CADTH Reimbursement Review
Amivantamab (Rybrevant)
Sponsor: Janssen Inc.
Therapeutic area: Non–small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ATE
average treatment effect
ATO
average treatment effect for the overlap population
ATT
average treatment effect on the treated
BICR
blinded independent central review
CBR
clinical benefit rate
CI
confidence interval
CR
complete response
CRISP
Clinical Research Platform Into Molecular Testing, Treatment and Outcome Registry of (Non-)Small Cell Lung Carcinoma Patients
DCO
data cut-off
DLT
dose-limiting toxicity
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EGFR
epidermal growth factor receptor
EQ-5D-5L
5-level EQ-5D
ESME
Epidemiological Strategy and Medical Economics
EU
European Union
HR
hazard ratio
HRQoL
health-related quality of life
ILD
interstitial lung disease
IO
immunotherapy
IPD
individual patient data
IPW
inverse probability weighting
IRR
infusion-related reaction
ITC
indirect treatment comparison
LCC
Lung Cancer Canada
LCC-MAC
Lung Cancer Canada – Medical Advisory Committee
MET
mesenchymal-epithelial transition
MTD
maximally tolerated dose
NGS
next-generation sequencing
nNGM
national Network Genomic Medicine
NSCLC
non–small cell lung cancer
NSCLC-SAQ
Non-small Cell Lung Cancer Symptom Assessment Questionnaire
OH-CCO
Ontario Health – Cancer Care Ontario
OR
odds ratio
ORR
overall response rate
OS
overall survival
PD
progressive disease
PFS
progression-free survival
PGIC
Patient Global Impression of Change
PGIS
Patient Global Impression of Severity
PHE
Public Health England
PR
partial response
PRO
patient-reported outcome
QoL
quality of life
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumors
RP2D
recommended phase II dose
RR
response rate
SAE
serious adverse event
SD
stable disease
SET
safety evaluation team
SLR
systematic literature review
SOC
standard of care
TEAE
treatment-emergent adverse event
TKI
tyrosine kinase inhibitor
TTF
time to treatment failure
TTNT
time to next treatment
VEGFi
vascular endothelial growth factor inhibitor
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Amivantamab (Rybrevant) for injection 50 mg/mL concentrate for solution for IV infusion |
Indication | For the treatment of adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations whose disease has progressed on, or after, platinum-based chemotherapy |
Reimbursement request | As per indication |
Health Canada approval status | NOC/c |
Health Canada review pathway | Advance consideration under NOC/c, Project Orbis |
NOC date | March 30, 2022 |
Sponsor | Janssen Inc. |
EGFR = epidermal growth factor receptor; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; NSCLC = non–small cell lung cancer.
Introduction
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer deaths in Canada. In 2022, it was estimated that there would be 30,000 new cases of lung cancer diagnosed, and 20,700 deaths from lung cancer in Canada. It is estimated that 1 in 18 men, and 1 in 20 women, will die of lung cancer.1 Lung cancer is classified as either non–small cell lung cancer (NSCLC) or small cell lung cancer, with NSCLC accounting for approximately 88% of cases in Canada.2 Lung cancer symptoms may be nonspecific. The most common symptoms include unspecific cough, chest and shoulder pain, hemoptysis, weight loss, dyspnea, hoarseness, bone pain, and fever.3
Approximately 15% of Canadians with NSCLC have an epidermal growth factor receptor (EGFR)–activating mutation in the region encoding the tyrosine kinase domain.4-6 The most common activating EGFR mutations arise from exon 19 deletions or exon 21 L858R point substitutions, accounting for 85 to 90% of EGFR mutations.5-11 Exon 20 insertion mutations are the third most common EGFR mutations, occurring in less than or equal to 12% of all EGFR mutations globally, though a Canadian-based retrospective observational cohort study suggested that this may only occur in approximately 4% of NSCLC EGFR mutations in Canada.12 It has been estimated that patients with EGFR exon 20 insertion make up between 0.1% and 4.0% of all NSCLC cases globally and approximately 0.4% to 0.6% of overall NSCLC cases in Canada.13,14
First-line standard of care (SOC) for patients with locally advanced or metastatic NSCLC harbouring EGFR exon 20 insertion mutations remains platinum-based chemotherapy (cisplatin or carboplatin),6 generally in combination with pemetrexed, followed by pemetrexed maintenance, though gemcitabine, vinorelbine, or paclitaxel are approved but rarely used. Following failure of first-line platinum-doublet chemotherapy, second-line treatment options are limited to single-agent non–platinum-based chemotherapeutic drugs, primarily docetaxel. For lack of a better option, EGFR tyrosine kinase inhibitors (TKIs), immunotherapy (IO), or platinum rechallenge may be used based on chart reviews and real-world evidence collected in Canada. The sponsor suggested that variability in treatment patterns exists in Canadian clinical practice, with patients receiving EGFR TKIs, platinum-based chemotherapy, or IOs in combination with platinum-based chemotherapy or chemotherapy in the front-line setting. Patients who progress on first-line therapy may receive an alternative drug class (often either EGFR TKI or IO) and, if progression continues, the terminal therapy may be supportive care or docetaxel, if not previously received, due to its toxicity.15 The sponsor suggested that, based on a retrospective population-based cohort study in patients with metastatic NSCLC with EGFR mutations (n = 6,666) receiving second-line therapy in Alberta, Canada, real-world median overall survival (OS) of patients with EGFR exon 20 insertion mutations was 8.1 months (95% confidence interval [CI], 6.0 to not reached) compared to patients with EGFR exon 19 deletions (median OS = 17.8; 95% CI, 13.7 to 20.1) or exon 21 L858R mutations (median OS = 14.9; 95% CI, 11.4 to 21.9).12,15
Amivantamab is indicated by Health Canada for the treatment of adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations whose disease has progressed on, or after, platinum-based chemotherapy.16 The Health Canada Notice of Compliance with conditions was granted on March 30, 2022, based on the October 2020 data cut-off (DCO), through Project Orbis. Conditions for authorization are pending the results of confirmatory and other ongoing trials including the phase III PAPILLON trial, which aims to evaluate the efficacy of amivantamab in combination with chemotherapy in the first-line treatment of locally advanced or metastatic EGFR exon 20 insertion NSCLC, and the final report for CHRYSALIS to verify the clinical benefit of amivantamab.15 The sponsor is requesting that amivantamab be reimbursed as per the indication from Health Canada. Amivantamab has not been previously reviewed by CADTH.
The objective of the current report is to review the beneficial and harmful effects of amivantamab (1,050 mg for patients weighing less than 80 kg, and 1,400 mg for patients weighing greater than or equal to 80 kg) for the treatment of adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations whose disease has progressed on, or after, platinum-based chemotherapy.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, Lung Cancer Canada (LCC) and the Lung Health Foundation, submitted patient group input for this review. LCC gathered data through phone interviews with 4 EGFR-positive NSCLC patients, 2 of whom have EGFR exon 20 insertion mutations. All 4 patients were diagnosed with stage IV NSCLC and are currently part of the clinical trial for amivantamab in Ontario. The Lung Health Foundation obtained input from patients with lung cancer via an online survey (2 respondents) and telephone interviews (3 respondents), none of whom had experience with the treatment under review.
Input from LCC highlighted that conventional TKIs have poor efficacy in a majority of EGFR exon 20 insertion subtypes and therefore treatment options for these patients are limited, indicating a significant unmet need in this population. LCC respondents recalled their initial distress at receiving a stage IV NSCLC diagnosis. They cited various psychosocial effects, including anxiety and depression; however, symptoms such as a persistent cough and back pain were often mild. Respondents to both patient groups reported that future lung cancer treatments should be effective in curing their disease rather than slowing progression (though delayed disease progression remains important), provide additional treatment options with improved management of disease symptoms, and have minimal or manageable side effects so that they can participate in regular activities while on treatment and allow them to maintain quality of life (QoL). Regarding their experience with the treatment under review, the most common adverse events (AEs) were skin related, including inflammation of the nail bed, rashes on the face and legs, acne, and dry and sensitive skin. Other side effects that were reported include severe dry mouth, slight vision deterioration, severe muscle pain the first few days after treatment, fatigue, chronic constipation, and yeast infections. Some patients stated that although the side effect burden was more than what they had experienced with other targeted therapies, they would not consider discontinuing treatment because the hope of survival outweighed the side effects experienced. Two patients who began amivantamab as a first-line treatment stated that when they were feeling well, they were able to participate in physical activities and work they previously enjoyed and that they hoped to be able to continue to maintain their independence, functionality, and health-related quality of life (HRQoL). Two patients who were able to take amivantamab as a third-line treatment expressed the relief of having an additional treatment option after exhausting other alternatives. According to the Lung Health Foundation input, respondents found the psychosocial effects of having a disease with a poor prognosis challenging, as was maintaining relationships with families and friends. Patients noted that side effects related to some previous treatments severely affected their ability to participate in daily activities and their HRQoL.
Clinician Input
Input From Clinical Experts Consulted by CADTH
In patients with NSCLC with EGFR exon 20 insertion mutations, the clinical experts emphasized that not all patients benefit from current therapies, stating that there is a need for molecularly targeted therapies in this patient population due to the limited activity of EGFR TKIs or IO therapy in patients with exon 20 insertion mutations.
Currently, the mainstay of treatment for patients with EGFR exon 20 insertion mutations is platinum-doublet chemotherapy, most commonly cisplatin or carboplatin plus pemetrexed, followed by maintenance pemetrexed in first-line therapy. Docetaxel or pemetrexed would be used in second-line therapy following failure of platinum-based chemotherapy. According to the clinical experts, unless patients have private insurance, EGFR TKIs are not funded as second-line therapy, and activity with IO or TKIs is limited. The clinical experts believe that amivantamab would be used as monotherapy in patients with exon 20 insertion mutations following failure of platinum-doublet chemotherapy, displacing the current second-line and third-line options, though it would be reasonable to use amivantamab in any subsequent line of therapy. The clinical experts expressed that there is an unmet need for effective new treatment options that would improve QoL, delay progression or prolong survival, and reduce disease-related symptoms through demonstrated response to treatment.
Per the indication for amivantamab, patients are required to have confirmed EGFR exon 20 insertion mutations, though the clinical experts highlighted that there is no data to identify certain subgroups that do not benefit from therapy with amivantamab. The experts noted that treatment with amivantamab should be discontinued due to disease progression or unacceptable toxicity that cannot be managed. The experts noted that the trial allowed treatment beyond progression, where patients who originally had meaningful improvement before progression may still experience some benefit. The clinical experts noted that patients with NSCLC are typically under the care of expert medical and thoracic oncologists, and that, given the method of administration and potential for adverse reactions, patients should receive treatment in cancer-specific institutions under the supervision of an appropriately trained cancer specialist or unit with expertise in treating lung cancers and experience in managing infusion reactions, which are common and may be severe. Due to the difficulty in obtaining robust survival estimates in this rare population, tumour response or disease stabilization resulting in improvements in disease-related symptoms are the most meaningful outcomes. The clinical experts noted that, in clinical practice, response would be assessed every 9 to 12 weeks.
Clinician Group Input
Clinician group input was received from 2 groups: LCC – Medical Advisory Committee (LCC-MAC), with 21 clinicians contributing to the submission; and from the Ontario Health – Cancer Care Ontario (OH-CCO) Lung Cancer Drug Advisory Committee, with input from 5 clinicians. Based on global estimates, the LCC-MAC input noted that approximately 200 to 1,000 patients are diagnosed with EGFR exon 20 insertion NSCLC in Canada each year and experience resistance to the first-generation and second-generation EGFR TKIs. The clinician group also highlighted the rarity of the indication, stating that less than 2% of all EGFR exon 20 insertion NSCLC patients would be candidates for second-line amivantamab therapy, given the high number of patients considered to be too sick to receive first-line or second-line therapy for locally advanced or metastatic NSCLC. The clinician groups highlighted the poor prognosis for patients with locally advanced or metastatic NSCLC EGFR exon insertion due to the lack of effective targeted therapies, and emphasized a significant unmet need for novel targeted therapies that prolong progression-free survival (PFS) and improve HRQoL. Clinician groups stated that the ideal treatment for these patients should directly inhibit the driver mutation; be well tolerated, with a predictable and low toxicity profile; have a durable response; and correlate with an improvement in QoL. Both clinician groups stated that patients most likely to respond to amivantamab are those with an EGFR exon 20 insertion mutation. The clinician groups also stated that next-generation sequencing (NGS) is routinely conducted in all patients with advanced NSCLC with a non-squamous and squamous histology, and without a smoking history. In terms of place in therapy, the OH-CCO input notes that amivantamab would be used after all standard therapies acceptable to the patient have failed, usually following platinum-doublet chemotherapy with or without immunotherapy and maintenance pemetrexed, or after docetaxel therapy. The LCC-MAC also notes that such targeted therapies against driver mutations should ideally be offered in a first-line setting for maximal efficacy and that clinical trials are ongoing in this setting. Both submissions stated that a clinically meaningful end point is a radiological response to treatment. The submissions indicated that the drug should be administered at a cancer centre, infusion clinic, or hospital (outpatient) setting by a specialist or personnel experienced in administering chemotherapy drugs.
Drug Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, considerations for initiation of therapy, considerations for continuation or renewal of therapy, considerations for discontinuation of therapy, considerations for prescribing of therapy, generalizability, funding algorithm issues, care provision issues, and system and economic issues. Refer to Table 3 for more details.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
One study was included in the review. Study EDI1001 (CHRYSALIS) is an ongoing, phase I/Ib, single-arm, open-label, multicentre study of amivantamab as monotherapy in advanced NSCLC that includes both a dose-escalation phase (part 1) to determine the recommended phase II dose (RP2D) of amivantamab, and a dose-expansion phase (part 2) in which patients were treated with the RP2D of 1,050 mg amivantamab (1,400 mg for patients weighing greater than or equal to 80 kg). Treatment in part 2 was delivered once weekly for the first 4 weeks and then once every 2 weeks starting at week 5 until disease progression or unacceptable toxicity. A total of 489 patients across part 1 and part 2 received treatment with amivantamab monotherapy. Within part 2, individual cohorts were defined by mutation and previous treatment. Of interest to this review was cohort D, which enrolled a total of 153 patients with EGFR exon 20 insertion mutations whose disease has progressed on or after platinum-based chemotherapy. The primary outcome of the CHRYSALIS trial was overall response rate (ORR) per investigator and blinded independent central review (BICR) assessment in the primary efficacy population (patients who had undergone greater than or equal to 3 postbaseline disease assessments; N = 81), with secondary outcomes consisting of clinical benefit rate (CBR) and best overall response, PFS, OS, duration of response (DOR), and time to treatment failure (TTF).17
In the safety analysis set (patients who received ≥ 1 dose of amivantamab; N = 153), most patients were diagnosed with stage IV adenocarcinoma (78.9%). The median age of patients enrolled in cohort D was 61.0 years, and the majority of patients were Asian (62.1%), female (61.4%), and had an Eastern Cooperative Oncology Group (ECOG) performance status of 1 (72.5%); 36 (23.5%) patients had brain metastases at baseline. The median number of prior lines of therapy was 2, and all patients received prior platinum-based chemotherapy (mostly carboplatin; 65.4%).17 Baseline characteristics for the primary efficacy population and the additional efficacy population were consistent with the safety analysis set.
Efficacy Results
At the time of the March 30, 2021, DCO, the median follow-up was 14.5 months. In the primary efficacy population (N = 81), a total of 31 (38.3%; 95% CI, 27.7% to 49.7%) patients achieved an ORR per investigator assessment, and 35 (43.2%; 95% CI, 32.2% to 54.7%) patients achieved ORR per BICR. Best response to treatment per investigator assessment consisted of only partial responses (PRs) (31; 38.3%), while 12 (14.8%) patients had a best response of progressive disease (PD). The results of subgroup analyses by age group, sex, race, smoking history, and prior IO therapy were consistent with the primary analyses. Among the 31 responders, the median duration of treatment was 14.03 months, and the median DOR was 12.45 months (95% CI, 6.54 to 16.13).17
As of the March 30, 2021, DCO, the median PFS was 8.25 months (95% CI, 5.49 to 12.32), and a total of 57 (70.4%) PFS events had occurred. The progression-free rate for amivantamab per investigator assessment was 75% (95% CI, 64% to 83%) at 3 months, 58% (95% CI, 47% to 68%) at 6 months, and 40% (95% CI, 29% to 50%) at 12 months. The progression-free rate for amivantamab per investigator assessment was 75% (95% CI, 0.64 to 0.83) at 3 months, 58% (95% CI, 0.47 to 0.68) at 6 months, and 40% (95% CI, 0.29 to 0.50) at 12 months. The median OS was estimated at 22.77 months (95% CI, 17.48 to not estimable). The estimated survival rates were 90% (95% CI, 81% to 95%) at 6 months, 74% (95% CI, 63% to 83%) at 12 months, and 41% (95% CI, 21% to 59%) at 24 months.17
Harms Results
All patients in cohort D of the CHRYSALIS trial experienced 1 or more treatment-emergent adverse events (TEAEs). The most frequent TEAEs were infusion-related reactions (IRRs), paronychia, rash, dermatitis acneiform, hypoalbuminemia, and stomatitis. Grade 3 or higher TEAEs were reported in 64 (41.8%) patients, with the most frequent consisting of pulmonary embolism, hypokalemia, diarrhea, dyspnea, hypoalbuminemia, paronychia, pneumonia, IRRs, and neutropenia. A total of 44 (28.8%) patients experienced serious adverse events (SAEs); the most frequently reported SAEs were pneumonia, dyspnea, pulmonary embolism, back pain, muscular weakness, and pneumonitis.17
IRRs were the most frequent reason for infusion modifications, reported in 90 (58.8%) patients. Dose reductions were reported in 22 (14.4%) patients, mostly due to dermatitis acneiform and paronychia, while dose interruptions were reported in 55 (35.9%) patients, mainly due to IRRs (15.0%). In total, 18 (11.8%) patients withdrew from treatment with amivantamab due to TEAEs, most frequently pneumonia, IRRs, pleural effusion, and pneumonitis.17
Overall, 11 (7.2%) patients treated with amivantamab experienced a TEAE leading to death, with respiratory, thoracic, and mediastinal disorders the most common TEAE leading to death in 6 (3.9%) patients. As of the March 30, 2021, DCO, 45 (29.4%) patients in the CHRYSALIS trial died, primarily due to PD (31; 20.3%).17
The most frequent notable harm associated with amivantamab included IRRs (97; 63.4%), most of which were nonserious (grade 1 or 2) and occurring mainly on day 1 of cycle 1 (66.3%), and rash events (130; 85.0%), which were also mostly grade 1 or 2, and occurred mostly during the first treatment cycle. Other notable harms included interstitial lung disease (ILD) (6; 3.9%), paronychia (81; 52.9%), and ophthalmologic disorders (19; 12.4%).17
Table 2: Summary of Key Results From Pivotal and Protocol Selected Studies (Investigator Assessed; March 30, 2021, DCO)
Study outcomes | CHRYSALIS cohort D (investigator assessment) | CHRYSALIS cohort D (BICR assessment) |
|---|---|---|
Efficacy outcomes (primary efficacy population, N = 81) | ||
ORR (CR + PR) | 31 (38.3) | 35 (43.2) |
95% CI | (27.7 to 49.7) | (32.2 to 54.7) |
Best overall response | ||
CR | 0 (0.0) | 3 (3.7) |
PR | 31 (38.3) | 32 (39.5) |
SD | 37 (45.7) | 35 (43.2) |
PD | 12 (14.8) | 9 (11.1) |
Not evaluable/unknown | 1 (1.2) | 2 (2.5) |
Median DOR (95% CI) | 12.45 (6.54 to 16.13) | 11.04 (6.90 to NE) |
Median PFS (95% CI) | 8.25 (5.49 to 12.32) | 8.31 (5.52 to 11.07) |
Event | 57 (70.4) | 54 (66.7) |
Censored | 24 (29.6) | 27 (33.3) |
Median OS (95% CI) | 22.77 (17.48 to NE) | NA |
Event | 31 (38.3) | NA |
Censored | 50 (61.7) | NA |
Harms outcomes (safety analysis set, N = 153) | ||
AEs | 153 (100.0) | NA |
SAEs | 44 (28.8) | NA |
WDAEs | 18 (11.8) | NA |
Deaths | 11 (7.2) | NA |
Notable harms, n (%) | ||
IRRs | 97 (63.4) | NA |
Rash | 130 (85.0) | NA |
Interstitial lung disease | 6 (3.9) | NA |
Paronychia | 81 (52.9) | NA |
Ophthalmologic disorders | 19 (12.4) | NA |
AE = adverse event; BICR = blinded independent central review; CI = confidence interval; CR = complete response; DCO = data cut-off; DOR = duration of response; IRR = infusion-related reaction; NA = not applicable; NE = not estimable; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; SAE = serious adverse event; SD = stable disease; WDAE = withdrawal due to adverse event.
Source: CHRYSALIS Clinical Study Report interim analysis (March 30, 2021).17
Critical Appraisal
CHRYSALIS was a first-in-human phase I/Ib clinical study with the primary purposes of determining the RP2D (part 1) and subsequently assessing the safety of the selected dose in part 1 and estimating the clinical activity of amivantamab (part 2). A phase I/b or phase II trial may not accurately predict the harm or effectiveness of treatments. The clinical experts consulted by CADTH noted that, despite the high unmet need, conducting a randomized controlled trial (RCT) in this setting with a targeted therapy, such as amivantamab, compared to the available therapies currently used in Canadian clinical practice, would likely not be feasible. No inferential statistical testing was performed for the efficacy outcomes in cohort D; thus, no P values were reported. Point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. The threshold for a positive study outcome for cohort D was observing a 95% CI for ORR with a lower limited larger than 12%. Interpretation of time‐to‐event end points such as OS or PFS is limited in single‐arm studies; because all patients in cohort D received the same treatment, the extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear. The results for patient-reported outcomes (PROs) were inconclusive given the small sample size (N = 36); the noncomparative, open-label design of the trial; the substantial decline in patients available to provide assessments over time; and the descriptive nature of the analysis.
The clinical experts conducted by CADTH indicated that although the inclusion and exclusion criteria were appropriate, they hypothesized that the exclusion criteria may have been restrictive, selecting for ideal patients who were less severely ill. The clinical experts also noted that the baseline characteristics of the included population were generally reflective of Canadian clinical practice; however, there was a high proportion of patients who were Asian (62.1%) enrolled, though this was not considered likely to affect the generalizability of the results per the clinical experts.
The noncomparative design of the CHRYSALIS trial precludes the ability to assess the relative therapeutic benefit or safety of amivantamab against currently available therapies in Canadian clinical practice. As noted previously, the clinical experts consulted by CADTH agreed that direct randomized comparisons between amivantamab and currently used therapies are unlikely to take place for advanced or metastatic NSCLC with exon 20 insertion that has progressed after platinum-based chemotherapy. In the absence of a direct comparison of amivantamab with relevant treatment options, the sponsor submitted an adjusted treatment comparison using external control arms derived from real-world data sources. The results of the adjusted treatment comparison favoured amivantamab for ORR, PFS, and OS compared to treatment of physician’s choice. The CADTH review team identified several limitations (e.g., concerns regarding heterogeneity across the study designs and populations and the inability to adjust for important potential confounders and prognostic variables across each cohort) and concluded that no firm conclusions could be drawn about how amivantamab compared to other relevant treatment options. However, the clinical experts consulted by CADTH anticipated that based on the CHRYSALIS results and on poor results with existing treatment options in clinical practice, amivantamab would likely offer improved and clinically meaningful benefits compared with currently available therapies.
Indirect Comparisons
Adjusted Treatment Comparison
Description of Studies
The sponsor submitted an adjusted treatment comparison that compared the efficacy of amivantamab from individual patient data (IPD) from cohort D (N = 81) of the single-arm CHRYSALIS trial to current treatments using an external control arm derived from real-world data sources from the US, the UK, Germany, and France. The primary objective of the sponsor-submitted adjusted treatment comparison was to compare the efficacy (ORR, OS, PFS, time to next treatment [TTNT]) of amivantamab in the CHRYSALIS trial (cohort D) to current treatments from real-world settings in patients with advanced EGFR NSCLC with exon 20 insertion mutations following platinum-based chemotherapy.18
Amivantamab was compared to both a pooled basket of treatments, labelled as physician’s choice, and individual treatment classes (TKI, IO, non–platinum-based chemotherapy, vascular endothelial growth factor inhibitor [VEFGi] plus chemotherapy–, and other–based regimens). Data from the 4 European data sources were pooled to create a European Union (EU) cohort, and collectively compared against amivantamab. Data from the 3 US data sources were also pooled to create the US cohort. Additionally, all data sources were combined to create a US plus EU cohort.18
Efficacy Results
In all comparisons, CHRYSALIS was the index trial, consisting of the primary efficacy population (N = 81). In the comparison to physicians’ choice, the EU plus US cohort included 349 patients, with 206 from the US cohort and 143 from the EU cohort. In the comparison of CHRYSALIS to the different treatment classes in the EU plus US cohort, there were 60, 89, 76, 58, and 66 patients in the TKI, IO, non–platinum-based chemotherapy, VEFGi plus chemotherapy, and other groups, respectively.18
For the primary outcome of ORR, the adjusted ORR was 38.3% for amivantamab compared to █████ for physicians’ choice for the EU plus US cohort (odds ratio [OR] = █████████████████████]; relative risk ratio = ██████████████████████). The adjusted ORR for amivantamab versus physicians’ choice in the US cohort was 38.3% versus █████ (OR = ██████████████████████; relative risk ratio = █████████████████████), and 38.3% versus █████ in the EU cohort (OR = ████████████████████; relative risk ratio = ██████████████████████). Results for the individual treatment classes were consistent with the primary analysis; however, they were hindered by small sample sizes.18
For OS, amivantamab was favoured over physicians’ choice in the EU plus US cohort (hazard ratio [HR] = █████████████████████████), US cohort (HR = █████████████████████████), and EU cohort (HR = ████████████████████████) after adjustment. The median OS for amivantamab was at 22.77 months (95% CI, 17.48 to not estimable) compared to █████ months (██████████████████), █████ months (██████████████████), and █████ months (█████████████████) from the EU plus US cohort, the US cohort, and the EU cohort, respectively. Compared to the individual treatment classes, amivantamab was favoured after adjustment in all cases.18
For PFS, amivantamab was favoured over physicians’ choice in the EU plus US cohort (HR = █████████████████████████), US cohort (HR = ████████████████████████), and EU cohort (HR = █████████████████████████) after adjustment. The median PFS for amivantamab was 8.25 months (95% CI, 5.49 to 12.32) compared to ████ months (████████████████), ████ months ██████████████████, and ████ months ██████████████████ from the EU plus US cohort, the US cohort, and the EU cohort, respectively.18
Critical Appraisal
The choice to conduct an adjusted treatment comparison of amivantamab and external real-world data cohorts as a comparator arm was justified by the lack of a comparator arm for the CHRYSALIS trial. Data derived from 7 international real-world data sources were used for the comparison with amivantamab; therefore, there is a high risk of selection bias. Moreover, the methods and reasons for selecting these specific databases were unknown. Data analyzed retrospectively from databases and medical records are more prone to unique biases (e.g., selection bias, confounding, limited data availability) compared with those collected from prospective interventional studies that cannot be fully controlled for. In general, comparisons with externally generated cohorts also suffer from the potential of missing information.
There was notable heterogeneity in the populations included in the adjusted treatment comparison. Inclusion and exclusion criteria from cohort D of the CHRYSALIS trial were applied to all real-world data sources to select the appropriate population. Any criteria that could not be applied to patients from a given data source due to missing data were omitted, which may have resulted in unaccounted-for differences in patient populations. It was unclear how many potential patients were excluded following the application of inclusion and exclusion criterion from CHRYSALIS. Appropriately, all patients in the analysis had confirmation of EGFR exon 20 insertion mutation positivity. However, due to limited data availability, ECOG performance status, which was noted as an important prognostic factor by the clinical experts consulted by CADTH, was not included in the inverse probability weighting (IPW) adjustment for the EU plus US cohort or the EU cohort, potentially resulting in some unaccounted-for heterogeneity. No consideration or covariate adjustment was given to follow-up duration, and the time of assessment for end points was unknown. As a result, it is uncertain whether the follow-up times between CHRYSALIS and the real-world data sources are comparable, and may also contribute to heterogeneity, especially for survival analyses.
Multiple comparative analyses were performed for the CHRYSALIS population versus the various data sources. Individual real-world data sources from Europe and from the US were pooled to increase sample size. The sponsor noted that pooling of the EU and US cohorts into a single combined cohort was possible due to the high consistency between the results and a comparable treatment distribution of the EU and US cohorts; however, the observable differences in baseline characteristics of populations between the EU and US cohorts, as well as the significant missing data between databases, may have resulted in significant heterogeneity in the population, though this was not explored and remains uncertain. Additionally, amivantamab was compared to both a pooled basket of treatments (physicians’ choice), and various treatment class regimens (TKIs, IOs, non-platinum-based chemotherapy, VEGFi, and other). Pooling of treatments for the physicians’ choice group assumes equivalence of treatment benefit; however, it is unclear how exclusion of treatments irrelevant to the Canadian context such as VEGFis would have affected the results, because this was not explored.
The results of the adjusted treatment comparisons were consistent across end points and statistical methodologies, generally favouring amivantamab over physicians’ choice across end points, as well as for the individual treatment classes. However, there was notable imprecision in all cases, as demonstrated by the moderately to severely wide 95% CIs, though the reason for this imprecision was unknown and may be due to small sample sizes and unexplored heterogeneity.
In summary, given the phase I/Ib nature of the CHRYSALIS trial and the lack of a comparator arm, the ability to make definitive conclusions on the comparative efficacy of amivantamab was limited. Comparisons using the external control arms derived from real-world data sources were subject to substantial uncertainty and risk of bias due to the methods of data collection, small sample sizes, and high degree of heterogeneity due to pooling assumptions, as well as the limited data availability for important confounders and potential prognostic factors, including ECOG performance status in the EU and EU plus US cohorts. Additionally, outcomes important to patients including HRQoL and AEs were not analyzed in the adjusted treatment comparisons; thus, the comparative efficacy of amivantamab on these outcomes remains uncertain.
Other Relevant Evidence
No long-term extension studies or other relevant studies were included in the sponsor’s submission to CADTH.
Conclusions
One phase I/Ib, single-arm, open-label trial (CHRYSALIS; cohort D) provided evidence for the efficacy and safety of amivantamab in adult patients with metastatic or unresectable NSCLC who harboured EGFR exon 20 insertion mutations and failed on, or progressed after, platinum-based chemotherapy. The CHRYSALIS trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR greater than 12%) in cohort D. The clinical experts consulted by CADTH felt that the achieved ORR per investigator assessment of 38.3% (43.2% per BICR) (March 30, 2021, DCO date) was clinically meaningful for the target population and durable (median DOR 12.45 months, 95% CI, 6.54 to 16.13). In the opinion of the clinical experts, the observed responses appeared higher than what is seen with currently used therapies in the target setting. There was uncertainty around the magnitude of the clinical benefit given the limitations in the evidence from the noncomparative phase I/Ib clinical trial. While time-to-event end points, OS, and PFS, appeared supportive of the observed ORR, the nonrandomized design of the CHRYSALIS trial made interpreting the PFS and OS events attributable to amivantamab challenging. The CADTH clinical assessment identified limitations with the sponsor’s adjusted treatment comparison (including small sample sizes, heterogeneity across study designs and pooled populations, and the inability to adjust for important potential confounders and prognostic variables), which substantially limited the ability to interpret the relative treatments effects observed between amivantamab and other treatments. The results for HRQoL and symptom severity were exploratory outcomes and remained inconclusive due to a number of important limitations. Harms associated with amivantamab were largely consistent with treatments based on EGFR inhibition and were considered manageable according to the clinical experts consulted by CADTH. Overall, the ability to draw firm conclusions on the magnitude of clinical benefit and safety of amivantamab was limited given the limitations in the evidence.
Introduction
Disease Background
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer deaths in Canada. Survival from lung cancer of all stages and types is poor, with an overall 5-year net survival rate of 19%.1 In 2022, it was estimated that there would be 30,000 new cases of lung cancer diagnosed and 20,700 deaths from lung cancer in Canada. It is estimated that 1 in 18 men, and 1 in 20 women, will die of lung cancer.1
Lung cancer is classified as either NSCLC or small cell lung cancer, with NSCLC accounting for approximately 88% of cases in Canada.2 NSCLC is further classified into 3 main histologic subtypes: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma.
To determine prognosis and treatment, NSCLC is staged using the American Joint Committee on Cancer staging criteria, which involves TNM (tumour, node, metastasis) classification of the disease based on the size and spread of the primary tumour (T), lymph node involvement (N), and occurrence of metastasis (M).19 Approximately half of all lung cancer cases in Canada are stage I to stage III at diagnosis.2 Early-stage NSCLC is often asymptomatic.3,19 If patients do present with symptoms, they are often unspecific and difficult to directly attribute to a lung cancer diagnosis. The most common symptoms include unspecific cough, chest and shoulder pain, hemoptysis, weight loss, dyspnea, hoarseness, bone pain, and fever.3 Diagnostic procedures include imaging of the lungs, sputum cytology, and tissue biopsy.
Approximately 15% of Canadians with NSCLC have an EGFR-activating mutation in the region encoding the tyrosine kinase domain.4-6 EGFR mutations are more frequently observed in those who have never smoked, people of Asian descent, patients with adenocarcinoma, and females.4,20 The most common activating EGFR mutations arise from exon 19 deletions or exon 21 L858R point substitutions, accounting for 85% to 90% of EGFR mutations.5-9 Exon 20 insertion mutations are the third most common EGFR mutations, occurring in less than or equal to 12% of all EGFR mutations, though a Canadian-based retrospective observational cohort study suggested that this may only occur in approximately 4% of NSCLC EGFR mutations in Canada,12 It has been estimated that patients with EGFR exon 20 insertion make up between 0.1% and 4% of all NSCLC cases globally and approximately 0.4% to 0.6% of overall NSCLC cases in Canada.13-15
Standards of Therapy
Progress has been made in treating EGFR-mutated NSCLC with the introduction of EGFR TKIs. First-, second-, and third-generation EGFR TKIs have activity in NSCLC tumours harbouring sensitizing EGFR gene mutations that demonstrate improved efficacy compared with chemotherapy in delaying disease progression in first-line treatment of locally advanced or metastatic NSCLC.6 However, though similar to other EGFR mutations in biology and epidemiology, patients with advanced NSCLC whose tumours harbour uncommon EGFR mutations (i.e., EGFR exon 20 insertion or de novo exon 20 T790M mutations) are TKI resistant as a result of an altered enzyme active site that sterically hinders TKI binding, resulting in low response rates (0% to 9%) with approved EGFR TKIs.6,21-26 As such, the first-line SOC in Canada for patients with EGFR exon 20 insertion mutations remains platinum-based chemotherapy (cisplatin or carboplatin), generally in combination with pemetrexed followed by pemetrexed maintenance, although gemcitabine, vinorelbine, paclitaxel, or docetaxel may be used,6 with a median OS of 16 months, compared with 39 months in EGFR TKI-sensitive disease treated with TKIs.24,27-32
Following failure of first-line platinum-doublet chemotherapy, second-line treatment options are limited to non–platinum-based chemotherapeutic drugs, primarily docetaxel. After first-line platinum-based chemotherapy, it is estimated that only about 50% of patients go on to subsequent lines of therapy.33 For lack of a better option, EGFR TKIs, immunotherapy, or platinum rechallenge may be used based on chart reviews and real-world evidence collected in Canada. The sponsor suggested that variability in treatment patterns exists in Canadian clinical practice, with patients receiving EGFR TKIs, platinum-based chemotherapy, or IOs in combination with platinum-based chemotherapy or chemotherapy in the front-line setting. Patients who progress on first-line therapy may receive an alternative drug class (often either EGFR TKI or IO) and, if progression continues, the terminal therapy may be supportive care or docetaxel, if not previously received, due to its toxicity.15 The sponsor suggested that, based on a retrospective population-based cohort study in patients with metastatic NSCLC with EGFR mutations (n = 6,666) receiving second-line therapy in Alberta, Canada, real-world median OS of patients with EGFR exon 20 insertion mutations was 8.1 months (95% CI, 6.0 to not reached) compared to patients with EGFR exon 19 deletions (median OS = 17.8; 95% CI, 13.7 to 20.1) or exon 21 L858R mutations (median OS = 14.9; 95% CI, 11.4 to 21.9).12,15
Drug
Amivantamab is indicated by Health Canada for the treatment of adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations whose disease has progressed on, or after, platinum-based chemotherapy.16 The Health Canada Notice of Compliance with Conditions was granted on March 30, 2022, based on the October 2020 DCO, through Project Orbis. The conditions for authorization include the submission of the phase III PAPILLON study, which aims to evaluate the efficacy of amivantamab in combination with chemotherapy in the first-line treatment of locally advanced or metastatic EGFR exon 20 insertion NSCLC and is expected to complete in late 2025, and the final study report for CHRYSALIS, including efficacy results on at least 129 patients who have been followed for at least 6 months from the date of initial response. Additional progress reports of other ongoing trials are also required, as well as ongoing safety monitoring.15 The sponsor is requesting that amivantamab be reimbursed as per the indication from Health Canada. Amivantamab has not been previously reviewed by CADTH.
Amivantamab is a bispecific antibody that binds to the extracellular domains of the EGFR and mesenchymal-epithelial transition (MET) receptors, disrupting EGFR and MET signalling functions through blocking ligand binding and enhancing degradation of these receptors. The presence of EGFR and MET on the surface of tumour cells also allows for targeting of these cells for destruction by immune effector cells, such as natural killer cells and macrophages, through antibody-dependent cellular cytotoxicity and trogocytosis mechanisms, respectively.16
Amivantamab is administered via IV infusion once weekly via split infusion on days 1 and 2 of cycle 1, and then on days, 8, 15, and 22, followed by every 2 weeks starting at week 5 (cycle 2). The recommended dose of amivantamab is based on patient body weight at baseline. If patients weigh less than 80 kg, the recommended dose of amivantamab is 1,050 mg, representing 3 vials of amivantamab. In patients weighing greater than or equal to 80 kg at baseline, the recommended dose of amivantamab is 1,400 mg, representing 4 vials of amivantamab. Antihistamines, antipyretics, and glucocorticoids must be administered before the initial infusion (week 1, days 1 and 2) to reduce the risk of IRRs. For subsequent doses, antihistamines and antipyretics must be administered before all infusions, and glucocorticoids administered as necessary. Amivantamab should be used until disease progression or unacceptable toxicity.16
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Two patient groups, LCC and the Lung Health Foundation, submitted patient group input for this review. LCC gathered data through phone interviews with 4 EGFR-positive NSCLC patients, 2 of whom have EGFR exon 20 insertion mutations. All 4 patients were diagnosed with stage IV NSCLC and are currently enrolled in the clinical trial for amivantamab in Ontario. The Lung Health Foundation obtained input from patients with lung cancer via an online survey (2 respondents) and telephone interviews (3 respondents) conducted between September and December 2021, none of whom had experience with the treatment under review.
Input from LCC highlighted that conventional TKIs have poor efficacy in a majority of EGFR exon 20 insertion subtypes and therefore treatment options for these patients are limited, indicating a significant unmet need in this population. LCC respondents recalled their initial distress at receiving a stage IV NSCLC diagnosis, citing various psychosocial effects including anxiety and depression; given their mild symptoms of a persistent cough and back pain. Respondents to both patient groups reported that future lung cancer treatments should be effective in curing their disease rather than slowing progression (though delayed disease progression remains important), provide additional treatment options with improved management of disease symptoms, and have minimal or manageable side effects so that they can participate in regular activities while on treatment and allow them to maintain QoL. Regarding their experience with the treatment under review, the most common AEs were skin related, including inflammation of the nail bed, rashes on the face and legs, acne, and dry and sensitive skin. Other side effects that were reported include severe dry mouth, slight vision deterioration, severe muscle pain the first few days after treatment, fatigue, chronic constipation, and yeast infections. Some patients stated that although the side effect burden was more than what they had experienced with other targeted therapies, they would not consider discontinuing treatment because the hope of survival outweighed the side effects experienced. Two patients who began amivantamab as a first line of treatment stated that when they were feeling well, they were able to participate in physical activities and work they previously enjoyed and that they hoped to be able to continue to maintain their independence, functionality, and HRQoL. Two patients who were able to take amivantamab as a third-line treatment expressed the relief of having an additional treatment option after exhausting other alternatives. According to the Lung Health Foundation input, respondents found the psychosocial effects of having a disease with a poor prognosis challenging, as was maintaining relationships with families and friends. Patients noted that side effects related to some previous treatments severely affected their ability to participate in daily activities and their HRQoL.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of NSCLC.
Unmet Needs
The clinical expert stated that although EGFR exon 20 insertion mutations are considered the third most common mutation, they are quite rare, effective current therapies are limited, and there is a need for molecularly targeted therapies in this patient population. The clinical experts emphasized that not all patients benefit from current therapies. Current second-line treatments after failure of platinum-based chemotherapy consist of docetaxel, though EGFR TKIs or IO may be used as a last line of therapy if available. However, they have limited activity for exon 20 insertion mutations.
The clinical experts expressed that there is an unmet need for effective new treatment options that improve QoL, delay progression or prolong survival, and reduce disease-related symptoms through demonstrated response to treatment.
Place in Therapy
Unless there is an appropriate clinical trial available, platinum-doublet chemotherapy, most commonly cisplatin or carboplatin plus pemetrexed followed by maintenance pemetrexed, is first-line SOC in patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. Following failure of first-line therapy, docetaxel or pemetrexed would be used, and patients may receive nivolumab, pembrolizumab, or atezolizumab as another line of therapy. The clinical experts stated that the ORR to first-line platinum-based doublet is approximately 30% and is only about 10% for second-line therapy. In general, unless patients have private insurance, EGFR TKIs are not funded as second-line therapy, and activity with IO or TKIs is limited.
The clinical experts believe that amivantamab would be used as monotherapy in patients with exon 20 insertion mutations following failure of platinum-doublet chemotherapy, displacing the current second- and third-line options, though it would also be reasonable to use amivantamab in any subsequent line of therapy. It was also noted that there are currently no data to support combinations of amivantamab with other drugs in this patient population; however, clinical trials investigating combinations are ongoing. The clinical experts also highlighted that there are several drugs currently in development for this patient population, some of which are unavailable in Canada, though optimal sequencing of this class of drugs and activity following failure or resistance are unclear.
Patient Population
Per the indication for amivantamab, patients are required to have locally advanced or metastatic NSCLC with confirmed EGFR exon 20 insertion mutations. Genetic testing for EGFR mutations is standard across Canada, most commonly through polymerase chain reaction or NGS. In line with the trial criteria, patients with brain metastases must have received treatment for these before commencing amivantamab. The clinical experts highlighted that there is no data to identify certain subgroups that do not benefit from therapy with amivantamab.
Assessing Response to Treatment
The clinical experts stated that the most meaningful outcomes in this patient population are delayed progression and improved OS; however, given that these outcomes are difficult to measure in individual patients, tumour response (or shrinkage) or disease stabilization resulting in improvements in disease-related symptoms is the most meaningful outcome in clinical practice.
In the clinical trial, response was assessed every 6 weeks. The clinical experts noted that, in clinical practice, response would be assessed every 9 to 12 weeks.
Discontinuing Treatment
The clinical experts suggested that treatment with amivantamab should be discontinued due to disease progression or unacceptable toxicity that cannot be managed. The experts noted that the trial allowed treatment beyond progression, in which patients who originally had meaningful improvement before progression may still experience some benefit. The experts also noted that patients with evidence of benefit in their systemic disease who develop central nervous system progression should have their central nervous system metastases treated appropriately and then be allowed to continue treatment with amivantamab if they are otherwise well.
Prescribing Conditions
The clinical experts noted that patients with NSCLC are typically under the care of expert medical and thoracic oncologists. The experts highlighted that given the method of administration and potential for adverse reactions, patients should receive treatment with amivantamab in cancer-specific institutions or infusion clinics under the supervision of an appropriately trained cancer specialist or unit with expertise in treating lung cancers and experience in managing infusion reactions, which are common and may be severe.
Additional Considerations
The clinical experts highlighted the paucity of available randomized data in this population given the rarity of EGFR exon 20 insertion mutations, clinical equipoise, and the strong unmet need.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
Clinician group input was received from 2 groups: LCC-MAC, with 21 clinicians contributing to the submission; and the OH-CCO Lung Cancer Drug Advisory Committee, with input from 5 clinicians. Based on global estimates, the LCC-MAC input noted that approximately 200 to 1,000 patients are diagnosed with EGFR exon 20 insertion NSCLC in Canada each year and experience resistance to the first-generation and second-generation EGFR TKIs. The clinician group also highlighted the rarity of the indication, stating that less than 2% of all EGFR exon 20 insertion NSCLC patients would be candidates for second-line amivantamab therapy, given the high number of patients considered to be too sick to receive first-line or second-line therapy for locally advanced or metastatic NSCLC. The clinician groups highlighted the poor prognosis for patients with locally advanced or metastatic NSCLC EGFR exon 20 insertion due to the lack of effective targeted therapies, and emphasized a significant unmet need for novel targeted therapies that prolong PFS and improve HRQoL. Clinician groups stated that the ideal treatment for these patients should directly inhibit the driver mutation; be well tolerated, with a predictable and low toxicity profile; have a durable response; and correlate with an improvement in QoL. Both clinician groups stated that patients most likely to respond to amivantamab are those with an EGFR exon 20 insertion mutation, and stated that NGS is routinely conducted in all patients with advanced NSCLC with a non-squamous and squamous histology, without a smoking history. In terms of place in therapy, the OH-CCO input notes that amivantamab would be used after all standard therapies acceptable to the patient have failed, usually following platinum-doublet chemotherapy with or without immunotherapy and maintenance pemetrexed, or after docetaxel therapy. The LCC-MAC also notes that such targeted therapies against driver mutations should ideally be offered in a first-line setting for maximal efficacy and that clinical trials are ongoing in this setting. Both submissions stated that a clinically meaningful end point is radiological response to treatment. The submissions indicated that the drug should be administered at a cancer centre, infusion clinic, or hospital (outpatient) setting by a specialist or personnel experienced in administering chemotherapy drugs.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 3.
