CADTH Reimbursement Review
Pralsetinib (Gavreto)
Sponsor: Hoffman-La Roche Ltd.
Therapeutic area: RET fusion–positive non–small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ALT
alanine transferase
AST
aspartate aminotransferase
BICR
blinded independent central review
CCO
Cancer Care Ontario
CI
confidence interval
CNS
central nervous system
CR
complete response
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
ESS
effective sample size
HR
hazard ratio
HRQoL
health-related quality of life
IPD
individual patient data
IPW
inverse probability weighting
ITC
indirect treatment comparison
LCC
Lung Cancer Canada
MID
minimally important difference
NSCLC
non–small cell lung cancer
OS
overall survival
PD-1
programmed cell death protein 1
PD-L1
programmed death ligand 1
PFS
progression-free survival
PR
partial response
RCT
randomized controlled trial
RECIST 1.1
Response Evaluation Criteria in Solid Tumors Version 1.1
RP2D
recommended phase II dose
SAE
serious adverse event
SD
standard deviation
SLR
systematic literature review
SMD
standardized mean difference
SOC
standard of care
WHO PS
WHO Performance Status
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Pralsetinib (Gavreto), 400 mg (four 100 mg oral tablets once daily) |
Indication | For the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC |
Reimbursement request | For the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC; treatment should continue as long as the patient is deriving clinical benefit from therapy or until unacceptable toxicity |
Health Canada approval status | NOC/c |
Health Canada review pathway | Standard review |
NOC date | June 30, 2021 |
Sponsor | Hoffman-La Roche Ltd. |
NOC/c = Notice of Compliance with conditions; NSCLC = non–small cell lung cancer.
Introduction
Lung cancer is the most frequently diagnosed cancer in Canada and the leading cause of cancer-related deaths,1 with more than 29,600 new diagnoses and 21,000 disease-related deaths projected in 2021.1 Lung cancers are classified into 2 types based on histology: small cell lung cancer and non–small cell lung cancer (NSCLC), the latter being the most common histology. Patients may experience worsening coughs, chest pain, hemoptysis, malaise, weight loss, dyspnea, and hoarseness at clinical presentation or upon chest imaging.1,2 The adjusted 5-year net survival estimate in Canada for all forms of lung cancers is 22%,1 and the anticipated 5-year survival is approximately 25% for patients with NSCLC and 7% for patients with stage IV disease.3 Unfortunately, almost 50% of NSCLC diagnoses in Canada are made at stage IV, with only about 23.1% of cases diagnosed at early-stage I.1 Abnormal RET receptor activation by rearrangement or mutation is recognized as an oncogenic driver for many cancers, including NSCLC. These alterations are commonly associated with patients with adenocarcinoma histology, younger patients (usually ≤ 60 years), and those with non-smoking or light-smoking status.4
Pralsetinib is an orally available, highly selective inhibitor of the RET receptor tyrosine kinase. It is available in 100 mg capsules. Pralsetinib received a Notice of Compliance with conditions from Health Canada on June 30, 2021, for the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC.
The objective of this review is to perform a systematic review of the beneficial and harmful effects of pralsetinib 400 mg oral tablets for the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
One patient advocacy group, Lung Cancer Canada (LCC), provided input regarding patients’ experiences, values, and preferences related to RET fusion–positive NSCLC and its treatment. The group was able to gather information from 4 patients with RET fusion–positive NSCLC who had received pralsetinib treatment and 1 caregiver from 4 countries (Canada, the US, Ireland, and Norway) in March 2022.
Patients and caregivers emphasized the consequences of delayed diagnosis due to mild and unspecific symptoms such as lower back pain, weight loss, coughing, and shortness of breath. As a result, patients are often diagnosed at an advanced or metastatic stage for which the prognosis is relatively poor. Patients reported that chemotherapy has limited long-term effectiveness due to toxicity. Patients experienced harsh side effects, such as fatigue, hair loss, and blood clots, which have negative impacts on patients’ functionality and quality of life and create additional burdens on patients.
Patients who had experience with pralsetinib indicated that the drug was effective in shrinking tumour size, producing less-severe side effects and improving functionality. For all 4 patients, pralsetinib treatment allowed them to continue working or doing household chores and conduct their daily lives with autonomy and dignity. The most frequently reported side effect was fatigue, which happened during onboarding and the initial weeks of treatment. Patients also reported other general side effects such as dry mouth, anemia, constipation, loss of appetite, and itchiness and/or dry skin. One patient was re-hospitalized due to liver function conditions and had a severe headache. Patients reported alleviation of the side effects once their dosages were reduced.
Outcomes important to patients were treatment effectiveness in managing symptoms, stopping or delaying disease progression, settling patients into long-term remission for improved survivorship, having manageable side effects, maintaining independence and functionality that would minimize the burden on their caregivers and family members, and improved quality of life. Detailed information of the patient group input is provided in the Stakeholder Input document.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
One clinical expert with experience in the diagnosis and management of NSCLC highlighted the differences in patients with RET fusion–positive NSCLC and the broader NSCLC population, with some of the key differences being that patients with RET fusion–positive NSCLC are younger and less likely to have a history of tobacco use. The expert noted that single-drug immunotherapy has limited activity in this population, and chemotherapy, while it is as effective in RET fusion patients as in the broader NSCLC population, does not have activity in the brain. Pralsetinib, a targeted oral therapy, is an option with good response rates and activity in the brain. The expert also noted the potential to reduce hospital burden through oral administration. This is in contrast to IV administration of immunotherapy and chemotherapy, which is more likely to require in-person or hospital care for adverse events (AEs). The clinical expert noted that radiographic assessments would generally be conducted every 8 to 12 weeks, with clinical assessments every 3 to 4 weeks, and patients would be discontinued from treatment in the presence of unacceptable AEs, patient preference, and symptomatic disease progression, with the exception of oligoprogression amenable to local intervention. Detailed information on the clinical expert input is provided in the Stakeholder Input section in the main body of the report.
Clinician Group Input
Clinician group input on the review of pralsetinib for the treatment of RET fusion–positive locally advanced unresectable or metastatic NSCLC was received from 2 groups: LCC and the Ontario Health – Cancer Care Ontario (CCO) Lung Cancer Drug Advisory Committee. The input was generally consistent with that provided by the clinical expert. The submission from the CCO committee suggested that patients with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 3 or greater would be least suitable for treatment with pralsetinib, whereas the clinical expert supported extending access to pralsetinib to patients with an ECOG PS of 2 or 3. The submission from LCC highlighted pandemic considerations and the potential for a reduced patient footprint in cancer centres with an oral therapy such as pralsetinib. Detailed information of the clinician group input is provided in the Stakeholder Input document.
Drug Program Input
The drug programs provide input on each drug reviewed through CADTH’s reimbursement review process by identifying issues that may affect their ability to implement a recommendation. The drug plans identified implementation issues related to relevant comparators; considerations for initiation, prescribing, and discontinuation of therapy; generalizability; funding algorithms; care provision; system issues; and economic considerations. The clinical expert consulted by CADTH for this review weighed evidence from the included study and other clinical considerations to provide responses to the drug plan’s implementation questions. Detailed information of the drug program input is provided in the Drug Program Input section in the main body of the report.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
The ARROW trial (N = 281; safety population at the November 6, 2020, data cut-off) is an ongoing phase I and II, open-label, single-arm study of pralsetinib in RET fusion–positive locally advanced or metastatic NSCLC patients. The primary objective of the phase II portion of the study was to determine the efficacy (measured by the overall response rate [ORR]) and safety of pralsetinib 400 mg once daily. The phase II portion of the study and the 400 mg once daily dosage are the focus of this report, as they represent the Health Canada–approved indication. Intracranial ORR, duration of response (DOR), progression-free survival (PFS), overall survival (OS), and health-related quality of life (HRQoL) were secondary end points in the trial. There was no predefined duration of treatment; patients with progressive disease could remain on treatment if the investigator determined that it was in the best interest of the patient to do so. Two unplanned interim clinical data cut-offs are presented in this report. The first is the November 18, 2019, data cut-off presented in a provided clinical study report5 and the November 6, 2020, data cut-off that was summarized in a European Medicines Agency report.6 The efficacy population in both analyses was a subset of patients who had been enrolled at the time of data cut-off to allow for an appropriate amount of time for patients to achieve an ORR. The respective cut-off dates were July 11, 2019, and May 22, 2020. A safety analysis was provided for all patients who had been enrolled up to each data cut-off date. At the November 6, 2020, data cut-off, the median age was 60 years, with similar proportions of each sex (52.4% female and 47.6% male); 51.9% of patients were White and 39.5% of patients were Asian.
Efficacy Results
Key efficacy outcomes are summarized in Table 2.
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies
Outcomes | ARROW November 18, 2019, data cut-off Pralsetinib efficacy population N = 132 | ARROW November 6, 2020, data cut-off Pralsetinib efficacy population N = 233 |
|---|---|---|
Overall survival | ||
Overall survival median follow-up time (95% CI)a | |||||||||||||||||||| | 17.1 (13.7 to 19.6) |
Median overall survival (95% CI)a | |||||||||||||||||||| | NR |
Deaths, n (%) | |||||||||||||||||||| | 57 (24.5) |
Censored, n (%) | |||||||||||||||||||| | 176 (75.5) |
HRQoL (EORTC QLQ-C30 global health status) | ||
Baseline, mean (SD); n | |||||||||||||||||||| | NR |
Week 24, mean (SD); n | |||||||||||||||||||| | NR |
Change from baseline to week 24, mean (SD); n | |||||||||||||||||||| | NR |
Progression-free survival | ||
Median progression-free survival, months (95% CI) | |||||||||||||||||||| | 16.4 (11.0 to 24.1) |
Patients with event, n (%) | |||||||||||||||||||| | 102 (43.8) |
Censored, n (%) | |||||||||||||||||||| | 131 (56.2) |
Overall response rate | ||
Overall response rate, n (%) | |||||||||||||||||||| | 150 (64.4) |
95% CIb | |||||||||||||||||||| | (57.9 to 70.5) |
Intracranial overall response rate | ||
Overall response rate, n (%) | |||||||||||||||||||| | 7 (70.0) |
95% CIb | |||||||||||||||||||| | (34.8 to 93.3) |
Duration of response | ||
Median duration of response, months (95% CI)a; n | |||||||||||||||||||| | 22.3 (14.7 to NR); 150 |
Harms, N (safety analysis population) | |||||||||||||||||||| | 281 |
Patients with ≥ 1 adverse event | |||||||||||||||||||| | 279 (99.3) |
Patients with ≥ 1 serious adverse event | |||||||||||||||||||| | 166 (59.1) |
Patients who stopped treatment due to AEs | |||||||||||||||||||| | 55 (19.6)c |
Deaths | |||||||||||||||||||| | 35 (12.5) |
Notable harm – pneumonitis | ||
Grade 3, 4, or 5 | |||||||||||||||||||| | 6 (2.1) |
SAE | |||||||||||||||||||| | 13 (4.6) |
Dose reduction | |||||||||||||||||||| | 18 (6.4) |
Dose interruption | |||||||||||||||||||| | 27 (9.6) |
Treatment discontinuation | |||||||||||||||||||| | 7 (2.5) |
Death due to AE | |||||||||||||||||||| | 0 |
AE = adverse event; CI = confidence interval; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HRQoL = health-related quality of life; NR = not reported; SAE = serious adverse event; SD = standard deviation.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
a95% CI based on the Greenwood formula.
b95% CI based on an exact binomial distribution using the Clopper-Pearson method.
cPatients who discontinued treatment.
Source: ARROW Clinical Study Report,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Overall Survival and Progression-Free Survival
At the November 18, 2019, data cut-off, the median OS follow-up time was 10.5 months (95% confidence interval [CI], 9.7 to 13.1); the median OS had not been reached. At data cut-off, 19.7% of patients had died and 80.3% were censored. Median PFS was 12.7 months (95% CI, 9.1 to not estimable) with 62.1% of patients censored at data cut-off.
At the November 6, 2020, data cut-off, the median OS follow-up time was 17.1 months (95% CI, 13.7 to 19.6); the median OS had not been reached. At data cut-off, 24.5% of patients had died and 75.5% were censored. Median PFS was 16.4 months (95% CI, 11.0 to 24.1), with 56.2% of patients censored at data cut-off.
Health-Related Quality of Life
Baseline mean global health status scores on the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) was |||||||||||| recorded from |||||| total patients. At the |||||| time point, the mean global health status score, recorded from ||||||| patients was ||||||| corresponding to a mean change from baseline of ||||||||||||||, meeting the published minimally important difference (MID) for a moderate improvement. Data for HRQoL were only available at the November 18, 2019, data cut-off.
Overall Response Rate, Intracranial Overall Response Rate, and Duration of Response
At the November 18, 2019, data cut-off, the ORR was |||||||||||||||||||||. Among patients who achieved a response (|||||||), the median DOR ||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In ||||||| patients with measurable intracranial lesions, the ORR rate was |||||||||||||||||||||). At the November 6, 2020, data cut-off, the ORR was 64.4% (95% CI, 57.9 to 70.5). Among patients who achieved a response (150 of 233), the median DOR was 22.3 months (95% CI, 14.7 to not reported). In the 10 patients with measurable intracranial lesions, the ORR was 70.0% (95% CI, 34.8 to 93.3). Additional subgroups reported for patients who received prior systemic therapy, prior platinum therapy, prior nonplatinum therapy, and no prior systemic therapy, along with analysis of the measurable disease population, showed results similar to those of the primary analysis.
Harms Results
At the November 18, 2019, data cut-off, ||||||| of a total of ||||||| patients in the safety analysis set experienced at least 1 AE. The most common AEs were increased aspartate transaminase (AST) (|||||||), constipation (|||||||), anemia (|||||||), and increased alanine transaminase (ALT) (|||||||). At the November 6, 2020, data cut-off, 99.3% of the 281 patients in the safety analysis set experienced at least 1 AE. The most common were anemia (45.9%), increased AST (44.8%), constipation (42.0%), hypertension (34.2%), and increased ALT (32.7%).
At the November 18, 2019, data cut-off, pneumonitis was reported as a grade 3, 4, or 5 AE by ||||||| of patients and a serious adverse event (SAE) by ||||||| of patients, resulting in a dose reduction in ||||||| of patients, a dose interruption in 8.4% of patients, and treatment discontinuation by ||||||| of patients. There were |||||||||||||| attributed to pneumonitis at the November 18, 2019, data cut-off. At the November 6, 2020, data cut-off, pneumonitis was reported as a grade 3, 4, or 5 AE by 2.1% of patients and an SAE by 4.6% of patients, resulting in a dose reduction in 6.4% of patients, a dose interruption in 9.6% of patients, and treatment discontinuation by 2.5% of patients. No deaths attributable to pneumonitis were reported at the November 6, 2020, data cut-off.
Critical Appraisal
The most important limitation with the ARROW trial stems from the single-arm design. This design increased the risk of bias in estimating treatment effects due to the potential for confounding related to unidentified prognostic factors and treatment-effect modifiers that could affect the activity of the study drug. While RET fusion patients are considered rare, accounting for 1% to 2% of all NSCLC patients, a phase III randomized trial is currently being conducted for pralsetinib in this patient population.
The results for the primary end point of ORR rejected the null hypothesis for response, and the clinical expert consulted described the response rates and duration of responses as impressive. No pre-specified interim analyses were planned in the statistical analysis plan for ARROW, increasing the potential for bias and type I error with successive ad hoc data cut-off analyses.
Patients recruited to the treatment-naive group were initially required to be deemed unsuitable for standard of care (SOC) chemotherapy, but the inclusion criteria were later amended to allow all treatment-naive patients. This amendment may have biased the results against pralsetinib if the patients recruited before the amendment had a worse prognosis compared to the average first-line patient. Important protocol deviations further increased the level of uncertainty, given that 16 patients at the November 6, 2020, data cut-off did not have measurable disease at baseline and 1 had inconclusive evidence of RET fusion. Patients who did not have measurable disease would be unlikely to record a response, biasing the ORR results against pralsetinib; however, OS and PFS would be unaffected. Results of subgroup analyses of the post-eligibility revision group, as well as the measurable-disease-only group, were similar to those of the primary analysis.
Uncertainty remains regarding the long-term effects of pralsetinib on secondary outcomes such as PFS, OS, and HRQoL given the lack of a comparator and the immaturity of the survival data as the median OS was not reached. The HRQoL results, which are important to patients, appear to be positive, reaching the MID for a moderate improvement; however, the number of patients in the analysis is low because this measure was added to the protocol through an amendment after initiation of the study, and patient numbers were further reduced as the time points progressed. There is potential for selection bias over time given that long-term survivors in the trials tend to be healthier patients. The absence of a comparator arm and open-label design introduces reporting bias, and the impact of pralsetinib on patient-reported outcomes in relation to other therapies is unknown.
According to the clinical expert consulted by CADTH, the demographic and disease characteristics of the ARROW population were reflective of the Canadian population of patients with RET fusion–positive NSCLC.
Indirect Comparisons
Description of Studies
In the absence of direct comparative evidence from trials, the aim of each analysis was to compare the efficacy (OS and PFS) of pralsetinib in patients with RET fusion–positive locally advanced or metastatic NSCLC versus patients with wild-type NSCLC receiving comparators of interest. The studies identified for comparators of interest were KEYNOTE-042 (pembrolizumab monotherapy),7 KEYNOTE-189 (pembrolizumab plus platinum-based chemotherapy plus pemetrexed),8 IMpower132 (platinum-based chemotherapy plus pemetrexed),9 OAK (second-line docetaxel),10 CheckMate 057 (second-line nivolumab),11 and GOIRC 02 to 200612 pooled with NVALT713 (carboplatin plus pemetrexed). The IMpower132 and OAK studies were chosen due to the availability of individual patient data (IPD), allowing a propensity-score weighting method to be applied to adjust for differences in study populations for the first-line platinum-based chemotherapy plus pemetrexed and second-line docetaxel comparisons. All other comparisons were naive unadjusted analyses that did not account for differences in population characteristics.
Efficacy Results
Propensity-Score Weighted Analysis
The adjusted hazard ratio (HR) for OS in the comparison of pralsetinib versus platinum-based chemotherapy plus pemetrexed was |||||||||||||||||||||. The adjusted HR for OS in the comparison of pralsetinib versus docetaxel was ||||||||||||||||||||||||||||. The adjusted HR for PFS in the comparison of pralsetinib versus platinum-based chemotherapy plus pemetrexed was||||||||||||||||||||||||||||. The adjusted HR for PFS in the comparison of pralsetinib versus docetaxel was ||||||||||||||||||||||||||||.
Naive Comparisons
The HRs for OS and PFS for the naive comparisons of pralsetinib versus first-line pembrolizumab monotherapy, first-line pembrolizumab plus pemetrexed-platinum, second-line nivolumab, and second-line pemetrexed plus carboplatin all favoured pralsetinib.
Critical Appraisal
A key limitation of the indirect treatment comparison (ITC) submitted by the sponsor comes from the single-arm design of the ARROW study, precluding any connected network of trials and resulting in a reliance on unanchored comparisons. For 2 comparisons, first-line platinum-based chemotherapy plus pemetrexed and second-line docetaxel, the sponsor had access to IPD and was able to conduct propensity-score weighting to adjust for between-trial differences in population characteristics. The methodology for choosing the prognostic factors to adjust for relied on data availability in place of a rigorous literature search. The analysis assumed the presence of RET fusion was not a predictive factor and therefore not included in the model. While a lack of evidence available in patients with RET fusion–positive NSCLC required this assumption, patients who are RET fusion–positive tend to be younger, less likely to smoke, and have mostly nonsquamous histology. Patients who are RET fusion–positive are more likely to respond to targeted RET therapy and less likely to respond to immunotherapy. The sponsor provided evidence from Hess et al. (2021)14 suggesting that, before the introduction of RET inhibitors, there was no relationship between RET status and outcomes in an adjusted model. However, the clinical expert consulted for this review suggested that the presence of RET fusion is a positive predictor for the efficacy of RET-targeted therapy and a negative predictor for the effect of immunotherapy. A methodology to adjust for prognostic factors other than RET fusion status was used; however, it was not possible to account for all differences in patient characteristics. With regard to the naive comparisons specifically, no adjustments were made. Patients with positive or negative RET fusion status are therefore expected to respond differently to pralsetinib and it is unclear how similar the patient populations in the comparator studies are to those enrolled in the ARROW trial, despite the adjustments in propensity-score weighted analysis. Once adjusted, the trial populations were vastly reduced in size (||||||||||||||||||||| in the case of the OAK trial), likely a result of the initial imbalance in baseline covariates.
As IPD were available for only 2 comparisons in the sponsor-submitted ITC, the remaining comparisons were unadjusted naive comparisons (no adjustments for between-trial differences in population characteristics were made). This introduces major uncertainty to the results, given that the prognostic factors identified by the sponsor as having an impact on treatment effects remained heterogenous for the naive comparisons. Conclusions cannot be drawn based upon the naive comparisons and conclusions drawn from the propensity-score weighted analysis are uncertain.
With these limitations in mind, all results were directionally consistent and in line with the clinical expert’s expectations that pralsetinib is likely superior to the comparators included in the ITC analysis.
An additional ITC identified from the literature search, Popat et al. (2022),15 compared first-line patients receiving pralsetinib in the ARROW trial to synthetic control arms sourced from 3 real-world populations. The first involved patients with RET fusion–positive NSCLC receiving a basket of best alternative therapy (most commonly pembrolizumab plus chemotherapy). The remaining 2 real-world populations comprised patients with wild-type NSCLC receiving pembrolizumab monotherapy and pembrolizumab plus chemotherapy, respectively. The analysis used inverse probability weighting (IPW) where possible to adjust for differences in prognostic factors. The results indicate that patients given pralsetinib received a statistically significant benefit in OS and PFS compared to the chosen comparators, which is consistent with the expectations of the clinical expert consulted for this review; however, the same limitations are present as in the sponsor-submitted ITC. The analysis is an unanchored ITC relying on a limited number of prognostic factors and a small effective sample size (ESS) compared to the original sample sizes of the populations.
Other Relevant Evidence
Description of Studies
The CADTH review team identified an ongoing phase III, randomized, open-label study, AcceleRET-Lung, comparing pralsetinib to a physician’s choice of platinum chemotherapy–based regimen based on SOC treatments for first-line treatment of patients with RET fusion–positive metastatic NSCLC who have not previously received systemic anticancer therapy for metastatic disease. No results are currently available, as this trial is actively recruiting patients. The estimated primary completion date (on which the last participant in a clinical study will be examined or receive an intervention to collect final data for the primary outcome measure) and study completion date (on which the last participant in a clinical study will be examined or receive an intervention or treatment to collect final data for the primary outcome measures, secondary outcome measures, and AEs) are September 30, 2023, and December 31, 2024, respectively.
Conclusions
The evidence supporting the funding request for pralsetinib was derived from an ongoing phase I and II, open-label, single-arm study, ARROW. The ORR observed in the ARROW trial, based on unplanned interim analysis results, suggested favourable tumour response in both treatment-naive and treatment-experienced patients and was consistent with further follow-up analysis. The ORR and DOR, including central nervous system (CNS) ORR, were considered clinically meaningful by the clinical expert consulted for the review. The ability to draw conclusions from time-to-event end points of PFS and OS were affected by the immaturity of the data and the single-arm design of the trial. The safety profile of pralsetinib in the ARROW trial was considered by the clinical expert consulted for this review to be an improvement compared to SOC chemotherapy and immunotherapy. According to expert clinical opinion, the differences in safety profiles compared with selpercatinib highlight the benefits to patients that come with additional treatment options. The ITC submitted to inform the comparative effects of pralsetinib was associated with limitations that prevented drawing conclusions from the results, and uncertainty remains in the comparative effectiveness and safety of pralsetinib.
Introduction
Disease Background
Lung cancer is the most frequently diagnosed cancer in Canada and the leading cause of cancer-related deaths in males and females,1 with more than 29,600 new diagnoses (12.5% of new cancer cases in males and 13.3% new cases in females) and 21,000 disease-related deaths (24.2% of cancer deaths in males and 25.8% in females) projected in 2021.1 The adjusted 5-year net survival estimate in Canada for all forms of lung cancers is 22%1 and the anticipated 5-year survival is approximately 25% for patients with NSCLC and 7% for patients with stage IV disease.3 Smoking is an established risk factor for developing lung cancer, accounting for more than 72% of newly diagnosed cases in Canada.1,2
Lung cancers are classified into 2 types based on histology: small cell lung cancer and NSCLC. The latter is the most common histology, and is further categorized based on cell type: adenocarcinomas, squamous cell carcinomas, or large cell carcinomas. The clinical expert shared information from their jurisdiction’s provincial data showing that up to 80% of all NSCLC cases are classified as nonsquamous and suggested that this figure could be higher in areas with lower tobacco use.
Early diagnosis improves prognoses and offers the best chance at optimal therapy. Diagnosis is based on symptom presentation2,16; patients may experience worsening coughs, chest pain, hemoptysis, malaise, weight loss, dyspnea, and hoarseness at clinical presentation or upon chest imaging.1,2 In advanced or metastatic disease, patients experience additional symptom burdens, such as trouble breathing, chronic coughing and chest pain, pain in bones or the spine, yellowing of the skin or eyes, weakness or numbness of arms or legs, fatigue and unexplained weight loss depression, insomnia, and pain.17,18 Staging at diagnosis is key in determining disease prognosis and facilitates treatment selection.2,18 Diagnosis at an advanced stage is a significant contributing factor to early mortality and challenging for disease management in real-world practice. Unfortunately, almost 50% of NSCLC diagnoses in Canada are made at stage IV, with only about 23.1% of cases diagnosed at early-stage I.1
The expression of oncogenic driver mutations in tumours plays a vital role in patient response to treatment when there is an accessible targeted therapy specific to that mutation.18 Several predictive driver mutations identified in recent years, including mutations in EGFR, ROS1, KRAS, ALK, BRAF V600E, and others have greatly influenced treatment strategies in practice, improved patient quality of life, and increased OS for patients.18-20 The RET protein is a transmembrane tyrosine kinase receptor encoded by the RET gene and is known to play a substantial role in the development and maintenance of many systems, including the enteric nervous and genitourinary systems in neonates.21 Abnormal RET receptor activation by rearrangement or mutation was recognized as an oncogenic driver for many cancers, including NSCLC. These alterations are commonly associated with patients with adenocarcinoma histology, younger patients (usually ≤ 60 years), and those with no or minimal history of tobacco use.4 Prevalence estimates from studies show that only about 1% to 2% of NSCLC cases are RET fusion–positive.22 Testing for driver mutations at initial diagnosis using molecular techniques such as next-generation sequencing or polymerase chain reaction amplification is available across jurisdictions in Canada.18,19
Standards of Therapy
The clinician experts and clinician groups consulted for this review outlined similar treatment goals for patients with advanced or metastatic disease, including improvement in median OS, rapidity and prolonged improvement in cancer-related symptoms and improvement in quality of life (given that patients with advanced and metastatic disease experience greater symptom burden), reduced treatment-related toxicity, prevention, and treatment of brain metastasis.
Expert opinion from the clinician groups and drug plans consulted during the CADTH review emphasized the importance of treatment combinations funded in practice for patients without confirmed RET fusion. For the treatment-naive population, first-line treatment combinations with platinum plus pemetrexed and pembrolizumab were identified as the most preferred in patients with programmed death ligand 1 (PD-L1) expression below 50%. For those patients with PD-L1 expression of 50% or greater, options include single-drug pembrolizumab or platinum plus pemetrexed and pembrolizumab, with the latter often favoured for patients who are nonsmokers, have high disease (or symptom) burdens, or who have a known oncogene driver mutation associated with poor outcomes when treated with immunotherapy alone. For patients who progressed on prior systemic therapy, treatment options with second-line platinum plus pemetrexed are most preferred if they had received pembrolizumab in the first-line therapy. Second-line anti–PD-L1 therapy, using pembrolizumab, nivolumab, or atezolizumab, is favoured for those who received platinum plus pemetrexed as first-line therapy, and second-line docetaxel for those who progressed on platinum plus pemetrexed and pembrolizumab.
The drug plans consulted for this review identified publicly funded options for patients with advanced unresectable or metastatic NSCLC who are treatment-naive, including pembrolizumab single-drug therapy for patients with PD-L1 expression greater than or equal to 50%, pembrolizumab plus pemetrexed plus platinum-based chemotherapy, or platinum-based chemotherapy based on histology. Nivolumab in combination with ipilimumab plus platinum doublet chemotherapy is under consideration for listing in provinces. In the second-line setting, the drug plans noted that funded options may include immune checkpoint inhibitors if no prior programmed cell death protein 1 (PD-1) inhibitor was administered to the patient (either pembrolizumab, nivolumab, atezolizumab depending on patient’s PD-L1 status), or chemotherapy if a prior PD-1 inhibitor (docetaxel or pemetrexed) was administered. The LCC clinician group pointed out that evidence from some cohort studies indicates that patients with RET fusion–positive NSCLC are sensitive to pemetrexed. The group therefore noted that, in the absence of any randomized data, a combination of pemetrexed and platinum will likely be the most efficacious therapy in patients with RET fusion–positive NSCLC who had received only pembrolizumab as first-line therapy.
The clinician expert consulted identified treatments similar to those outlined by the clinician group and drug plans. The expert mentioned that the most preferred therapy used in the first-line setting across jurisdictions in Canada is a triplet therapy of platinum, pemetrexed, and pembrolizumab regardless of the PD-L1 tumour proportion score. While they acknowledged that single-drug immunotherapies are approved and available in practice for those with a PD-L1 tumour proportion score of 50% or greater, they added that studies have reported poor response rates to immunotherapy when given alone in patients with RET fusion–positive NSCLC. The expert indicated that patients with RET fusion–positive NSCLC (most likely with adenocarcinoma histology) have been shown to respond to platinum and pemetrexed combinations, and other platinum doublets would be considered inferior for any adenocarcinoma patient.
The clinical expert added that, beyond the first line (after administration of a triple therapy), single-drug docetaxel is the typical SOC, although there is limited evidence for outcomes specific to patients with RET fusion–positive NSCLC using docetaxel. If a patient received pembrolizumab in the first line, a doublet combination of platinum and pemetrexed may be administered, and if they received platinum plus pemetrexed in the first line, they may likely receive immunotherapy (e.g., pembrolizumab, nivolumab, or atezolizumab) in the second line. However, if the patient was tested for RET fusion, they may likely be placed on docetaxel after platinum-pemetrexed doublet therapy in the second line rather than immunotherapy (based on reports of low response rates to immunotherapy in patients positive for RET fusion. The experts added that gemcitabine and vinorelbine are available as therapies in the third line and beyond. Other nonsystemic options outlined included radiation and surgical interventions employed as aggressive modalities in patients with oligometastatic disease or as palliative interventions with the goal of alleviating symptoms.
Drug
Pralsetinib is an orally available, highly selective, adenosine triphosphate–competitive, small-molecule inhibitor of RET receptor tyrosine kinase. It is available in 100 mg capsules. Pralsetinib received a Notice of Compliance with conditions from Health Canada on June 30, 2021, for the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC. The sponsor is requesting reimbursement for the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC, specifically noting that treatment should continue as long as the patient is deriving a clinical benefit from therapy or until unacceptable toxicity, in line with the Health Canada product monograph.
Pralsetinib has obtained regulatory approval, including the FDA (September 4, 2020) and the European Medicines Agency (November 18, 2021) and is marketed in other regulatory jurisdictions for indications similar to those outlined in the Canadian product monograph. Market approval in Canada was granted based on evidence generated from the ARROW trial, a phase I and II open-label trial in patients 18 years and older with RET-altered NSCLC, medullary thyroid cancer, and other RET-altered solid tumours. The recommended dose is 400 mg, taken as four 100 mg capsules, once daily.
Table 3: Key Characteristics of Pralsetinib
Characteristic | Pralsetinib |
|---|---|
Mechanism of action | Orally available, highly selective, ATP-competitive small-molecule inhibitor of the RET receptor tyrosine kinase |
Indicationa | For the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC |
Route of administration | Oral |
Recommended dose | 400 mg (four 100 mg oral tablets once daily) |
Serious adverse effects or safety issues |
|
Other | A validated test is required before treatment to identify RET fusion status |
ATP = adenosine triphosphate; NSCLC = non–small cell lung cancer.
aHealth Canada–approved indication
Source: Pralsetinib product monograph.23
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
One patient advocacy group, LCC, provided input regarding patients’ experiences, values, and preferences related to RET fusion–positive NSCLC and its treatment. The group was able to gather information from 4 patients with RET fusion–positive NSCLC who had received pralsetinib treatment and 1 caregiver in March 2022; the respondents were from Canada, the US, Ireland, and Norway.
Patients and caregiver respondents emphasized the consequences of a delay in diagnosis due to mild and unspecific symptoms such as lower back pain, weight loss, coughing, and shortness of breath. As a result, patients are often diagnosed at an advanced or metastatic stage for which the prognosis is relatively poor. Patients reported that chemotherapy has limited long-term effectiveness due to toxicity. Patients experienced harsh side effects, such as fatigue, hair loss, and blood clots, which have negative effects on patients’ functionality and quality of life and create additional burdens on patients.
Patients who had experience with pralsetinib indicated that the drug was effective in shrinking the tumour size, resulted in less-severe side effects, and improved functionality. For all 5 patients, the benefits of pralsetinib treatment allowed them to continue working or doing household chores and conduct their daily lives with autonomy and dignity. The most frequently reported side effect was fatigue, which happened during onboarding and the initial weeks of treatment. Patients also reported other general side effects, such as dry mouth, anemia, constipation, loss of appetite, and itchiness and/or dry skin. One patient was re-hospitalized due to liver function conditions and had a severe headache. Patients reported alleviation of the side effects once their dosages were reduced.
Outcomes important to patients are treatment effectiveness in managing symptoms, stopping or delaying disease progression, settling patients into long-term remission for improved survivorship, having manageable side effects, maintaining patients’ independence and functionality (thereby minimizing the burden on their caregivers and family members), and improving quality of life. Detailed information of the patient group input is provided in the Stakeholder Input document.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by a clinical specialist with expertise in the diagnosis and management of locally advanced and metastatic NSCLC.
Unmet Needs
At the time of the pralsetinib submission, the clinical expert noted that no reimbursed therapies for NSCLC that specifically target RET fusion are available in Canada . Although the clinical expert noted that selpercatinib has received a positive recommendation with conditions from CADTH, the time from a final recommendation to actual reimbursement by different provincial jurisdictions can be considerable. At present, Eli Lilly offers a patient support program to facilitate access to selpercatinib for Canadian patients.
The clinical expert explained that patients are treated with chemotherapy and immunotherapy that require IV infusions and come with significant adverse effects requiring intensive supportive care and hospital stays. Furthermore, not all patients respond to chemotherapy in combination with immunotherapy (the response rate in the KEYNOTE-189 trial with platinum-pemetrexed-pembrolizumab was 48%).8 Because single-drug immunotherapy has limited utility in the RET fusion population and chemotherapy does not have activity in the brain, patients receiving chemotherapy who develop brain metastasis will require brain radiation, which carries a significant risk of toxicity.
Place in Therapy
The clinical expert highlighted that pralsetinib would be given as a single drug as an alternative to selpercatinib. As targeted therapies, they would ideally be used in first-line therapy, with chemotherapy and immunotherapy shifted to the second or later line. The clinical expert reiterated that patients should receive either pralsetinib or selpercatinib, but not both. It would be inappropriate to begin patients on chemotherapy and immunotherapy, which, according to the clinical expert, would be less effective, more toxic, and carry a larger burden on the health care system given that IV therapies must be administered in clinics and are more likely to require in-person or in-hospital supportive care for AEs. Additionally, when considering attrition between lines of therapy, the clinical expert highlighted the importance of offering either pralsetinib or selpercatinib in the first line so that the largest number of patients with RET fusion–positive NSCLC are able to gain the expected benefits from the therapy.
The clinical expert explained that if patients have already received or are currently receiving treatment other than a RET-specific tyrosine kinase inhibitor due to a lack of availability of pralsetinib and selpercatinib at the time of initiation of first-line therapy, pralsetinib or selpercatinib should be used in the next line of therapy upon progression.
Patient Population
According to the clinical expert, all patients with incurable RET fusion–positive NSCLC can be expected to respond to pralsetinib. The clinical expert indicated that patients who are candidates for curative-intent therapy should not be offered pralsetinib. Multiple molecular tests are used to detect RET fusion: immunohistochemistry, fluorescence in situ hybridization, reverse transcriptase polymerase chain reaction amplification, and next-generation sequencing using either DNA or both DNA and RNA. Although testing technology has become more affordable in Canada, access to next-generation sequencing can still vary across jurisdictions, according to the clinical expert.
Assessing Response to Treatment
The clinical expert noted that radiographic assessments of patients receiving pralsetinib would generally be conducted every 8 to 12 weeks, or sooner if new symptoms or physical findings suggest progression. Clinical assessments of the presence and severity of symptoms and AEs would be conducted every 3 to 4 weeks.
According to the clinical expert, clinically meaningful responses to treatment would include improved survival, improved time to progression of disease, reduced symptom burden, increased functioning, and improved quality of life.
Discontinuing Treatment
According to the clinical expert, the factors that should be considered when deciding to discontinue treatment with pralsetinib are the presence of unacceptable adverse effects, patient preference, and symptomatic disease progression, with the exception of oligoprogression amenable to local intervention to achieve disease control or progression in the CNS amenable only to brain-targeted therapy such as radiation.
Prescribing Conditions
The clinical expert noted that the prescribing criteria should be consistent with those of selpercatinib. That is, pralsetinib should be prescribed by clinicians with expertise in NSCLC and should not be prescribed in combination with other systemic anticancer drugs.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Clinician group input on the review of pralsetinib for the treatment of RET fusion–positive locally advanced unresectable or metastatic NSCLC was received from 2 groups: LCC and the CCO Lung Cancer Drug Advisory Committee. The input was generally consistent with that supplied by the clinical expert. The submission from the CCO committee suggested that patients with an ECOG PS of 3 or greater would be least suitable for treatment with pralsetinib, whereas the clinical expert supported extending access to pralsetinib to patients with an ECOG PS of 2 or 3. The submission from LCC highlighted pandemic considerations and the potential for a reduced patient footprint in cancer centres that offer an oral therapy such as pralsetinib. Details of the clinician group input is provided in the Stakeholder Input document.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Comments from the drug plans (response not required):
| No response. For pERC consideration. |
The pERC recently reviewed and issued a draft recommendation for selpercatinib for the treatment of metastatic RET fusion–positive NSCLC. What is the comparative efficacy of pralsetinib vs. selpercatinib? | There is no evidence to suggest that one drug is more efficacious than the other. In practice, the side-effect profile of either drug would be compared to the medical history of the patient to determine the most suitable option. Beyond side-effect considerations, the 2 drugs are considered equivalent. |
Considerations for initiation of therapy | |
Initial eligibility criteria of the ARROW trial included patients with an ECOG PS of 0 to 2. Following a protocol amendment, eligibility was limited to patients with and ECOG PS of 0 or 1. Should patients with an ECOG PS of 2 or greater be eligible for pralsetinib? | Yes. Access to pralsetinib should be extended to patients with an ECOG PS of 2 or 3 as pralsetinib is a well-tolerated therapy with a significant likelihood of improving symptom burden, and therefore improving the ECOG PS. |
Initial eligibility criteria of the ARROW trial limited enrolment to patients who were previously treated with standard of care or who were treatment-naive and not candidates for available standard therapies. After the enrolment cut-off for efficacy analysis, a protocol amendment expanded eligibility to include treatment-naive patients regardless of whether they were candidates for standard therapies. Should pralsetinib be used in patients who are treatment-naive as well as those who have been previously treated? | Yes. All patients with RET fusion–positive NSCLC should be treated with pralsetinib, regardless of whether they have been pre-treated, as pralsetinib is more effective and less toxic than chemotherapy and immunotherapy checkpoint inhibitors. Based on those principles, it is most appropriate to use pralsetinib in the first line, or in the next line of therapy after progression on a current line of therapy. The only exception would be in a patient who had previous treatment with selpercatinib, in which case treatment with pralsetinib would not be appropriate. |
In the ARROW trial, patients with untreated CNS metastases were permitted if they had no progressive neurologic symptoms. Patients requiring corticosteroids for management of CNS disease must have been on a stable dose for 2 weeks or more before initiating pralsetinib. Should patients with stable CNS metastases be eligible for pralsetinib? | Pralsetinib is a drug with CNS activity. In the updated results from the ARROW trial, 10 patients had a brain metastasis. Seven of the 10 patients had responses in the brain (70%), 3 of which were complete responses. The remaining 3 patients had stable CNS disease, giving pralsetinib a 100% rate of disease control in the CNS. Pralsetinib is therefore an ideal drug for any patient with brain metastasis. |
Should the funding criteria for pralsetinib be aligned to that of selpercatinib? | Yes. They are highly comparable in terms of both efficacy and incidence of significant toxicity. Both should not be used in a single patient, but equal access to both should be offered to facilitate a choice for patients and oncologists and enhance the ability to provide best care. For example, there are some differences in adverse-effect profiles in which the option to use either drug would be important. Because selpercatinib is associated with a risk of developing a prolonged QT interval, while pralsetinib has no clinically relevant or significant effect on QT interval prolongation, pralsetinib would be a more appropriate choice in a patient with RET fusion–positive NSCLC and a pre-existing prolonged QT or who requires the use of concomitant medications that can prolong the QT interval. Pralsetinib can also cause pneumonitis, making selpercatinib a more appropriate choice in a patient with pre-existing limited pulmonary reserves or who already has pneumonitis from a different cause, such as palliative chest radiation. |
Considerations for discontinuation of therapy | |
In the trial, treatment after disease progression was allowed if this was the best medical interest of the patient as determined by the treating physician. What should the discontinuation criteria be for pralsetinib? | Unacceptable toxicity, clinical progression not amenable to local therapies such as radiation, and patient choice are appropriate discontinuation criteria. |
Considerations for prescribing of therapy | |
Comments from the drug plans (response not required):
| No response. For pERC consideration. |
Should prescribing criteria for pralsetinib align with selpercatinib? | Yes. The prescribing criteria should align with selpercatinib with the exception that pralsetinib should not be prescribed if the patient has previously progressed on selpercatinib. Intolerance to selpercatinib would not preclude the use of pralsetinib. |
Generalizability | |
Should patients currently receiving systemic therapy but whose disease has not yet progressed switch to pralsetinib? | No. Unless there is unacceptable toxicity or the patient decides they no longer want to receive treatment with a current line of therapy on which there has not been progression, that line of therapy should continue until progression, after which it would be appropriate to switch to pralsetinib. |
Funding algorithm | |
Comments from the drug plans (response not required):
| No response. For pERC consideration. |
| Pralsetinib and selpercatinib should not be sequenced. Pralsetinib, if funded, would be an alternative to selpercatinib. There are no significant differences in efficacy between selpercatinib and pralsetinib to suggest a superior option between the 2 on the basis of expected outcomes. However, in clinical circumstances, the differential adverse-effect profiles in the context of each patient may be critical in the choice between pralsetinib and selpercatinib. |
Care provision | |
Comments from the drug plans (response not required):
| No response. For pERC consideration. |
System and economic issues | |
Comments from the drug plans (response not required):
| No response. For pERC consideration. |
CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; NSCLC = non–small cell lung cancer; PD-1 = programmed cell death protein 1; PD-L1 = programmed death ligand 1; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The clinical evidence included in the review of pralsetinib is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes a summary of key ongoing clinical trials.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of pralsetinib 400 mg oral tablets for the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC Subgroups:
|
Intervention | Pralsetinib: 400 mg (four 100 mg oral tablets once daily) |
Comparator | First line:
Second line:
Metastatic only:
|
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, mortality, notable harms (pneumonitis/interstitial lung disease, hypertension, hepatoxicity, hemorrhagic events) |
Study designs | Published and unpublished phase III and phase IV randomized controlled trialsb |
AE = adverse event; ECOG PS = Eastern Cooperative Oncology Group Performance Status; SAE = serious adverse event; vs. = versus; WDAE = withdrawal due to adverse event;
aComparator identified by the drug programs.
bIf no phase III or phase IV trial, then phase II published and unpublished.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.24
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in EndNote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Gavreto (pralsetinib). Clinical trials registries searched included the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Appendix 1 provides detailed search strategies. The initial search was completed on March 31, 2022. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee on August 10, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.25 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Appendix 1 provides for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Findings From the Literature
One study identified from the literature met criteria for inclusion in the systematic review (Figure 1). The included study identified from other relevant sources is summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Figure 1: Flow Diagram for Inclusion and Exclusion of Studies
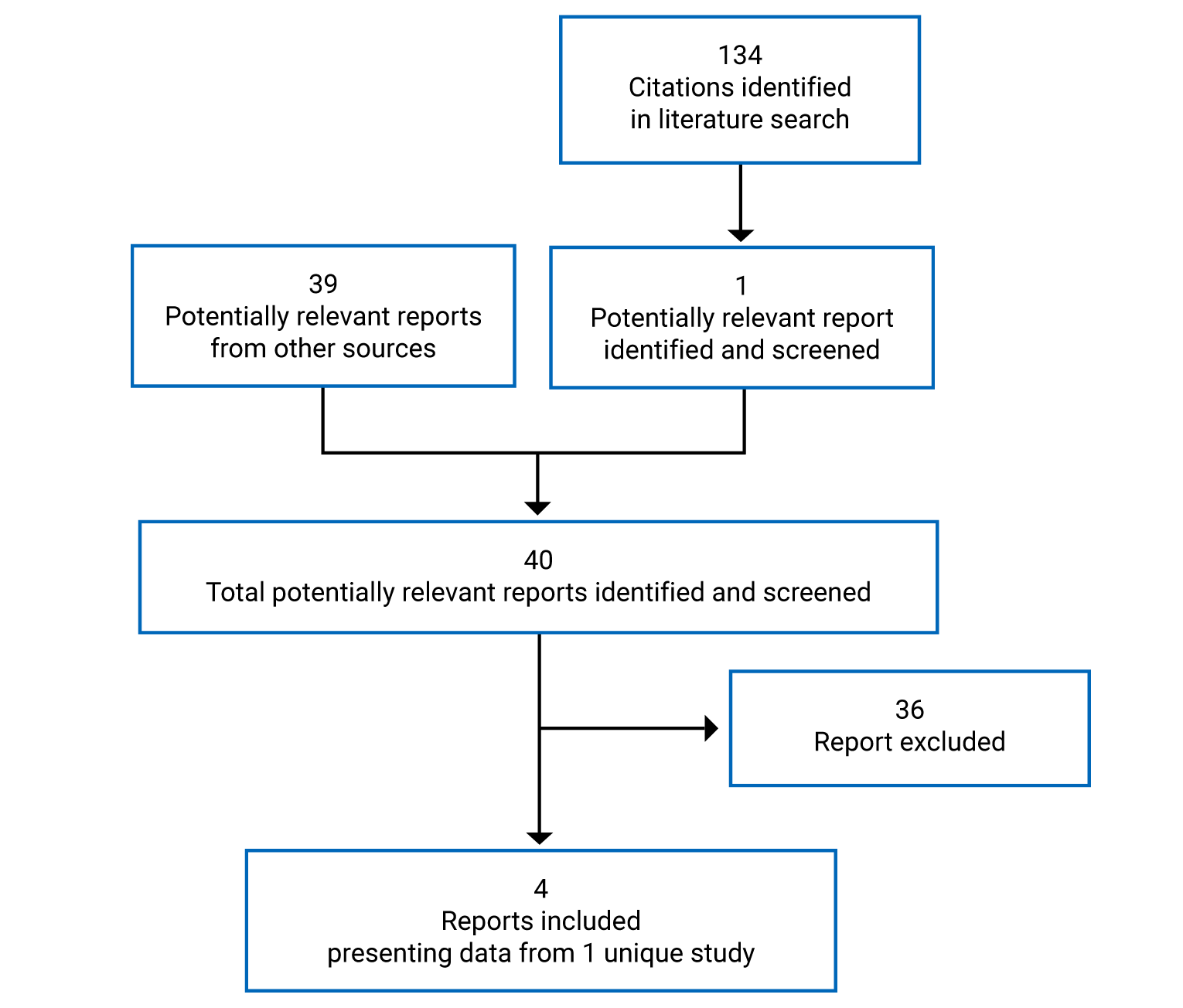
Source: ARROW Clinical Study Report,5 European Medicines Agency Pralsetinib Public Assessment Report,6 Gainor et al. (2021),26 Health Canada Reviewers Report.27
Table 6: Details of Included Studies
Characteristic | ARROW |
|---|---|
Designs and populations | |
Study design | Phase I and II, open-label study in patients with thyroid cancer, NSCLC, and other solid tumours. Phase I dose-escalation study to determine the MTD, followed by a phase II expansion to assess the clinical efficacy and further define safety and tolerability |
Locations | 53 centres (22 in Europe, 17 in the US, and 14 in Asia) |
Patient enrolment dates | First patient enrolled March 17, 2017 |
Final enrolment of all tumour types and all doses (N) | 528 |
RET fusion–positive, enrolled at 400 mg q.d. at the November 18, 2019, data cut-off (N) | |||||||||||||| |
RET fusion–positive, enrolled at 400 mg q.d. at the November 6, 2020, data cut-off (N) | 281 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs (phase II only) | |
Intervention | Pralsetinib 400 mg q.d. oral |
Comparator(s) | NA |
Duration (phase II only) | |
Phase | |
Screening | 4 weeks |
Treatment phase | There was no predefined maximum duration of treatment; patients received pralsetinib until precluded by toxicity, noncompliance, withdrawal of consent, death, or closure of the study by the sponsor; patients with progressive disease could remain on treatment if, in the opinion of the investigator, the patient has benefited from the pralsetinib therapy, and it was clearly in the best medical interest of the patient to remain on treatment |
Follow-up | Following completion of the end-of-study visit patients were followed up every 3 months for OS and patients without progressive disease were evaluated every 3 months until documentation of progressive disease |
Outcomes (phase II only) | |
Primary end point |
|
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publications | Gainor et al. (2021)24 |
CBR = clinical benefit rate; CNS = central nervous system; DCR = disease control rate; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; MTD = maximum tolerated dose; NA = not applicable; NSCLC = non–small cell lung cancer; ORR = overall response rate; OS = overall survival; q.d. = once daily; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1; ULN = upper limit of normal.
aSpecific to NSCLC cohorts within the ARROW trial.
