4 (5.1 to 10.1) | 8.0 (6.8 to 10.2) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.09 (0.84 to 1.42) | ████████████ | ||
Jaundice | ||||
Event/n (%) | 106/304 (34.9) | 101/322 (31.4) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 8.9 (7.4 to 11.1) | 8.4 (7.7 to 11.2) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.00 (0.76 to 1.32) | ████████████ | ||
Pain | ||||
Event/n (%) | 92/299 (30.8) | 94/320 (29.4) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 10.9 (7.9 to NC) | 11.2 (7.7 to 13.7) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.93 (0.70 to 1.25) | ████████████ | ||
Anxiety | ||||
Event/n (%) | 100/302 (33.1) | 91/317 (28.7) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 10.9 (7.2 to 12.0) | 9.1 (7.8 to 12.7) | ████████████ | ████████████ |
HR or OR (95% CI)a | 0.99 (0.74, 1.32) | ████████████ | ||
Tiredness | ||||
Event/n (%) | 144/297 (48.5) | 152/317 (47.9) | ████████████ | ████████████ |
Median TTD, months (95% CI) | 3.5 (2.4 to 4.5) | 3.7 (2.9 to 5.7) | ████████████ | ████████████ |
HR or OR (95% CI)a | 1.04 (0.82 to 1.31) | ████████████ | ||
BOR = best overall response; CI = confidence interval; Cis = cisplatin; D = durvalumab; EORTC QLQ-BIL21 = European Organisation for Research and Treatment of Cancer 21-Item Cholangiocarcinoma and Gallbladder Cancer Quality of Life Questionnaire; Gem = gemcitabine; HR = hazard ratio; IA −2 = second interim analysis; N/A = not applicable; NC = not calculable; OR = odds ratio; PBO = placebo; PRO = patient-reported outcome; TTD = time to deterioration.
aHRs are presented for time to deterioration. HRs < 1 favour durvalumab plus gemcitabine plus cisplatin. ORs are presented for improvement rates based on BOR. ORs > 1 favour durvalumab plus gemcitabine plus cisplatin.
Source: TOPAZ-1 CSR (IA-2).23
EQ-5D-5L
The impact of treatment and disease state on health state utility, assessed with the EQ-5D-5L, was an exploratory outcome of the TOPAZ-1 trial. ███████████ ███ █████ ███████████ █ █████ ██ ██████ ███ ███ ████ ██████ ████ █████████ █████ █████ ██████ ██████████████ ███████ ███████████████ ██ ████████████ ████ ███████ ██ ██ ████ ██████ ██████████████ ███████████ █████████ ████ ██ ███████████ ██████ ██████████ ████ ██ ██████████ ████ ██████ ████ ████████████ █████ ███ ███████ ███████ ██████████████ █████ ██████████ ███ ███ ████████ ████ █████████ ██████ ██████████ ████████ ███████████████ ███████ ████████ ██ █████████ ████████ █████████ ██████████████ ███████████████ ██████. Results for change from baseline in EQ-5D-5L index score are summarized in Figure 8. ███ ███████████████ ████████████ ██████████████████ █████ ██████████ ███████ █████ ████████████████████ ██████████ ███████████ ████ ███████ ██ █ ███ ███████ █████ ████ ████ ████████ ██████ ██████ ███████████ █ ██ ██████ ███ █████ █ ██ ██████████ ███████ █████████ ████ █████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██████████ ███████████ ██████ ██ █████████████. Results for change from baseline in EQ-VAS are summarized in Figure 9. ███ ███ ████ █████ █████ ██████████ █████ ███████████ ███████ ███████████████ ██ ███ ██ █████ ████ ████████████ ██████ █████ ██ ██ ████ ██████ ████████ ███████████ ███████████ ██████████████ ███ ██████ ████████ ██████████ ██ ███ ██ ███████ ████ ████████ ███ ████████ ██ ██ █████ ███ ███████ ████ ████ ████ ███ █████████████.
Symptom Severity
PGI-S of Cancer Symptoms
PGI-S was an exploratory outcome of the TOPAZ-1 trial. ██████████████ █████████ █████ █ ██ ████████ ██ ███████ ██████ ███ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ███████ ███ ██████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██████ ████ ██████ ████████ ██ ██ ███████ ██████ ██████ █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████ ████ ██ ████ ██ ██████ ██████ ████ ███ ████ ███████████████ ███████████████████ ██ ████████ ██████████████████ ███████████ ████ ██████████████ ███████ █████████████████████ ████ ██████████ ███████ ███████ ████ ███ ████ █████ ██████████████ █████ █████ █ ██ ██████████ ███████ █████ ████ █████████ ████ ████ ██████ ██ ████ ██ ██████ ██████████ ███████████████████ █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ████ ██████ ███████████ █ ██ ██████████ ███████ █████ ████ ████ █████ ████ ██████████ ██ ████ ██ ██████ ██████████ ████ ███████ █ ██ ██████████ ███████ █████████ ███ ██████ ████ ██████████ ██ ████ ██ ██████ ████ ██████ ███████████ █ ██ ██████████ ███████ █████████ ███ ██████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██████████████.
Treatment Tolerability
PRO-CTCAE
Treatment tolerability, assessed with the PRO-CTCAE, was an exploratory outcome of the TOPAZ-1 trial. █████ █ ██ ████ ██ ████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ █████████ █ ██ ██ █████ ███ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ █████ █████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ████████ ██ ██ █ ███ ███ █ ██ █████ █████ ███████ █████████ █ ████████ ████ ████ ██████ ██ ████ ██ ██████ ██████ ████ ██ ██ ███████ █ ██ ███████ ███ ███████ ███ ██████ █████████ ████ ██████████ ██ ████ ██ ██████ ███ ████ ███ ██ ██ ██ █████ █ ██ ██████████ ███████ █████ ████ ██████ ███ ████ █████ █████ ██ ████ ██ ██████ ██████████ ██ ██ ███ ████ █ ██ ███████ ███ ███████ █████████ █████ ████ ████ ██████████ ██ ████ ██ ██████ █ ███████ ██ ██ ███ ██████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █████ █████ ███████████ █ ██ ██████ ████ ███████ ███ ██████ █████████ ████ ████ ██████ ██ ████ ██ █████ █ ███ ██ █████ █████ ██████ █ ██ ██████████ ███████ █████████ █ ████████ ███ █ ██████████ ██ ████ ██ ██████ ██ ████████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ █ █ ██████ ████ ██████ ████ ███████ ██ █████ ██████ ██████ ███ ████ ████ █████ ████████ ██████████ █████ █████████ ██████ ██████.
Harms
Only harms identified in the review protocol are reported here. Refer to Table 22 and Table 23 for detailed harms data from IA-2 and the 6.5-month update.
Adverse Events
At both IA-2 and the 6.5-month update, TEAEs were reported in 99.4% and 98.8% of patients in the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups, respectively. The most frequently reported TEAEs in the durvalumab plus gemcitabine and cisplatin group included anemia (163 [48.2%]), nausea (137 [40.5%]), constipation (109 [32.2%]), and neutropenia (107 [31.7%]). The most frequently reported TEAEs in the placebo plus gemcitabine and cisplatin group were anemia (154 [45.0%]), nausea (119 [34.8%]), decreased neutrophil count (106 [31.0%]), and neutropenia (102 [29.8%]). Nausea was the only TEAE that had a greater than 5% difference between durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin (40.5% versus 34.8%). AEs with known association to durvalumab were reported at a higher rate with durvalumab plus gemcitabine and cisplatin than with placebo plus gemcitabine and cisplatin, including rash (11.8% versus 7.9%), pruritis (12.7% versus 8.5%), and pyrexia (20.4% versus 17.0%).23
As of the 6.5-month update, grade 3 or 4 AEs were reported at a similar frequency in the 2 treatment groups, with a total of 250 (74.0%) and 257 (75.1%) in the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups, respectively. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █ ██ ████ ███ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █████ █████ ██████.
█████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ██ ██ ████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ██ ██████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████ ████ ██ ████ ██ ██████ ██████████ ██ █ ████ ████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ █ █ ██████ ██ █████ ████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██ ████ █ ███ ██████ ██████
Serious Adverse Events
SAEs at the 6.5-month follow-up were consistent with those in IA-2. SAEs were well balanced across treatment groups and were reported for ████████ ████████ ███████ patients in the durvalumab plus gemcitabine and cisplatin group and ███ █████ ███████ ████████ patients in the placebo plus gemcitabine and cisplatin group.23,24
█████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ██ ██ █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ███ ███ ████ ███ ████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ █████ █ ██ ██████████ ███████ █████████ █████████ ████████ █████████████████████
Withdrawals Due to Adverse Events
At IA-2 of the TOPAZ-1 study, 44 (13.0%) patients in the durvalumab plus gemcitabine and cisplatin group and 52 (15.2%) patients in the placebo plus gemcitabine and cisplatin group discontinued treatment because of AEs. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ██ ██ ██ █ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ █ ██ ████ ███ ██████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ██ █████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██████.
At the 6.5-month update, 1 patient in the durvalumab plus gemcitabine and cisplatin group was removed from the dataset, as the withdrawal due to asthenia was considered to be related to disease progression by the investigator, for a total of 43 (12.7%) patients who discontinued due to AEs. █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █████ █████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ███ ███ ██████████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ █ ██ ███ ████ ██ ████ ██ ██████ ███ ███████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██ ███ █████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ███ ███████ ██ ████ ██ ██████ ██████████ ███████ ████████ ██████████████.
Mortality
At IA-2, of the 341 patients who were randomized to receive durvalumab plus gemcitabine and cisplatin, 12 (3.6%) patients experienced fatal AEs. There were 14 (4.1%) patients who experienced fatal AEs in the placebo plus gemcitabine and cisplatin group due █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ████ ████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ████ ██ ██ ██ ████ ███████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ███ ██████ █ ████ ███████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ████████ ███████████ ██████████.
At the 6.5-month update, 1 additional AE leading to death was reported in the durvalumab plus gemcitabine and cisplatin group, for a total of 13 (3.8%); AE-related deaths in the placebo plus gemcitabine and cisplatin remained at 14 (4.1%). As of the 6.5-month update, █████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ████ ███████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ███ █ ██ █ ███ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ██ ██████ ███████████ ██████ ████████████.
Table 22: Summary of Harms (Safety Analysis Set)
Harms | IA-2 (DCO: August 11, 2021) | 6.5-month update (DCO: February 25, 2022) | ||
|---|---|---|---|---|
D + Gem and Cis (N = 338) | PBO + Gem and Cis (N = 342) | D + Gem and Cis (N = 338) | PBO + Gem and Cis (N = 342) | |
Any AE, n (%)a | ||||
Patients with any AE | 336 (99.4) | 338 (98.8) | ██████████ | █████████████ |
Anemia | 163 (48.2) | 153 (44.7) | ██████████ | █████████████ |
Nausea | 136 (40.2) | 117 (34.2) | ██████████ | █████████████ |
Constipation | 108 (32.0) | 99 (28.9) | ██████████ | █████████████ |
Neutropenia | 107 (31.7) | 102 (29.8) | ██████████ | █████████████ |
Fatigue | 91 (26.9) | 90 (26.3) | ██████████ | █████████████ |
Neutrophil count decrease | 91 (26.9) | 106 (31.0) | ██████████ | █████████████ |
Decreased appetite | 87 (25.7) | 79 (23.1) | ██████████ | █████████████ |
Platelet count decrease | 70 (20.7) | 79 (23.1) | ██████████ | █████████████ |
Pyrexia | 68 (20.1) | 56 (16.4) | ██████████ | █████████████ |
Vomiting | 62 (18.3) | 62 (18.1) | ██████████ | █████████████ |
Diarrhea | 57 (16.9) | 51 (14.9) | ██████████ | █████████████ |
Abdominal pain | 47 (13.9) | 58 (17.0) | ██████████ | █████████████ |
Asthenia | 48 (14.2) | 48 (14.0) | ██████████ | █████████████ |
Thrombocytopenia | 43 (12.7) | 45 (13.2) | ██████████ | █████████████ |
Pruritus | 38 (11.2) | 28 (8.2) | ██████████ | █████████████ |
Rash | 38 (11.2) | 27 (7.9) | ██████████ | █████████████ |
Abdominal pain upper | 35 (10.4) | 30 (8.8) | ██████████ | █████████████ |
WBC count decrease | 37 (10.9) | 46 (13.5) | ██████████ | █████████████ |
Insomnia | 32 (9.5) | 36 (10.5) | ██████████ | █████████████ |
ALT increased | 29 (8.6) | 35 (10.2) | ██████████ | █████████████ |
Patients with ≥ 1 SAE, n (%)b | ||||
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
Patients who stopped treatment due to AEs, n (%)c | ||||
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
Deaths, n (%) | ||||
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
█████████████ | ██████████ | ██████████ | ██████████ | █████████████ |
AE = adverse event; ALT = alanine transaminase; Cis = cisplatin; D = durvalumab; DCO = data cut-off; Gem = gemcitabine; GI = gastrointestinal; IA-2 = second interim analysis; IRR = infusion-related reactions; PBO = placebo; SAE = serious adverse event; WBC = white blood cell.
aFrequency of ≥ 10% of patients in either treatment group.
bReported for ≥ 2% of patients in either treatment group.
cReported for 2 or more patients in either treatment group.
Sources: TOPAZ-1 CSR (IA-2 and 6.5-month update).23,24
Notable Harms
Notable harms of interest to this review, summarized in Table 23, included imAEs, IRRs, infections, and GI events.
Immune-Mediated AEs
At IA-2, imAEs were identified for 43 (12.7%) patients in the durvalumab plus gemcitabine and cisplatin group and 16 (4.7%) patients in the placebo plus gemcitabine and cisplatin group. The most common imAEs with durvalumab plus gemcitabine and cisplatin were hypothyroidism (20 [5.9%] versus 5 [1.5%]) and dermatitis or rash (12 [3.6%] versus 1 [0.3%]). Grade 3 or 4 imAEs occurred in 8 (2.4%) and 5 (1.5%) patients treated with durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin, respectively.23
█████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████ █ ███ ██ █████ ████ █ ██ ██████████ ███████ █████████ ██ ███████ ████ ██████████ ██ ████ ██ ██████ ████████ ██ ██ █████████ █ ██ ██████████ ███████ █████ ████ █████ ████ ████ ██████████ ██ ████ ██ ██████ ███ ███ ████ ██ ██ ██ █████ █ ██ ███████ ███ ███████ █████ ████ ████ █████ ████ ██████████ ██ ████ ██ ██████ █ ████ █████ ████ ███████ █ ██ ██████████ ███████ ██████ ███ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ██████ ██ ██ █████████ █ ██ ███ ███████ ████ ███ ████ █████ █████████ ████ ██████████ ██ ████ ██ ██████ ███ ████ ███ ██████ ███ █████ ██████████ ████████ ██████
Infusion-Related Reactions
█████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ███ █████ █ ██ ██████ ████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███████ ████████ ███████
Infections
█████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ██ ██████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ █ ████ ███ ██ █████████ █ ██ ████ ██████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █ ██ ███ ████ █ ██ ████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █ ███ █ ██ ███ ███████████ █ ██ ██ ████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ █ █████ ██ ███ █████ ███████████ █ ██ ███████ ███ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██ ████ ███ ███████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██ ████ ██ ██ ██████ ███████████ █ ██ █████ █████ ████ ███ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █ █████ ████ ███████████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ █ █████████ ███████████ █ ██ ████████ ██ ███████ █████████ █████████ ████ ██ ██ ███ ███ ██ ████ ██ █ █████ ██████████ ███████████ █ ██ ████████ ██ ███████ █████████ █████████ ████ █ █ ████████ ██ ████ ██ ██████ ██████████ ███████████ █ ██ ███ ███████ ███████ █████████ █████████ ████ ██ ████████ ██ ████ ██ ██████ ██████████ █████ █ ██ ██████████ ███████ █████████ █████████ ██ ██ ████ █████ █████ ██████
GI Events
Given the mechanism of action of durvalumab, GI events were considered of interest to this review. Any GI AEs are reported in Table 22, and GI imAEs are reported in Table 23.
█████ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██████████ ███ ██ ████ ██ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ██ ██ ████ ███ █████ ███ █ ██ ██████████ ███████ ███ ██████ ██ ███████ ████ ████ █████ █ ██ ████ ██ ██████ █████ █████ ███ ██████ ██ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██ ████ ███ ███████ ███████ ████ █ ██ ██ ████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██ ████ ████ ██████ ██████ █████ █ ██ ██████████ ███████ ██████ ███ ████ █████ ████ ██████████ ██ ████ ██ ██████ ██ ████████ ███ ██████████ █████ ████ ██████████ ██████
██ ███ █ ██ ██████████ ███████ █████████ █████████ ████ ███████ ███ ██ ████ ██ ██████ ██████████ ████ ██ █████ █ ██ ██████████ ███████ █████████ ███ ██████ ████ █████ █████ ██ ████ ██ ██████ ██████████ ██ ████ █████ █ ██ ██████████ ███████ █████████ ████ █████ ████ ██████████ ██ ████ ██ ██████ ██████████ ██ ████ ██ ███ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ████ █ █████ █████ ███ ███ █ ██ ██████████ ███████ █████████ █████████ ████ ██████████ ██ ████ ██ ██████ ██ ████████ ███████████████ █████ █████ █████ █████ █████
Table 23: Summary of Notable Harms (Safety Analysis Set)
Notable harms | IA-2 (DCO: August 11, 2021) | 6.5-month update (DCO: February 25, 2022) | ||
|---|---|---|---|---|
D + Gem and Cis (N = 338) | PBO + Gem and Cis (N = 342) | D + Gem and Cis (N = 338) | PBO + Gem and Cis (N = 342) | |
imAEs, n (%) | ||||
Any imAE | ██████████ | ██████████ | ██████████ | ██████████ |
Pneumonitis AEs | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
GI AEs | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
Hepatic AEs | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
Endocrinopathy AEs | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
Dermatitis or rash AEs | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
Pancreatic AEs | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
Myositis AEs | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
IRRs, n (%) | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
Infections, n (%) | ||||
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
██████████ | ██████████ | ██████████ | ██████████ | ██████████ |
AE = adverse event; ALT = alanine aminotransferase; Cis = cisplatin; D = durvalumab; DCO = data cut-off; Gem = gemcitabine; GI = gastrointestinal; IA-2 = interim analysis 2; ILD = interstitial lung disease; imAE = immune-mediated adverse event; IRR = infusion-related reaction; PBO = placebo.
Sources: TOPAZ-1 CSR (IA-2 and 6.5-month update).23,24
Critical Appraisal
Internal Validity
TOPAZ-1 was an international, double-blind, phase III RCT. Appropriate methods of randomization, treatment allocation, and stratification were employed using IVRS or IWRS. The methods for randomization were considered by the CADTH review team to result in a low risk of bias, as patients were randomly assigned a treatment group using technology that ensured that allocation was concealed until treatment assignment. Patients did not differ with regard to baseline demographic or disease characteristics, indicating that randomization was successful. Most patients discontinued treatment due to PD, per RECIST criteria, with approximately 13% and 10% more patients in the placebo plus gemcitabine and cisplatin group than in the durvalumab plus gemcitabine and cisplatin group experiencing disease progression at IA-2 and the 6.5-month follow-up, respectively. There was potential for unblinding in the TOPAZ-1 study because of differences in rates of disease progression and known treatment-related toxicities associated with durvalumab, such as imAEs. Overall, withdrawals and discontinuations due to AEs were similar across treatment groups and, therefore, likely did not result in unblinding; methods to maintain blinding were also appropriate, although the number of patients who were unblinded for various reasons was not reported.
The original study protocol was amended 6 times. It was unclear how many patients were enrolled at each protocol amendment, so the impact of potential biases due to protocol amendments remains unknown. Overall, important protocol deviations in the TOPAZ-1 study were low, although they occurred at a slightly higher frequency in the durvalumab plus gemcitabine and cisplatin group, primarily due to randomization issues that led patients to receive alternative treatment to that which they were randomized; this occurring only for patients in the durvalumab group (2.3% versus 0.0%). Given the low proportion of patients for which this deviation occurred, the CADTH review team considered the impact to be negligible.
Two interim analyses and 1 final analysis were planned for this study. At IA-2, the study met its primary end point (██ ████ ██ ██ ███ ███████ ███████████ █████) and was considered the final analysis by the Independent Data Monitoring Committee. Trials that stop early for benefit may show a higher or better treatment effect estimate in the intervention group, and this must be considered.25,26 With only ██ ████ ██ ████████ ████ ██████ ████ █ █████████, a risk of overestimation may be present, although the presence and extent is uncertain. The 6.5-month follow-up was performed at 76.9% maturity for OS, which demonstrated consistent benefit, compared to IA-2, although comparisons between DCOs were not formally conducted. Additionally, the primary efficacy outcome of OS was based on an assumed 4-month improvement on a control of 11.7 months and an assumed average OS HR of 0.745. The rationale for the choice of 4-month improvement or average OS HR was also unclear; therefore, it was remains uncertain whether these assumptions were appropriate.
The primary outcome for analysis at IA-1 was ORR and DOR, to support early registration. Both outcomes were measured by investigator assessment and BICR. After IA-1, all outcome analyses were only assessed by the investigator, and BICR was not conducted at IA-2. Although unlikely to affect objective end points such as OS, where the event is not subjective, given the use of RECIST criteria for other study end points, there is the potential for exaggeration or bias in estimated the treatment effect when reviewed only by the investigator. Considering the double-blind nature of in the TOPAZ-1 study, the risk of bias is likely low, but remains uncertain due to a lack of BICR assessment at the final analysis (IA-2).
Acceptable methods to account for multiplicity were conducted in the TOPAZ-1 trial for the primary efficacy end point of OS (using the family-wise error rate) and the selected key secondary end point of PFS (using a hierarchical testing procedure). Other secondary outcomes, including ORR, DOR, EORTC QLQ-C30 and EORTC QLQ-BIL21, and exploratory outcomes, including PGI-S, PRO-CTCAE, and EQ-5D-5L, were not controlled for multiplicity, so need to be interpreted in light of the increased risk of type I error. Missing dates were imputed, per the SAP, although missing safety data were generally not imputed. The amount of missing data was generally not reported for most efficacy outcomes; therefore, the impact of missing data on efficacy outcomes remains unknown.
