CADTH Reimbursement Review
Lurbinectedin (Zepzelca)
Sponsor: Jazz Pharmaceuticals Inc.
Therapeutic area: Metastatic small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AD
aggregated data
AE
adverse event
ALT
alanine aminotransferase
AST
asparagine aminotransferase
BIC
Bayesian information criterion
CAV
cyclophosphamide plus doxorubicin plus vincristine
CI
confidence interval
CNS
central nervous system
CrI
credible interval
CTFI
chemotherapy-free interval
DIC
deviance information criterion
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
ES
extensive stage
G-CSF
granulocyte colony-stimulating factor
HR
hazard ratio
HRQoL
health-related quality of life
IA
investigator assessment
IPD
individual patient data
IQR
interquartile range
ITC
indirect treatment comparison
KM
Kaplan-Meier
LCC
Lung Cancer Canada
LDH
lactate dehydrogenase
LHF
Lung Health Foundation
LS
limited stage
MAIC
matching-adjusted indirect comparison
NMA
network meta-analysis
OL
open label
OR
odds ratio
ORR
objective response rate
OS
overall survival
PFS
progression-free survival
PS
performance status
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
SAE
serious adverse event
SCA
synthetic control arm
SCLC
small cell lung cancer
SD
standard deviation
STC
simulated treatment comparison
TTNT
time to next treatment
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Lurbinectedin (Zepzelca) for injection, lyophilized powder, 4 mg/vial, IV infusion |
Indication | Treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-containing therapy |
Reimbursement request | As per indication |
Health Canada approval status | NOC/c |
Health Canada review pathway | Advance consideration under NOC/c |
NOC date | September 29, 2021 |
Sponsor | Jazz Pharmaceuticals, Inc. |
NOC/c = Notice of Compliance with conditions; SCLC = small cell lung cancer.
Source: Sponsor’s submission for lurbinectedin (Zepzelca).1
Introduction
Small cell lung cancer (SCLC) accounts for 10% to 15% of all lung cancers.2 SCLC is classically staged as limited-stage (LS) or extensive-stage (ES) disease; approximately two-thirds of patients present with metastatic ES disease at diagnosis and approximately one-quarter present with stage III LS disease.2,3 The initial symptoms of SCLC are nonspecific and include cough, chest pain, trouble breathing, wheezing, hoarseness, loss of appetite, weight loss, and fatigue.4 The physical, emotional, and social toll of an SCLC diagnosis negatively impacts patient health-related quality of life (HRQoL).5 For patients with metastatic ES disease, median overall survival (OS) is less than 1 year, and the 5-year survival rate is approximately 5%.6,7 The sponsor estimated that there would be 521 patients per year (as of 2022) receiving second-line or third-line therapy for SCLC (238 with platinum-resistant disease, defined as a chemotherapy-free interval [CTFI] shorter than 90 days) who would be eligible to receive lurbinectedin.
According to the clinical experts consulted by CADTH for this review, standard first-line systemic therapy for patients with LS (stage III or earlier) or ES (metastatic) SCLC is a platinum-containing drug (cisplatin or carboplatin) plus etoposide for 4 to 6 cycles.8 Since 2021, standard first-line therapy for patients with ES disease has included platinum doublet therapy plus durvalumab.9 Second-line treatment options in Canada for patients with ES or LS disease include rechallenge with platinum plus etoposide (if progression occurs after an interval of approximately 3 months from the last dose of first-line chemotherapy), topotecan, and cyclophosphamide plus doxorubicin and vincristine (CAV). Third-line treatment options include topotecan and CAV (if not used as second-line therapy), as well as irinotecan with or without a platinum-containing drug. According to the clinical experts consulted by CADTH for this review, response rates decrease as line of therapy advances, and many of the second-line and third-line treatment options are difficult to tolerate; therefore, there is a high need for additional treatment options. The clinical experts stated that the goal of treatment for stage III or metastatic SCLC is to prolong survival while maintaining HRQoL.
Lurbinectedin is an alkylating drug that is indicated for the treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-containing therapy. Lurbinectedin received advance consideration from Health Canada under a Notice of Compliance with conditions; these conditions were to conduct timely, well-designed studies to verify the clinical benefit of the drug; to provide appropriate educational materials; and to comply with any postmarket surveillance commitments and advertising, labelling, and distribution requirements placed on the drug. The drug is supplied as a 4 mg vial and administered at a dose of 3.2 mg/m2 by IV infusion over 60 minutes, repeated every 21 days.
The objective of this report was to perform a systematic review of the beneficial and harmful effects of lurbinectedin (3.2 mg/m2 by IV infusion over 60 minutes, repeated every 21 days) for the treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-containing therapy.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups provided input for this review: Lung Cancer Canada (LCC); and the Lung Health Foundation (LHF), previously known as the Ontario Lung Association. LCC conducted phone interviews with 2 patients in Canada with SCLC (1 with localized and 1 with metastatic disease) and environmental scans with 1 patient and 2 caregivers of patients in the US with metastatic SCLC in March 2022; all had experience with lurbinectedin. LHF conducted an online survey (2 respondents; no demographic or disease information collected) and phone interviews (3 patients in Canada with lung cancer; type and stage not reported) from September to December 2021 and collected input from 2 additional individuals (1 registered nurse and 1 certified respiratory educator); none had experience with lurbinectedin. Patients highlighted the nonspecific early symptoms of SCLC, which led to delays in diagnosis, as well as the physical (e.g., shortness of breath, cough, fatigue, pain), emotional, and social toll of an SCLC diagnosis. Patients acknowledged that although existing treatments for SCLC (e.g., surgery, radiation, chemotherapy, targeted therapy, immunotherapy) prolong survival and delay disease progression, the side effects of currently available second-line and third-line chemotherapies for metastatic SCLC (e.g., nausea, fatigue, weight, and hair loss) were sometimes severe and negatively impacted HRQoL, employment, and the ability to perform activities of daily living. Patients identified an unmet need for additional second-line treatment options for metastatic SCLC that can prolong survival, delay disease progression, manage cancer symptoms, and maintain HRQoL with minimal side effects. Patients emphasized that stopping or delaying disease progression was the most important factor when choosing treatments, and they were more receptive to the potential side effects of efficacious therapies. Patients who had experience with lurbinectedin felt that the drug had reduced or stabilized tumour size, delayed disease progression, helped them continue or resume activities of daily living, including employment, and led to more manageable side effects and a shorter recovery time compared with other SCLC therapies they had received.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Two clinical specialists with expertise in the diagnosis and management of stage III and metastatic SCLC provided input for this review. According to the clinical experts, patients with stage III or metastatic SCLC manifest rapid responses to first-line chemotherapy, but these are not sustained. Currently available second-line chemotherapy options (e.g., topotecan, CAV, irinotecan) have significant drawbacks, including toxicity and inconvenience (e.g., topotecan has a dosage regimen of 5 consecutive days of IV treatment every 3 weeks). The clinical experts stated that lurbinectedin would be used as second-line or third-line therapy for patients with stage III or metastatic SCLC after first-line platinum plus etoposide therapy and potential rechallenge; if progression following first-line therapy occurred after a relatively long interval (e.g., 6 to 12 months), many clinicians would rechallenge with platinum plus etoposide as a second-line option before using lurbinectedin. The clinical experts emphasized that all patients with ES SCLC need additional treatment options to prolong survival and maintain HRQoL. According to the clinical experts, the patient population best suited to treatment with lurbinectedin includes patients with ES SCLC who progress after treatment with platinum plus etoposide with or without durvalumab; patients with poor performance status (e.g., Eastern Cooperative Oncology Group [ECOG] performance status [PS] score of 3 or greater) or limited organ function are least suitable for treatment with lurbinectedin. According to the clinical experts, assessment of response to lurbinectedin would involve imaging scans (approximately every 3 months), clinical improvement, and laboratory markers. Clinically meaningful responses to treatment would be manifested by an improvement in symptoms and improvement or stabilization in HRQoL. The clinical experts stated that lurbinectedin should be discontinued when disease progression or unacceptable toxicities occur or by patient choice. The clinical experts stated that lurbinectedin would be administered in an outpatient setting and would be ordered by a medical oncologist.
Clinician Group Input
Two clinician groups, the LCC Medical Advisory Committee (10 medical oncologists, 2 respirologists, 1 thoracic surgeon, and 1 pathologist) and the Ontario Health-Cancer Care Ontario Lung Cancer Drug Advisory Committee (5 medical oncologists), provided input for this review. No major contrary views were presented. Clinician groups echoed the high unmet need for additional efficacious second-line treatment options for stage III and metastatic SCLC that have fewer side effects and are more convenient to administer. The clinician groups noted that some clinicians would perform imaging evaluations slightly more frequently than others (every 2 to 3 cycles or 6 to 9 weeks versus every 3 months), and that in addition to improvement or stabilization of symptoms and HRQoL, clinically meaningful responses to lurbinectedin would be manifested as tumour shrinkage observed on imaging scans. In addition, the clinician groups noted that it was not yet clear if re-treatment with platinum plus etoposide would be the preferred option for patients with platinum-sensitive disease who have treatment-free periods beyond some cut-off (e.g., 6 months).
Drug Program Input
The Provincial Advisory Group identified the following jurisdictional implementation issues: relevant comparators, considerations for initiation of therapy, considerations for prescribing of therapy, funding algorithm, care provision issues, and system and economic issues. The clinical experts consulted by CADTH for this review weighed evidence from the included study and other clinical considerations to provide responses to drug program implementation questions.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
One phase II, multicentre, OL, basket trial (study B-005)10 designed to evaluate the efficacy and safety of lurbinectedin in previously treated patients with advanced solid tumours provided evidence for this review; only data for the SCLC cohort (N = 105) are described in this report. The primary objective of the study was to assess the objective response rate (ORR) per investigator assessment (IA) of lurbinectedin in patients with advanced SCLC who had received 1 prior line of systemic therapy. Secondary objectives included ORR per independent review committee (IRC), duration of response (DOR) per IA and IRC, clinical benefit rate per IA and IRC, progression-free survival (PFS) per IA and IRC, and OS. Adult patients (aged 18 years and older) with SCLC who had received 1 previous line of systemic therapy for advanced disease and met the eligibility criteria were enrolled at 1 of 26 sites, primarily in Europe (predominantly Spain; no sites in Canada). Patients were treated with lurbinectedin (3.2 mg/m2 on day 1 of a 21-day treatment cycle) until disease progression or unacceptable toxicity.
Adult patients (aged 18 years and older) with SCLC, an ECOG PS score of 2 or lower, and measurable disease who had received 1 prior line of systemic therapy for advanced disease were eligible if they did not have central nervous system (CNS) involvement identified by CT or MRI, did not have serious comorbidities, and had not received chemotherapy in the previous 3 weeks. The median age at study entry was 60 years. Most patients (56.2%) had an ECOG PS score of 1, roughly one-third (36.2%) had an ECOG PS score of 0, and only 7.6% (8 patients) had an ECOG PS score of 2. Most patients (93.3%) had ES disease at study entry; only 2 patients (1.9%) had nonmetastatic disease at study entry. Nearly all patients (93.3%) had received 1 line of prior systemic therapy (platinum-containing drugs: 100.0%; etoposide: 99.0%); only 7.6% of patients had received prior immunotherapy. Based on their CTFIs, 42.9% of patients had platinum-resistant disease (CTFI < 90 days, including both refractory disease [CTFI < 30 days = 20.0%] and resistant disease [CTFI 30 to 89 days = 22.9%]), whereas 57.1% of patients had platinum-sensitive disease (CTFI ≥ 90 days, including both sensitive disease [CTFI 90 to 179 days = 38.1%] and very sensitive disease [CTFI ≥ 180 days = 19.0%]).
Lurbinectedin (3.2 mg/m2) was administered as a 1-hour IV infusion on day 1 of a 3-week treatment cycle. The dose was capped at a body surface area of 2.0 m2 (6.4 mg). Treatment was continued until disease progression (per IA), unacceptable toxicity, a treatment delay of 3 weeks or longer (except in the case of clear clinical benefit with sponsor’s approval), the requirement for more than 2 dose reductions, intercurrent illness of sufficient magnitude to preclude safe continuation of the study, a major protocol deviation that could affect the risk-to-benefit ratio for the participating patient, investigator decision, noncompliance with study requirements, or patient refusal. During the treatment period, tumour response was evaluated for all original sites of disease involvement at baseline, every 2 cycles until cycle 6, and then every 3 cycles thereafter.
Efficacy Results
Key efficacy results of study B-005 are summarized in Table 2.
Overall Survival
Median OS was 9.3 months overall (95% confidence interval [CI], 6.3 to 11.8 months). Median OS among patients with a CTFI shorter than 90 days and 90 days or longer was 5.0 months (95% CI, 4.1 to 6.3 months) and 11.9 months (95% CI, 9.7 to 16.2 months), respectively.
Progression-Free Survival
Median PFS per IA was 3.5 months overall (95% CI, 2.6 to 4.3 months). Median PFS per IA among patients with a CTFI shorter than 90 day and 90 days or longer was 2.6 months (95% CI, 1.3 to 3.9 months) and 4.6 months (95% CI, 2.8 to 6.5 months), respectively.
Median PFS per IRC was 3.5 months overall (95% CI, 2.6 to 4.2 months). Median PFS per IRC among patients with a CTFI shorter than 90 days and 90 days or longer was 1.4 months (95% CI, 1.3 to 3.5 months) and 4.3 months (95% CI, 3.0 to 6.3 months), respectively.
Objective Response Rate
The ORR per IA was 35.2% overall (95% CI, 26.2% to 45.2%). The ORR per IA among patients with a CTFI shorter than 90 days and 90 days or longer was 22.2% (95% CI, 11.2% to 37.1%) and 45.0% (95% CI, 32.1%, 58.4%), respectively.
The ORR per IRC was 30.5% overall (95% CI, 21.9%, 40.2%). The ORR per IRC among patients with a CTFI shorter than 90 days and 90 days or longer was 13.3% (95% CI, 5.1%, 26.8%) and 43.3% (95% CI, 30.6%, 56.8%), respectively.
Duration of Response
Median DOR per IA in patients who had a confirmed complete response or partial response as best overall response was 5.3 months overall (95% CI, 4.1 to 6.4 months). Median DOR per IA among patients with a CTFI shorter than 90 days and 90 days or longer was 4.7 months (95% CI, 2.6 to 5.6 months) and 6.2 months (95% CI, 3.5 to 7.3 months), respectively.
Median DOR per IRC in patients who had a confirmed complete response or partial response as best overall response was 5.1 months overall (95% CI, 4.9 to 6.4 months). Median DOR per IRC among patients with a CTFI shorter than 90 days and 90 days or longer was 4.8 months (95% CI, 2.4 to 5.3 months) and 5.3 months (95% CI, 4.9 to 7.0 months), respectively.
Harms Results
Key harms results of study B-005 are summarized in Table 2. Adverse events (AEs) occurred in most patients (98.1%), serious adverse events (SAEs) occurred in 32.4% of patients, AEs leading to dose reduction occurred 26.3% of patients, and withdrawal due to AEs occurred in 3.8% of patients. Sixty-six patients (62.9%) died during the study, all due to progressive disease. Among CADTH protocol-defined notable harms, the most common myelosuppression-associated AEs in study B-005 were anemia (95.2%), lymphopenia (85.7%), leukopenia (79.0%), neutropenia (71.4%), and thrombocytopenia (43.8%). Febrile neutropenia occurred in 4.8% of patients. The most common hepatotoxicity-associated AEs were alanine aminotransferase (ALT) increase (71.8%), gamma-glutamyl transferase increase (65.0%), asparagine aminotransferase (AST) increase (44.7%), and alkaline phosphatase increase (33.0%). Peripheral neuropathy and peripheral sensory neuropathy occurred in 2 patients (1.9%).
Table 2: Summary of Key Results From Study B-005 (Treated Patients)
Outcome | Overall N = 105 | CTFI < 90 days N = 45 | CTFI ≥ 90 days N = 60 |
|---|---|---|---|
OS (months) | |||
Events, n (%) | 66 (62.9) | 37 (82.2) | 29 (48.3) |
OS, median (95% CI)a | 9.3 (6.3 to 11.8) | 5.0 (4.1 to 6.3) | 11.9 (9.7 to 16.2) |
PFS (months) | |||
Investigator assessment | |||
Events, n (%) | 90 (85.7) | 41 (91.1) | 49 (81.7) |
PFS, median (95% CI)a | 3.5 (2.6 to 4.3) | 2.6 (1.3 to 3.9) | 4.6 (2.8 to 6.5) |
Independent review committee | |||
Events, n (%) | 81 (77.1) | 37 (82.2) | 44 (73.3) |
PFS, median (95% CI)a | 3.5 (2.6 to 4.2) | 1.4 (1.3 to 3.5) | 4.3 (3.0 to 6.3) |
ORR, % (95% CI) | |||
Investigator assessment | 35.2 (26.2 to 45.2) | 22.2 (11.2 to 37.1) | 45.0 (32.1 to 58.4) |
Independent review committee | 30.5 (21.9 to 40.2) | 13.3 (5.1 to 26.8) | 43.3 (30.6 to 56.8) |
DOR (months) | |||
Investigator assessment | |||
Responders, N | 37 | 10 | 27 |
Events, n (%) | 29 (78.4) | 9 (90.0) | 20 (74.1) |
DOR, median (95% CI)a | 5.3 (4.1 to 6.4) | 4.7 (2.6 to 5.6) | 6.2 (3.5 to 7.3) |
Independent review committee | |||
Responders, N | 32 | 6 | 26 |
Events, n (%) | 22 (66.8) | 5 (83.3) | 17 (65.4) |
DOR, median (95% CI)a | 5.1 (4.9 to 6.4) | 4.8 (2.4 to 5.3) | 5.3 (4.9 to 7.0) |
Harms, n (%) | |||
AEs | 103 (98.1) | NR | NR |
SAEs | 34 (32.4) | NR | NR |
AEs leading to dose reduction | 25 (26.3) | NR | NR |
WDAEs | 4 (3.8) | NR | NR |
Deaths | 66 (62.9) | NR | NR |
Notable harms, n (%) | |||
Myelosuppression | |||
Febrile neutropenia | 5 (4.8) | NR | NR |
Iron deficiency anemia | 1 (1.0) | NR | NR |
Anemia | 100 (95.2) | NR | NR |
Lymphopenia | 90 (85.7) | NR | NR |
Leukopenia | 83 (79.0) | NR | NR |
Neutropenia | 75 (71.4) | NR | NR |
Thrombocytopenia | 46 (43.8) | NR | NR |
Hepatotoxicity | |||
Hepatomegaly | 2 (1.9) | NR | NR |
Hepatic pain | 1 (1.0) | NR | NR |
ALT increase (n = 103) | 74 (71.8) | NR | NR |
GGT increase (n = 103) | 67 (65.0) | NR | NR |
AST increase (n = 103) | 46 (44.7) | NR | NR |
AP increase (n = 103) | 34 (33.0) | NR | NR |
Bilirubin increase (n = 103) | 10 (9.7) | NR | NR |
CPK increase (n = 103) | 7 (6.8) | NR | NR |
Peripheral neuropathy | |||
Neuropathy peripheral | 2 (1.9) | NR | NR |
Peripheral sensory neuropathy | 2 (1.9) | NR | NR |
AE = adverse event; ALT = alanine aminotransferase; AP = alkaline phosphatase; AST = asparagine aminotransferase; CI = confidence interval; CPK = creatine phosphokinase; CTFI = chemotherapy-free interval; DOR = duration of response; GGT = gamma-glutamyl transferase; IA = investigator assessment; IRC = independent review committee; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Notes: Treatment-emergent AEs reported in this table were defined as any untoward medical occurrence after administration of the first dose of study drug and within 30 days of the last dose of study drug. AEs were coded using MedDRA version 21.0 and graded according to NCI-CTCAE version 4.
For biochemical parameters that were measured in fewer than 105 patients, the denominator is indicated.
aFrom Kaplan-Meier analysis.
Source: B-005 Clinical Study Report.11
Critical Appraisal
The major limitations of study B-005 were its single-arm, noncomparative design and its relatively small size, with associated uncertainty in the estimation of effect sizes. Other potential internal validity concerns included potential for bias in outcome assessment (e.g., tumour response) because of the single-arm, OL study design and the descriptive nature of efficacy analyses and absence of formal statistical hypothesis testing (other than ORR per IA).
The clinical experts consulted by CADTH for this review considered the demographic and disease characteristics of patients in study B-005 to be broadly reflective of adult patients with advanced SCLC who have received prior platinum-based doublet therapy in Canada. However, the clinical experts felt that patients in study B-005 were slightly younger, in better health, and more treatment-seeking than the general population of SCLC patients. Patients with CNS involvement, who would be expected to have worse prognoses, were excluded from study B-005; however, patients with a short CTFI, who are excluded from many trials due to their poor prognoses, were included. Although the study assessed lurbinectedin in the second-line treatment setting, the clinical experts felt that the study results could be generalized to the third-line treatment setting as well. Stabilization of HRQoL and cancer symptoms, which were identified as important outcomes to patients and goals of treatment by the clinical experts consulted by CADTH for this review, were not assessed in study B-005.
Indirect Comparisons
Description of Studies
Three sponsor-submitted indirect treatment comparisons (ITCs) are included in this CADTH report. The first ITC study evaluated the treatment landscape and comparative efficacy of lurbinectedin in the treatment of patients in Alberta with advanced SCLC following exposure to platinum-based therapy, who were diagnosed with SCLC (any stage) and who initiated a post–platinum-based systemic therapy to build a synthetic control arm (SCA) to evaluate aggregate trial-level data for comparison to the phase II B-005 study.12
The second ITC was a simulated treatment comparison (STC) that facilitated the indirect comparison of lurbinectedin (using individual patient data from the B-005 trial) to topotecan IV (using aggregated data from the von Pawel et al. [2014]13 randomized controlled trial [RCT]) in patients with relapsed or refractory SCLC.14
The third ITC was a matching-adjusted indirect comparison (MAIC) created to be used with a network meta-analysis (NMA) to compare the efficacy and safety of lurbinectedin with competing interventions (including topotecan IV and carboplatin plus etoposide) among patients with SCLC receiving second-line treatment with respect to ORR, DOR, OS, and PFS, as well as hematological AEs of grade 3 and 4 (including anemia, thrombocytopenia, neutropenia, and febrile neutropenia).15
Efficacy Results
The first ITC, the SCA analysis, was descriptive in nature, but also provides a comparison of lurbinectedin (Study B-005) against the SCA stratified by CTFI and stage at initial diagnosis. The median OS in this adjusted population analysis of the SCA reached 5.8 months (95 CI, 5.1 to 6.9 months), and the median OS in the B-005 trial was 9.3 months (95% CI, 6.3 to 11.8). The unadjusted OS reached a median of 6.58 months (95% CI, 5.75 to 7.46 months). The SCA was subsequently updated to align more closely with the B-005 trial population by excluding patients who developed brain metastasis after diagnosis but before initiating post–platinum-based therapy. The median OS in this adjusted population analysis of the updated SCA reached 6.1 months (95% CI, 5.4 to 7.7 months) and the unadjusted OS reached a median of 6.7 months (95% CI, 6.0 to 7.7 months).
In the second ITC, the STC evaluated OS and PFS. Adjusted estimates showed the median OS was 10.0 months (95% CI, 8.5 to 11.6 months) and 7.8 months (95% CI, 6.6 to 8.5 months) in the lurbinectedin and topotecan trials, respectively, with a mean difference of 2 months (95% CI, 0.4 to 4.0 months). For PFS, adjusted estimates were obtained, with a median PFS of 3.4 months (95% CI, 3.0 to 3.9 months) and 3.5 months (95% CI, 2.9 to 4.2 months) in the lurbinectedin and topotecan trials, respectively, with a mean difference of –0.10 months (95% CI, –0.89 to 0.69 months).
The third ITC provided results of an MAIC and an NMA that evaluated OS, PFS, ORR, DOR, and harms.
For OS, from the MAIC evaluation, the hazard ratio (HR) for lurbinectedin versus carboplatin plus etoposide was 0.42 (95% CI, 0.27 to 0.65). In the base-case NMA for OS, lurbinectedin had an HR of 0.43 (95% credible interval [CrI], 0.26 to 0.70) against IV topotecan, and 0.42 (95% CrI, 0.30 to 0.58) against carboplatin plus etoposide.
█████ █ █████████ █ █ █ ██ █ ███ █ ███ █ ████████████ █ █████ █ ██████████ █ ███ █ ████████ █ █████████ █ ██ █ ███████ █ ███ █ █████ █ ██ █ █ █ ██ █ ████████████ █ ██ █ ██ █ █ █ █ █████ █ █ █ █████ █ █ █ █ █ ████ █ ██████ █ ███ █ █ █ ██████
For ORR, when assessing the MAIC base case, no evidence of difference was detected between lurbinectedin and carboplatin plus etoposide (███ █ ████ █ ███ █ █ ████ █ █ █ █ ██ █ ███ █ █ █ ████). In the NMA, no evidence of difference was detected between lurbinectedin and IV topotecan (odds ratio [OR] = 2.36; 95% CrI, 0.89 to 6.23) or between lurbinectedin and carboplatin plus etoposide (OR = 0.85; 95% CrI, 0.40 to 1.83).
███ █ ██ █ █████ █ ██ █ █ █ ██ █ ███ █ ████ █ █ █ ███████ █ ██████ █ ███████ █ ████████ █ ███████ █ ████ █ ████████████ █ ██ █ █ █████ █ ██ █ █ █ ███ █ █████ █ ███ █ ██ █ ███ █ █ █ ███ █ ███████ █ ████ █ ██ █ █████ █ ██ █ ██████████ █ ███ █ ████████ █ ██ █ █ █ ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ████████
Harms Results
Harms were only directly evaluated in the third ITC (MAIC and NMA).
Anemia of grade 3 or 4 in MAIC estimates had lower odds in the lurbinectedin arm compared with the carboplatin plus etoposide arm (█ █ █ ████ █ ██ █ ██ █ ███ █ █ █ ████). The NMA estimates also showed lower odds in the lurbinectedin arm compared with the carboplatin plus etoposide arm (OR = 0.22; 95% CrI, 0.08 to 0.61) and the IV topotecan arm (OR = 0.21; 95% CrI, 0.06, 0.74), with consistent results in the sensitivity analyses.
MAIC estimates for grade 3 or 4 thrombocytopenia also showed lower odds in the lurbinectedin arm than in the carboplatin plus etoposide arm (█ █ █ ████ █ ██ █ ██ █ ███ █ █ █ ████), and results were similar across sensitivity analyses. Similarly, in the NMA assessing thrombocytopenia of grade 3 or 4, the base-case analysis showed lower odds (OR = 0.23; 95% CrI, 0.08 to 0.69), and results were consistent with the sensitivity analyses.
However, in the MAIC, the odds of neutropenia of grade 3 or 4 (█ █ █ ████ █ ██ █ ██ █ ███ █ █ █ █████) was higher with lurbinectedin compared with carboplatin plus etoposide, but this effect was the opposite when lurbinectedin was compared with IV topotecan in the group of patients with any platinum sensitivity (sensitivity analysis) (█ █ █ ████ █ ██ █ ██ █ ███ █ █ █ ████) and when observing the patients with sensitive disease, involving a CTFI longer than 90 days (█ █ █ ████ █ ██ █ ██ █ ███ █ █ █ ████). In the NMA, the results were similar, with increased odds in the lurbinectedin arm compared with the carboplatin plus etoposide arm (OR = 7.05; 95% CrI, 3.09 to 16.11), but not in the lurbinectedin arm compared with the IV topotecan arm (OR = 1.19; 95% CrI, 0.45 to 3.17). The reason for these differences in neutropenia rates was deemed to be explained by differences in the requirements for prophylaxis with granulocyte colony-stimulating factor (G-CSF) across studies.