Source: ARROW Clinical Study Report,5 Gainor et al. (2021).26
Description of Studies
The ARROW trial is an ongoing phase I and II, multi-centre, multi-cohort, single-arm open-label study of pralsetinib in patients with thyroid cancer, NSCLC, and other solid tumours with oncogenic RET fusions, consisting of a dose-escalation phase (phase I) and a dose-expansion phase (phase II). The first patient was enrolled on March 17, 2012, and the trial had 53 participating centres in the Europe, the US, and Asia at the time of the November 18, 2019, data cut-off. At that point, 179 patients had been recruited specifically in the NSCLC groups, and at the time of the November 6, 2020, data cut-off, 281 patients had been recruited in the NSCLC groups. Final enrolment in all groups and all tumour types was 528 patients. A schematic of the study design of both phases is shown in Figure 2.
Phase I: Dose Escalation
The objective of the dose-escalation phase was to determine the maximum tolerated dose and the recommended phase II dose (RP2D), along with safety and efficacy. The maximum tolerated dose was determined based on isotonic regression and was the dose for which the isotonic estimate of the toxicity rate, defined as a dose-limiting toxicity (grade 3 or greater AE), was closest to the target of 30%, although the RP2D could be chosen at a lower dose if clinical data warranted. The phase I portion of the study was completed on April 3, 2018, with the RP2D determined to be 400 mg once daily. Patients included in the phase I portion who had begun treatment at 400 mg once daily and met inclusion criteria for the phase II expansion were pooled in the final analysis. The focus of this report will be on the phase II expansion at the RP2D of 400 mg once daily, given that this is the Health Canada–approved dose. Phase I results for patients receiving doses other than 400 mg will not be expanded on further.
Phase II: Dose Expansion at Recommended Phase II Dose
The phase II portion of the ARROW trial is ongoing and includes patients from phase I who had been treated at the 400 mg once daily dosage in addition to newly recruited patients. Patients were recruited into 7 groups based on type of cancer and prior treatment history, as shown in Figure 2. As only groups 1 and 2 included patients with RET fusion–positive locally advanced and metastatic NSCLC, which is the indication of interest for this review, this report focuses solely on these groups, and results relating to patients in other groups are not expanded on further.
The primary objective of the phase II expansion was to determine the clinical efficacy of pralsetinib in patients with RET fusion–positive NSCLC, as measured by ORR, and to further define the safety and tolerability of pralsetinib at the 400 mg once daily dose. Two clinical data cut-offs are presented in this report, both of which were unplanned interim analyses conducted to support regulatory approval: the November 18, 2019, data cut-off presented in a provided clinical study report5 and the November 6, 2020, data cut-off that was summarized in a European Medicines Agency report.6 The efficacy population in both analyses was a subset of patients who had been enrolled at the time of data cut-off (July 11, 2019 and May 22, 2020, respectively) to allow for an appropriate amount of time for patients to achieve an ORR. Safety analysis was provided for all patients that had been enrolled up to each data cut-off.
There was no predefined duration of treatment; patients with progressive disease could remain on treatment if the investigator determined that it was in the best interest of the patient to do so.
Figure 2: ARROW Study Schematic
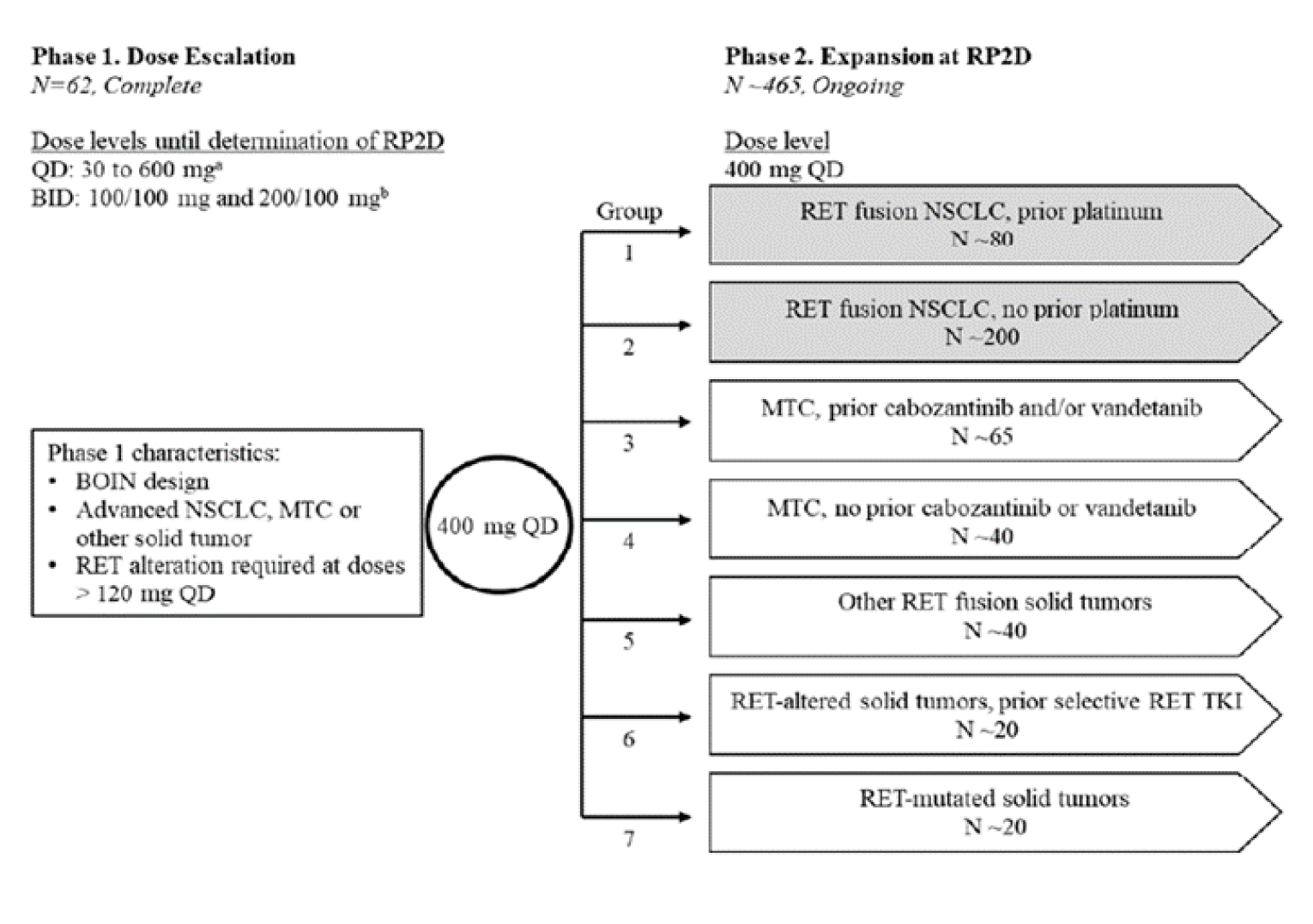
BID = twice daily; BOIN = Bayesian optimal interval; MTC = medullary thyroid cancer; NSCLC = non–small cell lung cancer; QD = once daily; RP2D = recommended phase II dose; TKI = tyrosine kinase inhibitor.
Source: ARROW Clinical Study Review.5
At the November 6, 2020, data cut-off a total of 41 patients (17.6%) had a major protocol deviation. Of these patients, 16 did not have measurable disease at baseline according to a blinded independent central review (BICR) and 1 patient had inconclusive evidence of RET fusion. Two of these 17 patients ultimately achieved a response.
Populations
Inclusion and Exclusion Criteria
Patients in the ARROW study, specifically those in the locally advanced and metastatic NSCLC groups, were required to be 18 years of age or older with an oncogenic RET fusion or mutation (excluding synonymous, frameshift, and nonsense mutations) and pathologically documented, definitively diagnosed, locally advanced or metastatic NSCLC. Patients with prior treatment experience were required to have been treated with a platinum-based chemotherapy, while patients recruited into the treatment-naive patients could not have received any prior systemic therapy. Prior platinum chemotherapy was permitted in the neo-adjuvant or adjuvant setting provided that the last dose was 4 months or more before the first dose of pralsetinib. Prior to the July 2019 protocol amendment, patients recruited into the treatment-naive group were required to have been deemed unsuitable for systemic therapy. Following the amendment, this requirement was removed. Patients were also required to have an ECOG PS of 0 or 1, and eligibility was adjusted following a July 2018 amendment to exclude patients with an ECOG PS of 2. Patients were required to have nonresectable and measurable disease as defined by the Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1).
Patients were excluded from the ARROW trial if a primary driver mutation other than RET was known to be present or if they had a primary CNS tumour or CNS metastases associated with progressive neurologic symptoms or that required increasing doses of corticosteroids to control. Patients with uncontrolled grade 3 or 4 congestive heart failure or clinically symptomatic interstitial lung disease were also excluded from the trial.
Baseline Characteristics
Baseline characteristics are shown in Table 7. There were 132 patients in the efficacy population at the November 18, 2019, data cut-off. The median age was ||||||||||||||, with slightly more females (||||||| compared with ||||||| males). Most patients were either Asian ||||||| or White |||||||. The efficacy population at the November 6, 2020, data cut-off included 233 patients. The median age in these patients remained 60 years, with similar proportions of the sexes (52.4% female and 47.6% male). At the data cut-off, 51.9% of patients were White and 39.5% of patients were Asian.
Baseline disease characteristics for the ARROW efficacy population are shown in Table 8. Almost all patients had tumours classified as adenocarcinomas, |||||||||||||| at the November 18, 2019, and November 6, 2020, data cut-offs, respectively. At the November 18, 2019, and November 6, 2020, data cut-offs, ||||||| and 37.3% of patients had a history of or current CNS metastasis, respectively. Nonlung lesion locations were evenly distributed at both data cut-offs. Few patients at either data cut-off presented with stage III disease, |||||||||||||| and 2.5% at the November 18, 2019, and November 6, 2020, data cut-offs, respectively. Most patients at both data cut-offs were never smokers, ||||||| and 62.2% at the November 18, 2019, and November 6, 2020, data cut-offs, respectively.
A summary of prior antineoplastic therapies is shown in Table 9. At the November 18, 2019, data cut-off, ||||||| of patients had received prior chemotherapy, ||||||| had received prior PD-1 and/or PD-L1 inhibitors, and |||||||||||||| had received prior multikinase inhibitors. At the November 6, 2020, data cut-off, 59.2% of patients had received prior chemotherapy, 29.6% had received prior PD-1 and/or PD-L1 inhibitors, and 18.9% had received prior multikinase inhibitors.
Table 7: Baseline Characteristics ARROW Efficacy Population
Characteristic | ARROW November 18, 2019, data cut-off Pralsetinib efficacy population N = 132 | ARROW November 6, 2020, data cut-off Pralsetinib efficacy population N = 233 |
|---|---|---|
Age, years | ||
Mean (SD) | ||||||||||||||||||||| | 59.2 (12.20) |
Median (range) | ||||||||||||||||||||| | 60.0 (26 to 87) |
< 65, n (%) | ||||||||||||||||||||| | 145 (62.2) |
≥ 65, n (%) | ||||||||||||||||||||| | 88 (37.8) |
Sex, n (%) | ||
Male | ||||||||||||||||||||| | 111 (47.6) |
Female | ||||||||||||||||||||| | 122 (52.4) |
Ethnicity, n (%) | ||
Hispanic or Latino | ||||||||||||||||||||| | 9 (3.9) |
Not Hispanic or Latino | ||||||||||||||||||||| | 201 (86.3) |
Not reported | ||||||||||||||||||||| | 6 (2.6) |
Unknown | ||||||||||||||||||||| | 17 (7.3) |
Race, n (%) | ||
Asian | ||||||||||||||||||||| | 92 (39.5) |
Native Hawaiian or other Pacific Islander | ||||||||||||||||||||| | 2 (0.9) |
White | ||||||||||||||||||||| | 121 (51.9) |
Unknown | ||||||||||||||||||||| | 16 (6.9) |
Other | ||||||||||||||||||||| | 2 (0.9) |
Body mass index, kg/m2 | ||
Mean (SD) | ||||||||||||||||||||| | NR |
Median, (range) | ||||||||||||||||||||| | NR |
Body surface area, m2 | ||
Mean (SD) | ||||||||||||||||||||| | NR |
Median (range) | ||||||||||||||||||||| | NR |
NR = not reported; SD = standard deviation.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Table 8: Baseline Disease Characteristics
Characteristic | ARROW November 18, 2019, data cut-off Pralsetinib efficacy population N = 132 | ARROW November 6, 2020, data cut-off Pralsetinib efficacy population N = 233 |
|---|---|---|
ECOG Performance Status | ||
0 | ||||||||||||||||||||| | 78 (33.5) |
1 | ||||||||||||||||||||| | 149 (63.9) |
2 | ||||||||||||||||||||| | 6 (2.6) |
Histology type | ||
Adenocarcinoma | ||||||||||||||||||||| | 224 (96.1) |
Squamous | ||||||||||||||||||||| | 3 (1.3) |
Undifferentiated | ||||||||||||||||||||| | 1 (0.4) |
Other | ||||||||||||||||||||| | 5 (2.1) |
CNS metastasis (history or current) | ||||||||||||||||||||| | 87 (37.3) |
Target/nontarget lesion location | ||
Lung | ||||||||||||||||||||| | 195 (83.7) |
Bone | ||||||||||||||||||||| | 86 (36.9) |
Mediastinal adenopathy | ||||||||||||||||||||| | 92 (39.5) |
CNS (brain) | ||||||||||||||||||||| | 64 (27.5) |
Liver | ||||||||||||||||||||| | 52 (22.3) |
Pleural | ||||||||||||||||||||| | 49 (21.0) |
Hilar adenopathy | ||||||||||||||||||||| | 32 (13.7) |
TNM stage at screening | ||
Stage IIBa | ||||||||||||||||||||| | 1 (0.4) |
Stage IIIA | ||||||||||||||||||||| | 1 (0.4) |
Stage IIIB | ||||||||||||||||||||| | 3 (1.3) |
Stage IIIC | ||||||||||||||||||||| | 1 (0.4) |
Stage IV | ||||||||||||||||||||| | 109 (46.8) |
Stage IVA | ||||||||||||||||||||| | 40 (17.2) |
Stage IVB | ||||||||||||||||||||| | 72 (30.9) |
Stage IVC | ||||||||||||||||||||| | 6 (2.6) |
Smoking history | ||
Never smoker | ||||||||||||||||||||| | 145 (62.2) |
Former | ||||||||||||||||||||| | 78 (33.5) |
Current | ||||||||||||||||||||| | 6 (2.6) |
Unknown | ||||||||||||||||||||| | 4 (1.7) |
RET alteration | ||
RET fusion | ||||||||||||||||||||| | NR |
KIF5B | ||||||||||||||||||||| | 164 (70.4) |
CCDC6 | ||||||||||||||||||||| | 41 (17.6) |
NCOA4 | ||||||||||||||||||||| | 1 (0.4) |
Other | ||||||||||||||||||||| | 27 (11.6) |
CNS = central nervous system; ECOG = Eastern Cooperative Oncology Group; NR = not reported; TNM = tumour node metastasis.
aPatient had recurrent non–small cell lung cancer and was considered unfit for surgery.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Table 9: Prior Antineoplastic Therapies
Prior therapy | ARROW November 18, 2019, data cut-off Pralsetinib efficacy population N = 132 | ARROW November 6, 2020, data cut-off Pralsetinib efficacy population N = 233 |
|---|---|---|
Patients with any prior antineoplastic therapy | ||||||||||||||||||||| | NR |
Chemotherapy | ||||||||||||||||||||| | 138 (59.2) |
Platinum-based chemotherapy | ||||||||||||||||||||| | 136 (58.4) |
PD-1/PD-L1 inhibitors | ||||||||||||||||||||| | 69 (29.6) |
MKIs | ||||||||||||||||||||| | 44 (18.9) |
Cabozantinib or vandetanib | ||||||||||||||||||||| | NR |
Other MKI except cabozantinib and vandetanib | ||||||||||||||||||||| | NR |
Others | ||||||||||||||||||||| | NR |
Prior radiation therapy | ||||||||||||||||||||| | 90 (38.6) |
Prior cancer-related surgeries or procedures | ||||||||||||||||||||| | 116 (49.8) |
MKI = multikinase inhibitor; NR = not reported; PD-1 = programmed cell death protein 1; PD-L1 = programmed death ligand 1.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Characteristic | ARROW November 18, 2019, data cut-off Pralsetinib efficacy population N = 132 | ARROW November 6, 2020, data cut-off Pralsetinib efficacy population N = 233 |
|---|---|---|
Patients with any ongoing medical history | 130 (98.5) | NR |
Hypertension | 45 (34.1) | NR |
Cough | 35 (26.5) | NR |
Dyspnea | 33 (25.0) | NR |
Fatigue | 34 (25.8) | NR |
Back pain | 26 (19.7) | NR |
Menopause | 25 (18.9) | NR |
Anxiety | 26 (19.7) | NR |
Decreased appetite | 17 (12.9) | NR |
Headache | 16 (12.1) | NR |
Anemia | 17 (12.9) | NR |
Constipation | 28 (21.2) | NR |
Gastro-esophageal reflux disease | 18 (13.6) | NR |
Hypothyroidism | 16 (12.1) | NR |
Nausea | 20 (15.2) | NR |
Insomnia | 22 (16.7) | NR |
NR = not reported.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
The medical history for the efficacy population at the November 18, 2019, data cut-off is summarized in Table 10. The most common conditions in patients’ medical histories were hypertension (34.1%), cough (26.5%), dyspnea (25.0%), and fatigue (25.8%). Medical history was not available for the November 6, 2020, data cut-off.
Interventions
Patients received pralsetinib orally as 100 mg capsules at a dosage of 400 mg once daily. As the ARROW trial was a single-arm phase I and II trial, there was no randomization and all patients enrolled into the phase II dose expansion were assigned the RP2D determined in phase I of the trial (400 mg once daily). Dose reductions by 100 mg increments, but to no lower than a 100 mg total dose, were permitted in the case of grade 3 or higher AEs. If reductions were required below this level, the patient was discontinued from treatment. Dose interruptions were permitted for 4 weeks; however, if the AE that led to discontinuation did not resolve to grade 2 or lower the patient was discontinued from treatment. Temporary interruptions up to 2 weeks were permitted for patients requiring surgery or other procedures.
Prohibited concomitant medications in the ARROW trial included strong inhibitors and inducers of CYP3A4, any investigational drug other than pralsetinib, any antineoplastic agent other than pralsetinib, and neutrophil growth factor, unless the patient experienced dose-limiting toxicity (grade 4) associated with neutropenia.
Use of concomitant medications in the ARROW trial at the November 18, 2019, data cut-off is summarized in Table 11. The most commonly reported concomitant medications were other analgesics and antipyretics |||||||||, opioids (|||||||||), drugs for constipation (|||||||||), and drugs for peptic ulcer and gastro-esophageal reflux disease (|||||||||). Concomitant medication information was not available for the November 6, 2020, data cut-off.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 12. These end points are further summarized below. A detailed discussion and a critical appraisal of the outcome measures are provided in Appendix 4.
Table 11: Concomitant Medications
Concomitant medication | ARROW November 18, 2019, data cut-off Pralsetinib efficacy population N = 132 | ARROW November 6, 2020, data cut-off Pralsetinib efficacy population N = 233 |
|---|---|---|
Patients with any concomitant medication | ||||||||| | NR |
Other analgesics and antipyretics | ||||||||| | NR |
Opioids | ||||||||| | NR |
Drugs for constipation | ||||||||| | NR |
Drugs for peptic ulcer and gastro-esophageal reflux disease | ||||||||| | NR |
Corticosteroids for systemic use, plain | ||||||||| | NR |
antithrombotic agents | ||||||||| | NR |
Beta-lactam antibacterials, penicillins | ||||||||| | NR |
Other beta-lactam antibacterials | ||||||||| | NR |
Anxiolytics | ||||||||| | NR |
IV solutions | ||||||||| | NR |
Quinolone antibacterials | ||||||||| | NR |
Selective calcium channel blockers with mainly vascular effects | ||||||||| | NR |
Vitamin B12 and folic acid | ||||||||| | NR |
Antiemetics and antinauseants | ||||||||| | NR |
Other antibacterials | ||||||||| | NR |
Potassium | ||||||||| | NR |
Blood and related products | ||||||||| | NR |
Vitamin A and D (including combinations of the 2) | ||||||||| | NR |
High-ceiling diuretics | ||||||||| | NR |
Antiepileptic | ||||||||| | NR |
NR = not reported.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Table 12: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | ARROW |
|---|---|
Overall survival | Secondary |
Health-related quality of life | Exploratory |
Progression-free survival | Secondary |
Overall response rate | Primary |
Overall response rate – intracranial response | Exploratory |
Duration of response | Secondary |
Overall survival was defined as the time from the first dose of pralsetinib to the date of death due to any cause. Patients who were still alive or lost to follow-up at the time of data cut-off were censored according to the rules listed in Table 13 at the last known time alive.
The EORTC QLQ-C30, a 30-item, patient-reported, cancer-specific questionnaire using 4- and 7-point Likert scales was used to measure HRQoL. All scales and single-item measures are scored from 0 to 100. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” and “very much”), with scores on these items ranging from 1 to 4. For the 2 items that form the global HRQoL scale, the response format is a 7-point Likert-type scale with anchors at 1 (“very poor”) and 7 (“excellent”). Validity, responsiveness, and reliability have been demonstrated in NSCLC patients. An MID of 5 to 10 points corresponded with a small clinical change, 10 to 20 points with a moderate clinical change, and greater than 20 points with a large clinical change.
Progression-free survival was defined as the time from the first dose of pralsetinib to the date of first documented progressive disease or death due to any cause. Patients without documented progression at the time of data cut-off were censored according to the rules listed in Table 13.
The ORR was defined, according to RECIST 1.1 or a response assessment in neuro-oncology if appropriate, as the proportion of patients with a complete response (CR) or partial response (PR) for at least 2 assessments separated by at least 28 days with no documented progressive disease in between. The ORR was analyzed according to a BICR. The ORR in brain or CNS lesions were also assessed according to RECIST v1.1.
The DOR was analyzed within the population of patients with confirmed CR or PR. It was defined as the time from first documented CR or PR to the date of first documented progressive disease or death due to any cause. Patients still in response at the time of data cut-off were censored according to the rule listed in Table 13.
Table 13: Censoring Rules in ARROW
Situation | Date of progression or censoring | FDA censoring rule |
|---|---|---|
No baseline assessments and alive after 2 scheduled assessments (at least 128 days) | Date of first dose of treatment | Censored |
Progression documented between scheduled visits | Date of radiological assessment showing progression | Event |
No progression | Date of last radiological assessment with evidence of no progression (or first dose date if no assessment) | Censored |
New antineoplastic/nonprotocol treatment started before progression | Date of last radiological assessment with evidence of no progression before the start of new antineoplastic treatment | Censored |
Death before the second scheduled post-baseline assessment if the first scheduled post-baseline assessment is not progressive disease (defined as 128 days after first dose) | Date of death | Event |
Death between scheduled assessments | Date of death | Event |
Death or progression after missing 2 or more consecutively scheduled disease assessments (2 more missed scheduled assessments defined by at least 128 days if before EOT visit, 197 days if after EOT visit) | Date of last radiological assessment with evidence of no progression before death/progression | Censored |
EOT = end of trial.
Statistical Analysis
Twelve protocol amendments were reported for the ARROW trial. Of these 12, the following were particularly noteworthy and associated with a consequential impact. Protocol amendment 4.1, made July 25, 2018, removed eligibility for patients with an ECOG PS of 2 to enrol in the ARROW trial. Protocol amendment 9, made July 3, 2019, removed the need for patients enrolled in the treatment-naive group to be deemed unsuitable for SOC chemotherapy.
Sample-size calculations were conducted separately for treatment-experienced and treatment-naive patient groups in the ARROW study. For treatment-experienced patients with NSCLC, approximately 80 patients would provide greater than 95% power at the 2-sided significance level of 0.05, for which the null hypothesis was an ORR of 0.23 compared to the alternative hypothesis of an ORR of 0.5. For treatment-naive patients, approximately 170 patients would provide greater than 90% power to test the null hypothesis of an ORR of 0.48 compared to the alternative hypothesis of an ORR of 0.61. No interim analyses were planned for the ARROW trial and no adjustments for multiplicity applied to multiple end points or multiple data cut-offs were analyzed.
Patients enrolled in phase I and treated at the RP2D were pooled together with the appropriate phase II patient groups for analyses. A description of the statistical analysis models used in the ARROW trial is provided in Table 14.
Table 14: Statistical Analysis of Efficacy End Points
End point | Statistical model | Sensitivity analyses |
|---|---|---|
Primary end point | ||
Overall response rate | Two-sided 95% CI based on Clopper-Pearson exact binomial distribution | Sensitivity analysis conducted for the response-evaluable population who had no major protocol violations |
Secondary end points | ||
Overall survival, progression-free survival, duration of response | Analyzed using Kaplan-Meier methods with estimated median and 2-sided 95% confidence interval provided; results at specific time points analyzed with standard error according to the Greenwood formula | NA |
NA = not applicable.
Analysis Populations
Efficacy population from the November 18, 2019, data cut-off (N = 132) included all patients who received a dose of pralsetinib on or before July 11, 2019. The efficacy population from the November 6, 2020, data cut-off (N = 233) included all patients who received a dose of pralsetinib on or before May 22, 2020.
The safety population included all patients who had received 1 or more doses of pralsetinib 400 mg once daily at the time of data cut off. At the November 18, 2019, data cut-off, the safety population included 179 patients; at the November 6, 2020, data cut-off, the population had grown to 281 patients.
The responder analysis set included all patients with a confirmed response within the efficacy population. The responder analysis set was used to determine a DOR. At the November 18, 2019, data cut-off set this included 75 patients; at the November 6, 2020, data cut-off, the set had grown to 150 patients.
The response-evaluable population was used for a sensitivity analysis conducted on a subset of patients who did not have major protocol violations (including incomplete baseline imaging), evidence of RET mutation, or a known primary driver other than RET. At the November 18, 2019, data cut-off, the response-evaluable population was 163 patients; at the November 6, 2020, data cut-off the population had grown to 216 patients. As this was not a predefined subgroup analysis, the results are not shown in this report.
Results
Patient Disposition
A summary of patient disposition in the ARROW trial at the November 18, 2019, and November 6, 2020, data cut-offs is presented in Table 15. At the November 18, 2019, data cut-off, 404 patients with all tumour types had been enrolled and received treatment with pralsetinib. Of these, 179 patients had RET fusion–positive NSCLC and received treatment at 400 mg once daily, representing the safety analysis population. From the safety population, 3 patients were originally enrolled in the phase I portion of the trial. The efficacy population included 132 of these patients who had received their first dose on or before July 11, 2019. At the time of data cut-off, 49.2% patients had discontinued treatment, most commonly citing disease progression; however, 18.2% of patients discontinued due to AEs. Discontinuation of the study occurred in 44.7% of patients, with death and disease progression being the most common reasons, and withdrawal of consent the next most common reason (7.6%).
At the November 6, 2020, data cut-off, 587 patients with all tumour types had been screened, and 521 enrolled and received treatment. Of these, 281 patients had RET fusion–positive NSCLC and received treatment at 400 mg once daily, representing the safety population. The efficacy population included 233 of these patients who had received treatment on or before May 22, 2020. At the time of data cut-off, 52.8% of patients had discontinued treatment, mostly due to disease progression; however, 14.6% discontinued due to AEs and 43.8% had discontinued from the study, with death and disease progression being the most common reasons, and withdrawal of consent the next most common reason (6.9%).
Exposure to Study Treatments
Treatment exposure in the ARROW trial safety population is summarized in Table 16. At the November 18, 2019, data cut-off, the median duration of exposure was ||||||||||||||||||) weeks with a median relative dose intensity, defined as actual dose divided by initial assigned dose times 100, of ||||||||||||||||||. At the November 6, 2020, data cut-off, the median duration of exposure was 7.89 (range = 0.3 to 28.4) months with a median relative dose intensity of 92.1% (range = 27% to 100%).
Disposition | ARROW | |
|---|---|---|
November 18, 2019, data cut-off | November 6, 2020, data cut-off | |
Screened, N | ||||||||| | 587 |
Enrolled and received treatment, N | ||||||||| | 521 |
RET fusion–positive NSCLC enrolled and received 400 mg q.d., N | ||||||||| | 281 |
RET fusion–positive NSCLC enrolled and received 400 mg q.d. before efficacy enrolment cut-off,a N | ||||||||| | 233 |
Continuing treatment, N (%) | ||||||||| | 110 (47.2) |
Discontinued from treatment, N (%) | ||||||||| | 123 (52.8) |
Reason for discontinuation, N (%) | ||
Disease progression | ||||||||| | 74 (31.8) |
Adverse events | ||||||||| | 34 (14.6) |
Withdrew consent | ||||||||| | 10 (4.3) |
Investigator’s decision | ||||||||| | 3 (1.3) |
Lost to follow-up | ||||||||| | 0 |
Administrative/other | ||||||||| | 2 (0.9) |
Continuing study follow-up, N (%) | ||||||||| | 131 (56.2) |
Discontinued from study, N (%) | ||||||||| | 102 (43.8) |
Reason for discontinuation, N (%) | ||
Disease progression | ||||||||| | 25 (10.7) |
Adverse events | ||||||||| | 2 (0.9) |
Death | ||||||||| | 55 (23.6) |
Withdrew consent | ||||||||| | 16 (6.9) |
Investigator’s decision | ||||||||| | 0 |
Initiation of another antineoplastic agent | ||||||||| | 2 (0.9) |
Lost to follow-up | ||||||||| | 2 (0.9) |
Efficacy population, N | ||||||||| | 233 |
Safety, N | ||||||||| | 281 |
Responder analysis set, N | ||||||||| | 150 |
NSCLC = non–small cell lung cancer; q.d. = once daily.
aThe safety analysis includes all patients who received pralsetinib at a dosage of 400 mg once daily. Safety analysis conducted for all pralsetinib doses is not shown in this report.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Table 16: Treatment Exposure in ARROW Safety Population
Detail | ARROW November 18, 2019, data cut-off Pralsetinib safety population (N = 179) | ARROW November 6, 2020, data cut-off Pralsetinib safety population (N = 281) |
|---|---|---|
Duration of exposure, weeks | ||
Median (range) | |||||||||||||||||| | 7.89a (0.3 to 28.4) |
Relative dose intensity (%) | ||
Median (range) | |||||||||||||||||| | 92.1 (27 to 100) |
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
aDuration of exposure measured in months.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Appendix 3 provides detailed efficacy data.
Overall Survival
Overall survival in the ARROW trial at the November 18, 2019, and November 6, 2020, data cut-offs is summarized in Table 17. At the November 18, 2019, data cut-off the median OS follow-up time was 10.5 (95% CI, 9.7 to 13.1) months. In the efficacy population, 19.7% of patients had died and median OS had not been reached. Kaplan-Meier estimates of OS at |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| respectively. Results were similar when analysis was conducted separately for treatment-experienced and treatment-naive patients.
At the November 6, 2020, data cut-off the median, the OS follow-up time was 17.1 (95% CI, 13.7 to 19.6) months. In the efficacy population, 24.5% of patients had died and the median OS had not been reached. Kaplan-Meier estimates of OS at 3, 6, 12, and 24 months were 96.0% (95% CI, 93.5 to 98.6), 87.6% (95% CI, 83.2 to 92.0), 76.0% (95% CI, 69.9 to 82.0) and 66.0% (95% CI, 57.9 to 74.1), respectively. Results were similar when treatment-experienced and treatment-naive patients were analyzed separately. A separate analysis for OS was conducted on the safety population of all patients who had received a dose of pralsetinib up to the data cut-off. In this population of 281 patients, the median follow-up time was 13.2 month (95% CI, 12.1 to 15.5) and the median OS was not reached.
Health-Related Quality of Life
The HRQoL as measured by global health status on the EORTC QLQ-C30 at the November 18, 2019, data cut-off is summarized in Table 18. Of the 150 patients included in the HRQoL analysis set, baseline results were available for |||||||||. The mean baseline value was ||||||||||||||||||. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The HRQoL results from the November 6, 2020, data cut-off were not available.
Table 17: Overall Survival Analysis in the ARROW Trial
Detail | ARROW November 18, 2019, data cut-off | ARROW November 6, 2020, data cut-off | ||||
|---|---|---|---|---|---|---|
Pralsetinib efficacy population N = 132 | Pralsetinib prior systemic treatment N = 103 | Pralsetinib no prior systemic therapy N = 29 | Pralsetinib efficacy population N = 233 | Pralsetinib prior systemic treatment N = 158 | Pralsetinib no prior systemic therapy N = 75 | |
Deaths | ||||||||| | ||||||||| | ||||||||| | 57 (24.5) | 45 (28.5) | 12 (16.0) |
Censored | ||||||||| | ||||||||| | ||||||||| | 176 (75.5) | 113 (71.5) | 63 (84.0) |
Alive | ||||||||| | ||||||||| | ||||||||| | 158 (67.8) | 100 (63.3) | 58 (77.3) |
Lost to follow-up | ||||||||| | ||||||||| | ||||||||| | 2 (0.9) | 1 (0.6) | 1 (1.3) |
Withdrawal of consent | ||||||||| | ||||||||| | ||||||||| | 16 (6.9) | 12 (7.6) | 4 (5.3) |
Kaplan-Meier estimates, months (95% CI)a | ||||||
OS follow-up time | ||||||||| | ||||||||| | ||||||||| | 17.1 (13.7 to 19.6) | 20.1 (19.0 to 21.5) | 12.8 (11.1 to 15.0) |
Median OS | ||||||||| | ||||||||| | ||||||||| | NR (NR to NR) | NR (NR to NR) | NR (NR to NR) |
Kaplan-Meier estimates of overall survival at time points, % (95% CI)a | ||||||
3 months | ||||||||| | ||||||||| | ||||||||| | 96.0 (93.5 to 98.6) | 96.1 (93.0 to 99.2) | 96.0 (91.6 to 100.0) |
6 months | ||||||||| | ||||||||| | ||||||||| | 87.6 (83.2 to 92.0) | 85.6 (79.9 to 91.3) | 91.7 (85.4 to 98.1) |
9 months | ||||||||| | ||||||||| | ||||||||| | 80.9 (75.5 to 86.2) | 75.8 (68.6 to 82.9) | 91.7 (85.4 to 98.1) |
12 months | ||||||||| | ||||||||| | ||||||||| | 76.0 (69.9 to 82.0) | 72.5 (64.9 to 80.0) | 82.3 (71.9 to 92.8) |
18 months | ||||||||| | ||||||||| | ||||||||| | 69.8 (62.5 to 77.1) | 67.4 (58.9 to 75.9) | 74.0 (59.3 to 88.6) |
24 months | ||||||||| | ||||||||| | ||||||||| | 66.0 (57.9 to 74.1) | 63.2 (54.0 to 72.4) | 74.0 (59.3 to 88.6) |
CI = confidence interval; NA = not applicable; NR = not reached.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
a95% CI based on the Greenwood formula.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Table 18: EORTC QLQ-C30 Global Health Status in the ARROW Trial
Detail | ARROW November 18, 2019, data cut-off, Pralsetinib HRQoL analysis set N = 150 | |||
|---|---|---|---|---|
Score | Change from baseline | |||
N | Mean (SD) | N | Mean (SD) | |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
HRQoL = health-related quality of life; SD = standard deviation
Note: The November 18, 2019, was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
Source: ARROW Clinical Study Review.5
Progression-Free Survival
Progression-free survival in the ARROW trial at the November 18, 2019, and November 6, 2020, data cut-offs is summarized in Table 19. At the November 18, 2019, data cut-off, in the efficacy population, ||||||||| of patients had experienced an event and median PFS was |||||||||||||||||| months. Kaplan-Meier estimates of PFS at |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| respectively. Results were similar when analyses were conducted separately for treatment-experienced and treatment-naive patients.
At the November 6, 2020, data cut-off, 43.8% of patients in the efficacy population had experienced an event and the median PFS was 16.4 months (95% CI, 11.0 to 24.1). Kaplan-Meier estimates of PFS at 3, 6, 12, and 24 months were 90.7% (95% CI, 86.9 to 94.5), 75.1% (95% CI, 69.3 to 80.9), 56.0% (95% CI, 48.9 to 63.1), and 42.1% (95% CI, 33.2 to 51.0), respectively. Results were similar when analyses were conducted separately for treatment-experienced and treatment-naive patients. A separate analysis for PFS was conducted on all 281 patients in the safety population who had received a dose of pralsetinib up to the data cut-off. In this population, 37.7% of patients had experienced an event and the median PFS was 16.4 (95% CI, 11.0 to 24.1) months.
Table 19: Progression-Free Survival Analysis in the ARROW Trial
Detail | ARROW November 18, 2019, data cut-off | ARROW November 6, 2020, data cut-off | ||||
|---|---|---|---|---|---|---|
Pralsetinib efficacy population N = 132 | Pralsetinib prior systemic treatment N = 103 | Pralsetinib no prior systemic therapy N = 29 | Pralsetinib efficacy population N = 233 | Pralsetinib prior systemic treatment N = 158 | Pralsetinib no prior systemic therapy N = 75 | |
Censored | ||||||||| | ||||||||| | ||||||||| | 131 (56.2) | 83 (52.5) | 48 (64.0) |
Patients with event | ||||||||| | ||||||||| | ||||||||| | 102 (43.8) | 75 (47.5) | 27 (36.0) |
PD | ||||||||| | ||||||||| | ||||||||| | 77 (33.0) | 55 (34.8) | 22 (29.3) |
Death without PD before first scheduled assessment | ||||||||| | ||||||||| | ||||||||| | 8 (3.4) | 5 (3.2) | 3 (4.0) |
Death without PD before second scheduled assessment | ||||||||| | ||||||||| | ||||||||| | 11 (4.7) | 8 (5.1) | 3 (4.0) |
Death without PD after second scheduled assessment | ||||||||| | ||||||||| | ||||||||| | 14 (6.0) | 12 (7.6) | 2 (2.7) |
Kaplan-Meier estimates, months (95% CI)a | ||||||
Median PFS | ||||||||| | ||||||||| | ||||||||| | 16.4 (11.0 to 24.1) | 16.4 (10.7 to 24.1) | 13.0 (9.1 to NR) |
Kaplan-Meier estimates of PFS at time points, % (95% CI)a | ||||||
3 months | ||||||||| | ||||||||| | ||||||||| | 90.7 (86.9 to 94.5) | 90.8 (86.1 to 95.4) | 90.5 (83.8 to 97.2) |
6 months | ||||||||| | ||||||||| | ||||||||| | 75.1 (69.3 to 80.9) | 72.7 (65.5 to 79.9) | 80.2 (70.9 to 89.6) |
9 months | ||||||||| | ||||||||| | ||||||||| | 64.7 (58.2 to 71.2) | 62.5 (54.6 to 70.5) | 69.5 (58.1 to 80.9) |
12 months | ||||||||| | ||||||||| | ||||||||| | 56.0 (48.9 to 63.1) | 56.3 (48.0 to 64.5) | 52.6 (37.7 to 67.5) |
18 months | ||||||||| | ||||||||| | ||||||||| | 46.7 (38.8 to 54.7) | 46.5 (37.5 to 55.5) | 47.8 (31.6 to 64.1) |
24 months | ||||||||| | ||||||||| | ||||||||| | 42.1 (33.2 to 51.0) | 41.6 (31.8 to 51.3) | 47.8 (31.6 to 64.1) |
CI = confidence interval; NA = not applicable; NR = not reached; PD = progressive disease; PFS = progression-free survival.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
a95% CI based on the Greenwood formula.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Overall Response Rate
Table 20 summarizes the ORR results at the November 18, 2019, and November 6, 2020, data cut-offs from the ARROW trial. At the November 18, 2019, data cut-off, in the efficacy population, the ORR was ||||||||||||||| including ||||||||| of patients with a CR. The ORR was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||) compared with patients |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
At the November 6, 2020, data cut-off, the ORR in the efficacy population was 64.4% (95% CI, 52.7 to 68.4), including 4.7% of patients with a CR. The ORR was higher in patients with no prior systemic therapy (72.0%; 95% CI, 60.4 to 81.8) compared with patients with prior systemic therapy (60.8%; 95% CI, 52.7 to 68.4). These results successfully reject the pre-specified null hypothesis for treatment effect at both data cut-offs. A summary of the maximum change from baseline in target lesions for patients who had not received prior systemic therapy is shown in Figure 3, and in Figure 4 for patients who had received prior platinum therapy.