A Cox proportional hazards model was used for OS and PFS outcomes to examine the assumption of proportionality. Proportionality was assumed initially; however, there was violation of the proportional hazards assumption at approximately 6 months of treatment, when there was a delayed separation of the curves for durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin. The sponsor noted that the likely reason for violation was the delayed mechanism of action of immunotherapy, which was considered reasonable by the CADTH review team. In a posthoc analysis, the piecewise HR after separation of the curves (i.e., after 6 months) was consistent with the primary analysis.
Predefined sensitivity analyses were conducted to rule out attrition bias and to determine the effect of covariates on HR estimates. Predefined subgroup analyses were conducted to examine the consistency of the primary and secondary analysis results across subgroup levels. Overall, the results of sensitivity and prespecified subgroup analyses were generally aligned with the overall analysis for OS, PFS, and ORR; however, it should be noted that the sensitivity analysis for potential attrition bias on OS was not statistically significant, increasing uncertainty in the results for OS. Additionally, sensitivity and subgroup analyses were not adjusted for multiplicity or missing data, and should be considered to be supportive of the overall effect of durvalumab.
For secondary HRQoL end points of EORTC QLQ-C30 and EORTC QLQ-BIL21, clinically meaningful change was defined as an increase or decrease of 10 points, indicating a clinically meaningful improvement or decline in HRQoL. This rationale was supported by findings of the study by Osoba et al. (1998)45 for the EORTC QLQ-C30; however, no rationale or MID was identified for the EORTC QLQ-BIL21, and no justification for a 10-point threshold was provided by the sponsor. The population included for HRQoL measures was made up of the PRO analysis set, which was added at the SAP amendment on June 7, 2021. As such, the PRO analysis set is only a subset of the FAS, and all patients were enrolled in the study before this change, although the impact of this remains unknown. All other PRO end points (PGI-S, PRO-CTCAE, and EQ-5D-5L) were exploratory and should be treated as such. Moreover, no statistical tests were conducted, and 95% CIs for effect estimates (HR or OR) were too imprecise, precluding the ability to draw strong conclusions about HRQoL or treatment tolerability. Additionally, for all time points for all HRQoL and PRO end points, there was a notable amount of missing data; thus, there was a high risk of attrition bias for these outcomes.
External Validity
In discussion with the clinical experts consulted by CADTH, the inclusion and exclusion criteria for the TOPAZ-1 trial were generally as expected for patients with locally advanced or metastatic BTC, although that histologic diagnosis is not always confirmed, they noted. In line with immunotherapy exclusion criteria, because of an elevated risk of imAEs, patients with active or prior documented autoimmune or inflammatory disorders were appropriately excluded. It was considered that patients included in the study may be a less sick population for various reasons, including ECOG PS, and the clinical experts noted that patients with an ECOG PS of 2 would also be eligible to receive durvalumab plus gemcitabine and cisplatin, provided they have adequate organ function. Baseline characteristics were well balanced, although the clinical experts consulted by CADTH highlighted some characteristics that were not reflective of clinical practice in Canada, primarily that there was an overrepresentation of Asian patients in the TOPAZ-1 trial (56.4%), which is not representative of what would be seen in Canada. This was a product of where the trial was conducted, as no Canadian sites were included, and most patients were enrolled from Asian countries (n = 374; 54.6%). Additionally, the experts thought there was a lower proportion of patients with locally advanced disease, although they did note that this proportion would be dependent on referral patterns, and patients with locally advanced and metastatic disease would receive the same treatment regardless. Regarding BTC subtypes, the experts felt that the proportion of patients with IHCC was higher and the proportion with EHCC was lower than is seen in the real world, which may have a subtle effect on prognosis.
The chosen comparator of standard platinum chemotherapy, consisting of gemcitabine plus cisplatin, in the TOPAZ-1 trial aligns with the recommended SOC guidelines in Canada, although there may be other options available, such as carboplatin or a single-drug regimen of gemcitabine. The median duration of treatment in the durvalumab plus gemcitabine and cisplatin and the placebo plus gemcitabine and cisplatin groups was 7.33 months and 5.77 months, respectively, reflecting the dosing schedule of the chemotherapy backbone of gemcitabine and cisplatin, which is only given for up to 8 cycles (approximately 6 months). The experts noted that, in some cases, clinicians may treat beyond 8 cycles if patients are tolerating treatment. The experts also stated that a single-drug regimen of gemcitabine may be continued beyond the 8 cycles, especially in the event of renal toxicities.
Outcomes of the TOPAZ-1 trial were clinically relevant and important to both clinicians and patients. A key aspect of new treatments, as noted by clinicians and patients, should be the extension or prolongation of life. The addition of durvalumab to the SOC resulted in a statistically significant improvement in OS and PFS, and the experts noted that the median OS for the comparator of placebo plus gemcitabine and cisplatin was in line with what is currently seen in clinical practice. However, the experts considered the median PFS for the comparator arm to be higher than expected, although, coupled with the assumption that patients may be less sick than patients seen in clinical practice, the elevated median PFS was considered reasonable. Maintenance of QoL was also considered a highly important outcome for new treatments, but the trial was not powered for this outcome, the results were not tested statistically, and there was a high risk of attrition bias owing to large amounts of missing data, especially at later time points. Further, HRQoL was not included in the statistical hierarchy and the effect estimates were imprecise, precluding the ability to draw conclusions for this outcome. Additionally, the relationship between improved survival and QoL was not evaluated, so it remains unclear whether patients with improved survival have improved QoL as well.
Subsequent anticancer treatment was evenly reported for durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin. The clinical experts consulted by CADTH noted that the proportion of patients receiving subsequent anticancer therapy was higher than expected, although likely reflected the location of patients enrolled in the trial. It was hypothesized that the elevated proportion of patients who received subsequent anticancer therapy was due to the baseline ECOG PS; patients likely remained fit enough for subsequent lines of therapy. As previously noted, this was not considered reflective of clinical practice in Canada; therefore, the proportion of patients receiving subsequent anticancer therapy and the type of subsequent anticancer therapy received may not be generalizable to the population in Canada.
Indirect Evidence
No indirect evidence was included in the sponsor’s submission to CADTH. A focused literature search for indirect treatment comparisons dealing with Imfinzi or durvalumab or biliary cancer was run in MEDLINE All (1946–) on August 11, 2022. No limits were applied to the search. A total of 50 citations were identified, and 1 met the inclusion criteria for this review.
An SLR and NMA were conducted by Feng et al. (2022),27 comparing the OS and PFS of different first-line treatment regimens for patients with advanced BTC using methods of a random-effects NMA. The NMA consisted of a comprehensive list of 19 different treatment regimens from 17 unique studies, including best supportive care, gemcitabine monotherapy, gemcitabine and cisplatin, gemcitabine and oxaliplatin, S-1 chemotherapy, S-1 chemotherapy and cisplatin, gemcitabine and S-1 chemotherapy, vandetanib, gemcitabine and vandetanib, fluorouracil and folinic acid (known as FUFA), oxaliplatin plus irinotecan and infusional fluorouracil (known as FOLFIRINOX), oxaliplatin, capecitabine and oxaliplatin (known as XELOX), erlotinib plus gemcitabine and oxaliplatin, cetuximab plus gemcitabine and oxaliplatin, panitumumab plus gemcitabine and oxaliplatin, cediranib plus gemcitabine and cisplatin, ramucirumab plus gemcitabine and cisplatin, merestinib plus gemcitabine and cisplatin, and durvalumab plus gemcitabine and cisplatin. Based on the CADTH review protocol, only comparisons involving the drug regimen under review (durvalumab plus gemcitabine and cisplatin) and relevant Canadian comparators were of interest (Table 4). As such, only comparisons between durvalumab plus gemcitabine and cisplatin and gemcitabine and cisplatin alone were of interest to this review (data for which came from an abstract of the pivotal TOPAZ-1 trial). There was insufficient evidence to draw conclusions for the outcome of OS (HR, 0.27; 95% CI, 0.06 to 1.29), whereas for PFS, the results produced by the NMA for comparisons of interest were in favour of durvalumab plus gemcitabine and cisplatin (HR, 0.22; 95% CI, 0.08 to 0.62).27 Although a large evidence base was available for gemcitabine and cisplatin, there was notable heterogeneity in the populations from the studies included in the NMA, including differences for age, ECOG PS, and primary tumour location, there were different proportions of patients with IHCC, EHCC, and GBC, and some trials included patients with AoV cancer. This heterogeneity likely resulted in notable imprecision, given the wide 95% CIs, and uncertainty in the comparative efficacy estimates.
Other Relevant Evidence
No long-term extension studies or other relevant studies were included in the sponsor’s submission to CADTH.
Discussion
Summary of Available Evidence
One phase III, double-blind, international RCT, TOPAZ-1, was included in this review. The TOPAZ-1 trial consisted of 685 patients with locally advanced or metastatic BTC, randomized in a 1:1 ratio to receive durvalumab (n = 341) 1,500 mg or placebo (n = 344) delivered by IV infusion every 3 weeks in combination with gemcitabine 1,000 mg/m2 and cisplatin 25 mg/m2 every 3 weeks on days 1 and 8, for up to 8 cycles, until disease progression or unacceptable toxicity. The primary end point of the TOPAZ-1 trial was OS, and secondary end points were PFS, ORR, DOR, and HRQoL.
Baseline characteristics of the TOPAZ-1 trial were well balanced in the 2 groups, with 11.1% and 16.6% of patients, respectively, in the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group having locally advanced disease, and 88.9% and 83.1% of patients, respectively, having metastatic disease. The median age of all patients was 64 years, 50.4% of patients were male, and 56.4% of patients were Asian. The majority of patients had IHCC (████ ████), followed by GBC (███ █ ████), and EHCC (███ ███ ██). The TOPAZ-1 trial may have enrolled a healthier group of patients; 49.1% and 50.9% of patients, respectively, had an ECOG PS of 0 or 1 and most had initially unresectable disease (███ █████). As of the most recent DCO (February 25, 2022), the median follow-up of the TOPAZ-1 trial was ██████ █████ ███ ████ ██ ████████ ██ █ ██ █████ ███ ███ ████ for the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups for OS.
Interpretation of Results
Efficacy
As previously mentioned, the inclusion and exclusion criteria for the TOPAZ-1 trial were generally as expected for patients with locally advanced or metastatic BTC and resulted in well balanced demographic and baseline characteristics across groups. The clinical experts consulted by CADTH noted that the proportion of patients with IHCC in the TOPAZ-1 trial was higher than is seen in North America, and the proportion with EHCC was lower than is seen in the real world. The experts hypothesized that this may have a subtle effect on prognosis. Subgroup analyses were generally consistent with the primary analysis, although because of the exploratory nature of subgroup analyses, small sample sizes, and the lack of control for multiplicity, no conclusions can be drawn, and results for subgroup analyses should only be viewed as supportive.
TOPAZ-1 met its primary end point at IA-2, demonstrating improved survival with the addition of durvalumab to the existing SOC, based on a median OS of 12.8 months (95% CI, 11.1 to 14.0 months) with durvalumab plus gemcitabine and cisplatin and 11.5 months (95% CI, 10.1 to 12.5 months) with placebo plus gemcitabine and cisplatin (HR, 0.80; 95% CI, 0.66 to 0.97). There is some additional uncertainty concerning the OS estimates, given that the sensitivity analysis accounting for attrition bias was no longer statistically significant. Similar OS results were observed with an additional 6.5 months of follow-up (12.9 months [95% CI, 11.6 to 14.1 months] versus 11.3 months [95% CI, 10.1 to 12.5 months]; HR, 0.76 [95% CI, 0.64 to 0.91]). As noted by the clinical experts consulted by CADTH, the observed median OS for the SOC arm was consistent with what has been demonstrated in previous trials,36 and with what is observed in clinical practice. According to patients and clinicians, prolonged survival remains the most important outcome of treatment for BTC. The median OS benefit with the addition of durvalumab to SOC was about 1.3 months and 1.6 months, respectively, at IA-2 and the 6.5-month follow-up. The clinical importance of the modest survival gains afforded by treatment with durvalumab compared with SOC is subjective and may vary from patient to patient. Considering that the TOPAZ-1 trial was stopped early for efficacy based on an overall maturity of OS of █████ █ ██ ██████████ ███████ █████████ █ █ █ ██████ ██ ████████, the possibility of a potentially overestimated treatment effect must be considered, and the true magnitude of the OS benefit is uncertain.25,26 According to the clinical experts consulted by CADTH, it is worth noting that the OS rates at 18 months and 24 months suggest that more patients achieve a longer OS benefit, reflecting the mechanism of action of immunotherapy.
The key secondary end point of the TOPAZ-1 trial was PFS. The median PFS for the placebo plus gemcitabine and cisplatin group was considered higher than what would be seen in clinical practice, per the experts consulted by CADTH. Overall, the median PFS was 7.2 months (95% CI, 6.7 to 7.4 months) with durvalumab and 5.7 months (95% CI, 5.6 to 6.7 months) with placebo. In line with the dosing regimen for gemcitabine and cisplatin, treatment is most often stopped after 8 cycles, which translates to roughly 6 months; thus, it appears that most patients progress after the prescribed SOC regimen, whereas durvalumab offered an additional 1.5 months without PD. Considering that the experts felt the patients included in the TOPAZ-1 trial were healthier than those seen in clinical practice, the magnitude of PFS benefit may also be greater in the trial than in clinical practice, although the presence and extent of any difference is unknown or cannot be predicted.
Objective response rate was also a secondary end point of the TOPAZ-1 trial. The ORR was in favour of durvalumab plus gemcitabine and cisplatin (26.7% versus 18.7%; OR, 1.60 [95% CI, 1.11 to 2.31]), which the clinical experts felt was in line with clinical practice, and durvalumab demonstrated some disease activity; however, the P value was not controlled for multiplicity, and must be interpreted in light of the increased risk of type I error. At IA-1, ORR was the primary end point to support early registration, and results were assessed by the investigator and BICR. Results for ORR were consistent between outcome assessors, but BICR was not performed for any end points beyond IA-1. Although unlikely to affect results for the primary end point of OS, other subjective end points, including PFS, ORR, HRQoL and AEs, may be associated with some bias because of the lack of BICR assessment, although the double-blind nature of the TOPAZ-1 trial may reduce the possibility of this bias.
Kaplan-Meier curves for OS and PFS generally displayed delayed separation due to the mechanism of action of durvalumab, which resulted in nonproportional hazards, although there were no concerns from the review team on this matter. In a posthoc analysis, the piecewise HR after separation of the curves (i.e., after 6 months) was consistent with the primary analysis. As previously mentioned, results for sensitivity analyses and subgroup analyses for OS, PFS, and ORR were performed and were generally in line with the primary analysis. One sensitivity analysis for OS that examined censoring patterns to identify attrition, although consistent with the primary analysis, was no longer statistically significant (█████ █ ██ ██████████ ███████ ███████ ██ █ ██ ██████ ██ ██████ ██ ████ ████), adding some uncertainty to the results for OS. Results for subgroup and sensitivity analyses should be interpreted with caution, as the study was not powered for any of the individual subgroup evaluations and no adjustments were made for multiplicity; therefore, results should only be viewed as supportive.
HRQoL and treatment tolerability comprised secondary end points of the TOPAZ-1 trial, assessed using the EORTC QLQ-C30 and EORTC QLQ-BIL21, and exploratory end points were assessed with the PGI-S, PRO-CTCAE, and EQ-5D-5L. Patients cited maintenance of QoL as an important outcome of treatment; however, the TOPAZ-1 study was not powered to evaluate these outcomes. Additionally, results from the HRQoL and PRO analyses were not controlled for multiplicity and must be interpreted in light of the possibility of type I error, and there was a high risk of attrition bias. Overall, effect estimates for these outcomes were imprecise, and no conclusions on HRQoL can be drawn. The clinical experts consulted by CADTH noted that durvalumab has been available for some time and is anecdotally well tolerated, so they would not expect durvalumab to be associated with notable deterioration. However, HRQoL end points were based on the PRO analysis set, which was a subset of the total population and was added after the enrolment of all patients. At baseline, HRQoL measures were completed by most patients in the PRO analysis set; however, the number of patients that completed the measures significantly dropped at later times of assessment, so there is a high risk of attrition bias. Because of the low sample sizes at later times of assessment, it remains unclear whether patients who experience the additional survival benefit of durvalumab also have an improved QoL, which is important, given the modest survival improvement, and, overall, the results for HRQoL may not be generalizable to patients in Canada.
Harms
The overall incidence of harms reported in the TOPAZ-1 trial was well balanced, with the exception of the known AEs of special interest associated with durvalumab. Known risks associated with durvalumab include imAEs and GI events such as diarrhea and colitis. From the patients’ perspective, a reduction in side effects from current treatment was considered highly important; however, in the durvalumab plus gemcitabine and cisplatin and placebo plus gemcitabine and cisplatin groups, the overall rates of AEs (99.4% versus 98.8%) and SAEs █████ ██████ █████ ███ █████ were similar. Only the preferred term of nausea was associated with a greater than or equal to 5% difference between the 2 treatment groups. Tolerability of treatment was also an outcome measured in the TOPAZ-1 trial (PRO-CTCAE, QLQ-BIL21 item 49); however, the analysis had the same issues as in the HRQoL analyses. Most initial AEs were experienced █████ █████████ ██ after treatment in both groups, although median time to onset for grade 3 or 4 AEs was ████ █ ██████████ █████ ██████ ██████ ██████ ███ ██ █████ days for the durvalumab plus gemcitabine and cisplatin group and the placebo plus gemcitabine and cisplatin group, respectively, which may reflect the delayed mechanism of action of durvalumab. Both withdrawals due to AEs and deaths due to AEs were also similar, occurring in 12.7% and 15.2% of patients, respectively, in the durvalumab plus gemcitabine and cisplatin group and in 3.8% and 4.1% of patients, respectively, in the placebo plus gemcitabine and cisplatin group. As previously mentioned, imAEs and IRRs are known AEs of special interest associated with durvalumab plus gemcitabine and cisplatin, and infections and GI events are common AEs observed in patients with BTC, per the clinical experts; therefore, the higher incidence of AEs for durvalumab plus gemcitabine and cisplatin than for placebo plus gemcitabine and cisplatin was not a concern, but the patient group emphasized the need for treatments with fewer side effects. According to the clinical experts consulted by CADTH, the AEs observed in the TOPAZ-1 study are manageable in clinical practice.
Conclusions
Patients and clinicians highlighted the need for new effective treatments that prolong life, maintain QoL, and reduce the side effects associated with current treatments. In the randomized, phase III TOPAZ-1 trial, durvalumab plus gemcitabine and cisplatin demonstrated a statistically significant, albeit modest, improvement in median OS compared with placebo plus gemcitabine and cisplatin, which is the current SOC in Canada for patients with locally advanced or metastatic BTC. However, the clinical importance of the modest survival gain afforded by treatment with durvalumab compared with SOC is subjective and may vary from patient to patient, although the OS rates may also suggest that patients achieve longer OS benefit. Analyses of secondary outcomes, including PFS and ORR, supported the efficacy of adding durvalumab to the SOC. Because the findings for survival and response outcomes were based on an interim analysis, the benefits of durvalumab relative to SOC may be overestimated, although the presence and extent of any overestimation is uncertain. No definitive conclusions could be drawn about the impact of treatment with durvalumab compared with SOC on HRQoL or on time to deterioration, owing to limitations of the analyses, including a lack of statistical testing and risk of attrition bias. The safety of durvalumab is well known, considering its use in other indications; however, aside from the AEs specific to durvalumab, the frequency of AEs and SAEs was similar in the 2 treatment groups. Overall, although there are limitations associated with stopping the trial at IA-2 and surrounding the statistical analysis for secondary end points, the TOPAZ-1 study demonstrated improvement in OS with the addition of durvalumab to gemcitabine and cisplatin. However, given the modest gain in OS observed in the trial, the clinical benefit is likely to be incremental, and there remains uncertainty about the magnitude of clinical benefit in the locally advanced or metastatic BTC population in Canada.
References
1.Bridgewater JA, Goodman KA, Kalyan A, Mulcahy MF. Biliary tract cancer: epidemiology, radiotherapy, and molecular profiling. Am Soc Clin Oncol Educ Book. 2016;35(36):e194-203. PubMed
2.Valle JW, Kelley RK, Nervi B, Oh DY, Zhu AX. Biliary tract cancer. Lancet. 2021;397(10272):428-444. PubMed
3.National Cancer Institute. Bile duct cancer (cholangiocarcinoma) treatment (PDQ®): health professional version. 2022: https://www.cancer.gov/types/liver/hp/bile-duct-treatment-pdq. Accessed 2022 Aug 17.
4.Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Semin Liver Dis. 2004;24(2):115-125. PubMed
5.Vogel A, Bridgewater J, Edeline J, et al. Biliary tract cancer: ESMO clinical practice guideline for diagnosis, treatment and follow up. Ann Oncol. 2022;S0923-7534(22):04699-04693. https://www.esmo.org/guidelines/guidelines-by-topic/gastrointestinal-cancers/biliary-cancer. Accessed 2022 Nov 10.
6.Canadian Cancer Statistics Advisory Committee, Canadian Cancer Society, Statistics Canada, Public Health Agency of Canada. Canadian cancer statistics 2021. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf. Accessed 2022 Oct 6.
7.Zamani Z, Fatima S. Biliary tract cancer. [Updated 2022 Jul 17]. StatPearls. Treasure Island (FL): StatPearls Publishing; 2022: https://www.ncbi.nlm.nih.gov/books/NBK560550/. Accessed 2022 Sep 16.
8.Wistuba, II, Gazdar AF. Gallbladder cancer: lessons from a rare tumour. Nat Rev Cancer. 2004;4(9):695-706. PubMed
9.Cholangiocarcinoma and gallbladder cancer. Clinical practice guideline GI-010, version 5. Edmonton (AB): Alberta Health Services; 2019 Mar: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-gi010-biliary.pdf. Accessed 2022 Aug 17.