Table 3: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
CHRYSALIS is a phase I/Ib multicentre, open-label, multicohort study that assessed the safety and efficacy of amivantamab in adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations that had progressed on or after platinum-based chemotherapy (cohort D). There was no comparator arm in the CHRYSALIS trial. Currently funded options for patients with exon 20 insertion mutations who progress on or after treatment with platinum-based chemotherapy include IO (nivolumab, pembrolizumab, atezolizumab) docetaxel or pemetrexed. Some jurisdictional funding policies require patients with an EGFR mutation to have progressed during or after therapy with both a TKI and chemotherapy before being able to access an IO drug. | No response required. For pERC consideration. |
Considerations for initiation of therapy | |
In the CHRYSALIS trial, for patients who achieved CR, treatment could be interrupted after 2 additional cycles of amivantamab were administered. Re-treatment could be considered. If treatment is stopped after confirmation of CR, can amivantamab be restarted at time of progression? | Patients with advanced lung cancer generally do not experience CR to treatment. In the CHRYSALIS trial, per BICR, 3 patients experienced a CR with amivantamab. Regardless, it was the opinion of the clinical experts that in patients with confirmed CR, for whom treatment is stopped, amivantamab can be restarted at the time of progression, providing there are no contraindications. |
Patients with untreated brain metastases were excluded from the CHRYSALIS trial. Should patients with CNS involvement be eligible for treatment with amivantamab? Is there evidence to inform the safety and efficacy of amivantamab in this patient population? | In the CHRYSALIS trial, patients with untreated brain metastases were excluded. In total, 23.5% of patients had treated brain metastases at baseline. Therefore, patients with treated brain metastases should be eligible for treatment with amivantamab and patients with untreated brain metastases should be treated before starting amivantamab. |
Considerations for continuation or renewal of therapy | |
In the CHRYSALIS trial, response was assessed by CT, MRI, or other imaging or examination at least every 6 weeks after the first dose of amivantamab. In clinical practice, how should response to treatment with amivantamab be assessed? | Assessment of disease every 6 weeks is a clinical trial–imposed period. In clinical practice, patients should be assessed for response every 9 to 12 weeks, depending on disease stability (e.g., initial imaging may be at 6 to 9 weeks and if patient is responding, imaging may be done every 12 weeks). |
Considerations for discontinuation of therapy | |
In the CHYRSALIS trial, treatment beyond progression was allowed in the case of continued clinical benefit. What are the discontinuation criteria for amivantamab? | Discontinuation criteria would be in line with the CHRYSALIS trial and consist of clear, objective disease progression, especially with worsening of disease-related symptoms, or loss of clinical benefit. |
In the CHRYSALIS trial, if there was a delay due to toxicity resulting in 2 consecutive missed doses of amivantamab (i.e., 28 days), then subjects were required to discontinue the drug. Patients were only permitted to restart treatment after missing 2 consecutive doses if there was clear clinical benefit, approval was obtained from the sponsor, and any necessary adjustments to dosing and infusion protocol were made. Should patients be permitted to continue treatment with amivantamab if 2 consecutive doses are missed due to toxicity? | Yes, patients who missed 2 consecutive doses due to toxicity should be eligible to continue treatment with amivantamab, provided the toxicity experienced was not life-threatening. Clinical judgment should be used. |
Considerations for prescribing of therapy | |
Administration rates for amivantamab follow an escalation schedule for the first few doses (rates vary for 1,050 mg and 1,400 mg doses). These escalating infusion-rate schedules will require additional monitoring by nursing. Target doses are administered over 2 hours at a fixed rate. The administration of the first dose is split over 2 days. This affects the availability of resources in the chemotherapy treatment room and pharmacy. | No response required. For pERC consideration. |
Generalizability | |
Should patients with ECOG performance status ≥ 2 be eligible? | The clinical experts noted that patients with ECOG performance status of 0 or 1 are generally a restriction of clinical trials. In clinical practice, patients with ECOG performance status of 2 are often treated using the same treatment algorithms. The use of amivantamab should be allowed in patients with ECOG performance status ≤ 2, but clinical judgment should be used. |
Should patients who are being treated with another drug and have not progressed be eligible to switch to amivantamab, assuming all other reimbursement criteria for treatment with amivantamab are met? | Yes, in line with other lung cancer treatments, patients who have failed platinum-based chemotherapy and are being treated with other drugs, even if they have not experienced disease progression, should be eligible to switch to amivantamab because the currently available treatments are generally less effective. |
Funding algorithm | |
Drug may change place in therapy of comparator drugs, or drugs reimbursed in subsequent lines. | No response required. For pERC consideration. |
If patients are eligible to receive both amivantamab or IO therapy, is there evidence to support the order of sequencing of these medications? | Results from the CHRYSALIS trial suggest that amivantamab is superior to IO therapies in exon 20 insertion NSCLC; therefore, IO therapy should be considered as a last line of therapy option after amivantamab. |
Care provision issues | |
Amivantamab is available as 350 mg vials. Recommended doses and dose adjustments correspond to available vial size and should minimize wastage. The product monograph indicates a need to withdraw a volume from the infusion bag equal to the volume of drug being added and that the volume in the infusion bag should be 250 mL. It requires extra work to ensure a final volume of exactly 250 mL. | No response required. For pERC consideration. |
Premedications are required to prevent infusion-related reactions Additional therapies (e.g., emollient creams, topical corticosteroids, oral or IV antibiotics, oral steroids) may be required for the management of skin toxicities. | No response required. For pERC consideration. |
The product monograph indicates that the presence of an EGFR exon 20 insertion mutation is required to be determined using a validated test, though the manufacturer submission indicates no companion diagnostics. What method of testing should be used for detection of EGFR exon 20 insertion mutations? When should testing for the EGFR exon 20 insertion mutation occur? | In line with current Canadian guidelines, patients with NSCLC should receive testing for genetic mutations at diagnosis using either PCR or NGS. |
System and economic issues | |
There are confidential prices for IO comparators (nivolumab, pembrolizumab, atezolizumab) | No response required. For pERC consideration. |
BICR = blinded independent central review; CNS = central nervous system; CR = complete response; ECOG = Eastern Cooperative Oncology Group; EGFR = epidermal growth factor receptor; IO = immuno-oncology; NGS = next-generation sequencing; NSCLC = non–small cell lung cancer; PCR = polymerase chain reaction; pERC = pan-Canadian Oncology Drug Review Expert Review Committee; TKI = tyrosine kinase inhibitor.
Clinical Evidence
The clinical evidence included in the review of amivantamab (Rybrevant) is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the Systematic Review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of amivantamab (50 mg/mL) 1,050 mg for patients weighing less than 80 kg, and 1,400 mg for patients weighing greater than or equal to 80 kg for the treatment of adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations whose disease has progressed on, or after, platinum-based chemotherapy.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 4. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 4: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations Subgroups:
|
Intervention | Amivantamab, 1,050 mg for patients weighing < 80 kg, and 1,400 mg for patients weighing ≥ 80 kg (50 mg/mL concentrate for solution for infusion) |
Comparator |
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase II, III, and IV RCTs |
AE = adverse event; DOR = duration of response; EGFR = epidermal growth factor receptor; HRQoL = health-related quality of life; ILD = interstitial lung disease; IO = immuno-oncology; IRR = infusion-related reactions; NSCLC = non–small cell lung cancer; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SAE = serious adverse event; TKI = tyrosine kinase inhibitor; TTNT = time to next treatment; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.34
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974—) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was amivantamab. The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the EU) Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on May 19, 2022. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee on September 14, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.35 Included in this search were the websites of regulatory agencies (the US FDA and the European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for indirect treatment comparisons (ITCs) dealing with EGFR-mutated NSCLC was run in MEDLINE All (1946–) on May 18, 2022. No limits were applied. The literature search for ITCs identified 93 articles; however, no articles evaluated the efficacy and safety of amivantamab in patients with NSCLC with EGFR exon 20 insertion.
Findings From the Literature
A total of 60 studies were identified from the literature, with 1 study selected for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 5. At the time of this review, the CADTH literature search did not identify any phase III studies that are planned or currently enrolling patients with EGFR exon 20 insertion NSCLC in the post–platinum-based chemotherapy setting. A list of excluded studies is presented in Appendix 2.
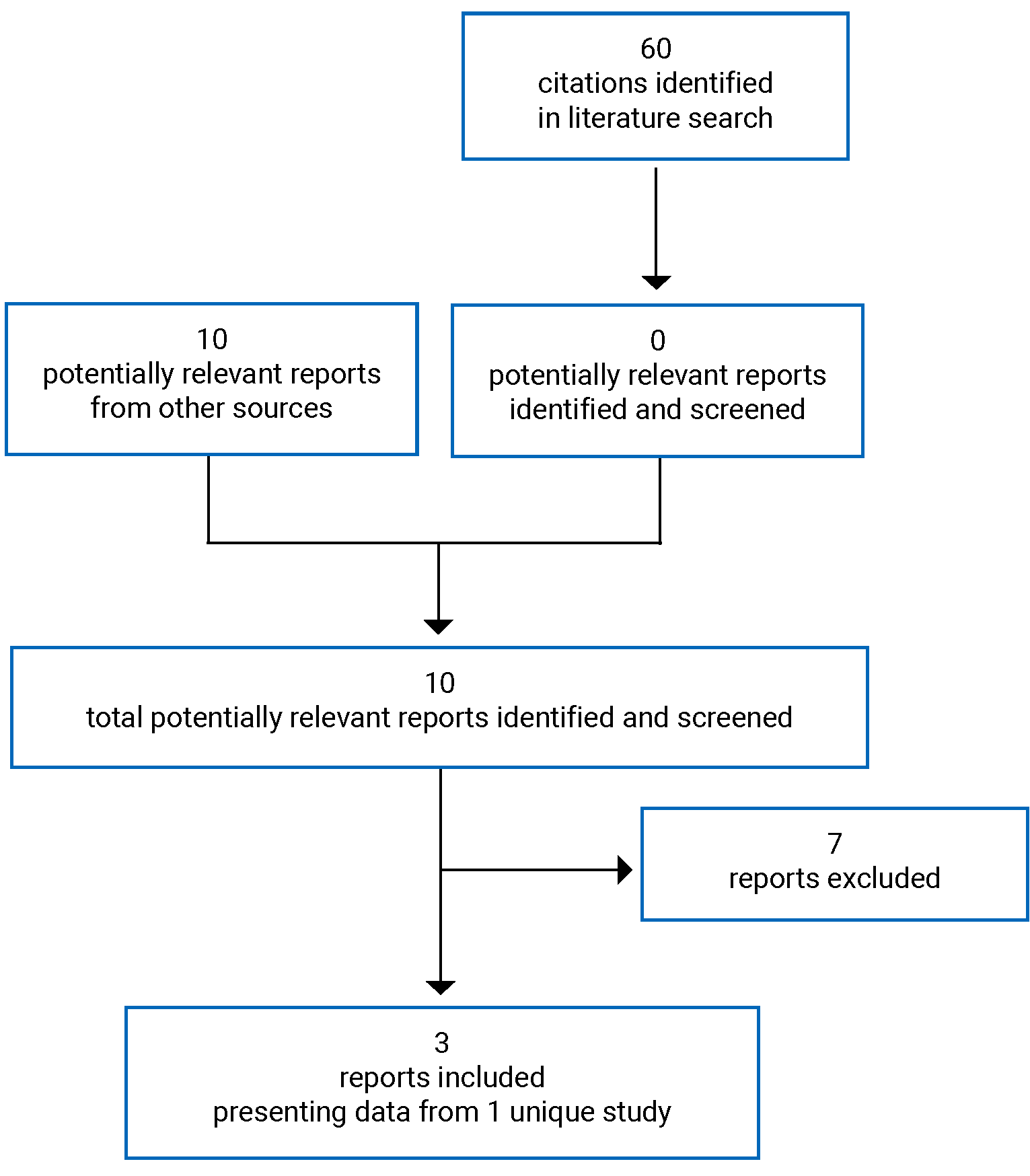
Table 5: Details of Included Studies
Detail | CHRYSALIS |
|---|---|
Designs and populations | |
Study design | Phase I/Ib, open-label, single-arm, dose-escalation study |
Locations | Australia, Canada, China, France, Japan, Korea, Spain, Taiwan, US, UK |
Patient enrolment dates | Cohort D start date: August 2, 2018 Cohort D end date: September 29, 2020 (last patient enrolled) Study end date: 6 months after the last patient on study treatment completes therapy with amivantamab and has at least 6 months of follow-up |
Data cut-off dates |
|
Enrolled (N) |
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Part 1 (dose escalation): Amivantamab (50 mg/mL) once weekly IV infusion in planned dose cohorts of 140 mg, 350 mg, 700 mg, 1,050 mg, 1,400 mg, and 1,750 mg Part 2 (dose expansion): Amivantamab (50 mg/mL) 1,050 mg (< 80 kg) or 1,400 mg (≥ 80 kg) once weekly IV infusion for cycle 1, followed by every 2 weeks beginning at cycle 2 |
Comparator(s) | Not applicable |
Duration | |
Phase | |
Screening | 28 days before first administration of amivantamab |
Treatment | Extending from cycle 1, day 1 until 30 ( + 7) days after the last dose of study drug (or start of subsequent anticancer therapy) |
Follow-up | Until disease progression or treatment discontinuation |
Outcomes | |
Primary efficacy end point | ORR (cohort D); Defined as the proportion of patients with a best overall response of a confirmed CR or PR based on RECIST v1.1 criteria |
Secondary and exploratory end points | Secondary (cohort D):
Exploratory:
Safety: AEs, laboratory abnormalities, monitoring of viral signs, ECGs, and physical examinations |
Notes | |
Publications | Park et al., 2021 |
AE = adverse event; CBR = clinical benefit rate; CHF = congestive heart failure; CR = complete response; ctDNA = circulating tumour DNA; DBP = diastolic blood pressure; DCO = data cut-off; DOR = duration of response; DVT = deep vein thrombosis; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group; EGFR = epidermal growth factor receptor; EQ-5D-5L = 5-level EQ-5D; ILD = interstitial lung disease; LLN = lower limit of normal; LVEF = left ventricular ejection fraction; MET = mesenchymal-epithelial transition; MI = myocardial infarction; MUGA = multigated acquisition; NSCLC-SAQ = Non–small Cell Lung Cancer Symptom Assessment Questionnaire; NYHA = New York Heart Association; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PGIC = Patient Global Impression of Change; PGIS = Patient Global Impression of Severity; PR = partial response; QTcF = corrected QT interval using Fridericia formula; RECIST = Response Evaluation Criteria in Solid Tumors; SBP = systolic blood pressure; SOC = standard of care; TKI = tyrosine kinase inhibitor; TTF = time to treatment failure.
Source: CHRYSALIS Clinical Study Report interim analysis 1.36
Description of Studies
One study was included in the review. Study EDI1001 (CHRYSALIS) is an ongoing, phase I/Ib, open-label, single-arm, multicentre study of amivantamab as monotherapy in patients with advanced NSCLC that includes both a dose-escalation phase (part 1), and a dose-expansion phase (part 2). The primary objective of part 1 of the CHRYSALIS study was to determine the maximally tolerated dose (MTD), if 1 existed (part 1 monotherapy dose escalation only), and the RP2D for patients with NSCLC treated with amivantamab. For part 2, the primary objective was to determine the safety, tolerability, and antitumour activity of amivantamab at the RP2D, and to estimate the antitumour activity of amivantamab at the RP2D in selected populations of patients with documented EGFR or MET mutation(s) who have progressed after treatment with SOC.36
A diagram of the study design for the monotherapy cohorts is provided in Figure 3. In part 1, a 3 plus 3 design was utilized to determine the MTD and the RP2D regimen(s) for amivantamab in patients with advanced NSCLC. Initially, 3 to 6 patients received amivantamab to achieve at least 3 dose-limiting toxicities (DLTs). If none of the first 3 DLT evaluable patients experienced a DLT, dose escalation proceeded with a new cohort. If 1 out of the first 3 DLT evaluable patients experienced a DLT, at least 3 additional patients were enrolled and treated at that dose level for safety. Under these circumstances, all additional patients in the cohort must have completed cycle 1 of treatment and if there was no occurrence of DLT in any additional DLT evaluable patients (i.e., less than 33% of patients at the current dose level have experienced DLT), then further dose escalation may have proceeded. Otherwise, the dose level with less than 33% of patients experiencing a DLT was considered the MTD.
Figure 2: Dose-Escalation Scheme of the CHRYSALIS Trial
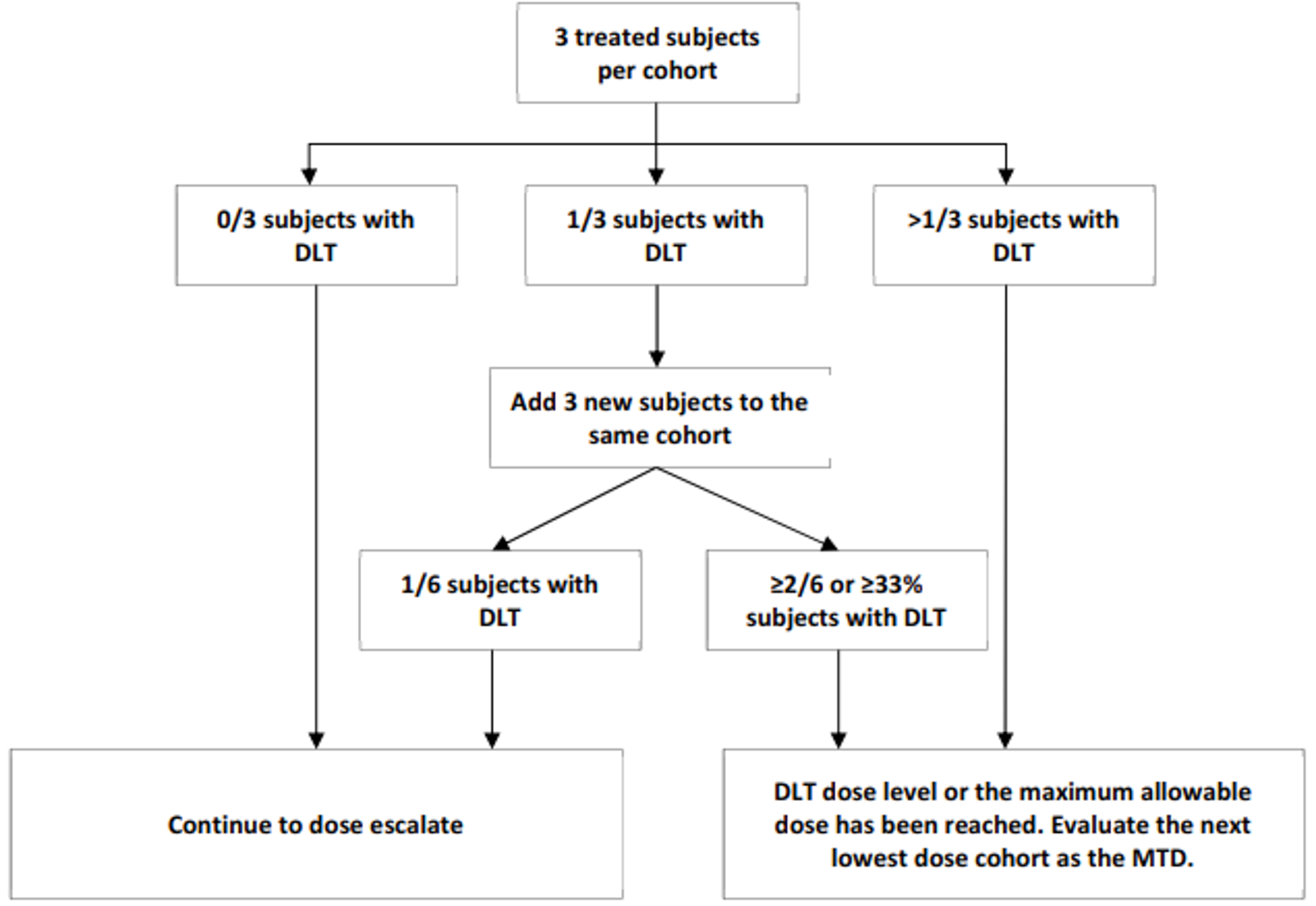
DLT = dose-limiting toxicity; MTD = maximally tolerated dose.
Source: CHRYSALIS Clinical Study Report interim analysis 1.36
In part 1, the starting dose of amivantamab was 140 mg, administered as an IV infusion once weekly for 4 weeks (cycle 1), then every 2 weeks thereafter during subsequent cycles until DLT. In the absence of DLT, dose escalation included planned dose cohorts of 140 mg, 350 mg, 700 mg, 1,050 mg, 1,400 mg, and 1,750 mg. Only toxicities that occurred during the period from the start of the first amivantamab infusion on cycle 1, day 1 through day 28 were used for the purpose of defining a DLT. Dose escalation was stopped when the MTD, defined as the highest dose level at which less than 33% of patients treated at that level experienced a DLT, was reached.36
Intrapatient dose escalation was allowed in part 1, with patients able to move from a lower dose level to the next higher dose level, if that dose has been previously declared safe according to the safety review process, after consultation with the sponsor’s medical monitor and in the absence of disease progression. Patients must tolerate at least 2 cycles at the dose at which they were enrolled (and at least 1 cycle at subsequent dose levels, if applicable) and receive approval from the medical monitor before being escalated to the next dose level.
In part 2, patients with advanced or metastatic NSCLC who had a previously diagnosed activating EGFR and/or MET mutation, measurable disease, and disease progression following prior systemic anticancer therapy were enrolled into 2 separate molecularly defined tumour subgroups (Cohorts A and B). Cohorts A and B were closed with study amendment 4 (further defined in the following section), and Cohorts C, D, MET-1, and MET-2 were added with Amendment 4.36 Note that only cohort D is of interest to this review because its patient population aligns with the requested reimbursement criteria. The current analysis presents the results of amivantamab monotherapy after platinum-based chemotherapy in patients who harboured EGFR exon 20 insertion mutations. Results for other cohorts will not be summarized and appraised.
Figure 3: Design of the CHRYSALIS Study: Monotherapy Cohorts
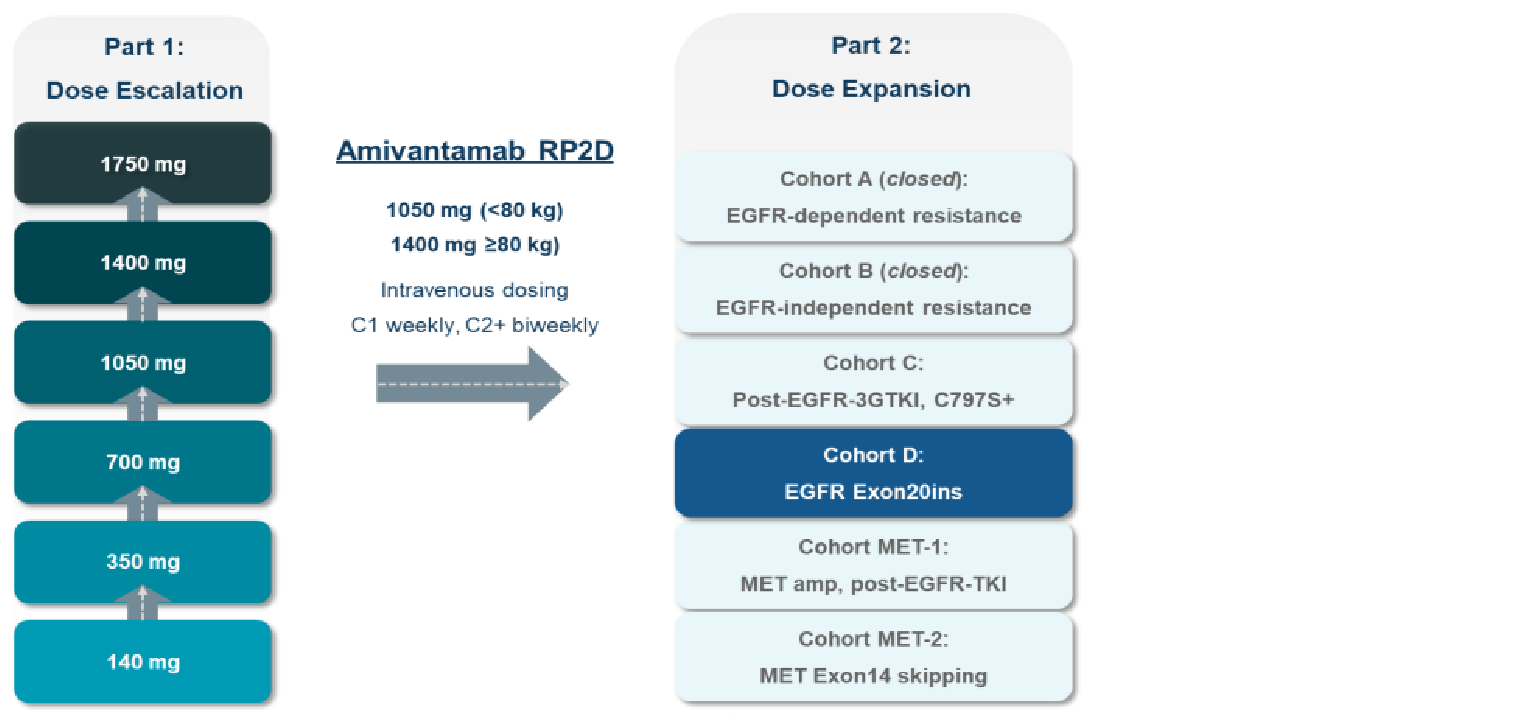
EGFR = epidermal growth factor receptor; MET = mesenchymal-epithelial transition; RP2D = recommended phase II dose; TKI = tyrosine kinase inhibitor.
Source: Sponsor submission.15
Across part 1 and part 2, patients with previously diagnosed activating EGFR exon 20 insertion mutation who had not been previously treated with a TKI with known activity in exon 20 insertion disease (e.g., poziotinib) but who had received treatment with a platinum-based chemotherapy regimen were enrolled into cohort D, in which patients were treated with the RP2D identified in part 1. This cohort was closed globally to further enrolment on May 29, 2020, except for in Japan and China,17,36 though the reason was not provided.15
The end of study was planned to occur 6 months after the last patient had completed study treatment with 6 or more months of follow-up.15
As of the first interim analysis, with a clinical cut-off of June 8, 2020 (median follow-up of 5.1 months), a total of 362 patients received at least 1 dose of amivantamab across part 1 and part 2, including 114 patients enrolled into cohort D. The primary efficacy population included 81 patients, and the safety analysis set consisted of 114 patients. There was 1 Canadian study site, which enrolled 6 patients into cohort D.36
At the third interim analysis, with a clinical cut-off of March 30, 2020 (median follow-up of 14.5 months), a total of 489 patients were enrolled into the CHRYSALIS trial, including 153 patients in cohort D, 81 patients in the primary efficacy population, 124 patients in the additional efficacy population, and 153 patients in the safety analysis set. As of this DCO, there was 1 Canadian study site that enrolled 21 patients.17 Details on the populations evaluated in cohort D of the CHRYSALIS trial are summarized in Table 6 and Figure 4. Data from the March 30, 2020, DCO are included in the main body of this report and data from the second interim analysis, October 8, 2020, are included in Appendix 3.
Table 6: Populations Evaluated in Cohort D of the CHRYSALIS Trial
Population | Definition | Data cut-off | ||
|---|---|---|---|---|
June 2020a | October 2020b | March 2021c | ||
Primary efficacy | Received a first dose of amivantamab with ≥ 3 efficacy assessments in the June 2020 cohort; included all patients who received the first dose of amivantamab as monotherapy on or before February 5, 2020 | 81 | 81d | 81e |
Expanded efficacy | Received a first dose of amivantamab with ≥ 3 efficacy assessments | NA | 114f | 124g |
Primary safety | Received a first dose of amivantamab within the June 2020 cohort | 114 | NA | NA |
Expanded safety | Received a first dose of amivantamab | NA | 129 | 153 |
NA = not applicable.
aN = 81 had a median follow-up time of 6.5 months; N = 114 had a median follow-up time of 5.1 months.
bN = 81 had a median follow-up time of 9.7 months; N = 114 had a median follow-up time of 8.3 months
cN = 81 had a median follow-up time of 14.5 months; N = 124 had a median follow-up time of 11.9 months.
dAll patients had greater than or equal to 6 months of follow-up from the onset of response, or had discontinued treatment as of the data cut-off.
eAll patients had greater than or equal to 6 months of follow-up from the onset of response, or had discontinued treatment as of the data cut-off.
fThis population of 114 patients had each initiated amivantamab therapy by June 4, 2020. All patients had greater than or equal to 6 months of follow-up from the onset of response, or had discontinued treatment as of the data cut-off.
gThis population of 124 patients had greater than or equal to 6 months of follow-up from the last-patient-enrolled date (September 29, 2020) at the data cut-off. Also, all patients had greater than or equal to 6 months of follow-up from the onset of response, or had discontinued treatment as of the data cut-off.
Figure 4: Visual Representation of Patient Populations Evaluated in Cohort D of the CHRYSALIS Trial
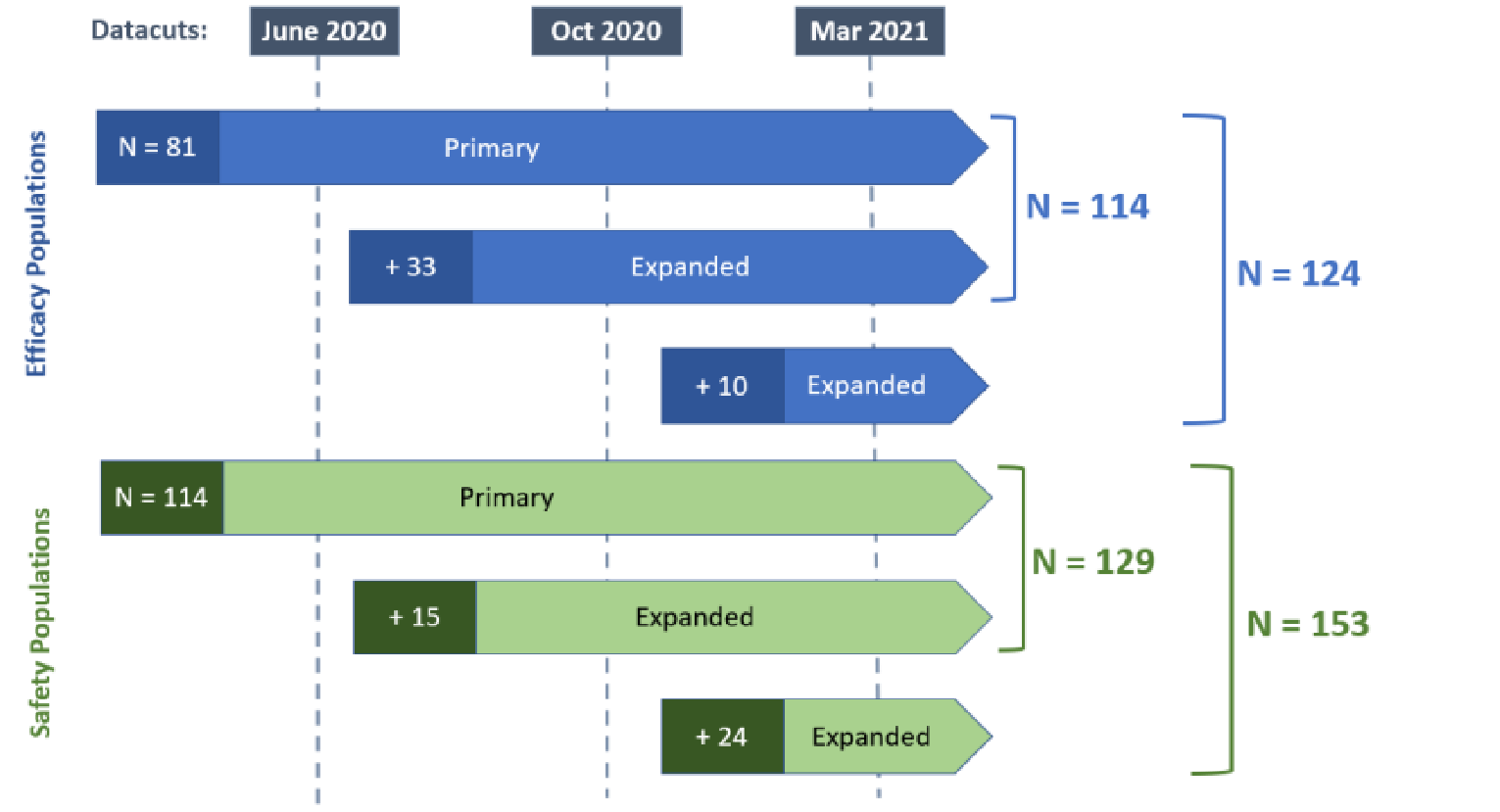
Source: Sponsor submission.15
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria for the CHRYSALIS trial are summarized in Table 5. Briefly, eligible patients consisted of adults (aged 18 years or older) diagnosed with metastatic or unresectable NSCLC who had either progressed after prior SOC therapy (prior platinum-based chemotherapy for cohort D) for metastatic disease, or were ineligible for or had refused all other currently available therapeutic options. For part 2, cohort D, patients also had to have marketed TKI-resistant mutations such as exon 20 insertion mutations, and specifically for cohort D, patients not previously treated with a TKI with known activity in exon 20 insertion disease (e.g., poziotinib) were eligible. Patients were also required to have evaluable disease (part 1) or measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 (part 2), and ECOG performance status less than or equal to 1. Patients were excluded if they had untreated or active brain metastases and a history of ILD.36
Baseline Characteristics
The baseline characteristics of patients in cohort D of the CHRYSALIS trial are summarized in Table 7. At baseline, per the March 30, 2021, DCO, 153 and 81 patients made up the safety analysis set and efficacy analysis set, respectively. In the safety analysis set, the median age of patients enrolled was 61.0 years, with the majority of patients younger than 65 years (62.1%). Most patients were Asian (62.1%) and female (61.4%), and had an ECOG performance status of 1 (72.5%). Most patients had stage III (8.6%) or IV (78.9%) disease, adenocarcinoma (96.1%), and no history of smoking (61.4%). The median number of prior lines of therapy was 2, with as many as 10 lines of therapy received. All patients (100%) had received prior platinum-based chemotherapy (mostly carboplatin; 65.4%), as well as non–platinum-based chemotherapy (mostly pemetrexed; 60.8%).36
Baseline characteristics for the primary efficacy population and the additional efficacy population were consistent with the safety analysis set.
Table 7: Summary of Baseline Characteristics for Cohort D (Safety Analysis Set; March 30, 2021, DCO)
Characteristic | Safety analysis set (N = 153) | Primary efficacy population (N = 81) | Additional efficacy population (N = 124) | |
|---|---|---|---|---|
Age | ||||
Mean (SD) | 61.2 (10.0) | 62.3 (9.96) | ███████████ | |
Median (range) | 61.0 (35 to 84) | 62.0 (42 to 84) | ███████████ | |
< 65 | 95 (62.1) | 48 (59.3) | ████████ | |
≥ 65 | 58 (37.9) | 33 (40.7) | ████████ | |
< 75 | 141 (92.2) | 74 (91.4) | █████████ | |
≥ 75 | 12 (7.8) | 7 (8.6) | ███████ | |
Sex, n (%) | ||||
Female | 94 (61.4) | 48 (59.3) | ████████ | |
Male | 59 (38.6) | 33 (40.7) | ████████ | |
Race, n (%) | ||||
Asian | 95 (62.1) | 40 (49.4) | ████████ | |
Black or African American | 3 (2.0) | 2 (2.5) | ██████ | |
White | 45 (29.4) | 30 (37.0) | ████████ | |
Not reported | 10 (6.5) | 9 (11.1) | ███████ | |
Weight, kg | ||||
Mean (SD) | 63.7 (15.4) | 67.49 (16.784) | █████████████ | |
Median (range) | 59.9 (35.4 to 115.0) | 62.5 (35.4 to 115.0) | ████████████████ | |
BMI, mean (SD), kg/m2 | 23.8 (4.5) | 25.0 (4.90) | ██████████ | |
ECOG performance status, n (%) | ||||
0 | 41 (26.8) | 26 (32.1) | ████████ | |
1 | 111 (72.5) | 54 (66.7) | ████████ | |
2 | 1 (0.7) | 1 (1.2) | ██████ | |
Smoking history, n (%) | ||||
Yes | 59 (38.6) | 38 (46.9) | █████████ | |
No | 94 (61.4) | 43 (53.1) | █████████ | |
NSCLC subtype, n (%) | ||||
Adenocarcinoma | 147 (96.1) | 77 (95.1) | █████████ | |
Squamous cell carcinoma | 4 (2.6) | 3 (3.7) | ██████ | |
Other | 2 (1.3) | 1 (1.2) | ██████ | |
Stage at initial diagnosis, n (%) | ||||
IA/IB | 10 (6.6) | 7 (8.6) | ███████ | |
IIA/IIB | 9 (5.9) | 5 (6.1) | ██████ | |
IIIA/IIIB | 13 (8.6) | 8 (9.8) | ███████ | |
IV | 120 (78.9) | 61 (75.3) | ████████ | |
Location of metastasis,a n (%) | ||||
Bone | 66 (43.1) | 35 (43.2) | ████████ | |
Liver | 14 (9.2) | 8 (9.9) | ████████ | |
Brain | 36 (23.5) | 18 (22.2) | ████████ | |
Lymph node | 77 (50.3) | 43 (53.1) | ████████ | |
Adrenal gland | 10 (6.5) | 3 (3.7) | ██████ | |
Other | 80 (52.3) | 45 (55.6) | ████████ | |
Number of prior lines of therapy | ||||
Mean (SD) | 2.2 (1.4) | 2.3 (1.41) | █████████ | |
Median (range) | 2.0 (1 to 10) | 2.0 (1 to 7) | █████████ | |
1 | 60 (39.2) | 31 (38.3) | ████████ | |
2 | 47 (30.7) | 24 (29.6) | ████████ | |
3 | 23 (15.0) | 11 (13.6) | ████████ | |
4 | 12 (7.8) | 7 (8.6) | ██████ | |
5+ | 11 (7.3) | 8 (9.8) | ███████ | |
Prior therapy, n (%) | ||||
Platinum-based chemotherapy | 153 (100.0) | 81 (100.0) | ██ | |
Carboplatin | 100 (65.4) | NR | ██ | |
Cisplatin | 80 (52.3) | NR | ██ | |
Non–platinum-based chemotherapy | 153 (100.0) | NR | ██ | |
Pemetrexed | 93 (60.8) | NR | ██ | |
Pemetrexed disodium | 38 (24.8) | NR | ██ | |
Paclitaxel | 32 (20.9) | NR | ██ | |
Docetaxel | 26 (17.0) | NR | ██ | |
Gemcitabine | 19 (12.4) | NR | ██ | |
EGFR TKI: third generation (osimertinib [mesylate]) | 13 (8.5) | 7 (9) | ██ | |
EGFR TKI: second generation (afatinib) | 15 (9.8) | 6 (7) | ██ | |
EGFR TKI: first generation (gefitinib, erlotinib [hydrochloride]) | 10 (6.5) | 6 (7) | ██ | |
Immunotherapy | 65 (42.5) | 37 (46) | ██ | |
Pembrolizumab | 34 (22.2) | NR | ██ | |
Nivolumab | 14 (9.2) | NR | ██ | |
BMI = body mass index; DCO = data cut-off; ECOG = Eastern Cooperative Oncology Group; EGFR = epidermal growth factor receptor; NR = not reported; NSCLC = non–small cell lung cancer; SD = standard deviation; TKI = tyrosine kinase inhibitor.
aPatients can be counted in more than 1 category.
Source: CHRYSALIS Clinical Study Report interim analysis (March 30, 2021);17 sponsor submission.15
Interventions
Amivantamab
Amivantamab was administered via IV infusion at a dose of 1,050 mg for less than 80 kg and 1,400 mg for greater than 80 kg of body weight and was given once weekly for the first 4 weeks (i.e., cycle 1) and once every 2 weeks in all subsequent 28-day cycles, over a minimum 60-minute infusion. Amivantamab was supplied as a sterile liquid in either 3 mL or 7 mL glass vials of 50 mg/mL for dilution for IV infusion. To minimize the risk of IRRs, the first dose was split over 2 days (cycle 1, days 1 and 2), required steroid premedication, and was administered using an accelerated infusion strategy.36
In part 1, amivantamab was given in doses of 140 mg, 350 mg, 700 mg, 1,050 mg, and 1,400 mg, as assigned to each dose-escalation cohort, dosed according to the 28-day cycle. After identifying body weight as a primary covariate explaining interindividual pharmacokinetic variability, the recommended RP2D for part 2 was determined to be 1,050 mg for patients weighing less than 80 kg and 1,400 mg for patients weighing greater than or equal to 80 kg, administered at the dosing schedule. Treatment was administered until disease progression, unacceptable toxicity, or withdrawal of consent. Patients who continued to receive clinical benefit despite documented disease progression were permitted to remain on study treatment at the discretion of the investigator.36
Protocol defined treatment discontinuation criteria included:
documented clinical or radiographic (RECIST v1.1) disease progression
unacceptable toxicity
general or specific changes in the patient’s condition that render the patient unacceptable for further treatment in the judgment of the investigator
pregnancy
intercurrent illness that prevents further administration of treatment
refusal of further treatment with study drug
receipt of concurrent (nonprotocol) systemic anticancer treatment
noncompliance with study drug or procedure requirements.
Prior and Concomitant Therapy
Prophylactic and predose medications were to be administered to all patients to prevent or lessen the severity of IRRs, rash, and nausea. Prophylactic treatment for IRRs included:36
IV administration of a glucocorticoid (i.e., dexamethasone 10 mg or methylprednisolone 40 mg) 45 to 60 minutes before study drug infusion on cycle 1, day 1, and 2
administration of antihistamine (diphenhydramine 25 to 50 mg or equivalent) given either IV 15 to 30 minutes or orally 30 to 60 minutes before each amivantamab infusion
administration of paracetamol (650 to 1,000 mg or equivalent) given either IV 15 to 30 minutes or orally 30 to 60 minutes before each amivantamab infusion.
Optional prophylactic treatments included glucocorticoid (IV or oral) given from cycle 1, day 8 and onward; histamine H2 antagonist (ranitidine 50 mg or equivalent) given IV at any cycle; or antiemetics (ondansetron 16 mg IV or 8 mg oral or equivalent) given at any cycle.36
Postinfusion medications could have been prescribed and continued for up to 48 hours after infusion of study drug if clinically indicated for the management of IRRs or other infusion-related symptoms. These included IV or oral glucocorticoids, antihistamines, antipyretics (paracetamol), opiates (meperidine), and antiemetics.36
Dose Modifications
In addition to prophylactic and reactive treatment regimen for rash, investigators were instructed to consider reducing the dose for grade 2 events and to interrupt treatment for grade 3 events (or grade 2 events that did not resolve after 2 weeks) until the event improved to less than or equal to grade 1. If rash worsened or did not improve after 2 weeks, treatment discontinuation was recommended.36
Investigators were instructed to withhold amivantamab for patients with drug-related toxicity (of any grade) whose symptoms were intolerable (per investigator assessment) until the toxicity returned to less than or equal to grade 1 or baseline. Investigators were instructed to interrupt dosing in the event of grade 3 or 4 toxicity. Following resolution of the event(s) (i.e., return to baseline or to less than or equal to grade 1 for nonhematologic toxicity), dosing could have been restarted according to the following guidance:36
Interruption of less than or equal to 7 days: restart at current dose level (grade 3 toxicity) or at next lower dose level (grade 4 toxicity)
Interruption of greater than 7 to less than or equal to 28 days: restart at next lower dose level
Investigators were instructed to consider permanently discontinuing amivantamab for patients whose dose was withheld for more than 28 days (i.e., 2 consecutive doses) due to toxicity, and for patients with grade 4 toxicity whose treatment was interrupted for greater than 7 to less than or equal to 28 days unless the patient was considered to be benefiting from the drug and with agreement from the sponsor’s medical monitor.36
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized in the following sections.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | CHRYSALIS cohort D |
|---|---|
ORR | Primary |
DOR | Secondary |
PFS | Secondary |
OS | Secondary |
HRQoL | Secondary |
Symptom severity | Secondary |
TTF | Secondary |
Safety | Secondary |
DOR = duration of response; HRQoL = health-related quality of life; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; TTF = time to treatment failure.