Table 21 summarizes the intracranial ORR results at the November 18, 2019, and November 6, 2020, data cut-offs from the ARROW trial. For the 9 response-evaluable patients with measurable intracranial metastasis at baseline at the November 18, 2019, data cut-off, the ORR was ||||||||||||||||||||||||||||||||||||, including ||||||||| of these patients achieving a CR. Among the ||||||||| patients who did not have a history of CNS metastasis at study entry, ||||||||| developed CNS metastasis while on pralsetinib 400 mg once daily at the November 18, 2019, data cut-off. At the November 6, 2020, data cut-off, for the 10 patients with measurable intracranial metastasis at baseline, the ORR was 70.0% (95% CI, 34.8 to 93.3), including 30.0% of patients achieving a CR.
Table 20: Overall Response Rate Analysis in the ARROW Trial
Detail | ARROW November 18, 2019, data cut-off | ARROW November 6, 2020, data cut-off | ||||
|---|---|---|---|---|---|---|
Pralsetinib efficacy population N = 132 | Pralsetinib prior systemic treatment N = 103 | Pralsetinib no prior systemic therapy N = 29 | Pralsetinib efficacy population N = 233 | Pralsetinib prior systemic treatment N = 158 | Pralsetinib no prior systemic therapy N = 75 | |
Overall response rate, n (%) | ||||||||| | ||||||||| | ||||||||| | 150 (64.4) | 96 (60.8) | 54 (72.0) |
95% CIa | ||||||||| | ||||||||| | ||||||||| | (57.9 to 70.5) | (52.7 to 68.4) | (60.4 to 81.8) |
Complete response, n (%) | ||||||||| | ||||||||| | ||||||||| | 11 (4.7) | 7 (4.4) | 4 (5.3) |
Partial response, n (%) | ||||||||| | ||||||||| | ||||||||| | 139 (59.7) | 89 (56.3) | 50 (66.7) |
Stable disease, n (%) | ||||||||| | ||||||||| | ||||||||| | 61 (26.2) | 47 (29.7) | 14 (18.7) |
Progressive disease, n (%) | ||||||||| | ||||||||| | ||||||||| | 13 (5.6) | 8 (5.1) | 5 (6.7) |
NE, n (%) | ||||||||| | ||||||||| | ||||||||| | 9 (3.9) | 7 (4.4) | 2 (2.7) |
CI = confidence interval; NE = not evaluable.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
a95% CI based on exact binomial distribution using Clopper-Pearson method.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Table 21: Intracranial Overall Response Rate Analysis in the ARROW Trial
Detail | ARROW November 18, 2019, data cut-off | ARROW November 6, 2020, data cut-off | ||||
|---|---|---|---|---|---|---|
Pralsetinib N = 9 | Pralsetinib prior systemic treatment N = 9 | Pralsetinib no prior systemic therapy N = 0 | Pralsetinib N = 10 | Pralsetinib prior systemic treatment N = 10 | Pralsetinib no prior systemic therapy N = 0 | |
Overall response rate, n (%) | ||||||||| | ||||||||| | ||||||||| | 7 (70.0) | 7 (70.0) | NA |
95% CIa | ||||||||| | ||||||||| | ||||||||| | (34.8 to 93.3) | (34.8 to 93.3) | NA |
Complete response, n (%) | ||||||||| | ||||||||| | ||||||||| | 3 (30.0) | 3 (30.0) | NA |
Partial response, n (%) | ||||||||| | ||||||||| | ||||||||| | 4 (40.0) | 4 (40.0) | NA |
Stable disease, n (%) | ||||||||| | ||||||||| | ||||||||| | 3 (30.0) | 3 (30.0) | NA |
Progressive disease, n (%) | ||||||||| | ||||||||| | ||||||||| | 0 | 0 | NA |
NE, n (%) | ||||||||| | ||||||||| | ||||||||| | 0 | 0 | NA |
CI = confidence interval; NA = not applicable; NE = not evaluable.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
a95% CI based on exact binomial distribution using the Clopper-Pearson method.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Figure 3: Maximum Change From Baseline in Patients Who Had Not Received Prior Systemic Therapy, November 6, 2020, Data Cut-Off
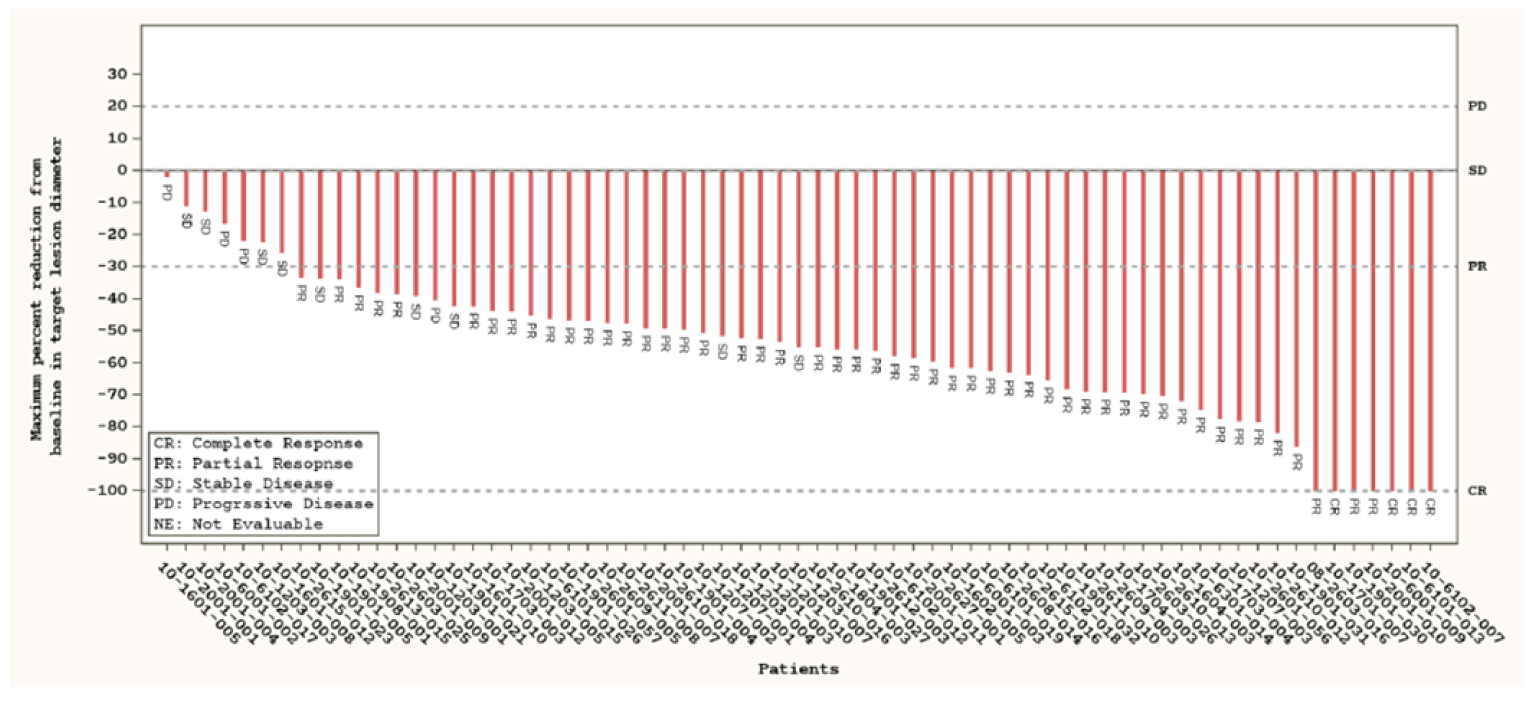
Source: European Medicines Agency Pralsetinib Public Assessment Report.6
Figure 4: Maximum Change From Baseline in Patients Who Received Prior Platinum Therapy, November 6, 2020, Data Cut-off
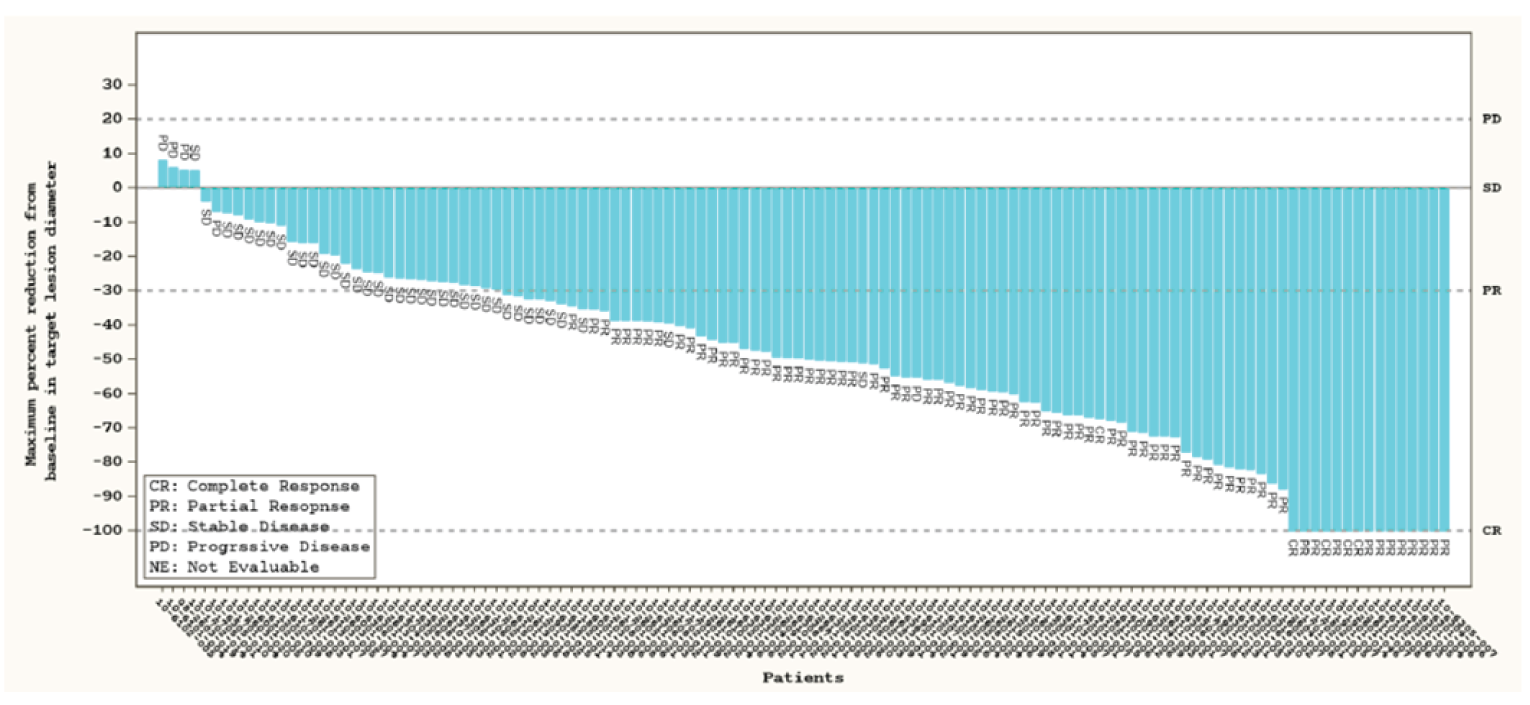
Source: European Medicines Agency Pralsetinib Public Assessment Report.6
The ORR was also reported in a post hoc subgroup of only patients with measurable disease at baseline. A further subgroup analysis of these patients was conducted to report the ORR in treatment-naive patients before the eligibility criteria were revised and after revision. These results, along with the subgroups for prior platinum therapy and prior nonplatinum therapy are presented in Appendix 3 along with DOR results. The ORR and DOR results in all patient subgroups are similar to those of the primary analysis.
The ORR results by pre-specified subgroups of ECOG PS and history of CNS metastasis from the November 6, 2020, data cut-off are summarized in Table 22. The analysis was stratified by prior platinum experience or no prior systemic therapy. In the group with prior platinum experience, the ORR was 67.6% (95% CI, 50.2 to 82.0) and 56.4% (95% CI, 45.8 to 66.6) for an ECOG PS of 0 and 1, respectively. In patients with no prior systemic therapy the ORR was 83.9% (95% CI, 66.3 to 94.5) and 62.8% (95% CI, 46.7 to 77.0) for an ECOG PS of 0 and 1, respectively. When considering the history of CNS metastasis, patients with prior platinum experience had an ORR of 55.6% (95% CI, 41.4 to 69.1) with a history of CNS metastasis and 61.0% (95% CI, 49.6 to 71.6) without a history of CNS metastasis. In patients with no prior systemic therapy, these values were 68.0% (95% CI, 46.5 to 85.1) and 74.0% (95% CI, 59.7 to 85.4), respectively.
Duration of Response
Table 23 summarizes the DOR results at the November 18, 2019, and November 6, 2020, data cut-offs for the ARROW trial. At the November 18, 2019, data cut-off, in the responder analysis set of 75 patients, the median DOR |||||||||||||||||||||||||||||||||||| with ||||||||| of patients having had experienced an event. The Kaplan-Meier estimate of DOR at 12 months was |||||||||||||||||||||||||||. At the November 6, 2020, data cut-off, the median DOR in the responder analysis set of 150 patients was 22.3 (95% CI, 14.7 to NR) months, with 32.7% of patients having had experienced an event. The Kaplan-Meier estimate of DOR at 12 months was 63.8% (95% CI, 54.5 to 73.0). The observed DOR included 32.0% of patients who had a response longer than or equal to 12 months. The Kaplan-Meier curve for DOR from the November 6, 2020, data cut-off is shown in Figure 5.
Table 22: ORR Subgroup Analysis by ECOG PS and CNS Metastasis at Baseline
Detail | ARROW November 6, 2020, data cut-off | |||
|---|---|---|---|---|
Pralsetinib prior platinum | Pralsetinib no prior systemic therapy | |||
N | ORR, % (95% CI) | N | ORR, % (95% CI) | |
ECOG PS 0 | 37 | 67.6 (50.2, 82.0) | 31 | 83.9 (66.3, 94.5) |
ECOG PS 1 | 94 | 56.4 (45.8, 66.6) | 43 | 62.8 (46.7, 77.0) |
ECOG PS 2 | 5 | 40.0 (5.3, 85.3) | 1 | 100 (2.5, 100) |
History of CNS/brain metastasis | 54 | 55.6 (41.4, 69.1) | 25 | 68.0 (46.5, 85.1) |
No history of CNS/brain metastasis | 82 | 61.0 (49.6, 71.6) | 50 | 74.0 (59.7, 85.4) |
CI = confidence interval; CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ORR = overall response rate.
Note: The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
Source: European Medicines Agency Pralsetinib Public Assessment Report.6
Table 23: Duration of Response Analysis (Responder Analysis Set)
Detail | ARROW November 18, 2019, data cut-off | ARROW November 6, 2020, data cut-off |
|---|---|---|
Pralsetinib (N = 75) | Pralsetinib (N = 150) | |
Patients with event | ||||||||| | 49 (32.7) |
Censored | ||||||||| | 101 (67.3) |
Kaplan-Meier estimates, months (95% CI)a | ||
Median duration of response | ||||||||| | 22.3 (14.7 to NR) |
Kaplan-Meier estimate of duration of response at time points, % (95% CI)a | ||
3 months | ||||||||| | 100.0 (100.0 to 100.0) |
6 months | ||||||||| | 84.3 (78.1 to 90.5) |
9 months | ||||||||| | 73.2 (65.3 to 81.2) |
12 months | ||||||||| | 63.8 (54.5 to 73.0) |
18 months | ||||||||| | 52.9 (42.2 to 63.6) |
24 months | ||||||||| | 44.1 (26.0 to 62.2) |
Observed duration of response | ||
< 3 months | ||||||||| | 8 (5.3) |
≥ 3 to < 6 months | ||||||||| | 40 (26.7) |
≥ 6 months | ||||||||| | 102 (68.0) |
≥ 9 months | ||||||||| | 67 (44.7) |
≥ 12 months | ||||||||| | 48 (32.0) |
≥ 18 months | ||||||||| | 26 (17.3) |
≥ 24 months | ||||||||| | 2 (1.3) |
CI = confidence interval; NA = not applicable; NR = not reached.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
a95% CI based on the Greenwood formula.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Figure 5: Duration of Response (Responder Analysis Set; November 6, 2020, Data Cut-Off)
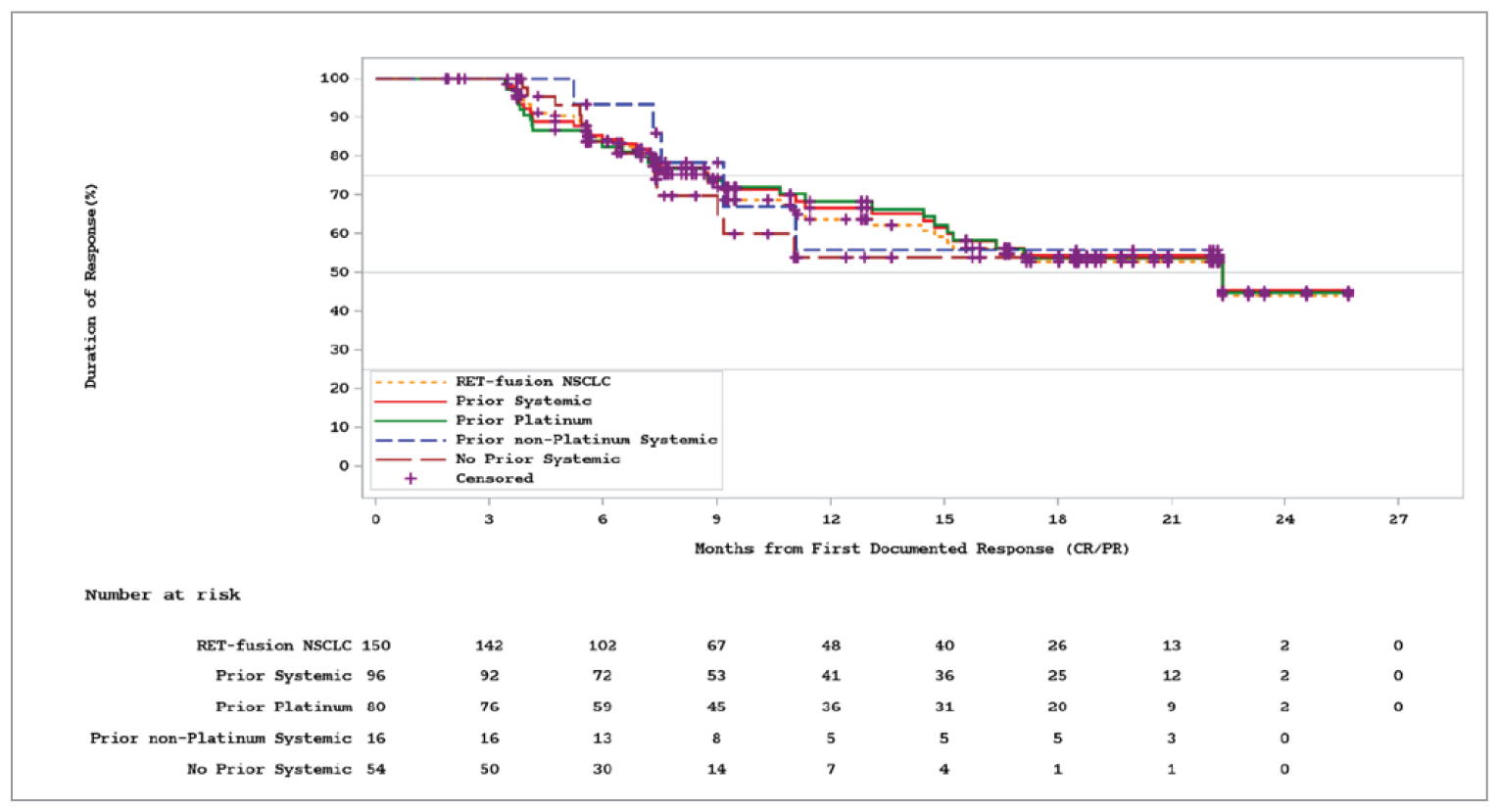
NSCLC = non–small cell lung cancer.
Source: Sponsor submission.4
Harms
Only those harms identified in the review protocol are reported. Table 24 provides detailed harms data.
Detail | ARROW November 18, 2019, data cut-off ||||||||| | ARROW November 6, 2020, data cut-off N = 281 |
|---|---|---|
Patients with ≥ 1 adverse event | ||
n (%) | ||||||||| | 279 (99.3) |
Most common events,a n (%) | ||
Increased AST | ||||||||| | 126 (44.8) |
Constipation | ||||||||| | 118 (42.0) |
Anemia | ||||||||| | 129 (45.9) |
Diarrhea | ||||||||| | 70 (24.9) |
Increased ALT | ||||||||| | 92 (32.7) |
Hypertension | ||||||||| | 96 (34.2) |
Fatigue | ||||||||| | 67 (23.8) |
Pyrexia | ||||||||| | 72 (25.6) |
Increased blood creatinine | ||||||||| | 62 (22.1) |
Cough | ||||||||| | 65 (23.1) |
Neutropenia | ||||||||| | 61 (21.7) |
Decreased neutrophil count | ||||||||| | 81 (28.8) |
Dry mouth | ||||||||| | 47 (16.7) |
Peripheral edema | ||||||||| | 42 (14.9) |
Decreased white blood cell count | ||||||||| | 72 (25.6) |
Increased blood creatine phosphokinase | ||||||||| | 53 (18.9) |
Dyspnea | ||||||||| | 47 (16.7) |
Pneumonia | ||||||||| | 44 (15.7) |
Patients with ≥ 1 serious adverse event | ||
n (%) | ||||||||| | 166 (59.1) |
Most common events,b n (%) | ||
Pneumonia | ||||||||| | 33 (11.7) |
Disease progression | ||||||||| | 21 (7.5) |
Pneumonitis | ||||||||| | 13 (4.6) |
Sepsis | ||||||||| | 8 (2.8) |
Anemia | ||||||||| | 9 (3.2) |
Hypertension | ||||||||| | 4 (1.4) |
Pyrexia | ||||||||| | 8 (2.8) |
Urinary tract infection | ||||||||| | 6 (2.1) |
Dyspnea | ||||||||| | 6 (2.1) |
Pleural effusion | ||||||||| | 6 (2.1) |
Patients who stopped treatment due to adverse events | ||
n (%) | ||||||||| | 55 (19.6) |
Most common events,a n (%) | ||
Disease progression | ||||||||| | 10 (3.6) |
Pneumonia | ||||||||| | 7 (2.5) |
Pneumonitis | ||||||||| | 7 (2.5) |
Dyspnea | ||||||||| | 2 (0.7) |
Sepsis | ||||||||| | 3 (1.1) |
Deaths | ||
n (%) | ||||||||| | 35 (12.5) |
Most common events,d n (%) | ||
Disease progression | ||||||||| | 14 (5.0) |
Pneumonia | ||||||||| | 4 (1.4) |
Notable harms | ||
Pneumonitis, n (%) | ||
Grade 3/4/5 | ||||||||| | 6 (2.1) |
Serious adverse event | ||||||||| | 13 (4.6) |
Dose reduction | ||||||||| | 18 (6.4) |
Dose interruption | ||||||||| | 27 (9.6) |
Treatment discontinuation | ||||||||| | 7 (2.5) |
Death due to adverse event | ||||||||| | 0 |
Hypertension, n (%) | ||
Grade 3/4/5 | ||||||||| | 45 (16.0) |
Serious adverse event | ||||||||| | 4 (1.4) |
Dose reduction | ||||||||| | 12 (4.3) |
Treatment interruption | ||||||||| | 24 (8.5) |
Treatment discontinuation | ||||||||| | 1 (0.4) |
Death due to adverse event | ||||||||| | 0 |
Hepatoxicity, n (%) | ||
Grade 3 | ||||||||| | NR |
Grade 4 | ||||||||| | NR |
Serious adverse event | ||||||||| | NR |
Dose reduction | ||||||||| | NR |
Treatment discontinuation | ||||||||| | NR |
Death due to adverse event | ||||||||| | NR |
Hemorrhagic events, n (%) | ||
Grade 3 | ||||||||| | NR |
Grade 4 | ||||||||| | NR |
Serious adverse event | ||||||||| | NR |
Dose reduction | ||||||||| | NR |
Treatment discontinuation | ||||||||| | NR |
Death due to adverse event | ||||||||| | NR |
ALT = alanine transaminase; AST = aspartate transaminase.
aFrequency greater than 15% at either data cut-off.
bFrequency greater than 2% at either data cut-off.
cPatients who discontinued treatment.
dFrequency greater than 1%.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Adverse Events
At the November 18, 2019, data cut-off, ||||||||| of a total of ||||||||| patients in the safety analysis set experienced at least 1 AE. The most common were increased AST (|||||||||), constipation (|||||||||), anemia (|||||||||), and increased ALT |||||||||. At the November 6, 2020, data cut-off, 99.3% of the 281 patients in the safety analysis set experienced at least 1 AE. The most common were anemia (45.9%), increased AST (44.8%), constipation (42.0%), hypertension (34.2%), and increased ALT (32.7%).
Serious Adverse Events
At the November 18, 2019, data cut-off, ||||||||| of patients experienced at least 1 SAE. The most common were pneumonia (||||||||| and disease progression (|||||||||). At the November 6, 2020, data cut-off, 59.1% of patients had experienced at least 1 SAE. The most common were consistent with the earlier data cut-off, with pneumonia reported in 11.7% of patients and disease progression reported in 7.5%.
Withdrawals Due to Adverse Events
At the November 18, 2019, data cut-off, ||||||||| of patients had discontinued treatment due to an AE. The most common AEs to result in treatment discontinuation were disease progression (|||||||||), pneumonia (|||||||||), and pneumonitis (|||||||||). At the November 6, 2020, data cut-off, 19.6% of patients had discontinued treatment due to an AE. The most common AEs to result in treatment discontinuation were pneumonia (3.6%), disease progression (2.5%), and pneumonitis (2.5%).
Mortality
At the November 18, 2019, data cut-off, ||||||||| of patients had died as a result of an AE. The most common AEs to result in death were disease progression (||||||||| and pneumonia (|||||||||). At the November 6, 2020, data cut-off, 12.5% of patients had died as a result of an AE. The most common AEs to result in death were disease progression (5.0%) and pneumonia (1.4%).
Notable Harms
Detailed information on notable harms was available for pneumonitis and hypertension. At the November 18, 2019, data cut-off, pneumonitis was reported as a grade 3, 4, or 5 AE by ||||||||| of patients and an SAE by ||||||||| of patients, resulting in a dose reduction in ||||||||| of patients, a dose interruption in of patients, and treatment discontinuation by ||||||||| of patients. There were |||||||||||||||||||||||||||||||||||| at the November 18, 2019, data cut-off. At the November 6, 2020, data cut-off, pneumonitis was reported as a grade 3, 4, or 5 AE by 2.1% of patients and an SAE by 4.6% of patients, resulting in a dose reduction in 6.4% if patients, a dose interruption in 9.6% of patients, and treatment discontinuation by 2.5% of patients. No deaths were attributed to pneumonitis at the November 6, 2020, data cut-off.
At the November 18, 2019, data cut-off, hypertension was reported as a grade 3, 4, or 5 AE by ||||||||| of patients and an SAE by ||||||||| of patients, resulting in a dose reduction in ||||||||| of patients, a dose interruption in ||||||||| of patients, and treatment discontinuation by ||||||||| of patients. There were ||||||||||||||||||||||||||||||||||||||||||||| at the November 18, 2019, data cut-off. At the November 6, 2020, data cut-off, hypertension was reported as a grade 3, 4, or 5 AE by 16.0% of patients and an SAE by 1.4% of patients, resulting in a dose reduction in 4.3% if patients, a dose interruption in 8.5% of patients, and treatment discontinuation by 0.4% of patients. No deaths were attributed to hypertension at the November 6, 2020, data cut-off.
Summary data for hemorrhage and hepatoxicity were not reported.
Critical Appraisal
Internal Validity
The ARROW study is an ongoing phase I and II, open-label, single-arm assessment of the clinical efficacy and safety of pralsetinib in patients with RET fusion–positive locally advanced or metastatic thyroid cancer, NSCLC, or other solid tumours. Given that ARROW is a single-arm trial, the most important limitation is the lack of a comparator arm. This design increases the risk of bias in estimating treatment effects due to the potential for confounding related to unidentified prognostic factors and treatment-effect modifiers that could affect the activity of the study drug. CADTH, as well as the clinical expert consulted for this review, acknowledge that patients with RET fusion–positive NSCLC are rare, representing 1% to 2% of all NSCLC cases, increasing the difficulty of conducting randomized controlled trials. However, the sponsor has initiated a phase III randomized controlled trial (RCT) assessing first-line pralsetinib in RET fusion–positive metastatic NSCLC against physician’s-choice platinum-based chemotherapy, with results estimated to be available for the primary end point in December 2024.
The primary objective investigated in the phase II portion of the ARROW study was the ORR as measured by RECIST 1.1. The trial used standardized imaging protocols across study sites and centralized reading to ensure quality and consistency across the trial for imaging end points. The FDA considers the ORR a surrogate measurement when assessing treatment response in advanced or metastatic NSCLC patients, and it may not correlate well with survival, unless the effect size of the ORR is large, and the responses are durable.28 The ARROW trial reported a null hypothesis of an ORR of 0.23 and an alternative hypothesis of an ORR of 0.5 in treatment-experienced patients, and a null hypothesis of 0.48 and alternative hypothesis of 0.61 in treatment-naive patients, both of which were confirmed to be reasonable and clinically significant by the clinical expert consulted for this review. Although the trial’s sample size was not large enough to satisfy the reported power calculations at the time of both data cut-offs, the observed results exceeded both alternative hypotheses and the lack of power is therefore not of concern.
While there was a lack of formal statistical hypothesis testing, there was also no pre-specified interim analyses planned in the statistical analysis plan for the ARROW trial, increasing the potential for bias and type I error with successive ad hoc data cut analyses. Furthermore, extensive protocol amendments affecting the conduct of the trial after patients had first been randomized may have biased the results and increased uncertainty by increasing the heterogeneity of the patient population. Patients recruited to the treatment-naive group were initially required to be deemed unsuitable for SOC chemotherapy, but this was later amended to allow all treatment-naive patients. This may have biased the results against pralsetinib if the patients recruited before this amendment had a worse prognosis compared to the average first-line patient. Important protocol deviations further increased the uncertainty, given that 16 patients (6.9% of the efficacy population) at the November 6, 2020, data cut-off did not have measurable disease at baseline and 1 had inconclusive evidence of RET fusion. This was due to eligibility for the trial being based on investigator assessments that were ultimately not confirmed by a BICR. The sponsor did provide sensitivity analyses removing these patients from the analysis and reported results similar to those of the primary analysis; however, the ad hoc removal of patients due to protocol deviations means the results should be interpreted with caution.
Time-to-event outcomes of PFS and OS were immature and, given the lack of comparator arm, uncertainty remains regarding the long-term effects of pralsetinib on PFS and OS. The HRQoL outcome was evaluated using the EORTC QLQ-C30, which has been validated in NSCLC patients and is considered appropriate for advanced or metastatic forms of the disease. Although this was an exploratory outcome investigated in the ARROW trial, the clinician expert and clinician groups noted that findings for quality of life are clinically significant because patients with advanced disease usually experience greater symptom burdens due to disease progression. Although the HRQoL results are directionally positive, uncertainty remains regarding these findings because the number of patients who completed the questionnaires at baseline represented a smaller subset of the overall population due to the use of a protocol amendment in addition to attrition in later weeks of the trial. There is potential for selection bias over time given that long-term survivors in the trials tend to be healthier patients. In the absence of a comparator arm and an open-label design, which introduces reporting bias, the impact of pralsetinib on patient-reported outcomes in relation to other therapies is unknown.
External Validity
The outcomes assessed in the ARROW trial (ORR, DOR, OS, PFS, and HRQoL) are standard in oncology and considered clinically meaningful. According to the clinical expert consulted by CADTH, the demographic and disease characteristics of the ARROW population were reflective of the Canadian population of patients with RET fusion–positive NSCLC. Patients are more likely to have never smoked, be younger, have adenocarcinoma histology, and present with stage 4 disease. Few patients with stage III disease were included in the ARROW trial; however, the clinical expert indicated that if a patient with stage III disease was ineligible for curative therapy they would be treated and expected to respond just as well as a patient with stage IV disease. Patients with an ECOG PS of 2 were originally included in the trial population but later removed from the inclusion criteria. There are therefore few patients in the trial with an ECOG PS of 2, calling into question the generalizability of the study results to patients with an ECOG PS of 2 or greater. The clinical expert indicated that pralsetinib should be offered to patients with an ECOG PS of 2 or 3, given the likelihood of improving symptom burden and ECOG PS.
Pralsetinib dosing and drug administration interval in the trial align with Health Canada’s indication and are therefore generalizable to the Canadian setting. All outcomes evaluation in the trial and considered in this review (ORR, DOR, HRQoL, OS, and PFS) were clinically relevant, important to patients, and used in clinical practice. The duration of follow-up was sufficient for the assessment of the primary outcome ORR; however, longer-term outcomes of PFS and OS are difficult to interpret given the immaturity of the data. Although there was no predefined maximum duration of treatment in the ARROW trial, the most recent data cut-off had a median survival follow-up of 17.1 months. As such, the generalizability of long-term safety and efficacy conclusions is uncertain beyond what is seen at the November 6, 2020, data cut-off. The clinical expert consulted for this review did not anticipate that extending treatment beyond the follow-up times in the trial would present any issues of concern, noting that treatment would be discontinued in the event of clinical (not radiographic) progression, unacceptable toxicity, or patient preference. (Refer to Table 4).
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
This section provides a summary and appraisal of the indirect evidence submitted by the sponsor comparing pralsetinib to other treatments used in RET fusion–positive locally advanced or metastatic NSCLC.
A focused literature search for network meta-analyses dealing with NSCLC was run in MEDLINE All (1946–) on March 31, 2022. No search limits were applied.
Description of Indirect Comparison
The sponsor noted that, due to the lack of available evidence for comparators of interest in the population with RET fusion–positive NSCLC, an ITC comparing pralsetinib evidence in patients with RET fusion–positive NSCLC to evidence for comparators of interest in wild-type NSCLC was conducted. Given that the pivotal trial for pralsetinib, ARROW, is a single-arm trial, unanchored comparisons were required. For comparators for which IPD were available, propensity-score analyses were conducted to adjust for differences in population characteristics between the trials. For comparators for which IPD were not available, naive comparisons, with no adjustments for differences in population characteristics, were made.
One published ITC, Popat et al. (2022),15 compared first-line patients from the ARROW trial receiving pralsetinib against relevant comparators through the use of synthetic control arms from real-world population sets. The authors used IPW to adjust for differences in patient characteristics.
Methods of Sponsor-Submitted Indirect Treatment Comparison
Objectives
In the absence of direct comparative evidence from trials, the aim of each analysis was to compare the efficacy (OS and PFS) of pralsetinib in patients with RET fusion–positive locally advanced or metastatic NSCLC versus patients with wild-type NSCLC receiving comparators of interest.
Study Selection Methods
The index trial was based on IPD from the November 6, 2020, data cut-off of the phase I and II ARROW trial; specifically, the safety analysis set (N = 281) that included all patients who had received a dose of pralsetinib up to the data cut-off. To identify evidence for relevant comparators and identify evidence from RCTs, a systematic literature review (SLR) was conducted. The selection criteria for inclusion in the ITC are summarized in Table 25.
Table 25: Study Selection Criteria and Methods for Indirect Treatment Comparisons
Characteristic | Sponsor-submitted indirect treatment comparison |
|---|---|
Population | Adult patients with stage III/IV wild-type NSCLC, regardless of treatment line |
Intervention | First line:
Second line:
|
Comparator | Placebo or any intervention of interest |
Outcome |
|
Study design |
|
Publication characteristics | Studies published in 2017 or later (pre-2017 studies were identified from a pre-existing SLR) |
Databases searched | Embase, MEDLINE, Evidence-Based Medicine Reviews |
Selection process | Citations screened by single analyst and independently checked by a second analyst. Discrepancies resolved by consensus. |
Data extraction process | Data extraction conducted by single analyst and independently checked by a second analyst. |
Quality assessment | Not reported |
CNS = central nervous system; NSCLC = non–small cell lung cancer; SLR = systematic literature review; RCT = randomized control trial.
Source: Sponsor-submitted indirect treatment comparison report.29
The SLR identified 131 unique studies; the resulting list was refined to determine inclusion in the comparative-analysis ITC. For example, 112 of the 131 studies reported data for either first-line platinum-based chemotherapy plus pemetrexed or paclitaxel (68 studies) or second-line docetaxel (44 studies). The sponsor determined that, where possible, studies with IPD available would be prioritized and included in the analysis. As such, the IMpower132 and OAK trials were selected for these comparators, respectively. If no IPD were available for a comparator, studies were chosen using a sequential decision tree based on whether the population aligned with the ARROW trial in terms of histology and PD-L1 status (RET status was assumed not to be predictive); whether pooled analysis were available, and which study had the largest number of patients enrolled. A summary of the included studies is provided in Table 26.
ITC Analysis Methods
In the absences of direct trial evidence for relevant comparators or RCTs to form a connected network, 2 methods for generating comparative efficacy estimates for pralsetinib versus comparators of interest were used. Only the PFS and OS outcomes were analyzed. For comparisons for which IPD were available for the comparator trials (the IMpower132 trial for platinum-based chemotherapy plus pemetrexed in the first-line setting and the OAK trial for docetaxel in the second-line setting) a propensity-score method was used. For comparisons for which no IPD were available for the comparator trials, naive comparisons were generated for first-line pembrolizumab monotherapy, pembrolizumab in combinations with platinum-based chemotherapy plus pemetrexed, and second-line nivolumab, and pemetrexed plus carboplatin.
Propensity-Score Method
The propensity-scoring approach is based on methodology outlined in National Institute for Health and Care Excellence Decision Support Unit guidance (Technical Support Document 17).30 Using IPW, the approach attempts to control for the sampling bias whereby the probability of being assigned a treatment in 1 trial versus another is not random, but a function of observable covariates. The method adjusts the population of the comparator trial based on these covariates. Appropriate covariates must be chosen as inputs for the statistical model. The sponsor consulted expert clinical input and selected the following 7 prognostic factors (from the characteristics that were reported in both comparator studies) to be included in the analysis:
age (continuous)
gender (male or female)
presence of CNS metastasis (yes or no)
ECOG PS (0 or 1)
race (Asian or non-Asian)
histology (adenocarcinoma or other)
smoking status (never or current/former).
Table 26: Characteristics of Included Studies in the Indirect Treatment Comparison
Study name: | IPD available? | Study design | Inclusion and exclusion criteria | N | Median age | Smoking status | PD-L1 expression |
|---|---|---|---|---|---|---|---|
ARROW: Pralsetinib | Yes | Phase I/II, open-label, international | Locally advanced or metastatic RET fusion–positive NSCLC ECOG 0 or 1 | Prior therapy: 165 No prior therapy: 116 | Prior therapy: 59 No prior therapy: 62.5 | Prior therapy: 33.7% current/former No prior therapy: 39.8% current/former smoker | NR |
First line | |||||||
KEYNOTE-042:7 Pembrolizumab monotherapy | No | Phase III open-label, international, RCT | Locally advanced or metastatic NSCLC without EGFR or ALK ECOG 0 or 1 PD-L1 ≥ 1% unstable or untreated CNS metastasis excluded | 638 | 63 | 78% current/former smoker | 1% to 19%: 35% 20% to 49%: 18% ≥ 50%: 49% |
KEYNOTE-189:8 Pembrolizumab plus platinum-based chemotherapy in combination with pemetrexed | No | Phase III double-blind, international, RCT | Locally advanced or metastatic nonsquamous NSCLC without EGFR or ALK ECOG 0 or 1 symptomatic CNS metastasis excluded | 410 | 65 | 88.3% current/former smoker | < 1%: 31.0% ≥ 1%: 63.4% ≥ 50%: 32.2% |
IMpower132:9 Platinum-based chemotherapy in combination with pemetrexed | Yes | Phase III open-label, international RCT | Stage IV nonsquamous NSCLC without EGFR or ALK ECOG 0 or 1 | 286 | 63 | 89.5% | ≥ 1% to < 50%: 43.5% |
Second line | |||||||
OAK:10 Docetaxel | Yes | Phase III, open-label, international RCT | Squamous or nonsquamous NSCLC ECOG 0 or 1 Patients received ≥ 1 platinum-based combination therapy Patients with treated asymptomatic CNS metastases were eligible | 425 | 64 | 83% current/former smoker | ≥ 1%: 52% ≥ 5%: 32% |
CheckMate 057:11 Nivolumab monotherapy | No | Phase III open-label international RCT | Stage IIIB/IV nonsquamous NSCLC ECOG 0 or 1 Prior treatment with platinum-based therapy Stable CNS metastasis where eligible | 287 | 61 | 79% current/former smoker | ≥ 1%: 53.2% ≥ 5%: 41.1% ≥ 10%: 37.2% |
GOIRC 02 to 2006:12 Pemetrexed plus carboplatin | NO | Phase II open-label, RCT from Italy | Unresectable stage IIIB/IV NSCLC ECOG 0,1, or 2 Disease progression after 1 first-line treatment with platinum-based chemotherapy | 119 | 64 | NR | NR |
NVALT7:13 Pemetrexed plus carboplatin | NO | Phase II open-label, RCT | NSCLC Evidence of disease progression after cytotoxic treatment, which should have included a platinum compound | NR (240 patients across 2 treatment arms) | 62 | NR | NR |
CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; NR = not reported; NSCLC = non–small cell lung cancer; RCT = randomized control trial.
Source: Sponsor-submitted indirect treatment comparison report.29
Weights were obtained by fitting a logistic regression model, with treatment assignment being a function of these 7 covariates. A Cox regression model was then fitted to the pooled IPD for OS and PFS (although there was no mention of whether the proportional hazards assumption was violated) using the weights obtained from the propensity-scoring analyses to estimate the weighted treatment effect between pralsetinib and the comparator of interest. The sponsor provided absolute mean differences of covariates pre- and post-weighting as well as the distribution of propensity-score weights as a diagnostic to check the weighting.
In contrast to the naive comparisons, for the propensity-scoring analysis PFS was based on investigator assessment, not a BICR. This was because the most recent data cut-off for the IMpower132 study only reported investigator-assessed PFS and, to be consistent, the sponsor also used investigator-assessed PFS for the ARROW and OAK trials. If patients were missing covariate data in any trial (ARROW, IMpower132, or OAK), they were removed from the analysis (9 were removed for the IMpower132 comparison and 15 removed for the OAK comparison). Additional patients were removed from analysis before propensity-score weighting, including patients in the ARROW trial with an ECOG PS of 2 (1 patient in the ARROW trial), those with ALK- or EGFR-driver mutations in the IMpower132 study (5 patients) as they would not have been eligible to enrol in the ARROW trial, and patients not treated as intended in IMpower132 (12 patients).
Naive Comparisons
In the absence of IPD for comparators of interest, naive comparisons were conducted. The was no adjustment for differences between trial populations and no selective exclusion of patients from the analyses. Data for OS and PFS were recreated from published Kaplan-Meier curves using the Guyot method,31 and virtual IPD were created for the comparator arms. A Cox regression model was fitted to the IPD to estimate naive HRs. General conclusions from the naive comparisons are commented on in the following section; however, given the uncertainty in conducting naive comparisons, detailed results are not provided in this report.