10.Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001;33(6):1353-1357. PubMed
11.Xiao Y, Cattelan L, Lagacé F, et al. Epidemiologic trends and geographic distribution of patients with gallbladder and extrahepatic biliary tract cancers in Canada. HPB (Oxford). 2021;23(10):1541-1549. PubMed
12.Flemming JA, Zhang-Salomons J, Nanji S, Booth CM. Increased incidence but improved median overall survival for biliary tract cancers diagnosed in Ontario from 1994 through 2012: a population-based study. Cancer. 2016;122(16):2534-2543. PubMed
13.Meza-Junco J, Montano-Loza AJ, Ma M, Wong W, Sawyer MB, Bain VG. Cholangiocarcinoma: has there been any progress? Can J Gastroenterol. 2010;24(1):52-57. PubMed
14.Marcano-Bonilla L, Mohamed EA, Mounajjed T, Roberts LR. Biliary tract cancers: epidemiology, molecular pathogenesis and genetic risk associations. Chin Clin Oncol. 2016;5(5):61. PubMed
15.Hezel AF, Deshpande V, Zhu AX. Genetics of biliary tract cancers and emerging targeted therapies. J Clin Oncol. 2010;28(21):3531-3540. PubMed
16.Nakeeb A, Pitt HA, Sohn TA, et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg. 1996;224(4):463-473; discussion 473-465. PubMed
17.Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557-588. PubMed
18.PrPEMAZYRE™ (pemigatinib): tablets, 4.5 mg, 9 mg, and 13.5 mg, Oral [product monograph]. Wilmington (DE): Incyte Corporation; 2021 Sep 8: https://pdf.hres.ca/dpd_pm/00062968.PDF. Accessed 2022 Sep 16.
19.CADTH reimbursement recommendation: pemigatinib (Pemazyre) for cholangiocarcinoma. Can J Health Technol. 2022;2(4):1-15. https://www.cadth.ca/sites/default/files/DRR/2022/PC0252%20Pemazyre%20-%20CADTH%20Final%20Rec.pdf. Accessed 2022 Sep 16.
20.Imfinzi (durvalumab for injection): concentrate for solution for infusion, 50 mg / mL, intravenous [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2022.
21.CADTH reimbursement review: durvalumab (Imfinzi) for extensive-stage small cell lung cancer. Can J Health Technol. 2021;1(9):1-125. https://www.cadth.ca/sites/default/files/DRR/2021/PC0234-combined-final.pdf. Accessed 2022 Oct 6.
22.pan-Canadian Oncology Drug Review final clinical guidance report: Durvalumab (Imfinzi) for non-small cell lung cancer. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pcodr/Reviews2019/10131DurvalumabNSCLC_fnCGR_NOREDACT_Post_03May2019_final.pdf. Accessed 2022 Oct 6.
23.Clinical study report: durvalumab-D933AC00001. A phase III randomized, double-blind, placebo-controlled, multi-regional, international study of durvalumab in combination with gemcitabine plus cisplatin versus placebo in combination with gemcitabine plus cisplatin for patients with first-line advanced biliary tract cancers (TOPAZ-1) [internal sponsor's report]. Gaithersburg (MD): AstraZeneca Pharmaceuticals LP; 2022 Feb 21.
24.Clinical study report addendum (6.5 month update): durvalumab-D933AC00001. A phase III randomized, double-blind, placebo-controlled, multi-regional, international study of durvalumab in combination with gemcitabine plus cisplatin versus placebo in combination with gemcitabine plus cisplatin for patients with first-line advanced biliary tract cancers (TOPAZ-1) (6.5 Month Update) [internal sponsor's report]. Gaithersburg (MD): AstraZeneca Pharmaceuticals LP; 2022 May 20.
25.Guyatt GH, Briel M, Glasziou P, Bassler D, Montori VM. Problems of stopping trials early. BMJ. 2012;344:e3863. PubMed
26.Bassler D, Briel M, Montori VM, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA. 2010;303(12):1180-1187. PubMed
27.Feng L, Wang Y, Xu H, Yi F. Comparison of different first-line systemic therapies in advanced biliary tract cancer based on updated random controlled trials: a systematic review and network meta-analysis. Biomed Res Int. 2022;2022:1720696. PubMed
28.Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 Suppl(0):S2-6.
29.European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182-236. PubMed
30.Zhang Y, Yang C, Huang M. Global, regional, and national burden of biliary tract cancer from 1990 to 2017 based on the 2017 Global Burden of Disease study. Cancer Epidemiol. 2021;73:101949. PubMed
31.Shin HR, Oh JK, Masuyer E, et al. Comparison of incidence of intrahepatic and extrahepatic cholangiocarcinoma: focus on East and South-Eastern Asia. Asian Pac J Cancer Prev. 2010;11(5):1159-1166. PubMed
32.Benavides M, Antón A, Gallego J, et al. Biliary tract cancers: SEOM clinical guidelines. Clin Transl Oncol. 2015;17(12):982-987. PubMed
33.Charbel H, Al-Kawas FH. Cholangiocarcinoma: epidemiology, risk factors, pathogenesis, and diagnosis. Curr Gastroenterol Rep. 2011;13(2):182-187. PubMed
34.Randi G, Malvezzi M, Levi F, et al. Epidemiology of biliary tract cancers: an update. Ann Oncol. 2009;20(1):146-159. PubMed
35.Ghidini M, Pizzo C, Botticelli A, et al. Biliary tract cancer: current challenges and future prospects. Cancer Manag Res. 2019;11:379-388. PubMed
36.Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273-1281. PubMed
37.Lamarca A, Palmer DH, Wasan HS, et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021;22(5):690-701. PubMed
38.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
39.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jul 28.
40.Oh DY, He AR, Qin S, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evidence. 2022;1(8):EVIDoa2200015.
41.Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab for injection), concentrate for solution for infusion, 50 mg / mL, intravenous [internal sponsor's package]. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Jul 13.
42.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
43.Friend E, Yadegarfar G, Byrne C, et al. Development of a questionnaire (EORTC module) to measure quality of life in patients with cholangiocarcinoma and gallbladder cancer, the EORTC QLQ-BIL21. Br J Cancer. 2011;104(4):587-592. PubMed
44.Kaupp-Roberts SD, Yadegarfar G, Friend E, et al. Validation of the EORTC QLQ-BIL21 questionnaire for measuring quality of life in patients with cholangiocarcinoma and cancer of the gallbladder. Br J Cancer. 2016;115(9):1032-1038. PubMed
45.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
46.Trask PC, Dueck AC, Piault E, Campbell A. Patient-reported outcomes version of the common terminology criteria for adverse events: methods for item selection in industry-sponsored oncology clinical trials. Clin Trials. 2018;15(6):616-623. PubMed
47.Dueck AC, Mendoza TR, Mitchell SA, et al. Validity and reliability of the US National Cancer Institute's Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). JAMA Oncol. 2015;1(8):1051-1059. PubMed
48.U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. Washington (DC): U.S. Department of Health and Human Services; 2017: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5x7.pdf. Accessed 2022 Oct 7.
49.Gail M, Simon R. Testing for qualitative interactions between treatment effects and patient subsets. Biometrics. 1985;41(2):361-372. PubMed
50.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
51.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
52.Fayers PM, Aaronson NK, Bjordal K, et al. The EORTC QLC-C30 scoring manual. 3rd ed. Brussels (BE): European Organisation for Research and Treatment of Cancer; 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2022 Sep 14.
53.Snyder CF, Blackford AL, Sussman J, et al. Identifying changes in scores on the EORTC-QLQ-C30 representing a change in patients' supportive care needs. Qual Life Res. 2015;24(5):1207-1216. PubMed
54.Kluetz PG, Chingos DT, Basch EM, Mitchell SA. Patient-reported outcomes in cancer clinical trials: measuring symptomatic adverse events with the National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). ASCO Educational Book. 2016(36):67-73. https://ascopubs.org/doi/abs/10.1200/EDBK_159514. Accessed 2022 Sep 16.
55.van Reenen M, Janssen B, Oppe M, Kreimeier S, Greiner W, Stolk E. EQ-5D-Y User Guide. Rotterdam (NL): EuroQoL Research Foundation; 2020 Sep: https://euroqol.org/publications/user-guides. Accessed 2022 Sep 16.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: August 10, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
(imfinzi* or durvalumab* or medi 4736 or medi4736 or 28X28X9OKV).ti,ab,kf,ot,hw,rn,nm.
biliary tract neoplasms/ or exp bile duct neoplasms/ or gallbladder neoplasms/
exp Cholangiocarcinoma/
((bile duct* or biliary or hepatobiliary or bile tract* or hepatic duct* or hepatocellular* or choledochus*) adj5 (cancer* or tumor* or tumour* or carcinoma* or neoplas* or malignan* or adenocarcinoma*)).ti,ab,kf,ot.
((gall bladder* or gallbladder* or gall duct*) adj5 (cancer* or tumor* or tumour* or carcinoma* or neoplas* or malignan* or adenocarcinoma*)).ti,ab,kf,ot.
((cholangio* or ampulla*) adj5 (cancer* or tumor* or tumour* or carcinoma* or neoplas* or malignan* or adenocarcinoma*)).ti,ab,kf,ot.
cholangiocarcinoma*.ti,ab,kf,ot.
or/2-7
1 and 8
9 use medall
*durvalumab/
(imfinzi* or durvalumab* or medi 4736 or medi4736).ti,ab,kf,dq.
or/11-12
exp biliary tract cancer/
((bile duct* or biliary or hepatobiliary or bile tract* or hepatic duct* or hepatocellular* or choledochus*) adj5 (cancer* or tumor* or tumour* or carcinoma* or neoplas* or malignan* or adenocarcinoma*)).ti,ab,kf,dq.
((gall bladder* or gallbladder* or gall duct*) adj5 (cancer* or tumor* or tumour* or carcinoma* or neoplas* or malignan* or adenocarcinoma*)).ti,ab,kf,dq.
((cholangio* or ampulla*) adj5 (cancer* or tumor* or tumour* or carcinoma* or neoplas* or malignan* or adenocarcinoma*)).ti,ab,kf,dq.
cholangiocarcinoma*.ti,ab,kf,dq.
or/14-18
13 and 19
20 use oemezd
(conference review or conference abstract).pt.
21 not 22
10 or 23
remove duplicates from 24
Clinical Trials Registries
ClinicalTrials.gov
Produced by the U.S. National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search terms – (Imfinzi or durvalumab) and (Biliary Tract Cancer or Bile Duct Cancer or Bile Duct Neoplasm or Hepatobiliary Cancer or Hepatobiliary Neoplasm or Cholangiocarcinoma or Gall Bladder Cancer or Ampullary Adenocarcinoma)]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms – Imfinzi, durvalumab, biliary cancer, hepatobiliary, gall bladder]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – imfinzi, durvalumab, cholangiocarcinoma, biliary cancer, hepatobiliary cancer, gall bladder cancer, bile duct cancer, ampullary adenocarcinoma]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – imfinzi, durvalumab, cholangiocarcinoma, biliary cancer, hepatobiliary cancer, gall bladder cancer, bile duct cancer, ampullary adenocarcinoma]
Grey Literature
Search dates: August 11-12, 2022
Keywords: imfinzi, durvalumab, cholangiocarcinoma, biliary cancer, hepatobiliary cancer, gall bladder cancer, bile duct cancer, ampullary adenocarcinoma
Limits: no limits applied
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search.
Appendix 2: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Figure 3: Kaplan-Meier Plot of OS (FAS)
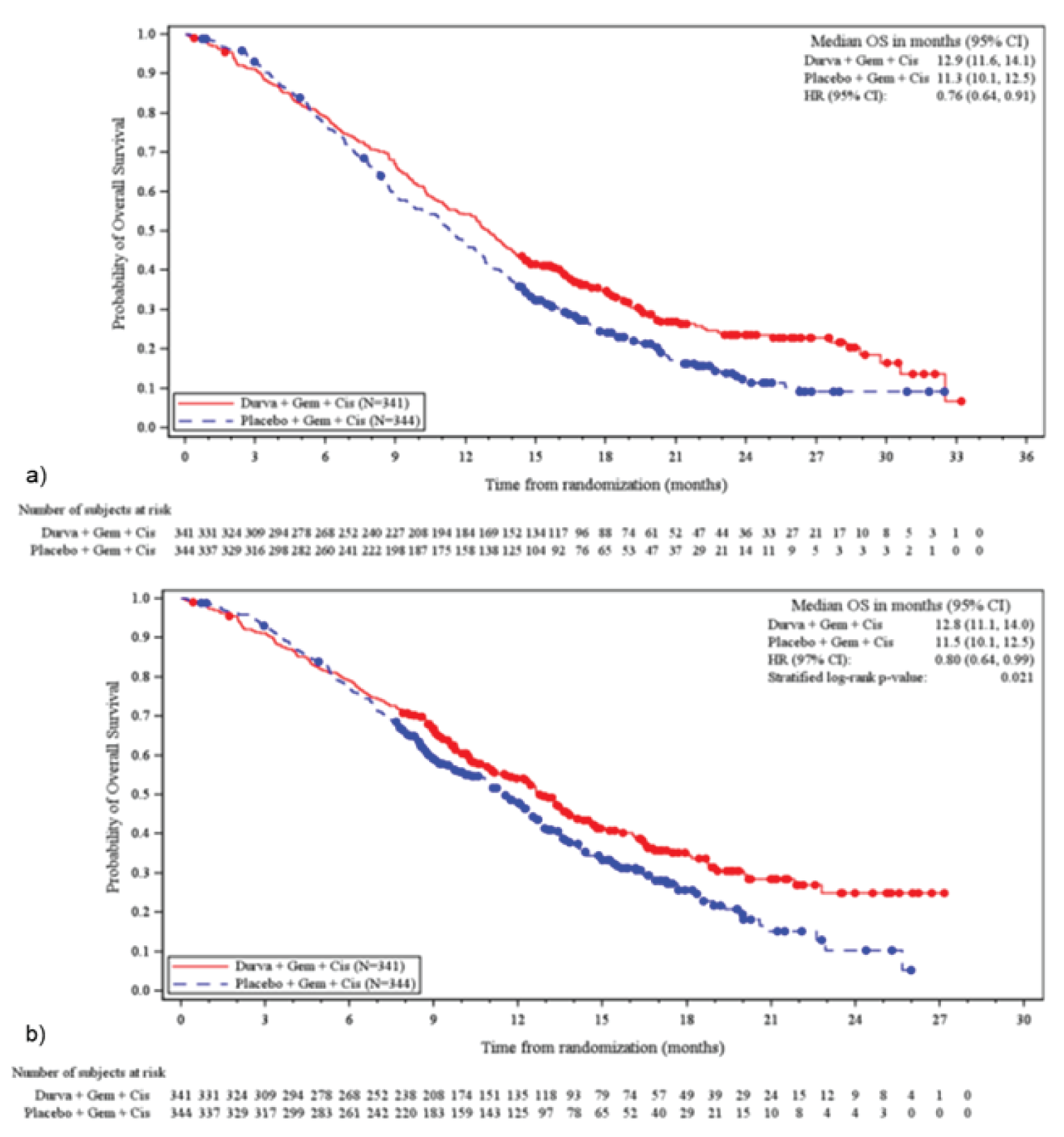
CI = confidence interval, Cis = cisplatin; Durva = durvalumab; DCO = data cut-off; FAS = full analysis set; Gem = gemcitabine; HR = hazard ratio; OS = overall survival.
a) 6.5-month update; b) IA-2
Figure 4: Forest Plot of OS by Subgroup (FAS)
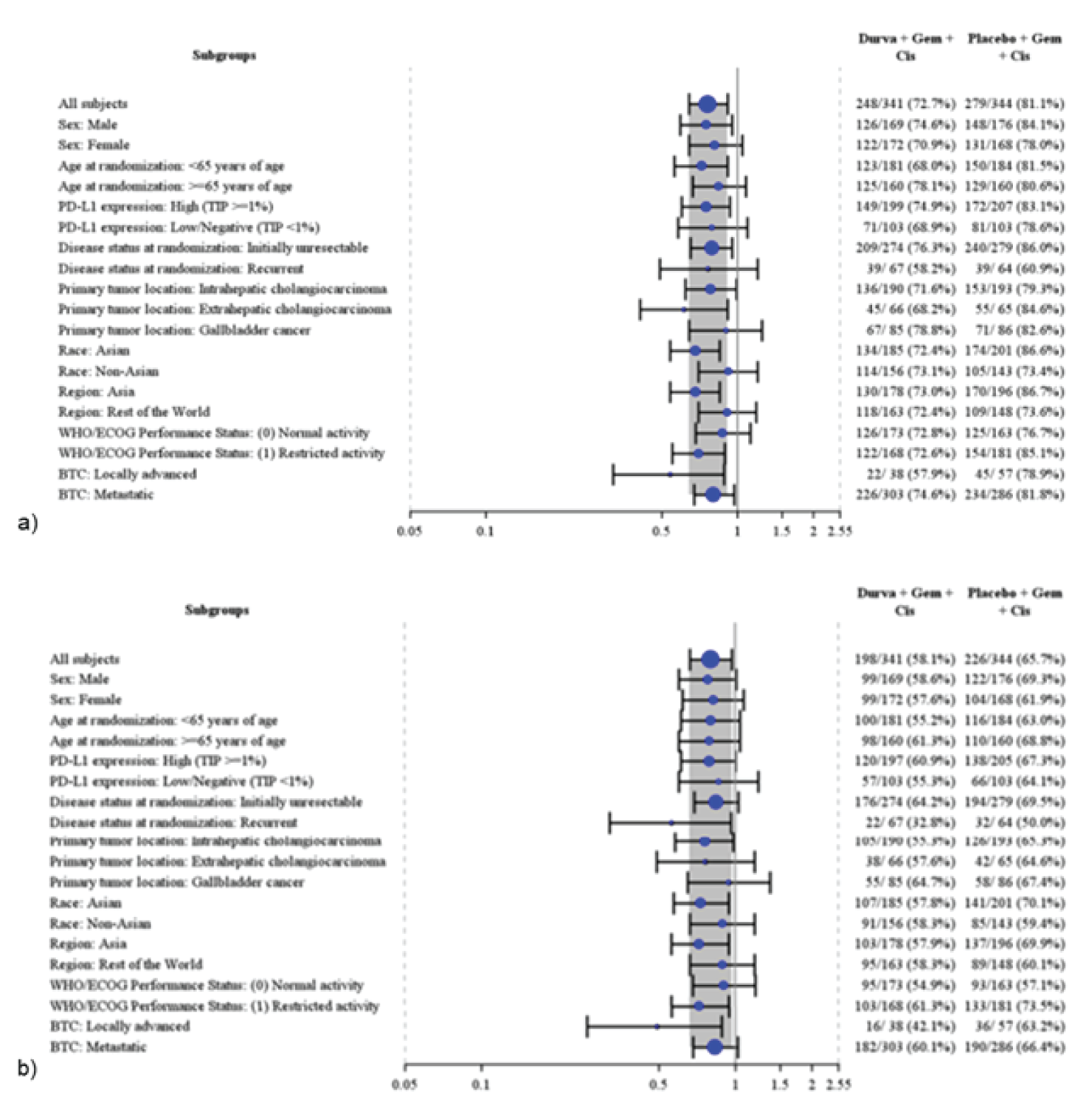
BTC = biliary tract cancer; CI = confidence interval; Cis = cisplatin; Durva = durvalumab; eCRF = electronic case report form; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; FUP = follow-up; Gem = gemcitabine; IVRS = Interactive Voice Response System; PD-L1 = programmed cell death ligand-1; TIP = tumour and/or immune cell positivity; WHO = World Health Organization.
a) Subgroup analysis of OS at the 6.5-month update; b) Subgroup analysis of OS at IA-2
Figure 5: Kaplan-Meier Plot of PFS (FAS)
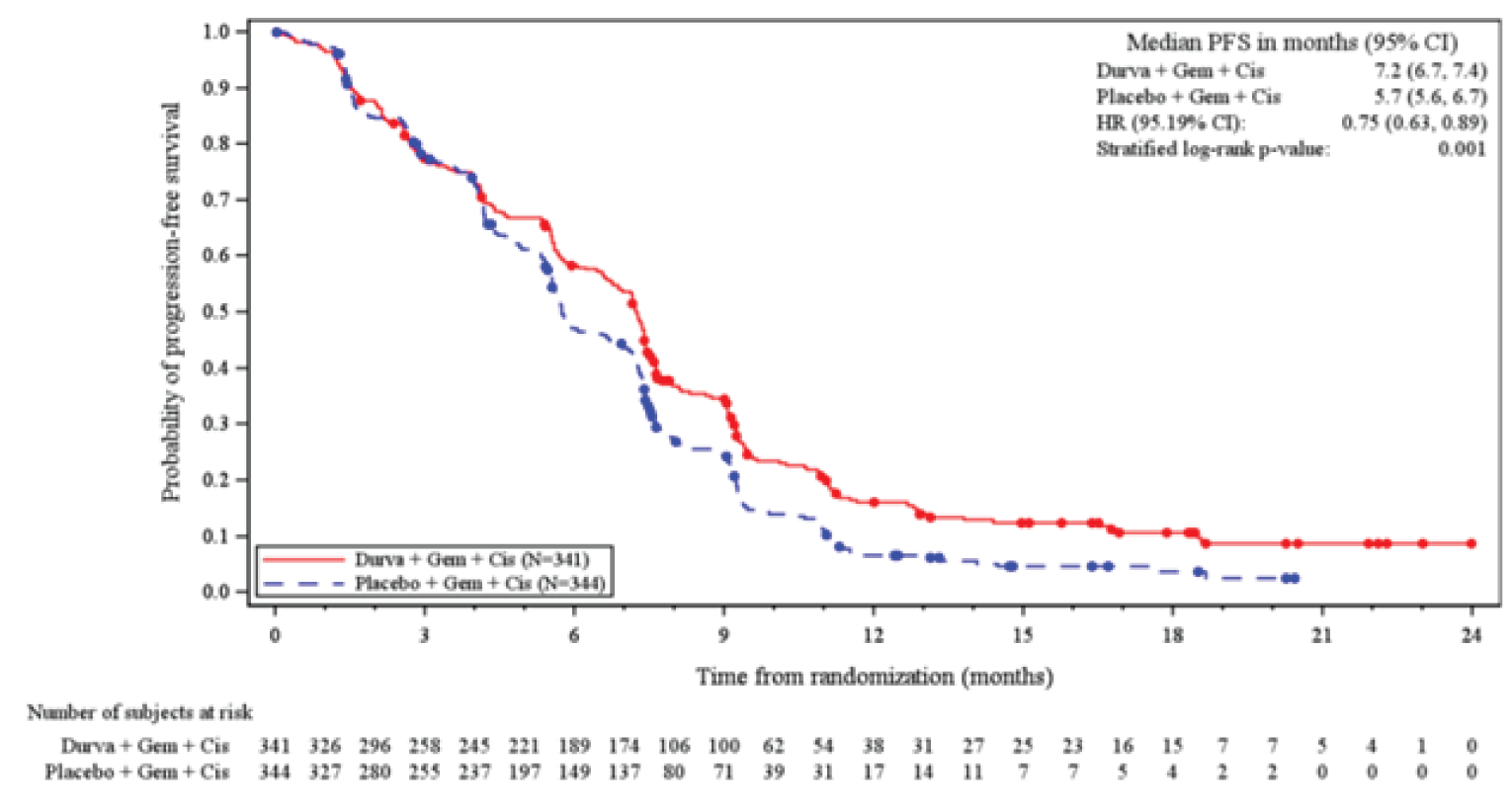
BTC = biliary tract cancer; CI = confidence interval; Cis = cisplatin; Durva = durvalumab; eCRF = electronic case report form; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; FUP = follow-up; Gem = gemcitabine; IVRS = Interactive Voice Response System; PD-L1 = programmed cell death ligand-1; TIP = tumour and/or immune cell positivity; WHO = World Health Organization.