Primary Efficacy End Point
The primary outcome of the CHRYSALIS trial was ORR, defined as the proportion of patients with a best overall response of a confirmed complete response (CR) or PR as assessed by the investigator and BICR based on RECIST v1.1 criteria. Best overall response was recorded from the start of the study drug until disease progression, withdrawal of consent, or start of a subsequent anticancer therapy, whichever came first.36
Disease assessments were performed every 6 (plus or minus 1) weeks during part 1 and part 2.36
Secondary Efficacy End Points
The secondary outcomes of the CHRYSALIS trial for cohort D were based on investigator and BICR and included:36
CBR was defined as the percentage of patients achieving a best overall response of confirmed CR, confirmed PR, or durable stable disease (SD; duration of at least 11 weeks) as defined by RECIST v1.1. CBR was not defined as an outcome of interest in the CADTH review protocol. Results for CBR are presented in Appendix 3.
DOR was defined as the time from first documentation of a PR or CR to the date of first documented evidence of disease progression or death due to any cause, whichever occurred first. Patients who were progression-free and alive or had unknown status were censored at last tumour assessment, and patients who started a subsequent anticancer therapy in the absence of progression were censored at the last disease assessment before the start of subsequent therapy.
PFS was defined as the time from the first dosing date to the first date of disease progression or death due to any cause, whichever occurred first. Patients who were progression-free and alive or have unknown status were censored at last tumour assessment. Patients with no postbaseline disease assessment were censored on day 1. Patients who started a subsequent anticancer therapy in the absence of progression were censored at the last disease assessment before the start of subsequent therapy, and patients whose diseases have not progressed and who were still alive at the end of the study or clinical cut-off were censored at the last adequate disease assessment.
OS was defined as the time interval from the first dosing date to the date of death from any cause. If the patient was alive or the vital status was unknown, then the patient’s data were censored at the date the patients was last known to be alive.
TTF was defined as the time interval from the first dosing date to study drug discontinuation for any reason. Patients who are treatment failure-free or have unknown status was censored at last tumour assessment, and patients with no postbaseline disease assessment were censored on day 1.
Four PRO assessments were included in this study with implementation of Amendment 7. Instruments included the Non–small Cell Lung Cancer Symptom Assessment Questionnaire (NSCLC-SAQ), the Patient Global Impression of Severity (PGIS) and Patient Global Impression of Change (PGIC), and the 5-level EQ-5D (EQ-5D-5L). The NSCLC-SAQ contains 7 items that assess cough, pain, dyspnea, fatigue, and poor appetite over a 7-day recall period. The EQ-5D-5L is a validated tool to measure health status and health utility, including mobility, self-care, usual activities, pain, discomfort, and anxiety/depression on a scale of 0 (worst) to 100 (best). The PGIS and PGIC are single-item questionnaires with PGIS scores ranging from 1 (not at all severe) to 5 (very severe), and PGIC scores ranging from 1 (a lot better now) to 7 (a lot worse now).15 Due to the limited follow-up time and small sample, PRO data were only included as supportive data in the sponsors’ clinical package.36
Safety End Points
Safety analyses were performed based on the safety analysis set. Safety monitoring was the same for both part 1 and part 2 and included evaluation of incidence, severity, and type of AEs and laboratory abnormalities (hematology, clinical chemistry), which were graded per the National Cancer Institute Common Terminology Criteria for Adverse Events, Version 4.03. Other safety measures include monitoring of vital signs (temperature, pulse rate, blood pressure, respiratory rate, pulse oximetry), ECGs, and physical examinations.
Statistical Analysis
Sample Size and Power Calculation
For part 1, cohorts of 3 to 6 patients were typically treated at each dose level based on the 3 plus 3 dose-escalation scheme (described previously). For part 2, the maximum total sample size at an RP2D was set to be approximately 460 patients. This included approximately 40 patients in Cohort A, 20 patients in Cohort B, and up to 100 patients each if sufficient efficacy was observed in Cohorts C, D, MET-1, and MET-2 at an RP2D of amivantamab monotherapy.36
The sample size calculation for CHRYSALIS was based on the primary end point of ORR, assuming a null hypothesis of ORR for amivantamab per RECIST v1.1 of less than or equal to 15%, and alternative hypothesis of ORR greater than or equal to 30%. With a 1-sided alpha of 2.5%, and a power of 87.5%, the total number of patients needed for each cohort was 86 response-evaluable patients. Assuming a nonevaluable rate of 15%, approximately 100 patients were to be enrolled within each cohort, though the number of patients could be expanded beyond 100 patients (maximum of approximately 150) to further characterize activity for subpopulations within a cohort.36
The sample size consideration for the subgroup in cohort D who were required to have had previous therapy with a combination platinum-doublet chemotherapy regimen was based on the null hypothesis of ORR for amivantamab per RECIST v1.1 of less than or equal to 12%, and the alternative hypothesis of ORR greater than 25% based on response rates for platinum-doublet or single-drug chemotherapy regimens. To have a power of 80% to reject the null hypothesis with a 1-sided alpha of 2.5%, at least 60 patients were required to be enrolled in the subgroup of patients with exon 20 insertion mutations previously treated with platinum chemotherapy; approximately 100 patients were targeted for enrolment to characterize the activity of amivantamab in this population.36
Interim Analyses
For cohorts C, D, MET-1, and MET-2 in part 2, within each cohort, a 2-stage design was employed.
A planned interim monitoring for futility was carried out separately for cohorts C, D, MET-1, and MET-2 when there were at least 30 response-evaluable patients. With 30 patients evaluable for response within cohorts C, D, MET-1, and MET-2, if 5 or fewer responses were observed, the null hypothesis (ORR ≤ 15%) was to be accepted and enrolment for the cohort may have been terminated for futility by the safety evaluation team (SET). Otherwise, additional patients were enrolled for total 100 patients in the cohort for the final analysis. This stopping criterion led to a probability of early termination of at least 71.1% for Cohorts C, D, MET-1, and MET-2.15 No further information or results of the interim monitoring for futility was provided.
The first interim analysis was conducted based on a clinical cut-off date of June 8, 2020, representing 6.5 months of follow-up for the primary efficacy population. According to the sponsor, hypothesis testing for the primary end point was conducted at the June 8, 2020, DCO.37 The second interim analysis considered an addendum to the first interim analysis had a clinical cut-off date of October 8, 2020, and had a median follow-up of 9.7 months. The third interim analysis was based on a clinical cut-off of March 30, 2021, representing 14.5 months of follow-up. According to the sponsor, both the June 8, 2022, and October 8, 2020, DCO dates were based on agreements with the FDA.
The primary efficacy population (described below) represented the same group of patients followed up over time. The safety analysis set increased with additional enrolment beyond the cut-off for the primary efficacy population.
Analysis Populations
Results of efficacy and safety data are presented for the following populations in this report:17,36,38
Primary efficacy population: Includes exon 20 insertion plus prior chemotherapy patients (N = 81) enrolled in part 1 (dose escalation) or part 2 (dose expansion) who have received their first dose with RP2D amivantamab monotherapy by February 5, 2020. These 81 consecutively treated patients had undergone 3 or more postbaseline disease assessments or discontinued treatment for any reason, including disease progression/death, before the clinical cut-off of June 8, 2020.
Additional efficacy population:
Includes exon 20 insertion plus prior chemotherapy patients (N = 114) enrolled in part 1 and part 2 who had received prior platinum-based chemotherapy and were treated at the RP2D, and had undergone 3 or more postbaseline disease assessments or discontinued treatment for any reason, including disease progression/death, as of the clinical cut-off of October 8, 2020. All patients had initiated treatment before June 8, 2020, and this population was inclusive of the 81 patients in the primary efficacy population.38
Includes exon 20 insertion plus prior chemotherapy patients (N = 124) enrolled in part 1 or part 2 who had received prior platinum-based chemotherapy and 1 or more dose of amivantamab monotherapy at the RP2D, and who had 6 or more months of follow-up from the last-patient-enrolled date (September 29, 2020) at this clinical cut-off of March 30, 2021. This population was inclusive of the patient efficacy set described previously (i.e., N = 114).
Primary safety population: Includes exon 20 insertion plus prior chemotherapy at RP2D patients (N = 153) with EGFR exon 20 insertion NSCLC enrolled in part 1 or part 2 who had received prior platinum-based chemotherapy and 1 or more doses of amivantamab monotherapy at the RP2D as of the March 30, 2021, clinical cut-off.
Efficacy Analyses
All efficacy analyses were performed using the primary and the additional efficacy populations. In addition to the investigator assessment, scans were centrally collected for potential assessment of response by BICR using RECIST v1.1 criteria.36
Primary Efficacy End Point
The primary efficacy analysis of ORR with confirmed best overall responses was performed approximately 12 weeks after the last patient received the first infusion or at the end of study.15 The observed ORR and its 95% 2-sided exact CI were presented based on the primary efficacy population. Assessment of responses was performed according to RECIST v1.1 criteria by the investigator. Subsequently, assessment of efficacy within exon 20 insertion patients was completed by BICR.36
The following response criteria (according to RECIST v1.1) were acceptable: CR, PR, SD, PD, and not evaluable. A response of PR or CR must have been confirmed by repeat assessments 4 or more weeks from the initial observation. For a response to qualify as SD, follow-up measurements must have met SD criteria at least once at a minimum interval not less than 6 weeks after the first dose of study drug. If symptomatic deterioration (on the basis of global deterioration of health status) was recorded as the basis for determining disease progression, every effort was to be made to document radiographic progression even after discontinuation of treatment for symptomatic deterioration, but before initiation of subsequent anticancer therapy.36
Subgroup Analyses
Prespecified subgroup analyses for the primary end point of ORR (and 95% CI) per investigator assessments were analyzed for the following subgroups of the exon 20 insertion at RP2D efficacy populations: age (aged < 65 years versus ≥ 65 years and < 75 years versus ≥ 75 years), sex (male versus female), race (those who identified as Asian versus those who did not), baseline ECOG performance status (0 versus ≥ 1), smoking history (yes versus no), prior immunotherapy (yes versus no), and key exon 20 insertion variants.36 Only subgroups identified a priori in the CADTH review protocol (Table 4) were presented in the results section of this report.
Secondary Efficacy End Points
Secondary outcomes of the CHRYSALIS trial included CBR, DOR, PFS, OS, and TTF. CBR and its 95% 2-sided exact CI were presented based on the efficacy analysis set. DOR was summarized descriptively using the Kaplan-Meier method for responders in efficacy analysis set. The median PFS, OS, and TTF and 95% CI were estimated using the Kaplan-Meier method.
Safety Analyses
Safety monitoring was the same for both part 1 and part 2 and was based on the safety analysis population. The baseline value for safety assessment is defined as the value collected at the time closest to, but before, the first dose of study drug. Exposure to investigational product and reasons for discontinuation of study drug were tabulated. Unless otherwise specified, no inferential statistical analyses were performed in analyzing the safety data.36
Safety data obtained during the study were reviewed on a routine basis by the SET, consisting of participating principal investigators, the sponsor’s medical monitor and clinical pharmacologist, and the sponsor’s safety management team chair. Among the SET’s responsibilities were the recommendation of modification(s) to the study drug dose, schedule, and/or regimen in the dose-escalation phase (part 1); the RP2D regimen(s) to be investigated in the dose-expansion phase (part 2); and continued enrolment or termination of cohorts in the dose-expansion phase (part 2).36
Protocol Amendments and Deviations
There were 10 global amendments to the original protocol dated October 15, 2015, as well as several country-specific amendments. Key global amendments are summarized in Table 9. Patient enrolment into cohort D initiated with Amendment 4 on March 9, 2018, in which 6 additional amendments followed enrolment.
In Amendment 5 (September 6, 2018), timing of assessment for response was updated in accordance with RECIST v1.1: assessed at baseline, at week 6 (plus 1), and then every 6 (plus 1) weeks until disease progression by imaging, start of new anticancer therapy, or withdrawal of consent, as opposed to baseline, week 6, week 15, and then every 8 weeks, which may have affected the assessment of time to response or time to progression; however, given that the number of patients enrolled before this amendment was not provided, the effect remains uncertain.
Amendment 6 (May 29, 2019) clarified that characterization of tumour tissues for EGFR and MET via NGS was to be conducted during the prescreening period and of circulating tumour DNA via NGS during the screening period. As such, the target group was better identified from this point, though it was uncertain how many patients were enrolled before this change.
Table 9: Key Changes Implemented with Global Protocol Amendments for CHRYSALIS
Amendment number (date) | Key changes |
|---|---|
Amendment 1 (April 14, 2016) |
|
Amendment 2 (December 12, 2016) |
|
Amendment 3 (May 31, 2017) | Based upon preliminary data suggesting an early tumour response in a patient with an EGFR mutation treated in the 700 mg cohort of part 1, the protocol amendment was implemented to:
|
Amendment 4 (March 9, 2018) | This primary purpose of this amendment was to establish that the 1,400 mg dose level was determined as safe by the SET, and to add a 1,750 mg dose level to the planned dose-escalation scheme for part 1. In addition, this amendment:
|
Amendment 5 (September 6, 2018) | The primary purpose of this amendment was to enact the SET decision to limit eligibility for Cohort C (part 2) to those patients with a demonstrable EGFR or MET mutation conferring resistance to treatment with previous TKI. In addition, the protocol was amended to:
|
Amendment 6 (May 29, 2019) |
|
Amendment 7 (August 19, 2019) |
|
Amendment 8 (January 27, 2020) |
|
Amendment 9 (April 30, 2020) |
|
Amendment 10 (August 16, 2021) |
|
ALK = anaplastic lymphoma kinase; CBR = clinical benefit rate; ECG = electrocardiogram; EGFR = epidermal growth factor receptor; ILD = interstitial lung disease; IRR = infusion-related reaction; MET = mesenchymal-epithelial transition; NGS = next-generation sequencing; NSCLC = non–small cell lung cancer; ORR = overall response rate; OS = overall survival; RP2D = recommended phase II dose; SET = safety evaluation team; SOC = standard of care; TKI = tyrosine kinase inhibitor; WT = wild type.
Source: CHRYSALIS Clinica Study Report interim analysis 1;3 CHRYSALIS CSR interim analysis (March 30, 2021).17
A total of 28 major protocol deviations occurred in 25 (16.3%) patients in cohort D of the CHRYSALIS trial (Table 10). A total of 6 (3.9%) patients developed withdrawal criteria but were not withdrawn. All 6 patients had confirmed disease progression and continued study treatment before obtaining sponsor approval for treatment beyond progression.
None of these deviations led to exclusion of data from the safety or efficacy analyses or were considered to have affected the conduct or integrity of the study.
Table 10: Summary of Major Protocol Deviations (Safety Analysis Set; March 30, 2021, DCO)
Protocol deviations | Cohort D (exon 20 insertion prior chemotherapy) (N = 153) |
|---|---|
Patients with major protocol deviations, n (%) | 25 (16.3) |
Developed withdrawal criteria but not withdrawn | 6 (3.9) |
Entered but did not satisfy criteria | 5 (3.3) |
Received a disallowed concomitant treatment | 1 (0.7) |
Received wrong treatment or incorrect dose | 7 (4.6) |
Other | 9 (5.9) |
DCO = data cut-off.
Source: CHRYSALIS Clinical Study Report interim analysis (March 30, 2021).17
Results
Patient Disposition
As of the March 30, 2021, DCO, a total of 489 patients received amivantamab monotherapy in the CHRYSALIS trial, of which 380 patients received amivantamab monotherapy at RP2D (n = 30 in part 1, and n = 350 in part 2), and 153 patients with exon 20 insertion who had received prior platinum-based chemotherapy received monotherapy at RP2D (n = 5 in part 1, and n = 148 in part 2). For cohort D, a total of 43 (28.1%) patients completed the study (patients are considered to have completed the study if the patient died before the end of study), 95 (62.1%) were still in the study, and 15 (9.8%) had terminated the study prematurely. The most common reason for premature termination was withdrawal by the patient (12; 7.8%).17
As of the March 30, 2021, DCO, 56 (36.6%) patients were still receiving amivantamab and 97 (63.4%) had discontinued treatment. The most common reason for treatment discontinuation was PD (73; 47.7%). The primary efficacy analysis population consisted of 81 patients enrolled in cohort D, while the safety analysis set consisted of 153 patients.17
Table 11: Patient Disposition (March 30, 2021, DCO)
Disposition | CHRYSALIS |
|---|---|
All treated (all RP2D + non-RP2D) | 489 |
All treated at RP2D | 380 |
All treated cohort D (exon 20 insertion) at RP2D | 153 |
Study disposition (cohort D safety analysis set, N = 153) | |
Ongoing on study, N (%) | 95 (62.1) |
Completed study participation, N (%)a | 43 (28.1) |
Terminated study participation prematurely, N (%) | 15 (9.8) |
Treatment disposition (cohort D safety analysis set, N = 153) | |
Ongoing on treatment, N (%) | 56 (36.6) |
Discontinued study treatment, N (%) | 97 (63.4) |
Reason for discontinuation, N (%) | |
Progressive disease | 73 (47.7) |
Adverse event | 12 (7.8) |
Withdrawal by patient | 7 (4.6) |
Physician decision | 2 (1.3) |
Death | 3 (2.0) |
Primary efficacy analysis set, N | 81 |
Additional efficacy analysis set, N | 124 |
Primary safety analysis set, N | 153 |
DCO = data cut-off; RP2D = recommended phase II dose.
aA patient is considered to have completed the study if the patient died before the end of study.
Source: CHRYSALIS Clinical Study Report interim analysis 1 and 2.17,36
Exposure to Study Treatments
The extent of exposure for patients treated with amivantamab in cohort D is summarized in Table 12. The median duration of treatment with amivantamab was 5.6 months (range, 0.03 to 23.89), with 71 (46.4%) patients having received treatment for 6 or more months. The median number of treatment cycles was 7.0, with 52 (34.0%) patients having received treatment for 10 or more cycles.
The median relative dose intensity, defined as the ratio of total received dose versus total prepared dose, was 100%. Infusion modifications were reported in 93 (60.8%) patients, with most amivantamab modifications during infusion due to IRRs (90; 58.8%). Dose reductions were reported for 22 (14.4%) patients, mostly due to dermatitis acneiform (7; 4.6%) and paronychia (6; 3.9%).17 Dose interruptions occurred in 55 (35.9%) patients due to AEs. Details on the duration of infusions for amivantamab in the safety analysis population are summarized in Table 36, Appendix 3.
Overall, 8 (5.2%) patients missed scheduled doses of amivantamab due to COVID-19–related issues.17
Table 12: Summary of Treatment Exposure (Safety Analysis Set; March 30, 2021, DCO)
Treatment exposure | CHRYSALIS cohort D (N = 153) |
|---|---|
Duration of exposure, monthsa | |
Mean (SD) | 7.28 (5.81) |
Median (range) | 5.55 (0.03 to 23.89) |
< 2 months | 31 (20.3) |
2 to < 4 months | 26 (17.0) |
4 to < 6 months | 25 (16.3) |
≥ 6 months | 71 (46.4) |
Total number of cyclesb | |
Mean (SD) | 8.5 (6.24) |
Median (range) | 7.0 (1, 27) |
Relative dose intensity (%) | |
Mean (SD) | 99.21 (6.55) |
Median (Range) | 100 (20.0 to 100.0) |
Dose modifications, reductions, or interruptions | |
Infusion modificationc | 93 (60.8) |
Infusion aborted | 14 (9.2) |
Infusion interrupted | 88 (57.5) |
Infusion rate decreased | 82 (53.6) |
AE | 91 (59.5) |
IRRs | 90 (58.8) |
Non-IRRs | 2 (1.3) |
Other | 7 (4.6) |
Dose reduced compared to prior infusion | 25 (16.3) |
Dose interruption (due to AEs)d | 55 (35.9) |
Infusion skipped (and not made up)e | 66 (43.1) |
COVID-19 related | 8 (5.2) |
Cycle delay | 30 (19.6) |
Infusion delayed within the cycle | 11 (7.2) |
AE = adverse event; IRR = infusion-related reaction; SD = standard deviation.
aTreatment duration is defined as the duration from the date of the first dose of study drug to the date of last dose of study drug plus 1 divided by 30.4375.
bA patient is considered as treated in a cycle if the patient received any nonzero dose of study drug in that cycle.
cInfusion modification of study drug is based on infusion interrupted, infusion rate decreased, and infusion aborted.
dExcludes infusion-related reactions.
eWhen infusion skipped and COVID-19 protocol deviation or AE occur concurrently, the infusion skipped is considered to be COVID-19 related.
Source: CHRYSALIS Clinical Study Report interim analysis (March 30, 2021).17
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported below. Refer to Appendix 3 for detailed efficacy data.
Clinical Response
Overall Response Rate
ORR was the primary end point of the CHRYSALIS trial. Results for ORR for both efficacy populations as assessed by the investigator and BICR are summarized in Table 13. In the primary efficacy population (N = 81), a total of 31 (38.3%; 95% CI, 27.7% to 49.7%) patients achieved an ORR per investigator assessment, and 35 (43.2%; 95% CI, 32.2% to 54.7%) patients achieved an ORR per the BICR assessment.17
According to the investigators, no patients achieved a CR, and all patients who contributed to ORR were partial responders (31; 38.3%). In the BICR assessment, there were 3 (3.7%) patients who achieved a CR, and 32 (39.5%) who had PR. Most patients had SD in both the investigator and BICR assessments (37 [45.7%] and 35 [43.2%], respectively).17 Results for CBR are presented in Table 34, Appendix 3.
Results for the additional efficacy analysis set (N = 124) were consistent with the primary analysis (Table 13).
Results for the October 8, 2020, DCO for the primary efficacy population (N = 81; median follow-up = 9.7 months) and the additional efficacy population (N = 114; median follow-up = 8.3 months) are available in Table 35, Appendix 3. Results at the October 8, 2020, DCO were overall consistent with the results at the March 30, 2021, DCO.38
Results at the June 8, 2020, DCO date are not presented in this report but were, overall, consistent with the results at the March 30, 2021, DCO.
Table 13: Summary of ORR Based on RECIST v1.1 in Patients With Measurable Disease at Baseline — Investigator and BICR Assessment (March 30, 2021, DCO)
Outcome | Cohort D primary efficacy population (N = 81) | Cohort D additional efficacy population (N = 124) | ||
|---|---|---|---|---|
Investigator assessed | BICR | Investigator assessed | BICR | |
ORR | ||||
ORR (confirmed CR + confirmed PR), n (%) | 31 (38.3) | 35 (43.2) | ████████ | ████████ |
95% CI | (27.7 to 49.7) | (32.2 to 54.7) | ████████ | ████████ |
BOR, n (%) | ||||
CR | 0 (0.0) | 3 (3.7) | ██████ | ██████ |
PR | 31 (38.3) | 32 (39.5) | ████████ | ████████ |
SD | 37 (45.7) | 35 (43.2) | ████████ | ████████ |
PD | 12 (14.8) | 9 (11.1) | ████████ | ████████ |
NE or unknown | 1 (1.2) | 2 (2.5) | ██████ | ██████ |
BICR = blinded independent central review; BOR = best overall response; CI = confidence interval; CR = complete response; NE = not evaluable; ORR = overall response rate; PD = progressive disease; PR = partial response; RECIST = Response Evaluation Criteria in Solid Tumors; SD = stable disease.
Source: CHRYSALIS Clinical Study Report interim analysis (March 30, 2021).17
Subgroup Analysis
Subgroup analyses for ORR in the CHRYSALIS trial as assessed by the investigator and BICR for the primary and additional efficacy populations are summarized in Table 14. Results for the subgroup analyses were generally consistent with the primary analysis.
Table 14: Subgroup Analysis of ORR Based on RECIST v1.1 in Patients With Measurable Disease at Baseline (March 30, 2021, DCO)
Subgroup | Cohort D primary efficacy population (N = 81) | Cohort D additional efficacy population (N = 124) | ||||||
|---|---|---|---|---|---|---|---|---|
Investigator assessed | BICR | Investigator assessed | BICR | |||||
n of N | ORR (95% CI) | n of N | ORR (95% CI) | n of N | ORR (95% CI) | n of N | ORR (95% CI) | |
Overall | 31 of 81 | 38.3 (27.7 to 49.7) | 35 of 81 | 43.2 (32.2 to 54.7) | ██████ | █████████ | ██████ | █████████ |
Age | ||||||||
< 65 | 20 of 48 | 41.7 (27.6 to 56.8) | 23 of 48 | 47.9 (33.3 to 62.8) | █████ | ███████ | █████ | █████████ |
≥ 65 | 11 of 33 | 33.3 (18.0 to 51.8) | 12 of 33 | 36.4 (20.4 to 54.9) | █████ | ███████ | █████ | █████████ |
< 75 | 28 of 74 | 37.8 (26.8 to 49.9) | 33 of 74 | 44.6 (33.0 to 56.6) | ██████ | ███████ | ██████ | █████████ |
≥ 75 | 3 of 7 | 42.9 (9.9 to 81.6) | 2 of 7 | 28.6 (3.7 to 71.0) | ████ | ███████ | ████ | █████████ |
Sex | ||||||||
Male | 13 of 33 | 39.4 (22.9 to 57.9) | 16 of 33 | 48.5 (30.8 to 66.5) | █████ | ███████ | █████ | █████████ |
Female | 18 of 48 | 37.5 (24.0 to 52.6) | 19 of 48 | 39.6 (25.8 to 54.7) | █████ | ███████ | █████ | █████████ |
Race | ||||||||
Asian | 14 of 40 | 35.0 (20.6 to 51.7) | 18 of 40 | 45.0 (29.3 to 61.5) | █████ | ███████ | █████ | ████████ |
Other | 13 of 32 | 40.6 (23.7 to 59.4) | 13 of 32 | 40.6 (23.7 to 59.4) | █████ | ███████ | █████ | ████████ |
Smoking history | ||||||||
Yes | 12 of 38 | 31.6 (17.5 to 48.7) | 15 of 38 | 39.5 (24.0 to 56.6) | █████ | ███████ | █████ | ████████ |
No | 19 of 43 | 44.2 (29.1 to 60.1) | 20 of 43 | 46.5 (31.2 to 62.3) | █████ | ███████ | █████ | ████████ |
Prior IO | ||||||||
Yes | 16 of 38 | 42.1 (26.3 to 59.2) | 19 of 38 | 50.0 (33.4 to 66.6) | █████ | ███████ | █████ | ████████ |
No | 15 of 43 | 34.9 (21.0 to 50.9) | 16 of 43 | 37.2 (23.0 to 53.3) | █████ | ███████ | █████ | ████████ |
BICR = blinded independent central review; CI = confidence interval; DCO = data cut-off; IO = immunotherapy; ORR = overall response rate.
Source: CHRYSALIS Clinical Study Report interim analysis (March 30, 2021).17
Progression-Free Survival
Results for PFS are summarized in Table 15 and displayed graphically in Figure 5, Appendix 3. At the time of the DCO (March 30, 2021), a total of 57 PFS events had occurred in the primary efficacy population. With a median follow-up of 14.5 months, the median PFS was 8.25 months (95% CI, 5.49 to 12.32) based on the investigator assessment, and 8.31 months (95% CI, 5.52 to 11.07) based on the BICR assessment. The PFS rate with amivantamab at 3, 6, and 12 months per investigator assessment was 75% (95% CI, 64% to 83%), 58% (95% CI, 47% to 68%), and 40% (95% CI, 29% to 50%), respectively.17
With a median follow-up of 11.9 months, the median PFS for the additional efficacy population (N = 124) per investigator and BICR assessment was ███████████████████████████████████████████████████████████), respectively.17 The PFS rate for the additional efficacy population was consistent with the primary efficacy population.
Results for the October 8, 2020, DCO for the primary efficacy population (N = 81; median follow-up: 9.7 months) and the additional efficacy population (N = 114; median follow-up: 8.3 months) are available in Table 35, Appendix 3. Results at the October 8, 2020, DCO were overall consistent with the results at the March 30, 2021, DCO.38
Results at the June 8, 2020, DCO date are not presented in this report but were overall consistent with the results at the March 30, 2021, DCO date.
Table 15: Summary of PFS per Investigator and BICR Assessment (March 30, 2021, DCO)
Progression-free survival | Cohort D primary efficacy population (N = 81) | Cohort D additional efficacy population (N = 124) | ||
|---|---|---|---|---|
Investigator assessed | BICR | Investigator assessed | BICR | |
Event | 57 (70.4) | 54 (66.7) | ████████ | ████████ |
Censored | 24 (29.6) | 27 (33.3) | ████████ | ████████ |
Time to event (months) | ||||
Median (95% CI) | 8.25 (5.49 to 12.32) | 8.31 (5.52 to 11.07) | █████████████ | █████████████ |
PFS rate, % (95% CI) | ||||
3 month | 0.75 (0.64 to 0.83) | 0.79 (0.68 to 0.87) | █████████████ | █████████████ |
6 month | 0.58 (0.47 to 0.68) | 0.61 (0.49 to 0.71) | █████████████ | █████████████ |
9 month | 0.44 (0.33 to 0.54) | 0.47 (0.35 to 0.58) | █████████████ | █████████████ |
12 month | 0.40 (0.29 to 0.50) | 0.36 (0.25 to 0.47) | █████████████ | █████████████ |
15 month | 0.31 (0.21 to 0.42) | 0.26 (0.16 to 0.37) | █████████████ | █████████████ |
18 month | 0.21 (0.10 to 0.34) | 0.17 (0.07 to 0.32) | █████████████ | █████████████ |
21 month | 0.21 (0.10 to 0.34) | 0.17 (0.07 to 0.32) | █████████████ | █████████████ |
24 month | 0.21 (0.10 to 0.34) | 0 (NE to NE) | █████████████ | ████████ |
27 month | 0 (NE to NE) | — | ████████ | █ |
BICR = blinded independent central review; CI = confidence interval; NE = not estimable; PFS = progression-free survival.
Source: CHRYSALIS CSR interim analysis (March 30, 2021).17
Results for treatments received postprogression are summarized in Table 37, Appendix 3.
Overall Survival
Results for OS as of the March 30, 2021, DCO are summarized in Table 16 and displayed graphically in Figure 6, Appendix 3. With a median follow-up of 14.5 months for the primary efficacy population (N = 81), and 11.9 months for the additional efficacy population (N = 124), the median OS was 22.77 months (95% CI, 17.48 to not estimable) in both the primary and additional efficacy populations. The estimated 6-, 12-, and 24-month survival rates for the primary efficacy population were 90% (95% CI, 81 to 95), 74% (95% CI, 63 to 83), and 41% (95% CI, 21 to 59), respectively. The estimated 6-, 12-, and 24-month survival rates for the additional efficacy population were ████████████████████████████████████████████████████████, respectively.17
Results for the October 8, 2020, DCO for the primary efficacy population (N = 81; median follow-up: 9.7 months) and the additional efficacy population (N = 114; median follow-up: 8.3 months) are available in Table 35, Appendix 3. Results at the October 8, 2020, DCO were overall consistent with the results at the March 30, 2021, DCO.38
Results at the June 8, 2020, DCO date are not presented in this report but were overall consistent with the results at the March 30, 2021, DCO date.
Table 16: Summary of OS (March 30, 2021, DCO)
OS | Cohort D primary efficacy population (N = 81) | Cohort D additional efficacy population (N = 124) |
|---|---|---|
Event | 31 (38.3) | ████████ |
Censored | 50 (61.7) | ████████ |
Time to event (months) | ||
Median (95% CI) | 22.77 (17.48 to NE) | ███████████████ |
OS rate, % (95% CI) | ||
3 month | 0.94 (0.86 to 0.97) | ███████████████ |
6 month | 0.90 (0.81 to 0.95) | ███████████████ |
9 month | 0.80 (0.69 to 0.87) | ███████████████ |
12 month | 0.74 (0.63 to 0.83) | ███████████████ |
15 month | 0.67 (0.55 to 0.76) | ███████████████ |
18 month | 0.62 (0.49 to 0.72) | ███████████████ |
21 month | 0.54 (0.39 to 0.67) | ███████████████ |
24 month | 0.41 (0.21 to 0.59) | ███████████████ |
27 month | 0.41 (0.21 to 0.59) | ███████████████ |
30 month | 0.41 (0.21 to 0.59) | ███████████████ |
CI = confidence interval; NE = not estimable; OS = overall survival.
Chinese patients enrolled beyond the initial global cohort enrolment are excluded.
Source: CHRYSALIS CSR interim analysis (March 30, 2021).17
Duration of Response
Results for DOR based on investigator and BICR assessment for both the primary (N = 81) and additional efficacy populations (N = 124) are summarized in Table 17. Among the 31 responders as assessed by the investigator in the primary efficacy population, the median duration of treatment was 14.03 months and the median DOR was 12.45 months (95% CI, 6.54 to 16.13), and 21 (67.7%) patients had a DOR greater than or equal to 6 months. Per BICR, the median duration of treatment was 13.90 months, the median DOR assessed by BICR was 11.04 months (95% CI, 6.90 to not estimable), and 60% of patients had a DOR greater than or equal to 6 months.17
Results for DOR in the additional efficacy population were generally consistent with the primary analysis; however, the BICR assessment was associated with a ███████████████████████████████████████████████████████████████████████████████████████████████████████████████. The median duration of study treatment was also ████████████████████████████████████████████████████████████ per the investigator and BICR assessment.17
Results for the October 8, 2020, DCO for the primary efficacy population (N = 81; median follow-up: 9.7 months) and the additional efficacy population (N = 114; median follow-up: 8.3 months) are available in Table 35, Appendix 3. Results at the October 8, 2020, DCO were overall consistent with the results at the March 30, 2021 DCO.38
Table 17: Summary of DOR in Responders per Investigator and BICR Assessment (March 30, 2021, DCO)
DOR | Cohort D primary efficacy population (N = 81) | Cohort D additional efficacy population (N = 124) | ||
|---|---|---|---|---|
Investigator assessed | BICR | Investigator assessed | BICR | |
Responders, n (%) | 31 (38.3) | 35 (43.2) | ████████ | ████████ |
Event, n (%) | 18 (58.1) | 19 (54.3) | ████████ | ████████ |
Censored, n (%) | 13 (41.9) | 16 (45.7) | ████████ | ████████ |
Time to event (months) | ||||
Median DOR (95% CI) | 12.45 (6.54 to 16.13) | 11.04 (6.90 to NE) | ████████████ | ████████████ |
Duration of response ≥ 6 months, n (%) | 21 (67.7) | 21 (60.0) | ████████ | ████████ |
Duration of study treatment (months)a | ||||
Mean (SD) | 13.71 (5.235) | 13.14 (6.115) | ██████████ | ██████████ |
Median (range) | 14.03 (3.3 to 23.9) | 13.90 (2.8 to 23.9) | ████████████ | ████████████ |
BICR = blinded independent central review; CI = confidence interval; DCO = data cut-off; DOR = duration of response; NE = not estimable; SD = standard deviation.
aTreatment duration is defined as the duration from the date of the first dose of study drug to the date of last dose of study drug plus 1 divided by 30.4375.
Source: CHRYSALIS CSR interim analysis (March 30, 2021).17
Health-Related Quality of Life
Four patient-reported HRQoL assessments were used in the CHRYSALIS trial, including the NSCLC-SAQ, PGIS, PGIC, and EQ-5D-5L. The Clinical Study Report for the CHRYSALIS trial did not include results of HRQoL due to limited sample size and short duration of follow-up. Data on HRQoL and PROs from the CHRYSALIS trial were included as additional information within the sponsor’s submission. As of the March 30, 2021, DCO, 36 patients had available HRQoL data at baseline. Results were summarized descriptively.
For the NSCLC-SAQ, the mean total score at baseline was 7.4. Change from baseline at each cycle is shown in Figure 7, Appendix 3, with initial decreases in total score observed until cycle 4, followed by changes from baseline upwards of 1.5 points until cycle 13.
Results for the EQ-5D-5L visual analogue scale for the PRO population are summarized in Figure 8, Appendix 3. No observable trend was demonstrated, with score improving (increasing) until cycle 5, followed by a decrease (worsening) until cycle 10 before increasing again.
Symptom Severity
Symptom severity was measured via the PGIS and PGIC. Results were summarized descriptively. Results of the 36 patients who had evaluable PRO data from the PGIS and PGIC are summarized in Figure 9 and Figure 10, Appendix 3, respectively. Minimal changes were observed across measures in terms of symptom severity and change in symptoms.
Time to Treatment Failure
TTF, defined as the time interval from the first dosing date to study drug discontinuation for any reason, was a secondary outcome of the CHRYSALIS trial. Results for TTF in both the primary and additional efficacy populations are summarized in Table 18. As of the March 30, 2021, DCO, the median TTF was 7.7 months (95% CI, 5.62 to 10.61) months, and the 6-, 12-, and 24-month TTF rates were 60% (95% CI, 49 to 70), 37% (95% CI, 27 to 47), and 7% (95% CI, 1 to 22), respectively.17
Results for the additional efficacy population (N = 124) were consistent with the primary efficacy population.
Table 18: Summary of TTF per Investigator Assessment (March 30, 2021, DCO)
TTF | Cohort D primary efficacy population (N = 81) | Cohort D additional efficacy population (N = 124) |
|---|---|---|
Event, n (%) | 62 (76.5) | ████████ |
Censored, n (%) | 19 (23.5) | ████████ |
Time to event (months) | ||
Median (95% CI) | 7.72 (5.62 to 10.61) | ███████████████ |
TTF rate, % (95% CI) | ||
3 month | 0.78 (0.67 to 0.85) | ███████████████ |
6 month | 0.60 (0.49 to 0.70) | ███████████████ |
9 month | 0.44 (0.33 to 0.55) | ███████████████ |
12 month | 0.37 (0.27 to 0.47) | ███████████████ |
15 month | 0.33 (0.22 to 0.43) | ███████████████ |
18 month | 0.30 (0.19 to 0.41) | ███████████████ |
21 month | 0.13 (0.05 to 0.26) | ███████████████ |
24 month | 0.07 (0.01 to 0.22) | ███████████████ |
27 month | 0 (NE to NE) | ████████ |
CI = confidence interval; DCO = data cut-off; NE = not estimable; TTF = time to treatment failure.
Chinese patients enrolled beyond the initial global cohort enrolment are excluded.
Source: CHRYSALIS CSR interim analysis (March 30, 2021).17
Harms
Only those harms identified in the review protocol are reported below. Refer to Table 19 for detailed harms data.
Adverse Events
The incidence of TEAEs in the CHRYSALIS trial is summarized in Table 19. Overall, 100% of patients experienced at least 1 TEAE in the CHRYSALIS trial as of the March 30, 2021, DCO. The most frequently reported TEAEs with amivantamab included IRRs (97; 63.4%), paronychia (81; 52.9%), rash (66; 43.1%), dermatitis acneiform and hypoalbuminemia (60 each; 39.2%), and stomatitis (34; 22.2%).17
A total of 64 (41.8%) patients experienced 1 or more TEAE that was greater than or equal to grade 3. The most frequently reported greater than or equal to grade 3 TEAEs included pulmonary embolism (7; 4.6%), hypokalemia (6; 3.9%), diarrhea, dyspnea, hypoalbuminemia, paronychia, and pneumonia (5 each; 3.3%), and IRRs and neutropenia (4 each; 2.6%).17
Serious Adverse Events
The incidence of SAEs is summarized in Table 19. A total of 44 (28.8%) patients experienced SAEs. The most common SAEs with amivantamab were pneumonia (5; 3.3%), dyspnea and pulmonary embolism (4 each; 2.6%), and back pain, muscular weakness, and pneumonitis (3 each; 2.0%).17
Grade 4 SAEs were reported for 3 (2.0%) patients, and grade 5 (fatal) SAEs were reported in 11 (7.2%) patients.17
Withdrawals Due to Adverse Events
As of the March 30, 2021, DCO, a total of 18 (11.8%) patients withdrew from treatment with amivantamab due to TEAEs. The most common reason for withdrawals due to AEs was pneumonia (4; 2.6%), and IRRs, pleural effusion, and pneumonitis (2 each; 1.3%).17
Mortality
Deaths occurring within the treatment period (up to 30 days after last dose of amivantamab) were reported as grade 5 TEAEs regardless of whether death resulted from PD, TEAEs, or any other cause. TEAEs resulting in death during the study are summarized in Table 19. Overall, 11 (7.2%) patients treated with amivantamab experienced a TEAE leading to death, with respiratory, thoracic, and mediastinal disorders the most common TEAE leading to death. As of the March 30, 2021, DCO, 45 (29.4%) patients died. The most common cause of death on study was PD (31; 20.3%).17
Notable Harms
Notable harms of IRRs, ILD, skin disorders, paronychia, and ophthalmologic disorders are summarized in Table 19.
Infusion-Related Reactions
IRRs occurred for 97 (63.4%) patients treated with amivantamab. The majority of IRRs were grade 1 (16; 10.5%) or 2 (77; 50.3%) severity, and grade 3 IRRs were reported for only 4 (2.6%) patients, 2 of which were considered serious.17
The median time to onset of greater than or equal to grade 1 TEAEs was 47.0 minutes. The grade 3 IRRs had a median time to onset of 327.5 minutes after the first dose of amivantamab. IRRs occurred almost exclusively on cycle 1, day 1 (66.32%), decreasing for cycle 1, day 2 (3.51%).17
IRRs were managed prophylactically as described previously. IRRs leading to infusion modification of an ongoing infusion were reported for 90 (59.5%) patients; 11 (7.2%) patients had the cycle 1, day 1 infusion aborted due to an AE; and IRRs leading to discontinuation of amivantamab were reported for 2 (1.3%) patients. Postinfusion medications to manage IRRs and symptoms were uncommon, with postinfusion administration of systemic antihistamines (9; 5.9%), systemic corticosteroids (4; 2.6%), and paracetamol (3; 2.0%).17
Interstitial Lung Disease
AEs of ILD were reported in 6 (3.9%) patients: 5 (3.3%) with pneumonitis and 1 (0.7%) with ILD. Of these, 2 were grade 1, 3 were grade 2, and 1 was grade 3 (pneumonitis). Of the ILD events, 4 were considered serious. Amivantamab was discontinued due to TEAEs of pneumonitis in 2 patients.17
The median time to first onset of ILD from the first dose of amivantamab was 50.5 days.17
Skin Reactions
Skin and subcutaneous tissue disorders were the most frequently reported class of TEAEs in the CHRYSALIS trial. Rash events (grouped term) occurred in 130 (85.0%) patients. Rash and dermatitis acneiform were the most frequently reported skin reactions, occurring in 66 (43.1%) and 60 (39.2%) patients, respectively.17
In general, rash events were of grade 1 (96; 73.8%) or 2 (34; 26.2%) severity and nonserious. Grade 3 TEAEs included acne and rash (2 each; 1.3%), and dermatitis acneiform, erythema, papular rash, and toxic epidermal necrolysis (1 each; 0.7%). Of the grade 3 TEAEs, 2 (1.3%) patients had rash that was considered serious, and 1 (0.7%) patient had toxic epidermal necrolysis that was considered serious. Rash events resulted in dose interruption and in dose reduction for 14 (10.8%) patients each.17
Most of the rash events occurred within cycle 1, with the median time to first onset of 11.5 minutes, and a median time of onset for greater than or equal to grade 3 rash events of 28.5 minutes.17
Of the 130 patients with rash events, the most frequent treatments included systemic antibacterials (83; 63.8%), most frequently tetracyclines (71; 54.6%), dermatological corticosteroid preparations (59; 45.4%), and systemic corticosteroids (55; 42.3%).17
Paronychia
Paronychia was an AE of clinical importance in the CHRYSALIS trial. It was reported for 81 (52.9%) patients. Most events were grade 1 or 2. Grade 3 events were reported for 5 (3.3%) patients, none of which were serious. One patient discontinued treatment with amivantamab due to grade 2 paronychia.17
Ophthalmologic Disorders
Ophthalmologic disorders were considered an EGFR-mediated event in the CHRYSALIS trial. Overall, eye disorders were reported for 19 (12.4%) patients. The most frequently reported ophthalmologic TEAEs were dry eye (4; 2.6%) and eyelid edema (3; 2.0%). All ophthalmologic TEAEs were considered grade 1 or 2, and none were serious.
