CADTH Reimbursement Review
Tebentafusp (Kimmtrak)
Sponsor: Medison Pharma Canada Inc.
Therapeutic area: Unresectable or metastatic uveal melanoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ATT
average treatment effect of the treated
BOR
best overall response
CI
confidence interval
CNS
central nervous system
CR
complete response
CRS
cytokine release syndrome
DCR
disease control rate
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EQ VAS
EQ visual analogue scale
EQ-5D-5L
5-Level EQ-5D
ESS
effective sample size
gp
glycoprotein
HLA
human leukocyte antigen
HR
hazard ratio
HRQoL
health-related quality of life
ICR
independent central review
IPD
individual patient data
IPTW
inverse probability of treatment weighting
ITC
indirect treatment comparison
ITT
intention-to-treat
KM
Kaplan-Meier
LDH
lactate dehydrogenase
LS
least squares
MAIC
matching-adjusted indirect comparison
MC
Melanoma Canada
MID
minimally important difference
mUM
metastatic uveal melanoma
NR
not reported
OR
odds ratio
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
PR
partial response
QoL
quality of life
RAS
rash analysis set
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
SD
standard deviation
SLR
systematic literature review
SYSF
Save Your Skin Foundation
TEAE
treatment-emergent adverse event
UAIC
unadjusted indirect comparison
ULN
upper limit of the normal range
UM
uveal melanoma
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Tebentafusp (Kimmtrak), 100 mcg/0.5 mL, solution, IV infusion |
Indication | For the treatment of human leukocyte antigen (HLA)-A*02:01-positive adults with unresectable or metastatic uveal melanoma |
Reimbursement request | Per indication |
Health Canada approval status | Notice of Compliance |
Health Canada review pathway | Priority review, Project Orbis |
NOC date | June 7, 2022 |
Sponsor | Medison Pharma Canada Inc. |
NOC = Notice of Compliance.
Introduction
Uveal melanoma (UM) is a rare subset of melanoma that arises from the uveal tract (which consists of the choroid, ciliary body, and iris) in the eye.1 It is estimated that there were 3.75 new cases of UM per million in Canada each year from 1992 to 2010, with an increase of 0.074 new cases per million individuals annually.2 At diagnosis, most patients have localized disease and approximately 50% of patients are symptomatic (vision loss or disturbances).1 About 50% of patients will progress to metastatic disease,3 with metastasis most commonly in the liver (93%).1 Survival of patients with metastatic uveal melanoma (mUM) is not favourable; the estimated 1-year survival rate is 43% to 52% in the first-line setting, and about 37% in the second-line setting or later.4,5 The leading cause of death is parenchymal liver failure.6
Patient with a limited number of metastatic lesions in the liver alone (oligometastatic disease) may benefit from local therapies. Treatment modalities include radiofrequency ablation, stereotactic radiation, and surgical resection. In contrast, patients with mUM who have a larger number of metastatic lesions and/or disease external to the liver are usually prescribed systemic therapies, according to the clinical expert consulted by CADTH. The most commonly prescribed systemic therapies in the first-line setting are immunotherapies (off-label), administered as monotherapies (i.e., nivolumab or pembrolizumab alone) or in combination (i.e., ipilimumab plus nivolumab). Ipilimumab monotherapy is generally given in later-line settings, according to the clinical expert. Systemic chemotherapies have a limited role in the treatment of mUM in Canada because of toxicity and a low response rate, the clinical expert explained. Given that there are poor survival benefits and a poor tumour response to all available systemic treatments, the standard of care for mUM is enrolment in a clinical trial, if available.7,8 Of note, funding for systemic therapies for mUM is usually assessed on a case-by-case basis by the jurisdictions.
Tebentafusp is a glycoprotein (gp)100 peptide-human leukocyte antigen (HLA)-A*02:01 directed T-cell receptor CD3 bispecific T-cell engager, which has been shown, in vitro, to activate polyclonal T-cells to release inflammatory cytokines and cytolytic proteins, resulting in the direct lysis of UM tumour cells. It is the first treatment approved by Health Canada specifically for the treatment of HLA-A*02:01-positive adults with unresectable UM or mUM. The recommended dose of tebentafusp, administered by IV infusion, is 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15, and 68 mcg once every week thereafter. Per the Health Canada product monograph, it is recommended that patients be treated until unacceptable toxicity or disease progression occur.9 The first 3 infusions should be administered in an appropriate health care setting, where the patient can be monitored during the infusion and for at least 16 hours after to mitigate the risk of cytokine release syndrome (CRS).9 Only patients with a positive HLA-A*02:01 genotype status are eligible for treatment with tebentafusp.9
The objective of this report was to perform a systematic review of the beneficial and harmful effects of tebentafusp (100 mcg/0.5 mL) solution administered by IV infusion for the treatment of HLA-A*02:01-positive adults with unresectable UM or mUM.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from a clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups provided input for this reimbursement review: Melanoma Canada (MC) and Save Your Skin Foundation (SYSF). MC gathered information from patients with UM and their caregivers (n = 19) using an online survey. SYSF collected responses from patients with ocular melanoma (n = 38) using patient interviews, patient roundtables, and online surveys. Most of the SYSF respondents (n = 33) resided in Canada. The majority of respondents to the MC and SYSF requests for input were diagnosed with early-stage or primary disease.
MC respondents mentioned that the diagnosis of UM affects their day-to-day life and quality of life (QoL), and reported that the most common issues are loss of vision, vision impairment, fear or anxiety, depression, and fatigue. SYSF respondents mentioned that their balance is affected, which causes huge physical and psychological deterioration.
Five MC respondents and 10 SYSF respondents indicated that they had experience with tebentafusp through clinical trials or a Health Canada program for compassionate access. According to the submissions by MC and SYSF, 3 patients (2 MC respondents and 1 SYSF respondents) who had access to tebentafusp indicated that the drug was effective in slowing down disease progression, and another 2 patients could not comment on the effectiveness because it was too early for them to tell. The side effects frequently reported by patients from both groups were skin rash, fever, fatigue, cognitive impairment, gastrointestinal issues, nausea, muscle, joint pain, and headaches. Most patients described the side effects as short-term, tolerable, and manageable, and reported that, in their experience, the benefits of the treatments outweighed the negative side effects. Only 1 patient from SYSF said the side effects were not manageable. Patients from both groups reported that they had to travel long distances to access the drug and had financial difficulties.
Patients from both groups expressed their desire for an effective treatment that can preserve vision and provide a good QoL and longer survival. In addition, patients interview by MC indicated that they would like to know of improvements in earlier diagnosis and the detection of metastasis.
Clinician Input
Input From Clinical Expert Consulted by CADTH
The clinical expert highlighted that mUM is an aggressive disease with poor survival and no currently available therapies that predictably improve outcomes. The clinical expert expressed that a shift in the current treatment paradigm is anticipated, in which tebentafusp is used in the first-line setting or later, and is supported by clinical trial and anecdotal evidence.
The clinical expert indicated that only HLA-A*02:01-positive patients with mUM are expected to benefit from tebentafusp, owing to its unique mechanism of action. However, the clinical expert was unable to identify which HLA-A*02:01-positive patients are most likely to benefit from tebentafusp, noting that there is currently no good clinical or biologic predictor of response to tebentafusp. The clinical expert indicated that patients with a poor Eastern Cooperative Oncology Group Performance Status [ECOG PS], (3 or above) are generally not eligible for treatment in clinical practice as they are unlikely to benefit.
The clinical expert strongly suggested that patients be allowed to continue treatment as long as they continue to derive clinical benefits from tebentafusp, noting that there is some evidence from a posthoc analysis of Study 202 showing that patients with radiographic progression on tebentafusp can continue to benefit from treatment beyond progression.10 The clinical expert highlighted that, given the complexity of the clinical considerations involved, treatment response and the decision to discontinue treatment should be left to the discretion of the attending oncologist, based on assessments of history and physical examinations (every 3 to 4 weeks), laboratory tests (weekly), and imaging (every 12 to 16 weeks) findings. According to the clinical expert, treatment discontinuation is generally considered in clinical practice when a patient develops intolerable toxicities or clear evidence of significant progression, which is indicated by a decline in ECOG PS, an increase in pain, rising lactate dehydrogenase (LDH) levels, and marked radiographic progression.
The clinical expert recommended that tebentafusp initially be prescribed only by specialist physicians experienced in the use of tebentafusp and familiar with the management of CRS, noting that the risk of CRS is highest with the first 3 or 4 doses and substantially lower with subsequent doses. The clinical expert noted that once a pattern of use is established and the risk of CRS is absent, subsequent infusions can be performed in a community clinic setting.
Clinician Group Input
CADTH received input from 1 clinician group — the Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee — based on responses from 6 clinicians. The Skin Cancer Drug Advisory Committee provides timely evidence-based clinical and health-system guidance on drug-related issues in support of Cancer Care Ontario’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. No major contrary views from those provided by the clinical expert consulted by CADTH for this review were presented.
The clinician group stated that UM, which is distinct from cutaneous melanoma, is a disease that has a low tumour mutational burden and a low 1-year survival rate (50%) in the metastatic setting. None of the current systemic treatments for UM have been proven to have an overall survival (OS) benefit, which is a major unmet need in patients with mUM. The clinician group noted that tebentafusp has demonstrated an improvement in OS in clinical trials. The clinician group noted that the treatment goal is to improve OS and QoL. The clinician group explained that tebentafusp would be the first-line treatment of choice for HLA-A*02:01-positive patients with mUM, although they did not comment on the use of tebentafusp in second-line settings or later. The clinician group stated that HLA-A*02:01-positive patients with mUM who do not meet the exclusion criteria of Study 202 would be suitable for tebentafusp. They noted that ongoing ECOG PS, tumour size, and Response Evaluation Criteria in Solid Tumours (RECIST) progression requirements are the clinical outcomes used to determine whether a patient is responding. They indicated that treatment response is considered clinically meaningful when tumour size is reduced by at least 20% and performance status improves, but added that treatment may be continued in some patients with radiographic progression (new and/or increasing target lesions) if clinical benefits are observed. The clinician group noted that toxicity or symptomatic disease progression without clinical benefit would be considered when the decision to discontinue tebentafusp is made. The clinician group indicated that oncologists with experience in the inpatient management of side effects related to tebentafusp are required because of the toxicities that commonly occur after the first 3 doses.
Drug Program Input
The drug programs expressed an interest in understanding treatment eligibility for patients who received prior systemic treatments in the advanced or metastatic setting, who have central nervous systemic (CNS) metastases, or who have an ECOG PS of 2 or greater, as well as identifying appropriate discontinuation criteria.
The clinical expert consulted by CADTH indicated that the use of tebentafusp in patients who have received prior therapies in the advanced or metastatic setting is appropriate, and noted that there is a high unmet need for effective systemic treatments in the first-line setting and beyond and that the use of tebentafusp in these settings aligns with clinical trial and anecdotal evidence. The clinical expert indicated that patients with CNS metastases should be eligible for treatment if CNS metastases have been controlled with radiation or surgery. The clinical expert noted that in clinical practice, patients with an ECOG PS of 3 or worse are not eligible for systemic treatment because they are unlikely to benefit from treatment.
In terms of discontinuation criteria, clinical trial evidence has shown that patients may continue to derive clinical benefits from tebentafusp despite initial radiographic progression, the clinical expert explained. In general, treatment discontinuation may be warranted in patients with clear evidence of significant progression, suggested by a decline in ECOG PS, an increase in pain, rising LDH levels, and marked progression on imaging, the clinical expert noted. However, given that the clinical considerations involved are complex, the decision to discontinue treatment should be left to the discretion of attending oncologist, the clinical expert explained.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Study
Study 202 met the inclusion criteria for the CADTH Systematic Review.10 Study 202 was a phase III, open-label, randomized, active-controlled study that aimed to compare the efficacy and safety of tebentafusp with investigator’s choice of treatment (pembrolizumab, ipilimumab, or dacarbazine) in HLA-A*02:01-positive adults with mUM who had not received prior therapy in the metastatic setting (N = 378). Patients were randomized in a 2:1 ratio to receive IV infusions of tebentafusp (20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15, and weekly thereafter) or IV infusions of dacarbazine (1,000 mg/m2), ipilimumab (3 mg/kg), or pembrolizumab (2 mg/kg per dose [up to 200 mg] or a 200 mg flat dose) every 3 weeks. Treatments were continued until disease progression per RECIST 1.1, unacceptable toxicity, or completion of a maximum of 4 doses of ipilimumab. Patients receiving tebentafusp, pembrolizumab, or ipilimumab were permitted to continue treatment after initial disease progression if prespecified criteria were met that indicated clinical benefit and tolerance of the study drugs.
Study 202 aimed to establish the superiority of tebentafusp to investigator’s choice of therapy through the coprimary end points of OS in the rash analysis set (RAS), which consisted of patients receiving tebentafusp who developed a rash in the first week after treatment and all patients in the investigator’s-choice arm, and OS in the intention-to-treat (ITT) analysis set. The key secondary end points were progression-free survival (PFS) and best overall response (BOR) (evaluated statistically as objective response rate [ORR]). Other secondary outcomes included health-related quality of life (HRQoL) (5-Level EQ-5D [EQ-5D-5L] and European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 [EORTC QLQ-C30]), disease control rate (DCR), duration of response (DOR), and safety, all of which were measured without control for multiplicity. The first interim OS analysis and the final PFS analysis were performed with a data cut-off date of October 13, 2020. An informal updated OS analysis and the primary ORR analysis were performed with a data cut-off date of August 12, 2021, to fulfill regulatory requirements in Europe.
The baseline patient characteristics were balanced between treatment arms. Overall, the mean age of patients was 62.1 (standard deviation [SD] = 11.6) years. Approximately half of the patients were female. The majority of patients were white, had an ECOG PS of 0, and had liver metastases. Most patients had not undergone surgery for metastatic disease (91.3%). A small proportion of patients (3.7%) had received prior antineoplastic systemic treatments (in any setting), and 40.2% of patients had received prior local radiotherapy. In the investigator’s-choice arm, the majority of patients were assigned pembrolizumab (81.7%), whereas 12.7% received ipilimumab and 5.6% received dacarbazine.
Efficacy Results
The key efficacy results from Study 202 are summarized in Table 2.
Overall Survival
In the first interim OS analysis (median duration of follow-up of 14.1 months), the median OS in the RAS was 27.4 (95% confidence interval [CI], 20.2 to not reported [NR]) months in the tebentafusp arm and 16.0 (95% CI, 9.7 to 18.4) months in the investigator’s-choice arm, with a hazard ratio [HR] of 0.38 (95% CI, 0.25 to 0.56) and a P value of < 0.0001 in favour of tebentafusp. The median OS in the ITT analysis set was 21.7 (95% CI, 18.6 to 28.6) months in the tebentafusp arm and 16.0 (95% CI, 9.7 to 18.4) months in the investigator’s-choice arm, with an HR of 0.51 (95% CI, 0.37 to 0.71) and a P value of < 0.0001 in favour of tebentafusp. The results of the informal updated OS analysis were consistent with those of the first interim analysis.
Progression-Free Survival
In the final PFS analysis (median follow-up duration of 11.4 months), the median PFS in the ITT analysis set was 3.3 (95% CI, 3.0 to 5.0) months in the tebentafusp arm and 2.9 (95% CI, 2.8 to 3.0) months in the investigator’s-choice arm, with an HR of 0.73 (95% CI, 0.58 to 0.94) and a P value of 0.0139 in favour of tebentafusp.
Health-Related Quality of Life
As of the data cut-off date of October 13, 2020, the change from baseline in EORTC QLQ-C30 score was generally stable and similar between treatment arms at most time points for almost all domains. With respect to the fatigue scale (higher scores indicate worse symptoms), the difference in least square (LS) mean change in score from baseline at end of treatment between tebentafusp versus investigator’s choice was −9.259. This analysis was not adjusted for multiplicity.
The baseline mean EQ visual analogue scale (VAS) (higher scores indicate better HRQoL) was 81.0 (SD = 16.4) in the tebentafusp arm and 80.4 (SD = 18.3) in the investigator’s-choice arm. The mean change from baseline at end of treatment was –10.1 (SD = 22.53) in the tebentafusp arm and –11.7 (SD = 21.40) in the investigator’s-choice arm. The difference between arms was not tested statistically.
Duration of Response
As of the data cut-off date of August 12, 2021, 26 (10.3%) patients receiving tebentafusp and 6 (4.76%) patients receiving investigator’s choice of therapy had achieved a complete response (CR) or partial response (PR). Among these patients, the median DOR was 9.9 (95% CI, 5.6 to 22.1) months in the tebentafusp arm and 9.7 (95% CI, 2.7 to NR) months in the investigator’s-choice arm. The difference between arms was not tested statistically.
Objective Response Rate
In the primary ORR analysis (data cut-off date of August 12, 2021), the ORR in the ITT analysis set was 10.3% (95% CI, 6.9% to 14.8%) in the tebentafusp arm and 4.8% (95% CI, 1.8% to 10.1%) in the investigator’s-choice arm, corresponding to an odds ratio (OR) of 2.26 (95% CI, 0.91 to 5.61) and a P value of 0.684. Because of imprecision, the results for ORR are inconclusive.
Disease Control Rate
As of the data cut-off date of August 12, 2021, the DCR in the ITT analysis set was 45.6% (95% CI, 39.4% to 52.0%) in the tebentafusp arm and 27.0% (95% CI, 19.5% to 35.6%) in the investigator’s-choice arm, corresponding to an OR of 2.34 (95% CI, 1.45 to 3.76). This analysis was not adjusted for multiplicity.
Table 2: Summary of Key Efficacy Results From Study 202
Outcome | Data cut-off date: October 13, 2020 | Data cut-off date: August 12, 2021 | ||
|---|---|---|---|---|
Tebentafusp (N = 252)a | Investigator’s choice (N = 126) | Tebentafusp (N = 252)a | Investigator’s choice (N = 126) | |
Coprimary end point: OS (RAS) | ||||
Death, n (%) | 43 (28.9) | 63 (50.0) | NR | NR |
OS (months),b median (95% CI) | 27.4 (20.2 to NR) | 16.0 (9.7 to 18.4) | NR | NR |
HRc (95% CI) | 0.38 (0.25 to 0.56) | NR | ||
P valued | < 0.0001 | NR | ||
Coprimary end point: OS (ITT) | ||||
Death, n (%) | 87 (34.5) | 63 (50.0) | 127 (50.4) | 79 (62.7) |
OS (months),b median (95% CI) | 21.7 (18.6 to 28.6) | 16.0 (9.7 to 18.4) | 21.7 (19.1 to 26.0) | 16.7 (11.8 to 19.3) |
HRc (95% CI) | 0.51 (0.37 to 0.71) | 0.58 (0.44 to 0.77) | ||
P valued | < 0.0001 | 0.0001e | ||
Key secondary end point: PFS (ITT) | ||||
PFS event, n (%) | 198 (78.6) | 97 (77.0) | NR | NR |
PFS (months),b median (95% CI) | 3.3 (3.0 to 5.0) | 2.9 (2.8 to 3.0) | NR | NR |
HRc (95% CI) | 0.73 (0.58 to 0.94) | NR | ||
P valued | 0.0139 | NR | ||
HRQoL: EQ VAS (ITT) | ||||
Baseline EQ VAS score, mean (SD) | 81.0 (16.4) | 80.4 (18.3) | NR | NR |
Change from baseline in EQ VAS score, mean (SD) | ||||
Cycle 3 day 1 | 0.4 (14.69) | –0.8 (14.28) | NR | NR |
Cycle 9 day 11 | –0.9 (19.81) | –3.3 (13.30) | NR | NR |
End of treatment | –10.1 (22.53) | –11.7 (21.40) | NR | NR |
Duration of responsef (complete + partial) | ||||
PFS events, n (%) | 9 (39.1) | 4 (66.7) | 16 (61.5) | 4 (66.7) |
Duration of responseg (months), median (95% CI) | 9.9 (5.4 to NR) | 9.7 (2.7 to NR) | 9.9 (5.6 to 22.1) | 9.7 (2.7 to NR) |
Key secondary end point: objective response rate (ITT) | ||||
Objective response, n (%) | 23 (9.1) | 6 (4.8) | 26 (10.3) | 6 (4.8) |
Odds ratio (95% CI)h | 1.98 (0.79 to 4.97) | 2.26 (0.91 to 5.61) | ||
P value | NR | 0.0684 | ||
CI = confidence interval; EQ VAS = EQ visual analogue scale; HRQoL = health-related quality of life; ITT = intention to treat; NR = not reported; OS = overall survival; PFS = progression-free survival; RAS = rash analysis set; SD = standard deviation.
aN = 252 refers to the intention-to-treat analysis set sample size; N = 149 refers to the RAS sample size.
bBased on Kaplan-Meier estimate.
cHR was estimated using a Cox proportional hazards model stratified by LDH status.
dP value based on a log-rank test of the Kaplan-Meier curve stratified by LDH status.
eAccording to the sponsor, the OS analysis at the data cut-off date of August 12, 2021, was informal; as such, the P value should be considered exploratory.
fBased on the patient population that had achieved a CR or PR (at the data cut-off date of October 13, 2020, there were 23 patients in the tebentafusp arm and 6 in the investigator’s-choice arm; at the data cut-off date of August 12, 2021, there were 26 patients in the tebentafusp arm and 6 in the investigator’s-choice arm).
gDuration of response is defined as the time from the date of first documentation of partial response or better to the date of first documentation of progressive disease or death due to any cause, whichever comes first.
hOdds ratio was calculated using Cochran-Mantel-Haenszel test stratified by LDH status.
Source: Study 202 Clinical Study Report.11
Harms Results
The key harms outcomes from Study 202 are summarized in Table 3.
As of the data cut-off date of October 13, 2020, treatment-emergent adverse events (TEAE) were reported in all patients in the tebentafusp arm and in 94.6% of patients in the investigator’s-choice arm. The most common TEAEs (tebentafusp arm versus investigator’s-choice arm) were pyrexia (76.0% versus 7.2%), pruritus (69.0% versus 23.4%), rash (55.1% versus 16.2%), and fatigue (51.0% versus 35.1%). Serious TEAE were reported in 28.2% of patients in the tebentafusp arm and 23.1% of patients in the investigator’s-choice arm. The most common serious TEAE in the tebentafusp arm was CRS (9.8% versus 0.0% in the investigator’s-choice arm). The proportion of patients who discontinued treatment due to TEAEs was 3.3% in the tebentafusp arm and 6.6% in the investigator’s-choice arm. There were 84 deaths (34.3%) and 57 deaths (51.4%) reported in the tebentafusp arm and the investigator’s-choice arm, respectively. The majority of deaths in both arms was attributed to disease progression.
The proportion of patients who reported CRS (or presentations related to CRS) and dermatological was notably higher in the tebentafusp arm than in the investigator’s-choice arm. The most common notable harms of any grade (30% or greater) were pyrexia, pruritus, rash, fatigue, nausea, chills, hypotension, dry skin, headache, and maculopapular rash, all of which were mostly grade 1 or 2.
Table 3: Summary of Key Harms Results From Study 202 (SAS)
Harms | Study 202 (data cut-off date: October 13, 2020) | |||
|---|---|---|---|---|
Tebentafusp (N = 245) | Investigator’s choice (N = 111) | Tebentafusp (N = 245) | Investigator’s choice (N = 111) | |
Harms, n (%) | Any grade | Grade 3 or higher | ||
Patients with ≥ 1 TEAEa | 245 (100.0) | 105 (94.6) | 133 (54.3) | 40 (36.0) |
Patients with ≥ 1 serious TEAEsa | 69 (28.2) | 26 (23.4) | NR | NR |
Patients who discontinued treatment due to a TEAEa | 8 (3.3) | 7 (6.3) | NR | NR |
Deaths | 8 (3.3) | 7 (6.3) | NA | NA |
Notable harms, n (%) | Any grade | Grade 3 or higher | ||
Pyrexia | 187 (76.3) | 8 (7.2) | 9 (3.7) | 1 (0.9) |
Pruritus | 169 (69.0) | 26 (23.4) | 11 (4.5) | 0 |
Rash | 135 (55.1) | 18 (16.2) | 23 (9.4) | 0 |
Fatigue | 125 (51.0) | 39 (35.1) | 13 (5.3) | 0 |
Nausea | 120 (49.0) | 29 (26.1) | 5 (2.0) | 1 (0.9) |
Chills | 117 (47.8) | 4 (3.6) | 1 (0.4) | 0 |
Hypotension | 95 (38.8) | 3 (2.7) | 8 (3.3) | 0 |
Dry skin | 77 (31.4) | 4 (3.6) | 0 | 0 |
Headache | 75 (30.6) | 11 (9.9) | 1 (0.4) | 1 (0.9) |
Maculopapular rash | 75 (30.6) | 9 (8.1) | 21 (8.6) | 0 |
Vomiting | 73 (29.8) | 10 (9.0) | 3 (1.2) | 0 |
Diarrhea | 61 (24.9) | 22 (19.8) | 3 (1.2) | 3 (2.7) |
Erythema | 60 (24.5) | 1 (0.9) | 1 (0.4) | 0 |
Cytokine release syndromeb | 51 (20.8) | 0 | 2 (0.8) | 0 |
Skin exfoliation | 51 (20.8) | 2 (1.8) | 1 (0.4) | 0 |
Hair colour changes | 48 (19.6) | 0 | 1 (0.4) | 0 |
Vitiligo | 40 (16.3) | 4 (3.6) | 0 | 0 |
Hypoxia | 4 (1.6) | 0 | 2 (0.8) | 0 |
NA = not applicable; NR = not reported; SAS = safety analysis set; TEAE = treatment-emergent adverse event.
aTEAE was defined as an AE that had an onset date on or after the date of the first dose or pretreatment TEAEs that increase in severity on or after the date of the first dose up to an including 90 days after the date of the last dose.
bCRS was reported by the investigator based on Lee et al. (2014)12 grading criteria.
Source: Study 202 Clinical Study Report.11
Critical Appraisal
The overall design of Study 202 was appropriate for the objectives of the study. There was no particular concern with the methods of randomization or allocation concealment. The open-label design was considered reasonable by the clinical expert., There is potential for reporting bias on tumour response (ORR, DCR, BOR, DOR) and subjective harms outcomes because these outcomes were based on the investigator’s assessment, although the extent and direction of bias are unclear. The statistical analyses were generally appropriate, with proper processes in place to preserve power in the interim and final OS analyses and to account for multiplicity for the coprimary end points and key secondary end points using a hierarchal approach. DCR, DOR, and HRQoL outcomes, however, were not adjusted for multiplicity and were considered exploratory because of the increased risk of a type I error. It should be noted that the OS analyses were interim, and interim analyses are typically associated with a risk of overestimation of the treatment effects in favour of the experimental intervention (i.e., tebentafusp).13 Considering that the OS analyses were based on a relatively small number of events, the OS results are prone to imprecision. The OS analysis in the RAS should also be interpreted with caution because of the risk of confounding, resulting from the absence of randomization in the comparison, although the direction of bias could not be determined. There is also uncertainty in the HRQoL outcomes related to potential reporting and attrition bias and because the instruments used (EORTC QLQ-C30 and EQ-5D-5L) had not been validated in patients with mUM.
In terms of generalizability, a limitation to note is that the studies included patients who had received no prior therapies in the metastatic setting; therefore, the applicability of trial results to patients who had received prior therapies in the metastatic setting is unclear. The treatments included in the comparator arm account for a small proportion of systemic treatments prescribed for mUM in Canada, which increases uncertainty about the generalizability of study results. The clinical expert consulted by CADTH noted that the impact on generalizability is likely to be small because the efficacy of immunotherapies is considered similar by clinicians. However, this opinion was based on a small retrospective cohort study and is associated with some uncertainties. The OS benefits of tebentafusp were considered clinically meaningful by the clinical expert, who noted, however, that PFS and tumour response outcomes have limited clinical relevance because tumour response is poorly correlated with OS in patients with mUM receiving systemic therapy in general. The clinical relevance of HRQoL outcomes was also uncertain because the instruments used are not routinely administered in clinical practice, although they are able to capture some of the most common HRQoL concerns (e.g., anxiety, depression, fatigue) reported by patients. With respect to safety, specifically CRS, the clinical expert expected the study findings to be generalizable to clinical practice, provided that tebentafusp is administered in appropriate treatment settings, as specified in the product monograph, but noted that CRS generally occurs after the first 3 to 4 infusions and is manageable if proper supportive care is provided.
Indirect Comparisons
Description of Study
One indirect treatment comparison (ITC) was submitted by the sponsor and included in this review.14 No additional ITCs were identified in the literature. The sponsor performed an unanchored matching-adjusted indirect comparison (MAIC) to estimate OS and PFS with tebentafusp compared with ipilimumab plus nivolumab in patients with mUM who had received no prior therapy in the metastatic setting, based on data from the GEM-1402 comparator trial,15 which was identified in a systematic literature review (SLR), and the Study 202 index trial. Of note, the GEM-1402 trial enrolled patients with mUM regardless of HLA status and Study 202 enrolled HLA-A*02:01-positive patients with mUM. The sponsor submitted a supplementary analysis of the prognostic value of HLA-A*02:01 for OS to support the MAIC analyses.16
Efficacy Results
The ITC included 237 patients in the tebentafusp arm from Study 202, and the effective sample size (ESS) of the ipilimumab plus nivolumab arm from the GEM-1402 trial was 115.9. The MAIC analysis between tebentafusp and ipilimumab plus nivolumab showed results in favour of tebentafusp with respect to both OS (HR, 0.507; 95% CI, 0.324 to 0.793) and PFS (HR, 0.647; 95% CI, 0.445 to 0.941).
Of the 80 patients included in the supplementary HLA status analysis, 43 were HLA-A*02:01-positive and 37 were HLA-A*02:01-negative. Median OS was 45.9 months (range, NR) in HLA-A*02:01-positive patients and 45.2 months (range, NR) in HLA-A*02:01-negative patients, with an HR of 0.82 (95% CI, 0.36 to 1.88).
Harms Results
The ITC did not assess safety outcomes.
Critical Appraisal
A key limitation of the ITC was that the selection criteria for the SLR were not determined a priori, increasing the risk of selection bias for comparator trials included in the MAIC analysis. In addition, patient population heterogeneity, specifically with respect to HLA-A*02:01 status, could be a potential source of confounding, given that the comparator trial included patients regardless of HLA-A*02:01 status. The sponsor-submitted supplementary HLA analysis did not allow the CADTH review team to rule out the possible confounding effect of HLA status on OS, given that the study was based on a small observational cohort and results are subject to imprecision (a wide 95% CI for HR with respect to OS). Further, time since primary diagnosis, a covariate identified in the multivariate analysis, was excluded from the adjustment because it was not reported in the comparator trial, which may contribute to the uncertainty of the results. There is also concern about a loss of precision in the results, given the significant reduction in the ESS. Overall, the direction of bias of the limitations noted could not be determined, and the results of the MAIC should be interpreted with caution because of the potential biases. There is also an evidence gap, in that HRQoL and harms outcomes, as well as the efficacy of tebentafusp in treatment-experienced patients, were not addressed in this analysis.
Other Relevant Evidence
This section includes 2 additional studies from the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the Systematic Review. The first was a phase I/II, multicentre, open-label study (Study 102) that analyzed the efficacy and safety of tebentafusp in HLA-A*02:01-positive patients with mUM who had received 1 or 2 prior lines of therapy in the metastatic or advanced setting.17 The second study was observational and compared patients receiving tebentafusp from Study 202 with patients receiving ipilimumab plus nivolumab from the GEM-1402 trial in a first-line mUM setting after propensity score weighting.18 The analysis was based on the same studies that informed the sponsor-submitted MAIC and was submitted to CADTH after the sponsor had obtained individual patient data (IPD) from the GEM-1402 trial. The study aimed to address limitations of the MAIC analysis related to the use of aggregate data from the GEM-1402 trial.
Study 102
The Study 102 was a phase I/II study that analyzed the efficacy and safety of tebentafusp in HLA-A*02:01-positive patients with mUM who had received 1 or 2 prior lines of therapy in the advanced or metastatic setting.17 The primary end point for the phase I part was the incidence (number) of dose-limiting toxicities, whereas the primary end point for the phase II, single-arm, dose-expansion part was ORR. The secondary end points for the phase II part were OS, PFS, DOR, DCR, and BOR.
Efficacy Results
In the phase II expansion cohort (data cut-off date of March 20, 2020), the ORR of 4.7% (95% CI, 1.8% to 10.0%) was based on 6 of the 127 patients who received tebentafusp and achieved a PR; no patients achieved a CR. After a median follow-up of 19.6 (95% CI, 16.0 to 22.2) months, median OS was 16.8 (95% CI, 12.9 to 21.3) months. Median PFS was 2.8 (95% CI, 2.0 to 3.7) months, according to RECIST 1.1 assessment by independent central review (ICR). Median DOR (CR or PR) was 8.7 (95% CI, 5.6 to 24.5) months, according to assessment of RECIST 1.1 by ICR. DCR (CR, PR, or stable disease) was 22.8% (95% CI, 15.7% to 31.2%) at 24 weeks or beyond. The most frequently observed BOR was disease progression (47.2%), followed by stable disease (44.9%) and PR (4.7%).
Harms Results
As in Study 202, all patients in Study 102 experienced AEs of any grade. In addition, the phase II expansion part of Study 102 and Study 202 had comparable rates of TEAEs of grade 3 or higher (59.1% versus 54.3%). In the phase II expansion part of Study 102, 9 patients (7.1%) experienced TEAEs leading to tebentafusp discontinuation, a proportion comparable to that of Study 202 (6.6%). The most frequently reported any-grade TEAEs were pyrexia (81.1%), pruritus (68.5%), nausea (67.7%), chills (66.1%), and hypotension (41.7%). These TEAEs were also observed in the tebentafusp arm in Study 202. In the phase II expansion part of Study 102, 86 serious TEAEs were reported in 42 (33.1%) patients, a proportion comparable to that of Study 202 (28.2%). Serious TEAEs were CRS, reported in 4 (3.1%) patients, and sepsis, alanine transaminase increase, rash maculopapular, and hypotension, reported in 3 (2.4%) patients each. No deaths related to TEAEs or the study drug were observed.
Critical Appraisal
The noncomparative design of Study 102, with no statistical testing, is the key limitation. The lack of direct comparative data means there is uncertainty regarding the magnitude of effects obtained for the efficacy outcomes. Although the clinical expert consulted noted that the efficacy outcomes of tebentafusp in Study 102 were clinically meaningful, demonstrated the activity of the drug, and were compatible with the Study 202 phase III trial, CADTH review team noted that in the absence of a comparative arm, the findings from the efficacy and safety analysis are uncertain, as the single-arm design does not allow for conclusions to be drawn about the comparative efficacy of tebentafusp or for symptoms of underlying mUM disease to be differentiated from treatment-related AEs.
Propensity Score Analysis (IPTW Approach)
The sponsor-submitted observational study compared patients in the Study 202 tebentafusp arm with patients in the GEM-1402 trial who received ipilimumab plus nivolumab in a first-line metastatic setting.18 The study was not randomized, and propensity score weighting with the inverse probability of treatment weighting (IPTW) approach was used in an attempt to adjust for confounding.
Efficacy
The ESS of the GEM-1402 trial was 34.4 after weighting, compared to a sample size of 45.0 before weighting. The patient characteristics were generally balanced between the tebentafusp and ipilimumab plus nivolumab cohorts after weighting.
In the primary analysis, the median OS of the tebentafusp cohort was 21.7 months (SD = NR), and the weighted median OS of the ipilimumab plus nivolumab cohort was 12.6 months (SD = NR). The HR between tebentafusp and ipilimumab plus nivolumab with respect to OS was 0.430 (95% CI, 0.287 to 0.643), in favour of tebentafusp.
Harms Results
The study did not assess safety outcomes.
Critical Appraisal
The IPTW approach improved on the MAIC by leveraging IPD from the GEM-1402 trial; however, many of the limitations of the MAIC analysis also apply to the current analysis. Specifically, the lack of a priori selection criteria for the SLR is a potential source of selection bias for comparator studies. It is also unclear whether all known or unknown confounding factors have been adequately adjusted for. Heterogeneity in HLA status was noted in the comparator groups and considered in the supplementary analysis submitted by the sponsor that assessed the impact of HLA status on OS, the CADTH review was unable to confidently rule out confounding effects related to the difference in HLA status between the cohorts. Last, outcomes that are of interest to stakeholders, such as HRQoL, harms, and the efficacy of tebentafusp in patients who received prior systemic therapy in the metastatic setting, were not addressed in this analysis.
Conclusions
Tebentafusp demonstrated a statistically significant and clinically meaningful improvement in OS compared with investigator’s choice of therapy (pembrolizumab, ipilimumab, dacarbazine) in HLA-A*02:01-positive adults with mUM who had received no prior therapy in the metastatic setting, based on the phase III pivotal study. Tebentafusp led to a statistically significant improvement in PFS but not in ORR, although these outcomes are of limited clinical relevance in the treatment of mUM, according to the clinical expert consulted by CADTH. Analyses of other secondary end points showed results in favour of tebentafusp (DCR) or results that were comparable between treatment arms (DOR, HRQoL), but no conclusion can be drawn because of the lack of statistical testing or control for multiplicity. The comparator treatments account for a small proportion of systemic treatments prescribed for mUM in Canada, which increases uncertainty about the generalizability of the study results. Results of a phase II, single-arm study suggested clinical activity of tebentafusp in patients who had received prior therapy in the metastatic setting, but they are subject to limitations related to the noncomparative study design. No definitive conclusions can be drawn from the sponsor-submitted MAIC or propensity score analysis about the relative efficacy of tebentafusp compared with ipilimumab plus nivolumab because of significant limitations of the analyses. CRS and dermatological AEs were common with tebentafusp but were generally grade 1 or 2, transient, and manageable with supportive care.
Introduction
Disease Background
UM is a rare subset of melanoma that accounts for 2.9% of all melanoma cases in Canada.2 UM refers to melanomas that arise from the uveal tract (choroid, ciliary body, and iris) in the eye. It is the most common primary intraocular malignancy in adults, representing 95% of all ocular melanomas.1 It is estimated that 3.75 new cases of UM developed per million people each year in Canada from 1992 to 2010, with an increase of 0.074 new cases per million individuals annually.2
UM is most commonly diagnosed in Caucasians [as cited] (97.8%), and the mean age at diagnosis is approximately 58 years.19,20 Light eye and skin colour, skin and iris nevi (dark spots), UV light exposure, and melanocytosis (hyperpigmentation) of the eye or skin near the eye are known risk factors for UM.1
Visual symptoms, such as flashes, floaters, and vision loss, are present in approximately 50% of patients with UM; other patients are often asymptomatic and identified during routine eye examination.1 Most patients have localized disease at diagnosis, and about 50% of patients will eventually progress to metastatic disease,3 with metastases most commonly involving the liver (93%), lung (24%), bone (16%), and skin or subcutaneous tissues (11%).1 The survival of patients with mUM is unfavourable; it has an estimated 1-year survival rate of 43% to 52% in the first-line treatment setting, and about 37% in the second-line setting or beyond.4,5 Parenchymal liver failure is the leading cause of death in patients with mUM.6
The diagnosis of UM involves clinical evaluations of the history and physical status of the patient and an eye examination using imaging techniques such as biomicroscopy, dilated fundus exam, ultrasound, optical coherence tomography, and fluorescein angiography.7,21 Metastatic disease is diagnosed with biopsy (if clinically appropriate), and imaging is used for baseline staging and the evaluation of specific signs and symptoms.7
Standards of Therapy
The primary tumour in the eye may be treated with brachytherapy, external electron beam therapy, or enucleation (removal of the eye), according to the clinical expert. The clinical expert noted that the goal is cessation of growth and slow shrinkage of the tumour, which denotes a long-term local clinical response. Brachytherapy controls the local tumour in 90% of cases; in the remaining 10%, enucleation is used as salvage therapy, according to the clinical expert. OS is identical with brachytherapy and enucleation, so the former is generally preferred, the clinical expert noted, and enucleation is reserved for large or invasive tumours that cannot be treated with brachytherapy.
Treatments for mUM can be broadly divided into local therapies and systemic therapies.1
Local therapies, which include surgical resection, radiofrequency ablation, stereotactic radiation, and infusion of radioactive beads or chemotherapy into the liver through the hepatic arteries, are generally used in patients with limited or oligometastatic disease with small number of lesions in the liver, according to the clinical expert consulted by CADTH.
Systemic therapies are usually prescribed for patients with a larger number of metastatic lesions and/or disease external to the liver. Immunotherapies (off-label) are commonly prescribed systemic treatments for mUM, either as monotherapies (i.e., nivolumab, or pembrolizumab alone) or in combination (i.e., ipilimumab plus nivolumab) in the first-line setting. According to the clinical expert, ipilimumab monotherapy is generally used in later-line settings in Canada and systemic chemotherapies are rarely prescribed because of the poor response rate and poor toxicity tolerance. The clinical expert noted that UM tumours typically have a low ORR (approximately 10% to 20%) to immunotherapies because of their low tumour mutation burden; as such, the standard of care for mUM is enrolment in a clinical trial when available and clinically appropriate.7,8 Funding for systemic therapies for mUM is usually assessed on a case-by-case basis by the jurisdictions.
In patients with mUM and multiple metastases restricted to the liver, hepatic artery infusion may be considered. Therapeutic drugs include chemotherapy or radiolabelled microbeads.22
Drug
Tebentafusp is a bispecific T-cell engager comprising 2 domains: a T-cell-receptor-targeting domain that binds to gp100 peptides presented by HLA-A*02:01 on UM tumour cell surface; and an anticluster of differentiation 3 (CD3) effector domain that binds and activates T-cells.23 Tebentafusp forms an immune synapse between UM cells and T-cells, thereby redirecting and activating T-cells to release inflammatory cytokines and cytolytic proteins that cause lysis of UM tumour cells.23 HLA-A*02:01 expression is required in patients receiving tebentafusp, as the T-cell-receptor-targeting domain is specific to the HLA-A*02:01-gp100280 to 288 complex.23
This is the first CADTH review for tebentafusp. Tebentafusp was granted a Health Canada Notice of Compliance for the indication of treatment of HLA-A*02:01-positive adults with unresectable UM or mUM on June 7, 2022. A positive HLA-A*02:01 genotype status is required for patients to be eligible for treatment with tebentafusp. The recommended dose of tebentafusp, per the Health Canada product monograph, is 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15, and 68 mcg once every week thereafter.9 Patients should be treated until unacceptable toxicity or disease progression occur.9 The first 3 infusions of tebentafusp should be administered in an appropriate health care setting by IV infusion over 15 to 20 minutes.9 Patients should be monitored during the infusion and for at least 16 hours after the infusion is complete.9
The sponsor is seeking reimbursement of tebentafusp per the indication.
The key characteristics of tebentafusp and the comparator drugs are summarized in Table 4.