Critical Appraisal
The results from all ITCs have uncertainty due to imprecision in effect estimates, risk of confounding, and risk of bias in the body of evidence (e.g., violation of proportional hazards, intransitivity, poor overlap of covariates in the MAIC weighting process, use of observational data from the single-arm, nonrandomized trial connected through a MAIC to each NMA) with sparsity of the formed network used for the NMAs. The ORR and DOR were also uncertain, with no evidence of better ORR odds for lurbinectedin compared with carboplatin plus etoposide or to topotecan IV and incomplete evidence for evaluating the DOR. The maturity of data for evaluating long-term outcomes was also uncertain.
Generalizability issues arose because some arms included in the third ITC evaluated drugs not used in Canada (e.g., oral topotecan) and some variables considered important by clinical experts could not be included.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Conclusions
Evidence from study B-005 suggested that administration of lurbinectedin in patients with SCLC who received 1 prior line of platinum-containing chemotherapy resulted in objective responses in some patients that persisted for several months. In the absence of a control group, PFS and OS results could not be interpreted, and there was no direct evidence to inform the relative efficacy of lurbinectedin compared with other treatment options. Indirect evidence (3 sponsor-submitted ITCs) suggested that lurbinectedin treatment may result in improved OS and/or PFS compared with IV topotecan, and compared with carboplatin plus etoposide, albeit with a high risk of bias (due to unanchored comparisons and limited ability to adjust for variability in prognostic factors and treatment-effect modifiers) and a sparse dataset. In study B-005, the main toxicity of lurbinectedin, reversible myelosuppression, was considered acceptable and manageable with dose reductions and appropriate transfusion and growth-factor support. The indirect evidence also suggested that lurbinectedin was associated with lower frequencies of grade 3 or 4 anemia and thrombocytopenia compared with oral topotecan and carboplatin plus etoposide, again with high uncertainty. The indirect evidence was aligned with some outcomes identified as important to patients with SCLC, who are seeking additional second-line and third-line treatment options that prolong survival, delay disease progression, and maintain HRQoL and that have acceptable toxicity profiles.
Introduction
Disease Background
SCLC accounts for 10% to 15% of all lung cancers and is characterized by rapid growth, early dissemination, and high rates of acquired drug resistance.2 Smoking tobacco is the strongest risk factor for SCLC and contributes to most SCLC diagnoses.2 SCLC is classically staged as LS or ES disease; although there is no direct correspondence with tumour, node, and metastasis staging, in most patients, LS disease is stage III and ES disease is stage IV (metastatic).3 Approximately two-thirds of patients present with metastatic ES disease at diagnosis, approximately one-quarter present with stage III LS disease, and very few patients (less than 5%) present with stage 0 to II disease.2,3 The initial symptoms of SCLC are nonspecific and include cough, chest pain, trouble breathing, wheezing, hoarseness, loss of appetite, weight loss, and fatigue.4 Nevertheless, the physical, emotional, and social toll of a SCLC diagnosis negatively impacts patient HRQoL.5
Based on estimates of the population of Canada,16 the annual incidence of lung cancer (97 per 100,000 population),6 the proportion of SCLC among all lung cancers (12.1%),6,17 the proportions of patients diagnosed with LS (stage III) and ES (metastatic) disease (25% and 67%, respectively),6 the proportions of patients (any stage) receiving first-line platinum-containing chemotherapy (54.6%) and subsequent lines of systemic therapy (28.4%),1,12 the sponsor calculated that in 2022 there would be 420 patients receiving second-line therapy and 101 patients receiving third-line therapy in Canada, outside of Quebec, who would be eligible to receive lurbinectedin. In this calculation, it was assumed that SCLC incidence was equivalent to prevalence, as most patients discontinue treatment and/or die in the year after diagnosis. Approximately 80% of patients were estimated to have platinum-sensitive disease (CTFI ≥ 90 days), whereas approximately 20% were estimated to have platinum-resistant disease (CTFI < 90 days).10,12 For patients with metastatic ES stage, median OS is less than 1 year, and the 5-year survival rate is approximately 5%.6,7 Most patients with LS disease will relapse after potentially curative first-line therapy, with a median OS of approximately 2 years and a 5-year survival of approximately 25%.6,7
According to the clinical experts consulted by CADTH for this review, after the development of symptoms or an abnormal chest X-ray, patients are referred to a cancer centre, where the diagnosis of SCLC can be made by a team of specialists (pulmonologist, medical oncologist, radiation oncologist, pathologist, radiologist, and thoracic surgeon) based on biopsy and imaging findings.
Standards of Therapy
According to the clinical experts consulted by CADTH for this review, standard first-line systemic therapy for patients with LS (stage III or earlier) or ES (metastatic) SCLC is a platinum-containing drug (cisplatin or carboplatin) plus etoposide for 4 to 6 cycles.8 Patients with LS disease are treated with chemotherapy combined with thoracic radiation and prophylactic cranial irradiation as part of potentially curative first-line therapy. Until recently, thoracic radiation and prophylactic cranial irradiation were optionally given to patients with ES disease who had stable disease or better after chemotherapy. However, since 2021, standard first-line therapy for patients with ES disease and without contraindications has included durvalumab (added starting at the first or second chemotherapy cycle and continuing until progression);9 no thoracic radiation or prophylactic cranial irradiation is given in patients receiving immunotherapy.
The clinical experts consulted by CADTH for this review explained that patients with LS disease who relapse 3 months or more after potentially curative first-line therapy receive the standard first-line treatment in the metastatic setting (platinum doublet plus durvalumab), as well as other second-line options; those who relapse more rapidly would typically receive other, non–platinum-based, second-line options. Second-line treatment options available to patients with ES or LS disease in Canada include rechallenge with platinum plus etoposide (if progression occurs more than 3 months after the last dose of first-line chemotherapy), topotecan (used off-label in patients who progress in the 60 days after initiation of first-line therapy), and CAV. Third-line treatment options include topotecan and CAV (if not used as second-line therapy) and irinotecan with or without a platinum-containing drug. Oral etoposide may also be used in a small number of patients. According to the clinical experts consulted by CADTH for this review, ORRs and DOR decrease with as the line of therapy advances, and many of the second-line and third-line treatment options are difficult to tolerate; therefore, there is a high need for additional treatment options. The clinical experts stated that the goal of treatment for stage III or metastatic SCLC is to prolong survival while maintaining HRQoL.
Drug
Key characteristics of lurbinectedin are shown in Table 3. Lurbinectedin is an alkylating drug with a mechanism of action that involves binding to DNA, inhibition of transcription, and induction of apoptosis. The Health Canada–recommended dosage is 3.2 mg/m2 by IV infusion over 60 minutes, repeated every 21 days until disease progression or unacceptable toxicity. Lurbinectedin is indicated for the treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-containing therapy. The drug is not approved in Canada for other indications and has not been previously reviewed by CADTH. Lurbinectedin received advance consideration from Health Canada under a Notice of Compliance with conditions; these conditions were to conduct timely, well-designed studies to verify the clinical benefit of the drug, to provide appropriate educational material, and to comply with any postmarket surveillance commitments and advertising, labelling, and distribution requirements placed on the drug. The sponsor’s reimbursement request is aligned with the Health Canada indication.
Table 3: Key Characteristics of Chemotherapy Drugs for the Treatment of Patients With SCLC
Characteristic | Lurbinectedin | Cisplatin or carboplatin plus etoposide | Topotecan | CAV | Irinotecan with or without cisplatin or carboplatin |
|---|---|---|---|---|---|
Mechanism of action | Alkylating drug that binds to DNA, inhibits transcription, and results in apoptosis | Cisplatin and carboplatin: bifunctional alkylating drugs that cross-link DNA Etoposide: topoisomerase II inhibition | Topoisomerase II inhibition | Cyclophosphamide: bifunctional alkylating drug that cross-links DNA Doxorubicin: DNA synthesis inhibition Vincristine: microtubule inhibitor | Irinotecan: topoisomerase I inhibition Cisplatin and carboplatin: refer to third column |
Indicationa | Treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-containing therapy | Cisplatin: metastatic testicular cancer, metastatic ovarian cancer, advanced bladder cancer Carboplatin: advanced ovarian carcinoma Etoposide: SCLC, malignant lymphoma, NSCLC, testicular cancer | Metastatic ovarian cancer, SCLC | Cyclophosphamide: malignant lymphomas (various), multiple myeloma, leukemias (various), mycosis fungoides Doxorubicin: various neoplasms Vincristine: various neoplasms | Irinotecan: colon and rectal cancer Cisplatin and carboplatin: refer to third column |
Route of administration | IV | Cisplatin and carboplatin: IV Etoposide: IV or oral | IV | Cyclophosphamide: IV or oral Doxorubicin: IV or intravesical Vincristine: IV | Irinotecan: IV Cisplatin and carboplatin: refer to third column |
Recommended dose | 3.2 mg/m2 over 60 minutes every 21 days | Carboplatin: 400 mg/m2 every 4 weeksb Cisplatin: 50 mg/m2 to 75 mg/m2 every 3 to 4 weeks, or 15 mg/m2 to 20 mg/m2 daily for 5 days every 3 to 4 weeksc Etoposide IV: 50 mg/m2 to 100 mg/m2 daily for 5 days every 4 weeksd Etoposide oral: 100 mg/m2 to 200 mg/m2 daily for 5 days every 3 to 4 weekse | 1.5 mg/m2 daily for 5 days every 3 weeks | Cyclophosphamide IV: 10 mg/kg to 50 mg/kg (1.5 g/m2 to 1.8 g/m2) administered as 10 mg/kg to 20 mg/kg per day for 2 to 5 days (adult loading); 10 mg/kg to 15 mg/kg every 7 to 10 days, or 3 mg/kg to 5 mg/kg twice weekly (adult maintenance)f Cyclophosphamide oral: 1 mg/kg to 5 mg/kg per day Doxorubicin: 60 mg/m2 to 75 mg/m2 every 21 days or 20 mg/m2 weekly (IV)f Vincristine:1.4 mg/m2 once per week (adults)f | Irinotecan: 350 mg/m2 every 3 weeks or 125 mg/m2 once weekly for 4 weeks followed by a 2-week restg Cisplatin and carboplatin: refer to third column |
Serious adverse effects or safety issues | Myelosuppression, hepatotoxicity, peripheral neuropathy | Carboplatin: myelosuppression, peripheral neuropathy, hepatotoxicity, nephrotoxicity, cardiovascular toxicity, infusion reactions Cisplatin: Myelosuppression, infusion reactions, infections, neurotoxicity, nephrotoxicity, cardiovascular toxicity Etoposide: myelosuppression, cardiovascular toxicity, nausea and vomiting, alopecia, infusion reactions | Myelosuppression, neutropenic colitis, gastrointestinal perforation, interstitial lung disease | Cyclophosphamide: secondary malignancy, cardiac toxicity, QT prolongation and ventricular tachyarrhythmia, hepatotoxicity, myelosuppression, urotoxicity, nephrotoxicity, pulmonary toxicity, infusion reactions, drug-drug interactions, infection Doxorubicin: cardiomyopathy, decreased LVEF, congestive heart failure, secondary malignancies, tissue necrosis, myelosuppression, hepatotoxicity Vincristine: alopecia, neuromuscular changes including sensory impairment, paresthesia, neuropathic pain, motor difficulties, constipation | Irinotecan: severe early and late diarrhea; typhlitis, ulcerative and ischemic colitis, ileus, and intestinal perforation, myelosuppression, infections, thromboembolic events, hyperglycemia; hepatotoxicity, infusion reactions |
CAV = cyclophosphamide, doxorubicin, and vincristine; LVEF = left ventricular ejection fraction; NSCLC = non–small cell lung cancer; SCLC = small cell lung cancer.
aHealth Canada–approved indications.
bIn the SCLC protocol, carboplatin is dosed at an area under the concentration, taking renal function into account, on day 1 of a 21-day treatment cycle.
cIn the SCLC protocol, cisplatin is dosed at 25 mg/m2 per day on days 1 to 3 of a 21-day treatment cycle.
dIn the SCLC protocol, IV etoposide is dosed at 100 mg/m2 per day on days 1 to 3 of a 21-day treatment cycle.
eIn the SCLC protocol, oral etoposide is often dosed at 50 mg/m2 to 100 mg/m2 per day over 3 to 5 days of a 21-day week treatment cycle. However, other schedules, including more prolonged administration, are also used.
fIn the SCLC protocol, the CAV regimen is administered at doses of 1,000 mg/m2 cyclophosphamide, 50 mg/m2 doxorubicin, and 1.4 mg/m2 vincristine, all on day 1 of a 21-day cycle.
gIn the SCLC protocol, irinotecan is often dosed at 50 mg/m2 weekly on a 3-week or 4-week cycle, but other variations are also used.
Sources: Product monographs for lurbinectedin,18 carboplatin,19 cisplatin,20 etoposide,21 topotecan,22 cyclophosphamide,23 doxorubicin,24 vincristine,25 and irinotecan.26
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The original patient group submissions can be found at the end of this report.
Two patient groups provided input for this review: LCC and the LHF, previously known as the Ontario Lung Association. LCC conducted phone interviews with 2 patients in Canada with SCLC (1 with localized and 1 with metastatic disease) and environmental scans with 1 patient and 2 caregivers of patients with metastatic SCLC in the US in March 2022; all had experience with lurbinectedin. The LHF conducted an online survey (2 respondents; no demographic or disease information collected) and phone interviews (3 patients in Canada with lung cancer; type and stage not reported) from September to December 2021, and collected input from 2 additional individuals (1 registered nurse and 1 certified respiratory educator); none had experience with lurbinectedin. Patients highlighted the nonspecific early symptoms of SCLC that led to delays in diagnosis, as well as the physical (e.g., shortness of breath, cough, fatigue, pain), emotional, and social toll of an SCLC diagnosis. Patients acknowledged that although existing treatments for SCLC (e.g., surgery, radiation, chemotherapy, targeted therapy, immunotherapy) prolong survival and delay disease progression, the side effects of currently available second-line and third-line chemotherapies for metastatic SCLC (e.g., nausea, fatigue, weight, and hair loss) were sometimes severe and negatively impacted HRQoL, employment, and the ability to perform activities of daily living. Patients identified an unmet need for additional second-line treatment options for metastatic SCLC that can prolong survival, delay disease progression, manage cancer symptoms, and maintain HRQoL and that have minimal side effects. Patients emphasized that stopping or delaying disease progression was the most important factor in choosing treatments, and they were more receptive to potential side effects of efficacious therapies. Patients who had experience with lurbinectedin felt that the drug had reduced or stabilized tumour size, delayed disease progression, helped them continue or resume activities of daily living, including employment, and had more manageable side effects and a shorter recovery time compared with other SCLC therapies they had received.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of stage III and metastatic SCLC.
Unmet Needs
The clinical experts consulted by CADTH for this review stated that patients with stage III or metastatic SCLC generally manifest rapid responses to first-line chemotherapy (although these are not usually sustained) and that there are currently no biomarkers for response to immunotherapy. Brain and bone metastases are common in this population. Currently available second-line chemotherapy options (e.g., topotecan, CAV) have significant drawbacks, including toxicity and inconvenience (e.g., topotecan has a dosage regimen of 5 consecutive days of IV treatment every 3 weeks).
Place in Therapy
According to the clinical experts consulted by CADTH for this review, lurbinectedin would be used as second-line or third-line therapy for stage III or metastatic SCLC (after first-line platinum plus etoposide therapy and potential rechallenge). The clinical experts stated that if progression occurred a relatively long interval after first-line therapy (e.g., 6 to 12 months), many clinicians would rechallenge with platinum plus etoposide as a second-line option before using lurbinectedin.
Patient Population
The clinical experts consulted by CADTH for this review emphasized that all patients with ES SCLC need additional treatment options to prolong survival and maintain HRQoL. The patient population best suited to treatment with lurbinectedin would be patients with ES SCLC who progress after treatment with platinum plus etoposide with or without durvalumab. Such patients are followed by a medical oncologist and would be identified at the time of progression. Patients with a poor performance status (e.g., ECOG PS score of 3 or greater) or limited organ function would be least suitable for treatment with lurbinectedin. Apart from these factors, it is not currently possible to identify patients who would be most likely to respond to lurbinectedin.
Assessing Response to Treatment
According to the clinical experts consulted by CADTH for this review, assessment of response to lurbinectedin therapy would involve imaging scans (CT or MRI), clinical improvement, and laboratory markers (e.g., liver function tests, lactate dehydrogenase levels, carcinoembryonic antigen levels). Clinically meaningful responses to treatment would manifest as improvement in symptoms and improvement or stabilization of HRQoL. Response to treatment is assessed with clinical examinations, imaging scans, laboratory assessments, and evaluation of patient-reported outcomes. All of these except imaging would be assessed on the same schedule as treatment cycle length (3 weeks). Imaging would be assessed before each cycle via chest X-ray when appropriate and with imaging scans approximately every 3 months.
Discontinuing Treatment
The clinical experts stated that lurbinectedin should be discontinued in patients who progress according to Response Evaluation Criteria in Solid Tumours (RECIST) criteria, when unacceptable toxicities occur, or by patient choice.
Prescribing Conditions
The clinical experts stated that lurbinectedin would be given in an outpatient setting and would be ordered by a medical oncologist.
Additional Considerations
The clinical experts emphasized the unmet need for better second-line and third-line treatment options for patients with stage III or metastatic SCLC.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The original clinician group submissions can be found in the Stakeholder Input section at the end of this report.
Two clinician groups, the LCC Medical Advisory Committee (10 medical oncologists, 2 respirologists, 1 thoracic surgeon, and 1 pathologist) and the Ontario Health-Cancer Care Ontario Lung Cancer Drug Advisory Committee (5 medical oncologists), provided input for this review. No major contrary views were presented. Clinician groups echoed the unmet need for additional efficacious second-line treatment options for patients with stage III or metastatic SCLC that have fewer side effects and are convenient to administer. The clinician groups noted that some clinicians would perform imaging evaluations slightly more frequently than others (every 2 to 3 cycles [6 to 9 weeks] versus every 3 months) and that in addition to improvement or stabilization of symptoms and HRQoL, clinically meaningful responses to lurbinectedin would be manifested as tumour shrinkage observed on imaging scans. In addition, the clinician groups noted that it was not yet clear if re-treatment with platinum plus etoposide would be the preferred option for patients with platinum-sensitive disease who have treatment-free periods beyond some cut-off (e.g., 6 months).
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Implementation issues | Clinical experts’ response |
|---|---|
Relevant comparators | |
PAG noted that the B-005 trial was a single-arm, phase II, basket trial and no comparators were included. Relevant comparators depend on whether relapsed disease is considered platinum-sensitive (cisplatin plus etoposide, carboplatin plus etoposide) or platinum-resistant (topotecan, CAV, cisplatin plus irinotecan, carboplatin plus irinotecan). | For pERC consideration. |
Considerations for initiation of therapy | |
PAG noted that the B-005 trial included patients with platinum-sensitive and platinum-resistant or refractory disease. Patients had received 1 or 2 prior lines of therapy; 100% of patients had received platinum-containing drugs and 98% had received etoposide. According to the sponsor, the place in therapy for lurbinectedin would be in the second-line setting (patients with platinum-sensitive, platinum-resistant, and platinum-refractory disease) and in the third-line setting (patients who have received more than 1 prior systemic therapy for advanced SCLC, including at least 1 platinum-containing drug). 1. Is the place in therapy for lurbinectedin suggested by the sponsor appropriate in clinical practice? 2. Would lurbinectedin be used preferentially as second-line therapy for patients with platinum-sensitive disease instead of rechallenge with platinum plus etoposide? If so, could platinum plus etoposide rechallenge be used in later lines after lurbinectedin? | The clinical experts felt that the sponsor’s proposed place in therapy for lurbinectedin was appropriate. The clinical experts further explained that patients with LS or stage III disease and patients with ES or metastatic disease who progress during or after platinum doublet therapy (potentially curative in the former group) could be candidates for lurbinectedin. In both groups of patients, the CTFI would guide the selection of the most appropriate therapy; the longer the CTFI, the more likely clinicians would choose to rechallenge with platinum doublet. The clinical experts stated that for patients with LS or stage III SCLC who progress or relapse long after potentially curative platinum doublet therapy, it would not be appropriate to use lurbinectedin as first-line chemotherapy in the metastatic setting. According to the clinical experts, in the second-line setting, many clinicians would choose to rechallenge patients with platinum-sensitive tumours with platinum doublet therapy before using lurbinectedin, especially in patients with longer CTFIs. However, in some platinum-sensitive patients, other options (including lurbinectedin) may be more appropriate. The clinical experts emphasized that although the B-005 study was in the second-line setting, few patients are treated in the third line and beyond, and in these patients, the available treatment options are inconvenient, difficult to tolerate, and not effective in all patients. The clinical experts stated that lurbinectedin would be equally useful in the second-line and third-line settings. The experts noted that for patients with platinum-sensitive tumours, rechallenge with platinum plus etoposide after second-line lurbinectedin would be appropriate. |
The product monograph states that lurbinectedin should not be used in patients with serum albumin levels < 30 g/L. Lurbinectedin is highly protein bound (≥ 95%). 1. What is the clinical rationale for this recommendation regarding albumin level? 2. How common is hypoalbuminemia in this population? 3. Should patients with albumin levels < 30 g/L be excluded from lurbinectedin in clinical practice? | The clinical experts agreed that very high protein binding could potentially be related to pharmacokinetics or pharmacodynamics, biodistribution, and safety. However, the clinical experts felt that they did not have the expertise to answer this question conclusively. Although albumin levels are not routinely measured in patients with SCLC before they initiate treatment, the clinical experts speculated that many patients would have albumin levels of approximately 30 g/L, but few would have levels significantly below this threshold. The clinical experts emphasized that in practice, patients with albumin levels < 30 g/L would probably not be excluded from receiving lurbinectedin and that albumin levels would not be a part of clinical decision-making because lurbinectedin is a palliative drug with a relatively short treatment duration and clinicians could reduce doses to mitigate unacceptable toxicities. |
In the B-005 trial, only 8% of patients had received prior immunotherapy. The addition of durvalumab to first-line platinum plus etoposide was recently recommended for reimbursement, although funding is not yet available. This combination may become the new standard of care. 1. Should patients with prior immunotherapy in earlier lines be eligible to receive lurbinectedin? | The clinical experts explained that the addition of durvalumab to first-line platinum plus etoposide is a relatively recent development and that patients who received prior immunotherapy should be eligible to receive lurbinectedin. |
The B-005 trial excluded patients with known CNS involvement. 1. Should patients with CNS involvement be eligible for lurbinectedin? Is there clinical evidence to inform the efficacy and safety of lurbinectedin in this patient subpopulation? | The clinical experts stated that patients with known CNS involvement could be considered eligible for lurbinectedin, although they acknowledged that there is currently no clinical evidence to inform the efficacy and safety of lurbinectedin in this subpopulation. The clinical experts were unaware of any evidence regarding the CNS penetration of lurbinectedin. The experts emphasized that CNS involvement in patients with SCLC is dealt with by radiation, not chemotherapy. |
Considerations for prescribing of therapy | |
PAG noted that lurbinectedin dosing is 3.2 mg/m2 IV over 60 minutes every 21 days. Recommended dose reduction levels are 2.6 mg/m2 and 2 mg/m2. | For pERC consideration. |
Funding algorithm | |
PAG noted that lurbinectedin may change the places in therapy of comparator drugs and of drugs reimbursed in subsequent lines of therapy. | For pERC consideration. |
Care provision issues | |
PAG noted that lurbinectedin is supplied as a 4 mg vial. At 3.2 mg/m2, each dose will likely require more than 1 vial per preparation; therefore, drug wastage is anticipated. Vial sharing would only be possible if multiple patients were scheduled to be treated together at centres close to a hazardous sterile compounding pharmacy facility. The beyond-use date of lurbinectedin vials after reconstitution is 6 hours, and the final preparation must be used no more than 24 hours after compounding, per the product monograph. | For pERC consideration. |
PAG noted that lurbinectedin undergoes hepatic metabolism via CYP3A4 and, thus, there is the potential for drug-drug, drug-herb, and drug-food interactions that require assessment and potential intervention and monitoring. Additional pharmacy resources would be used to assess potential interactions. | For pERC consideration. |
System and economic issues | |
PAG noted that all relevant chemotherapy comparators have existing confidential negotiated prices in place. | For pERC consideration. |
CAV = cyclophosphamide plus doxorubicin and vincristine; CTFI = chemotherapy-free interval; CNS = central nervous system; CYP3A4 = cytochrome P450 family 3 subfamily A member 4; ES = extensive stage; LS = limited stage; PAG = Provincial Advisory Group; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee.
Clinical Evidence
The clinical evidence included in the review of lurbinectedin is presented in 2 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor. No other relevant evidence was identified for this review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of lurbinectedin (3.2 mg/m2 by IV infusion over 60 minutes, repeated every 21 days) for the treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-containing therapy.
Methods
Studies selected for inclusion in the Systematic Review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Adult patients (aged ≥ 18 years) with stage III or metastatic SCLC who have progressed on or after platinum-containing therapy. Subgroups:
|
Intervention | Lurbinectedin 3.2 mg/m2 by IV infusion over 60 minutes, repeated every 21 days |
Comparators |
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published and unpublished phase II, III, and IV RCTs |
AE = adverse event; CTFI = chemotherapy-free interval; DOR = duration of response; HRQoL = health-related quality of life; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event; SCLC = small cell lung cancer; WDAE = withdrawal due to adverse event.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.27
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974–) via Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Zepzelca and lurbinectedin. The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.27 No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.28 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
See Appendix 1 for the detailed search strategies. The initial search was completed on March 24, 2022. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Review Expert Review Committee (pERC) on July 13, 2022.
The searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
Three reports of a single study were identified from the literature10,29,30 and 1 report of the same study was identified from other sources11 for inclusion in the Systematic Review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Table 6: Details of the Included Study (B-005)
Detail | B-005 study |
|---|---|
Designs and populations | |
Study design | Phase II, OL, basket trial |
Locationsa | 26 sites in Belgium (n = 3 patients), France (n = 20), Italy (n = 2), Spain (n = 59), Switzerland (n = 7), the UK (n = 3), and the US (n = 11) |
Patient enrolment dates | October 21, 2015, to October 15, 2018b |
Data cut-off | January 15, 2019 |
Enrolled (N)a | 105 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | 3.2 mg/m2 lurbinectedin on day 1 of a 3-week treatment cycle (1-hour IV infusion)c |
Comparator(s) | None |
Duration | |
Phase | |
Screening | 4 weeks |
OL treatment | Until disease progression, unacceptable toxicity, treatment delay > 3 weeks (except in case of clear clinical benefit with sponsor’s approval), need for > 2 dose reductions, intercurrent illness of sufficient magnitude to preclude safe continuation of the study, major protocol deviation that may affect the risk-to-benefit ratio for the participating patient, investigator decision, noncompliance with study requirements, or patient refusal |
Follow-up | Until disease progression, start of new anticancer therapy, death, or data cut-off |
Outcomes | |
Primary end point | ORR per IA |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publications | Trigo et al. (2020)10 Subbiah et al. (2020)30 Fernández-Teruel et al. (2021)29 |
ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = asparagine aminotransferase; CNS = central nervous system; CPK = creatine phosphokinase; DBIL = direct bilirubin; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group performance status; IA = investigator assessment; IRC = independent review committee; NCI-CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; OL = open label; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PK = pharmacokinetics; RBC = red blood cell; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SCLC = small cell lung cancer; TBIL = total bilirubin; ULN = upper limit of normal.
Note: One additional report was included (B-005 Clinical Study Report).
aNumbers of patients reflect only those with SCLC.
bDate of first and last patient registrations.
cDose was capped at a body surface area of 2.0 m2 (6.4 mg).