Results of the Sponsor-Submitted Indirect Treatment Comparison
Results of Propensity-Scoring Method
The population characteristics for the first-line comparison between pralsetinib and platinum-based chemotherapy plus pemetrexed before and after IPW adjustments conducted in the IMpower132 study are summarized in Table 27. Residual imbalances are evident in gender (||||||||||||||||||||||||||||||||||||||||||||||||||||||) and CNS metastasis (||||||||||||||||||||||||||||||||||||||||||||||||||||||).
The population characteristics for the second-line comparison between pralsetinib and docetaxel before and after IPW adjustments conducted on the OAK study are summarized in Table 28. There is a residual imbalance in ECOG PS of 1 (||||||||||||||||||||||||||||||||||||).
The adjusted and unadjusted HRs for OS results using IPW analysis for the IMpower132 and OAK studies compared with the ARROW trial are summarized in Table 29. All adjusted and unadjusted HRs favoured pralsetinib for OS. The adjusted HR for OS for the pralsetinib versus platinum-based chemotherapy plus pemetrexed comparison was |||||||||||||||||||||||||||. The adjusted HR for OS for the pralsetinib versus docetaxel comparison was |||||||||||||||||||||||||||.
The adjusted and unadjusted HRs for the PFS results using IPW analysis for the IMpower132 and OAK studies compared with ARROW are summarized in Table 30. All adjusted and unadjusted HR favoured pralsetinib for PFS. The adjusted PFS HR for the pralsetinib versus platinum-based chemotherapy plus pemetrexed comparison was |||||||||||||||||||||||||||. The adjusted PFS HR for the pralsetinib versus docetaxel comparison was |||||||||||||||||||||||||||.
Table 27: Patient Characteristics Before and After IPW With IMpower132 — First Line
Detail | First-line pralsetinib | First-line platinum-based chemotherapy plus pemetrexed | |
|---|---|---|---|
ARROW | IMpower132 – Unadjusted | IMpower132 – Adjusted | |
Number of patients | ||||||||| | ||||||||| | ||||||||| |
Age (mean) | ||||||||| | ||||||||| | ||||||||| |
Gender (%): male | ||||||||| | ||||||||| | ||||||||| |
ECOG PS (%): 1 | ||||||||| | ||||||||| | ||||||||| |
CNS metastasis (%): yes | ||||||||| | ||||||||| | ||||||||| |
Smoking status (%): never | ||||||||| | ||||||||| | ||||||||| |
Histology (%): adenocarcinoma | ||||||||| | ||||||||| | ||||||||| |
Race (%): Asian | ||||||||| | ||||||||| | ||||||||| |
CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IPW = inverse probability weighting.
Source: Sponsor-submitted indirect treatment comparison report.29
Table 28: Patient Characteristics Before and After IPW With OAK — Second Line
Detail | Patients with prior systemic therapy: pralsetinib | Patients previously treated: docetaxel | |
|---|---|---|---|
ARROW | OAK – unadjusted | OAK – adjusted | |
# patients | ||||||||| | ||||||||| | ||||||||| |
Age (mean) | ||||||||| | ||||||||| | ||||||||| |
Gender (%): male | ||||||||| | ||||||||| | ||||||||| |
ECOG PS (%): 1 | ||||||||| | ||||||||| | ||||||||| |
CNS metastasis (%): yes | ||||||||| | ||||||||| | ||||||||| |
Smoking status (%): never | ||||||||| | ||||||||| | ||||||||| |
Histology (%): adenocarcinoma | ||||||||| | ||||||||| | ||||||||| |
Race (%): Asian | ||||||||| | ||||||||| | ||||||||| |
CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; IPW = inverse probability weighting.
Source: Sponsor-submitted indirect treatment comparison report.29
Table 29: Adjusted and Unadjusted Overall Survival Results in the IPW Analysis
Detail | Median, months (95% CI) for pralsetinib | Median, months (95% CI) for comparator | Hazard ratio (95% CI) |
|---|---|---|---|
First line | |||
Pralsetinib vs. platinum-based chemotherapy plus pemetrexed | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| |
Second line | |||
Pralsetinib vs. docetaxel | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| |
CI = confidence interval; IPW = inverse probability weighting; NE = not estimable.
Source: Sponsor-submitted indirect treatment comparison report.29
Table 30: Adjusted and Unadjusted Progression-Free Survival Results in the IPW Analysis
Detail | Median, months (95% CI) for pralsetinib | Median, months (95% CI) for comparator | Hazard ratio (95% CI) |
|---|---|---|---|
First line | |||
Pralsetinib vs. platinum-based chemotherapy plus pemetrexed | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| |
Second line | |||
Pralsetinib vs. docetaxel | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| |
CI = confidence interval; IPW = inverse probability weighting; NE = not estimable.
Source: Sponsor-submitted indirect treatment comparison report.29
Results of the Naive Comparisons
The unadjusted naive comparisons for OS and PFS all favoured pralsetinib over first-line pembrolizumab monotherapy, pembrolizumab in combinations with platinum-based chemotherapy plus pemetrexed, and second-line nivolumab and pemetrexed plus carboplatin. Given the uncertainty in unadjusted naive comparisons across heterogeneous populations, detailed results are not shown in this report.
Critical Appraisal of Sponsor-Submitted Indirect Treatment Comparison
The sponsor submitted a single ITC report that included comparisons of interest for pralsetinib against pembrolizumab monotherapy, pembrolizumab plus platinum-based chemotherapy in combination with pemetrexed, and platinum-based chemotherapy in combination with pemetrexed in treatment-naive patients, as well as comparisons against docetaxel, nivolumab, and pemetrexed plus carboplatin in treatment-experienced patients. Comparator trials were identified via an SLR that selected publications to be included in the analysis. Given the lack of evidence for comparators of interest in patients with RET fusion–positive NSCLC specifically, the decision was made to compare with wild-type NSCLC, under the assumption that RET fusion is not a predictive or prognostic factor. The CADTH review team acknowledges that this assumption was required to provide an estimate of comparative efficacy; however, it is associated with key limitations. Patients who are RET fusion–positive tend to be younger, less likely to smoke, and have mostly nonsquamous histology. The sponsor provided evidence from Hess et al. (2021)14 suggesting that, before the introduction of RET inhibitors, there was no relationship between RET status and outcomes in an adjusted model. However, the clinical expert consulted for this review suggested that the presence of RET fusion is a positive predictor of the efficacy of RET-targeted therapy and a negative predictor for the effect of immunotherapy. A methodology to adjust for prognostic factors other than RET status was used; however, all differences in patient characteristics could not be accounted for. With regard to the naive comparisons specifically, no adjustments were made. Patients with a positive or negative RET fusion status are expected to respond differently to pralsetinib and it is unclear how similar the patient populations in the comparator studies are to those enrolled in the ARROW trial, despite the adjustments.
As the index trial for pralsetinib (ARROW) is a single-arm trial with no comparator arm, no connected network could be formed. The sponsor noted that, due to differences in population characteristics between the patients with RET fusion–positive NSCLC in ARROW, who were likely to be younger, have a nonsquamous histology, and no or minimal beforebacco use compared with the wild-type NSCLC patients in the comparator trials, a matched-adjusted indirect comparison would result in an ESS too small to draw informative conclusions from the results. The sponsor chose to selectively use studies for which they had access to IPD to conduct propensity-score weighting to adjust for between-trial differences in population characteristics. Selecting comparator studies for an arbitrary reason such as availability of IPD can introduce selection bias; however, the ability to adjust for important population characteristics adds informative power to the results in contrast to the naive comparisons also presented. Additionally, propensity-score weighting with IPD from both trials comes with an advantage over matched-adjusted indirect comparisons in that the comparator trial population is adjusted to better reflect the characteristics of the index trial, which is a better representation of the requested reimbursement population.
A key limitation with methods centred on adjusting for prognostic factors is that identification of all prognostic factors that affect treatment effects is unlikely. A rigorous SLR validated by clinical opinion is the preferred method of identifying prognostic factors. The sponsor consulted clinical opinion and selected 7 prognostic factors based on availability in the reported data, which is not a robust method of identification, although the clinical expert consulted by CADTH considered the 7 prognostic factors to be appropriate. After the comparator trials had been adjusted using the IPW methodology, the reductions in overall sample size were quite large (from 510 to 142 in the case of the OAK trial), likely due to the imbalance in baseline covariates. The distribution of propensity-scoring weights were generally concentrated around 0 and 1; however, a small number of inflated weights of 14 and 15 added to the uncertainty of the results.
As IPD were available for only 2 comparisons in the sponsor-submitted ITC, the remaining comparisons were unadjusted naive comparisons i.e., no adjustments for between-trial differences in population characteristics were made. This introduces a high risk of bias in the results, given that the important prognostic factors, identified by the sponsor as having an impact on treatment effects, remained heterogenous for the naive comparisons. Given these limitations, conclusions cannot be drawn based upon the naive comparisons, and conclusions drawn from the propensity-score weighted analysis are uncertain.
With these limitations in mind, all results were directionally consistent and in line with the clinical expert’s expectations that pralsetinib is likely a better option for patients than the comparators included in the ITC analysis.
Indirect Treatment Comparison Published by Popat et al. (2022)15
Methods
The ITC published by Popat et al. (2022)15 compared the efficacy of pralsetinib in patients with RET fusion–positive advanced NSCLC against SOC therapies. The authors analyzed the first-line cohort from the ARROW study (n = 116) in comparison to real-world data from synthetic control arms from the Flatiron Clinico-Genomic Database (CGDB) and Enhanced Datamart (EDM). Given the limited number of real-world data sources specific to patients with RET fusion–positive advanced NSCLC, only the CGDB population was RET fusion–positive (n = 10) and treated with a basket of best available therapy (the most common of which was pembrolizumab plus chemotherapy). Two Flatiron EMD populations, both with wild-type NSCLC, were considered. One population was treated with pembrolizumab monotherapy (n = 686) and the other was treated with pembrolizumab plus chemotherapy (n = 1,270). To account for the heterogeneity between the ARROW first-line population and the wild-type NCSLC patients in the Flatiron EMD, IPW adjustments were made to the EMD populations. Characteristics included in the IPW model were sex, ECOG PS, time from initial diagnosis, stage at diagnosis, age, smoking history, and race. Metastasis to the CNS was not included in the IPW adjustments due to differing methods of recording metastasis. The small sample size of the CGDB population precluded any adjustments based on the IPW methodology.
Population Characteristics
For the unadjusted comparison between the CGDB population and the ARROW first-line population, heterogeneity (standardized mean difference [SMD] > 0.6) in patient characteristics were noted in sex (20% male versus 47.4% male), ECOG PS (50% ECOG PS 0 and 30% ECOG PS 1 versus 30.2% ECOG PS 0 and 69.0% ECOG PS 1), and race (60% White versus 49.1% White). All characteristics had an SMD greater than 0.1.
Prior to IPW adjustments, there was heterogeneity between the ARROW first-line population compared to the pembrolizumab monotherapy and pembrolizumab plus chemotherapy populations, most notably with respect to smoking history (39.4% in ARROW versus 91.5% and 90.1%, respectively). Following IPW adjustments, there were residual differences in the pembrolizumab monotherapy comparison versus ARROW first-line population (SMD > 0.1), in age (48.3% younger than 65 years versus 59.6% younger than 65 years), smoking history (48.9% versus 39.4%), and race (56.7% versus 49.5%). Metastasis to the CNS was not adjusted for and therefore remained at an SMD of 0.241. The ESS following IPW adjustment was 115 from an original 686. For the comparison of pembrolizumab plus chemotherapy versus the ARROW first-line population, following IPW adjustments, all characteristics had an SMD of less than 0.1, except for CNS metastasis that was not included in the IPW model and therefore remained at an SMD of 0.383. The ESS following IPW adjustment was 217 from an original 1,270.
Results
Table 31 summarizes the HRs for PFS and OS in the comparisons of pralsetinib versus best alternative therapy in the CGDB RET fusion–positive population, pembrolizumab monotherapy in the EDM population, and pembrolizumab plus chemotherapy in the EDM population. The HRs for PFS favoured pralsetinib in all comparisons, with statistically significant findings against pembrolizumab monotherapy (HR = 0.47; 95% CI, 0.31 to 0.70) and against pembrolizumab plus chemotherapy (HR = 0.50; 85% CI, 0.36 to 0.70). The HRs for OS favoured pralsetinib in all comparisons, with statistically significant findings against pembrolizumab monotherapy (HR = 0.33; 95% CI, 0.18 to 0.61) and against pembrolizumab plus chemotherapy (HR = 0.36; 95% CI, 0.21 to 0.64).
Table 31: Comparative Efficacy Results Reported by Popat et al. (2022)
Detail | Pralsetinib vs. CGDB RET fusion–positive best alternative therapy N = 10 | Pralsetinib vs. Flatiron EDM pembrolizumab monotherapy N = 686 ESS = 115 | Pralsetinib vs. Flatiron EDM pembrolizumab plus chemotherapy N = 1,270 ESS = 217 |
|---|---|---|---|
Progression-free survival | |||
Hazard ratio (95% CI) | 0.71 (0.32, 1.55) | 0.47 (0.31, 0.70) | 0.50 (0.36, 0.70) |
Overall survival | |||
Hazard ratio (95% CI) | 0.45 (0.16, 1.25) | 0.33 (0.18, 0.61) | 0.36 (0.21, 0.64) |
CGDB = Clinico-Genomic Database; CI = confidence interval; EDM = Enhanced Datamart; ESS = effective sample size; vs. = versus.
Source: Popat et al. (2022).15
Critical Appraisal
The ITC presented by Popat et al. (2022)15 utilizes synthetic control arms based on real-world data to estimate comparative efficacy for pralsetinib in comparison to a basket of best alternative therapy, pembrolizumab monotherapy, and pembrolizumab plus chemotherapy. Criteria for selection of real-world datasets were unclear in the publication and the presence of selection bias of available real-world populations is therefore uncertain. There are limitations to comparisons made between single-arm clinical trials and observational real-world data, 1 of which is seen in the heterogenous patient characteristics between the patients in the ARROW trial and real-world patient characteristics, particularly the lack of RET fusion–positive status in the pembrolizumab populations and increased smoking history, which was above 90% in the pembrolizumab populations and 39.4% in the ARROW population. The authors of the ITC conducted IPW adjustments to account for the differences in patient characteristics and achieved mostly balanced characteristics for the comparison against pembrolizumab plus chemotherapy (except for CNS metastasis, which was described by the clinical expert consulted for this review as an important prognostic factor in patients with RET fusion–positive NSCLC). The IPW methodology adjusts populations based on chosen prognostic factors. It is unclear in the publication how this list of prognostic factors was chosen, but the clinical expert considered the list to be appropriate. However, the inability to adjust based on CNS metastasis is a major limitation given its importance as a prognostic factor. Accordingly, IPW adjustments substantially reduced both pembrolizumab populations, producing an ESS much smaller than the original population in both cases (pembrolizumab monotherapy = 683 reduced to 115, and pembrolizumab plus chemotherapy = 1,270 reduced to 217), introducing uncertainty to the results.
The analysis conducted by Popat et al. showed statistically significant improvements in PFS and OS for patients receiving pralsetinib as compared to pembrolizumab and pembrolizumab plus chemotherapy. Pralsetinib showed a numerical benefit over a basket of best alternative therapy; however, despite the fact that this analysis was conducted in patients with RET fusion–positive NSCLC, the small sample size resulted in such wide CIs that it was difficult to draw any conclusions from the analysis.
To account for some of the limitations of the analysis, the authors conducted various sensitivity analyses. First, to assess the impact of missing ECOG PS inputs in the pembrolizumab populations, multiple imputation rules were tested with all analyses maintaining statistically significant OS and PFS results. Sensitivity analysis to include metastases in the IPW model despite differences in recording definitions maintained statistically significant OS and PFS results. Quantitative bias analysis was provided, with the authors suggesting that the results show that it is implausible for systematic differences in unmeasured prognostic factors to reverse the findings. Despite the limitations to the published ITC, the results are aligned with the expectation of the consulted clinical expert that there is likely to be a benefit for patients receiving pralsetinib over pembrolizumab monotherapy and pembrolizumab plus chemotherapy.
Other Relevant Evidence
In addition to the pivotal ARROW trial, a phase I and II study with no comparator arm, the AcceleRET-Lung study was considered as an other relevant study for this report. The CADTH review team identified AcceleRET-Lung as an ongoing phase III, randomized, open-label study that met systematic review inclusion criteria with the exception that no results are currently available. In addition, Health Canada issued a Notice of Compliance with conditions for pralsetinib and requested the final report, including datasets of an ongoing study, to verify and further characterize the clinical benefit of pralsetinib for the treatment of treatment-naive patients with RET fusion–positive metastatic NSCLC to be shared. For these reasons, the study is summarized in the following section. The study sponsor is F. Hoffmann-La Roche, Ltd.
AcceleRET-Lung
The AcceleRET-Lung trial is a phase III, randomized, multi-centre, open-label study comparing pralsetinib to a physician’s choice of platinum chemotherapy–based regimens based on SOC treatments for the first-line treatment of patients with RET fusion–positive metastatic NSCLC who have not previously received systemic anticancer therapy for metastatic disease. Patients will be enrolled in approximately 97 sites in across the Americas (including Canada), Asia, Europe, and Oceania.32-34 Table 32 provides more details.
Table 32: Details of Other Relevant Studies — AcceleRET-Lung
Characteristic | AcceleRET-Lung |
|---|---|
Study design | Phase III, multi-centre, randomized, open-label study |
Locations | Approximately 97 sites across the Americas (including Canada), Asia, Europe, and Oceania |
Patient enrolment date | July 24, 2020 |
Estimated primary completion datea | September 30, 2023 |
Estimated study completion dateb | December 31, 2024 |
Randomized (N) | Planned: 226 Enrolled: NA (currently recruiting) |
Inclusion criteria |
|
Exclusion criteria |
|
Intervention | Pralsetinib: 400 mg once daily, oral |
Comparator(s) | Active comparator: platinum-based chemotherapy with or without pembrolizumab
|
Primary end points | Progression-free survivalc |
Secondary end points | Overall response ratec Overall survivalc Number of participants with adverse events and serious adverse eventsd Changes in ECOG PSd Duration of responsec Clinical benefit ratec Disease control ratec EORTC QLQ-C30e EORTC QLQ-LC13e Time to intracranial progression Intracranial overall response rate |
Exploratory end points | Identification of potential biomarkers of antineoplastic activity and resistance |
Notes | Ongoing study, results are not available at the time of this review |
Publications | AcceleRET-Lung,32 Besse et al. (2021),33 and Popat et al. (2022)34 |
CNS = central nervous system; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; NSCLC = non–small cell lung cancer; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1.
aThe date on which the last participant in a clinical study was examined or received an intervention to collect final data for the primary outcome measure. Whether the clinical study ended according to the protocol or was terminated does not affect this date. For clinical studies with more than a primary outcome measure with different completion dates, this term refers to the date on which data collection is completed for all the primary outcome measures.
bThe date on which the last participant in a clinical study was examined or received an intervention or treatment to collect final data for the primary outcome measures, secondary outcome measures, and adverse events (that is, the last participant's last visit).
cEstimated at up to 32 months.
dBaseline, at every 21 day cycle visit until progressive disease or death (estimated 32 months).
eFrom baseline until progressive disease or death (estimated 32 months).
Source: AcceleRET-Lung,32 Besse et al. (2021),33 and Popat et al. (2022).34
This study is currently recruiting participants, with a planned enrolment of approximately 226 patients. The estimated primary completion date (on which the last participant in a clinical study will be examined or receive an intervention to collect final data for the primary outcome measure) and study completion date (on which the last participant in a clinical study will be examined or receive an intervention or treatment to collect final data for the primary outcome measures, secondary outcome measures, and AEs) are September 30, 2023, and December 31, 2024, respectively. Patients are to be randomized in a 1:1 ratio to receive oral pralsetinib (400 mg once daily) or SOC. The SOC will be 1 of 6 platinum-based chemotherapy treatment regimens (with or without pembrolizumab) determined by the treating investigator based on histology. For nonsquamous histology, the treatment regimen will be carboplatin or cisplatin plus pemetrexed (with vitamin supplementation) with or without pembrolizumab followed by optional pemetrexed (with vitamin supplementation) maintenance with or without pembrolizumab; for squamous histology, the treatment regimen will be either carboplatin or cisplatin plus gemcitabine, or carboplatin with paclitaxel plus nab-paclitaxel and pembrolizumab followed by maintenance pembrolizumab.32-34
Stratification factors include intended use of pembrolizumab, history of brain metastases, and ECOG PS. Key eligibility criteria include no prior systemic treatment for advanced and/or metastatic NSCLC; RET fusion–positive tumour by local or central assessment; no additional actionable oncogenic drivers; no prior selective RET inhibitor; and measurable disease as defined by RECIST 1.1. Patients with CNS metastases were included if they were asymptomatic and on a stable dose of corticosteroids. Crossing over to receive pralsetinib upon disease progression will be permitted for patients randomized to the SOC arm.32-34
The primary end point is PFS (defined by RECIST 1.1) assessed by a BICR, whereas the secondary end points include ORR, OS, DOR, disease control rate, clinical benefit rate, time to intracranial progression, intracranial ORR, safety and tolerability, and quality of life evaluations. Identification of potential biomarkers of antineoplastic activity and resistance is an exploratory end point.32-34
Discussion
Summary of Available Evidence
The pivotal trial submitted for this review, ARROW (N = 281; safety population at the November 6, 2020, data cut-off), is an ongoing phase I and II, open-label, single-arm study of pralsetinib in patients with RET fusion–positive locally advanced or metastatic cancer, including NSCLC. The first phase of the study was a dose-escalation study to establish the fixed dose for the second phase. The primary objective of the second phase of the study was to determine the efficacy (as measured by ORR) and safety of pralsetinib (400 mg once daily). The ORR was analyzed at the November 6, 2020, data cut-off in a subset efficacy population of 233 patients with NSCLC who had received their first dose of pralsetinib on or before May 22, 2020, with a median follow-up of 17.1 months. The average age was 59.2 years; 68% of patients had received prior systemic therapy, while 32% had not received any prior systemic therapy.
The sponsor submitted an ITC report aiming to demonstrate the efficacy of pralsetinib compared to relevant treatments. An SLR was conducted to identify evidence for comparators of interest. Given the lack of evidence for comparators of interest in patients with RET fusion–positive NSCLC, pralsetinib was compared to trials in wild-type NSCLC. For comparisons for which IPD were available to the sponsor (platinum-based chemotherapy plus pemetrexed in the first line and docetaxel in the second line), a propensity-scoring method was used to adjust for differences in identified prognostics factors across study populations. For all other comparisons for which IPD were not available to the sponsor, naive unadjusted comparisons were made for first-line pembrolizumab monotherapy, pembrolizumab in combinations with platinum-based chemotherapy plus pemetrexed, second-line nivolumab, and pemetrexed plus carboplatin. Key limitations included the unanchored nature of the indirect comparisons, the inability to adjust for all known and unknown prognostic factors, as well as the use of naive comparisons for all but 2 comparisons.
A published ITC from Popat et al. (2022)15 was identified that compared first-line patients receiving pralsetinib in the ARROW trial to synthetic control arms sourced from 3 real-world populations. In the first, patients with RET fusion–positive NSCLC received either a basket of best alternative therapy (most commonly pembrolizumab plus chemotherapy). The remaining 2 real-world populations were patients with wild-type NSCLC receiving pembrolizumab monotherapy and pembrolizumab plus chemotherapy. The analysis used IPW methodology where possible to adjust for differences in prognostic factors. The results found that patients given pralsetinib received a statistically significant benefit in OS and PFS compared to the chosen comparators, which is consistent with the expectations of the clinical expert consulted for this review. However, the same limitations are present as in the sponsor-submitted ITC. The analysis is an unanchored ITC relying on a limited number of prognostic factors and a small ESS compared to the original sample sizes of the populations.
Interpretation of Results
Efficacy
The patient input and the clinical expert consulted for this review identified several important goals of treatment for locally advanced and metastatic NSCLC, including prolonging a patient’s life, reducing symptom burden, delaying progression, and improving HRQoL. They confirmed that the outcomes (ORR, OS, PFS, DOR, CNS ORR, and HRQoL) investigated in the ARROW trial were appropriate and align with the needs of patients, caregivers, and clinicians in practice.
The clinical expert consulted for this review considered the ORR responses to be clinically meaningful to patients in practice. The expert emphasized that, based on their experience, the ORR is larger and DOR longer than what would be expected with other therapies offered as SOC. The expert noted that there is limited evidence that patients with locally advanced or metastatic RET fusion–positive NSCLC benefit from currently available single-drug immunotherapy, and not all patients respond to chemotherapy plus immunotherapy, indicating an unmet need for this patient population. The ITC was submitted to inform these comparisons of pralsetinib with SOC chemotherapy and immunotherapy; however, there were major limitations with this analysis stemming from the single-arm design of the ARROW trial, necessitating an unanchored comparison. Propensity-score weighting was used to account for between-trial differences for first-line platinum-based chemotherapy plus pemetrexed and second-line docetaxel, but naive unadjusted comparisons were made for all other comparisons. Given these limitations, conclusions cannot be drawn from the naive comparisons and should be drawn instead, but with caution, from the propensity-score weighted analysis. However, all results for PFS and OS from the ITC were directionally in favour of pralsetinib and aligned with the clinical expert’s expectation that pralsetinib is likely to be a beneficial option over SOC chemotherapy and immunotherapy for this patient population. Furthermore, the ITC published by Popat et al. (2022) reports a statistically significant benefit for OS and PFS from pralsetinib over pembrolizumab monotherapy and pembrolizumab plus chemotherapy, although this analysis comes with important limitations as well.
The median OS was not reached in the ARROW trial and is therefore considered immature. The sponsor provided analysis based on the methodology published by Anderson et al. (2008)35 investigating the correlation of survival based on the ORR within the ARROW trial. The results suggest that responders in the ARROW trial were more likely to be alive at 2, 3, and 4 months. While this analysis provides important context and adds to the evidence base, it must be considered alongside systematic reviews such as those published by Haslam et al. (2019)36 and Cooper et al. (2020)37 that show surrogate end points, such as ORR, may be unreliable indicators of long-term OS.
The HRQoL outcomes in the ARROW trial were limited by low patient numbers due to the measure being added to the protocol as an amendment after the trial had begun and the further loss of patients over time. The results were consistent and positive, reaching the MID threshold for a moderate improvement (10 to 20 points) at some time points. Health-related quality of life is an important outcome for patients and, although the results from ARROW are promising, their interpretation is limited by the single-arm design of the trial, low patient numbers, and loss of patients to study discontinuation for reasons other than mortality follow-up.
The ability to affect CNS and/or brain metastasis outcomes is an important factor when comparing pralsetinib to SOC chemotherapy and immunotherapy. A common site for NSCLC metastasis is the CNS and/or brain, which come with burdensome symptoms for patients and worse prognosis. Although SOC chemotherapy and immunotherapy has little to no activity in the brain, given that pralsetinib can cross the blood-brain barrier, it is expected to benefit patients with CNS and/or brain metastasis. This expectation is further supported by the ARROW trial, in which 10 patients had evaluable intracranial lesions, and the ORR was 70% for these specific lesions, including 30% of patients who reported a CR. The clinical expert consulted for this review suggested that there may be a protective element to pralsetinib and the prevention of CNS and/or brain metastasis, and this was supported by the ARROW trial: of the patients treated at 400 mg once daily who did not have a history of CNS metastasis at baseline, none developed CNS metastasis throughout the trial. However, due to the single-arm nature of the trial, this should be considered speculatory.
It is noted that selpercatinib, a similar targeted therapy for patients with RET fusion–positive NSCLC, has recently received a positive recommendation with conditions from CADTH. Although there is no evidence available comparing the 2 therapies, the clinical expert consulted for this review stated that there was no evidence to suggest a significant difference between pralsetinib and selpercatinib in terms of efficacy.
Harms
In the ARROW trial, pralsetinib appeared to have a tolerable safety profile, according to the clinical expert consulted for this review. The proportion of patients who discontinued treatment due to an AE was 19.6%, although this number is likely inflated due to the fact that disease progression was characterized as an AE and was the most commonly cited AE that resulted in discontinuations. Pneumonitis, a key AE of special interest for pralsetinib, was reported as a SAE by 4.6% of patients at the November 6, 2020, data cut-off. At that same data cut-off, 2.1% of patients reported grade 3, 4, or 5 pneumonitis, and no deaths due to pneumonitis were reported. The clinical expert noted that, given the assumption of similar efficacy between pralsetinib and selpercatinib, treatment decisions for patients would likely be determined by the differing safety profile between the 2 therapies, with pneumonitis or de novo poor pulmonary reserves, for example, indicating that a patient may be better suited for selpercatinib. The submitted ITC did not conduct analysis on safety end points; however, input from patient groups and clinical opinion suggests that pralsetinib, as a targeted oral therapy, offers reduced toxicity compared to IV SOC chemotherapies and immunotherapies.
Conclusions
The evidence supporting the funding request for pralsetinib was derived from an ongoing phase I and II, open-label, single-arm study, ARROW. The ORR observed in the ARROW trial, based on unplanned interim analysis results, suggested favourable tumour response in both treatment-naive and treatment-experienced patients and was consistent with further follow-up analysis. The ORR and DOR, including CNS ORR, were considered clinically meaningful by the clinical expert consulted for this review. The ability to draw conclusions from time-to-event end points of PFS and OS was limited by the immaturity of the data and the single-arm design of the trial. The safety profile of pralsetinib in the ARROW trial was considered by the clinical expert to be an improvement compared to SOC chemotherapy and immunotherapy. In the opinion of the clinical expert, the differences in the safety profile compared with selpercatinib highlight the benefits to patients that come with additional treatment options. The ITC submitted to inform the comparative effects of pralsetinib were associated with limitations that prevented drawing conclusions from the results. Uncertainty therefore remains in the comparative effectiveness and safety of pralsetinib.
References
1.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics 2021. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf. Accessed 2022 Apr 26.
2.National Cancer Institute. Non-small cell lung cancer treatment (PDQ®) – health professional version. 2022; https://www.cancer.gov/types/lung/hp/non-small-cell-lung-treatment-pdq. Accessed 2022 Apr 26.
3.Cancer.Net. Lung cancer - non-small cell: statistics. 2022; https://www.cancer.net/cancer-types/lung-cancer-non-small-cell/statistics. Accessed 2022 Apr 26.
4.Drug Reimbursement Review sponsor submission: Gavreto (pralsetinib), 100 mg capsules, oral [internal sponsor's package]. Mississauga (ON): Hoffmann-La Roche Ltd.; 2022 Mar 9.
5.Clinical Study Report: BLU-667-1101. A phase 1/2 study of the highly-selective RET inhibitor, BLU-667, in patients with thyroid cancer, non-small cell lung cancer (NSCLC) and other advanced solid tumors [internal sponsor's report]. Cambridge (MA): Blueprint Medicines; 2020 Feb 26.
6.Committee for Medicinal Products for Human Use. Assessment report: Gavreto (pralsetinib). (European public assessment report). Amsterdam (NL): European Medicines Agency; 2021: https://www.ema.europa.eu/en/documents/assessment-report/gavreto-epar-public-assessment-report_en.pdf. Accessed 2022 May 3.
7.Mok TSK, Wu Y-L, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. The Lancet. 2019;393(10183):1819-1830. PubMed
8.Gadgeel S, Rodríguez-Abreu D, Speranza G, et al. Updated analysis from KEYNOTE-189: pembrolizumab or placebo plus pemetrexed and platinum for previously untreated metastatic nonsquamous non–small-cell lung cancer. J Clin Oncol. 2020;38(14):1505-1517. PubMed
9.Nishio M, Barlesi F, West H, et al. Atezolizumab plus chemotherapy for first-line treatment of nonsquamous NSCLC: results from the randomized phase 3 IMpower132 trial. J Thorac Oncol. 2021;16(4):653-664. PubMed
10.Gandara DR, von Pawel J, Mazieres J, et al. Atezolizumab treatment beyond progression in advanced NSCLC: results from the randomized, phase III OAK study. J Thorac Oncol. 2018;13(12):1906-1918. PubMed
11.Borghaei H, Gettinger S, Vokes EE, et al. Five-year outcomes from the randomized, phase III trials checkmate 017 and 057: nivolumab versus docetaxel in previously treated non-small-cell lung cancer. J Clin Oncol. 2021;39(7):723-733. PubMed
12.Ardizzoni A, Tiseo M, Boni L, et al. Pemetrexed versus pemetrexed and carboplatin as second-line chemotherapy in advanced non-small-cell lung cancer: results of the GOIRC 02-2006 randomized phase II study and pooled analysis with the NVALT7 trial. J Clin Oncol. 2012;30(36):4501-4507. PubMed
13.Smit EF, Burgers SA, Biesma B, et al. Randomized phase II and pharmacogenetic study of pemetrexed compared with pemetrexed plus carboplatin in pretreated patients with advanced non-small-cell lung cancer. J Clin Oncol. 2009;27(12):2038-2045. PubMed
14.Hess LM, Han Y, Zhu YE, Bhandari NR, Sireci A. Characteristics and outcomes of patients with RET-fusion positive non-small lung cancer in real-world practice in the United States. BMC Cancer. 2021;21(1):28. PubMed
15.Popat S, Liu SV, Scheuer N, et al. Addressing challenges with real-world synthetic control arms to demonstrate the comparative effectiveness of pralsetinib in non-small cell lung cancer. Nat Commun. 2022;13(1):3500. PubMed
16.Lung Cancer Canada. Lung cancer staging in Canada. 2020; https://www.lungcancercanada.ca/Lung-Cancer/Staging.aspx. Accessed 2022 Apr 26.
17.Iyer S, Roughley A, Rider A, Taylor-Stokes G. The symptom burden of non-small cell lung cancer in the USA: a real-world cross-sectional study. Support Care Cancer. 2014;22(1):181-187. PubMed
18.NCCN clinical practice guidelines in oncology (NCCN guidelines): non-small cell lung cancer. Version 3.2022. Plymouth Meeting (PA): National Comprehensive Cancer Network (NCCN); 2022: https://www.nccn.org/. Accessed 2022 Apr 26.
19.Planchard D, Popat S, Kerr K, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Lugano (CH): European Society for Medical Oncology; 2020: https://www.esmo.org/content/download/347819/6934778/1/ESMO-CPG-mNSCLC-15SEPT2020.pdf. Accessed 2022 Jul 19.
20.Ellis PM, Vella ET, Ung YC. Systemic treatment for patients with advanced non-small cell lung cancer. (Guideline 7-10, version 3). Toronto (ON): Cancer Care Ontario; 2016: https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/31811 Accessed 2022 Jul 19.
21.Mahato AK, Sidorova YA. RET receptor tyrosine kinase: role in neurodegeneration, obesity, and cancer. Int J Mol Sci. 2020;21(19):7108. PubMed
22.Cascetta P, Sforza V, Manzo A, et al. RET inhibitors in non-small-cell lung cancer. Cancers (Basel). 2021;13(17):01.
23.Gavreto (pralsetinib capsules): capsules, 100 mg, oral. Protein kinase inhibitor (L01EX) [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Jul 12.
24.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
25.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Mar 23.
26.Gainor JF, Curigliano G, Kim DW, et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): a multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021;22(7):959-969. PubMed
27.Health Canada reviewer's report: Gavreto (pralsetinib) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Gavreto (pralsetinib), 100 mg capsules, oral. Mississauga (ON): Hoffmann-La Roche Ltd.; 2022 Mar 9.
28.Clinical trial endpoints for the approval of non-small cell lung cancer drugs and biologics: guidance for industry. Silver Spring (MD): U.S. Food and Drug Administration; 2015: https://www.fda.gov/media/116860/download. Accessed 2022 Apr 26.
29.Wild type (WT) non-small cell lung cancer (NSCLC): updated clinical systematic literature review (SLR) and comparative analyses of pralsetinib with comparators of interest [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Gavreto (pralsetinib), 100 mg capsules, oral. Mississauga (ON): Hoffmann-La Roche Ltd.; 2022 Mar 9.
30.Faria R, Hernández Alava M, Manca A, Wailoo AJ. NICE DSU Technical Support Document 17: the use of observational data to inform estimates of treatment effectiveness in technology appraisal: methods for compariatve individual patient data. London (GB): National Institute for Health and Care Excellence; 2015: https://www.sheffield.ac.uk/media/34204/download?attachment. Accessed 2022 Jul 19.
31.Guyot P, Ades AE, Ouwens MJNM, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12(1):9. PubMed
32.Hoffmann-La Roche. NCT04222972: A study of pralsetinib versus standard of care for first-line treatment of advanced non-small cell lung cancer (NSCLC) (AcceleRET-Lung). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/study/NCT04222972. Accessed 2022 Jun 9.
33.Besse B, Felip E, Kim ES, et al. PUL01.02 AcceleRET lung: a phase 3 study of first-line pralsetinib in patients with RET-fusion+ advanced/metastatic NSCLC. J Thorac Oncol. 2021;16(1):S44-S45.
34.Popat S, Felip E, Kim ES, et al. AcceleRET Lung: A phase 3 study of first-line pralsetinib in patients with RET fusion–positive advanced/metastatic NSCLC. J Clin Oncol. 2022;40(16_suppl):TPS9159-TPS9159.
35.Anderson JR, Cain KC, Gelber RD. Analysis of survival by tumor response and other comparisons of time-to-event by outcome variables. J Clin Oncol. 2008;26(24):3913-3915. PubMed
36.Haslam A, Hey SP, Gill J, Prasad V. A systematic review of trial-level meta-analyses measuring the strength of association between surrogate end-points and overall survival in oncology. Eur J Cancer. 2019;106:196-211. PubMed
37.Cooper K, Tappenden P, Cantrell A, Ennis K. A systematic review of meta-analyses assessing the validity of tumour response endpoints as surrogates for progression-free or overall survival in cancer. Br J Cancer. 2020;123(11):1686-1696. PubMed
38.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
39.Letellier ME, Dawes D, Mayo N. Content verification of the EORTC QLQ-C30/EORTC QLQ-BR23 with the International Classification of Functioning, Disability and Health. Qual Life Res. 2015;24(3):757-768. PubMed
40.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-1028. PubMed
41.Larsson M, Ljung L, Johansson BB. Health-related quality of life in advanced non-small cell lung cancer: correlates and comparisons to normative data. Eur J Cancer Care. 2012;21(5):642-649. PubMed
42.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-117. PubMed
43.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
44.Fayers PM, Aaronson NK, Bjordal K, et al. The EORTC QLQ-C30 Scoring Manual. 3rd ed. Brussels (BE): European Organisation for Research and Treatment of Cancer; 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2022 Jul 19.
45.Osoba D, Zee B, Pater J, Warr D, Kaizer L, Latreille J. Psychometric properties and responsiveness of the EORTC quality of Life Questionnaire (QLQ-C30) in patients with breast, ovarian and lung cancer. Qual Life Res. 1994;3(5):353-364. PubMed
46.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
47.Maringwa JT, Quinten C, King M, et al. Minimal important differences for interpreting health-related quality of life scores from the EORTC QLQ-C30 in lung cancer patients participating in randomized controlled trials. Support Care Cancer. 2011;19(11):1753-1760. PubMed
48.Revicki D, Hays RD, Cella D, Sloan J. Recommended methods for determining responsiveness and minimally important differences for patient-reported outcomes. J Clin Epidemiol. 2008;61(2):102-109. PubMed
49.Van Der Weijst L, Surmont V, Schrauwen W, Lievens Y. Real life data on patient-reported outcomes and neuro-cognitive functioning of lung cancer patients: the PRO-Long study. Front Oncol. 2021;11:685605. PubMed
50.Smit EF, van Meerbeeck JP, Lianes P, et al. Three-arm randomized study of two cisplatin-based regimens and paclitaxel plus gemcitabine in advanced non-small-cell lung cancer: a phase III trial of the European Organization for Research and Treatment of Cancer Lung Cancer Group--EORTC 08975. J Clin Oncol. 2003;21(21):3909-3917. PubMed
51.Sarna L, Swann S, Langer C, et al. Clinically meaningful differences in patient-reported outcomes with amifostine in combination with chemoradiation for locally advanced non-small-cell lung cancer: an analysis of RTOG 9801. Int J Radiat Oncol Biol Phys. 2008;72(5):1378-1384. PubMed
52.Rutkowski J, Szymanik M, Blok M, Kozaka J, Zaucha R. Prospective evaluation of anxiety, depression and quality of life in medically inoperable early stage non-small cell lung cancer patients treated with stereotactic ablative radiotherapy. Rep Pract Oncol Radiother. 2017;22(3):217-222. PubMed
53.Rittmeyer A, Gorbunova V, Vikstrom A, et al. Health-related quality of life in patients with advanced nonsquamous non-small-cell lung cancer receiving bevacizumab or bevacizumab-plus-pemetrexed maintenance therapy in AVAPERL (MO22089). J Thorac Oncol. 2013;8(11):1409-1416. PubMed
54.Ponce Aix S, Talbot D, Govindan R, et al. Quality of life with second or third line nab-paclitaxel-based regimens in advanced non-small-cell lung cancer. Fut Oncol. 2020;16(12):749-762. PubMed
55.Movsas B, Scott C, Langer C, et al. Randomized trial of amifostine in locally advanced non-small-cell lung cancer patients receiving chemotherapy and hyperfractionated radiation: radiation therapy oncology group trial 98-01. J Clin Oncol. 2005;23(10):2145-2154. PubMed
56.Mathieu D, Campeau MP, Bahig H, et al. Long-term quality of life in early-stage non-small cell lung cancer patients treated with robotic stereotactic ablative radiation therapy. Pract Radiat Oncol. 2015;5(4):e365-373. PubMed
57.Hui R, Ozguroglu M, Villegas A, et al. Patient-reported outcomes with durvalumab after chemoradiotherapy in stage III, unresectable non-small-cell lung cancer (PACIFIC): a randomised, controlled, phase 3 study. Lancet Oncol. 2019;20(12):1670-1680. PubMed
58.Hechtner M, Eichler M, Wehler B, et al. Quality of life in NSCLC survivors - a multicenter cross-sectional study. J Thorac Oncol. 2019;14(3):420-435. PubMed
59.Fiteni F, Anota A, Bonnetain F, et al. Health-related quality of life in elderly patients with advanced non-small cell lung cancer comparing carboplatin and weekly paclitaxel doublet chemotherapy with monotherapy. Eur Respir J. 2016;48(3):861-872. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: March 31, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq=# | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(pralsetinib* or pratinib* or gavreto* or BLU-667 or BLU667 or BLU-123244 or BLU123244 or BLU-3244 or BLU3244 or C-683 or C683 or CS-3009 or CS3009 or RG-6396 or RG6396 or X-581238 or X581238 or WHO-11004 or WHO11004 or 1WPE73O1WV).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*pralsetinib/
(pralsetinib* or pratinib* or gavreto* or BLU-667 or BLU667 or BLU-123244 or BLU123244 or BLU-3244 or BLU3244 or C-683 or C683 or CS-3009 or CS3009 or RG-6396 or RG6396 or X-581238 or X581238 or WHO-11004 or WHO11004).ti,ab,kf,dq.
3 or 4
5 use oemezd
(conference abstract or conference review).pt.