Table 4: Key Characteristics of Tebentafusp, Pembrolizumab, Nivolumab, and Ipilimumab
Characteristic | Tebentafusp | Pembrolizumaba | Nivolumaba | Ipilimumaba |
|---|---|---|---|---|
Mechanism of action | ImmTAC (bispecific fusion protein) Forms an immune synapse between T-cells and UM tumour cells to redirect and activate T-cells, leading to lysis of HLA-A*02:01-positive UM tumour cells | PD-1 immune checkpoint inhibitor (mAb) Enhances antitumour response by T-cells through blockade of PD-1 | PD-1 immune checkpoint inhibitor (mAb) Enhances antitumour response by T-cells through blockade of PD-1 | CTLA-4 immune checkpoint inhibitor (mAb) Enhances antitumour response by T-cells through blockade of CTLA-4 |
Indication for mUMb | For the treatment HLA-A*02:01-positive adults with unresectable UM or mUM | None (off-label) | None (off-label as monotherapyc or in combination with ipilimumab) | None (off-label as monotherapy or in combination with nivolumab) |
Route of administration | IV infusion | IV infusion | IV infusion | IV infusion |
Recommended dose | 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15, and 68 mcg once every week thereafter |
| Monotherapy:
In combination with ipilimumab:
| Monotherapy:
In combination with nivolumab:
|
Serious AEs or safety issues |
| Immune-mediated AEs | Immune-mediated AEs | Immune-mediated AEs |
Other |
| Continue treatment until disease progression or unacceptable toxicityd | Continue treatment as long as clinical benefit is observed or until treatment is no longer toleratedd | Continue treatment for a maximum of 4 doses or until toxicity, whichever occurs firstd |
AE = adverse event; CTLA-4 = cytotoxic T-lymphocyte-associated antigen 4; ImmTAC = immune mobilizing monoclonal T-cell receptors against cancer; mAb = monoclonal antibody; mUM = metastatic uveal melanoma; PD-1 = programmed cell death 1 protein; UM = uveal melanoma.
aOff-label treatment. The Health Canada indication is for the treatment of unresectable or metastatic melanoma. Patients with UM were excluded from the pivotal trials that led to approval of the indication.
bHealth Canada–approved indication.
cIpilimumab monotherapy is used in second- or later-line settings in the treatment of mUM in Canada.
dOff-label dosing. Dosing shown is approved for the treatment of melanoma (cutaneous).
Sources: Tebentafusp product monograph,9 Keytruda product monograph,24 Opdivo product monograph,25 Yervoy product monograph.26
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The original patient group submissions can be found on CADTH’s website.
Two patient groups provided input for this review: MC and SYSF. MC (formerly the Melanoma Network of Canada) is an organization based in Canada that advocates on behalf of patients with melanoma, coordinates educational and prevention strategies, and assists in funding for melanoma research. SYSF is a national patient-led not-for-profit group committed to the fight against nonmelanoma skin cancers, melanoma, and ocular melanoma through nationwide education, advocacy, and awareness initiatives.
MC gathered information from an online survey. A total of 19 patients with UM and 15 caregivers responded to the survey. There were 9 patient respondents from Ontario, 2 from Nova Scotia, 2 from British Columbia, 3 from Alberta, 1 from Prince Edward Island, 1 from Newfoundland, and 1 from outside Canada. The majority of respondents had early-stage disease or did not know their staging. SYSF obtained information and feedback from patient interviews, virtual patient roundtables, and online surveys. Input was gathered from 38 patients with ocular melanoma, the majority of whom were diagnosed with primary ocular melanoma (28 patients). There were 9 respondents from British Columbia, 9 from Ontario, 8 from Alberta, 3 from Saskatchewan, 2 from Newfoundland, 1 from Manitoba, 1 from Quebec, 2 from outside Canada, and 3 did not identify their province of residence.
Respondents from MC mentioned that the diagnosis of UM affected their day-to-day life and QoL. Loss of vision or vision impairment (77.78%) was reported to be the major issue affecting their daily life and QoL, followed by fear or anxiety (61.11%), depression (38.89%) and fatigue (27.78%). In addition, respondents from SYSF mentioned that their balance was affected, which caused serious physical and psychological deterioration. The respondents also indicated that they had very few symptoms leading up to their diagnosis and were concerned about the risk of metastasis.
Respondents from MC and SYSF indicated that they had experience with a variety of treatments, such as surgery, immunotherapies, radiation, and brachytherapy. According to the MC respondents, the frequently reported issues with current treatments were loss of vision or vision impairment (82.35%), fatigue (35.29%), disrupted sleep (29.41%), post-traumatic stress (23.53%), negative impact to family or social life (23.53%), pain (17.65%), and headaches (17.65%).
There were 5 patients from MC and 10 patients from SYSF who indicated that they had experience with tebentafusp through clinical trials or a Health Canada program for compassionate access. According to the MC and SYSF submissions, 3 patients (2 from MC and 1 from SYSF) who had experience with tebentafusp indicated that the drug had shown effectiveness in slowing disease progression, and another 2 patients could not comment on the effectiveness as it was too early for them to tell. Patients from both groups reported a variety of side effects: skin rash, fever, fatigue, cognitive impairment, stomach-related issues, nausea, muscle and joint pain, and headaches. Most tebentafusp-experienced patients described the side effects as short-term, tolerable, and manageable, and reported that the benefits of treatments outweighed the negative side effects. Only 1 patient from SYSF reported that the side effects were not manageable. In addition, patients from both groups reported that they had to travel long distances to get access to the drug and had financial difficulties.
Patients from both groups expressed their desire for an effective treatment that can preserve vision and provide a good QoL and longer survival. In addition, patients from MC indicated that they would like to see improvements in earlier diagnosis and detection of metastasis.
Clinician Input
Input From Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of unresectable UM or mUM.
Unmet Needs
The clinical expert consulted by CADTH noted that mUM is an aggressive disease associated with poor survival and that available systemic treatments for mUM have not demonstrated improvement in OS, PFS, or ORR. Therefore, the clinical expert noted that the development of novel therapies for mUM is a priority. The clinical expert also noted that UM tumours in general have a low ORR to immune checkpoint inhibitors (approximately 10% to 20%) because of the low tumour mutation burden, although the ORR is slightly higher in atypical UM. This predicts a poor response to immunotherapy, per the clinical expert. Chemotherapies are associated with a low ORR (about 5% to 15%) and considerable toxicity, according to the clinical expert.
Place in Therapy
The clinical expert indicated that the mechanism of action of tebentafusp is unique among current therapies for UM, in that the drug exploits the biology of the UM cell. The clinical expert explained that patients must have an HLA-A*02:01-positive status for tebentafusp to be effective, owing to the unique mechanism of action, which leaves only 45% of all patients with mUM (mainly of European origin) eligible for tebentafusp.
The clinical expert considered tebentafusp to be a major advance in the therapy of UM, and noted that although it is not curative, it will likely fulfill the unmet need for an effective treatment that improves survival. A shift in the current treatment paradigm is anticipated by the clinical expert. The clinical expert noted that in melanoma clinics where tebentafusp can be accessed through compassionate drug programs, tebentafusp is currently the preferred first-line systemic treatment for mUM. The clinical expert stated that tebentafusp should be used as a first-line treatment, if possible, but that its use in the second-line setting and beyond is also rational, particularly in patients for whom access to compassionate supplies of tebentafusp was not possible in the past and who therefore had to be treated with other therapies.
Patient Population
The clinical expert indicated that only HLA-A*02:01-positive patients with mUM are expected to benefit from tebentafusp. The HLA subtype can be determined with a routine HLA genotype testing, which is available in larger treatment centres.
The clinical expert indicated that there is currently no good clinical or biologic predictor of response to tebentafusp. The clinical expert noted that patients with low-volume disease tend to respond better to tebentafusp than patients with large metastases, although this was based on anecdotal evidence. In addition, patients with a better performance status are also more likely to benefit from treatment in general in clinical practice, according to the clinical expert.
The clinical expert indicated that patients with a poor performance status are least suitable for treatment with tebentafusp, noting that patients with an ECOG PS of 3 or above are generally not eligible for treatment in clinical practice.
Assessing Response to Treatment
According to the clinical expert, OS is considered the most important and clinically meaningful outcome in patients with mUM, whereas PFS and ORR have limited relevance, as survival does not corelate with ORR in patients with mUM receiving tebentafusp. The clinical expert noted that a posthoc analysis of Study 202 showed that patients with progression on tebentafusp could still benefit from treatment beyond progression.10 The clinical expert, therefore, concluded that patients should be allowed to continue treatment as long as they derive clinical benefits from tebentafusp, and treatment response should be left to the discretion of the attending oncologist based on assessments of history and physical examinations (every 3 to 4 weeks), laboratory tests (weekly), and imaging (every 12 to 16 weeks) findings.
Discontinuing Treatment
The clinical expert highlighted that the decision to discontinue treatment should be left to the discretion of the attending oncologist, given the complexity of the considerations involved. In clinical practice, treatment discontinuation is generally considered in the presence of intolerable toxicities or when there is clear evidence of significant progression, as indicated by a decline in performance status, an increase in pain, rising LDH levels, and marked radiographic progression, according to the clinical expert.
Prescribing Conditions
The clinical expert emphasized that the first 3 or 4 infusions of tebentafusp should be given in hospital because of the risk of CRS, which usually presents with rash, fever or chills, hypotension, and, in rare cases, compromise of pulmonary, renal, or hepatic function. After cycle 4, the risk of CRS is substantially lower, and most patients have no side effects from tebentafusp thereafter.
The clinical expert recommended that initially tebentafusp should only be prescribed by a specialist physician experienced in the use of tebentafusp and familiar with the management of CRS. Once a pattern of use is established and the risk of CRS is absent, subsequent infusions can be performed in a community clinic setting.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The original clinician group submissions can be found on CADTH’s website.
CADTH received input from 1 clinician group, Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee, based on responses from 6 clinicians. The Skin Cancer Drug Advisory Committee provides timely evidence-based clinical and health-system guidance on drug-related issues in support of Cancer Care Ontario’s mandate, including the Provincial Drug Reimbursement Programs and the Systemic Treatment Program. No major views in contrast to those provided by the clinical expert consulted by CADTH for this review were presented.
The clinician group stated that UM is a disease that has a low tumour mutational burden and a low 1-year survival rate (50%) in the metastatic setting, which is distinct from cutaneous melanoma. None of the current systemic treatments for UM have been proven to have an OS benefit, which is a major unmet need in patients with mUM. The clinician group noted that tebentafusp has demonstrated an improvement in OS in clinical trials. The clinician group noted that the treatment goal is to improve OS and QoL. The clinician group explained that tebentafusp would be the first-line treatment of choice for HLA-A*02:01-positive patients with mUM, although did not comment on the use of tebentafusp in second-line settings or beyond. The clinician group stated that HLA-A*02:01-positive patients with mUM who do not meet the exclusion criteria of Study 20210 would be suitable for tebentafusp. They noted that ongoing performance status, tumour size, and RECIST progression requirements are the clinical outcomes used to determine whether a patient is responding to treatment. They indicated that treatment response is considered clinically meaningful in the presence of a reduction in tumour size of at least 20% and improved performance status, and noted that treatment can be continued in some patients with radiographic progression (new and/or increasing target lesions) if clinical benefits are observed. Toxicity and symptomatic disease progression without clinical benefit would be considered when deciding whether to discontinue tebentafusp, according to the clinician group. They indicated that oncologists with experience in the inpatient management of side effects related to tebentafusp are required because of the known toxicities that commonly occur after the first 3 doses.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparator | |
The IMCgp100 to 202 trial (Study 202) compared tebentafusp with investigator’s choice of pembrolizumab, ipilimumab, or dacarbazine.10 Immunotherapy (e.g., pembrolizumab, nivolumab, ipilimumab) and chemotherapy (e.g., dacarbazine, temozolomide, paclitaxel-carboplatin) are funded for cutaneous melanoma in most jurisdictions. Funding for systemic therapies for UM is usually assessed on a case-by-case basis and would usually include therapies used for cutaneous melanoma. Other options for patients with UM include enrolment in clinical trials, where available. | This is a comment from the drug programs to inform pERC deliberations. |
Initiation of therapy | |
Patients require confirmation of HLA-A*02:01-positive status to be eligible for tebentafusp. HLA typing would be required for all patients diagnosed with UM. Access to HLA typing may differ by jurisdiction. Study 202 required HLA testing by central assay.10 | This is a comment from the drug programs to inform pERC deliberations. |
Study 202 included patients who had not received any prior therapy for advanced or metastatic disease but were permitted to have received prior (neo)adjuvant therapy.10 The funding request is not specific to the use of tebentafusp in the first-line setting. Should patients who received prior therapies in the advanced or metastatic setting be eligible for tebentafusp? Is there clinical evidence to inform the efficacy and safety of tebentafusp in this patient population? | The clinical expert agreed that patients who received prior therapies in the advanced or metastatic setting should be eligible for tebentafusp. The clinical expert noted that treatment options are limited. Although immunotherapy is a reasonable first option, the response to immunotherapy is poor, so clinical trials are preferred for patients in the first-line setting and beyond, according to the clinical expert. The clinical expert concluded that there is an unmet need for effective treatment options in the first-line setting and beyond, and noted that if tebentafusp is available in the first-line setting and beyond, clinicians would prescribe this drug. The clinical expert noted that in Study 102,27 in which tebentafusp was used as a second-line or later treatment in patients with mUM, patients appeared to benefit from tebentafusp, although the clinical expert acknowledged that this is a phase II trial which is subject to limitations. In addition, the clinical expert reported that clinical experience has shown that patients receiving tebentafusp through compassionate drug programs in a second-line setting also benefit; therefore, the clinical expert felt that there is no justification to exclude patients who had prior therapies from receiving tebentafusp. The clinical expert explained that access to tebentafusp is particularly important for patients who did not have access to compassionate supplies of tebentafusp in the past and, therefore, had to receive other therapies. |
Study 202 excluded patients with symptomatic CNS metastases.10 Should patients with CNS involvement be eligible for tebentafusp? Is there clinical evidence to inform the efficacy and safety of tebentafusp in this patient population? | The clinical expert noted that CNS metastases are rare in patients with UM and tend to occur only after a prolonged period of existing metastatic disease. The clinical expert noted that, historically, patients with CNS metastasis would have been excluded from systemic therapy. However, with the use of stereotactic radiosurgery, which has been shown to be an effective localized treatment for limited (≤ 10) metastatic lesions, there is no reason to withhold tebentafusp from patients with CNS metastases if the metastases are controlled with radiation or surgery. |
Discontinuation of therapy | |
It is not clear how long patients can be treated because response evaluation may not follow the RECIST-based assessment. What discontinuation criteria should be applied] to tebentafusp? | The clinical expert noted that a posthoc exploratory analysis of patients in Study 202 who had disease progression as their best overall response showed that patients who received tebentafusp had longer OS than patients in the investigator’s-choice arm,11 suggesting that patients with evidence of progression on tebentafusp may still benefit from treatment beyond progression. The clinical expert, therefore, felt that the decision to discontinue treatment should be left to the discretion of the attending oncologist. The clinical expert indicated that in clinical practice, treatment discontinuation is generally considered in patients with clear evidence of significant progression, suggested by a decline in performance status, increasing pain, rising LDH levels, and marked progression on imaging. |
Prescribing of therapy | |
Tebentafusp is available as a 100 mcg/0.5 mL vial. Doses are administered weekly by IV infusion over 15 to 20 minutes, and follow an escalation schedule in which dose 1 is 20 mcg, dose 2 is 30 mcg, dose 3 and beyond is 68 mcg. Weekly treatment continues until disease progression or unacceptable toxicity. | This is a comment from the drug programs to inform pERC deliberations. |
The first 3 infusions of tebentafusp should be administered in an appropriate health care setting by IV infusion over 15 to 20 minutes, per manufacturer’s product monograph.9 An observation period of up to 16 hours is required after each of the first 3 doses to provide monitoring and management of potential CRS.9 If the patient experiences no CRS of grade 2 or greater after 3 doses, subsequent doses can be administered on an outpatient basis, with the observation period reduced to 30 minutes.9 Administration of tebentafusp, in particular the first 3 doses in an inpatient setting, would represent a significant increase in health-system resources over other comparators. It is noted that the patient population eligible for tebentafusp is small. | This is a comment from the drug programs to inform pERC deliberations. |
Clinicians and/or facilities with experience monitoring and managing CRS are required for the administration of tebentafusp. In some jurisdictions, systemic treatments administered in the inpatient setting are outside the scope of drug plan budgets. Will coverage of inpatient treatment be addressed? | For pERC consideration. |
Generalizability | |
Study 202 only enrolled patients with an ECOG PS of 0 or 1.9 Should patients with ECOG PS of 2 or greater be eligible for tebentafusp? | The clinical expert noted that patients with an ECOG PS of 2 or greater may benefit from tebentafusp in clinical practice but, in general, patients with a very poor performance status are less likely to benefit from systemic treatments. Clinicians generally do not treat a patient with an ECOG PS of 3 or worse, according to the clinical expert. |
Care provision issues | |
The preparation of tebentafusp is complex and intense and will require considerable pharmacy resources to prepare each dose. Preparations use very small volumes from each drug vial (dose 1 uses 20 mcg from the 100 mcg vial); drug wastage will occur with each weekly dose. The manufacturer’s monograph specifies “do not prepare more than 1 dose from the vial,” so vial sharing will not be possible. Preparations require integration of human albumin, which will not be readily available in most sterile compounding pharmacy facilities (it would usually be requested from the blood bank, introducing an additional step to this preparation). The volume of human albumin required is small, so wastage is expected to occur. The use of human albumin, as a biologic drug, requires additional decontamination of the biologic safety cabinet during the preparation process. Because the human albumin volume and drug volume are so low, each time the drug is prepared, there are dozens of steps to ensure appropriate mixing. The manufacturer outlines the recommended methodological preparation process, which will require considerable pharmacy resources for each weekly dose. Although the administration time of each dose is short (15 to 20 minutes), the observation time required after doses 1 to 3 is considerable (16 hours). Doses 1 to 3 must be administered in an inpatient setting. Therefore, use of health-system resources for the preparation, administration, and monitoring of tebentafusp is significant. | For pERC consideration. |
The potential for CSR requires monitoring and management on an inpatient basis for doses 1 to 3. | This is a comment from the drug programs to inform pERC deliberations. |
For patients diagnosed with nonmetastatic UM, should HLA typing be evaluated at diagnosis? What is the anticipated turnaround time for HLA testing results? | The clinical expert noted that, for patients with nonmetastatic UM, initial HLA testing is not usually done in major treatment centres because the turnaround is about 1 week; in a peripheral centre, however, testing might be advisable to prescreen patients. |
Careful coordination and transfer of care between inpatient and outpatient care teams and facilities will be required to ensure the continuity of weekly treatments for each patient. As a rarely used drug, it is not anticipated that many facilities would have this drug in regular stock, so adequate communication and preparation time will be required to coordinate appropriate care for each patient. | This is a comment from the drug programs to inform pERC deliberations. |
System and economic issues | |
The potential drug acquisition cost per patient is high. The health-system resource use for tebentafusp is considerable relative to comparators. | This is a comment from the drug programs to inform pERC deliberations. |
Tebentafusp requires specialized clinicians for administration, preparation, and monitoring, so treatment is likely to be limited to larger centres. This introduces the potential need for travel, which has an additional impact to daily life, and the potential for increased expenses for eligible patients. Drug wastage is quite significant, as the standard dose is considerably less than the vial size (68 mcg vs 100 mcg) and vial sharing is unlikely to occur. In some jurisdictions, wastage is not reimbursed by the drug plan, so hospitals may not be able to absorb the wastage cost. | This is a comment from the drug programs to inform pERC deliberations. |
CNS = central nervous system; CRS = cytokine release syndrome; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HLA = human leukocyte antigen; LDH = lactate dehydrogenase; mUM = metastatic uveal melanoma; OS = overall survival; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee; RECIST = Response Evaluation Criteria in Solid Tumours; UM = uveal melanoma.
Clinical Evidence
The clinical evidence included in the review of tebentafusp is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes additional relevant studies that were considered to address important gaps in the evidence included in the Systematic Review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of tebentafusp (100 mcg/0.5 mL) IV infusion for the treatment of HLA-A*02:01-positive adults with unresectable UM or mUM.
Methods
Studies selected for inclusion in the Systematic Review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 6: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | HLA-A*02:01-positive adults with unresectable UM or mUM Subgroups:
|
Intervention | Tebentafusp 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15, and 68 mcg once every week thereafter by IV infusion until unacceptable toxicity or disease progression |
Comparators |
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published and unpublished phase III and IV RCTs |
AE = adverse event; CNS = central nervous system; HLA = human leukocyte antigen; HRQoL = health-related quality of life; RCT = randomized controlled trial; SAE = serious adverse event; UM = uveal melanoma; WDAE = withdrawal due to adverse event.
aOff-label treatment. The Health Canada indication is for the treatment of unresectable or metastatic melanoma. Patients with UM were excluded from the pivotal trials that led to approval of the indication.
bIpilimumab monotherapy is used in a second-line setting or beyond for mUM in Canada.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.28
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was tebentafusp. The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on May 18, 2022. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on November 9, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.29 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
One study was identified from the literature for inclusion in the Systematic Review (Figure 1).10,11,30 The included study is summarized in Table 7. A list of excluded studies is presented in Appendix 2.
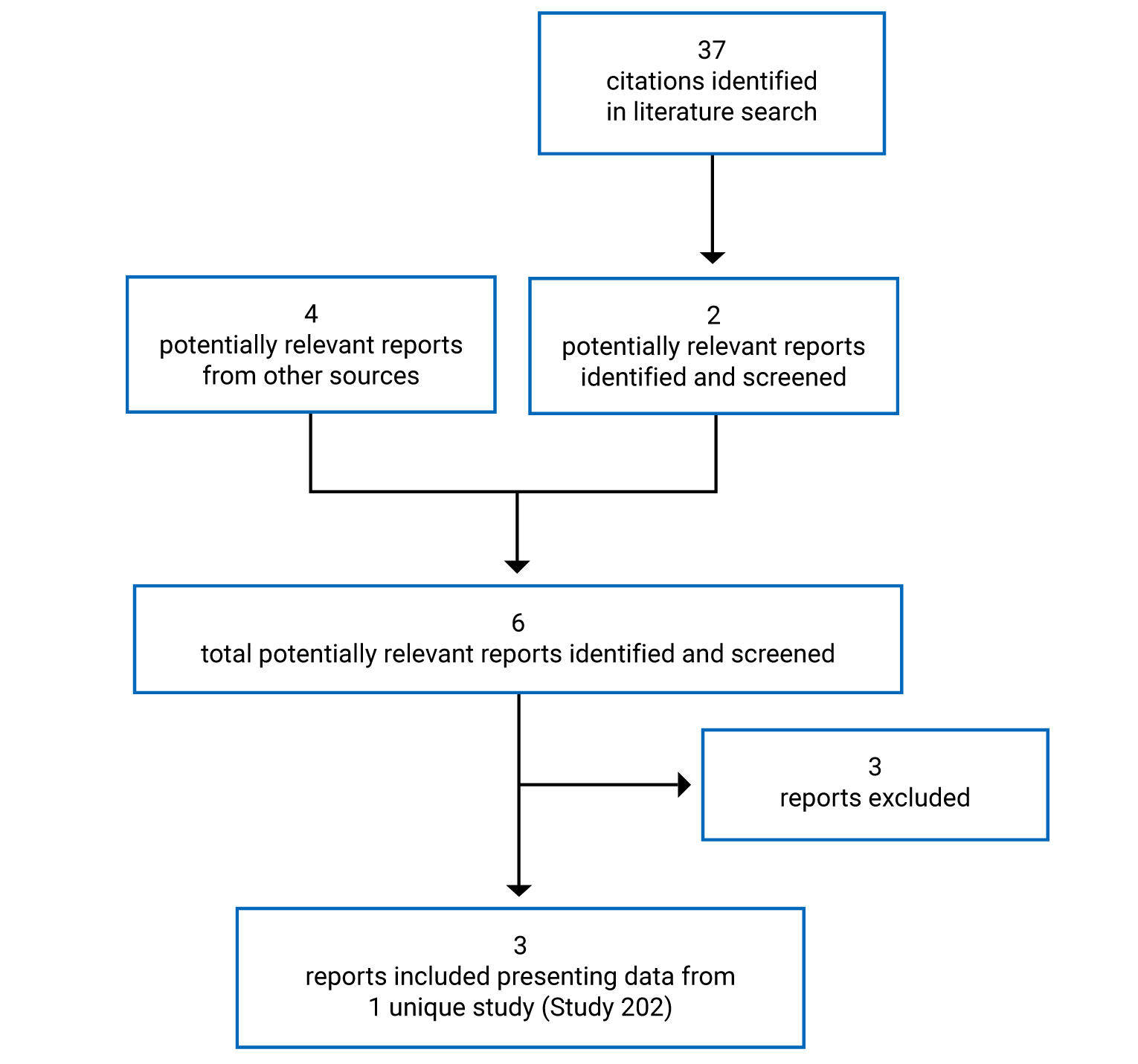
Table 7: Details of Included Study
Criteria | Study 202 |
|---|---|
Design and population | |
Study design | Phase III, multicentre, open-label, randomized, active-controlled trial |
Locations | US (24 sites), Canada (2 sites), Europe (29 sites), and Australia (3 sites) |
Study duration | Start date: October 4, 2017 (ongoing) Expected completion date: June 2025 |
Data cut-off date | First interim analysis: October 13, 2020 (updated analysis: August 12, 2021) |
Randomized (N) | 378 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Tebentafusp IV infusion of 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15, and 68 mcg once every week thereafter |
Comparator(s) | Investigator’s choice of 1 of the following:
|
Duration | |
Phase | |
Prescreening | NR |
Screening | Up to 4 weeks |
Treatment | Until completion of 4 planned doses (ipilimumab only), disease progression, or intolerable toxicity |
Follow-up |
|
Outcomes | |
Coprimary end points | Overall survival in rash analysis set and intention-to-treat analysis set |
Secondary and exploratory end points | Key secondary:
Other secondary:
Exploratory:
|
Notes | |
Publications | Nathan et al. (2021)10 |
AE = adverse event; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HLA = human leukocyte antigen; mUM = metastatic uveal melanoma; NR = not reported; SAE = serious adverse event.
Note: One additional report from the FDA was included.30
aHistologically or cytologically confirmed mUV.
bPrior surgical resection of oligometastatic disease was allowed. Prior neoadjuvant or adjuvant therapy was allowed, provided it was administered in the curative setting in patients with localized disease. Patients must not have been re-treated with an investigator’s-choice therapy that was administered as adjuvant or neoadjuvant treatment. Additionally, patients who received nivolumab as prior adjuvant or neoadjuvant treatment should not have received pembrolizumab as investigator’s choice of therapy.
cPatients with brain metastases are eligible if lesions have been treated with localized therapy and there is no evidence of progression for at least 4 weeks before the study.
Source: Study 202 Clinical Study Report.11
Description of Studies
One study (Study 202) met the inclusion criteria for the CADTH Systematic Review. Study 202 was a phase III, open-label, randomized, active-controlled study that aimed to compare the efficacy and safety of tebentafusp with investigator’s choice (pembrolizumab, ipilimumab, or dacarbazine) in HLA-A*02:01-positive adults with mUM who had not received prior therapy in the metastatic setting (N = 378). The study was initiated on October 4, 2017, and is currently ongoing at 58 cites (2 in Canada) in 14 countries.
Study 202 consisted of 4 phases: prescreening, screening, treatment, and follow-up. Patients who were identified as HLA-A*02:01-positive in the prescreening phase entered a screening phase of up to 28 days to assess study eligibility. Upon entering the treatment phase, eligible patients were randomized to either tebentafusp or investigator’s choice (pembrolizumab, ipilimumab, or dacarbazine) in a 2:1 ratio. Randomization was stratified by baseline LDH level (higher than the upper limit of normal [ULN] range, defined as 250 units/L, or lower than or equal to the ULN) using the Interactive Response Technology system. Patients received study treatments until completion of 4 planned doses (ipilimumab only), disease progression, or intolerable toxicity, and they were subsequently monitored in the follow-up phase for safety, survival, and disease progression (in patients who discontinued treatment for reasons other than progressive disease).
This report presents the primary analysis of Study 202 as of the data cut-off date of October 13, 2020, which corresponded to the first interim analysis of OS and the final analysis of PFS. An informal updated analysis of OS and the primary analysis of ORR (data cut-off date of August 12, 2021), which were conducted to fulfill regulatory requirements in Europe, are also presented.
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria of Study 202 are summarized in Table 7. The study enrolled HLA-A*02:01-positive adults, aged 18 years and older, with mUM, an ECOG PS of 0 or 1, and no prior systemic therapy (chemotherapy, immunotherapy, or targeted therapy) or prior liver-directed therapy (chemotherapy, radiotherapy, or embolization) in the metastatic or advanced setting. Patients with prior surgical resection of oligometastatic disease were allowed. Patients who received prior neoadjuvant or adjuvant therapy for localized disease were allowed if the therapy was administered in the curative setting. Patients who had symptomatic or untreated CNS metastases or who were receiving systemic immunosuppressive medications were excluded.
Baseline Characteristics
Baseline patient demographics, disease characteristics, and prior cancer treatments were, overall, balanced between treatment arms, as shown in Table 8. Approximately half of the patients were female and most were white. Overall, the mean age of the study population was 62.1 (SD = 11.6) years. The majority of patients had an ECOG PS of 0 (73.3%) and liver metastasis, with melanoma initially located in the choroid (75.7%) and no metastasis at diagnosis (92.3%). One-third of patients had an elevated baseline LDH. Most patients had received no prior surgery for metastatic disease (91.3%). A small proportion of patients (3.7%) had received prior antineoplastic systemic treatments (in any setting) and 40.2% of patients had received prior local radiotherapy (in any setting).
Table 8: Summary of Baseline Characteristics (ITT Population)
Characteristic | Study 202 | |
|---|---|---|
Tebentafusp (N = 252) | Investigator’s choice (N = 126) | |
Demographics | ||
Age, years | ||
Mean (SD) | 61.3 (11.9) | 63.6 (10.7) |
Median (range) | 63.5 (23 to 92) | 65.5 (25 to 88) |
Sex, n (%) | ||
Female | 124 (49.2) | 64 (50.8) |
Male | 128 (50.8) | 62 (49.2) |
Race, n (%) | ||
White | 222 (88.1) | 107 (84.9) |
Not reported | 23 (9.1) | 14 (11.1) |
Not allowed, per local regulatory | 5 (2.0) | 3 (2.4) |
Unknown | 1 (0.4) | 1 (0.8) |
American Indian or Alaska Native | 0 | 1 (0.8) |
Other | 1 (0.4) | 0 |
ECOG PS, n (%) | ||
0 | 192 (76.2) | 85 (67.5) |
1 | 49 (19.4) | 31 (24.6) |
2 | 0 | 1 (0.8) |
Missing | 11 (4.4) | 9 (7.1) |
Disease characteristics | ||
Site of initial uveal melanoma, n (%) | ||
Iris | 3 (1.2) | 5 (4.0) |
Ciliary body | 25 (9.9) | 13 (10.3) |
Choroid | 193 (76.6) | 93 (73.8) |
Unknown | 30 (11.9) | 14 (11.1) |
Missing | 1 (0.4) | 1 (0.8) |
Stage of initial diagnosis, n (%) | ||
I | 48 (19.0) | 14 (11.1) |
II | 89 (35.3) | 40 (31.7) |
III | 56 (22.2) | 34 (27.0) |
IV | 23 (9.1) | 7 (5.6) |
Missing | 36 (14.3) | 31 (24.6) |
Metastasis observed at initial diagnosis, n (%) | ||
Yes | 17 (6.7) | 10 (7.9) |
No | 234 (92.9) | 115 (91.3) |
Missing | 1 (0.4) | 1 (0.8) |
Location of metastasis, n (%) | ||
Hepatic only | 131 (51.9) | 59 (46.8) |
Extrahepatic only | 9 (3.6) | 10 (7.9) |
Hepatic and extrahepatic | 111 (44.0) | 55 (43.7) |
Missing | 1 (0.4) | 2 (1.6) |
Baseline LDH level, n (%) | ||
LDH ≤ ULN (250 units/L) | 162 (64.3) | 80 (63.5) |
LDH > ULN (250 units/L) | 90 (35.7) | 46 (36.5) |
Largest metastatic lesion at baseline, n (%) | ||
≤ 3 cm | 139 (55.2) | 70 (55.6) |
3.1 to 8.0 cm | 92 (36.5) | 46 (36.5) |
≥ 8.1 cm | 21 (8.3) | 10 (7.9) |
Prior cancer treatments | ||
Prior surgery for metastatic disease, n (%) | ||
Yes | 24 (9.5) | 9 (7.1) |
No | 228 (90.5) | 117 (92.9) |
Prior local therapy, n (%) | ||
Radiotherapy | 100 (39.7) | 52 (41.3) |
Other | 8 (3.2) | 5 (4.0) |
Prior antineoplastic systemic drugs, n (%) | 11 (4.4) | 3 (2.4) |
Sunitiniba | 9 (3.6) | 2 (1.6) |
Nivolumab | 1 (0.4) | 0 |
Fotemustine | 0 | 1 (0.8) |
Other | 1 (0.4) | 0 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITT = intention to treat; LDH = lactate dehydrogenase; SD = standard deviation; ULN = upper limit of normal.
Note: Data cut-off date of October 13, 2020.
aSunitinib or sunitinib malate.
Source: Study 202 Clinical Study Report.11
Interventions
Patients in the tebentafusp arm received 20 mcg on day 1, 30 mcg on day 8, and 68 mcg on day 15 and weekly thereafter by IV infusion over 15 minutes.
Patients in the investigator’s-choice arm received 1 of the following 3 treatments selected by the investigator(s) before randomization:
dacarbazine 1,000 mg/m2 by IV infusion on day 1 of each 21-day cycle
ipilimumab 3 mg/kg by IV infusion over 90 minutes on day 1 of each 21-day cycle for a maximum of 4 doses
pembrolizumab at either 2 mg/kg per dose (up to 200 mg) or a flat dose of 200 mg (where approved locally) by IV infusion over 30 minutes on day 1 of each 21-day cycle.
No blinding of interventions occurred. All treatments were continued until radiographic progression per RECIST 1.1 (except for patients receiving ipilimumab, for which a maximum of 4 doses were given), or unacceptable toxicity. Exceptions were applied to tebentafusp, pembrolizumab, and ipilimumab, which could be continued after initial disease progression when all of the following prespecified criteria were met:
no signs or symptoms indicating clinically significant progressive disease
no decline in ECOG PS
no impending threat to vital organs or critical anatomic sites requiring urgent alternative medical intervention or no cases in which continuation of the study therapy would have prevented the institution of such an intervention
the absence of any of the study treatment discontinuation criteria (i.e., completion of 4 doses of ipilimumab; initiation of alternative anticancer therapy; unacceptable toxicity; patient met 1 or more of the exclusion criteria or failed to meet all of the inclusion criteria and continuation of the study treatment would pose a safety risk; consent withdrawal; or pregnancy).
In patients who received treatment beyond initial progression, treatments were discontinued in the presence of further progression, which is defined as any 1 of the following:
an additional increase in tumour burden of at least 20% (sum of the diameters of both target and new measurable lesions) accompanied by an absolute increase of at least 5 mm
unequivocal progression of nontarget lesions
new nonmeasurable lesions.
In the tebentafusp arm, inpatient safety monitoring was required for at least 16 hours after each of the first 3 doses; outpatient administration was permitted for subsequent doses unless the patient experienced hypotension of grade 2 or higher after the third dose. In the investigator’s-choice arm, no extended postinfusion monitoring was required.
Patients who experienced an infusion reaction to prior doses were given premedication to manage potential infusion reactions (e.g., acetaminophen, antihistamine). Concomitant medications and therapies that were necessary for the supportive care and safety of such patients were generally allowed (e.g., antidiarrheals, antiemetics, electrolyte supplementation), and palliative radiotherapy, hematopoietic colony-stimulating growth factors, anticoagulants, antihypertensives, bisphosphonates, and denosumab were allowed at the discretion of the investigator(s).
Anticancer therapy (other than study treatments) and most monoclonal antibody therapies were prohibited from the study. Immunosuppressive medications were prohibited, with the exception of systemic corticosteroids, which were permitted for the management of infusion reactions, immune-mediated toxicities, and other toxicities (e.g., hypotension, CRS).
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 9. These end points are subsequently summarized. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 9: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study 202 |
|---|---|
Overall survival | Primary |
Progression-free survival | Key secondary |
HRQoL | Secondary |
Duration of response | Secondary |
Best overall response and objective response ratea | Key secondary |
Disease control rate | Secondary |
Harms (AEs, SAEs, WDAE, mortality, notable harms) | Secondary |
AE = adverse event; HRQoL = health-related quality of life; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aThe sponsor considered the definition of best overall response and objective response rate to be synonymous. Objective response rate was the end point that was evaluated statistically.
The coprimary end points of Study 202 were OS in the ITT population and OS in the RAS (i.e., patients who developed rash in the week after tebentafusp treatment). OS was defined as the time between the date of randomization and the date of death from any cause in an individual patient. OS was followed continuously while the patients received study treatment and every 3 months during the follow-up phase. The inclusion of the RAS in the OS analysis was informed by phase II trial data that suggested rash (an on-target but off-tumour AE) might be associated with a clinical benefit,17 per the sponsor.
The key secondary end points of Study 202 were PFS and BOR (evaluated statistically as ORR). Other secondary efficacy end points included HRQoL measured with EORTC QLQ-C-30 and EQ-5D-5L scores, DOR, and DCR. Tumour response end points were based on investigator assessments, according to RECIST 1.1, conducted every 3 months during the treatment phase and the follow-up phase. HRQoL end points were based on assessments conducted every other cycle until cycle 5 and then every fourth cycle during the treatment period, and conducted every 3 months during the follow-up period. Secondary end points were defined as follows:
PFS was defined as the time from randomization to the date of first documented progression (based on RECIST 1.1) or death from any cause, whichever occurred first.
HRQoL was measured with the EORTC QLQ-C-30 and EQ-5D-5L instruments. Detailed information on the scoring and validity of the EORTC QLQ-C30 and EQ-5D-5L instruments is provided in Appendix 4.
EORTC QLQ-C30 is a patient-reported outcome instrument designed to assess the HRQoL of cancer patients by the evaluation of functional, symptom, global health status/QoL domains.31 Outcome scores are computed using a linear transformation of the raw score such that scores range from 0 to 100. A higher score represents a higher (better) level of functioning and a higher (worse) level of symptoms. A higher score for global health status/QoL represents a better level of global health status. The validity, reliability, responsiveness, and minimally important difference (MID) estimate of the EORTC QLQ-C30 instrument have not been evaluated in patients with mUM. An MID estimate of 10 points was identified in 2 studies of various cancer types32,33 and was a commonly used threshold for the assessment of a clinically meaningful change in HRQoL, although other MIDs have been reported by some studies33,34 (refer to Table 40 in Appendix 4 for more details).
EQ-5D-5L is a generic patient-reported outcome instrument that measures overall health status in patients by generating an index-based summary score on 5 health dimensions (mobility, self-care, daily activities, pain, and anxiety) based upon societal preference weights.35 The EQ-5D-5L also uses a vertical VAS for patients to rate their health, with 0 indicating the best health you can imagine and 100 indicating the worst health you can imagine. The validity, reliability, responsiveness, and MID estimates of the EQ-5D-5L instrument have not been evaluated in patients with mUM. In a study of patients with various cancer types, MID estimates of 7 to 12 have been reported for EQ VAS.36
DOR was calculated only for patients who achieved a CR or PR and was defined as the time from the date of first documented response (based on RECIST 1.1) to the date of first progression or death.
ORR was defined as the number of randomized patients with at least 1 visit response of CR or PR (based on RECIST 1.1) divided by the number of randomized patients and presented as a percentage for each treatment arm in the ITT population.
DCR was defined as the proportion of patients with a BOR of CR or PR or stable disease (based on RECIST 1.1) at 24 weeks or more after randomization to the study drug and before any progressive disease event.
BOR was defined as the best response (CR, PR, stable disease, progressive disease, or not evaluable, based on RECIST 1.1) from randomization until progression, death, or the start of new anticancer therapy.
Harms
The harms outcomes were measured with TEAEs, death, safety laboratory parameters, vital signs, electrocardiogram results, physical examination, and extent of exposure. The outcomes were based on safety assessments conducted once every cycle in the investigator’s-choice arm, and 3 times every cycle in the tebentafusp arm (except in cycle 1, where 6 assessments were conducted) during the treatment phase and safety follow-up phase.
Statistical Analysis
A summary of the statistical analyses of efficacy outcomes in Study 202 is shown in Table 11.
Sample Size and Power Calculation
The sample-size calculation was based on the primary end point of OS in the ITT population. Assuming 33 months of nonuniform recruitment and a 10% dropout rate, the study required a sample size of 369 patients in the ITT population, to be randomized in a 2:1 ratio to the tebentafusp arm and the investigator’s-choice arm, to achieve at least 250 death events to detect an HR of 0.645 with respect to OS at a 2-sided alpha of 0.045 with a power of 89% using a log-rank test.
In the RAS, 164 death events were needed to detect an HR of 0.531 with respect to OS at a 2-sided alpha of 0.005 with a power of 89% using a log-rank test, assuming 50% of patients in the tebentafusp arm developed a rash in the first week after treatment.
Interim and Final Analyses
A group sequential approach was used to account for 2 formal interim analyses and 1 final analysis. The first and second interim analyses were planned to occur after 150 and 200 death events, respectively, in the ITT population; and the final analysis will be conducted after 250 death events have occurred in the ITT population. The interim analyses were reviewed by an independent data monitoring committee. The primary end point of OS was tested at an overall alpha of 0.05 (2-sided), with 10% allocated to the RAS analysis (i.e., alphaRAS = 0.005) and 90% allocated to the ITT analysis (i.e., alphaITT = 0.045), based on the O’Brien-Fleming boundary as implemented using the Lan-DeMets approach. The stopping boundaries for OS are outlined in Table 10. If the stopping boundaries were crossed at the first interim analysis, then the alpha in RAS was carried over to the ITT analysis, resulting in an alphaITT of 0.05. PFS and BOR were not tested on an interim basis. A final PFS analysis was planned to occur after 274 progression events, and a BOR (evaluated statistically as ORR) analysis was planned to occur after all randomized patients had been followed for approximately 9 months (i.e., 3 planned assessments).
Table 10: Stopping Boundaries for OS in the Interim and Final Analyses
Analysis set (allocated alpha) | Analysis | OS events (information fraction, %) | Lower Z-score boundary | Upper Z-score boundary | Nominal alpha (2-sided) | Cumulative alpha |
|---|---|---|---|---|---|---|
RAS (alphaRAS = 0.005) | First interim | 99 (60) | –3.732 | 3.732 | 0.0002 | < 0.001 |
Second interim | 131 (80) | –3.198 | 3.198 | 0.0014 | 0.001 | |
Final | 164 (100) | –2.838 | 2.838 | 0.0045 | 0.005 | |
ITT (alphaITT = 0.045) | First interim | 150 (60) | –2.724 | 2.724 | 0.006 | 0.006 |
Second interim | 200 (80) | –2.336 | 2.336 | 0.019 | 0.021 | |
Final | 250 (100) | –2.073 | 2.073 | 0.038 | 0.045 | |
ITT (alphaITT = 0.05) | First interim | 150 (60) | –2.669 | 2.669 | 0.008 | 0.008 |
Second interim | 200 (80) | –2.289 | 2.289 | 0.022 | 0.024 | |
Final | 250 (100) | –2.031 | 2.031 | 0.042 | 0.050 |
ITT = intention to treat; OS = overall survival; RAS = rash analysis set.