Source: B-005 Clinical Study Report.11
Description of Studies
Study B-00510 was a phase II, single-arm, multicentre, OL, basket trial designed to evaluate the efficacy and safety of lurbinectedin in previously treated patients with advanced solid tumours. Only data for the SCLC cohort (N = 105) are described in this report. The primary objective of the study was to assess ORR by IA of lurbinectedin in patients with advanced SCLC who had received 1 prior line of systemic therapy. Secondary objectives included ORR per IRC, DOR per IA and IRC, clinical benefit rate per IA and IRC, PFS per IA and IRC, and OS.
Adult patients (aged 18 years and older) with SCLC who had received 1 previous line of systemic therapy for advanced disease and met the eligibility criteria were enrolled at 26 sites (primarily in Europe, predominantly Spain; no sites in Canada) from October 21, 2015, to October 15, 2018. Patients were treated with lurbinectedin (3.2 mg/m2 on day 1 of a 21-day treatment cycle) until disease progression or unacceptable toxicity. The data cut-off date was January 15, 2019, at which point the study was ongoing and patients were still being treated and followed. For SCLC patients, survival follow-up was every 6 months until death or study termination. The study was funded by PharmaMar, a Spanish pharmaceutical company that entered into an exclusive licensing agreement with the sponsor in 2019 for commercialization of lurbinectedin in North America.
Populations
Inclusion and Exclusion Criteria
Adult patients aged 18 years and older with SCLC, an ECOG PS score of 2 or less, and measurable disease who had received 1 prior line of systemic therapy for advanced disease were eligible if they had not been previously treated with lurbinectedin or trabectedin and did not have known CNS involvement identified on CT or MRI. In addition, patients with serious comorbidities (e.g., cardiac problems, breathing difficulties, infections, wounds) and patients who had received chemotherapy within 3 weeks of cycle 1 day 1 were excluded.
Baseline Characteristics
The baseline demographic characteristics of patients in the B-005 study are shown in Table 7. More than 3-quarters of patients were white, 60.0% were male, and the median age was 60 years. Most patients (56.2%) had an ECOG PS of 1, roughly 36.2% had an ECOG PS score of 0, and only 7.6% (8 patients) had an ECOG PS score of 2. Almost all patients (92.4%) were current or former smokers. Approximately one-third (30.5%) of patients were diagnosed with LS disease; the rest (69.5%) were diagnosed with ES disease. However, only 7 patients (6.7%) had LS disease at study entry and only 2 patients (1.9%) had nonmetastatic disease at study entry. Almost all patients (98.1%) had lung disease at baseline, most (81.9%) had lymph node involvement, and nearly half (41.0%) had liver involvement. Very few patients had received prior potentially curative or palliative surgery (1.0% each), whereas most had received external radiotherapy (71.4%) or prophylactic cranial irradiation (58.1%). Nearly all patients (93.3%) had received 1 line of prior systemic therapy (100.0% received platinum-containing drugs and 99.0% received etoposide), whereas only 7.6% of patients had received prior immunotherapy. Based on their CTFIs, 42.9% of patients had platinum-resistant disease (CTFI < 90 days, including both refractory disease [CTFI < 30 days = 20.0%] and resistant disease [CTFI 30 to 89 days = 22.9%]), whereas 57.1% of patients had platinum-sensitive disease (CTFI ≥ 90 days, including both sensitive disease [CTFI 90 to 179 days = 38.1%] and very sensitive disease [, CTFI ≥ 180 days = 19.0%]).
Table 7: Summary of Baseline Demographic and Disease Characteristics in Study B-005 (Treated Patients)
Characteristic | Study B-005 |
|---|---|
Sex, n (%) | |
Male | 63 (60.0) |
Female | 42 (40.0) |
Age (years) | |
Median (range) | 60 (40 to 83) |
18 to 40, n (%) | 2 (1.9) |
41 to 64, n (%) | 66 (62.9) |
≥ 65, n (%) | 37 (35.2) |
Race, n (%) | |
White | 79 (75.2) |
Othera | 24 (22.9) |
Black or African American | 1 (1.0) |
Asian | 1 (1.0) |
ECOG PS, n (%) | |
0 | 38 (36.2) |
1 | 59 (56.2) |
2 | 8 (7.6) |
Weight (kg), median (range) | 71.0 (46.0 to 138.3) |
Height (cm), median (range) | 167 (150 to 183) |
BSA (m2), median (range) | 1.8 (1.4 to 2.6) |
Albumin (g/dL), median (range) | 4.1 (3.1 to 5.1) |
LDH (× ULN) | |
Median (range)b | 0.9 (0.2 to 12.8) |
Abnormal (> 1 × ULN), n (%)b | 47 (45.2) |
Smoker status, n (%) | |
Former or current | 97 (92.4) |
Never | 8 (7.6) |
Stage at diagnosis, n (%) | |
Limited | 32 (30.5) |
Early | 3 (2.9) |
Locally advanced | 29 (27.6) |
Extended | 73 (69.5) |
Stage at study entry, n (%) | |
Limited | █ █████ |
Nonmetastatic | █ █████ |
Extended | █ █ ██████ |
Number of sites at baseline | |
Median (range) | 3 (1 to 6) |
< 3 sites, n (%) | 26 (24.8) |
≥ 3 sites, n (%) | 79 (75.2) |
Sites of disease at baseline, n (%) | |
Lung | 103 (98.1) |
Lymph nodes | 86 (81.9) |
Liver | 43 (41.0) |
Adrenal | 27 (25.7) |
Bone | 27 (25.7) |
Pleura | 21 (20.0) |
Peritoneum | 5 (4.8) |
Soft tissue | 4 (3.8) |
Kidney | 3 (2.9) |
Pancreas | 3 (2.9) |
Pericardial | 2 (1.9) |
CNSc | 1 (1.0) |
Sum of target lesions size, n (%) | |
> 50 mm | 74 (70.5) |
> 100 mm | 36 (34.3) |
Bulky disease (1 lesion > 50 mm) | 34 (32.4) |
History or current presence of CNS involvement, n (%)c | 4 (3.8) |
Paraneoplastic syndrome, n (%)d | 9 (8.6) |
Time from diagnosis to registration (months), median (range) | 8.2 (2.1 to 20.0) |
Prior surgery, n (%) | |
Curative | 1 (1.0) |
Palliative | 1 (1.0) |
Prior radiotherapy, n (%) | |
External (including IMRT) | 75 (71.4) |
IMRT | 14 (13.3) |
Prophylactic cranial irradiation, n (%) | 61 (58.1) |
Lines of medical anticancer therapy | |
1 line | 98 (93.3) |
2 linese | 7 (6.7) |
Prior anticancer drugs, n (%) | |
Platinum compounds | 105 (100.0) |
Etoposidef | 104 (99.0) |
Immunotherapyg | 8 (7.6) |
PARPih | 2 (1.9) |
Best response to last prior platinum, n (%) | |
Complete response | 9 (8.6) |
Partial response | 70 (66.7) |
Stable disease | 19 (18.1) |
Progressive disease | 4 (3.8) |
Unknown or not available | 3 (2.9) |
Time to progression from last prior therapy (months), median (range) | 6.5 (1.4 to 17.8) |
Time to progression from last prior platinum (months), median (range) | 6.9 (1.4 to 17.8) |
Time from last progressive disease before study entry (weeks), median (range) | 1.6 (0.0 to 10.0) |
Platinum resistance or sensitivity, n (%) | |
Resistant disease (CTFI < 90 days) | 45 (42.9) |
Sensitive disease (CTFI ≥ 90 days) | 60 (57.1) |
CTFI (months), median (range) | 3.5 (0.0 to 16.1) |
0 to 89 days, n (%) | 45 (42.9) |
< 30 days (refractory), n (%) | 21 (20.0) |
30 to 89 days (resistant), n (%) | 24 (22.9) |
≥ 90 days, n (%) | 60 (57.1) |
90 to 179 days (sensitive), n (%) | 40 (38.1) |
≥ 180 days (very sensitive), n (%) | 20 (19.0) |
BSA = body surface area; CNS = central nervous system; CTFI = chemotherapy-free interval; ECOG PS = Eastern Cooperative Oncology Group performance status; IMRT = intensity-modulated radiotherapy; LDH = lactate dehydrogenase; PARPi = poly(ADP-ribose) polymerase inhibitor; ULN = upper limit of normal.
aPatients recruited in France and Belgium did not have race information available due to ethical requirements in those countries.
bOne patient had no LDH data at baseline.
cOne patient with CNS metastases at baseline was treated (reported as a protocol deviation). The other 3 patients did not have CNS involvement at study entry.
dSyndrome of inappropriate antidiuretic hormone secretion (n = 6), Cushing’s syndrome (n = 2), and paraneoplastic syndrome not specified (n = 1).
eFive patients were treated with nivolumab, 1 patient was treated with carboplatin plus etoposide (rechallenge plus atezolizumab), and 1 patient was treated with an investigational drug.
fOne patient received platinum plus gemcitabine as first-line therapy.
gFive patients received nivolumab as second-line therapy, 2 patients received platinum and etoposide plus atezolizumab as first-line therapy, and 1 patient received platinum and etoposide plus atezolizumab as second-line therapy.
hTwo patients received carboplatin plus etoposide plus veliparib as first-line therapy.
Source: B-005 Clinical Study Report.11
Interventions
Lurbinectedin (3.2 mg/m2) was administered as a 1-hour IV infusion on day 1 of a 3-week treatment cycle. The dose was capped at a body surface area of 2.0 m2 (6.4 mg). Infusions were administered at hospitals and cancer centres in an outpatient setting. Treatment was continued until disease progression (per IA), unacceptable toxicity, a treatment delay of 3 weeks or longer (except in the case of clear clinical benefit with sponsor’s approval), the need for more than 2 dose reductions, intercurrent illness of sufficient magnitude to preclude safe continuation of the study, major protocol deviation that may affect the risk-to-benefit ratio for the participating patient, investigator decision, noncompliance with study requirements, or patient refusal.
All patients received antiemetic prophylaxis before each lurbinectedin infusion, including corticosteroids, serotonin antagonists, dexamethasone, and metoclopramide. Allowed medications included therapies for pre-existing and treatment-emergent medical conditions (including pain management), blood products and transfusions, bisphosphonates, secondary prophylaxis and/or symptomatic treatment for emesis, G-CSF for therapy and for secondary prophylaxis, erythropoietin, and anticoagulation therapy. Use of other antineoplastic therapies (except somatostatin analogues for neuroendocrine tumours), radiotherapy, other investigational drugs, aprepitant and related drugs, immunosuppressive therapies (other than corticosteroids for antiemetic prophylaxis and/or pain control), and primary G-CSF prophylaxis was forbidden.
To continue treatment, patients had to fulfill the following re-treatment criteria at each cycle: ECOG PS score of 2 or less, hemoglobin of 8 g/dL or greater, absolute neutrophil count of 1.5 × 109/L or greater, platelet count of 100 × 109/L or greater, AST and ALT of 3.0 × upper limit of normal (ULN) or lower, total bilirubin of 1.5 × ULN or lower or direct bilirubin of 1.0 × ULN, albumin of 3 g/dL or higher, serum creatinine of 1.5 × ULN or lower, or creatinine clearance of 30 mL/min or higher. In addition, active infections (including sepsis) and/or bleeding (any grade) had to be absent, and the following AEs must have resolved before re-treatment: grade 1 or lower creatinine phosphokinase elevation, grade 1 or lower nonhematological drug-related AEs (except isolated increased gamma-glutamyl transferase and/or alkaline phosphatase, grade 2 asthenia, constipation, alopecia, peripheral neuropathy, or nonoptimally treated nausea and/or vomiting). Patients who did not meet these requirements on day 1 of a cycle were reassessed at least every 2 to 3 days; after a maximum delay of 3 weeks, patients were withdrawn from the study. Dose reduction was implemented after recovery from the following AEs: grade 3 or higher treatment-related nonhematological toxicity (except grade 3 or 4 nausea and/or vomiting not optimally treated, grade 3 asthenia lasting 3 days or less, grade 3 diarrhea lasting 2 days or less or not optimally treated, grade 3 transient ALT and/or AST elevations that were rapidly reversible and did not lead to subsequent delays, and nonclinically relevant biochemical abnormalities), grade 4 thrombocytopenia, or grade 3 thrombocytopenia with grade 3 or 4 bleeding, grade 4 neutropenia, any grade of febrile neutropenia or neutropenia associated with infection and/or sepsis, and frequent or prolonged (longer than 1 week) dose delays due to any treatment-related AEs. Dose reduction occurred stepwise from 3.2 mg/m2 to 2.6 mg/m2 and then to 2.0 mg/m2; up to 2 dose reductions were allowed per patient, and dose was not re-escalated under any circumstances. Patients who experienced grade 3 or 4 hypersensitivity reactions were withdrawn from treatment. Patients who continued to experience treatment-related toxicity and/or frequent dose delays could continue receiving the study medication if objective clinical benefit was adequately documented by the investigator, and upon agreement with the sponsor.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized subsequently.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study B-005 outcome | Definition |
|---|---|---|
OS | Secondary | OS was defined as the period of time from the date of first infusion to the date of death or last contact in the case of patients lost to follow-up or alive at the data cut-off |
PFS per IA and IRC | Secondary | PFS was defined as the period of time from the date of first infusion to the date of disease progression by RECIST 1.1, death from any cause, or last tumour evaluation; PFS was censored at the date of first infusion for patients with no posttreatment tumour assessment, at the date of initiation of subsequent anticancer therapy before documented disease progression, or at the last available tumour assessment for patients who were progression-free |
ORR per IA | Primary | ORR was defined as the percentage of patients with a confirmed response (complete response or partial response) by RECIST 1.1 |
ORR per IRC | Secondary | |
DOR per IA and IRC | Secondary | DOR was defined as the time between the date of confirmed response (complete response or partial response) by RECIST 1.1. and the date when disease progression, recurrence, or death were documented; DOR was censored at the date of initiation of subsequent anticancer therapy before documented disease progression or recurrence, or at the last available tumour assessment for patients who were progression-free or recurrence-free |
DOR = duration of response; IA = investigator assessment; IRC = independent review committee; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: B-005 Clinical Study Report.10
OS, PFS, ORR, and DOR are standard and broadly accepted outcome measures in oncology trials for the treatment of SCLC (refer to Table 8 and Table 9 for outcome definitions and censoring rules). Involvement of all measurable sites of disease, as well as nonmeasurable sites of disease, was evaluated by CT or MRI at baseline (in the 28 days before cycle 1 day 1). During the treatment period, tumour response was evaluated for all original sites of disease involvement at baseline every 2 cycles until cycle 6, and then every 3 cycles thereafter. Imaging evaluations were repeated at the end-of-treatment visit, if not previously done, and if the reason for treatment discontinuation was not disease progression. The same imaging modality was used for each patient throughout the study. Anonymized copies of all scans were sent to the sponsor and subsequently for evaluation by an IRC. Whether or not the IRC was blinded to the nature of the study and intervention was not stated.
Tumour response was assessed using RECIST 1.1.31 Progressive disease was defined as a predefined increase in the sum of diameters of target lesions (at least 20%), taking as reference the smallest sum of diameters on study, in target lesions or new nontarget lesions; the sum must also have demonstrated an absolute increase of at least 5 mm. Partial response was defined as at least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters. Complete response was defined as the disappearance of all target lesions with a reduction of the short axis of any pathological lymph nodes to less than 10 mm. Stable disease was defined as neither sufficient shrinkage (compared to baseline) to qualify for partial response nor sufficient increase (taking as reference the smallest sum diameters while on study) to qualify for progressive disease. An initial indication of response per IA was confirmed 4 or more weeks later (also by IA).
Decisions to discontinue protocol therapy due to progressive disease were made by the investigator, based on local imaging scans and clinical evaluation to assess clinical deterioration in the absence of radiological evidence. Patients who discontinued treatment without documented progressive disease continued to have radiological assessments every 2 months for the first 6 months, and every 3 months thereafter, until progressive disease, start of new antitumour therapy, death, or the date of study termination. Following disease progression, patients were followed up for survival on the same schedule (in person or by phone call) until death.
Harms outcomes included treatment-emergent AEs, SAEs, AEs requiring dose reduction, withdrawals due to AEs, and deaths. AEs that began or worsened on or after the start of protocol therapy were captured until 30 days after the last dose of study drug. AEs were defined as any untoward medical occurrence and were coded according to the Medical Dictionary for Regulatory Activities (MedDRA) version 21.032 and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.33
Statistical Analysis
Statistical analyses in study B-005 are summarized in Table 9. The sample size for each tumour type (9 cohorts in total) was based on an adaptive design. Up to 25 evaluable patients for each tumour type were to be recruited to test the null hypothesis that 1% of patients or fewer with each tumour type achieve an objective response to lurbinectedin and the alternative hypothesis that at least 10% of patients achieve an objective response. The variance of the standardized test was based on the null hypothesis. The type I error associated with this 1-sided test was 0.025 and the type II error was 0.2 using a normal approximation (approximately 0.3 if using an exact binomial distribution); hence, the statistical power was 80% using a normal approximation (approximately 70% if using an exact binomial distribution). Under these assumptions, if the number of patients who achieve a confirmed objective response was 2 or more, this would allow rejection of the null hypothesis.
An interim analysis to reject H0 (nonbinding) or to reject H1 (futility) was planned after the recruitment of 15 evaluable patients in each cohort; if the number of responding patients was 2 or more at the interim analysis, then recruitment had to continue up to 25 evaluable patients. A gamma family boundary would be used to control for the type I error, the parameter to reject H0 was fixed as –1 and the parameter to reject H1 was fixed as 0. If none of the first 15 evaluable patients in a specific tumour cohort had a confirmed response, the alternative hypothesis would be rejected, according to boundaries and sample size assumptions, and recruitment would be stopped. If the number of responding patients was already 2 or more at the interim analysis, then H0 could be rejected and the study would have sufficient power to be stopped. In contrast, if there was 1 confirmed response, recruitment would be continued to up to 25 evaluable patients for the relevant tumour type.
In the SCLC and endometrial carcinoma cohorts, the analysis at 25 evaluable patients would serve as the second interim analysis to decide on continuation of recruitment. Two confirmed responses would be required to expand accrual up to 100 (SCLC) and 50 (endometrial carcinoma) evaluable patients. For the SCLC cohort, expanded recruitment was not preplanned and occurred as a result of 2 protocol amendments: the first, in July 2016, permitted enrolment of up to 50 patients based on results of the PM1183-A-003-10 study, which showed responses to lurbinectedin plus doxorubicin; and the second, in March 2017, permitted enrolment of up to 100 patients based on early results of study B-005 and to support the ATLANTIS trial. For the SCLC cohort, a type I or II error would be controlled for with a gamma family boundary (–1 to reject H0, 0 to reject H1). For the endometrial carcinoma cohort, a type I or II error would be controlled for with a gamma family boundary (–1 to reject H0, –3 to reject H1)
The sample size for the SCLC cohort of was to be increased to 100 evaluable patients if the success boundary (at least 2 confirmed responses) was reached in the first 25 evaluable patients. Type I and type II errors were to be controlled for with a gamma family boundary (–1 to reject H0, 0 to reject H1). These additional patients were to be recruited to test the null hypothesis that 15% of patients or less achieve objective responses to lurbinectedin and the alternative hypothesis that at least 30% of patients achieve objective responses. The variance of the standardized test was based on the null hypothesis. The type I error associated with this 1-sided test was 0.025 and the type II error was 0.051 (normal approximation; approximately 0.05 if exact binomial distribution); hence, statistical power was 95% (normal approximation; approximately 95% if exact binomial distribution). Under these assumptions, if the number of patients who achieve a confirmed objective response was at least 23, the null hypothesis could be rejected. With the sample size of 100 evaluable SCLC patients, the CIs for outcomes would be narrower, and their half-widths would be confined to ± 15%.
For the primary analysis of ORR per IA among all treated patients, the ORR and its exact 95% binomial CI were calculated. The same approach was used for the secondary outcome of ORR per IRC. PFS per IA and IRC, DOR per IA and IRC, and OS functions among all treated patients were estimated using the Kaplan-Meier product limit method; 2-sided 95% CIs for median PFS, DOR, and OS were obtained with log-log transformation. PFS and OS rates at fixed time points and their 95% CIs were derived from a Kaplan-Meier analysis. No hypothesis tests of secondary outcomes were conducted, and no multiplicity adjustment was performed.
No adjustment factors were included in the statistical analyses. Missing data were accounted for in the Kaplan-Meier analyses of OS, PFS, and DOR with censoring. For analyses of ORR, patients with unknown or missing response data were classified as nonresponders. A sensitivity analysis of ORR per IA was conducted among all evaluable patients. Subgroup analyses by CTFI (using a 90-day cut-off, as well as by refractory [CTFI < 30 days], resistant [CTFI 30 to 89 days], sensitive [CTFI 90 to 179 days], and very sensitive [CTFI ≥ 180 days] disease) were prespecified for all outcomes; however, hypothesis tests for deviation from threshold values were not conducted and multiplicity was not taken into account. In addition, prespecified subgroup analyses for efficacy by sex (male versus female), age (younger than 65 years versus 65 years and older), race (white versus other), number of prior lines of therapy (1 versus 2 or more), body surface area (1.8 m2 or less versus greater than 1.8 m2), ECOG PS (0 versus 1 versus 2), geographic area (Europe versus US), and alpha-1 acid glycoprotein level were performed for the primary efficacy end point (ORR by IA).
Table 9: Statistical Analysis of Efficacy End Points in Study B-005
End point | Statistical model | Sensitivity analyses | Handling of missing data |
|---|---|---|---|
OS (all treated patients) | Median OS and OS rates at prespecified time points (6 and 12 months), each with 2-sided 95% CIs, from KM methodology | None | Censoring |
PFS per IA and IRC (all treated patients) | Median PFS and PFS rates at prespecified time points (4 and 6 months), each with 2-sided 95% CIs, from KM methodology | None | Censoring |
ORR per IA and IRC (all treated patients) | ORR and its exact binomial 95% CI were calculated | All evaluable patients (ORR per IA) | Complete case analysis |
DOR per IA and IRC (all responding patients) | Median DOR with 2-sided 95% CI from KM methodology | None | Censoring |
CI = confidence interval; DOR = duration of response; IA = investigator assessment; IRC = independent review committee; KM = Kaplan-Meier; ORR = objective response rate; OS = overall survival; PFS = progression-free survival.
Source: B-005 Clinical Study Report.10
Analysis Populations
The set of all included patients refers to all patients recorded in the trial database, regardless of whether they received the study drug; this set was not used for any efficacy analyses. The set of all treated patients was defined as all included patients who received at least 1 partial or complete infusion of lurbinectedin.
The set of all evaluable patients (for efficacy) was defined as all included patients who had received at least 1 complete infusion of lurbinectedin and either had at least 1 postbaseline tumour assessment per RECIST 1.1 or were categorized as treatment failures. Patients who discontinued treatment because of treatment-related toxicity before tumour assessment was performed, patients who died early from malignant disease, and patients in whom treatment was withdrawn because of clinical progression and/or symptomatic deterioration with no tumour assessments were classified as treatment failures and considered nonevaluable for objective tumour response (not included in the denominator for evaluation of response); these patients were still considered evaluable for efficacy. Separate sets of evaluable patients were defined per IA and per IRC.
The set of all responding patients was defined as all evaluable patients who had a confirmed complete response or partial response as best overall response according to RECIST 1.1. Separate sets of responding patients were defined per IA and per IRC. All treated patients were considered evaluable for safety.
Results
Patient Disposition
Patient disposition in study B-005 is summarized in Table 10. In total, 110 patients were screened for participation in the study, of whom 105 (95.5%) were treated with lurbinectedin. The 5 patients who were not treated did not meet various eligibility criteria (e.g., ECOG PS, albumin levels, CNS involvement, dyspnea, and ALT levels). Most patients (89.5%) discontinued lurbinectedin during the study; the most common reason for treatment discontinuation was disease progression (80.0%). Four patients (3.8%) discontinued treatment due to investigator decision and 2 patients (1.9%) refused treatment. Only 2 patients (1.9%) discontinued treatment due to treatment-related AEs. Only 1 patient (0.9%) was lost to follow-up. In total, 104 (94.5%) of patients were considered evaluable for efficacy per IA and 98 (89.1%) were considered evaluable for efficacy per IRC.
Major protocol deviations (those that might affect the risk-to-benefit ratio of the clinical trial by study investigators) in study B-005 are summarized in Table 11. Fifteen patients (13.6%) had assessments not performed per protocol (primarily missing biochemical and/or coagulation tests required for lurbinectedin re-treatment), 10 patients (9.1%) had protocol deviations related to treatment noncompliance (primarily failure to reduce or delay doses in patients meeting protocol requirements), 9 patients (8.2%) had protocol deviations related to eligibility (primarily missing data on hematological and/or organ function and a washout period from prior monoclonal antibody therapy of less than 4 weeks), and 2 patients (1.8%) had protocol deviations related to issues with the informed-consent form.
Table 10: Patient Disposition in Study B-005
Patient disposition | Study B-005 |
|---|---|
Included, N | 110 |
Treated, n (%) | 105 (95.5) |
Discontinued treatment, n (%) | 94 (89.5) |
Reason for treatment discontinuation, n (%) | |
Progressive diseasea | 84 (80.0) |
Investigator decisionb | 4 (3.8) |
Death (disease-related) | 2 (1.9) |
Treatment-related AEc | 2 (1.9) |
Patient refusald | 2 (1.9) |
Status at data cut-off, n (%) | |
Deathe | █ █ ██████ |
Under follow-up for survival | █ █ ██████ |
Ongoing treatment | █ █ ██████ |
Patient refusald | █ █████ |
Lost to follow-upf | █ █████ |
Evaluable for efficacy per IA, n (%)g | 104 (94.5) |
Evaluable for efficacy per IRC, n (%)h | 98 (89.1) |
Evaluable for safety, n (%) | 105 (95.5) |
AE = adverse event; IA = investigator assessment; IRC = independent review committee.
aIncludes 2 patients with symptomatic deterioration or clinical disease progression (without radiological evidence of progressive disease).
bInvestigator decision was related to persistent asthenia not meeting preplanned criteria for treatment discontinuation (n = 2), clinical deterioration (n = 1), and not specified (n = 1).
cOne patient had grade 4 febrile neutropenia, grade 3 thrombocytopenia, and grade 3 anemia reported as SAEs, and 1 patient had worsening of peripheral neuropathy.
dOne patient withdrew consent and 1 patient refused treatment because of bone radiotherapy for pain control but allowed follow-up.
eAll deaths were due to disease progression.
fOne patient was lost to follow-up on February 1, 2016, with stable disease as best response to treatment and progressive disease at last assessment (January 15, 2016).
gTwo patients were treated but not evaluable due to early death, 2 patients were treated but did not have radiologically documented progressive disease (symptomatic deterioration or clinical progressive disease), and 1 patient was treated but refused to undergo disease measurement after having received 2 cycles of lurbinectedin.
hSeven patients were not evaluable by RECIST per IRC because of missing imaging data at baseline or during treatment.
Source: B-005 Clinical Study Report.10
Table 11: Major Protocol Deviations in Study B-005 (Included Patients)
Category | Study B-005 N = 110 |
|---|---|
Assessment not performed per protocol, n (%) | █ █ ██████ |
Treatment noncompliance, n (%) | █ █ █████ |
Eligibility, n (%) | █ █████ |
Informed-consent-form issue, n (%) | █ █████ |
Source: B-005 Clinical Study Report.10
Exposure to Study Treatments
Exposure to lurbinectedin in study B-005 is summarized in Table 12. The median number of cycles administered was 4 (range, 1 to 24 cycles), and the median time on treatment was 14.0 weeks (range, 1.1 to 85.0 weeks). The median relative dose intensity was 97.4% (range, 65.2% to 104.3%). Lurbinectedin was administered during clinic visits so adherence was not a relevant consideration.