Dot indicates a censored observation. PFS based on investigator assessments according to RECIST 1.1. Patients who had not progressed or died at the time of analysis were censored at the time of the latest date of assessment from their last evaluable RECIST 1.1 assessment prior to the 2 missed visits and therefore excluded from the number of events.
Source: TOPAZ-1 CSR (IA-2)23
Figure 6: PFS by Subgroups of Interest (FAS)
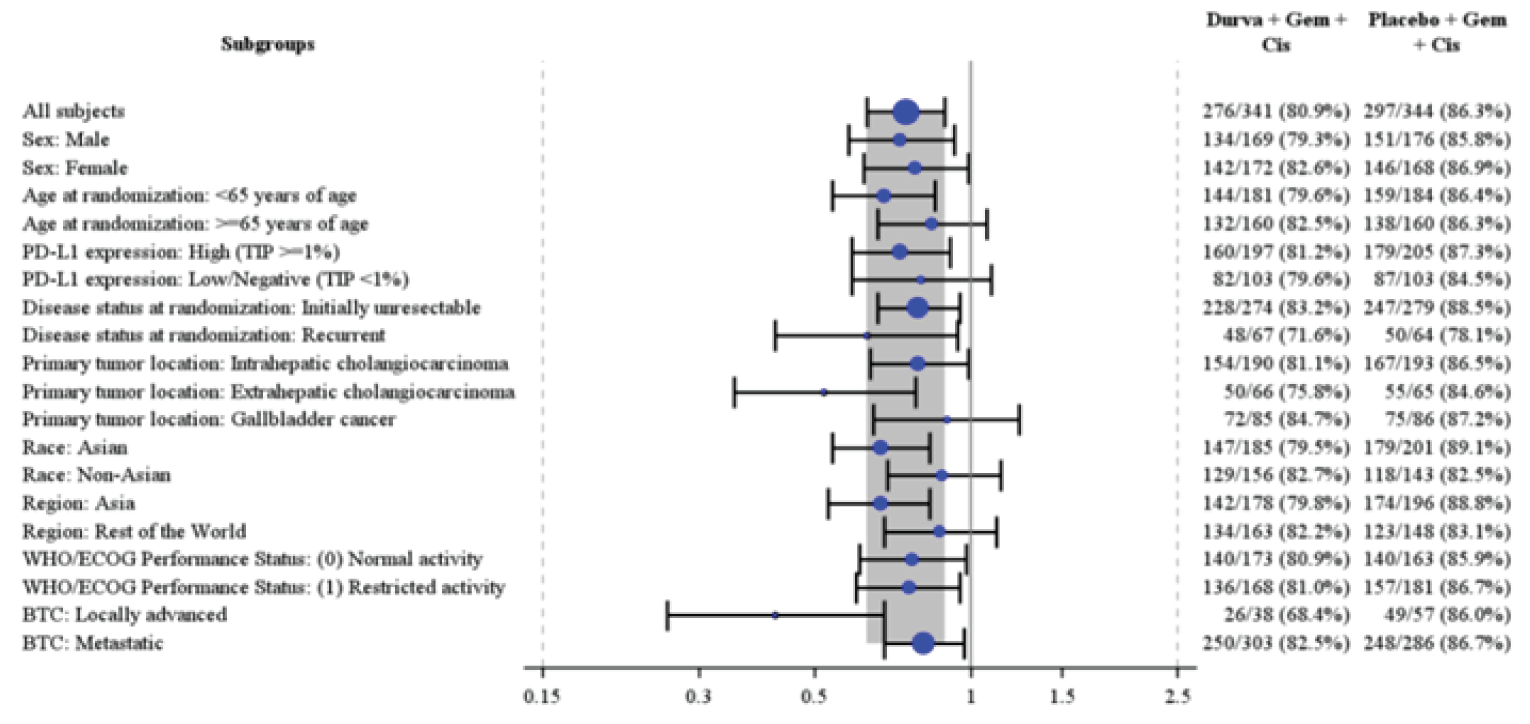
BTC = biliary tract cancer; Cis = cisplatin; Durva = durvalumab; ECOG = Eastern Cooperative Oncology Group; Gem = gemcitabine; PD-L1 = programmed cell death ligand-1; TIP = tumour and/or immune cell positivity; WHO = World Health Organization.
The overall analysis was performed using a stratified Cox proportional hazards model, adjusting for disease status (initially unresectable or recurrent) and primary tumour location (intrahepatic cholangiocarcinoma, extrahepatic cholangiocarcinoma or gallbladder cancer). Profile likelihood methods were used to calculate CIs. Stratification factors were from the eCRF. Estimates for all subgroup categories were from an unstratified Cox model with treatment as the only covariate.
Size of circle is proportional to the number of events. Grey band represents the 95% CI for the overall (all patients) hazard ratio.
Hazard ratio (Durva plus Gem plus Cis vs Placebo plus Gem plus Cis) and 95% CI. A HR < 1 favours Durva plus Gem plus Cis
Source: TOPAZ-1 CSR (IA-2)23

BTC = biliary tract cancer; CI = confidence interval; Cis = cisplatin; Durva = durvalumab; eCRF = electronic case report form; ECOG = Eastern Cooperative Oncology Group; FAS = full analysis set; FUP = follow-up; Gem = gemcitabine; IVRS = Interactive Voice Response System; PD-L1 = programmed cell death ligand-1; TIP = tumour and/or immune cell positivity; WHO = World Health Organization.
Note: This figure has been redacted.
Source: TOPAZ-1 CSR (IA-2)23

Cis = cisplatin; Durva = durvalumab; Gem = gemcitabine
Baseline is defined as last evaluable assessment on or prior to first dose start time. Only subjects who have a baseline EQ-5D-5L assessment are included.
Note: This figure has been redacted.
Source: TOPAZ-1 CSR (IA-2)23

Cis = cisplatin; Durva = durvalumab; Gem = gemcitabine
Baseline is defined as last evaluable assessment on or prior to first dose start time. Only subjects who have a baseline EQ-5D-5L assessment are included.
Note: This figure has been redacted.
Source: TOPAZ-1 CSR (IA-2)23
Appendix 3: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and minimal important difference):
EORTC QLQ-BIL21
PRO-CTCAE
PGI-S
EORTC QLQ-C30
EQ-5D-5L
Findings
Table 25: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-BIL21 | A disease-specific module to be used in addition to the EORTC QLQ-C30 to assess HRQoL in patients with cholangiocarcinoma and gallbladder cancer.43 It consists of 21 questions, with 18 items grouped into 5 scales: eating symptoms (4 items), jaundice symptoms (3 items), tiredness (3 items), pain symptoms (4 items), and anxiety symptoms (4 items).43 The remaining 3 items are single-item assessments of treatment side effects, difficulties with drainage bags/tubes, and concerns about weight loss.43 The questions have been translated according to QoL group guidelines into Chinese Mandarin, Italian, German, Dutch, Spanish, and Hindi.43,44 Patients complete the questionnaire based on a 1-week recall period by rating each item on a 4-point Likert scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much).43 The scores are then transformed linearly to a 0 to 100 scale to yield scale scores using EORTC guidelines, with higher scores indicating more severe symptoms.43,44 The single-item assessment of difficulties with drainage bags and tubes was considered irrelevant by 29 patients.44 The study authors noted that not all patients experience drains during their treatment and that perhaps there should be a ‘not applicable’ option for responding to that item.44 | An international study was conducted to validate the EORTC QLQ-BIL21 in patients with biliary tract cancer.44 The study included 172 adult patients with cholangiocarcinoma and 91 patients with gallbladder cancer who had an expected minimum survival of 3 months and were undergoing treatment.44 Patients completed the EORTC QLQ-C30 and QLQ-BIL21, and KPS was recorded at ≤ 1 month before treatment and 2 months later. The analysis included 478 questionnaires.44 Patients were assigned to 1 of 3 groups based on the treatment received: surgical treatment; chemotherapy, radiotherapy, photodynamic, or laser therapy; or supportive care only. Validity: All items demonstrated item-scale convergence (construct) validity (Pearson’s r > 0.4, prespecified).44 For discriminant validity, no items had r > 0.70, indicating no items correlated with scales outside of the scale they were placed in. Known groups (construct) validity was demonstrated by the greater EORTC QLQ-BIL21 mean scores in patients with KPS < 70 at baseline compared to patients with KPS > 70 for all scales, with the exception of the jaundice and weight loss scales (N = 238 to 256).44 Reliability: Internal consistency was acceptable (alpha ≥ 0.70) for all multiitem scales at baseline (Cronbach alpha = 0.71 to 0.87) and when the assessment time points were pooled (Cronbach alpha = 0.71 to 0.89).44 Of note, Cronbach alpha ranged from 0.61 to 0.93 at the 2-month assessment, with a coefficient of 0.61 and 0.68 for the jaundice and pain scale, respectively.44 Test-retest reliability was acceptable (ICC ≥ 0.7050) for all scales, with ICC ranging from 0.81 to 0.96, in 67 clinically stable patients across all intervention groups in 2 weeks.a,44 Responsiveness: Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. | Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. |
EORTC QLQ-C30 | A multidimensional, cancer-specific, patient-reported measure used to assess HRQoL in response to treatment in clinical trials.51 The core questionnaire consists of 30 items that make 5 multiitem functional scales (physical [5 items], role [2 items], emotional [4 items], cognitive [2 items], and social [2 items] functioning), 3 multiitem symptom scales (fatigue [3 items], nausea/vomiting [2 items], and pain [2 items]), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea and financial impact), and a 2-item global QoL scale.51 Patients complete the questionnaire based on a 1-week recall period by rating most items on a 4-point Likert-type scale (1 = not at all; 2 = a little; 3 = quite a bit; 4 = very much). For the 2 items in the global QoL scale, the response format is a 7-point Likert-type scale (1 = very poor; 7 = excellent).52 Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation. A decline in the symptom scale score reflects an improvement, whereas an increase in the function and quality of life scale scores reflect an improvement.52 According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale, the score for that scale can still be computed if there are responses for at least one-half of the items. In calculating the scale score, missing items are ignored.52 | The psychometric properties of the EORTC QLQ-C30 were evaluated in the validation study for the EORTC QLQ-BIL21 described above.44 Validity: All items demonstrated item-scale convergence (construct) validity (Pearson’s r > 0.4, prespecified).44 Although the study44 stated known group comparison was performed for EORTC QLQ-30, results were not reported. Reliability: Internal consistency was acceptable (alpha ≥ 0.7044) for all scales, except for the physical function (alpha = 0.47), cognitive function (alpha = 0.65), and nausea/vomiting (alpha = 0.67) scales at baseline.44 Test-retest reliability was demonstrated by the ICCs that ranged from 0.52 to 0.92 in 67 clinically stable patients across all intervention groups in 2 weeks.a,44 Responsiveness: Although the study44 stated responsiveness to clinical change over time of the EORTC QLQ-30, results were not reported. | Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. In patients with breast cancer (n = 246) and small cell lung cancer (n = 111) who were in RCTs of chemotherapy, an anchor-based approach using global ratings of change measured by a subjective significance questionnaire was performed to determine the importance of change in HRQoL scales to patients.45 In general, patients who reported ‘a little,’ ‘moderate,’ and ‘very much’ absolute change corresponded to a mean change in EORTC QLQ-C30 score of 5 to 10, 10 to 20, and > 20, respectively.b,45 In patients with newly diagnosed breast (63%) and colorectal (37%) cancer who received surgery in Canada, an anchor-based approach using the SCNS-SF34 was performed to estimate a clinically important change in the EORTC QLQ-C30 scales (N = 193).53 In patients who reported improvement in supportive care needs, the mean change in EORTC QLQ-C30 score ranged from 9.8 (global health) to 32.3 (role function). In patients who reported worsening, the mean change in EORTC QLQ-C30 score ranged from –9.3 to –21.2 (emotional function); note that results were mixed as some scales did not change in the hypothesized direction or no changes were observed. As a result, the authors suggested a 10-point change in the EORTC QLQ-C30 to represent change in supportive care needs.c,53 |
PRO-CTCAE | A patient-reported outcome version of the CTCAE measurement system developed by NCI used to evaluate symptomatic AEs of treatments in cancer clinical trials from the patient perspective. It was designed as a companion to the CTCAE, consisting of 124 distinct items representing 78 symptomatic AEs that are common in oncology clinical trials.54 Each item is presented in its plain language term for that AE.47 Patients select the response that best describes their experience over the past 7 days using a 5-point Likert scale; response options capture the presence, frequency, severity, or interference with activities depending on the symptom.54 | Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. The psychometric properties of the 124 items in the PRO-CTCAE item library were evaluated in 940 adult patients with diversity in cancer type (e.g., breast, aerodigestive tract, genitourinary, lung, colorectal, and lymphoma/myeloma) and treatment received (e.g., chemotherapy and radiotherapy) in the US. Each patient was presented with a maximum of 58 symptomatic AEs depending on the diagnosis.47 Validity: Convergent (construct) validity (Pearson correlation ≥ 0.10) was demonstrated in 107 PRO-CTCAE items with the EORTC QLQ-C30 summary score. PRO-CTCAE items that were likely to impact physical functioning were found to have the strongest correlation with the physical functioning scales (e.g., shortness of breath severity: r = 0.47). Similar results were observed between PRO-CTCAE items and conceptually related EORTC QLQ-C30 emotional, role, cognitive and social functioning scales. All PRO-CTCAE items were found to have strong correlations with analogous EORTC QLQ-C30 symptom scales/items (r > 0.69).47 Known groups (construct) validity was demonstrated by the higher mean scores in patients with ECOG PS 2 to 4 versus 0 to 1 group for 94 PRO-CTCAE items.47 Reliability: Test-retest reliability was acceptable for 36 of the 49 prespecified items demonstrated by ICC ≥ 0.70. The ICCs ranged from 0.53 to 0.96 in 80 patients between 1 to 3 days.47 Responsiveness: Responsiveness to change was demonstrated by comparing change between the first and second visit in 27 items selected a priori and according to GIC response (worse, unchanged, or improved). The SRM in patients who reported worsening and improvement ranged from 0.03 to 0.40 and –0.30 to 0.09, respectively.47 Pearson correlations between PRO-CTCAE item changes and corresponding EORTC QLQ-C30 scale changes for all 27 items ranged from 0.10 to 0.56.47 | Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. |
PGI-S | A single item used to assess how a patient perceives their overall severity of symptoms at the time of assessment.23 A 6-point scale is used (1 = no symptoms; 2 = very mild; 3 = mild; 4 = moderate; 5 = severe; 6 = very severe).23 | Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. | Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. |
EQ-5D-5L | A generic measure of health status comprised of 2 parts. The descriptive system assesses health in 5 dimensions (mobility, self-care, usual activities, pain/discomfort and anxiety/depression).55 Each dimension has 5 increasing levels of severity/response (no problems, slight problems, moderate problems, severe problems and unable to/extreme problems). A unique health state profile is generated as a 5-digit code (e.g., 12345 indicates no problems with mobility, slight problems with self-care, moderate problems with usual activities, severe pain or discomfort and extreme anxiety or depression).55 The health state can be converted to a summary index score based on societal (countries/regions) preference weights for the health state. Index scores range from less than 0 (negative values represent worse than dead, which is represented by 0) to 1 (full health), with higher scores representing higher health utility.55 Patient’s perceived health status on that day is also rated using the VAS, ranging from 0 (worst imaginable health) to 100 (best imaginable health).55 | Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. | Not identified for patients with biliary tract cancer, including cholangiocarcinoma and gall bladder cancer. |
AE = adverse event; ECOG = Eastern Cooperative Oncology Group; EORTC = European Organization for Research and Treatment of Cancer; GIC = Global Impression of Change; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; KPS = Karnofsky performance status; MID = minimal important difference; NCI = National Cancer Institute; PGI-S = Patient Global Impression of Severity; PRO-CTCAE = Patient-Reported Outcomes – Common Terminology Criteria for Adverse Events; PS = performance status; QLQ-BIL21 = 21-Item Cholangiocarcinoma and Gallbladder Cancer Quality of Life Questionnaire; QLQ-C30 = 30-Item Core Quality of Life Questionnaire; QoL = quality of life; RCT = randomized clinical trial; SCNS-SF34 = Supportive Care Needs Survey-Short Form-34; SRM = standardized response means; VAS = visual analogue scale.
aPatients who received intravenous chemotherapy at the time were excluded from the test-retest assessment.
bResults should be interpreted with caution given the use of a previous version of the questionnaire and the patient population was composed of patients with breast and small cell lung cancer only.
cResults should be interpreted with caution given the limited sample sizes (ranging from 8 to 58) for each estimate/analysis, the unknown validity of using single items of the SCNS-SF34 as anchors (individual items determined by the study authors to be relevant to the EORTC QLQ-C30 physical function, role function, emotional function, global health, pain, and fatigue scales were used as anchors and mean changes in these 6 scales associated with improvement, worsening, and no change in the corresponding SCNS-SF34 items were calculated53), and the patient population being composed of patients with breast or colorectal cancer only.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BTC
biliary tract cancer
CUA
cost-utility analysis
EQ-ED-5L
5 Level EQ-ED
FOLFIRI
5-fluorouracil, leucovorin, and irinotecan
FOLFOX
5-fluorouracil, folinic acid (leucovorin), and oxaliplatin
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
LY
life-year
OS
overall survival
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
QoL
quality of life
RDI
relative dose intensity
TTD
time to treatment discontinuation
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Durvalumab (Imfinzi), vial for IV infusion |
Submitted price | Durvalumab, 50 mg/mL, single-use vial for IV solution: $7.82 per mg ($939 per 120 mg vial, $3,911 per 500 mg vial) |
Indication | In combination with gemcitabine-based chemotherapy, for the treatment of patients with locally advanced or metastatic BTC |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review, Project Orbis (Type C) |
NOC date | September 29, 2022 |
Reimbursement request | Per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Previously reviewed: Yes Indication: the treatment of patients with locally advanced, unresectable non–small cell lung cancer after curative-intent, platinum-based chemoradiation therapy for a maximum of 12 months Recommendation date: May 3, 2019 Recommendation: reimburse with clinical criteria and/or conditions Indication: the first-line treatment of adults with extensive-stage small cell lung cancer, in combination with etoposide and either carboplatin or cisplatin Recommendation date: July 27, 2021 Recommendation: reimburse with clinical criteria and/or conditions |
BTC = biliary tract cancer; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adults receiving first-line treatment for locally advanced or metastatic BTC |
Treatment | Durvalumab + gemcitabine and cisplatin |
Comparator | Gemcitabine and cisplatin |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (20 years) |
Key data source | A phase III, randomized, double-blind, placebo-controlled clinical trial assessing the safety and efficacy of durvalumab + gemcitabine and cisplatin compared with gemcitabine and cisplatin for patients with advanced BTC |
Submitted results | Durvalumab + gemcitabine and cisplatin was associated with an ICER of $426,083 per QALY compared with gemcitabine and cisplatin (incremental costs = $171,414; QALYs = 0.40) |
Key limitations |
|
CADTH reanalysis results |
|
BTC = biliary tract cancer; EQ-5D-5L = 5 Level EQ-5D; ICER = incremental cost-effectiveness ratio; LY = life-year; OS = overall survival; QALY = quality-adjusted life-year; WTP = willingness to pay.
Conclusions
The CADTH Clinical Review showed that compared with gemcitabine and cisplatin, durvalumab (Imfinzi) plus gemcitabine and cisplatin was associated with a statistically significant improvement in overall survival (OS) and progression-free survival (PFS) in adults receiving first-line treatment for locally advanced or metastatic biliary tract cancer (BTC), based on the TOPAZ-1 trial. However, the clinical importance of the modest survival gain afforded by treatment with durvalumab plus gemcitabine and cisplatin compared with gemcitabine and cisplatin alone is subjective. Measures of health-related quality of life (HRQoL) were prespecified in the protocol and suggested no detriment to quality of life (QoL) with durvalumab plus gemcitabine and cisplatin. However, no definitive conclusions could be drawn about the impact of treatment with durvalumab plus gemcitabine and cisplatin compared with gemcitabine and cisplatin alone on time to deterioration and HRQoL because of limitations of the analyses, including a lack of statistical testing and attrition bias in the TOPAZ-1 trial. The long-term clinical effectiveness associated with durvalumab treatment beyond the trial period (approximately ██ months) was also highly uncertain.