Source: Study 202 Statistical Analysis Plan.37
Multiplicity Adjustment
Hypothesis testing was formally performed on the coprimary end points of OS in the RAS analysis and OS in the ITT analysis, and the key secondary end points of PFS and BOR (evaluated statistically as ORR) in the ITT analysis. The end points were tested in a sequential order using the graphical approach, as outlined in Figure 2, to control for multiplicity. In Figure 2, the arrows specify the alpha transfer path. Once the null hypothesis for an end point was rejected (i.e., deemed statistically significant) at its assigned significance level, its significance level was transferred to subsequent end point(s), following the arrow. The fraction indicated at each node denotes the fraction of alpha allocated to the end point, and the number on the arrows denote the weights for transferring significance level. OS in the RAS was tested first, and if the test was statistically significant, the significance was transferred to test OS in the ITT analysis set. If OS in both RAS and ITT analyses were statistically significant, the significance was transferred to test PFS in the ITT analysis set, and so on.
Figure 2: Multiplicity Strategy Involving Sequential Testing of OS, PFS, and BOR
BOR = best overall response; H1 = null hypothesis for OS in the RAS; H2 = null hypothesis for OS in the ITT analysis set; H3 = null hypothesis for PFS in ITT; H4 = null hypothesis for BOR (evaluated statistically as ORR) in ITT; ITT = intention to treat; ORR = objective response rate; PFS = progression-free survival; RAS = rash analysis set.
Notes: The fraction (or whole number) at the bottom of each node represents the allocated weight for alpha in the testing sequence.
This figure was adapted in consultation with the sponsor to correct errors identified in the allocation fractions.
Source: Study 202 Statistical Analysis Plan.37
Statistical Analysis for Coprimary Efficacy End Points
In the primary analyses of the coprimary end points of OS in the RAS and ITT set, the difference in OS between the tebentafusp arm and the investigator’s-choice arm were tested using a 2-sided log-rank test stratified by LDH level (LDH > 250 units/L versus LDH ≤ 250 units/L). The HR and the 95% CI were based on a Cox proportional hazards model stratified by LDH level using the Efron approach for handling ties. OS was analyzed using the Kaplan-Meier (KM) method. The median follow-up time for OS and the 95% CI were presented based on the reverse KM method. OS rate every 6 months was derived from KM estimates.
A sensitivity analysis of OS, which aimed to examine the impact of misstratification, was planned based on strata determined by the central LDH results if discrepancies of more than 10% were observed in LDH results compared to strata recorded at the time of randomization in the Interactive Response Technology used in the primary analysis.
Exploratory subgroup analyses were conducted with respect to OS. The ECOG PS subgroup (ECOG PS of 0 versus 1) was relevant to this review. The HR and 95% CI between treatment arms were estimated using the Cox proportional hazards model using Efron’s approach for handling ties, adjusted for treatment, subgroup, and treatment-by-subgroup interaction term. No adjustment for type I error was involved in the subgroup analysis.
No imputation was performed to account for missing OS data in general (except for partial dates).
Statistical Analysis for Secondary Efficacy End Points
The secondary efficacy analyses were performed in the ITT population and were based on tumour response per RECIST 1.1 as assessed by the investigator. PFS was analyzed using the same methodology as was used in the primary analysis of OS. For ORR and DCR, the difference between treatment arms was analyzed using the Cochran-Mantel-Haenszel test stratified by LDH level, and OR, along with 95% CI, were reported. Descriptive data were provided for DOR based on KM estimates. The median duration of follow-up for DOR and the 95% CI were presented based on the reverse KM method.
Descriptive statistics were used to report EQ-5D-5L and EORTC QLQ-C30 outcomes. For each of the EORTC QLQ-C30 domain, the LS mean change from baseline in scale score was estimated in each treatment arm using the mixed model repeated measures approach with treatment group (binary), time point (categorical), LDH status (binary), and baseline score (continuous) as covariates, and the difference between treatment groups was reported.
Two sensitivity analyses for PFS were conducted to assess:
evaluation-time bias (when scans are not performed as scheduled in the protocol), in which the midpoint between the time of progression and the previous evaluable tumour assessment were analyzed using a stratified log-rank test stratified by LDH level
attrition bias, in which the PFS analysis was repeated with actual PFS event times (as opposed to censored times) for patients who progressed or died in the absence of progression immediately after 2 or more nonevaluable or missed tumour assessments.
Exploratory subgroup analyses were conducted with respect to PFS using the same methodology as was used in subgroup analyses of OS, outlined previously. The ECOG PS subgroup (ECOG PS of 0 versus 1) was relevant to this review.
No imputation was performed to account for missing data in any secondary efficacy end points in general (except for partial dates).
Safety Analyses
The safety analyses were performed on the safety analysis set and were summarized using descriptive statistics.
Table 11: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
Overall survival in RAS and ITT | Estimated using the KM methods; 2-sided log-rank test to assess between-group difference; Cox proportional hazard models to estimate hazard ratio and 95% CI | LDH levels (> 250 units/L vs. ≤ 250 units/L) based on strata recorded at the time of randomization in the IRT | Stratification based on central LDH results; conducted only if discrepancies in LDH results were > 10% between central LDH and strata recorded by IRT at randomization |
Progression-free survival in ITT | Estimated using the KM methods; 2-sided log-rank test to assess between-group difference; Cox proportional hazard models to estimate hazard ratio and 95% CI | LDH levels | Evaluation-time bias and attrition bias analyses |
EORTC QLQ-C30 in ITT | MMRM to estimate LS mean change from baseline in scale score in each treatment arm | Treatment group (binary), time point (categorical), LDH status (binary), and baseline score (continuous) | Not performed |
EQ-5D-5L in ITT | Descriptive statistics | NA | Not performed |
Objective response rate in ITT | CMH test to assess difference between treatment arms | LDH levels | Not performed |
Disease control rate in ITT | CMH test to assess difference between treatment arms | LDH levels | Not performed |
Best overall response in ITT | Descriptive statistics | NA | Not performed |
Duration of response in ITT | Estimated using the KM method; summarized with descriptive statistics | NA | Not performed |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = 5-Level EQ-5L; IRT = Interactive Response Technology; ITT = intention to treat; KM = Kaplan-Meier; LDH = lactate dehydrogenase; LS = least squares; MMRM = mixed model repeated measures; NA = not applicable; RAS = rash analysis set.
Source: Study 202 Statistical Analysis Plan.37
Analysis Populations
The ITT analysis set included all patients randomized in the study and was analyzed by treatment assignment. The ITT set was used for all demographic characteristics, baseline characteristics, and efficacy analyses.
The RAS included all patients randomized to tebentafusp who developed a rash in the first week after treatment and all patients randomized to the investigator’s-choice arm. It was prespecified that the results (demographic characteristics, baseline characteristics, efficacy, and safety) of the RAS analysis would only be reported if the RAS analysis crossed the stopping boundaries for OS but the ITT analysis did not.
The safety analysis set included all patients who received at least 1 full or partial dose of tebentafusp or investigator’s choice of therapy and was analyzed by the initial treatment received. The safety set was used for safety analyses of the study.
The pharmacokinetic analysis set was not reviewed in this report because it was of limited relevance.
Results
Patient Disposition
A summary of patient disposition is shown in Table 12. Of the 447 patients screened, 378 were randomized (252 to the tebentafusp arm and 126 to the investigator’s-choice arm). In the investigator’s-choice arm, the number of patients who received pembrolizumab, ipilimumab, and dacarbazine were 103 (81.7%), 16 (12.7%), and 7 (5.6%), respectively. As of the data cut-off date, the proportion of patients who discontinued the study was numerically higher in the investigator’s-choice arm (54.8%) than in the tebentafusp arm (38.1%), mostly due to death (50.0% versus 34.5%). The proportion of patients who discontinued the study drug was numerically higher in the investigator’s-choice arm (79.4%) than in the tebentafusp arm (68.3%), with the primary reason being disease progression (61.9% versus 61.1%).
Disposition | Study 202 | |
|---|---|---|
Tebentafusp | Investigator’s choice | |
Screened, n | 447 | |
Randomized, n | 252 | 126a |
Treated, n (%) | 245 (97.2) | 111 (88.1) |
Discontinued from study, n (%) | 96 (38.1) | 69 (54.8) |
Death | 87 (34.5) | 63 (50.0) |
Lost to follow-up | 1 (0.4) | 1 (0.8) |
Withdrawal of consent | 8 (3.2) | 5 (4.0) |
Discontinued study drug, n (%) | 172 (68.3) | 100 (79.4) |
Progressive disease | 154 (61.1) | 78 (61.9) |
Patient decision | 7 (2.8) | 3 (2.4) |
Adverse event | 6 (2.4) | 5 (4.0) |
Death | 3 (1.2) | 1 (0.8) |
Completed 4 doses of study treatment | 0 | 10 (7.9)b |
Alternative anticancer treatment | 1 (0.4) | 1 (0.8) |
Investigator’s decision | 1 (0.4) | 2 (1.6) |
ITT analysis set, n (%) | 252 (100.0) | 126 (100.0) |
Rash analysis set, n (%) | 149 (59.1) | 126 (100.0) |
Safety analysis set, n (%) | 245 (97.2) | 111 (88.1) |
ITT = intention to treat.
Note: Data cut-off date of October 13, 2020.
aThe number of patients receiving pembrolizumab (n = 103), ipilimumab (n = 16), or dacarbazine (n = 7).
bTen patients received a maximum of 4 doses of ipilimumab, per protocol.
Source: Study 202 Clinical Study Report.11
Protocol Deviation
A numerically higher proportion of patients reported at least 1 type of important protocol deviation in the tebentafusp arm (39.3%) than in the investigator’s-choice arm (28.6%). The most common protocol deviation in the tebentafusp arm and investigator’s-choice arm was related to trial procedures (22.6% versus 11.1%). The investigators attributed the imbalance in protocol deviations to the more frequent dosing and the intensive schedule of visits in the tebentafusp arm, and stated that the protocol deviations were not expected to affect interpretation of the results.
Exposure to Study Treatments
Exposure to study treatments is summarized in Table 13. The median duration of treatment was 163.0 (range, 1 to 1,016) days in the tebentafusp arm and 65.0 (range, 1 to 658) days in the investigator’s-choice arm. The median relative dose intensity was 100.0% in the tebentafusp and investigator’s-choice arms (range, 95% to 100% and 100% to 100%, respectively).
Table 13: Study Drug Exposure (Safety Analysis Set)
Drug exposure | Study 202 | ||
|---|---|---|---|
Tebentafusp (N = 245) | Investigator’s choice (N = 111) | ||
Duration of exposure (days)a | |||
Mean (SD) | 219.5 (191.6) | 118.9 (130.3) | |
Median (range) | 163.0 (1 to 1,016) | 65.0 (1 to 658) | |
Relative dose intensity (%)b | |||
Mean (SD) | 99.9 (0.4) | 100.0 (0.0) | |
Median (range) | 100.0 (95 to 100) | 100.0 (100.0 to 100.0) | |
SD = standard deviation.
Note: Data cut-off date of October 13, 2020.
aDate of last study drug administration – date of first study drug administration + 1.
b(Total actual dose received intensity / total planned dose intensity) * 100.
Source: Study 202 Clinical Study Report.11
Treatment Beyond Initial Progression
The proportion of patients who continued the assigned study treatment beyond initial disease progression was 43.3% in the tebentafusp arm and 14.3% in the investigator’s-choice arm, whereas the median duration of treatment beyond initial progression was 1.9 (range, 0.1 to 28.8) months and 1.1 (range, 0.10 to 13.0) months, respectively.
Concomitant Medications
A summary of common concomitant medications used during the study is shown in Table 15. The most frequently used concomitant medications (tebentafusp arm versus investigator’s-choice arm) were analgesics (89.7% versus 41.3%), systemic antihistamine (86.1% versus 18.3%), blood substitutes and perfusion solutions (56.7% versus 14.3%), topical corticosteroids (54.0% versus 14.3%), and anti-inflammatory and antirheumatic products (47.2% versus 29.4%). The proportion of patients who received these medications was notably higher in the tebentafusp arm than in the investigator’s-choice arm, including psycholeptics (32.9% versus 20.6%).
Subsequent Anticancer Treatments
A summary of anticancer treatments used after discontinuation of the study treatment is shown in Table 16. The most common subsequent anticancer treatment was systemic therapy, reported in 43.3% of patients in the tebentafusp arm and 43.7% of patients in the investigator’s-choice arm. The median duration of subsequent systemic therapy was 110 (range, 70 to 653) days in the tebentafusp arm and 70 (range, 1 to 721) days in the investigator’s-choice arm.
Table 14: Treatment Beyond Initial Progression (ITT Analysis Set)
Treatment | Tebentafusp (N = 252) | Investigator’s choice (N = 126) |
|---|---|---|
Patients receiving treatment beyond progression,a n (%) | 109 (43.3) | 18 (14.3) |
Duration of treatment beyond progression (months) | ||
Mean (SD) | 3.5 (4.6) | 2.3 (3.2) |
Median (range) | 1.9 (0.1 to 28.8) | 1.1 (0.10 to 13.0) |
ITT = intention to treat; SD = standard deviation.
aDuration of treatment beyond RECIST 1.1 progression is the time from RECIST 1.1 progression to the date of last dosing recorded at the time of the data cut-off date.
Source: Study 202 Clinical Study Report.11
Table 15: Common Concomitant Medications (in > 30% of Patients) (ITT Analysis Set)
Drug class | Study 202 | |
|---|---|---|
Tebentafusp (N = 252) | Investigator’s choice (N = 126) | |
Analgesics, n (%) | 226 (89.7) | 52 (41.3) |
Systemic antihistamine, n (%) | 217 (86.1) | 23 (18.3) |
Blood substitutes and perfusion solutions, n (%) | 143 (56.7) | 18 (14.3) |
Topical corticosteroids, n (%) | 136 (54.0) | 18 (14.3) |
Anti-inflammatory and antirheumatic products, n (%) | 119 (47.2) | 37 (29.4) |
Drugs for acid-related disorders, n (%) | 109 (43.3) | 46 (36.5) |
Antithrombotic drugs, n (%) | 88 (34.9) | 40 (31.7) |
Corticosteroids for systemic use, n (%) | 86 (34.1) | 37 (29.4) |
Drugs acting on the renin-angiotensin system, n (%) | 81 (32.1) | 37 (29.4) |
Psycholeptics, n (%) | 83 (32.9) | 26 (20.6) |
Lipid-modifying drugs, n (%) | 65 (25.8) | 38 (30.2) |
ITT = intention to treat.
Note: Data cut-off date of October 13, 2020.
Source: Study 202 Clinical Study Report.11
Table 16: Subsequent Anticancer Treatments (ITT Analysis Set)
Subsequent anticancer treatments | Tebentafusp (N = 252) | Investigator’s choice (N = 126) |
|---|---|---|
Systemic therapy, n (%) | 109 (43.3) | 55 (43.7) |
Chemotherapy | 26 (10.3) | 16 (12.7) |
Immunotherapy | 99 (39.3) | 39 (31.0) |
Targeted therapy | 6 (2.4) | 8 (6.3) |
Radiotherapy, n (%) | 18 (7.1) | 16 (12.7) |
Local therapy other than radiotherapy, n (%) | 15 (6.0) | 16 (12.7) |
Surgery, n (%) | 1 (0.4) | 2 (1.6) |
Other, n (%) | 2 (0.8) | 2 (1.6) |
ITT = intention to treat.
Note: Data cut-off date of October 13, 2020.
Source: Study 202 Clinical Study Report.11
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 3 for detailed efficacy data.
Survival Outcomes
Overall Survival
The coprimary end points of OS in the ITT analysis set and the RAS are summarized Table 17 and Table 18, respectively. A KM plot of OS in the ITT analysis set is shown in Figure 3.
Table 17: OS Outcomes (ITT Analysis Set)
Outcome | Study 202 first interim OS analysis (data cut-off date: October 13, 2020) | Study 202 updated OS analysis (data cut-off date: August 12, 2021) | ||
|---|---|---|---|---|
Tebentafusp | Investigator’s choice | Tebentafusp | Investigator’s choice | |
N | 252 | 126 | 252 | 126 |
Death, n (%) | 87 (34.5) | 63 (50.0) | 127 (50.4) | 79 (62.7) |
OS (months),a median (95% CI) | 21.7 (18.6 to 28.6) | 16.0 (9.7 to 18.4) | 21.7 (19.1 to 26.0) | 16.7 (11.8 to 19.3) |
Hazard ratiob (95% CI) | 0.51 (0.37 to 0.71) | 0.58 (0.44 to 0.77) | ||
P valuec | < 0.0001 | 0.0001d | ||
Median follow-up time (months),a median (95% CI) | 14.1 (12.7 to 15.6) | 22.4 (20.7 to 24.0) | ||
Survival probability,a % (95% CI) | ||||
6 months | 88.8 (84.1 to 92.2) | 78.1 (69.6 to 84.6) | 89.1 (84.5 to 92.4) | 77.6 (69.1 to 81.4) |
12 months | 73.2 (66.4 to 78.8) | 58.5 (48.3 to 67.3) | 73.0 (66.9 to 78.1) | 59.1 (49.8 to 67.3) |
24 months | 44.8 (34.9 to 54.2) | 20.3 (9.1 to 34.7) | 46.2 (38.8 to 53.3) | 23.7 (14.5 to 34.3) |
CI = confidence interval; ITT = intention to treat; OS = overall survival.
aBased on Kaplan-Meier estimate.
bHazard ratio was estimated using a Cox proportional hazards model stratified by LDH status.
cP value based on log-rank test of the Kaplan-Meier curve stratified by LDH status.
dAccording to the sponsor, the OS analysis at the data cut-off date of August 12, 2021, was informal; as such, the P value should be considered exploratory.
Source: Study 202 Clinical Study Report.11
Table 18: OS Outcomes (RAS Analysis Set)
Outcome | Study 202 first interim OS analysis (data cut-off date: October 13, 2020) | |
|---|---|---|
Tebentafusp | Investigator’s choice | |
N | 149 | 126 |
Death, n (%) | 43 (28.9) | 63 (50.0) |
OS (months),a median (95% CI) | 27.4 (20.2 to NR) | 16.0 (9.7 to 18.4) |
Hazard ratiob (95% CI) | 0.38 (0.25 to 0.56) | |
P valuec | < 0.0001 | |
Median follow-up time (months),a median (95% CI) | 15.2 (13.3 to 17.0) | |
Survival probability,a % (95% CI) | ||
6 months | 95.8 (90.8 to 98.1) | 78.1 (69.6 to 84.6) |
12 months | 82.9 (74.6 to 88.7) | 58.5 (48.3 to 67.3) |
24 months | 54.7 (42.3 to 65.5) | 20.3 (9.1 to 34.7) |
CI = confidence interval; NR = not reported; OS = overall survival; RAS = rash analysis set.
Note: The RAS included all patients randomized to tebentafusp who developed a rash in the first week after treatment and all patients randomized to the investigator’s-choice arm.
aBased on Kaplan-Meier estimate.
bHazard ratio was estimated using a Cox proportional hazards model stratified by LDH status.
cP value based on log-rank test of the Kaplan-Meier curve stratified by LDH status.
Source: Study 202 Clinical Study Report.11
The primary analysis of OS (data cut-off date of October 13, 2020) in the ITT analysis set was based on a median duration of follow-up of 14.1 (95% CI,12.7 to 15.6) months when 150 death events occurred (87 events [34.5%] in the tebentafusp arm; 63 events [50.0%] in the investigator’s-choice arm). The KM estimate for the median OS in the ITT analysis set was 21.7 (95% CI, 18.6 to 28.6) months in the tebentafusp arm and 16.0 (95% CI, 9.7 to 18.4) months in the investigator’s-choice arm, with an HR of 0.51 (95% CI, 0.37 to 0.71; P < 0.0001) in favour of tebentafusp. Results of the updated analysis (data cut-off date of August 12, 2021) were consistent with those of the primary analysis, and showed that the KM estimate for the median OS in the ITT analysis set was 21.7 (95% CI, 19.1 to 26.0) months in the tebentafusp arm and 16.7 (95% CI, 11.8 to 19.3) months in the investigator’s-choice arm, with an HR of 0.58 (95% CI, 0.44 to 0.77).
The primary analysis of OS (data cut-off date of October 13, 2020) in the RAS, which included all patients randomized to tebentafusp who developed a rash in the first week after treatment and all patients randomized to the investigator’s-choice arm, was based on a median duration of follow-up of 15.2 (95% CI, 13.3 to 17.0) months when 106 death events occurred (43 patients [28.9%] in the tebentafusp arm; 63 patients [50.0%] in the investigator’s-choice arm). The KM estimate for the median OS in the RAS was 27.4 (95% CI, 20.2 to NR) months in the tebentafusp arm and 16.0 (95% CI, 9.7 to 18.4) months in the investigator’s-choice arm, with an HR of 0.38 (95% CI, 0.25 to 0.56; P < 0.0001) in favour of tebentafusp.
Figure 3: KM Plot of OS (ITT) — First Interim Analysis (Data Cut-Off Date of October 13, 2020)
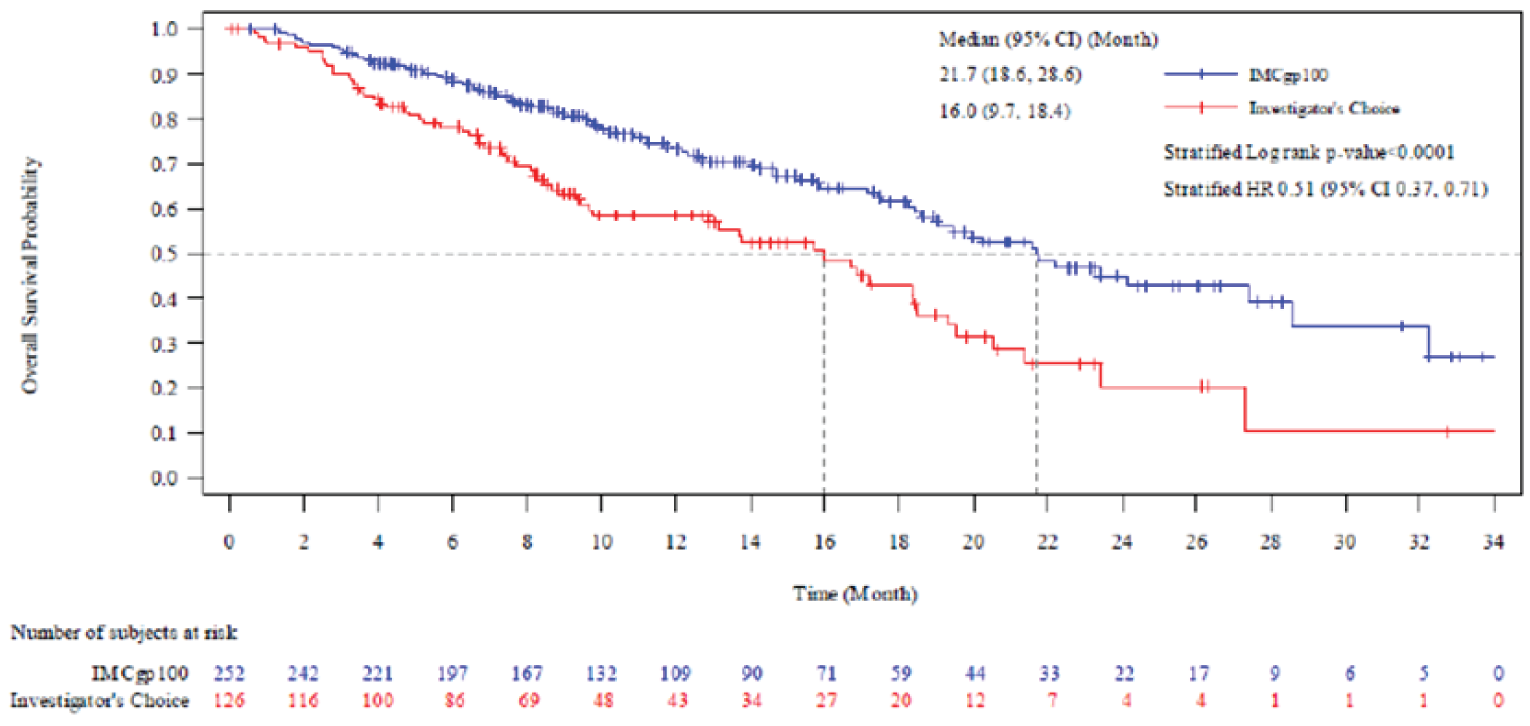
CI = confidence interval; HR = hazard ratio; KM = Kaplan-Meier; IMCgp100 = tebentafusp; ITT = intention to treat; OS = overall survival.
Source: Study 202 Clinical Study Report.11
The prespecified sensitivity analysis of OS in the ITT analysis set (data cut-off date of October 13, 2020) did not occur because the predefined criteria for conducting such an analysis (if discrepancies were more than 10% between the LDH stratum used at randomization and the central LDH stratum) was not met.
In an exploratory subgroup analysis based on ECOG PS (data cut-off date of October 13, 2020), the HR between the tebentafusp arm and the investigator’s-choice arm with respect to OS in the ITT analysis set was 0.48 (95% CI, 0.33 to 0.72) in the ECOG PS of 0 subgroup and 0.72 (95% CI, 0.39 to 1.36) in the ECOG PS of 1 subgroup (Table 35 in Appendix 3).
Progression-Free Survival
PFS was a key secondary end point, and the final PFS analysis was available at the data cut-off date of October 13, 2020 (Table 19). A KM plot of PFS is presented in Figure 4.
The final PFS analysis was conducted based on a median follow-up duration of 11.4 (95% CI, 11.1 to 16.6) months and a total of 295 PFS events (198 events [78.6%] in the tebentafusp arm; 97 events [77.0%] in the investigator’s-choice arm). The KM estimate for the median PFS was 3.3 (95% CI, 3.0 to 5.0) months in the tebentafusp arm and 2.9 (95% CI, 2.8 to 3.0) months in the investigator’s-choice arm, with an HR of 0.73 (95% CI, 0.58 to 0.94; P = 0.0139) in favour of tebentafusp.
Outcome | Study 202 final PFS analysis (data cut-off date: October 13, 2020) | |
|---|---|---|
Tebentafusp (N = 252) | Investigator’s choice (N = 126) | |
PFS event, n (%) | 198 (78.6) | 97 (77.0) |
Disease progression | 183 (72.6) | 83 (65.9) |
Death | 15 (6.0) | 14 (11.1) |
PFS (months),a median (95% CI) | 3.3 (3.0 to 5.0) | 2.9 (2.8 to 3.0) |
Hazard ratiob (95% CI) | 0.73 (0.58 to 0.94) | |
P valuec | 0.0139 | |
Median follow-up time (months), a median (95% CI) | 11.4 (11.1 to 16.6) | |
PFS probability,a % (95% CI) | ||
6 months | 30.9 (25.0 to 37.0) | 18.9 (12.0 to 27.2) |
12 months | 14.1 (9.5 to 19.5) | 6.2 (2.3 to 13.0) |
24 months | 9.2 (5.1 to 14.8) | 0.0 (NR) |
CI = confidence interval; ITT = intention to treat; NR = not reported; PFS = progression-free survival.
aBased on Kaplan-Meier estimate.
bHazard ratio was estimated using a Cox proportional hazards model stratified by LDH status.
cP value based on log-rank test of the Kaplan-Meier curve stratified by LDH status.
Source: Study 202 Clinical Study Report.11
Figure 4: KM Plot of PFS (ITT) — Final Analysis (Data Cut-Off Date of October 13, 2020)
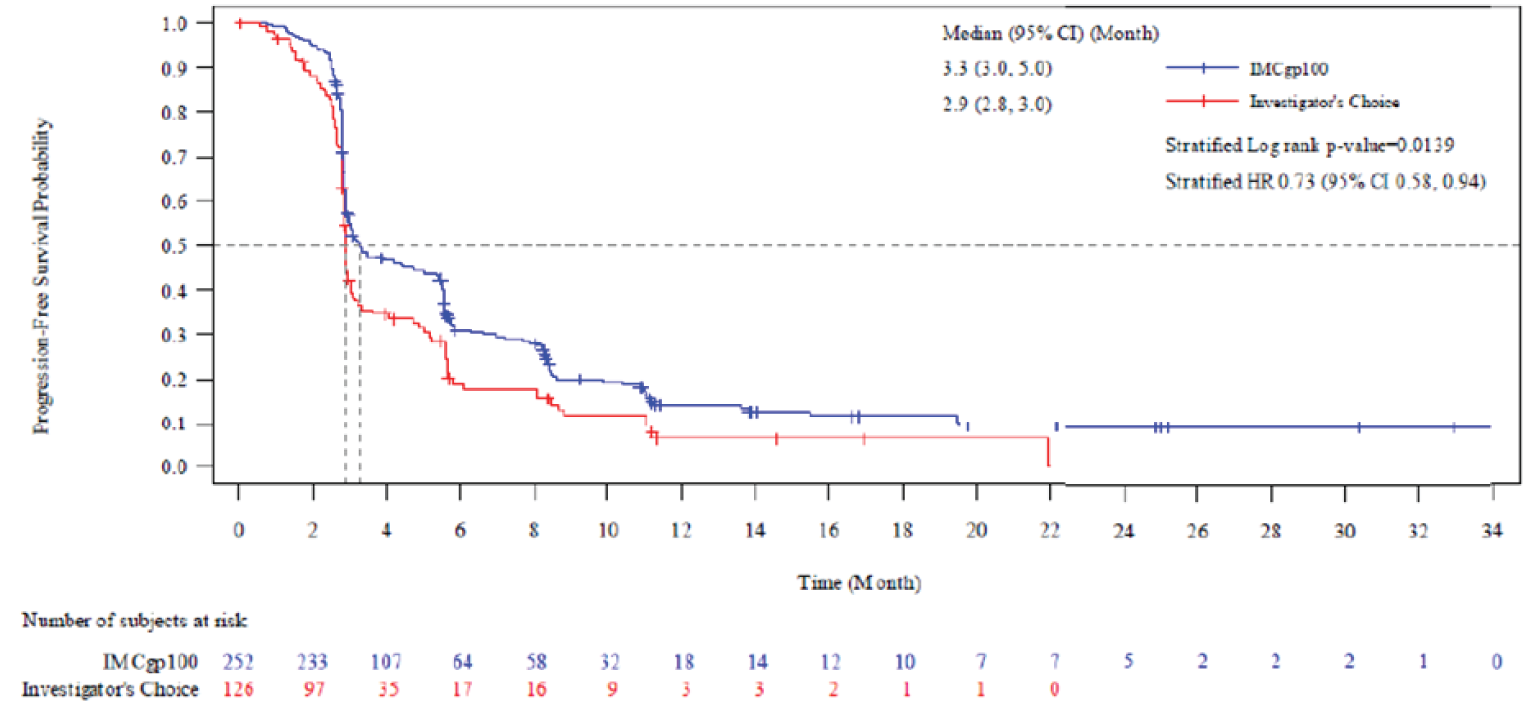
CI = confidence interval; HR = hazard ratio; IMCgp100 = tebentafusp; ITT = intention to treat.
Source: Study 202 Clinical Study Report.11
The results of 2 sensitivity analyses of PFS that assessed evaluation-time bias and attrition bias were consistent with those of the primary analysis (Table 36 in Appendix 3).
In an exploratory subgroup analysis of PFS by ECOG PS, the HR between the tebentafusp arm and the investigator’s-choice arm was 0.68 (95% CI, 0.51 to 0.91) in the ECOG PS of 0 subgroup and 0.86 (95% CI, 0.53 to 1.41) in the ECOG PS of 1 subgroup (Table 37 in Appendix 3).
Health-Related Quality of Life
EORTC QLQ-C30
EORTC QLQ-C30 score was a secondary end point. Compliance rates were above 50% through cycle 29 in the tebentafusp arm, whereas in the investigator’s-choice arm, compliance rates declined to less than 50% after cycle 17.
As of the data cut-off date of October 13, 2020, the mean global health status/QoL scale score at baseline was 76.1 (SD = 20.2) in the tebentafusp arm (n = 173) and 74.9 (SD = 20.4) in the investigator’s-choice arm (n = 65). At the end of treatment, the LS mean change in score from baseline was –10.7 (SD = NR) in the tebentafusp arm (n = 76) and –11.0 (SD = NR) in the investigator’s-choice arm (n = 34). The LS mean difference between treatment groups was 0.334 (SD = NR) (Figure 5).
The mean fatigue score at baseline was 20.9 (SD = 20.5) in the tebentafusp arm (n = 173) and 22.9 (SD = 24.6) in the investigator’s-choice arm (n = 65). The LS mean change in score from baseline trended toward deterioration in both arms, and was 10.9 (SD = NR) in the tebentafusp arm (n = 76) and 20.1 (SD = NR) in the investigator’s-choice (n = 34) arm by the end of treatment, with a LS mean difference between groups of –9.259 (SD = NR) (Figure 6).
Analyses of all other EORTC QLQ-30 function and symptom scores were generally consistent with those of the global health status/QoL scale, where the change in score from baseline was stable and similar between treatment arms at most time points.
EQ-5D-5L
EQ-5D-5L score was a secondary outcome. Compliance rates were above 50% through cycle 29 in the tebentafusp arm, whereas in the investigator’s arm, compliance rates declined to below 50% at most time points after cycle 17.
As summarized in Table 20, the mean baseline EQ VAS score was 81.0 (SD = 16.4) in the tebentafusp arm (n = 194) and 80.4 (SD = 18.3) in the investigator’s-choice arm (n = 78). As of the data cut-off date of October 13, 2020, the mean change in EQ VAS score from baseline to end of treatment was –10.1 (SD = 22.53) in the tebentafusp arm (n = 95) and –11.7 (SD = 21.40) in the investigator’s-choice arm (n = 45).
Results of the EQ-5D-5L descriptive system are summarized in Table 38 in Appendix 3. As of the data cut-off date of October 13, 2020, the proportion of patients reporting “no problems” in EQ-5D-5L domains at end of treatment in the tebentafusp arm versus the investigator’s-choice arm, respectively, was 74.8% versus 71.2% for mobility, 89.2% versus 91.5% for self-care, 63.1% versus 61.0% for usual activities, 49.5% versus 40.7% for pain or discomfort, and 55.6% versus 42.4% for anxiety or depression.
Tumour Response Outcomes
Duration of Response
The DOR (secondary end point) in patients who achieved a CR or PR is summarized in Table 21. As of the data cut-off date of August 12, 2021, the median DOR was 9.9 (95% CI, 5.6 to 22.1) months in the tebentafusp arm (n = 26) and 9.7 (95% CI, 2.7 to NR) months in the investigator’s-choice arm (n = 6).
ORR, DCR, BOR
ORR, DCR, and BOR are summarized in Table 22. As of the data cut-off date of August 12, 2021, the prespecified time point for the ORR analysis (i.e., all randomized patients had been followed for at least 3 assessments) was met. The ORR was 10.3% (95% CI, 6.9% to 14.8%) in the tebentafusp arm and 4.8% (95% CI, 1.8% to 10.1%) in the investigator’s-choice arm, corresponding to an OR of 2.26 (95% CI, 0.91 to 5.61; P = 0.684). The DCR was 45.6% (95% CI, 39.4% to 52.0%) in the tebentafusp arm and 27.0% (95% CI, 19.5% to 35.6%) in the investigator’s-choice arm, corresponding to an OR of 2.34 (95% CI, 1.45 to 3.76). In both the tebentafusp and investigator’s-choice arms, the most frequently observed BOR was disease progression (52.0% versus 64.3%), followed by stable disease (35.3% versus 22.2%), PR (9.9% versus 4.8%), and CR (0.4% versus 0%).
Figure 5: LS Mean Change From Baseline in Mean Global Health Status/QoL Scale Score Over Time (ITT Analysis Set) — Data Cut-Off Date of October 13, 2020
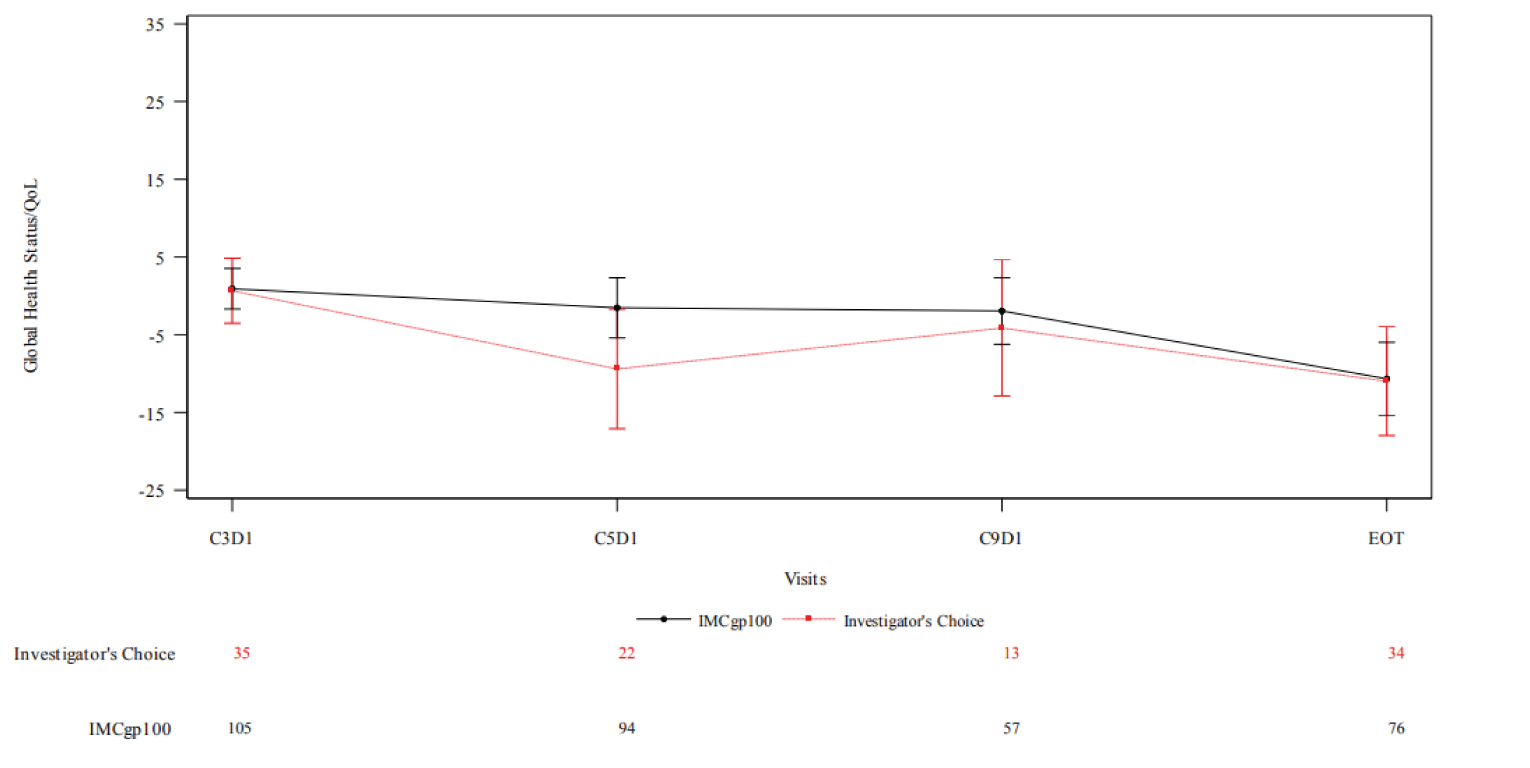
C3D1 = cycle 3 day 1; C5D1 = cycle 5 day 1; C9D1 = cycle 9 day 1; EOT = end of treatment; IMCgp100 = tebentafusp; ITT = intention to treat; LS = least squares; QoL = quality of life.
Note: The LS mean change from baseline in scale score was computed using the mixed model repeated measures approach, with treatment group (binary), time point (categorical), LDH status (binary), and baseline score (continuous) as covariates.
Source: Study 202 Clinical Study Report.11
Figure 6: LS Mean Change From Baseline in Mean Fatigue Scale Score Over Time (ITT Analysis Set) — Data Cut-Off Date of October 13, 2020
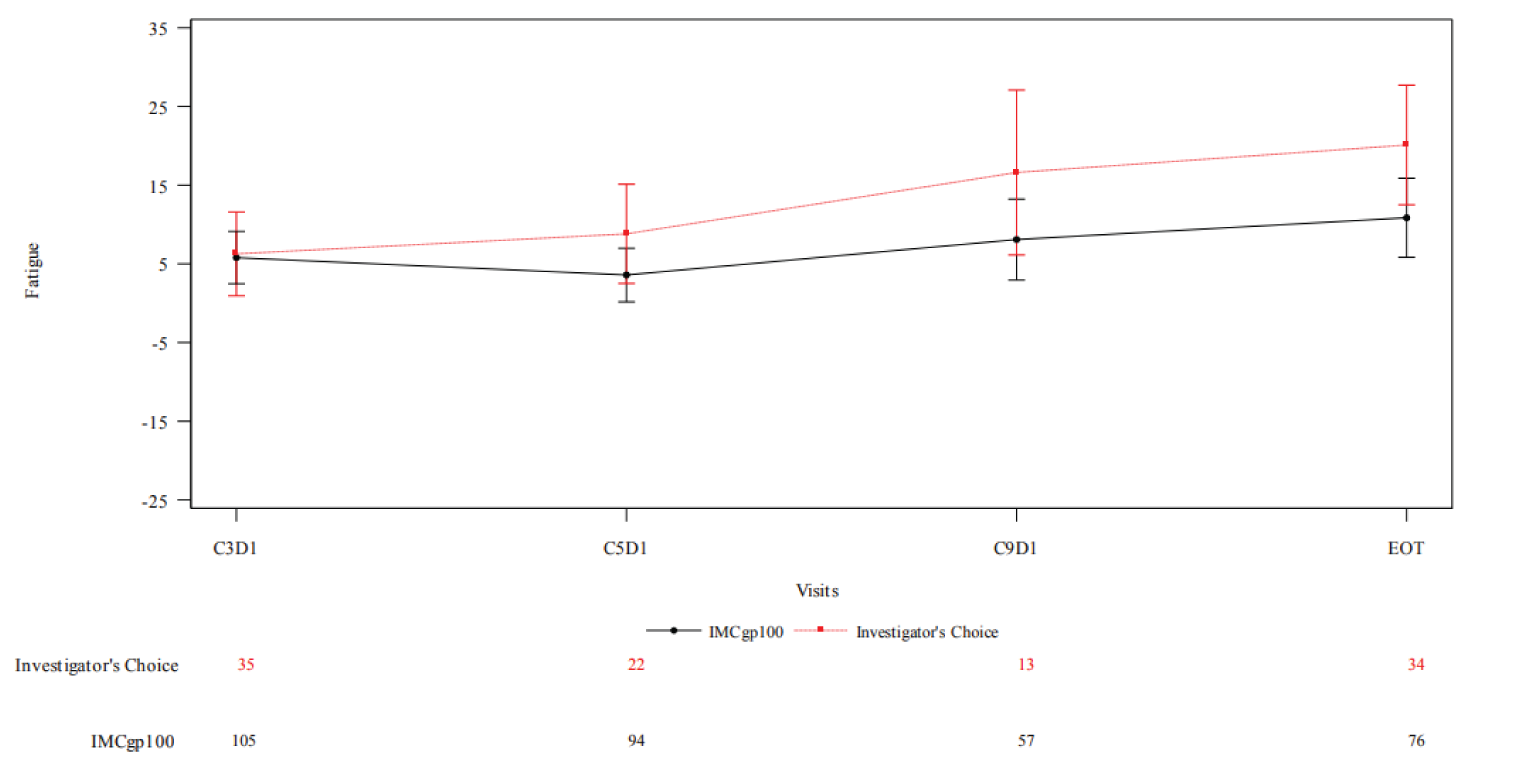
C3D1 = cycle 3 day 1; C5D1 = cycle 5 day 1; C9D1 = cycle 9 day 1; EOT = end of treatment; IMCgp100 = tebentafusp; ITT = intention to treat; LS = least squares.