For OS analyses, 66 patients (62.9%) died and 39 (37.1%) were censored; the median follow-up time was 17.1 months (95% CI, 8.9 to 22.5 months). For PFS analyses per IA, 90 patients (85.7%) experienced disease progression or death and 15 (14.3%) were censored; the median follow-up time was 14.5 months (95% CI, 14.5 months to not reached). For PFS analyses per IRC, 81 patients (77.1%) experienced disease progression or death and 24 (22.9%) were censored; the median follow-up time was 16.2 months (95% CI, 6.0 months to not reached).
Table 12: Treatment Exposure in Study B-005 (Treated Patients)
Parameter | Study B-005 N = 105 |
|---|---|
Total cycles administered | 618 |
Number of cycles administered per patient | |
Median (range) | 4 (1 to 24) |
1 cycle, n (%) | █ █ █████ |
2 cycles, n (%) | █ █ ██████ |
3 cycles, n (%) | █ █████ |
4 cycles, n (%) | █ █ ██████ |
5 cycles, n (%) | █ █████ |
≥ 6 cycles, n (%) | █ █ ██████ |
Time on treatment (weeks)a, median (range) | 14.0 (1.1 to 85.0) |
Cumulative dose (mg/m2), median (range) | ███ █ ████ █ █████ |
Dose intensity (mg/m2 per week)b, median (range) | 1.0 (0.7 to 1.1) |
Relative dose intensity (%)c, median (range) | 97.4 (65.2 to 104.3) |
aCalculated as date of last infusion plus 30 days, or date of death or subsequent therapy (whichever comes first) minus date of first infusion.
bCalculated as the cumulative dose divided by the number of weeks of treatment.
cCalculated as the ratio of absolute dose intensity divided by the intended dose intensity.
Source: B-005 Clinical Study Report.10
Concomitant transfusion and growth-factor support in study B-005 were as follows: 9 patients (8.6%) received transfusion support (red blood cells: 7.6%; platelets: 2.9%), 2 patients (1.9%) received erythropoietin, and 23 patients (21.9%) received G-CSF (secondary prophylaxis: 8.6%; therapeutic: 11.4%; both secondary prophylaxis and therapeutic: 1.9%).
Anticancer therapies received subsequent to study treatment discontinuation in study B-005 are summarized in Table 13. Forty-seven patients (44.8%) received subsequent systemic anticancer therapy and 20 patients (19.0%) received subsequent radiotherapy. The most common subsequent systemic drugs were carboplatin (15.2%), etoposide (14.3%), paclitaxel (12.4%), and topotecan (12.4%).
Table 13: Subsequent Therapies in Study B-005 (Treated Patients)
Therapy | Study B-005 N = 105 |
|---|---|
Type of therapy, n (%) | |
Medical | 47 (44.8) |
Radiotherapy | 20 (19.0) |
Surgery | 1 (1.0) |
Number of subsequent drugs in medical therapies, n (%) | |
Median (range) | 0 (0 to 7) |
0 | 58 (55.2) |
1 | 15 (14.3) |
2 | 12 (11.4) |
≥ 3 | 20 (19.0) |
Drugs used as subsequent medical therapy, n (%) | |
Carboplatin | 16 (15.2) |
Etoposide | 15 (14.3) |
Paclitaxel | 13 (12.4) |
Topotecan | 13 (12.4) |
Investigational druga | 11 (10.5) |
Gemcitabine | 7 (6.7) |
Irinotecan | 6 (5.7) |
Cyclophosphamide | 6 (5.7) |
Monoclonal antibodies | 5 (4.8) |
Doxorubicin | 5 (4.8) |
Vincristine | 4 (3.8) |
Ipilimumab | 3 (2.9) |
Lomustine | 2 (1.9) |
Cisplatin | 1 (1.0) |
Docetaxel | 1 (1.0) |
Epirubicin | 1 (1.0) |
Oxaliplatin | 1 (1.0) |
Vinorelbine | 1 (1.0) |
aThree patients received lurbinectedin as further treatment and 2 patients received rovalpituzumab tesirine. Seven patients received immunotherapy: 3 received ipilimumab and nivolumab, 1 received nivolumab monotherapy, 1 received nivolumab with chemotherapy, 1 received pembrolizumab monotherapy, and 1 received atezolizumab with chemotherapy.
Source: B-005 Clinical Study Report.10
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here.
Datasets Analyzed
Data for all patients were reviewed for outcome assessment per IA and per IRC. Of the 105 included patients, 104 were evaluable for efficacy per IA (1 patient withdrew consent before the first postbaseline tumour assessment). Of the 105 included patients, 98 were evaluable for efficacy per IRC (1 patient withdrew consent before the first postbaseline tumour assessment; 2 patients died during the first cycle before tumour assessments were done; 2 patients had symptomatic deterioration or clinical progressive disease during the first cycle before tumour assessments were done; and 2 patients did not have baseline imaging available for IRC review).
Of the 105 included patients, 100 were evaluable for tumour response per IA (1 patient withdrew consent before the first postbaseline tumour assessment; 2 patients died during the first cycle before tumour assessments were done; and 2 patients had symptomatic deterioration or clinical progressive disease during the first cycle before tumour assessments were done). Of the 105 included patients, 98 were evaluable for tumour response per IRC for the same reasons as for evaluability for efficacy.
Overall Survival
OS results among all treated patients in study B-005 are summarized in Table 14, Figure 2, and Figure 3. Median OS was 9.3 months overall (95% CI, 6.3 to 11.8 months). Median OS among patients with a CTFI shorter than 90 days and 90 days or longer was 5.0 months (95% CI, 4.1 to 6.3 months) and 11.9 months (95% CI, 9.7 to 16.2 months), respectively. Median OS among patients with a CTFI shorter than 30 days, 30 to 89 days, 90 to 179 days, and 180 days or longer was 4.7 months (95% CI, 1.6 to 6.3 months), 6.2 months (95% CI, 4.1 to 7.6 months), 11.8 months (95% CI, 7.8 to 15.8 months), and 16.2 months (95% CI, 9.6 months to not calculable), respectively.
Table 14: OS in Study B-005 (Treated Patients)
Parameter | Overall | CTFI < 90 days | CTFI ≥ 90 days | CTFI < 30 days | CTFI 30 to 89 days | CTFI 90 to 179 days | CTFI ≥ 180 days |
|---|---|---|---|---|---|---|---|
n | 105 | 45 | 60 | ██ | ██ | ██ | 20 |
Patients with events, n (%) | 66 (62.9) | 37 (82.2) | 29 (48.3) | █ █ ██████ | █ █ ██████ | █ █ ██████ | 9 (45.0) |
Patients censored, n (%) | 39 (37.1) | 8 (17.8) | 31 (51.7) | █ ██████ | █ ██████ | █ █ ██████ | 11 (55.0) |
Median OS (95% CI), monthsa | 9.3 (6.3 to 11.8) | 5.0 (4.1 to 6.3) | 11.9 (9.7 to 16.2) | █ █ ██████ | █ █ ██████ | █ █ ██████ | 16.2 (9.6 to NC) |
OS at 6 months, % (95% CI)a | 67.1 (57.6 to 76.7) | 45.8 (30.4 to 61.3) | 83.6 (73.7 to 93.5) | █ █ ██████ | █ █ ██████ | █ █ ██████ | 89.7 (76.2 to 100.0) |
OS at 12 months, % (95% CI)a | 34.2 (23.2 to 45.1) | 15.9 (3.6 to 28.2) | 48.3 (32.5 to 64.1) | █ █ ██████ | █ █ ██████ | █ █ ██████ | 60.9 (35.7 to 86.2) |
CI = confidence interval; CTFI = chemotherapy-free interval; NC = not calculable; OS = overall survival.
aFrom Kaplan-Meier analysis.
Source: B-005 Clinical Study Report.10
Figure 2: Kaplan-Meier Plot of OS in Study B-005 (Treated Patients)
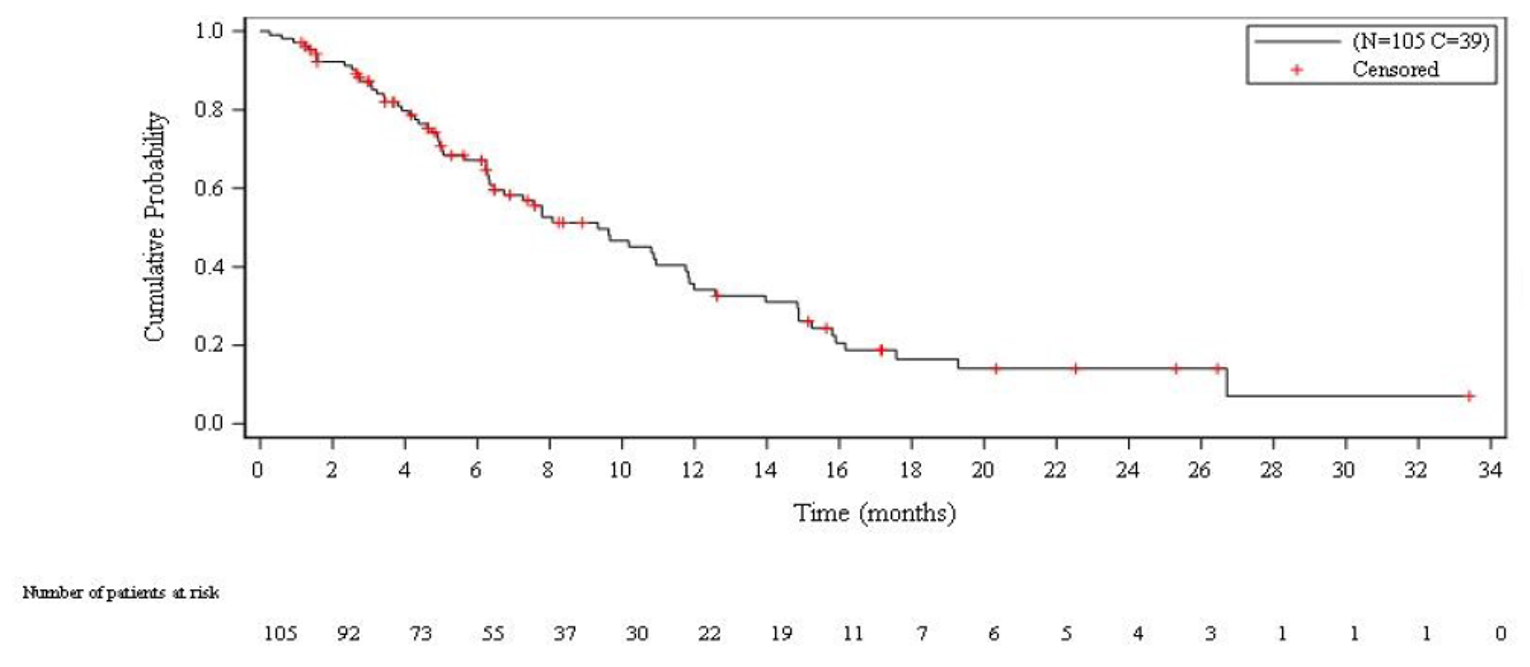
OS = overall survival.
Source: B-005 Clinical Study Report.10
Figure 3: Kaplan-Meier Plot of OS in Study B-005 (Treated Patients With a CTFI < 90 Days or ≥ 90 Days)
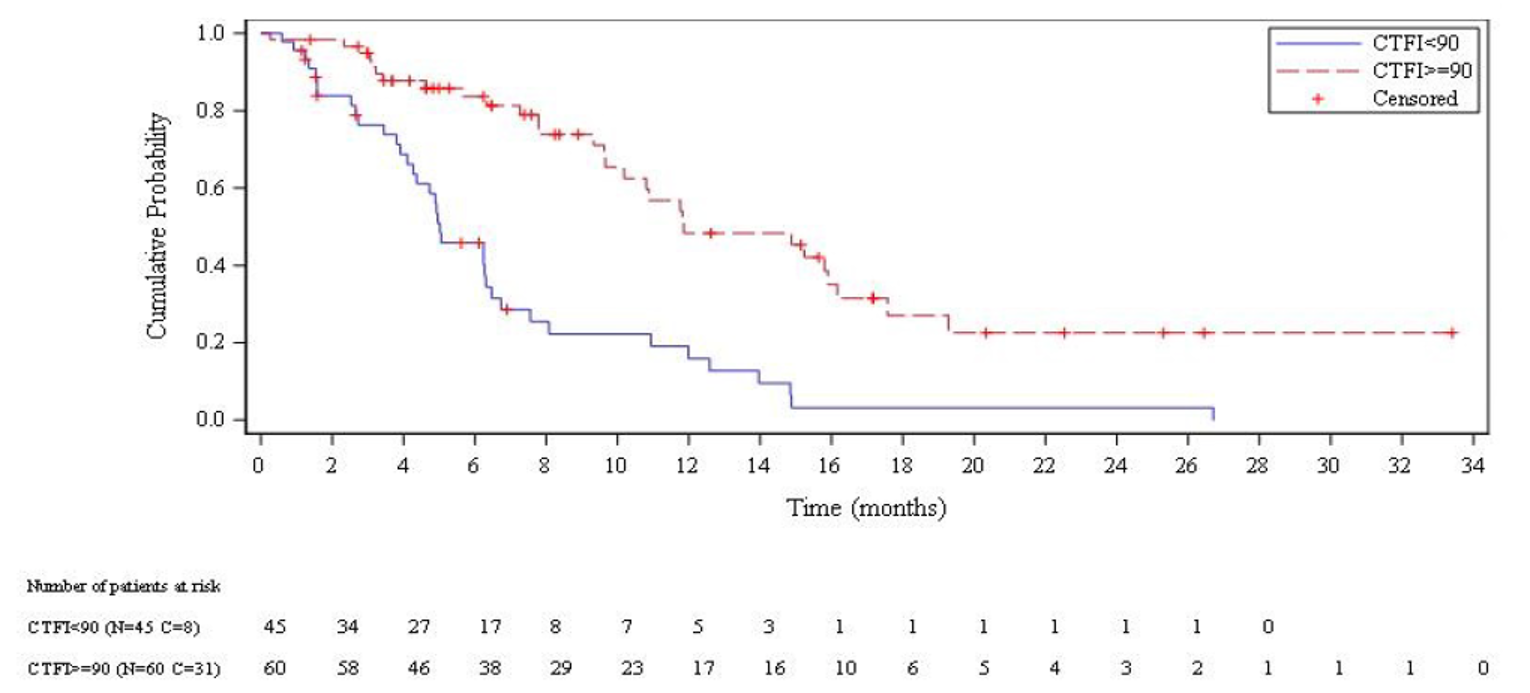
CTFI = chemotherapy-free interval; OS = overall survival.
Source: B-005 Clinical Study Report.10
Health-Related Quality of Life
HRQoL was not assessed in study B-005.
Progression-Free Survival
PFS results per IA and IRC among all treated patients in study B-005 are summarized in Table 15, Figure 4, and Figure 5. Median PFS per IRC was 3.5 months overall ███ █ ██ █ ██ █ █ █ ██ █ ███████. Median PFS per IRC among patients with CTFI less than 90 days and 90 or more days was ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████ and ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████, respectively. Median PFS per IRC among patients with CTFI less than 30 days, 30 to 89 days, 90 to 179 days, and 180 or more days was ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████ █ ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████ █ ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████ █ ██ █ ██ █ █████ █ ███ █ ██ █ ██ █ █████ █ █ █ ██ █ ████████ respectively.
Table 15: PFS in Study B-005 (Treated Patients)
Parameter | Overall | CTFI < 90 days | CTFI ≥ 90 days | CTFI < 30 days | CTFI 30 to 89 days | CTFI 90 to 179 days | CTFI ≥ 180 days |
|---|---|---|---|---|---|---|---|
n | 105 | 45 | 60 | ██ | ██ | ██ | 20 |
Patients with events per IA, n (%) | 90 (85.7) | 41 (91.1) | 49 (81.7) | █ █ ███████ | █ █ ███████ | █ █ ███████ | 18 (90.0) |
███████ █ ███ █ █████ █ ██ █ ███ █ █ ███ | █ █ ██████ | █ █ ██████ | █ █ ██████ | █ █ ██████ | █ █ ██████ | █ █ ██████ | █ █ ██████ |
Patients censored per IA, n (%) | 15 (14.3) | 4 (8.9) | 11 (18.3) | █ ███████ | █ ██████ | █ ███████ | 2 (10.0) |
Patients censored per IRC, n (%) | 24 (22.9) | 8 (17.8) | 16 (26.7) | █ ██████ | █ ██████ | █ █ ██████ | 4 (20.0) |
Median PFS (95% CI) per IA, monthsa | 3.5 (2.6 to 4.3) | 2.6 (1.3 to 3.9) | 4.6 (2.8 to 6.5) | █ █ ██ █ ████ | ██ █ ████ █ █████ | ██ █ ████ █ ████ | 4.6 (2.6 to 7.3) |
█████ █ ██ █ ███ █ ██ █ ██ █ ███ █ ███████ | ██ █ ██ █ ████ | ██ █ ██ █ ████ | ██ █ ██ █ ████ | ██ █ ████ █ ████ | ██ █ ████ █ ████ | ██ █ ████ █ ███ | █ █ █ █ ███ |
PFS at 4 months per IA, % (95% CI)a | 46.6 (36.7 to 56.5) | 29.1 (15.3 to 42.8) | 59.9 (47.1 to 72.7) | ███ █ ██ █ ██████ | ███ █ ██ █ █████ | ███ █ ██ █ ████ | █ █ █ █ ███ |
██ █ █ █ █ █████ █ ██ █ ███ █ █ ███ █ ████ | ██ █ ██ █ ███ | ██ █ ██ █ ███ | ██ █ ██ █ ███ | ███ █ ███ █ █████ | █ █ █ █ ███ | █ █ █ █ ███ | █ █ ██ █ ███ |
PFS at 6 months per IA, % (95% CI)a | 32.9 (23.3 to 42.5) | 18.8 (6.8 to 30.9) | 43.5 (30.1 to 56.9) | ██ █ ██ █ █████ | ██ █ ██ █ ████ | ██ █ ██ █ ████ | ██ █ █ █ ███ |
██ █ █ █ █ █████ █ ██ █ ███ █ █ ███ █ ████ | ██ █ ██ █ ████ | ██ █ ██ █ ████ | ██ █ ██ █ ███ | ██ █ ███ █ ████ | ██ █ ██ █ ███ | ██ █ ██ █ ████ | ██ █ █ █ ███ |
CI = confidence interval; CTFI = chemotherapy-free interval; IA = investigator assessment; IRC = independent review committee; PFS = progression-free survival.
aFrom Kaplan-Meier analysis.
Source: B-005 Clinical Study Report.10
Figure 4: Kaplan-Meier Plot of PFS in Study B-005 (Treated Patients)
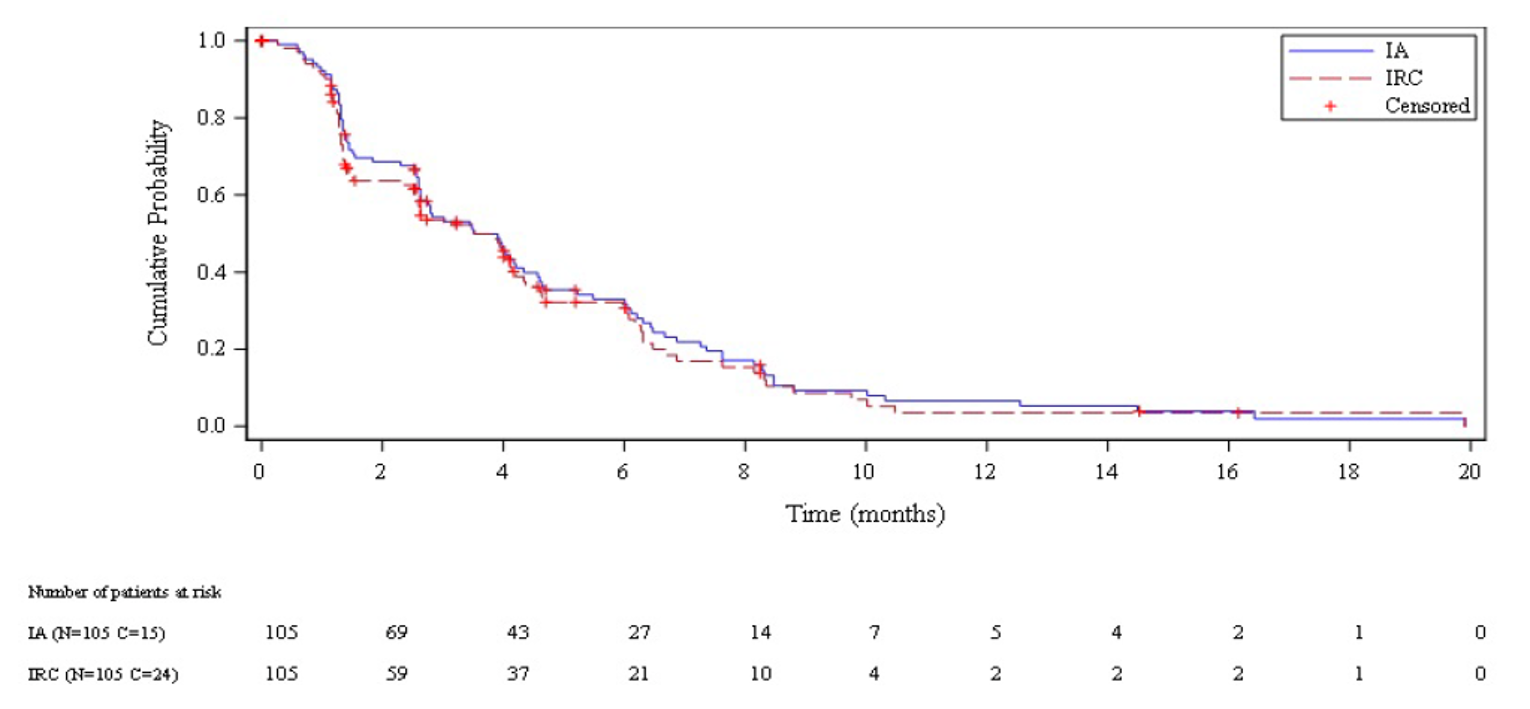
IA = investigator assessment; IRC = independent review committee; PFS = progression-free survival.
Source: B-005 Clinical Study Report.10
Figure 5: Kaplan-Meier Plot of PFS per IRC in Study B-005 (Treated Patients With a CTFI < 90 Days or ≥ 90 Days)
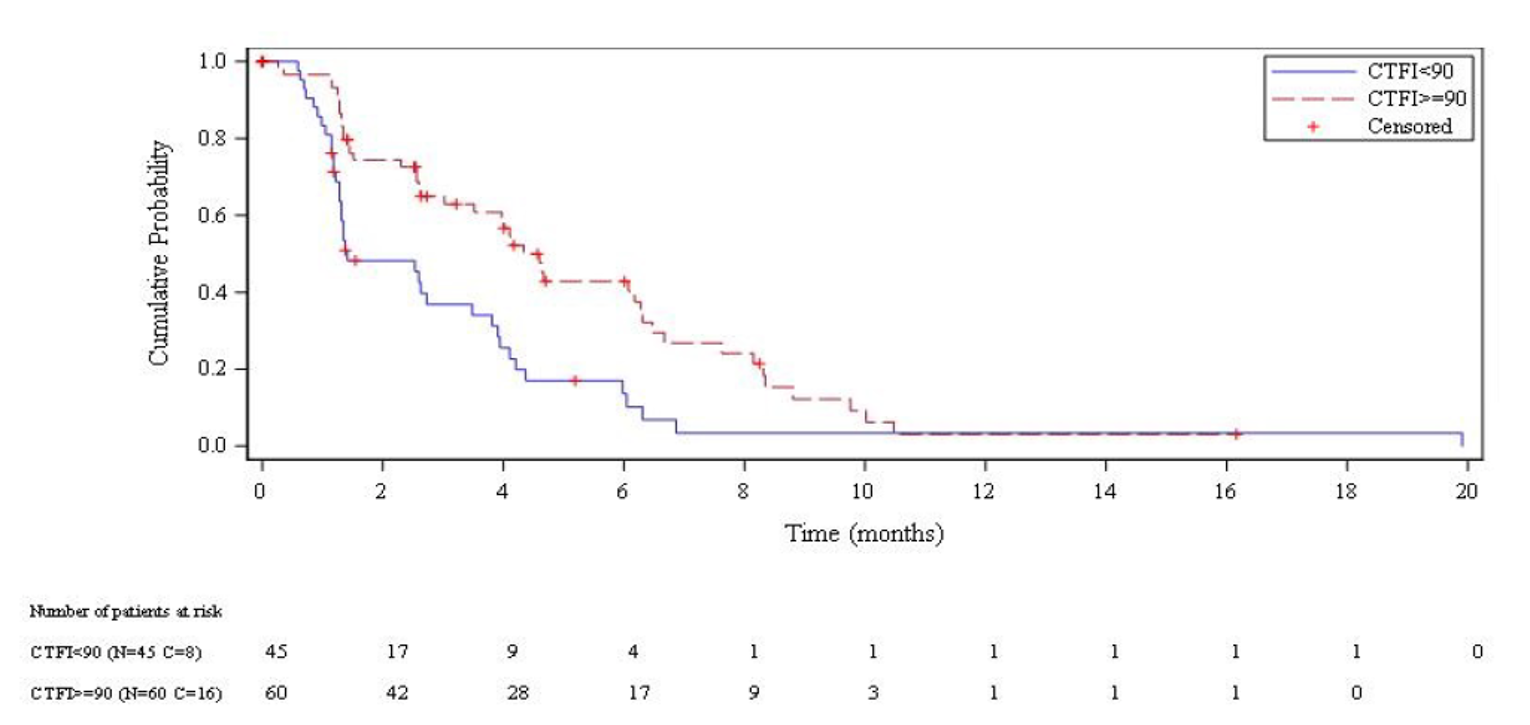
CTFI = chemotherapy-free interval; IRC = independent review committee; PFS = progression-free survival.
Source: B-005 Clinical Study Report.10
Objective Response Rate
ORR results per IA and IRC among all treated patients in study B-005 are summarized in Table 16. The ORR per IA was 35.2% overall (95% CI, 26.2% to 45.2%). The null hypothesis that no more than 15% of patients achieved objective responses to lurbinectedin was rejected, as the number of patients with objective responses exceeded the minimum number needed (23 of 100 patients). The ORR per IA among patients with a CTFI shorter than 90 days and 90 or more days was 22.2% (95% CI, 11.2% to 37.1%) and 45.0% (95% CI, 32.1%, 58.4%), respectively. The ORR per IA among patients with a CTFI shorter than 30 days, 30 to 89 days, 90 to 179 days, and 180 or more days was ████ █ ███ █ ██ █ ███ █ █ █ ██████ █ ████ █ ███ █ ██ █ ████ █ █ █ ██████ █ ████ █ ███ █ ██ █ ████ █ █ █ ███████ and 60.0% (95% CI, 36.1%, 80.9%), respectively. All confirmed responses were partial responses. A sensitivity analysis of ORR per IA among all evaluable patients showed results similar to the primary analysis of ORR per IA among all included and treated patients. A subgroup analysis showed that ORR per IA among all treated patients who had received 1 line of prior systemic therapy (n = 98) was ████ █ ███ █ ██ █ ████ █ █ █ ██████, whereas among all treated patients who had received 2 or more lines of prior systemic therapy (n = 7), it was ████ █ ███ █ ██ █ ███ █ █ █ ██████.
The ORR per IRC was 30.5% overall (95% CI, 21.9% to 40.2%). The ORR per IRC among patients with a CTFI shorter than 90 days and 90 or more days was 13.3% (95% CI, 5.1% to 26.8%) and 43.3% (95% CI, 30.6% to 56.8%), respectively. The ORR per IRC among patients with a CTFI shorter than 30 days, 30 to 89 days, 90 to 179 days, and 180 or more days was 9.5% (95% CI, 1.2% to 30.4%), 16.7% (95% CI, 4.7% to 37.4%), 40.0% (95% CI, 24.9% to 56.7%), and 50.0% (95% CI, 27.2%, 72.8%), respectively.