Table 19: Summary of Harms (Safety Analysis Set; March 30, 2021, DCO)
Harms | Cohort D safety analysis set (N = 153) |
|---|---|
TEAEs, n (%) | |
Patients with ≥ 1 TEAEs | 153 (100.0) |
Skin and subcutaneous tissue disorders | 136 (88.9) |
Rash | 66 (43.1) |
Dermatitis acneiform | 60 (39.2) |
Pruritus | 24 (15.7) |
Dry skin | 21 (13.7) |
Injury, poisoning, and procedural complications | 102 (66.7) |
IRR | 97 (63.4) |
Gastrointestinal disorders | 114 (74.5) |
Nausea | 38 (24.8) |
Constipation | 36 (23.5) |
Stomatitis | 34 (22.2) |
Vomiting | 21 (13.7) |
Diarrhea | 21 (13.7) |
Infections and infestations | 107 (69.9) |
Paronychia | 81 (52.9) |
Metabolism and nutrition disorders | 92 (60.1) |
Hypoalbuminemia | 60 (39.2) |
Decreased appetite | 27 (17.6) |
Hypocalcemia | 16 (10.5) |
Respiratory, thoracic, and mediastinal disorders | 88 (57.5) |
Dyspnea | 30 (19.6) |
Cough | 26 (17.0) |
General disorders and administration-site conditions | 96 (62.7) |
Peripheral edema | 35 (22.9) |
Fatigue | 30 (19.6) |
Pyrexia | 26 (17.0) |
Musculoskeletal and connective tissue disorders | 73 (47.7) |
Back pain | 25 (16.3) |
Myalgia | 18 (11.8) |
Investigations | 63 (41.2) |
Increased ALT | 34 (22.2) |
Increased AST | 25 (16.3) |
Increased blood alkaline phosphatase | 16 (10.5) |
Nervous system disorders | 50 (32.7) |
Dizziness | 18 (11.8) |
Headache | 11 (7.2) |
Blood and lymphatic system disorders | 36 (23.5) |
Anemia | 20 (13.1) |
Psychiatric disorders | 29 (19.0) |
Insomnia | 16 (10.5) |
SAEs, n (%) | |
Patients with ≥ 1 SAEs | 44 (28.8) |
Respiratory, thoracic, and mediastinal disorders | 17 (11.1) |
Dyspnea | 4 (2.6) |
Pulmonary embolism | 4 (2.6) |
Pleural effusion | 2 (1.3) |
Pneumonitis | 3 (2.0) |
Respiratory failure | 2 (1.3) |
Infections and infestations | 13 (8.5) |
Pneumonia | 5 (3.3) |
Respiratory tract infection | 2 (1.3) |
Sepsis | 2 (1.3) |
Cellulitis | 2 (1.3) |
Musculoskeletal and connective tissue disorders | 7 (4.6) |
Back pain | 3 (2.0) |
Muscular weakness | 3 (2.0) |
Injury, poisoning, and procedural complications | 5 (3.3) |
IRR | 2 (1.3) |
Thoracic vertebral fracture | 2 (1.3) |
Gastrointestinal disorders | 4 (2.6) |
Diarrhea | 2 (1.3) |
Skin and subcutaneous tissue disorders | 2 (1.3) |
Rash | 2 (1.3) |
Cardiac disorders | 5 (3.3) |
General disorders and administration-site conditions | 2 (1.3) |
Nervous system disorders | 2 (1.3) |
WDAEs, n (%) | |
Patients with ≥ 1 AEs leading to discontinuation | 18 (11.8) |
Infections and infestations | 7 (4.6) |
Pneumonia | 4 (2.6) |
Respiratory, thoracic, and mediastinal disorders | 6 (3.9) |
Pleural effusion | 2 (1.3) |
Pneumonitis | 2 (1.3) |
Injury, poisoning, and procedural complications | 3 (2.0) |
IRR | 2 (1.3) |
Skin and subcutaneous tissue disorders | 2 (1.3) |
Deaths, n (%) | |
Deaths during study | 45 (29.4) |
Progressive disease | 31 (20.3) |
AE | 9 (5.9) |
Other | 5 (3.3) |
Deaths during treatment | 12 (7.8) |
AE | 8 (5.2) |
Progressive disease | 4 (2.6) |
Patients with 1 or more AEs leading to deatha | 11 (7.2) |
Infections and infestations | 3 (2.0) |
Respiratory, thoracic, and mediastinal disorders | 6 (3.9) |
Respiratory failure | 2 (1.3) |
Dyspnea | 2 (1.3) |
Cardiac disorders | 1 (0.7) |
General disorders and administration-site conditions | 1 (0.7) |
Notable harms, n (%) | |
IRRs | 97 (63.4) |
Rash | 130 (85.0) |
Interstitial lung disease | 6 (3.9) |
Paronychia | 81 (52.9) |
Ophthalmologic disorders | 19 (12.4) |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; DCO = data cut-off; IRR = infusion-related reaction; SAE = serious adverse event; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
Note: Deaths during treatment are presented for patients who died within 30 days of last study drug dose.
aAEs leading to death are based on AE outcome of fatal. Patients are counted only once for any given event, regardless of the number of times they experienced the event. AEs are coded using MedDRA (Medical Dictionary for Regulatory Activities) Version 23.0.
Source: CHRYSALIS CSR interim analysis (March 30, 2021).17
Critical Appraisal
Internal Validity
Primary purpose of phase I/Ib design: CHRYSALIS was a first-in-human phase I/Ib clinical study with the primary purposes of determining the RP2D (part 1) and subsequently assessing the safety of the selected dose in part 1 and estimating the clinical activity of amivantamab (part 2). A phase I/Ib or phase II trial may not accurately predict harm and/or effectiveness of treatments. There are numerous examples of phase III trials in which the results did not support the phase II trial results.39 There are currently no randomized phase III trials under way for this review’s target population. The clinical experts consulted by CADTH noted that, despite the high unmet need, conducting an RCT in this setting with a targeted therapy, such as amivantamab, compared with the available therapies currently used in Canadian clinical practice would likely not be feasible. According to the clinical experts, developing phase III RCTs is hindered by the overall low number of patients who meet the current indication and because equipoise between amivantamab and other chemotherapy drugs does not exist. A phase III RCT, PAPILLON, is currently under way; it is investigating the efficacy and safety of amivantamab in combination with carboplatin-pemetrexed compared to carboplatin-pemetrexed alone for the first-line treatment of patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. After first-line therapy for locally advanced or metastatic NSCLC, it is estimated that only about 50% of patients go on to subsequent lines of therapy.33
Limited interpretation of time-to-event end points: The single-arm, nonrandomized design of the CHRYSALIS trial makes interpreting the efficacy and safety events attributable to amivantamab challenging, because all patients received the same treatment. While ORR may be directly attributable to the drug’s antitumour activity, interpreting PFS and OS events is significantly limited. The extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear.40 The FDA’s multidisciplinary review for the assessment of amivantamab in the present indication included the following reviewer comment: “FDA considers time-to-event end points to be uninterpretable in a single-arm study.”41 Consequently, the efficacy outcomes contributing to the FDA’s accelerated approval of amivantamab in the current setting included response outcomes and no survival end points.41
Open-label design: The CHRYSALIS trial had an open-label design whereby the investigator and the study participants are aware of their treatment status, which increases the risk of detection bias and performance bias. Overall, the magnitude and direction of this bias remains unclear. However, to mitigate the impact of this bias, the response outcomes were also assessed by BICR using standardized criteria (i.e., based on RECIST 1.1 with confirmation of a CR and PR at least 4 weeks after the initial assessment). Discordance between investigator assessment and BICR for responders and nonresponders was 22.22%, suggesting some disagreement between investigator and central review, with sites providing slightly more conservative interpretations of the scans. Given the open-label design of the trial, it is likely that the BICR assessment of response provided more unbiased response assessments compared with the investigator’s assessments. FDA and Health Canada assessments of response focused on the BICR assessments of response.41,42 Furthermore, subjective outcomes (i.e., adverse outcomes and PROs) may be biased due to the open-label design. For example, if study personnel and patients knew that the treatment was amivantamab, this could have influenced the reporting of harms. In addition, the nonrandomized design of the CHRYSALIS trial makes interpreting the safety events attributable to amivantamab challenging, because all patients received the same treatment. Overall, the magnitude and direction of this bias remain unclear.
Statistical analyses: No inferential statistical testing was performed for the efficacy outcomes in cohort D; thus, no P values were reported. Point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. The threshold for a positive study outcome for cohort D was observing a 95% CI for ORR with a lower limited larger than 12%. This threshold for tumour activity was based on historical response rates observed in phase III trials in patients who were treated with single-drug chemotherapy upon failure of platinum-based chemotherapy and was overall considered acceptable in signalling promising treatment effect.42 Results for ORR appeared consistent with the sample size assumptions, and the study recruited the intended number of patients.
Small sample size: A limited number of patients were included in the primary (N = 81) and additional (N = 124) efficacy sets of cohort D. The magnitude of the treatment-effect estimates observed in a small study sample may not be replicable in a larger study sample.
Subgroup analysis: Methodological issues limited the ability to interpret the results from subgroup analyses. Wide CIs reflected uncertainty in the effect estimates, and small sample sizes limited the generalizability to a broader population.
HRQoL and symptom severity assessments: PROs were a late inclusion in the trial; they were included as an amendment and were not part of the regulatory submission package. Due to the small sample size (N = 36), a substantial decline in patients available to provide assessments over time, and the descriptive nature of the analysis, the potential effect of amivantamab on HRQoL and symptom severity remains inconclusive.
Multiple protocol amendments: There were multiple amendments to the protocol of the CHRYSALIS study, including changes to the primary and secondary end points and that the overall population cohorts enrolled were discontinued or altered (Amendments 3 and 4), though no patients were believed to have been enrolled into cohort D before the change in end points, with the exception of HRQoL outcomes. Because multiple revisions to the protocol are commonly seen in exploratory phase I studies, this does not necessarily raise concerns with regards to the conduct of the study. It was noted in the Health Canada reviewer report that the assessment of protocol amendments did not raise major concerns in regards to interpreting the results of cohort D.42
External Validity
The CHRYSALIS trial was an international, multicentre study that included sites in Australia, Canada, China, France, Japan, Korea, Spain, Taiwan, the US, and the UK. One Canadian treatment centre enrolled 6 patients as of the June 2020 interim analysis, and 21 at the March 30, 2021, DCO. The treatment regimen used in part 2 of the CHRYSALIS trial aligns with the Health Canada recommended dose of 1,050 mg for patients weighing less than or equal to 80 kg, and 1,400 mg for patients weighing greater than or equal to 80 kg. The clinical experts conducted by CADTH indicated that although the inclusion and exclusion criteria were appropriate, they hypothesized that the exclusion criteria may have been restrictive, selecting for ideal, less severely ill patients. In line with the exclusion criteria, patients with untreated central nervous system metastases were not enrolled. Overall, 23.5% of patients enrolled in cohort D had previously treated brain metastases at baseline. The clinical experts consulted by CADTH agreed that results for amivantamab are likely generalizable to patients with treated brain metastases.
The clinical experts also noted that the baseline characteristics of the included population was generally reflective of Canadian clinical practice; however, there was a high proportion of patients who were Asian (62.1%) enrolled. The clinical experts noted that the higher proportion of patients who were Asian in the trial may be due to the fact that people of Asian descent are genetically more susceptible to mutations involving EGFR. Mutations of EGFR in NSCLC adenocarcinoma occur in 40% to 50% of patients of Asian descent compared to 15% of patients from Western countries.43,44 Additionally, in part 2, cohort D was closed globally to further enrolment on May 29, 2020, except to complete country-specific enrolment requirements in Japan and China, likely resulting in a greater proportion of patients who were Asian enrolled in the trial. Furthermore, the clinical experts did not anticipate seeing differential treatment effects between patients who were Asian and those who were not. Therefore, the high proportion of patients of Asian descent enrolled in cohort D is unlikely to affect the generalizability of the results.
Noncomparative design: The noncomparative design of the CHRYSALIS trial precludes the ability to assess the relative therapeutic benefit or safety of amivantamab against currently available therapies in Canadian clinical practice. As noted previously, the clinical experts consulted by CADTH agreed that direct randomized comparisons between amivantamab and currently used therapies are unlikely to take place for advanced or metastatic NSCLC with exon 20 insertion that has progressed after platinum-based chemotherapy. In the absence of a direct comparison of amivantamab with relevant treatment options, the sponsor submitted an adjusted treatment comparison using external control arms derived from real-world data sources. The results of the adjusted treatment comparison favoured amivantamab for ORR, PFS, and OS in comparison with treatment of physician’s choice. The CADTH review team identified several limitations (e.g., concerns regarding heterogeneity across the study designs and populations, and the inability to adjust for important potential confounders and prognostic variables across each cohort) and concluded that no firm conclusions could be drawn about how amivantamab compared to other relevant treatment options. However, the clinical experts consulted by CADTH anticipated that, based on the CHRYSALIS results and on poor results with existing treatment options in clinical practice, amivantamab would offer improved and clinically meaningful clinical benefits compared to currently available therapies.
Relevance of trial efficacy outcomes: The primary outcome in the CHRYSALIS trial was ORR and 1 of the secondary outcomes was DOR. According to the clinical experts consulted by CADTH, ORR and durability of response are clinically meaningful end points for patients with advanced or metastatic NSCLC who have progressed on prior therapy. Responses in this patient population are important because of accompanying delay in the worsening of symptoms and a slower decline in ECOG performance status. According to the clinical experts, it is rare to see CRs to treatment in this setting. While the clinical experts agreed that, based on the available evidence, it was not possible to firmly conclude whether the antitumour activity expressed as responses would translate into clinical benefits in terms of PFS and OS, they felt that durable responses could potentially delay tumour progression and result in prolonged survival benefit in this patient population.
Indirect Evidence
A focused literature search for ITCs dealing with EGFR-mutated NSCLC was run in MEDLINE All (1946–) on May 18, 2022. No limits were applied. The literature search for ITCs identified 93 articles; however, no articles evaluated the efficacy and safety of amivantamab in patients with NSCLC with EGFR exon 20 insertion.
Methods of the Adjusted Treatment Comparison
Objectives and Methods for the Summary of the Sponsor-Submitted Adjusted Treatment Comparison
Due to the lack of direct evidence comparing amivantamab to relevant treatment options for the management of advanced EGFR-mutated NSCLC with exon 20 insertion, the sponsor submitted an adjusted treatment comparison of trial data against real-world evidence that was used to inform the pharmacoeconomic analysis, and an additional report by Minchom et al. (2022) comparing amivantamab with real-world therapies in the US.15,18 The objective of this section is to summarize and critically appraise the methods and findings of the sponsor-submitted adjusted treatment comparison of amivantamab and relevant drug comparators for the treatment of adult patients with advanced EGFR-mutated NSCLC with exon 20 insertion following platinum-based chemotherapy.
Description of the Sponsor-Submitted Adjusted Treatment Comparison
The sponsor submitted an adjusted treatment comparison that compared the efficacy of amivantamab from IPD from cohort D of the single-arm CHRYSALIS trial to current treatments using an external control arm derived from real-world data sources from the US, the UK, Germany, and France. Data from the real-world sources were identified by applying inclusion criteria from the CHRYSALIS trial.
An additional report by Minchom et al. (2022) was submitted as part of the sponsors’ clinical data package, which followed a similar methodology as the submitted adjusted treatment comparison. The primary objective of this study was to evaluate the effectiveness of amivantamab versus physicians’ choice in real-world settings among patients with advanced NSCLC harbouring EGFR exon 20 insertion who failed platinum-based chemotherapy. Similar to the adjusted treatment comparison, cohort D from the CHRYSALIS trial was used as the index trial for comparison. The external control cohort comprised patients in 3 real-world data sources from the US (ConcertAI, COTA, and Flatiron Health Inc.) who met clinically relevant eligibility criteria for CHRYSALIS. One of the key differences between the primary report and the report by Minchom et al. (2022) were the dates of data collection for the US cohorts (report by Minchom et al. (2022) versus the primary report for the adjusted treatment comparison: 2011 to 2020 versus 2009 to 2020), which affected the number of patients included from the US data sources (N = 125 versus N = 206). As well, the primary report used a DCO for CHRYSALIS of March 30, 2021, that represented the most mature data available, while the report by Minchom et al. (2022) used data from the October 8, 2020, DCO, with a refresh of the OS data from April 19, 2021. Results comparing amivantamab to the external cohorts for the report by Minchom et al. were overall consistent with those from the submitted ITC for all outcomes of ORR (amivantamab versus external cohorts: 40% versus 16%), PFS (median = 8.3 months versus 2.9 months; HR = 0.47 [95% CI, 0.34 to 0.65]), and OS (median = 22.8 months versus 12.8 months; HR = 0.49 [95% CI, 0.31 to 0.77]).15 Given the similarities in design and methodology, as well as the overlap with the US cohort from the submitted adjusted treatment comparison and results trending in the same direction, this additional report will not be further summarized.
Methods of the Sponsor-Submitted Adjusted Treatment Comparison
Objectives
The primary objective of the sponsor-submitted adjusted treatment comparison was to conduct adjusted treatment comparisons on the efficacy (ORR, OS, PFS, TTNT) of amivantamab in the CHRYSALIS trial (cohort D) to current treatments from real-world settings in patients with advanced EGFR-mutated NSCLC with exon 20 insertion following platinum-based chemotherapy.
Study Selection Methods
The index trial in all analyses was based off IPD from cohort D of the CHRYSALIS trial (n = 81). This cohort included adult patients with EGFR-mutated NSCLC with exon 20 insertion who had progressed on, or after, receiving platinum-based therapy. All efficacy analyses included patients who received the RP2D dose of amivantamab as monotherapy and had undergone at least 3 scheduled postbaseline disease assessments or discontinued treatment for any reason, including disease progression/death, before the clinical cut-off.
An external control arm for cohort D of the CHRYSALIS trial was generated via real-world data sources by applying the inclusion and exclusion criteria from CHRYSALIS to patients from the following real-world data sources:
Flatiron Health Spotlight: US
ConcertAI: US
COTA: US
Public Health England (PHE): UK
The national Network Genomic Medicine (nNGM): Germany
The Clinical Research Platform Into Molecular Testing, Treatment and Outcome Registry of (Non-)Small Cell Lung Carcinoma Patients (CRISP): Germany
Epidemiological Strategy and Medical Economics (ESME): France
Any criteria that could not be applied to patients due to missing data were omitted from the list of inclusion and exclusion criteria applied to that data source.
The index date for patients in cohort D of the CHRYSALIS trial was the date on which the first dose of amivantamab was received. For patients from the real-world data sources, the index date was the start of any line of therapy at the start of which inclusion and exclusion criteria were met. Only treatment lines in which patients received EGFR exon 20 insertion testing before initial treatment were included. Some real-world patients satisfied inclusion criteria at multiple times during their follow-up. Therefore, to achieve an unbiased comparison in this situation, patients receiving qualifying treatment in more than 1 line setting were included multiple times in an analysis, once for each qualifying line setting. Correlation of outcomes across treatments within each patient was accounted for using the robust sandwich estimator.
All data sources were pooled to create an EU plus US cohort, representing the base-case analysis due to the increased sample size. The Flatiron, ConcertAI, and COTA databases were combined and presented for a pooled US population analysis, and the PHE, nNGM, ESME, and CRISP were pooled and are presented as an EU cohort. Data collection occurred at the following time periods for each database: 2009 and 2020 for US databases, 2016 for PHE, 2013 to 2021 for nNGM, and 2015 to 2021 for CRISP and ESME.
The primary patient population consisted of patients enrolled in cohort D of the CHRYSALIS trial (n = 81), with locally advanced or metastatic EGFR-mutated NSCLC with an exon 20 insertion, who had received their first dose of amivantamab monotherapy on or before February 5, 2020, and were treated with the RP2D (i.e., patients that had undergone at least 3 scheduled postbaseline disease assessments or discontinued treatment for any reason, including disease progression/death, before the clinical cut-off). Data from the latest DCO of CHRYSALIS (March 30, 2021) were used for all analysis sets.
Treatments across comparator real-world data sources were variable. Comparative analyses of amivantamab versus the whole population of patients treated with physicians’ choice (i.e., comparison versus a single basket of treatments) were carried out. Additional comparisons versus relevant treatments classes were also conducted: TKI-based regimens, IO-based regimens, non-platinum-based chemotherapy regimens, VEGFi-based regimens, and other regimens (platinum-based chemotherapy, investigational drugs, drugs not considered to be part of the SOC, and treatments that included a combination of the other treatment classes).
End points of interest for the adjusted treatment comparison were ORR, PFS, OS, and TTNT. For the adjusted treatment comparison, ORR was defined as the proportion of all patients who achieved a best response of partial response or better; OS was defined as the interval between index date and date of death; PFS was defined as the interval between the index date and the date of disease progression or death, whichever occurred first; and TTNT was defined as the interval between the index date and initiation of subsequent systemic anti-cancer therapy or death, whichever occurred first. In CHRYSALIS, ORR and PFS were assessed by both investigator and BICR. Because only investigator results are available in the real-world data sources, this was the key method of assessment for these end points and was used as the base case. For completeness, ORR and PFS end points with CHRYSALIS results based on BICR assessment were presented as sensitivity analyses. No ORR or PFS data were available from the PHE cohort, and no ORR data were available from ESME.
Adjusted Treatment Comparison Analysis Methods
Multiple methodological approaches were implemented to adjust for differences in observed baseline characteristics between the CHRYSALIS cohort and the real-world data sources, which were considered potential confounders. Adjusted comparative analyses were implemented using IPW and covariate adjustment for the comparison of amivantamab versus physicians’ choice. In cases where IPW was considered the primary analysis, covariate adjustment results were used to demonstrate consistency in results. Covariate adjustment was considered when IPW did not achieve a good covariate balance, led to extreme weights which overrepresent a small portion of patients in the treatment group, or when IPW estimates were unstable due to small sample size.
When comparing amivantamab to treatment classes in the pooled EU and pooled US cohorts, the whole real-world data population was compared to CHRYSALIS with covariate adjustment adjusting for treatment class and baseline characteristics (as opposed to comparing the populations of each treatment class from real-world evidence separately to CHRYSALIS via IPW methods). This was due to the number of observations available for the individual treatment classes often being low, meaning that IPW was not feasible or stable. However, due to the larger sample size of the EU plus US cohort, IPW methods were feasible and so are presented in the report alongside covariate adjustment results. Comparisons versus treatment classes are not presented for individual data sources, as the results were underpowered due to the small sample sizes. Analysis results are presented as an effect measure with a 2-sided 95% CI and corresponding P value.
In CHRYSALIS, ORR and PFS were assessed by both investigator and BICR. Because only investigator results are available in the real-world data sources, this was the key method of assessment for ORR and was used as the base case. For completeness, ORR and PFS end points with CHRYSALIS results based on BICR assessment were presented as sensitivity analyses.
IPW: Propensity Score–Based Adjusted Analysis
IPW is a propensity score–based method used to mimic randomization by creating a balance between 2 treatment groups with respect to prognostic baseline covariates. Where IPW was conducted, the following weighting schemes were applied:
The average treatment effect on the treated (ATT) approach generated a comparative arm reflecting the population enrolled in CHRYSALIS by reweighting the real-world cohort to match the amivantamab patients of CHRYSALIS. ATT-based estimates represent the relative treatment effect in the CHRYSALIS population, and for these analyses, a scaled ATT approach was taken. To maintain the original sample size for the weighted populations and to properly reflect the associated uncertainty, the ATT weights were multiplied by the ratio of the original sample size versus the sum of the ATT weights, making the sum of these recalculated weights equal to the original sample size. This approach is referred to as the “ATT approach” throughout the report.
The average treatment effect approach estimated the ATE across both cohorts, because it weights up both propensity score distributions toward the middle. Weights are assigned to patients in the amivantamab cohort and the real-world cohort, creating a more similar distribution of the covariates between the 2 cohorts.
The average treatment effect for the overlap population (ATO) approach was more weighted toward patients whose characteristics could appear with substantial probability in either population. This approach downweighs patients at both extremes of the distributions.
The ATT approach was the primary analysis and was preferred because it preserved the sample size of the amivantamab population. This also matched the outcome of interest being the real-world data treatment’s adjusted effect on amivantamab’s treatment effect rather than treatment effect on the entire population. Furthermore, the sponsor considered the ATT approach to be preferred over the ATE approach due to the higher degree of heterogeneity in the real-world population, as that data came from multiple databases.
Multivariable Regression Approach with Direct Adjustment for Covariates
Covariate adjustment based on a multivariable regression (Cox regression for time-to-event end points and logistic regression for binary end points) was considered as an alternative to propensity score–based adjustment in adjusting for covariate imbalance and potential confounding. This was of particular use when comparing CHRYSALIS to real-world data sources with small sample sizes (physicians’ choice or individual treatment classes). Small sample sizes often led to sudden drops in the Kaplan-Meier estimates after IPW adjustment, which lacked clinical validity and indicated the requirement for an alternative approach. With the multivariable regression approach, the treatment effects were estimated using a multivariable model that included all relevant prognostic variables as covariates together with the treatment group indicator. An advantage of covariate adjustment over the propensity score approach described in the previous section is that it provides a predictive model (including treatment) for the risk (hazard) of the outcome, which gives insight as to which covariates have the strongest influence on risk.
Statistical Analysis
For the binary end point (ORR) adjusted treatment effects, in terms of OR and the corresponding 95% CIs, were generated using logistic regression models. For the IPW approach, a weighted logistic regression model including treatment only was used. For covariate adjustment, an unweighted logistic regression model that includes treatments and relevant covariates was used. To estimate treatment effects in terms of response rate (RR) ratio, the same framework was implemented using a generalized linear model with the appropriate link (instead of logistic regression).
For each time-to-event end point (PFS, OS, TTNT), the following approaches were considered:
Unadjusted comparison without inclusion of potential confounders was used.
The IPW approach provided weights for estimating the treatment effect of amivantamab versus comparators in a weighted Cox proportional hazards model, to estimate the treatment effect in terms of the HR with 95% Wald-type CI and corresponding P values. A robust sandwich variance estimator was also used. Kaplan-Meier curves were generated, based on which median survival with 95% CI was reported for each treatment group.
The covariate adjustment approach used a multivariable Cox proportional hazards model, including treatment and prognostic variables as covariates, with a robust sandwich variance estimator.
Handling of Missing Data and Data Pooling
No imputation method was applied to account for missing data, except for partial dates. If a substantial amount of data were missing for a specific covariate, that covariate was not included in the logistic regression model for propensity score weight generation. For both the EU and EU plus US cohorts, individual data sources were excluded from end point comparisons if no data were available. As previously mentioned, for both cohorts, no ORR data were included from PHE and ESME and no PFS data were included for PHE.
Data from the 4 European data sources (CRISP, nNGM, ESME, and PHE) were pooled to create an EU cohort and collectively compared against amivantamab using the same methods (IPW and covariate adjustment) as for the individual data sources’ analyses. Data from the 3 US data sources (Flatiron, ConcertAI, and COTA) were pooled to create the US cohort in the same manner as for the EU cohort. Direct access to IPD allowed the pooling of data from CRISP, nNGM, and PHE; however, this was not possible for ESME data, which were only remotely available on the servers of the data owners. Only aggregated outcomes data were made available by ESME. For the comparison versus physician’s choice, aggregated outcomes data from ESME were used to reconstruct the unadjusted and ATT-weighted IPD outcome data, which were then combined with the unadjusted and ATT-weighted IPD, respectively, from the other data sources. This is only feasible for the comparison versus physicians’ choice, and not versus treatment classes, for which, in ESME, no IPW-based analyses were performed. Furthermore, adjusted comparisons versus treatment classes that required access to pooled IPD with baseline characteristics (i.e., covariate adjustment and pairwise IPW adjustment per treatment class) were not possible when including ESME. Therefore, for the pooled EU and pooled EU plus US cohorts, comparisons versus treatment classes always excluded ESME.
Due to the high consistency between the results and a comparable treatment distribution of the EU and US cohorts, data from all available data sources were pooled to create an EU plus US cohort. The large sample size of the EU plus US cohort enabled ATT weighting adjustment to be applied for comparisons with individual treatment classes, which is consistent with the preferred approach taken for the comparison between amivantamab and physicians’ choice.
For the US cohort, because multiple real-world data sources were used, some patients were captured multiple times due to overlap of the data sources. Deduplication was used in these instances. For the US cohort, patients in Flatiron were removed from ConcertAI and COTA, and patients in ConcertAI were removed from COTA.
Confounding Factors
A systematic literature review (SLR) was conducted to identify potential prognostic patient and disease characteristic confounders in NSCLC. A comprehensive list of potential confounders was validated and narrowed down by clinical experts. The following confounders were considered relevant to each end point: ECOG performance status, number of prior lines of treatment, overall number of metastatic locations, localization of metastases, age, Asian ethnicity, body mass index, EGFR co-mutation TP53, baseline anemia, smoking history, cancer stage at initial diagnosis, surgery, gender, rebiopsy, programmed cell death 1 ligand 1 (PD-L1) status, liver insufficiency, and renal insufficiency. Some potential confounders were identified by the SLR but could not be considered in confounder adjustment for the present analyses because they were not available at baseline from the relevant data sources, though these confounders were not reported. Other potential confounders identified by the SLR were considered irrelevant by the clinical experts so were not included in confounder adjustment. These included neutrophil-lymphocyte ratio, platelet-lymphocyte ratio, and leukocyte-relevant telomere length.
Because patients in all cohorts may not be comparable and/or exchangeable due to lack of randomization, all comparative analyses were adjusted for imbalances in prognostic baseline characteristics between both treatment cohorts. The characteristics adjusted for in each real-world data source were based on the confounders identified by the SLR, clinical expert opinion, and data availability. The resulting characteristics adjusted for in each real-world data source analysis are presented in Table 20, while baseline characteristics excluded from comparative analyses are summarized in Table 21.
Table 20: Baseline Characteristics Adjusted for in Comparative Analyses
Baseline characteristics | EU plus US cohort | US cohort | EU cohort | PHE | nNGM | CRISP | ESME |
|---|---|---|---|---|---|---|---|
Age | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
Gender | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
Race (Asian) | NA | NA | NA | NA | NA | NA | NA |
Smoking history | NA | NA | NA | NA | NA | NA | Yes |
Cancer stage at initial diagnosis | NA | Yes | NA | NA | Yes | NA | NA |
Number of metastatic locations | NA | Yes | NA | NA | Yes | Yes | Yes |
Brain metastasis | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
Prior lines of treatment | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
ECOG | NA | Yes | NA | Yes | NA | NA | NA |
Hemoglobin | NA | Yes | NA | NA | NA | NA | NA |
Liver metastasis | NA | NA | Yes | Yes | Yes | Yes | Yes |
Body mass index | NA | NA | NA | Yes | NA | Yes | NA |
CRISP = Clinical Research Platform Into Molecular Testing, Treatment and Outcome Registry of (Non-)Small Cell Lung Carcinoma Patients; ECOG = Eastern Cooperative Oncology Group; ESME = Epidemiological Strategy and Medical Economics; EU = European Union; NA = not available; nNGM = national Network Genomic Medicine; PHE = Public Health England.
Source: Sponsor-submitted indirect treatment comparison.18
Table 21: Baseline Characteristics Excluded From Comparative Analyses and Their Justification
Data source | Characteristics excluded | Justification |
|---|---|---|
EU + US cohort | All characteristics not common to the pooled sources | High rate of missingness |
US cohort | Smoking | Not prognostic |
Race (Asian) | Not prognostic, high rate of missingness | |
EU cohort | All characteristics not common to the pooled data sources | High rate of missingness |
PHE | Histology grade | Not prognostic |
nNGM | Smoking | Not prognostic |
ECOG | High rate of missingness | |
CRISP | Smoking | Not prognostic, high rate of missingness |
ECOG | High rate of missingness | |
ESME | Gender | Not prognostic |
Smoking | Not prognostic | |
ECOG | High rate of missingness |
CRISP = Clinical Research Platform Into Molecular Testing, Treatment and Outcome Registry of (Non-)Small Cell Lung Carcinoma Patients; ECOG = Eastern Cooperative Oncology Group; ESME = Epidemiological Strategy and Medical Economics; EU = European Union; nNGM = national Network Genomic Medicine; PHE = Public Health England.
Source: Sponsor-submitted indirect treatment comparison.18
Results of the Sponsor-Submitted Adjusted Treatment Comparison
Summary of Included Studies
Unadjusted baseline and IPW-adjusted characteristics of treatment lines of patients from CHRYSALIS across the pooled data sources compared to physicians’ choice are summarized in Table 22 and compared to treatment class in Table 23.
In the comparison to physicians’ choice, the CHRYSALIS trial included 81 patients, while the EU plus US cohort included 349 patients, with 206 from the US cohort and 143 from the EU cohort. There was some variation in the baseline characteristics of patients in CHRYSALIS and the individual cohorts, and there was a high degree of missing baseline characteristics overall. All common variables across these data sources were included in the adjustment. Before adjustment, age range and gender were generally similar between the CHRYSALIS trial and the overall cohort; however, there was a greater variation in the prior number of lines of treatment received, with a lower proportion of patients receiving 1 prior line of treatment (38.3% versus 44.4%) in CHRYSALIS, and a higher proportion with 4 or more prior lines of treatment (18.5% versus 9.7%). Additionally, more patients did not have brain metastases at baseline in CHRYSALIS compared to the EU plus US cohort (77.8% versus 62.2%). Baseline characteristics were well balanced after IPW. There were further individual differences between the baseline characteristics of the CHRYSALIS trial and the US cohort, including the presence of brain metastases (22.2% versus 38.8%), the number of metastatic locations (1 location: 40.7% versus 29.6%; 2 locations: 37.0% versus 19.4%; 3 locations: 16% versus 21.4%; and 4 locations: 6.2% versus 22.8%), and normal or high hemoglobin levels (58% versus 46.6%); and the EU cohort, including prior lines of treatment (1 line: 38.3% versus 51.0%, and 4-plus lines: 18.5% versus 4.9%), and the presence of metastases in the brain (22.2% versus 36.4%) and liver (9.9% versus 22.4%). Baseline characteristics were well balanced after IPW.
In the comparison of CHRYSALIS to the different treatment classes in the EU plus US cohort, there were 81 patients in CHRYSALIS, and 60, 89, 76, 58, and 66 patients in the TKI, IO, non–platinum-based chemotherapy, VEGFi plus chemotherapy, and “other” groups, respectively. There were some baseline differences between age groups, particularly for the “other” treatment group, where there were more patients aged below 55 years, and more patients aged above 60 years. The same differences listed previously for the comparison between CHRYSALIS and physicians’ choice with regards to prior lines of treatment and the presence of brain metastases before adjustment applied to the comparison between CHRYSALIS and individual treatment classes.
Table 22: Baseline Characteristics of Treatment Lines for Patients in CHRYSALIS and the EU Plus US Cohort, US Cohort, and EU Cohort Before and After Adjustment (Physicians’ Choice)
Characteristic | CHRYSALIS (N = 81) | EU + US cohorta (N = 349) | US cohort (N = 206) | EU cohort (N = 143)a | |||
|---|---|---|---|---|---|---|---|
Before adjustment | IPW ATT weightedb | Before adjustment | IPW ATT weightedc | Before adjustment | IPW ATT weightedd | ||
Age groups, n (%) | |||||||
≤ 55 | 19 (23.5) | 97 (27.8) | 77 (22.1) | NR | NR | 44 (30.8) | 29 (20.0) |
55 to ≤ 60 | 16 (19.8) | 54 (15.5) | 72 (20.6) | NR | NR | 17 (11.9) | 30 (20.8) |
> 60 | 46 (56.8) | 198 (56.7) | 200 (57.3) | NR | NR | 82 (57.3) | 85 (59.2) |
< 60 | 33 (40.7) | NR | NR | 77 (37.4) | 79 (38.3) | NR | NR |
60 to 70 | 26 (32.1) | NR | NR | 63 (30.6) | 70 (33.8) | NR | NR |
≥ 70 | 22 (27.2) | NR | NR | 66 (32.0) | 58 (27.9) | NR | NR |
Gender, n (%) | |||||||
Male | 33 (40.7) | 137 (39.3) | 143 (41.1) | 79 (38.3) | 87 (42.3) | 58 (40.6) | 60 (41.8) |
Female | 48 (59.3) | 212 (60.7) | 206 (58.9) | 127 (61.7) | 119 (57.7) | 85 (59.4) | 83 (58.2) |
ECOG PS, n (%) | |||||||
0 | 26 (32.1) | NR | NR | 61 (29.6) | 62 (30.2) | NR | NR |
1 | 55 (67.9) | NR | NR | 145 (70.4) | 144 (69.8) | NR | NR |
Prior lines of treatment, n (%) | |||||||
1 | 31 (38.3) | 155 (44.4) | 133 (38.1) | 82 (39.8) | 82 (39.9) | 73 (51.0) | 53 (37.3) |
2 | 24 (29.6) | 108 (30.9) | 105 (30.1) | 64 (31.1) | 62 (30.1) | 44 (30.8) | 44 (30.5) |
3 | 11 (13.6) | 52 (14.9) | 46 (13.1) | 33 (16.0) | 27 (12.9) | 19 (13.3) | 19 (13.3) |
4+ | 15 (18.5) | 34 (9.7) | 65 (18.7) | 27 (13.1) | 35 (17.0) | 7 (4.9) | 27 (18.9) |
Brain metastasis, n (%) | |||||||
Yes | 18 (22.2) | 132 (37.8) | 77 (22.1) | 80 (38.8) | 44 (21.5) | 52 (36.4) | 31 (21.7) |
No | 63 (77.8) | 217 (62.2) | 272 (77.9) | 126 (61.2) | 162 (78.5) | 91 (63.6) | 112 (78.3) |
Liver metastasis, n (%) | |||||||
Yes | 8 (9.9) | NR | NR | NR | NR | 32 (22.4) | 14 (9.7) |
No | 73 (90.1) | NR | NR | NR | NR | 111 (77.6) | 129 (90.3) |
Number of metastatic locations, n (%) | |||||||
1 | 33 (40.7) | NR | NR | 61 (29.6) | 85 (41.1) | NR | NR |
2 | 30 (37.0) | NR | NR | 40 (19.4) | 75 (36.6) | NR | NR |
3 | 13 (16.0) | NR | NR | 44 (21.4) | 34 (16.3) | NR | NR |
4 | 5 (6.2) | NR | NR | 47 (22.8) | 12 (6.0) | NR | NR |
Missing | 0 (0.0) | NR | NR | 14 (6.8) | 0 (0.0) | NR | NR |
Hemoglobin, n (%) | |||||||
Normal/high | 47 (58.0) | NR | NR | 96 (46.6) | 122 (59.0) | NR | NR |
Low | 34 (42.0) | NR | NR | 110 (53.4) | 84 (41.0) | NR | NR |
Cancer stage at initial diagnosis, n (%) | |||||||
I | 7 (8.6) | NR | NR | 23 (11.2) | 20 (9.7) | NR | NR |
II | 5 (6.2) | NR | NR | 10 (4.9) | 14 (6.8) | NR | NR |
IIIA | 4 (4.9) | NR | NR | 13 (6.3) | 10 (4.9) | NR | NR |
IIIB/IV | 65 (80.2) | NR | NR | 160 (77.7) | 162 (78.7) | NR | NR |
ATT = average treatment effect on the treated; ECOG = Eastern Cooperative Oncology Group; EU = European Union; IPW = inverse probability weighting; NR = not reported; PS = performance score.
aExcluding Epidemiological Strategy and Medical Economics.
bBaseline characteristics included in adjustment: prior lines of treatment, brain metastases, age, gender.
cBaseline characteristics included in adjustment: prior lines of treatment, brain metastases, age, ECOG performance status, number of metastatic locations, hemoglobin, gender, and cancer stage at diagnosis.
dBaseline characteristics included in adjustment: prior lines of treatment, brain metastases, liver metastases, age, gender.
Source: Sponsor-submitted indirect treatment comparison.18
Table 23: Baseline Characteristics of Treatment Lines for Patients in CHRYSALIS and the EU Plus US Cohort Before and After Adjustment (Treatment Class)
Characteristic | CHRYSALIS (N = 81) | TKI (N = 60) | IO (N = 89) | Non-platinum CT (N = 76) | VEGFi + CT (N = 58) | Other (N = 66) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Before adjustment | IPW ATT weighted | Before adjustment | IPW ATT weighted | Before adjustment | IPW ATT weighted | Before adjustment | IPW ATT weighted | Before adjustment | IPW ATT weighted | ||
Age groups, n (%) | |||||||||||
≤ 55 | 19 (23.5) | ██████ | ██████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
55 to ≤ 60 | 16 (19.8) | ██████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
> 60 | 46 (56.8) | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
Gender, n (%) | |||||||||||
Male | 33 (40.7) | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
Female | 48 (59.3) | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
Prior lines of treatment, n (%) | |||||||||||
1 | 31 (38.3) | ███████ | ██████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
2 | 24 (29.6) | ███████ | ██████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
3 | 11 (13.6) | ██████ | ██████ | ███████ | ███████ | ███████ | ███████ | ██████ | ███████ | ███████ | ███████ |
4+ | 15 (18.5) | ██████ | ██████ | ██████ | ███████ | ███████ | ███████ | ██████ | ██████ | ██████ | ███████ |
Brain metastasis, n (%) | |||||||||||
Yes | 18 (22.2) | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
No | 63 (77.8) | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ | ███████ |
ATT = average treatment effect on the treated; CT = chemotherapy; EU = European Union; IO = immuno-oncology; IPW = inverse probability weighting; TKI = tyrosine kinase inhibitor; VEGFi = vascular endothelial growth factor inhibitor.
Source: Sponsor-submitted indirect treatment comparison.18
Results
Results using the ATT IPW approach were considered the base-case analysis across cohorts. Results for analyses using the ATE and ATO IPW approaches were not described in detail in the sponsor-submitted report.
Overall Response Rate
Results for the observed (unadjusted) and adjusted ORR per investigators assessment in all cohorts versus physicians’ choice is summarized in Table 24. Compared to the EU plus US cohort, the adjusted (via the IPW ATT approach adjustment) ORR was 38.3% versus █████ for amivantamab and physicians’ choice, respectively. The OR following adjustment via the IPW ATT approach was ████████████████████), with an RR of ████████████████████). Results using covariate adjustment based on multivariable proportional hazards regression were consistent with the primary analysis, with an adjusted OR for amivantamab versus physicians’ choice of ████████████████████), and a corresponding RR of ████████████████████).
Results for the US and EU cohort were similar. In the comparison against physicians’ choice, ORR adjusted via the IPW ATT approach was █████ in the US cohort and █████ in the EU cohort, versus 38.3% with █████████████████ US cohort, the IPW ATT approach resulted in an OR of █████████████████████ and RR of █████████████████████, while the EU cohort resulted in an OR of █████████████████████ and an RR of ██████████████████████
Results for analyses using the ATE and ATO IPW approaches were only reported for the US cohort, in which they were consistent with the ATT approach.
Table 24: Unadjusted and Adjusted ORR, OR, and RR for ORR for CHRYSALIS vs. EU Plus US Cohort, US Cohort, and EU Cohort (Amivantamab vs. Physicians’ Choice)
Method | ORR amivantamab | ORR PC | OR (95% CI) | RR (95% CI) |
|---|---|---|---|---|
CHRYSALIS vs. EU + US cohort | ||||
Unadjusted | ||||
Amivantamab vs. PC | 38.3% | █████ | ███████████████ | — |
Adjusted | ||||
IPW: ATT approach | 38.3% | █████ | ███████████████ | ███████████████ |
Covariate adjustment | 36.5% | █████ | ███████████████ | ███████████████ |
CHRYSALIS vs. US cohort | ||||
Unadjusted | ||||
Amivantamab vs. PC | 38.3% | █████ | ███████████████ | — |
Adjusted | ||||
IPW: ATT approach | 38.3% | █████ | ███████████████ | ███████████████ |
Covariate adjustment | 38.6% | █████ | ███████████████ | ███████████████ |
CHRYSALIS vs. EU cohort | ||||
Unadjusted | ||||
Amivantamab vs. PC | 38.3% | █████ | ███████████████ | — |
Adjusted | ||||
IPW: ATT approach | 38.3% | █████ | ███████████████ | ███████████████ |
Covariate adjustment | 33.9% | █████ | ███████████████ | ███████████████ |
ATT = average treatment effect among the treated; CI = confidence interval; EU = European Union; IPW = inverse probability weighting; OR = odds ratio; ORR = overall response rate; PC = physician’s choice; RR = response rate; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison.18
Results for ORR comparing amivantamab to individual treatment classes are summarized in Table 25. The sponsor considered the results for this analysis exploratory and underpowered due to the small sample sizes in individual treatment classes.
Table 25: Adjusted ORR, OR, and RR for ORR for CHRYSALIS vs. EU Plus US Cohort, US Cohort, and EU Cohort (Amivantamab vs. Individual Treatment Classes)
Method | ORR amivantamab | OR (95% CI) | RR (95% CI) |
|---|---|---|---|
CHRYSALIS vs. EU + US cohort (IPW: ATT Approach) | |||
Amivantamab (N = 81) | 38.3% | — | — |
TKI (N = 60) | ████ | ██████████████████ | ██████████████████ |
IO (N = 89) | █████ | ████████████████ | ███████████████ |
Non–platinum-based chemotherapy (N = 76) | █████ | ███████████████ | ███████████████ |
VEGFi + chemotherapy (N = 58) | █████ | ███████████████ | ███████████████ |
Other (N = 66) | █████ | ███████████████ | ███████████████ |
CHRYSALIS vs. US cohort (covariate adjustment approach) | |||
Amivantamab (N = 81) | 40.0% | — | — |
TKI (N = 34) | ████ | ████████████████ | ████ |
IO (N = 55) | ████ | ████████████████ | ████ |
Non–platinum-based chemotherapy (N = 52) | █████ | ████████████████ | ████ |
VEGFi + chemotherapy (N = 29) | █████ | ███████████████ | ████ |
Other (N = 36) | █████ | ███████████████ | ████ |
CHRYSALIS vs. EU cohort (covariate adjustment approach) | |||
Amivantamab (N = 81) | 32.3% | — | — |
TKI (N = 26) | ████ | ██ | ██ |
IO (N = 34) | █████ | ███████████████ | ███████████████ |
Non–platinum-based chemotherapy (N = 24) | █████ | ███████████████ | ███████████████ |
VEGFi + chemotherapy (N = 29) | █████ | ████████████████ | ███████████████ |
Other (N = 30) | █████ | ████████████████ | ████████████████ |
ATT = average treatment effect among the treated; CI = confidence interval; EU = European Union; IO = immuno-oncology; IPW = inverse probability weighting; NE = not estimable; OR = odds ratio; ORR = overall response rate; RR = response rate; TKI = tyrosine kinase inhibitor; VEGFi = vascular endothelial growth factor inhibitor.
aGeneralized linear model for RR did not converge. RR is derived as the ratio of the overall response rates generated from the logistic regression model for the odds ratios.