6 not 7
2 or 8
remove duplicates from 9
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search terms: (Gavreto OR pralsetinib) AND RET fusion–positive non-small cell lung cancer (NSCLC)]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms: (Gavreto OR pralsetinib) AND RET fusion–positive non-small cell lung cancer (NSCLC)]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms: (Gavreto OR pralsetinib) AND RET fusion–positive non-small cell lung cancer (NSCLC)]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms: (Gavreto OR pralsetinib) AND RET fusion–positive non-small cell lung cancer (NSCLC)]
Grey Literature
Search dates: March 25 – March 30, 2022
Keywords: (Gavreto OR pralsetinib) AND RET fusion–positive non-small cell lung cancer (NSCLC)
Limits: No limits
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for Exclusion |
|---|---|
Gainor JF, Curigliano G, Kim DW, et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): a multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021;22(7):959-969. | Duplicate study |
Griesinger F, Curigliano G, Thomas M, et al. Safety and efficacy of pralsetinib in RET fusion-positive NSCLC including as first-line therapy: update from the ARROW trial. Manuscript in preparation. | Duplicate study |
Correction to Lancet Oncol 2021; 22: 959-69. Lancet Oncol. 2021;22(8):e347. doi:10.1016/S1470-2045(21)00392-2 | Duplicate study |
Zhou Q, Wu Y, Chang J, et al. MA02.02 efficacy and safety of pralsetinib in chinese patients with advanced RET fusion+ non-small cell lung cancer. J Thorac Oncol. 2021;16(10):S889-S890. | Duplicate study |
Zhou Q, Wu Y, Chang J, et al. MA02.02 efficacy and safety of pralsetinib in chinese patients with advanced RET fusion+ non-small cell lung cancer. Presented at the 2020 WCLC; Sep 8-14, 2021. | Duplicate study |
Curigliano G, Gainor JF, Griesinger F, et al. Safety and efficacy of pralsetinib in patients with advanced RET fusion-positive non-small cell lung cancer: Update from the ARROW trial. J Clin Oncol. 2021;39(15). | Duplicate study |
Curigliano G, Gainor JF, Griesinger F, et al. Safety and efficacy of pralsetinib in patients with advanced RET fusion-positive non-small cell lung cancer: Update from the ARROW trial. Presented at ASCO 2021; Jun 4-8, 2021. | Duplicate study |
Subbiah V, Cassier PA, Siena S, et al. Clinical activity and safety of the RET inhibitor pralsetinib in patients with RET fusion-positive solid tumors: Update from the ARROW trial. J Clin Oncol. 2021;39(15). | Duplicate study |
Subbiah V, Cassier PA, Siena S, et al. Clinical activity and safety of the RET inhibitor pralsetinib in patients with RET fusion‒positive solid tumors: update from the ARROW trial. Presented at ASCO 2021; June 4-8, 2021. | Duplicate study |
Zhou Q, Wu Y, Chang J, et al. JICC01.14 efficacy and safety of pralsetinib in chinese patients with advanced RET fusion+ non-small cell lung cancer after platinum-based chemotherapy. J Thorac Oncol. 2021;16(3):S100. | Duplicate study |
Zhou Q, Wu Y, Chang J, et al. FP14.17 efficacy and safety of pralsetinib in chinese patients with advanced RET fusion+ non-small cell lung cancer after platinum-based chemotherapy. J Thorac Oncol. 2021;16(3):S235. | Duplicate study |
Zhou Q, Wu Y, Chang J, et al. FP14.17 efficacy and safety of pralsetinib in chinese patients with advanced RE fusion+ non-small cell lung cancer after platinum-based chemotherapy. Presented at the 2020 WCLC; Jan 28-31, 2021. | Duplicate study |
Gainor J, Curigliano G, Doebele RC, et al. OA05.02 analysis of resistance mechanisms to pralsetinib in patients with RET fusion-positive non-small cell lung cancer (NSCLC) from the ARROW study. J Thorac Oncol. 2021;16(1):S5. | Duplicate study |
Gainor J, Curigliano G, Doebele RC, et al. OA05.02 analysis of resistance mechanisms to pralsetinib in patients with RET fusion-positive non-small cell lung cancer (NSCLC) from the ARROW study. Presented at the 2020 WCLC; Jan 28-31, 2021. | Duplicate study |
Gainor JF, Curigliano G, Kim D-, et al. MO01.38 registrational dataset from the phase 1/2 ARROW trial of pralsetinib (BLU-667) in patients with advanced RET fusion+ non-small-cell lung cancer (NSCLC). J Thorac Oncol. 2021;16(1):S31-S32. | Duplicate study |
Gainor JF, Curigliano G, Doebele R, et al. Analysis of resistance mechanisms to pralsetinib (BLU-667) in patients with RET fusion–positive non-small cell lung cancer (NSCLC) from the ARROW study. Presented at NACLC 2020; October 16-17, 2020. | Duplicate study |
Gainor JF, Curigliano G, Kim D, et al. Registrational dataset from the phase I/II ARROW trial of pralsetinib (BLU-667) in patients (pts) with advanced RET fusion+non-small cell lung cancer (NSCLC). J Clin Oncol. 2020;38(15). | Duplicate study |
Gainor JF, Curigliano G, Kim D, et al. Registrational dataset from the phase I/II ARROW trial of pralsetinib (BLU-667) in patients (pts) with advanced RET fusion+non-small cell lung cancer (NSCLC). Presented at ASCO 2020 in Chicago, IL; May 29 – Jun 2, 2020. | Duplicate study |
Subbiah V, Hu MI-, Gainor JF, et al. Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+ solid tumors. J Clin Oncol. 2020;38(15). | Duplicate study |
Subbiah V, Hu MI-, Gainor JF, et al. Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+solid tumors. Presented at ASCO 2020 in Chicago, IL; May 29 – Jun 2, 2020. | Duplicate study |
Curigliano G, Cappuzzo F, Siena S, et al. Registrational dataset from the phase 1/2 arrow trial of pralsetinib (BLU-667) in patients (PTS) with advanced ret fusion+non-small-cell lung cancer (NSCLC). Tumori. 2020;106(2):6-7. | Duplicate study |
Curigliano G, Cappuzzo F, Siena S, et al. Registrational dataset from the phase 1/2 arrow trial of pralsetinib (BLU-667) in patients (PTS) with advanced ret fusion+non-small-cell lung cancer (NSCLC). Presented at the 22nd National Congress of Italian Association of Medical Oncology; Oct 30 – Nov 1, 2020. | Duplicate study |
Lee DH, Subbiah V, Gainor JF, et al. Treatment with pralsetinib (formerly BLU-667), a potent and selective RET inhibitor, provides rapid clearance of ctDNA in patients with RET-altered non-small cell lung cancer (NSCLC) and medullary thyroid cancer (MTC). Ann Oncol. 2019;30:ix122. | Duplicate study |
Lee DH, Subbiah V, Gainor JF, et al. Treatment with pralsetinib (formerly BLU-667), a potent and selective RET inhibitor, provides rapid clearance of ctDNA in patients with RET-altered non-small cell lung cancer (NSCLC) and medullary thyroid cancer (MTC). Presented at the 5th ESMO Asia Congress; Nov 22-24, 2019. | Duplicate study |
Curigliano G, Subbiah V, Gainor JF, et al. Treatment with BLU-667, a potent and selective RET inhibitor, provides rapid clearance of ctDNA in patients with RET-altered non-small cell lung cancer (NSCLC) and thyroid cancer. Ann Oncol. 2019;30:v790. | Duplicate study |
Curigliano G, Subbiah V, Gainor JF, et al. Treatment with BLU-667, a potent and selective RET inhibitor, provides rapid clearance of ctDNA in patients with RET-altered non-small cell lung cancer (NSCLC) and thyroid cancer. Presented at ESMO 2019 in Barcelona, Spain; Sep 27-Oct 1, 2019. | Duplicate study |
Evans E, Hu W, Cao F, Hoeflich K, Dorsch M. P2.03-44 BLU-667 demonstrates robust activity in RET-fusion driven intracranial tumor models. J Thorac Oncol. 2019;14(10):S701. | Duplicate study |
Evans E, Hu W, Cao F, Hoeflich K, Dorsch M. P2.03-44 BLU-667 demonstrates robust activity in RET fusion driven intracranial tumor models. Presented at the 2019 WCLC in Barcelona, Spain; Sep 7-10, 2019. | Duplicate study |
Gainor JF, Lee DH, Curigliano G, et al. Clinical activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients (pts) with advanced RET fusion+non-small cell lung cancer (NSCLC). J Clin Oncol. 2019;37. | Duplicate study |
Gainor JF, Lee DH, Curigliano G, et al. Clinical activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients (pts) with advanced RET fusion+non-small cell lung cancer (NSCLC). Presented at the ASCO 2019 in Chicago, IL; May 31-Jun 4, 2019. | Duplicate study |
Brubaker J. Discovery of BLU-667 for RET-driven cancers. Presented at AACR 2019 in Atlanta GA; Mar 29-Apr 4, 2019. | Duplicate study |
Rahal R, Maynard M, Hu W, et al. BLU-667: A highly selective RET inhibitor to target RET-driven NSCLC. Clin Cancer Res. 2018;24(17). | Duplicate study |
Rahal R, Maynard M, Hu W, et al. BLU-667: A highly selective RET inhibitor to target RET-driven NSCLC. Presented at the 5th AACR-IASLC International Joint Conference in San Diego, CA; Jan 8-11, 2018. | Duplicate study |
Subbiah V, Gainor JF, Rahal R, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 2018;8(7):836-849. | Duplicate study |
Subbiah V, Taylor M, Lin J, et al. Highly potent and selective RET inhibitor, BLU-667, achieves proof of concept in a phase I study of advanced, RET-altered solid tumors. Cancer Res. 2018;78(13). | Duplicate study |
Subbiah V, Taylor M, Lin J, et al. Highly potent and selective RET inhibitor, BLU-667, achieves proof of concept in a phase I study of advanced, RET-altered solid tumors. Presented at the 2018 AACR in Chicago, IL; Apr 14-18, 2018. | Duplicate study |
Appendix 3: Detailed Outcome Data
Table 35: Measurable Disease Population, ORR, and DOR
Detail | ARROW November 6, 2020, Data Cut-off | ||||||
|---|---|---|---|---|---|---|---|
Pralsetinib Measurable Disease Population N = 216 | Pralsetinib Prior systemic treatment N = 148 | Pralsetinib Prior platinum N = 126 | Pralsetinib Prior nonplatinum N = 22 | Pralsetinib No prior systemic therapy: All patients N = 68 | Pralsetinib No prior systemic therapy: Pre-protocol amendmenta N = 43 | Pralsetinib No prior systemic therapy: Post-protocol amendmenta N = 25 | |
ORR, % | 69 | 64 | 62 | 73 | 79 | 74 | 88 |
95% CIa | 62, 75 | 55, 71 | 53, 70 | 50, 89 | 68, 88 | 59, 87 | 69, 98 |
CR, % | 4 | 3 | 4 | 0 | 6 | 9 | 0 |
PR, % | 64 | 60 | 58 | 73 | 74 | 65 | 88 |
Stable disease, % | 23 | 28 | 29 | 18 | 13 | 16 | 8 |
PD, % | 5 | 5 | 4 | 9 | 4 | 7 | 0 |
NE, % | 4 | 4 | 5 | 0 | 3 | 2 | 4 |
DOR, Months (95% CI) | 22.3 (15, NR) | 22.3 (15.1, NR) | 22.3 (15.1, NR) | NR (9.2, NR) | NR (9.0, NR) | 11.0 (7.4, NR) | NR (NR, NR) |
CI = confidence interval; CR = complete response; NR = not reached; ORR = overall response rate; PD = progressive disease; PR = partial response.
Note: The November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, data cut-off was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
aInclusion criteria for treatment-naive patients was amended part way through the trial to allow for patients to enrol that are eligible for standard of care therapy, prior to this amendment, treatment-naive patients could enrol only if deemed unsuitable for standard of care therapy.
b95% CI based on exact binomial distribution using Clopper-Pearson method.
Note that this table has not been copy-edited.
Source: Sponsor Submission4
Table 36: ORR Analysis in the ARROW Trial, Platinum Therapy Subgroups
Detail | ARROW November 18, 2019, Data Cut-off | ARROW November 6, 2020, Data Cut-off | ||
|---|---|---|---|---|
Pralsetinib Prior platinum therapy N = 92 | Pralsetinib Prior nonplatinum therapy N = 11 | Pralsetinib Prior platinum therapy N = 136 | Pralsetinib Prior nonplatinum therapy N = 22 | |
ORR, n (%) | |||||||||||| | |||||||||||| | 80 (58.8) | 16 (72.7) |
95% CIa | |||||||||||| | |||||||||||| | 50.1, 67.2 | 49.8, 89.3 |
CR, n (%) | |||||||||||| | |||||||||||| | 7 (5.1) | 0 |
PR, n (%) | |||||||||||| | |||||||||||| | 73 (53.7) | 16 (72.7) |
Stable disease, n (%) | |||||||||||| | |||||||||||| | 43 (31.6) | 4 (18.2) |
PD, n (%) | |||||||||||| | |||||||||||| | 6 (4.4) | 2 (9.1) |
NE, n (%) | |||||||||||| | |||||||||||| | 7 (5.1) | 0 |
CI = confidence interval; CR = complete response; ORR = overall response rate; PD = progressive disease; PR = partial response.
Note: November 18, 2019, data cut-off was an unplanned interim analysis with requirement for patients to have been enrolled on or before July 11, 2019. The November 6, 2020, was an unplanned interim analysis, generated in support of the European Medicines Agency submission, with a requirement for patients to have been enrolled on or before May 22, 2020.
a95% CI based on exact binomial distribution using Clopper-Pearson method.
Source: ARROW Clinical Study Review,5 European Medicines Agency Pralsetinib Public Assessment Report.6
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
European Organization for Research and Treatment of Cancer, 30 Item Core Quality of Life Questionnaire (EORTC QLQ C-30)
Findings
Table 37: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ C-30 | A 30-item, patient-reported, cancer-specific, HRQoL questionnaire using 4- and 7-point Likert scales.38 | Validity: Content validity: When mapping to the World Health Organization’s ICF framework, 25 of the 30 items in the EORTC QLQ-C30 were endorsed by the experts.39 Convergent validity: The strongest correlations were observed (before and during treatment) between physical functioning, role functioning, and fatigue scales, with an r ranging between 0.54 and 0.63. in patients with nonresectable lung cancer.38 Known-groups validity: Patients with nonresectable lung cancer who had better ECOG PS scores at the pre-treatment stage reported higher physical, cognitive, and role functioning and overall QoL scores, as well as significantly lower symptom scores (ANOVA: n = 295, P < 0.001 to P < 0.05).38 Criterion validity/ clinical validity: For WHO PS, interaction effects were observed for global health status/QoL, and physical, role and social functions (P < 0.0001). For a standardized 6-minute walk test, strongest interaction effect was seen with physical, role, and social functioning, fatigue, and global QoL (P < 0.0001).40 Concurrent validity: EORTC QLQ C-30 demonstrated a strong correlation (- 0.75) between emotional functioning and the HADS anxiety scale, along with a substantial correlation (- 0.47) with global QoL.40 Reliability: Internal consistency reliability was adequate in patients with nonresectable lung cancer. Cronbach alpha coefficients for the global QoL were 0.86 before treatment and 0.89 during treatment, which can be considered a good reliability.38 Responsiveness: Between-group differences over time were observed for global quality of life (P < 0.01), physical functioning (P < 0.001), role functioning (P < 0.001), fatigue (P < 0.01), and nausea/vomiting (P < 0.05) scale.38 | Patients with NSCLC, breast cancer and SCLC,
Patients with breast cancer, followed by lung, prostate, gastrointestinal, renal cell, and other advanced cancers
|
ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; HADS = Hospital Anxiety and Depression Scale; HRQoL= health-related quality of life; MID = minimal important difference; NSCLC = non–small cell lung cancer; SCLC = small cell lung cancer; WHO PS = WHO Performance Status.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30)
Description and Scoring
The EORTC QLQ-C30 is one of the most commonly used patient-reported outcomes (PRO) measures in oncology clinical trials.43 It is a multi-dimensional, cancer-specific, evaluative measure of HRQoL. This standardized, patient self-administered questionnaire has been designed to evaluate the quality of life of patients with cancer participating in clinical trials.38
The EORTC QLQ-C30 is composed of both multi-item scales and single-item measures. These include 5 functional scales (physical, role, emotional, cognitive, and social functioning), 3 symptom scales (fatigue, nausea/vomiting, and pain), and 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties). This instrument also includes a global health status and overall quality of life section.40
The EORTC QLQ-C30 uses a 1-week recall period to assess functional status and symptoms. All scales and single-item measures are scored from 0 to 100. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 to 4. For the 2 items that form the global HRQoL scale, the response format is a 7-point Likert-type scale with anchors at 1 = “very poor” and 7 = “excellent.” Raw scores for each scale are computed as the average of the items that contribute to a particular scale. Scale sum scores are transformed such that a high score on the functional scales represents a high/healthy level of functioning, a high score on the symptom scales represents a high level of symptomatology, and a high score on the global health status/HRQoL scale represents a high HRQoL.44
According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale (i.e., the participant did not provide a response), the score for the scale can still be computed if there are responses for at least half of the items. The values for missing items are interpolated with the average of the respondent-completed items.44
Assessment of Validity
One study39 assessed the content validity of the EORTC QLQ-C30 based on the opinions of 21 experts. When mapping to WHO’s International Classification of Functioning (ICF) framework, 25 of the 30 items in the EORTC QLQ-C30 were endorsed by the experts: 15 items mapping to impairment of body function, 7 mapping to activity limitations/participation restrictions, and 1 item mapping to both components. There were only 2 items of the EORTC QLQ-C30 tapped content outside of functioning: Item 29 mapping to perceived health and item 30 mapping to global quality of health. The authors stated that the fact that most items from the EORTC QLQ-C30 can be linked to the ICF framework means that the instrument’s content reflects functioning, which is a key component of HRQoL.39
Aaronson et al.38 tested construct validity of EORTC QLQ-C30 in these patients with nonresectable lung cancer (of 287 patients with reported histologic types, 63.1% had NSCLC) undergoing either radiotherapy or chemotherapy. These patients were recruited from an international field trial of 305 patients in 13 countries, including Canada. The construct validity of the instrument was evaluated using the correlations among the EORTC QLQ-C30 scales and the known-groups comparison method. While assessing the correlations, a substantial correlation (Pearson’s r ≥ 0.40) was expected among the conceptually related scales, such as physical functioning and fatigues, whereas lower correlations (Pearson’s r < 0.40) were expected among the scales with less commonality with each other, such as, cognitive functioning and nausea/vomiting. In the known-groups comparison method, the ability of the questionnaire scores to differentiate between the patient subgroups with different clinical status was evaluated. For convergent validity, the strongest correlations were observed (before and during treatment) between physical functioning, role functioning, and fatigue scales, with an r ranging between 0.54 and 0.63. Based on the known-groups approach, patients with better ECOG PS scores at the pre-treatment stage reported significantly higher physical, cognitive, and role functioning and overall QoL scores, as well as significantly lower symptom scores (ANOVA: n = 295, P < 0.001 to P < 0.05), compared with patients with poorer PS scores. In addition, statistically significant group differences were observed as expected for all functional and symptom scores, according to the on treatment ECOG PS grouping variable (ANOVA: n = 265, P < 0.001 to P < 0.05), and for 5 out of 6 functional scales and 5 out of 7 symptom measures, based on toxicity ratings as group variable (ANOVA: n = 244, P < 0.001 to P < 0.05). Similarly, statistically significant group differences were observed in pre-treatment when patients having less weight loss reported better QoL scores as expected (ANOVA: n = 295, P < 0.001 to P < 0.05).38
Nicklasson et al.40 conducted construct, criterion, and concurrent validity tests of EORTC QLQ-C30 with 112 Swedish patients diagnosed with advanced lung cancer or pleural mesothelioma, including 85 (76%) patients with NSCLC, not amenable to curative or life prolonging treatment. Construct validity was examined by multitrait analysis, based on the definition of item convergent validity as a correlation of ≥.4 between an item and its own hypothesized scale, and of scaling error as the case when an item correlated >1 standard error better with another scale than its own hypothesized scale. Criterion validity/ clinical validity was assessed by variance and correlation with an array of clinical parameters, including performance status, 6-min walk test, spirometry, tumour stage, and blood tests. Concurrent validity was evaluated by established scales for emotional distress (Hospital Anxiety and Depression Scale or HADS) and pain (Brief Pain Inventory or BPI). Correlations were designated as strong (>0.60), substantial (>0.40) or moderate (>0.20).40
While assessing the criterion validity/ clinical validity based on WHO Performance Status (WHO PS), significant interaction effects were observed for global health status/QoL, and physical, role and social functions (P < 0.0001). For a standardized 6-minute walk test, strongest interaction effect was seen with physical, role, and social functioning, fatigue, and global QoL (P < 0.0001). In a correlation analysis employing walking distance (> 200m, n = 58) as a continuous variable, a strong correlation (r = 0.77) with physical functioning, and substantial correlation (r > 0.4) with role functioning, fatigues, and global health status/QoL was observed. With spirometry, a correlation (r = not reported) with global health status/QoL was observed such that patients with an FEV1 predicted value <50% (n = 27) scored worse than did patients with an FEV1 predicted value ≥50% (n = 61).40
While assessing the concurrent validity, a strong correlation (- 0.75) was observed between emotional functioning and the HADS anxiety scale, along with a substantial correlation (- 0.47) with global QoL. In addition, the HADS depression scale correlated substantially (>0.40) with all functioning scales, appetite loss and fatigue. On the other hand, the BPI intensity subscale (BPI-I) correlated strongly (r = 0.72) with the QLQ-C30 pain scale, moderately but significantly (>0.40) with functioning scales (except physical and social functioning), global QoL, and the remaining symptom scales (except nausea/vomiting). The BPI function subscale (BPI-F) correlated substantially (>0.40) with all functioning scales, global QoL, dyspnea and pain measures.40
Assessment of Reliability
Aaronson et al.38 tested reliability/internal consistency in the same population as described above in the validity section. The reliability coefficients (Cronbach alpha) for the global QoL were 0.86 before treatment and 0.89 during treatment, which can be considered a good reliability.
Nicklasson et al.40 performed reliability testing in the same population as described in the validity section above. Reliability of the global health status/QoL scale showed an internal consistency of 0.70 or higher, which is an accepted threshold for group comparisons.
Responsiveness to Change
Aaronson et al.38 measured the responsiveness in the context of improvement or deterioration of health status, which was estimated based on at least one level upward or downward shift on the ECOG PS scale. Statistically significant changes in EORTC QLQ-C30 scores were tested using repeated-measures ANOVA, as a function of observed changes in PS. Using this repeated-measures ANOVA with divided patient samples based on ECOG PS, statistically significant between-group differences over time were observed for global quality of life (P < 0.01), physical functioning (P < 0.001), role functioning (P < 0.001), fatigue (P < 0.01), and nausea/vomiting (P < 0.05) scale. No changes were noted in QLQ-C30 scores among those patients whose performance status had remained unchanged.38
Table 38: Responsiveness of EORTC QLQ-C30 Scores Over Timea
EORTC QLQ-C30 scale | Improved ECOG (n = 34) | Deteriorated ECOG (n = 79) | ||||||
|---|---|---|---|---|---|---|---|---|
Pre-treatment | On-treatment | Pre-treatment | On-treatment | |||||
Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
Global Health Statusb | 53.3 | 21.8 | 62.9 | 19.4 | 56.2 | 25.5 | 50.5 | 25.0 |
Physical Functioningc | 58.1 | 27.1 | 67.5 | 22.6 | 67.8 | 27.6 | 54.7 | 32.0 |
Role Functioningc | 55.9 | 36.4 | 67.6 | 34.6 | 60.1 | 38.7 | 44.3 | 39.2 |
Fatigueb | 43.1 | 27.6 | 40.1 | 26.0 | 42.6 | 25.7 | 53.2 | 27.7 |
Nausea and Vomitingd | 11.8 | 20.7 | 14.7 | 20.8 | 9.9 | 18.4 | 26.4 | 29.2 |
ECOG = Eastern Cooperative Oncology Group; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire – Core 30; SD = standard deviation
aBased on repeated measures ANOVA. Statistical tests for group x time interaction with 3 groups (improved, deteriorated, and unchanged ECOG PS) and 2 assessment points (pre-treatment and on-treatment). N = 262 due to missing ECOG PS ratings. Mean for the unchanged ECOG group are not presented. b
bP < 0.01
cP < 0.001
dP < 0.05
Source: Aaronson et al.38
One study by Osoba et al.45 aimed to assess the responsiveness of the EORTC QLQ-C30 in 160 lung cancer patients who received chemotherapy. The QLQ-C30 showed responsiveness to changes in disease state and treatment to chemotherapy in the expected direction. Patients with metastatic disease and those who received chemotherapy had diminished scores in the domains of physical and social role functions, and global quality of life, and had greater fatigue and nausea and vomiting compared with before chemotherapy.45
Minimal Important Difference
For use in clinical trials, scores on the EORTC QLQ-C30 can be compared between groups of patients or within a group of patients over time. One study conducted in breast cancer and small-cell lung cancer patients in 1998 estimated a clinically relevant change in score on any scale of the EORTC QLQ-C30 to be 10 points.46 The estimate was based on a study that used an anchor-based approach to estimating the MID in which patients who reported “a little” change (for better or worse) on the subjective significance questionnaire had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately 5 to 10 points. Participants who reported a “moderate” change had corresponding changes in the EORTC QLQ-C30 of about 10 to 20 points, and those who reported being “very much” changed had corresponding changes of more than 20 points.46
In 2014, a Canadian study estimated the MID for EORTC QLQ-C30 in 369 patients with advanced cancer who completed the questionnaire at baseline and 1 month post-radiation.42 The most common cancer type was breast cancer, followed by lung, prostate, gastrointestinal, renal cell, and other cancers. The MID was estimated using both anchor- and distribution-based methods for improvement and deterioration. Two anchors of overall health and overall QoL were used, both taken directly from the EORTC QLQ-C30 (questions 29 and 30) where patients rated their overall health and QoL themselves. Improvement and deterioration were categorized as an increase or decrease by 2 units to account for the natural fluctuation of patient scoring. With these 2 anchors, the estimated MIDs across all EORTC QLQ-C30 scales ranged from 9.1 units to 23.5 units for improvement, and from 7.2 units to 13.5 units for deterioration. Distribution-based estimates were closest to 0.5 SD.42
Maringwa et al.47 estimated MIDs based on anchor-based method by pooling data from 2 RCTs on EORTC. Total 812 patients with palliative, locally advanced, and/or metastatic NSCLC undergoing treatment were enrolled. As for anchors chosen, physician-rated WHO PS and weight change were used based on their relevance to patients with NSCLC. Effect size of 0.2 SD, 0.5 SD, and threshold of 1 standard error of mean (SEM) of HRQoL scores have been reported as distribution-based MIDs to compare with the anchor-based MIDs.
MID estimates for improvement (i.e., 1 category change in PS, 5 - <20% weight gain) were: 9 and 4 for physical functioning, 14 and 7 for role functioning, 5 and 7 for social functioning, 14 and 5 for fatigue, 16 and 2 for pain, and 9 and 4 for global health status. The respective MID estimates for deterioration (i.e., 1 category change in PS, 5 - <20% weight loss) were: 4 and 6 for physical functioning, 5 for role functioning, 7 and 9 for social functioning, 6 and 11 for fatigue, 3 and 7 for pain, and 4 for global health status.47 MID estimates based on anchor-based and distribution-based methods are shown in Table 39.
Table 39: Summary of MIDs for the EORTC QLQ-C30 Subscale
EORTC QLQ-C30 scales | MID for improvement (anchor-based) | MID for deterioration (anchor-based) | MID (distribution-based) | MID (distribution-based) | MID (distribution-based) |
|---|---|---|---|---|---|
PS, weight gain | PS, weight loss | SEM | 0.5 SD | 0.25 SD | |
Global Health Status | 9, 4 | 4, 4 | 9 | 11 | 4 |
Physical Functioning | 9, 5 | 4, 6 | 7 | 12 | 5 |
Role Functioning | 14, 7 | 5, 5 | 14 | 17 | 6 |
Social Functioning | 5, 7 | 7, 9 | 10 | 14 | 6 |
Fatigue | 14, 5 | 6, 11 | 11 | 13 | 5 |
Pain | 16, 2 | 3, 7 | 12 | 16 | 6 |
EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire – Core 30; MID = minimal important difference; PS = performance status; SD = standard deviation; SEM = standard error of mean.
Source: Maringwa et al.47
The limitation of MID estimation performed by Maringwa et al.47 is poor correlations between changes in either anchor (WHO PS or weight) and QLQ-C30. For example, for changes in global health status scores and changes in both anchors, the correlations coefficients range from 0.10 to 0.14 in absolute values. The Spearman rank correlation of at least 0.30 is suggested to be acceptable association.48
MIDs and/or clinically significant/relevant differences were also applied in other studies to assess changes in HRQoL among 50 patients with locally advanced and metastatic NSCLC in Belgium,49 480 patients with advanced NSCLC (stage IIIB and stage IV) in Europe, South Africa and Egypt,50 138 patients with NSCLC (stage IIa-IIIb) in the US and Canada.51 Fifty-one medically inoperable, early NSCLC patients in Poland,52 376 patients with advanced nonsquamous NSCLC in Europe, Russia, Turkey and United Arab Emirates,53 240 patients with NSCLC in US, Canada, the UK, and Europe,54 120 NSCLC patients in the US and Canada.55 Forty-five early-stage NSCLC patients in US and Canada,56 334 patents with advanced NSCLC in Sweden,41 713 stage III, unresectable NSCLC in North and South America (including Canada), Asia, Australia, Europe, UK, and South Africa,57 717 NSCLC survivors in Germany,58 and 451 elderly patients with advanced NSCLC in France.59 A 10-point change in score within a patient over time was considered the threshold of MCIDs and/or clinically significant/relevant differences in all of these studies, except for Rutkowski et al.,52 where the clinically meaningful improvement/clinical relevance were considered to be greater than 7%, for Larsson et al.,41 where clinically relevant differences were considered small for 5 to 10 points changes, moderate for 11 to 19 points changes, and large for greater than 20-point changes, based on the Osoba et al. study,46 and for Fiteni et al.,59 where a 5-point decrease was used as the MCID.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
NSCLC
non–small cell lung cancer
OS
overall survival
PD-L1
programmed death ligand 1
PBC
platinum-based chemotherapy
pERC
CADTH pan-Canadian Oncology Drug Review Expert Review Committee
PFS
progression-free survival
QALY
quality-adjusted life-year
SLR
systematic literature review
TTD
time to discontinuation
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Pralsetinib (Gavreto), 100 mg oral capsule |
Submitted price | Pralsetinib, 100 mg: $102.06 per capsule |
Indication | For the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | June 30, 2021 |
Reimbursement request | For the treatment of adult patients with RET fusion–positive locally advanced unresectable or metastatic NSCLC; treatment should continue as long as the patient is deriving clinical benefit from therapy or until unacceptable toxicity |
Sponsor | Hoffman-La Roche Ltd. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; NSCLC = non–small cell lung cancer.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model |
Target populations | Adult patients with metastatic RET fusion–positive NSCLC not previously treated with an RET inhibitor, assessed in the following subgroups:
|
Treatment | Pralsetinib |
Comparators | Treatment-naive: pembrolizumab plus pemetrexed plus carboplatin/cisplatin (triple therapy), pembrolizumab alone, PBC (carboplatin/cisplatin) plus pemetrexed Treatment-experienced: docetaxel, nivolumab, PBC plus pemetrexed (cisplatin) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | 20 years |
Key data source |
|
Submitted results | Treatment-naive: sequential ICER for pralsetinib = $165,789 per QALY gained vs. PBC plus pemetrexed (incremental costs: $206,531; incremental QALYs: 1.25) Treatment-experienced: sequential ICER for pralsetinib = $187,319 per QALY gained vs. docetaxel (incremental costs: $299,749; incremental QALYs: 1.60) |
Key limitations |
|
CADTH reanalysis results |
Scenario analyses considering the sponsor’s optimistic OS benefits with pralsetinib suggested price reductions in excess of 60% and 75% in the treatment-naive and exposed settings, respectively, were necessary for pralsetinib to be considered cost-effective at a $50,000 per QALY threshold when excluding testing costs |
ICER = incremental cost-effectiveness ratio; NSCLC = non–small cell lung cancer; OS = overall survival; PBC = platinum-based chemotherapy; PFS = progression-free survival; QALY = quality-adjusted life-year; vs. = versus.
Conclusions
The CADTH clinical review concluded that data from the pivotal trial were inadequate to interpret overall survival (OS) and progression-free survival (PFS) findings due to the single-arm trial design and immaturity of the data. Evidence generated from the sponsor-submitted indirect treatment comparison (ITC) was insufficient to make comparisons across therapies due to a significant amount of uncertainty arising from patient heterogeneity between trials that was not accounted for.
Given that OS and PFS are key components within a partition survival model, the lack of interpretability of this information for pralsetinib directly affects any conclusions that may be drawn from the economic evaluation. The absence of robust comparative evidence limits any conclusions that can be drawn regarding the cost-effectiveness of pralsetinib. CADTH conducted exploratory reanalyses to determine the impact of addressing identified key limitations. CADTH revised the sponsor’s model to assume equal OS for each comparator within each subgroup, selected alternative PFS extrapolation distributions, revised drug costs to reflect dosing and stopping rules in alignment with product monographs and expected use in practice, and revised subsequent therapy use to reflect clinical practice. Not all of the concerns with the sponsor’s submission could be addressed, and these outstanding limitations (e.g., PFS benefits), may bias results in favour of pralsetinib.
Based on CADTH’s exploratory reanalysis, the incremental cost-effectiveness ratio (ICER) for pralsetinib was in excess of $1 million per quality-adjusted life-year (QALY), in treatment-naive and treatment-experienced populations regardless of whether the cost of testing was included. Under the assumption of equal OS, results are largely driven by the drug acquisition costs for pralsetinib. In the treatment-naive setting, pralsetinib was associated with an ICER of $3,063,599 per QALY ($4,108,183 per QALY including testing) versus triple therapy. A price reduction of at least 80% (or 90% with inclusion of full testing costs) would be required to achieve cost-effectiveness at a threshold of $50,000 per QALY based on the sequential analysis. In the treatment-experienced setting, pralsetinib was associated with an ICER of $1,567,170 per QALY ($1,726,230 per QALY including testing) versus docetaxel. A price reduction of at least 96% (or 99% with inclusion of full testing costs) is required for pralsetinib to be considered cost-effective in treatment-experienced patients at a threshold of $50,000 per QALY in this setting. Scenario analyses considering the sponsor’s optimistic OS benefits from pralsetinib suggested price reductions in excess of 60% and 75% in the treatment-naive and treatment-experienced settings, respectively, were necessary for pralsetinib to be considered cost-effective at a threshold of $50,000 per QALY when excluding testing costs. Scenario analyses considering equal post-progression survival across all comparators suggested price reductions in excess of 70% and 85% in the treatment-naive and treatment-experienced settings, respectively, were necessary for pralsetinib to be considered cost-effective at a threshold of $50,000 per QALY when excluding testing costs.
Given the absence of comparative clinical evidence for pralsetinib and the submitted model structure, the exploratory results and subsequent price reductions may remain biased in favour of pralsetinib. As such, and given the submitted clinical information, the true cost-effectiveness of pralsetinib in comparison to relevant treatment comparators in both the treatment-naive and treatment-experienced setting is highly uncertain. CADTH was also unable to assess cost-effectiveness of pralsetinib in comparison with selpercatinib, the other therapy for metastatic RET fusion–positive non–small cell lung cancer (NSCLC) that recently received a positive listing recommendation from the CADTH pan-Canadian Oncology Drug Review Expert Review Committee (pERC), at the time of this review.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received patient input from Lung Cancer Canada, a national organization that provides resources for lung cancer education, provides patient support, and supports research and advocacy. The input was based on interviews with 5 patients with RET-positive NSCLC and caregivers with experience with pralsetinib in Canada, the US, Ireland, and Norway. Patients identified improved functionality in life, delayed disease progression and improved survivorship, reduced symptoms of disease such as shortness of breath, manageable treatment side effects, and maintenance of independence as treatment goals. Patients receiving chemotherapy as current standard of care noted that it is not viable as a long-term treatment option due to side effects such as weakness and fatigue leading to decreased functionality. Among those receiving immunotherapy, patients reported fewer side effects compared to chemotherapy, but added that access is limited by the need to travel to hospitals or clinics. Patients noted that targeted therapies for those with targetable mutations such as RET fusion are preferred in the second-line setting for managing and treating symptoms of NSCLC. Four patients reported treatment benefits and improved quality of life following treatment with pralsetinib. However, the interviewed patients commented that fatigue was the most common side effect of pralsetinib, followed by dry mouth, anemia, constipation, loss of appetite, and itchiness. One patient reported being hospitalized for higher liver function. Respondents noted that an oral targeted therapy option offers greater benefits compared with standard IV treatment due to the ease of administration, minimal travel requirement for treatment, and decreased patient and caregiver burden.
CADTH received registered clinician input from the Ontario Health – Cancer Care Ontario Lung Cancer Drug Advisory Committee and the Lung Cancer Canada Medical Advisory Committee. Both groups stated that first-line therapies include platinum-pemetrexed doublet chemotherapy, platinum-pemetrexed plus pembrolizumab (depending on programmed death ligand 1 [PD-L1] status), and pembrolizumab alone. Second-line therapies include platinum-pemetrexed for those receiving pembrolizumab as first-line therapy, anti–PD-L1 therapy, including pembrolizumab, nivolumab, and atezolizumab for those receiving platinum-pemetrexed as first-line therapy, followed by docetaxel for those progressing on platinum-pemetrexed and pembrolizumab. Upon treatment failure of the previous options, best supportive care would be administered. Both clinician groups stated that pralsetinib would be used in any line of therapy, depending on time of RET fusion diagnosis.
Drug plan input included concern regarding the lack of a comparator in the phase I and II ARROW trial. At present, no publicly funded treatments that specifically target RET fusion for advanced NSCLC are available in Canada; however, selpercatinib is a recently reviewed comparator of interest. Drug plans also noted the uncertainty surrounding eligibility for pralsetinib related to treatment-naive status, an Eastern Cooperative Oncology Group Performance Status of greater than 2, and the presence of central nervous system metastases. Drug plans also emphasized the uncertainty surrounding treatment discontinuation, funding criteria, treatment sequencing, and clinical preference, specifically for pralsetinib use in comparison to selpercatinib. Last, RET fusion testing is required to identify eligible patients for pralsetinib, which was highlighted by drug plans.
Several of these concerns were addressed in the sponsor’s model:
The sponsor compared pralsetinib to first-line (pembrolizumab, platinum-based chemotherapy [PBC] plus pemetrexed plus pembrolizumab, and PBC alone) and second-line therapies (docetaxel, nivolumab, and PBC) for the treatment of RET fusion–positive NSCLC.
In addition, CADTH addressed some of these concerns as follows:
CADTH explored the impact of including the costs of RET fusion testing in the pharmacoeconomic evaluation.
CADTH was unable to address in its reanalysis the concerns raised in stakeholder input regarding:
Treatment sequencing, beyond consideration of subsequent therapy costs, and clinical preference for pralsetinib in consideration of selpercatinib.
Economic Review
The current review is for pralsetinib (Gavreto) for adult patients with metastatic RET fusion–positive NSCLC, both those receiving a first-line treatment and those who have received prior therapy non–RET targeted therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of pralsetinib compared to alternative therapeutic options both as a first-line treatment for treatment-naive patients and as a second-line treatment for treatment-experienced adult patients with metastatic RET fusion–positive NSCLC. The target population was aligned with the Health Canada indication and the reimbursement request.
Pralsetinib is available as 100 mg oral capsules for use as monotherapy with a recommended dosage of 400 mg once daily until loss of clinical benefit or unacceptable toxicity.1 For treatment-naive patients, the submission identified 3 comparators: triple therapy (pembrolizumab, pemetrexed, and PBC), pembrolizumab alone, and PBC (cisplatin-carboplatin) plus pemetrexed. Triple therapy was assumed to be given at a dose of 75 mg/m2 for cisplatin and 500 mg/m2 for pemetrexed every 3 weeks.2 Pembrolizumab was assumed to be given at a dose of 200 mg for up to 2 years or until loss of clinical benefit or unacceptable toxicity.2 Pemetrexed plus PBC was assumed to be given at an area-under-the-curve dose of 5 mg/mL for carboplatin or 75 mg/m2 for cisplatin and 500 mg/m2 for pemetrexed administered every 3 weeks.2 All comparators were administered by IV, and treatment was assumed to continue until progressive disease or unacceptable toxicity for all comparators in lieu of product monograph–specified dose scheduling.
For second-line use, 3 comparators were identified: docetaxel, nivolumab, and PBC (cisplatin) plus pemetrexed. PBC plus pemetrexed was assumed to be given at a dose of 75 mg/m2 for cisplatin and 500 mg/m2 for pemetrexed administered every 3 weeks.2 Nivolumab alone, which was assumed to represent all possible immunotherapies given to treatment-experienced patients, was given as 3 mg/kg of body weight once every 2 weeks until disease progression, and docetaxel alone would be given at a dose of 75 mg/m2 until disease progression or unacceptable toxicity.2 Dosing schedules were based on the sponsor’s assumption in lieu of product monograph–recommended treatment-stopping rules.
Pralsetinib is dispensed at a cost of $102 per 100 mg capsule, leading to an average daily cost of $408 and a 30-day cycle cost of $12,426.2 For treatment-naive therapy, the cost of triple therapy per cycle was $18,709, the cost per cycle of pembrolizumab alone was $12,755, and the cost per cycle of PBC plus pemetrexed was $6,015.2 For treatment-experienced therapy, the cost per cycle for docetaxel was $289, the cost per cycle of nivolumab was $10,204, and the cost per cycle of PBC plus pemetrexed was $5,578.2 No administration costs were applicable to pralsetinib due to oral administration, and IV infusion costs were $207 per model cycle for all comparators except nivolumab, which incurred a per-cycle administration cost of $155.2 The sponsor’s analysis assumed no drug wastage for IV products.
Outcomes modelled included QALYs and life-years over a lifetime time horizon of 20 years. A base-case analysis was conducted from the Canadian public health care system perspective, with costs and outcomes discounted at 1.5% annually. The cycle length was 1 month with a half-cycle correction.
Model Structure
The sponsor submitted a partitioned survival model that consisted of 3 mutually exclusive health states: progression-free, post-progression, and death. Within the model, the proportions of patients occupying each state were estimated on a monthly basis and costs (relating to treatment, treatment-related adverse events [AEs] and disease state) and utilities (relating to AEs and disease states) were allocated.
The model does not incorporate the transition of patients between health states, but rather the proportion of patients who are progression-free, and the proportion who are alive at each time point are estimated independently using PFS and OS curves. A figure of the sponsor’s model structure can be found in Appendix 3 (Figure 1).
Model Inputs
The modelled populations included 2 patient cohorts: previously treated (treatment-experienced) patients with RET fusion–positive NSCLC and treatment-naive patients with RET fusion–positive NSCLC. The cohort of patients with RET fusion–positive advanced NSCLC consisted of those from the pivotal ARROW trial (mean age of 60 years; 48% male), whereas the treatment-experienced cohort was used to compare second-line therapies and was assumed to have the same characteristics as the subset of 165 patients stratified by prior treatment within the unrestricted efficacy population of the ARROW trial.3,4 The treatment-naive cohort was used to compare first-line therapies and was assumed to have the same characteristics as the subset of 116 patients stratified by treatment-naive status within the unrestricted efficacy population of the ARROW trial.3,4
The sponsor used parametric survival modelling to extrapolate the PFS and OS data beyond the limited time horizon of the trial data collected from the single-arm, ongoing phase I and II ARROW trial for patients treated with pralsetinib, stratified by line of therapy. For the treatment-naive population, an exponential distribution was selected for OS and a log-normal distribution was selected for PFS based on best statistical fit and clinical plausibility. For the treatment-experienced population, the parametric distribution selected for first-line therapy was also applied in the second-line setting to ensure consistency between projections.
In the complete absence of head-to-head comparisons with any comparators used in the first-line or second-line setting, the sponsor conducted an ITC to assess the relative efficacy of pralsetinib versus all comparators for PFS, time to discontinuation (TTD), and OS, stratified by line of therapy. Hazard ratios to inform the relative treatment effects of relevant comparators to pralsetinib were derived primarily from unadjusted, naive ITC analyses of wild-type NSCLC clinical data identified via the sponsor’s systematic literature review (SLR), including the KEYNOTE-042 (pembrolizumab monotherapy), KEYNOTE-189 (triple therapy), and IMpower132 (PBC plus pemetrexed or paclitaxel) trials.5 The sponsor’s comparisons with docetaxel and dual therapy did attempt to adjust for prognostic factors, but the analyses were unanchored. These hazard ratios were then applied to the extrapolated OS and PFS curves from the ARROW trial to estimate the treatment efficacy of comparators in the treatment-naive population with regard to OS, PFS, and TTD. Similarly, hazard ratios for pralsetinib versus comparators were derived from the OAK (docetaxel), LUME-Lung 1 (docetaxel and nintedanib), CheckMate 057 (nivolumab), and GOIRC 02 to 2006 and NVALT7 (PBC plus pemetrexed or paclitaxel) trials and applied to OS and PFS curves from the ARROW trial to estimate the relative efficacy of second-line comparators.5 This methodology allowed for an artificial comparison of pralsetinib to first- and second-line therapies in patients with RET fusion–positive advanced NSCLC. It was assumed that the relative treatment effects would be sustained for the time horizon of the model, including the period after treatment discontinuation.
Health-related quality of life data by health state estimated using the EQ-5D utility instrument and ED-5D Visual Analogue Scale were taken from a study of patients with advanced NSCLC in Canada, Europe, Australia, and Turkey.6 Disutilities due to age and sex were applied using a regression algorithm described by Ara and Brazier.7 Disutilities due to AEs were incorporated as utility decrements sourced from the literature based on the duration of the AE.2
Costs included in the model included those associated with drug acquisition, health services, treatment of AEs, and terminal care. Drug acquisition costs and dosing were consistent with those reported in the Overview section, with drug costs for pralsetinib obtained from the sponsor’s submission. Costs of comparator treatments were obtained from IQVIA DeltaPA data and Association Québécoise des Pharmaciens Propriétaires prices where Ontario wholesale prices were not available.8 Treatment administration costs were applied for IV treatments only.9 The base-case analysis did not include costs of screening and identifying patients with RET fusion–positive NSCLC; however, the cost of 1 test ($400) per patient with RET fusion–positive NSCLC was included in a scenario analysis.10 Costs of subsequent treatment after failing first- or second-line therapy were applied as a total weighted cost for 2 model cycles, with the distribution of treatments specific to the initial intervention received based on Canadian clinical expert opinion. Health services costs associated with health states included outpatient visits, chest X-rays, CT scans, electrocardiograms, community nurse visits, general practitioner home visits, therapist visits, and clinical nurse specialist visits.11 The frequency of resource use in the progression-free and post-progression states was based on a physician survey of NSCLC resource use in the UK setting and assumed to be applicable to Canadian patients.12 Costs related to AEs were obtained from the Ontario Case Costing Initiative and applied in the first cycle based on the proportion of patients experiencing AEs from the ARROW trial.13 Terminal care costs were based on a published value for patients with NSCLC receiving palliative care.14
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic analyses were broadly similar. The probabilistic findings are presented in the following section.