Table 16: ORR in Study B-005 (Treated Patients)
Parameter | Overall | CTFI < 90 days | CTFI ≥ 90 days | CTFI < 30 days | CTFI30 to 89 days | CTFI 90 to 179 days | CTFI ≥ 180 days |
|---|---|---|---|---|---|---|---|
n | 105 | 45 | 60 | ███ | ███ | ██ | 20 |
Complete response (IA), n | 0 | 0 | 0 | ███ | ███ | ███ | 0 |
Complete response (IRC), n | 0 | 0 | 0 | ███ | ███ | ███ | 0 |
Partial response (IA), n (%) | 37 (35.2) | 10 (22.2) | 27 (45.0) | █ ███████ | █ ███████ | █ █ ███████ | 12 (60.0) |
Partial response (IRC), n (%) | █ █ ██████ | █ ██████ | █ █ ██████ | █ █████ | █ ██████ | █ █ ██████ | 10 (50.0) |
Stable disease (IA),a n (%) | 35 (33.3) | 13 (28.9) | 22 (36.7) | █ █ ███████ | █ ███████ | █ █ ███████ | 7 (35.0) |
Stable disease (IRC),a n (%) | █ █ ██████ | █ █ ██████ | █ █ ██████ | █ ██████ | █ ██████ | █ █ ██████ | 6 (30.0) |
Stable disease ≥ 4 months (IA) | 10 (9.5) | 3 (6.7) | 7 (11.7) | █ ██████ | █ ██████ | █ ███████ | █ █████ |
Stable disease ≥ 4 months (IRC) | █ █████ | █ █████ | █ █████ | █ █████ | █ █████ | █ █████ | █ ██████ |
Stable disease < 4 months (IA) | 25 (23.8) | 10 (22.2) | 15 (25.0) | █ ███████ | █ ██████ | █ ███████ | █ ██████ |
Stable disease < 4 months (IRC) | █ █ ██████ | █ █ ██████ | █ █ ██████ | █ ██████ | █ ██████ | █ ██████ | █ ██████ |
Progressive disease (IA), n (%) | 28 (26.7) | 18 (40.0) | 10 (16.7) | █ ███████ | █ █ ███████ | █ ███████ | 1 (5.0) |
Progressive disease (IRC), n (%) | █ █ ██████ | █ █ ██████ | █ █ ██████ | █ ██████ | █ █ ██████ | █ █ ██████ | 4 (20.0) |
Not evaluable (IA),b n (%) | 5 (4.8) | 4 (8.9) | 1 (1.7) | █ ██████ | █ ██████ | █ ██████ | 0 |
Not evaluable (IRC),b n (%) | █ █████ | █ ██████ | █ █████ | █ ██████ | █ █████ | █ █████ | 0 |
ORR per IA, % (95% CI) | 35.2 (26.2 to 45.2) | 22.2 (11.2 to 37.1) | 45.0 (32.1 to 58.4) | ██ █ ██ █ █████ | ██ █ ███ █ █████ | ██ █ ███ █ █████ | 60.0 (36.1 to 80.9) |
ORR per IRC, % (95% CI) | █ █ █ ██ | █ █ █ ██ | █ █ █ ██ | ██ █ ████ █ █████ | ██ █ ████ █ █████ | ██ █ ████ █ █████ | 50.0 (27.2 to 72.8) |
CI = confidence interval; CTFI = chemotherapy-free interval; IA = investigator assessment; IRC = independent review committee; ORR = objective response rate.
aStable disease category includes 4 patients with partial response not confirmed.
bOne patient was not evaluable because of patient refusal to have disease measurement. Four patients were not evaluable per RECIST: 2 patients had early death considered related to malignant disease occurring 28 days and 18 days after the first infusion, respectively; and 2 patients had symptomatic deterioration due to progressive disease during cycle 1 without any radiological assessment performed.
Source: B-005 Clinical Study Report.10
Duration of Response
DOR results per IA and IRC among all responding patients in study B-005 are summarized in Table 17, Figure 6, and Figure 7. Median DOR per IRC was 5.1 months overall (95% CI, 4.9 to 6.4 months). Median DOR per IRC among patients with CTFIs shorter than 90 days and 90 or more days was ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ██████ █ ██ █ ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████, respectively. Median DOR per IRC among patients with a CTFI shorter than 30 days, 30 to 89 days, 90 to 179 days, and 180 or more days was ██ █ █████ █ ███ █ ██ █ ██ █ ███████████ █ ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████ █ ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████ █ ██ █ ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████, respectively.
Table 17: DOR in Study B-005 (Responding Patients)
Parameter | Overall | CTFI < 90 days | CTFI ≥ 90 days | CTFI < 30 days | CTFI 30 to 89 days | CTFI 90 to 179 days | CTFI ≥ 180 days |
|---|---|---|---|---|---|---|---|
Investigator assessment | |||||||
n | 37 | 10 | 27 | ██ | ██ | ███ | 12 |
Patients with events, n (%) | 29 (78.4) | 9 (90.0) | 20 (74.1) | █ ███████ | █ ████████ | █ █ ███████ | 10 (83.3) |
Patients censored, n (%) | 8 (21.6) | 1 (10.0) | 7 (25.9) | █ ███████ | █ ██████ | █ ███████ | 2 (16.7) |
Median DOR (95% CI), monthsa | 5.3 (4.1 to 6.4) | 4.7 (2.6 to 5.6) | 6.2 (3.5 to 7.3) | █ █ █ █ ███ | ██ █ ████ █ █████ | ██ █ ███ █ █████ | 5.5 (2.9 to 11.2) |
DOR of 4 months or longer among responders, % (95% CI)a | █ █ █ ███ | █ █ █ ███ | █ █ █ ██| | █ █ █ ███ | ██ █ ████ █ ██████ | ██ █ ███ █ █████ | ██ █ ██ █ ████ |
DOR of 6 months or longer among responders, % (95% CI)a | █ █ █ █ ██ | █ █ █ ███ | █ █ █ ███ | █ █ █ █ ████ | ██ █ ████ █ ████ | ██ █ ██ █ █████ | ██ █ ██ █ ████ |
DOR of 12 months or longer among responders, % (95% CI)a | █ █ █ █ █████ | ████ █ ████ | █ █ █ █ ████ | ██ █ ██ █ ████ | ██ █ ██ █ ████ | ██ █ ██ █ █████ | ██ █ █ █ ████ |
Independent review committee | |||||||
n | 32 | █|| | ██ | █|| | █|| | ██ | ██ |
Patients with events, n (%) | █ █ ██████ | █ ██████ | █ █ ██████ | █ █ ██████ | █ ███████ | █ ██████ | █ ██████ |
Patients censored, n (%) | █ █ ██████ | █ ██████ | █ ██████ | █ ██████ | █ █████ | █ ██████ | █ ██████ |
Median DOR (95% CI), monthsa | 5.1 (4.9 to 6.4) | █ █ ███ █ ████ | █████ █ ████ | ██ █ ███ █ ███ | ██ █ ████ █ ████ | ██ █ ██ █ ████ | ██ █ ██ █ ████ |
DOR of 4 months or longer among responders, % (95% CI)a | █ █ ███ █ ████ | █ █ █ ███ | █ █ ███ █ ████ | ██ █ ███ █ █████ | ██ █ ███ █ █████ | ██ █ ███ █ █████ | ██ █ █ █ █████ |
DOR of 6 months or longer among responders, % (95% CI)a | █ █ █ ███ | █ █ ███ █ ███ | █ █ █ ███ | ██ █ ████ █ ███ | ██ █ ████ █ ███ | ██ █ ███ █ █████ | ██ █ ██ █ ████ |
DOR of 12 months or longer among responders, % (95% CI)a | █ █ █ █ ██ | ██ █ ██ █ ███ | █ █ █ ██ | ██ █ ███ █ ███ | ██ █ ███ █ ███ | ██ █ ██ █ ████ | █ █ ██ █ ███ |
CI = confidence interval; CTFI = chemotherapy-free interval; DOR = duration of response; NC = not calculable.
aFrom Kaplan-Meier analysis.
Source: B-005 Clinical Study Report.10
Figure 6: Kaplan-Meier Plot of DOR in Study B-005 (Treated Patients)
DOR = duration of response; IA = investigator assessment; IRC = independent review committee.
Source: B-005 Clinical Study Report.10
Figure 7: Kaplan-Meier Plot of DOR per IRC in Study B-005 (Treated Patients With a CTFI < 90 Days or ≥ 90 Days)
CTFI = chemotherapy-free interval; DOR = duration of response; IRC = independent review committee.
Source: B-005 Clinical Study Report.10
Cancer Symptoms
Cancer symptoms were not assessed in study B-005.
Harms
Only harms identified in the review protocol are reported here. Refer to Table 18 for detailed harms data.
Adverse Events
Overall, 103 patients (98.1%) experienced AEs in study B-005. The most common AEs were fatigue (76.2%), nausea (37.1%), decreased appetite (33.3%), constipation (31.4%), dyspnea (29.5%), vomiting (21.9%), and diarrhea (20.0%).
Serious Adverse Events
SAEs occurred in 34 patients (32.4%) in study B-005. The most common SAEs were febrile neutropenia (4.8%), neutropenia (3.8%), and thrombocytopenia (3.8%).
Adverse Events Leading to Dose Reduction
AEs leading to dose reduction occurred in 25 patients (26.3%) in study B-005. The most common AEs leading to dose reduction were related to hematological toxicities (17.9%) or both hematological and nonhematological toxicities (4.2%).
Withdrawals Due to Adverse Events
Four patients (3.8%) withdrew from lurbinectedin treatment in study B-005 because of AEs. One patient discontinued treatment after the development of grade 4 febrile neutropenia, grade 3 thrombocytopenia, and grade 3 anemia. Another patient discontinued treatment because of peripheral neuropathy. For the remaining 2 patients, discontinuation was not considered to be treatment-related.
Mortality
Overall, 66 patients (62.9%) died during study B-005. All deaths were related to disease progression. Two deaths were associated with AEs (both grade 5 dyspnea related to progressive disease).
Notable Harms
Among CADTH protocol-defined notable harms, the most common myelosuppression-associated AEs in study B-005 were anemia (95.2%), lymphopenia (85.7%), leukopenia (79.0%), neutropenia (71.4%), and thrombocytopenia (43.8%). Febrile neutropenia occurred in 4.8% of patients. The most common hepatotoxicity-associated AEs were ALT increase (71.8%), gamma-glutamyl transferase increase (65.0%), AST increase (44.7%), and AP increase (33.0%). Peripheral neuropathy and peripheral sensory neuropathy occurred in only 2 patients (1.9%).
Table 18: Summary of Harms in Study B-005 (Treated Patients)
Parameter | B-005 N = 105 |
|---|---|
Patients with ≥ 1 AE | |
n (%) | 103 (98.1) |
Common AEs, n (%)a | |
Anemia | 100 (95.2) |
Lymphopenia | 90 (85.7) |
Creatinine increase (n = 104) | 86 (82.7)c |
Leukopenia | 83 (79.0) |
Hyperglycemia (n = 103) | 79 (76.7) |
Fatigue | 80 (76.2) |
ALT increase (n = 103) | 74 (71.8) |
Neutropenia | 75 (71.4) |
GGT increase (n = 103) | 67 (65.0) |
AST increase (n = 103) | 46 (44.7) |
Thrombocytopenia | 46 (43.8) |
Hyponatremia (n = 104) | 40 (38.5) |
Nausea | 39 (37.1) |
Decreased appetite | 35 (33.3) |
AP increase | 34 (33.0) |
Hypoalbuminemia (n = 100) | 33 (33.0) |
Constipation | 33 (31.4) |
Hypomagnesemia (n = 96) | 29 (30.2) |
Dyspnea | 31 (29.5) |
Vomiting | 23 (21.9) |
Hyperkalemia | 21 (20.2) |
Diarrhea | 21 (20.0) |
Hypokalemia (n = 104) | 20 (19.2) |
Cough | 19 (18.1) |
Back pain | 15 (14.3) |
Pyrexia | 13 (12.4) |
Hypercalcemia (n = 98) | 11 (11.2) |
Bilirubin increase (n = 103) | 10 (9.7) |
Chest pain | 10 (9.5) |
Patients with ≥ 1 SAE | |
n (%) | 34 (32.4) |
Common SAEs, n (%)b | |
Febrile neutropenia | 5 (4.8) |
Neutropenia | 4 (3.8) |
Thrombocytopenia | 4 (3.8) |
Anemia | 3 (2.9) |
General physical health deterioration | 3 (2.9) |
Pneumonia | 3 (2.9) |
Upper respiratory tract infection | 3 (2.9) |
Diarrhea | 2 (1.9) |
Lung infection | 2 (1.9) |
Hyponatremia | 2 (1.9) |
Patients with AEs leading to dose reduction | |
n (%) | 25 (26.3) |
Reasons for dose reduction, n (%) | |
Hematological toxicity | 17 (17.9) |
Nonhematological toxicity | 3 (3.2) |
Both hematological and nonhematological toxicity | 4 (4.2) |
Nontreatment related | 1 (1.0) |
Patients with WDAEs | |
n (%) | 4 (3.8) |
Deaths | |
n (%) | 66 (62.9) |
Deaths due to AEs | |
n (%) | 2 (1.9) |
Notable harms | |
Myelosuppression | |
Febrile neutropenia | 5 (4.8) |
Iron deficiency anemia | 1 (1.0) |
Anemia | 100 (95.2) |
Lymphopenia | 90 (85.7) |
Leukopenia | 83 (79.0) |
Neutropenia | 75 (71.4) |
Thrombocytopenia | 46 (43.8) |
Hepatotoxicity | |
Hepatomegaly | 2 (1.9) |
Hepatic pain | 1 (1.0) |
ALT increase (n = 103) | 74 (71.8) |
GGT increase (n = 103) | 67 (65.0) |
AST increase (n = 103) | 46 (44.7) |
AP increase (n = 103) | 34 (33.0) |
Bilirubin increase (n = 103) | 10 (9.7) |
CPK increase (n = 103) | 7 (6.8) |
Peripheral neuropathy | |
Neuropathy peripheral | 2 (1.9) |
Peripheral sensory neuropathy | 2 (1.9) |
AE = adverse event; ALT = alanine aminotransferase; AP = alkaline phosphatase; AST = asparagine aminotransferase; CPK = creatine phosphokinase; GGT = gamma-glutamyl transferase; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Notes: Treatment-emergent AEs reported in this table were defined as any untoward medical occurrence after administration of the first dose of study drug and in the 30 days after the last dose of study drug. AEs were coded using MedDRA version 21.0 and graded according to NCI-CTCAE version 4.
For biochemical parameters that were measured in fewer than 105 patients, the denominator is indicated.
aAEs with frequency ≥ 10% are listed.
bSAEs occurring in ≥ 2 patients are listed.
cMost of these were not clinically significant creatinine increases; creatinine values were within normal range and grade 1 was due to creatinine increase > 1.5 × baseline, according to the NCI-CTCAE v.4 severity calculation. Using NCI-CTCAE v.3, only 25 patients (24.0%) would have a grade 1 or 2 creatinine increase.
Source: B-005 Clinical Study Report.10
Critical Appraisal
Internal Validity
Study B-00510 was a single-arm, phase II, basket trial in which a relatively small number of adult patients with SCLC (N = 105) were treated with OL second-line lurbinectedin following progression on or after platinum doublet therapy. In the absence of a randomized comparison with an alternative treatment, effect estimates are at high risk of bias, and causal inferences around the magnitude of the effect of the intervention cannot be made. The study results cannot be directly compared with those of prior or subsequent studies.
Because investigators (and potentially the IRC) were aware of treatment in this single-arm study, a potential source of bias in study B-005 was observer bias in outcome assessment (for ORR, PFS, and DOR per IA), as well as in investigator decisions to discontinue treatment due to progressive disease. Ten patients (9.5%) evaluated as having stable disease or a tumour response per IA but with progressive disease according to IRC review remained on protocol therapy; in addition, 4 patients who were discontinued from therapy after progressive disease per IA had stable disease per IRC. Protocol measures meant to control for bias in the interpretation of end points included standardized protocols for image acquisition, interpretation, and measurement of response and progression, and independent review of all images by an IRC. However, the organizational details and algorithm of the IRC were not provided. According to the clinical experts consulted by CADTH for this review, bias in favour of the drug would be more likely among investigators participating in single-arm oncology trials. Estimates of ORR per IRC were, in general, more conservative than those per IA. It was unclear whether the IRC was blind to the study and treatment, so the potential for observer bias in IRC review was unclear. Very few patients (2.8%) discontinued the study for reasons other than death after disease progression, so attrition biases were likely minimal.
The clinical experts consulted by CADTH for this review acknowledged that the protocol deviations in study B-005 related to missed assessments for lurbinectedin re-treatment, and failure to reduce or delay doses could have theoretically contributed to higher estimates of the drug’s efficacy. However, the experts emphasized that the impacts of these protocol deviations would be minor and unlikely to affect interpretation of the study results.
The outcomes used in study B-005 (OS, PFS, ORR, DOR) are standard in oncology trials and tumour responses were objectively evaluated using RECIST 1.1. Thus, there were no major concerns regarding the validity or measurement properties of outcomes, although in the absence of a comparator arm, interpretation of the efficacy results was limited. However, the analysis of OS would be influenced by subsequent therapies in the third line and later, which were received by nearly half (44.8%) of the patients. In addition, the potential for patient and/or investigator observer bias in harms outcome reporting is inherent in any single-arm, OL trial.
Several statistical issues should be noted when interpreting the results of study B-005. First, the OS data were relatively immature (median follow-up of 17.1 months, 37.1% censoring) and the results may differ in the final analysis when the study is completed. Second, although statistical methods were overall appropriate, all efficacy analyses were descriptive and no formal statistical hypothesis testing was conducted, except for the primary outcome of ORR per IA. For outcomes other than OS, missing data (PFS, 14.3% to 22.9% censoring; ORR, 4.8% to 6.7% not evaluable; DOR, 21.6% to 31.3% censoring) were primarily due to patients being progression-free and/or recurrence-free at the time of assessment, or to rapid progression or death without tumour assessments (for ORR). Missing data were accounted for appropriately using Kaplan-Meier methodology. Subgroup analyses by CTFI were prespecified but exploratory, with no formal hypothesis testing and no adjustment for multiplicity.
External Validity
According to the clinical experts consulted by CADTH for this review, the eligibility criteria for study B-005 would be expected to result in the recruitment of adult patients with stage III or metastatic SCLC representative of the Canadian patient population undergoing second-line treatment for advanced disease. However, the clinical experts noted that in practice, approximately 25% of patients with advanced SCLC undergoing second-line therapy would be expected to have CNS involvement; these patients generally have worse prognoses and were not included in the study. The expectations of the clinical experts with regard to CNS involvement were not aligned with the small number of screening failures in the study (5 of 110 patients), and the reasons for this discrepancy were unclear. The clinical experts also emphasized that study B-005 included patients with a short CTFI (platinum-refractory and platinum-resistant) who are excluded from many trials, which they felt was laudable; these patients generally have worse prognoses than those with platinum-sensitive disease. The clinical experts stated that the baseline characteristics of the B-005 study population were similar to those of patients with SCLC in Canada undergoing second-line chemotherapy for advanced disease, although trial participants were somewhat younger, with better performance status and overall health compared with the general population (as is the case in most oncology trials). The clinical experts also noted the relatively high proportion of patients in the study (approximately half) who went on to receive other systemic anticancer therapies in the third line and beyond, which they explained was higher than they would expect in clinical practice and potentially reflected the recruitment of a treatment-seeking trial population in better health than the general population.
The clinical experts consulted by CADTH for this review felt that the absence of Canadian sites in study B-005, which was conducted primarily in Europe, would not limit generalizability to patients in Canada. Moreover, although the study was conducted in the second-line setting, the clinical experts did not see any major issues with generalizing the study results to the third-line setting, although they noted the generally decreasing effectiveness of treatment with advancing lines of therapy.
Doses of lurbinectedin administered in study B-005 were aligned with the Health Canada–approved dosing. The clinical experts stated that the transfusion and growth-factor support received by patients in study B-005 was generally appropriate, and as expected. However, the clinical experts stated that because secondary G-CSF prophylaxis is not typically given in Canada in the metastatic setting, some harms outcomes (e.g., hematological toxicities) may have been underestimated compared with real-world clinical practice in Canada.
Input from patient groups for this review suggested that patients with SCLC desired new second-line treatment options that prolonged survival (OS) and delayed disease progression (PFS) while maintaining HRQoL and controlling disease symptoms, with an acceptable toxicity profile. ORR and DOR were not specifically mentioned as an outcome important to patients.
The duration of follow-up in study B-005 was adequate for assessment of ORR, DOR, and PFS ████ █ ████ █ ███ █ ████ █ ██ █ █ ███ █ █████ █ ███ █ █████ █ ███ █ ████ █ ██ █ ███ █ ████ █ ██████. Although OS data were not fully mature (median follow-up 17.1 months, 37.1% censoring), the duration of follow-up was probably adequate for estimation of median OS because of the relatively short survival expectations in this patient population.
According to the clinical experts, patients in study B-005 underwent imaging scans more frequently (every 2 cycles until cycle 6, and every 3 cycles thereafter) than many patients treated in clinical practice in Canada would. However, the clinical experts did not feel that this would limit the generalizability of the study findings.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The objective of this section is to summarize and appraise evidence from ITCs for the relative effects and safety of lurbinectedin for the treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-containing therapy. The aim is to fill a gap created by the absence of trials directly comparing lurbinectedin to comparators relevant in the Canadian landscape, as stated in the protocol of this CADTH report.
A focused literature search for NMAs dealing with SCLC was run in MEDLINE All (1946–) on March 23, 2022. No limits were applied to the search. No published ITCs were found in the literature search from CADTH comparing lurbinectedin to comparators of interest based on inclusion criteria in this clinical report.
Three ITCs were submitted by the sponsor and are reviewed in this section: an SCA analysis, an STC, and an MAIC and NMA.
Description of Indirect Comparisons
The SCA analysis12 aimed to evaluate the treatment landscape and efficacy of lurbinectedin in the treatment of patients with advanced SCLC after exposure to platinum-based therapy in Alberta. This study consisted of 2 phases: the first phase evaluated a real-world cohort of all individuals in Alberta diagnosed with SCLC at any stage from 2004 to 2019 and initiated a post–platinum-based systemic therapy and a second phase consisted of building (from the first phase population) an SCA for comparison with the SCLC cohort in the phase II B-005 (single-arm) study.
The second ITC14 was an STC to facilitate the indirect comparison of lurbinectedin (using individual patient data [IPD] from the B-005 study) to topotecan IV (using aggregated data [AD] from the von Pawel et al. [2014] RCT13) in patients with relapsed or refractory SCLC.
The third submitted ITC15 was an MAIC combined with an NMA to evaluate the efficacy and safety of lurbinectedin versus relevant comparators among patients with SCLC receiving second-line treatment with respect to ORR, DOR, OS, and PFS, as well as hematological AEs of grade 3 or 4 (including anemia, thrombocytopenia, neutropenia, and febrile neutropenia).
Synthetic Control Arm
Objectives
The objectives in the first phase of the study include defining the population of patients with SCLC who initiated a post–platinum systemic therapy and characterizing OS, time to next treatment (TTNT) or death, and frequency of hospitalization and emergency department visits in the 6 months after therapy (as proxy measures of SAEs) in this population.
For the second phase — the synthetic control, feasibility, and summary-level analyses — the primary objectives included:
conduct a feasibility assessment to define trial eligibility and evaluate the distribution of prognostic variables in the patient population
if feasible, identify a well-characterized SCA population in alignment with the SCLC cohort of the B-005 study
estimate standardized rates for OS and TTNT in the SCA population that can be used as benchmarks for the B-005 study.
Patient Selection From Databases
In the first phase of the study, a streamlined process was used to access various provincial administrative databases maintained by the Alberta government using an Alberta unique lifetime identifier assigned to each person at birth or upon becoming a resident of the province, which captures all demographic information, health care encounters, and electronic medical records in the province. The database linkage and statistical analyses were conducted by a research group in charge of the study that de-identified and analyzed all data. The following databases were used and linked:
Alberta Cancer Registry: captures information on all individuals diagnosed with cancer in Alberta (300,000+ cases), information related to cancer, and information on the date and cause of death and last known date of follow-up
ARIA electronic medical records: a database of electronic medical records for all 17 provincial cancer centres (2 tertiary, 4 regional, and 11 community centres), covering 4.5 million residents of Alberta
Discharge Abstract Database: captures information each time an individual is discharged from an inpatient hospital bed and information on the date and the duration of the hospitalization
National Ambulatory Care Reporting System database: captures information on all inpatient and outpatient use of ambulatory care services
Practitioner Claims database: captures physician and allied practitioner claims used for reimbursement and shadow-billing purposes that have been processed by the Alberta government
population registry: includes information related to birth and death dates as well as migration out of province for all residents with Alberta Health Care Insurance Plan coverage.
The data collected and analyzed are population-based. This system of databases and registries captures more than 99% of cancers diagnosed in Alberta. Any loss to follow-up was expected to be noninformative because of the reliance on vital statistics and the population registry to identify the last known date of follow-up. In addition, data were linked via a chart review with administrative data at the population level due to the implementation of provincial electronic medical records in Alberta.
Potentially eligible subjects were identified from the Alberta Cancer Registry and then selected for inclusion based on the availability of administrative data sources with relevant data of interest. Because this is a population-based registry that captures all cases of cancer diagnosed in Alberta, no specific sampling techniques were employed, and all eligible individuals were included. Various administrative data algorithms were used to approximate different eligibility criteria, such as disease progression, adequate laboratory measures, impending need for radiotherapy, and high-grade toxicity.
The first phase of the study included patients aged 18 years and older who were diagnosed with SCLC (any stage) in Alberta from 2004 to 2019. Patients were followed until the end of 2020.
The second phase of this investigation involved a subset of the population identified in the first phase with the following additional eligibility criteria applied to approximate the eligibility criteria used in the B-005 trial:
pretreatment with only 1 previous treatment line of systemic therapy (immunotherapy was allowed, combined with chemotherapy or alone)
evidence of disease progression and adequate hematologic, renal, and liver function based on patients’ receipt of subsequent systemic therapy (defined as a systemic drug not in the original treatment regimen or a gap of more than 90 days between subsequent treatment dispensations)
time from the end date of prior systemic treatment to initiation of subsequent treatment of at least 3 weeks for patients initially treated with chemotherapy alone, or 4 weeks for patients initially treated with immunotherapy or radiotherapy
evidence of prior or concurrent malignant disease
high-grade toxicity on first treatment (e.g., at least 1 hospitalization or emergency department visit)
absence of brain metastases at initial diagnosis.
Patients who received radiation therapy after the end of the initial systemic treatment regimen and before subsequent systemic treatment were excluded from the second-phase analysis.
Outcomes
OS was defined as the time of initiation of subsequent systemic therapy administered after initial systemic therapy (used as proxy for disease progression) until death from any cause. Individuals lost to follow-up were censored. Meanwhile, TTNT was defined as the time of initiation of subsequent systemic therapy administered after exposure to platinum-based therapy until death from any cause or until initiation of subsequent systemic therapy. Individuals lost to follow-were censored.
High-grade AEs were defined as causing 1 or more hospitalizations or emergency department visits in the 6 months after initiation of systemic therapy. For the health care resource-use outcomes, the total number and mean encounters for each patient were reported in each year of follow-up, starting from the initiation of a subsequent line of therapy for hospitalizations, ambulatory care encounters, and physician visits.