Source: Sponsor-submitted indirect treatment comparison.18
Results of the sensitivity analysis for ORR using BICR from CHRYSALIS were consistent with the base-case analysis, though the point estimates were generally greater in the sensitivity analysis in favour of amivantamab.
Overall Survival
Results for IPW ATT–adjusted HR for OS for amivantamab versus physicians’ choice, and versus treatment classes in the EU plus US cohort, US cohort, and EU cohorts, are summarized in Table 26 and Table 27, respectively. In all cases, amivantamab was favoured over physicians’ choice for OS in the EU plus US cohort (HR = ████████████████████████), US cohort (HR = ████████████████████████), and EU cohort (HR = 0 ███████████████████████). The median OS for amivantamab was estimated at 22.77 months (95% CI, 17.48 to not estimable) compared to ███████████████████████████████ for the physicians’ choice group from the EU plus US cohort, ███████████████████████████████ for the physicians’ choice group from the US cohort, and ██████████████████████████████ for the physicians’ choice group from the EU cohort.
Compared to the individual treatment classes (Table 27), amivantamab was favoured after adjustment in all cases, with HRs ranging from ██████████ for the EU plus US cohort, ██████████ for the US cohort, and ██████████ for the EU cohort.
Table 26: OS for CHRYSALIS vs. EU Plus US Cohort, US Cohort, and EU Cohort Minus IPW ATT (Amivantamab vs. Physicians’ Choice)
OS | Amivantamab (N = 81) | EU + US cohort | US cohort | EU cohort |
|---|---|---|---|---|
PC (N = 401) | PC (N = 206) | PC (N = 195) | ||
Unadjusted | ||||
Events | 31 (38.27) | ██████████ | ██████████ | ██████████ |
Censored | 50 (61.73) | ██████████ | █████████ | █████████ |
Median OS (95% CI) | 22.77 (17.48 to NE) | ██████████████ | ██████████████ | █████████████ |
HR (95% CI) vs. PC | — | ██████████████ | ██████████████ | ██████████████ |
P value | — | ███████ | ███████ | ███████ |
Adjusted (IPW – ATT) | ||||
Events | 31 (38.27) | ██████████ | ██████████ | ██████████ |
Censored | 50 (61.73) | ██████████ | █████████ | █████████ |
Median OS (95% CI) | 22.77 (17.48 to NE) | ██████████████ | ██████████████ | █████████████ |
HR (95% CI) vs. PC | — | █████████████ | ██████████████ | █████████████ |
P value | — | ███████ | ██████ | ███████ |
ATT = average treatment effect among the treated; CI = confidence interval; EU = European Union; HR = hazard ration; IPW = inverse probability weighting; NE = not estimable; OS = overall survival; PC = physicians’ choice; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison.18
Table 27: HR for OS for CHRYSALIS vs. EU Plus US Cohort, US Cohort, and EU Cohort (Amivantamab vs. Treatment Classes)
Models | Amivantamab vs. | ||||
|---|---|---|---|---|---|
TKI | IO | Non–platinum-based chemotherapy | VEGFi + chemotherapy | Others | |
CHRYSALIS vs. EU + US cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ███████████ | ███████████ | ██████████ | ██████████ | ███████████ |
sATT adjustment | |||||
HR (95% CI) | ██████████ | ███████████ | ███████████ | ███████████ | ███████████ |
Covariate adjustment | |||||
HR (95% CI) | ██████████ | ███████████ | ███████████ | ███████████ | ███████████ |
CHRYSALIS vs. US cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ██████████ | ███████████ | ██████████ | ███████████ | ███████████ |
Covariate adjustment | |||||
HR (95% CI) | ██████████ | ███████████ | ███████████ | ███████████ | ███████████ |
CHRYSALIS vs. EU cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ███████████ | ███████████ | ███████████ | ███████████ | ███████████ |
Covariate adjustment | |||||
HR (95% CI) | ███████████ | ███████████ | ███████████ | ███████████ | ███████████ |
CI = confidence interval; EU = European Union; HR = hazard ratio; IO = immuno-oncology; OS = overall survival; sATT = scaled average treatment effect among the treated; TKI = tyrosine kinase inhibitor; VEGFi = vascular endothelial growth factor inhibitor; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison.18
Progression-Free Survival
Results for adjusted HR for PFS for amivantamab versus physicians’ choice, and versus treatment classes in the EU plus US cohort, US cohort, and EU cohorts, are summarized in Table 28 and Table 29, respectively. In all cases for PFS, amivantamab was favoured over physicians’ choice in the EU plus US cohort (HR = █████████████████████████), US cohort (HR = ███████████████████████████ and EU cohort (HR = ████████████████████████). The median PFS for amivantamab was 8.25 months (95% CI, 5.49 to 12.32) compared to ████████████████████████████ for the physicians’ choice group from the EU plus US cohort, ████████████████████████████ for the physicians’ choice group from the US cohort, and ████████████████████████████ for the physicians’ choice group from the EU cohort.
Compared to the individual treatment classes (Table 29), amivantamab was favoured after adjustment in nearly all cases, with HRs ranging from ██████████ for the EU plus US cohort, ██████████ for the US cohort, and ██████████ for the EU cohort; however, amivantamab was not favoured in 3 cases in the EU cohort compared to IOs (HR = █████████████████████), non–platinum-based chemotherapy (HR = 0.67; 95% CI, 0.37 to 1.20), and other therapy (HR = █████████████████████).
Table 28: PFS for CHRYSALIS vs. EU Plus US Cohort, US Cohort, and EU Cohort Minus IPW ATT (Amivantamab vs. Physicians’ Choice)
PFS | Amivantamab | PC – EU + US cohort | PC – US cohort | PC – EU cohort |
|---|---|---|---|---|
Unadjusted | ||||
N | 81 | 388 | 206 | 182 |
Events | 57 (70.37) | ██████████ | ██████████ | ██████████ |
Censored | 24 (29.63) | █████████ | █████████ | █████████ |
Median PFS (95% CI) | 8.25 (5.49 to 12.32) | ███████████████ | ███████████████ | ███████████████ |
HR (95% CI) vs. PC | — | ███████████████ | ███████████████ | ███████████████ |
P value | — | ███████ | ███████ | ███████ |
Adjusted (IPW – ATT) | ||||
N | 81 | 384 | 206 | 174 |
Events | 57 (70.37) | ██████████ | ██████████ | ██████████ |
Censored | 24 (29.63) | █████████ | █████████ | █████████ |
Median PFS (95% CI) | 8.25 (5.49 to 12.32) | ██████████████ | ███████████████ | ██████████████ |
HR (95% CI) vs. PC | — | ██████████████ | ███████████████ | ██████████████ |
P value | — | ███████ | ███████ | ██████ |
ATT = average treatment effect among the treated; CI = confidence interval; EU = European Union; HR = hazard ratio; IPW = inverse probability weighting; PC = physicians’ choice; PFS = progression-free survival; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison.18
Table 29: HR for PFS for CHRYSALIS vs. EU Plus US Cohort, US Cohort, and EU Cohort (Amivantamab vs. Treatment Classes)
Models | Amivantamab vs. | ||||
|---|---|---|---|---|---|
TKI | IO | Non–platinum-based chemotherapy | VEGFi + chemotherapy | Others | |
CHRYSALIS vs. EU + US Cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ███████████ |
sATT adjustment | |||||
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
Covariate adjustment | |||||
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
CHRYSALIS vs. US Cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
Covariate adjustment | |||||
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
CHRYSALIS vs. EU Cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
Covariate adjustment | |||||
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
CI = confidence interval; EU = European Union; HR = hazard ratio; IO = immuno-oncology; PFS = progression = free survival; sATT = scaled average treatment effect among the treated; TKI = tyrosine kinase inhibitor; VEGFi = vascular endothelial growth factor inhibitor; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison.18
Results of the sensitivity analysis for PFS using BICR from CHRYSALIS were consistent with the base-case analysis.
Time to Next Treatment
Results for adjusted HR for TTNT for amivantamab versus physicians’ choice, and versus treatment classes in the EU plus US cohort, US cohort, and EU cohorts, are summarized in Table 30 and Table 31, respectively. In all cases for TTNT, amivantamab was favoured over physicians’ choice in the EU plus US cohort (HR = ████████████████████████), the US cohort (HR = ████████████████████████), and the EU cohort (HR = ████████████████████████). The median TTNT for amivantamab was 12.42 months (95% CI, 7.66 to 18.79), ████████████████████████████ for physicians’ choice in the EU plus US cohort, ████████████████████████████ for physicians’ choice in the US cohort, and ████████████████████████████ for physicians’ choice in the EU cohort.
Compared to the individual treatment classes (Table 31), amivantamab was favoured over all other treatment options, with HR ranging from 0.35 to 0.58 for the EU plus US cohort, 0.27 to 0.61 for the US cohort, and 0.36 to 0.54 for the EU cohort.
Table 30: TTNT for CHRYSALIS vs. EU Plus US Cohort, US Cohort, and EU Cohort Minus IPW ATT (Amivantamab vs. Physicians’ Choice)
TTNT | Amivantamab (N = 81) | EU + US cohort PC (N = 401) | US cohort PC (N = 206) | EU cohort PC (N = 195) |
|---|---|---|---|---|
Unadjusted | ||||
Events | 50 (61.73) | ██████████ | ██████████ | ██████████ |
Censored | 31 (38.27) | █████████ | █████████ | █████████ |
Median TTNT (95% CI) | 12.42 (7.66 to 18.79) | ███████████████ | ███████████████ | ███████████████ |
HR (95% CI) vs. PC | — | ███████████████ | ███████████████ | ███████████████ |
Adjusted (IPW – ATT) | ||||
Events | 50 (61.73) | ██████████ | ██████████ | ██████████ |
Censored | 31 (38.27) | █████████ | █████████ | █████████ |
Median TTNT (95% CI) | 12.42 (7.66 to 18.79) | ███████████████ | ███████████████ | ███████████████ |
HR (95% CI) vs. PC | — | ███████████████ | ███████████████ | ███████████████ |
ATT = average treatment effect on the treated; CI = confidence interval; EU = European Union; HR = hazard ratio; IPW = inverse probability weighting; PC = physicians’ choice; PFS = progression-free survival; TTNT = time to next treatment; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison.18
Table 31: HR for TTNT for CHRYSALIS vs. EU Plus US Cohort, US Cohort, and EU Cohort (Amivantamab vs. Treatment Classes)
Models | Amivantamab vs. | ||||
|---|---|---|---|---|---|
TKI | IO | Non–platinum-based chemotherapy | VEGFi + chemotherapy | Others | |
CHRYSALIS vs. EU + US cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ████████████ | ██████████ | ████████████ | ████████████ | ████████████ |
sATT adjustment | |||||
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
Covariate adjustment | |||||
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
CHRYSALIS vs. US cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
Covariate adjustment | |||||
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
CHRYSALIS vs. EU cohort | |||||
Unadjusted | |||||
N | ██ | ██ | ██ | ██ | ██ |
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
Covariate adjustment | |||||
HR (95% CI) | ████████████ | ████████████ | ████████████ | ████████████ | ████████████ |
sATT = scaled average treatment effect among the treated; CI = confidence interval; EU = European Union; HR = hazard ratio; IO = immuno-oncology; TKI = tyrosine kinase inhibitor; TTNT = time to next treatment; VEGFi = vascular endothelial growth factor inhibitor; vs. = versus.
Source: Sponsor-submitted indirect treatment comparison
Critical Appraisal of the Sponsor-Submitted Adjusted Treatment Comparison
The choice to conduct an adjusted treatment comparison of amivantamab and external real-world data cohorts as a comparator arm was justified by the lack of a comparator arm for the CHRYSALIS trial. Data derived from 7 international real-world data sources were used for the comparison with amivantamab. The methods and reasons for selecting these databases were not reported; thus, there is a risk of selection bias because the patients may not be representative of Canadian patients. Additionally, these data sources represented a broad population from different regions. There may also be differences in clinical practice by region given that recruitment into various databases occurred at different time points, though the direction of potential bias is unclear. Additionally, line-of-therapy definitions may not always have been similar to that of CHRYSALIS. Comparisons of trial evidence with real-world data may result in greater uncertainty given the difference in measurements, reporting, and study design. As such, the quality of effectiveness estimates from this nonrandomized study was considered according to the checklist in the National Institute for Health and Health Care Excellence Decision Support Unit Technical Document 17 (NICE DSU TSD 17).45
There were important differences in the design of the cohorts that limit the ability to draw strong conclusions about the efficacy of amivantamab compared with other treatments. CHRYSALIS was a phase I/Ib, single-arm trial, whereas the comparators were derived from real-world cohorts. Data analyzed retrospectively from databases and medical records are more prone to biases that cannot be fully controlled for (e.g., selection bias, confounding bias, limited data availability) compared to those collected from prospective interventional studies (such as RCTs and single-arm trials). The population for the CHRYSALIS trial used in the adjusted treatment comparison included adult patients with EGFR-mutated NSCLC with exon 20 insertion mutations after failure of platinum-based therapy, using the March 30, 2021, DCO, which was consistent with the indicated population for amivantamab. Inclusion and exclusion criteria from CHRYSALIS were then applied to patients from all real-world data sources to select the appropriate population. Any criteria that could not be applied to patients from a given data source due to missing data were omitted from the list of inclusion and exclusion criteria applied to that data source, which may have resulted in unaccounted-for differences in patient populations. Appropriately, given the application of inclusion and exclusion criteria from CHRYSALIS, patients in the analysis all had confirmation of EGFR exon 20 insertion mutation positivity, though it was unclear when testing was conducted for the real-world data sources. As well, it was unclear how many potentially eligible patients were excluded because their eligibility status could not be confirmed due to missing data. The index, or baseline, date for real-world data cohorts was the start of any line of therapy at the start of which inclusion and exclusion criteria were met; thus, patients in the real-world data sources may have received qualifying treatment in more than 1 line of therapy during their follow-up and may be included multiple times in the analyses. Correlation of outcomes across treatment lines for the same patient was accounted for using a robust sandwich estimator to reduce the bias in favour of amivantamab, though scenario analyses exploring scenarios in which patients were only included in 1 line of treatment were not conducted.
Multiple comparative analyses (IPW and covariate adjustment) were performed for the CHRYSALIS population versus the pooled European data sources, the pooled US data sources and versus the pooled, combined EU plus US cohort. The choice to conduct the analyses using the ATT approach was justified, considering the preservation of sample size for the CHRYSALIS trial. The results for ATE and ATO analyses were described as consistent with the ATT approach, though these were not reported in detail. Individual real-world data sources from Europe and from the US were also pooled to increase sample size. The sponsor noted that pooling of the EU and US cohorts into a single combined cohort was possible due to the high consistency between the results and a comparable treatment distribution of the EU and US cohorts. However, there were observable differences in baseline characteristics of populations between the EU and US cohorts, as well as the potential for unknown systematic differences across cohorts due to significant missing data between databases. As a result, significant heterogeneity may have been present in the populations, though this was not explored and remains uncertain. Propensity scoring for the IPW analyses was appropriate, demonstrating good balance on adjusted covariates, where available. Amivantamab was also compared to both a pooled basket of treatments (physician’s choice) and various treatment class regimens (TKIs, IOs, non–platinum-based chemotherapy, VEGFi, and other). In some instances, comparisons of amivantamab to individual treatment classes were not feasible due to the reduced sample sizes. As noted by the clinical experts consulted by CADTH, not all these treatment classes, particularly the VEGFis (N = 58), are relevant to Canadian clinical practice. Given that pooling of treatments for the physicians’ choice group assumes equivalence of treatment benefit, it is unclear how inclusion of treatments irrelevant to the Canadian context, such as VEGFis, would have affected the results. Analysis of all end points by treatment class was also conducted, which favoured amivantamab for all treatment comparisons, though these analyses were limited by small sample sizes and wide 95% CIs.
In general, comparisons with externally generated cohorts are limited by the availability of information important to the analysis. No imputation method was applied to account for missing data, except for partial dates, and if a substantial amount of data was missing, then covariates were not included in the propensity score models, which may affect the results of the comparisons, though the direction of this impact would depend on what information was missing and remains unclear. At baseline, before adjustment, there was a substantial amount of missing baseline clinical and demographic characteristics across the individual US and EU databases, resulting in heterogeneous patient populations between CHRYSALIS and the EU and US real-world cohorts, particularly for the factors of age and the number of prior lines of treatment received. However, individual EU and US pooling seemed more appropriate based on data availability. A comprehensive list of potential prognostic factors and treatment-effect modifiers was identified via systematic literature search, as well as in consultation with clinical experts; however, not all variables were available in each database. The clinical experts consulted by CADTH noted that ECOG performance status is an important prognostic factor in these patients. However, due to limited availability in the real-world cohorts, and given that only those variables that were common across data sources were included in the adjustment, ECOG performance status was not a variable within the propensity scoring adjustment in the IPW-weighted EU plus US cohort or the EU cohort, which may result in some unaccounted-for heterogeneity in the populations. Baseline characteristics were presented both before and after weighting for all analyses. Following adjustment of available baseline characteristics, the populations were generally similar; however, important factors were not accounted for, including smoking status and race in the US cohort; ECOG performance status, smoking status, and race in the EU cohort; and smoking status, ECOG performance status, and race in the combined EU plus US cohort; therefore, the comparisons were not balanced for some important confounders and were not mutually randomizable populations. Incremental summaries of the effect of adding covariates to a covariate adjustment model suggested minimal influence for most covariates, with the possible exception of ECOG and smoking history in some datasets, though the estimates themselves were very imprecise. However, despite adjustment for important covariates, a potential risk of residual confounding bias remains. Additionally, PHE and ESME databases were excluded from some analyses because no ORR or PFS data were available from the PHE cohort and no ORR data were available from ESME; thus, it was unclear whether this may have affected the results, given potential heterogeneity across populations.
The data collection period and setting of the included real-world data sources varied, with enrolment for some beginning as far back as 2009, whereas the enrolment for cohort D of the CHRYSALIS trial began in 2018 and was completed in September 2020. Overall, there has been minimal change in the management of EGFR exon 20 insertion NSCLC; therefore, it is unlikely that there was much bias due to historical comparisons. Apart from the median follow-up for efficacy in the CHRYSALIS trial of 14.5 months at the March 30, 2021, DCO, no consideration or adjustment was given to follow-up duration and the time of assessment for various end points was unknown. As a result, it is uncertain whether the follow-up times between CHRYSALIS and the real-world data sources are comparable, and may also contribute to heterogeneity, especially for survival analyses.
The efficacy outcomes measured in the adjusted treatment comparison were important to patients and physicians, and the models selected were appropriate for these outcomes. Investigator-assessed outcomes from CHRYSALIS were used in the base case, given that independent review of outcomes in the real-world data sources was not possible. Sensitivity analyses using BICR results from CHRYSALIS were also conducted, which were consistent with the results of the analyses using investigator assessment. No other sensitivity or subgroup analyses were conducted for relevant subgroups or to evaluate differences for potential treatment-effect modifiers or prognostic factors. No safety outcomes were included in the analysis.
Overall, the results of the adjusted treatment comparisons were consistent across end points and statistical methodologies, generally favouring amivantamab over physicians’ choice, as well as for the individual treatment classes, with 95% CIs that most often did not include the null threshold. However, there was notable imprecision in all cases, as demonstrated by the moderately to severely wide 95% CIs, though the reason for this imprecision was unknown and may be due to small sample sizes and unexplored heterogeneity. Furthermore, given the phase I/Ib nature of the CHRYSALIS trial and the lack of a comparator arm, the ability to make definitive conclusions on the comparative efficacy of amivantamab was limited. Comparisons using the external control arms derived from real-world data sources were subject to substantial uncertainty and risk of bias due to the methods of data collection, small sample sizes, high degree of heterogeneity due to pooling assumptions, and limited data availability of important confounders and potential prognostic factors, including ECOG performance status in the EU and the EU plus US cohorts. Outcomes important to patients, including HRQoL and AEs, were not analyzed in the adjusted treatment comparisons; thus, the comparative efficacy of amivantamab on these outcomes remains unknown.
Other Relevant Evidence
No long-term extension studies or other relevant studies were included in the sponsor’s submission to CADTH.
Discussion
Summary of Available Evidence
One ongoing, phase I/Ib, open-label study (CHRYSALIS) was included in this review. The review for amivantamab was based on cohort D of the CHRYSALIS trial, which consisted of 153 patients treated with amivantamab monotherapy at the RP2D of 1,050 mg (1,400 mg for patients weighing greater than or equal to 80 kg) with EGFR exon 20 insertion mutations who had progressed after prior platinum-based chemotherapy for metastatic disease. Patients were excluded if they had untreated or active brain metastases and a history of ILD. The primary end point of the CHRYSALIS trial was ORR, with secondary end points of CBR, DOR, PFS, OS, TTF, and safety. Exploratory end points included HRQoL and symptom severity.
At baseline, patients included in the CHRYSALIS (cohort D) trial were primarily Asian (62.1%) and female (61.4%), and had an ECOG performance status of 1 (72.5%), with a median age of 61.0 years. Most patients had stage III or IV (87.5%) disease, and the median number of prior lines of therapy was 2. All patients had received prior platinum-based chemotherapy with either carboplatin-containing or cisplatin-containing regimens. The median follow-up of the CHRYSALIS trial as of the March 30, 2021, DCO was 14.5 months.
One sponsor-submitted ITC was summarized and critically appraised. Two analysis scenarios were conducted comparing amivantamab to physicians’ choice, and to individual treatment classes using real-world data from 7 European and American databases. The primary end point for all comparisons was ORR. Other relevant outcomes included OS, PFS, and TTNT. Outcomes specifically important to patients, including AEs and HRQoL, were not assessed.
Interpretation of Results
Efficacy
As of the March 30, 2021, DCO, the median follow-up of the CHRYSALIS trial in the primary efficacy population was 14.5 months. The investigator-assessed ORR of 38.3% (43.2% per BICR) in the CHRYSALIS trial (March 30, 2021, DCO) was considered clinically meaningful and appeared to be favourable compared to currently available therapies, according to the clinical experts consulted by CADTH and the clinician input received by LCC-MAC. Responses in this patient population are important because of accompanying delay in the worsening of symptoms and a slower decline in ECOG performance status. Input provided by the patient advocacy groups highlighted improved management of disease symptoms as an important treatment goal for patients. No inferential statistical testing was performed for the efficacy outcomes in cohort D, and point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. The lower bound of the 95% CI for both analyses (ORR per investigator and per BICR) was greater than 12%, thus meeting the prespecified statistical threshold for a positive study outcome. The experts also noted that in clinical practice, CRs are rare in patients with lung cancer; therefore, it is reflective of real-world treatment that most patients involved in the calculation of ORR only had PRs (3 patients had CR, as assessed by BICR), and were therefore not concerned about the low rate of CR per BIRC observed in cohort D. This view was echoed by the clinician input received by LCC-MAC, which noted that a clinically meaningful end point in the population of reimbursement is stable radiological response, especially if it is durable. Additionally, the median follow-up time was considered appropriate by the clinical experts for determining response to treatment, and the response was considered durable. Results were consistent across DCO dates (i.e., June 8, 2020; October 8, 2020; and March 30, 2021) and across efficacy sets with different follow-up times (i.e., N = 81, N = 114, and N = 124), supporting consistent antitumour activity of amivantamab. The median PFS for amivantamab was 8.25 months (95% CI, 5.49 to 12.32) per investigator assessment (BIRC = 8.31 months; 95% CI, 5.52 to 11.07). Given the typical short duration of PFS noted by the clinical experts using current therapies, the clinical experts agreed that the median PFS observed with amivantamab appeared to be favourable. The clinical experts noted that the median follow-up duration of CHRYSALIS was appropriate for the outcome of PFS. As of the March 30, 2021, DCO, 45 (29.4%) patients had died in the safety population, most commonly from PD (31; 20.3%). The median OS was estimated at 22.77 months at a median follow-up time of 14.5 months, which was considered immature for estimating OS, according to the clinical experts. Due to the single-arm nature of the CHRYSALIS trial, the ability to interpret the results for PFS and OS was significantly limited.
There are currently no randomized phase III trials under way for this review’s target population. The clinical experts consulted by CADTH noted that, despite the high unmet need, conducting an RCT in this setting with a targeted therapy, such as amivantamab, compared to the available therapies currently used in Canadian clinical practice would likely not be feasible. According to the clinical experts, developing phase III RCTs is hindered by the overall low number of patients who meet the current indication and because equipoise between amivantamab and other chemotherapy drugs does not exist. As previously mentioned, the majority of patients within cohort D of the CHRYSALIS trial were Asian (62%). This is primarily reflective of where the trial was conducted, as well as the indication given that EGFR exon 20 insertion mutations are far more common in patients of Asian descent than patients from Western countries. The results of exploratory subgroup analyses for patients who identified as Asian and those who did not were consistent with the primary analysis, though per BICR, 10% more patients who were Asian achieved ORR than compared to investigator assessment, which may be reflective of variation in interpretation between radiologists by region, as opposed to an actual variation in response. Overall, in consultation with the clinical experts, it was believed that regardless of the higher proportion of patients who were Asian, the generalizability of the results to Canadian patients should not be affected, given that race is not believed to impact the efficacy of amivantamab.
Maintenance of or improved HRQoL was cited as an important outcome to patients; however, HRQoL and other PROs were added as an amendment to the study protocol and were not included in the regulatory submission. Due to the small sample size of only 36 patients included in the analyses, and a substantial decline in patients available to provide assessments over time, the effect of amivantamab on HRQoL remains inconclusive.
In the absence of comparative evidence, the sponsor submitted an adjusted treatment comparison of amivantamab compared to currently available treatment regimens, creating an external cohort using retrospectively collected real-world data from Europe and the US. The adjusted treatment comparison of the phase I/Ib, single-arm CHRYSALIS trial with patients from the real-world databases had small sample sizes, high clinical heterogeneity in the matching and pooling of cohorts due to limited data availability, and inability to adjust for important confounding and prognostic factors across all cohorts. The results of the adjusted treatment comparisons suggested that amivantamab was favoured over physicians’ choice as well as over individual treatment classes consisting of IO-based regimens, TKI-based regimens, and non–platinum-based regimens for ORR, OS, and PFS. Though results were consistently in favour of amivantamab, estimates of treatment effect had moderate-to-wide CIs, suggesting imprecision and uncertainty in the results. Given the limitations that were identified in the adjusted treatment comparison, it remained uncertain whether amivantamab provided additional benefits versus currently available therapies. The clinical experts consulted by CADTH anticipated that, based on the CHRYSALIS results and on poor results with existing treatment options in clinical practice, amivantamab would likely offer improved and clinically meaningful benefits compared with currently available therapies.
Harms
Amivantamab represents a treatment with a novel mechanism of action that acts as a bispecific antibody for both EGFR and MET mutations in NSCLC. This first-in-class mechanism of action results in some uncertainty around the long-term safety of amivantamab, particularly for EGFR-specific AEs such as rash, paronychia, and ILD. The product monograph for amivantamab also highlights several clinically relevant side effects associated with amivantamab, including IRRs, due to the method of administration and mechanism of action, and ophthalmologic disorders associated with inhibition of EGFR.16
Analysis of safety was based on 153 patients enrolled in cohort D of the CHRYSALIS trial who received treatment with amivantamab monotherapy. All patients in cohort D experienced at least 1 TEAE, of which 64 (41.8%) patients experienced 1 or more TEAEs that was greater than or equal to grade 3, and 44 (28.8%) experienced serious TEAEs. A total of 18 (11.8%) patients withdrew from treatment due to AEs, and 11 (7.2%) experienced death due to AEs.
Notable harms of interest to this review included IRRs, ILDs, skin disorders, paronychia, and ophthalmologic disorders, which are mainly reflective of the IV method of administration and are understood to be side effects associated with EGFR-targeting therapies. IRRs were the most common TEAE experienced by patients in the CHRYSALIS study (63.4%). To minimize the occurrence of IRRs in the CHRYSALIS study, the study protocol was modified to deliver amivantamab as a split-dose infusion over day 1 and day 2 of the first cycle, when IRRs are most likely to occur. Additionally, prophylactic preinfusion medication was required to prevent or reduce any IRRs. Aside from potential underestimation in the frequency or severity of IRRs due to split-dose and prophylactic medication administration, there were no clinical concerns, because this is a routine manageable practice to administer amivantamab according to the clinical experts consulted by CADTH. More than half of the patients in the CHRYSALIS trial required either infusion modifications (93; 60.8%) consisting of infusion rate reductions (82; 53.6%) or infusion interruptions (88; 57.5%).
As part of the exclusion criteria for this study, patients with a history of ILD or pneumonitis were excluded from participation, which was considered appropriate. Interstitial lung disease and ILD-like AEs have been associated with EGFR TKI use. Overall, the frequency of ILD was low, generally occurring 50 days after the first dose of amivantamab. There were a total of 4 serious ILD events, only 1 of which was grade 3 (pneumonitis). All ILD events were managed according to the prescribing information in the product monograph.
Rash and other dermatologic side effects are also frequently reported with EGFR TKIs. It is known that EGFR is widely expressed in the epidermis, with EGFR inhibition disrupting the epidermal integrity, inducing a cytokine reaction. Overall, rash events (grouped term) were common, occurring in 130 (85.0%) patients. In most cases, the reactions were mild and nonserious, resolving after dose interruption or reduction, though additional medications were often required, and prior experience with EGFR TKIs has resulted in specific guidelines on the management of rash TEAEs. Paronychia was also considered a clinically meaningful TEAE due to its association with EGFR TKIs, and occurred in 52.9% of patients. Paronychia reactions were generally mild and manageable, with only 1 patient discontinuing treatment due to grade 2 paronychia. The clinical experts noted that paronychia can be quite painful for patients.
Overall, in consultation with the clinical experts, the safety profile of amivantamab was considered similar to currently available targeted therapies in NSCLC, including TKIs and IOs.
Patient groups cited minimal side effects of treatment as an important consideration for new therapies. According to patients who have experience with amivantamab, side effects related to some previous treatments severely affected their participation in daily activities and HRQoL, while amivantamab did not.
The sponsor-submitted adjusted treatment comparison did not assess safety outcomes.
Conclusions
One phase I/Ib, single-arm, open-label trial (CHRYSALIS; cohort D) provided evidence for the efficacy and safety of amivantamab in adult patients with metastatic or unresectable NSCLC who harboured EGFR exon 20 insertion mutations and failed on, or progressed after, platinum-based chemotherapy. The CHRYSALIS trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR > 12%) in cohort D. The clinical experts consulted by CADTH felt that the achieved ORR per investigator assessment of 38.3% (43.2% per BICR) (March 30, 2021, DCO date) was clinically meaningful for the target population and durable (median DOR 12.45 months, 95% CI, 6.54 to 16.13). In the opinion of the clinical experts, the observed responses appeared higher than what is seen with currently used therapies in the target setting. There was uncertainty around the magnitude of the clinical benefit given the limitations in the evidence from the noncomparative phase I/Ib clinical trial. While time-to-event end points, OS and PFS, appeared supportive of the observed ORR, the nonrandomized design of the CHRYSALIS trial made interpreting the PFS and OS events attributable to amivantamab challenging. The CADTH clinical assessment identified limitations with the sponsor’s adjusted treatment comparison (including small sample sizes, heterogeneity across study designs and pooled populations, and the inability to adjust for important potential confounders and prognostic variables), which substantially limited the ability to interpret the relative treatments effects observed between amivantamab and other treatments. The results for the HRQoL and symptom severity were exploratory outcomes and remained inconclusive due to a number of important limitations. Harms associated with amivantamab were largely consistent with treatments based on EGFR inhibition and were considered manageable, according to the clinical experts consulted by CADTH. Overall, the ability to draw firm conclusions about the magnitude of clinical benefit and safety of amivantamab was limited given the limitations in the evidence.
References
1.Canadian Cancer Society. Lung and bronchus cancer statistics. 2022; https://cancer.ca/en/cancer-information/cancer-types/lung/statistics. Accessed 2022 Jul 28.
2.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics: A 2020 special report on lung cancer. Toronto (ON): Canadian Cancer Society; 2020: https://cdn.cancer.ca/-/media/files/cancer-information/resources/publications/2020-canadian-cancer-statistics-special-report/2020-canadian-cancer-statistics-special-report-en.pdf?rev=15c66a0b05f5479e935b48035c70dca3&hash=3D51B0D0FB5C3F7E659F896D66495CE8. Accessed 2022 Jul 28.
3.Birring SS, Peake MD. Symptoms and the early diagnosis of lung cancer. Thorax. 2005;60(4):268-269. PubMed
4.Midha A, Dearden S, McCormack R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII). Am J Cancer Res. 2015;5(9):2892-2911. PubMed
5.Graham RP, Treece AL, Lindeman NI, et al. Worldwide frequency of commonly detected EGFR mutations. Arch Pathol Lab Med. 2018;142(2):163-167. PubMed
6.Melosky B, Banerji S, Blais N, et al. Canadian consensus: a new systemic treatment algorithm for advanced EGFR-mutated non-small-cell lung cancer. Curr Oncol. 2020;27(2):e146-e155. PubMed
7.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129-2139. PubMed
8.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497-1500. PubMed
9.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306-13311. PubMed
10.Vyse S, Huang PH. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct Target Ther. 2019;4(1):5. PubMed
11.Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288-300. PubMed
12.Boyne DJ, Jarada T, Yusuf A, et al. 51P - Testing patterns and outcomes of different EGFR positive metastatic non-small cell lung cancer (NSCLC) patients in a Canadian real-world setting. Ann Oncol. 2022;33(suppl_2):S27-S70.
13.Burnett H, Emich H, Carroll C, Stapleton N, Mahadevia P, Li T. Epidemiological and clinical burden of EGFR Exon 20 insertion in advanced non-small cell lung cancer: A systematic literature review. PLoS One. 2021;16(3):e0247620. PubMed
14.Riess JW, Gandara DR, Frampton GM, et al. Diverse EGFR exon 20 insertions and co-occurring molecular alterations identified by comprehensive genomic profiling of NSCLC. J Thorac Oncol. 2018;13(10):1560-1568. PubMed
15.Drug Reimbursement Review sponsor submission: RYBREVANT® (amivantamab) 50 mg/mL concentrate for solution for infusion [internal sponsor's package]. Toronto (ON): Janssen Inc.; 2022 Apr 21.
16.Rybrevant® (amivantamab for injection): 50 mg/mL concentrate for solution for infusion [product monograph]. Toronto (ON): Janssen Inc.; 2022 Mar 30.
17.Interim Clinical Study Report for Subjects With EGFR Exon 20 Insertion Mutations Treated With Amivantamab Monotherapy (30 March 2021 Cutoff): 61186372EDI1001. A Phase 1, First-in-Human, Open-Label, Dose Escalation Study of JNJ-61186372, a Human Bispecific EGFR and cMet Antibody, in Subjects with Advanced Non-Small Cell Lung Cancer [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2021 Oct 21.
18.Amivantamab for the treatment of adult patients with advanced non-small cell lung cancer with activating epidermal growth factor exon 20 insertion mutations, after failure of platinum-based therapy: adjusted treatment comparison versus standard of care from real-world evidence sources [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: RYBREVANT® (amivantamab) 50 mg/mL concentrate for solution for infusion. Toronto (ON): Janssen Inc.; 2022 Apr 21.
19.Detterbeck FC, Boffa DJ, Kim AW, Tanoue LT. The Eighth Edition Lung Cancer Stage Classification. Chest. 2017;151(1):193-203.
20.Ellis PM, Verma S, Sehdev S, Younus J, Leighl NB. Challenges to implementation of an epidermal growth factor receptor testing strategy for non-small-cell lung cancer in a publicly funded health care system. J Thorac Oncol. 2013;8(9):1136-1141. PubMed
21.Fang W, Huang Y, Hong S, et al. EGFR exon 20 insertion mutations and response to osimertinib in non-small-cell lung cancer. BMC Cancer. 2019;19(1):595. PubMed
22.Kate S, Chougule A, Joshi A, et al. Outcome of uncommon EGFR mutation positive newly diagnosed advanced non-small cell lung cancer patients: a single center retrospective analysis. Lung Cancer (Auckl). 2019;10:1-10. PubMed
23.Wu JY, Yu CJ, Shih JY. Effectiveness of treatments for advanced non-small-cell lung cancer with exon 20 insertion epidermal growth factor receptor mutations. Clin Lung Cancer. 2019;20(6):e620-e630. PubMed
24.Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015;16(7):830-838. PubMed
25.Kim TM, Ock CY, Kim M, et al. 1529P - Phase II study of osimertinib in NSCLC patients with EGFR exon 20 insertion mutation: A multicenter trial of the Korean Cancer Study Group (LU17-19). Ann Oncol. 2019;30:v628.
26.Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med. 2018;24(5):638-646. PubMed
27.Dersarkissian M, Bhak R, Lin H, et al. Real-World Treatment Patterns and Survival in Non-Small Cell Lung Cancer Patients with EGFR Exon 20 Insertion Mutations. J Thorac Oncol. 2019;14(10 Suppl):S681.
28.Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382(1):41-50. PubMed
29.Beau-Faller M, Prim N, Ruppert AM, et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: a multicentre observational study by the French ERMETIC-IFCT network. Ann Oncol. 2014;25(1):126-131. PubMed
30.Naidoo J, Sima CS, Rodriguez K, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer. 2015;121(18):3212-3220. PubMed
31.Yasuda H, Park E, Yun CH, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013;5(216):216ra177. PubMed
32.Lee CK, Davies L, Wu YL, et al. Gefitinib or erlotinib vs chemotherapy for EGFR mutation-positive lung cancer: individual patient data meta-analysis of overall survival. J Natl Cancer Inst. 2017;109(6). PubMed
33.Li Y, Appius A, Pattipaka T, Feyereislova A, Cassidy A, Ganti AK. Real-world management of patients with epidermal growth factor receptor (EGFR) mutation-positive non-small-cell lung cancer in the USA. PLoS One. 2019;14(1):e0209709. PubMed
34.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
35.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 May 6.
36.Interim Clinical Study Report: 61186372EDI1001. A Phase 1, First-in-Human, Open-Label, Dose Escalation Study of JNJ-61186372, a Human Bispecific EGFR and cMet Antibody, in Subjects with Advanced Non-Small Cell Lung Cancer [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2020 Oct 27.
37.Janssen Inc. response to July 12 DRR request for additional information regarding amivtantamab DRR review [internal additional sponsor's information]. Toronto (ON): Janssen Inc.; 2022.
38.Addendum to Interim Clinical Study Report dated 27 October 2020 Efficacy Analyses [08 October 2020 Cutoff]: 61186372EDI1001. A Phase 1, First-in-Human, Open-Label, Dose Escalation Study of JNJ-61186372, a Human Bispecific EGFR and cMet Antibody, in Subjects with Advanced Non-Small Cell Lung Cancer [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2020 Nov 24.
39.22 case studies where phase 2 and phase 3 trials had divergent results. Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2017: https://www.fda.gov/media/102332/download. Accessed 2022 Jul 28.
40.Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics: Guidance for Industry. Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2018: https://www.fda.gov/media/71195/download. Accessed 2022 Jul 28.
41.Center for Drug Evaluation and Research. Multi-disciplinary review and evaluation. Rybrevant (amivantamab for injection): 50 mg/mL concentrate for solution for infusion. Company: Janssen Biotech Inc. Application No.:761210. Approval date: 05/21/2021 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2020: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761210Orig1s000MultidisciplineR.pdf. Accessed 2022 Jul 28.
42.Health Canada reviewer's report: Rybrevant (amivantamab) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: amivantamab, 100 mg and 300 mg IV infusion. Toronto (ON): Janssen Inc.; 2022.
43.Jänne PA, Johnson BE. Effect of epidermal growth factor receptor tyrosine kinase domain mutations on the outcome of patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2006;12(14 Pt 2):4416s-4420s. PubMed
44.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175-180. PubMed
45.Faria R, Hernandez Alava M, Manca A, Wailoo A. NICE DSU technical support document 17: the use of observational data to inform estimates of treatment effectiveness in technology appraisal: methods for comparative individual patient data. Sheffield (UK): Decision Support Unit, ScHARR, University of Sheffield; 2015: https://www.sheffield.ac.uk/media/34204/download?attachment. Accessed 2022 Jul 28.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of Search: May 19, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type
Limits: Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(rybrevant* or amivantamab* or CNTO-4424 or CNTO4424 or JNJ-611* or JNJ611* or JNJ-6372 or JNJ6372 or 0JSR7Z0NB6).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*amivantamab/
(rybrevant* or amivantamab* or CNTO-4424 or CNTO4424 or JNJ-611* or JNJ611* or JNJ-6372 or JNJ6372).ti,ab,kf,dq.
3 or 4
5 not (conference abstract or conference review).pt.
6 use oemezd
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- rybrevant OR amivantamab OR “CNTO-4424” OR CNTO4424 OR “JNJ-611” OR JNJ611 OR “JNJ 61,186,372” OR JNJ61186372 OR “JNJ-6372” OR JNJ6372]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms -- rybrevant OR amivantamab OR “CNTO-4424” OR CNTO4424 OR “JNJ-611” OR JNJ611 OR “JNJ 61,186,372” OR JNJ61186372 OR “JNJ-6372” OR JNJ6372]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – rybrevant, amivantamab, CNTO-4424, CNTO4424, JNJ-611, JNJ611, JNJ 61,186,372, JNJ61186372, JNJ-6372, JNJ6372]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- rybrevant OR amivantamab OR “CNTO-4424” OR CNTO4424 OR “JNJ-611” OR JNJ611 OR “JNJ 61,186,372” OR JNJ61186372 OR “JNJ-6372” OR JNJ6372]
Grey Literature
Search Dates: May 6 to 19, 2022
Keywords: rybrevant, amivantamab, CNTO-4424, CNTO4424, JNJ-611, JNJ611, JNJ 61,186,372, JNJ61186372, JNJ-6372, JNJ6372, exon 20, ex20
Limits: None
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Park K, Haura EB, Leighl NB, et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non–Small Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS phase I Study. J Clin Oncol. 2021;39(30):3391 to 3402. | Duplicate study |
Park K, John T, Jong SWK, et al. Amivantamab (JNJ-61186372), an anti-EGFR-MET bispecific antibody, in patients with EGFR exon 20 insertion (exon20 insertion)-mutated non–small cell lung cancer (NSCLC). J Clin Oncol. 2020;38(suppl_15):9,512. | Duplicate study |
Leighl N, Shu C, Minchom A, et al. Amivantamab Monotherapy and in Combination with Lazertinib in Post-Osimertinib EGFR-mutant NSCLC: Analysis from the CHRYSALIS study. Ann Oncol. 2021;32(suppl_5):S949-S1039. | Duplicate study |
Sabari JK, Shu CA, Park K, et al. Amivantamab in Post-platinum EGFR exon 20 insertion Mutant Non–small Cell Lung Cancer. J Thorac Oncol. 2021;16(35):S108-S109. | Duplicate study |
Park K, Sabari JK, Haura EB, et al. 1247P Management of infusion-related reactions (IRRs) in patients receiving amivantamab. Ann Oncol. 2021;32(Supplement 5):S981-S982. | Duplicate study |
Minchom A, Viteri S, Bazhenova L, et al. Amivantamab compared with real-world therapies in patients with advanced non–small cell lung cancer harbouring EGFR exon 20 insertion mutations who progressed after platinum-based chemotherapy. Lung Cancer. 2022; doi: https://doi.org/10.1016/j.lungcan.2022.03.005 | Study design |
Minchom AR, Girard N, Bazhenova L, et al. Amivantamab compared with real-world therapies in patients with NSCLC with EGFR insertion mutations who have progressed after platinum-based chemotherapy. J Clin Oncol. 2021; 9(15_suppl): abstract 9,052. | Study design |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 34: Summary of CBR Based on RECIST v1.1 in Patients With Measurable Disease at Baseline — Investigator and BICR Assessment (March 30, 2021, DCO)
CBR | Cohort D Primary Efficacy Population (N = 81) | Cohort D Additional Efficacy Population (N = 124) | ||
|---|---|---|---|---|
Investigator Assessed | BICR | Investigator Assessed | BICR | |
CBRb (Confirmed CR + Confirmed PR + SD), n (%) | 59 (72.8) | 59 (72.8) | 93 (75.0) | 91 (73.4) |
95% CI | (61.8, 82.1) | (61.8, 82.1) | (66.4, 82.3) | (64.7, 80.9) |
CBR = clinical benefit rate; CR = complete response; PR = partial response; SD = stable disease
CBR is defined as the percentage of patients achieving confirmed complete or partial response, or durable SD (duration of at least 11 weeks).