Base-Case Results
In the first-line setting for treatment-naive patients, average costs were $114,236 for PBC, $171,074 for pembrolizumab, $317,273 for triple therapy, and $320,768 for pralsetinib. Average QALYs were 1.53 for PBC, 1.19 for pembrolizumab, 1.88 for triple therapy, and 2.78 for pralsetinib. In the sequential analysis, pembrolizumab was dominated by PBC, and triple therapy was extendedly dominated by pralsetinib, leaving pralsetinib and PBC on the cost-effectiveness frontier. The sequential ICER for pralsetinib versus PBC was $165,789 per QALY (Table 3).
In the second-line setting for treatment-experienced patients, average costs were $49,005 for docetaxel, $79,109 for PBC, $133,135 for nivolumab, and $348,754 for pralsetinib. Average QALYs were 0.78 for docetaxel, 0.70 for PBC, 0.96 for nivolumab, and 2.38 for pralsetinib. In the sequential analysis, PBC was dominated by docetaxel, and nivolumab was extendedly dominated by pralsetinib. The sequential ICER for pralsetinib versus docetaxel was $187,319 per QALY (Table 4).
Table 3: Summary of Sponsor’s Economic Evaluation Results for Treatment-Naive Patients
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Platinum-based chemotherapy | 114,236 | 1.53 | Reference |
Pralsetinib | 320,768 | 2.78 | 165,789 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
Table 4: Summary of Sponsor’s Economic Evaluation Results for Treatment-Experienced Patients
Drug | Total costs ($) | Total QALYs | Sequential ICER ($ per QALY) |
|---|---|---|---|
Docetaxel | 49,005 | 0.78 | Reference |
Pralsetinib | 348,754 | 2.38 | 187,319 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
Incremental costs for pralsetinib in both lines of therapy were due largely to higher drug costs, while incremental QALYs were due to greater time spent in the progression-free and disease-progressed states. Given that the trial informing the efficacy of pralsetinib in both subgroups had only a single arm, 100% of the incremental life-years and QALYs are derived from extrapolation methods as opposed to clinical trial evidence.
Additional results from the sponsor’s submitted economic evaluation are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
The sponsor conducted various sensitivity and scenario analyses involving the inclusion of the cost of one-time genetic testing (per patient identified as RET fusion–positive), alternative efficacy data for comparator treatments derived from the Flatiron Enhanced Datamart and Clinico-Genomic Database, an alternate time horizon, and various parametric distributions for OS and PFS. In these analyses, the ICER was most sensitive to a reduced time horizon of 5 years and extrapolation of OS. The ICER values across all scenario analyses in the first- and second-line settings varied from $111,292 to $261,536 per QALY and did not achieve cost-effectiveness at a $50,000 willingness-to-pay threshold regardless of the scenario analysis conducted.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Relative effect of pralsetinib in comparison with relevant comparators on PFS, OS, and TTD is highly uncertain: To estimate the relative effect of pralsetinib on the outcomes of interest in the model, the sponsor conducted an ITC of the included comparators. The ITC consisted primarily of a naive comparison, with adjusted estimates derived in comparison with docetaxel and PBC plus pemetrexed; however, these analyses were unanchored and did not account for all relevant prognostic factors. Furthermore, there is considerable uncertainty with respect to the relative effect of pralsetinib on delaying PFS and OS, as well as the relative time on treatment, given the absence of randomized controlled evidence, the small sample size of patients with RET fusion–positive NSCLC in the single-arm phase I and II ARROW trial used to interpolate a treatment effect or duration, and the reliance on data for comparators not relating to patients with RET fusion–positive NSCLC for PFS and OS. The CADTH clinical review was unable to draw conclusions regarding the relative effectiveness of pralsetinib and the comparators included in the sponsor’s SLR based on the naive comparisons conducted. For the 2 comparisons using the propensity-score weighted analysis (docetaxel in the treatment-experienced setting and PBC plus pemetrexed in the treatment-naive setting) and the analyses using the Flatiron data, conclusions should be made with caution. Consequently, the estimated effect size with pralsetinib is highly uncertain and the true cost-effectiveness of pralsetinib in this setting is unknown.
CADTH was unable to address this limitation and notes that relative effect estimates may have been overestimated. Alternative assumptions related to the relative effect of pralsetinib on OS were tested in scenario analyses and are addressed in detail in the discussion of the next limitation.
Overestimation of OS benefit associated with pralsetinib and inappropriate extrapolation beyond the trial period: The sponsor assumed that OS is independent of whether individuals are on treatment and whether individuals are in the progression-free or post-progression health states. As such, the model predicts a substantial survival benefit of 2 years for pralsetinib, including a significant portion (up to 49% of total life-years) incurred in the post-progression period. However, the clinical expert consulted by CADTH suggested that survival is linked to progression and therefore the transition probability to death should vary for patients within the post-progression or progression-free health states. The clinical expert consulted by CADTH noted there is no evidence, nor a plausible explanation via the mechanism of action of pralsetinib, to support this sustained post-progression survival benefit. Consequently, the sponsor’s partition survival model framework, which assumes survival is independent of progression, is inappropriate, and an alternative model framework that allowed for transitions dependent on being progression-free or progressed would have been more appropriate.
CADTH acknowledges clinician observations that there is the potential for an OS benefit with pralsetinib due to a potential impact on delaying treatment progression; however, the OS data from the ARROW trial was immature and noncomparative in nature, and there were significant limitations with the available ITC. The relative effect on OS is therefore highly uncertain. Furthermore, the clinical expert consulted by CADTH noted the sponsor’s long-term OS predictions with pralsetinib were overly optimistic and not expected to occur in clinical practice.
The gains in life expectancy for patients treated with pralsetinib estimated in the sponsor’s base case are due to the partition survival model framework and the sponsor’s selected parametric survival extrapolations, and they are not supported by available evidence. The true cost-effectiveness of pralsetinib in this setting is therefore unknown and the sponsor’s estimates are biased in favour of pralsetinib.
CADTH addressed this limitation by assuming equal OS across all comparators in exploratory reanalyses. This approach was the only available option, as the sponsor’s model was not flexible enough to assess alternative approaches to modelling post-progression survival, nor were more-robust estimates of the relative treatment effect available. An alternative set of calibrated hazard ratios was provided by the sponsor, which provided equal post-progression survival across all comparators; however, this does not address the assumption of independence between PFS and OS. More importantly, there remains an absence of comparative evidence to determine if an OS benefit exists. These alternative calibrated hazard ratios were considered in an exploratory scenario analysis. Limitations with the sponsor’s model structure could not be addressed.
Inappropriate extrapolation of PFS beyond the trial period: The sponsor’s model relies on extrapolation of observed Kaplan-Meier data for pralsetinib for PFS, OS, and TTD, and hazard ratios derived from their SLR applied to the pralsetinib survival extrapolation to determine the PFS, OS, and TTD of all comparators. However, the parametric extrapolations of PFS for pralsetinib in both the treatment-naive and treatment-experienced populations led to implausible PFS estimates for the target populations, particularly at the 5- and 10-year landmarks. For example, the estimate of 8% of treatment-naive patients being progression-free after 10 years, based on the generalized gamma distribution selected in the sponsor’s base case, does not meet face validity given the severe nature of the disease and the mean age of 60 years of patients in the ARROW trial. Based on clinical expert feedback, an exponential distribution was determined to be more plausible and led to 7% of patients remaining progression-free after 5 years and 0.4% of patients remaining progression-free after 10 years in the treatment-naive and treatment-experienced populations, respectively. These extrapolations were also more closely aligned with the sponsor’s estimated TTD curves, which typically are reliable indicators of PFS. The sponsor’s selected distribution resulted in an overestimated PFS benefit for both treatment-naive and treatment-experienced patients, a benefit that was not expected to occur in clinical practice.
CADTH selected the exponential distribution for PFS in both the treatment-naive and treatment-experienced patients in the exploratory reanalysis.
Incorrect dosing and exclusion of treatment-stopping rules for the calculation of comparator drug costs: The sponsor’s analysis included incorrect calculations of drug costs across several comparator treatments due to issues with the calculation of the required dose, as well as the exclusion of stopping rules. Pembrolizumab costs were calculated based on a flat dose and the drug was assumed to be used indefinitely; however, drug plan feedback indicated weight-based dosing up to 200 mg would be administered for up to a maximum of 2 years. The incorrect pembrolizumab costs affected the costs of triple therapy and pembrolizumab monotherapy. Nivolumab was incorrectly calculated to be administered as a double dose (480 mg) once per 28-day cycle, although the product monograph indicated 1 standard dose (up to 240 mg) per 14-day cycle. Dosing for nivolumab was also incorporated as a flat dose as opposed to weight-based dosing up to 240 mg administered for up to 2 years. For PBC, the sponsor assumed that treatment would continue indefinitely, although it is only used in clinical practice for a maximum of four to six 21-day cycles, followed by maintenance pemetrexed and pembrolizumab where relevant. Additionally, the dosing for PBC (cisplatin and carboplatin) were calculated inappropriately and did not consider body surface area or area under the curve to determine the required dose. The docetaxel costs used in the sponsor’s base case were per millilitre rather than per vial, underestimating this comparator cost. These issues are likely due to inefficient programming of the model that resulted in errors in calculations spread unnecessarily across multiple sheets. In consultation with drug plans and clinical experts, CADTH modified the expected dosing and stopping rules for all affected comparators, with all revisions available in Appendix 4 (Table 19). The sponsor’s implementation of comparator drug acquisition costs overestimated the costs associated with comparator regimens, except for docetaxel, biasing the results in favour of pralsetinib.
CADTH manually adjusted the expected dosing and stopping rules for all affected comparators (Table 19) to reflect each respective comparator’s product monograph, as well as clinical expert and drug plan input.
Subsequent therapy use post-progression: The sponsor’s estimated distribution of subsequent therapy use in both lines of therapy assessed in this submission did not align with the subsequent therapies expected to be used in clinical practice. The sponsor’s base case also included some subsequent therapies that were deemed irrelevant and excluded relevant therapies. Notably, the sponsor assumed that a substantial proportion of patients who were on a drug other than pralsetinib would receive pralsetinib in a subsequent line of therapy. However, pralsetinib is not a treatment option currently available in Canada and its inclusion as a subsequent therapy option is inappropriate under CADTH submission guidelines. Furthermore, the sponsor’s assumptions regarding the rest of the distribution of subsequent therapies by initial therapy lacked face validity according to the clinical expert consulted by CADTH. For example, the sponsor assumed that 80% of patients receiving docetaxel as second-line treatment would continue to receive docetaxel as third-line treatment. This is not plausible given that such patients would have previously discontinued docetaxel due to lack of treatment benefit or other reasons. Furthermore, the clinical expert noted that pembrolizumab plus PBC would be administered to approximately 65% of patients discontinuing pralsetinib in the first-line setting, but the sponsor did not include pembrolizumab plus PBC as a subsequent treatment option in the model. Instead, these patients were assumed to receive nivolumab or PBC plus pemetrexed. The distribution of subsequent therapies by line of therapy in the sponsor’s base case in comparison with those assumed in CADTH’s reanalyses is available in Table 6.
The sponsor also assumed that subsequent treatment only occurs for up to 2 model cycles, regardless of the therapy received, whereas the clinical expert consulted by CADTH for this review noted that patients on PBC or pembrolizumab are treated for several additional cycles. While CADTH acknowledges that the sponsor provided an option to alter the number of cycles of subsequent therapy, the ability to adjust for differing stopping rules or likely durations of therapy based on the subsequent therapy receive (i.e., platinum chemotherapy is administered for up to 4 or 6 cycles, whereas pembrolizumab can be administered for up to 2 years) was not incorporated in the submitted model. Overall, estimates of cost-effectiveness in the sponsor’s base case are biased in favour of pralsetinib due to inflated costs for subsequent-therapy comparators.
CADTH adjusted subsequent-therapy use to exclude pralsetinib, include pembrolizumab plus PBC, and reflect the expected distribution of subsequent therapy use in clinical practice. CADTH could not address limitations with the sponsor’s model assuming the same number of cycles regardless of the subsequent therapy received, which limits the interpretability of the cost-effectiveness estimates. CADTH assumed that subsequent therapy would be used for a mean of 5 cycles in the treatment-naive setting and 4 cycles in the treatment-experienced setting, based on clinical expert feedback.
Testing costs: Drug plan input noted that there is likely a need for increased testing to identify patients with RET fusion–positive NSCLC for treatment with pralsetinib. Additional testing is therefore likely required given that the product monograph for pralsetinib indicates that testing for RET fusion is required before starting pralsetinib. In a scenario analysis assessing the impact of testing costs, the sponsor’s model assumed that the costs associated with testing would be for 1 test ($400). However, given the positive-testing rate for RET fusion is only 1.5%, the testing cost should account for the total number of tests needed to identify a single patient who is RET fusion–positive ($400/0.015 = $26,667). As a result, the sponsor’s incorporation of genetic testing costs to identify patients who are RET fusion–positive was inappropriate and did not meet face validity, underestimating potential testing costs.
CADTH conducted analyses in which testing costs were and were not considered in the total costs associated with pralsetinib. In the scenarios in which testing costs were included, the testing costs in the sponsor’s model were adjusted to reflect the number of patients required to be tested to identify a single patient who is RET fusion–positive.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 5).
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Due to limitations with the available clinical evidence and the sponsor’s model structure, CADTH could not determine a base-case estimate of the cost-effectiveness of pralsetinib in treatment-naive or experienced adult patients with metastatic RET fusion–positive NSCLC not previously treated with an RET inhibitor.
CADTH undertook a series of scenario analyses to assess the impact of addressing other key limitations identified with the sponsor’s submission. The results of the CADTH exploratory analyses were derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These changes included assuming equal OS for each comparator within each subgroup, selection of an alternative PFS extrapolation distribution in both subgroups, revising drug costs to reflect true prices, aligning dosing and stopping rules with product monographs and clinical practice, and revising subsequent therapies and the assumed distribution of use to reflect clinical practice. These analyses are highly uncertain given the lack of comparative clinical effectiveness evidence to inform the PFS and OS benefits, if any, associated with pralsetinib.
Given the uncertainty over costs of testing for RET fusion, CADTH can only provide an exploratory analysis for both treatment-naive and treatment-experienced patients based on the inclusion and exclusion of testing costs.
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Adverse event rates for comparators are based on studies not specific to RET fusion–positive NSCLC. | Reasonable assumption not likely to affect results. |
Adverse events are resolved within the first model cycle through either treatment or discontinuation. | Uncertain, although not likely to affect results. |
Nivolumab alone was assumed to represent all possible immunotherapies (i.e., pembrolizumab monotherapy and atezolizumab) given to treatment-experienced patients, | Reasonable assumption not likely to largely affect results. The primary comparator in treatment-experienced patients is docetaxel; however, the consideration of all relevant treatment comparators in the base-case analysis is preferred. |
Time to discontinuation was modelled independently from PFS. | Uncertain. The sponsor’s model structure assumes that TTD and PFS are independent, based on the ARROW trial. However, TTD should be similar to PFS given that the discontinuation of pralsetinib according to the product monograph is dependent on disease progression or unacceptable toxicity. The likely treatment discontinuation in the real-world is uncertain; however, discontinuation is generally aligned with PFS in the CADTH exploratory analyses due to the revised PFS curves selected. |
Utilities were derived from patients with advanced NSCLC and assumed to be applicable to those with RET fusion–positive mutations. | Uncertain. The applicability of the sponsor’s utility values to the Canadian population of patients with RET fusion–positive NSCLC is unknown. |
NSCLC = non–small cell lung cancer; PFS = progression-free survival; TTD = time to discontinuation.
Table 6: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Overall survival benefit with pralsetinib | Assumed overall survival benefit for pralsetinib, including improved survival post-progression | Assumed equal overall survival for each comparator in comparison with pralsetinib (i.e., hazard ratio set to 1) |
2. Parametric survival extrapolation for pralsetinib PFS | Treatment-naive: generalized gamma Treatment-experienced: log-normal | Treatment-naive: exponential Treatment-experienced: exponential |
3. Dosing and stopping rules for comparator drugs | Incorrect dosing was applied for pembrolizumab, nivolumab, carboplatin, and cisplatin No stopping rules were applied to any comparator treatments Incorrect costing of docetaxel vials | Dosing based on weight, BSA, and/or GFR were applied where applicable to recalculate dosing of comparator products; dosing schedule of nivolumab was amended to reflect the product monograph to a 14-day dosing schedule as opposed to 28 days; all changes applied are documented in Appendix 4, Table 19 Stopping rules were applied to all comparator treatments as per their respective product monographs Corrected docetaxel vial costs |
4. Subsequent therapies 4.a. Distributions 4.b. Duration of subsequent treatment | Treatment-naive Pralsetinib: 40% nivolumab, 40% PBC plus pemetrexed Pembrolizumab: 30% pralsetinib, 50% PBC plus pemetrexed Triple therapy: 30% pralsetinib, 50% PBC plus pemetrexed PBC plus pemetrexed: 30% pralsetinib, 25% docetaxel, 25% nivolumab Treatment-experienced: 80% docetaxel for all comparators Subsequent treatment was assumed to occur for 2 cycles | Treatment-naive Pralsetinib: 15% PBC plus pemetrexed, 65% triple therapy Pembrolizumab: 80% PBC plus pemetrexed Triple therapy: 80% docetaxel PBC plus pemetrexed: 75% docetaxel, 5% nivolumab Note: Triple therapy added as option, pralsetinib removed as option Treatment-experienced: 80% docetaxel except patients failing second-line docetaxel who would discontinue treatment Subsequent treatment was assumed to occur for 5 cycles following first-line treatment and for 4 cycles following second-line treatment |
5. Genetic testing | Underestimated costs of testing for RET fusion status, accounting for a single test ($400) | Provided analysis including comprehensive genetic testing. Rather than just the price of a single test ($400), CADTH included the costs of testing to identify a single patients who has RET fusion based on a testing positive rate of 1.5% ($26,667) |
CADTH exploratory analyses | 1 + 2 + 3 + 4 ( + 5 to explore upper limit of potential testing costs) | |
BSA = body surface area; GFR = glomerular filtration rate; PBC = platinum-based chemotherapy; PFS = progression-free survival.
Table 7: Summary of Stepped Analysis of CADTH Reanalysis — Treatment-Naive Patients
Stepped analysis | ICER ($ per QALY): pralsetinib vs. comparator | Sequential analysis ($ per QALY) |
|---|---|---|
Sponsor’s base case (probabilistic) | vs. PBC plus pemetrexed: 165,789 vs. pembrolizumab: 94,163 vs. triple therapy: 3,911 | Pembrolizumab vs. PBC + pemetrexed: dominated Pralsetinib vs. PBC plus pemetrexed: 165,789 Triple therapy subject to extended dominance |
CADTH reanalysis 1 | vs. PBC plus pemetrexed: 886,551 vs. pembrolizumab: 611,218 vs. triple therapy: dominant | Pralsetinib vs. PBC plus pemetrexed: 886,551 Pembrolizumab and triple therapy dominated by PBC plus pemetrexed |
CADTH reanalysis 2 | vs. PBC plus pemetrexed: 162,020 vs. pembrolizumab: 97,247 vs. triple therapy: 10,298 | Pralsetinib vs. PBC plus pemetrexed: 162,020 Pembrolizumab dominated by PBC plus pemetrexed Triple therapy subject to extended dominance |
CADTH reanalysis 3 | vs. PBC plus pemetrexed: 150,844 vs. pembrolizumab: 115,735 vs. triple therapy: 83,773 | Pralsetinib vs. PBC plus pemetrexed: 150,844 Pembrolizumab dominated by PBC plus pemetrexed Triple therapy subject to extended dominance |
CADTH reanalysis 4 | vs. PBC plus pemetrexed: 155,376 vs. pembrolizumab: 93,215 vs. triple therapy: 15,123 | Pralsetinib vs. PBC plus pemetrexed: 155,376 Pembrolizumab dominated by PBC plus pemetrexed Triple therapy subject to extended dominance |
CADTH exploratory analysis (1 + 2 + 3 + 4, probabilistic) | vs. PBC plus pemetrexed: 1,626,594 vs. pembrolizumab: 1,481,688 vs. triple therapy: 3,063,599 | Triple therapy vs. PBC + pemetrexed: 1,261,851 Pralsetinib vs. triple therapy: 3,063,599 Pembrolizumab dominated by PBC plus pemetrexed |
CADTH exploratory analysis (1 + 2 + 3 + 4 + 5, probabilistic) | vs. PBC plus pemetrexed: 1,842,863 vs. pembrolizumab: 1,709,056 vs. triple therapy: 4,108,183 | Triple therapy vs. PBC + pemetrexed: 1,263,383 Pralsetinib vs. triple therapy: 4,108,183 Pembrolizumab dominated by PBC plus pemetrexed |
ICER = incremental cost-effectiveness ratio; PBC = platinum-based chemotherapy; QALY = quality-adjusted life-year; vs. = versus.
Note: All stepped analyses conducted deterministically.
Table 8: Summary of Stepped Analysis of CADTH Reanalysis — Treatment-Experienced Patients
Stepped analysis | ICER ($ per QALY): pralsetinib vs. comparator | Sequential analysis ($ per QALY) |
|---|---|---|
Sponsor’s base case (probabilistic) | vs. docetaxel: 187,319 vs. nivolumab: 151,546 vs. PBC plus pemetrexed: 160,274 | Pralsetinib vs. docetaxel: 187,319 PBC plus pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
CADTH reanalysis 1 | vs. docetaxel: 1,130,713 vs. nivolumab: 916,291 vs. PBC plus pemetrexed: 1,008,219 | Pralsetinib vs. docetaxel: 1,130,713 PBC plus pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
CADTH reanalysis 2 | vs. docetaxel: 195,963 vs. nivolumab: 160,278 vs. PBC plus pemetrexed: 167,735 | Pralsetinib vs. docetaxel: 195,963 PBC plus pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
CADTH reanalysis 3 | vs. docetaxel: 178,651 vs. nivolumab: 162,207 vs. PBC plus pemetrexed: 157,528 | Pralsetinib vs. docetaxel: 178,651 PBC plus pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
CADTH reanalysis 4 | vs. docetaxel: 185,982 vs. nivolumab: 150,687 vs. PBC plus pemetrexed: 159,135 | Pralsetinib vs. docetaxel: 185,982 PBC plus pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
CADTH exploratory analysis (1 + 2 + 3 + 4, probabilistic) | vs. docetaxel: 1,567,170 vs. nivolumab: 1,487,336 vs. PBC plus pemetrexed: 1,413,900 | Pralsetinib vs. docetaxel: 1,567,170 PBC plus pemetrexed and nivolumab subject to extended dominance |
CADTH exploratory analysis (1 + 2 + 3 + 4 + 5, probabilistic) | vs. docetaxel: 1,726,230 vs. nivolumab: 1,679,844 vs. PBC plus pemetrexed: 1,571,655 | Pralsetinib vs. docetaxel: 1,726,230 PBC plus pemetrexed and nivolumab subject to extended dominance |
ICER = incremental cost-effectiveness ratio; PBC = platinum-based chemotherapy; QALY = quality-adjusted life-year; vs. = versus.
Note: All stepped analyses are conducted deterministically.
Based on the sponsor’s analysis, the CADTH exploratory analyses found that pralsetinib is not cost-effective in either treatment-naive or treatment-experienced patients at a willingness-to-pay threshold of $50,000 per QALY.
For treatment-naive patients, the incremental cost per QALY gained (ICER) for pralsetinib was $1,626,594 versus PBC plus pemetrexed, $1,481,688 versus pembrolizumab, and $3,063,599 versus triple therapy if the costs of testing were excluded. Including the costs of testing increased the ICERs to $1,842,863 versus PBC plus pemetrexed, $1,709,056 versus pembrolizumab, and $4,108,183 versus triple therapy. In a sequential analysis, the ICER for triple therapy versus PBC plus pemetrexed was $1,261,851 without testing and the ICER for pralsetinib versus triple therapy was $3,063,599. Including the costs of testing increased the ICERs to $1,842,863 and $4,108,183, respectively. Pembrolizumab was dominated by PBC plus pemetrexed regardless of the inclusion of testing costs. Incremental costs for pralsetinib were primarily due to higher drug costs and incremental QALYs were due to assumptions of greater time in the progression-free state (Appendix 4).
For treatment-experienced patients, the incremental cost per QALY gained (ICER) for pralsetinib was $1,567,170 versus docetaxel, $1,487,336 versus nivolumab, and $1,413,900 versus PBC plus pemetrexed if the costs of testing were excluded. Including the costs of testing increased the ICERs to $1,726,230 versus docetaxel, $1,679,844 versus nivolumab, and $1,571,655 versus PBC plus pemetrexed. In a sequential analysis, the ICER for pralsetinib versus docetaxel was $1,567,170 without testing and $1,726,230 with testing. PBC plus pemetrexed and nivolumab were subject to extended dominance regardless of the inclusion of testing. Incremental costs for pralsetinib were primarily due to higher drug costs, and incremental QALYs were due to assumptions of greater time in the progression-free state (Appendix 4).
These analyses are based on publicly available prices of the comparator treatments. Furthermore, these analyses are based on potentially optimistic assumptions relating to the relative effectiveness of pralsetinib given the lack of available comparative effectiveness evidence. The estimated ICERs, although exploratory in nature, may therefore be optimistic and favour pralsetinib.
Scenario Analysis Results
Based on the CADTH exploratory analysis, a price reduction greater than 80% is required for the ICER to be reduced to $50,000 per QALY in the treatment-naive setting when excluding testing costs (Table 9). A price reduction greater than 90% is required when additional testing costs are considered in this setting. Similarly, a price reduction for pralsetinib of at least 96% is required for the ICER to be reduced to $50,000 per QALY in the treatment-experienced setting excluding testing costs. A price reduction greater than 99% is required when additional testing costs are considered in this setting.
When the sponsor’s estimates of OS benefit with pralsetinib are included within the CADTH exploratory reanalysis, the estimated price reduction required is greater than 60% in the treatment-naive setting, and greater than 75% in the treatment-exposed setting, not including testing costs. When post-progression survival was considered equal across all comparators, the price reductions required were 70% and 85% in the treatment-naive and experienced settings, respectively, not included testing costs.
These analyses are based on publicly available prices of the comparator treatments. Furthermore, they are based on potentially optimistic assumptions relating to the relative effectiveness of pralsetinib given the lack of comparative clinical effectiveness data. The estimated required price reductions may therefore be optimistic and favour pralsetinib.
Table 9: CADTH Price-Reduction Analyses for Treatment-Naive Patients
Price reduction | Sequential ICERs for pralsetinib vs. relevant comparators on cost-effectiveness frontier | ||
|---|---|---|---|
Sponsor base case | CADTH exploratory analysis | ||
Analysis excluding testing | Analysis including testing | ||
No price reduction | $165,789 vs. PBC plus pemetrexed | $3,063,599 vs. triple therapy | $4,108,183 vs. triple therapy |
10% | $146,100 vs. PBC plus pemetrexed | $2,086,239 vs. triple therapy | $3,135,399 vs. triple therapy |
20% | $126,411 vs. PBC plus pemetrexed | $1,108,878 vs. triple therapy | $2,162,615 vs. triple therapy |
30% | $106,721 vs. PBC plus pemetrexed | $1,033,029 vs. PBC plus pemetrexed | $1,189,832 vs. triple therapy |
40% | $87,032 vs. PBC plus pemetrexed | $835,174 vs. PBC plus pemetrexed | $1,050,246 vs. PBC plus pemetrexed |
50% | $67,343 vs. PBC plus pemetrexed | $637,319 vs. PBC plus pemetrexed | $852,092 vs. PBC plus pemetrexed |
60% | $47,653 vs. PBC plus pemetrexed | $439,464 vs. PBC plus pemetrexed | $653,938 vs. PBC plus pemetrexed |
70% | $27,964 vs. PBC plus pemetrexed | $241,609 vs. PBC plus pemetrexed | $455,783 vs. PBC plus pemetrexed |
80% | $8,272 vs. PBC plus pemetrexed | $43,754 vs. PBC plus pemetrexed | $257,629 vs. PBC plus pemetrexed |
90% | Dominant | Dominant | $59,475 vs. PBC plus pemetrexed |
ICER = incremental cost-effectiveness ratio; PBC = platinum-based chemotherapy; vs. = versus.
Table 10: CADTH Price-Reduction Analyses for Treatment-Experienced Patients
Price reduction | Sequential ICERs for pralsetinib vs. relevant comparators on cost-effectiveness frontier | ||
|---|---|---|---|
Sponsor base case | CADTH exploratory analysis | ||
Analysis excluding testing | Analysis including testing | ||
No price reduction | $187,319 vs. docetaxel | $1,567,170 vs. docetaxel | $1,726,230 vs. docetaxel |
10% | $170,172 vs. docetaxel | $1,409,420 vs. docetaxel | $1,568,016 vs. docetaxel |
20% | $153,024 vs. docetaxel | $1,251,669 vs. docetaxel | $1,409,801 vs. docetaxel |
30% | $135,877 vs. docetaxel | $1,093,919 vs. docetaxel | $1,251,586 vs. docetaxel |
40% | $118,729 vs. docetaxel | $936,168 vs. docetaxel | $1,093,371 vs. docetaxel |
50% | $101,582 vs. docetaxel | $778,418 vs. docetaxel | $935,157 vs. docetaxel |
60% | $84,435 vs. docetaxel | $620,668 vs. docetaxel | $776,942 vs. docetaxel |
70% | $67,287 vs. docetaxel | $462,917 vs. docetaxel | $618,727 vs. docetaxel |
80% | $50,140 vs. docetaxel | $305,167 vs. docetaxel | $460,513 vs. docetaxel |
90% | $32,992 vs. docetaxel | $147,417 vs. docetaxel | $302,298 vs. docetaxel |
ICER = incremental cost-effectiveness ratio; vs. = versus.
Additionally, CADTH conducted a series of scenario analyses to explore the impact of alternative assumptions on the cost-effectiveness of pralsetinib:
Wastage was considered in the calculation of drug costs.
TTD was assumed to be equal to PFS.
The sponsor’s original OS assumptions were applied.
OS benefit halved relative to sponsor’s base case.
Treatment benefit related to PFS was removed.
Time horizon was shortened to 10 years.
Post-progression survival was set to be equal across all comparators using calibrated hazard ratios provided by the sponsor.
The results of these analyses are presented in Appendix 4 (Table 17 and Table 18). The scenario analysis involving the removal of treatment benefit due to delayed progression had the largest effect on the cost-effectiveness estimates, in which pralsetinib was dominated by nivolumab in the treatment-experienced setting. Given the uncertainty in the available clinical evidence, the relative effectiveness of pralsetinib in delaying progression remains unknown and these results were substantially different from the sponsor’s estimates and those of the CADTH exploratory analysis.
Issues for Consideration
Additional costs of testing for RET fusion status would strongly affect the estimated costs associated with pralsetinib. It is currently unknown whether the rate of testing would increase if pralsetinib were to be funded.
As noted by drug plans, selpercatinib recently received a positive listing recommendation from pERC and would be a key treatment comparator to pralsetinib once listed. However, the comparative efficacy, and therefore the cost-effectiveness, of pralsetinib versus selpercatinib is unknown. CADTH notes that differences in the submitted models (i.e., selpercatinib model could run as a functional Markov model; pralsetinib model lacked flexibility to do so) and inputs led to differences in the approaches that could be taken to address limitations identified in each review, as well as the results observed.
The clinical expert consulted by CADTH noted that nivolumab in combination with ipilimumab plus PBC plus pemetrexed may be a relevant comparator and was not assessed in the sponsor’s submission. The CADTH reanalysis could not address this exclusion in reanalysis and the relative cost-effectiveness of pralsetinib versus this comparator is also unknown.
Overall Conclusions
The CADTH clinical review concluded that data from the pivotal trial were inadequate to interpret OS and PFS findings due to the single-arm trial design and immaturity of the data. Evidence generated from the ITC was insufficient to make comparisons across therapies due to a significant amount of uncertainty arising from patient heterogeneity between trials that was not accounted for.
Given that OS and PFS are key components within a partition survival model, the lack of interpretability of this information for pralsetinib directly affects any conclusions that may be drawn from the economic evaluation. Further, the absence of robust comparative evidence limits any conclusions that can be drawn regarding the cost-effectiveness of pralsetinib. CADTH conducted exploratory reanalyses to determine the impact of addressing identified key limitations. CADTH revised the sponsor’s model to assume equal OS for each comparator within each subgroup, selected alternative PFS extrapolation distributions, revised drug costs to reflect dosing and stopping rules in alignment with product monographs and expected use in practice, and revised subsequent therapy use to reflect clinical practice. Not all of the concerns with the sponsor’s submission could be addressed, and these outstanding limitations (e.g., PFS benefits), may bias results in favour of pralsetinib.
Based on CADTH’s exploratory reanalysis, the ICER for pralsetinib was in excess of $1 million per QALY in treatment-naive and experienced populations and regardless of the inclusion of testing. Under the assumption of equal OS, results are driven largely by the drug acquisition costs for pralsetinib. In the treatment-naive setting, pralsetinib was associated with an ICER of $3,063,599 per QALY ($4,108,183 per QALY including testing) versus triple therapy. A price reduction of at least 80% (or 90% with inclusion of full testing costs) would be required to achieve cost-effectiveness at a threshold of $50,000 per QALY based on the sequential analysis. In the treatment-experienced setting, pralsetinib was associated with an ICER of $1,567,170 per QALY ($1,726,230 per QALY including testing) versus docetaxel. A price reduction of at least 96% (or 99% with inclusion of full testing costs) is required for pralsetinib to be considered cost-effective in treatment-experienced patients at a threshold of $50,000 per QALY in this setting. Scenario analyses considering the sponsor’s optimistic OS benefits with pralsetinib suggested price reductions in excess of 60% and 75% in the treatment-naive and treatment-experienced settings, respectively, were necessary for pralsetinib to be considered cost-effective at a threshold of $50,000 per QALY when excluding testing costs. Scenario analyses considering equal post-progression survival across all comparators suggested price reductions in excess of 70% and 85% in the treatment-naive and treatment-experienced settings, respectively, were necessary for pralsetinib to be considered cost-effective at a threshold of $50,000 per QALY when excluding testing costs.
Given the absence of comparative clinical evidence for pralsetinib and the submitted model structure, the exploratory results and subsequent price reductions may remain biased in favour of pralsetinib. As such, and given the submitted clinical information, the true cost-effectiveness of pralsetinib in comparison to relevant treatment comparators in both the treatment-naive and treatment-experienced settings is highly uncertain. CADTH was also unable to assess the cost-effectiveness of pralsetinib in comparison with selpercatinib, the other metastatic RET fusion–positive NSCLC therapy, which recently received a positive listing recommendation from pERC, at the time of this review.
References
1.Gavreto (pralsetinib capsules): capsules, 100 mg, oral. Protein kinase inhibitor (L01EX) [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Jul 12.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Gavreto (pralsetinib), 100 mg capsules, oral [internal sponsor's package]. Mississauga (ON): Hoffmann-La Roche Ltd.; 2022 Mar 9.
3.Clinical Study Report: BLU-667-1101. A phase 1/2 study of the highly-selective RET inhibitor, BLU-667, in patients with thyroid cancer, non-small cell lung cancer (NSCLC) and other advanced solid tumors [internal sponsor's report]. Cambridge (MA): Blueprint Medicines; 2020 Feb 26.
4.Committee for Medicinal Products for Human Use. Assessment report: Gavreto (pralsetinib). (European public assessment report). London (GB): European Medicines Agency; 2021 Sep 16: https://www.ema.europa.eu/en/documents/assessment-report/gavreto-epar-public-assessment-report_en.pdf. Accessed 2022 May 3.
5.Wild type (WT) non-small cell lung cancer (NSCLC): updated clinical systematic literature review (SLR) and comparative analyses of pralsetinib with comparators of interest [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Pralsetinib, 400 mg QD oral. Oxfordshire (United Kingdom): F. Hoffmann – La Roche Ltd; 2021 Oct 4. 2021.
6.Chouaid C, Agulnik J, Goker E, et al. Health-related quality of life and utility in patients with advanced non-small-cell lung cancer: a prospective cross-sectional patient survey in a real-world setting. J Thorac Oncol. 2013;8(8):997-1003. PubMed
7.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-518. PubMed
8.DeltaPA. [Ottawa (ON)]: IQVIA; 2021: https://www.iqvia.com/. Accessed 2022 May 3.
9.Ng R, Hasan B, Mittmann N, et al. Economic analysis of NCIC CTG JBR.10: a randomized trial of adjuvant vinorelbine plus cisplatin compared with observation in early stage non-small-cell lung cancer--a report of the Working Group on Economic Analysis, and the Lung Disease Site Group, National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(16):2256-2261. PubMed
10.Johnston KM, Sheffield BS, Yip S, Lakzadeh P, Qian C, Nam J. Comprehensive genomic profiling for non-small-cell lung cancer: health and budget impact. Curr Oncol. 2020;27(6):e569-e577. PubMed
11.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2021: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 May 3.
12.National Institute for Health and Care Excellence. Single technology appraisal. Osimertinib for treating metastatic EGFR and T790M mutation-positive non-small-cell lung cancer [ID874]. (Committee Papers) 2016; https://www.nice.org.uk/guidance/ta653/documents/committee-papers-2. Accessed 2022 May 3.
13.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 May 3.
14.de Oliveira C, Pataky R, Bremner KE, et al. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):809. PubMed
15.Baracos V, Reiman T, Mourtzakis M, Gioulbasanis I, Antoun S. Body composition in patients with non-small cell lung cancer: a contemporary view of cancer cachexia with the use of computed tomography image analysis. Am J Clin Nutr. 2010;91:1133S-1137S. PubMed
16.Carboplatin injection bp (carboplatin injection), sterile solution, 10 mg/mL (50 mg, 150 mg, 450 mg, 600 mg of carboplatin per vial); Antineoplastic agent[product monograph]. Kirkland (QC): Pfizer Canada ULC; 2019 Dec 16.
17.Opdivo (nivolumab for injection), intravenous infusion, 10 mg nivolumab/mL 40 mg and 100 mg single-use vials; Antineoplastic [product monograph]. Montreal (QC): Bristol-Myers Squibb Canada Co.; 2019 Mar 13.
18.Keytruda (pembrolizumab) solution for infusion 100 mg/4 mL vial; Antineoplastic agent, monoclonal antibody [product monograph]. Kirkland (QC): Merck Canada Inc.; 2022 Jul 5.
19.Pemetrexed, pemetrexed disodium for injection, 100 mg or 500 mg pemetrexed per vial, sterile lyophilized powder; Antineoplastic agent [product monograph]. Toronto (ON): Apotex Inc.; 2016 Jun 1.
20.Tecentriq (atezolizumab) concentrate for solution for infusion, 60 mg/mL, 1200 mg/20 mL single use vials; Antineoplastic agent[product monograph]. Mississauga (ON): Hoffman-La Roche Limited; 2019 Aug 8.
21.GlobalRPh. Carboplatin AUC calculator. 2021; https://globalrph.com/medcalcs/carboplatin-auc-calculator/. Accessed 2021 Dec 13.
22.Budget Impact Analysis [internal sponsor's report]. In: Gavreto (pralsetinib), 100 mg capsules, oral [internal sponsor's package]. Mississauga (ON): Hoffmann-La Roche Ltd.; 2022 Mar 9.
23.Offin M, Guo R, Wu SL, et al. Immunophenotype and response to immunotherapy of RET-rearranged lung cancers. JCO Precis Oncol. 2019;3. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 11: CADTH Cost Comparison Table for RET Fusion–Positive Non–Small Cell Lung Cancer
Treatment | Strength / concentration | Form (Vial size if single-use) | Price | Recommended dosagea | Average daily cost | 28-day cost |
|---|---|---|---|---|---|---|
Pralsetinib (Gavreto) | 100 mg | Capsule | $102.0600 | 400 mg once daily | $408.24 | $11,431 |
First-line therapies | ||||||
Monotherapies | ||||||
Pembrolizumab | 25 mg/mL | 4 mL solution for IV injection | $4,400.0000 | 2 mg/kg to 200 mg per 3 weeks | $419.05 | $11,733 |
Combination regimens | ||||||
Carboplatin | 10 mg/mL | 5 mL 15 mL 45 mL 60 mL | $70.0000 $210.0000 $600.0000 $775.0000 | AUC 5 mg/mL per 3 weeksb | $31.90 | $893 |
Cisplatin | 1 mg/mL | 50 mL 100 mL Solution for IV injection | $323.0000 $646.0000 | 75 mg/m2 per 3 weeks | $46.14 | $1,292 |
Pemetrexed | 25 mg/mL | 100 mg 500 mg Powder for IV injection | $429.0000 $2,145.0000 | 500 mg/m2 per 3 weeks | $204.29 | $5,720 |
Carboplatin + pemetrexed + pembrolizumab | $655.24 | $18,347 | ||||
Cisplatin + pemetrexed + pembrolizumab | $669.48 | $18,745 | ||||
Carboplatin + pemetrexed | $236.19 | $6,613 | ||||
Cisplatin + pemetrexed | $250.43 | $7,012 | ||||
Second-line therapies | ||||||
Monotherapies | ||||||
Atezolizumab | 60 mg/mL | 20 mL solution for IV infusion | $6,776.0000 | 1,200 per 3 weeks | $322.67 | $9,035 |
Docetaxel | 10 mg/mL | 80 mL 160 mL Solution for IV injection | $1,850.0000 $925.0000 | 75 to 100 mg/m2 per 3 weeks | $75.98 to $101.31 | $2,128 to $2,837 |
Nivolumab | 10 mg/mL | 4 mL 10 mL Solution for IV infusion | $782.2200 $1,955.5600 | 3 mg/kg to 240 mg per 2 weeks | $335.24 | $9,387 |
Pembrolizumab | 25 mg/mL | 4 mL solution for IV injection | $4,400.0000 | 2 mg/kg to 200 mg per 3 weeks | $419.05 | $11,733 |
Combination regimens | ||||||
Carboplatin | 10 mg/mL | 5 mL 15 mL 45 mL 60 mL | $70.0000 $210.0000 $600.0000 $775.0000 | AUC 5 mg/mL per 3 weeksb | $31.90 | $893 |
Cisplatin | 1 mg/mL | 50 mL 100 mL Solution for IV injection | $323.0000 $646.0000 | 75 mg/m2 per 3 weeks | $46.14 | $1,292 |
Pemetrexed | 25 mg/mL | 100 mg 500 mg Powder for IV injection | $429.0000 $2,145.0000 | 500 mg/m2 per 3 weeks | $204.29 | $5,720 |
Carboplatin + pemetrexed | $236.19 | $6,613 | ||||
Cisplatin + pemetrexed | $250.43 | $7,012 | ||||
GFR = glomerular filtration rate; IV = intravenous.