Note: The LS mean change from baseline in scale score was computed using the mixed model repeated measures approach, with treatment group (binary), time point (categorical), LDH status (binary), and baseline score (continuous) as covariates.
Source: Study 202 Clinical Study Report.11
Table 20: EQ VAS Results (ITT Analysis Set)
Outcome | Study 202 (data cut-off date: October 13, 2020) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Tebentafusp | Investigator’s choice | |||||||||
Baseline | C3D1 | C9D1 | C17D1 | EOT | Baseline | C3D1 | C9D1 | C17D1 | EOT | |
N | 194 | 160 | 85 | 27 | 111 | 78 | 58 | 14 | 6 | 59 |
EQ VAS score, mean (SD) | 81.0 (16.4) | 81.9 (13.75) | 80.6 (18.43) | 78.3 (17.30) | 73.3 (19.75) | 80.4 (18.3) | 78.0 (19.55) | 82.9 (14.08) | 78.0 (29.39) | 71.3 (20.28) |
Change from baseline | ||||||||||
N | 194 | 136 | 71 | 22 | 95 | 78 | 45 | 14 | 6 | 45 |
Change from baseline in EQ VAS score, mean (SD) | Reference | 0.4 (14.69) | –0.9 (19.81) | –10.2 (20.93) | –10.1 (22.53) | Reference | –0.8 (14.28) | –3.3 (13.30) | –8.5 (33.82) | –11.7 (21.40) |
C3D1 = cycle 3 day 1; C9D1 = cycle 9 day 1; C17D1 = cycle 17 day 1; EOT = end of treatment; EQ VAS = EQ visual analogue scale; ITT = intention to treat; SD = standard deviation.
Source: Study 202 Clinical Study Report.11
Table 21: DOR (All Patients Who Achieved a CR or PR in the ITT Analysis Set)
Event | Study 202 (data cut-off date: October 13, 2020) | Study 202 (data cut-off date: August 12, 2021) | ||
|---|---|---|---|---|
Tebentafusp (N = 23) | Investigator’s choice (N = 6) | Tebentafusp (N = 26) | Investigator’s choice (N = 6) | |
Progression-free survival events, n (%) | 9 (39.1) | 4 (66.7) | 16 (61.5) | 4 (66.7) |
Progressive disease | 9 (39.1) | 4 (66.7) | 16 (61.5) | 4 (66.7) |
Death | 0 | 0 | 0 | 0 |
DORa (months), median (95% CI) | 9.9 (5.4 to NR) | 9.7 (2.7 to NR) | 9.9 (5.6 to 22.1) | 9.7 (2.7 to NR) |
Probability of neither progressive disease nor death, % (95% CI) | ||||
6 months | 60.6 (34.2 to 7.2) | 50.0 (11.1 to 80.4) | 66.9 (44.6 to 81.9) | 50.0 (11.1 to 80.4) |
12 months | 46.8 (21.8 to 68.4) | 50.0 (11.1 to 80.4) | 45.3 (24.9 to 63.7) | 50.0 (11.1 to 80.4) |
Median follow-up time (months), median (95% CI) | 10.8 (2.8 to 13.8) | 9.3 (2.8 to NR) | 16.8 (11.1 to NR) | 16.6 (9.3 to NR) |
CI = confidence interval; CR = complete response; DOR = duration of response; ITT = intention to treat, NR = not reported; PR = partial response.
Note: Only patients who achieved a CR or PR were included.
aDOR is defined as the time from the date of first documentation of PR or better to the date of first documentation of progressive disease or death from any cause, whichever comes first.
bCalculated using the reverse Kaplan-Meier method.
Source: Study 202 Clinical Study Report.11
Table 22: ORR, DCR, and BOR (ITT)
Event | Study 202 (data cut-off date: October 13, 2020) | Study 202 primary analysis (data cut-off date: August 12, 2021) | ||
|---|---|---|---|---|
Tebentafusp (N = 252) | Investigator’s choice (N = 126) | Tebentafusp (N = 252) | Investigator’s choice (N = 126) | |
ORR | ||||
Objective response, n (%) | 23 (9.1) | 6 (4.8) | 26 (10.3) | 6 (4.8) |
95% CIa | 5.9 to 13.4 | 1.8 to 10.1 | 6.9 to 14.8 | 1.8 to 10.1 |
Odds ratio (95% CI)b | 1.98 (0.79 to 4.97) | 2.26 (0.91 to 5.61) | ||
P value | NR | 0.0684 | ||
DCR | ||||
Disease controlled, n (%) | 115 (45.6) | 34 (27.0) | 115 (45.6) | 34 (27.0) |
95% CIa | 39.4 to 52.0 | 19.5 to 35.6 | 39.4 to 52.0 | 19.5 to 35.6 |
Odds ratio (95% CI)b | 2.33 (1.45 to 3.75) | 2.34 (1.45 to 3.76) | ||
P valuec | NR | 0.0004 | ||
BOR | ||||
Complete response, n (%) | 1 (0.4) | 0 | 1 (0.4) | 0 |
Partial response, n (%) | 22 (8.7) | 6 (4.8) | 25 (9.9) | 6 (4.8) |
Stable diseased, n (%) | 92 (36.5) | 28 (22.2) | 89 (35.3) | 28 (22.2) |
Disease progression, n (%) | 131 (52.0) | 78 (61.9) | 131 (52.0) | 81 (64.3) |
Not evaluable, n (%) | 6 (2.4) | 14 (11.1) | 6 (2.4) | 11 (8.7) |
BOR = best overall response; CI = confidence interval; DCR = disease control rate; ITT = intention to treat; NR = not reported; ORR = objective response rate.
a95% CIs were calculated using the exact Clopper-Pearson method.
bOdds ratio was calculated using the Cochran-Mantel-Haenszel test, stratified by LDH status.
cNot controlled for multiplicity.
dStable disease was classified based on assessments made at least 77 days after the date of randomization.
Source: Study 202 Clinical Study Report.11
Posthoc OS Analysis in Patients With a BOR of Progressive Disease
A posthoc analysis of landmark OS on day 100 was conducted in patients who had a BOR of disease progression to understand the role of treatment beyond progression. The proportion of patients who were alive and had a best response of progressive disease by day 100 was 41.7% in the tebentafusp arm and 42.1% in the investigator’s-choice arm. In these patients, the HR of OS between the tebentafusp arm and the investigator’s-choice arm was 0.43 (95% CI, 0.27 to 0.68). A postlandmark KM plot of OS in patients with a BOR of progressive disease is shown in Figure 10 in Appendix 3.
Harms
Only harms identified in the review protocol are reported here. Refer to Table 23 for detailed harms data as of the data cut-off date of October 13, 2020.
Adverse Events
All patients in the tebentafusp arm and 94.6% of patients in the investigator’s-choice arm reported at least 1 TEAE. TEAEs of grade 3 or higher occurred in 54.3% of the tebentafusp arm and 36.0% of the investigator’s-choice arm. The most common TEAEs of any grade were (tebentafusp versus investigator’s choice) pyrexia (76% versus 7.2%), pruritus (69.0% versus 23.4%), rash (55.1% versus 16.2%), and fatigue (51.0% versus 35.1%).
Serious Adverse Events
Serious TEAEs were reported in 28.2% of patients in the tebentafusp arm and 23.1% of patients in the investigator’s-choice arm. The most common serious TEAE was CRS, reported in 9.8% of patients in the tebentafusp arm and no patients in the investigator’s-choice arm.
Withdrawal Due to Adverse Events
The proportion of patients who discontinued treatment due to TEAEs was 3.3% in the tebentafusp arm and 6.6% the investigator’s-choice arm. No specific AE was identified to account for the majority of treatment discontinuations.
Mortality
Eighty-four deaths (34.3%) and 57 deaths (51.4%) were reported in the tebentafusp arm and the investigator’s-choice arm, respectively. The majority of deaths in the tebentafusp and investigator’s-choice arms were attributed to disease progression (32.7% versus 46.8%). No specific AE was identified to account for the majority of deaths in either arm.
Notable Harms
The proportion of patients who reported CRS (or presentations related to CRS) and dermatological AEs was notably higher in the tebentafusp arm than in the investigator’s-choice arm, with the most common notable harms (30% or greater) being pyrexia, pruritus, rash, fatigue, nausea, chills, hypotension, dry skin, headache, and maculopapular rash. The most commonly reported notable harms of grade 3 or higher in the tebentafusp arm were rash (9.4%), maculopapular rash (8.6%), fatigue (5.3%), and pruritus (4.5%).
Table 23: Summary of Harms (Safety Analysis Set)
Harms | Tebentafusp (N = 245) | Investigator’s choice (N = 111) |
|---|---|---|
Patients with ≥ 1 TEAEa | ||
n (%) | 245 (100.0) | 105 (94.6) |
Most common events,b n (%) | ||
Pyrexia | 187 (76.3) | 8 (7.2) |
Pruritus | 169 (69.0) | 26 (23.4) |
Rash | 135 (55.1) | 18 (16.2) |
Fatigue | 125 (51.0) | 39 (35.1) |
Nausea | 120 (49.0) | 29 (26.1) |
Chills | 117 (47.8) | 4 (3.6) |
Hypotension | 95 (38.8) | 3 (2.7) |
Dry skin | 77 (31.4) | 4 (3.6) |
Headache | 75 (30.6) | 11 (9.9) |
Maculopapular rash | 75 (30.6) | 9 (8.1) |
Vomiting | 73 (29.8) | 10 (9.0) |
Peripheral edema | 66 (26.9) | 3 (2.7) |
Diarrhea | 61 (24.9) | 22 (19.8) |
Abdominal pain | 60 (24.5) | 17 (15.3) |
Erythema | 60 (24.5) | 1 (0.9) |
AST increase | 56 (22.9) | 11 (9.9) |
Arthralgia | 53 (21.6) | 18 (16.2) |
ALT increase | 51 (20.8) | 12 (10.8) |
Cytokine release syndromec | 51 (20.8) | 0 |
Skin exfoliation | 51 (20.8) | 2 (1.8) |
Upper abdominal pain | 50 (20.4) | 14 (12.6) |
Patients with ≥ 1 grade 3 or higher TEAE | ||
n (%) | 133 (54.3) | 0 (36.0) |
Most common events,d n (%) | ||
Rash | 23 (9.4) | 0 |
Maculopapular rash | 21 (8.6) | 0 |
Hypertension | 21 (8.6) | 3 (2.7) |
Fatigue | 13 (5.3) | 1 (0.9) |
AST increase | 13 (5.3) | 1 (0.9) |
Lipase increase | 10 (4.1) | 6 (5.4) |
Patients with ≥ 1 serious TEAE | ||
n (%) | 69 (28.2) | 26 (23.4) |
Most common events,e n (%) | ||
Cytokine release syndromec | 24 (9.8) | 0 |
Pulmonary embolism | 1 (0.4) | 3 (2.7) |
Abdominal pain | 2 (0.8) | 3 (2.7) |
Hyperbilirubinemia | 2 (0.8) | 3 (2.7) |
Rash | 6 (2.4) | 0 |
Pyrexia | 6 (2.4) | 2 (1.8) |
Hypotension | 5 (2.0) | 0 |
Patients who discontinued treatment due to TEAEa | ||
n (%) | 8 (3.3) | 7 (6.3) |
Deaths | ||
n (%) | 84 (34.3) | 57 (51.4) |
Death due to progressive disease, n (%) | 80 (32.7) | 52 (46.8) |
Death due to AE, n (%) | 2 (0.8) | 3 (2.7) |
Death due to subdural hematoma, n (%) | 0 | 1 (0.9) |
Death due to unknown reason, n (%) | 2 (0.8) | 1 (0.9) |
Notable harms | ||
Pyrexia, n (%) | 187 (76.3) | 8 (7.2) |
Pruritus, n (%) | 169 (69.0) | 26 (23.4) |
Rash, n (%) | 135 (55.1) | 18 (16.2) |
Fatigue, n (%) | 125 (51.0) | 39 (35.1) |
Nausea, n (%) | 120 (49.0) | 29 (26.1) |
Chills, n (%) | 117 (47.8) | 4 (3.6) |
Hypotension, n (%) | 95 (38.8) | 3 (2.7) |
Dry skin, n (%) | 77 (31.4) | 4 (3.6) |
Headache, n (%) | 75 (30.6) | 11 (9.9) |
Maculopapular rash, n (%) | 75 (30.6) | 9 (8.1) |
Vomiting, n (%) | 73 (29.8) | 10 (9.0) |
Diarrhea, n (%) | 61 (24.9) | 22 (19.8) |
Erythema, n (%) | 60 (24.5) | 1 (0.9) |
Cytokine release syndrome,c n (%) | 51 (20.8) | 0 |
Skin exfoliation, n (%) | 51 (20.8) | 2 (1.8) |
Hair colour changes, n (%) | 48 (19.6) | 0 |
Vitiligo, n (%) | 40 (16.3) | 4 (3.6) |
Hypoxia, n (%) | 4 (1.6) | 0 |
Notable harms, grade 3 or higher in ≥ 1% of patients | ||
Rash, n (%) | 23 (9.4) | 0 |
Maculopapular rash, n (%) | 21 (8.6) | 0 |
Fatigue, n (%) | 13 (5.3) | 0 |
Pruritus, n (%) | 11 (4.5) | 0 |
Pyrexia, n (%) | 9 (3.7) | 1 (0.9) |
Hypotension, n (%) | 8 (3.3) | 0 |
Nausea, n (%) | 5 (2.0) | 1 (0.9) |
Vomiting, n (%) | 3 (1.2) | 0 |
Diarrhea, n (%) | 3 (1.2) | 3 (2.7) |
AE = adverse event; ALT = alanine transaminase; AST = aspartate transaminase; TEAE = treatment-emergent adverse event.
Note: Data cut-off date of October 13, 2020.
aTEAE was defined as an AE that had an onset date on or after the date of first dose or pre-treatment TEAEs that increase in severity on or after the date of first dose up to and including 90 days after the date of last dose of the study drug.
bFrequency ≥ 20%.
cCRS was reported by the investigator based on Lee et al. (2014)12 grading criteria.
dFrequency ≥ 5%.
eFrequency ≥ 2%.
Source: Study 202 Clinical Study Report.11
Critical Appraisal
Internal Validity
Study 202 was a phase III, open-label, randomized controlled trial (RCT). The methods of randomization, which involved stratification by LDH level and the use of an Interactive Response Technology system for randomized assignment, were considered appropriate. There was generally no notable imbalance in baseline patient characteristics between treatment groups, which suggests that randomization was likely successful. A notably higher proportion of patients received concomitant analgesics, systemic antihistamines, blood substitutes and perfusion solutions, topical corticosteroids, and anti-inflammatory and antirheumatic products in the tebentafusp arm than the investigator’s-choice arm. The clinical expert noted this to be reasonable, as these medications are routinely used for the management of CRS and rash associated with tebentafusp and were not expected to confound the study results.
The CADTH review team and the clinical expert considered the open-label design to be reasonable, given that the distinct dosing regimens and toxicity profiles of the interventions could likely allow investigators and patients to make inferences about treatment assignment regardless of blinding. It is unlikely that the reporting of OS was influenced by knowledge of treatment assignment, considering the objective nature of this outcome; however, subjective outcomes reported by patients, such as HRQoL and AEs, might be subject to reporting bias. In addition, tumour response outcomes (PFS, ORR, BOR, DCR, DOR) were based on investigator assessment; no independent blinding committee was involved in the assessment process to help mitigate the biases associated with the open-label study design. Therefore, there is a risk of bias associated with the measurement of these outcomes, although the extent and direction of bias are unclear.
The primary analysis presented constitutes the first interim analysis of OS and the final analysis of PFS, which were conducted as planned a priori based on the number of OS or PFS events that occurred. Stopping rules for interim analyses of OS were prespecified in the protocol and alpha levels were properly accounted for to preserve power. A graphical approach involving sequential testing of the coprimary end point of OS in the RAS and ITT populations, and key secondary end points (PFS and BOR [evaluated statistically as ORR]), was appropriately used to account for multiplicity. No formal hypothesis testing was carried out for other secondary outcomes (DCR, DOR, HRQoL), so these results are considered exploratory. There is an increased risk of false-positive conclusions for outcomes that were not controlled for multiplicity. No conclusion could be drawn regarding the subgroup effects of ECOG PS on OS, considering the subgroup analysis was exploratory in nature, and there was a lack of sample-size consideration and control for multiplicity.
With respect to the Cox proportional hazards model used for OS and PFS analyses, no assessment of the proportional hazards assumption was reported; therefore, it is unknown whether there were any violations of the assumption or whether adjustments were involved to address any potential violations. The censoring rules and the method of handling missing data used were, overall, considered appropriate.
There are 2 limitations to note with respect to the OS analyses. First, there is typically a risk of overestimating the treatment effect in favour of the experimental intervention (i.e., tebentafusp) in an interim analysis.13 Considering that the interim OS analyses were based on a relatively small number of events, the OS results are prone to imprecision. Second, the results of the OS analysis of the RAS and the posthoc exploratory landmark OS analysis in patients with a BOR of progressive disease should be interpreted with caution, given that no randomization was involved in the comparison. The results were subject to confounding by the potential differences in prognostic factors and treatment-effect modifiers between the treatment arms; however, the direction of bias is inconclusive.
It should also be noted that 16 patients in the investigator’s-choice arm were allowed to cross over to the tebentafusp arm between the data cut-off dates of October 13, 2020, and August 12, 2021. No adjustments were made to account for the crossover in the informal updated analysis of OS or the primary BOR (evaluated statistically as ORR) analysis (data cut-off date of August 12, 2021), according to the sponsor. The treatment crossover might have biased the findings in favour of the investigator’s-choice arm, rather than the tebentafusp arm, and therefore is not expected to have had a major impact on interpretation of OS findings in this study, specifically with respect to the direction of OS benefits.
The EORTC QLQ-C30 and EQ-5D-5L scales used for HRQoL assessment in this study are commonly used in oncology trials, although the validity, reliability, and responsiveness of these instruments have not been studied in patients with UM. There are also uncertainties about the HRQoL outcomes because of the large amount of missing data as a result of treatment discontinuation and poor questionnaire compliance rates (above 50% in the tebentafusp arm throughout the study but declined to less than 50% in the investigator’s-choice arm at most time points after cycle 17). There is a risk that reporting bias results from patients who remained in the trial affected the interpretability of HRQoL trends over time. Also, no data imputation was performed to account for the missing data.
External Validity
The clinical expert noted that the inclusion and exclusion criteria of Study 202 were generally reflective of the eligibility criteria in clinical practice for systemic treatments; however, patients with prior therapy in the metastatic setting were not captured in the study. This was a limitation, and the clinical expert noted that tebentafusp should not only be offered in the first-line setting, but also in the second-line setting and beyond, based on clinical trial and anecdotal evidence. The clinical expert also indicated that clinical practice is usually less restrictive than clinical trials with respect to ECOG PS and comorbidities. The baseline characteristics of enrolled patients were generally similar to the Canadian patient population, per the clinical expert.
The dosing of tebentafusp in Study 202 was consistent with the recommended dosing in the product monograph. Of note, although the product monograph recommended continuation of treatment until disease progression or unacceptable toxicity, the clinical expert explained that continuation of tebentafusp beyond initial progression until no clinical benefits are observed is more appropriate in clinical practice. The dosing approach of tebentafusp in Study 202 was consistent with the approach used in clinical practice and, therefore, would not undermine the external validity of the results.
In the investigator’s-choice arm, patients were assigned to pembrolizumab (81.7%), ipilimumab (12.7%), or chemotherapy (5.6%). According to the clinical expert, ipilimumab and chemotherapy are not the standard of care for patients with mUM who are naive to systemic treatment. Further, the clinical expert indicated that pembrolizumab only accounts for approximately 10% of systemic treatments prescribed in the first-line setting. The clinical expert indicated that ipilimumab plus nivolumab is currently used in most patients with mUM receiving systemic treatments. However, this regimen was not included in the comparator arm. The clinical expert commented that the potential impact on generalizability is likely to be small, noting that a retrospective study showed no significant difference in response rates between single-drug and combination immunotherapy regimens,38 although the study results might be subject to bias, owing to the small sample size and retrospective design.
OS is an important clinical outcome, according to clinician and patient input. The clinical expert noted that given the data available, the OS benefits of tebentafusp were considered clinically meaningful, although a longer follow-up (2 to 3 more years) would help provide further confidence in the OS benefits of tebentafusp. PFS and tumour response outcomes were noted to be less clinically important by the clinical expert, as tumour response is poorly correlated with OS in patients with mUM who receive systemic therapy in general. Further, the PFS benefit of tebentafusp was small in Study 202, with a median extension in PFS of 0.4 months compared with investigator’s choice. Improvement in HRQoL was highly valued by patients with mUM. The clinical expert noted that the EORTC QLQ-C-30 and EQ-5D-5L scales are not routinely used in clinical practice, so the generalizability of HRQoL findings in Study 202 could not be ascertained. It is worth noting that these HRQoL instruments capture some of the most common HRQoL impacts reported by patients, including fear, anxiety, depression, fatigue, and decline in physical and psychological functioning; symptoms such as loss of vision and balance were not assessed. Prevention of vision loss was noted to be an important outcome by the patient group; however, this outcome was not assessed in the study. The clinical expert noted that prevention of vision loss is not an expected outcome of tebentafusp.
The product monograph recommends that the first 3 doses of tebentafusp be administered in a health care setting that would allow a patient to be monitored during the infusion and for 16 hours after the infusion is complete, whereas subsequent doses can be administered in an ambulatory setting in the absence grade 2 or worse hypotension, which reflects the treatment settings in Study 202. The clinical expert indicated that the safety findings of tebentafusp are generalizable to clinical practice, provided tebentafusp is administered in appropriate treatment settings, as specified in the product monograph, and noted that, based on clinical experience, CRS generally occurs after the first 3 or 4 infusions and is generally manageable if proper supportive care is provided.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Because of a lack of studies directly comparing tebentafusp with systemic treatments other than pembrolizumab, ipilimumab, and dacarbazine monotherapies for mUM, a review of indirect evidence was undertaken.
A focused literature search for ITCs dealing with UM was run in MEDLINE All (1946–) on May 18, 2022. No limits were applied to the search. Three published ITCs were identified, none of which met the selection criteria in the review protocol (Table 6). One sponsor-submitted ITC was included in this review. The sponsor also submitted a supplementary analysis of the prognostic value of HLA-A*02:01 status for OS to support the MAIC analyses.
Description of Indirect Comparisons
The sponsor submitted 1 report that included 3 MAIC analyses of survival results in patients with previously untreated mUM who received ipilimumab plus nivolumab versus tebentafusp, versus pembrolizumab, and versus the investigator’s-choice arm in Study 202. The comparison between tebentafusp and ipilimumab plus nivolumab is of interest for this review and will be addressed in this report.
Methods of Sponsor-Submitted ITC
Objectives
The primary objective of the sponsor-submitted ITC was to compare OS between tebentafusp and ipilimumab plus nivolumab in patients with previously untreated mUM.
Study Selection Methods
To identify the clinical evidence for tebentafusp and relevant comparator interventions, an SLR was conducted using 7 electronic databases and 2 trial registries, along with handsearches of review articles, reference lists of included full-text publications, and free-text keyword searches in internet search engines. The selection criteria of the SLR are summarized in Table 24. Of note, the SLR that was originally designed to fulfill regulatory requirements in Europe and the inclusion criteria were wider than intended for the ITC, specifically the cutaneous melanoma population, and outcomes such as ORR, DCR, AEs, and QoL were not of interest for the sponsor-submitted ITC. It was inferred that the selection criteria could have been refined to align with the objectives of the analysis, although no details were stated in the sponsor-submitted ITC report.
Table 24: Study Selection Criteria and Methods for the Sponsor-Submitted Systematic Review for ITC
Criteria | Sponsor-submitted systematic review for ITC |
|---|---|
Population | Adults with advanced or metastatic CMa or UM |
Intervention | Tebentafusp |
Comparator | All other therapeutic interventions for CMa or UM
|
Outcome |
|
Study design |
|
Exclusion criteria |
|
Databases searched | EMBASE, MEDLINE, CENTRAL, WHO ICTRP, CDSR, DARE, HTAD, Epistemonikos, Clinicaltrials.gov up to March 2020 (updated search in September 2021) |
Selection process | Articles screened independently by 2 reviewers with a third reviewer to resolve discrepancies |
Data extraction process | Single reviewer extraction with quality check by a second reviewer |
Quality assessment | Cochrane RoB 2.0 tool was used for RCTs (single reviewer) Cochrane ROBINS-I tool was used for single-arm trials (single reviewer) |
CDSR = Cochrane Database of Systematic Reviews; CENTRAL = Cochrane Central Register of Controlled Trials; CM = cutaneous melanoma; DARE = Database of Abstracts of Reviews of Effects; HTAD = Health Technology Assessment Database; ITC = indirect treatment comparison; RCT = randomized controlled trial; UM = uveal melanoma; WHO ICTRP = WHO International Clinical Trials Registry.
aThe SLR protocol was initially designed to fulfill regulatory requirements and included patients with advanced or metastatic CM and outcomes including overall response rate, disease control rate, adverse events, and quality of life. These elements, however, were not of interest for the sponsor-submitted ITC.
Source: Sponsor-submitted SLR for ITC.39
The criteria included studies published before March 2020 and the search was updated September 2021. Article screening was performed independently by 2 reviewers at 2 stages (title and abstracts, and then full papers), and a third reviewer helped resolve disagreements. Reasons for exclusion were documented. Data extraction was performed by 1 reviewer and quality checked by a second reviewer.
Quality assessment of the selected studies was conducted by 1 reviewer to assess the risk of bias using the Cochrane RoB 2.0 tool for RCTs, and Cochrane ROBINS-I tool for single-arm trials
ITC Analysis Methods
A summary of the analysis methods for the MAIC is shown in Table 25. An unanchored MAIC approach was selected for the indirect comparison between tebentafusp and ipilimumab plus nivolumab, owing to the lack of a common comparator. The covariates used for match adjustment were informed by a multivariate analysis of OS in Study 202. Adaptations were applied to the covariates to account for the limitations of available data and to allow matching to be implemented. Note that time since primary diagnosis was an identified covariate but could not be used in matching as it was not reported in the GEM-1402 trial. IPD from Study 202 were then weighted to match baseline characteristics in the comparator trial (GEM-1402) using weights estimated with the method of moments described in the Signorovitch et al. (2012)40,41 studies, and weights were examined for extreme values. The ESS was calculated using the methods described in the Pillippo et al. (2016)42 study. The covariate characteristics of patients before and after weighting were presented. A weighted Cox proportional hazards model was fit, with the MAIC weights applied to produce the efficacy end points.
Results of an unadjusted indirect comparison (UAIC) and MAIC were presented.
The sponsor noted that potential issues with ESS calculations and modelling could arise because of the notably smaller proportion of patients with extrahepatic disease only in Study 202 than in the GEM-1402 trial (3.6% versus 21.2%); as such, 2 exploratory sensitivity analyses were conducted to assess the impact of using alternative definitions for the disease location covariate for matching.
Note that OS was the prespecified outcome of interest in the MAIC analyses, and the PFS analysis was added subsequent to observing the results for OS.
Table 25: ITC Analysis Methods
Variable | Sponsor-submitted MAIC |
|---|---|
ITC methods | Unanchored MAIC |
Covariates used for match adjustment |
|
Outcomes | Overall survival Progression-free survival (exploratory) |
Sensitivity analyses | 2 sensitivity analyses:
|
Subgroup analysis | None |
ALP = alkaline phosphatase; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITC = indirect treatment comparison; LDH = lactate dehydrogenase; MAIC = matching-adjusted indirect comparison.
Source: Sponsor-submitted ITC Technical Report.14
Results of Sponsor-Submitted ITC
Summary of Included Studies
After removal of duplicates, 1,531 citations were screened by title and abstract; of these, 336 citations were reviewed by full-text screening and eventually 60 studies met the selection criteria. Two published studies of ipilimumab plus nivolumab were identified —Pelster et al. (2021)43 and GEM-140215 — both of which were single-arm studies of patients with mUM. Study 20210 (data cut-off date of October 13, 2020) was the index trial, which contained IPD for the tebentafusp group.
The GEM-1402 trial was selected by the sponsor as the comparator study for the MAIC because the trial had a larger sample size and reported more key covariates for matching than the Pelster et al. (2021) study. In addition, the sponsor noted that the GEM-1402 trial was a multicentre study that contained treatment-naive patients only, similar to Study 202, whereas the Pelster et al. (2021) study was a single-centre study in which only 57% of patients were treatment-naive. The characteristics of Study 202 and the GEM-1402 trial are summarized in Table 26. The risk of bias (per ROBINS-I appraisal by a single reviewer) in the GEM-1402 trial was noted to be low in the sponsor’s assessment.
Results
Patient characteristics from Study 202 (unadjusted and match-adjusted) and the GEM-1402 trial are summarized in Table 27. The ESS for tebentafusp after match adjustment was 115.93 (48.9% of the original sample size from Study 202).
Table 26: Summary of Included Studies in the MAIC
Study details | Study 20210 (index trial) | GEM-140215 (comparator trial) |
|---|---|---|
Study design | Phase III, multicentre, open-label, RCT | Phase II, multicentre, open-label, single-arm |
Sample size | 379 (252 in tebentafusp arm) | 52 |
Population | HLA-A*02:01-positive adults with unresectable UM or mUM who had received no prior therapy in the metastatic setting
| Adults with mUM who were systemic treatment-naive
|
Intervention | Tebentafusp IV infusion 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15, and 68 mcg once every week thereafter | Nivolumab (1 mg/kg once every 3 weeks) and ipilimumab (3 mg/kg once every 3 weeks) for 4 doses, followed by nivolumab (3 mg/kg once every 2 weeks) |
Comparison | Dacarbazine, ipilimumab, or pembrolizumab | None |
Outcomes of interest | ||
OS | Median OS (IPD available): Definition of OS: time from randomization to death from any cause, with those still alive censored at last date of known alive status | Median OS: Definition of OS: time from first dose to death from any cause, with those still alive censored at date of last reported contact |
PFS | Median PFS (IPD available): Definition of PFS: time from randomization to the date of first documented progression (based on investigator-assessed RECIST 1.1) or death from any cause, whichever occurred first | Median PFS: Definition of PFS: time from the first nivolumab dose to progressive disease (based on investigator-assessed RECIST 1.1) or death from any cause |
Median follow-up | 14.1 months (data cut-off date: October 13, 2020) | 13.4 months (data cut-off date: July 9, 2019) |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; HLA = human leukocyte antigen; IPD = individual patient data; mUM = metastatic uveal melanoma; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; RECIST = Response Evaluation Criteria in Solid Tumours; UM = uveal melanoma.
Sources: Study 202 Clinical Study Report,11 Piulats et al. (2020).15
Table 27: Summary of Model Covariates in Unadjusted Indirect Comparison and MAIC
Characteristic | Tebentafuspa | Ipilimumab + nivolumab | |
|---|---|---|---|
Study 202 unadjusted | Study 202 match-adjusted | GEM-1402 | |
N (or ESS) | 237 | (115.93) | 52 |
Median age (years) | 63.0 | 59.7 | 59.1 |
Female, n (%)b | 118 (49.2) | 80.8 (44.2) | 23 (44.2) |
Normal LDH level, n (%)b | 156 (43.0) | 114.6 (62.8) | 27 (62.8) |
Normal ALP level, n (%)b | 189 (78.7) | 155.4 (85.1) | 40 (85.1) |
Extrahepatic disease only, n (%)b | 9 (3.8) | 38.6 (21.2) | 11 (21.2) |
Hepatic and extrahepatic disease, n (%)b | 106 (44.2) | 66.7 (36.5) | 19 (36.5) |
ECOG PS of 0, n (%)b | 191 (79.6) | 154.5 (84.6) | 44 (84.6) |
ALP = alkaline phosphatase; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ESS = effective sample size; LDH = lactate dehydrogenase.
Note: Study 202 data cut-off date: October 13, 2020; GEM-1402 data cut-off date: July 9, 2019).
aMissing values for any variables were excluded from the analysis.
bMissing data in the GEM-1402 trial were excluded from the calculation of proportions.
Source: Sponsor-submitted Indirect Treatment Comparison Technical Report.14
UAIC and MAIC (primary analysis) of OS and PFS between tebentafusp and ipilimumab plus nivolumab are summarized in Table 28. The KM curves of OS and PFS from MAIC analyses are shown in Figure 7 and Figure 8, respectively.
The MAIC analysis between tebentafusp and ipilimumab plus nivolumab showed results in favour of tebentafusp with respect to OS (HR, 0.507; 95% CI, 0.324 to 0.793) and PFS (HR, 0.647; 95% CI, 0.445 to 0.941). Results of the sensitivity analyses, which assessed the impact of using alternative definitions for the disease location covariate for matching, were consistent with the primary analyses (Table 39).
Table 28: OS and PFS Results in Unadjusted Indirect Comparison and MAIC (Primary Analysis)
Event | Tebentafusp | Ipilimumab plus nivolumab | |
|---|---|---|---|
Study 202 unadjusted | Study 202 match-adjusted | GEM-1402 | |
OS | |||
N (or ESS) | 237 | (115.93) | 52 |
Death event, n (%) | 82 (34.6) | 61.4 (53.0) | 39 (75.0) |
OS (months), median | 21.7 | 21.6 | 12.1 |
12-month OS probability (%) | 74.7 | 78.6 | 51.2 |
Hazard ratio (95% CI) vs. ipilimumab + nivolumab, robust SE | 0.514 (0.35 to 0.756) | 0.507 (0.324 to 0.793) | Reference |
PFS | |||
N (or ESS) | 237 | (115.93) | 52 |
PFS event, n | 190 | 139.1 | 51 |
PFS (months), median | 3.3 | 4.8 | 3.1 |
12-month PFS probability (%) | 14.7 | 16.5 | 15.4 |
Hazard ratio (95% CI) vs. ipilimumab + nivolumab, robust SE | 0.717 (0.525 to 0.978) | 0.647 (0.445 to 0.941) | Reference |
CI = confidence interval; ESS = effective sample size; OS = overall survival; PFS = progression-free survival; SE = standard error.
Source: Sponsor-submitted Indirect Treatment Comparison Technical Report.14
Sponsor-Submitted Supplementary HLA Analysis
The sponsor submitted a supplementary analysis (N = 80) assessing the comparative OS in patients with mUM who were HLA-A*02:01-positive (n = 43) versus patients with mUM who were HLA-A*02:01-negative (n = 37).16 The OS data were sourced from a clinical dataset published in the Liu et al. (2018)44 study. With respect to statistical analysis, the sponsor stated that “Survival analysis was carried out using R package survminer v0.4.9 and Cox log-rank test was used to assess differences between the survival curves. Univariate Cox proportional hazards methods (R package survival version 3.2-11) were used to model the prognostic importance of potential predictors of survival. Tests for correlation (2-sided) were conducted using R stats package 4.1.” The median OS was 45.9 months (range, NR) in HLA-A*02:01-positive patients and 45.2 months (range, NR) in HLA-A*02:01-negative patients, with an HR of 0.82 (95% CI, 0.36 to 1.88). The OS curves of HLA-A*02:01-positive and HLA-A*02:01-negative patients with mUM are shown in Figure 12 in Appendix 3.
Critical Appraisal of Sponsor-Submitted ITC
The MAIC was conducted based on studies identified by an SLR. The selection criteria of the SLR were broad and, as a result, many studies that were not of interest for the MAIC were identified in the search. It appeared that the selection process was refined to select studies that align with the MAIC study objectives, but there is a lack of transparency on how the process was carried out. There is potential for selection bias because the criteria for selecting the comparator trial was not decided a priori. The search approach was comprehensive, involving multiple electronic databases, clinical trial registries, and a supplementary manual search. The study selection and data extraction processes were considered appropriate, involving at least 2 independent reviewers. Reasons for study exclusion were reported. A quality assessment was performed for all included trials. Because the risk of bias appraisals were performed by a single reviewer without verification or consensus, there is an increased risk of error and bias in the appraisals. The comparator trial was noted to have a low risk of bias, based on the assessment by the sponsor.
Figure 7: KM Plot of OS From MAIC (Primary Analysis)
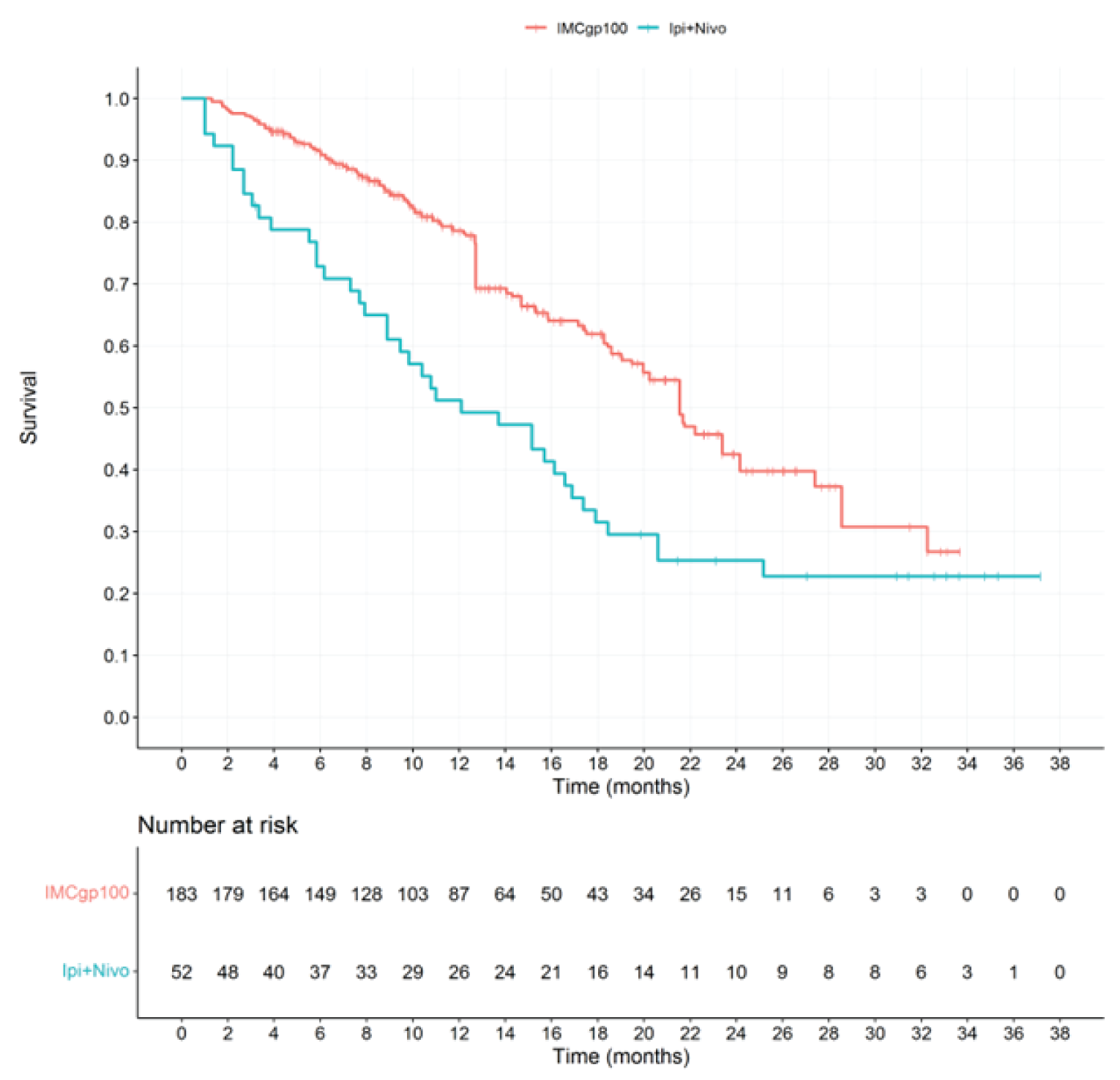
IMCgp100 = tebentafusp; Ipi = ipilimumab; KM = Kaplan-Meier; MAIC = matching-adjusted indirect comparison; Nivo = nivolumab; OS = overall survival.
Source: Sponsor-submitted Indirect Treatment Comparison Technical Report.14
An important limitation of the MAIC is that the patient population in the index trial (i.e., HLA-A*02:01-positive patients with mUM) was narrower than the comparator trial (patients with mUM regardless of HLA status). It is not possible to account for such difference in the adjustment process because only aggregate data were available for the comparator trial. It is unclear what proportion of patients in the comparator trial was HLA-A*02:01-positive. The sponsor submitted a supplementary analysis exploring the impact of HLA-A*02:01 status on disease prognosis in patients with mUM; however, CADTH was unable to draw any definitive conclusions, given that the study was based on a small observational cohort and results are subject to imprecision, considering the wide 95% CI. It is also not clear if the methodologies of the analysis were robust from the brief description of statistical analysis provided. As such, results of the MAIC should be interpreted with caution, owing to potential confounding by the heterogeneity in patient population between the trials. All other inclusion and exclusion criteria, as well as the median duration of follow-up, were generally similar in the trials. There was inconsistency in the definition of PFS and OS between the trials, although the impact on study findings is unlikely to be substantial.