Analysis Methods
For the first phase, baseline demographics were summarized and reported for the identified patient population using summary statistics (i.e., mean or median for continuous variables and proportion for categorical variables). Baseline characteristics reported were stratified by CTFI (CTFI < 90 days and CTFI ≥ 90 days). Kaplan-Meier plots of the survival function for OS and TTNT were provided, along with the median, 6-month interval survival, and corresponding 95% CI. High-grade AEs were assessed using (as a surrogate) the number and proportion of patients experiencing 1 or more hospitalizations or emergency department visits in the 6 months after the initiation of subsequent systemic therapy. For health care resource-use outcomes, the total count and the mean number of events per patient were reported in each year of follow-up, starting from the initiation of subsequent systemic therapy (i.e., number of hospitalizations, days hospitalized, encounters with ambulatory care services, and physician visits).
For the second phase, an SCA was identified by applying the aforementioned eligibility criteria to approximate that of the lurbinectedin trial (Study B-005). Baseline characteristics of the SCA were estimated and compared with those in the lurbinectedin trial. The magnitudes of differences in the mean and prevalence of baseline characteristics were assessed using standardized differences; values less than 0.1 were judged to be indicative of the balance achieved in a randomized trial, and values greater than 0.1 were considered to reflect a clinically meaningful imbalance. OS, TTNT, and high-grade AEs were estimated in the SCA using the methods described in the first phase. The study authors adjusted 2 prognostic variables (based on input from clinical experts): the stage at initial diagnosis (LS versus ES); and CTFI (90 days or shorter versus longer than 90 days). Estimates of median OS, median TTNT, and proportion who experienced a high-grade AE were generated within strata defined by stage and CTFI. The strata-specific estimates were pooled using weights defined by the distribution observed in the lurbinectedin trial where estimates can be provided for the lurbinectedin trial and where there is a sufficient sample size in the SCA. The 95% CI for the pooled estimates were generated using the percentile bootstrap method (1,000 iterations). Also, bootstrapping was used to obtain P values that assessed the 1-sided null hypothesis that OS in the SCA was equal to or greater than that of the B-005 trial.
Results of the SCA Analysis
For the first phase, an overview is presented of all individuals in Alberta who were diagnosed with SCLC (any stage) from 2004 to 2019 and who initiated a post–platinum-based systemic therapy (platinum regimen refers to carboplatin-containing or cisplatin-containing regimens). In total, 3,721 individuals were diagnosed with SCLC during the study period, 2,031 (55%) of whom initiated platinum-containing therapy. Of the 2,031 individuals who initiated platinum-based therapy, 577 (28%) subsequently initiated a post–platinum-based systemic therapy regimen.
The baseline characteristics of patients in the first and second phases (SCA) are presented in Table 19.
Table 19: Baseline Characteristics of Patients With SCLC Who Initiated Post–Platinum-Based Therapy in the SCA
Variable | Overall and per-strata values | Patients included in the SCA |
|---|---|---|
N | 577 | 224 |
Age at initiation of post–platinum-based treatment, years, mean (SD) | 65.48 (8.90) | 65.04 (8.69) |
Sex, n (%) | ||
Female | 294 (51.0) | 114 (50.9) |
Male | 283 (49.0) | 110 (49.1) |
Stage at initial diagnosis, n (%) | ||
ES | 335 (58.1) | 128 (57.1) |
LS | 236 (40.9) | 96 (42.9) |
Missing | 6 (1.0) | 0 |
Number of metastatic sites at diagnosis, median (IQR) | 1.00 (0.00 to 2.00) | 1.00 (0.00 to 2.00) |
CTFI (days), median (IQR) | 171.00 (104.00 to 280.00) | 194.00 (117.75 to 301.00) |
CTFI, n (%) | ||
CTFI < 90 days | 117 (20.3) | 35 (15.6) |
CTFI 90+ days | 460 (79.7) | 189 (84.4) |
Front-line platinum-based therapy, n (%) | ||
Cisplatin + etoposide | 335 (58.1) | NR |
Carboplatin + etoposide | 226 (39.2) | NR |
Other platinum regimen | 16 (2.8) | NR |
Cisplatin | NR | 137 (61.2) |
Carboplatin | NR | 87 (38.8) |
Post–platinum-based regimen, n (%) | ||
Carboplatin + etoposide | 297 (51.5) | 121 (54.0) |
Cisplatin + etoposide | 102 (17.7) | 46 (20.5) |
Etoposide monotherapy | 57 (9.9) | 18 (8.0) |
CAV | 53 (9.2) | 20 (8.9) |
Other platinum regimen | 26 (4.5) | 19 (8.5) |
Other | 24 (4.2) | NR |
Topotecan | 18 (3.1) | NR |
CAV = cyclophosphamide + doxorubicin + vincristine; CTFI = chemotherapy-free interval; ES = extensive stage; IQR = interquartile range; LS = limited stage; NR = not reported; SCA = synthetic control arm; SCLC = small cell lung cancer; SD = standard deviation.
Source: Sponsor-submitted ITC1 report.12
In terms of OS in the first phase of the study, the median reached 6.67 months (95% CI, 6.08 to 7.2 months).
AEs were assessed indirectly as the proportion of patients who were hospitalized or who had at least 1 emergency department visit in the 6 months after initiation of post–platinum-based therapy. This was used as a proxy measure for serious treatment-related AEs. Overall, among the 577 included patients, 300 (52.0%) experienced at least 1 hospitalization in the 6 months after initiation of post–platinum-based therapy, 364 (63.1%) experienced at least 1 emergency department visit, and 388 (67.2%) experienced at least 1 hospitalization or emergency department visit.
Phase 2 originated from the initial 577 patients in the first phase who received post–platinum-based therapy. Of these, 224 (39%) were eligible for inclusion in the SCA for comparison with the phase II B-005 trial. An overview of the baseline characteristics of these patients is presented in Table 19. The vast majority of individuals had 1 prior line of therapy (fewer than 10 patients had 2 prior lines of therapy), and nearly all received platinum-based therapy in combination with etoposide as their first platinum-based therapy (fewer than 10 patients received platinum in combination with an alternative systemic drug). The majority of other post–platinum-based therapies were other platinum combinations (fewer than 10 patients received topotecan post–platinum-based therapy).
Overall Survival
OS estimates for the SCA are presented here as overall and as stratified by the CTFI and by stage at initial diagnosis. Estimates of OS simultaneous stratification (by both CTFI and stage) could not be obtained due to small cell counts (i.e., fewer than 10 patients in the SCA had a CTFI of less an 90 days and were LS at initial diagnosis).
Survival estimates are presented unadjusted and after adjustment for differences in the distribution of CTFI and stage at initial diagnosis. Specifically, the strata-specific estimates from the SCA were pooled using the weights estimated from the trial. This standardization provides an estimate of the outcome in the SCA if the SCA had the same distribution of CTFI and stage at initial diagnosis as the trial.
For the overall population, the unadjusted OS reached a median of 6.58 months (95% CI, 5.75 to 7.46 months). The median OS did not differ significantly between patients stratified in the ES group (OS = 5.75 months; 95% CI, 4.90 to 6.81 months) and those stratified in the LS group (OS = 7.63 months; 95% CI, 6.25 to 9.83 months). However, it differed when comparing the group of patients with a CTFI shorter than 90 days (median OS = 4.31 months; 95% CI, 3.29 to 6.74 months) and those with a CTFI of 90 days or more (median OS = 6.87 months; 95% CI, 3.29 to 6.74 months; P = 0.011).
In the adjusted population analysis, the OS reached a median of 5.8 months (95 CI, 5.1 to 6.9 months), whereas the median OS in the B-005 trial was of 9.3 months (95% CI, 6.3 to 11.8 months).
When comparing estimates of OS between the SCA and the phase II B-005 trial, the results favoured the B-005 trial arm, whether adjusted or unadjusted (see Table 20).
The SCA was subsequently updated to align more closely with the B-005 trial population by excluding patients who developed brain metastasis after diagnosis but before initiating post–platinum-based therapy. In the updated SCA, the unadjusted OS reached a median of 6.7 months (95% CI, 6.0 to 7.7 months). In the adjusted population analysis, the median OS reached 6.1 months (95% CI, 5.4 to 7.7 months). CADTH was unable to independently verify the methodological details of the analysis.
Table 20: Comparison of OS Between the Phase II B-005 Trial and the SCA (Unadjusted and Adjusted for CTFI and for Stage at Initial Diagnosis)
Analysis statistic | Estimate (95% CI) |
|---|---|
Median OS (months) | |
Trial B-005 | 9.3 (6.3 to 11.8) |
SCA unadjusted | 6.6 (5.6 to 7.4) |
SCA adjusted | 5.8 (5.1 to 6.9) |
6-month survival (%) | |
Trial B-005 | 67.1 (57.6 to 76.7) |
SCA unadjusted | 54.8 (48.0 to 61.5) |
SCA adjusted | 49.1 (40.6 to 57.9) |
12-month survival (%) | |
Trial B-005 | 34.2 (23.2 to 45.1) |
SCA unadjusted | 22.7 (17.5 to 28.4) |
SCA adjusted | 19.0 (13.4 to 25.4) |
CI = confidence interval; CTFI = chemotherapy = free interval; OS = overall survival; SCA = synthetic control arm.
Source: Sponsor-submitted ITC1 report.12
Unadjusted and adjusted estimates were reported for the proportion of individuals who were hospitalized or admitted to the emergency department within 6 months after initiation of post–platinum-based therapy in the SCA. The adjusted analysis (stratified by stage and CTFI) showed that the proportion of individuals who were hospitalized or who had 1 or more emergency department visits in the months after initiation of post–platinum-based therapy was 64.9% (95% CI, 56.9% to 72.5%). The unadjusted analysis showed similar results (60.7%; 95% CI, 54.5% to 67.0%).
Critical Appraisal of the SCA Analysis
This submitted analysis assesses the treatment landscape of patients with advanced SCLC after exposure to platinum-based therapy in Alberta, and creates an SCA for comparison against the SCLC cohort in the phase II B-005 (single-arm) study.
There were several linked databases used for the first phase of the study. Although these seemed appropriate and thorough, it is possible that information on some patient groups might have been missing or underrepresented, and no specific techniques for sampling were used. Overall, the set of patients seemed generalizable to the population of patients with SCLC in Alberta and likely in the rest of the Canadian provinces and territories, according to the clinical experts consulted by CADTH. However, there are no data on other important patient and disease characteristics, such as ethnicity, socioeconomic status, and smoking status, that would help establish the generalizability of the results.
In the second phase of this analysis, the authors compared an SCA of patients selected from the first phase population with the SCLC cohort in the B-005 trial. In the SCA, the authors were unable to exclude patients who developed a brain metastasis after diagnosis but before initiation of post–platinum-based therapy, which may have resulted in an underestimation of survival in the SCA and a bias in favour of lurbinectedin. However, in the updated SCA analysis, this bias was accounted for by the exclusion of patients who developed brain metastases after diagnosis but before the initiation of post–platinum-based therapy.
Administrative data algorithms were used to assess and select SCA patients in alignment with B-005 eligibility criteria, such as disease progression, adequate laboratory measures, impending need for radiotherapy, and high-grade toxicity. The use of different administrative data algorithms may introduce potential inaccuracies in patient characteristics and outcomes to the analyses, which could result in misclassification. There is a lack of data on performance status and the presence of brain metastases at the time of disease progression. It was not possible to restrict selection to patients with an ECOG PS of 2 or less or who did not develop a brain metastasis after diagnosis, as was done in the B-005 trial, which also has the potential to produce a bias in favour of lurbinectedin.
The authors state that due to confidentiality regulations, they had to rely on aggregate-level data in the B-005 study during the second phase, so they could not adjust for more than 2 variables simultaneously, which could have implications on selection bias when estimating any treatment effects.
As the comparisons between the SCA and the B-005 trial rely on observational data in a nonrandomized fashion, there is high risk of bias related to residual confounding (unobserved or unmeasured variables that could have an effect on outcomes).
Simulated Treatment Comparison
The second ITC described in this CADTH report consists of an STC submitted by the sponsor14 and performed per the general guidelines of the Decision Support Unit Technical Support Document 18.34 Furthermore, the authors used the information to assess the feasibility of conducting a population-matched ITC and to evaluate whether the important assumptions necessary for a matching are met.
Objectives
The main objective was to facilitate an ITC of lurbinectedin to topotecan in patients with SCLC who progress on or after platinum-containing therapy.
Study Selection Methods
A targeted literature search was used to obtain the relevant study with which to compare lurbinectedin with IV topotecan. It was determined that 1 study (von Pawel et al. [2014]13) represented the most recent data for the topotecan arm. No specific search strategy, study selection, data extraction, or study quality assessment was described in the report.
For the lurbinectedin arm, IPD from the B-005 study were used.10 The study design and eligibility criteria of the lurbinectedin and topotecan trials are presented in Table 21.
Table 21: Study Designs, Eligibility Criteria, and Population Baseline Characteristics of the Lurbinectedin and Topotecan Trial Arms
Criteria | Lurbinectedin trial (B-005) | Topotecan trial |
|---|---|---|
Study design | ||
Indication | Second-line treatment for SCLC | Second-line treatment for SCLC |
Design | Phase II basket trial, safety, and efficacy | Phase III trial, safety, and efficacy |
Randomization | No (single arm, open label) | Yes (2:1 ratio) |
Treatment | Lurbinectedin | Amrubicin, topotecan |
Dosing | 3.2 mg/m2 administered as a 1-hour IV infusion once every 3 weeks | Amrubicin: 40 mg/m2 administered as a 5-minute IV infusion once daily on days 1 to 3 of a 21-day cycle Topotecan: 1.5 mg/m2 administered as a 30-minute IV infusion once daily on days 1 to 5 of a 21-day cycle |
Response | RECIST (version 1.1) | RECIST (version 1.0) |
Progression | Independent review committee | Determined by investigator |
Prophylactic hematopoietic growth factors | Primary prophylaxis not allowed; secondary prophylaxis for neutropenia allowed | Mandated in all cycles |
Prophylactic antibiotic | Recommended for patients at high risk for infectious complications | Recommended for patients at high risk for infectious complications |
Adverse events | CTCAE 4.0 | CTCAE 3.0 |
Enrolment start date | October 2015 | December 2007 |
Enrolment end date | January 2019 | January 2010 |
Inclusion criteria | ||
Age | ≥ 18 years | ≥ 18 years |
Disease | Pathologically proven diagnosis of SCLC | Histologically or cytologically confirmed SCLC |
Progression | Documentation of progression after first-line platinum-containing chemotherapy | Documentation of progression after first-line platinum-containing chemotherapy |
ECOG PS | 0 to 2 | 0 to 1 |
Organ function | Adequate function of kidneys and liver | Adequate organ function |
Treatment | An interval of > 3 weeks between any previous treatment; pre-treatment with only 1 previous chemotherapy-containing treatment line | NA |
Toxicities | Grade 1 or lower | NA |
Bone marrow function | Adequate | Adequate |
Exclusion criteria | ||
Radiotherapy | Impending need for radiotherapy | Chest radiotherapy ≤ 28 days before treatment |
Treatment | Previous lurbinectedin or trabectedin | Previous anthracycline, topotecan, or irinotecan |
Metastasis | Previous or concurrent malignant disease unless in complete remission for 5 years | Prior brain metastasis |
CNS | Known CNS involvement | Symptomatic CNS metastases |
Subtypes | NA | Mixed or combined subtypes of SCLC were ineligible |
Medical conditions | Concomitant unstable or serious medical condition in the previous year | NA |
CNS = central nervous system; CTCAE = Common Terminology Criteria for Adverse Events; ECOG PS = Eastern Cooperative Oncology Group performance status; NA = not applicable; RECIST = Response Evaluation Criteria for Solid Tumours; SCLC = small cell lung cancer.
Source: Sponsor-submitted STC report.14
ITC Analysis Methods
After the main trials were selected, the feasibility of conducting an STC was assessed and the assumptions necessary for matching were evaluated. Because the included studies lacked a common control arm, the authors concluded that an unanchored STC was required.
The first step of the STC process included an outcome (regression) model, which was constructed for the lurbinectedin arm with covariates selected from a literature search for effect modifiers, prognostic variables, and an analysis of imbalances between studies.
The following covariates and evaluations were identified by the authors in the literature as prognostic factors of the outcomes:
Stage — SCLC was classified as LS or ES disease. Patients with ES disease have a considerably worse 5-year prognosis of survival (2.2%) than those with LS disease (28.5%)
Sex — Females were observed to have a higher 5-year prognosis of survival than males (4.7% to 7.7%)
Age — Older age at diagnosis was negatively correlated with a worse 5-year prognosis of survival for patients (< 45 years = 13.4%; < 65 years = 8.3%; ≥ 65 years = 4.9%).
ECOG PS — Even allowing for sex and stage of disease, ECOG PS scores have been shown to be independently prognostic of survival, with higher scores observed to be associated with a shorter median survival.
The following covariate was identified in the literature as a treatment-effect modifier:
Response to prior first-line therapy — patients who, in response to a first-line platinum-containing therapy, achieve a CTFI of 90 days or longer are deemed to be sensitive as opposed to resistant (CTFIs shorter than 90 days) and are shown to achieve longer median OS and median PFS in the clinical trials of resistant or refractory SCLC.
For survival outcomes, a systematic model exploration was performed. Model analyses included tests of proportional hazards and accelerated failure time models for their model fit, visual comparability and comparisons of reasonable predictive ability, and estimates from the original Kaplan-Meier curve. Seven parametric model distributions were tested, both with full and stepwise covariate inclusion (exponential, Weibull, Gompertz, log-normal, log-logistic, generalized gamma, and simple gamma). Modelling was performed in statistical software R version 4.02, using the flexsurvreg survival function.
In the second step of the STC process, a prediction of transformed outcomes on treatment with lurbinectedin in the comparator trial using the outcome model was performed. Once lurbinectedin treatment-effect outcomes were predicted among the simulated comparator population, the authors proceeded to step 3 in the STC, which is a final ITC between the adjusted lurbinectedin outcome estimates and the published comparator results. The difference in efficacy between interventions is calculated as the difference between adjusted lurbinectedin and the comparator, and standard errors were calculated (step 4) by sampling randomly from the asymptotic normal distribution of the maximum-likelihood estimate, and then taking quantiles to calculate the CIs for the transformed outcome.
To provide evidence that absolute outcomes could be predicted with sufficient accuracy in relation to the relative treatment effects and present an estimate of the likely range of residual systematic error (step 5), during exploration to find the optimal models, sample methods were explored to estimate the likely range of residual systematic error. Model accuracy was considered with several model-fitting scenarios of prognostic variables and effect modifiers and was assessed with statistical fit and visual inspection of the curve with to regard consistency of estimates between models.
For step 6 — the transportation of the effect estimate (the difference between adjusted lurbinectedin and topotecan) into the target population for the decision — the authors described the representativeness of the matched comparison as observed in the differences between study designs, inclusion and exclusion criteria of each study, and baseline characteristic distributions (Table 22).
For the seventh (and final) step, model-fit statistics were used to confirm whether models were reasonably constructed after each parametric regression. Fit statistics included the Akaike information criterion and Bayesian information criterion (BIC), where lower values signify better fit prediction models. As adding additional covariates generally tends to improve model fit, BIC is assessed to take into account that it penalizes models for over-fitting. Log-likelihood and chi-square distribution values were also used to compare model significance between similar models.
Table 22: Patient Baseline Characteristics of the Lurbinectedin and Topotecan Trial Arms
Characteristics | Lurbinectedin trial (B-005) N = 105 | Topotecan trial N = 213 |
|---|---|---|
Sex, n (%) | ||
Male | 63 (60) | 127 (60) |
Female | 42 (40) | 86 (40) |
Age (years), median (IQR) | 60 (54 to 68) | 61 (30 to 81) |
ECOG PS, n (%) | ||
0 | 38 (36) | 72 (34) |
1 | 59 (56) | 137 (64) |
2 | 8 (8) | 4 (2) |
3 | 0 | 0 |
Abnormal LDH (> upper limit of normal), n (%) | 47 (45) | NR |
Smoking status, n (%) | ||
Former or current | 97 (92) | 189 (89) |
Never | 8 (8) | 24 (11) |
Disease stage | ||
Limited | 7 (7) | 26 (12)a |
Extensive | 98 (93) | 187 (88)a |
Time from SCLC diagnosis in months, median (IQR) | NR | 8.4 (1.6 to 49.6) |
Number of tumour sites at baseline, median (IQR) | 3 (1 to 6) | NR |
Patients with ≥ 3 tumour sites at baseline, n (%) | 79 (75) | NR |
CNS involvement, n (%) | 4 (4) | NR |
Patients with 1 previous line of therapy, n (%) | 98 (93) | NR |
Patients with 2 previous lines of therapy, n (%) | 7 (7) | NR |
Sensitive to prior first-line of therapy, n (%) | 60 (57) | 117 (55) |
Refractory to prior first line of therapy, n (%) | 45 (43) | 96 (45) |
Median chemotherapy-free interval, months, median (IQR) | 3.5 (1.9 to 5.1) | 3.5 (NR) |
CTFI < 90 days, n (%) | 45 (43) | 93 (44) |
CTFI ≥ 90 days, n (%) | 60 (57) | 120 (56) |
CNS = central nervous system; CTFI = chemotherapy-free interval; ECOG PS = Eastern Cooperative Oncology Group performance status; IQR = interquartile range; LDH = lactate dehydrogenase; NA = not applicable; SCLC = small cell lung cancer.
aMeaningful imbalances as assessed by the standardized difference and proportion of the variance of ≥ 0.2 (20%).
Source: Sponsor-submitted STC report.14
Baseline characteristics obtained from each arm trial (Table 22) were assessed and compared to address important imbalances. The standardized difference and proportion of the variance as explained by group variance were used to describe imbalances, considering a standardized difference of 0.2 (20%) to denote meaningful imbalances in baseline covariates.
The outcomes matched and evaluated in this comparison include survival analysis end points: median OS and median PFS.
OS is a time-to-event outcome defined as time from randomization to death from any cause. Each study estimated and reported median OS. The outcomes model to fit median OS used parametric survival analysis to estimate and adjust for imbalanced effect modifiers and prognostic variables. Similarly, PFS is a time-to-event end point defined as the time from randomization to death or progression, whichever happens first. PFS was measured using documentation of radiographic disease progression or death from any cause at the time of data cut-off. Each study estimated and reported median OS and PFS.
The PFS and OS outcomes models for the STC used parametric survival analysis to estimate median OS and PFS and adjust for imbalanced effect modifiers and prognostic variables. Each of the 7 parametric distributions were tested, presented based on the best statistical fit, and validated through visual inspection. The effects of the covariates estimated in the outcomes model were applied to calculate a conditional median (prediction) using the means and/or proportions of the matched comparators’ imbalanced baseline characteristics. The results are interpreted as a mean difference, with values greater than 0 favouring lurbinectedin and denoting longer median PFS, and values less than 0 favouring topotecan and denoting shorter median PFS.
Results of the STC
Characteristics and Differences of Included Studies
The 2 studies included in the ITC had similar definitions for OS and PFS, and these were the 2 outcomes addressed in the body of evidence. However, as described in Table 21, there were discrepancies observed in the ascertainment of end points; for example, in the topotecan trial, PFS was determined by the investigator (unblinded), whereas in the lurbinectedin trial, it was determined by the IRC (with IA available as well). Another example is the phase of the study; the topotecan trial is a phase III, randomized study, whereas B-005 is single-arm, OL study.
For eligibility criteria, both the lurbinectedin and topotecan trials had similar inclusion criteria, with 3 exceptions. Overall, there were differences in the ECOG PS inclusion criteria, with a maximum PS of 2 in the lurbinectedin trial and of 1 in the topotecan trial. The time from diagnosis and its impact on CTFI was also different; 20% of patients in the lurbinectedin arm had a CTFI shorter than 30 days, whereas no patients in the topotecan arm did. Therefore, the STC authors restricted the lurbinectedin sample to patients with a CTFI longer than 30 days to match the topotecan arm. For the ECOG inclusion threshold, despite differences in the topotecan study eligibility criteria, the distributions of patients with a ECOG PS of 2 were similar in the topotecan and lurbinectedin trials.
The lurbinectedin trial reported the stage of disease at diagnosis, whereas the topotecan publication reported the stage of disease at study enrolment. The stage of disease at study enrolment for lurbinectedin patients could retroactively be assigned from the B-005 Clinical Study Report, using information on the number of patients with disease restricted to ipsilateral hemithorax at study entry.
When assessing differences in baseline patient characteristics between the lurbinectedin and topotecan trials (as assessed by the standardized difference), potential clinically meaningful imbalances were observed in ECOG PS and disease stage at diagnosis.
Clinical Efficacy End Points
Results for OS and PFS are presented in Table 23.
For OS, unadjusted estimates showed a median of 10.9 months (95% CI, 7.8 to 14.9 months) and 7.8 months (95% CI, 6.6 to 8.5 months) in the lurbinectedin and topotecan trials, respectively, with a mean difference of 3.1 months (95% CI, –0.6 to 6.8 months). When adjusted estimates were obtained, median OS was 10.0 months (95% CI, 8.5 to 11.6 months) and 7.8 months (95% CI, 6.6 to 8.5 months) in the lurbinectedin and topotecan trials, respectively, with a mean difference of 2.0 months (95% CI, 0.4 to 4.0 months).
For PFS, unadjusted estimates showed a median of 4.1 months (95% CI, 2.8 to 5.2 months) and 3.5 months (95% CI, 2.9 to 4.2 months) in the lurbinectedin and topotecan trials, respectively, with a mean difference of 0.60 months (95% CI, –0.76 to 1.96). When adjusted estimates were obtained, median PFS was 3.4 months (95% CI, 3.0 to 3.9 months) and 3.5 months (95% CI, 2.9 to 4.2 months) in the lurbinectedin and topotecan trials, respectively, with a mean difference of –0.10 months (95% CI, –0.89 to 0.69 months).
When testing model fitness, the most suitable distribution for modelling OS was determined to be the log-logistic. This was validated after consideration of each of the statistical indices and further validated with visual inspection. For PFS, the most suitable distribution for modelling PFS was determined to be the generalized gamma, because it had the smallest BIC. This was also validated after consideration of each of the statistical indices and further validated with visual inspection of each of the 7 curves overlaid on the Kaplan-Meier curve. There was nonconvergence in exponential, Weibull, log-normal, log-logistic, Gompertz, and simple gamma as a result of time-varying hazards.
Table 23: OS and PFS in the STC of Lurbinectedin and Topotecan
Characteristic | Lurbinectedin arm (B-005) N = 105 | Topotecan arm N = 213 | Mean difference lurbinectedin vs. topotecan |
|---|---|---|---|
Covariate distribution after restriction to CTFI > 30 days, n (%) | |||
Sex (male),% | 58 | 60 | NA |
Median age (years) | 61 | 61 | NA |
ECOG PS (0), % | 37 | 34 | NA |
SCLC stage (extensive), % | 92 | 88 | NA |
CTFI (≥ 90 days), % | 71 | 56 | NA |
OS, monthsa | |||
Unadjusted, median (95% CI) | 10.9 (7.8 to 14.9) | 7.8 (6.6 to 8.5) | 3.1 (–0.6 to 6.8) |
Adjusted, median (95% CI) | 10.0 (8.5 to 11.6) | 7.8 (6.6 to 8.5) | 2.0 (0.4 to 4.0) |
PFS, months a | |||
Unadjusted, median (95% CI) | 4.1 (2.8 to 5.2) | 3.5 (2.9 to 4.2) | 0.60 (–0.76 to 1.96) |
Adjusted, median (95% CI) | 3.4 (3.0 to 3.9) | 3.5 (2.9 to 4.2) | –0.10 (–0.89 to 0.69) |
CI = confidence interval; CTFI = chemotherapy-free interval; ECOG PS = Eastern Cooperative Oncology Group performance status; NA = not applicable; OS = overall survival; PFS = progression-free survival; SCLC = small cell lung cancer; STC = simulated treatment comparison.
aValues are median except for the last column.