Source: CHRYSALIS CSR Interim Analysis (March 30, 2021)17
Table 35: Summary of Efficacy Outcomes From the October 2020 DCO
Efficiency outcome | Cohort D Primary Efficacy Population (N = 81) | Cohort D Additional Efficacy Population (N = 114) | ||
|---|---|---|---|---|
Investigator Assessed | BICR | Investigator Assessed | BICR | |
ORR | ||||
ORR (Confirmed CR + Confirmed PR), n (%) | 29 (35.8) | 32 (39.5) | NR (35.1%) | NR (39.5%) |
95% CI | (25.4, 47.2) | (28.8, 51.0) | (26.4, 44.6) | (30.4, 49.1) |
BOR | ||||
CR | 0 (0.0) | 3 (3.7) | NR | NR |
PR | 29 (35.8) | 29 (35.8) | NR | NR |
SD | 39 (48.1) | 39 (48.1) | NR | NR |
PD | 12 (14.8) | 8 (9.9) | NR | NR |
Not Evaluable/Unknown | 1 (1.2) | 2 (2.5) | NR | NR |
Duration of Response | ||||
Median (95% CI) | 11.20 (6.34, 13.14) | 11.14 (6.90, NE) | 11.20 (6.34, 13.14) | 10.84 (5.55, NE) |
PFS | ||||
Median (95% CI) | 8.25 (5.49, 10.61) | 8.28 (6.51, 10.87) | 7.16 (5.55, 8.84) | 6.87 (5.49, 9.66) |
OS | ||||
Median (95% CI) | 22.77 (14.59, NE) | 22.77 (14.59, NE) | ||
BOR = best overall response; BICR = blinded independent central review; CR = complete response; CI = confidence interval; NE = not estimable; NR = not reported; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; SD = stable disease.
Source: CHRYSALIS CSR Interim Analysis October 202038
Figure 5: Kaplan-Meier Plot of PFS (Primary Efficacy Population; March 30, 2021, DCO)
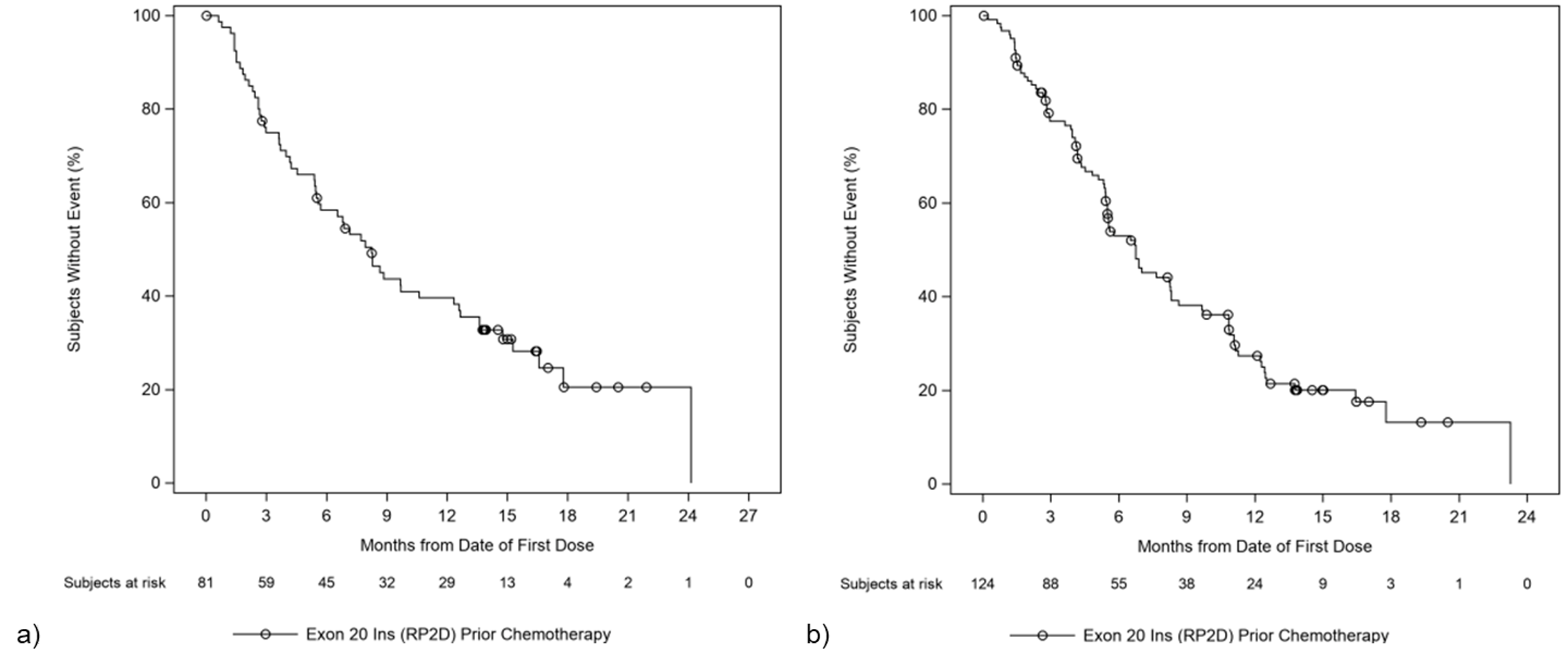
PFS = progression-free survival; RP2D = recommended phase II dose.
a) Kaplan-Meier plot of PFS per investigator assessment; b) Kaplan-Meier plot of PFS per BICR.
Source: CHRYSALIS CSR Interim Analysis (March 30, 2021)17
Figure 6: Kaplan-Meier Plot of OS per Investigator Assessment (Primary Efficacy Population; March 30, 2021, DCO)
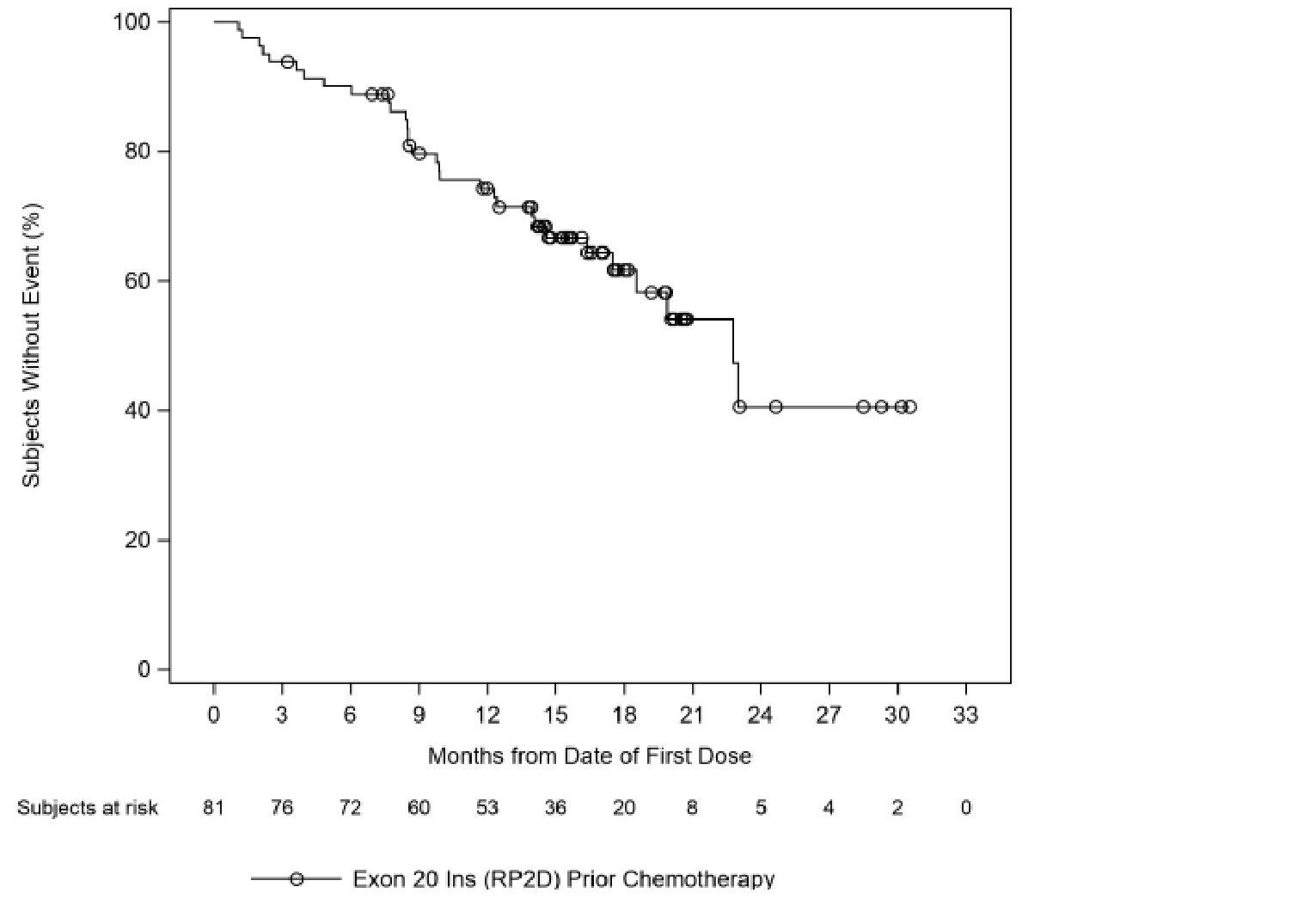
OS = overall survival; RP2D = recommended phase II dose.
Source: CHRYSALIS CSR Interim Analysis (March 30, 2021)17
Table 36: Duration of Amivantamab Infusions (March 30, 2021, DCO)
Duration of Infusions (Hours) | Safety Analysis Set (N = 153) |
|---|---|
First Infusion | |
N | 153 |
Mean (SD) | 5.34 (1.492) |
Median (Range) | 4.98 (0.7, 9.7) |
Second Infusion | |
N | 151 |
Mean (SD) | 4.77 (0.795) |
Median (Range) | 4.55 (2.3, 7.1) |
All Subsequent Infusions | |
N | 2,573 |
Mean (SD) | 2.38 (0.601) |
Median (Range) | 2.28 (0.7, 24.0) |
Source: Sponsor Submission15
Table 37: Postprogression and Subsequent Treatments (March 30, 2021, DCO)
Treatment | Cohort D | ||
|---|---|---|---|
Safety Analysis Set (N = 153) | Primary Efficacy Population (N = 81) | Additional Efficacy Population (N = 124) | |
Duration of postprogression treatment (months)a | |||
N | NR | 21 | 38 |
Mean (SD) | NR | 2.738 (3.3078) | 2.688 (2.7388) |
< 2 months | NR | 12 (57.1) | 19 (50.0) |
2 to < 4 months | NR | 4 (19.0) | 9 (23.7) |
4 to < 6 months | NR | 2 (9.5) | 7 (18.4) |
≥ 6 months | NR | 3 (14.3) | 3 (7.9) |
Subsequent Treatment | |||
Patients with Subsequent Treatment | 66 (43.1) | NR | NR |
Patients with subsequent anticancer therapy | 48 (31.4) | NR | NR |
Antineoplastic Agents | 40 (26.1) | NR | NR |
Platinum Compounds | 15 (9.8) | NR | NR |
Monoclonal Antibodies | 22 (14.4) | NR | NR |
Folic Acid Analogues | 4 (2.6) | NR | NR |
Taxanes | 28 (18.3) | NR | NR |
Protein Kinase Inhibitors | 13 (8.5) | NR | NR |
Pyrimidine Analogues | 11 (7.2) | NR | NR |
Vinca Alkaloids and Analogues | 2 (1.3) | NR | NR |
Other Antineoplastic Agents | 2 (1.3) | NR | NR |
Podophyllotoxin Derivatives | 1 (0.7) | NR | NR |
Patients with subsequent surgery/procedure | 9 (5.9) | NR | NR |
Patients with subsequent radiotherapy | 34 (22.2) | NR | NR |
aPostprogression treatment duration is defined as the duration from the earliest date of progression to the date of last dose of study drug plus1 divided by 30.4375.
Source: Sponsor Submission.15
Figure 7: Mean Baseline and Change From Baseline in NSCLC-SAQ Total Score in the PRO Population (N = 36)
NSCLC-SAQ = Non-Small Lung Cancer Symptom Assessment Questionnaire.
Source: Sponsor Submission.15
Figure 8: EQ-5D VAS: Patient Perceived Health in the PRO Population (N = 36)
EQ-5D-5L = 5-level EQ-5D.
Source: Sponsor Submission.15
Figure 9: Frequency of Response to PGIS in the PRO Population (N = 36)
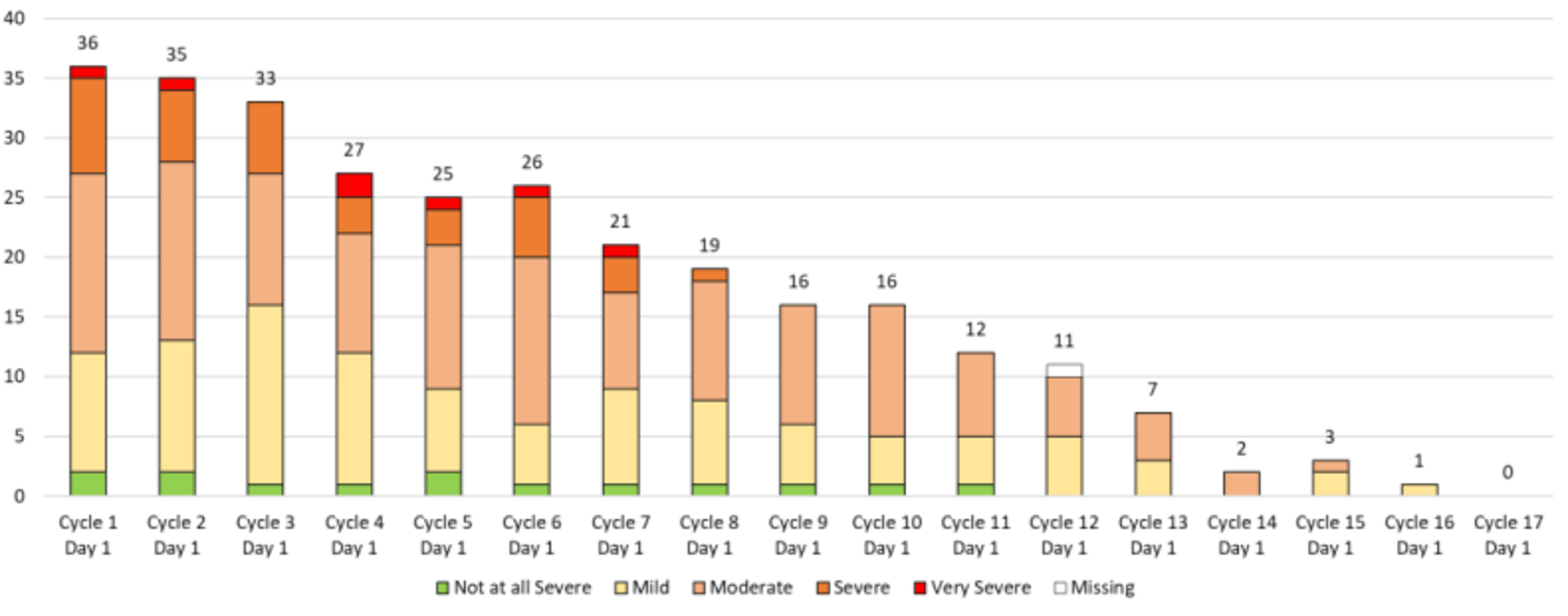
PGIS = Patient Global Impression of Severity.
Source: Sponsor Submission.15
Figure 10: PGIC Scores Over Time in the PRO Population (N = 36)
PGIC = Patient Global Impression of Change.
Source: Sponsor Submission.15
Pharmacoeconomic Review
Abbreviations
AE
adverse event
EGFR
epidermal growth factor receptor
ICER
incremental cost-effectiveness ratio
IO
immunotherapy
IRR
infusion-related reaction
KM
Kaplan-Meier
LY
life-year
NPBC
non–platinum-based chemotherapy
NSCLC
non–small cell lung cancer
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
RWE
real-world evidence
TKI
tyrosine kinase inhibitor
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Amivantamab (Rybrevant), liquid concentrate for IV infusion, 50 mg/mL |
Submitted price | Amivantamab, 350 mg/7 mL vial: $1,676.00 |
Indication | For the treatment of adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations whose disease has progressed on, or after, platinum-based chemotherapy |
Health Canada approval status | NOC/c |
Health Canada review pathway | Project Orbis, advance consideration under NOC/c |
NOC date | March 30, 2022 |
Reimbursement request | As per indication |
Sponsor | Janssen Inc. |
Submission history | Previously reviewed: No |
EGFR = epidermal growth factor receptor; NOC/c = Notice of Compliance with Conditions; NSCLC = non–small cell lung cancer.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target population | Adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations whose disease has progressed on or after platinum-based chemotherapy |
Treatment | Amivantamab |
Comparators | 3 categories of therapy:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (10 years) |
Key data source |
|
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
EGFR = epidermal growth factor receptor; ICER = incremental cost-effectiveness ratio; NOC/c = Notice of Compliance with conditions; NPBC = non–platinum-based chemotherapy; NSCLC = non–small cell lung cancer; IO = immuno-oncology; LY = life-year; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; RWE = real-world evidence; TKI = tyrosine kinase inhibitor; vs. = versus.
Conclusions
The CADTH Clinical Review highlighted the high degree of uncertainty associated with the results of CHRYSALIS. The single-arm, open-label, nonrandomized, phase I/Ib design of the CHRYSALIS trial makes interpreting the efficacy and safety events attributable to amivantamab challenging, because all patients received the same treatment. While objective response rate (ORR) may be directly attributable to the drug’s antitumour activity, interpreting progression-free survival (PFS) and overall survival (OS) events is significantly limited. The extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear. The CADTH assessment of the sponsor-submitted adjusted treatment comparison identified several key limitations including small sample sizes, heterogeneity across study designs and pooled populations, and the inability to adjust for all potential confounders and prognostic variables, all of which significantly limited the ability to interpret the relative treatments effects observed between amivantamab and other treatments. Overall, it was noted that the phase I nature of the CHRYSALIS trial limits the ability to make firm conclusions on comparative efficacy given the short duration of follow-up, which results in immature data, as well as the small sample size of the CHRYSALIS trial and comparator cohorts.
Due to the high degree of unresolved uncertainty with the clinical data, CADTH was unable to derive an economic base case.
CADTH performed an exploratory analysis in which a Weibull parametric extrapolation was used for the OS data for amivantamab. Based on CADTH sequential reanalysis, amivantamab is associated with an incremental cost-effectiveness ratio (ICER) of $253,131 per quality-adjusted life-year (QALY) compared to non–platinum-based chemotherapy (NPBC), with a 0% probability of being cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY. This estimate is based on the sponsor’s clinical assumptions, which predict an additional 0.79 life-years (LYs) and 0.60 QALYs for those receiving amivantamab compared with NPBC. A price reduction of at least 77% would be required for amivantamab to be considered cost-effective compared to NPBC at this threshold.
If the comparative clinical data — particularly the PFS and OS data — are considered sufficiently robust, then the exploratory analysis performed by CADTH may be informative. However, in light of the available clinical information, CADTH’s results are based on assumptions (e.g., survival extrapolations, which result in OS benefits for amivantamab both preprogression and postprogression) that cannot be validated at this time and hence are associated with a large degree of uncertainty. As such, additional price reductions may be required to ensure the cost-effectiveness of amivantamab.
Due to the limitations with the clinical evidence and substantial uncertainty associated with the comparative clinical effects of amivantamab with relevant comparators, CADTH performed a scenario cost comparison analysis in which only drug acquisition costs were included. Results of this analysis suggest that amivantamab would require a price reduction of 86.4% to achieve cost parity with the least costly comparator, docetaxel.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was received from 2 groups, the Lung Health Foundation and Lung Cancer Canada. Lung Cancer Canada conducted 4 telephone interviews with patients living in Ontario, Canada, who were part of the amivantamab clinical trial. All 4 of the interviewed patients were epidermal growth factor receptor (EGFR) positive and 2 had EGFR exon 20 insertion mutations. The Lung Health Foundation conducted 3 phone interviews with Canadian lung cancer patients and received 2 responses to an online survey (demographic data not collected). Patients interviewed by the Lung Health Foundation reported previous treatment with surgery, radiation, chemotherapy, targeted therapy, and immunotherapy. The input indicated that most patients struggled with lingering side effects, including fatigue, nausea, weight loss, and hair loss, and that side effects from chemotherapy were specifically noted to affect patients’ quality of life, ability to work, and daily activities. The patient input noted a specific unmet need for targeted therapy to the exon 20 insertion mutation, which current chemotherapy does not address. The patients interviewed by Lung Cancer Canada were enrolled in the amivantamab clinical trial and had experience with the drug under review. Some patients experienced stability in tumours and metastases on amivantamab, while others experienced shrinkage. The most common adverse events (AEs) reported by patients were skin-related, including inflammation of the nail bed, rashes, acne, and dry and sensitive skin. All patients noted that side effects are manageable and that the potential benefits outweigh the negatives.
Clinician input was received from 2 groups, the Ontario Health – Cancer Care Ontario Lung Cancer Drug Advisory Committee and the Lung Cancer Canada Medical Advisory Committee. Clinicians indicated that current standard of care for patients in first-line therapy is platinum-based doublet chemotherapy, followed by docetaxel in second-line therapy, though response rates are poor. EGFR tyrosine kinase inhibitors (TKIs) may also be used for patients with EGFR mutations but patients with EGFR exon 20 insertion mutations are resistant to EGFR TKIs and have a low response rate to immunotherapy. Feedback from Ontario Health suggested that amivantamab would be used after all standard therapies acceptable to the patient have failed, suggested to be platinum-based doublet chemotherapy with maintenance pemetrexed and, potentially, docetaxel as well. The input from Lung Cancer Canada highlighted that targeted therapies against driver mutations should ideally be offered in a first-line setting, and that clinical trials in this setting are ongoing.
Drug plan input received for this review noted that additional resource use would be required with amivantamab due to its split first dose and escalating infusion-rate schedule. Additional labour will be required for drug preparation, and premedications are required to prevent infusion-related reactions (IRRs). The plans noted the presence of confidential prices for nivolumab, pembrolizumab, and atezolizumab.
The following concern was addressed in the sponsor’s model:
the sponsor-included AEs, including IRRs.
CADTH was unable to address the following concerns raised from the stakeholder input:
CADTH was unable to incorporate the presence of confidential, negotiated prices for immuno-oncology (IO) drugs.
The cost of preinfusion medications was not considered by the sponsor.
The model only considers the cost-effectiveness of amivantamab in a platinum-treated population.
Economic Review
The current review is for amivantamab (Rybrevant) for adult patients with locally advanced or metastatic non–small cell lung cancer (NSCLC) with activating EGFR exon 20 insertion mutations whose disease has progressed on, or after, platinum-based chemotherapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of amivantamab compared to IO drugs, TKIs, and NPBC. The modelled population is consistent with the Health Canada indication and the reimbursement request, and these populations are aligned with the sponsor’s CHRYSALIS trial population.1-3
Amivantamab is supplied in single-use vials containing 350 mg amivantamab in 7 mL solution (50 mg/mL).2 The recommended dose of amivantamab is 1,050 mg (3 vials) for patients weighing less than 80 kg and 1,400 mg (4 vials) for patients weighing greater than or equal to 80 kg.2 The recommended dosing schedule for amivantamab is once weekly for the first 4 weeks (first dose split on days 1 and 2) and every 2 weeks starting at week 5.2 Amivantamab should be reconstituted and administered by a health care professional with appropriate medical support to manage IRRs if they occur. Premedications consisting of antihistamines, antipyretics, and glucocorticoids should be administered to reduce the risk of IRRs. Amivantamab should be administered until disease progression or unacceptable toxicity.2 The cost for amivantamab is $1,676.00 per vial, leading to costs per dose of $5,028 and $6,704 for patients weighing less than 80 kg or greater than or equal to 80 kg, respectively.3 The cost in the first cycle is $20,112 for patients weighing less than 80 kg and $26,816 for patients weighing greater than or equal to 80 kg; the cost in subsequent cycles is $10,056 and $13,408, respectively.3
The sponsor selected 3 therapy classes as comparators: IO drugs consisting of atezolizumab (33.3%), nivolumab (33.3%), and pembrolizumab (33.3%); EGFR TKIs consisting of gefitinib (50%), afatinib (25%), and osimertinib (25%); and NPBC consisting of docetaxel (100%). Costs and effects for these comparators were weighted by these proportions, which were derived based on Canadian market share estimates and clinician input.3 The costs per cycle calculated by the sponsor were $10,052 for IO drugs, $3,471 for TKIs, and $2,775 for NPBC.3
The clinical outcomes of interest were QALYs and LYs over a lifetime horizon (10 years). Discounting (1.5% per annum) was applied to both costs and outcomes and a cycle length of 4 weeks was used along with a half-cycle correction. The base-case perspective was that of the Canadian publicly funded health care payer.
Model Structure
The sponsor submitted a partitioned survival model (PSM) consisting of 3 mutually exclusive health states: preprogression, postprogression, and death. All patients entered the model in the preprogression state and received amivantamab or a comparator. The allocation of patients into health states is based on treatment-specific PFS and OS functions. The proportion of patients in the preprogression health state followed the PFS curve for amivantamab from the CHRYSALIS trial, and the PFS curve generated from Kaplan-Meier (KM) data from a sponsor-commissioned real-world evidence (RWE) study for comparators.3 In the CHRYSALIS trial, PFS was derived based on investigator assessment and independent review committee assessment; in the RWE study, PFS was defined as human abstraction of physician evaluation of tumour progression.3 The proportion of patients in the postprogression health state was equal to the difference between the OS and the PFS curves. Patients in the postprogression state can receive subsequent-line therapy. Patients transitioning into the death state remained there until the end of the model time horizon. A figure of the sponsor’s model structure is available in Figure 1, Appendix 3.
Model Inputs
The target population of the economic evaluation was based on cohort D of the phase I/Ib single-arm CHRYSALIS trial, which included NSCLC patients with activating EGFR exon 20 insertion mutations whose disease had progressed on or after platinum-based chemotherapy. The mean age of the population was 62.3 years, mean weight was 67.5 kg, and 41% were male.1 The key clinical inputs (i.e., PFS, OS, and treatment discontinuation) for amivantamab were obtained from the primary efficacy population (n = 81), while safety data were informed from the expanded safety population (n = 153) of the trial. Data were based on the March 30, 2021, data cut-off date.4
In the absence of direct comparative evidence, comparative effectiveness data were derived from a sponsor-commissioned adjusted treatment comparison of CHRYSALIS versus an RWE cohort (n = 349). The sponsor-commissioned RWE study cohort was created using various RWE sources from the US (Flatiron Health Spotlight, ConcertAI, COTA) and Europe (Public Health England, 2 from Germany, and 1 from France). The adjusted treatment comparison used inverse probability of treatment weights derived from a propensity score model to account for variables including, but not limited to, age, sex, race, smoking history, Eastern Cooperative Oncology Group (ECOG) performance status, and time from initial diagnosis to advanced diagnosis.3 It was assumed that unobserved confounders did not have an impact on the comparator treatment-effect estimate. The covariates used to estimate the weights in the analysis of the European plus US cohort included age, gender, presence of brain metastases, and number of previous lines of therapy in the metastatic disease setting.3 For PFS, OS, and time to next treatment, time-to-event analyses were performed using weighted Cox proportional hazard models. KM curves were generated and used to estimate median time-to-event estimates for each treatment group.5
Extrapolations of the PFS, OS, and time to treatment discontinuation data were performed for amivantamab and comparators. For amivantamab, the PFS, OS, and time to treatment discontinuation data were fit with a lognormal, gamma, and exponential parametric fit, respectively. Full details of the parametric extrapolations are available in Table 10, Appendix 3.3
Regarding the safety of amivantamab, the model included grade 3 or 4 AEs occurring in 5% or more of patients in cohort D of CHRYSALIS, as well as grade 1 to 2 IRRs.4 Product monographs for individual treatments were used to inform safety data, with AE incidence rates for selected index drugs used to represent the corresponding treatment class.3
Utility values were derived from Labbé et al., who performed a longitudinal cohort study of metastatic lung cancer; the values obtained were from the EGFR-mutated NSCLC population.6 The publication reported a utility value of 0.81 for progression-free disease and 0.70 for postprogression disease, which were employed in the sponsor’s model. Disutilities due to AEs were derived from various sources and were also applied in the model as a one-time disutility decrement.3
The economic model included costs related to drugs (acquisition, administration), AEs, monitoring and disease management, subsequent treatment, and terminal care. Dosing for amivantamab was as previously described, with weighted acquisition costs for the initial and subsequent cycles of $21,850 and $10,925, respectively, based on data from CHRYSALIS, which found that 74.1% of patients weighed less than 80 kg at baseline. Drug acquisition costs for comparators are as previously described. An administration cost of $201 per hour of chair time was assumed based on a published study;7 amivantamab administrations were assumed to require 2 to 6 hours of chair time, while comparators required 1 hour. Monitoring and disease management costs consisted of costs for outpatient visits, chest radiography, CT scans, nurse visits, and routine blood work, which were obtained from the Ontario Schedule of Benefits for Physician and Laboratory Services.8,9 Resource utilization frequency was assumed to be the same across all treatments, but differed for patients in the preprogression and postprogression health states. Costs for most AEs were derived from the Ontario Case Costing Initiative, while the cost of IRRs and febrile neutropenia came from published sources.7,10,11 Half of modelled patients were assumed to receive subsequent therapy upon progression, which consisted of IO drugs, TKIs, and NPBC, though patients were assumed not to receive the same therapy twice. Duration of subsequent therapy ranged from 2.8 months for NPBC to 4.2 months for IO drugs and amivantamab.12 Finally, a one-off terminal care cost of $53,008 was applied based on a published study.13
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in the following sections.
Base-Case Results
In the sponsor’s base case, amivantamab was associated with an estimated cost of $233,445 and 1.81 QALYs over a lifetime horizon. In sequential analysis, amivantamab was associated with an ICER of $210,591 compared to NPBC (incremental cost: $142,316, incremental QALYs: 0.68). IO drugs were dominated by NPBC in the sponsor’s base case, resulting in fewer QALYs and higher cost. In the sponsor’s sequential analysis, amivantamab had a 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY. Results of the base case suggest that 0.34 incremental QALYs were accrued after the maximum follow-up in CHRYSALIS of 30 months — that is, approximately 51% of the incremental benefit was obtained after the trial period. Less than 1% of patients on amivantamab remained alive after 10 years. Additional results from the sponsor’s submitted economic evaluation base case are available in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
EGFR TKIs | 84,532 | 1.15 | 0.85 | Reference |
NPBC | 91,129 | 1.52 | 1.13 | 23,594 vs. EGFR TKIs |
Amivantamab | 233,445 | 2.42 | 1.81 | 210,591 vs. NPBC |
EGFR = epidermal growth factor receptor; ICER = incremental cost-effectiveness ratio; LY = life-year; NPBC = non–platinum-based chemotherapy; QALY = quality-adjusted life-year; TKI = tyrosine kinase inhibitor; vs. = versus.
Note: The submitted analyses are based on the publicly available prices of comparators and may not reflect confidential, negotiated prices. Only treatments on the cost-effectiveness frontier are reported in this table.
Source: Sponsor’s pharmacoeconomic submission.3
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses involving an expanded efficacy population from CHRYSALIS (n = 114), definition of progression (based on independent review committee or investigator assessed), amivantamab PFS and OS extrapolations, treatment discontinuation, 5-year time horizon, and vial sharing. Results were generally similar among the scenarios tested, though sequential analysis was not performed on the scenarios. The ICER for amivantamab versus NPBC was most influenced by the choice of OS extrapolation for amivantamab, with ICERs ranging from $144,147 to $237,700 per QALY depending on the extrapolation chosen. The analysis with a 5-year time horizon resulted in an ICER of $238,659.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The clinical data for amivantamab is based on a single-arm study with evidence for clinical response. The clinical efficacy of amivantamab was assessed in a single-arm, open-label, nonrandomized, phase I/Ib trial. As noted in the CADTH Clinical Review, the design of the trial makes interpreting the efficacy and safety events attributable to amivantamab challenging, because all patients received the same treatment. The lack of a control arm increases the risk of bias in the estimation of treatment effect due to the potential for confounding related to fluctuations in health status and other unidentified prognostic factors that could affect subjectively assessed outcomes. Patient selection and lack of randomization may have also introduced bias. Although the sponsor attempted to minimize the risk of bias by using an independent review committee assessment for key study outcomes, the open-label, single-arm design can increase the risk of bias in reporting of outcomes that are subjective in measurement and in interpretation, such as response and AEs. Additionally, the primary and secondary outcomes for cohort D were not controlled for multiple testing, and must be considered with respect to type I error and should be viewed as supportive evidence for the overall effect of amivantamab. While ORR may be directly attributable to the drug’s antitumour activity, interpreting other efficacy outcomes that rely on tumour biology, disease prognosis, and patients’ performance status — including PFS, OS, and duration of response — is significantly limited. The extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear. Finally, the median duration of follow-up was 14.5 months for the primary efficacy population, which was considered appropriate for the primary end point of ORR but was immature for survival outcomes such as OS. As such, the true benefit of amivantamab on survival outcomes is uncertain.
CADTH was not able to address the limitations associated with the submitted clinical data. The clinical uncertainty directly affects the confidence that can be drawn from the results of the economic model.
The comparative clinical efficacy of amivantamab with relevant comparators is highly uncertain. Due to the lack of head-to-head evidence comparing amivantamab to relevant comparators in a randomized controlled trial, the sponsor submitted an adjusted treatment comparison that derived comparator information from an RWE cohort to inform the pharmacoeconomic model (PFS, OS, time to next treatment). In general, comparing trial evidence to RWE will result in greater uncertainty given that clinical trials and RWE measure different components; clinical trials measure treatment effects under controlled circumstances, while RWE aligns better with how treatments are used and assessed in clinical practice. As outlined in the CADTH Clinical Review, there were numerous limitations with the adjusted treatment comparison, which add considerable uncertainty to the analysis. The RWE data were analyzed retrospectively from electronic medical records and databases; these sources are more prone to biases that cannot be fully controlled for (e.g., selection bias, confounding, limited data availability) compared with those collected from prospective, interventional studies that cannot be fully controlled for. Inclusion and exclusion criteria from CHRYSALIS were applied to real-world data sources to select the appropriate population; however, any criteria that could not be applied to patients due to missing data were omitted from the list of criteria applied to that data source, which may have resulted in unaccounted-for differences in patient populations. No imputation method was applied to account for missing data, and if a substantial amount of data was missing, covariates were not included, which may affect the results of the comparisons. The sponsor performed propensity score matching without accounting for some important factors including smoking status, ECOG, and race; therefore, the comparisons were not balanced for confounders and were not mutually randomizable populations. It was assumed that unobserved confounders did not have an impact on the comparator treatment effect estimate, which is highly uncertain.
Furthermore, the model results for NPBC do not align with published literature, further increasing the uncertainty with the sponsor’s analysis. The sponsor’s model predicts a median survival of 13 months for patients treated with NPBC. This is substantially higher than historical data for previously treated platinum chemotherapy patients with NSCLC who then receive docetaxel (median survival 7.0 months).14 The clinical experts consulted by CADTH acknowledged that advances in technology over the past 2 decades could be expected to improve OS in these patients, and suggested that a median survival of between 8 and 10 months would be reasonable for patients receiving docetaxel. This estimate is lower than the sponsor’s model predictions.
CADTH was unable to address the limitations associated with the sponsor’s adjusted treatment comparison and notes that considerable uncertainty remains in the analysis that could not be resolved. CADTH tested alternate OS extrapolations for NPBC and noted that results varied little, with none resulting in median survival estimates that aligned with clinical expert opinion.
The OS extrapolation for amivantamab is uncertain. The sponsor extrapolated the OS data from CHRYSALIS for amivantamab from a median follow-up of 14.5 months out to the 10-year model time horizon.1 As stated in the CADTH Clinical Review, these survival results were immature. The sponsor’s model predicted a median survival of 23 months for amivantamab, which, while aligned with the KM data from CHRYSALIS, is substantially higher than survival results for the comparators, even despite the overestimates for comparators described above. As shown in Figure 2, Appendix 3, patients in CHRYSALIS experienced a median PFS of approximately 8 months — that is, amivantamab is predicted to delay disease progression by 8 months. If that result is combined with clinical experts’ maximum predicted survival for this population (10 months), the theoretical maximum survival for patients on amivantamab is 18 months, which is lower than the 23 months predicted by the sponsor’s model. Ultimately, given the aforementioned limitations regarding the single-arm trial design and adjusted treatment comparison, OS results for amivantamab are substantially higher than that expected from current therapy with no robust comparative data to support these results.
As part of an exploratory analysis, CADTH chose a Weibull extrapolation for the OS of amivantamab, which was deemed reasonable by clinical experts, to explore the impact of an alternate survival assumption.
The sponsor’s model structure is not appropriate. Health Canada gave amivantamab, for patients with NSCLC with EGFR exon 20 insertion mutations, a Notice of Compliance with conditions in March 2022, pending the final study results of CHRYSALIS.15 Furthermore, the product monograph for amivantamab stated that the “clinical effectiveness of Rybrevant is based on ORR and duration of response from a single-arm trial in patients with activating EGFR exon 20 insertion mutations.”2 Given that PFS and OS for amivantamab have not been established in this patient population and that there is no robust evidence to confirm that response measures are a prognostic marker of PFS or OS, the sponsor’s PSM (which incorporated PFS and OS data based on the progression-free and postprogression health states) was not appropriate or supported by the available evidence. A model based on response rates may have been more appropriate based on the available data, though the output of such a model would still be constrained by the quality of the data used to inform it, which may have similar limitations to those described in the appraisal of the clinical evidence.
CADTH could not address this limitation due to the submitted model structure.
Safety outcomes were based on a naive comparison. The sponsor included grade 3 or 4 AEs occurring in 5% of more of patients in cohort D of CHRYSALIS along with grade 1 to 2 IRRs.4 However, because safety was not captured in the RWE study, product monographs for individual treatments were used to inform safety data, which were compared directly to AE rates in CHRYSALIS. This represents an uncontrolled, naive comparison of safety, because rates of AEs were not included in the adjusted treatment comparison. It is uncertain whether the metric used to define AEs (e.g., treatment-emergent or treatment-related) would be similar in all product monographs or whether additional experience gained with these treatments since publication of the product monograph might result in some mitigation of AE rates observed at the time of product approval.
As part of a scenario analysis, CADTH set rates of AEs for all comparators equal to amivantamab.
One additional limitation was identified but was not considered to be a key limitation. The sponsor did not include the cost of preinfusion medications in its analysis despite the stipulation in the product monograph that these medications must be administered between 15 and 60 minutes before amivantamab.2 These medications, which consist of diphenhydramine, acetaminophen/paracetamol, and dexamethasone/methylprednisolone, would represent additional drug acquisition costs that are only incurred for patients on amivantamab; however, the cost of these drugs is minimal and unlikely to affect the base case.16
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
PFS in CHRYSALIS was defined by a blinded independent review committee. | Clinical experts consulted by CADTH considered this reasonable. |
Unobserved confounders in the adjusted treatment comparison did not have an impact on the comparator treatment-effect estimate. | Not appropriate and highly uncertain. The sponsor used a propensity score model to account for age, sex, race, smoking history, ECOG performance status, and time from initial to advanced diagnosis. Unobserved data were not adjusted for and missing data were not imputed, increasing uncertainty in the analysis. |
Time-to-next-treatment data for comparators were used to represent time to discontinuation. | Uncertain whether these parameters are equivalent. |
Amivantamab was associated with significant infusion time and overhead costs. | Appropriate, based on the product monograph and drug plan input. Amivantamab infusions were associated with 2 to 6 hours of chair time, depending on the dose received, compared to a maximum of 1 hour for comparators. |
ECOG = Eastern Cooperative Oncology Group; PFS = progression-free survival.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Due to the myriad of limitations outlined previously involving both the direct and indirect clinical evidence, CADTH was unable to derive a base case.
CADTH undertook a correction to the sponsor’s model to address a dosing error, and an exploratory analysis involving 1 change to the sponsor’s OS assumptions for amivantamab (refer to Table 5).
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
1.Dosing of docetaxel | To achieve a dose of 180 mg docetaxel: 9 vials of 20 mg each = $2,081 | To achieve a dose of 180 mg docetaxel: 1 vial each of 160 mg and 20 mg = $1,239 |
Changes to derive the CADTH exploratory analysis | ||
1.Amivantamab OS parametric extrapolation | Gamma | Weibull |
CADTH exploratory analysis | Reanalysis 1 | |
OS = overall survival.
The results of the CADTH exploratory analysis suggested that amivantamab is associated with higher costs and QALYs than NPBC, the next comparator on the frontier (Table 6). The ICER for amivantamab compared to NPBC was $253,131, indicating that amivantamab is not cost-effective at a $50,000 WTP threshold, and had a 0% probability of being cost-effective at this threshold. Results are driven by the high drug acquisition and administration cost of amivantamab. Full results are available in Appendix 4.
Table 6: Summary of the CADTH Exploratory Analysis
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor’s base case (corrected) | |||
NPBC | 81,409 | 1.13 | Reference |
Amivantamab | 232,972 | 1.81 | 224,274 |
CADTH exploratory analysis | |||
NPBC | 81,409 | 1.13 | Reference |
Amivantamab | 233,131 | 1.73 | 253,131 |
ICER = incremental cost-effectiveness ratio; NPBC = non–platinum-based chemotherapy; QALY = quality-adjusted life-year.
Note: Immuno-oncology drugs and epidermal growth factor receptor tyrosine kinase inhibitors were dominated in the sponsor’s results and CADTH exploratory analysis and, as such, do not appear on the efficiency frontier. Full results are available in Appendix 4.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s results and the CADTH exploratory analysis. The CADTH exploratory analysis suggested that a price reduction of 77% would be required to achieve cost-effectiveness of amivantamab relative to NPBC at a $50,000 per QALY threshold (Table 7).
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for amivantamab vs. NPBC ($/QALY) | |
|---|---|---|
Price reduction | Sponsor’s base case (corrected) | CADTH exploratory analysis |
No price reduction | 224,274 | 253,131 |
10% | 200,770 | 226,627 |
20% | 177,269 | 200,129 |
30% | 153,767 | 173,632 |
40% | 130,265 | 147,135 |
50% | 106,764 | 120,638 |
60% | 83,262 | 94,141 |
70% | 59,760 | 67,643 |
75% | 48,009 | 54,395 |
77% | 43,309 | 49,095 |
ICER = incremental cost-effectiveness ratio; NPBC = non–platinum-based chemotherapy; QALY = quality-adjusted life-year; vs. = versus.
CADTH undertook 1 scenario analysis involving setting adverse event rates equal to amivantamab for all comparators. This scenario, detailed in Table 15, Appendix 4, resulted in similar results as the CADTH exploratory analysis. In addition, in light of the uncertainty about the magnitude of clinical benefit, CADTH performed a cost comparison analysis in which drug acquisition costs only were considered. Drug costs from the CADTH cost table were multiplied by the median PFS from the CHRYSALIS trial, 8.3 months, and compared to the least costly option within a given drug class.1 The sponsor’s base case assumptions about the proportion of patients weighing less than 80 kg were not modified. Results of this analysis are available in Table 15, Appendix 4. This analysis suggests that a price reduction of 86.4% would be required to achieve cost parity with docetaxel, the least costly comparator in this disease space.
Issues for Consideration
CHRYSALIS was a phase I/Ib study that included a dose-escalation component in which amivantamab was trialled at doses ranging from 350 mg to 1,750 mg.1 The experts noted that, based on the findings of the trial, once patients received 700 mg or greater, the efficacy and safety results tended to plateau, suggesting that 700 mg may be sufficient for disease management. This has implications for the cost of amivantamab, which is priced per 350 mg vial — that is, the cost may be lower if patients receive a lower dose than recommended.
Drug plan input highlighted the additional resource use required with amivantamab due to its split first dose and escalating infusion-rate schedule. Additional labour will also be required for drug preparation, and premedications are required to prevent IRRs. This may put a strain on resources (e.g., staff, pharmacists, overhead) in some settings.
Two other targeted therapies — poziotinib and mobecertinib — have been recently submitted to the FDA for adult patients with NSCLC with exon 20 insertion mutations.17,18 The cost-effectiveness of amivantamab versus these other drugs is unknown.
Overall Conclusions
The CADTH Clinical Review highlighted the high degree of uncertainty associated with the results of CHRYSALIS. The single-arm, open-label, nonrandomized, phase I/Ib design of the CHRYSALIS trial makes interpreting the efficacy and safety events attributable to amivantamab challenging, because all patients received the same treatment. While ORR may be directly attributable to the drug’s antitumour activity, the ability to interpret PFS and OS events is significantly limited. The extent to which the observed survival is due to the natural history of the tumour or the intervention remains unclear. The CADTH assessment of the sponsor-submitted adjusted treatment comparison identified several key limitations, including small sample sizes, heterogeneity across study designs and pooled populations, and the inability to adjust for all potential confounders and prognostic variables, all of which significantly limited the ability to interpret the relative treatments effects observed between amivantamab and other treatments. Overall, it was noted that the phase I nature of the CHRYSALIS trial limits the ability to make firm conclusions about comparative efficacy given the short duration of follow-up, which resulted in immature data, and the small sample size of the CHRYSALIS trial and comparator cohorts.
Therefore, there exists a high degree of unresolved uncertainty with the clinical data, preventing CADTH from deriving an economic base case. In addition to the clinical uncertainty, the sponsor’s PSM model structure was not appropriate given that PFS and OS have not been established in this patient population, and there is no robust evidence to confirm that response measures (such as ORR) are a prognostic marker of PFS or OS.