Figure 8: KM Plot of PFS From MAIC (Primary Analysis)
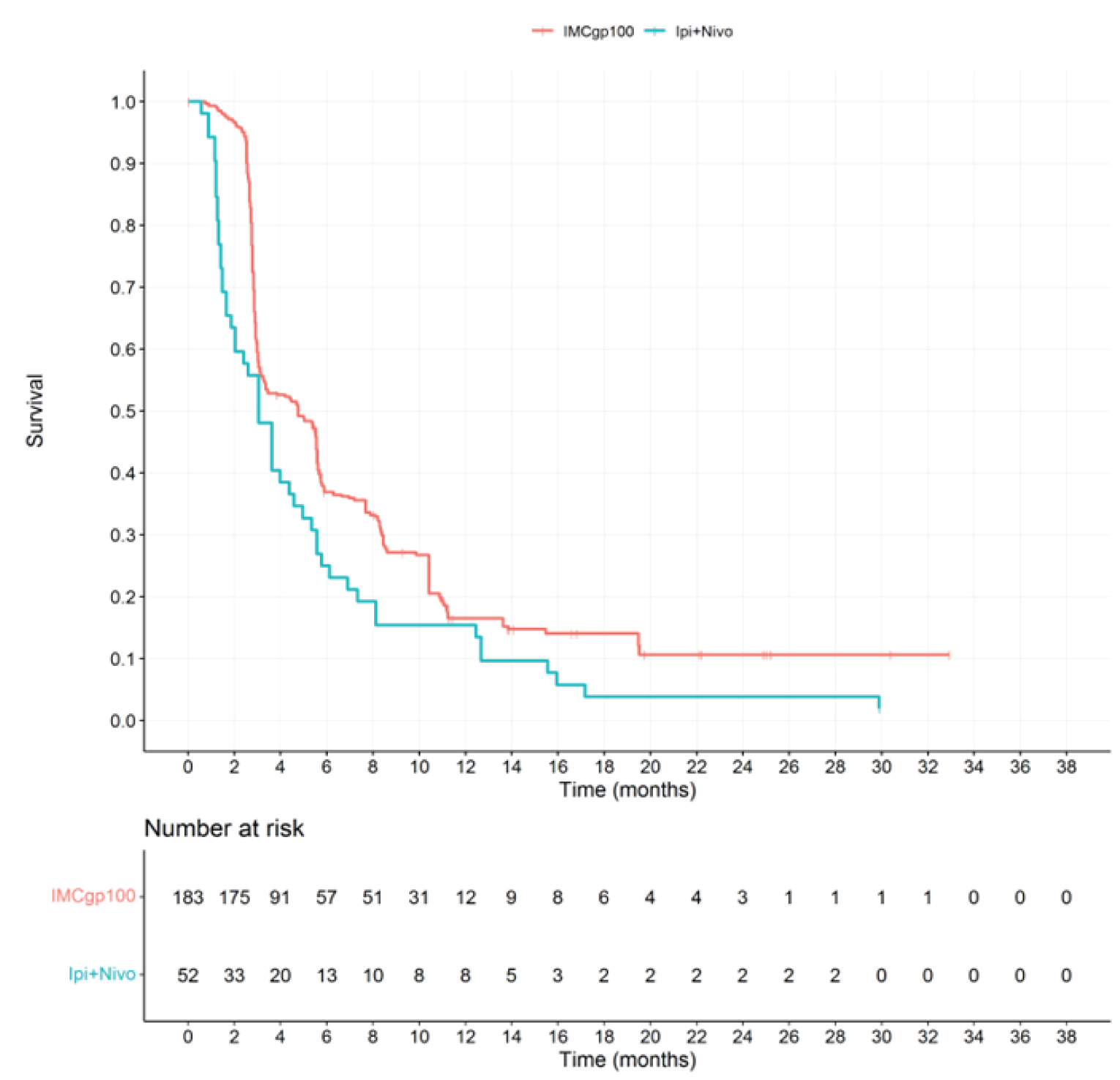
IMCgp100 = tebentafusp; Ipi = ipilimumab; KM = Kaplan-Meier; MAIC = matching-adjusted indirect comparison; Nivo = nivolumab; PFS = progression-free survival.
Source: Sponsor-submitted Indirect Treatment Comparison Technical Report.14
The choice to conduct an unanchored MAIC was appropriately justified by the lack of a common comparator across the included trials. The prognostic and treatment-effect modifying factors that were adjusted for in the weighting process were identified from a multivariate analysis of OS results in Study 202. The methods of the multivariate analysis were not available for assessment by the CADTH review team; as such, there is uncertainty about the appropriateness of the identified factors and the direction of potential bias. Another point to note is that time since primary diagnosis, a covariate identified in the multivariate analysis, was excluded from adjustment because it was not reported in the comparator trial.
Unadjusted and match-adjusted baseline covariates were reported. Rescaling of extreme weights was involved after an examination of weight distribution, and the distribution of rescaled weights was reported. There was a notable reduction in ESS, by approximately 51.1%, after the weighting process, which suggest poor population overlap. A significant reduction in sample size can contribute to imprecision, increasing uncertainty of the results. A similar issue was noted with the sensitivity analyses, which further limits the ability to draw any conclusion from the MAIC. With respect to the UAIC, the result should be considered exploratory because naive direct comparisons are subject to significant confounding and bias, owing to the lack of control for systematic differences between trials.45 It should also be noted that the PFS results should be interpreted with caution because the posthoc analysis was performed after results of the OS analysis were available and was subject to potential bias.
With respect to external validity, ipilimumab plus nivolumab was a relevant comparator as it accounts for most first-line systemic treatment prescribed for mUM in Canada, according to the clinical expert. A limitation to note is that the analysis was restricted to patients who were treatment-naive in the metastatic setting. The clinical expert consulted by CADTH noted that use of tebentafusp in patients with mUM who had received prior systemic therapy was anticipated; thus, the findings of the MAIC may have limited generalizability to this specific patient population. Further, the MAIC assessed OS and PFS end points only, and other efficacy outcomes that were of interest to the stakeholders, such as HRQoL and harms, were not investigated.
Other Relevant Evidence
This section includes 2 additional studies from the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the Systematic Review.
The first study (Study 102) was a phase I/II, multicentre, open-label study that analyzed the efficacy and safety of tebentafusp in HLA-A*02:01-positive patients with mUM who had received 1 or 2 prior lines of therapy in the metastatic or advanced setting. Although the study did not meet the Systematic Review inclusion criteria, as it is not a phase III study, the CADTH review team summarized the study design and results of Study 102 to provide supporting evidence for tebentafusp in patients with mUM who had received prior therapies.
The second study was an observational study that compared patients receiving tebentafusp from Study 202 with patients receiving ipilimumab plus from the GEM-1402 trial after propensity score weighting. The analysis was based on the same studies that informed the sponsor-submitted MAIC and was submitted to CADTH after the sponsor had obtained IPD from the GEM-1402 trial. The study aimed to address the limitations of the MAIC analysis related to the use of aggregate data from the GEM-1402 trial.
Study 102
The characteristics of Study 102 are presented in Table 29. Study 102 was designed as a phase I/II study of tebentafusp administered on a weekly basis with an intrapatient escalation dosing regimen. The intrapatient escalation regimen consisted of fixed low doses of tebentafusp at cycle 1 day 1 (20 mcg) and cycle 1 day 8 (30 mcg), followed by an escalated cohort specified weekly dose administered at cycle 1 day 15 and beyond. According to this regimen, all patients in the study received 2 weekly doses of tebentafusp at a dose level below the identified recommended phase II dose of 50 mcg, and then dose escalation commenced at the third weekly dose (cycle 1 day 15), with the goal of achieve a long-term dosing regimen at a dose higher than that identified for the recommended phase II dose. Doses of 54 mcg, 64 mcg, 73 mcg, 68 mcg were evaluated in the phase I dose-escalation cohorts. The dose-escalation regimen identified the intrapatient escalation regimen of 68 mcg was selected for the phase II dose-expansion cohort. The overview of the study designs for Study 102 is shown in Figure 9. The primary end point for phase I was the incidence (number) of dose-limiting toxicity, whereas ORR was the primary end point for the phase II, single-arm, dose-expansion cohort. The key secondary end points for phase II — OS, PFS, DOR, DCR, and BOR — were detailed in this report, according the prespecified CADTH systematic review protocol. Patients were eligible for the phase II expansion component if they were HLA-A*02:01-positive, had an ECOG PS of 0 or 1, had previously received systemic treatment for mUM, and had measurable disease according to RECIST 1.1. The exclusion criteria were generally consistent with those used in Study 202.
Study 102 is ongoing, with an estimated completion date of March 20, 2024. The evidence summarized in this section is from a planned interim analysis with a data cut-off date of March 20, 2020, and contains results of the phase II expansion cohort only. Results of the phase I study were not of interest to this review, owing to the dose-finding nature and the small sample size.
Table 29: Details of Study 102
Details | Study 102 |
|---|---|
Design and population | |
Study design | Phase I/II, multicentre, open-label study |
Locations | 26 study centres in 5 countries (Canada, Germany, Spain, UK, and US) |
Patient enrolment date | January 19, 2017 |
Data cut-off date | March 20, 2020 |
Estimated study completion datea | March 20, 2024 |
Enrolment size | 127 in phase II |
Key inclusion criteria |
|
Key exclusion criteria |
|
Interventions (phase II) | |
Week 1 | Tebentafusp 20 mcg |
Week 2 | Tebentafusp 30 mcg |
Week 3 onward | Expansion cohort: tebentafusp 68 mcg |
Outcomes (phase II) | |
Primary end point | ORRb,c |
Key secondary end points | OSd PFSd,e DORd DCRf |
Notes | |
Publications | Sacco et al. (2020)17 |
CNS = central nervous system; DCR = disease control rate; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HLA = human leukocyte antigen; mUM = metastatic uveal melanoma; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumours.
aThe date on which the last participant in a clinical study was examined or received an intervention or treatment to collect final data for the primary outcome measures, secondary outcome measures, and adverse events (that is, the last participant's final visit).
bUp to 38 months.
cORR is defined as the percentage of participants with measurable disease with at least 1 visit response of CR or PR that is confirmed at least 4 weeks later, as defined in RECIST 1.1 and assessed by an investigator. The denominator in the calculation of ORR is the number of participants in the full analysis set with measurable disease at baseline.
dUp to 49 months.
ePFS is defined as the time (in months) from first dose of study drug until the date of disease progression or death (from any cause in the absence of disease progression), as assessed by RECIST 1.1 by the investigator for phase I and by the ICR for phase II.
f24 weeks.
Sources: Study 102 Clinical Study Report;27 Study 102 Registration.46
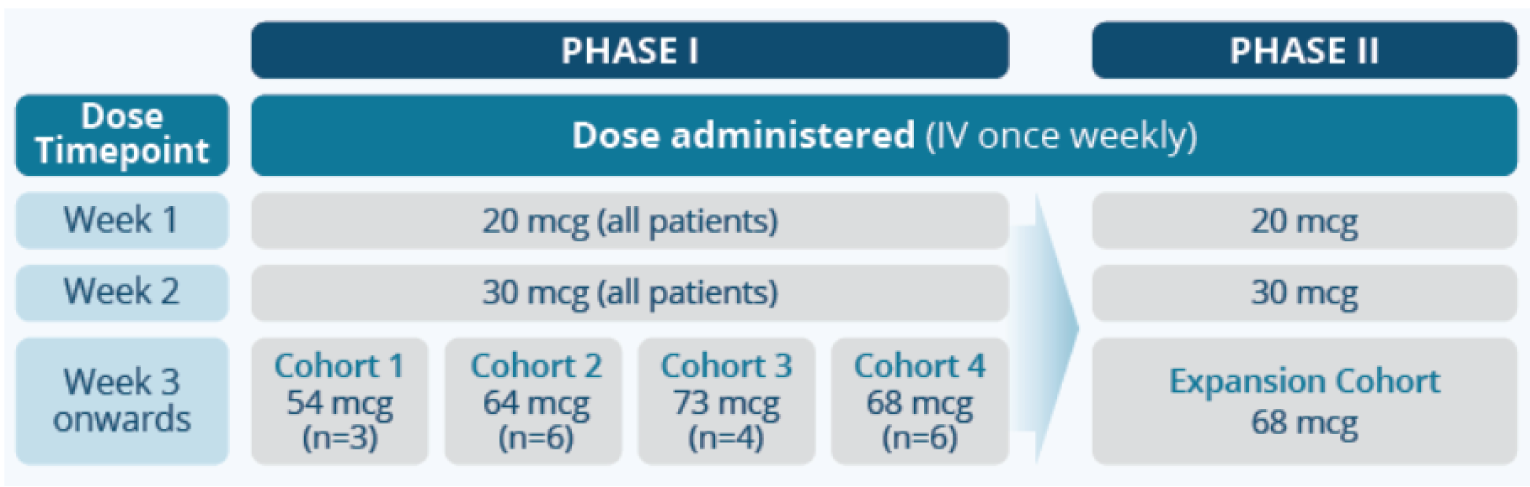
Baseline Characteristics
In the phase II expansion cohort, the majority of patients were white (99.2%) and approximately half were female (49.6%), with a mean age of 61.0 (SD = 10.93) years. There was a higher proportion of patients who were younger than 65 years (63.0%) than who were 65 years or older (37.0%). Most patients (70.1%) had an ECOG PS of 0 at baseline. A total of 58.3% of patients had baseline LDH greater than the ULN, 80.3% of patients had a baseline absolute lymphocyte count of at least 1.0 × 109/L, and 70.9% of patients had a baseline alkaline phosphatase level no higher than the ULN. Of the 127 patients in the phase II dose-expansion cohort, at initial diagnosis, most patients (32.3%) had stage II disease, followed by stage III (22.0%), stage IV (16.5%), and stage I (8.7%). The median time from primary diagnosis to metastatic disease was 2.96 years and the median time from primary diagnosis to study entry was 4.42 years. Approximately two-thirds of patients had received 1 prior anticancer regimen and 33.9% of patients had received at least 2 prior therapies. The most common prior treatments were radiotherapy (75.6%), immunotherapy (73.2%), and liver-directed therapy (44.9%). At study entry, per the investigator’s assessment, 117 patients had at least 1 measurable metastatic lesion in the liver; the largest lesion in 43 (33.9%) patients was no larger than 3 cm in diameter and in 69 (54.3%) patients was at least 3 cm. The baseline characteristics of patients in Study 102, phase II are summarized Table 30.
Table 30: Baseline Characteristics of Patients in Study 102, Phase II (Full Analysis Set)
Characteristic | Study 102, phase II (n = 127) |
|---|---|
Age | |
Mean (SD) | 61.0 (10.93) |
Median (range) | 61.0 (25 to 88) |
< 65, n (%) | 80 (63.0) |
≥ 65, n (%) | 47 (37.0) |
Sex, n (%) | |
Female | 63 (49.6) |
Male | 64 (50.4) |
Race, n (%) | |
White | 126 (99.2) |
Other | 1 (0.8) |
Ethnicity, n (%) | |
Hispanic or Latino | 4 (3.1) |
Not Hispanic or Latino | 123 (96.9) |
ECOG PS, n (%) | |
0 | 89 (70.1) |
1 | 38 (29.9) |
Baseline LDH, n (%) | |
≤ ULN | 53 (41.7) |
> ULN | 74 (58.3) |
Missing | 0 |
Baseline ALC, n (%) | |
< 1.0 × 109/L | 25 (19.7) |
≥ 1.0 × 109/L | 102 (80.3) |
Baseline ALP, n (%) | |
≤ ULN | 90 (70.9) |
> ULN | 37 (29.1) |
Stage at initial diagnosis, n (%) | |
Stage I | 11 (8.7) |
Stage II | 41 (32.3) |
Stage III | 28 (22.0) |
Stage IV | 21 (16.5) |
Missing | 26 (20.5) |
Time from primary diagnosis to metastatic disease (years) | |
Mean (SD) | 4.8 (4.9) |
Median (range) | 2.96 (0.0 to 28.1) |
Time from primary diagnosis to study entry (years) | |
Mean (SD) | 6.25 (5.5) |
Median (range) | 4.42 (0.6 to 28.5) |
Largest liver metastasis (investigator assessment),a n (%) | |
Diameter < 3 cm | 44 (34.6) |
Diameter ≥ 3 cm | 73 (57.5) |
No liver metastases | 10 (7.9) |
Largest liver metastasis (ICR),b n (%) | |
Diameter < 3 cm | 43 (33.9) |
Diameter ≥ 3 cm | 69 (54.3) |
No liver metastases | 15 (11.8) |
Number of prior anticancer therapy regimens, n (%) | |
1 | 84 (66.1) |
2 | 36 (28.3) |
3 | 2 (1.6) |
4 | 4 (3.1) |
5 | 1 (0.8) |
Prior oncology treatments, n (%) | |
Systemicc | 106 (83.5) |
Immunotherapy | 93 (73.2) |
Checkpoint inhibitor (PD-1/PD-L1) | 83 (65.4) |
Checkpoint inhibitor (CTLA4) | 39 (30.7) |
Other immunotherapy | 5 (3.9) |
Chemotherapy | 14 (11.0) |
Targeted therapy | 11 (8.7) |
Other | 6 (4.7) |
Liver-directed therapyc,d | 57 (44.9) |
Radiotherapyc | 96 (75.6) |
Brachytherapy | 79 (62.2) |
Surgeryc | 52 (40.9) |
Enucleation | 40 (31.5) |
Other | 21 (16.5) |
ALC = absolute lymphocyte count; ALP = alkaline phosphatase; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ICR = independent central review; LDH = lactate dehydrogenase; PD-1 = programmed cell death 1 protein; PD-L1 = programmed cell death 1 ligand 1; SD = standard deviation; ULN = upper limit of normal.
aLiver metastases measurements based on investigator assessment.
bLiver metastases measurements based on ICR.
cMedically reviewed therapy types based on the coded term, reported term, electronic Case Report Form therapy class, and reason for therapy. Includes therapies for primary and metastatic disease only. Also includes liver-directed chemotherapeutic drugs.
dLiver-directed therapies include the following subcategories: ablation, bland embolization, chemoembolization, immunoembolization, perfusion, radiation, and radioembolization.
Source: Study 102 Clinical Study Report.27
Statistical Analysis
Study 102 data were summarized using descriptive statistics. Categorical data were presented as frequencies and percentages. For continuous data, mean, SD, 95% CI (where applicable), median, minimum, and maximum were presented. The primary end point of the phase II dose expansion, ORR, was estimated according to the number and proportion of patients with an objective response (BOR of CR or PR), based on ICR assessment according to RECIST 1.1. The associated 2-sided 95% CI for the true ORR using the exact Clopper-Pearson method was also presented. This was based on the full analysis set, which included all patients assigned to treatment. The key secondary end points of the phase II dose expansion identified by the CADTH review protocol — OS, PFS, DOR, DCR, and BOR — were derived and reported based on ICR data.
Results
Patient Disposition
A summary of patient disposition is shown in Table 31. A total of 148 patients were screened and 127 patients were enrolled in the phase II dose-expansion part of the study. Of the 127 (100.0%) patients who received the study drug, 106 (83.5%) discontinued the study drug and 21 (16.5%) remained on treatment. The primary reason for treatment discontinuation was disease progression (70.1%). As of the data cut-off date of March 20, 2020, 53 (41.7%) patients remained on study and 74 (58.3%) patients were reported to have ended the study. Death was the primary cause of study discontinuation (54.3%).
Efficacy
The summary of results in this section focuses on the primary and key secondary outcomes of Study 102, phase II (OS, PFS, DOR, DCR, and BOR) to assess the effects of tebentafusp on patients with mUM who had received 1 or 2 prior lines of therapy (shown in Table 32).
For ORR, 6 of 127 patients achieved a PR (4.7%; 95% CI, 1.8% to 10.0%); no patients achieved a CR. After a median follow-up of 19.6 (95% CI, 16.0 to 22.2) months, the median OS was 16.8 (95% CI, 12.9 to 21.3) months. The median PFS was 2.8 (95% CI, 2.0 to 3.7) months, per the RECIST 1.1 assessment by ICR. The median DOR (CR or PR) per the RECIST 1.1 assessment by ICR, was 8.7 (95% CI, 5.6 to 24.5) months. The DCR (CR, PR, or stable disease) was 22.8% (95% CI, 15.7% to 31.2%) at 24 weeks and beyond. Of the 127 patients who received the study drug, the most frequently observed BOR was disease progression (47.2%), followed by stable disease (44.9%), and PR (4.7%).
Table 31: Patient Disposition in Study 102, Phase II (Full Analysis Set)
Disposition | Study 102, phase II |
|---|---|
Patients screened | 148 |
Patients treated | 127 |
Discontinued study, n (%) | 74 (58.3) |
Died | 69 (54.3) |
Lost to follow-up | 2 (1.6) |
Withdrawal | 1 (0.8) |
Other | 2 (1.6) |
Discontinued treatment, n (%) | 106 (83.5) |
Disease progression | 89 (70.1) |
Patient or physician decision | 8 (6.3) |
Adverse events | 6 (4.7) |
Other | 2 (1.6) |
Other cancer | 1 (0.8) |
Note: Data cut-off date of March 20, 2020.
Source: Study 102 Clinical Study Report.27
Table 32: Summary of Key Outcomes in Study 102, Phase II (Full Analysis Set)
Outcomes | Study 102, phase II (N = 127) |
|---|---|
Primary outcome | |
ORR,a n (%) | 6 (4.7) |
95% CIb | 1.8 to 10.0 |
Secondary outcomes | |
OS (months), median (95% CI) | 16.8 (12.9 to 21.3) |
PFS (months),c median (95% CI) | 2.8 (2.0 to 3.7) |
DOR (months),d median (95% CI) | 8.7 (5.6 to 24.5) |
DCR at ≥ 24 weeks,e n (%) | 28 (33.1) |
95% CIb | 15.7 to 31.2 |
Best overall responsec | |
Complete response,f n (%) | 0 |
Partial response,f n (%) | 6 (4.7) |
Stable disease,f n (%) | 57 (44.9) |
Disease progression,f n (%) | 60 (47.2) |
Not evaluable,f n (%) | 4 (3.1) |
CI = confidence interval; DCR = disease control rate; DOR = duration of response; ORR = objective response rate; OS = overall survival; PFS = progression-free survival.
Note: Data cut-off date of March 20, 2020.
aORR is defined as the percentage of participants with measurable disease with at least 1 visit response of CR or PR that is confirmed at least 4 weeks later, as defined in RECIST 1.1 and assessed by ICR. The denominator in the calculation of the ORR is the number of participants in the full analysis set with measurable disease at baseline.
b95% CIs were calculated using the exact Clopper-Pearson method.
cAssessed by RECIST 1.1 by ICR.
dDOR is defined as the time (in months) from the date of first documented objective response (CR or PR) until the date of documented disease progression or death from cause in the absence of disease progression, as assessed by ICR.
eParticipants with a BOR of CR or PR or stable disease recorded at least 24 weeks (± 1 week) after commencement of study drug and before any progressive disease event, as assessed by ICR.
fResponse confirmation at least 4 weeks after initial evaluation, as defined in RECIST 1.1 and assessed by an ICR.
Source: Study 102 Clinical Study Report.27
Harms
As in Study 202, all patients in Study 102 experienced an AE of any grad. In addition, the Study 102 phase II expansion and Study 202 had comparable rates of grade 3 or higher TEAEs (59.1% versus 54.3%). In the Study 102 phase II expansion, 9 patients (7.1%) experienced TEAEs leading to tebentafusp discontinuation, a proportion comparable to that of Study 202 (6.6%). The most frequently reported TEAEs of any grade were pyrexia (81.1%), pruritus (68.5%), nausea (67.7%), chills (66.1%), and hypotension (41.7%). These TEAEs were also observed in the tebentafusp arm of Study 202. A total of 86 serious TEAEs were reported in 42 (33.1%) patients, a proportion comparable to that of Study 202 (28.2%). The serious TEAEs were CRS in 4 (3.1%) patients and sepsis, elevated alanine transaminase, rash maculopapular, and hypotension in 3 (2.4%) patients each. No deaths related to TEAEs were observed in Study 102.
Critical Appraisal
Study 102 was designed as a phase I/II, open-label, single-arm study to determine the optimal drug dosage(s) and to generate hypotheses for the phase III trial (Study 202). The primary end point of ORR and the key secondary end points, such as PFS and DOR, were assessed by ICR based on RECIST 1.1. The use of ICR assessments reduced potential bias in the estimation of treatment effects related to the investigator or patient knowing the intervention received, which could affect the subjectively assessed outcomes, such as response and subjective AEs.
However, the noncomparative design of the Study 102, with no statistical testing, is the key limitation. The lack of a randomized control group means there is uncertainty regarding the magnitude of effects obtained for the efficacy outcomes and their relationship to the study drug. Although the clinical expert consulted explained that the efficacy outcomes of tebentafusp in Study 102 were clinically meaningful, demonstrated the activity of the drug, and were comparable with the Study 202 phase III trial, the CADTH review team notes that in the absence of a comparative arm, the findings obtained from the efficacy and safety analysis are uncertain, as the single-arm design does not allow for conclusions to be drawn about the comparative efficacy for tebentafusp or the differentiation of symptoms of the underlying mUM disease from treatment-related AEs.
Study 102 included patients who had received prior therapy in the metastatic setting, which is supported by the clinical expert’s comments, based on clinical trial and anecdotal evidence, that tebentafusp should not only be offered in first-line setting, but also in the second-setting and beyond. The clinical expert also indicated that clinical practice is usually less restrictive with respect to ECOG PS and comorbidities. The baseline characteristics of Study 102 were suggestive of an overrepresentation of patients with a higher functioning status (ECOG PS of 0) and may compromise the representativeness of the study sample to the general population of adults with mUM.
Propensity Score Analysis (IPTW Approach)
This observational study compared patients in the tebentafusp arm of Study 202 to patients who received ipilimumab plus nivolumab in the GEM-1402 trial.18 The study was not randomized, and propensity score weighting was used in an attempt to adjust for confounding.
Methods
Objectives
The primary objective of this analysis was to compare OS between tebentafusp and ipilimumab plus nivolumab in patients with previously untreated mUM.
Patient Selection Criteria
Key selection criteria of the 2 cohorts included in the study are summarized in Table 26. As mentioned in the MAIC section, although both studies enrolled patients with unresectable UM or mUM who had received no prior therapy in the metastatic setting, Study 202 enrolled HLA-A*02:01-positive patients only, whereas the GEM-1402 trial enrolled patients regardless of HLA status. All other selection criteria were generally consistent between the studies.
Propensity Score Development
The patient characteristics that were used to balance the cohorts for effect estimation included time from diagnosis to metastasis and the same covariates used in the MAIC analysis, including median age, sex, baseline LDH (normal versus elevated), baseline alkaline phosphatase (normal versus elevated), disease location (hepatic only, extrahepatic only, hepatic and extrahepatic), and ECOG PS, all of which were informed by a multivariate analysis of OS in Study 202. These covariates were used as predictors in a logistic regression model in which tebentafusp treatment was the binary outcome used to estimate the probability of receiving tebentafusp for each patient in the GEM-1402 trial.
Statistical Methods
The propensity score analysis weighted patients using the IPTW approach to estimate treatment effects. In the primary analysis, the average treatment effect of the treated (ATT) method was used, in which all patients who received tebentafusp were assigned a weight of 1, and patients who received ipilimumab plus nivolumab received a weight calculated as the propensity score of treatment divided by 1 minus the propensity score of treatment. The distribution of weights was assessed visually, using box plots for outliners.
Two additional analyses that used different methods to calculate weights were conducted. One analysis used the average treatment effect of the control weights, in which the roles of tebentafusp and ipilimumab plus nivolumab were reversed in the previously described propensity score equation, whereas the second analysis used the average treatment effect of the combined population weights, in which patients who received tebentafusp were assigned a weight of 1 divided by the propensity score, and patients who received ipilimumab plus nivolumab received a weight of 1 divided by 1 minus the propensity score.
After weighting, the median OS and OS curves were estimated using the KM method. The HR and 95% CI were calculated using an IPTW Cox regression model. Variance was calculated using robust sandwich estimation. A sensitivity analysis using a multivariate Cox regression model was conducted.
Patient with missing data for at least 1 relevant covariate were excluded from the primary analysis. A sensitivity analysis using multiple imputation of missing baseline covariate values was performed.
Results
Based on visual inspection of the boxplots, there were no clear weight outliers and no extreme weights. Patient characteristics before and after propensity score weighting are summarized in Table 33. The ESS of the GEM-1402 trial was 34.4 after weighting, compared to a sample size of 45 before weighting. The patient characteristics were generally balanced in the tebentafusp and ipilimumab plus nivolumab cohorts after weighting.
In the primary analysis, median OS of the tebentafusp cohort was 21.7 months (SD = NR), and the weighted median OS of the ipilimumab plus nivolumab cohort was 12.6 months (SD = NR). The HR between tebentafusp and ipilimumab plus nivolumab with respect to OS was 0.430 (95% CI, 0.287 to 0.643) in favour of tebentafusp. A KM plot of OS for the tebentafusp cohort and the weighted ipilimumab plus nivolumab cohort in the primary analysis is shown in Figure 10. Results of the sensitivity analyses were consistent with those of the primary analysis.
Table 33: Patient Characteristics Before and After Weighting (Propensity Score Analysis [IPTW])
Characteristic | Ipilimumab + nivolumab | Tebentafusp | |
|---|---|---|---|
GEM-1402 before weighting | GEM-1402 after weighting | Study 202 | |
N (or ESS) | 45 | (34.4) | 237 |
Median age, years (SD) | 59.3 (13.3) | 61.8 (30.0) | 61.2 (12.0) |
Male, n (%) | 23 (51.1) | 110.6 (46.3) | 120 (50.6) |
Elevated LDH level, n (%) | 19 (42.2) | 82.1 (34.4) | 84 (35.4) |
Elevated ALP level, n (%) | 7 (15.6) | 50.9 (21.3) | 51 (21.5) |
Extrahepatic disease only, n (%) | 10 (22.2) | 8.5 (3.6) | 9 (3.8) |
Hepatic disease only (%) | 20 (44.4) | 124.2 (52.0) | 123 (51.9) |
Hepatic and extrahepatic disease, n (%) | 15 (33.3) | 106.1 (44.4) | 105 (44.3) |
ECOG PS of 0, n (%) | 38 (84.4) | 196.0 (82.1) | 188 (79.3) |
Time from diagnosis to metastasis (years) (SD) | 4.7 (4.6) | 4.1 (9.6) | 4.0 (4.4) |
ALP = alkaline phosphatase; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ESS = effective sample size; IPTW = inverse probability of treatment weights; LDH = lactate dehydrogenase; SD = standard deviation.
Note: Study 202 data cut-off date: October 13, 2020; GEM-1402 data cut-off date: July 9, 2019.
Source: Sponsor-submitted Propensity Score Analysis Statistical Report.47
Critical Appraisal
The propensity score analysis (IPTW) and the MAIC analysis conducted by the sponsor share some common features and, as such, much of the appraisal of the MAIC analysis with respect to study selection, covariate identification, and external validity also applies to the current analysis.
As previously noted, the GEM-1402 trial was identified from an SLR. There is potential for selection bias in the SLR, owing to the lack of a priori selection criteria for the comparator trial. Other methods of the SLR were generally appropriate, with proper literature search, study selection, and data extraction methods in place. The GEM-1402 trial was noted to have a low risk of bias based on a quality assessment conducted by the sponsor; however, a single reviewer was involved in the process with no verification, which increases the risk of error and bias in appraisal.
The sponsor’s justification for an IPTW approach over other propensity score approaches (matching, stratification, and covariate adjustment) was that IPTW is better suited for analyzing a small sample size than matching approaches, and for analyzing time-to-event end points. The sponsor noted that for time-to-event analysis, there is potential bias in KM estimates and marginal HRs with other propensity score approaches.
Figure 10: KM Plot of OS After Weighting (Primary Propensity Score Analysis [IPTW])
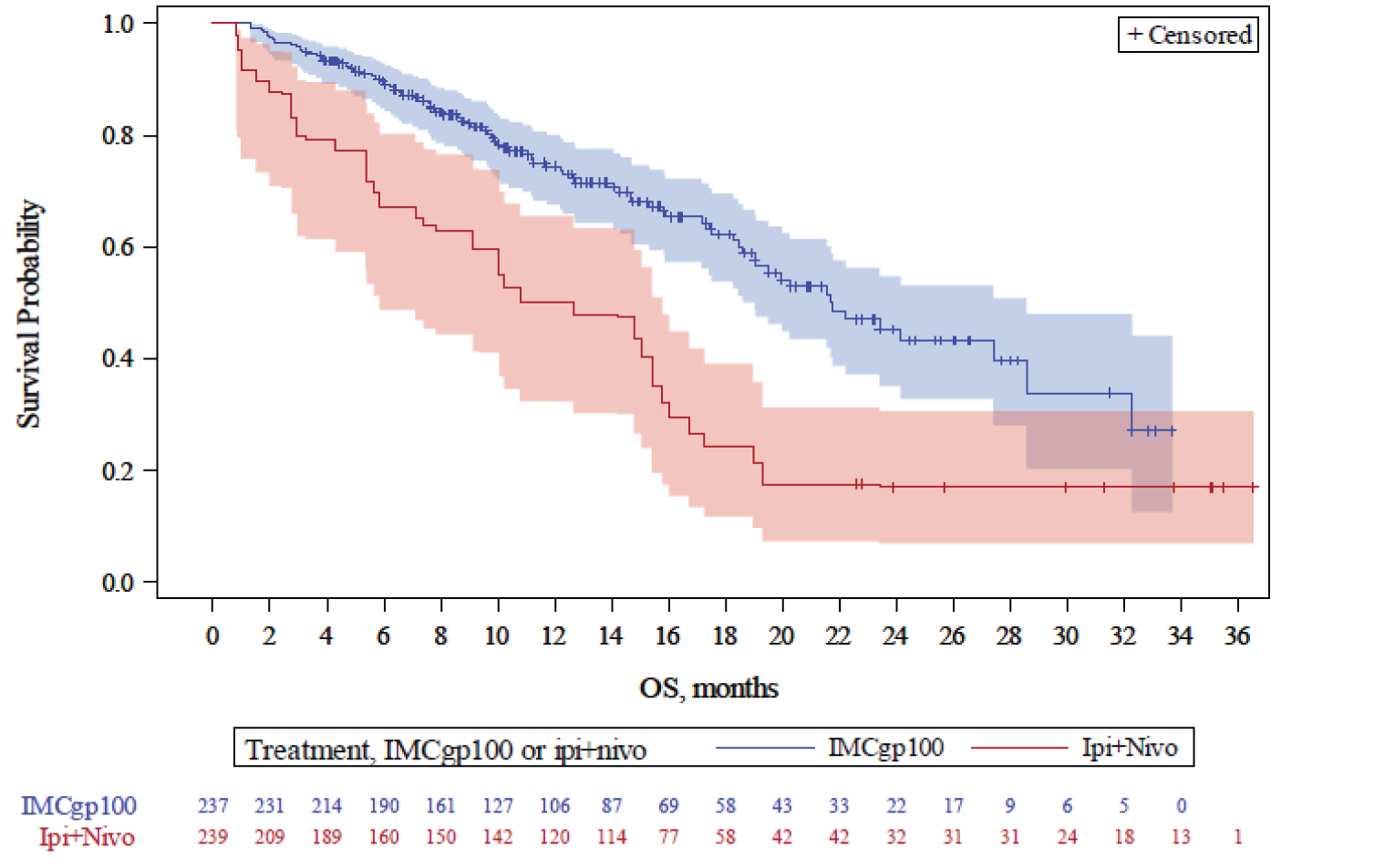
IMCgp100 = tebentafusp; Ipi = ipilimumab; IPTW = inverse probability of treatment weighting; KM = Kaplan-Meier; Nivo = nivolumab; OS = overall survival.
Source: Sponsor-submitted Propensity Score Analysis Statistical Report.47
The covariates used for propensity score calculation were informed by a multivariate analysis of OS in Study 202 conducted by the sponsor. Because the methods of the multivariate analysis were not available for assessment by the CADTH review team, there is uncertainty as to whether the identified factors were appropriate or comprehensive. Further, Austin (2014)48 suggested that the identification of potentially prognostically important covariates should be based on clinical expertise or an SLR of available literature, rather than statistical testing in the study sample. It should also be noted that there is heterogeneity in the HLA status of patients in Study 202 (HLA-A*02:01-positive patients with mUM) and the GEM-1402 trial (patients with mUM regardless of HLA status). It remains unclear to the CADTH review team after a review of the sponsor-submitted supplementary analysis that assessed the impact of HLA status on OS whether HLA status is or is not a possible confounder of OS, given that the analysis was based on a small observational cohort and the results are subject to imprecision. Overall, it is uncertain if all known and unknown confounding factors have been adequately accounted for in the weighting process, which is a key assumption of a propensity score model.48
There is a reasonable overlap in the distribution of propensity scores between the treatment groups. The weights were calculated with the IPTW approach using the ATT method in the primary analysis. Additional analyses using alternative methods (ATT stabilized, average treatment effect, and average treatment effect of the control) were conducted, which showed results consistent with the primary analysis. The distribution of generated weights was examined, and no extreme outlying weight was identified. Baseline covariates before and after weighting were reported and were similar between the comparison groups. Some patients were excluded from the primary analysis because of missing data for baseline covariates. The sponsor conducted a sensitivity analysis using multiple imputation, which suggested results similar to the primary analysis. Of note, no formal hypothesis testing was conducted, which renders the trial findings exploratory.
It should also be noted that the analysis was informed with interim OS data from Study 202, which adds uncertainty to the results; there is typically a risk of overestimation of the treatment effects in an interim analysis.13
The generalizability of study results is expected to be similar to that of Study 202. The analysis was restricted to patients with mUM who were treatment-naive in the metastatic setting. The findings may have limited generalizability to patients who had received prior systemic therapy, which is a relevant patient population, according to the clinical expert, because tebentafusp will likely be used in previously treated patients. In addition, OS was the only assessed outcome; HRQoL and harms outcomes that were of interest to the stakeholders were not assessed.
Discussion
Summary of Available Evidence
This report summarizes the evidence on tebentafusp based on 1 phase III RCT, 1 ITC, 1 phase I/II study, and 1 propensity score (IPTW-based) analysis.
Study 202, a phase III, open-label RCT, was included in the CADTH Systematic Review. The study aimed to demonstrate the superiority of tebentafusp to investigator’s choice of therapy (pembrolizumab, ipilimumab, or dacarbazine) in HLA-A*02:01-positive adults with mUM who had received no prior therapy in the metastatic setting (N = 378) through the coprimary end points of OS in the RAS and ITT analysis set, and the key secondary end points of PFS and BOR (evaluated statistically as ORR) in the ITT analysis set. In the primary analysis (data cut-off date of October 13, 2020), the mean age of the study population was 62.1 (SD = 11.6) years. Approximately half of the patients were female. The majority of patients were white, had an ECOG PS of 0, and had liver metastases at baseline. Most patients had undergone no prior surgery for metastatic disease (91.3%) and had received no prior antineoplastic systemic treatments (in any setting) (96.3%). Prior radiotherapy was reported in 40.2% of patients.
One sponsor-submitted ITC was summarized and critically appraised. Given the lack of direct comparative evidence between tebentafusp and ipilimumab plus nivolumab, the most commonly prescribed immunotherapy for mUM in Canada, the sponsor performed an unanchored MAIC to estimate the comparative OS and PFS benefits between the interventions in patients with mUM who had received no prior therapy in the metastatic setting. The analysis was informed by an SLR that identified the open-label, single-arm GEM-1402 trial of ipilimumab plus nivolumab as the comparator trial.
Study 102, a phase I/II, open-label study, was also summarized and appraised as supporting evidence. The phase II, single-arm expansion cohort is the only evidence to date for tebentafusp in HLA-A*02:01-positive patients with mUM who had received 1 or 2 prior therapies (chemotherapy, immunotherapy, or targeted therapy) in the metastatic setting (N = 127 in the phase II expansion cohort). The primary end point in the phase II expansion cohort was ORR, and the secondary end points included OS, PFS, DOR, DCR, and safety. At baseline, the mean age of patients was 60.4 (SD = 10.93) years. The majority of patients were white, had an ECOG PS of 0, liver metastasis, and an LDH level that was above the ULN. The majority of patients (66.1%) had received 1 prior anticancer therapy, whereas others had received 2 or more prior anticancer therapies. Of note, 83.5% of patients received prior systemic therapies (73.2% received immunotherapy, 11.0% received chemotherapy, and 8.7% received targeted therapy. Other common prior therapies included radiotherapy (75.6%) and liver-directed therapy (44.9%).
The report also includes an observation study that used propensity score weighting (IPTW approach) to compare OS between tebentafusp and ipilimumab plus nivolumab based on IPD from the same studies that informed the sponsor-submitted MAIC. The study was submitted to CADTH after the sponsor had obtained IPD from the GEM-1402 trial. The study aimed to address the limitations of the MAIC analysis related to the use of aggregate data from the GEM-1402 trial.
Interpretation of Results
Efficacy
In the pivotal trial, Study 202, the coprimary end point of OS in the RAS and OS in the ITT analysis set were met in the first interim OS analysis (data cut-off date of October 13, 2020), supporting the superiority of tebentafusp to investigator’s choice of therapy in the first-line treatment setting for mUM with respect to OS, the most clinically relevant outcome noted by patients and clinicians. The clinical expert consulted by CADTH considered the magnitude of the OS benefits of tebentafusp in the ITT analysis to be clinically meaningful, but noted that a longer follow-up (2 to 3 more years) would help provide further confidence in the OS benefits of tebentafusp. OS analysis in the RAS, which aimed to assess the comparative OS benefits of tebentafusp with investigator’s choice therapy in patients who developed a rash in the first week after treatment with tebentafusp, however, should be interpreted with caution because of the potential for confounding related to the lack of randomization in the comparison, although the extent and direction of any bias cannot be determined. No conclusion could be drawn regarding the subgroup effects of ECOG PS on OS or from the posthoc landmark OS analysis among patients with a BOR of progressive disease, owing to the exploratory nature, lack of control for multiplicity, and the risk of confounding in the absence of randomization in the comparisons.
The key secondary end point of PFS was also met, in favour of tebentafusp. No statistically significant difference was noted in the ORR analysis between treatment arms; however, considering the wide 95% CI of the OR, the results are subject to serious imprecision, rendering the comparative effect of tebentafusp versus investigator’s choice therapy on ORR inconclusive. In addition, tumour response was assessed by investigators using RECIST 1.1 and, considering the open-label design, PFS and ORR, along with other secondary tumour response outcomes (DOR, DCR) might be subject to reporting bias; however, the extent and the direction of the bias is unclear. The clinical expert noted that PFS and ORR outcomes were of limited clinical relevance in the treatment of mUM because there is generally a poor correlation between tumour response and survival in patients with mUM receiving systemic treatments. The DOR in patients who achieved a CR or PR were numerically similar between the treatment arms; however, the difference between treatment arms was not statistically tested, which precludes the drawing of conclusions regarding the effect of tebentafusp on DOR. The DCR analysis suggested results in favour of tebentafusp; however, these results should be interpreted with caution because of the lack of control for multiplicity and, thus, an increased risk of a type I error.