In its reanalysis, CADTH extrapolated OS with durvalumab using a spline odds (1 knot) function, assumed a relative dose intensity (RDI) of 100% for all treatments, and used age-adjusted estimates of health state utility. CADTH’s base case was aligned with the sponsor’s results, with both analyses suggesting that first-line durvalumab treatment is associated with higher costs and increased QALYs, but is not cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY. Based on publicly available list prices for all comparators, a price reduction of at least 93% would be required for durvalumab to be cost-effective at this WTP threshold.
The cost-effectiveness of durvalumab plus gemcitabine and cisplatin is sensitive to the uncertainty in long-term effectiveness of durvalumab treatment. The sponsor submitted OS data with multiple data cut-off points; using the most recent data cut-off increased the incremental cost-effectiveness ratio (ICER). Approximately 68.9% of the long-term survival benefit associated with durvalumab was predicted through extrapolation beyond the trial period. Given the high degree of uncertainty in long-term survival, a higher price reduction may be warranted. The cost-effectiveness of durvalumab plus gemcitabine and cisplatin in subsequent lines of treatment is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process and, specifically, contains information that pertains to the economic submission.
Patient input was received from the Canadian Cancer Survivor Network, with the participation of Canadian Liver Foundation, Canadian Organization for Rare Disorders, Cholangiocarcinoma Foundation, Colorectal Cancer Resource and Action Network, Gastrointestinal Society, and Regroupement québécois des maladies orphelines. The perspectives of caregivers and patients with BTC were collected with online surveys and interviews in Canada, the US, the UK, and Spain. Approximately 62% of patients had late stage (IV) or metastatic disease. Patients with BTC reported several negative impacts on QoL, including abdominal pain, loss of appetite or weight loss, nausea or vomiting, itching, dark urine, fever, and jaundice. Current treatments noted to be available to patients include radiation, surgery, targeted therapy, immunotherapy, gemcitabine and cisplatin, and a combination of 5-fluorouracil, folinic acid (leucovorin), and oxaliplatin (known as FOLFOX) or 5-fluorouracil, leucovorin, and irinotecan (known as FOLFIRI). Patients reported several side effects related to current treatments, including diarrhea, nausea or vomiting, bruising or bleeding, hair loss, muscle weakness, tingling and numbness, tiredness, difficulty sleeping, and swelling of arms and legs. Patients who had experience with durvalumab reported side effects such as constipation, decreased platelet count, and fatigue. Treatment goals include maintained QoL, delayed onset of symptoms, decreased side effects, prolonged life, and disease cure. Caregivers of patients with BTC reported encountering issues with mental health, management of medications, and management of side effects.
Clinician input was received from the Canadian Gastrointestinal Oncology Evidence Network and Ontario Health. The clinician input noted that the current standards of care for patients with locally advanced or metastatic BTC are gemcitabine and cisplatin and gemcitabine and carboplatin. Patients with BTC were reported to have poor prognosis; approximately 50% of untreated patients die within 90 days of presentation, and those who receive treatment survive less than a year. The clinicians noted that clinically meaningful treatment goals include prolonged life, delayed disease progression, and improved QoL. A clinically meaningful treatment response was described as maintenance or improvement in QoL and prolonged survival, measured through improvement in symptoms and response on CT imaging. The clinicians recommended assessing clinical response every 3 weeks and radiographic response every 2 to 3 months. The clinician input also recommended that treatment be discontinued in cases of progression on imaging, poor tolerance that cannot be improved with dose delays or reductions, and patient preference to stop treatment.
The drug plans noted concerns with choice of backbone chemotherapy (potential substitution of carboplatin or oxaliplatin for patients with compromised renal function or gemcitabine monotherapy for patients with poor performance status). The plans also noted considerations for initiation, continuation, and discontinuation of therapy, as well as the feasibility of adoption and sustainability of durvalumab treatment.
Several of these concerns were addressed in the sponsor’s model:
Clinical benefits (i.e., delaying disease progression and prolonging OS) and adverse events (AEs) associated with durvalumab plus gemcitabine and cisplatin were considered.
The budget impact associated with the reimbursement of durvalumab plus gemcitabine and cisplatin in first-line therapy was considered.
In addition, CADTH explored uncertainty about durvalumab uptake on the estimated budget impact.
CADTH was unable to address the impact of treatment interruptions and re-treatment on the cost-effectiveness or budget impact of durvalumab.
Economic Review
The current review is for durvalumab used in the first-line treatment of adults with locally advanced or metastatic BTC.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) comparing durvalumab plus gemcitabine and cisplatin with gemcitabine and cisplatin for the first-line treatment of adults with locally advanced or metastatic BTC. The Health Canada indication does not specify the line of treatment for the patient population in the metastatic setting. The modelled population was therefore not aligned with the Health Canada indication or the reimbursement request.
Durvalumab is available as a solution for IV infusion (50 mg/mL single-use vial). The recommended dose is 1,500 mg every 3 weeks for 8 cycles in combination with gemcitabine and cisplatin, followed by 1,500 mg every 4 weeks as monotherapy. At the submitted price of $939 per 2.4 mL, or $3,911 per 10 mL vial, the cost of durvalumab was $11,577 per cycle. In combination with gemcitabine and cisplatin, the cost of the durvalumab regimen was $12,769 per cycle. The recommended doses of gemcitabine (1,000 mg/m2) and cisplatin (25 mg/m2) were based on doses administered to patients in the TOPAZ-1 trial. This resulted in a per-cycle cost of $905 and $287, respectively, for a treatment duration of 8 cycles. The sponsor assumed no vial sharing and RDI when estimating drug-acquisition costs.
The submitted model reported both quality-adjusted life-years (QALYs) and life-years (LYs) over a lifetime time horizon (20 years) in the modelled population. The base-case analysis was conducted from the perspective of the public health care payer in Canada. Both costs and health outcomes were discounted at an annual rate of 1.5%.
Model Structure
The sponsor submitted a partitioned survival model (PSM) that included 3 health states: progression-free, postprogression, and death. The model’s cycle length was 1 week. All patients began in the progression-free health state, and were assumed to be stable or responding to therapy, as defined by the PFS measure assessed in the TOPAZ-1 trial (Response Evaluation Criteria in Solid Tumours Version 1.1 criteria). During each cycle, patients in the progression-free health state remained in the state, transitioned to death, or transitioned to the postprogression health state. The proportion of patients in the postprogression state was calculated by subtracting the proportion of patients alive and progression-free (based on the PFS curve) from the proportion of patients alive (based on the OS curve). Patients in the postprogression health state either remained in this state or transitioned to death.
Model Inputs
The modelled patient cohort comprised previously untreated patients 18 years and older with unresectable locally advanced or metastatic BTC whose baseline characteristics mainly reflected the pivotal TOPAZ-1 trial.1 The median age of patients in the model was 64 years, with a body surface area of ████ m2 and a creatinine clearance of ████ mL/min.
Key clinical efficacy inputs (i.e., OS and PFS) and time to treatment discontinuation (TTD) for durvalumab plus gemcitabine and cisplatin and gemcitabine and cisplatin alone were derived from the TOPAZ-1 trial. The sponsor submitted trial data with multiple cut-off points; the February 2022 data cut-off was used for OS and the August 2021 data cut-off was used for PFS and TTD. PFS, OS, and time on treatment (TOT) outcomes were extrapolated beyond the trial duration by fitting parametric survival models to the trial data (maximum duration of follow-up was approximately ████ years for OS and ████ years for PFS and TTD). Model selection was based on statistical fit (Akaike information criterion, Bayesian information criterion, visual inspection of goodness of fit to observed data, clinical plausibility, and external validation against real-world data). The sponsor selected a log-logistic distribution to extrapolate OS for patients on durvalumab plus gemcitabine and cisplatin and gemcitabine and cisplatin alone. For the long-term extrapolations of PFS, the spline hazards 3 knots distributions were selected for both treatment arms. Further, OS was capped using general population mortality rates. The sponsor assumed that the efficacy of durvalumab plus gemcitabine and cisplatin persisted for the entire time horizon. The sponsor fitted log-logistic and spline odds 3 knots distributions to extrapolate TTD curves for durvalumab and gemcitabine and cisplatin, respectively, based on treatment discontinuation data from the pivotal TOPAZ-1 trial.
Health state utility values were based on a mixed-effects models fitted with 5 Level EQ-5D-5L (EQ-5D-5L) data collected in the TOPAZ-1 trial, which were adapted to the population living in Canada using Canadian tariffs.2 Patients with grade 3 and 4 AEs accrued both costs and disutilities. The sponsor obtained disutilities and duration of AEs using published literature on diseases comparable to BTC, such as advanced cholangiocarcinoma, neutropenia, and thrombocytopenia.3 AEs were assumed to occur during the first treatment cycle only.
The model included costs related to drug acquisition, administration, monitoring, AEs, and terminal care. Drug-acquisition costs were applied in the model based on the aforementioned TTD curves. Per the trial protocol, treatment with gemcitabine and cisplatin was stopped after 8 cycles in both arms. Patients were assumed to accrue the cost of subsequent treatments after disease progression, which included second-line treatments such as FOLFOX, FOLFIRI, capecitabine plus oxaliplatin, nivolumab monotherapy, and pembrolizumab monotherapy. The distribution of subsequent treatments and mean treatment duration was obtained from the TOPAZ trial. Unit costs were obtained from the IQVIA DeltaPA database and dosing schedules from the TOPAZ-1 trial, the Cancer Care Ontario regimen formulary, and published literature.1,2,4,-10 The sponsor estimated disease management costs based on the frequency of health care resource use in the ICES-HOPE study, and applied these costs to modelled health states.11 AE costs and terminal care costs were applied as 1-time costs at the beginning of the first cycle and death, respectively.12,13
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations), and the deterministic and probabilistic results were similar. The probabilistic findings are presented here. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
Durvalumab plus gemcitabine and cisplatin were associated with an ICER of $426,083 per QALY compared with gemcitabine and cisplatin over a 20-year time horizon (Table 3). In the sponsor’s base case, durvalumab had a 0% probability of being a cost-effective strategy at a WTP threshold of $50,000 per QALY. The majority (66%) of incremental QALYs associated with durvalumab were accrued beyond the trial follow-up and were based on the sponsor’s extrapolations of the trial data. Furthermore, more than one-third (37%) of QALYs were accrued in the postprogression health state.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug regimen | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|
Gemcitabine and cisplatin | 88,259 | Reference | 1.08 | Reference | Reference |
Durvalumab + gemcitabine and cisplatin | 259,673 | 171,414 | 1.48 | 0.40 | 426,083 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.11
Sensitivity and Scenario Analysis Results
The sponsor performed scenario analyses by considering alternative parametric survival models, assuming vial sharing, and adopting a micro-costing approach to estimate health care resource use. Cost-effectiveness results were robust to changes in most parameters and assumptions. The scenarios with the greatest impact on the ICER were alternative parametric models for OS. Compared with gemcitabine and cisplatin, the estimated ICERs of durvalumab plus gemcitabine and cisplatin increased to $694,344 per QALY when a gamma distribution was used to predict long-term OS.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The population in the sponsor’s pharmacoeconomic evaluation was not aligned with the Health Canada indication: The population for the sponsor’s base case was adults receiving treatment for locally advanced or metastatic BTC in the first-line setting, which does not reflect the Health Canada–indicated population. The product monograph for durvalumab does not specify line of therapy, and the clinical evidence in the TOPAZ-1 trial is only available for durvalumab use in the first-line setting.1,14 The use of durvalumab in subsequent lines of therapy was not considered in the sponsor’s submission. As such, the effectiveness and cost-effectiveness of durvalumab in subsequent lines of therapy for these patients is unknown. Clinical expert feedback received by CADTH for this review noted that the expected place of durvalumab is in the first-line setting, given that there are limited treatments available for this patient population. CADTH focused this review on durvalumab treatment in the first-line setting only.
CADTH could not address this limitation. The cost-effectiveness of durvalumab plus gemcitabine and cisplatin in subsequent lines of therapy is unknown.
The long-term clinical efficacy of durvalumab treatment is uncertain: The sponsor submitted a PSM in which the long-term clinical efficacy of durvalumab was based on the sponsor’s extrapolations of PFS and OS, using efficacy data from the TOPAZ-1 trial. In the intention-to-treat analysis using the February 2022 data cut-off for OS and the August 2021 data cut-off for PFS, median OS and PFS increased by ███ months and ███ months, respectively, for patients treated with durvalumab plus gemcitabine and cisplatin, compared with gemcitabine and cisplatin alone. However, in the sponsor’s pharmacoeconomic analysis, the long-term extrapolation of OS and PFS resulted in an incremental gain of 5.75 months and about 2.28 months, respectively. Approximately 68.9% of LYs derived from durvalumab treatment were accrued through extrapolation beyond the trial duration. The results of this cost-effectiveness analysis are sensitive to the sponsor’s extrapolation assumptions, adding meaningful uncertainty to the magnitude of effectiveness.
The sponsor assumed that the treatment effect of durvalumab is maintained over the time horizon of 20 years. The clinical experts consulted by CADTH for this review noted that there may be a persistent treatment effect beyond the trial follow-up, but there was insufficient evidence to support a long-term treatment effect. The Kaplan-Meier PLOT of OS using the February 2022 data cut-off appeared to show a merging of the OS curves near the end of the trial follow-up, indicating no difference in OS beyond approximately ██ months of follow-up. As such, the magnitude and duration of a treatment effect of durvalumab that endures beyond the end of the trial period is highly uncertain. CADTH noted that including long-term follow-up data that relied on the most recent (February 2022) data cut-off, instead of the data cut-off of August 2021, decreased the estimated long-term OS associated with durvalumab.
In its reanalysis, CADTH used Kaplan-Meier data cut-off in February 2022 to estimate OS associated with durvalumab and adopted a conservative estimate of long-term treatment effect of durvalumab using the sponsor-provided spline odds (1 knot) function.
The model structure may overestimate comparative efficacy: Results from the sponsor's model suggested that durvalumab plus gemcitabine and cisplatin was associated with longer survival after progression. Nearly half the incremental LYs (48%) associated with durvalumab were accrued in the postprogression health state. Although the pivotal TOPAZ-1 trial showed a statistically significant impact of durvalumab on PFS and OS, there was no clear mechanism by which durvalumab would continue to provide clinical benefit after progression. Clinical expert feedback received for this review also did not propose any potential mechanism that could explain a postevent survival benefit. The sponsor’s use of a PSM introduces structural assumptions about the relationship between PFS and OS that likely do not accurately reflect causal relationships within the disease pathway. These assumptions may produce a postprogression survival bias that favours durvalumab. Because of the assumed independence between OS and PFS end points in a PSM, extrapolations for each end point may reflect within-trial trends in the rates of progression and death.
CADTH was unable to determine the extent to which the implied postprogression benefit was due to the effect of treatment rather than to structural bias within the PSM, and could not address this in its reanalysis.
RDI may not accurately reflect actual drug costs: The sponsor’s base case incorporated reduced doses of durvalumab, gemcitabine, and cisplatin using RDIs obtained from the TOPAZ-1 trial. The use of RDI is problematic, as dose delays, missed doses, dose reductions to manage toxicity, and subsequent dose re-escalations have different impacts on drug costs. Consistent with previous reviews, given the inability to link reduced dose intensity with outcomes, the CADTH base case does not incorporate RDIs.
In its reanalysis, CADTH assumed an RDI of 100% for durvalumab, gemcitabine, and cisplatin. CADTH explored the impact of assuming the sponsor’s adopted RDIs in a scenario analyses.
Health utilities are uncertain: The sponsor derived health state utilities using EQ-5D-5L data collected as an exploratory end point in the TOPAZ trial. However, the CADTH clinical appraisal noted that the results of the analysis were not controlled for multiplicity and, thus, need to be interpreted in light of the possibility of type I error and should only be viewed as supportive of the overall effect of durvalumab. As such, the estimated health utilities and QALYs are highly uncertain. The sponsor also assumed that age does not have an impact on health utility. Given that patients survive longer with the addition of durvalumab to gemcitabine and cisplatin treatment than with gemcitabine and cisplatin alone, the sponsor’s approach estimates QALYs in favour of durvalumab by excluding the impact of increasing age on health utility.
In its reanalysis, CADTH adopted an age-based cap adjustment of health utilities.
Additionally, the key assumptions outlined in Table 4 were made by the sponsor and have been appraised by CADTH.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Data cut-off points | Multiple data cut-off points of the clinical evidence of OS in the TOPAZ-1 trial were available. CADTH tested the use of multiple cut-off points for OS and found it had limited impact on the results. |
Relevant comparators | Acceptable. The clinical expert consulted for this review identified gemcitabine + carboplatin as a comparator, but noted that this treatment is not commonly used for patients with the indication of interest. |
Disutilities | The sponsor obtained disutilities for AEs from disease areas that the sponsor assumed were comparable to BTC using published literature. CADTH anticipates that this uncertainty would have a minimal impact on the results. |
Treatment duration | The clinical experts noted that durvalumab treatment may be stopped at disease progression or continued at the physician’s discretion if a clinical benefit persists, per TOPAZ-1 trial protocol. CADTH tested the uncertainty in treatment duration of durvalumab and found it had a minimal impact on the results. The clinical experts also noted that gemcitabine and cisplatin may be given for a shorter time period (4 to 6 cycles). Given that durvalumab is added to gemcitabine and cisplatin and that no difference is anticipated in the treatment duration of backbone chemotherapy, alone or in combination with durvalumab, variations in treatment duration of gemcitabine and cisplatin would have a minimal impact on the results. |
AE = adverse event; BTC = biliary tract cancer; OS = overall survival.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CADTH’s base case adopted the spline odds (1 knot) function, 100% RDI, and age-based utilities with a cap.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Distribution used to estimate OS with durvalumab treatment | Log-logistic | Spline odds (1 knot) |
2. Relative dose intensity | < 100% (varies by drug) | 100% |
3. Health utilities | No age-based adjustment | Age-based utilities with a cap |
CADTH base case | 1 + 2 + 3 | |
OS = overall survival.
In the CADTH base case, durvalumab plus gemcitabine and cisplatin was associated with estimated total costs of $258,395 and total QALYs of 1.30, compared with total costs of $89,298 and total QALYs of 1.04 for patients receiving gemcitabine and cisplatin alone. The ICER associated with durvalumab was $665,692 per QALY for adults receiving first-line treatment for locally advanced or metastatic BTC. The probability of cost-effectiveness at a $50,000 per QALY WTP threshold was 0%. Nearly half the incremental QALYs (46%) associated with durvalumab plus gemcitabine and cisplatin, compared with gemcitabine and cisplatin alone, were accrued beyond the trial follow-up period and based on the sponsor’s extrapolation of trial data. Approximately 19% of QALYs were accrued in the postprogression health state. Results of the stepped reanalysis are available in Table 6, with full disaggregated results available in Table 11 of Appendix 4.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Gemcitabine + cisplatin | 88,259 | 1.08 | Reference |
Durvalumab + gemcitabine + cisplatin | 259,673 | 1.48 | 426,083 | |
Sponsor’s base case (deterministic) | Gemcitabine + cisplatin | 87,781 | 1.05 | Reference |
Durvalumab + gemcitabine + cisplatin | 258,095 | 1.44 | 433,828 | |
CADTH reanalysis 1 | Gemcitabine + cisplatin | 87,781 | 1.05 | Reference |
Durvalumab + gemcitabine + cisplatin | 254,222 | 1.31 | 638,712 | |
CADTH reanalysis 2 | Gemcitabine + cisplatin | 88,982 | 1.05 | Reference |
Durvalumab + gemcitabine + cisplatin | 261,490 | 1.44 | 439,416 | |
CADTH reanalysis 3 | Gemcitabine + cisplatin | 87,781 | 1.04 | Reference |
Durvalumab + gemcitabine + cisplatin | 258,095 | 1.43 | 438,794 | |
CADTH base case (1 + 2 + 3, probabilistic) | Gemcitabine + cisplatin | 89,298 | 1.04 | Reference |
Durvalumab + gemcitabine + cisplatin | 258,395 | 1.30 | 665,692 | |
CADTH base case (1 + 2 + 3, deterministic) | Gemcitabine + cisplatin | 88,982 | 1.04 | Reference |
Durvalumab + gemcitabine + cisplatin | 257,586 | 1.30 | 659,175 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: All stepped analyses were conducted deterministically.
Scenario Analysis Results
In scenario analyses, CADTH explored the impact of assuming RDIs, as in the sponsor’s base case. The inclusion of RDI did not meaningfully change the ICER.
CADTH undertook a price-reduction analysis based on the sponsor's base case and CADTH's base case (Table 7). The results show that a price reduction of 93% is required for durvalumab plus gemcitabine and cisplatin to be considered cost-effective at a WTP threshold of $50,000 per QALY.
Table 7: CADTH Price-Reduction Analyses
Analysis | ICERs for durvalumab + gemcitabine and cisplatin vs. gemcitabine and cisplatin ($/QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
None | $433,828 | $659,175 |
10% | $394,243 | $583,935 |
20% | $351,783 | $519,392 |
30% | $309,324 | $454,850 |
40% | $266,865 | $390,308 |
50% | $224,405 | $325,765 |
60% | $181,946 | $261,223 |
70% | $139,486 | $196,681 |
80% | $97,027 | $132,138 |
90% | $54,567 | $67,596 |
100% | $12,108 | $3,054 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Issues for Consideration
The clinical evidence for the treatment efficacy of durvalumab is only available in the first-line setting. The clinical experts consulted for this review noted that some patients who have started treatment with gemcitabine and cisplatin but have not progressed may be treated with durvalumab. However, the effectiveness and cost-effectiveness of durvalumab in subsequent lines of therapy is unknown. Should durvalumab be added to backbone chemotherapy in subsequent lines of therapy, the budget impact may be an underestimate.
Overall Conclusions
The CADTH Clinical Review found that, compared with gemcitabine and cisplatin, durvalumab plus gemcitabine and cisplatin was associated with a statistically significant improvement in OS and PFS in adults receiving first-line treatment for locally advanced or metastatic BTC, based on the TOPAZ-1 trial. However, the clinical importance of the modest survival gain afforded by treatment with durvalumab plus gemcitabine and cisplatin, compared with gemcitabine and cisplatin alone, is subjective. Measures of HRQoL were prespecified in the protocol and suggested no detriment to QoL with durvalumab plus gemcitabine and cisplatin. However, no definitive conclusions could be drawn on the impact of treatment with durvalumab compared with gemcitabine and cisplatin alone on time to deterioration and HRQoL because of limitations of the analyses, including a lack of statistical testing and attrition bias. The long-term clinical effectiveness associated with durvalumab treatment beyond the trial period (approximately ██ months) was also highly uncertain.