Note: All prices are from the IQVIA Delta PA database (accessed April 21st, 2022), unless otherwise indicated, and do not include dispensing fees. Costs are based on patient characteristics reported in the literature including a weight of 71 kg, body surface area of 1.84 m2, and glomerular filtration rate of 73 mL/minute.15 Vial sharing was not considered, and wastage was assumed to occur where applicable.
aRecommended dosages are per the respective product monographs.16-20
bDose is calculated as = target AUC × (GFR + 25).21
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | The model programming is not fully transparent which made it difficult to fully appraise and address identified limitations. CADTH identified several programming errors. For example, the sheet for calculating the pairwise ICERs were incorrectly programmed in the treatment-experienced setting. |
Model structure is adequate for decision problem | No | The states are appropriate but assumptions relating to the independence of the overall survival and progression-free survival lacks validity. A Markov model structure would have more accurately reflected the disease pathway by explicitly modelling the relationship between PFS and OS. Please refer to the key limitations section. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | There are numerous errors in the incorporation of cost data into the model. Upon the manual modification of several cells specifically regarding treatment costs and dosing, the cells reset to the sponsor’s original value after running the model probabilistically. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The submission was not adequately organized, and reporting was not clear. For example, the calculation of drug costs was done inappropriately and not described in the accompanying pharmacoeconomic report. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 13: Sponsor’s Disaggregated Results — Treatment-Naive Patients
Parameter | Pralsetinib | Pembrolizumab | Triple therapy | PBC + pemetrexed |
|---|---|---|---|---|
LYs | ||||
Progression-Free | 2.63 | 0.98 | 1.60 | 0.93 |
Post-Progression | 2.02 | 0.94 | 1.43 | 1.66 |
Total (undiscounted) | 4.65 | 1.92 | 3.03 | 2.59 |
QALYs | ||||
Progression-Free | 1.72 | 0.67 | 1.12 | 0.64 |
Post-Progression | 1.06 | 0.52 | 0.77 | 0.89 |
Total | 2.78 | 1.19 | 1.88 | 1.53 |
Discounted costs of study treatment ($) | ||||
Acquisition | 245,480.32 | 115,887.64 | 248,264.64 | 52,770.25 |
Administration | 0.00 | 1,880.93 | 2,746.98 | 1,816.08 |
Adverse events | 7,433.08 | 635.10 | 7,125.42 | 632.67 |
Total | 252,913.40 | 118,403.67 | 258,137 | 55,219 |
General disease management costs ($) | ||||
Progression-Free | 13,462.67 | 5,155.91 | 8,334.06 | 4,925.31 |
Post-Progression | 16,658.14 | 8,068.54 | 11,973.38 | 13,957.25 |
Total | 30,121 | 13,224 | 20,307 | 18,883 |
Other costs ($) | ||||
Subsequent treatment | 3,945.75 | 4,247.92 | 4,202.23 | 5,282.02 |
End of Life | 33,787.57 | 35,198.22 | 34,625.84 | 34,852.70 |
Total | 37,733 | 39,446 | 38,828 | 40,135 |
Total costs ($) | ||||
Total | 320,768 | 171,074 | 317,272 | 114,236 |
LYs = life-years; PBC = platinum-based chemotherapy; QALY = quality-adjusted life-year.
Note: Discounted disaggregated results were not made available from the sponsor for state-specific LYs or QALYs.
Source: Sponsor’s pharmacoeconomic submission.2
Table 14: Sponsor’s Disaggregated Results — Treatment-Experienced Patients
Parameter | Pralsetinib | Docetaxel | Nivolumab | PBC + pemetrexed |
|---|---|---|---|---|
LYs | ||||
Progression-Free | 2.73 | 0.51 | 0.76 | 0.51 |
Post-Progression | 1.10 | 0.74 | 0.76 | 0.59 |
Total | 3.83 | 1.25 | 1.52 | 1.10 |
QALYs | ||||
Progression-Free | 1.79 | 0.36 | 0.53 | 0.36 |
Post-Progression | 0.60 | 0.43 | 0.43 | 0.34 |
Total | 2.38 | 0.78 | 0.96 | 0.70 |
Discounted costs of study treatment ($) | ||||
Acquisition | 275,061 | 1,677 | 85,657 | 32,464 |
Administration | 0.00 | 1,200 | 1,303 | 1,205 |
Adverse events | 18,558 | 3,531 | 2,348 | 4,098 |
Total | 293,619 | 6,408 | 89,309 | 37,767 |
General disease management costs ($) | ||||
Progression-Free | 14,011 | 2,787 | 4,105 | 2,800 |
Post-Progression | 9,283 | 6,567 | 6,659 | 5,257 |
Total | 23,294 | 9,354 | 10,764 | 8,057 |
Other costs ($) | ||||
Subsequent treatment | 174 | 283 | 231 | 253 |
End of Life | 31,666 | 32,959 | 32,832 | 33,032 |
Total | 31,840 | 33,242 | 33,063 | 33,286 |
Total costs ($) | ||||
Total | 348,754 | 49,005 | 133,135 | 79,109 |
LYs = life-years; PBC = platinum-based chemotherapy; QALY = quality-adjusted life-year.
Note: Discounted disaggregated results were not made available from the sponsor for state-specific LYs.
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Exploratory Analysis
Table 15: CADTH Exploratory Analysis Disaggregated Results — Treatment-Naive Patients
Parameter | Pralsetinib | Pembrolizumab | Triple therapy | PBC + pemetrexed |
|---|---|---|---|---|
LYs | ||||
Progression-Free | 1.96 | 0.89 | 1.31 | 0.86 |
Post-Progression | 2.70 | 3.77 | 3.35 | 3.81 |
Total | 4.66 | 4.66 | 4.66 | 4.66 |
QALYs | ||||
Progression-Free | 1.32 | 0.61 | 0.94 | 0.59 |
Post-Progression | 1.39 | 1.98 | 1.75 | 2.00 |
Total | 2.71 | 2.60 | 2.69 | 2.59 |
Discounted costs of study treatment ($) | ||||
Acquisition | 245,909 | 71,452 | 169,841 | 50,272 |
Administration | 0 | 1,772 | 2,760 | 1,811 |
Adverse events | 7,433 | 635 | 7,125 | 633 |
Total | 253,342 | 73,860 | 179,726 | 52,715 |
General disease management costs ($) | ||||
Progression-Free | 10,267 | 4,725 | 6,938 | 4,546 |
Post-Progression | 22,036 | 31,180 | 27,529 | 31,476 |
Total | 32,304 | 35,905 | 34,467 | 36,021 |
Other costs ($) | ||||
Subsequent treatment | 7,126 | 6,365 | 1,791 | 2,640 |
End of Life | 33,783 | 33,783 | 33,783 | 33,783 |
Total | 40,909 | 40,148 | 35,573 | 36,423 |
Testing costs ($) | ||||
Total | 26,667 | 0 | 0 | 0 |
Total costs ($) | ||||
Total (without testing costs) | 326,555 | 149,912 | 249,766 | 125,159 |
Total (with testing costs) | 353,222 | 149,912 | 249,766 | 125,159 |
LYs = life-years; PBC = platinum-based chemotherapy; QALY = quality-adjusted life-year.
Note: Discounted disaggregated results were not made available from the sponsor for state-specific LYs.
Table 16: CADTH Exploratory Analysis Disaggregated Results — Treatment-Experienced Patients
Parameter | Pralsetinib | Docetaxel | Nivolumab | PBC + pemetrexed |
|---|---|---|---|---|
LYs | ||||
Progression-Free | 2.00 | 0.51 | 0.74 | 0.51 |
Post-Progression | 1.83 | 3.33 | 3.10 | 3.33 |
Total | 3.83 | 3.83 | 3.83 | 3.83 |
QALYs | ||||
Progression-Free | 1.36 | 0.35 | 0.52 | 0.36 |
Post-Progression | 0.96 | 1.79 | 1.66 | 1.79 |
Total | 2.32 | 2.14 | 2.17 | 2.15 |
Discounted costs of study treatment ($) | ||||
Acquisition | 275,044 | 12,675 | 67,055 | 34,881 |
Administration | 0 | 1,193 | 2,493 | 1,204 |
Adverse events | 18,558 | 3,531 | 2,348 | 4,098 |
Total | 293,602 | 17,399 | 71,897 | 40,183 |
General disease management costs ($) | ||||
Progression-Free | 10,567 | 2,751 | 3,989 | 2,759 |
Post-Progression | 15,104 | 27,998 | 25,956 | 27,985 |
Total | 25,671 | 30,749 | 29,945 | 30,744 |
Other costs ($) | ||||
Subsequent treatment | 1,846 | 0 | 4,360 | 5,135 |
End of Life | 31,657 | 31,657 | 31,657 | 31,657 |
Total | 33,504 | 31,657 | 36,017 | 36,792 |
Testing costs ($) | ||||
Total | 26,667 | 0 | 0 | 0 |
Total costs ($) | ||||
Total (without testing costs) | 352,776 | 79,805 | 137,859 | 107,719 |
Total (with testing costs) | 379,443 | 79,805 | 137,859 | 107,719 |
LYs = life-years; PBC = platinum-based chemotherapy; QALY = quality-adjusted life-year.
Note: Discounted disaggregated results were not made available from the sponsor for state-specific LYs.
Scenario Analyses
Table 17: Summary of Scenario Analyses — Treatment-Naive Patients
Scenario | ICER ($ per QALY): pralsetinib versus comparator | Sequential analysis ($ per QALY) |
|---|---|---|
CADTH exploratory analysis | Versus PBC + pemetrexed: 1,626,594 Versus pembrolizumab: 1,481,688 Versus triple therapy: 3,063,599 | Triple therapy versus PBC + pemetrexed: 1,261,851 Pralsetinib versus triple therapy: 3,063,599 Pembrolizumab dominated by PBC + pemetrexed |
1. Wastage | Versus PBC + pemetrexed: 1,581,818 Versus pembrolizumab: 1,272,175 Versus triple therapy: 772,718 | Pralsetinib versus PBC + pemetrexed: 1,581,818 Pembrolizumab dominated by PBC + pemetrexed Triple therapy subject to extended dominance |
2. TTD set equal to PFS | Versus PBC + pemetrexed: 1,854,044 Versus pembrolizumab: 1,706,830 Versus triple therapy: 3,495,983 | Triple therapy versus PBC + pemetrexed: 1,402,898 Pralsetinib versus triple therapy: 3,495,983 Pembrolizumab dominated by PBC + pemetrexed |
3. Sponsor’s OS assumptions | Versus PBC + pemetrexed: 168,253 Versus pembrolizumab: 126,487 Versus triple therapy: 97,563 | Pralsetinib versus PBC + pemetrexed: 168,253 Pembrolizumab dominated by PBC + pemetrexed Triple therapy subject to extended dominance |
4. OS benefit from sponsor’s base case halved | Versus PBC + pemetrexed: 240,821 Versus pembrolizumab: 167,874 Versus triple therapy: 148,995 | Pralsetinib versus PBC + pemetrexed: 240,821 Pembrolizumab dominated by PBC + pemetrexed Triple therapy subject to extended dominance |
5. Removal of PFS benefit | Versus PBC + pemetrexed: dominated Versus pembrolizumab: dominated Versus triple therapy: dominated | Triple therapy versus PBC + pemetrexed: 2,737.861 Pralsetinib dominated by triple therapy Pembrolizumab dominated by PBC + pemetrexed |
6. Time horizon of 10 years | Versus PBC + pemetrexed: 1,618,831 Versus pembrolizumab: 1,471,849 Versus triple therapy: 2,913,758 | Triple therapy versus PBC + pemetrexed: 1,273,221 Pralsetinib versus triple therapy: 2,913,758 Pembrolizumab dominated by PBC + pemetrexed |
7. Equal post-progression survival | Versus PBC + pemetrexed: 282,322 Versus pembrolizumab: 255,792 Versus triple therapy: 166,520 | Pralsetinib versus PBC + pemetrexed: 282,322 Triple therapy subject to extended dominance Pembrolizumab dominated by PBC + pemetrexed |
ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; TTD = time to discontinuation.
Note: All scenario analyses were conducted deterministically; the sponsor’s deterministic and probabilistic results were aligned. Testing was not considered in the CADTH scenario analyses.
Table 18: Summary of Scenario Analyses — Treatment-Experienced Patients
Scenario | ICER ($ per QALY): pralsetinib versus Comparator | Sequential analysis ($ per QALY) |
|---|---|---|
CADTH exploratory analysis (probabilistic) | Versus docetaxel: 1,567,170 Versus nivolumab: 1,487,336 Versus PBC + pemetrexed : 1,413,900 | Pralsetinib versus docetaxel: 1,567,170 PBC + pemetrexed and nivolumab subject to extended dominance |
1. Wastage | Versus docetaxel: 1,551,892 Versus nivolumab: 1,471,122 Versus PBC + pemetrexed : 1,386,499 | Pralsetinib versus docetaxel: 1,551,892 PBC + pemetrexed and nivolumab subject to extended dominance |
2. TTD set equal to PFS | Versus docetaxel: 1,727,576 Versus nivolumab: 1,581,982 Versus PBC + pemetrexed : 1,515,700 | Pralsetinib versus docetaxel: 1,727,576 PBC + pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
3. Sponsor’s OS assumptions | Versus docetaxel: 189,896 Versus nivolumab: 172,047 Versus PBC + pemetrexed : 165,663 | Pralsetinib versus docetaxel: 189,896 PBC + pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
4. OS benefit from sponsor’s base case halved | Versus docetaxel: 239,113 Versus nivolumab: 225,327 Versus PBC + pemetrexed : 202,414 | Pralsetinib versus docetaxel: 239,113 PBC + pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
5. Removal of PFS benefit | Versus docetaxel: 144,479,829 Versus nivolumab: dominated Versus PBC + pemetrexed : 204,115,344 | Nivolumab versus docetaxel: 25,249,528 Pralsetinib dominated by nivolumab PBC + pemetrexed dominated by docetaxel |
6. Time horizon of 10 years | Versus docetaxel: 1,569,890 Versus nivolumab: 1,487,475 Versus PBC + pemetrexed : 1,415,493 | Pralsetinib versus docetaxel: 1,569,890 PBC + pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
7. Equal post-progression survival | Versus docetaxel: 299,721 Versus nivolumab: 281,818 Versus PBC + pemetrexed : 271,577 | Pralsetinib versus docetaxel: 299,721 PBC + pemetrexed dominated by docetaxel Nivolumab subject to extended dominance |
ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; TTD = time to discontinuation.
Note: All scenario analyses were conducted deterministically; the sponsor’s deterministic and probabilistic results were aligned. Testing was not considered in the CADTH scenario analyses.
Detailed Inputs Applied in CADTH Exploratory Reanalysis
The following tables include the revised values used for calculating dosing and treatment duration in the derivation of drug acquisition costs for all comparators.
Table 19: Summary of Drug Acquisition Cost Revisions
Comparator | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Treatment-naive setting | ||
1. Pembrolizumab (monotherapy and in triple therapy) | Flat dosing of 200 mg per 3 weeks No stopping rule applied | Weight-based dosing (2 mg/kg) up to 200 mg per 3 weeks Treatment up to 2 years |
2. Cisplatin (triple therapy and PBC + pemetrexed) | Incorrect cost per pack ($270 for 100 mg and $135 per 50 mg) No stopping rule applied | Cost per pack derived from DeltaPA data ($646 per 100 mg and $323 per 50 mg) Treatment up to 3 months (4 treatment cycles) |
3. Carboplatin (PBC + pemetrexed) | Flat dosing of 700 mg per 3 weeks No stopping rule applied | Dosing calculated as target AUC x (GFR + 25) based on product monograph dose of AUC 5mg/mL per 3 weeks |
Treatment-experienced setting | ||
1. Docetaxel | Incorrect cost per pack ($121.60 per 80 mg and $243.20 per 160 mg) No stopping rule applied | Cost per pack derived from DeltaPA data ($925 per 80 mg and $1,850 per 160 mg) Treatment up to 2 years |
2. Nivolumab | Flat dose of 480 mg per 4 weeks No stopping rule applied | Weight-based dosing (3 mg/kg) up to 240 mg per 2 weeks Until progression, treatment up to a maximum of 2 years |
3. Cisplatin (PBC + pemetrexed) | Incorrect cost per pack ($270 for 100 mg and $135 per 50 mg) No stopping rule | Cost per pack derived from DeltaPA data ($646 per 100 mg and $323 per 50 mg) Treatment up to 3 months (4 treatment cycles) |
AUC = area under the curve; GFR = glomerular filtration rate.
Note: Recommended dosages in CADTH exploratory reanalysis are per the respective product monographs or clinical expert opinion.16-19
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 20: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor-submitted budget impact analysis (BIA) assessed the introduction of pralsetinib for the treatment of adults with metastatic RET fusion–positive non–small cell lung cancer, examined separately in the first-line and second-line settings. The analysis was taken from the perspective of Canadian public drug plans using an epidemiology-based approach, with only drug acquisition costs included. A 3-year time horizon was used, from 2023 to 2025, with 2022 as a base year. The population size was derived using the incidence of lung cancer, followed by applying the proportion of those patients with NSCLC, metastatic cancer, RET fusion–positive, and finally eligibility for therapies to the derive the relevant Canadian population. Population growth rates were applied per year in the derivation of the target population.
In the first-line setting, the reference case scenario included the comparators pembrolizumab in combination with pemetrexed and PBC (carboplatin or cisplatin), PBC in combination with pemetrexed, and pembrolizumab alone, with anticipated market distribution based on PD-L1 TPS status. In the second-line setting, the reference case scenario included the comparators docetaxel, nivolumab, and PBC plus pemetrexed, depending on what was received as first-line treatment and PD-L1 TPS status. Clinical trial participation was included in both the first and second-line setting. The new drug scenarios included the same comparators with the addition of pralsetinib. Key inputs to the BIA are documented in Table 21.
Figure 1: Sponsor’s Estimation of the Size of the Eligible Population
Source: Sponsor’s budget impact analysis submission.22
Table 21: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) | |
|---|---|---|
Target population | Treatment-naivea | Treatment experienced |
Sponsor’s calculated target population | 3,333 / 3,418 / 3,503 | 1,133 / 1,162 / 1,191 |
Market Uptake (3 years) | ||
Uptake (reference scenario) | ||
RET Fusion–Positive | ||
Pralsetinib | 0% / 0% / 0% | 0% / 0% / 0% |
PBC + pemetrexed | 0% / 0% / 0% | 0% / 0% / 0% |
Clinical trials | 0% / 0% / 0% | 0% / 0% / 0% |
PD-L1 <1% | ||
PBC + pemetrexed + pembrolizumab | 55% / 55% / 55% | 0% / 0% / 0% |
PBC + pemetrexed | 35% / 35% / 35% | 0% / 0% / 0% |
Nivolumab | 0% / 0% / 0% | 0% / 0% / 0% |
Docetaxel | 0% / 0% / 0% | 70% / 70% / 70% |
Clinical trials | 10% / 10% / 10% | 30% / 30% / 30% |
PD-L1 1% - 49% | ||
PBC + pemetrexed + pembrolizumab | 70% / 70% / 70% | 0% / 0% / 0% |
PBC + pemetrexed | 20% / 20% / 20% | 0% / 0% / 0% |
Nivolumab | 0% / 0% / 0% | 0% / 0% / 0% |
Docetaxel | 0% / 0% / 0% | 70% / 70% / 70% |
Clinical trials | 10% / 10% / 10% | 30% / 30% / 30% |
PD-L1 ≥50% | ||
PBC + pemetrexed + pembrolizumab | 5% / 5% / 5% | 0% / 0% / 0% |
PBC + pemetrexed | 10% / 10% /10% | 55% / 55% / 55% |
Pembrolizumab | 80% / 80% / 80% | 0% / 0% / 0% |
Docetaxel | 0% / 0% / 0% | 30% / 30% / 30% |
Clinical trials | 5% / 5% / 5% | 15% / 15% / 15% |
Uptake (new drug scenario) | ||
RET Fusion–Positive | ||
Pralsetinib | 40% / 50% / 60% | 10% / 10% / 10% |
PBC + pemetrexed | 50% / 40% / 30% | 50% / 50% / 50% |
Clinical trials | 10% / 10% / 10% | 40% / 40% / 40% |
PD-L1 <1% | ||
PBC + pemetrexed + pembrolizumab | 55% / 55% / 55% | 0% / 0% / 0% |
PBC + pemetrexed | 35% / 35% / 35% | 0% / 0% / 0% |
Nivolumab | 0% / 0% / 0% | 0% / 0% / 0% |
Docetaxel | 0% / 0% / 0% | 70% / 70% / 70% |
Clinical trials | 10% / 10% / 10% | 30% / 30% / 30% |
PD-L1 1% - 49% | ||
PBC + pemetrexed + pembrolizumab | 70% / 70% / 70% | 0% / 0% / 0% |
PBC + pemetrexed | 20% / 20% / 20% | 0% / 0% / 0% |
Nivolumab | 0% / 0% / 0% | 0% / 0% / 0% |
Docetaxel | 0% / 0% / 0% | 70% / 70% / 70% |
Clinical trials | 10% / 10% / 10% | 30% / 30% / 30% |
PD-L1 ≥50% | ||
PBC + pemetrexed + pembrolizumab | 5% / 5% / 5% | 0% / 0% / 0% |
PBC + pemetrexed | 10% / 10% /10% | 55% / 55% / 55% |
Pembrolizumab | 80% / 80% / 80% | 0% / 0% / 0% |
Docetaxel | 0% / 0% / 0% | 30% / 30% / 30% |
Clinical trials | 5% / 5% / 5% | 15% / 15% / 15% |
Cost of treatment (per patient) | ||
Cost of regimen over first year of treatment Pralsetinib | $149,008 | |
First-line: PBC + pemetrexed + pembrolizumab PBC + pemetrexed Pembrolizumab | $129,046 $34,288 $87,535 | |
Second-line: Nivolumab Docetaxel PBC + pemetrexed | $19,210 $2,120 $26,694 | |
PBC = platinum-based chemotherapy; RET = rearranged during transfection.
Note: PBC consisted of either carboplatin or cisplatin.
aFor patients in the second-line setting who are RET targeted therapy treatment-naive, the market share uptake in the new drug scenario is identical to that of first-line, treatment-naive patients.
Summary of the Sponsor’s Budget Impact Analysis Results
The estimated budget impact of funding pralsetinib for the treatment of adults with metastatic RET fusion–positive NSCLC was $2,032,018 in year 1, $2,562,343 in year 2, and $3,387,687 in year 3. The 3-year total budget impact was $7,982,047.
For first-line treatment, the budget impact was -$1,060,372 in year 1, $803,867 in year 2, and $2,330,523 in year 3, for a 3-year total of $2,074,018. For second-line treatment, the budget impact was $3,092,390 in year 1, $1,758,476 in year 2, and $1,057,163 in year 3, for a 3-year total of $5,908,029.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Market uptake of pralsetinib underestimated: Given the lack of available treatments for adult patients with metastatic RET fusion–positive NSCLC, the clinical expert consulted by CADTH noted that the market share estimates for pralsetinib in the new drug scenario were likely underestimated. Clinician and patient input indicated that uptake of pralsetinib would be rapid and immediate if it was to be made available given there are at present no other treatment options available for this patient population.
CADTH increased the market shares of pralsetinib in the base case to 70% starting in all 3 years, as anticipated by clinical experts consulted by CADTH.
Proportion of patients continuing to second-line therapy: The sponsor assumed that 40% of patients not responding in the first-line setting would continue to second-line treatment. However, this assumption does not align with the feedback received from the clinical expert consulted by CADTH, who anticipated that an estimated proportion of 60% of patients treated in the first-line setting would continue to second-line therapies. This led to an underestimation of the target population.
CADTH assumed that 60% of patients would continue to second-line treatment following progression from first-line treatment.
The sponsor’s assumption regarding participation in clinical trials as a relevant comparator with market share is inappropriate: The sponsor assumed patient participation in clinical trials captured 10% to 40% of market share and accrued no costs. Participation in clinical trials is uncertain and the sponsor’s inclusion of clinical trials in the market mix artificially decreases the estimated population size, disregarding the treatment costs incurred by drug plans and underestimating the budget impact. The clinical expert consulted for this review noted that patients in clinical trials would be eligible for the modelled treatment options.
In reanalysis, CADTH removed clinical trials from the market mix; the market share of clinical trials was re-distributed evenly to available treatment options.
Inappropriately derived target population: The sponsor did not specifically select for the RET fusion–positive patient population in the derivation of their target population in the reference scenario. RET fusion–positive patients were only accounted for in the new drug scenario. Consequently, the population size is significantly larger than expected and includes patients who would not be eligible for pralsetinib. Incremental costs estimated form the sponsor’s model were not affected, but the interpretability of the sponsor’s estimates by reference and new drug scenarios are affected. The population-specific costs are therefore unavailable from the sponsor’s model, and only the incremental costs are applicable for the reimbursement of pralsetinib.
CADTH could not address this limitation in reanalysis due to limitations with the sponsor’s model.
Exclusion of testing costs: Drug plan input noted that there is likely a need for increased testing in order to identify RET fusion–positive patients for treatment with pralsetinib. Therefore, additional testing is likely required given that the product monograph for pralsetinib indicates that testing for RET fusion is required prior to starting pralsetinib. The sponsor’s base case assumed the majority of jurisdictions would include RET fusion testing as part of existing screening and no costs related to screening would be incurred. This is assumption could not be verified by CADTH.
As part of a scenario analysis, CADTH applied a cost associated with testing for RET-fusion testing for all patients.
Uncertainty in the derivation of drug acquisition costs: The sponsor included drug acquisition costs based on treatment duration calculated using mean PFS for each treatment comparator. However, there is uncertainty surrounding duration of therapy of pralsetinib and all comparators, as this is derived using data from various clinical trials, and may not align with time to treatment discontinuation as estimated in the sponsor’s base case. CADTH notes that duration of PFS is a key driver of the sponsor’s estimated budget impact of pralsetinib.
CADTH could not address this limitation in reanalysis and notes that treatment duration remains an area of uncertainty in the CADTH base case.
Incorrect drug acquisition costs for comparators: As previously noted in the pharmacoeconomic model, the drug acquisition costs for docetaxel and cisplatin were incorrect, with the per mL cost used rather than the per vial cost. This underestimated some of the costs of comparators in the BIA.
CADTH included the correct cost per pack as per CADTH’s updated pharmacoeconomic model.
Uncertainty in distribution of patients by PD-L1 status: The distribution of patients by PD-L1 TPS status was used to determine patient eligibility and distribution by relevant comparators. Upon consultation with clinical experts, the PD-L1 proportion breakdown of patients used by the sponsor did not align with expectations; particularly for patients with a PD-L1 TPS > 50%. The clinical expert consulted by CADTH noted that 30% was likely an overestimate and a more accurate estimate is 19%.23 As such, the subsequent breakdown is more likely to be 58% for TPS <1%; 23% for TPS 1% to 49%; and 19% for TPS ≥50%.23
As part of a scenario analysis, CADTH assessed the impact of the alternate PD-L1 TPS status distribution.
CADTH Reanalyses of the Budget Impact Analysis
As part of the base case CADTH revised the anticipated uptake of pralsetinib, increased the proportion of patients continuing to second-line therapy, and removed clinical trial participation as a comparator taking up market share (Table 22).
Table 22: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Market uptake of pralsetinib underestimated | 40% / 50% / 60% in years 1 to 3 | 70% / 70% / 70% in years 1 to 3 |
2. Eligibility for second-line treatment | 40% | 60% |
3. Clinical trial participation | 10% to 40% of patients were assumed to participate in clinical trials | No participation in clinical trials. Market shares were evenly re-distributed to relevant comparators |
4. Incorrect drug costs for docetaxel and cisplatin | Cisplatin: $135.00 per 50 mg pack; $270.00 per 100 mg pack Docetaxel: $121.60 per 80 mg pack; $243.20 per 160 mg pack | Cisplatin: $323.00 per 50 mg pack; $646.00 per 100 mg pack Docetaxel: $925.00 per 80 mg pack; $1,850.00 per 160 mg pack |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
PBC = platinum-based chemotherapy.
The results of the CADTH step-wise reanalyses are presented in summary format in Table 23 and a more detailed breakdown is presented in Table 24. Based on the CADTH base case, the budget impact of the reimbursement of pralsetinib for the treatment of metastatic RET+ NSCLC is expected to be $8,114,211 in year 1, $7,589,974 in year 2, and $6,515,821 in year 3, for a 3-year total of $22,220,006.
For first-line treatment, the budget impact was $3,248,344 in year 1, $4,411,113 in year 2, and $4,521,154 in year 3, for a 3-year total of $12,108,611. For second-line treatment, the budget impact was $4,865,867 in year 1, $3,178,861 in year 2, and $1,994,667 in year 3, for a 3-year total of $10,039,395.
The scenario in which testing costs were included resulted in a 3-year budget impact of $33,128,411. The scenario where alternate PD-L1 proportions were applied did not impact the budget impact.
Table 23: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $7,982,047 |
CADTH reanalysis 1 – market shares | $17,998,107 |
CADTH reanalysis 2 – patients eligible for second-line treatment | $9,545,926 |
CADTH reanalysis 3 – clinical trial participation as market shares | $10,374,323 |
CADTH reanalysis 4 – incorrect drug costs for docetaxel and cisplatin | $7,438,060 |
CADTH base case | $22,220,006 |
Table 24: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $290,104,961 | $293,437,891 | $300,883,504 | $308,408,066 | $902,729,460 |
New drug | $290,104,961 | $295,469,908 | $303,445,847 | $311,795,752 | $910,711,507 | |
Budget impact | $0 | $2,032,018 | $2,562,343 | $3,387,687 | $7,982,047 | |
CADTH base case | Reference | $331,244,931 | $335,050,506 | $343,551,986 | $352,143,611 | $1,030,746,103 |
New drug | $331,244,931 | $343,164,717 | $351,141,960 | $358,659,432 | $1,052,966,109 | |
Budget impact | $0 | $8,114,211 | $7,589,974 | $6,515,821 | $22,220,006 | |
CADTH sensitivity analysis— testing cost | Reference | $331,244,931 | $335,050,506 | $343,551,986 | $352,143,611 | $1,030,746,103 |
New drug | $331,244,931 | $346,711,093 | $354,777,788 | $362,385,633 | $1,063,874,514 | |
Budget impact | $0 | $11,660,587 | $11,225,802 | $10,242,022 | $33,128,411 | |
CADTH sensitivity analysis— PD-L1 status | Reference | $331,244,931 | $335,050,506 | $343,551,986 | $352,143,611 | $1,030,746,103 |
New drug | $331,244,931 | $343,164,717 | $351,141,960 | $358,659,432 | $1,052,966,109 | |
Budget impact | $0 | $8,114,211 | $7,589,974 | $6,515,821 | $22,220,006 |
Stakeholder Input
Patient Input
Lung Cancer Canada
About Lung Cancer Canada
Lung Cancer Canada is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research and advocacy. Lung Cancer Canada is a member of the Global Lung Cancer Coalition and is the only organization in Canada focused exclusively on lung cancer.
https://www.lungcancercanada.ca/
Lung Cancer Canada is registered with CADTH.
Information Gathering
Data Collection
The information discussed throughout this submission consists of the thoughts and experiences of RET-positive non-small cell lung cancer patients and caregivers, conducted through phone interviews and environmental scans. All data was sourced in March 2022.
Demographic Data
RET-fusion-positive non-small cell lung cancer (NSCLC) is a relatively rare mutation, with only 1-2% of lung cancer patients exhibiting this mutation; thus, it was difficult to find Canadian patients who had RET and were also on pralsetinib, as it is a new drug given NOC/c by Health Canada in July 2021. All of the patients discussed are RET-positive and have experience with pralsetinib. Specific treatment experience can be found under the Experience With Drug Under Review section.
Name | Gender | Patient/ or caregiver | Type of lung cancer | Stage at diagnosis | Age | Location | Source |
|---|---|---|---|---|---|---|---|
||||||||| | Male | Patient | RET-positive NSCLC | Stage 4 | 62 | Canada (ON) | Phone interview |
||||||||| | Male | Patient | RET-positive NSCLC | Stage 4 | 79 | USA (Iowa) | Phone Interview |
||||||||| | Female | Caregiver | RET-positive NSCLC | Stage 4 | 52 | Ireland | Phone Interview |
||||||||| | Male | Patient | RET-positive NSCLC (adenocarcinoma) | Stage 4 | 45 | Norway | Phone Interview |
||||||||| | Unknown | Patient | RET-positive NSCLC | Stage 4 | Unknown | USA | Environmental Scan |
Disease Experience
Throughout his entire adult life, ||||||||| had always been a gym fanatic and was extremely active, going to the gym 4 days per week for 2 hours at a time doing intense workouts and building up a huge muscle mass. He was also working full-time in sales and marketing for a large company and had a wonderful life with his wife and two sons. He never thought anything could slow him down, and lung cancer was the last thing on his mind. In 2018, he had some mild lower back pain that wasn’t initially too severe, but over the course of 5 months between October 2018 to February 2019, it got continuously worse. After visiting 5 different doctors while on a business trip in Dubai, he was told they were stress fractures and was prescribed pain killers and physiotherapy. However, when he returned home to India, he had an MRI done which did not look good, leading to more tests that eventually confirmed ||||||||| was diagnosed with stage 4 non-small cell lung cancer (NSCLC) on Valentine’s Day, 2019. His initial prognosis was only 3-6 months as he had metastases everywhere in his body, including his bones, ribs, shoulder, femur, hip, vertebrae, liver and both lungs; to say he was in pain was “an extreme understatement’ – he had difficulty standing up for long, yet even sitting and lying down was uncomfortable. He began treatment, alternating chemotherapy and radiation, which helped clear some tumors, but others stayed stubborn. His oncologist then recommended a clinical trial for a new treatment – pralsetinib. He started on pralsetinib in March 2021 and has been on it ever since. Details on |||||||||’s experience with each treatment are further explored in this submission.
Before diagnosis, ||||||||| was also an active person and in good health, walking 4 times per week and also playing a full 18-hole round of golf three times a week for the last 7-8 years after he retired from his sales position of 30+ years at Xerox. However, in 2000, ||||||||| had also been diagnosed with stage 3 lung cancer after his friend, an endocrinologist, noticed some spots in his CT scans. He had surgery to remove his lower-left lobe, which freed him of cancer and required no further treatment. 21 years later in July 2021, his cancer unfortunately returned after he noticed he was getting easily fatigued, short of breath, and couldn’t play the full 18-holes without needing to stop and catch his breath every few minutes. His scans revealed both lungs were full of cancerous cells, though it hadn’t yet spread into the bloodstream elsewhere in his body, which was the silver lining. After discovering he was RET-positive through a liquid biopsy, his oncologist immediately eliminated surgery and radiation, and instead started him on pralsetinib by October. With the incredible support of his wife and 2 strong daughters, his family was initially concerned about a second cancer diagnosis, but remained optimistic, which in turn helped ||||||||| stay positive throughout his journey. ML has continued to be on pralsetinib ever since October 2021 and has an incredible quality of life in that there’s nothing he cannot do now that he wasn’t able before.
||||||||| husband had always been careful about his health after being diagnosed with Type II diabetes a few years ago, and about a year before he was diagnosed, he started on a new injection medication to help manage his diabetes, which also turned out to be an appetite suppressant and he often vomited after most meals. However, in late August/early September 2021, he started vomiting more frequently after meals and lost a lot of weight. His physician took him off the injection, in which the washout period took 5 weeks, and during this time, he only became sicker and sicker with each day, even developing a new cough. ||||||||| did her own research as they do have family history of cancer, and with her husband’s symptoms getting worse with frequent vomiting, a new cough, and pain in his lower back, she had a gut feeling it could be cancer. At this point, he could barely eat without vomiting and even needed help getting up from his chair since he was so weak. On December 6th just after ||||||||| was done getting CAT scans at the hospital, they were only 2 blocks from the hospital before they were called back to return immediately to be admitted. Scans showed metastases in his liver, spine, and neck in addition to the primary tumour in his lungs, which diagnosed him with stage 4 NSCLC. However, within 2.5 weeks, her husband’s health has declined so much that ||||||||| was called to the hospital with her children, as physicians didn’t think he’d survive more than 2 days, which was extremely terrifying for their family. They started him on emergency chemotherapy 2 days before Christmas, but the next day, biomarker testing confirmed he had the RET mutation, so on Christmas Eve, he was immediately started on pralsetinib, which ||||||||| recalls as the “best Christmas present their family has ever gotten”. Within a few days of starting pralsetinib, he quickly came off oxygen, was able to walk around the hospital, and his appetite had returned that he was even eating 2 dinners, which was such a drastic change from being so ill just a few days prior. While LCC was speaking to |||||||||, she repeatedly remarked pralsetinib as being a “literal miracle drug for her husband and their family”, and they haven’t looked back since.
Genetic alterations in the RET proto-oncogene drive 1-2% of non-small-cell lung cancers (NSCLCs), with a global cancer burden of over 10,000 cases each year. NSCLCs are the most common type of lung cancer, covering 80-85% of lung cancer cases. Currently, the standard of care in Canada for NSCLC patients with RET-fusion mutations include the use of chemotherapy or immunotherapy for first line treatment. However, targeted therapy has since emerged as an important mean of disease management for NSCLC patients with a targetable mutation, including RET. This form of treatment has greatly improved patient outcomes and quality of life and is now a treatment option that is some patients’ only hope. It has seen incredible success and has allowed patients like |||||||||, |||||||||, and ||||||||| a chance at survivorship and a livelihood that is nearly comparable to before diagnosis, something that they may never had thought would be possible before.
The development of pralsetinib is a positive step forward and represents a fundamental change in the treatment of RET-fusion and non-small cell lung cancer, as this agent has shown to be clinically beneficial, RET-specific, and is well tolerated by patients. Pralsetinib is the second therapy of its kind approved in Canada for RET-fusion-positive targeted therapies, as the discovery of RET mutations in lung cancer is so new, where not much research or treatment has been publicly available. As a result, this is one of the first opportunities for Canada to have a publicly funded targeted therapy for RET-fusion, and there is an incredible amount of potential for pralsetinib to be able to drive the pathway for future lung cancer treatment for thousands of Canadians. Lung Cancer Canada strongly encourages CADTH to take this into consideration for pralsetinib to be reimbursed as it would lead the pathway to new developments, new treatments, improvements in accessibility, and better affordability for lung cancer patients across the country.
Experiences With Currently Available Treatments
The current standard of care for RET-fusion-positive NSCLC patients is chemotherapy or immunotherapy. Chemotherapy has been a long-standing and well-documented standard of care for lung cancer patients and has seen some benefits. However, it is limited as a viable long-term treatment option due to its harsh side effects, which often creates additional burdens on patients, leading to decreased functionality and increased dependence on caregivers in daily activities without bringing much benefit or efficacy in treating disease. Thus, they are typically only used as a source of initial first-line treatment. Immunotherapy carries fewer toxic effects compared to chemotherapy; however, the requirement to travel to hospitals or medical clinics creates barriers to access for patients, especially those who live in rural communities. Targeted therapy has since emerged as another important treatment option for those with targetable mutations, such as RET-fusion, particularly in second-line and beyond. Targeted therapies have been met with much greater success in lung cancer patients, that they essentially overcome the limited benefits that chemotherapy and radiation are able to provide, whist being able to also manage and treat the symptoms that patients experience with NSCLC. This has made them extremely valuable to patients.
Chemotherapy: Living in Norway, then 43-year-old ||||||||| had always maintained a healthy lifestyle, was sporty and did a lot of fitness throughout his life, so when he suddenly started becoming exhausted and also developed a cough that didn’t seem to go away in throughout the spring and summer of 2019, it was very odd. Doctors first thought it was just an allergy at first, but after a few blood tests, they had found he had a lung embolism, and further scans confirmed stage 4 NSCLC with mets in his thorax and stomach. He started first-line treatment with 10 cycles of chemotherapy/Immunotherapy (carboplatin, pemetrexed, and pembrolizumab) in November 2019, the standard of care in Norway. By spring 2020, scans showed limited efficacy of this treatment, though he didn’t have many side effects other than occasional fatigue and the general feeling of being unwell. It did require him to change his lifestyle slightly and wasn’t able to do any high-intensity fitness he was used to. However, in September 2020 when he went to Germany for a second opinion, he also had biomarker testing done, which revealed he had the RET+ mutation, which quickly led his physicians to get pralsetinib available for him within a few weeks. ||||||||| started treatment with pralsetinib through the German hospital the same month and continued on it for 9 months until June 2021.
Within a month of being diagnosed, ||||||||| started chemotherapy treatment in March 2019 while living in India for 8-10 sessions, which was hard on him due to its toxic and harsh side effects. He was constantly fatigued, very weak, and even had difficulty finding a comfortable position because of the extent of his metastases when diagnosed. He couldn’t stand up for long, lying down hurt his back, and sitting was also painful because of the spread to his spine near his coccyx. However, he alternated between radiation and chemotherapy during this time, which was effective at clearing many tumours while others were stubborn. His physician then recommended immunotherapy, which he started soon after his chemo treatments were complete. ||||||||| attributes the nature of his active lifestyle before diagnosis that contributed to his strength to endure a total of 24 sessions of chemotherapy throughout his cancer journey
Immunotherapy: After ||||||||| 9-month treatment with pralsetinib, by June 2021, it was discovered that there were new lesions in his thorax and brain, which was evidence he became immune to pralsetinib, despite having great efficacy earlier on. In December 2021, he went back on a combination of chemotherapy and immunotherapy for 4 cycles (atezolizumab, bevacizumab, paclitaxel, carboplatin) every 3 weeks, which led to quite severe side effects. He lost his hair for the first time during this treatment, which was hard mentally for him as his first line chemo/IO did not have this impact. After fulfilling the 4 cycles by February 2022, he is currently continuing on with the immunotherapy atezolizumab and bevacizumab every 3 weeks to this day. ||||||||| continues to do very well and is living each day to the fullest.
In 2020, ||||||||| started pembrolizumab immunotherapy treatment after completing first-line chemotherapy/radiation, which was again effective at clearing some tumours yet others stayed stubborn. He also did not recall any significant side effects from the 12 sessions of pembrolizumab. He completed several treatments before ||||||||| ultimately decided to move to Canada with his wife for better treatment, landing in August 2020. He did 3-4 rounds of chemotherapy and radiation again, which eventually did not work, and was put on blood thinners after a blood clot in his leg was discovered in February 2021. At this time, his oncologist in Canada suggested the pralsetinib clinical trial, which he was very fortunate to have access to. ||||||||| started on pralsetinib in March 2021 and has been on it ever since.
Improved Outcomes
There have been many incredible advancements in recent years for lung cancer treatment that have changed the paradigm for patients. With RET being a relatively new discovery in lung cancer research, there has not been many previous opportunities for the development and refinement of new targeted therapy treatments for RET-fusion, until now. It has been seen that RET-targeted therapies, including pralsetinib, have been met with incredible success that gives patients their livelihoods back, allows them to hope for a better tomorrow and plan further down the line for a possible future. These outcomes play a huge role in the goals that patients have in their treatment decisions, including:
Improved management of their symptoms of non-small cell lung cancer
Allowing patients to have a full and worthwhile quality of life
Having manageable side effects
Allowing patients to live longer and maintain their independence and functionality so minimize the burden on their caregivers and loved ones
Delaying disease progression and settling patients into long-term remission for improved survivorship
Experience With Drug Under Review
Table 2: Experience With Drug Under Review
Patient | Diagnosis date | Drug access method | Period on pralsetinib | Duration on pralsetinib | Line of treatment with pralsetinib | Currently on pralsetinib? |
|---|---|---|---|---|---|---|
||||||||| | February 2019 | Pharmaceutical Compassionate Access Program | March 2021 – present | 1 year | 4th line | Yes |
||||||||| | July 2021 | Pharmaceutical Compassionate Access, Medicare (USA) | October 2021 – present | 5 months | 1st line | Yes |
||||||||| | December 2021 | Pharmaceutical Compassionate Access | December 2021 – present | 3 months | 1st line | Yes |
||||||||| | November 2019 | Pharmaceutical Compassionate Access | October 2020 – August 2021 | 10 months | 2nd line | No |
||||||||| | Unknown | Unknown | September 2021 – present | 6 months | Unknown | Yes |
Pralsetinib is effective at treating disease and shrinking tumours
Pralsetinib works as a highly selective oral RET kinase inhibitor that has shown incredibly promising results from the ARROW clinical study. In treatment-naïve patients, overall response rate was seen to be 70%, 11% of which were complete responses (Gainor et al., 2021). Median progression-free survival was 9.1 months and median overall survival for this group was 13.6 months (Gainor et al., 2021). The majority of study participants did have previous exposure to actinium chemotherapy, where ORR was 61%, 6% being complete responses, and median progression-free-survival was 17.1 months (Gainor et al., 2021). The relatively long duration of progression-free survival time is critical for patients with such advanced disease to have this opportunity to improve their survivorship, maximize their quality of life, and be able to continue their daily lives with autonomy and dignity, while also helping treat their disease.
At diagnosis, ||||||||| had both lungs full of cancer cells and had trouble with shortness of breath, even though he was a very active person who consistently exercised multiple times per week. He had no spread anywhere else in his body yet, which was a very good sign, so when the liquid biopsy revealed he had the RET mutation, he was immediately started on pralsetinib in October 2021 and has been on it ever since. His most recent CAT scan in January 2022 revealed a dramatic and remarkable reduction in cancer cells, to the point where his oncologist noted he had nearly no evidence of disease (NED). He continues to be on the 200 mg dosage to this day in April 2022 and continues to see positive results from it.
||||||||| started pralsetinib in October 2020 after getting a DNA biomarker test done while in Germany that revealed he had the RET mutation. After only 3 days of taking the drug, he felt a dramatic change in himself, and it was already working. Scans after the first 3 months showed the therapy was working remarkably well; it had reduced many of his metastases in addition to his main tumour in the lung. Pralsetinib continued to work very well for him and shrunk the tumours even further, and since he had been getting the targeted therapy from his physician in Germany, he had to travel there from his home in Norway every month to pick up the drugs, despite COVID-19 restrictions, which he had virtually no issues with at all regardless of his disease. Unfortunately, after 9 months on the treatment, there were two new lesions discovered in his thorax, 4-8 mets in his brain, and his main tumour had slightly increased in size. He was gradually eased off treatment in July/August of 2021 and was treatment-free until December 2021, when he started chemotherapy and immunotherapy again, which he has been on ever since.