HRQoL, a secondary end point in the study, was noted to be an important outcome of interest by patients and clinicians. The EORTC QLQ-C30 analysis showed that the change in score from baseline at most time points for all domains was stable and numerically similar between treatment arms, based on the commonly used MID estimate of 10 points for cancer patients. Of note, the sponsor considered a LS mean difference of fatigue score between treatment arms of –9.259 points by end of treatment to be statistically significant and clinically significant in the Clinical Study Report, although the MID used was not reported. Given the lack of control for type I error in HRQoL outcomes, the EORTC QLQ-C30 analyses should be considered exploratory. For the same reason, results of the EQ VAS analysis, which suggested a numerically similar change in score from baseline at all time points between treatment arms, should be considered exploratory. It should be noted that there is a significant amount of missing data in HRQoL outcomes, owing to deaths and poor questionnaire completion rates, increasing uncertainty of the results because of potential attrition bias. Also, considering that these instruments have not been validated in patients with mUM and that they are not routinely administered to patients with mUM in clinical practice, per the clinical expert, it is impossible to draw any conclusion about the effect of tebentafusp on HRQoL from available data.
Further, the generalizability of study results is uncertain because the treatments included in the comparator arm account for a small proportion of systemic therapies for mUM prescribed in Canada. Of note, 2 commonly prescribed immunotherapy regimens, ipilimumab plus nivolumab and nivolumab monotherapy, were not included in the comparator arm. The impact on generalizability was likely to be small because, as the clinical expert noted, immunotherapy regimens are similar with respect to tumour response rates; however, this opinion was formed based on a small retrospective cohort. The lack of direct evidence comparing tebentafusp with ipilimumab plus nivolumab and with nivolumab represents an evidence gap in the treatment of mUM.
In an effort to address the evidence gap, the sponsor conducted an MAIC, which suggested that tebentafusp demonstrated OS and PFS benefits over ipilimumab plus nivolumab in patients with mUM who had received no prior therapies in the metastatic setting; however, the results are highly uncertain considering the limitations of the analysis. Specifically, there was a risk of selection bias in the SLR in the absence of a priori selection criteria. The comparator trial included patients with mUM regardless of HLA-A*02:01 status. The heterogeneity in study populations between trials could have potentially confounded the study results. Further, time since primary diagnosis, a covariate identified in the multivariate analysis, was excluded from adjustment because it was not reported in the comparator trial, which may contribute to the uncertainty of the results. There is also concern with a loss of precision in the results, given a significant reduction in the ESS. Overall, the direction of bias of the aforementioned limitations could not be determined, and the results of the MAIC should be interpreted with caution. There is also an evidence gap in that HRQoL and harms outcomes, as well as the efficacy of tebentafusp in treatment-experienced patients, were not addressed in this analysis. The IPTW analysis improved upon the MAIC by using IPD from the comparator trial; however, many of the limitations of the MAIC analysis (risk of selection bias in the SLR, confounding adjustment, and lack of assessment of HRQoL, harms, and efficacy in a treatment-experience population) remained unaddressed. As such, the CADTH review team considered the results of this analysis to be uncertain.
The phase II, single-arm expansion cohort of Study 102 provided noncomparative evidence of tebentafusp treatment in HLA-A*02:01-positive patients with mUM, the majority (83.5%) of whom had received prior systemic therapies. The clinical expert stated that the study demonstrated activity of tebentafusp in the study population and that the results were compatible with the pivotal study, but noted that it is a challenge to draw any definitive conclusions, given the phase II, single-arm study design. The CADTH review team agreed that the noncomparative study design was a key limitation of the study.
Harms
In the pivotal study, tebentafusp is associated with a notably higher frequency of CRS (or presentations related to CRS) and dermatological AEs than investigator’s choice of therapy, with the most common AEs (> 50%) being pyrexia, pruritus, rash, and fatigue in the tebentafusp arm. However, it should be noted that most of the AEs were grade 1 or 2. In addition, the proportion of patients who discontinued treatment due to a TEAE (3.3%) and the proportion of patients who died due to an AE (0.8%) were both low in the tebentafusp arm. The harms of tebentafusp in patients with mUM who had received prior therapies in phase II of Study 102 were consistent with the harms observed in the pivotal study.
Although CRS is a common AE of tebentafusp, in the clinical experience of the clinical expert consulted by CADTH, it is usually transient, and the risk is substantially lower after 3 or 4 infusions. In addition, CRS is generally manageable with monitoring and supportive care (e.g., saline infusion, antihistamine, acetaminophen) in an inpatient setting for initial doses, and admission to the intensive care unit is rarely required, according to the clinical expert. This is consistent with the perspective of patients included in the patient group input, who reported that the side effects of tebentafusp are generally short-term, tolerable, and manageable. The clinical expert added that the recommended dosing of tebentafusp, which involves titration of initial doses, allows patients to build up tolerance to the treatment, and dose adjustments can be implemented to address CRS during the titration process.
Conclusions
Tebentafusp demonstrated a statistically significant and clinically meaningful improvement in OS compared with investigator’s choice of therapy (pembrolizumab, ipilimumab, or dacarbazine) in HLA-A*02:01-positive adults with mUM who had received no prior therapy in the metastatic setting, based on the phase III pivotal study. Tebentafusp resulted in a statistically significant improvement in PFS but not in ORR, although these outcomes are of limited clinical relevance in the treatment of mUM, per the clinical expert. Analyses of other secondary end points showed results in favour of tebentafusp (DCR) or comparable results between treatment arms (DOR, HRQoL), but no conclusions can be drawn because of the lack of statistical testing or control for multiplicity. The comparator treatments account for a small proportion of systemic treatments prescribed for mUM in Canada, which increases uncertainty in the generalizability of the study results. A phase II, single-arm study suggested clinical activity of tebentafusp in patients who had received prior therapy in the metastatic setting, but the results are subject to limitations arising from the noncomparative study design. No definitive conclusion can be drawn from the sponsor-submitted MAIC and propensity score analyses on the relative efficacy of tebentafusp versus ipilimumab plus nivolumab because of significant limitations with the analysis. CRS and dermatological AEs were common with tebentafusp, but were generally grade 1 or 2, transient, and manageable with supportive care.
References
1.Carvajal RD, Harbour JW. Management of metastatic uveal melanoma. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2022 Jun 2.
2.Ghazawi FM, Darwich R, Le M, et al. Uveal melanoma incidence trends in Canada: a national comprehensive population-based study. Br J Ophthalmol. 2019;103(12):1872-1876. PubMed
3.Damato BE, Dukes J, Goodall H, Carvajal RD. Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers (Basel). 2019;11(7):11. PubMed
4.Khoja L, Atenafu EG, Suciu S, et al. Meta-analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: an international rare cancers initiative (IRCI) ocular melanoma study. Ann Oncol. 2019;30(8):1370-1380. PubMed
5.Rantala ES, Hernberg M, Kivela TT. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res. 2019;29(6):561-568. PubMed
6.Nathan P, Cohen V, Coupland S, et al. Uveal Melanoma UK National Guidelines. Eur J Cancer. 2015;51(16):2404-2412. PubMed
7.NCCN Guidelines. Melanoma: uveal, version 2.2022. Plymouth Meeting (PA): National Comprehensive Cancer Network (NCCN); 2022: https://www.nccn.org/. Accessed 2022 Jun 2.
8.Guideline Resource Unit, Cancer Care Alberta. Uveal Melanoma. (Clinical practice guideline CU-015 – version 2). Edmonton (AB): Alberta Health Services; 2021: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-cu015-uveal-melanoma.pdf. Accessed 2022 Jun 2.
9.Kimmtrak (tebentafusp): 100 mcg / 0.5 mL solution for intravenous infusion [product monograph]. Dublin (IE): Immunocore Ireland Limited; 2022 Jun 7.
10.Nathan P, Hassel JC, Rutkowski P, et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N Engl J Med. 2021;385(13):1196-1206. PubMed
11.Clinical Study Report: IMCgp100-202. A phase II randomized, open-label, multi-center study of the safety and efficacy of IMCgp100 compared with investigator’s choice in HLA-A*0201 positive patients with previously untreated advanced uveal melanoma [internal sponsor's report]. Oxfordshire (GB): Immunocore Ltd; 2021 Apr 13.
12.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188-195. PubMed
13.Guyatt GH, Briel M, Glasziou P, Bassler D, Montori VM. Problems of stopping trials early. BMJ. 2012;344:e3863. PubMed
14.Match-adjusted indirect comparison of tebentafusp vs ipilimumab+nivolumab in patients with previously untreated uveal melanoma statistical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kimmtrak (tebentafusp), 100mcg/0.5 mL for intraveous infusion. Dublin (IE): Immunocore; 2021 Dec 7.
15.Piulats JM, Espinosa E, de la Cruz Merino L, et al. Nivolumab Plus Ipilimumab for Treatment-Naive Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J Clin Oncol. 2021;39(6):586-598. PubMed
16.TCGA UVM overall survival and metastasis free survival. Patients stratified by possession of HLA*A02:01 allele [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kimmtrak (tebentafusp), 100 mcg / 0.5 mL solution for intravenous infusion, 100mcg/0.5 mL for Intraveous infusion. Clonshaugh (Dublin): Imunocore; 2022 Jul 28.
17.Sacco JJ, Carvajal R, Butler MO, Shoushtari AN, Hassel JC. 64MO - A phase (ph) II, multi-center study of the safety and efficacy of tebentafusp (tebe) (IMCgp100) in patients (pts) with metastatic uveal melanoma (mUM). Ann Oncol. 2020;31:S1442-S1443.
18.Propensity score analysis comparing IMCgp100 or pembrolizumab (from study IMCgp100-202) to ipilimumab+nivolumab (from GEM-1402 study) in previously untreated uveal melanoma patients [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kimmtrak (tebentafusp), 100 mcg / 0.5 mL solution for intravenous infusion, 100mcg/0.5 mL for intraveous infusion. Dublin (IE): Immunocore; 2022 Jun 16.
19.Krantz BA, Dave N, Komatsubara KM, Marr BP, Carvajal RD. Uveal melanoma: epidemiology, etiology, and treatment of primary disease. Clin Ophthalmol. 2017;11:279-289. PubMed
20.Rodriguez A, Duenas-Gonzalez A, Delgado-Pelayo S. Clinical presentation and management of uveal melanoma. Mol Clin Oncol. 2016;5(6):675-677. PubMed
21.Harbour JW, Shih HA. Initial management of uveal and conjunctival melanomas. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2022: http://www.uptodate.com. Accessed 2022 May 10.
22.Sato T. Locoregional management of hepatic metastasis from primary uveal melanoma. Semin Oncol. 2010;37(2):127-138. PubMed
23.Drug Reimbursement Review sponsor submission: Kimmtrak (tebentafusp), 100 mcg / 0.5 mL solution for intravenous infusion [internal sponsor's package]. Toronto (ON): Medison Pharma Canada Inc.; 2022 Apr 20.
24.Keytruda (pembrolizumab): solution for infusion 100 mg/4 mL vial [product monograph]. Kirkland (QC): Merck Canada Inc; 2022 May 4. Accessed 2022 Jun 2.
25.Opdivo (nivoluman): intravenous solution (10mg/mL) 40 and 100 mg single-use vials [product monograph]. Montreal (QC): Bristol-Myers Squibb Canada Co; 2022 Feb 28. Accessed 2022 Jun 2.
26.Yervoy (ipilimumab): intravenous infusion, 5 mg/mL in 10 mL and 40 mL vials [product monograph]. Montreal (QC): Bristol-Myers Squibb Canada Co; 2022 March 3. Accessed 2022 Jun 2.
27.Clinical Study Report: IMCgp100-102. A phase 1/2, open-label, multi-center study of the safety and efficacy of IMCgp100 using the intra-patient escalation dosing regimen in patients with advanced uveal melanoma [internal sponsor's report]. Oxfordshire (GB): Immunocore Ltd; 2021 Feb 8.
28.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 Guideline Statement. J Clin Epidemiol. 2016;75:40-46. PubMed
29.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jul 07.
30.Center for Drug Evaluation Research. Multidiscipline review(s). Kimmtrak (tebentafusp) intravenous infusion. Company: Immunocore, LLC. Application No.: 761228 . Approval date: 01/25/2022 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2022: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761228Orig1s000MultidisciplineR.pdf. Accessed 2022 Jun 23.
31.Fayers PM, Aaronson NK, Bjordal K, et al. The EORTC QLC-C30 scoring manual (3rd Edition). Brussels (BE): European Organisation for Research and Treatment of Cancer (EORTC); 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2021 Apr 23.
32.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
33.Snyder CF, Blackford AL, Sussman J, et al. Identifying changes in scores on the EORTC-QLQ-C30 representing a change in patients' supportive care needs. Qual Life Res. 2015;24(5):1207-1216. PubMed
34.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-117. PubMed
35.Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727-1736. PubMed
36.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. PubMed
37.Statistical analysis plan: a phase II randomized, open-label, multi-center study of the safety and efficacy of IMCgp100 compared with investigator choice in HLA-A*0201 positive patients with previously untreated advanced uveal melanoma [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Kimmtrak (tebentafusp), 100 mcg / 0.5 mL solution for intravenous infusion [internal sponsor's package]. Toronto (ON): Medison Pharma Canada Inc. 2020 Oct 28.
38.Kelly D, Rose AAN, Muniz TP, et al. Development of a Metastatic Uveal Melanoma Prognostic Score (MUMPS) for Use in Patients Receiving Immune Checkpoint Inhibitors. Cancers (Basel). 2021;13(14). PubMed
39.Tebentafusp for the treatment of HLA-A*02:01-positive metastatic uveal melanoma - clinical systematic literature review [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kimmtrak (tebentafusp), 100mcg/0.5 mL for intraveous infusion. Abingdon (GB): Immunocore; 2021 Oct 19.
40.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-947. PubMed
41.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28(10):935-945. PubMed
42.Phillippo D, Ades T, Dias S, Palmer S, Abrams K, Welton N. NICE DSU Technical Support Document 18: Methods for population-adjusted indirect comparisons in submissions to NICE. Vol 18. Sheffield (UK): Decision Support Unit, ScHARR, University of Sheffield; 2016: https://research-information.bris.ac.uk/ws/portalfiles/portal/94868463/Population_adjustment_TSD_FINAL.pdf. Accessed 2022 Jun 24.
43.Pelster MS, Gruschkus SK, Bassett R, et al. Nivolumab and Ipilimumab in Metastatic Uveal Melanoma: Results From a Single-Arm Phase II Study. J Clin Oncol. 2021;39(6):599-607. PubMed
44.Liu J, Lichtenberg T, Hoadley KA, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173(2):400-416 e411. PubMed
45.Kim H, Gurrin L, Ademi Z, Liew D. Overview of methods for comparing the efficacies of drugs in the absence of head-to-head clinical trial data. Br J Clin Pharmacol. 2014;77(1):116-121. PubMed
46.Immunocore. NCT02570308: A phase I/II open-label, multi-center study of the safety and efficacy of IMCgp100 using the intra-patient escalation dosing regimen in patients with advanced uveal melanoma. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/study/NCT02570308. Accessed 2022 Jul 4.
47.Propensity score analysis comparing IMCgp100 (from study IMCgp100-202) to ipilimumab+nivolumab (from GEM-1402 study) in previously untreated uveal melanoma patients statistical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kimmtrak (tebentafusp), 100 mcg / 0.5 mL solution for intravenous infusion, 100mcg/0.5 mL for intraveous infusion. Dublin (IE): Immunocore; 2022 Jun 16.
48.Austin PC. The use of propensity score methods with survival or time-to-event outcomes: reporting measures of effect similar to those used in randomized experiments. Stat Med. 2014;33(7):1242-1258. PubMed
49.Luo N, Fones CS, Lim SE, Xie F, Thumboo J, Li SC. The European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-c30): validation of English version in Singapore. Qual Life Res. 2005;14(4):1181-1186. PubMed
50.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. J Patient Rep Outcomes. 2021;5(1):4. PubMed
51.Richardson J, Iezzi A, Khan MA, Chen G, Maxwell A. Measuring the Sensitivity and Construct Validity of 6 Utility Instruments in 7 Disease Areas. Med Decis Making. 2016;36(2):147-159. PubMed
52.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-Defined Estimates of the Minimally Important Difference for EQ-5D-5L Index Scores. Value Health. 2017;20(4):644-650. PubMed
53.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
54.Herdman M, Kerr C, Pavesi M, et al. Testing the validity and responsiveness of a new cancer-specific health utility measure (FACT-8D) in relapsed/refractory mantle cell lymphoma, and comparison to EQ-5D-5L. J Patient Rep Outcomes. 2020;4(1):22. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 18, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Humans
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
(Kimmtrak* or tebentafusp* or imcgp100* or imcgp 100* or immtac gp* or WHO10664 or WHO 10664 or N658GY6L3E).ti,ab,kf,ot,hw,nm,rn.
1 use medall
*tebentafusp/
(Kimmtrak* or tebentafusp* or imcgp100* or imcgp 100* or immtac gp* or WHO10664 or WHO 10664).ti,ab,kf,dq.
3 or 4
5 use oemezd
(conference review or conference abstract).pt.
6 not 7
2 or 8
remove duplicates from 9
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search terms -- Kimmtrak, tebentafusp]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- Kimmtrak, tebentafusp]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- Kimmtrak, tebentafusp]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- Kimmtrak, tebentafusp]
Grey Literature
Search dates: May 1-15, 2022
Keywords: Kimmtrak, tebentafusp, melanoma
Limits: none
Updated: Search updated prior to the meeting of CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC).
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Carvajal RD, Nathan P, Sacco JJ, et al. Phase I Study of Safety, Tolerability, and Efficacy of Tebentafusp Using a Step-Up Dosing Regimen and Expansion in Patients With Metastatic Uveal Melanoma. J Clin Oncol. 2022:JCO2101805. | Study Design |
Sacco J., Carvajal R., Butler M, et al. A Phase (ph) II, Multi-Center Study of the Safety and Efficacy of Tebentafusp (tebe) (IMCgp100) in Patients (pts) with Metastatic Uveal Melanoma (mUM). Ann Oncol. 2020. 2020 Dec 1. | Study Design |
Clinical Study Report: IMCgp100-102. A phase 1/2, open-label, multi-center study of the safety and efficacy of IMCgp100 using the intra-patient escalation dosing regimen in patients with advanced uveal melanoma [internal sponsor’s report]. Oxfordshire (GB): Immunocore, Ltd.; 2021 Feb 8. | Study Design |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 36: Subgroup Analysis of OS by ECOG PS (ITT) - Primary Analysis (Data Cut-Off Date of October 13, 2020)
Outcome | Tebentafusp | Investigator’s choice |
|---|---|---|
ECOG = 0 | ||
N | 192 | 85 |
Death, n (%) | 59 (30.7) | 42 (49.4) |
Overall survival (months)a, median (95% CI) | 24.1 (20.2 to 32.3) | 16.9 (13.7 to 19.3) |
Hazard Ratiob (95% CI) | 0.48 (0.33 to 0.72) | |
ECOG = 1 | ||
N | 49 | 31 |
Death, n (%) | 24 (49.0) | 18 (58.1) |
Overall survival (months)a, median (95% CI) | 17.4 (7.6 to 19.5) | 8.3 (4.7 to 20.5) |
Hazard Ratiob (95% CI) | 0.72 (0.39 to 1.36) | |
P value for interaction (ECOG = 0 vs 1)c | 0.28 | |
CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; ITT = intention to treat.
aBased on Kaplan-Meier estimate.
bEstimated using the Cox proportional hazards model with Efron’s approach for handling ties adjusted for treatment, subgroup, and treatment-by-subgroup interaction term.
cP value < 0.1 suggests there is an interaction between treatment and the subgroup. The p value was not adjusted for multiplicity.
Source: Study 202 Clinical Study Report11
Table 37: Sensitivity Analyses of PFS Outcomes (ITT) — Final Analysis (Data Cut-Off Date of October 13, 2020)
Outcome | Tebentafusp | Investigator’s choice |
|---|---|---|
Sensitivity analysis – Evaluation-time bias | ||
N | 252 | 126 |
PFS event, n (%) | 200 (79.4) | 101 (80.2) |
Death | 17 (6.7) | 18 (14.3) |
Disease progression | 135 (53.6) | 53 (42.1) |
Disease progression on first scan and first scan outside of correct time windows | 30 (11.9) | 19 (15.1) |
Disease progression occurred at a scan performed outside of the protocol-scheduled time points | 18 (7.1) | 11 (8.7) |
PFS (months)a, median (95% CI) | 3.0 (2.9 to 4.4) | 2.9 (2.8 to 2.9) |
Hazard Ratiob (95% CI) | 0.77 (0.60 to 0.98) | |
P valuec | 0.0308 | |
Sensitivity analysis – Attrition bias | ||
N | 252 | 126 |
PFS event, n (%) | 195 (77.4) | 89 (70.6) |
Death | 13 (5.2) | 9 (7.1) |
Disease progression | 182 (72.2) | 80 (63.5) |
PFS (months)a, median (95% CI) | 3.3 (3.0 to 5.0) | 2.9 (2.8 to 3.0) |
Hazard Ratiob (95% CI) | 0.74 (0.58 to 0.96) | |
P valuec | 0.0208 | |
CI = confidence interval; ITT = intention to treat; PFS = progression-free survival.
aBased on Kaplan-Meier estimate.
bHazard ratio was estimated using a Cox proportional hazards model stratified by LDH status.
cP value based on log-rank test of the Kaplan-Meier curve stratified by LDH status. P value was not adjusted for multiplicity.
Source: Study 202 Clinical Study Report.11
Table 38: Subgroup Analysis of PFS by ECOG PS (ITT Analysis Set) — Final Analysis (Data Cut-Off Date of October 13, 2020)
Outcome | Tebentafusp | Investigator’s choice |
|---|---|---|
ECOG = 0 | ||
N | 192 | 85 |
Progression-free survival event, n (%) | 148 (77.1) | 68 (80.0) |
Progression-free survival (months)a, median (95% CI) | 4.2 (3.0 to 5.6) | 2.9 (2.8 to 3.1) |
Hazard Ratiob (95% CI) | 0.68 (0.51 to 0.91) | |
ECOG = 1 | ||
N | 49 | 31 |
Progression-free survival event, n (%) | 43 (87.8) | 27 (87.1) |
Progression-free survival (months)a, median (95% CI) | 2.9 (2.8 to 3.4) | 2.8 (2.6 to 3.3) |
Hazard Ratiob (95% CI) | 0.86 (0.53 to 1.41) | |
CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; ITT = intention to treat.
aBased on Kaplan-Meier estimate.
bEstimated using the Cox proportional hazards model with Efron’s approach for handling ties adjusted for treatment, subgroup, and treatment-by-subgroup interaction term.
Source: Study 202 Clinical Study Report.11
Table 39: EQ-5D-5L Descriptive System Results (ITT) — Data Cut-Off Date of October 13, 2020
EQ-5D dimension | Tebentafusp | Investigator’s choice | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Baseline | C3D1 | C9D1 | C17D1 | EOT | Baseline | C3D1 | C9D1 | C17D1 | EOT | |
N | 194 | 160 | 85 | 27 | 111 | 78 | 58 | 14 | 6 | 59 |
Mobility, n (%) | ||||||||||
No problems | 167 (86.1) | 138 (86.3) | 70 (82.4) | 20 (74.1) | 83 (74.8) | 62 (79.5) | 46 (79.3) | 11 (78.6) | 4 (66.7) | 42 (71.2) |
Slight problems | 20 (10.3) | 15 (9.4) | 7 (8.2) | 5 (18.5) | 19 (17.1) | 12 (15.4) | 10 (17.2) | 3 (21.4) | 2 (33.3) | 7 (11.9) |
Moderate problems | 3 (1.5) | 6 (3.8) | 7 (8.2) | 2 (7.4) | 7 (6.3) | 4 (5.1) | 2(3.4) | 0 | 0 | 7 (11.9) |
Severe problems | 2 (1.0) | 1 (0.6) | 1 (1.2) | 0 | 2 (1.8) | 0 | 0 | 0 | 0 | 3 (5.1) |
Extreme problems | 2 (1.0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Self-care, n (%) | ||||||||||
No problems | 184 (94.8) | 148 (92.5) | 79 (92.9) | 25 (92.6) | 99 (89.2) | 74 (94.9) | 54 (93.1) | 14 (100) | 6 (100) | 54 (91.5) |
Slight problems | 8 (4.1) | 9 (5.6) | 5 (5.9) | 1 (3.7) | 9 (8.1) | 3 (3.8) | 4(6.9) | 0 | 0 | 3 (5.1) |
Moderate problems | 0 | 2 (1.3) | 1 (1.2) | 0 | 1 (0.9) | 1 (1.3) | 0 | 0 | 0 | 2 (3.4) |
Severe problems | 0 | 0 (0.6) | 0 | 1 (3.7) | 1 (0.9) | 0 | 0 | 0 | 0 | 0 |
Extreme problems | 2 (1.0) | 0 | 0 | 0 | 1 (0.9) | 0 | 0 | 0 | 0 | 0 |
Usual activities, n (%) | ||||||||||
No problems | 158 (81.4) | 113 (70.6) | 62 (72.9) | 18 (66.7) | 70 (63.1) | 61 (78.2) | 43 (74.1) | 11 (78.6) | 5 (83.3) | 36 (61.0) |
Slight problems | 28 (14.4) | 34 (21.3) | 14 (16.5) | 6 (22.2) | 27 (24.3) | 13 (16.7) | 10 (17.2) | 3 (21.4) | 1 (16.7) | 11 (18.6) |
Moderate problems | 7 (3.6) | 11 (6.9) | 6 (7.1) | 1 (3.7) | 12 (10.8) | 4 (5.1) | 5(8.6) | 0 | 0 | 11(18.6) |
Severe problems | 1 (0.5) | 2 (1.3) | 3 (3.5) | 2 (7.4) | 1 (0.9) | 0 | 0 | 0 | 0 | 1 (1.7) |
Extreme problems | 0 | 0 | 0 | 0 | 1 (0.9) | 0 | 0 | 0 | 0 | 0 |
Pain or discomfort, n (%) | ||||||||||
No problems | 115 (59.3) | 105 (65.6) | 47 (55.3) | 14 (51.9) | 55 (49.5) | 50 (64.1) | 33 (56.9) | 10 (71.4) | 4 (66.7) | 24 (40.7) |
Slight problems | 60 (30.9) | 41 (25.6) | 28 (32.9) | 9 (33.3) | 39 (35.1) | 17 (21.8) | 18 (31.0) | 3 (21.4) | 2 (33.3) | 23 (39.0) |
Moderate problems | 16 (8.2) | 12 (7.5) | 9 (10.6) | 3 (11.1) | 12 (10.8) | 11 (14.1) | 6 (10.3) | 1 (7.1) | 0 | 10 (16.9) |
Severe problems | 3 (1.5) | 2 (1.3) | 1 (1.2) | 1 (3.7) | 5 (4.5) | 0 | 1 (1.7) | 0 | 0 | 1 (1.7) |
Extreme problems | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.7) |
Anxiety or depression, n (%) | ||||||||||
No problems | 98 (50.5) | 101 (63.1) | 49 (57.6) | 15 (55.6) | 45 (40.5) | 37 (47.4) | 39 (67.2) | 8 (57.1) | 5 (83.3) | 25 (42.4) |
Slight problems | 56 (28.9) | 43 (26.9) | 24 (28.2) | 8 (29.6) | 42 (37.8) | 29 (37.2) | 18 (31.0) | 3 (21.4) | 1 (16.7) | 17 (28.8) |
Moderate problems | 32 (16.5) | 15 (9.4) | 8 (9.4) | 4 (14.8) | 18 (16.2) | 8 (10.3) | 0 | 2 (14.3) | 0 | 11 (18.6) |
Severe problems | 4 (2.1) | 1 (0.6) | 3 (3.5) | 0 | 3 (2.7) | 3 (3.8) | 1 (1.7) | 1 (7.1) | 0 | 4 (6.8) |
Extreme problems | 4 (2.1) | 0 | 1 (1.2) | 0 | 3 (2.7) | 1 (1.3) | 0 | 0 | 0 | 2 (3.4) |
C3D1 = Cycle 3 day 1; C9D1 = Cycle 9 day 1; C17D1 = Cycle 17 day 1; EOT = end of treatment; ITT = intention to treat.
Source: Study 202 Clinical Study Report.11
Table 40: Sensitivity Analyses of OS and PFS in Unadjusted Indirect Comparison and MAIC
Outcome | Tebentafusp | Ipilimumab plus nivolumab | |
|---|---|---|---|
Study 202 Unadjusted | Study 202 Match-adjusted | GEM-1402 | |
Overall survival sensitivity analysis – disease location (pooled categories) | |||
N or ESS | N = 237 | ESS = 210.61 | N = 52 |
Death event, n | 82 | 73.4 | 39 |
Overall survival (months), median | 21.7 | 23.4 | 12.1 |
12-month overall survival probability (%) | 74.7 | 76.4 | 51.2 |
Hazard ratio (95% CI) versus ipilimumab plus nivolumab, robust SE | 0.514 (0.35 to 0.756) | 0.476 (0.313 to 0.724) | REF |
Overall survival sensitivity analysis - largest metastatic liver lesion | |||
N or ESS | N = 237 | ESS = 101.61 | N = 52 |
Death event, n | 82 | 57.1 | 39 |
Overall survival (months), median | 21.7 | 21.6 | 12.1 |
12-month overall survival probability (%) | 74.7 | 79.6 | 51.2 |
Hazard ratio (95% CI) versus ipilimumab plus nivolumab, robust SE | 0.514 (0.35 to 0.756) | 0.495 (0.314 to 0.781) | REF |
Progression-free survival sensitivity analysis – disease location (pooled categories) | |||
N or ESS | N = 237 | ESS = 210.61 | N = 52 |
Progression-free survival event, n | 190 | 178.4 | 51 |
Progression-free survival (months), median | 3.3 | 3.4 | 3.1 |
12-month progression-free survival probability (%) | 14.7 | 14.9 | 15.4 |
Hazard ratio (95% CI) versus ipilimumab plus nivolumab, robust SE | 0.717 (0.525, 0.978) | 0.702 (0.498 to 0.989) | REF |
Progression-free survival analysis - largest metastatic liver lesion | |||
N or ESS | N = 237 | ESS = 101.61 | N = 52 |
Progression-free survival event, n | 190 | 130.7 | 51 |
Progression-free survival (months), median | 3.3 | 4.8 | 3.1 |
12-month progression-free survival probability (%) | 14.7 | 16.8 | 15.4 |
Hazard ratio (95% CI) versus ipilimumab plus nivolumab, robust SE | 0.717 (0.525, 0.978) | 0.645 (0.441 to 0.944) | REF |
CI = confidence interval; ESS = effective study size; SE = standard error.
Source: Sponsor-submitted Indirect Treatment Comparison Technical Report.14
Figure 11: KM Plot of Exploratory Postlandmark (Day 100) OS in Patients With BOR of Progressive Disease
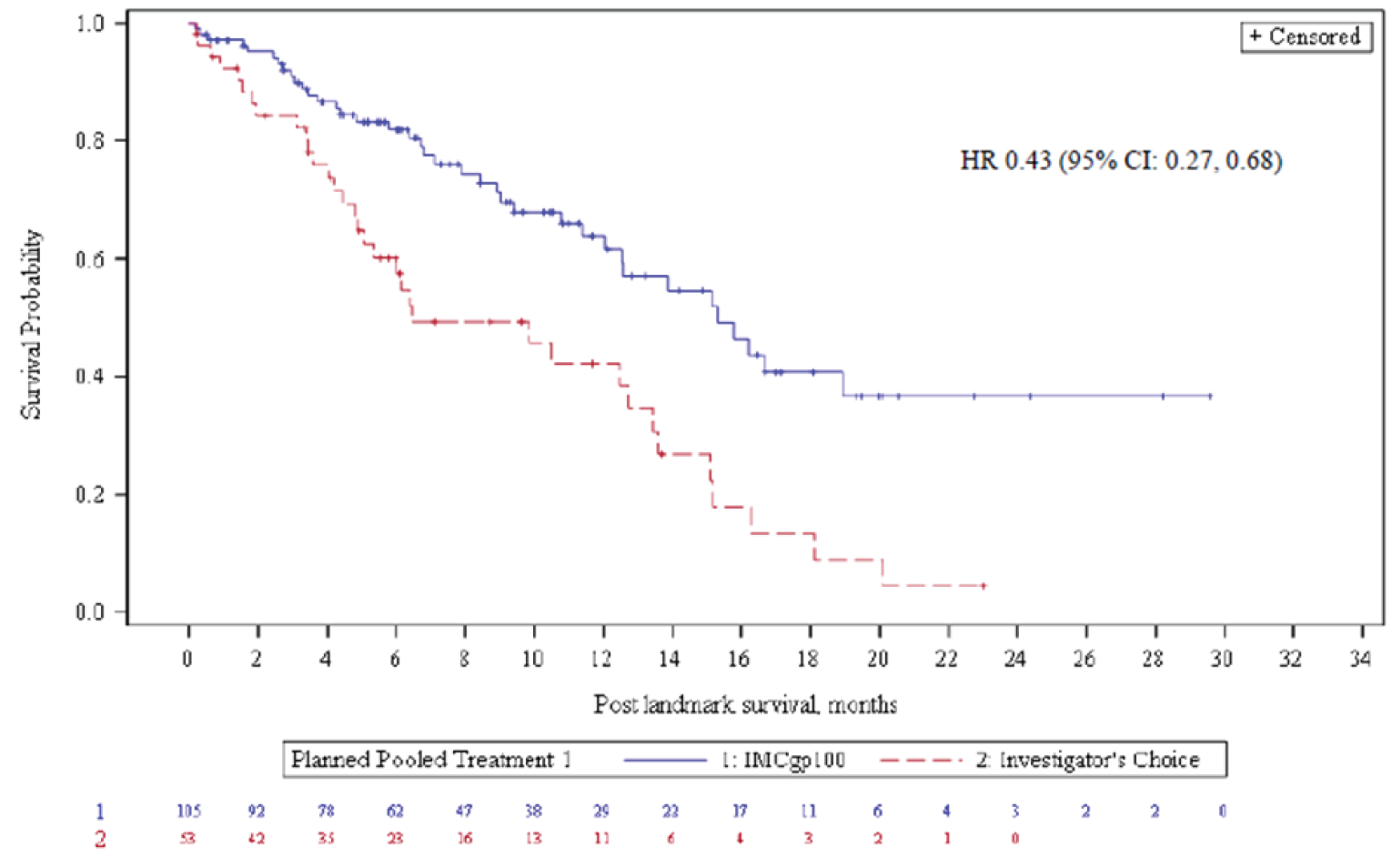
Source: Study 202 Clinical Study Report11
Figure 12: OS Curves of HLA-A*02:01-Positive and HLA-A*02:01-Negative Patients With mUM
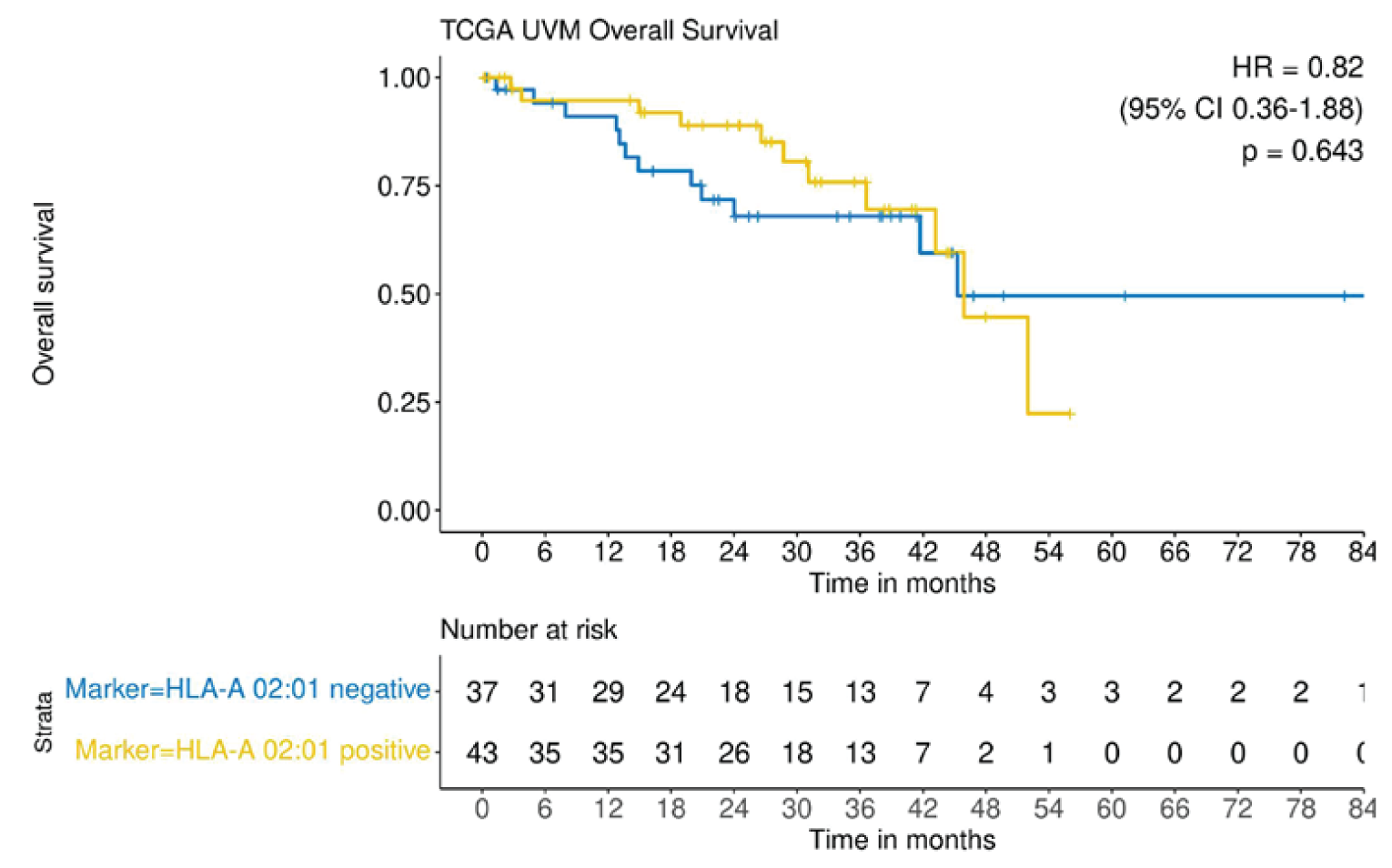
CI = confidence interval; HLA = human leukocyte antigen; HR = hazard ratio; mUM = metastatic uveal melanoma; OS = overall survival; TCGA = The Cancer Genome Atlas; UVM = uveal melanoma.
Source: Sponsor-submitted HLA status analysis.16
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30)
EQ-5D-5L
Findings
Table 41: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | A 30-item cancer-specific measure of HRQoL consisting of 5 multi-item functional scales (physical, role, emotional, cognitive and social functioning), 3 multi-item symptom scales (fatigue, nausea/vomiting and pain), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea and financial impact), and a 2-item quality of life scale.31 | Validity: Convergent validity was assessed using Spearman’s correlations between EORTC QLQ-C30 and SF-36 scales. Results showed moderate to strong correlations between EORTC QLQ-C30 and SF-36 scales, ranging from 0.35 to 0.67 across the assessed scales in patients in Singapore with various cancers.49 The EORTC QLQ-C30 demonstrated weak to strong interscale correlations (Spearman’s correlations r ranged from 0.07 to 0.73) in Kenyan patients with cancer.50 Known-groups validity: Generally, patients with cancer who had mild severity of symptoms, early stage of disease, and without presence of comorbid conditions reported higher scores in the 6 domains of EORTC QLQ-C30 (p values ranged from 0.015 to 0.970) in patients in Singapore with various cancer.49 Reliability: Internal consistency reliability was adequate in patients in Singapore and Kenya with various cancers. Cronbach alpha coefficients ranged from 0.19 to 0.95.49,50 Responsiveness: No relevant evidence of responsiveness found in patients with uveal melanoma. The validity, reliability, and responsiveness of the EORTC QLQ-C30 instrument have not been evaluated in patients with mUM. | Two studies estimated an MID of 10 points based on analysis in patients with breast cancer, small-cell lung cancer, and colorectal cancer.32,33 Another study in Canadian patients with various cancer types estimated MIDs for improvement to be between 10 to 32 points using SCNS-SF34 as the anchor and 9.1 to 23.5 using overall health and overall QoL as anchors. While the MID for deterioration was estimated to range from 9 to 21 points using SCNS-SF34 as the anchor and 7.2 to 13.5 using overall health and overall QoL as anchors.33,34 There have been no studies estimating the MIDs of the EORTC QLQ-C30 for patients with mUM. |
EQ-5D-5L | Generic preference based HRQoL instrument, consisting of a VAS and a composite index score of 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression.35 | Validity: Convergent validity: The EQ-5D-5L showed good convergent validity, reporting moderate to strong correlations with SF-36, the Capabilities Instrument, and the Subjective Well-Being Instrument of the UK Office of National Statistics, preference measures of VAS (r ranged from 0.43 to 0.66) in patients with a chronic disease including cancer.51 The EQ-5D-5L was able to discriminate between known groups based on healthy respondents and respondents with a chronic disease including Cancer, showing statistically significantly differences between groups in the mean index scores in patients.51 Reliability: No relevant evidence of reliability found in patients with uveal melanoma. Responsiveness: No relevant evidence of responsiveness found in patients with uveal melanoma. The validity, reliability, and responsiveness of the EQ-5D-5L instrument have not been evaluated in patients with mUM. | EQ-5D-5L: McClure et al. (2017) obtained a Canadian-specific simulation-based instrument-defined MID of 0.037.52 EQ-5D VAS: In patients with advanced (stage III or IV) cancer of the bladder, brain, breast, colon or rectum, head or neck, liver or pancreas, kidney, lung, lymphoma, ovary, or prostate, estimates of the MID ranged from 8 to 12 based on the ECOG performance status, and from 7 to 10 based on FACT quality of life questionnaire quintiles.36 There have been no studies estimating the MIDs of the EQ-5D-5L for patients with mUM. |
ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ VAS = EuroQol Visual Analogue Scale; EQ-5D-5L = EuroQol 5-dimensional 5-level questionnaire; FACT = Functional Assessment of Cancer Therapy; HRQoL= Health-related quality of life; MID = minimal important difference; MIPI = Mantle Cell Lymphoma International Prognostic Index; mUM = metastatic uveal melanoma; QoL = quality of life; SCNS-SF34 = Supportive Care Needs Survey-Short Form-34; SF-36 = Short Form 36 Health Survey; TOI = Trial Outcome Index; VAS = visual analogue scale.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30)
Description and Scoring
The EORTC QLQ-C30 is one of the most commonly used patient-reported outcomes measures in oncology clinical trials. It is a multidimensional, cancer-specific, evaluative measure of health-related quality of life (HRQoL). It was designed specifically for the purpose of assessing changes in participants’ HRQoL in clinical trials, in response to treatment.53 The core questionnaire of the EORTC QLQ-C30 consists of 30 questions that are scored to create 5 multi-item functional scales (physical, role, emotional, cognitive and social functioning), 3 multi-item symptom scales (fatigue, nausea/vomiting and pain), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea and financial impact), and a 2-item quality of life (QoL) scale. A higher score on the global health status and the functional assessments represents QoL improvement.31
The EORTC QLQ-C30 uses a 1-week recall period in assessing function and symptoms. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 to 4. For the 2 items that form the global quality of life scale, however, the response format is a 7-point Likert-type scale, with anchors between 1 (very poor) and 7 (excellent).31
Raw scores for each scale are computed as the average of the items that contribute to a particular scale. This scaling approach is based upon the assumption that it is appropriate to provide equal weighting to each item that comprises a scale. There is also an assumption that, for each item, the interval between response options is equal (for example, the difference in score between “not at all” and “a little” is the same as “a little” and “quite a bit,” at a value of 1 unit). Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, higher symptoms on the symptom scales, and better quality of life (i.e., higher scores simply reflect higher levels of response on that scale). Thus, a decline in score on the symptom scale would reflect an improvement, whereas an increase in score on the function and quality of life scale would reflect an improvement.31
Assessment of Validity, Reliability, and Responsiveness
Validity
There have been no studies evaluating the psychometric properties of the EORTC QLQ-C30 for patients with mUM. One cross-sectional study aimed to validate the EORTC QLQ-30 in 57 convenience sample of cancer patients in Singapore.49 Most patients had breast and colorectal cancer, leukemia, lung cancer, lymphoma, or germ cell tumour, and other cancers were also reported. Convergent validity was assessed using spearman’s correlations between EORTC QLQ-C30 and Short Form-36 (SF-36) scales, hypothesizing moderate to strong correlation (defined as correlation coefficient of 0.35–0.5, and >0.5, respectively) between scales of these 2 instruments measuring similar dimensions of HRQoL. Results showed moderate to strong correlations between EORTC QLQ-C30 and SF-36 scales, ranging from 0.35 to 0.67 across the assessed scales. In addition, a recent cross-sectional study in Kenya was conducted to evaluate the convergent validity of the EORTC QLQ-C30, using the English or Kiswahili version in a mix of 100 cancer patients.50 Most patients had breast cancer, followed by prostate, Kaposi sarcoma, lung, and other cancers. The study assessed convergent validly by examining the interscale correlations among the subscales of EORTC QLQ-C30. The interscale correlations were weak to strong and the absolute magnitude ranged from 0.07 to 0.73. Notably, with the exception of cognitive functioning, emotional functioning, nausea and vomiting, dyspnea, appetite loss, constipation, and diarrhea, the global health status (GHS) correlated moderately with the remaining subscales (r ≥ 0.30).50
Known-groups approach was used to compare 6 EORTC QLQ-C30 scale scores between patients reporting mild and severe symptoms, as well as by stage of disease and presence of comorbid conditions. With the exception of emotional functioning, the remaining 5 scales showed better scores in patients with mild symptoms than those with severe symptoms (P<0.05). Patients in early stages of cancer (or with no comorbid conditions) generally had better EORTC QLQ-C30 scores than those in advanced disease stages (or with comorbid conditions); however, none of these differences were statistically significant.49
Reliability
The Singaporean cross-sectional study described above also assessed internal consistency reliability by calculating Cronbach alpha coefficient for all the EORTC QLQ-C30 scales. Cronbach alpha coefficient was ≥ 0.70 for 6 of the 9 assessed the EORTC QLQ-C30 scales; cognitive functioning, physical functioning, and nausea and vomiting had a Cronbach alpha coefficient ranging from 0.19 to 0.68.49
The same study investigating convergent validity of the EORTC QLQ-C30 in Kenyan assessed the internal consistency of each scale of the questionnaire using Cronbach alpha coefficient. The EORTC QLQ-C30 demonstrated adequate scale internal consistency (Cronbach alpha coefficient values ranged from 0.72 to 0.95) with the exception of the Cognitive Function scale (Cronbach alpha coefficient = 0.37).50
Responsiveness
No literature was identified that assessed the responsiveness of EORTC QLQ-C30 in patients with uveal melanoma.