CADTH identified several limitations with the sponsor’s submitted pharmacoeconomic analysis. This included a misalignment between the modelled population and the Health Canada–indicated population, a lack of long-term efficacy data for durvalumab, overestimation of postprogression survival that lacked face validity, the use of RDI estimates that introduced important uncertainties in treatment costs, and methodological issues related to the way health state utilities were incorporated into the model.
CADTH undertook a reanalysis by adopting a spline odds (1 knot) function to extrapolate durvalumab OS, assuming an RDI of 100% for all treatments, and using age-adjusted estimates of health state utility. CADTH’s base case was aligned with the sponsor’s results, with both analyses suggesting that durvalumab treatment is associated with higher costs and increased QALYs, and is not cost-effective at a $50,000 per QALY WTP threshold as a first-line treatment. Based on publicly available list prices for all comparators, a price reduction of at least 93% would be required for durvalumab to be cost-effective at this WTP threshold. CADTH's estimates of cost-effectiveness may be biased in favour of durvalumab, as its reanalysis was unable to address the critical limitation regarding the apparent postprogression survival benefit because of constraints introduced by the submitted model structure.
The cost-effectiveness of durvalumab plus gemcitabine and cisplatin is sensitive to the uncertainty in long-term effectiveness of durvalumab treatment. The sponsor submitted OS data with multiple data cut-off points; using the most recent data cut-off increased the ICER. Approximately 68.9% of the long-term survival benefit associated with durvalumab was predicted in the period beyond the trial period through extrapolation. Given the modest magnitude of benefit and the high degree of uncertainty in long-term survival, a higher price reduction may be warranted. Further, the population in the sponsor’s pharmacoeconomic evaluation does not reflect the full Health Canada indication. The effectiveness and cost-effectiveness of durvalumab plus gemcitabine and cisplatin in subsequent lines of treatment is unknown.
References
1.Clinical study report: durvalumab-D933AC00001. A phase III randomized, double-blind, placebo-controlled, multi-regional, international study of durvalumab in combination with gemcitabine plus cisplatin versus placebo in combination with gemcitabine plus cisplatin for patients with first-line advanced biliary tract cancers (TOPAZ-1) [internal sponsor's report]. Gaithersburg (MD): AstraZeneca Pharmaceuticals LP; 2022 Feb 21.
2.Xie F, Pullenayegum E, Gaebel K, et al. A Time Trade-off-derived Value Set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. PubMed
3.National Institute for Health and Care Excellence. Pemigatinib for treating relapsed or refractory advanced cholangiocarcinoma with FGFR2 alterations [ID3740]: Committee Papers [sponsor submitted citation]. (Technology appraisal guidance TA722) 2021; https://www.nice.org.uk/guidance/ta722/documents/committee-papers.
4.DeltaPA. [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/. Accessed 2022 Aug 1.
5.Cancer Care Ontario. MFOLFOX6 - Provider Monograph [sponsor submitted citation]. 2019; https://www.cancercareontario.ca/en/drugformulary/regimens/monograph/58361.
6.Cancer Care Ontario. Regimen Monograph: FOLFIRI Regimen. Folinic Acid (Leucovorin)-Fluorouracil-Irinotecan [sponsor submitted citation]. 2019; https://www.cancercareontario.ca/sites/ccocancercare/files/FOLFIRI_GI_NET_P.pdf.
7.Cancer Care Ontario. Regimen Monograph: XELOX Regimen. Capecitabine (Xeloda®)-Oxaliplatin [sponsor submitted citation]. 2022; https://www.cancercareontario.ca/en/system/files_force/XELOX_GI_COLSBA_A.pdf.
8.Committee for Medicinal Products for Human Use. Teysuno: EPAR - Product Information [sponsor submitted citation]. (European public assessment report). Amsterdam (NL): European Medicines Agency; 2022: https://www.ema.europa.eu/en/documents/product-information/teysuno-epar-product-information_en.pdf.
9.H. Lee Moffitt Cancer Center and Research Institute. NCT02829918: Study of Nivolumab in Patients with Advanced Refractory Biliary Tract Cancers [sponsor submitted citation]. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/NCT02829918.
10.Keytruda (pembrolizumab): solution for infusion, 100 mg/4 mL vial [product monograph, sponsor submitted citation]. Kirkland (QC): Merck Canada Inc.; 2022: https://www.merck.ca/en/wp-content/uploads/sites/20/2021/04/KEYTRUDA-PM_E.pdf.
11.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab for injection), concentrate for solution for infusion, 50 mg / mL, intravenous. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Jul 13.
12.Yu M, Guerriere DN, Coyte PC. Societal costs of home and hospital end-of-life care for palliative care patients in Ontario, Canada. Health Soc Care Community. 2015;23(6):605-618. PubMed
13.Canadian Institute for Health Information. Patient cost estimator [sponsor submitted citation]. 2022; https://www.cihi.ca/en/patient-cost-estimator.
14.Imfinzi (durvalumab for injection): concentrate for solution for infusion, 50 mg / mL, intravenous [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2022.
15.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Imfinzi (durvalumab for injection), concentrate for solution for infusion, 50 mg / mL, intravenous. Mississauga (ON): AstraZeneca Canada Inc.; 2022 Jul 13.
16.Statistics Canada. Annual Demographic Estimates: Canada, Provinces and Territories, 2021 (Total Population only) 2021 [sponsor submitted citation]. 2021; https://www150.statcan.gc.ca/n1/pub/91-215-x/91-215-x2021001-eng.htm.
17.Indigenous Services Canada. Non-Insured Health Benefits Program: First Nations and Inuit Health Branch: Annual Report 2018/2019 [sponsor submitted citation]. 2020; https://www.sac-isc.gc.ca/eng/1584392581890/1584393350542.
18.Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000) [sponsor submitted citation]. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701.
19.B. C. Government. BC PharmaCare formulary search [sponsor submitted citation]. 2022; https://pharmacareformularysearch.gov.bc.ca.
20.Saskatchewan Drug Plan: search formulary [sponsor submitted citation]. 2022; https://formulary.drugplan.ehealthsask.ca/SearchFormulary.
21.Ontario Ministry of Health. Ontario Drug Programs Reference Manual [sponsor submitted citation]. 2020; https://www.health.gov.on.ca/en/pro/programs/drugs/resources/odp_reference_manual.pdf.
22.Reference Guide for Alberta pharmacies [sponsor submitted citation]. Edmonton (AB): Alberta Blue Cross; 2014: https://www.ab.bluecross.ca/pdfs/82477_pharmacy_reference_guide.pdf.
23.Information for Pharmacists. Updated - Claims Submission Procedure: Pharmaceutical Distribution Rate Changes [sponsor submitted citation]. Winnipeg (MB): Manitoba Health, Seniors and Active Living Non-Insured Benefits Branch; 2019: https://www.gov.mb.ca/health/pharmacare/profdocs/csp_pdrc.pdf.
24.Markup Policies in Public Drug Plans, 2018/19 [sponsor submitted citation]. Ottawa (ON): Patented Medicine Prices Review Board Canada; 2019: http://www.pmprb-cepmb.gc.ca/CMFiles/NPDUIS/refdocs/ReferenceDoc_MarkupPolicies_EN_2018-19.pdf.
25.Markup Policies in Public Drug Plans, 2019/20. Ottawa (ON): Patented Medicine Prices Review Board; 2021: http://www.pmprb-cepmb.gc.ca/view.asp?ccid=1312&lang=en. Accessed 2022 Sep 23.
26.Manitoba Health. Changes to Dispensing Fees. https://www.gov.mb.ca/health/pharmacare/dispensing_fees.html. Accessed 2022 Sep 23.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Locally Advanced and Metastatic BTC
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Average 28-day cost ($) |
|---|---|---|---|---|---|---|
Durvalumab | 50 mg / mL | 2.4 mL (120 mg) 10 mL (500 mg) Vial IV infusion | 938.6700 3,911.1100 | 1,500 mg q.3.w. for 8 cycles, followed by 1,500 mg every q.4.w. as monotherapya | Initial 8 doses: 558.73 Maintenance doses: 419.05 | Initial 8 doses: 15,644 Maintenance doses: 11,733 |
Durvalumab + gemcitabine/cisplatin | Initial 8 doses: 623.07 Maintenance doses: 483.39 | Initial 8 doses: 17,446 Maintenance doses: 13,535 | ||||
Durvalumab + gemcitabine/carboplatin | Initial 8 doses: 657.12 to 667.12 Maintenance doses: 517.44 to 527.44 | Initial 8 doses: 18,399 to 18,679 Maintenance doses: 14,488 to 14,768 | ||||
Chemotherapy | ||||||
Cisplatin | 1 mg/mL | 50 mg 100 mg Vial for IV infusion | 135.0000 270.0000 | 25 mg/m2 on Day 1 and 8 Q3W | 12.86 | 360 |
Gemcitabine | 40 mg/mL | 1,000 mg 2,000 mg Vial for IV infusion | 270.3000 540.6000 | 1,000 mg/m2 on Day 1 and 8 Q3W | 51.43 | 1,440 |
Gemcitabine/cisplatin | 64.34 | 1,802 | ||||
Carboplatin | 10 mg/mL | 50 mg 150 mg 450 mg 600 mg Vial for IV infusion | 70.0000 210.0000 599.9985 775.0020 | AUC 5 to 6 on Day 1 Q3W | 46.90 to 56.90 | 1,313 to 1,593 |
Gemcitabine | 40 mg/mL | 200 mg 1,000 mg 2,000 mg Vial for IV infusion | Not availableb 270.3000 540.6000 | 1,000 mg/m2 on Day 1 and 8 Q3W | 51.49 | 1,442 |
Gemcitabine/carboplatin | 98.39 to 108.39 | 2,755 to 3,035 | ||||
q.3.w. = every 3 weeks; q.4.w. = every 4 weeks
aFor individuals with weight ≤ 30 kg, the recommended dosing is 20 mg/kg q.3.w. for 8 cycles, followed by monotherapy at 20 mg/kg q.4.w. until weight increases to greater than 30 kg.14 The clinical experts noted this dosage would not be commonly administered given it is rare to find adults weighing ≤ 30 kg.
bOnly expired price was available from IQVIA Delta PA (accessed August 2022).4
Note: All prices are from wholesale prices from IQVIA Delta PA (accessed August 2022),4 unless otherwise indicated, and do not include dispensing fees. All cost calculations for agents with weight or height-based dosing was calculated using the mean body surface area of 1.8 m2 and mass of 75 kg. Daily and 28-day costs include drug wastage. The recommended dosage was obtained from the TOPAZ-1 trial.1
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | See CADTH appraisal. |
Model has been adequately programmed and has sufficient face validity | No | The model predicted a high postprogression survival benefit and a decreased ICER when treatment waning of durvalumab was assumed. Both observations lacked face validity. The latter also indicated that the sponsor’s embedded option to include treatment waning was not adequately programmed. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Multiple data cut-off points were available, which created confusion in aligning CADTH’s clinical and pharmacoeconomic appraisal. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of Sponsor’s Base Case
Parameter | Durvalumab + Gemcitabine/Cisplatin | Gemcitabine/Cisplatin | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 1.80 | 1.32 | 0.48 |
Progression-free | 0.85 | 0.56 | 0.29 |
Postprogression | 0.95 | 0.76 | 0.19 |
Discounted QALYs | |||
Total | 1.47 | 1.07 | 0.40 |
Progression-free | 0.73 | 0.48 | 0.25 |
Postprogression | 0.74 | 0.59 | 0.15 |
Discounted costs | |||
Total | $259,673 | $88,259 | $171,414 |
Drug acquisition in first-line | $173,988 | $7,934 | $166,054 |
Drug administration in first-line | $12,846 | $9,162 | $3,684 |
Adverse event | $5,768 | $6,010 | -$242 |
Disease management | $44,468 | $37,469 | $6,999 |
Drug acquisition in subsequent line | $11,104 | $15,593 | -$4,489 |
Drug administration in subsequent line | $4,060 | $4,562 | -$502 |
Terminal care | $7,439 | $7,528 | -$89 |
ICER ($/QALY) | $426,083 | ||
ICER = incremental cost-effectiveness ratio; LY= life-year; QALY = quality-adjusted life-year
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Durvalumab + Gemcitabine/Cisplatin | Gemcitabine/Cisplatin | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 1.63 | 1.31 | 0.32 |
Progression-free | 0.83 | 0.56 | 0.27 |
Postprogression | 0.81 | 0.76 | 0.05 |
Discounted QALYs | |||
Total | 1.30 | 1.04 | 0.25 |
Progression-free | 0.67 | 0.45 | 0.22 |
Postprogression | 0.62 | 0.59 | 0.04 |
Discounted costs | |||
Total | $258,395 | $89,298 | $169,097 |
Drug acquisition in first-line | $177,123 | $8,907 | $168,216 |
Drug administration in first-line | $12,800 | $9,156 | $3,644 |
Adverse event | $5,795 | $6,038 | –$243 |
Disease management | $39,994 | $37,505 | $2,488 |
Drug acquisition in subsequent line | $11,201 | $15,661 | –$4,460 |
Drug administration in subsequent line | $4,025 | $4,522 | –$497 |
Terminal care | $7,458 | $7,509 | –$51 |
ICER ($/QALY) | $665,692 | ||
ICER = incremental cost-effectiveness ratio; LY= life-year; QALY = quality-adjusted life-year
Detailed Results of CADTH Base Case
Scenario Analyses
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results — Scenario Analyses
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
CADTH base case | Gemcitabine/cisplatin | 89,298 | 1.04 | Reference |
Durvalumab + gemcitabine/cisplatin | 258,395 | 1.30 | 665,692 | |
Scenario 1: Assuming sponsor’s adopted RDI | Gemcitabine/cisplatin | 87,781 | 1.04 | Reference |
Durvalumab + gemcitabine/cisplatin | 254,192 | 1.30 | 650,602 | |
Scenario 2: Removing age-based cap on health utilities | Gemcitabine/cisplatin | 88,982 | 1.05 | Reference |
Durvalumab + gemcitabine/cisplatin | 257,586 | 1.31 | 647,244 | |
Scenario 3: Assuming age-based multiplier adjustment to health utilities | Gemcitabine/cisplatin | 88,982 | 1.05 | Reference |
Durvalumab + gemcitabine/cisplatin | 257,586 | 1.31 | 650,798 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; RDI = relative dose intensity
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Takeaways
Key Takeaways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA)15 estimating the incremental budget impact of reimbursing durvalumab + gemcitabine/cisplatin for use by patients aged 18 years and older with locally advanced or metastatic biliary tract cancer (BTC) in first-line therapy. The BIA was undertaken from the perspective of the public drug plans in Canada over a 3-year time horizon (2024 – 2026), and the sponsor’s estimates from across Canada reflect the aggregated results from provincial budgets (excluding Quebec). Key inputs to the BIA are documented in Table 14.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Annual growth rate | 0.50% |
Annual incidence rate of BTC | ██████ per █████████ persons |
Percentage eligible for first-line treatment | ██████% |
Number of patients eligible for drug under review | ███ / ███ / ███ |
Market Uptake (3 years)a | |
Uptake (reference scenario) Gemcitabine + Cisplatin | 100% / 100% / 100% |
Uptake (new drug scenario) Durvalumab Gemcitabine + Cisplatin | ███% / ███% / ███% ███% / ███% / ███% |
Cost of treatment (per patient)b | |
Cost of treatment over week Durvalumab Gemcitabine Cisplatin | $3,858.97 $288.12 $90.00 |
BTC = biliary tract cancer
aThe sponsor estimated market share using internal data such as market research, product demand studies, clinical expert opinion, real-world evidence data, and competition assessments. However, the data used to estimate market share of durvalumab was not submitted to CADTH.
bOnly includes drug-acquisition costs.
The sponsor estimated the number of eligible patients for durvalumab treatment using an epidemiologic approach with data obtained from various sources including: Statistics Canada population estimates and the sponsor-commissioned ICES-HOPE study.15-17 The sponsor adopted an average annual population growth rate of 0.50% in estimating the total number of BTC cancer cases over the time horizon.18 The sponsor considered comparator in first-line setting to be gemcitabine/cisplatin. Although the sponsor included the number of progressed patients on subsequent treatments, which included FOLFOX, FOLFIRI, capecitabine + oxaliplatin, nivolumab monotherapy, pembrolizumab monotherapy and gemcitabine/cisplatin, the cost of subsequent treatments was not included in the sponsor’s base case. The sponsor included drug-acquisition costs of durvalumab, gemcitabine and cisplatin as well as mark-up fees, which were obtained from IQVIA DeltaPA database and published literature, respectively.4,19,20 Drug dosages were adjusted by relative dose intensities; both were obtained from the TOPAZ-1 trial.1,21-24 Total costs were calculated by multiplying drug cost per administration with treatment duration, which was modelled using time to treatment discontinuation curves. The sponsor assumed no drug wastage in estimating treatment costs.
Summary of the Sponsor’s BIA Results
The sponsor estimated the net 3-year budget impact of introducing durvalumab + gemcitabine/cisplatin for first-line treatment of patients aged 18 years and older with locally advanced or metastatic BTC to be $125,469,933 (Year 1: $32,462,792; Year 2: $42,860,434; Year 3: $50,146,707).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The population in the sponsor’s BIA was not aligned with the Health Canada indication: The estimated population included adult patients receiving treatment for locally advanced or metastatic BTC in first-line setting, which does not reflect the full Health Canada–indicated population. Clinical expert feedback received by CADTH for this review noted that the expected place of durvalumab is in first-line setting given there are limited treatments available for this patient population. As such, CADTH focused this review on durvalumab treatment in the first-line setting only.
CADTH could not address this limitation.
Market share of durvalumab is underestimated: The clinical expert consulted for this review by CADTH anticipated a potential rapid and higher uptake of durvalumab for patients with locally advanced or metastatic BTC. The clinical experts noted there have been no new treatments for the indication of interest for a long time and clinicians are attuned to durvalumab. Further, clinicians, nurses and pharmacists have been exposed to administration of durvalumab for other indications and, as such, the adoption of durvalumab is anticipated to be quick. Should durvalumab be reimbursed by public drug plans, the market share of durvalumab, and therefore, budget impact would be higher than estimated by the sponsor.
In CADTH reanalysis, durvalumab has a market share of 70% in year 1 and 74% in year 2.
Use of relative dose intensity (RDI) to estimate actual drug costs is not appropriate: The sponsor’s base case incorporated reduced dose intensities for durvalumab, gemcitabine and cisplatin using data from the TOPAZ-1 trial. The consideration of RDI is problematic as this parameter can be influenced by several factors. The dose received by a patient may differ from the full planned dose of the drug due to dose delays, missed doses, dose reductions to manage toxicity, or subsequent dose re-escalation; each of these have differing impacts on drug costs. The CADTH reanalysis of the CUA assumes 100% RDI and the BIA and CUA should be aligned.
In the CADTH reanalysis, 100% RDI was assumed. CADTH explored the impact of assuming sponsor’s adopted RDIs in a scenario analysis.
Markups and dispensing fees are uncertain: The sponsor included markups but excluded dispensing fees in estimating the drug-acquisition costs. The sponsor assumed that IV infusions are administered in a hospital setting and, as such, do not require a dispensing fee. The estimation of markups and dispensing fees for the hospital pharmacies is highly uncertain. It is unclear whether there is a cost associated with pharmacist’s time for the chair time required for IV administrations, rather than a dispensing fee. Further, in some provinces, such as Manitoba, there is no separate mark-up policy from the pharmacy dispensing fees, which makes the calculation of markups and dispensing fees uncertain.25,26 Although the inclusion of markups and dispensing fees is appropriate, the uncertainty in these estimates make the comparison of drug costs and the budget impact uncertain.
In CADTH reanalysis, the markups and dispensing fees were excluded. CADTH explored the impact of including markups and dispensing fees in a scenario analysis.
Treatment duration is uncertain: The sponsor used time to treatment discontinuation curves from TOPAZ-1 trial to model treatment duration. In the trial, patients were allowed to continue treatment beyond progression at the discretion of the investigator and patient as long as they are deemed to be receiving clinical benefit. However, the product monograph for durvalumab and clinical experts consulted by CADTH noted durvalumab treatment should be continued until disease progression or unacceptable toxicity.
CADTH explored the impact of adopting treatment duration based on progression-status in a scenario analysis.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by adopting a relative dose intensity of 100%, increasing the market share of durvalumab and excluding markups and dispensing fees.
Table 15: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Relative dose intensity | <100% (varies by drug) | 100% |
2. Market share of durvalumab (year 1 / year 2 / year 3) | ███% / ███% / ███% | 70% / 74% / 78% |
3. Markups | Included | Excluded |
CADTH base case | Reanalysis 1 + 2 + 3 | |
In the CADTH reanalysis, the 3-year budget impact of reimbursing durvalumab + gemcitabine/cisplatin from the public drug plan perspective for first-line treatment of patients aged 18 years and older with locally advanced or metastatic BTC increased to $135,947,567 (Year 1: $42,645,066; Year 2: $45,307,337; Year 3: $47,995,164).
The results of the CADTH step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $125,469,933 |
CADTH reanalysis 1 | $127,291,847 |
CADTH reanalysis 2 | $142,041,870 |
CADTH reanalysis 3 | $118,367,861 |
CADTH base case | $135,947,567 |
BIA = budget impact analysis
CADTH also conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 17. The scenario analyses involved:
Assuming sponsor’s estimated RDI.
Including markups and dispensing fees.
Assuming treatment duration based on progression-status.