Source: Sponsor-submitted STC report.14
Critical Appraisal of the STC
The most notable appraisal point of this ITC is the unanchored nature of the comparison. Even after adjustment for prognostic variables and effect modifiers, the consistency of the absolute-effects assumption in an STC cannot be validated. Unaccounted patient covariates and the risk of residual confounding is present and generates uncertainty in the effect estimates. It is unclear what process was used to choose the comparator trial. The identification of covariates to include in the adjustments was obtained through a literature review, but more information from clinical experts could reinforce the appropriate use of these variables.
The lurbinectedin and topotecan trials had dissimilarities in their design and populations (i.e., differences in eligibility criteria, design, ECOG PS, and disease stage) that can also produce bias. The lurbinectedin study sample size is small and, therefore, there will be limitations in the ability to detect differences in the comparative efficacy of lurbinectedin and a comparator (i.e., imprecision in the effect estimates).
Ascertainment by investigator in the topotecan trial was unblinded and could systematically bias the ascertainment of progression and, as a consequence, the effect estimates from the STC. The direction of this bias is unknown. The IRC is a strength of the lurbinectedin study; a committee of independent reviewers was used to standardize assessments across trial sites and, therefore, more accurately assign the date of progression.
Only OS and PFS could be assessed in this ITC, but HRQoL and AEs are considered important and valued end points to patients, clinicians, and other decision-makers. Although the population included is overall generalizable to the Canadian landscape, the differences in the ECOG PS (stricter in the topotecan trial) mean that patients might have a more stable status and/or fewer toxicities, which adds some uncertainty in the applicability of results of this STC.
Combined MAIC and NMA
Objectives
The objective of this ITC was to compare the efficacy and safety of lurbinectedin versus relevant comparators among patients with SCLC receiving second-line treatment with respect to ORR, DOR, OS, PFS, and specific hematological AEs of grade 3 or 4 (anemia, thrombocytopenia, neutropenia, and febrile neutropenia).
Study Selection Methods
Systematic Literature Search
First, a systematic literature review of clinical efficacy and safety was conducted to capture any relevant published information available on the second-line treatment of SCLC. A comprehensive database search of MEDLINE, Embase, and the Cochrane Register of Controlled Clinical Trials was conducted to identify RCTs, nonrandomized prospective trials, and single-arm trials eligible for inclusion. Searches of the US National Institutes of Health Clinical Trial Registry and 3 relevant conferences (American Society of Clinical Oncology, American Association for Cancer Research, and the International Association for the Study of Lung Cancer World Conference on Lung Cancer) were also performed. Two reviewers, working independently, conducted abstract selection, full-text selection, and data extraction. RCTs that reported data on tumour response, DOR, survival (overall or progression-free), or AEs were included. Nonrandomized trials or single-arm trials were included only when comparative RCTs were unavailable for an intervention of interest. Results of the systematic literature review provided the foundation from which to assess the feasibility of performing an ITC.
After screening and excluding single-arm and non-RCT studies with available comparative studies for an intervention of interest, 20 citations (12 full-text publications, 3 conference abstracts, 3 registry listings, and 2 client-provided documents) evaluating 10 unique trials were included. The 10 trials consisted of 5 RCTs, 2 single-arm trials, 2 RCTs with only 1 arm of interest, and 1 trial that included both randomized and nonrandomized cohorts with only 1 arm of interest.
Feasibility Assessment
A feasibility assessment was conducted to determine the appropriateness of proceeding with an NMA. The feasibility assessment process included 5 steps: a determination of whether the RCT evidence for the interventions of interest formed 1 connected network for the overall population and each outcome of interest and an assessment of the distribution of trial characteristics across the network; an assessment of the distribution of treatments; an exploration of the distribution of baseline patient characteristics within and between comparisons to identify factors that may bias indirect estimates (i.e., identify effect modifiers); an assessment of outcome definitions and outcome availability; and an assessment of the variability in observed relative treatment effects.
The full evidence network of the included trials is shown in Figure 8: the lurbinectedin single-arm basket trial (study B-005); 4 RCTs with data available in publications (Baize [2020], O’Brien [2006], Eckardt [2007], von Pawel [2001]); 1 RCT with data available in the form of a registry listing [United Therapeutics (2017)]); 2 RCTs with only 1 arm of interest (Owonikoko [2019], CheckMate 331), 1 trial with both a randomized and nonrandomized cohort with only 1 arm of interest (CheckMate 032); and 1 single-arm trial (Smyth [1994]). Figure 8 also describes platinum-sensitivity status by study.
Figure 8: Network of Trials Included in the Feasibility Assessment
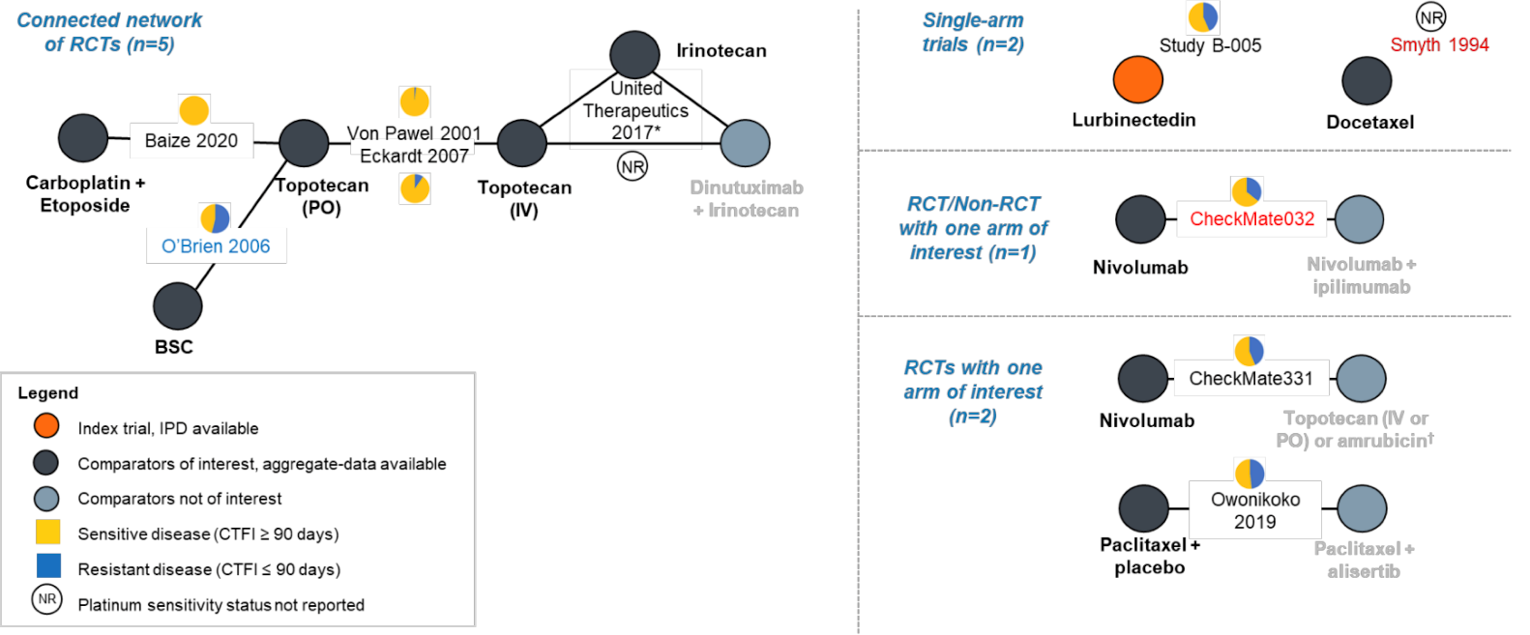
CTFI = chemotherapy-free interval; IPD = individual patient data; NR, not reported; PO = orally; RCT = randomized controlled trial.
Note: red text = trials excluded from analyses; blue text = trial excluded from base case but included in sensitivity analysis.
* United Therapeutics (2017) is a 2-part study; only part 2 is a relevant RCT (part 1 = dose escalation).
† Investigator’s choice: intervention considered not relevant, given that results are not stratified by treatment.
Source: Sponsor-submitted MAIC and NMA report.15
Among the 10 trials included in the feasibility assessment, there were some differences in overall study design, with a mix of RCTs (either OL or double-blind) and single-arm trials included in the evidence base. Eight of the 10 trials were international studies, Baize (2020) was conducted at multiple centres exclusively in France, and Smyth (1994) did not report study location. All trials included patients with limited or extensive disease, except for Smyth (1994), which was restricted to extensive disease only. Three trials were designed to enrol patients with sensitive disease (CTFI ≥ 90 days) (Baize [2020], Eckardt [2007], von Pawel [2001]), 6 trials recruited a mix of patients with sensitive (CTFI ≥ 90 days) and resistant (CTFI < 90 days) disease (O’Brien [2006], United Therapeutics [2017], Owonikoko [2019], CheckMate 331, CheckMate 032, study B-005), and the population type was unknown in 1 trial (Smyth [1994]). All trials focused on the second-line setting, except for CheckMate 032 (second-line and beyond) and Smyth (1994) (first-line and second-line).
Of the 10 trials in the evidence base, 5 RCTs formed a connected network (Baize [2020], O’Brien [2006], von Pawel [2001], Eckardt [2007], and United Therapeutics [2017]), 5 trials were disconnected (study B-005, Smyth [1994], Owonikoko [2019], CheckMate 032, and CheckMate 331)., The network of trials included in the final base-case ITC for patients with platinum-sensitive disease is depicted in Figure 9.
Figure 9: Network of Trials Included in the Base-Case ITC for Patients With Platinum-Sensitive Disease
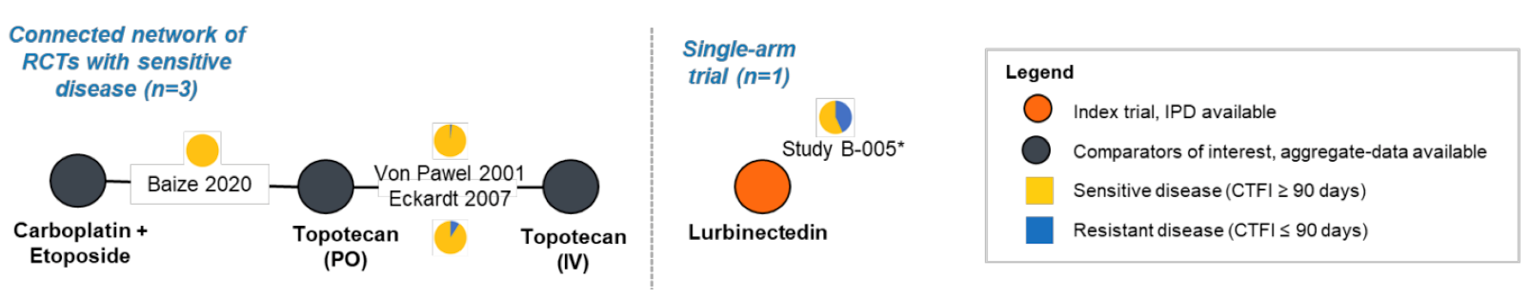
CTFI = chemotherapy-free interval; IPD = individual patient data; ITC = indirect treatment comparison; PO = orally; RCT = randomized controlled trial.
* Based on the CTFI ≥ 90 days subgroup.
Source: Sponsor-submitted MAIC and NMA report.15
ITC Analysis Methods
After feasibility was confirmed, an unanchored MAIC was conducted to inform the NMA. Once study B-005 was connected to either Baize (2020) or Eckardt (2007), the MAIC-based relative treatment effect (i.e., effect of lurbinectedin relative to topotecan) was considered as if it was a direct estimate of the relative treatment effect (Figure 10).
Figure 10: Evidence Networks for Base-Case Analysis in Patients With Platinum-Sensitive Disease
ITC = indirect treatment comparison; MAIC = matching-adjusted indirect comparison; PO = orally.
Note: (A.) Network for ORR, OS, grade 3 or 4 anemia, grade 3 or 4 thrombocytopenia, and grade 3 or 4 neutropenia. (B.) Network for PFS and grade 3 or 4 for CTFI ≥ 90-day subgroup (n = 60). Orange node: index trial with IPD. Yellow in pie charts: sensitive disease (CTFI ≥ 90 days). Blue in pie charts: resistant disease (CTFI ≤ 90 days). Trial names in red italic represent trials used to connect study B-005 to the network via an MAIC.
* Based on the CTFI ≥ 90 days subgroup
Source: Sponsor-submitted MAIC and NMA report.15
The studies included in this network are described in Table 24.
Table 24: Summary of Study Characteristics of Included Trials in the Base-Case Analysis
Trial | Line of therapy | Interventions | Study phase | N | Location | Study date | Baseline stratification factors | Median follow-up (months) |
|---|---|---|---|---|---|---|---|---|
Baize (2020) | Second | Arm 1: Topotecan (oral) Arm 2: Carboplatin + etoposide (IV) | Phase III, OL | 164 | France | 2013 to 2017 | ECOG PS (0 or 1 vs. 2); response to first-line (PR vs. CR) | 22.7 |
Study 396 (Eckardt [2007]) | Second | Arm 1: Topotecan (oral) Arm 2: Topotecan (IV) | Phase III, OL | 309 | International | 1999 to 2001 | Sex, CTFI (≤ 6 months or > 6 months); presence of liver disease (yes vs. no) | NR |
Study 065 (von Pawel [2001]) | Second | Arm 1: Topotecan (oral) Arm 2: Topotecan (IV) | Phase II, OL | 106 | International | NR | Disease stage (LS vs. ES); CTFI (3 to 6 months or ≥ 6 months); presence of liver disease (yes vs. no) | NR |
Study B-005 (Trigo [2020]) | Second | Arm 1: Lurbinectedin (IV) | Phase II, OL, single-arm | 105 | International | 2015 to 2021 | NA | 17.1 |
CR = complete response; CTFI = chemotherapy-free interval; ECOG PS = Eastern Oncology Cooperative Group performance status; ES = extensive stage; LS = limited stage; NA = not applicable; NR = not reported; OL = open label; PR = partial response.
Source: Sponsor-submitted MAIC and NMA report.15
Unanchored MAIC
Since the intervention of interest is represented by 1 single-arm study, it cannot be connected to the network of RCT evidence, and there is no relative treatment-effect estimate to incorporate in an NMA. To address these deficiencies, for each of the end points of interest, an unanchored MAIC was conducted comparing study B-005 with the reference group of a trial that was already part of the network. This contrast was then connected to the network and the estimated treatment differences used in NMAs estimating relative treatment effects for all treatments in the network.
For the MAIC, propensity score weights were used to adjust for differences between the population in the index trial (i.e., study B-005) and the population in the external AD trial that was already part of the network and was used to link study B-005 to the network. The estimation of these propensity weights was performed using a modified likelihood reweighting approach, which estimates weights from a logistic regression model. The method-of-moments approach, outlined by Signorovitch, was used to balance the mean covariate values across populations. The weighting scheme was based only on the covariates included and was therefore independent of the outcome. To evaluate the distribution of the patient characteristics and the effect and appropriateness of the weighting process, the weights were rescaled relative to the unit weights of the original IPD dataset based on sample size, which facilitated the interpretation of the distribution of weights.
Potentially important treatment-effect modifiers were identified from a literature search that produced an evidence base of relevant publications. These initial variables included platinum sensitivity (sensitive versus resistant), disease stage (limited versus extensive), ECOG PS score (0 versus 1 versus 2), and race (Asian versus non-Asian).
Later, MAIC weights were generated for the index trial to match each of the external trials. Based on the availability of baseline characteristics from each study, the covariates of interest for the MAIC models were age, sex, ECOG PS score, and disease stage.
A measure of the extent of overlap between the index trial and the external AD trial of interest was represented by the effective sample size, an adjustment of the sample size that accounts for the weighting of the observations and the resulting correlations between estimated responses. Large reductions in the effective sample size are not desirable because they imply a poor overlap between IPD and AD studies and introduce uncertainty.
Differences between the index trial and the external trial for each covariate were measured using the standardized mean difference, which ranges from 0% to 100%, and allows for comparisons of covariates measured in different units (e.g., age in years versus percentage of males). For a dichotomized covariate, the standardized mean difference for naive comparisons was calculated. A large standardized difference denotes that there is poor overlap between the 2 trials. For MAIC, the patient weights were calculated such that the weighted covariate mean of the index trial matched the reported covariate mean of the external trial. An exact match equated to a difference of 0; however, not all MAIC weight models resulted in differences of 0 for each covariate. The standardized difference for weighted comparisons for binary covariates was therefore calculated; a covariate was considered balanced when the absolute standardized difference was less than or equal to 10%.
To quantify the improvement after applying MAIC weights across all covariates of interest, 2 diagnostic measures based on standardized difference were used: overall improvement across the covariates of interest; and the number of covariates with improved balance using 10% as the cut point. An external trial with a larger overall improvement, after applying weights, indicated its covariates had poorer overlap with the index trial, compared with another external trial with a smaller overall improvement. Based on this assessment, the external trial with the most overlap with the index trial population was selected for the base-case analysis.
Once the index trial (B-005) was connected to an external trial by applying MAIC weights, outcomes for the index treatment were predicted for the target population by reweighting the observed outcomes from the index trial. Treatment comparisons were then made based on differences between the weighted averages from the index trial and the observed outcome from the AD study evaluating the competitor in the target population. These treatment comparisons were then considered pseudodirect comparisons in the NMA.
Network Meta-Analysis
Once study B-005 was connected to either the Baize (2020) or Eckardt (2007) studies, the MAIC-based relative treatment effect (i.e., effect of lurbinectedin relative to topotecan) was considered as if it was a direct estimate of relative treatment effect.
Because there were no closed loops in any evidence networks, it was not feasible to assess the consistency between direct and indirect comparisons in the current NMA.
The deviance information criterion (DIC) (a measure of model fit that penalizes model complexity), in addition to an assessment of overall stability, was used to guide the identification of the appropriate model (i.e., fixed or random effects). A more complex model results in a better fit to the data, demonstrating a smaller residual deviance. Normal noninformative prior distributions of the parameters were used with a mean of 0 and a variance of 10,000. Both fixed and random-effects models were considered for each analysis. For the random-effects models, 1 parameter for the between-study heterogeneity was used, assuming that the between-study heterogeneity was the same for each intervention relative to the overall reference treatment of choice. Given the limited evidence base, random-effects models lead to unstable estimates; therefore, fixed-effects models were preferred.
For time-to-event data, the proportional hazard assumption regarding time-to-event outcomes for each individual trial was assessed using the Grambsch and Therneau test and visual inspection of log-log plots. The NMA of reported HRs in terms of OS (assuming proportional hazards between treatments) was performed using a regression model with a contrast-based normal likelihood for the log HR (and corresponding standard error) of each trial (or comparison) in the network. If a Kaplan-Meier curve was not presented in the AD study and a median survival was reported instead, the median survival was estimated from the IPD of the lurbinectedin study, before and after applying weights. Because a detailed distribution from AD was not available, no formal test was performed to assess the relative treatment effect.
For binary outcomes (ORR and grade 3 or 4 AEs), the number of patients with the event and the number without were used to determine the probability of an event in the target population. The NMA could then be performed based on the proportion of patients experiencing the event of interest using a logistic regression model with a binomial likelihood and logit link. Relative treatment effects were expressed as ORs. A regression model with a contrast-based normal likelihood for the log OR (and corresponding standard error) of each comparison in the network was performed. Similarly, safety outcomes NMAs were performed using OR as input data.
The base-case analyses included patients with platinum-sensitive disease (defined as CTFI longer than 90 days), and sensitivity analyses were based on patients with any platinum-sensitivity status. The outcomes assessed were OS, PFS, ORR, DOR (only MAIC performed because only median DOR reported in external studies), and harms (specific hematological AEs of grade 3 or 4 anemia, thrombocytopenia, neutropenia, and febrile neutropenia).
Results of the Combined MAIC and NMA
The baseline patient characteristics of interest for the MAIC were age, sex, ECOG PS, and disease stage; an additional variable of platinum sensitivity was considered when the full population of study B-005 was considered. The distribution of these variables is shown for the included studies in Table 25.
Table 25: Summary of Baseline Patient Characteristics in Study B-005 and RCTs in the Connected Network for the Platinum-Sensitive Disease Population
Treatment | Line of therapy | N | Age, median | Male, n (%) | ECOG PS 0, n (%) | ECOG PS 1 n (%) | ECOG PS 2, n (%) | LS disease stage n (%) | ES disease stage n (%) | Resistant to 1L n (%) | Sensitive to 1L n (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
Baize (2020) | |||||||||||
Carboplatin + etoposide (IV) | 2L | 81 | 64.0 | 50 (62.0) | 28 (35.0) | 48 (59.0) | 5 (6.0) | 29 (36.0) | 52 (64.0) | 0 (0.0) | 81 (100) |
Eckardt (2007) | |||||||||||
Topotecan (IV) | 2L | 151 | 62.0 | 96 (63.6) | 35 (23.2) | 98 (64.9) | 18 (11.9) | 45 (29.8) | 106 (70.2) | 13 (8.6) | 137 (90.7) |
von Pawel (2001) | |||||||||||
Topotecan (IV) | 2L | 54 | 58.2 | 43 (79.6) | 18 (33.3) | 21 (38.9) | 15 (27.8) | 14 (25.9) | 39 (72.2) | 1 (1.8) | 53 (98.1) |
Study B-005 | |||||||||||
Lurbinectedin IV (treated population) | 2L | 105 | 60.0 | 63 (60.0) | 38 (36.2) | 59 (56.2) | 8 (7.6) | 32 (30.5) | 73 (69.5) | 45 (42.9) | 60 (57.1) |
Lurbinectedin IV (subgroup CTFI > 90 days) | 2L | 60 | 59.0 | 35 (58.3) | 27 (45.0) | 30 (50.0) | 3 (5.0) | 25 (41.7) | 35 (58.3) | 0 (0.0) | 60 (100.0) |
1L = first-line; 2L = second-line; ECOG PS = Eastern Cooperative Oncology Group performance status; ES = extensive stage; LS = limited stage, RCT = randomized controlled trial.
Source: Sponsor-submitted MAIC and NMA report.15
Overall Survival
The MAIC-adjusted HRs of OS for lurbinectedin compared with other treatments from an external trial of interest are presented in Table 26.
In the base-case NMA for OS (study B-005 sensitive subgroup connected to Baize [2020]), lurbinectedin had an HR of 0.43 (95% CrI, 0.26 to 0.70) against IV topotecan and of 0.42 (95% CrI, 0.30 to 0.58) against carboplatin plus etoposide. When study B-005 was connected to the Eckardt (2007) study (i.e., sensitivity analyses), the relative treatment effects of these comparisons were smaller (HR closer to 1) and no longer statistically significant whether lurbinectedin was compared to IV topotecan (HR = 0.75; 95% CrI, 0.56 to 1.03) or carboplatin plus etoposide (HR = 0.74; 95% CrI, 0.50 to 1.09).
Results from the random-effects models had similar HR estimates similar to the corresponding fixed-effect models, whereas the CrIs were much wider, as expected due to the small sample sizes that studies provided to the networks. The DIC for the random-effects models were slightly greater than those for the fixed-effect models (e.g., in the base case, DIC was 5.31 for the fixed-effects model versus 6.66 for the random-effects model).
Table 26: Results of MAIC for OS for Lurbinectedin (Study B-005) Versus Comparators (Baize [2020] or Eckardt [2007])
Study (treatment) | N | HR (95% CI) | ESS |
|---|---|---|---|
MAIC, base case (patients with sensitive disease)a | |||
Baize (2020) (carboplatin + etoposide)b | 77 | Reference | NA |
Study B-005 (lurbinectedin)b | 60 | 0.42 (0.27 to 0.65) | 37.98 |
MAIC, sensitivity analysis 1 and 3 (patients with any platinum-sensitivity status) | |||
Eckardt (2007) (IV topotecan)a,b | 151 | Reference | NA |
Study B-005 (lurbinectedin)a,b | 105 | ███ █ █████ █ █████ | █████ |
MAIC, sensitivity analysis 2 (patients with sensitive disease [CTFI ≥ 90 days]) | |||
Eckardt (2007) (IV topotecan)b | 151 | Reference | NA |
Study B-005 (lurbinectedin)b | 60 | ███ █ █████ █ █████ | █████ |
CI = confidence interval; CTFI = chemotherapy-free interval; ESS = effective sample size; HR = hazard ratio; MAIC = matched adjusted indirect comparison; NA = not applicable; OS = overall survival.
aConsidered as pseudodirect evidence.
bUsed as validation for NMA.
Source: Sponsor-submitted MAIC and NMA report.15
Progression-Free Survival
Among the connected RCTs, Baize (2020) reported PFS, whereas von Pawel (2001) and Eckardt (2007) only reported time to progression. The definitions for PFS and time to progression differed. Only studies that reported PFS were analyzed. The proportional hazard assumption was violated for Baize (2020) and Eckardt (2007). Fractional polynomial models were fitted to estimate time-varying HRs, instead of using the reported constant HR for the NMA. Weighted number at risk and weighted number of events from study B-005 and IPD reconstructed from external trial Kaplan-Meier curves were prepared as input for the NMA (rather than an MAIC), but further details were not provided.
The time-varying HR estimates indicated that carboplatin plus etoposide was associated with a higher HR for PFS than lurbinectedin at first (i.e., HR greater than 1), but the HR curve had a steep decline and by month 3, the direction of the HR changed (i.e., was less than 1), as shown in Table 27. From 6 months onward, PFS was longer with lurbinectedin than carboplatin plus etoposide, although HR estimates beyond 12 months were based on model extrapolation.
Table 27: Estimated PFS HRs Over Time for Lurbinectedin Versus Competing Interventions From Fixed-Effect Fractional Polynomial NMA
Lurbinectedin comparator | Time-varying HR (95% CrI) | |||||
|---|---|---|---|---|---|---|
3 months | 6 months | 9 months | 12 months | 18 months | 24 months | |
Carboplatin plus etoposide | █ █ █ █ ██ | █ █ ██ █ ██ | █ █ ██ █ ██ | █ █ ██ █ ██ | █ █ █ █ ██ | █ █ ██ █ ██ |
CrI = credible interval; HR = hazard ratio; NMA = network meta-analysis; PFS = progression-free survival.
Note: Cells shaded in grey indicate estimates based on model extrapolations.
Source: Sponsor-submitted MAIC and NMA report.15
Objective Response Rate
The MAIC-adjusted ORs of the ORR for lurbinectedin versus other treatments from an external trial of interest are presented in Table 28.
Table 28: Results of MAIC for ORR for Lurbinectedin (Study B-005) Versus Relevant Comparators (Baize [2020] or Eckardt [2007])
Study (treatment) | N | Number of events | Event rate (%) | OR (95% CI) | ESS |
|---|---|---|---|---|---|
MAIC, base case (patients with sensitive disease [CTFI ≥ 90 days]) | |||||
Baize (2020) (carboplatin + etoposide) | 79 | 39 | 49 | Reference | NA |
Study B-005 (lurbinectedin) | 60c | 27.98 | 47 | 0.90 (0.41 to 1.95) | 37.98 |
MAIC, sensitivity analysis 1 (patients with any platinum-sensitivity status) | |||||
Eckardt (2007) (IV topotecan)a | 151 | 33 | 22 | Reference | NA |
Study B-005 (lurbinectedin)a | 105d | 43.72 | 42 | ███ █ ██ █ ████ | 56.92 |
MAIC sensitivity analysis 2 (patients with sensitive disease [CTFI ≥ 90 days]) | |||||
Eckardt (2007) (IV topotecan) | 151 | 33 | 22 | Reference | NA |
Study B-005 (lurbinectedin)b | 60c | 26.32 | 44 | ███ █ ██ █ ████ | 44.86 |
CI = confidence interval; CTFI, chemotherapy-free interval; ESS = effective sample size; MAIC = matching-adjusted indirect comparison; OR = odds ratio; ORR = objective response rate.
Note: For response outcomes, OR > 1 favours lurbinectedin.
aConsidered to be pseudodirect evidence.
bUsed as validation for NMA.
cOne of 60 patients was not evaluable and was assumed to have no response.
dFive of 105 patients were not evaluable and were assumed to have no response.