Minimal Important Difference (MID)
No relevant literature was identified that estimated the MIDs of EORTC QLQ-C30 in patients with mUM. For use in clinical trials, scores on the EORTC QLQ-C30 can be compared between different groups of patients or within a group of patients over time. One study conducted in breast cancer and small-cell lung cancer patients in 1998 estimated a clinically relevant change in score on any scale of the EORTC QLQ-C30 to be 10 points.32 The estimate was based on a study that used an anchor-based approach to estimating the MID in which patients who reported “a little” change (for better or worse) on the subjective significance questionnaire had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately five to 10 points. Participants who reported a “moderate” change had corresponding changes in the EORTC QLQ-C30 of about 10 to 20, and those who reported being “very much” changed had corresponding changes of more than 20.32
More recently in 2015, a Canadian study estimated the MIDs of EORTC QLQ-C30 scales using data from 193 newly diagnosed breast and colorectal cancer patients.33 The Supportive Care Needs Survey-Short Form-34 (SCNS-SF34) was used as an anchor; mean changes in EORTC QLQ C-30 scales associated with improvement, worsening, and no-change in supportive care based on the SCNS-SF34 was then calculated. MIDs were assessed for the following scales: Physical function, role function, emotional function, global health/QoL (i.e., GHS), pain, and fatigue. For improvement, MIDs associated with a statistically significantly improved supportive care needs ranged from 10 to 32 points. For worsening, MIDs associated with a statistically significantly worsening of supportive care needs ranged from nine to 21 points. The range for unchanged supportive care needs was from 1-point worsening to 16-point improvement in EORTC QLQ C-30 score.33 Based on this, the authors suggested a 10-point change in EORTC QLQ C-30 score represented changes in supportive care needs, and therefore should be considered for clinical use.33
In 2014, another Canadian study estimated the MID for EORTC QLQ-C30 in 369 patients with advanced cancer, who completed the questionnaire at baseline and 1-month postradiation.34 The most common cancer type was breast cancer, followed by lung, prostate, gastrointestinal, renal cell, and others. MID was estimated using both anchor and distribution-based methods for improvement and deterioration. Two anchors of overall health and overall QoL were used, both taken directly from the EORTC QLQ-C30 (questions 29 and 30) where patients rated their overall health and QoL themselves. Improvement and deterioration were categorized as an increase or decrease by 2 units to account for the natural fluctuation of patient scoring. With these 2 anchors, the estimated MIDs across all EORTC QLQ C-30 scales ranged from 9.1 units to 23.5 units for improvement, and from 7.2 units to 13.5 units for deterioration. Distribution-based estimates were closest to 0.5 SD.34
EQ-5D-5L
Description and Scoring
The EQ-5D-5L is a generic measure of HRQoL. It was developed by the EuroQol Group as an improvement to the EQ-5D-3L to measure small and medium health changes and reduce ceiling effects.35,52 The instrument comprises 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension is rated on 5 levels: level 1 “no problems,” level 2 “slight problems,” level 3 “moderate problems,” level 4 “severe problems,” and level 5 “extreme problems” or “unable to perform.”35,52 A total of 3,125 unique health states are possible, with 55555 representing the worst health state and 11111 representing the best state. The corresponding scoring of EQ-5D-5L health states is based on a scoring algorithm that is derived from preference data obtained from interviews using choice-based techniques (e.g., time trade-off) and discrete choice experiment tasks.35,52 The lowest and highest score varies depending on the scoring algorithm used. The anchors are 0 (dead) and 1 (full health), however negative values are also allowed to represent health states that a population considers worse than death. As an example, a Canadian scoring algorithm results in a score of -0.148 for health state 55555 (worst health state) and a score of 0.949 for health state 11111 (best health state). Another component of the EQ-5D-5L is a visual analogue scale (EQ VAS), which asks respondents to rate their health on a visual scale from 0 (worst health imaginable) to 100 (best health imaginable).35
Assessment of Validity, Reliability, and Responsiveness
The validity, reliability, and responsiveness of the EQ-5D-5L instrument have not been evaluated in patients with mUM. Richardson et al.51 examined various instruments, including the EQ-5D-5L, in respondents who were healthy and who had a chronic disease (i.e., arthritis, asthma, cancer, depression, diabetes, hearing loss, and heart disease) through an online survey in Australia, Canada, Germany, Norway, the UK, and the US. For convergent validity, the EQ-5D-5L was strongly correlated with the physical component of the SF-36 in cancer patients (r = 0.66), moderately correlated with the psychosocial content of the mental component of the SF-36, the Capabilities Instrument, and the Subjective Well-Being Instrument of the UK Office of National Statistics (r = 0.50), and moderately correlated with preference measures of VAS and time trade-off on own health state (r = 0.43).51
The EQ-5D-5L was able to discriminate between known groups based on presence or absence of lymphoma symptoms, Eastern Cooperative Oncology Group (ECOG) performance score, and Mantle Cell Lymphoma International Prognostic Index (MIPI), showing statistically significantly differences between groups in the mean index scores.54 Richardson et al.51 also assessed the known-group validity, the mean EQ-5D-5L differed between healthy respondents and respondents with a chronic disease (0.88 in healthy, 0.18 in patients with cancer).
Minimal Important Difference
No relevant literature was identified that estimated the MIDs of EQ-5D-5L in patients with mUM. McClure et al. (2017) obtained the MID for the EQ-5D-5L by calculating the average absolute difference between the index score of the baseline health state and the index score of all single-level transitions from the baseline state.52 A single-level transition was defined as a change in a single dimension to the next worse/better level, while holding all other dimensions constant. Such single-level transitions across all 3,125 health states were averaged to arrive at MIDs for various countries, by applying country-specific scoring algorithms. For Canada, transitions between levels 3 and 4 were excluded from the average to form a constant distribution of MID values across the range of baseline scores. This analysis resulted in a Canadian-specific MID of 0.037.52 No estimates of the MID were identified for patients with MCL.
Pickard et al. (2007) estimated the MID of the EQ-5D VAS based on cross-sectional data collected from 534 patients with advanced (stage III or IV) cancer of the bladder, brain, breast, colon or rectum, head or neck, liver or pancreas, kidney, lung, lymphoma, ovary, or prostate.36 Using both anchor- based and distribution-based methods, estimates of the MID ranged from 8 to 12 based on the ECOG performance status, and from 7 to 10 based on FACT quality of life questionnaire quintiles.36
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
ECOG PS
Eastern Cooperative Oncology Group Performance Status
HLA
human leukocyte antigen
HRQoL
health-related quality of life
IC
investigator’s choice
ICER
incremental cost-effectiveness ratio
MAIC
matching-adjusted indirect treatment comparison
mUM
metastatic uveal melanoma
OS
overall survival
PFS
progression-free survival
QALY
quality-adjusted life-year
RECIST
Response Evaluation Criteria in Solid Tumours
TTD
time-to-treatment discontinuation
UM
uveal melanoma
WTP
willingness-to-pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Tebentafusp (Kimmtrak), vial for IV solution |
Submitted price | Tebentafusp, 100 mcg/0.5 mL, single-use vial for IV solution: $18,565 per vial |
Indication | For the treatment of HLA-A*02:01-positive adults with unresectable or metastatic UM |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | June 7, 2022 |
Reimbursement request | Per indication |
Sponsor | Medison Pharma Canada Inc. |
Submission history | Previously reviewed: No |
HLA = human leukocyte antigen; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target population | Adults with HLA-A*02:01-positive advanced (metastatic or unresectable) UM Base case: Previously untreated patients Scenario: Previously untreated and treated patients |
Treatment | Tebentafusp |
Comparators | Base case: Investigator’s choice (a basket of comparators consisting of ipilimumab, pembrolizumab, and chemotherapy with dacarbazine) Scenario: Nivolumab-ipilimumab combination therapy and ipilimumab or pembrolizumab monotherapy |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (38 years) |
Key data source | Base case: A phase III, randomized, open-label, multicentre clinical trial (Study 202) assessing the safety and efficacy of tebentafusp compared with investigator’s choice of therapy in HLA-A*0201-positive patients with previously untreated advanced metastatic or unresectable UM Scenario: A MAIC of tebentafusp relative to nivolumab-ipilimumab combination therapy in previously untreated patients, a single-arm, phase I/II, open-label, multicentre trial (Study 102), and single-arm studies evaluating the safety of ipilimumab or pembrolizumab in previously treated patients with UM |
Submitted results | Base case: Tebentafusp was associated with an ICER of $538,951 per QALY compared with investigator’s choice of comparator (incremental costs = $677,127; QALYs = 1.26) Scenario: Tebentafusp was associated with an ICER of $686,554 per QALY compared with nivolumab-ipilimumab combination therapy in previously untreated patients, and $992,816 per QALY compared with ipilimumab or pembrolizumab monotherapy in previously treated patients |
Key limitations |
|
CADTH reanalysis results |
|
HLA = human leukocyte antigen; ICER = incremental cost-effectiveness ratio; LY = life-year; MAIC = matching-adjusted indirect comparison; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; UV = uveal melanoma; willingness-to-pay.
Conclusions
The CADTH clinical review showed that, compared with investigator’s choice (IC) of therapy (pembrolizumab, ipilimumab, or dacarbazine), tebentafusp (Kimmtrak) was associated with a statistically significant improvement in overall survival (OS) in previously untreated human leukocyte antigen (HLA)-A*02:01-positive adults with metastatic uveal melanoma (mUM) in the pivotal study known as Study 202. Treatment with tebentafusp was associated with statistically significant improvements in progression-free survival (PFS) but not in the objective response rate; however, the clinical expert consulted by CADTH considered these outcomes to be of limited clinical relevance. The clinical review also showed health-related quality of life (HRQoL) outcomes to be comparable in the tebentafusp and IC arms of the pivotal study. The long-term clinical effectiveness associated with tebentafusp treatment beyond the trial period (approximately 7.3 months) remains highly uncertain.
CADTH undertook a reanalysis in which a compliance rate of 100% was assumed for tebentafusp and the cap on tebentafusp treatment costs at 24 months was removed. CADTH could not address the uncertainty related to the long-term clinical effectiveness associated with tebentafusp treatment because of a lack of clinical data over a longer follow-up period, a lack of direct comparative evidence relative to the frequently used treatment in Canadian clinical practice at the time of this review, and limitations with the sponsor’s approach to model utilities. CADTH’s base case was aligned with the sponsor’s results, in that both analyses suggested that tebentafusp treatment was associated with higher costs and improved QALYs but was not cost-effective at a $50,000 per quality-adjusted life-year (QALY) willingness-to-pay (WTP) threshold. Based on publicly available list prices for all comparators, a price reduction of at least 91% would be required for tebentafusp to be cost-effective at this WTP threshold. Approximately 72% of the long-term survival benefit associated with tebentafusp was estimated through extrapolation of the OS curve beyond the observation period of the trial. Given the high degree of uncertainty in the long-term effectiveness of tebentafusp treatment, a larger price reduction may be warranted.
The sponsor submitted a scenario analysis that assessed the cost-effectiveness of tebentafusp with nivolumab plus ipilimumab combination therapy using efficacy data from a sponsor-submitted matching-adjusted indirect comparison (MAIC). According to the CADTH clinical review, no firm conclusions could be made regarding the comparative clinical efficacy of tebentafusp relative to the combination of nivolumab plus ipilimumab in the previously untreated population. Treatment with tebentafusp is more costly than a maintenance dose of nivolumab plus ipilimumab combination therapy, and there was insufficient evidence to justify a price premium for tebentafusp above the nivolumab plus ipilimumab combination.
There is insufficient direct and indirect comparative evidence in previously treated patients with mUM, and the cost-effectiveness of tebentafusp in this population is unknown. The cost-effectiveness of tebentafusp in the full Health Canada–indicated population (all HLA-A*02:01-positive adults with unresectable uveal melanoma [UM] or mUM, irrespective of prior treatment) is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process, specifically, information that pertains to the economic submission.
Patient input was received from the Save Your Skin Foundation and Melanoma Canada, which collected perspectives from patients with uveal and ocular melanoma through online surveys in Canada. Patients with UM reported impaired vision, which severely affected quality of life, depth perception, ability to drive and work, personal life, and mental health. Patients also described experience with current treatments, including brachytherapy, radiation and/or surgery, enucleation, immunotherapy, nivolumab plus ipilimumab combination therapy, nivolumab monotherapy, and tebentafusp accessed through special access programs. Patient-reported side effects of tebentafusp included fatigue, cognitive impairment, fever, skin rash, muscle or joint pain, fever or flu-like symptoms, and gastrointestinal issues. There is no current treatment available for disease that has metastasized. Treatment goals included a treatment that prolongs survival, delays progression, and preserves vision.
Clinician input was received from the Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee. The input was discussed at the Drug Advisory Committee’s meeting. Current systemic treatments include dacarbazine, ipilimumab, first-line nivolumab plus ipilimumab combination therapy, nivolumab monotherapy, and pembrolizumab monotherapy. Clinicians noted that treatment goals with a new systemic therapy would include improved OS and quality of life. It was noted that tebentafusp would be used as a first-line treatment for HLA-A*02:01-positive patients with UM. Candidates eligible to receive tebentafusp would be identified using HLA-A*02:01 genotype testing. Most centres in Ontario have testing capability, but the HLA-A*02:01 genotype test is not currently reimbursed for UM. The clinicians defined a clinically meaningful response to treatment as a decrease in tumour size of more than 20% on CT scans (taken every 3 to 4 months) and improved Eastern Cooperative Oncology Group Performance Status (ECOG PS). Furthermore, clinicians noted that some patients with radiographic progression (new and/or increasing target lesions) could continue therapy, given that a clinical benefit exists. However, treatment should be discontinued in cases of toxicity or symptomatic disease progression without clinical benefit. The clinician input also noted that additional health care resources are required to safely administer the first 3 doses of tebentafusp, including access to physician care to manage drug toxicity, capacity for inpatient admission, and patient monitoring. In addition, access to a medical oncologist would be needed to diagnose, treat, and monitor patients in outpatient settings.
The drug plans noted that patients with UM may receive immunotherapy (e.g., pembrolizumab, nivolumab, ipilimumab) and chemotherapy (e.g., dacarbazine, temozolomide, paclitaxel plus carboplatin) through public coverage on a case-by-case basis or enrolment in clinical trials. The plans noted that additional health system resources are required for the preparation, administration, and monitoring of tebentafusp. The preparation of each dose requires small volumes of tebentafusp and human albumin, which are requested from the blood bank. Vial sharing is also not possible for tebentafusp, which creates potential for large amounts of drug wastage. Further, the first 3 doses of tebentafusp would be administered in inpatient settings, which may be outside the scope of drug plan budgets in some jurisdictions. Additional time would be required to coordinate the transfer of treatment from inpatient to outpatient care teams and facilities.
Several of these concerns were addressed in the sponsor’s model:
The cost of HLA-A*02:01 genotype testing and inpatient costs associated with the administration of tebentafusp were considered in the analysis.
Treatment continuation after disease progression for patients receiving tebentafusp was also included in the analysis.
In addition, CADTH addressed some of these concerns as follows:
CADTH assumed no vial sharing when estimating the budget impact of reimbursing tebentafusp for patients with mUM.
CADTH was unable to address the following concerns that arose from stakeholder input:
The cost of human albumin admixture required for tebentafusp preparation in Canada is unknown.
Economic Review
The current review is for tebentafusp for the treatment of HLA-A*02:01-positive adults with unresectable or mUM.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of tebentafusp compared with IC of dacarbazine, ipilimumab, or pembrolizumab for the treatment of previously untreated patients with HLA-A*02:01-positive unresectable or mUM in the base-case analysis.1 The Health Canada indication does not specify prior treatment status of the patient population in the metastatic setting.2 The sponsor submitted a request for deviation to consider previously treated patients in a scenario analysis only, which was granted by CADTH.
Tebentafusp is available as a solution for IV infusion (100 mg per 0.5 mL single-use vial). The recommended dose is escalated from 20 mcg on day 1, 30 mcg on day 8, 68 mcg on day 15, and a maintenance dose of 68 mcg once every week thereafter. At the submitted price of $18,565 per vial, the cost of tebentafusp was $18,565 per week. The sponsor assumed that tebentafusp treatment would not accrue costs after 24 months. In estimating the cost of IC of therapy, the sponsor weighted cost by the distribution of treatments used in the control arm of Study 202, which was 12.7% for ipilimumab, 81.7% for pembrolizumab, and 5.6% for chemotherapy with dacarbazine.3 The per cycle cost of these treatments was based on dosages administered to patients in Study 202. This resulted in a per cycle cost of $10,375 for the first 4 doses and $6,876 for subsequent doses. The difference in costs exists because after 4 doses of ipilimumab, treatment with ipilimumab was stopped. The sponsor assumed no vial sharing and a treatment adherence rate of 95% when estimating drug acquisition costs.
The submitted model reported both QALYs and life-years over a lifetime time horizon (38 years) in the modelled population. The base-case analysis was conducted from the perspective of the Canadian public health care payer. Both costs and health outcomes were discounted at an annual rate of 1.5%.
Model Structure
The sponsor submitted a partitioned survival model that included 3 health states: preprogression, postprogression, and death (Appendix 3). The modelled time cycle was 1 week. All patients began in the preprogression health state; patients in this state were assumed to be stable or responding to therapy, defined by the PFS measure assessed in Study 202(Response Evaluation Criteria in Solid Tumours [RECIST] 1.1). Patients in the preprogression health state remained in that state, transitioned to death, or transitioned to the postprogression health state. The proportion of patients in the postprogression state was calculated as the proportion of patients alive (based on the OS curve) minus the proportion of patients alive and progression-free (based on the PFS curve). Patients in the postprogression health state remained in that state or transitioned to death.
Model Inputs
The modelled cohort’s characteristics were based on Study 202, an open-label, randomized, phase III study comparing tebentafusp with IC of therapy (dacarbazine, ipilimumab, or pembrolizumab) in patients who had not received prior lines of therapy. The mean age of the patient cohort was 63 years, and 49.7% of the patients were female. The sponsor derived PFS, OS, and time-to-treatment discontinuation (TTD) curves using data from the control and intervention arms of Study 202, using an August 2021 data cut-off date. Outcomes for all treatments were not adjusted for crossover from the control arm to the intervention arm that occurred from October 2020 to September 2021 (n = 16).
PFS, OS, and TTD outcomes were extrapolated beyond the trial duration by fitting parametric survival models to the trial data. Model selection was based on statistical fit (Akaike information criterion, Bayesian information criterion), good fit to observed data, clinical plausibility, and external validation against real-world data). The sponsor used a Weibull distribution to extrapolate OS and a generalized gamma distribution to model PFS. OS was capped in the model using general population mortality rates.
In the study protocol, patients discontinued treatment around the time of disease progression in the control arm but could have stayed on tebentafusp treatment after progression. As such, the sponsor based treatment discontinuation on the modelled TTD curves rather than the PFS curves. The TTD curves were adjusted so that the number of patients receiving treatment was never higher than the number of patients alive, as estimated by the OS curves. Further, the sponsor used parametric curves to extrapolate the tails of the TTD curves from the point at which only 15% of patients remained at risk.
The TTD curve closely followed the PFS curve in the control arm, but was higher than the PFS curve in the tebentafusp arm. Given that tebentafusp treatment may be continued after disease progression, health state utility values were modelled on the TTD curves.
Health utility declined by 0.084 after treatment discontinuation, reflecting patient preferences collected using the 5-Level EQ-5D instrument in Study 202. The sponsor could not identify any published literature on health utility data in patients with mUM or on modelled health utility for preprogression and postprogression states. Instead, the sponsor modelled a gradual decrease in health utility as time to death decreased, based on sponsor-consulted clinical expert opinion and published literature.4 Utilities at baseline were adjusted for age.5
The model included costs related to drug acquisition, administration, monitoring, adverse events (AEs), and terminal care. Costs applied in the model were based on health states, using published literature and prices listed in public formularies.6-10 Patients were assumed to accrue costs of subsequent treatments after disease progression, which included immunotherapy (nivolumab plus ipilimumab combination, ipilimumab, pembrolizumab, and nivolumab) and chemotherapy with dacarbazine.
The sponsor’s model included AEs of grade 3 or higher that were prevalent in more than 3% of all patients, as well as endocrine disorders and colitis of any grade, using data from Study 202. AEs were assumed to occur during the first treatment cycle only. Patients with AEs accrued both costs and disutilities, which were obtained from published literature.11,12
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base case and 500 iterations for scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented here. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s base-case analysis, tebentafusp was associated with an incremental cost of $677,127 and 1.26 additional QALYs compared with IC over the lifetime horizon, resulting in an incremental cost-effectiveness ratio (ICER) of $538,951 per QALY (Table 3). In the sponsor’s base case, tebentafusp had a 0% probability of being the most cost-effective strategy at a WTP threshold of $50,000 per QALY. Nearly half (53%) of the incremental QALYs associated with tebentafusp treatment compared with IC were accrued in the postprogression health state. Nearly two-thirds (61%) of QALYs associated with tebentafusp were accrued beyond the trial follow-up and were based on the sponsor’s extrapolations of trial data.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total cost ($) | Incremental cost ($) | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|
Investigator’s choicea | 140,805 | Reference | 1.14 | Reference | Reference |
Tebentafusp | 817,932 | 677,127 | 2.40 | 1.26 | 538,951 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aInvestigator’s choice of treatment comprised dacarbazine, ipilimumab, or pembrolizumab.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analyses Results
The sponsor submitted a scenario analysis comparing the cost-effectiveness of tebentafusp with a treatment frequently used in Canada (nivolumab plus ipilimumab combination therapy) in previously untreated HLA-A*02 to 01-positive adults advanced UM. The efficacy data for tebentafusp and the combination of ipilimumab plus nivolumab was sourced from the MAIC and the study by Piulats et al. (2021), respectively.13,14 Overall, tebentafusp was associated with an ICER of $686,554 per QALY gained (incremental cost = $543,190; incremental QALY = 0.79).
The sponsor also provided a scenario analysis that compared the cost-effectiveness of tebentafusp with ipilimumab or pembrolizumab for the treatment of previously treated HLA-A*02:01-positive patients with unresectable or mUM. The OS and PFS curves for tebentafusp were obtained from Study 102, a phase I/II, open-label, single-arm trial that included patients with mUM who had received 1 or 2 prior lines of therapy.15 The sponsor derived simulated efficacy data for ipilimumab or pembrolizumab in previously treated patients using a hazard mapping approach and published single-arm studies that evaluated the safety of these comparators in previously treated patients with UM.16,17 Over the lifetime horizon, tebentafusp was associated with an ICER of $991,245 per QALY gained (incremental cost = $460,715; incremental QALY = 0.46).
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
Uncertainty about the long-term clinical efficacy of tebentafusp treatment: The sponsor submitted a partitioned survival model, in which the long-term clinical efficacy of tebentafusp is based on the sponsor’s extrapolations of PFS and OS using efficacy data from Study 202. In the intention-to-treat analysis, using the August 2021 data cut-off date, median OS and PFS were 5 months and about 12 days longer with tebentafusp than with IC, respectively. However, in the pharmacoeconomic analysis, the long-term extrapolation of survival advantage and PFS resulted in an incremental gain of 1.51 life-years and about 5 months, respectively. Approximately 72% of the OS benefit and 92% of the PFS benefit derived from tebentafusp treatment are accrued after the trial duration and, as a result, represent an outcome of modelling instead of real-world data. The results of this cost-effectiveness analysis are sensitive to the sponsor’s extrapolation assumptions, which were based on a small sample size and subject to the chance of bias, as well as limited generalizability. This adds uncertainty about the long-term clinical efficacy of tebentafusp treatment.
The sponsor used a log-logistic function to extrapolate the tail of the OS curve. Based on the sponsor’s extrapolated OS, about 16% of patients treated with tebentafusp are alive at the end of 5 years. Should the sponsor’s extrapolation of OS be an optimistic take on long-term survival, the long-term clinical efficacy associated with tebentafusp treatment may have been an overestimation.
The sponsor’s original pharmacoeconomic submission implied that a considerable proportion of the incremental benefit of tebentafusp (72%) would be realized after disease progression. CADTH asked that the sponsor provide justification for this apparent postprogression benefit. In response, the sponsor explained that the postprogression benefit was the result of continued tebentafusp treatment after disease progression and potential delayed effects of immunotherapy. The clinical expert consulted by CADTH found this explanation to be plausible. Regardless, a high degree of the postprogression benefit is based on the sponsor’s extrapolation of trial data and is associated with uncertainty because of the small sample size near the end of the trial’s follow-up period, meaning that the magnitude of the postprogression survival benefit remains uncertain.
In the scenario analysis, CADTH explored the impact of adopting a conservative extrapolation of the OS curve tail using the Weibull distribution.
Lack of direct comparative evidence to relevant comparator treatments: The clinical expert consulted by CADTH noted that, currently, nivolumab plus ipilimumab combination therapy is frequently prescribed for patients with mUM. The sponsor submitted a scenario analysis comparing the cost-effectiveness of tebentafusp with the nivolumab plus ipilimumab combination that used efficacy data from a sponsor-submitted MAIC. The CADTH clinical report identified several methodological limitations, including potential selection bias in the systematic literature review process, confounding due to heterogeneity in patient population, and loss of precision in results due to a reduction in effective sample size. The analysis was also restricted to previously untreated patients, so the results may not be generalizable to previously treated patients in the metastatic setting. As such, there was insufficient evidence to draw definitive conclusions from the sponsor-submitted MAIC about the relative efficacy of tebentafusp compared with the nivolumab plus ipilimumab combination.
CADTH could not address limitations related to the sponsor-submitted MAIC or the lack of direct comparative evidence. The cost-effectiveness of tebentafusp compared with nivolumab plus ipilimumab combination therapy is unknown.
Cost of tebentafusp is underestimated: The sponsor adopted a cap on drug acquisition costs for tebentafusp at 24 months. About 14% of patients remain on tebentafusp treatment at 24 months, based on the TTD curve, and continue to accrue health benefits. Asserting a benefit from treatment without considering drug acquisition costs is inappropriate and biases the results in favour of tebentafusp. In the drug’s product monograph, there is no cap imposed on treatment duration.2 Although treatment is recommended until unacceptable toxicity or disease progression in the product monograph, patients with evidence of progression continued treatment beyond progression in Study 202because they continued to derive clinical benefits from tebentafusp.2,3 CADTH solicited clinical expert input, which noted that treatment discontinuation in clinical practice would depend on a combination of clinical events and would be left to the discretion of the attending physician. As such, treatment discontinuation may vary among patients.
In the CADTH reanalysis, the cap on drug acquisition costs for tebentafusp was removed.
Use of compliance rate to estimate drug costs is not appropriate: The sponsor adopted a compliance rate of 95% to account for missed doses or treatment interruptions. This approach is problematic, as dose delays, missed doses, dose reductions to manage toxicity, and subsequent dose re-escalation have different impacts on drug costs. Tebentafusp and comparators are supplied as single-use vials and any unused product should be discarded.2,18-21 Tebentafusp also has a short shelf life; there is only a 4-hour window from the time of preparation to completion of infusion.2 It is unclear how the sponsor’s assumed compliance rate interacts with considerations about vial size and wastage. CADTH solicited clinical expert input, which noted that any planned missed doses due to vacation or AEs detected during clinical visits before dosing would not result in drug wastage because the dose would not have been prepared by pharmacies. Adopting a compliance rate of less than 100% to reflect treatment interruptions inappropriately reduces drug acquisition costs in the model.
In the CADTH reanalysis, a compliance rate of 100% was adopted.
In the comparator arm, the cost for dose 5 and subsequent doses is inaccurate: In the pharmacoeconomic analysis, the comparator arm is a basket of treatments used in the control arm of Study 202, distributed as follows: 12.7% for ipilimumab, 81.7% for pembrolizumab, and 5.6% for dacarbazine.3 Given that the treatment regimen recommends a total of 4 doses of ipilimumab, the cost of comparator arm should not include the cost of subsequent ipilimumab doses. As such, the cost of the comparator treatment should only be weighted by the distribution of pembrolizumab and dacarbazine for dose 5 and subsequent doses.
CADTH corrected treatment weighting by redistributing the weight assigned to ipilimumab equally over the other comparators in the control arm for doses 5 and subsequent doses (82.55% for pembrolizumab and 11.96% for dacarbazine).
Limitations associated with the sponsor’s chosen approach to model utilities: In estimating QALYs, the sponsor modelled health utilities based on TTD curves using data from Study 202. The sponsor’s approach assumes that patients who have progressed on tebentafusp experience the same health utility as patients who have not progressed. This assumption lacks face validity because, according to clinical expert input, HRQoL is worse for patients who have progressed than for patients who have not progressed. Patients with progressed disease exhibit symptoms such as pain, loss of appetite, decreased weight, low energy, and possible fractures resulting from bone metastasis. The sponsor’s approach may have overestimated the health utility of patients treated with tebentafusp in the postprogression health state, which biases the results in favour of tebentafusp. As noted in the CADTH guidelines,8 a more appropriate approach is to model health preferences (i.e., utilities) that reflect health states in the model and to combine health utilities to reflect a health state defined by a combination of health states.
CADTH could not address this limitation because of structural limitations of the sponsor’s model.
The key assumptions made by the sponsor have been appraised by CADTH and are shown in Table 4.
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Clinical effectiveness data used to inform the pharmacoeconomic submission included crossover between the intervention and comparator arms from October 2020 to September 2021. | CADTH notes that the most recent data cut-off date was used in the pharmacoeconomic analysis, but the analysis was not adjusted for crossover between the comparator arm and the tebentafusp arm. However, tebentafusp showed superiority at the first interim analysis, and only 16 patients had crossed over from the comparator arm to the tebentafusp arm from October 2020 to September 2021. The sponsor’s sensitivity analysis showed that using the October 2020 data cut-off date had a minor impact on the ICER. |
The sponsor assumed that patients on treatment have a higher health utility than patients off treatment. | This approach is unorthodox but did not meaningfully affect the ICER. The clinical expert consulted by CADTH found this assumption acceptable. |
The sponsor assumed that the quality of life of patients was maintained until 6 months before death, after which it gradually decreased until death (which had a utility value of 0). | This approach is unorthodox but did not meaningfully affect the ICER. The clinical expert consulted by CADTH found this assumption acceptable. |
The sponsor estimated the treatment effect on health utility using EQ-5D-5L data collected in Study 202. | Uncertain. Missing data related to deaths and poor questionnaire completion rates increase uncertainty of the results because of potential attrition bias. Further, the EQ-5D-5L instrument is not routinely administered to patients with mUM in clinical practice, per the clinical expert, and has not been validated in patients with mUM. |
Grade 3 AEs with a prevalence in more than 3% of all patients, as well as endocrine disorders and colitis of any grade, were included in the pharmacoeconomic model. | Uncertain. The sponsor assumed that AEs, except for endocrine disorders in the comparator arm, could occur only once during treatment. The use of health care resources to treat AEs was based on the opinion of clinical experts consulted by the sponsor. The sponsor assumed that pyrexia, pulmonary embolism, and colitis (any grade) would be treated only in the hospital setting. The inpatient cost of treating AEs was based on the Ontario Case Costing Initiative, which CADTH was unable to validate. The sponsor didn’t explicitly model disutility from AEs. In Study 202, 65.5% of patients in the tebentafusp arm and 27% of patients in the control arm had AEs. Treatment-related disutilities were obtained from published literature and based on the assumptions of clinical experts consulted by the sponsor. CADTH anticipates this limitation may have minimal impact on the cost-effectiveness of tebentafusp, given that AEs are transient. |
Treatment wanning. | Acceptable. Treatment effectiveness waning was not explicitly modelled. Given the gap between the OS curves of tebentafusp and IC decreases over time, some treatment waning is assumed. |
AE = adverse event; EQ-5D-5L = 5-Level EQ-5D; ICER = incremental cost-effectiveness ratio; mUM = metastatic uveal melanoma; OS = overall survival.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH corrected the sponsor’s model by estimating the cost of dose 5 in the comparator arm and subsequent doses using correct treatment weighting. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical expert. CADTH’s base case assumed a 100% compliance rate and removed the cap on tebentafusp treatment costs.
In the CADTH base case, tebentafusp was associated with estimated total costs and QALYs of $1,062,137 and 2.40, compared with total costs and QALYs of $147,736 and 1.14 for patients receiving IC of therapy. The ICER associated with tebentafusp compared with IC was $728,513 per QALY in the previously untreated population, and the probability of cost-effectiveness at a $50,000 per QALY WTP threshold was 0%. Results of the stepped reanalysis are shown in Table 6, with full disaggregated results shown in Table 14 in Appendix 4.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Treatment weighting to estimate cost of the comparator arm for dose 5 and beyond | Ipilimumab = 12.7% Pembrolizumab = 81.7% Dacarbazine = 5.6% | Ipilimumab = 0% Pembrolizumab = 88.05% Dacarbazine = 11.96% |
Changes to derive the CADTH base case | ||
1. Compliance rate | 95% | 100% |
2. Cap on tebentafusp treatment costs | Included at 24 months | Not included |
CADTH base case | — | 1 + 2 |
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case | ICa | 140,805 | 1.14 | Reference |
Tebentafusp | 817,932 | 2.40 | 538,951 | |
Sponsor’s corrected base case | ICa | 141,816 | 1.13 | Reference |
Tebentafusp | 819,028 | 2.37 | 543,152 | |
CADTH reanalysis 1 | ICa | 146,339 | 1.13 | Reference |
Tebentafusp | 857,966 | 2.37 | 570,754 | |
CADTH reanalysis 2 | ICa | 141,816 | 1.13 | Reference |
Tebentafusp | 1,003,934 | 2.37 | 691,453 | |
CADTH base case (1 + 2), deterministic | ICa | 146,339 | 1.13 | Reference |
Tebentafusp | 1,052,603 | 2.37 | 726,861 | |
CADTH base case (1 + 2), probabilistic | ICa | 147,736 | 1.14 | Reference |
Tebentafusp | 1,062,137 | 2.40 | 728,513 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Reanalysis is based on publicly available prices of the comparator treatments.
aInvestigator’s choice of treatment comprised dacarbazine, ipilimumab, or pembrolizumab.
Scenario Analysis Results
CADTH conducted a scenario analysis for the previously untreated population using a more conservative parametric function (Weibull) to extrapolate long-term OS in the tebentafusp arm. The ICER increased to $1,762,764 per QALY when compared to IC of therapy. Approximately 33% of the incremental life-years associated with tebentafusp was derived from the extrapolated period, compared to 72% in the base case. Full disaggregated results are shown in Table 17 in Appendix 4.
CADTH conducted a scenario analysis in the previously untreated population and found that tebentafusp was associated with an ICER of $722,268 per QALY gained when compared with nivolumab plus ipilimumab combination therapy. It is important to note that the estimated ICER relevant to the most frequently used treatment in the previously untreated population is highly uncertain, owing to a lack of direct comparative evidence and the previously identified methodological limitations of the sponsor’s MAIC.
CADTH conducted an additional scenario analysis in the previously treated population. In this analysis, tebentafusp was associated with an ICER of $1,054,187 per QALY compared with ipilimumab or pembrolizumab monotherapy. The cost-effectiveness of tebentafusp in the previously treated population could not be validated, owing to the lack of direct and indirect comparative evidence in this population.
A price reduction analysis was conducted based on the sponsor’s base case and CADTH’s reanalysis (Table 7). The results indicate that a price reduction of at least 91% is required for tebentafusp to be considered cost-effective at a WTP threshold of $50,000 per QALY.
Table 7: CADTH Price Reduction Analyses
Price reduction | ICERs for tebentafusp vs. investigator’s choicea ($/QALY) | |
|---|---|---|
Sponsor base case | CADTH reanalysis | |
No price reduction | $544,845 | $726,861 |
10% | $488,449 | $651,885 |
20% | $432,052 | $576,910 |
30% | $375,656 | $501,935 |
40% | $319,259 | $426,959 |
50% | $262,863 | $351,984 |
60% | $206,467 | $277,008 |
70% | $150,070 | $202,033 |
80% | $93,674 | $127,058 |
90% | $37,277 | $52,082 |
91% | $31,638 | $44,585 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aInvestigator’s choice of treatment comprised dacarbazine, ipilimumab, or pembrolizumab.
Issues for Consideration
Some patients with mUM in Canada currently receive tebentafusp through a compassionate access program. The circumstances surrounding compassionate access to tebentafusp are unclear, and no documentation was provided by the sponsor in this submission explaining the details of the program implementation. Should compassionate access cease and tebentafusp be reimbursed; the public health care payer will bear all costs.
Overall Conclusions
The CADTH clinical review showed that, compared with IC of therapy (pembrolizumab, ipilimumab, or dacarbazine), tebentafusp was associated with a statistically significant improvement in OS in previously untreated HLA-A*02:01-positive adults with mUM in the pivotal Study 202. Treatment with tebentafusp was associated with statistically significant improvements in PFS but not in the objective response rate; however, the clinical expert consulted by CADTH considered these outcomes to be of limited clinical relevance. The CLINICAL REVIEW also showed comparable HRQoL outcomes in the tebentafusp and IC arms of the pivotal study. The long-term clinical effectiveness associated with tebentafusp treatment beyond the trial period (approximately 7.3 months) remains highly uncertain.
CADTH identified several limitations of the sponsor’s submitted pharmacoeconomic analysis. These included a lack of long-term efficacy data for tebentafusp, the inappropriate inclusion of a cap on tebentafusp costs, the use of a compliance rate that introduced important uncertainties and likely underestimated treatment costs for tebentafusp, and methodological issues related to the way health state utilities were incorporated into the model. CADTH also identified an error related to the way the model reflected subsequent therapies.
CADTH undertook a reanalysis by adopting a compliance rate of 100% and removing the cap on tebentafusp treatment costs at 24 months. CADTH also corrected the error related to subsequent therapy. Although CADTH’s base case resulted in a higher ICER than the sponsor’s base case when tebentafusp was compared with IC of therapy ($728,513 per QALY) in the previously untreated population, both analyses suggested that tebentafusp treatment was associated with higher costs and more QALYs but was not cost-effective at a $50,000 per QALY WTP threshold compared with IC. Based on publicly available list prices for all comparators, a price reduction of at least 91% would be required to make tebentafusp an optimal treatment option at this WTP threshold. Approximately 72% of the long-term survival benefit associated with tebentafusp was estimated through extrapolation of the OS curve beyond the observation period of the trial. Given the high degree of uncertainty about the long-term effectiveness of tebentafusp treatment, a larger price reduction may be warranted.
The comparator considered in the base case, nivolumab plus ipilimumab combination therapy, is not currently frequently used in Canada for previously untreated patients. The sponsor submitted a scenario analysis that compared the cost-effectiveness of tebentafusp with nivolumab plus ipilimumab combination therapy using efficacy data from a sponsor-submitted MAIC. According to the CADTH clinical review, the MAIC was subject to important limitations, including a high risk of selection bias, confounding due to heterogeneity in patient populations, and a loss of precision in results due to a reduced effectiveness sample size. As such, no firm conclusions could be drawn regarding the comparative clinical efficacy of tebentafusp relative to the nivolumab plus ipilimumab combination therapy. Treatment with tebentafusp is more costly than the maintenance dose of nivolumab plus ipilimumab combination therapy, and there was insufficient evidence to justify a price premium for tebentafusp over nivolumab plus ipilimumab combination therapy in the previously untreated population.