CADTH performed an exploratory analysis in which a Weibull parametric extrapolation was used for the OS data for amivantamab. Based on CADTH sequential reanalysis, amivantamab is associated with an ICER of $253,131 per QALY compared to NPBC, with a 0% probability of being cost-effective at a WTP threshold of $50,000 per QALY. This estimate is based on the sponsor’s clinical assumptions, which predict an additional 0.79 LYs and 0.60 QALYs for those receiving amivantamab, compared to NPBC. A price reduction of at least 77% would be required for amivantamab to be considered cost-effective compared to NPBC at this threshold.
If the comparative clinical data — particularly the PFS and OS data — are considered sufficiently robust, then the exploratory analysis performed by CADTH may be informative. However, in light of the available clinical information, CADTH’s results are based on assumptions — for example, survival extrapolations that result in OS benefits for amivantamab in both the preprogression and postprogression states [(79 LYs gained), which cannot be validated at this time — and hence are associated with a large amount of uncertainty. As such, additional price reductions may be required to ensure the cost-effectiveness of amivantamab.
In light of the uncertainty with the available evidence, CADTH performed a scenario cost comparison analysis in which only drug acquisition costs were included, based on costs from the CADTH cost table and a median duration of treatment of 8.3 months. This analysis assumes equal efficacy and duration of treatment for all comparators. Results of this analysis suggest that amivantamab would require a price reduction of 86.4% to achieve cost parity with the least costly comparator, docetaxel. If amivantamab is deemed to be no more efficacious than other currently reimbursed alternatives, this price reduction will be required to ensure that costs to the health care system do not increase.
References
1.Park K, Haura EB, Leighl NB, et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J Clin Oncol. 2021;39(30):3391-3402. PubMed
2.RybrevantÒ (amivantamab for injection): 50 mg/mL concentrate for solution for infusion [product monograph]. Toronto (ON): Janssen Inc.; 2022 Mar 30.
3.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: RYBREVANT® (amivantamab) 50 mg/mL concentrate for solution for infusion. Toronto (ON): Janssen Inc.; 2022 Apr 21.
4.Interim Clinical Study Report for Subjects With EGFR Exon 20 Insertion Mutations Treated With Amivantamab Monotherapy (30 March 2021Cutoff): 61186372EDI1001. A Phase 1, First-in-Human, Open-Label, Dose Escalation Study of JNJ-61186372, a Human Bispecific EGFR and cMet Antibody, in Subjects with Advanced Non-Small Cell Lung Cancer [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2021 Oct 21.
5.Amivantamab for the treatment of adult patients with advanced non-small cell lung cancer with activating epidermal growth factor exon 20 insertion mutations, after failure of platinum-based therapy: adjusted treatment comparison versus standard of care from real-world evidence sources [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: RYBREVANT® (amivantamab) 50 mg/mL concentrate for solution for infusion. Toronto (ON): Janssen Inc.; 2022 Apr 21.
6.Labbé C, Leung Y, Lemes J, et al. Real-world EQ5D Health utility scores for patients with metastatic lung cancer by molecular alteration and response to therapy. Clin Lung Cancer. 2016;18(4):388-395. PubMed
7.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
8.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 Jun 10.
9.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2022 Jun 10.
10.Lathia N, Mittmann N, DeAngelis C, et al. Evaluation of direct medical costs of hospitalization for febrile neutropenia. Cancer. 2010;116(3):742-748. PubMed
11.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 Jun 10.
12.Park K, Bennouna J, Boyer M, et al. Sequencing of therapy following first-line afatinib in patients with EGFR mutation-positive non-small cell lung cancer. Lung Cancer. 2019;132:126-131. PubMed
13.De Oliveira C, Pataky R, Bremner KE, et al. Estimating the Cost of Cancer Care in British Columbia and Ontario: A Canadian Inter-Provincial Comparison. Healthc Policy. 2017;12(3):95-108. PubMed
14.Shepherd FA, Dancey J, Ramlau R, et al. Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol. 2000;18(10):2095-2103. PubMed
15.Health Canada. Notice of compliance (NOC) for Rybrevant. 2022; https://health-products.canada.ca/noc-ac/index-eng.jsp. Accessed 2022 Jun 15.
16.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Jun 10.
17.U.S. Food & Drug Administration (FDA). FDA grants accelerated approval to mobocertinib for metastatic non-small cell lung cancer with EGFR exon 20 insertion mutations. 2021; https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-mobocertinib-metastatic-non-small-cell-lung-cancer-egfr-exon-20. Accessed 2022 Jun 23.
18.Spectrum Pharmaceuticals. Press release: Spectrum Pharmaceuticals announces acceptance of new drug application filing for poziotinib. 2022; https://investor.sppirx.com/node/23976/pdf. Accessed 2022 Jun 23.
19.DeltaPA. Ottawa (ON): IQVIA; 2022: https://www.iqvia.com/. Accessed 2022 Jun 10.
20.OPDIVO® (nivolumab for injection): 10 mg nivolumab /mL, 40 mg and 100 mg single-use vials for intravenous infusion [product monograph]. Montreal (QC): Bristol-Myers Squibb Canada Co.; 2022 Feb 28: https://pdf.hres.ca/dpd_pm/00065021.PDF. Accessed 2022 Jun 10.
21.JAMP Gefitinib (gefitinib): 250 mg oral tablet [product monograph]. Boucherville (QC): JAMP Pharma Corporation; 2022 Mar 11: https://pdf.hres.ca/dpd_pm/00065057.PDF. Accessed 2022 Jun 10.
22.Apo-erlotinib (erlotinib hydrochloride): 25 mg, 100 mg, 150 mg tablets [product monograph]. Toronto (ON): Apotex Inc.; 2018 Dec 5: https://pdf.hres.ca/dpd_pm/00048641.PDF. Accessed 2022 Jun 10.
23.TAGRISSO® (osimertinib as osimertinib mesylate): 40 mg and 80 mg oral tablets [product monograph]. Mississauga (ON): AstraZeneca Canada inc.; 2022 Mar 01: https://pdf.hres.ca/dpd_pm/00064905.PDF. Accessed 2022 Jun 10.
24.Taro-Docetaxel (docetaxel for injection): 20 mg / mL, 80 mg / 4 mL, 160 mg / 8 mL single vial formulation, concentrated solution for intravenous infusion [product monograph]. Brampton (ON): Taro Pharmaceuticals Inc.; 2021 Aug 10: https://pdf.hres.ca/dpd_pm/00063672.PDF. Accessed 2022 Jun 10.
25.Giotrif (afatinib as afatinib dimaleate): 20, 30 and 40 mg tablets [product monograph]. Burlington (ON): Boehringer Ingelheim (Canada) Ltd.; 2019 Jun 6: https://www.boehringer-ingelheim.ca/sites/ca/files/documents/giotrifpmen.pdf. Accessed 2022 Jun 10.
26.Pemetrexed for injection (pemetrexed as pemetrexed disodium): 100 mg or 500 mg per vial, lyophilized powder [product monograph]. Mississauga (ON): Dr. Reddy’s Laboratories Canada Inc.; 2022 May 03: https://pdf.hres.ca/dpd_pm/00065681.PDF. Accessed 2022 Jun 10.
27.TECENTRIQ® (atezolizumab): 60 mg/mL, 1200 mg/20 mL and 840 mg/14 mL singal use vials, concentrate for solution for infusion [product monograph]. Mississauga (ON): Hoffman-La Roche Ltd.; 2022 Mar 29: https://pdf.hres.ca/dpd_pm/00065218.PDF. Accessed 2022 Jun 10.
28.KEYTRUDA® (pembrolizumab): 100 mg/4 mL vial, solution for infusion [product monograph]. Kirkland (QC): Merck Canada Inc.; 2022 May 04: https://pdf.hres.ca/dpd_pm/00065684.PDF. Accessed 2022 Jun 10.
29.Gemcitabine injection (as gemcitabine hydrochloride): 40 mg gemcitabine per mL, 200 mg/5 mL, 1 g/25 mL, 2 g/50 mL concentrate sterile solution for injection [product monograph]. Boucherville (QC): Sandoz Canada Inc.; 2014 Aug 14: https://pdf.hres.ca/dpd_pm/00026822.PDF. Accessed 2022 Jun 10.
30.VIZIMPRO™ (dacomitinib as dacomitinib monohydrate): 15mg, 30 mg, and 45 mg, oral tablets [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2021 Jul 29: https://pdf.hres.ca/dpd_pm/00062308.PDF. Accessed 2022 Jun 10.
31.Drug Reimbursement Review clinical guidance and pharmacoeconomic reports: nivolumab (Opdivo) for esophageal or gastroesophageal junction cancer. Can J Health Technol. 2022;2(3). https://www.cadth.ca/sites/default/files/DRR/2022/PC0253-Opdivo_combined.pdf. Accessed 2022 Jun 10.
32.Drug Reimbursement Review clinical guidance and pharmacoeconomic reports: pembrolizumab (Keytruda) for metastatic microsatellite instability-high/mismatch repairdeficient colorectal cancer. Can J Health Technol. 2021;1(9). https://www.cadth.ca/sites/default/files/DRR/2021/PC0235-Keytruda-Combined.pdf. Accessed 2022 Jun 10.
33.Boyne DJ, Jarada TN, Yussuf A, et al. 51P: Testing patterns and outcomes of different egfr-positive metastatic non-small cell lung cancer patients in a Canadian real-world setting. Poster presented at: 2022 European Lung Cancer Congress (ELCC). Prague (CZ); 2022 Mar 30 - Apr 02.
34.Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2022 Mar 07.
35.Brenner DR, Weir HK, Demers AA, et al. Appendix 4. In: Projected estimates of cancer in Canada in 2020. CMAJ. 2020;192(9):E199-E205. PubMed
36.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics: a 2020 special report on lung cancer. Toronto (ON): Canadian Cancer Society; 2020: www.cancer.ca/Canadian-Cancer-Statistics-2020-EN. Accessed 2022 Jun 10.
37.Bauml JM, Viteri S, Minchom A, et al. FP07.12 Underdiagnosis of EGFR Exon 20 Insertion Mutation Variants: Estimates from NGS-based Real-World Datasets. J Thorac Oncol. 2021;16(3 Suppl):S208-S209.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Locally Advanced or Metastatic NSCLC
Treatment | Strength / concentration | Form (Vial size if single-use) | Price ($) | Recommended dosagea | Daily cost ($)b | 28-day cost ($) |
|---|---|---|---|---|---|---|
Amivantamab (Rybrevant) | 50 mg/mL | 7 mL (350 mg) Solution for IV infusion | 1,676.0000c | Body weight < 80 kg: 1,050 mg weekly for 4 weeks, then every 2 weeks Body weight ≥ 80 kg: 1,400 mg weekly for 4 weeks, then every 2 weeks | First 28 days: 718.29 to 957.71 Thereafter: 359.14 to 478.86 | First 28 days: 20,112 to 26,816 Thereafter: 10,056 to 13,408 |
EGFR tyrosine kinase inhibitors | ||||||
Afatinib | 20 mg 30 mg 40 mg | Tablet | 73.3000 | 40 mg once daily | 73.30 | 2,052 |
Dacomitinib | 15 mg 30 mg 45 mg | Tablet | 116.6667 | 45 mg once daily | 116.67 | 3,267 |
Erlotinib | 25 mg 100 mg 150 mg | Tablet | 11.8666d 47.4666 71.2000 | 150 mg once daily | 71.20 | 1,994 |
Gefitinib | 250 mg | Tablet | 62.3050d | 250 mg once daily | 62.31 | 1,745 |
Osimertinib | 40 mg 80 mg | Tablet | 294.6763 | 80 mg once daily | 294.68 | 8,251 |
Immunotherapy drugs | ||||||
Atezolizumab | 60 mg/mL | 14 mL (840 mg) 20 mL (1,200 mg) Solution for IV infusion | 4,743.2000 6,776.0000 | 840 mg every 2 weeks; or, 1,200 mg every 3 weeks; or, 1,680 every 4 weeks | 322.67 to 338.80 | 9,035 to 9,486 |
Nivolumab | 10 mg/mL | 4 mL (40 mg) 10 mL (100 mg) Solution for IV infusion | 782.2200e 1,955.5600 | 3 mg/kg every 2 weeks; or, 240 mg every 2 weeks; or, 480 mg every 4 weeks | 335.24 | 9,387 |
Pembrolizumab | 25 mg/mL | 4 mL (100 mg) Solution for IV injection | 4,400.0000f | 200 mg every 3 weeks; or, 400 mg every 6 weeks | 419.05 | 11,733 |
Non-platinum-based chemotherapy | ||||||
Docetaxel | 20 mg/mL | 1 mL (20 mg) 4 mL (80 mg) 8 mL (160 mg) Solution for IV injection | 249.0000 497.0000 990.0000 | 100 mg/m2 every 3 weeks | 59.00 | 1,652 |
Gemcitabine | 40 mg/mL | 25 mL (1,000 mg) 50 mL (2000 mg) Solution for IV injection | 270.3000 540.6000 | 1,000 mg/m2 weekly 3 out of every 4 weeks | 57.92 | 1,622 |
Pemetrexed | 25 mg/mL | 100 mg 500 mg 1,000 mg Powder for reconstitution for IV injection | 50.0000 250.0000 429.0000 | 500 mg/m2 every 3 weeks | 20.43 | 572 |
EGFR = epidermal growth factor receptor; NSCLC = non–small cell lung cancer.
Note: All prices are from the IQVIA DeltaPA database (accessed June 2022),19 unless otherwise indicated, and do not include dispensing fees.
aRecommended doses are from the respective product monographs.20-30
bDaily and 28-day costs are based on the average patient from CHRYSALIS Cohort D with a body weight of 67.5 kg and body surface area of 1.74 m2.1
cSponsor-submitted price.3
dPrice from Ontario Drug Benefit formulary (accessed June 2022).16
ePrice from prior CADTH review of esophageal cancer.31
fPrice from prior CADTH review of colorectal cancer.32
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The OS and PFS data used in the model for amivantamab were immature. |
Model has been adequately programmed and has sufficient face validity | No | The sponsor’s base case survival estimates for NPBC were overestimated compared to published literature and clinical expert opinion. This limitation could not be resolved. |
Model structure is adequate for decision problem | No | As noted in the CADTH Appraisal of the Sponsor’s Economic Evaluation section, the PSM structure, which relies on estimates of PFS and OS, was not appropriate given the outcomes of the CHRYSALIS trial. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | CADTH was unable to derive a base case. The decision problem has not been addressed. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
NPBC = non-platinum-based chemotherapy; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 2: PFS, OS, and TTD Kaplan-Meier Curves for Amivantamab (CHRYSALIS; n = 81)
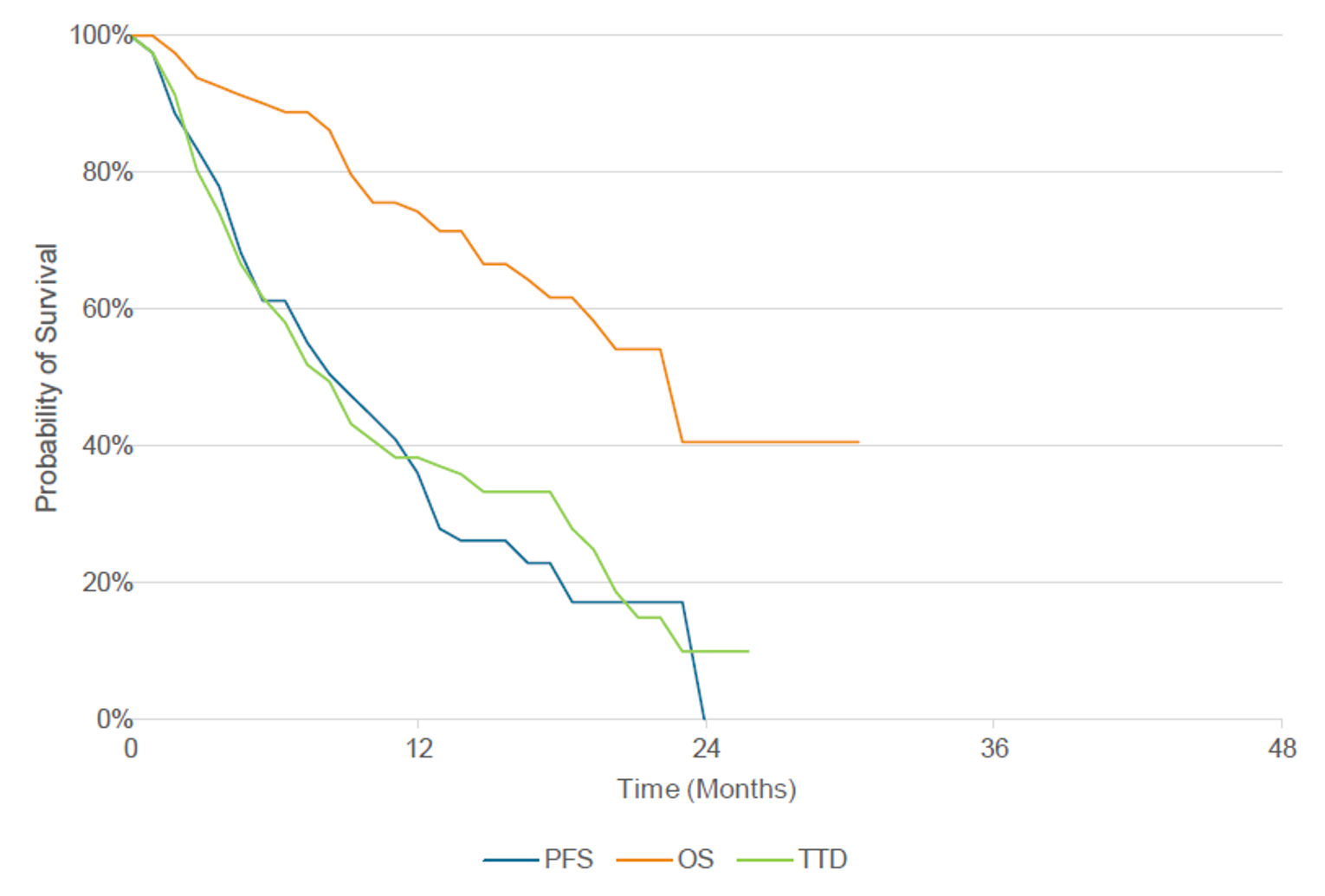
OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; TTD = time to treatment discontinuation.
Source: Sponsor’s pharmacoeconomic submission.3
Table 10: Predicted Survival and Reference Case Parametric Fittings for Amivantamab and Comparators
Survival curve | Predicted survival (months) | Reference case parametric fittings | |
|---|---|---|---|
Median | Mean | ||
PFS | |||
Amivantamab | 7.4 | 13.1 | Lognormal |
IO drugs | 2.8 | 5.5 | Log-logistic |
EGFR TKIs | 3.7 | 5.2 | Lognormal |
NPBC | 3.7 | 9.1 | Generalized gamma |
OS | |||
Amivantamab | 23.0 | 29.9 | Gamma |
IO drugs | 11.0 | 15.8 | Exponential |
EGFR TKIs | 11.0 | 13.9 | Gamma |
NPBC | 12.9 | 18.6 | Lognormal |
TTD | |||
Amivantamab | 8.3 | 12.5 | Exponential |
IO drugs | 4.6 | 7.4 | Log-logistic |
EGFR TKIs | 4.6 | 6.0 | Lognormal |
NPBC | 4.6 | 7.5 | Lognormal |
EGFR = epidermal growth factor receptor; IO = immunotherapy; NPBC = non-platinum-based chemotherapy; TKI = tyrosine kinase inhibitors.
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Results of the Sponsor’s Base Case
Parameter | Amivantamab | IO agents | EGFR TKIs | NPBC |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 2.42 | 1.30 | 1.15 | 1.52 |
Progression-free LYs | 1.06 | 0.45 | 0.43 | 0.73 |
Postprogression LYs | 1.35 | 0.85 | 0.72 | 0.79 |
Discounted QALYs | ||||
Total | 1.81 | 0.96 | 0.85 | 1.13 |
Progression-free QALYs | 0.86 | 0.37 | 0.35 | 0.59 |
Postprogression QALYs | 0.95 | 0.59 | 0.50 | 0.55 |
Disutilities due to AEs | 0.0017 | 0.0013 | 0.0009 | 0.0116 |
Discounted costs ($) | ||||
Total | 233,445 | 142,003 | 84,532 | 91,129 |
Drug acquisition | 158,824 | 81,824 | 22,782 | 23,272 |
Administration | 13,239 | 2,548 | 0 | 2,201 |
Monitoring | 403 | 238 | 195 | 243 |
AE management | 918 | 126 | 68 | 2,585 |
Disease management – preprogression | 1,810 | 771 | 734 | 1,235 |
Disease management – postprogression | 1,957 | 1,227 | 1,038 | 1,144 |
Terminal care | 50,451 | 51,930 | 52,083 | 51,523 |
Subsequent treatment | 5,844 | 3,339 | 7,631 | 8,927 |
Pairwise ICER of amivantamab vs. comparator ($/QALY) | NA | 107,956 | 155,861 | 210,591 |
AE = adverse event; EGFR = epidermal growth factor receptor; ICER = incremental cost-effectiveness ratio; LY = life-year; NPBC = non-platinum-based chemotherapy; QALY = quality-adjusted life-year; Ref. = reference; TKI = tyrosine kinase inhibitor.
Table 12: Probabilistic Cost-Effectiveness Sequential Analysis From Sponsor’s Base Case
Treatment | Cost ($) | QALYs | Incremental Cost ($) | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
EGFR TKIs | 84,532 | 0.85 | Ref. | Ref. | Ref. |
NPBC | 91,129 | 1.13 | 6,597 | 0.28 | 23,594 |
IO drugs | 142,003 | 0.96 | 50,874 | −0.17 | Dominated by NPBC |
Amivantamab | 233,445 | 1.81 | 142,316 | 0.68 | 210,591 |
EGFR = epidermal growth factor receptor; ICER = incremental cost-effectiveness ratio; IO = immunotherapy; LY = life-year; NPBC = non-platinum-based chemotherapy; QALY = quality-adjusted life-year; Ref. = reference; TKI = tyrosine kinase inhibitor.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Exploratory Analysis
Table 13: Disaggregated Summary of the CADTH Exploratory Analysis
Parameter | Amivantamab | IO agents | EGFR TKIs | NPBC |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 2.31 | 1.30 | 1.15 | 1.52 |
Progression-free LYs | 1.05 | 0.45 | 0.43 | 0.73 |
Postprogression LYs | 1.25 | 0.85 | 0.72 | 0.79 |
Discounted QALYs | ||||
Total | 1.73 | 0.96 | 0.85 | 1.13 |
Progression-free QALYs | 0.85 | 0.37 | 0.35 | 0.59 |
Postprogression QALYs | 0.88 | 0.59 | 0.50 | 0.55 |
Disutilities due to AEs | 0.0017 | 0.0013 | 0.0009 | 0.0116 |
Discounted costs ($) | ||||
Total | 233,131 | 141,308 | 83,782 | 81,409 |
Drug acquisition | 158,824 | 81,824 | 22,782 | 13,552 |
Administration | 13,239 | 2,548 | 0 | 2,201 |
Monitoring | 402 | 238 | 195 | 243 |
AE management | 918 | 126 | 68 | 2,585 |
Disease management – preprogression | 1,793 | 771 | 734 | 1,235 |
Disease management – postprogression | 1,815 | 1,227 | 1,038 | 1,144 |
Terminal care | 50,782 | 51,930 | 52,083 | 51,523 |
Subsequent treatment | 5,358 | 2,644 | 6,881 | 8,927 |
Pairwise ICER of amivantamab vs. comparator ($/QALY) | NA | 119,155 | 169,906 | 253,131 |
AE = adverse event; EGFR = epidermal growth factor receptor; ICER = incremental cost-effectiveness ratio; LY = life-year; NPBC = non-platinum-based chemotherapy; QALY = quality-adjusted life-year; Ref. = reference; TKI = tyrosine kinase inhibitor.
Table 14: Probabilistic Cost-Effectiveness Sequential Analysis From CADTH Exploratory Analysis
Treatment | Cost ($) | QALYs | Incremental Cost ($) | Incremental QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
NPBC | 81,409 | 1.13 | Ref. | Ref. | Ref. |
EGFR TKIs | 83,782 | 0.85 | 2,373 | −0.28 | Dominated by NPBC |
IO drugs | 141,308 | 0.96 | 59,899 | −0.17 | Dominated by NPBC |
Amivantamab | 233,131 | 1.73 | 151,722 | 0.60 | 253,131 |
EGFR = epidermal growth factor receptor; ICER = incremental cost-effectiveness ratio; IO = immunotherapy; LY = life-year; NPBC = non-platinum-based chemotherapy; QALY = quality-adjusted life-year; Ref. = reference; TKI = tyrosine kinase inhibitor.
Scenario Analyses
Table 15: Summary of Scenario Analyses Conducted on CADTH Exploratory Analysis
Drug | Total costs ($) | Total QALYs | Sequential ICER |
|---|---|---|---|
CADTH exploratory analysis | |||
NPBC | 81,409 | 1.13 | Ref. |
Amivantamab | 233,131 | 1.73 | 253,131 |
CADTH scenario 1: equal AE rates | |||
NPBC | 79,744 | 1.14 | Ref. |
Amivantamab | 233,131 | 1.73 | 260,191 |
CADTH scenario 2: cost comparisona | |||
NPBC (docetaxel) | 14,905 | NA | NA |
EGFR TKI (gefitinib) | 15,740 | NA | NA |
IO drugs (atezolizumab) | 81,516 | NA | NA |
Amivantamab | 109,498 | NA | NA |
AE = adverse event; EGFR = epidermal growth factor receptor; ICER = incremental cost-effectiveness ratio; IO = immunotherapy; LY = life-year; NA = not applicable; NPBC = non-platinum-based chemotherapy; QALY = quality-adjusted life-year; Ref. = reference; TKI = tyrosine kinase inhibitor.
aResults of the cost comparison indicate that amivantamab would require a price reduction of 86.4% to achieve cost parity with the least costly comparator, docetaxel.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 16: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted budget impact analysis (BIA) assessed the introduction of amivantamab for the treatment of post-platinum patients with EGFR Exon20ins mutated NSCLC. The analysis was taken from the perspective of the Canadian public drug plans using an epidemiology-based approach, with drug acquisition and administration costs included in the base case. A 3-year time horizon was used, from 2024 to 2026, with 2023 as a base year. The population size was estimated starting with incident lung cancer cases followed by a series of attritions. Key inputs to the BIA are documented in Table 17.
The reference case scenario included EGFR TKIs, IO drugs, and NPBC. The market share estimates were derived from Canadian chart review and a published conference proceeding.33 The new drug scenario included the same comparators along with amivantamab.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (Year 1 / Year 2 / Year 3) |
|---|---|
Target population | |
Population of CADTH-participating jurisdictions | 31,261,65334 |
Incidence of lung cancer | 61 per 100,00035 |
Proportion of new cases that are NSCLC | 88%36 |
Proportion of NSCLC that is Stage IV | 53%36 |
Proportion of NSCLC tested for EGFR mutations | 77.4%33 |
Proportion of NSCLC with EGFR mutations | 15.2%33 |
Proportion of EGFR mutations with Exon 20ins | |
Proportion of Exon 20ins that initiate first-line therapy | 72.2%33 |
Proportion of Exon 20ins that initiate second-line therapy | 61.5%33 |
Number of patients eligible for drug under review | ██ / ██ / ██ |
Market Uptake (3 years) | |
Uptake (reference scenario) EGFR TKIs IO drugs NPBC | 61% / 61% / 61% 13% / 13% / 13% 26% / 26% / 26% |
Uptake (new drug scenario) Amivantamab EGFR TKIs IO drugs NPBC | 45% / 55% / 50% 30% / 25% / 30% 5% / 0% / 0% 20% / 20% / 20% |
Cost of treatment (per patient) | |
Cost of treatment annually Amivantamab EGFR TKIs IO drugs NPBC | $152,054 $45,284 $130,988 $13,875 |
Duration of treatment in months (based on median PFS) | |
Amivantamab EGFR TKIs IO drugs NPBC | 8.3 3.9 2.9 4.1 |
EGFR = epidermal growth factor receptor; IO = immunotherapy; NPBC = non-platinum-based chemotherapy; NSCLC = non–small cell lung cancer; PFS = progression-free survival; TKI = tyrosine kinase inhibitors;
Summary of the Sponsor’s BIA Results
The estimated budget impact of funding amivantamab for the treatment of post-platinum patients with EGFR Exon 20ins mutated NSCLC was $1,757,694 in year 1, $2,118,425 in year 2, and $1,963,051 in year 3, for a 3-year total budget impact of $5,839,170.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The market share of amivantamab was underestimated in later years. The sponsor assumed market shares for amivantamab of 45%, 55%, and 50% in years 1, 2, and 3 of the BIA, respectively. These market share estimates do not meet face validity as they assume that the market share of amivantamab decreases in year 3. This implies that market share for amivantamab is being captured by an already existing product, which is unreasonable. Furthermore, the market shares themselves were felt to be underestimated by clinical experts, who expected the shift toward amivantamab to be more dramatic given the toxicities associated with TKIs and standard chemotherapy.
As part of the base case, CADTH assumed market shares for amivantamab of 45%, 65%, and 70% in years 1, 2, and 3, respectively. The extra market share was assumed to come from TKIs, whose share was noted by the experts noted to be higher than expected in both the reference and new drug scenarios.
Uncertainty regarding the final population size. The sponsor derived the population for the BIA using an epidemiology-based approach with a series of attritions, eventually resulting in about ██ incident patients per year that would potentially be eligible for amivantamab. This number was considered low by the clinical experts consulted by CADTH, and also considerably lower than the estimated annual incidence of 200 to 1000 patients provided by the clinician input from the Lung Cancer Canada Medical Advisory Committee. The experts considered an annual incidence of 100 patients to be more appropriate.
As part of a scenario analysis, CADTH increased the population size to 100 incident cases per year.
Subsequent therapy was not considered. The sponsor did not consider subsequent therapy in the BIA, despite including assumptions regarding subsequent therapy in their pharmacoeconomic model. This was an issue according to the clinical experts, who noted that patients would likely receive either a TKI, IO drug, or NPBC after progressing on amivantamab. The experts specifically stated that rates of chemotherapy usage would not be expected to change following administration of amivantamab. That is, amivantamab is expected to be used in addition to all currently approved therapies, meaning all costs associated with the drug would represent additional, incremental costs to the health care system.
As part of a scenario analysis, CADTH considered only the drug and administration costs for amivantamab.
CADTH Reanalyses of the BIA
Based on the identified limitations, CADTH’s base case included 1 correction to docetaxel dosing and 1 change to the market shares of amivantamab in the new drug scenario.
Table 18: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
1.Dosing of docetaxel | To achieve a dose of 180 mg docetaxel: 9 vials of 20 mg each = $2,081 | To achieve a dose of 180 mg docetaxel: One vial each of 160 mg and 20 mg = $1,239 |
Changes to derive the CADTH base case | ||
1.Market shares of amivantamab (Y1 / Y2 / Y3) | 45% / 55% / 50% | 45% / 65% / 70% |
CADTH base case | Reanalysis 1 | |
Y1 = year 1; Y2 = year 2; Y3 = year 3.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20. Based on the CADTH base case, the estimated budget impact of the reimbursement of amivantamab for the treatment of post-platinum patients with EGFR Exon20ins mutated NSCLC is expected to be $1,762,759 in year 1, $2,521,485 in year 2, and $2,782,311 in year 3, for a 3-year total of $7,066,555.
Scenario analyses conducted by CADTH indicate that the BIA results are sensitive to the population size, with the scenario assuming 100 incident patients per year resulting in a 3-year budget impact of $15,919,831. Results of the analysis in which only amivantamab costs are considered resulted in a 3-year budget impact of $8,403,136. Thus, the sponsor’s base case results may have underestimated the true budget impact.
Table 19: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case (corrected) | $5,854,479 |
CADTH reanalysis 1 and base case | $7,066,555 |
BIA = budget impact analysis.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|
Submitted base case (corrected) | Reference | 608,365 | 608,365 | 622,192 | 1,838,922 |
New drug | 2,371,124 | 2,371,855 | 2,590,422 | 7,693,401 | |
Budget impact | 1,762,759 | 2,123,490 | 1,968,231 | 5,854,479 | |
CADTH base case | Reference | 608,365 | 608,365 | 622,192 | 1,838,922 |
New drug | 2,371,124 | 3,129,850 | 3,404,503 | 8,905,477 | |
Budget impact | 1,762,759 | 2,521,485 | 2,782,311 | 7,066,555 | |
CADTH scenario analysis 1: increased population size | Reference | 1,382,648 | 1,382,648 | 1,382,648 | 4,147,945 |
New drug | 5,388,918 | 7,113,296 | 7,565,563 | 20,067,776 | |
Budget impact | 4,006,269 | 5,730,647 | 6,182,914 | 15,919,831 | |
CADTH scenario analysis 2: amivantamab costs only | Reference | 0 | 0 | 0 | 0 |
New drug | 2,082,379 | 3,007,881 | 3,312,876 | 8,403,136 | |
Budget impact | 2,082,379 | 3,007,881 | 3,312,876 | 8,403,136 | |
CADTH scenario analysis 3: 77% price reduction for amivantamab | Reference | 608,365 | 608,365 | 622,192 | 1,838,922 |
New drug | 767,692 | 813,782 | 853,589 | 2,435,062 | |
Budget impact | 159,327 | 205,416 | 231,397 | 596,140 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Lung Cancer Canada
About Lung Cancer Canada
Lung Cancer Canada is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research and advocacy. Lung Cancer Canada is a member of the Global Lung Cancer Coalition and is the only organization in Canada focused exclusively on lung cancer. https://www.lungcancercanada.ca/
Lung Cancer Canada is registered with CADTH.
Information Gathering
Data Collection
The information discussed throughout this submission consists of the thoughts and experiences of non-small cell lung cancer patients and caregivers, conducted through phone interviews by Lung Cancer Canada. All data was collected in May 2022.
Demographic Data
EGFR NSCLC is a relatively common mutation within non-small cell lung cancer (NSCLC); however, exon 20 insertion mutations as per this indication are very rare within the EGFR realm, making up between 0.1 – 4% of all NSCLC cases, and amivantamab is a relatively new therapy given NOC/c by Health Canada just in May 2022. All of the patients interviewed are EGFR positive, though only 2 of 4 patients are EFGR Exon 20 patients. All are currently part of the amivantamab clinical trial in Ontario.
Name | Gender | Patient/Caregiver | Stage at diagnosis | Type of lung cancer | Age | Location | Source |
|---|---|---|---|---|---|---|---|
SP | Female | Patient | Stage 4 NSCLC | EGFR Exon 20 | 63 | Canada (ON) | Phone interview |
LT | Female | Patient | Stage 4 NSCLC | EGFR Exon 20 | 76 | Canada (ON) | Phone Interview |
LC | Female | Patient | Stage 4 NSCLC | EGFR Exon 19 | 52 | Canada (ON) | Phone Interview |
JQ | Male | Patient | Stage 4 NSCLC | EGFR Exon 19 | 64 | Canada (ON) | Phone Interview |
Disease Experience
Prior to diagnosis, 63-year-old SP was extremely active and was always outdoors as she owns a landscaping business and routinely helps with gardening, landscaping, and designs. Living a healthy lifestyle and as a never-smoker, SP was constantly on-the-go and the thought of lung cancer never crossed her mind. However throughout early fall of 2021, SP developed a persistent cough and tried to treat it with a steroid puffer, but when things hadn’t improved over the next several weeks, something felt amiss. She insisted on an x-ray to rule out pneumonia, but instead, she was shocked to find out there was a mass in her lung, diagnosing her with stage 4 non-small-cell lung cancer, with EGFR Exon 20 insertion mutation. Her entire family, including her three adult children, was shocked to find the news, and it was extremely tough for SP to process herself as well, even until this day.
SP recalls, “it was incredibly shocking to hear the words ’lung cancer’ come out of the doctor’s mouth. I didn’t know what to do, it felt like time had stopped.” As hospital visits became more frequent once she started treatment with maintenance chemotherapy and amivantamab, she had to put her business on hold while still dealing with the emotional implications of her diagnosis. When patients like SP are handed a diagnosis, patients expect to hear what treatment options are available for them next. However, for patients with EGFR Exon 20 NSCLC, treatment options that are precise and specific to their disease are extremely limited, and there is a significant unmet need in this population. There are currently no targeted therapies specific to EGFR Ex20, until now. SP has started her treatment with amivantamab in December 2021 and continues to be on it as of May 2022 and has seen significant improvements in not only her physical health, but also her mental health.
8 years ago in early 2014, then 56-year-old JQ had some back pain and a cough that didn’t seem to go away for months, but simply thought it was due to his blood pressure medication. After he felt breathless after walking up one flight of stairs, he felt something was off and decided to head to his primary care doctor. A few tests and scans later, it was discovered he had lesions in his lower back, spine, and bones, which was traced back to the primary tumor in the lung, thus diagnosing JQ with stage 4 NSCLC in July 2014. After working as a physician in the army for 2 decades, he was faced with a number of patients with serious conditions, but when it became personal, it was hard to digest for not only himself but also his family.
JQ recalls, “When I was first given a diagnosis with stage 4 lung cancer, I felt like I had hit a dead end and didn’t think I’d even make it to the next Christmas, but then I did make it, and then to my next birthday, then Christmas, then each Christmas after that. It has been 8 Christmases and 8 birthdays since I got first diagnosed, and I feel eternally grateful I got this extra time.” Thanks to targeted therapies like amivantamab that are paving the way for patients to have increased quality of life and survivorship with advanced disease, JQ continues to be on amivantamab in May 2022 with a good quality of life and improvements in his disease and is hopeful he’ll continue to celebrate each birthday and each Christmas in the foreseeable future.
Lung cancer represents the highest incidence and prevalence of all cancers in Canada (in men and women), with an estimated 29,800 incident cases in 2020. Approximately 85% of cases are identified as the non-small cell subtype, and 15% of cases are small cell. Moreover, it is the leading cause of cancer-related mortality, with a 19% overall 5-year survival rate in 2020 (Burnett et al., 2021). Mutations in the epidermal growth factor receptor (EGFR) gene are among the most common targetable genomic drivers of non-small cell lung cancer (NSCLC). 90% of the EGFR mutations comprise of EGFR exon 19 deletion and exon 21 L858R mutation, while EGFR exon 20 insertions (EGFR Ex20Ins) are much less common, with a frequency of 0.1 - 4% in NSCLC (Burnett et al., 2021). Patients with EGFR Ex20 mutations have a poorer prognosis as conventional TKIs have poor efficacy in a majority of EGFR Ex20Ins subtypes. Chemotherapy remains the current standard of care for advanced NSCLC patients with this mutation. However, targeted therapy has since emerged as an important mean of disease management for NSCLC patients with a targetable mutation, including EGFR. This form of treatment has greatly improved patient outcomes and quality of life and is now a treatment option that is some patients’ only hope. It has seen incredible success and has allowed patients like SP and JQ a chance at survivorship and a livelihood that is nearly comparable to before diagnosis, something that they may never had thought would be possible before.
Amivantamab is a targeted therapy used to treat NSCLC for EGFR Exon 20 mutations and was given Notice of Compliance by Health Canada very recently in May 2022. It has shown promising results in efficacy and progression-free survival through the CHRYSALIS study, with median PFS of 8.3 months, overall response rate of 43.2%, and median overall survival of 22.8 months (Park et al., 2021). Time is extremely valuable for this subset of patients, and with the promising improvements amivantamab has seen to give patients within this population, it makes all the difference in their quality of life and livelihood. The treatment options for patients with this uncommon EGFR subtype are limited, with amivantamab being the first targeted therapy to be approved for this patient population in Canada, and one of only two Ex20Ins treatments approved by the US FDA (Park et al., 2021). Patients deserve treatment options that are effective beyond the current standard of care, and amivantamab has an incredible amount of potential to drive the pathway for future lung cancer treatment for thousands of Canadians. Lung Cancer Canada strongly encourages CADTH to take this into consideration for amivantamab to be reimbursed as it would lead the pathway to new developments, new treatments, improvements in accessibility, and better affordability for lung cancer patients across the country.
Experiences With Currently Available Treatments
The current standard of care for patients with NSCLC driven by EGFR exon 20 mutations is chemotherapy. Chemotherapy has been a long-standing and well-documented standard of care for lung cancer patients and has seen some benefits. However, it is limited as a viable long-term treatment option due to its nature as a systemic treatment with harsh and toxic side effects, which often creates additional burdens on patients, leading to decreased functionality and increased dependence on caregivers in daily activities without bringing much benefit or efficacy in treating disease. It also limits their ability to remain flexible in returning to work or continuing their hobbies, and thus, a more viable treatment option is necessary. Thus, chemotherapy typically only used as a first-line treatment option in this population, with limited mutation-specific options beyond first line.
Although the patients interviewed for this submission did not have experience on chemotherapy, most were offered chemotherapy as a back-up option should they not choose to take part in the amivantamab clinical trial. For example, JQ’s oncologist reassured him that chemotherapy was always an option, but rather as a last resort should other treatments not be effective. LC was offered a choice between chemotherapy or the amivantamab clinical trial and chose to move forward with amivantamab. SP is currently on combination amivantamab alongside maintenance chemotherapy; her experience is highlighted in Section 6. The experiences of patients on chemotherapy are well documented and discussed in previous Lung Cancer Canada submissions. It should be noted that patients treated with adjuvant chemotherapy still relapse, showing this form of treatment is not effective at preventing recurrence or keeping patients disease-free.
Some patients had experience with other targeted therapies for the EGFR mutation prior to treatment with amivantamab, though they do not have the specific Exon 20 mutation, rather other EGFR mutations. These therapies have been used to treat patients with other EFGR mutations and did have some success in keeping their disease stable but were limited in its effectiveness to target their mutations. Specifically for EGFR exon 20 mutations, there are no current treatment options that are precise and targeted to the genetic mutation, which leads to the significant unmet need within this group.
Improved Outcomes
There have been many incredible advancements in recent years for lung cancer treatment that have changed the paradigm for patients. While EGFR mutations being amongst the most common targetable drivers of non-small cell lung cancers, prevalent in roughly 38% of NSCLCs, EGFR Ex20 mutations are one of the rarest amongst this group, with exon 20 mutations accounting for only 3.8% of EGFR adenocarcinomas (Yoon et al., 2020). As a result, there have not been many opportunities for the development and refinement of new targeted therapy treatments for this specific subset of NSCLC patients, until now. It has been seen that targeted therapies for EGFR-driven NSCLC have been met with incredible success that gives patients their livelihoods back, allows them to hope for a better tomorrow and plan further down the line for a possible future. These outcomes play an integral role in the goals patients have in their treatment decisions, including:
Improved management of their disease symptoms of non-small cell lung cancer
Delaying disease progression and settling patients into long-term remission for improved survivorship
Allowing patients to have a full and worthwhile quality of life
Allowing patients to live longer and maintain their independence and functionality to minimize the caregiver burden
Having manageable side effects
Experience With Drug Under Review
Table 2: Experience With Drug Under Review
Patient | Diagnosis date | Drug access method | Period on amivantamab | Duration on amivantamab | Line of treatment with amivantamab | Currently on amivantamab? |
|---|---|---|---|---|---|---|
LC | December 2018 | Clinical Trial | December 2020 to present | 1.5 years | 3rd line | Yes |
JQ | July 2014 | Clinical Trial | January 2021 to present | 16 months | 3rd line | Yes |
SP | October 2021 | Clinical Trial | December 2021 to present | 6 months | 1st line | Yes |
LT | July 2021 | Clinical Trial | January 2022 to present | 4 months | 1st line | Yes |
Amivantamab is effective at treating patients’ disease.
By the time JQ started treatment with amivantamab, his disease had progressed in that he had a lesion in his brain and mets in his bones, in addition to the primary tumour in his lung. Although the treatment did not necessarily shrink the tumours, it was very effective at maintaining stability in his tumours and metastases, which is a feat in itself. The mets in his bones and lung have continued to be stable without any additional growth in the 14 months he has been on the treatment, which is a positive step. However, amivantamab does not cross the blood-brain barrier, so JQ has developed 6 metastases in his brain while on the therapy, compared to the one met when he started. He is hopeful he will be able to stay on the treatment for longer, while his oncologist continues to look into other treatments that are able to treat his brain mets.
Once SP was diagnosed in October 2021 with stage 4 EGFR Exon 20 insertion non-small cell lung cancer, she started on combination chemotherapy and amivantamab in December as her first line of treatment and has already seen improvements in her disease symptoms in the few months since. Within the first few sessions, she felt a noticeable difference as her cough is completely gone, no shortness of breath, and her tumours are responding well according to her oncologist.
LT was diagnosed July 2021 with stage 4 NSCLC with EGFR Exon 20 in both lungs, but fortunately did not have any metastases at diagnosis. She started on first-line treatment with amivantamab in January 2022, and after roughly 5 sessions, her scans have already showed shrinkage in the lung shadows within her scans. As this is her first treatment for her cancer, her oncologist is hopeful she can continue to stay on the treatment for the foreseeable future.
When LC started her treatment with amivantamab, the clinicians said she had responded to the treatment much quicker than most other participants. As of May 2022, about 1.5 years after starting her treatment, all of her tumours and metastases have shrunk and are much smaller than at treatment onset. They have all remained stable and relatively small ever since.
The side effects of amivantamab are manageable with minimal impact on daily life.
As per the CHRYSALIS clinical study, the most common treatment-related adverse events were rash, inflammation of the nailbed, hypoalbuminemia, swelling of the mouth lining, diarrhea, constipation, and nausea (Park et al., 2021). These side effects are all relatively manageable and do not impede the quality of life of patients as much as traditional systemic therapies do, as is the current standard of care for patients with EGFR Ex20 mutations after first-line treatment.