Price reduction of 93% for durvalumab.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $3,123,282 | $3,138,899 | $3,154,593 | $3,170,366 | $9,463,858 |
New drug | $3,123,282 | $35,601,691 | $46,015,027 | $53,317,073 | $134,933,791 | |
Budget impact | $0 | $32,462,792 | $42,860,434 | $50,146,707 | $125,469,933 | |
CADTH base case | Reference | $3,227,334 | $3,243,470 | $3,259,688 | $3,275,986 | $9,779,144 |
New drug | $3,227,334 | $45,888,536 | $48,567,024 | $51,271,150 | $145,726,711 | |
Budget impact | $0 | $42,645,066 | $45,307,337 | $47,995,164 | $135,947,567 | |
CADTH scenario analysis: sponsor’s estimated RDI | Reference | $2,946,493 | $2,961,225 | $2,976,031 | $2,990,911 | $8,928,168 |
New drug | $2,946,493 | $44,995,917 | $47,634,889 | $50,299,126 | $142,929,932 | |
Budget impact | $0 | $42,034,692 | $44,658,858 | $47,308,214 | $134,001,764 | |
CADTH scenario analysis: including markups and dispensing fees | Reference | $3,420,974 | $3,438,078 | $3,455,269 | $3,472,545 | $10,365,893 |
New drug | $3,420,974 | $48,641,849 | $51,481,046 | $54,347,419 | $154,470,313 | |
Budget impact | $0 | $45,203,770 | $48,025,777 | $50,874,874 | $144,104,421 | |
CADTH scenario analysis: estimating treatment duration based on progression-status | Reference | $4,841,000 | $4,865,205 | $4,889,531 | $4,913,979 | $14,668,716 |
New drug | $4,841,000 | $42,354,918 | $44,719,673 | $47,107,017 | $134,181,607 | |
Budget impact | $0 | $37,489,712 | $39,830,141 | $42,193,038 | $119,512,891 | |
CADTH scenario analysis: 93% price reduction for durvalumab | Reference | $3,227,334 | $3,243,470 | $3,259,688 | $3,275,986 | $9,779,144 |
New drug | $3,227,334 | $6,756,500 | $6,992,030 | $7,229,748 | $20,978,278 | |
Budget impact | $0 | $3,513,029 | $3,732,343 | $3,953,762 | $11,199,134 |
BIA = budget impact analysis
Stakeholder Input
Patient Input
Canadian Cancer Survivor Network
Partners of Canadian Cancer Survivor Network are: Canadian Liver Foundation, Canadian Organization for Rare Disorders, Cholangiocarcinoma Foundation, Colorectal Cancer Resource & Action Network, Gastrointestinal Society, Regroupement québécois des maladies orphelines.
About the Canadian Cancer Survivor Network
The Canadian Cancer Survivor Network (CCSN) is a national network of patients, families, survivors, friends, community partners, funders, and sponsors who have come together to take action to promote the very best standard of care, whether it be early diagnosis, timely treatment and follow-up care, support for cancer patients, or issues related to survivorship or quality of end-of-life care. https://survivornet.ca/
Information Gathering
The Canadian Cancer Survivor Network, along with its partners, utilized SurveyMonkey to create and collect quantitative data for the survey on durvalumab. All participating groups utilized their social media platforms to disseminate the survey to collect the responses. The Colorectal Cancer Resource & Action Network (CCRAN) gathered qualitative data between July 7 – July 22, 2022, by conducting seven interviews with patients (4) and caregivers (3) who have experience with durvalumab. The CCSN survey was open from July 18, 2022, to August 2, 2022, to obtain responses. From the 58 people who responded to the CCSN survey, 21 live in Canada, 35 live in the United States, 1 life in the UK, and 1 life in Spain. Out of the 58 respondents, 44 identify as female and 9 identify as male. Out of the 58 respondents, 12 patients have or are taking durvalumab, 25 have not taken durvalumab, and 17 are caregivers.
Disease Experience
The respondents were asked how their cancer was diagnosed; the survey resulted in 54 persons who provided responses:
Contrast-enhanced computed tomography (CT): 44
Magnetic resonance imaging (MRI): 38
Ultrasound (US): 30
Blood Tests: 37
Biopsy: 48
Other: 9 (Six patients underwent PET scans, one ablation, one pathology done on resected liver, and one patient underwent EGD & ERCP)
When asked what stage of biliary tract cancer they have, the survey provided 29 persons who gave responses, with the majority in late stage (4) and detailed as follows; three are in early stage (1), four are in middle stage (2 or 3), 18 are late stage (4) or metastatic, and four do not know what stage they are at currently.
Current treatments that were identified by the survey respondents include:
Radiation: 8
Surgical Therapy: 7
Targeted Therapy: 7
Immunotherapy: 13
Clinical Trials: 6
Gemcitabine plus Cisplatin: 26
Other: 10 (3 Folfox, 1 Folfox & Opdivo, 1 Xeloda, 1 Capecitabine adjuvant therapy, 1 Surgery in the US, 1 Abraxane, 1 Lobectomy, 1 series of OxaliplatinplusGemcitabine, Folfiri, microwave ablation, Cryoablation, and Chemoembolization).
Experiences With Currently Available Treatments
When asked about their stage of biliary tract cancer, among those with no experience with durvalumab, the survey provided 41 responses and these are broken down as follows; three are in early stage (1), nine are in middle stage (2 or 3), 26 are late stage (4) or metastatic, and four do not know what stage they are at currently.
When asked about their current quality of life and day-to-day living experience, the following responses were provided:
Itching: 7
Jaundice: 4
Light coloured, greasy stools: 4
Dark urine: 7
Abdominal (belly) pain: 19
Loss of appetite, weight loss: 14
Fever: 5
Nausea and vomiting: 10
Other: 17 (7 fatigue, 1 lower back pain, 1 GI issues, 1 inconsistent bowel, 1 radiating shoulder pain, 1 leg cramps, 1 cough, 1 skin rash, 2 neuropathy, 1 tinnitus, 1 foot pain due to skin issues and loss of nails, 1 ostomy).
Survey respondents indicated these adverse effects due to their current treatment:
Diarrhea: 11
Nausea, vomiting: 15
Infection: 3
Anemia: 7
Bruising, bleeding: 10
Kidney damage which may cause swelling, may require dialysis: 5
Hearing loss including ringing in ears: 8
Flu-like symptoms: 8
Rash: 5
Hair loss: 17
Muscle Weakness: 15
Blood in urine: 1
Feeling of “pins and needles” in arms and legs: 10
Numbness and tingling of the arms and legs: 12
Tiredness: 34
Difficulty sleeping: 17
Swelling of arms, legs: 10
Other: 15 (4 constipation, 1 leg jumping and pain, 1 cold sensitivity, 1 case of significant dry mouth, dry skin including cracks and peels, nail discoloration and became necrotic, hand-foot syndrome, 1 gums swelling & mouth ulcers, 1 sore mouth & change in taste, 1 cold clammy sweats, 1 neuropathy, 1 case of dry eyes, dry mouth, skin issues impacting feet and nails, and 1 case of feet feeling sore and strange at all times)
Survey respondents were asked if they have had any issues accessing any therapies; 41 responded as follows:
Limited availability in my community: 11
Financial hardship due to cost: 10
Travel costs associated with accessing therapy/treatment: 11
Supplies or issues with administration: 3
I haven’t had any issues accessing therapy: 19
Other: 6 (1 Initially insurance issues, 1 Language barrier, 1 Bile duct stents, high WBC, fever, and ERCPs cancelled chemotherapy, 1 Made comment about Imfinzi not being FDA approved so insurance wouldn’t pay).
When asked if any needs in their current therapy are not yet being met, 29 survey respondents said no, six said yes, and 10 said yes and gave some information as to why:
“Second opinions have recommended durvalumab and y90 radioembolization and oncologist is unwilling to pursue at this time.”
“Lack of access to drugs available in the US.”
“Doctors who are experienced.”
“I currently do not have a treatment plan for a reoccurrence. There is too much time between discovery of a reoccurrence and development of a plan of action.”
“Oncologist is hesitant to refer me to a larger cancer center.”
“Would love to try Imfinzi.”
Current chemo is no longer working. Moving on to clinical trial.”
“Insurance coverage.”
“Cure.”
“I have a IDH2 mutation, so treatments are extremely rare and there is no trial available in my province and not many in Canada.”
Improved Outcomes
When asked about the following issues that they would hope to see a new treatment address to manage their disease, 44 survey respondents replied:
Maintain quality of life: 40
Delay onset of symptoms: 23
Access to a new option for treatment: 29
Reduce side effects from current medications or treatments: 25
Ease of use: 21
Prolong life: 40
Provide a cure: 38
Other: 1 (Reduce tumour to the point it may be considered operable)
Survey respondents were asked to describe how much of an improvement would be needed from a new treatment to make it better than the current treatment that they are undergoing. The consensus among the 35 responses included prolonged life with similar side effects to current treatments, ease of access, quality of life to remain as normal as possible, reduce side effects and pain, and ultimately to find a cure.
Experience With Drug Under Review
When asked to compare their experience with durvalumab to other therapies in treating their biliary tract cancer, 17 respondents rated their experience as follows:
Symptom management: 2 Much Better; 14 Little or no difference
Side effects: 1 Much Better; 13 Little or no difference; 2 Much worse
Ease of use: 3 Much Better, 13 Little or no difference
Disease progression: 2 Much Better; 9 Little or no difference; 2 Much worse
Other: 3 Little or no difference
Survey respondents were asked what adverse effects were caused by taking durvalumab, 17 indicated the following:
Anemia: 3
White blood cell count decreased: 4
Platelet count decreased: 4
Fatigue: 13
Dermatitis/Rash:1
Thyroiditis: 1
Infection: 1
Constipation: 5
Decreased sodium level: 2
Decreased appetite: 3
Muscle/bone pain: 2
Other: 7 (1 Headache, fever, 1 Diarrhea, 3 cannot distinguish if the effects are from chemo or durvalumab)
Survey respondents were asked to describe the positive and negative effects of durvalumab:
“Just started so unsure of how this will go but on day 2, fever, congestion, and body aches worsened.”
“I receive Imfinzi every other treatment. I’ve not yet had labs or scans to determine its effectiveness. When I am treated with Imfinzi I need an extra recovery day because it causes great fatigue.”
“Worked for several months then stopped working. Switching to different treatment.”
“I have not really had any added side effects from adding Imfinzi. My latest CT did not really show any changes but hopefully the Imfinzi is helping keep the cancer from growing.”
“I added Imfinzi to Gem/Cis chemo during my first round of chemo. Completed 4 rounds of chemo (8 treatments) with Imfinzi every other chemo treatment (so 4 infusions total of Imfinzi) and my scans showed growth. I do have the FGFR2 mutation.”
“I have only been taking Imfinzi along with Cis/Gem for 4 months and had one set of scans. So far it looks like my disease has slowed progression and no new disease. It is hard to say the positive and negative effects because I have not been on it long enough. It seems much easier for my body to take than chemo.”
“Positive is an immunotherapy I am appropriate for. I don’t know the results. CT scan in a month. Negative is GI issues 3-4 days after therapy and chemo that I did not experience with gem/cis.”
“Imfinzi, in combination with chemotherapy, will not work for all patients but in those patients for whom it works, it has a durable benefit. This is an important outcome for patients facing a terminal diagnosis.”
Survey respondents were asked if they would recommend that durvalumab be made available to all patients who qualify for it. Of the 19 responses, there was a consensus that durvalumab should be made available to all who qualify for it. There were only two people who responded saying that they are unsure at this time.
“Yes, as there are few options available in Canada for patients that can no longer tolerate Gem/Cis or Folfox.”
“Absolutely. Cholangiocarcinoma is a death sentence. It may not work for everyone, but everyone should be able to try.”
“Yes. It is moving towards standard of care in the US. DISAPPOINTING Canada is so far behind.”
“Yes. There is Phase III data confirming a survival benefit. BTC is rare and has a very poor prognosis, any new therapies need to be embraced and studied in the larger population of Canadian cancer patients. We should provide patients with the therapies and make the effort to prospectively collect the outcomes.”
Survey respondents were asked to share about their own cancer journey:
“I stayed positive during this whole journey. Faith helped out and just staying physically active.”
“I wish chemo wasn’t as harsh.”
“Canada needs to adapt an integrative approach to cancer and be more willing to work with naturopaths as well. A wholistic approach to cancer care is needed including an opening to off label drugs that have shown to benefit cancer patients.”
“Surgical and radiation options in Canada are competitive, but systemic treatments are lagging.”
“That I wish doctors care about the patient, explaining what is happening, what to expect, and remember we are scared and looking to them for help.”
“Through our journey I found it difficult to get information. The Cholangiocarcinoma Foundation, which I stumbled on through an internet search, and its groups were my only source of real information and what to expect.”
“It is difficult to negotiate this disease treatment in a foreign language. I am sure a lot is lost in translation.”
“Worrying team around you don’t have specialist knowledge.”
“Include very sick patients into trials and give them treatment because most are at that late stage, and we have to do better for them other than excluding them and giving no hope.”
“I’d like to pursue aggressive treatments, but my oncologist is hesitant. It feels a bit frustrating when it seems as though she doesn’t want to do whatever we can to help me.”
“My doctor said if my cancer grows in any way, which it hasn’t, he is stopping my treatment and will let me die. That kind of care I get in Alberta.”
“It was only detected because of tests for other ailments.”
“Thankful for Imfinzi drug company for giving me a chance.”
“Currently on a clinical drug trial for the FGFR2 mutation and I am seeing shrinkage.”
“It was difficult. Mother suffered a lot and lost her appetite. She passed away May 6 of this year. Mom was diagnosed October 2021.”
“Getting appropriate and timely care for biliary tract cancer in BC has not been possible. Horrendous that there is a refusal to invest public money in us because of poor prognosis. Had I waited for care instead of going to the US, I assume I would be a lot sicker or dead now.”
“Patients need more treatment options and a chance at a longer survival.”
“It’s a challenge. Medical providers don’t get back to you or fail to schedule appointments. Not to mention insurance issues. Finding a GOOD doctor who knows what he/she is doing. Different medical facilities treat each patient differently, telling them there is nothing they can do for them, or they are too busy to treat you, while other will do whatever they can to help extend your life & quality of life.”
“Having a rare cancer and finding answer is very isolating. It would have been easier to find out information about my disease if BCCA had pointed me in the direction of the Cholangiocarcinoma Foundation right at the start of my diagnosis. I have a mixed hepatocellular/cholangiocarcinoma diagnosis and there is virtually no information on this cancer and only one study I could find out of China. It seems that the doctors are still trying to figure out how to treat this cancer. It is frustrating and depressing trying to find out any information. Most doctors treat it as CC but there are a few that think it should be treated as HCC - if they don't know how am I supposed to make treatment decisions? It would be nice if one of the drug companies did some research into this mixed cancer.”
As a caregiver for someone with biliary tract cancer, respondents selected the following as issues they encounter:
Fatigue: 17
Emotional Drain: 17
Anxiety/Worrying: 18
Management of medications: 15
Management of side effects: 15
Hours spent in medical appointments: 15
Monetary concerns (absence at work, driving expenses, etc.): 10
Lifestyle changes: 14
Inability to plan ahead: 15
Anger: 13
Feeling isolated (difficulty connecting with friends, geographical remoteness): 12
Feelings of “doom” due to challenging prognosis: 14
Feelings of helplessness: 14
Other: 7
“Sadness and depression from lack of support, doctors don’t care, and treatments aren’t explained.”
“Absolutely every point above, intensely magnified and I am a therapist. All my training and health-caregiving for 4 other humans previously did not help me emotionally or physically navigate this horrific diagnosis.”
“Language barrier.”
“Overwhelm! Exhaustion!”
“Neuropathy resulted in loss of driving. Huge impact on family.”
“I was very lucky to have noticed a small speck of blood when I coughed and had it seen to by my doctor.”
Caregivers were asked to describe how caring for someone with biliary tract cancer has affected their daily routine or lifestyle:
“Constant worry and advocacy. Being prepared at medical appointments so things are not missed.”
“Well, there is greater concern over his health, and I have reduced working hours to provide the attention needed. More than willing to do this however.”
“My life is all about him. No time for anything now. It’s caring for him, driving to treatments, calling insurance, being sad, feeling helpless, lost and alone.”
“I have no quality of life. Every day is life a freight train hitting me with a new issue, crisis to avert or attend to. Appointment, hospital stay, emergency, meds, vitals, meal prep based on today’s issue. Drains, shots, nurses, docs, fielding calls from friends, family, and colleagues. Loneliness, staying in hotels while husband is in hospital, driving the patient to each appointment, interstate travel, insurance company appeals. Waiting alone, taking care of my home and bills and repair-people alone, eating meals out of hospital vending machines, ignoring my own health issues. Not seeing friends for months, no restaurants, dates, vacations, or a break…”
“Constantly having to leave work early or go to work late to take care of them or for doctor appointments.”
“This cancer, and the medications used to manage symptoms, caused severe personality changes in my spouse. He became paranoid and suspicious of everything. His inability to cope resulted in uncontrollable spending which in turn saddled me with debt not just from treatment but the inability to accept a terminal diagnosis. My whole world shattered, and nothing is as it was.”
“Not being able to plan or travel. Unable to get travel insurance.”
“It has completely flipped my life and I’ll never be the same again. I value and respect all caregivers, but this was a whole different beast. The nature in which this disease took my loved one has forever changed me and caused me PTSD for life. It drives me to keep helping others though.”
“With all the medical appointments we have it is difficult to schedule other activities!”
“Upended it completely. Hard to work but have to.”
“My life is no longer my own. Time and energy revolve around my loved one. My life is consumed with anxiety and worry about the future or lack of future and being alone in my old age. We used to travel, now we just go to doctor’s appointments and take meds.”
“Changed our entire life. Everything is centered around this disease now.”
“I’m retired, but it takes so much time. Especially during Covid, tasks like grocery shopping which he could have done even while on chemo on the good days, I wouldn’t let him do because of risk of Covid.”
“Home life has become isolated from most friends, so life is quieter. I feel the need to watch over my wife and make sure she is ok. We used to travel and now we cannot because of the treatment.”
“It has changed our lives completely and redirected the course of our lives. We are still learning day by day how to work and live with this disease.”
“I have to take on most of the childcare, continue to work, and look after my husband. I have had to give up all extra-curricular activities at work and in my personal life.”
The caregivers were asked to describe the most challenging adverse effects related to their loved one and their current therapy or treatment. While some of the respondents mentioned fatigue, fear, hopelessness, and insomnia as adverse effects, some caregivers went into more detail:
“The reaction to the immune boosting shots after caused severe joint pain, constant hand and leg cramps, inability to properly rest or sleep, and weakness.”
“The narrow mindedness of our oncologist and the Canadian medical system. For example, shots he needs for WBCs are not covered for the chemo he is on, yet the chemo has caused this. Insurance has helped cover this, but the shots are $1,200 after every chemo treatment. Infuriating.”
“Making sure they get the right treatments and that it helps prolong their life, and hopefully find a cure or at least remission.”
“Recovery from liver resection and bile duct reconstruction nine weeks ago and the related complications.”
“IV lines, platelet counts, and dehydration.”
“Losing his ability to be independent. He went from once vibrant, to unable to stay awake for more than fifteen minutes at a time. Staying awake all night out of fear of going to sleep, to sleeping all the time. Eating, to not able to eat. Loss of memory and hearing. Angry all the time.”
“The language barrier.”
“Stents and ERCPs.”
“Infections and frequent hospitalizations.”
“Loss of appetite, weight loss, and mental changes.”
“Loss of ability to drive due to neuropathy.”
“Mental health day to day and planning the next step of treatment.”
“Discomfort, fear, and uncertainty about the future. Inability to plan ahead or make commitments.”
“No financial support.”
“Nutrition. Pain.”
“Fatigue and feeling discouraged about the prognosis and feeling unwell.”
Caregivers were asked if there was anything else that they would like to share about their experiences as a caregiver:
“Especially difficult during Covid. Recognize the strain this has also put on the healthcare system.”
“Don’t throw pamphlets at us to handle this. Provide support and understanding.”
“We need support and ways for that caregiver who gives up everything to still find financial support as the caregiver is responsible to take care of everything. How can you work when this is a full-time job caring for them? There is no support system if you don’t have family to help.”
“This is hard stuff.”
“Being a caregiver is incredibly difficult but also an honour to care for those we love so much. The most difficult part of it is that our life goes on but will never be the same. We are forever haunted by this disease by asking why.”
“Be prepared for a long and rough journey.”
“All therapy and treatment are for the patient. We are forgotten, but the stress and worry affect us too. Our lives are forever changed too.”
“This journey was very difficult. It brought me closer to my mother in prayer. I was with her every step of the way, and it hurt me to see her suffer. This journey was very painful for me. I love and miss my mother so much! I can say cancer sucks! Cholangiocarcinoma sucks!!”
“I wish there was a cure!’
“Just feeling so alone.”
“Limited treatment options.”
“New therapies provide hope and living with hope is probably the most important aspect of the cancer journey. Although the probability of success is low, we must continue to support research and the approval of new therapies to provide hope.”
Companion Diagnostic Test
Not applicable.
Anything Else?
The Canadian Cancer Survivor Network summarized seven interviews done by Colorectal Cancer Resource & Action Network that detail the experience of these individuals with durvalumab.
Respondent A
Respondent A is a 61-year-old woman who was diagnosed with metastatic biliary tract cancer in 2018. She is lives in Chambly, Quebec, is married, and has two adult children.
She was a regular cyclist and was diagnosed as a result of becoming very ill while cycling. When a CT scan and biopsy confirmed that the left lobe of her liver was full of cancer, treatment was initiated. She received 11 cycles of Cisplatin plus Gemcitabine, and the left lobe of her liver was removed in early December 2018. However, cancer returned in two lymph nodes in November 2019. She started in Oxaliplatin plus Gemcitabine in November 2019 until June 2020. She eventually experienced neuropathy and myelosuppression, so the Oxaliplatin was stopped. In September 2020, a scan showed that a third lymph node near the aorta was impacted, so she restarted the Cisplatin and Gemcitabine, which controlled the progression of the disease. She then had foundation medicine and Omnicure testing. She started Durvalumab plus Olaparib in March 2021 as part of a clinical trial at the Princess Margaret Hospital and had seven cycles from March 2021 to October 2021.