There is insufficient direct and indirect comparative evidence in the previously treated population of patients with mUM, and the cost-effectiveness of tebentafusp in this population is unknown. The cost-effectiveness of tebentafusp in the full Health Canada–indicated population (all HLA-A*02:01-positive adults with unresectable UM or mUM, irrespective of prior treatment) is unknown.
References
1.Drug Reimbursement Review pharmacoeconomic report: GENERIC NAME (BRAND NAME) for INDICATION. Ottawa (ON): CADTH; 1800: URL. Accessed 1800 Jan 1.
2.Guidance title. Supporting evidence: committee papers. London (UK): National Institute for Health and Care Excellence (NICE); 1800: URL. Accessed 1800 Jan 1.
3.Lutathera (lutetium (177Lu) oxodotreotide): 370 MBq/mL at calibration [product monograph]. Milburn (NJ): Advanced Accelerator Applications USA Inc.; 2021 May 13.
4.NICE. TA366 Pembrolizumab for advanced melanoma not previously treated with ipilimumab. 2015; https://www.nice.org.uk/guidance/ta366.
5.Szende A JB, Cabases J, eds. Self-Reported Population Health: An International Perspective based on EQ-5D. 2014; https://www.ncbi.nlm.nih.gov/books/NBK500356/.
6.pCODR. Pembrolizumab (Keytruda) for Metastatic Melanoma. 2015.
7.pCODR. Nivolumab (Opdivo) for Metastatic Melanoma. 2016.
8.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 1800 Jan 1.
9.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 1800 Jan 1.
10.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 1800 Jan 1.
11.NICE. TA319 Ipilimumab for previously untreated unresectable malignant melanoma. 2013; https://www.nice.org.uk/guidance/ta319.
12.NICE. TA384 Nivolumab for Treating Advanced (Unresectable or Metastatic) Melanoma. 2016; https://www.nice.org.uk/guidance/ta384.
13.Sponsor's NMA report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Kimmtrak (tebentafusp), 100 mcg / 0.5 mL solution for intravenous infusion. Toronto (ON): Medison Pharma Canada Inc.; 2022 Apr 20.
14.Piulats JM EE, de la Cruz Merino L, et al. Nivolumab Plus Ipilimumab for Treatment-Naïve Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J Clin Oncol. 2021. PubMed
15.Clinical Study Report: IMCgp100-102. A phase 1/2, open-label, multi-center study of the safety and efficacy of IMCgp100 using the intra-patient escalation dosing regimen in patients with advanced uveal melanoma [internal sponsor's report]. Oxfordshire (GB): Immunocore, Ltd.; 2021 Feb 8.
16.Karydis I CP, Wheater M, Arriola E, Szlosarek PW, Ottensmeier CH. Clinical activity and safety of Pembrolizumab in Ipilimumab pre-treated patients with uveal melanoma. 2016.
17.Maio M DR, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab in patients with pre-treated, uveal melanoma. Ann Oncol. 2013. PubMed
18.Inc. MC. KEYTRUDA® pembrolizumab PM 2022; https://www.merck.ca/en/wp-content/uploads/sites/20/2021/04/KEYTRUDA-PM_E.pdf.
19.Co. B-MSC. OPDIVO® nivolumab for injection PM 2022; https://www.bms.com/assets/bms/ca/documents/productmonograph/OPDIVO_EN_PM.pdf.
20.ULC PC. rDacarbazine for Injection BP PM 2019; https://www.pfizer.ca/sites/default/files/201902/Dacarbazine_PM_E_221511_25Jan2019.pdf.
21.Co. B-MSC. YERVOY® Ipilimumab for injection PM. 2022; https://www.bms.com/assets/bms/ca/documents/productmonograph/YERVOY_EN_PM.pdf.
22.DeltaPA. [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/. Accessed 1800 Jan 1.
23.CADTH. Nivolumab (Opdivo) in combination with Ipilimumab (Yervoy). 2021; https://www.cadth.ca/sites/default/files/DRR/2021/PC0229-Opdivo-Yervoy-combined.pdf.
24.pCODR. Nivolumab (Opdivo) plus Ipilimumab (Yervoy) for Advanced Renal Cell Carcinoma. 2018; https://www.cadth.ca/sites/default/files/pcodr/pcodr_opdivo_yervoy_rcc_fn_egr.pdf.
25.CADTH. Nivolumab (Opdivo). 2022; https://www.cadth.ca/sites/default/files/DRR/2022/PC0259%20Opdivo%20-%20Final%20CADTH%20Rec.pdf.
26.Sponsor's NMA report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Drug name, doses and administration. City (PROV): Sponsor name; 1800 Jan 1.
27.pCODR. pCODR Review Report: OPDIVA in combination with YERVOY for Metastatic Melanoma. 2017; https://www.cadth.ca/sites/default/files/pcodr/pcodr_opdivo_yervoy_metmela_fn_rec.pdf.
28.pCODR. pCODR Review Report: OPDIVO for Melanoma Adjuvant Treatment. 2019; https://www.cadth.ca/sites/default/files/pcodr/Reviews2019/10147NivolumabMelanoma%28Adjuvant%29_FnRec_ChairApproved_Post_07Mar2019_Final.pdf.
29.pCODR. pCODR Review Report: KEYTRUDA for Melanoma Adjuvant Treatment. 2019; https://www.cadth.ca/sites/default/files/pcodr/Reviews2019/10168PembrolizumabMAT_FnRec_31%20July%202019__Post_01Aug2019_final.pdf.
30.Ghazawi FM DR, Le M, et al. Uveal melanoma incidence trends in Canada: a national comprehensive population-based study. Br J Ophthalmol. 2019. PubMed
31.Tam VC KY, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol 2013. PubMed
32.NICE. British National Formulary (BNF). 2022; https://www.nice.org.uk/bnf-uk-only.
Appendix 1: Cost Comparison Tables
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for mUM
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Average Daily cost | Average 28-day cost |
|---|---|---|---|---|---|---|
Tebentafusp (Kimmtrak) | 100 mcg/0.5 mL | Vial IV infusion | 18,565.0000a | 20 mcg on Day 1, 30 mcg on Day 8, 68 mcg on Day 15, and 68 mcg once every week thereafter | 2,652.14 | 74,260 |
aSponsor’s submission1
Table 9: CADTH Cost Comparison Table for mUM (Not Indicated for mUM)
Treatment | Strength / concentration | Form | Price ($) | Recommended dosagea | Average Daily cost | Average 28-day cost |
|---|---|---|---|---|---|---|
Chemotherapy | ||||||
Dacarbazine (generic) | 6 mg/mL | 100 mL Vial IV infusion | 251.8200 | 1,000 mg/m2 on Day 1 every 3 weeks | 35.97 | 1,007 |
Immunotherapy | ||||||
Ipilimumab (Yervoy) | 5 mg/mL | 10 mL 40 mL Vial IV infusion | 5,8000.000b 23,200.00d | 3 mg/kg on Day 1 every 3 weeksc | 1,380.95 | 38,667 |
Pembrolizumab (Keytruda) | 25 mg/mL | 4 mL Vial IV infusion | 4,400.0000e | 2 mg/kg (or up to a maximum of 200 mg) on day 1 every 3 weeks | 419.05 | 11,733 |
Nivolumab (Opdivo) | 10 mg/mL | 4 mL 10 mL Vial IV infusion | 782.2200e 1,955.5600e | Monotherapy: 3 mg/kg (up to 240 mg) on Day 1 every 2 weeks Combination therapy with ipilimumab Initial dose: 1 mg/kg on Day 1 every 3 weeks for a total of 4 doses Maintenance dose: nivolumab monotherapy | Monotherapy: 335.24 Combination therapy with ipilimumab Initial dose: 74.50 | Monotherapy: 9,387 Combination therapy with ipilimumab Initial dose: 2,086 |
Nivolumab + Ipilimumab | Initial dose: 1,455.45 Maintenance dose: 335.24 | Initial dose: 40,753 Maintenance dose: 9,387 | ||||
Note: All prices are the wholesale prices from IQVIA Delta PA (accessed July 2022),22 unless otherwise indicated, and do not include dispensing fees. All cost calculations for agents with weight or height-based dosing was calculated using the mean body surface area of 1.8 m2 and mass of 75 kg. Daily and 28-day costs include drug wastage.
aRecommended dosages obtained from the IMC-gp100-102 trial.15
bCADTH review report (2021).23
cTreatment duration lasts for a total of 4 doses.
dCADTH review report (2018).24
eCADTH review report (2022).25
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | See CADTH appraisal section |
Model has been adequately programmed and has sufficient face validity | No | See CADTH appraisal section |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The model does not allow consideration of treatment waning. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of Sponsor’s Base Case — Previously Untreated Patients With mUM
Variable | Tebentafusp | Investigator’s choicea |
|---|---|---|
Total discounted costs ($) | $819,028.36 | $139,704.68 |
Drug costs | $703,161.89 | $58,669.68 |
Administration costs | $12,534.78 | $689.38 |
Subsequent therapy | $36,654.52 | $25,160.35 |
Health care resources – PFS | $24,451.37 | $12,681.84 |
Health care resources – PPS | $12,942.45 | $13,040.53 |
Health care resources – Death | $8,599.09 | $7,972.59 |
AE | $708.03 | $959.85 |
Friction | $0.00 | $0.00 |
Mortality | $19,976.24 | $20,530.46 |
Total QALYs | 2.37 | 1.13 |
PFS | 0.69 | 0.34 |
PD | 1.71 | 0.81 |
AE | -0.02 | -0.02 |
Total LYs | 3.06 | 1.56 |
PFS | 0.88 | 0.46 |
PPS | 2.18 | 1.09 |
aInvestigator’s choice of treatment was comprised of dacarbazine, ipilimumab, or pembrolizumab.
Table 12: Disaggregated Summary of Sponsor’s Scenario Analysis — Previously Untreated Patients With mUM
Variable | Tebentafusp | Nivolumab plus Ipilimumab |
|---|---|---|
Total discounted costs ($) | $770,571.65 | $227,845.77 |
Drug costs | $645,044.69 | $168,220.03 |
Administration costs | $14,499.62 | $1,947.00 |
Subsequent therapy | $47,119.56 | $958.82 |
Health care resources – PFS | $21,694.25 | $4,600.24 |
Health care resources – PPS | $12,786.19 | $22,382.00 |
Health care resources – Death | $8,796.91 | $7,258.32 |
AE | $647.25 | $2,215.93 |
Friction | $0.00 | $0.00 |
Mortality | $19,983.18 | $20,263.42 |
Total QALYs | 2.52 | 1.70 |
PFS | 0.65 | 0.57 |
PD | 1.89 | 1.17 |
AE | -0.02 | -0.03 |
Total LYs | 3.09 | 2.15 |
PFS | 0.79 | 0.72 |
PPS | 2.30 | 1.44 |
Table 13: Disaggregated Summary of Sponsor’s Scenario Analysis — Previously Treated Patients With mUM
Variable | Tebentafusp | Investigator’s Choicea |
|---|---|---|
Total discounted costs ($) | $639,262.96 | $178,806.10 |
Drug costs | $534,876.54 | $98,654.78 |
Administration costs | $10,262.60 | $818.29 |
Subsequent therapy | $35,743.92 | $24,025.26 |
Health care resources – PFS | $16,030.30 | $13,277.96 |
Health care resources – PPS | $13,016.08 | $13,036.33 |
Health care resources – Death | $8,101.90 | $7,258.04 |
AE | $705.01 | $1,031.54 |
Friction | $0.00 | $0.00 |
Mortality | $20,526.62 | $20,703.89 |
Total QALYs | 1.13 | 0.67 |
PFS | 0.43 | 0.34 |
PD | 0.72 | 0.35 |
AE | -0.02 | -0.02 |
Total LYs | 1.56 | 1.00 |
PFS | 0.58 | 0.48 |
PPS | 0.98 | 0.52 |
aInvestigator’s choice of treatment was comprised of ipilimumab or pembrolizumab monotherapy.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 14: Disaggregated Summary of CADTH’s Economic Evaluation Results — Previously Untreated Patients With mUM
Variable | Tebentafusp | Investigator’s Choicea |
|---|---|---|
Total discounted costs ($) | $1,052,603.49 | $146,339.19 |
Drug costs | $934,807.83 | $63,979.96 |
Administration costs | $12,534.78 | $689.38 |
Subsequent therapy | $38,583.71 | $26,484.58 |
Health care resources – PFS | $24,451.37 | $12,681.84 |
Health care resources – PPS | $12,942.45 | $13,040.53 |
Health care resources – Death | $8,599.09 | $7,972.59 |
AE | $708.03 | $959.85 |
Friction | $0.00 | $0.00 |
Mortality | $19,976.24 | $20,530.46 |
Total QALYs | 2.37 | 1.13 |
PFS | 0.69 | 0.34 |
PD | 1.71 | 0.81 |
AE | -0.02 | -0.02 |
Total LYs | 3.06 | 1.55 |
PFS | 0.88 | 0.46 |
PPS | 2.18 | 1.09 |
aInvestigator’s choice of treatment was comprised of dacarbazine, ipilimumab, or pembrolizumab.
Scenario Analyses
CADTH conducted a scenario analysis to assess the uncertainty surrounding the extrapolation of OS curve used in the sponsor’s model. Assuming a conservative extrapolation of OS increased the estimated ICER associated with tebentafusp compared with investigator’s choice of therapy (dacarbazine, ipilimumab, or pembrolizumab) in the previously untreated population.
Table 15: Summary of CADTH Scenario Analysis — Assuming Weibull Distribution on Previously Untreated Patients With mUM
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Sponsor's corrected base case | |||
Investigator’s Choicea | 139,704.68 | 1.13 | Reference |
Tebentafusp | 819,028.36 | 2.37 | 544,845 |
CADTH's base case | |||
Investigator’s Choicea | 63,979.96 | 1.13 | Reference |
Tebentafusp | 931,973.83 | 1.64 | 722,268 |
aInvestigator’s choice of treatment was comprised of dacarbazine, ipilimumab, or pembrolizumab.
CADTH found that tebentafusp treatment was associated with higher costs and improved QALYs compared with nivolumab-ipilimumab combination therapy in the previously untreated population and compared with ipilimumab or pembrolizumab in the previously treated population. Results of these scenario analyses are highly uncertain due to the previously identified limitations in the sponsor’s MAIC (for comparisons to nivolumab-ipilimumab in the previously untreated population) and the lack of direct or indirect evidence for the previously treated population.
Table 16: Summary of CADTH Scenario Analysis — Previously Untreated Patients With mUM
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Sponsor's corrected base case | |||
Nivolumab + Ipilimumab | 227,845.77 | 1.70 | Reference |
Tebentafusp | 770,571.65 | 2.52 | 686,554 |
CADTH's base case | |||
Nivolumab + Ipilimumab | 218,049.26 | 1.70 | Reference |
Tebentafusp | 807,753.01 | 2.52 | 722,268 |
Table 17: Summary of CADTH Scenario Analysis — Previously Treated Patients With mUM
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Sponsor's corrected base case | |||
Ipilimumab or pembrolizumab monotherapy | 178,806.10 | 0.67 | Reference |
Tebentafusp | 639,262.96 | 1.13 | 991,245 |
CADTH's base case | |||
Ipilimumab or pembrolizumab monotherapy | 185,262.94 | 0.67 | Reference |
Tebentafusp | 669,295.62 | 1.13 | 1,054,187 |
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 18: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA),26 assessing the expected budgetary impact of reimbursing tebentafusp for the treatment of HLA-A*02:01-positive adult patient with unresectable or metastatic uveal melanoma (mUM). The total budget impact was the sum of estimated budget impact in the previously treated and untreated populations. The analysis was done from the perspective of a Canadian public health care payer over a 3-year time horizon (2023 to 2025). Key inputs to the BIA are documented in Table 19.
The sponsor estimated the population size using an epidemiology-based approach, with data obtained from published literature to estimate the number of patients eligible for treatment with tebentafusp. The comparators included off-label treatments for mUM such as ipilimumab monotherapy, pembrolizumab monotherapy, nivolumab monotherapy and nivolumab-ipilimumab combination therapy. Comparators’ costs were obtained from previous CADTH review reports,27-29 and dosing regimens were obtained from product monograph and IMCgp100-202 trial.3,21 Total costs were calculated by multiplying drug cost per administration with the number of doses administered over the treatment duration. The sponsor estimated the duration of treatment with tebentafusp is twice as much as the comparators. The sponsor assumed no drug wastage in estimating treatment costs.
The sponsor assumed that if tebentafusp is reimbursed by public health care payer, patients currently receiving tebentafusp under compassionate use will continue to access the drug at no cost to payers for a limited time after the funding decision. As such, the sponsor excluded the treatment cost of patients currently receiving tebentafusp through compassionate use from the budget impact estimated in the first year of reimbursement.
Table 19: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Incidence of UM (per 1,000,000) | 4.71 / 4.79 / 4.86 a |
Proportion of unresectable or mUM | 50%b |
Percentage of patients to be reimbursed by public health care payer | 100% b |
Proportion of HLA-A*02:01 positive | 47% b |
Proportion of HLA-A*02:01 positive cases who fail on current therapy | 68%c |
Number of patients eligible for drug under review (previously untreated and treated patients) | 83 / 85 / 87 |
Number of patients eligible for drug under review (previously untreated patients) | 35 / 36 / 37 |
Number of patients eligible for drug under review (previously treated patients) | 47 /49 / 50 |
Number of patients currently receiving tebentafusp under compassionate use in Canadian Drug Review participating drug programs | 35 b |
Market Uptake (3 years) | |
Uptake (reference scenario) - previously untreated patients Tebentafusp Ipilimumab + nivolumab Pembrolizumab monotherapy Nivolumab monotherapy Ipilimumab monotherapy | 0% / 0% / 0% 76% / 76% / 76% 12% / 12% / 12% 12% / 12% / 12% 0% / 0% / 0% |
Uptake (reference scenario) - previously treated patients Tebentafusp Ipilimumab + nivolumab Pembrolizumab monotherapy Nivolumab monotherapy Ipilimumab monotherapy | 0% / 0% / 0% 54% / 54% / 54% 4% / 4% / 4% 4% / 4% / 4% 38% / 38% / 38% |
Uptake (new drug scenario) - previously untreated patients Tebentafusp Ipilimumab + nivolumab Pembrolizumab monotherapy Nivolumab monotherapy Ipilimumab monotherapy | 57% / 75% / 86% 33% / 19% / 11% 5% / 3% / 2% 5% / 3% / 2% 0% / 0% / 0% |
Uptake (new drug scenario) - previously treated patients Tebentafusp Ipilimumab + nivolumab Pembrolizumab monotherapy Nivolumab monotherapy Ipilimumab monotherapy | 50% / 40% / 30% 27% / 32% / 38% 2% / 2% / 3% 2% / 2% / 3% 19% / 23% / 27% |
Cost of treatment (per patient) | |
Cost of treatment over 21 days Tebentafusp – doses 1- 3 Tebentafusp – doses 4+ Ipilimumab + nivolumab Pembrolizumab monotherapy Nivolumab monotherapy Ipilimumab monotherapy | $21,906.70 $37,872.60 $28,985.43 $6,939.68 $6,939.66 $27,443.28 |
Administration cost (per patient)b | |
Tebentafusp Ipilimumab + nivolumab Pembrolizumab monotherapy Nivolumab monotherapy Ipilimumab monotherapy | $7,177.51 $2,001.86 $500.46 $750.70 $1,501.39 |
HLA = human leukocyte antigen; mUM = metastatic uveal melanoma.
aSource: Published literature30
bSource: Sponsor’s submission26
cSource: IMCgp100-202 trial3
dAdministration cost includes cost of intravenous infusion, inpatient administration, human albumin for admixture (20% concentration (200 mg per 1 mL, 50mL vial)) and companion diagnostic test (HLA 27 Typing, code L586).9,31,32
Summary of the Sponsor’s BIA Results
The sponsor estimated the net 3-year budget impact of introducing tebentafusp for the treatment of HLA-A*02:01-positive adult patient with unresectable or mUM to be $15,704,116. The sponsor estimated reimbursement of tebentafusp will result in cost savings of $1,907,254 in year 1. However, the budget impact would be cost-intensive in year 2 ($8,742,578) and year 3 ($8,868,578).
In a scenario analysis excluding the patients expected to receive tebentafusp through compassionate care, the sponsor estimated the net 3-year budget impact on public health care payer of reimbursing tebentafusp to be $25,749,638 (Year 1: $8,138,268; Year 2: $8,742,578; Year 3: $8,868,792). For previously untreated HLA-A*02:01-positive adult patient with mUM, the sponsor estimated the 3-year budget impact of introducing tebentafusp to be $15,257,907 (Year 1: $3,866,915; Year 2: $5,229,690; Year 3: $6,161,303). The estimated 3-year impact of introducing tebentafusp for the treatment of previously untreated HLA-A*02:01-positive adult patient with mUM was $10,491,730 (Year 1: $4,271,353; Year 2: $3,512,889; Year 3: $2,707,489).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The budget impact does not represent likely use of tebentafusp: In the sponsor’s submitted BIA, about 44% of eligible patients are currently receiving tebentafusp via compassionate access programs in Canadian Drug Review participating drug programs. The sponsor anticipates that these patients will continue to receive compassionate access to tebentafusp for a limited time only if tebentafusp is reimbursed by public plans. CADTH requested that the sponsor provide documentation confirming that compassionate access program will be provided an additional year beyond the funding decision. In response, the sponsor clarified that the BIA assumes that patients currently receiving tebentafusp through compassionate access will continue to receive it until public reimbursement is obtained. The clinical expert consulted for this review by CADTH noted that compassionate programs typically terminate as soon as public funding is available and many sponsors transfer patients currently receiving compassionate access to a publicly funded program. Should compassionate access cease and tebentafusp is reimbursed, the cost of tebentafusp treatment will be borne by the public health care payer.
In its current form, the sponsor’s base case estimate of budget impact is conditional on the funding decision such that compassionate access program will be continued for 44% of eligible population only if tebentafusp is reimbursed by public plans. Given that the sponsor will not continue compassionate access beyond approval, the cost of tebentafusp treatment will not be borne by public plans regardless of the reimbursement status. In this case, the reimbursement of tebentafusp will not have an impact on the total estimated budget impact.
In CADTH reanalysis, 0% of eligible patients were assumed to receive compassionate access to tebentafusp once reimbursed by public plans. CADTH explored the impact of continuing the compassionate access program in a scenario analysis.
Drug acquisition costs were underestimated: The sponsor assumed no drug wastage in estimating drug acquisition costs. The budget impact guidelines recommend consideration of drug costs such that costs reflect the number of drug units dispensed, rather than consumed. The product monographs note tebentafusp and comparators are available as a single-use product.2,18-21 This means any unused product would be discarded and the cost of drug wastage would be incurred by public health care payer. As such, the sponsor’s approach to calculating drug costs does not align with the budget impact guidelines and respective product monographs.
In CADTH reanalysis, drug wastage was included in estimating drug acquisition costs.
Market share of tebentafusp is underestimated: The clinical expert consulted for this review by CADTH anticipated a potential higher uptake of tebentafusp. Tebentafusp may become the standard of care for patients with mUM given there is a limited number of current treatment options with limited efficacy. This clinical expert noted that this change in practice will be abrupt once tebentafusp is funded, and the market share of tebentafusp may be higher than assumed in the sponsor’s submission. The clinical expert noted the market share of tebentafusp may increase to the range of 86% in year 1 and 2 for previously untreated patients. For previously treated patients, the clinical expert anticipated a market share in the range of 50% to 70% in year 1. There is uncertainty regarding prior treatments received by previously treated patient population. Should these patients have had prior treatment with tebentafusp, the market share of tebentafusp in year 2 and 3 may be in the range of 20% to 10%. However, given the limited efficacy of current treatment options, more patients would progress on the previous line of therapy and expected to switch to tebentafusp. As such, tebentafusp may continue to have a higher market share than comparators in the previously treated population.
In CADTH reanalysis, the market share of tebentafusp was increased to 86% in year 1 and 2 in previously untreated patients. In the previously treated population, the market share of tebentafusp was increased to 60% in year 1 based on feedback from clinical expert.
In scenario analysis, CADTH explored the impact of decreasing market share of tebentafusp to 20% in year 2 and 10% in year 3 in the previously treated population.
Treatment duration of tebentafusp is uncertain: In estimating the drug cost of tebentafusp, the sponsor adopted the median treatment duration of 8 cycles (24 doses) using data from IMCgp100-202 trial. However, treatment duration may vary for patients in clinical practice. In the IMCgp100-202 trial, treatment with tebentafusp was continued beyond initial progression for nearly half (43.3%) of the patients in the intervention arm. The median duration of treatment postdisease progression was 1.9 months for these patients. The clinical expert consulted by CADTH for this review noted that treatment with tebentafusp may be continued postdisease progression in clinical practice if patient is deriving benefit from the treatment. The decision to discontinue treatment would be at the discretion of the physician and depend on a combination of clinical events. Should treatment duration with tebentafusp be longer than assumed in clinical practice, the budget impact may have been underestimated.
In scenario reanalysis, CADTH explored the impact of extending treatment duration by 1.9 months for patients treated with tebentafusp.
Additional limitations were identified but not considered as key limitations. These included misalignment of relevant comparators between the sponsor-submitted pharmacoeconomic and budget impact analyses. The sponsor included dacarbazine and ipilimumab monotherapy as relevant comparators in the pharmacoeconomic analysis, but these comparators do not have a market share in the budget impact analysis. The sponsor excluded these comparators from the budget impact analysis based on limited uptake of these treatments in current Canadian practice. The clinical expert consulted by CADTH for this review agreed with the sponsor’s assertion. Further, the cost of human albumin required for admixture in preparation of tebentafusp was obtained from British National Formulary and may not represent the product cost in Canada.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by excluding the sponsor’s assumption on compassionate access, including drug wastage, and increasing the market share of tebentafusp.
Table 20: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Assuming compassionate access in the year of 2023 | Yes | No |
2. Drug wastage | Excluded | Included |
3.Market share in new drug scenario (year 1 / year 2 / year 3) | Previously untreated patients with mUM: Tebentafusp: 57% / 75% / 86% Ipilimumab + nivolumab: 33% / 19% / 11% Pembrolizumab monotherapy: 5% / 3% / 2% Nivolumab monotherapy: 5% / 3% / 2% Ipilimumab monotherapy: 0% / 0% / 0% Previously treated patients with mUM: Tebentafusp: 50% / 40% / 30% Ipilimumab + nivolumab: 27% / 32% / 38% Pembrolizumab monotherapy: 2% / 2% / 3% Nivolumab monotherapy: 2% / 2% / 3% Ipilimumab monotherapy: 19% / 23% / 27% | Previously untreated patients with mUM: Tebentafusp: 86% / 86% / 86% Ipilimumab + nivolumab: 11% / 11% / 11% Pembrolizumab monotherapy: 2% / 2% / 2% Nivolumab monotherapy: 2% / 2% / 2% Ipilimumab monotherapy: 0% / 0% / 0% Previously treated patients with mUM: Tebentafusp: 60% / 40% / 30% Ipilimumab + nivolumab: 22% / 32% / 38% Pembrolizumab monotherapy: 2% / 2% / 3% Nivolumab monotherapy: 2% / 2% / 3% Ipilimumab monotherapy: 15% / 23% / 27% |
CADTH base case | Reanalysis 1 + 2 + 3 | |
In the CADTH reanalysis, the 3-year budget impact of reimbursing tebentafusp for the treatment of HLA-A*02:01-positive adult patient with unresectable or mUM increased to $54,017,379 (Year 1: $20,229,773; Year 2: $17,499,596; Year 3: $16,288,010). For previously untreated patients, the 3-year budget impact was estimated to be $32,731,194 (Year 1: $10,613,612; Year 2: $10,909,080; Year 3: $11,208,502). For previously treated patients, the 3-year budget impact was estimated to be $21,286,185 (Year 1: $9,616,161; Year 2: $6,590,516; Year 3: $5,079,509).
The results were sensitive to uncertainty in treatment duration of tebentafusp. Extending treatment duration of tebentafusp by 1.9 months increased the estimated 3-year total budget impact to $77,670,205.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18.
Table 21: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total | ||
|---|---|---|---|
Previously untreated population | Previously treated population | Total population | |
Submitted base case | NA | NA | $15,704,116 |
Submitted base case, corrected | $15,257,907 | $10,491,730 | $25,749,638 |
CADTH reanalysis 1 | $27,756,838 | $19,683,492 | $47,440,330 |
CADTH reanalysis 2 | $17,992,306 | $11,346,001 | $29,338,307 |
CADTH base case | $32,731,194 | $21,286,185 | $54,017,379 |
BIA = budget impact analysis
NA: Budget impact for treated and untreated population that is adjusted for assumption of compassionate access was not provided.
CADTH also conducted additional scenario analyses to address remaining uncertainty using the CADTH base case. Results are provided in Table 22. The scenario analysis involved:
Extending treatment duration of tebentafusp is extended by additional 1.9 months
Decreasing market share of tebentafusp in the previously treated population to 20% in year 1 and 10% in year 2
Continuing compassionate access program in the first year of funding conditional on tebentafusp reimbursement status
Tebentafusp price reduction of 91%
Table 22: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $8,148,251 | $8,387,968 | $8,622,480 | $8,860,142 | $25,870,590 |
New drug | $8,148,251 | $6,480,714 | $17,365,058 | $17,728,934 | $41,574,706 | |
Budget impact | $0 | -$1,907,254 | $8,742,578 | $8,868,792 | $15,704,116 | |
Submitted base case, corrected | Reference | $8,148,251 | $8,387,968 | $8,622,480 | $8,860,142 | $25,870,590 |
New drug | $8,148,251 | $16,526,235 | $17,365,058 | $17,728,934 | $51,620,228 | |
Budget impact | $0 | $8,138,268 | $8,742,578 | $8,868,792 | $25,749,638 | |
CADTH base case | Reference | $8,204,894 | $8,446,250 | $8,682,389 | $8,921,700 | $26,050,340 |
New drug | $8,204,894 | $28,676,023 | $26,181,985 | $25,209,711 | $80,067,719 | |
Budget impact | $0 | $20,229,773 | $17,499,596 | $16,288,010 | $54,017,379 | |
CADTH scenario analysis: treatment duration of tebentafusp is extended by 1.9 months | Reference | $8,204,894 | $8,446,250 | $8,682,389 | $8,921,700 | $26,050,340 |
New drug | $8,204,894 | $37,557,490 | $33,840,269 | $32,322,785 | $103,720,545 | |
Budget impact | $0 | $29,111,240 | $25,157,880 | $23,401,085 | $77,670,205 | |
CADTH scenario analysis: market share of tebentafusp in the previously treated population (Year 2: 20%; Year 1: 10%) | Reference | $8,204,894 | $8,446,250 | $8,682,389 | $8,921,700 | $26,050,340 |
New drug | $8,204,894 | $28,676,023 | $22,886,727 | $21,823,371 | $73,386,122 | |
Budget impact | $0 | $20,229,773 | $14,204,338 | $12,901,671 | $47,335,782 | |
CADTH scenario analysis: continue compassionate access of tebentafusp in first year or funding | Reference | $8,204,894 | $8,446,250 | $8,682,389 | $8,921,700 | $26,050,340 |
New drug | $8,204,894 | $13,081,423 | $26,181,985 | $25,209,711 | $64,473,119 | |
Budget impact | $0 | $4,635,173 | $17,499,596 | $16,288,010 | $38,422,779 | |
CADTH scenario analysis: tebentafusp price reduction of 91% | Reference | $8,204,894 | $8,446,250 | $8,682,389 | $8,921,700 | $26,050,340 |
New drug | $8,204,894 | $4,854,994 | $5,641,660 | $6,131,697 | $16,628,350 | |
Budget impact | $0 | -$3,591,257 | -$3,040,729 | -$2,790,003 | -$9,421,989 |
BIA = budget impact analysis.
Note: Budget impact for total population, including previously treated and untreated patients, is presented.
Stakeholder Input
Patient Input
Save Your Skin Foundation
About the Save Your Skin Foundation
Save Your Skin Foundation (SYSF) is a national patient-led not-for-profit group dedicated to the fight against non- melanoma skin cancers, melanoma and ocular melanoma through nationwide education, advocacy, and awareness initiatives. SYSF provides a community of oncology patient and caregiver support throughout the entire continuum of care, from prevention and diagnosis to survivorship.
Information Gathering
Information was obtained through on-line surveys, virtual patient roundtables and one-on-one conversations. (28) patients have primary ocular melanoma (OM), (7) have metastatic OM, (1) diagnosed primary and metastatic OM at the same time, (2) are not sure.
There were (29) females and (9) males ranging between the age of 30 – 80+. The breakdown of respondents was (9) between 30-49, (10) between 50 – 59, (12) 60 – 69 (7) 70-79.
(16) respondents were retired, (9) working fulltime, (5) working PT, (3) not able to work because of health-related issues and (3) not employed.
There were (9) respondents from BC, (8) from Alberta, (3) from Saskatchewan, (1) from Manitoba, (9) from Ontario, (1) from QC, (2) from Newfoundland, and (2) from outside of Canada (USA).
Disease Experience
From information gathered we heard from patients that there are 2 distinct stages of OM. Primary and Metastatic. For both, all patients responded that they were in huge shock as OM is a rare cancer and in a rare spot in or behind the eyeball. They had very few if any symptoms leading up to diagnosis and were told that the tumours can grow quite large before being noticed and that large tumours have a higher risk of metastasis. All patients were horrified at being diagnosed, had all kinds of eye issues. Some patients had lost the eye completely while others had one eye vision or totally lost vision. They understood that they would never see like a normal person again. The care for the eye is horrific and painful if they were fortunate enough to have kept their eye. Their balance is affected which causes huge physical and psychological deterioration. They were also told by their physician that there is a 50/50 chance that their disease would metastasize, and the odds were against them. There were no treatment options available at the time, leaving NO hope for survivorship. There was almost 100% chance of death as it is a fatal cancer and progression is rapid. All patients described it as a terrifying time. “A dark space with literally and figuratively NO light.”
“Mentally it has been frustrating knowing the seriousness of this type of cancer” “Devastating, tumors all over. Mentally taxing and emotionally disruptive.” “Stressful, anxiety, worry and feel alone as it’s so rare”
“Getting a rare cancer like this is the toughest of battles. Half of the patients develop metastasis and die quickly. To date, there are no approved treatments. This cancer tries to destroy everything about you, and then your very life”
“Very unexpected, shocking, and life changing. Hoping on Tebe” “Emotional roller coaster and anxiety has intensified.”
“Mental anxiety every time as part of testing and waiting for results”
“Travelling for treatment is very expensive. The mental toll is more than I can stand sometime, always waiting for scan results to see if it is spread.”
“The worst experience of my life. I’ve lost over 1/3rd of my sight and what sight I have left is all warped. I’ve also had a lot of eye pain and aching.”
“It has been mentally, emotionally and physically draining.”
“I am only able to work part time. I’ve taken a financial hit. I struggle my ever-changing vision but a least I have some. I seem more tired and less focused. The anxiety and fear is overwhelming”
“The emotional toll was all-consuming initially but has subsided somewhat.”
Experiences With Currently Available Treatments
There are no available treatments for patients in the metastatic setting. Their only hope at this time is special access to Tebentafusp which is only available in two provinces with limited space. There is a desperate need for treatment options.
(8) patients had immunotherapy (IO), (2) had Nivo/Ipi, (1) had Nivo, (5) were unsure of which IO treatment they received. All patients that underwent IO therapy had typical IO side effects, fatigue etc. and (5) of the (8) progressed on IO and were then given Tebentafusp on special access.
Improved Outcomes
All patients said that they wanted longer survival and that this new treatment would give them HOPE. All patients, caregivers and loved ones said they would do and try anything for longer survival, even travelling outside their home provinces and re-locating if need be.
“There is currently No available approved treatment. So the promise of an effective treatment is the best news I have ever heard.”
“More availability in more sites in Canada for IO treatment like Tebentafusp – higher success rate”
“A cure would be fantastic, but of course we need more treatment for all HLA, Tebentafusp does not cure everyone.”
“Long term OS benefits and/or remission.”
“Any treatment that cures or gives substantial increase in life expectancy, ability to live a “normal” life.”
Experience With Drug Under Review
For the (10) patients that received Tebentafusp, (1) received through clinical trial, (5) through compassionate access,
(1) is a member of the Canadian Forces so costs were covered, (3) were not sure. All patients received varied numbers of treatment. Some are still on treatment. (1) has had 25 infusions, (1) has had 42 infusions and continuing.
Side effects were fatigue, cognitive impairment, fever, skin rash, and gastro issues. Only (1) patient said the side effects were not manageable. All patients said that the benefit of the treatment out weighted the experience of the side effects.
“I feel really good and have minimal issues, The lesions growth has slowed down”
(4) patients did not experience hardships accessing this drug therapy but the remainder did (see quotes below).
“Yes, had to travel to Toronto, lots of expenses”
“Dr said it is only available for stage 4. I have 3A with lesions in liver. Scary just waiting for stage 4 without any treatments.”
“Travelled across country to receive treatment. Speed of access was an issue – scans and biopsy were so SLOW, so went to the US for a genomic biopsy”
“The issues were being 4500 km from home and add COVID to the equation and it has been very hard for my wife and I. The cost of treatment will be covered but as this happened so quickly, we have paid $22,000 so far out of our savings.”
“I have been on Tebentafusp since June 2021 and the only bothersome side effect is the fatigue. I have tremendous hair loss and some skin discoloration but can live with looking awful.”
Companion Diagnostic Test
When first diagnosed with OM all patients agreed that a biopsy of the tumour tissue is extremely important right at diagnosis. As of now there are only three laboratories to have those tests done, one in Ontario (IMPACT), and one is the U.S. (CASTLE), and one in Liverpool, England. The benefit of the testing is to show statistical likelihood of development of metastasis, thereby, approximately half of those patients will be shown as low risk and will continue being scanned and monitored while patients at high risk will be more closely followed. Patients mentioned that in some provinces the cost of the test was covered and in other provinces it was self-pay. All patients interviewed wanted and/or wished they had had the test at time of diagnosis. All patients said that this test would help them be better informed to be able to make better treatment choices.
Additionally, all patients that are diagnosed with metastatic OM need blood marker testing for (HLA)-A*02:01. Unfortunately, only approximately half of metastatic patients have this blood marker, so for patients that do not, there are no options. All patients said that the HLA testing needs to be done as soon as possible as Tebentafusp is the only option at this time for a positive HLA marker, and time is of the essence.
Anything Else?
We asked patients and their family member or caregiver how important it was to them that they received this treatment, see responses below:
“The treatment so far saved my life”
“This treatment is actually the only hope for all of us with OM. Meaning life extension”
“Vital – my family members were worried sick. Access, information about speed of referral/scans etc. – effectiveness of treatment and HOPE.”
“This was the only treatment available to me as all other options had been attempted. I needed/wanted to do everything I could to prolong my life not only for me but for my family. It would be great to have a treatment that cures OM.”
When patients were asked if they were offered a treatment on clinical trial, would they consider taking it, all patients said YES.
Conflict of Interest Declaration — Save Your Skin Foundation
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Save Your Skin Foundation
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Clinician Input
Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
About the Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
This input was jointly discussed at a DAC meeting.
Current Treatments and Treatment Goals
Uveal melanoma is a disease that is distinct from cutaneous melanoma, with a low tumor mutational burden and a 1-year overall survival of approximately 50% in patients with metastatic uveal melanoma. There is no overall survival benefit with current systemic treatment. Currently patients receive dacarbazine, ipilimumab, first-line ipi-nivo, nivolumab, or pembrolizumab single-agent. For first line patients would normally receive ipi-nivo or monotherapy with an anti-PD1 for systemic therapy.
Tebentafusp is a bispecific protein consisting of an affinity-enhanced T-cell receptor fused to an anti-CD3 effector that can redirect T cells to target glycoprotein 100–positive cells. Recent randomized controlled trials demonstrate an improvement in overall survival with the use of Tebentafusp who are HLA-A*02:01 positive.
Patients would need HLA-A*02:01 genotype testing to be identified as a candidate to receive tebentafusp. Most Ontario centers have testing capability but it is currently not funded in Ontario for uveal melanoma.
Treatment goals with a new systemic therapy would be to improve overall survival and improved quality of life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Overall survival benefit with systemic treatment are lacking with the currently available treatments.
Recent randomized controlled trials demonstrate an improvement in overall survival with the use of Tebentafusp who are HLA-A*02:01 positive.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Tebentafusp would be first-line treatment for metastatic HLA-A*0201 positive uveal melanoma.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients best suited would need to be HLA-A*0201 positive and have metastatic uveal melanoma.
Patients least suited would match the clinical trial exclusion criteria.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Outcomes in clinical practice are aligned with the outcomes used in the clinical trial and include: ongoing performance status, tumour size, RECIST progression requirements (from CT scans).
CT scans every 3-4 months with >20% decrease in tumor size and improved performance status would be considered a clinically meaningful response to treatment.
Note: some patients with radiographic progression (new and/or increasing target lesions) may continue therapy if clinical benefit.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Toxicity or symptomatic disease progression without clinical benefit.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
A medical oncologist would need to diagnose, treat and monitor patients in hospital and outpatient clinic. Access to physician care to assist with toxicity, including capacity for inpatient admission and monitoring after administering the first three doses are required to administer Tebentafusp.
Additional Information
Tebentafusp requires weekly administration, including inpatient admission and monitoring for the first three doses, which requires more resource utilization. Median treatment cycles is for 4-6 months from the pivotal trial IMCgp100-202.
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Skin Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Frances Wright
Position: Ontario Health (CCO) Skin Cancer Drug Advisory Committee Lead
Date: 11-05-2022
Table 2: Conflict of Interest Declaration for Ontario Health (CCO) Skin Cancer Drug Advisory Committee — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Marcus Butler
Position: Ontario Health (CCO) Skin Cancer Drug Advisory Committee Member
Date: 11-05-2022
Table 3: Conflict of Interest Declaration for Ontario Health (CCO) Skin Cancer Drug Advisory Committee — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Tara Baetz
Position: Ontario Health (CCO) Skin Cancer Drug Advisory Committee Member
Date: 11-05-2022
Table 4: Conflict of Interest Declaration for Ontario Health (CCO) Skin Cancer Drug Advisory Committee — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Elaine McWhirter
Position: Ontario Health (CCO) Skin Cancer Drug Advisory Committee Member
Date: 11-05-2022
Table 5: Conflict of Interest Declaration for Ontario Health (CCO) Skin Cancer Drug Advisory Committee — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr. Teresa Petrella
Position: Ontario Health (CCO) Skin Cancer Drug Advisory Committee Member
Date: 11-05-2022
Table 6: Conflict of Interest Declaration for Ontario Health (CCO) Skin Cancer Drug Advisory Committee — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Dr. Xinni Song
Position: Ontario Health (CCO) Skin Cancer Drug Advisory Committee Member
Date: 11-05-2022
Table 7: Conflict of Interest Declaration for Ontario Health (CCO) Skin Cancer Drug Advisory Committee — Clinician 6
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.