Amongst the patients LCC interviewed for this submission, the aforementioned side effects correspond to what was reported by patients. The main adverse event patients experienced was issues with the skin, including inflammation of the nailbed, rashes on the face and legs, acne, and dry and sensitive skin. Other side effects that were reported include severe dry mouth, slight vision deterioration, severe muscle pain the first few days after treatment, fatigue, chronic constipation, and yeast infections. For SP specifically, she has had tinnitus for about 10 years even prior to her cancer diagnosis due to a bicycle accident, but amivantamab has caused the pitch to change throughout treatment, which was a tough adjustment. However, all patients mentioned the side effects they’ve had are manageable and do not have much impact their ability to go about their livelihoods. Some patients mentioned that although the side effect burden was more than what they had experienced with other targeted therapies in the past, they would not consider discontinuing treatment as the hope of survival outweighs the negatives.
Amivantamab has given patients additional treatment options when they’ve exhausted other options.
Both LC and JQ are currently being treated with amivantamab as their 3rd line of treatment, and both have used other targeted therapies in the past. When LC’s oncologist mentioned she had progressed on her 2nd cancer treatment with osimertinib in November 2020, chemotherapy was most likely the only other option left, and without treatment, LC would have only about 6 months left. LC recalls it felt like she had hit a dead end because of the lack of treatment options that were available for her specific EGFR mutation.
Similarly, for JQ, after he was also on osimertinib for nearly 5 years progression-free, it was hard to accept when he had progressed afterwards in late 2020. JQ mentions that amivantamab was a genuine lifesaver for himself and other patients who have no other option left. He qualified for the clinical trial after a biopsy revealed he had the cMET amplification, which he felt like a weight had been lifted off him because there was still a potential targeted treatment available. As the indication for this submission is for patients with advanced or metastatic disease, they are often faced with limited treatment options because of the lack of available targeted therapies in this patient population. Amivantamab will be the first drug in this exon 20 EGFR space, giving patients the opportunity to have a treatment that is specific and targeted to their disease. Having this opportunity is critical for specificity in treatment options, and there is a significant unmet need for novel targeted therapies that will prolong progression-free survival and improve quality of life for these patients. Amivantamab has the ability to change this.
Patients on amivantamab enjoy a quality of life and level of functionality that is similar to pre-diagnosis.
When LC was first diagnosed in late 2018, she was very weak and needed help from her son, who was about to finish grade 12 at the time. She was so ill that she could not take care of him or herself and needed to depend on her close friends and relatives for basic tasks around the house, including grocery shopping, cooking meals, cleaning, and doing the laundry. She recalls, “At my weakest, they became my strength. Without them, I don’t know how I would have gotten through that moment in time. Since then, they have stood by me – through all of the highs and lows”. When she started on amivantamab, she already started to feel better within 2 months. She has been able to maintain her functionality throughout all of her past cancer treatments, but amivantamab has further accelerated the activities she has been able to do. When she just started amivantamab, she was physically unwell and very weak and needed a lot of help from relatives and friends. She couldn’t drive herself to complete daily errands and required relatives’ help, but now she is able to travel to Toronto for treatments every 2 weeks via public transit and has started driving again while back in her hometown. She has returned to exercising at the gym every single day now after starting treatment, started swimming laps at the pool again, and return to the relatively active lifestyle she was used to. LC is grateful she never had any severe physical impairments that impeded her going about basic tasks but attributes the accelerated return to a quality of life she had before diagnosis from amivantamab.
As amivantamab is SP’s first line of treatment, the success she found with the treatment has allowed her to continue to maintain her independence throughout treatment and continues to be able to do all her activities of daily living that she could prior to diagnosis 7 months ago. As she was a self-employed landscaper, her days mainly consisted of physical labour outdoors, and although she was unable to do much in the first few days after her treatment due to the side effects, she is able to continue helping out with minor gardening and landscaping work as needed for her business and stays active outdoors whenever she feels well. SP is grateful she does not yet have any severe physical impairments because of her cancer so far and is hopeful amivantamab will continue to maintain her independence, functionality, and health-related quality of life.
Since starting the amivantamab clinical trial, JQ travels 4-5 hours across the province every 2 weeks for his treatment, which he coined as “the closest he’s gotten to travelling and being on vacation for the last 8 years”. After he developed further metastases in his brain while on amivantamab, he hasn’t been able to drive long distances and has his son drive each trip instead. However, he still continues to drive while in his hometown and can go about his daily life with some normalcy. He has absolutely no problems when sitting down and is able to go about his day-to-day life running errands like grocery shopping, cooking, cleaning the house, and doing physical activity with no issues. Although he can’t do more intense activities like mowing the lawn or shoveling the snow, JQ feels like amivantamab has given him a great quality of life nonetheless.
When LT was first diagnosed in July 2021, the only symptom of her disease she had was a persistent cough that never went away for months, and only insisted on going back to the doctors when she coughed up blood one day. She had been retired 20 years prior and was living a fulfilling life after immigrating to Canada in 1997, spending time with friends, family, travelling, attending dance and Zumba classes, and being active outdoors. After being diagnosed during the pandemic and subsequent treatment with amivantamab, she had to put a halt to the gym and dance activities, but because the nature of amivantamab does not impose side effects as harsh as other traditional systemic treatments, LT has been able to continue exercising at home and even continue working as a school bus driver every single day for over a decade even after retirement. She drives around the city every day, has no issues running errands like grocery shopping, taking public transit to meet with friends, shopping, and visiting her daughter and son-in-law nearly every weekend. The flexibility that amivantamab has as a targeted therapy with minimal and manageable side effects on patients like LC, LT, and SP allows patients to continue enjoying the hobbies they love and have a worthwhile quality of life.
Amivantamab has given patients a hope for the future and allowed them to make longer term goals.
When LC was diagnosed in late 2018, her son was just finishing grade 12 and as a single mother, she was immediately faced with thoughts about if he would be okay in the worst-case scenario. With a stage 4 diagnosis, these thoughts are unfortunately common in cancer patients with advanced disease like LC’s. However, after seeing the success and effectiveness amivantamab has had on her disease, her mindset has since been filled with hope and excitement. When asked by Lung Cancer Canada about her goals for the future, LC was excited to share she has been able to make short and long term goals alike – to see her son graduate from university next year, and further down the line, hopes that she has a shot at being there when he gets a full-time job, when he gets married, settles down and starts a family, and maybe even be there to meet her grandchildren.
SP has been on amivantamab for about half a year since December 2021, and when she was diagnosed with stage 4 disease, she was shocked as lung cancer was the last thing on her mind being a healthy and very active individual. With three adult children living their own lives, her entire family was shocked to say the least when she was told her prognosis wasn’t looking good. After about 6 months of starting treatment with amivantamab. SP noticed her mindset has improved since the initial shock but is still dealing with emotional implications of diagnosis and treatment. Although she doesn’t have any pressing hopes for her future at the moment, her motivation to keep going stems from “staying alive until the next treatment option”. Her youngest son will be getting married this fall, so she hopes to be around for that. Patients like SP and LC are able to maintain a relatively optimistic mindset thanks to amivantamab and allows patients to continue to have a sense of hope for the future.
Amivantamab has given some patients the flexibility to return to work, even after retirement.
76-year-old LT had already been retired for nearly 20 years when she was diagnosed in 2021, but that did not keep her away from her job for long. 5 years after she retired, she went back to her job as a school bus driver and has continued to drive and work every school day since for the last 16 years, even though her cancer diagnosis and journey. As amivantamab was her first line of treatment after diagnosis, the success and effectiveness she had with the therapy has allowed her to maintain her quality of life, independence, functionality, and livelihood in that she still drives the buses every morning and afternoon rush without needing much time off. LT repeatedly mentioned to LCC that she absolutely is still in love with her job and it’s also a great distraction from her disease while keeping her engaged in the community. It has allowed her to keep herself busy and distracted from the anxiety that would otherwise ensue if she continued to be at home all day, especially evident when schools were closed during the COVID-19 pandemic. LT foresees herself continuing to drive the school bus everyday as long as she is able to, and thanks to amivantamab, she’s able to maintain a relatively “normal” livelihood that she absolutely enjoys and finds worthwhile and fulfilling.
SP had to put her landscaping business on hold for two months at the beginning of treatment with amivantamab because her hospital visits were frequent but has since returned to working throughout her treatment when she’s able to. She did hire additional help to complete remaining contracts and is no longer taking any new projects but getting out of the house and tying up loose ends before her business is scheduled to close in June 2022 gives her a sense of relief.
Companion Diagnostic Test
Patients with EGFR Exon 20 insertions are identified using Next Generation Sequencing (NGS). NGS is routinely conducted in all patients with advanced NSCLC with a non-squamous and squamous histology, without a smoking history.
Anything Else?
There is a huge unmet need with this population of NSCLC EGFR Exon 20 mutation patients as there is currently no targeted treatment available, and rather the current standard of care continues to be systemic treatments like chemotherapy. This population is so rare and there are very few treatment options for these patients, yet patients are desperate for a treatment that works with specificity. Amivantamab is an exciting option for targeting this mutation and has the opportunity to be the first targeted therapy to be approved in this population. It is seen to be effective at treating disease, allows patients to continue living a worthwhile quality of life, has manageable side effects, and even allowed some to return to work. Clinical data from the CHRYSALIS study have shown promising results, with median progression-free survival at 8.3 months, overall response rate of 43.2%, and median overall survival of 22.8 months (Park et al., 2021). There are also ongoing clinical trials of amivantamab in NSCLC EGFR Ex20ins in the first line setting, which have been showing promising results as seen with first-line patients LT and SP above.
There is a significant unmet need for novel targeted therapies that will prolong progression-free survival and improve health-related quality of life for patients with EGFR Exon 20 insertions, but amivantamab has the opportunity to change this. Lung Cancer Canada strongly pushes CADTH to recognize this gap and barrier in adequate treatment options that are specific to these patients who deserve treatments that will work and increase the accessibility of these treatment for patients across Canada. LCC is hopeful that this may become the new standard of care in this disease for second line treatment and beyond, and patients in this setting are not faced at a “dead end” when treatment with other TKIs or chemotherapy are no longer effective.
References
Burnett, H., Emich, H., Carroll, C., Stapleton, N., Mahadevia, P., & Li, T. (2021). Epidemiological and clinical burden of EGFR Exon 20 insertion in advanced non-small cell lung cancer: A systematic literature review. PloS one, 16(3), e0247620. DOI: 10.1371/journal.pone.0247620
Park, K., Haura, E. B., Leighl, N. B., Mitchell, P., Shu, C. A., Girard, N., ... & Cho, B. C. (2021). Amivantamab in EGFR exon 20 insertion-mutated non-small-cell lung cancer progressing on platinum chemotherapy: Initial results from the CHRYSALIS phase I study. Journal of Clinical Oncology, 39 (30). 3391-3402. DOI: 10.1200/JCO.21.00662
Yoon, H. Y., Ryu, J. S., Sim, Y. S., Kim, D., Lee, S. Y., Choi, J., Park, S., Ryu, Y. J., Lee, J. H., & Chang, J. H. (2020). Clinical significance of EGFR mutation types in lung adenocarcinoma: A multi-centre Korean study. PloS one, 15(2), e0228925. DOI: 10.1371/journal.pone.0228925
Conflict of Interest Declaration — Lung Cancer Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 3: Financial Disclosures for Lung Cancer Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen Inc. (Johnson & Johnson Shared Services) | — | — | X | — |
Lung Health Foundation / The Ontario Lung Association
About Lung Health Foundation / The Ontario Lung Association
The Ontario Lung Association (now named Lung Health Foundation) is registered with the CADTH and pCODR (www.lunghealth.ca). The Lung Health Foundation (Ontario Lung Association) is a registered charity that assists and empowers people living with or caring for others with lung disease. It is a recognized leader, voice and primary resource in the prevention and control of respiratory illness, tobacco cessation and prevention, and its effects on lung health. The Foundation provides programs and services to patients and health-care providers, invests in lung research and advocates for improved policies in lung health. It is run by a board of directors and has approximately 46 employees, supported by thousands of dedicated volunteers.
Information Gathering
The information provided from the Lung Health Foundation in this submission was obtained from an online survey and three phone interviews that were conducted between September and December 2021. The interviews were with two female patients and one male patient living with lung cancer. All the patients interviewed were over the age of 50. One of the female patients is based in Ontario and the other patient is based in Manitoba. The male patient was from Quebec. There were 2 survey respondents and demographic data was not collected. Input from a Registered Nurse is also included based on information gathered from monthly support groups attended by patients and their caregivers. Input from a certified respiratory educator was also obtained for this submission. The individual reviewed sections related to disease experience, experiences with available treatments and outcomes.
Disease Experience
Patients interviewed expressed that they found it difficult to cope with a lung cancer diagnosis. Lung cancer is associated with a poor prognosis and is the leading cause of cancer related deaths (Canadian Cancer Statistics, 2021). The patients interviewed report that the symptoms they experience with lung cancer were, in most cases, mild and are often associated with other conditions which led to a late diagnosis. The symptoms reported were shortness of breath, fatigue and pain. One patient interviewed, reported that she had a lingering cough for over six months before she was screened for lung cancer. She had been considered low risk because she did not have a smoking history. Another patient interviewed reported that she received her diagnosis during the peak of the COVID-19 pandemic. Delays in getting diagnostic tests and starting treatment was a great source of distress for her. Patients found the psychosocial effects of having a disease with a poor prognosis challenging and they also struggled with side effects associated with some treatments.
Some of the psychosocial effects reported were anxiety (66%), distress (100%) and depression (66%). One patient reported that having lung cancer was particularly isolating because of the stigma associated with lung cancer. She withdrew from all activities because she did not want people to know that she was diagnosed with lung cancer. Another patient interviewed described having a challenging time maintaining relationships with families and friends. They felt short tempered and impatient. Physical and emotional intimacy were also reported to be a challenge.
The side effects related to some treatments severely impact day to day and quality of life. One of the patients interviewed reported that he struggled with the side effects of chemotherapy. Prior to starting treatment, he was active and played sports, but once he started chemotherapy, he was unable to participate in his usual activities. He reported having hair loss, loss of appetite, weight loss, poor sleep, difficulty breathing, and this severely impacted his quality of life. This was very challenging for him. He also reported that the hair loss impacted his self-esteem because he looked visibly ill.
Another patient interviewed, reported that she experienced neuropathy, difficulty swallowing, fatigue and scarring in her lungs resulting in breathing difficulties. This negatively impacted her quality of life and ability to work and care for her family.
Family members and caregivers of those living with lung cancer share the same psychosocial burdens as the patients. They also have the added responsibility of providing care. Being a caregiver affects their ability to work, their relationships with family and friends and their emotional well-being. Their independence and ability to travel and socialize are often impacted as well. Having to take time off work to drive those they are caring for to get groceries, run errands or attend medical appointments can be problematic for caregivers. Feelings of fatigue and emotional exhaustion are not uncommon.
Experiences With Currently Available Treatments
The treatments tried by the respondents’ included surgery, radiation, chemotherapy, targeted therapy and immunotherapy. The medications tried included Cisplatin, Docetaxel, Gefitinib, Entrectnib, Alectinib, Brigatinib, Opdivo+Yervoy and Tagrisso.
The benefits experienced with the treatments were prolonged life, delayed disease progression and a reduction in the severity of disease-related symptoms. Although these benefits were noted, most patients struggled with lingering side effects. Respondents who received surgery, reported deconditioning and chronic fatigue. Some of the side effects reported from radiation were fatigue, skin changes, hair loss and tissue scarring.
With oral and subcutaneous medications, the side effects reported included fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, hypothyroidism and neuropathy. Side effects from chemotherapy severely impacted the patients’ quality of life, ability to work and in some cases, the ability to perform activities of daily living.
When asked about challenges with access to treatment, the respondents reported that they struggled with the cost of treatments and navigating the healthcare system. Some respondents reported travelling several hours to access treatments and sometimes they needed to stay overnight in hotels. This added a financial burden to the treatment process.
Patients also found delays in treatment and diagnostic testing to be a great source of distress because lung cancer progresses quickly, and advanced disease is associated with poorer outcomes.
Improved Outcomes
Patients are looking for medications that are effective in curing lung cancer. Patients are most interested in medications that “cure versus treat”. Patients discussed the mental burden that comes from living with lung cancer. Although some therapies are effective in slowing down disease progression, they describe living in constant fear that the disease will eventually progress.
Lung cancer is often associated with a poor prognosis and patients describe that receiving a lung cancer diagnosis can feel like a “death sentence”. Many patients are diagnosed after the disease has progressed and are often left with limited treatment options. Patients and caregivers would like more treatment options available for advanced disease as well as a reimbursement criteria that allows their healthcare providers to have more flexibility when prescribing treatments.
Patients report that they have struggled with side effects related to some treatments. They reported that they had no symptoms from the actual cancer but struggled more with the side effects from treatment. Patients would like treatments with minimal side effects so that they can carry on with regular activities while on treatment. The importance of maintaining some quality of life cannot be overstated.
Experience With Drug Under Review
No patients within this evidence group submission had experience with the medication under review.
Companion Diagnostic Test
Not applicable.
Anything Else?
Not applicable.
Reference
Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada and the Public Health Agency of Canada. Canadian Cancer Statistics 2021. Toronto, ON: Canadian Cancer Society; 2021
Conflict of Interest Declaration — Lung Health Foundation / The Ontario Lung Association
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
Table 4: Financial Disclosures for Lung Health Foundation / The Ontario Lung Association
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen Inc. | — | — | X | — |
Clinician Input
Lung Cancer Canada Medical Advisory Committee
About the Lung Cancer Canada Medical Advisory Committee
Lung Cancer Canada (LCC) is a national charity with the purpose of increasing awareness about lung cancer, providing support and education to lung cancer patients and their families, to support research and to advocate for access to the best care for all lung cancer patients in all provinces and territories.
Through the LCC Medical Advisory Committee (MAC), we provide clinician input for submissions of new lung cancer drugs to the HTA process for many years. The LCC MAC consists of clinicians and key opinion leaders in the field of lung cancer across the countries.
Information Gathering
The information provided in this submission is from publicly available sources, primarily published manuscripts and conference presentations, together with clinical experience of the members from the MAC. This submission is entirely independent of the manufacturer (Janssen).
Current Treatments and Treatment Goals
Canadian NSCLC patients with an EGFR Exon 20 insertion are rare.
In 2022, lung cancer is again projected to be the leading cause of cancer deaths, accounting for 24.3% of cancer deaths.1 The projected number of new lung cancer cases in Canada in 2022 will be 30,000 (1); of these, approximately 88% will be diagnosed with non-small cell lung cancer (NSCLC).2,3
In Canada, approximately 15% of patients with NSCLC present with mutations in the epidermal-growth factor receptor (EGFR) gene.4 EGFR mutations include the common Exon 19 deletions and the L858R mutation, which make up 85 - 90% of the mutations we see. The remaining 10-15% of EGFR mutations consist of the rarer, uncommon mutations, which can be further subdivided into: “sensitizing” or “non-sensitizing”.
Sensitizing mutations, make up 90% of uncommon mutations, and are often compound, which means that they contain more than one EGFR mutation. Patients whose lung tumours contain sensitizing uncommon mutations can respond to the first and second-generation EGFR tyrosine kinase inhibitors (TKIs) currently used in clinical practice, including erlotinib and afatinib, and to third-generation EGFR TKI, osimertinib
Non-sensitizing mutations make up the remaining 10% of uncommon mutations, split between Exon 20 insertions and de novo T790M mutations. According to some estimates, patients with EGFR Exon 20 insertions make up between 0.1 – 4% of all NSCLC cases2,3, which means that there are few new patients diagnosed each year in Canada, approximately only 200-1000.
NSCLC patients with non-sensitizing uncommon mutations are resistant to the first and second-generation EGFR TKIs.5,6 One reason why EGFR TKIs do not work in patients with an EGFR Exon 20 insertion is because the small size of the binding pocket for the ATP Kinase interferes with the binding of the TKI. Efficacy data for third-generation EGFR TKI’s in patients EGFR Exon 20 insertions is still unknown and is being tested in clinical trials. These include trials with a double dose of osimertinib, a third generation EGFR TKI. While this trial is in a preliminary stage, the rationale for how the double dose will overcome the issue with the size of the binding pocket is unclear. In addition, several clinical trials are ongoing with poziotinib and mobocertinib. The results of these trials are not expected for some time.
The prognosis for patients with locally advanced or metastatic NSCLC EGFR Exon 20 insertions is poor, mostly because they have few treatment options and lack effective targeted therapies.
Several real-world studies, including one from Alberta7, have demonstrated that patients with EGFR Exon 20 insertions have much poorer outcomes in terms of median PFS (mPFS) and median overall survival (mOS) than patients with common EGFR mutations.5-7
Current treatments do not modify the underlying disease mechanisms. For patients with EGFR Exon 20 insertions, chemotherapy platinum doublet (CPD) is by default a standard of care and currently the only treatment used in the first line setting.8 The addition of immunotherapy to CPD makes little sense, as patients with EGFR mutations were excluded from the KEYNOTE189 trial because checkpoint inhibitors have not shown efficacy in the EGFR driver mutation.9
After patients progress on CPD, docetaxel may be used in the second line, however response rates are less than 10%..8
As mentioned previously, EGFR TKIs can be efficacious for patients whose tumours contain common EGFR mutations, but patients with EGFR Exon 20 insertions are resistant to EGFR TKIs and have low response to immunotherapy.10-14 However, as clinicians search for any treatment to help their patients, EGFR TKIs and immunotherapy are sometimes tried in desperation.
There is a significant unmet need for novel targeted therapies that will prolong PFS and improve health-related quality of life (HRQoL) for patients with EGFR Exon 20 insertions.
The ideal treatment for patients with an EGFR Exon 20 insertion is one that directly inhibits the driver mutation. The treatment should be well tolerated, with a predictable and low toxicity profile. The response should be durable and correlate with an improvement in quality of life. Oral medications are preferred over intravenous treatments.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
There is a significant unmet need for novel targeted therapies that will prolong PFS and improve health-related quality of life (HRQoL) for patients with EGFR Exon 20 insertions.
Currently available options:
The only currently available treatment for NSCLC patients with EGFR Exon 20 insertions is chemotherapy platinum doublet followed by docetaxel single agent chemotherapy. These agents have low efficacy and cause significant toxicity, which interferes with quality of life.8
As discussed previously, tumours with an EGFR Exon 20 insertion do not respond to the currently available EGFR TKIs.5,6
Single agent immunotherapy has demonstrated a dismal response rate of less than 5% in patients with EGFR mutations.(15) There is no evidence that patients with an EGFR Exon 20 insertion will have a different or better response compared to patients with other EGFR mutations.
Patients with EGFR mutations were excluded from first line trials, KEYNOTE024 (16), KEYNOTE189(9), EMPOWER-Lung 117.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The current submission is for amivantamab in the second line setting for NSCLC patients with EGFR Exon 20 insertions, who have progressed on, or after CPD. This agrees with the indication and trial results of the CHRYSALIS-D study.18,19
CHRISALIS cohort D (CHRYSALIS-D) was a single-arm, open-label, Phase I/Ib dose escalation study (NCT02609776).(19) Patients with advanced NSCLC with EGFR Exon 20 insertions treated with amivantamab post CPD (n=81), experienced an overall response rate (ORR) of 43.2%, mPFS of 8.3 months and a mOS of 22.8 months with a median 14.5 month follow up, as determined by a blinded independent central review.19
Amivantamab is a bispecific monoclonal antibody that binds both to the extracellular domain of EGFR and to the extracellular domain of the MET receptor, blocking the binding of both EGF and MET ligands to their receptors.20
As we have learned from the literature and clinical practice, targeted therapies against driver mutations should ideally be offered in a first line setting for maximal efficacy. Clinical trials of amivantamab in NSCLC patients with Exon 20 insertion, in the first line setting, are ongoing.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The NSCLC patients most likely to respond to amivantamab are those with an EGFR Exon 20 insertion. Patients need to have an adequate performance status to be considered for this therapy.
Patients with EGFR Exon 20 insertions are identified using Next Generation Sequencing (NGS). NGS is routinely conducted in all patients with advanced NSCLC with a non-squamous and squamous histology, without a smoking history.
At this time, it is not yet possible to identify those specific patients who are most likely to respond to this therapy. As with all targeted therapies, studies will be conducted to identify potential biomarkers.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Amivantamab directly helps address the unmet treatment need in this uncommon patient population as it is a targeted agent that significantly improves response rates, progression-free survival, and overall survival compared to real-world controls, while improving aspects of a patient’s HRQoL.
The outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials and include safety/side effect profiles, treatment response and clinical response which are evaluated at regular intervals. The intervals of evaluation in clinical practice are usually not as frequent as in clinical trials, where trial protocols need to be strictly adhered to. In general, patients in clinical practice are often seen and evaluated every 6-8 weeks with a variety of radiological imaging modalities, usually to evaluate baseline disease sites as well as a clinical evaluation to determine side effect profiles and general changes to HRQOL.
A clinically meaningful endpoint to treatment is a stable radiological response, especially if it is durable. In most patients, a radiological response to treatment is reflected by a clinical response, which is also durable.
The magnitude of response will vary across patients but should not vary across physicians.
HRQoL outcomes are often more subjective and are harder to evaluate. In the amivantamab trial, patient-reported outcome analyses show a modest initial improvement in overall symptom severity for cough and chest pain.21
Education of the drug side effect profile, for both physicians and patients, is beneficial for both and helps maintain patients on therapy to achieve the best results.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Factors for consideration when deciding to discontinue amivantamab are similar to any other cancer therapy: disease progression and lack of clinical benefit.
In addition, adverse events may require patients to discontinue amivantamab temporarily or more permanently in some cases. In CHRYSALIS-D, treatment-emergent adverse events (TEAEs) leading to amivantamab discontinuation were reported for 11.8% patients treated, with 5.2% considered to be treatment-related.22
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Ideally, this drug needs to be administered at a Cancer Centre or hospital, by personnel experienced in administering chemotherapy agents.
Amivantamab is an intravenous drug administered weekly for the first month (cycle) and then then every two weeks a month (cycle) until disease progression or unwanted toxicity.
Adverse reactions, particularly infusion reactions, can occur in the first week of the first cycle. In the amivantamab clinical trials, infusion reactions were very common, observed 63.4% of patients, with a median time to first onset of 47 minutes. The majority of infusion-related reactions were limited to the first infusion and were of Grade 1 (10.5%) or 2 (50.3%) severity.22 The discontinuation rate due to this side effect were low.
Infusion-related reactions can be prospectively managed by slow administration (delivering the dose over two days, 6 hours on day 1 and 4 hours on day 2) and by co-administering appropriate interventions including steroids and antihistamines.
As for all patients with advanced lung cancer, a medical oncology team is required to diagnose, treat and monitor patients.
In my clinical practice, this is doable.
None of my patients treated with amivantamab has yet experienced a grade three toxicity nor have they experienced an adverse event that have led to discontinuation. The pharmacy has developed a protocol, the nurses are experienced, and the patients well educated about the treatment and possible side effects. This has led to successful treatment delivery in our center.
Additional Information
Although the patients with EGFR Exon 20 insertions are very rare, we do encounter them in clinic. Similar to other EGFR mutated NSCLC patients, they are generally non-smokers. Patients with an EGFR Exon 20 insertion have few treatment options yet are desperate for treatment as most are young and have good performance status.
For these patients, none of the oral TKIs work. Immunotherapy, although not directly studied in this specific setting, is unlikely to work as well. As this population is so rare, adequately powered phase III clinical trials are unfeasible. This applies not only to oral TKIs and immunotherapy, but also to amivantamab. Additionally, this agent has been approved in the US, which reduces the potential participants for a phase III trial. With the incidence of EGFR Exon 20 insertion occurs in <4% of all newly diagnosed NSCLC patients, at least 30% of patients are too sick to even receive first-line CBD therapy, and about 50% of the remaining patients will get second-line therapy, <2% of patients will be candidates for amivantamab as second line therapy. Out of these potential patients, only about 50% will fulfill the stringent eligibility criteria to enroll into a study, making it impossible to generate overall survival or progression-free survival data through a phase III study within a reasonable timeframe. While waiting for the readout of a phase III study in this rare patient population, many patients would not receive the benefit of amivantamab.
Having the ability to identify the specific mutation, in this case an EGFR Exon 20 insertion, driving the tumour is good news, as it permits us to strategize on how to specifically inhibit that driver. Amivantamab is an exciting option for targeting this mutation. The ORR of 43.2% observed in the CHRYSALIS-D trial is exciting and encouraging. We are hopeful that this may become the new standard of care in this disease for second line treatment. Like other targeted therapies against driver mutations, we look forward to eventually offering this therapy to first line in the metastatic setting.
References
1. Brenner DR, Poirier A, Woods RR, Ellison LF, Billette JM, Demers AA, et al. Projected estimates of cancer in Canada in 2022. Cmaj. 2022;194(17):E601-e7.
2. Burnett H, Emich H, Carroll C, Stapleton N, Mahadevia P, Li T. Epidemiological and clinical burden of EGFR Exon 20 insertion in advanced non-small cell lung cancer: A systematic literature review. PLOS ONE. 2021;16(3):e0247620.
3. Riess JW, Gandara DR, Frampton GM, Madison R, Peled N, Bufill JA, et al. Diverse EGFR Exon 20 Insertions and Co-Occurring Molecular Alterations Identified by Comprehensive Genomic Profiling of NSCLC. J Thorac Oncol. 2018;13(10):1560-8.
4. Tan AC, Tan DSW. Targeted Therapies for Lung Cancer Patients With Oncogenic Driver Molecular Alterations. J Clin Oncol. 2022;40(6):611-25.
5. Girard N, Bazhenova L, Minchom A, Ignatius Ou S-H, Gadgeel SM, Trigo J, et al. WCLC Oral Presentation: Comparative clinical outcomes for patients with NSCLC harboring EGFR Exon 20 insertion mutations and common EGFR mutations. Oral Presentation 3390. 2020.
6. Girard N, Bazhenova L, Minchom A, Ou SI, Gadgeel SM, Trigo J, et al. MA04.07 Comparative Clinical Outcomes for Patients with NSCLC Harboring EGFR Exon 20 Insertion Mutations and Common EGFR Mutations. Journal Of Thoracic Oncology. 2021;16(3):S145-S6.
7. Janssen. Data on file. Health Outcomes and Healthcare Resource Utilization of Patients with Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutation Non-small Cell Lung Cancer (NSCLC) in Alberta. 2021.
8. Melosky B, Banerji S, Blais N, Chu Q, Juergens R, Leighl NB, et al. Canadian consensus: a new systemic treatment algorithm for advanced EGFR-mutated non-small-cell lung cancer. Curr Oncol. 2020;27(2):e146-e55.
9. Gadgeel S, Rodríguez-Abreu D, Speranza G, Esteban E, Felip E, Dómine M, et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J Clin Oncol. 2020;38(14):1505-17.
10. Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013;12(2):220-9.
11. Oxnard GR, Lo PC, Nishino M, Dahlberg SE, Lindeman NI, Butaney M, et al. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol. 2013;8(2):179-84.
12. Naidoo J, Sima CS, Rodriguez K, Busby N, Nafa K, Ladanyi M, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer. 2015;121(18):3212-20.
13. Vyse S, Huang PH. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduction and Targeted Therapy. 2019;4(1):5.
14. Janssen. Data on file. ONCO CAPPS: EGFR Positive and EGFR Exon 20 Mutation Advanced Non-Squamous NSCLC. January 2022. 2022.
15. Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin Cancer Res. 2016;22(18):4585-93.
16. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. New England Journal of Medicine. 2016;375(19):1823-33.
17. Sezer A, Kilickap S, Gümüş M, Bondarenko I, Özgüroğlu M, Gogishvili M, et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet. 2021;397(10274):592-604.
18. Janssen. Draft Product Monograph: Amivantamab. 2021.
19. Sabari JK, Shu CA, Park K, Leighl NB, Mitchell P, Kim S, et al. OA04.04 Amivantamab in Post-platinum EGFR Exon 20 Insertion Mutant Non-small Cell Lung Cancer. Journal of Thoracic Oncology. 2021;16(3):S108-S9.
20. Janssen. Phase 1B EDI1001 study protocol. 2019.
21. Janssen. CHRYSALIS PRO Analysis - Exon 20ins Expanded Efficacy Population (March 2021 CCO). 2021.
22. Janssen Research and Development. Data on file. Interim clinical study report for subjects with EGFR Exon 20 insertion mutationstreated with amivantamab monotherapy (30 March 2021 Cutoff). Protocol 61186372EDI1001; Phase 1 (CHRYSALIS). 2021.
Conflict of Interest Declarations — Lung Cancer Canada — Medical Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Barbara Melosky
Position: Medical Oncologist, BC Cancer
Date: May 13, 2022
Table 5: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 1
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
Novartis | Advisory Board | X | — | — | — |
Roche | Advisory Board | X | — | — | — |
Merck | Advisory Board | X | — | — | — |
Declaration for Clinician 2
Name: Dr. Shaqil Kassam
Position: Medical Oncologist, Southlake Regional Hospital
Date: May 13 ,2022
Table 6: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | X | — | — | — |
Merck | X | — | — | — |
BMS | X | — | — | — |
Takeda | X | — | — | — |
Novartis | X | — | — | — |
Ipsen | X | — | — | — |
Sanofi | X | — | — | — |
Pfizer | X | — | — | — |
Declaration for Clinician 3
Name: Dr. Ronald Burkes
Position: Medical Oncologist, Mount Sinai Health
Date: May 13, 2022
Table 7: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Geoffrey Liu
Position: Medical Oncologist, Princess Margaret Cancer Centre
Date: May 13, 2022
Table 8: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 4
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
Takeda Canada | Advisory Board, Health Technology Assessment Submission Advice, Speaker’s Bureau, past 10 years | — | — | X | — |
Takeda Canada | (To institution, not individual) Observational Study funding, past 10 years | — | — | — | X |
Hoffman La Roche | Advisory Board, Health Technology Assessment Submission Advice, past 10 years | — | — | X | — |
Pfizer | Advisory Board, Health Technology Assessment Submission Advice, part 10 years | — | — | X | — |
AstraZeneca | Advisory Board, Health Technology Assessment Submission Advice, Speaker’s Bureau, past 10 years, | — | — | X | — |
AstraZeneca | (To institution, not individual) Observational Study funding, past 10 years | — | — | — | X |
Bristol Myers Squibb | Advisory Board | X | — | — | — |
Boehringer Ingerheim | (To institution, not individual) Observational Study funding, past 10 years | — | — | X | — |
Abbvie | Advisory Board, past 10 years | — | X | — | — |
Merck | Advisory Board, Health Technology Assessment Submission Advice, past 10 years | — | X | — | — |
EMD Serono | Speaker’s Bureau, past 10 years | X | — | — | — |
Novartis | Advisory Board,past 10 years | — | — | X | — |
Glaxo Smith Kline | Advisory Board, past 10 years | — | X | — | — |
Declaration for Clinician 5
Name: Dr Catherine Labbé
Position: Head of Respiratory Medicine Service, Université de Laval
Date: May 13, 2022
Table 9: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Amgen | X | — | — | — |
Astra Zeneca | — | X | — | — |
Brystol-Myers Squibb | X | — | — | — |
Jazz Pharmaceuticals | X | — | — | — |
LEO Pharma | X | — | — | — |
Merck | X | — | — | — |
Pfizer | X | — | — | — |
Roche | X | — | — | — |
Sanofi Genzyme | X | — | — | — |
Declaration for Clinician 6
Name: Dr. Kevin Jao
Position: Medical Oncologist, Hôpital Sacré-Cœur, Montreal
Date: May 13, 2022
Table 10: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 6
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
Bristol-Myers Squibb | Advisory Role | X | — | — | — |
Declaration for Clinician 7
Name: Dr Nicole Bouchard
Position: Respirologist, Sherbrooke University Hospital
Date: May 13, 2022
Table 11: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 7
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
Astra Zeneca | Advisory Role/Conference | X | — | — | — |
Bristol-Myers Squibb | Advisory Role/Research | X | — | — | — |
Merck | Advisory Role /Research/Conference | X | — | — | — |
Bayer | Advisory Role | X | — | — | — |
Pfizer | Conference/Research | X | — | — | — |
Declaration for Clinician 8
Name: Dr. Quincy Chu
Position: Medical Oncologist, Cross Cancer Institute
Date: May 13, 2022
Table 12: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 8
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | X | — | — | — |
Amgen | X | — | — | — |
AnHeart | X | — | — | — |
Astellas | X | — | — | — |
Astra Zeneca | — | X | — | — |
BI | X | — | — | — |
BMS | X | — | — | — |
Eli Lilly | — | X | — | — |
Eisai | X | — | — | — |
J and J | — | X | — | — |
Jazz | X | — | — | — |
Merck | X | — | — | — |
Novartis | X | — | — | — |
Pfizer | X | — | — | — |
Roche | X | — | — | — |
Sanofi | — | X | — | — |
Takeda | X | — | — | — |
Merck KgaA- DSMB | — | — | — | — |
Astra Zeneca-research funding | — | — | X | — |
Declaration for Clinician 9
Name: Dr. Paul Wheatley-Price
Position: Medical Oncologist, The Ottawa Hospital; Associate Professor, Department of Medicine, University of Ottawa
Date: May 13, 2022
Table 13: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 9
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Sanofi | X | — | — | — |
Astra Zeneca | X | — | — | — |
Jazz Pharmaceuticals | X | — | — | — |
Amgen | X | — | — | — |
Janssen | X | — | — | — |
Novartis | X | — | — | — |
Merck | X | — | — | — |
BMS | X | — | — | — |
Roche | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Bayer | X | — | — | — |
Novartis | X | — | — | — |
Declaration for Clinician 10
Name: Dr. Donna Maziak
Position: Thoracic Surgeon, The Ottawa Hospital
Date: May 13, 2022
Table 14: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 10
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | X | — | — | — |
Declaration for Clinician 11
Name: Dr. Rosalyn Juergens
Position: Chair, LCC Medical Advisory Committee; Medical Oncologist, Juravinski Cancer Center
Date: April 1, 2022
Table 15: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 11
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Bristol Myers Squibb | X | — | — | — |
Astra Zeneca | — | X | — | — |
Merck Sharp and Dohme | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 12
Name: Dr Normand Blais
Position: Medical Oncologist, Hôpital Notre Dame du CHUM
Date: May 13, 2022
Table 16: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 12
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novartis | X | — | — | — |
Declaration for Clinician 13
Name: Dr. Lacey Pitre
Position: Medical Oncologist, Systemic Therapy Lead - Northeast Region, CCO/Ontario Health
Date: May 13, 2022
Table 17: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 13
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novartis Ribbon Program 2018 | X | — | — | — |
MERCK Oncology Speaker’s honoraria 2017 | X | — | — | — |
EMD Serono Speaker’s honoraria 2018 | X | — | — | — |
MERCK Oncology Speaker’s honoraria 2021 | X | — | — | — |
Astra Zeneca Speaker’s honoraria 2021 | X | — | — | — |
Astra Zeneca Speaker’s honoraria 2022 | X | — | — | — |
Fuse Health Advisory Board 2017 | X | — | — | — |
Novartis Advisory Board 2018 | X | — | — | — |
Astella’s Oncology Advisory Board 2016 | X | — | — | — |
Declaration for Clinician 14
Name: Dr Jeffrey Rothenstein
Position: Medical Oncologist, Lakeridge Health
Date: May 13, 2022
Table 18: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 14
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | X | — | — | — |
Declaration for Clinician 15
Name: Dr. Callista Phillips
Position: Medical Oncologist, Hamilton Health Sciences Center
Date: May 13, 2022
Table 19: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 15
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
Astra Zeneca | Advisory Board Stage 3 NSCLC | X | — | — | — |
Bayer | National Consultancy meeting and Train the Trainer- Larotrectenib in NTRK fusion positive cancers | X | — | — | — |
Roche | Lung regional Consultancy meeting | X | — | — | — |
Declaration for Clinician 16
Name: Dr Sunil Yadav
Position: Medical Oncologist, Saskatoon Cancer Centre
Date: May 13, 2022
Table 20: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 16
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
Bristol-Myers Squibb | Advisory Board | X | — | — | — |
Astra Zeneca | Advisory Board and Speaking | X | — | — | — |
Merck | Advisory Board and Speaking | — | — | X | — |
Roche | Advisory Board and Speaking | — | X | — | — |
Takeda | Advisory Board and Speaking | X | — | — | — |
Declaration for Clinician 17
Name: Dr. David Dawe
Position: Medical Oncologist, CancerCare Manitoba
Date: May 13, 20221
Table 21: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 17
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
AstraZeneca | Advisory boards | X | — | — | — |
Merck | Advisory Boards | X | — | — | — |
AstraZeneca | Research Grant | — | — | X | — |
Boehringer-Ingelheim | Honoraria | X | — | — | — |
Declaration for Clinician 18
Name: Dr. Stephanie Snow
Position: President, Lung Cancer Canada; Medical Oncologist, The QEII Health Sciences Center
Date: May 13, 2022
Table 22: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 18
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
Amgen | Advisory Role | X | — | — | — |
Astra Zeneca | Advisory Role | — | — | X | — |
Bayer | Advisory Role | — | X | — | — |
Boehringer Ingeiheim | Advisory Role | X | — | — | — |
Bristol-Myers Squibb | Advisory Role | — | — | X | — |
Eisai | Advisory Role | X | — | — | — |
Merck | Advisory Role | — | — | X | — |
Novartis | Advisory Role | X | — | — | — |
Pfizer | Advisory Role | X | — | — | — |
Purdue | Advisory Role | X | — | — | — |
Roche | Advisory Role | — | — | X | — |
Taiho | Advisory Role | X | — | — | — |
Takeda | Advisory Role | — | X | — | — |
Declaration for Clinician 19
Name: Dr. Parneet Cheema
Position: Medical Director, William Osler Health System
Date: May 13, 2022
Table 23: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 19
Company | Nature or description of activities or interests | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|---|
Bristol Myers Squibb | Advisory board/Honoraria | X | — | — | — |
Merck | Advisory board/Honoraria | X | — | — | — |
Astra Zeneca | Advisory board/Honoraria | X | — | — | — |
Roche | Advisory board/Honoraria | X | — | — | — |
Novartis | Advisory board/Honoraria | X | — | — | — |
Declaration for Clinician 20
Name: Dr. David Stewart
Position: Medical Oncologist, The Ottawa Hospital
Date: May 13, 2022
Table 24: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 20
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 21
Name: Dr Randeep Sangha
Position: Associate Professor, University of Alberta; Medical Oncologist, Cross Cancer Institute
Date: May 13, 2022
Table 25: COI Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 21
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee
About Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
This input was jointly discussed at a DAC meeting.
Current Treatments and Treatment Goals
The standard of care remains platinum-based chemotherapy. First-line platinum-based doublet chemotherapy +/- immunotherapy followed by docetaxel at the time of progression. In Ontario, most centers testing for this mutation is available.
Treatment goals would be tumor response, improvement in tumor related symptoms, quality of life, and prolonging life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 3, please describe goals (needs) that are not being met by currently available treatments.
Not all patients respond to the current treatments available. There is a lack of established molecular therapies for this molecularly defined subgroup of lung cancer patients.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Amivantamab would be used after all standard therapies acceptable to the patient have failed. Typically, this would be following platinum doublet chemotherapy and maintenance pemetrexed, although in some cases this would also be after docetaxel therapy.
Immune checkpoint inhibitors may also be used in these patients as combination first line therapy, or as second line therapy, as these patients were not explicitly tested for and excluded from pivotal trials, but it is expected that these drugs would be low efficacy and many patients and clinicians would forgo the toxicity of immunotherapy given the possibility of inferior efficacy.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients best suited would align with the study inclusion criteria (EGFR Exon 20 Insertion–Mutated NSCLC patients).
Patients least suited would align with the study exclusion criteria.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
In clinical practice the outcome that is used to determine if a patient is responding to treatment is clinical improvement and radiologic improvement. Clinical assessed every 4 weeks and radiologic assessment every 3 months.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Treatment continued until unequivocal disease progression or loss of clinical benefit, unacceptable toxicity, withdrawal of consent.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
A specialist is required in an infusion clinic or a hospital (outpatient) setting.
Additional Information
Not applicable.
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Ontario Health provided secretariat support to the DAC.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Dr. Donna Maziak
Position: Ontario Health Lung Cancer Drug Advisory Committee Lead
Date: 13/05/2022
Table 26: COI Declaration for OH-CCO Lung Cancer Drug Advisory Committee — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Peter Ellis
Position: Ontario Health Lung Cancer Drug Advisory Committee Member
Date: 13/05/2022
Table 27: COI Declaration for OH-CCO Lung Cancer Drug Advisory Committee — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Andrew Robinson
Position: Ontario Health Lung Cancer Drug Advisory Committee Member
Date: 13/05/2022
Table 28: COI Declaration for OH-CCO Lung Cancer Drug Advisory Committee — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Stephanie Brule
Position: Ontario Health Lung Cancer Drug Advisory Committee Member
Date: 13/05/2022
Table 29: COI Declaration for OH-CCO Lung Cancer Drug Advisory Committee — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr. Sara Kuruvilla
Position: Ontario Health Lung Cancer Drug Advisory Committee Member
Date: 13/05/2022
Table 30: COI Declaration for OH-CCO Lung Cancer Drug Advisory Committee — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.