Similarly, ||||||||| was diagnosed with stage IV NSCLC and started treatment with pralsetinib in September 2021 and has been on it ever since. His first 3-month scan showed a dramatic reduction in cancer cells and continues to see very positive results with pralsetinib.
When ||||||||| was diagnosed in early 2019, he already had stage 4 disease as his cancer had spread to numerous places in his liver and bones including ribcage, spine, shoulder, hip, and femur, and doctors gave him an initial prognosis of about 3-6 months. After treatments with chemotherapy, radiation, and immunotherapy did not work, pralsetinib was his 4th line of treatment, which he was extremely grateful to be able to access. He started pralsetinib in March 2021 and has continued to be on it to this day. 2 weeks prior to speaking with LCC, his scans revealed most tumours in his lungs and spine were gone, the 2 lesions in his liver had reduced to just about 2cm in diameter each, compared to 7cm and 10cm at diagnosis. It was a very remarkable change that has allowed ||||||||| to go from having trouble simply turning on his side in bed because of extreme pain, to being able to walk 3-4km everyday with no problem. He continues to be on pralsetinib in April 2022 and is doing very well.
||||||||| husband was hospitalized and incredibly weak prior to starting pralsetinib, to the point where ||||||||| was called to come to the hospital with her kids to prepare for the worst as physicians didn’t think he’d survive through the week. However, day after they found he had the RET mutation, he started pralsetinib on Christmas Eve 2021. Within a few days of taking the drug, he came off of oxygen quickly, was able to walk around the hospital hallways, his appetite returned, and ||||||||| recalls he “ate like a horse” and has never felt this good ever since being diagnosed earlier in the month. Within a. few weeks, scans showed his tumours were all either shrinking or stabilizing, and the pain in his back is completely gone. He hasn’t looked back ever since and was able to be released from the hospital 2 weeks after starting pralsetinib in mid-January, and continues to be doing well on it. .
Pralsetinib has manageable side effects that had much less impact on daily life in comparison to other treatment options
As per the ARROW study, the most common treatment-related adverse events were neutropenia, anemia, constipation, hypertension, leukopenia, fatigue, dry mouth, and diarrhea, amongst others (Gainor et al., 2021). These are all relatively minor side effects that carry much less burden to the patient in comparison to other available treatments that are used to treat patients with RET-fusion-positive NSCLC, such as chemotherapy, immunotherapy, and radiation.
Among the patients LCC had interviewed for this submission, fatigue was the most common side effect experienced for |||||||||, |||||||||, and |||||||||, particularly during onboarding and the first initial weeks of treatment. Dry mouth, anemia, constipation, loss of appetite, and itchiness/dry skin were also reported, which is in line with what is reported in the clinical trial. ||||||||| and ||||||||| had virtually no side effects other than occasional fatigue. However, most patients did have their dosages reduced to manage their side effects, particularly for |||||||||, who had initially pretty severe itchiness, dry skin, and loss of appetite, and |||||||||, who was re-hospitalized as his liver function was became continuously much higher than normal and also had a severe headache. However, once their dosages were reduced these adverse effects went away and have been doing well ever since.
For ||||||||| and |||||||||, pralsetinib has significantly improved both of their shortness of breath, which was their main disease symptom. Prior to treatment, ||||||||| went from stopping every 200 feet to catch his breath, to daily walks of 30 minutes without stopping. He feels much better physically, his breathing has improved dramatically, and is able to gradually increase his pace and distance with each walk. ||||||||| used to walk and play golf 4 times per week, which was virtually impossible to do prior to starting pralsetinib because his shortness of breath had worsened so much. However, in early March 2022, he pushed himself to get back outside for walks, and although he still continues to struggle stopping to catch his breath for 15 seconds every ½ mile, he’s able to walk half a block without much issue, and is getting stronger by the day.
Patients are able to enjoy a great quality of life and level of functionality similar to what they had before diagnosis
As discussed above, caregiver ||||||||| felt like she had hit a dead end when her husband was hospitalized, and doctors told her family to prepare for the worst. He could barely walk, had lost a lot of weight, was extremely ill and weak, was on oxygen, and couldn’t even go to the bathroom without someone helping him. Within a few days he started treatment with pralsetinib, he was back up and walking around hospital hallways, eating 2 dinners, and came off oxygen. When he was released from the hospital in mid-January 2022, he was slowly able to return to doing everything again – driving his 3 kids to school, cleaning the house, and even mow the grass in his lawn. It was such a drastic, 180-dregree change from just a few weeks ago, that ||||||||| mentions they sometimes forget he even has cancer. His wife ||||||||| remarks he is currently even better than before he was diagnosed, and she is happy he is able to return to a quality of life that is worthwhile. It has only been 3 months since ||||||||| has been taking pralsetinib, but he is doing extremely well, able to continue doing many of the hobbies he loves, socialize with friends, and has never looked back.
||||||||| has always been an extremely positive and optimistic individual, and especially after being diagnosed with cancer, he has endured many treatments that ultimately were not working, and even moved to Canada from India to access better treatment options. Prior to pralsetinib, he could barely even walk because of the metastases in his spine that made it extremely painful; he could barely even turn over on his side in bed because of the pain. He had difficulty standing up, lying down, and sitting, but he never gave up. Once he started pralsetinib in March 2021, he has been able to slowly get back to cleaning the house, cooking, and even going for 3-4km walks outside to maintain the physically active lifestyle he had before diagnosis. ||||||||| mentioned to LCC he is nearly back to his functionality as he had before diagnosis, and although he still has minor residual pain after walking for long periods, he is at a much better place than initially at diagnosis.
Throughout ||||||||| treatment with pralsetinib between October 2020 and Fall 2021, he had been travelling to Germany through COVID-19 for his monthly pick-up of the targeted therapy from his oncologist there. As his previous line of treatment was the standard of care with chemo/immunotherapy, it was very limiting in what he could do in his daily life, its side effects were exhausting, and was difficult for him to continue with more demanding daily activities like grocery shopping or cleaning the house, though he has had a lot of positive support from his family and friends, particularly his brother. Once he stated on pralsetinib, within 3 days he already felt a dramatic change in his mood, energy levels, and functionality, and within a few months, he was even able to start working again. He has continued to live an active and full life that is virtually the same as before diagnosis, driving out to meet friends, going out and shopping, and taking mini vacations when he flies out to Germany for an overnight stay when picking up medications. Though ||||||||| is no longer on pralsetinib, he continues to have no issues or barriers continuing the lifestyle he had while on pralsetinib, and is currently still doing all these activities independently with no issues.
Pralsetinib allowed patients to get back to the hobbies they enjoy
As mentioned previously, ||||||||| had always been a motivated gym fanatic and bodybuilder, working out 4 days a week for 2 hours each time; he could easily do 120 pullups, which gave him a huge muscle mass and very strong and fit body. Going to the gym was his favourite hobby as it was also a huge stress reliever, so after becoming so weak with several rounds of cancer treatments, it took a toll on him as it had hurt for him to even stand sometimes. However, he stayed very positive throughout his cancer journey, and with the success of pralsetinib, ||||||||| has started to document his journey on social media, and even took on a new hobby of painting. While he was in the hospital, he started to learn oil painting, which he now loves and is a newfound hobby of his. He has returned to walking 3-4km with ease and is hopeful he can soon get back to the high-intensity physical activities he used to love, including kickboxing, kung fu, and karate.
Similarly, ||||||||| had always been active prior to diagnosis and as a retiree of 8 years, he particularly enjoyed playing golf as well three times a week. After he noticed he wasn’t able to complete a full 18-hole round of golf without needing to catch his breath every few minutes, he cut down to only 9-holes, which was still very tiring for him. Since pralsetinib was his first-line of treatment, he has not necessarily had any severe setbacks during his cancer journey, and as of March 2022, his day-to-day life is pretty much as normal, thanks to the incredible efficacy of pralsetinib. He has since resumed walking, going for long-distance road trips to the cottage with his grandkids, and spending time with them. He notes to LCC that there’s nothing he cannot do right now that he couldn’t do before, and is hopeful he can soon return to getting back onto the green.
Pralsetinib has even allowed patients to return to work
With his previous treatments of chemotherapy/immunotherapy, ||||||||| wasn’t able to work at all because of the toll it took on his body and the mental fog he experienced, which made it hard for him to concentrate on anything. When he started pralsetinib, it worked incredibly well not only in shrinking his tumours and keeping his disease stable, but also for him mentally. He didn’t have the mental fog anymore, was able to start doing daily activities again around the house, and since he didn’t have to focus so much on his disease symptoms or his physical health, he was able to relay that attention elsewhere. He started working again a few months into his treatment with pralsetinib, which also made him feel better as a contributing taxpayer to the system rather than a patient charging the public system. Additionally, it saves the public system from another unemployed/sick leave person who needed the time and financial support, and was happy he was able to contribute back to society so others who needed it more are able to access the help they need. Even after he terminated pralsetinib and while currently on chemotherapy/immunotherapy again, he still continues to work without much issue, and is hopeful he can continue to work as long as possible.
Back in India, ||||||||| previously worked as a sales and marketing specialist at a company producing films and x-rays for 34 years, which required him to travel frequently across the Middle East and India; in fact, he was on a business trip in Dubai when his first symptoms of being in pain started prior to diagnosis in early 2019. Since being diagnosed, he has been unable to work at all in the last 3 years because of the demands with his cancer treatment and hasn’t earned a dollar during this time. However, since moving to Canada and being on pralsetinib, which has been working very well for him, he is looking to go back to work soon, depending on his oncologist’s recommendations.
||||||||| husband was so weak immediately prior to pralsetinib that physicians didn’t think he’d survive more than 2 days, but to their surprise, he regained his energy and functionality within days of starting treatment with pralsetinib. He has started working again in multiple roles, including their family farm in the countryside of Ireland, which has made ||||||||| very happy to see her husband doing well and being able to enjoy his time again. Pralsetinib has given patients like |||||||||, ||||||||| and ||||||||| the opportunities to go back to work with how well the drug has worked for them, allowing patients to focus on other aspects of life aside from their cancer.
Patients are able to look into the future and enjoy the extra time they’ve gained with pralsetinib
||||||||| never would have thought her husband would still be alive past Christmas 2021, let alone 3+ months later and doing so incredibly well. He was hospitalized in early December and physicians called ||||||||| and her children to the hospital 2 hours away from her hometown as they didn’t think he’d survive more than 2 days. Pralsetinib was their Christmas miracle for not only her family, but also their physicians, who were just as happy as they were when he got access to the drug and responded remarkably well within a few days. ||||||||| remarks that there are often days where her family forgets he even has cancer since he’s always on the go now, driving the kids to school, spending quality time with them, and just being there for the family. ||||||||| is incredibly grateful for the extra time they’ve gained thanks to pralsetinib, not to mention the excellent quality of life he currently has. They don’t have many goals for the future, but just wants to enjoy the time they have gained, and maybe even travel again after COVID restrictions subside. Pralsetinib has given families like ||||||||| the ability to enjoy their lives, the extra time they’ve gained, and actually envision a future.
Although ||||||||| has terminated treatment with pralsetinib and is currently on a different treatment, he attributes his incredible quality of life to pralsetinib, and it has given him the opportunity to recover from previous treatments and enjoy a great livelihood while on the drug. He was able to travel monthly to Germany, start working again, and spend time with friends. He was never given an initial prognosis from physicians, which in turn worked well for him as he doesn’t think it’s worthwhile to hand out timelines to patients, as many have exceeded these expectations and are still doing very well. ||||||||| believes he can still live many good years, and his oncologist has a plan in place for a next line of treatment in case his current regimen doesn’t work out. ||||||||| says to LCC, “It's about having as many cards as possible ahead of you and using each of them as long as possible”. As an avid skier in the wintertime and runner in the summer, ||||||||| is hopeful he’d be able to return to those activities soon.
Companion Diagnostic Test
Pralsetinib is indicated for RET-fusion on small cell lung cancer patients, in which biomarkers of their tumour are tested by next-generation sequencing (NGS) or FISH. Though RET is a relatively rare mutation in lung cancer, the availability of testing for the RET fusion mutation is nearly country wide, except for Newfoundland and PEI.
Anything Else?
With a diagnosis of advanced lung cancer, many patients are left shattered and unable to have goals or even plan for the future. For RET-positive NSCLC patients however, pralsetinib has helped give patients needed hope and the possibility to look towards the future. Pralsetinib has allowed patients faced with a life-changing diagnosis the ability and confidence to dream big again and return to activities and livelihoods that they enjoy. Pralsetinib has seen promising results for the patients interviewed and has given them an additional treatment option when for some, it seemed like the end of the road. Most patients had prior treatments with primarily chemotherapy that were not effective, or overtime, their cancer became resistant to these treatments. The success that pralsetinib has had on them has allowed patients like ||||||||||||||||||||||||||| to all have another chance at treating their cancer and one that is much more specific to their genetic mutation with RET.
It is important to remember that pralsetinib is a targeted therapy for those with the RET mutation, present in 1-2% of the lung cancer population. Its clinical efficacy is consistent to other targeted therapies, and due to the definition of targeted therapy, the population will remain small. The ARROW study and ongoing TAPISTRY clinical trial have showcased that pralsetinib is effective at treating patients’ disease, maintaining disease stability, and minimizing disease symptoms meanwhile with minimal side effects. The benefits of having an oral targeted therapy are numerous in comparison to standard IV treatment options, such as decreased patient and caregiver burden, ease of dosage, improved management of symptoms, and minimal travel requirement for treatment. These all lead to an incredible pathway to a better quality of life for these patients and allows them to make plans for the future without the pressing issues of their disease.
Per the results of the ARROW study, the median progression-free survival was 17.1 months, which is very promising and 2 of the patients interviewed by LCC are approaching or past the 12-month mark of treatment. Time is the most valuable asset to patients faced with advanced disease, and it is critical to have additional options in the current treatment paradigm as it can change the lives of patients across the country. Being able to broaden the treatment landscape for non-small cell lung cancer in Canada with the approval of pralsetinib is a critical step forward towards the future of patient care. This group of patients cannot afford to wait and deserve to have access to treatments that can help treat their disease and allow them to have a worthwhile quality of life that is meaningful. LCC hopes CADTH provides a positive recommendation for this submission.
Reference: Gainor, J.F., Curigliano, G., Kim, D.W., Lee, D.H., Besse, B., Baik, C.S., et. al. (2021). Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): a multi-cohort, open-label, phase 1/2 study. The Lancet Oncology, 22(7), 959-969. DOI: 10.1016/S1470-2045(21)00247-3
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 3: Financial Disclosures for Lung Cancer Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Roche | — | — | X | — |
Clinician Input
Lung Cancer Canada
About Lung Cancer Canada
Lung Cancer Canada (LCC) is a national charity with the purpose of increasing awareness about lung cancer, providing support and education to lung cancer patients and their families, to support research and to advocate for access to the best care for all lung cancer patients in all provinces and territories.
Through the LCC Medical Advisory Committee (MAC), we have been providing clinician input for submissions of new lung cancer drugs to the HTA process for many years. The LCC MAC is made up of clinicians and key opinion leaders in the field of lung cancer across the country.
Information Gathering
The feedback on the questions listed in this Clinician Group Input is based on current clinical data and practice guidelines, which will be referenced in the corresponding sections.
Current Treatments and Treatment Goals
In the last decade, our understanding of the pathogenesis of non-small cell lung cancer (NSCLC) has greatly evolved, where we now understand that a large proportion of NSCLC harbour oncogenic alterations that drive cancer pathogenesis. Many of these so-called driver mutations, which include RET fusions, serve as predictive biomarkers for response to precision targeted therapies. With this new understanding of the molecular landscape of NSCLC, a dichotomy has evolved in the treatment decision tree where some driver-driven lung cancers can be treated with precision targeted therapies, and those with other oncogenic alterations or unknown alterations are treated with non-precision treatments such as cytotoxic chemotherapy with/without immune checkpoint inhibitors (ICI).
Treatment naïve patients with NSCLC that harbour RET Fusions have typically been included in clinical trials using non-precision therapeutics, and as such the current treatment standards include:
First Line Therapy
Platinum/ Pemetrexed - doublet chemotherapy;
Platinum/Pemetrexed and pembrolizumab for those with PDL-1 expression <50%, and possibly those with PDL-1 expression > 50% who are non-smokers or have high disease burden requiring rapid tumour debulking;
Pembrolizumab alone for those with PDL-1 expression > 50%.
It should be noted that ICI such as pembrolizumab are not preferred in all patients, and can be relatively contraindicated in patients with uncontrolled autoimmune disorders requiring high doses of immunosuppressants, or have vital organ transplantations for the fear of exacerbating organ rejection.
For patients with wild-type (ie: proto-oncogene mutation free) NSCLC, the current standard of Chemotherapy and an Immune checkpoint inhibitor (ICI) such as Pembrolizumab has been shown to be superior than ICI free regimens. Sheng et al. performed a meta-analysis of 26 randomized trials that demonstrated improved mPFS for chemotherapy + pembrolizumab (HR=0.70; 95% CI: 0.58-0.80) without improvement in median overall survival (mOS; HR=0.90; 95% CI 0.79-1.05) [Sheng et al.Ther Adv Med Oncol 2021 May 29;13:]. Another meta-analysis of the Keynote (KN) suite of trials, 024, 042, 021, 189 and 407, also demonstrated an improvement in ORR for the chemotherapy and pembrolizumab combination over chemotherapy alone (Relative Risk: 1.6; 95% CI 1.2-2.2) and mPFS (HR=0.55; 95% CI: 0.32-0.94) while there was no difference detected in mOS (HR=0.76; 95% CI: 0.51-1.14) [Zhou et al. J Immunother Cancer 2019;7(1):120].
In the absence of other precision options, patient with RET-Fusion NSCLC have clinical characteristics that would result in Chemotherapy + ICI being chosen as their first line systemic therapy. Offin et al showed that only 19% of RET fusion positive NSCLC patients will have PDL-1 expression > 50%, and more commonly these individuals will be non-smokers. [J Clin Oncol Prec Oncol 2019;3:PO.18.00386]; thus, in these patients, platinum/pemetrexed and pembrolizumab would be considered standard of care in the Canadian context.
This then begs the question if Chemotherapy + ICI regimens improve outcome in RET-Fusion positive NSCLC as it does in wild type NSCLC, and if this combination can be considered the standard of care in this patient population. Hess et al. [BMC Cancer2021;21(1):28] reported comparably similar ORR (75% versus 60.5%, p=0.15), mPFS (6.6 months versus 5.7 months, p=1.0) and mOS NR vs. 14 months for RET fusion (N=9) and non-RET fusion (N=605) NSCLC patients treated with platinum/pemetrexed/pembrolizumab. After controlling for all covariates, there was no statistical difference for PFS or OS in the RET fusion positive and RET fusion negative cohorts. Similarly, by using two large de-identified real world databases, Bhandari et al. showed that clinical outcomes of patients with RET fusion positive NSCLC have similar efficacy outcomes to unselected populations when ICI-based therapies are used in the treatment naïve setting. [Bhandari et al. Immunotherapy 2021: 13(11): 893-904].
Subsequent Lines of Therapy
For the RET fusion NSCLC who progressed on prior systemic therapy, the options include:
Platinum/pemetrexed for those who had received pembrolizumab as first-line therapy,
Anti-PD(L)1 therapy, including pembrolizumab, nivolumab and atezolizumab, for those who had received platinum/pemetrexed as first-line therapy (but with the adoption of platinum/pemetrexed and pembrolizumab as first-line therapy, this represents a very small number of patients), and
Docetaxel for those who have progressed on platinum/pemetrexed and pembrolizumab.
RET fusion NSCLC is very sensitive to pemetrexed, [Gautschi et al. J Clin Oncol 2017;35(13):1403-1410], [Drilon et al[Ann Oncol 2016;27(7):1286-1291]. As such, pemetrexed/platinum is likely the most efficacious therapy in the RET fusion NSCLC patient who had received only pembrolizumab as first-line therapy, in the absence of any randomized data.
The efficacy of anti-PD(L)1 therapy is low. A majority of the retrospective series reported an ORR 0-20% and mPFS of 1.5-2.1 months [Mazieres et al. Ann Oncol 2019;30(8):1321-1328; Lee et al. Jpn J Clin Oncol 2020;5(5)”:594-560; Offin et al. J Clin Oncol Prec Oncol 2019;3: PO.18.00386] except for the outlier retrospective series by Guisier et al, where the ORR was 32.5%, mPFS was 7.6 months and 1-year OS was 89% among the 9 RET fusion NSCLC who received PD(L)1 therapy as second-line and beyond [Guisier et al. J Thorac Oncol 2020;15(6):628-636]. Furthermore, Tan et al.[J Thorac Oncol 2020;15(12):1928-1934] reported that RET fusion NSCLC patients who had or had not received immunotherapy at any time during their metastatic disease setting had similar mOS (37.7 months versus 49.3 months, p=0.53).
The clinical outcome of RET fusion NSCLC treated with single-agent docetaxel after prior systemic chemotherapy has not been reported. All in all, the ORR was 7%, mPFS of 10.6weeks and mOS of 7.5 months for unselected, previously treated, advanced or metastatic NSCLC [Shepherd et al. J Clin Oncol 2000;18(10):2095-2103]. Based on the subgroup analysis of CM057, KN010, and OAK, patients with EGFR or ALK aberration derived similar benefit from docetaxel and PD(L)1 therapy[Horn et al. J Clin Oncol 2017;35(35):3924-3933; Herbst et al. Lancet 2016;387(10027):1540-1550; Ritteyer et al. Lancet 2017;389(10066):255-265]. Thus, it is believed that patients with RET fusion NSCLC will benefit from docetaxel in a manner similar to patients with EGFR, ALK or unselected, previously treated, advanced or metastatic NSCLC.
The final question is whether RET fusion is a driver mutation for NSCLC. Driver mutation is a genomic alteration that provides a cancer cell with a fundamental growth advantage for its neoplastic transformation. By targeting the driver mutation, the therapy will alter the disease outcome. RET fusion resected NSCLC has similar median recurrence-free survival and mOS when compared toRET fusion negative patients. But in the metastatic setting, RET fusion NSCLC who had received multi-kinase inhibitor to RET had better mOS than those who did not (49.3 months versus 15.3 months, p<0.001)[Tan et al. J Thorac Oncol 2020;15(12):1928-1934]. Despite the modest anti-tumour activity of multi-kinase inhibitor to RET, Hedge et al.[ESMO Open 2020;5(5):e000799] reported a trend towards better mPFS with multi-kinase inhibitors over immunotherapy (9.3 months versus 3.4 months, p=0.16). These findings resemble that of ALK positive NSCLC and thus RET fusion is a driver mutation.
Treatment Goals
In the advanced or metastatic NSCLC setting, the goals of therapy are, in the order of priority:
Improvement in mOS: It should be keenly noted that a randomized trial with a crossover design, especially if there is a high crossover rate from the standard of care arm to the experimental arm, mOS may not be significantly improved as has been seen in PROFILE 1014 and the recently updated J-ALEX study [Solomon et al. J Clin Oncol 2018;36(22):2251-2258 and Yoshioka et al. PASCO 2021;39(15_Suppl):A9022]. The mOS from any non-comparative trials can be used for benchmarking with randomized data for potential major difference in OS outcome.
Improvement in ORR, and mPFS: As a majority of advanced or metastatic NSCLC are symptomatic at the time of initial diagnosis and at the time of progression from prior therapy, early and prolonged symptom improvement without disease progression radiologically will provide clinically relevant improvement in health-related quality-of-life.
Low toxicity profile: Incidences of Grade 2 toxicity experienced daily and Grade 3 or higher clinically important toxicity and dose reduction or dose discontinuation are especially important to consider for any systemic therapy. For one, constant grade 2 toxicity, such as nausea, vomiting, diarrhea, and so on, can negatively impact on the quality-of-life (QoL) of patients and oral medication adherence. The latter can further adversely affect the real life efficacy or effectiveness of an oral therapy. Second, as mentioned above, advanced or metastatic NSCLC patients have high symptom burden, which can further impair patient well-being in the setting of frequent and clinically significant toxicity.
Prevention or treatment of brain metastases: Up to 40% of advanced or metastatic NSCLC can present with brain metastases during their treatment journey. As reported by Peters et al. [Cancer Treat Rev. 2016;45(2):139-162],brain metastases have a negative impact on QoL and carry a poor prognosis. Only a small number of mNSCLC patients will be candidates for surgical resection and stereotactic brain radiation/gammaknife (GK). The majority will be treated with whole brain radiation (WBRT), which carries significant short-term and long-term toxicity, such as immediate memory loss, loss of higher cortical function and fatigue, can negatively impair the functional status, independence and QoL of patients. Therefore, brain penetrating systemic therapy, not only treat but also prevent/delay brain metastases, will improve the QoL and preserve functional status of mNSCLC patients.
Minimized resource utilization: Intravenous systemic therapy is given every 3-6 weeks, requiring resources for clinical assessment, laboratory investigation and drug administration for 1-3 hours, depending on the regimen used. But oral therapy can potentially reduce resources used, especially if there is a low incidence of grade 2 toxicity requiring clinical intervention and grade 3 or 4 toxicity. This is especially important in the Canadian setting due to clinic and chemotherapy daycare space constraints.
PANDEMIC Considerations on safety on systemic therapy: With the ongoing issue with COVID, oral therapy will reduce the patient footprint in cancer centres, which can reduce the chance of outbreak and exposure.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Gaps in current treatment goals 1 and 2: Improvement in OS, PFS and ORR:
At this time, there is no randomized data of pralsetinib versus standard therapy in the RET fusion mNSCLC setting. We do however have the first interim data from the ARROW [Gainor et al. Lancet Oncol 2021: 22: 959-969] a phase 1/2 study that reported the safety and efficacy of pralsetinib in 114 evaluable RET-Fusion positive- NSCLC patients with locally advanced or metastatic disease, 87 with previous platinum based chemotherapy exposure (median 2 prior lines, 45% with previous ICI exposure), and 27 treatment naïve patients.
In the treatment naïve patients (n-27) who were not candidates for standard platinum therapies, ORR was 70% (CI: 50-86) (n=19), 11% of which were complete responses; mPFS was 9.1 months (CI: 6.1-13.0); mOS not reached with median follow up of 13.6 months.
In the 87 patients with previous platinum exposure, ORR was 61% (CI: 50-71) (n=53), 6% of which were complete responses; mPFS was 17.1 months (CI: 8.3-22.1); mOS was not reached with a median follow up of 17.1 months.
Given that no comparator arm was included in the ARROW study, the final analysis of Keynote 189 [Rodriguez et al. Annals of Oncology 2021: 32(7):881-895] study that reported safety and efficacy for pembrolizumab vs placebo plus pemetrexed/platinum in treatment naïve patients with advanced nonsquamous NSCLC irrespective of PD-L1 expression serves as a good comparator trial as this pembrolizumab containing regimen is the current publicly funded standard of care for treatment naïve RET-Fusion positive NSCLC.
In this large (n=661) trial the experimental arm ORR was 48.3% (CI 43-53) (n=410), 1.2% of which were complete responses; mPFS of 9.0 months (CI:8.1-10.4); mOS was 22.0 months (CI: 19.5-24.5).
With all the caveats of comparing a smaller phase I/2 trial (Arrow) to a randomized phase 3 trial (Keynote 189), the efficacy of pralsetinib is at least comparable in the treatment naïve, and previously treated populations.
Table 4: Gaps in Current Treatment Goals #3: Low Toxicity Profile
Outcome | ARROW | KN024 | KN189 | ||
|---|---|---|---|---|---|
Chemotherapy | Pembrolizumab | Chemotherapy | Chemotherapy + pembrolizumab | ||
Treatment-related grade 3-4 toxicity (%) | 48% | 53.3% | 26.6% | 42.1% | 52.1% |
Treatment-related discontinuation of therapy (%) | 6% | 10.7% | 7.1% | 10.9% | 29.4% |
All cause related death (%) | N/A | NA | NA | 5.9% | 6.7% |
Treatment-related death (%) | 0% | 2.0% | 1.3% | NA | NA |
Unique toxicities have been reported with pralsetinib which differ from other selective RET inhibitors: [Gainer et al. Lancet Oncol 2021: 22: 959-969]
Grade 3+4 Pneumonia and Pneumonitis occurred 10% and 2% respectively.
All the above toxicity will require frequent clinical, laboratory and radiographic monitoring. Clinician, pharmacist and ultimately patient education and communication of these unique toxicities, and in particular pneumonitis, will be necessary during clinical adoption, especially in those with previous ICI exposure.
As a comparison, the clinical adoption of immune-related toxicity from PD(L)1 alone or in combination with chemotherapy also required additional clinical, laboratory and imaging follow-up, until these toxicities were routinely assessed and managed. The same applies to hyperlipidemia with lorlatinib, and pneumonitis with EGFR inhibitors.
Patient education on drug related toxicity and outpatient monitoring and management protocols will reduce the probability of toxicity leading to dose interruption, dose reduction, dose termination and mortality and morbidity from any therapy as well as health care utilization.
Gaps in current treatment goals #4: Prevention or treatment of brain metastases:
Based on the longitudinal CNS metastases data by Lee et al.[Jpn J Clin Oncol 2020;5(5):584-601], >60% of patients with metastatic RET fusion NSCLC developed CNS disease after 24 months of follow-up. Indeed in ARROW, 41% of patient had a history of or active brain metastases.
Pralsetinib is an intracranially active selective RET inhibitor. Arrow had 9 patients with measurable metastases, 56% (CI: 21-86) (n=5) of which had intracranial responses, 3 of which were complete responses. The estimated duration of intracranial response was 53% at 12 months (CI: 5-100). [Gainer et al. Lancet Oncol 2021: 22: 959-969]
Four retrospective studies [Baerz et al. Lung Cancer 2010;68:264-268; Bailon et al. Neuro Oncol 2012;14(4):491-495; Yu et al. Medicine 2019;98(3):e14110; and Barlesi et al Ann Oncol 2011;22(11):2466-2470] reported the intracranial ORR of 40% (CI : 38.4-41 and median intracranial PFS of 7.4-9.5 months with pemetrexed-based therapy for those who have untreated or progressing brain metastases. Equren-Santamaria et al. [Clin Cancer Res 2020;26:4186-4192] performed a meta-analysis of PD(L)1-based therapy in unselected mNSCLC with either asymptomatic or progressing brain metastases, and reported an intracranial ORR of 0-27%. Specifically, the prospective study by Goldberg et al. [JCO 2018:35(15_Suppl:2009)] reported an intracranial ORR of almost 30% in the 34 highly selected patients with PDL-1 >1% mNSCLC who had CNS metastases that measured <2 cm, were asymptomatic, and who did not require steroid.
Gaps in current treatment goals #5: Resource utilization:
Pralsetinib is an orally administered agent that will utilize no chemotherapy daycare services, but will require routine clinical assessments for toxicity and response. It should be noted that platinum/pemetrexed, PD(L)1, and their combinations will require more clinical and laboratory evaluation, and can be extremely resource intensive in the event an Immune related adverse event occurs. Management of said IRAE can necessitate multi-day and multi-disciplinary inpatient management, resources that have been limited during the current pandemic.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Single agent pralsetinib is the second selective RET inhibitor to be granted NOC, and to be considered for reimbursement by CADTH (See: Selpercatinib Project number: PC0261-00).
Pralsetinib has evidence to support its use in RET fusion positive NSCLC in both the treatment naïve (including patients ineligible for other systemic therapies), and treatment (platinum and ICI) exposed patient population. Pralsetinib can be considered a standard of care option for newly diagnosed RET fusion mNSCLC. The most optimal second-line therapy has not been established and would likely include platinum/pemetrexed or platinum/pemetrexed. For subsequent therapy, docetaxel, and anti-PD(L)1 therapy for those who have not received such agents in prior lines of therapy can be considered.
There is currently no data to dictate if selective RET inhibitors should be used in the first line setting or subsequent lines of therapy. Drawing from extensive experience from other oncogene addicted mNSCLC, pralsetinib should be offered to patients in the first line setting. To date, all available systemic therapy for mNSCLC, including chemotherapy, anti-PD(L)1 therapeutics and their combinations have not demonstrated better outcome or toxicity profile.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Pralsetinib is a highly potent oral, selective RET inhibitor that has shown efficacy in RET fusion-positive advanced NSCLC. RET fusions can be detected by several techniques, NGS rapidly becoming the Canadian Standard) in tissue or in the plasma by circulating tumour DNA. It is currently unknown if other RET alterations (such as point mutations) would benefit from pralsetinib as this cohort of patients is still being studied for efficacy.
In the current submission, pralsetinib would be best suited for patient with advanced NSCLC harbouring a RET fusion, this population comprises 1-2% of all advanced or metastatic NSCLC.
Pralsetinib would also be indicated in patients with asymptomatic brain metastases and have an ECOG status of 0-2, with or without prior systemic therapies.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
In clinical practice, the parameters used to document clinically meaningful response can include:
Improvement of lung cancer-related symptoms, improvement in subjective QOL with or without radiological response.
Evidence of reduction of documented sites of known disease at baseline, this can either be done clinically or through imaging modalities.
Like other oral tyrosine kinase inhibitors, imaging by CT/MRI for response to pralsetinib can be performed every 3 months or when clinically indicated. known sites of primary and metastatic disease can be performed every 3 months. Based on the longitudinal CNS metastases data by Lee et al. [Jpn J Clin Oncol 2020;5(5):594-601],>60% of RET fusion mNSCLC patients developed CNS disease at 24 months, the implementation of brain imaging (preferably by MRI) at initiation of therapy and routinely thereafter.
What factors should be considered when deciding to discontinue treatment with the drug under review?
In clinical practice, pralsetinib should continue until one or more of the following conditions is/are encountered:
Intolerable toxicity despite multiple dose reductions.
Patient preference to discontinue pralsetinib.
Exacerbation of concurrent medical condition(s) that will jeopardize the safety of the patient if pralsetinib is continued.
Disease progression lacking clinical benefit to the patient.
Disease progression (Intracranially or Extracranially) that is not amenable to local therapies.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Treatment with pralsetinib can be delivered through both academic and community cancer settings, like other orally administered tyrosine kinase inhibitors.
Additional Information
N/A
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Shaqil Kassam
Position: Medical Oncologist, Southlake Regional Hospital
Date: April 1, 2022
Table 5: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 1
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Roche | X | — | — | — |
Merck | X | — | — | — |
BMS | X | — | — | — |
Takeda | X | — | — | — |
Novartis | X | — | — | — |
Ipsen | X | — | — | — |
Sanofi | X | — | — | — |
Pfizer | X | — | — | — |
Declaration for Clinician 2
Name: Dr. Ronald Burkes
Position: Medical Oncologist, Mount Sinai Health
Date: April 1, 2022
Table 6: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 2
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Geoffrey Liu
Position: Medical Oncologist, Princess Margaret Cancer Centre
Date: April 1, 2022
Table 7: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 3
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Takeda Canada | Advisory Board, Health Technology Assessment Submission Advice, Speaker’s Bureau, past 10 years | — | — | X | — |
Takeda Canada | (To institution, not individual) Observational Study funding, past 10 years | — | — | — | X |
Hoffman La Roche | Advisory Board, Health Technology Assessment Submission Advice, past 10 years | — | — | X | — |
Pfizer | Advisory Board, Health Technology Assessment Submission Advice, part 10 years | — | — | X | — |
AstraZeneca | Advisory Board, Health Technology Assessment Submission Advice, Speaker’s Bureau, past 10 years, | — | — | X | — |
AstraZeneca | (To institution, not individual) Observational Study funding, past 10 years | — | — | — | X |
Bristol Myers Squibb | Advisory Board | X | — | — | — |
Boehringer Ingerheim | (To institution, not individual) Observational Study funding, past 10 years | — | — | X | — |
Abbvie | Advisory Board, past 10 years | — | X | — | — |
Merck | Advisory Board, Health Technology Assessment Submission Advice, past 10 years | — | X | — | — |
EMD Serono | Speaker’s Bureau, past 10 years | X | — | — | — |
Novartis | Advisory Board,past 10 years | — | — | X | — |
Glaxo Smith Kline | Advisory Board, past 10 years | — | X | — | — |
Declaration for Clinician 4
Name: Dr Catherine Labbé
Position: Head of Respiratory Medicine Service, Université de Laval
Date: April 1, 2022
Table 8: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 4
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Amgen | X | — | — | — |
Astra Zeneca | — | X | — | — |
Brystol-Myers Squibb | X | — | — | — |
Jazz Pharmaceuticals | X | — | — | — |
LEO Pharma | X | — | — | — |
Merck | X | — | — | — |
Pfizer | X | — | — | — |
Roche | X | — | — | — |
Sanofi Genzyme | X | — | — | — |
Declaration for Clinician 5
Name: Dr. Kevin Jao
Position: Medical Oncologist, Hôpital Sacré-Cœur, Montreal
Date: April 1, 2022
Table 9: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 5
Bristol-Myers Squibb | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Bristol-Myers Squibb | Advisory Role | X | — | — | — |
Declaration for Clinician 6
Name: Dr Nicole Bouchard
Position: Respirologist, Sherbrooke University Hospital
Date: April 1, 2022
Table 10: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 6
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Astra Zeneca | Advisory Role/Conference | X | — | — | — |
Bristol-Myers Squibb | Advisory Role/Research | X | — | — | — |
Merck | Advisory Role/ Research/ Conference | X | — | — | — |
Bayer | Advisory Role | X | — | — | — |
Pfizer | Conference/Research | X | — | — | — |
Declaration for Clinician 7
Name: Dr. Quincy Chu
Position: Medical Oncologist, Cross Cancer Institute
Date: April 1, 2022
Table 11: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 7
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to 50,000 | In excess of $50,000 | |
Abbvie | X | — | — | — |
Amgen | X | — | — | — |
AnHeart | X | — | — | — |
Astellas | X | — | — | — |
Astra Zeneca | — | X | — | — |
BI | X | — | — | — |
BMS | X | — | — | — |
Eli Lilly | — | X | — | — |
Eisai | X | — | — | — |
J and J | X | — | — | |
Jazz | X | — | — | — |
Merck | X | — | — | — |
Novartis | X | — | — | — |
Pfizer | X | — | — | — |
Roche | X | — | — | — |
Sanofi | — | X | — | — |
Takeda | X | — | — | — |
Merck KgaA- DSMB | — | — | — | — |
Astra Zeneca-research funding | — | — | X | — |
Declaration for Clinician 8
Name: Dr. Paul Wheatley-Price
Position: Medical Oncologist, The Ottawa Hospital; Associate Professor, Department of Medicine, University of Ottawa
Date: April 1, 2022
Table 12: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 8
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Sanofi | X | — | — | — |
Astra Zeneca | X | — | — | — |
Jazz Pharmaceuticals | X | — | — | — |
Amgen | X | — | — | — |
Janssen | X | — | — | — |
Novartis | X | — | — | — |
Merck | X | — | — | — |
BMS | X | — | — | — |
Roche | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Bayer | X | — | — | — |
Novartis | X | — | — | — |
Declaration for Clinician 9
Name: Dr. Zhaolin Xu
Position: Pathologist, QEII Health Sciences Centre
Date: April 1, 2022
Table 13: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 9
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to 50,000 | In excess of $50,000 | |
AstraZeneca | X | — | — | — |
Declaration for Clinician 10
Name: Dr. Donna Maziak
Position: Thoracic Surgeon, The Ottawa Hospital
Date: April 1, 2022
Table 14: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 10
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
No COI | X | — | — | — |
Declaration for Clinician 11
Name: Dr. Rosalyn Juergens
Position: Chair, LCC Medical Advisory Committee; Medical Oncologist, Juravinski Cancer Center
Date: April 1, 2022
Table 15: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 11
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Bristol Myers Squibb | X | — | — | — |
Astra Zeneca | — | X | — | — |
Merck Sharp and Dohme | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 12
Name: Dr Mahmoud Abdelsalam
Position: Medical Oncologist, The Moncton Hospital
Date: April 1, 2022
Table 16: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 12
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to 50,000 | In excess of $50,000 | |
BMS | — | X | — | — |
Declaration for Clinician 13
Name: Dr Normand Blais
Position: Medical Oncologist, Hôpital Notre Dame du CHUM
Date: April 1, 2022
Table 17: Conflict of Interest Declaration for Lung Cancer Canada — Clinician 13
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Novartis | X | — | — | — |
Ontario Health Cancer Care Ontario Lung Cancer Drug Advisory Committee
About Ontario Health Cancer Care Ontario Lung Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
This input was jointly discussed via Drug Advisory Committee meeting and email.
Current Treatments
The current treatment paradigm for RET positive metastatic/incurable NSCLC is chemotherapy (typically carboplatin and pemetrexed) with pembrolizumab (either given in combination or in sequence depending on PD-L1 status), followed by docetaxel, followed by best supportive care and death.
Radiation treatments are used for palliation of symptoms. Palliative care is essential as well.
Treatment Goals
The main goal of therapies is to delay death (improve length of life), and improve quality of life/delay deterioration in quality of life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Even with chemotherapy and immunotherapy, the survival is still quite poor, with a median of ~ 6-9 months in stage IV NSCLC in the real world. Virtually every patient with RET positive NSCLC will die from RET positive NSCLC (>90%). These deaths are preceded by a decrease in quality of life in the majority of patients, with increasing cancer related symptoms. There is a need for agents that control disease that are well tolerated.
Which patients have the greatest unmet need for an intervention such as the drug under review?
All patients with stage IV or advanced RET positive NSCLC. This is a "niche" population, approximately 1% of NSCLC patients. There are no known modifiable risk factors for RET fusion positive NSCLC, and the population tends to have more non-smokers than a general lung cancer population and be younger.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The drug would be used in any line of therapy - depending on when the results are known of RET fusion. The mechanism of action would complement additional standard treatments such as chemotherapy and immunotherapy, and would be used prior to, between, or after the other therapies. IT will not replace a therapy,and will not be used concurrently with other therapies (other than at times radiation and palliative care). The drug is one of the first treatment approved that targets the oncogenic driver, and does address in part the underlying disease process. It is expected if known RET positive that most clinicians and patients would prefer this as first line therapy if available, and shift other treatments to second, third, and fourth line (with resultant decreasing numbers). However, the key is that is approved as any line.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
No, it would not. Clinicians and patients can make this decision, and this is one of the first line recommended treatment approaches by all major oncology bodies.
How would this drug affect the sequencing of therapies for the target condition?
After this drug, cisplatin/pemetrexed/pembrolizumab/docetaxel will all be options down the road. In addition, radiation and palliative care.
Use in subsequent lines if used in first line would be extremely rare, except for situations where it's discovered there was perhaps an error in dosing etc. Use in subsequent lines, if a RET inhibitor has not been used in the first line, in special situations such as delay in NGS sequencing results wherein alternative available choices (such as chemotherapy or immunotherapy or the combination) may need to start first line more imminently, or in the context of transition of practice during the introduction of a RET inhibitor into the list of available therapies for RET positive patients).
Which patients would be best suited for treatment with the drug under review?
Stage IV or advanced RET fusion positive NSCLC. Any line. Performance status of 0,1,2.
How would patients best suited for treatment with the drug under review be identified?
Next generation sequencing of tumor or circulating DNA/RNA.
Which patients would be least suitable for treatment with the drug under review?
Patients with ECOG PS 3 or greater.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Yes - fusion testing.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Clinical judgement and assessment (history/physical) #1. ESAS scores etc. #2, Imaging (CT's typically) #3. Trials often place undue weight on protocolized imaging changes at extremely frequent intervals, but in general the outcomes are aligned.
What would be considered a clinically meaningful response to treatment?
Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth)
Stable or responding radiographic disease (as this disease typically worsens/grows and becomes symptomatic and life threatening, radiographic changes are meaningful.
How often should treatment response be assessed?
Clinical assessments every 4 wks are typical initially, and then every 8 wks if stable. Imaging as needed depending on clinical picture, although some will image every 3 months initially and then move to longer intervals.
What factors should be considered when deciding to discontinue treatment?
Significant symptomatic progression (#1); Significant toxicity (#2); Radiographic changes (#3) - diffuse progression.
What settings are appropriate for treatment with the drug under review?
Prescribed from Oncologist in community or hospital outpatient clinic.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
N/A
Additional Information
N/A
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Andrew Robinson
Position: OH-CCO Lung DAC Member
Date: 30-03-2022
Table 18: Conflict of Interest Declaration for Ontario Health Cancer Care Ontario Lung Cancer Drug Advisory Committee — Clinician 1
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Sara Kuruvilla
Position: OH-CCO Lung DAC Member
Date: 30-03-2022
Table 19: Conflict of Interest Declaration for Ontario Health Cancer Care Ontario Lung Cancer Drug Advisory Committee — Clinician 2
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.