Time to progression on the treatments she received before Durvalumab ranged from four to eleven months, while surgical resection kept her disease-free for eleven months. Side effects from previous treatments included neuropathy in her hands and feet throughout her treatment journey as well as myelosuppression, which she couldn’t really feel, severe nausea and vomiting in the beginning until the dosage of the chemo drugs was adjusted, and since her hemoglobin was low, she had breathing problems and was unable to exercise. One side effect that she found very difficult to control when on chemo was low white blood cells, making her immunocompromised.
Experience while on Durvalumab plus Olaparib
In general, she reported that she had no side effects while on Durvalumab plus Olaparib: “It has had no negative impact on my quality of life whatsoever. Nothing.” When asked to rate Durvalumab plus Olaparib on a scale of one to ten, she replied “I would give it a 10 out of 10!” She also stated that “I responded really well to Durva. My cancer was regressing and there was really nothing to worry about.” She said that the cancer in the three lymph nodes regressed. She reported that Durvalumab was easier to use – one-and-a-half hours versus for an eight-hour infusion for the other drugs. She also did not need to take anti-nausea drugs, and it gave her body a rest. She said, “I am here today because of Durva. I sincerely believe it,” adding, “Durva has a huge impact on targeting a specific mutation and has no toxicity. At least it has had no toxic effects on me.”
Respondent E
Respondent E is a 49-year-old man, married with three children from 8 to 13, living in Ottawa, Ontario. He was diagnosed with metastatic cholangiocarcinoma after experiencing digestive symptoms, reduced appetite, and mild pain and discomfort under his ribs. His CT scan showed a primary tumour in the liver and other tumours in the liver, lung, and abdomen. After his biopsy, a sample of his tumour was sent out for genetic profiling, and several mutations were discovered.
He reports that his initial symptoms resolved when he started chemo. His wife is a surgical oncologist, so she brought the data on his mutations to his treating oncologist. He then accessed Durva through the compassionate care program. They added Durva to his third cycle of Gem/Cis; he had three cycles of Durva and four of chemo. Gem/Cis resulted in tinnitus, low-grade nausea and mild constipation. His moderate fatigue was not disruptive. He has mild lightheadedness and a change in taste. As for Durva, he reported no other side effects, and after two cycles of Durva, the CT scan showed that the lung mets were no longer visible or had shrunk considerably, the liver mets regressed, as did the cancer in his lymph nodes in his abdomen.
He added, “I have been able to continue to work throughout it all. I am the CEO of an industry association for cement, so all good.” From one to ten, he rated Durva as a ten because it had not changed his quality of life. He was 100 per cent certain that it was worth accessing Durva “especially for a rare disease,” where any tool will help. He believes that it is “counter factual to not be able to access treatments that have promise because it adds to anxiety and discouragement that you couldn’t get through regulatory barriers...if you see something that helps you, it is discouraging if you can’t access it! If it helps you with outcomes, you should be able to access it. What adds insult to injury is that it is so expensive in the event you are required to pay for it to extend your survival.”
He then added: “It was wonderful. My tumour markers just plummeted, my CT scan showed a great response, and all my cancer symptoms are gone. I would say that this was a great success story.”
Respondent F
Respondent F is a married 51-year-old woman living in Watford, Ontario, with two young adult children. She was diagnosed after a long series of tests starting in 2020 and ending with a metastatic biliary tract cancer diagnosis in 2022, when she started chemo and was given compassionate access to Durva at the same time. Before that, she underwent surgical resection of her liver and then adjuvant therapy with Capecitabine, from which she reports having had bad foot syndrome to the point that she had difficulty walking, so her cycles were cut short.
Durva was made available to her on May 6, 2022, through the compassionate access program. At the time of this interview, she was in the middle of her fourth cycle. She reported that her white blood cells dropped after the first treatment, but other than that, she has reported few additional symptoms – mild constipation and a bit of nausea. She reported being able to continue full-time work as a registered nurse. She had no toxicity issues. She rated her quality of life while on Durva as being eight out of 10, remarking that “I am on chemo, so it cannot be perfect.” The fact that Durva was every three weeks in a one-hour infusion was quite convenient for her as well.
Respondent F said that everyone in her family was doing well, because she is doing well. Initially it was a lot to take in, but her physical health has not changed very much, which helps with her family’s ability to cope. As long as she is okay, her family is ok. She is grateful that she has had the opportunity to take Durva, which is fairly new in Canada for people with her disease. That is the biggest gift. “I was able to work, take care of my family, socialize, take care of my mother who requires me to be a caregiver to her, she lives far away from me, so I have to travel, and I get to do it, because I am feeling well enough.”
She continued: “There has been no change in standard of care treatment for so long, so the sooner we introduce therapies, the better in accessing new treatments, we could potentially enhance and improve life and outcomes for patients. We need to follow the science and stop the red tape. Listen to patients’ please. What if it were your loved one with this cancer? Wouldn’t you want them to access this drug therapy to live longer?”
Respondent G
Respondent G is a 50-year-old male from South Africa, married with two adult children. He was diagnosed with metastatic cholangiocarcinoma in late December 2021. He had several symptoms for six months before he was diagnosed, including weight loss, loss of appetite, and excessive diarrhea. He was started on Imfinzi plus chemo in the first week of 2022; however, he had to pay for Imfinzi himself, at a cost of $8,000 USD per cycle, while his insurance company paid for the chemo.
His most difficult symptom has been diarrhea, which has always been the biggest problem for him. He has meds for it, and he is able to control it. He also has pain below his right ribs, which might be disease-induced.
A recent CT scan showed great improvement in all areas of his body, so the protocol is working. His lungs improved by 20 per cent, his liver tumours have regressed, and the nodes have shrunk as well. He feels better all around; he can eat, he has energy, and aside from the financial impact, which is quite harsh and for which he has had to make financial sacrifices, the experience has been good for him.
His family has been very supportive – he and his wife took a year off to deal with his cancer. His wife is there every step of the way, and his sons and siblings have also been supportive. The fear is there, but she does not show it to him. He believes that together, they can beat this. But even though he can afford the therapy, others cannot. That is why he wants to help others, because it is sad when others succumb to this disease due to unaffordability.
He adds: “I have paid to date approximately $80,000 USD and that’s crazy. I should not have had to do that. Patients should not have to take out their credit cards and amass an exorbitant amount of money to stay alive. Please fund the medicine.”
Respondent A’s Caregiver
Respondent A’s caregiver was her husband. She reported that he was certainly impacted by her illness. To access Durvalumab, he drove Respondent A to Toronto for every treatment. He sat with her when she was accessing the eight-hour infusions; he researched her cancer; he took care of her when she had side effects. She said, “The uncertainty associated with a diagnosis such as mine can be so daunting; it eats away at the caregiver every day. It is terrible. He has been my mountain, not my rock. This journey of mine has affected every facet of his life, and our children’s lives, and in turn, it has affected everyone they know. This is the ripple effect of the cancer journey.”
Respondent E’s Caregiver
Respondent E’s caregiver was his wife. He said that it was the hardest on her, as she took on most of the burden. She did all the research, networking, reading the CTs, seeing what was out there. She helped him through the struggles of chemo, finding ways to alleviate the side effects. His illness was a bit of a shock.
His children were good, positive, and hopeful, and have a good outlook. They were confident as long as he and his wife were confident. His middle son was the most sensitive, so they had to deal with that.
Respondent B: Caregiver on Behalf of her Husband
The caregiver was caring for her 40-year-old husband diagnosed with Stage 4 cholangiocarcinoma in late January 2022. The couple had one teenage daughter, and they lived in Afton, Tennessee, United States. The caregiver reported that her husband had many symptoms before diagnosis, including back pain, stomach pain, ascites, and then COVID-19. The pain was hardest to control. Once diagnosed, he was started on Gem/Cis, which was very helpful until the ascites and back pain came back. That was when he first accessed Durvalumab, on April 18, 2022. She believes that Durva started at the perfect time because his cancer was progressing. But since starting Durva, she reported that he has been doing wonderfully, that the transformation in him has been incredible.
She reported that side effects while he was on Gen/Cis were quite typical – fatigue, neuropathy in his left foot, foot drop, and constipation. But he had good quality of life before the symptoms came back. Their insurance covered most of the cost of Durva, they paid the $2,500 difference. He has had a little bit of fatigue, but that was the only side effect he had from the Durva.
She stated, “The Durva helped a lot to resolve those symptoms. About one week after the second cycle, they disappeared completely. The ascites went away within two weeks of his first treatment, the back pain went away after the second treatment, and abdominal pain, too. Everything is gone!”
As for being his caregiver, there are definitely highs and lows associated with this journey. His cancer has really impacted their 14-year-old daughter, because she is so young and trying to process adult issues. His wife had to quit her job so she could be available to him. That was difficult; she used to be a claims adjuster, but it was the choice she made to leave her job and be near him to help him. She constantly looks for more trials, supports groups, medicines. She goes to all his appointments. She does the laundry, cooking, yard work, maintains the house. She does whatever he needs. She said that she has given up a lot for him and their daughter. That is what the cancer journey requires of the caregiver.
She believes that Durva saved him, took him from the grave. She stated that it was remarkable what Durva has done for him.
Respondent C: Caregiver on Behalf of her Father
The caregiver is the 32-year-old daughter of a 60-year-old man from Newmarket, Ontario, diagnosed after suffering from abdominal pain, lack of sleep, and difficulty eating. In early April 2022, he had blood tests, an ultrasound, and a CT scan, which identified a mass on his liver and peritoneal mets, regional liver lymph nodes, and a blood clot near his liver. His diagnosis was metastatic intrahepatic biliary tract cancer. Two weeks later he was started on Gem/Cis, and his oncologist said regulatory approval would take too long, so they could add Durva later. Subsequently, Durva was accessed in late June 2022 through the compassionate access program. At the time of the interview, he had received one cycle, and there were no side effects.
As a caregiver, she reported that this experience was life-changing, that it had turned their lives upside down. Her father’s journey and care have all been her responsibility, including research and advocacy. She takes him to the hospital because there is a language barrier – he doesn’t speak English, unfortunately. She provides emotional support, and she is the nutritional therapist. She is very close to him. She changed her life for him, and her husband supports her, but it has been challenging because she works full time as a financial controller at a tech start-up. Even worse, her dog was diagnosed with the same kind of cancer as her dad.
Accessing Durva gave them hope. It changed their outlook – her father felt hopeful again. She stated that “It is a blessing just receiving it, and I am really very, very grateful for my father to be receiving it.” She added, “I think that this cancer is so rare and deadly. The stats are dreadful and hopeless. Having Durva will increase the chances of survival and moving forward, when there is no hope, the mind stops fighting and your body follows. When there is hope, your body responds to that based on the research I have been doing. It is for the whole family too. Also, it is unfortunate to have a drug that works but is inaccessible to patients who need it. Let’s make it accessible!”
Respondent D: Caregiver on Behalf of his Wife
The caregiver is the husband of a 47-year-old woman from Morgan, Vermont, in the United States. The couple have five young adult children. She was diagnosed in November 2021 with metastatic intrahepatic cholangiocarcinoma after suffering from random episodes of vomiting and back pain for 18 months followed by terrible abdominal pain. A CT scan and PET scan revealed that she had an 8.8 cm tumour in her liver. She then had blood tests, a biopsy, enhanced imaging, a colonoscopy, and a gastroscopy, which showed that it had spread.
Treatment on Cis/Gen started on January 4, 2022; Durvalumab was added on February 15, 2022. After three rounds, she was taken off Gem/Cis, and left on Durva. She received eight cycles of chemo and six cycles of Durva. After the fifth cycle of chemo, she experienced neuropathy in her left arm, both hands, twitches in her feet, and weakness plus fatigue. The only side effect she had from Durva was a bit of fatigue.
She had a scan in April, which showed that her liver lesion went from 8.8 cm to 6 cm, and another scan showed stability. She is feeling better, which is a good indicator for her.
The patient herself provided the following: Her husband was very stressed, but he wanted to do it all, he took everything on himself. He became somewhat withdrawn, very protective, he did all the cleaning and cooking, and dealt with the side effects of her drugs, cleaned up every time she vomited. He became the online researcher regarding medicines and went to all her appointments. This was the crisis of a lifetime, and she has been in crisis. It consumed her. She found it very difficult to focus on everything else in her life. She drifts away from what she has to do so that she can heal her way.
Her caregiver replied that he knows that he neglects himself, but that is the price you pay for your loved one when they are fighting for their life.
He said that Durva has allowed them to celebrate time together. They are hoping that it will allow them to continue to celebrate more time together. They have had previous time together that money cannot buy. Gem/Cis by itself is horrible. Durva has already given them time that is so previous and valuable. Without it, this would have turned out much differently; they really believe that.
He continued that it’s important to make this drug available to patients who need it, not only for survival extension, but also because it provides hope. It keeps their spirits alive and motivated…” Patients want to live so desperately and want to be surrounded by like-minded people and experts who have a positive attitude and who can prescribe therapies that will help patients move forward in their journey.”
Conflict of Interest Declaration — Canadian Cancer Survivor Network
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for the Canadian Cancer Survivor Network
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca - 2021 | — | — | — | X |
AstraZeneca - 2022 | — | — | — | X |
Clinician Input
Canadian Gastrointestinal Oncology Evidence Network
About the Canadian Gastrointestinal Oncology Evidence Network
The Canadian GI Oncology Evidence Network (CGOEN) is a virtual and inclusive network of Canadian GI Oncology clinicians who contribute to the knowledge of GI cancer and its treatments, including participating in clinical trials, conducting observational research, and involvement in local/provincial and national clinical guideline development and health technology assessment.
Information Gathering
Information gathered for this submission was based on relevant data from the TOPAZ-1 trial and expert evidence-based review by Canadian gastrointestinal cancer specialists.
Current Treatments
Describe the current treatment paradigm for the disease
Biliary tract cancers (BTC) are a group of rare and aggressive cancers that occur in the bile ducts and gallbladder. BTC is relatively rare, and diagnosis usually occurs when the disease has already advanced -- when curative surgery not being feasible.
There have been a limited number of trials investigating new treatments for BTC, and for over a decade the standard of care for inoperable biliary tract cancer has been combined chemotherapy: Cisplatin plus gemcitabine (CisGem). CisGem remains the only currently available treatment option for patients with unresectable BTC.
The prognosis for patients with BTC is typically grim with approximately 50% of untreated patients dying within 90 days of presentation and with treated patients surviving less than a year.
The moderate survival benefit provided by CisGem demonstrates a significant need for more effective treatments in this setting.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Extending patients’ lives, delaying disease progression, and maintaining quality of life while on therapy are goals of current research into new treatments for the management of unresectable BTC.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
CisGem offers only modest survival benefit. There have not been any major advances in the first line treatment of BTC that has resulted in a survival benefit since CisGem.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Patients with advanced BTC have extremely limited treatment options with poor outcomes.
TOPAZ-1 enrolled a large, international patient population that was representative of the general population of patients with advanced BTC.
Thus, it is reasonable to conclude that all patients with unresectable BTC have the greatest unmet need for an intervention such as the drug under review.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Currently treatments for patients with BTC offer inadequate efficacy. The addition of durvalumab to chemotherapy would offer these patients improved efficacy. Importantly the TOPAZ-1 study also demonstrates that the addition of durvalumab to chemotherapy will not add additional toxicity.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Recognizing that durvalumab would be added to existing chemotherapy, there is no rationale to recommend that patients try other treatments.
How would this drug affect the sequencing of therapies for the target condition?
As there are no other available treatments for advanced BTC, the addition of durvalumab to CisGem would not affect sequencing of therapies.
Which patients would be best suited for treatment with the drug under review?
All patients with unresectable BTC.
How would patients best suited for treatment with the drug under review be identified?
BTC patients with unresectable disease would be referred to medical oncology for assessment for this therapy. Given that it is an unselected patient population, as long as the patient does not have a contraindication to treatment – most patients should be eligible for treatment.
Which patients would be least suitable for treatment with the drug under review?
Contraindication to immunotherapy, inadequate liver or renal function, poor ECOG performance 3 or greater.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
No – there was no biomarker that selected for patients.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
In clinical practice, the patient’s clinical condition and CT imaging are used to determine whether a patient is responding to treatment. If a patient is symptomatic from their cancer and a treatment results in improvement in the symptom then this may be an indication of response. The most objective measurement is CT imaging to compare the sizes of the primary cancer and metastases. CT imaging response is frequently used to assess outcomes in clinical trials.
What would be considered a clinically meaningful response to treatment?
A clinically meaningful response to treatment would be maintenance or improvement in quality of life and prolongation of survival.
How often should treatment response be assessed?
Treatment response and tolerance of the treatment should be assessed clinically every 3 weeks and response should be assessed radiographically with CT imaging every 2-3 months.
What factors should be considered when deciding to discontinue treatment?
Patients would discontinue treatment if there were clear evidence of cancer progression on imaging, poor tolerance of the treatment which cannot be improved with dose delays or reductions, or patient preference to stop treatment.
What settings are appropriate for treatment with the drug under review?
Any setting where standard chemotherapy is delivered.
Additional Information
While the survival benefit may seem modest, the treatment options for BTC in Canada are very limited. Even second line 5FU/Oxaliplatin provides a modest survival benefit and the majority of patients with BTC do not reach second line therapy as the disease rapidly progresses. In addition, second line 5FU/Oxaliplatin is poorly tolerated. All of the molecularly targeted drugs for BTC are not funded in Canada. Therefore, in order to maximize survival for these patients it is best to provide the optimal first line therapy.
Conflict of Interest Declarations — The Canadian Gastrointestinal Oncology Evidence Network
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input.
Declaration for Clinician 1
Name: Jennifer Knox
Position: Medical Oncologist, Princess Margaret Cancer Centre
Date: 04-08-2022
Table 2: COI Declaration for the Canadian Gastrointestinal Oncology Evidence Network — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche (research support) | — | — | — | X |
Ipsen (research support) | — | — | — | X |
Merck (research support) | — | — | — | X |
AZ (research support) | — | — | — | X |
Eisai (ad board/consulting) | X | — | — | — |
Roche (ad board/consulting) | X | — | — | — |
Ipsen (ad board/consulting) | X | — | — | — |
Merck (ad board/consulting) | X | — | — | — |
AZ (ad board/consulting) | X | — | — | — |
Declaration for Clinician 2
Name: Vincent Tam
Position: Medical Oncologist, Tom Baker Cancer Centre
Date: 29-07-2022
Table 3: COI Declaration for the Canadian Gastrointestinal Oncology Evidence Network — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca | — | — | X | — |
Incyte Biosciences | — | X | — | — |
Ipsen | — | X | — | — |
Eisai | — | X | — | — |
Roche | — | — | X | — |
Declaration for Clinician 3
Name: Ravi Ramjeesingh
Position: Medical Oncologist, Department of Medicine, Dalhousie University
Date: 28-07-2022
Table 4: COI Declaration for the Canadian Gastrointestinal Oncology Evidence Network — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Amgen | X | — | — | — |
Astra-Zeneca | X | — | — | — |
Eisai | X | — | — | — |
Incyte | X | — | — | — |
Ipsen | X | — | — | — |
Declaration for Clinician 4
Name: Petr Kavan
Position: Medical Oncologist, McGill University Health Centre
Date: 03-08-2022
Table 5: COI Declaration for the Canadian Gastrointestinal Oncology Evidence Network — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Incyte | X | — | — | — |
Declaration for Clinician 5
Name: Dr. Sharlene Gill
Position: Medical Oncologist, Professor of Medicine – BC Cancer, Vancouver
Date: 03-08-2022
Table 6: COI Declaration for the Canadian Gastrointestinal Oncology Evidence Network — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Howard Lim
Position: Medical Oncologist
Date: 08-08-2022
Table 7: COI Declaration for the Canadian Gastrointestinal Oncology Evidence Network — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Bayer | X | — | — | — |
Amgen | X | — | — | — |
BMS | — | X | — | — |
Lily | X | — | — | — |
Taiho | X | — | — | — |
Eisai | — | — | X | — |
Ipsen | X | — | — | — |
Declaration for Clinician 7
Name: Jennifer Spratlin
Position: MD FRCPC
Date: 08-08-2022
Table 8: COI Declaration for the Canadian Gastrointestinal Oncology Evidence Network — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Ontario Health Cancer Care Ontario Gastrointestinal Cancer Drug Advisory Committee
About the Ontario Health Cancer Care Ontario Gastrointestinal Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Information was jointly discussed via email and DAC meeting.
Current Treatments and Treatment Goals
The standard of care is CISGEM and CARBOGEM. The treatment goals would be prolonged life, delayed disease progression, and improved quality of life.
Treatment Gaps (Unmet Needs)
There is only one available regimen and the duration of response is poor. Therefore, new regimens are required.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Duravlaumab can be safely added to first line chemo and is well tolerated.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients best suited for the drug under review would be all patients who align with the clinical trial criteria.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Clinical and/or radiologic progression as per the discretion of the treating oncologist.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Treatment should be discontinued if there is disease progression and toxicity at the discretion of the treating oncologist.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
The setting would be hospital (outpatient clinic) and a specialist is required.
Additional Information
DAC is in agreement with weight-based dosing with a cap, since it is consistent with other disease site regimens. With IO, there is no dose-response relationship and with flat dose, we will be overtreating many.
The DAC suggests a duration of treatment until disease progression.
Conflict of Interest Declarations — Ontario Health Cancer Care Ontario Gastrointestinal Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes. Ontario Health provided secretariat function to the DAC.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Erin Kennedy
Position: Ontario Health (CCO) Gastrointestinal Cancer Drug Advisory Committee Lead
Date: 28-07-2022
Table 9: COI Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Suneil Khanna
Position: Ontario Health (CCO) Gastrointestinal Cancer Drug Advisory Committee Member
Date: 28-07-2022
Table 10: COI Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Michael Raphael
Position: Ontario Health (CCO) Gastrointestinal Cancer Drug Advisory Committee Member
Date: 28-07-2022
Table 11: COI Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Jim Biagi
Position: Ontario Health (CCO) Gastrointestinal Cancer Drug Advisory Committee Member
Date: 28-07-2022
Table 12: COI Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr. Rachel Goodwin
Position: Ontario Health (CCO) Gastrointestinal Cancer Drug Advisory Committee Member
Date: 28-07-2022
Table 13: COI Declaration for OH-CCO Gastrointestinal Cancer Drug Advisory Committee — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.