Source: Sponsor-submitted MAIC and NMA report.15
Similar to OS, because oral topotecan was the central node of the network, the ORs for lurbinectedin versus oral topotecan were used as input for the NMA. Results from the ORR fixed-effect NMA are presented.
In the base-case analysis (incorporating study B-005 sensitive subgroup via Baize [2020]), no evidence of difference was detected between lurbinectedin versus IV topotecan (OR = 2.36; 95% CrI, 0.89 to 6.23) or between lurbinectedin and carboplatin plus etoposide (OR = 0.85; 95% CrI, 0.40 to 1.83).
When study B-005 (in the any platinum-sensitivity subgroup) was connected to the network via Eckardt (2007) (i.e., sensitivity analyses), lurbinectedin demonstrated greater odds of an ORR than IV topotecan (OR = 2.71; 95% CrI, 1.46 to 4.96), but no difference was detected between lurbinectedin and carboplatin plus etoposide (OR = 0.96; 95% CrI, 0.38 to 2.46).
Similarly, lurbinectedin presented greater odds for an ORR in the sensitivity analysis of patients with platinum-sensitive disease connected via Eckardt (2007) when compared against IV topotecan (OR = 2.93; 95% CI, 1.54 to 5.6) but not against carboplatin plus etoposide (OR = 1.07; 95% CrI, 0.41 to 2.82).
When random-effects models were used to capture heterogenous between-trial treatment effects, the OR point estimates were comparable to the corresponding fixed-effect models; however, the CrIs were much wider, as expected. In addition, DICs for the random-effects models were slightly larger than those for the fixed-effect models (e.g., in the base case, DIC was 6.4 for the fixed-effect model versus 6.98 for the random-effects model).
Duration of Response
In the connected network of RCTs, only 3 trials reported median DOR (Baize [2020], von Pawel [2001] and Eckardt [2007]), with 1 providing 95% CIs and 2 providing ranges, but relative treatment-effect estimates (i.e., HR) or Kaplan-Meier curves were not available. Only an unanchored MAIC was performed for DOR, and only treatment-level data were reported, as no relative treatment effects could be estimated.
When the study B-005 sensitive subgroup was considered as the third arm of the Baize (2020) study, the median DOR for lurbinectedin was ██ █ █████ █ ███ █ ██ █ ███ █ █ █ ███ █ ███████, whereas the median DOR for carboplatin plus etoposide was ██ █ █████ █ ███ █ ██ █ ██ █ █ █ ██ █ ███████ with overlapping 95% CIs (see Table 29).
When the study B-005 full population was connected to Eckardt (2007) (sensitivity analysis 1), the median DOR for lurbinectedin was ███ █ █████ █ ███ █ ██ █ ███ █ █ █ ███ █ ███████. Similarly, when the study B-005 platinum-sensitive subgroup was connected to Eckardt (2007) (sensitivity analysis 2), the median DOR for lurbinectedin was ██ █ █████ █ ███ █ ██ █ ███ █ █ █ ███ █ ███████; the DOR estimate was similar regardless of the external study used to connect study B-005 to the network.
Harms
To present an overall description of the harms outcomes of interest in each study included in the NMAs, Table 30 depicts a summary of data across the trials included.
Table 29: Results of MAIC for DOR for Lurbinectedin (Study B-005) Versus Comparators (Baize [2020] or Eckardt [2007])
Adjustment | Study (treatment) | N | DOR (months), median | Measure of dispersion |
|---|---|---|---|---|
Base casea | ||||
No adjustment | Baize (2020) (carboplatin + etoposide) | ██ | ███ | ██ █ ██ █ ██ █ █ █ ███ |
Naive | Study B-005 (lurbinectedin) | ██ | ████ | ██ █ ██ █ ███ █ █ █ ████ |
MAIC | Study B-005 (lurbinectedin) | ████ | ████ | ██ █ ██ █ ███ █ █ █ ████ |
Sensitivity analysis 1b | ||||
No adjustment | Eckardt (2007) (IV topotecan) | ███ | ███ | ████ █ ██ █ █ █ ████ |
Naive | Study B-005 (lurbinectedin) | ██ | ████ | ██ █ █ █ ███ █ █ █ ████ |
MAIC | Study B-005 (lurbinectedin) | ████ | ████ | ██ █ █ █ ███ █ █ █ ████ |
Sensitivity analysis 2c | ||||
No adjustment | Eckardt (2007) (IV topotecan) | ███ | ███ | █████ █ ██ █ █ █ ████ |
Naive | Study B-005 (lurbinectedin) | ██ | ████ | ██ █ ██ █ ███ █ █ █ ████ |
MAIC | Study B-005 (lurbinectedin) | ████ | ████ | ██ █ ██ █ ███ █ █ █ ████ |
DOR = duration of response; MAIC = matching-adjusted indirect comparison.
Note: Only median DOR was available for Baize (2020) and Eckardt (2007), and comparative estimates could not be generated.
aPatients with sensitive disease (CTFI ≥ 90 days) and study B-005 connected via Baize (2020).
bPatients with any platinum-sensitivity status and study B-005 connected via Eckardt (2007).
cPatients with sensitive disease (CTFI ≥ 90 days) and study B-005 connected via Eckardt (2007).
Source: Sponsor-submitted MAIC and NMA report.15
Table 30: Summary of Harms Outcomes Across Trials Included in the NMA
Treatment | Analysis population, ITT or subgroup | N | Grade 3 or 4 anemia, n (%) | Grade 3 or 4 thrombocytopenia, n (%) | Grade 3 or 4 neutropenia, n (%) | Grade 3 or 4 febrile neutropenia, n (%) |
|---|---|---|---|---|---|---|
Baize (2020) | ||||||
Carboplatin + etoposide (IV) | ITT, CTFI ≥ 90 days | 81 | 20 (24.7) | 25 (30.9) | 11 (13.6) | 5 (6.0) |
O'Brien (2006) | ||||||
Best supportive care | ITT | 70 | NA | NA | NA | NA |
Eckardt (2007) | ||||||
Topotecan (IV) | ITT, CTFI ≥ 90 daysa | 151 | 46 (30.5) | 65 (43.0) | 130 (86.1) | NA |
von Pawel (2001) | ||||||
Topotecan (IV) | ITT, CTFI ≥ 90 daysa | 54 | (30.2) | (49.0) | (94.2) | NA |
Study B-005 (Trigo [2020] and Clinical Study Report; IPD) | ||||||
Lurbinectedin (IV) | Treated population | 105 | 9 (8.6) | 7 (6.7) | 48 (45.7) | 5 (4.8) |
Lurbinectedin (IV) | Sensitive subgroup, CTFI ≥ 90 days | 60 | 5 (8.3) | 4 (6.7) | 26 (43.3) | 1 (1.7) |
CTFI = chemotherapy-free interval; IPD = individual patient data; ITT = intention-to-treat; NA = not available; NMA = network meta-analysis.
Note: Either treatment-related or any hematological AEs were extracted.
aApproximately 10% of patients in both treatment groups had a CTFI < 90 days at study entry.
bApproximately 2% of patients in both treatment groups had a CTFI < 90 days at study entry.
Source: Sponsor-submitted MAIC and NMA report.15
MAIC Estimates
MAIC estimates of harms in the lurbinectedin arm compared to relevant comparators are presented in Table 31. When observing the MAIC estimates (ORs) for harms in the base-case analysis, lurbinectedin was associated with a lower incidence of grade 3 or 4 anemia compared with carboplatin plus etoposide (█ █ █ ████ █ ██ █ █ █ ████ █ ████). Similarly, the adjusted estimates for grade 3 or 4 thrombocytopenia for lurbinectedin versus carboplatin plus etoposide showed lower odds of harm (OR = 0.23; 95% CI, 0.06 to 0.95).
For grade 3 or 4 thrombocytopenia, lurbinectedin showed lower odds when compared with carboplatin plus etoposide (█ █ █ ████ █ ████ █ ███ █ █ █ ████), and odds were similar across sensitivity analyses.
However, lurbinectedin had higher odds of neutropenia grade 3 or 4 (█ █ █ ████ █ ██ █ █ █ ████ █ █████) when compared to carboplatin plus etoposide, but this effect was the opposite when lurbinectedin was evaluated against IV topotecan in the group of patients with any platinum sensitivity (sensitivity analysis) (█ █ █ ████ █ ██ █ █ █ ████ █ ████) and when the patients with sensitive disease (CTFI > 90 days) were observed (█ █ █ ████ █ ██ █ █ █ ████ █ ████). These differences in neutropenia rates were deemed by the authors to be related to differences in the requirements for prophylaxis with G-CSF across studies (i.e., primary prophylaxis with G-CSF was not permitted in study B-005 or in Eckardt [2007], but was recommended for all patients in Baize [2020]; thus, grade 3 or 4 neutropenia rates were lower in Baize [2020]).
Table 31: Results of MAIC for Harms of Lurbinectedin (Study B-005) Versus Comparators (Baize [2020] or Eckardt [2007])
Study (treatment) | N | Number of events | Event rate (%) | OR (95% CI) | ESS |
|---|---|---|---|---|---|
Grade 3 or 4 anemia | |||||
Baize (2020) (carboplatin + etoposide) | 81 | 20 | 25.0 | Reference | NA |
Study B-005 (lurbinectedin) | 60 | 4.1 | 7 | ███ █ ████ █ ████ | 37.98 |
Grade 3 or 4 thrombocytopenia | |||||
Baize (2020) (carboplatin + etoposide) | 81 | 25 | 31 | Reference | NA |
Study B-005 (lurbinectedin) | 60 | 5.6 | 9 | ███ █ ███ █ ████ | 37.98 |
Grade 3 or4 neutropenia | |||||
Baize (2020) (carboplatin + etoposide) | 81 | 11 | 14 | Reference | NA |
Study B-005 (lurbinectedin) | 60 | 31.6 | 53 | ███ █ ███ █ ████ | 37.98 |
CI = confidence interval; ESS = effective sample size; MAIC = matching-adjusted indirect comparison; NA = not available; NMA = network meta-analysis; OR = odds ratio.
Note: All analyses are base case.
Source: Sponsor-submitted MAIC and NMA report.15
NMA Estimates
The NMA estimates for grade 3 or 4 anemia in the base-case analysis showed lower odds of harm in the lurbinectedin arm than in the carboplatin plus etoposide arm (OR = 0.22; 95% CrI, 0.08 to 0.61) and than in the IV topotecan arm (OR = 0.21; 95% CrI, 0.06 to 0.74), with consistent results in the sensitivity analyses.
Similarly, for thrombocytopenia grade 3 or 4, the base-case analysis showed lower odds (OR = 0.23; 95% CrI, 0.08 to 0.69), and odds were consistent in the sensitivity analyses.
For the end point of neutropenia grade 3 or 4, higher odds were observed in the lurbinectedin arm than in the carboplatin plus etoposide arm (OR = 7.05; 95% CrI, 3.09 to 16.11), but not than in the IV topotecan arm (OR = 1.19; 95% CrI, 0.45 to 3.17). In the sensitivity analyses (patients with any platinum sensitivity and patients with platinum-sensitive disease), the odds of neutropenia were lower in the lurbinectedin arm than in the carboplatin plus etoposide arm, as described in the MAIC analysis.
Critical Appraisal of the Combined MAIC and NMA
A systematic literature process is defined in a separate technical report. Overall, the methods for the systematic literature search and study selection were well described and appropriate, including the search strategy, screening process, and quality assessment of individual studies.
This ITC represents 2 connected evidence syntheses. First, IPD from 2 different analysis populations from study B-005 (platinum-sensitive subgroup and full population of patients with any platinum-sensitivity status) were used to perform the unanchored MAICs to connect the lurbinectedin arm and AD from 1 of 2 other trials that included relevant comparators (either Baize [2020] or Eckardt [2007], depending on the outcome evaluated). Second, estimates of treatment effect from the MAIC were included in the NMA, with the rest of the studies by essentially treating it as direct evidence. The validity of this technique is unknown. The main concern is the inclusion of uncertainty due to lack of holding the transitivity assumptions. Sparsity of the network is also a concern, together with concerns about important differences between included studies.
In an unanchored MAIC, for the approach to be a valid comparison, the investigators would need to achieve balance on all prognostic factors and all effect modifiers between each arm of treatment by including all such factors in a weighting process to make the population similar for the evaluation of efficacy and safety end points. The effective sample size in different analyses presents moderate to large decreases from the original data, which suggests an inadequate overlap of covariate distribution between studies. The choice of covariates for the MAIC models was limited by the availability of baseline patient characteristics in the external RCTs. It was not feasible to adjust for covariates that were not reported or unknown in the external studies, and it was not feasible to adjust for differences in study design or location. These covariates of interest (age, sex, ECOG PS, disease stage, and platinum sensitivity) were selected based on those reported in the trials, rather than a comprehensive literature search and/or clinical expert advice. For instance, the presence of CNS metastases was considered important for inclusion in the overall MAIC analysis by experts consulted by CADTH.
There were also uncertainties due to concerns about violations of the proportional hazards assumptions for the end points, although the authors attempted to address this issue by using fractional polynomials to model in situations where the proportional hazards assumptions did not match.
In the NMA, the first issue is the addition of a single-arm trial through an unanchored MAIC, which increases risk of bias due to loss of randomization benefits. Further strong assumptions were made to form this connected network (e.g., disease stage comparable across trials, no significant differences between IA or IRC assessment for tumour response). Despite performing an extensive feasibility assessment to carefully select the trials for inclusion in the network, the assumption of consistency may not hold. Furthermore, there were no closed loops in any evidence networks, so it was not feasible to assess the consistency between direct and indirect comparisons.
The analysis of DOR was limited by data availability in the external trials; no relative treatment effect (i.e., HR) or Kaplan-Meier curves was available. Although median DORs were reported, standard error for median DOR was unavailable for the external RCTs in the connected network. Therefore, only an unanchored MAIC was performed for this outcome, as strong assumptions would be required to incorporate these data into an NMA (e.g., assumptions that the difference in median DOR is normally distributed and that the standard error can be estimated correctly based on the ranges).
The population of the MAIC from the IPD arm (study B-005) is, overall, generalizable to the target population for the indication in this CADTH submission (i.e., patients with SCLC receiving second-line treatment); however, as noted, the populations from the comparator arms had differences that did not align with the target population of interest, such as ECOG PS, disease stage, and platinum-sensitivity status. Furthermore, some comparators relevant to this CADTH review explained in the protocol of this report (i.e., CAV, irinotecan) were not included in this MAIC and NMA.
Harms data could be of different maturity between trials and have an impact on the certainty on these effect estimates. For example, some differences noted (heterogeneity) between the effect estimates in different sensitivity analyses were detected in the neutropenia grade 3 or 4 end point. However, duration of treatment (median number of cycles administered) was similar across studies. Last, no information on HRQoL was assessed in this ITC, which was an important outcome for this CADTH report.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Discussion
Summary of Available Evidence
One phase II, multicentre, single-arm, OL basket trial (B-005)31 designed to evaluate the efficacy and safety of lurbinectedin in previously treated adult patients with advanced solid tumours, including SCLC (N = 105), contributed evidence to this report. The study enrolled adult patients with SCLC who had received 1 prior line of chemotherapy for advanced disease, had an ECOG PS score of 2 or less, and did not have CNS involvement. Patients received OL lurbinectedin (3.2 mg/m2) until disease progression or unacceptable toxicity. The primary outcome of the study was ORR per IA; secondary outcomes included ORR per IRC, DOR per IA and IRC, PFS per IA and IRC, and OS. According to the clinical experts consulted by CADTH for this review, the baseline characteristics of the B-005 study population were broadly representative of patients with SCLC in Canada who have progressed on or after first-line platinum etoposide doublet therapy. The median age was 60 years, most patients had an ECOG PS of 0 (36.2%) or 1 (56.2%), and most patients had ES disease (93.3%) at study entry; according to the clinical experts consulted by CADTH for this review, the study population was younger and had better performance status than the general SCLC patient population, as expected for any clinical trial population. Proportions of patients with platinum-refractory or platinum-resistant disease (42.9% for CTFI < 90 days) and platinum-sensitive disease (57.1% for CTFI ≥ 90 days) were similar.
Three sponsor-submitted ITCs are included in this CADTH report. The first ITC evaluated treatment efficacy in patients with advanced SCLC after exposure to platinum-based therapy in Alberta, and used these data to build an SCA for comparison with the B-005 study population.12 The second ITC is an STC that facilitates the indirect comparison of lurbinectedin (using IPD from the B-005 trial) with topotecan IV (using AD from the von Pawel et al. [2014] RCT publication13) in patients with relapsed or refractory SCLC.14 The third ITC is a series of combined MAICs and NMAs to evaluate the efficacy and safety of lurbinectedin compared with competing interventions among patients with SCLC receiving second-line treatment with respect to ORR, DOR, OS, and PFS, as well as hematological AEs of grade 3 or 4.15
Interpretation of Results
Efficacy
There was no direct comparative evidence available to inform the relative efficacy of lurbinectedin compared with other treatment options in the second-line or third-line treatment of SCLC. According to the clinical experts consulted by CADTH for this review, the ORRs (35.2% per IA and 30.5% per IRC in the overall study population) and median DORs (5.3 months per IA and 5.1 months per IRC) observed in study B-005 were encouraging in the second-line treatment setting. The clinical experts emphasized that the decreasing efficacy of lurbinectedin (and any drug) with shorter CTFIs was expected, and that the observation of any objective responses lasting several months in patients with a CTFI shorter than 90 days was promising. Spontaneous tumour regression would not be expected in this patient population. In agreement with guidance from the FDA, the clinical experts explained that in the absence of a control group, interpretation of OS and PFS results was unclear.35 HRQoL and cancer symptoms, outcomes of importance to patients with SCLC, were not assessed in the study. The clinical experts stressed that despite the uncertainties regarding efficacy data from study B-005, available options for the second-line and third-line treatment of SCLC are not effective in many patients.
Regarding evidence from ITCs, the SCA analysis provides a comparison of lurbinectedin (study B-005) against the SCA based on real-world data adjusted by CTFI and stage at initial diagnosis, where a longer median OS in the lurbinectedin arm was observed. In the STC of lurbinectedin versus IV topotecan, adjusted estimates suggest longer median OS with lurbinectedin than with topotecan IV, although the results had limitations related to imprecision, risk of bias, and residual confounding. Results from the MAIC and NMA in the third ITC suggest that lurbinectedin improves median OS and PFS rates from 3 to 9 months compared with IV topotecan and carboplatin plus etoposide. However, results from the MAICs are limited by the imprecision, confounding, and risk of bias (e.g., violation of proportional hazards, intransitivity, poor overlap of covariates in the MAIC). ORR and DOR were uncertain, with no evidence of better ORR odds with lurbinectedin than with carboplatin plus etoposide or topotecan IV, and incomplete evidence for the evaluation the DOR. Overall, in the third ITC, there was too much uncertainty to draw conclusions because the NMA has limitations similar to those observed in the MAICs, which were used to include a single-arm trial in an already sparse network of studies.
Several phase III and phase IV studies investigating lurbinectedin for treatment of SCLC have been completed or are ongoing. The phase III ATLANTIS study is a randomized, OL trial of lurbinectedin plus doxorubicin versus investigator’s choice of CAV or topotecan in patients with SCLC who failed on prior line of platinum-containing chemotherapy (N = 613);36 the study did not meet its primary end point and no OS benefit was observed for the lurbinectedin plus doxorubicin combination (NCT02566993). However, lurbinectedin was dosed at 2 mg/m2 to mitigate toxicities of the combination regimen. The phase III LAGOON study is a randomized, OL trial comparing lurbinectedin monotherapy, lurbinectedin plus irinotecan, and investigator’s choice of irinotecan or topotecan in patients with SCLC who failed 1 line of platinum-containing chemotherapy (N = 705); results are not yet available (NCT05153239).37 The phase IV EMERGE-402 study is an ongoing real-world observational study of lurbinectedin monotherapy in any line among patients with ES SCLC (N = 300); results are not yet available (NCT04894591).38
Harms
The clinical experts consulted by CADTH for this review stressed that the potential importance of lurbinectedin for the treatment of patients with SCLC was tied closely to potentially improved tolerability compared with other treatment options in second-line and third-line settings. The clinical experts repeatedly stressed that the difficulties patients experience with available treatments in the second and subsequent lines, especially IV topotecan and CAV, are “terribly harsh.” Patient input also identified treatment options with improved toxicity profiles as important to patients with SCLC. Although roughly one-quarter of patients had dose reductions of lurbinectedin in study B-005, very few discontinued treatment due to AEs or unacceptable toxicity (3.8%). Moreover, SAEs related to hematological toxicities were relatively infrequent (neutropenia, febrile neutropenia, anemia, and thrombocytopenia all had frequencies of 5% or less), although the protocol did not include primary G-CSF prophylaxis. The clinical experts consulted by CADTH for this review felt that severe hematological toxicities occurred less frequently in patients receiving lurbinectedin in the B-005 study compared with their clinical experience with IV topotecan and CAV in the second-line and third-line settings.
Regarding the indirect evidence, harms were only evaluated through the MAIC and NMA. Overall, lurbinectedin was associated with a lower incidence of grade 3 or 4 anemia compared with carboplatin plus etoposide and a better profile for grade 3 to 4 thrombocytopenia. Lurbinectedin was associated with a greater incidence of grade 3 or 4 neutropenia in the base-case analysis. However, in the sensitivity analysis, a lower incidence of grade 3 or 4 neutropenia was observed with lurbinectedin compared with oral topotecan. These differences in neutropenia rates were deemed to be related to differences in the requirements for prophylaxis with G-CSF across studies. These harm-effect estimates suffer from the same limitations as the efficacy outcomes, although with better confidence in the generalizability of the results and ascertainment of the outcomes.
Conclusions
Evidence from study B-005 suggested that administration of lurbinectedin in patients with SCLC who received 1 prior line of platinum-containing chemotherapy resulted in objective responses in some patients that persisted for several months. In the absence of a control group, PFS and OS results could not be interpreted, and there was no direct evidence to inform the relative efficacy of lurbinectedin compared with other treatment options. Indirect evidence (3 sponsor-submitted ITCs) suggested that lurbinectedin treatment may result in improved OS and/or PFS compared with IV topotecan and carboplatin plus etoposide, albeit with a high risk of bias (due to unanchored comparisons and limited ability to adjust for variability in prognostic factors and treatment-effect modifiers) and a sparse dataset. In study B-005, the main toxicity of lurbinectedin, reversible myelosuppression, was considered acceptable and manageable with dose reductions and appropriate transfusion and growth-factor support. The indirect evidence also suggested that lurbinectedin was associated with lower frequencies of grade 3 or 4 anemia and thrombocytopenia compared with oral topotecan and carboplatin plus etoposide, again with high uncertainty. The indirect evidence was aligned with some outcomes identified as important to patients with SCLC, who are seeking additional second-line and third-line treatment options that prolong survival, delay disease progression, and maintain HRQoL while having acceptable toxicity profiles.
References
1.Drug Reimbursement Review sponsor submission: Zepzelca (lurbinectedin): lyphilized powder, 4 mg/vial, intravenous infusion [internal sponsor's package]. Mississauga (ON): Jazz Pharmaceuticals; 2022 Feb 24.
2.Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. 2021;7(1):3. PubMed
3.Kalemkerian GP, Gadgeel SM. Modern staging of small cell lung cancer. J Natl Compr Canc Netw. 2013;11(1):99-104. PubMed
4.Jackman DM, Johnson BE. Small-cell lung cancer. Lancet. 2005;366(9494):1385-1396. PubMed
5.Bennett BM, Wells JR, Panter C, Yuan Y, Penrod JR. The Humanistic Burden of Small Cell Lung Cancer (SCLC): A Systematic Review of Health-Related Quality of Life (HRQoL) Literature. Front Pharmacol. 2017;8:339. PubMed
6.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics: A 2020 special report on lung cancer. 2020; https://www.cancer.ca/Canadian-Cancer-Statistics-2020-EN. Accessed May 5, 2021.
7.Doherty J, Dawe DE, Pond GR, Ellis PM. The effect of age on referral to an oncologist and receipt of chemotherapy among small cell lung cancer patients in Ontario, Canada. J Geriatr Oncol. 2019;10(3):449-458. PubMed
8.Sun A, Durocher-Allen LD, Ellis PM, et al. Initial management of small-cell lung cancer (limited- and extensive-stage) and the role of thoracic radiotherapy and first-line chemotherapy: a systematic review. Curr Oncol. 2019;26(3):e372-e384. PubMed
9.Paz-Ares L, Dvorkin M, Chen Y, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394(10212):1929-1939. PubMed
10.Trigo J, Subbiah V, Besse B, et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: a single-arm, open-label, phase 2 basket trial. Lancet Oncol. 2020;21(5):645-654. PubMed
11.Clinical study report: Study number PM1183-B-005-14. A multicenter phase II clinical trial of lurbinectedin (PM01183) in selected advanced solid tumours - small cell lung cancer (SCLC) cohort [internal sponsor's report]. Madrid (Spain): Pharma Mar S.A.; 2019 October 29.
12.Oncology Outcomes. Evaluating the Treatment Landscape and Comparative Efficacy of Lurbinectedin in the Treatment of Small Cell Lung Cancer (SCLC) Following Exposure to Platinum Therapy in Alberta, Canada: Phase One and Two Results [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Lurbinectedin, 4mg single dose vial. Mississauga (ON): Jazz Pharmaceuticals; 2022. 2022.
13.von Pawel J, Jotte R, Spigel DR, et al. Randomized phase III trial of amrubicin versus topotecan as second-line treatment for patients with small-cell lung cancer. J Clin Oncol. 2014;32(35):4012-4019. PubMed
14.Purple Squirrel Economics. Simulated Treatment Comparisons of Survival Outcomes in Lurbinectedin and Topotecan Treated Relapsed / Refractory Small Cell Lung Cancer Patients. [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Lurbinectedin, 4 mg single dose vial. Mississauga (ON): Jazz Pharmaceuticals. 2021.
15.precisionHEOR. Systematic Literature Review and Indirect Treatment Comparison of Lurbinectedin for the Second-Line Treatment of Small Cell Lung Cancer: Technical Report. [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Lurbinectedin, 4 mg single dose vial. Mississauga (ON): Jazz Pharmaceuticals; 2022. 2021.
16.Canada S. Table 17-10-0005-01 Population Estimates on July 1st, by Age and Sex. 2021; https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1710000501. Accessed May 5, 2021.
17.Brenner DR, Weir HK, Demers AA, et al. Projected estimates of cancer in Canada in 2020. CMAJ. 2020;192(9):E199-E205. PubMed
18.PrZepzelcaTM (lurbinectedin): lyophilized powder, 4 mg/vial, intravenous injection [product monograph]. Dublin (IE): Jazz Pharmaceuticals; 2021 Sep 29.
19.PrCarboplatin injection BP: sterile solution, 10 mg/mL (50 mg, 150 mg, 450 mg, 600 mg of carboplatin per vial), antineoplastic agent [product monograph]. Kirkland, QC: Pfizer Canada; 2022 March 29.
20.PrCisplatin injection BP: sterile solution, 1 mg/mL (50 mg and 100 mg f carboplatin per vial), antineoplastic agent [product monograph]. Kirkland, QC: Pfizer Canada; 2020 April 3.
21.PrEtoposide injection USP: sterile solution, 20 mg/mL (100 mg/5 mL, 200 mg/10 mL, 500 mg/2 mL, 1 g/50 mL), antineoplastic agent [product monograph]. Boucherville, QC: Sandoz Canada Inc.; 2015 May 21.
22.PrTopotecan hydrochloride for injection: sterile solution, 4 mg topotecan (as topotecan hydrochloride) per 4 mL, antineoplastic agent [product monograph]. Kirkland, QC: Pfizer Canada Inc.; 2018 January 5.
23.PrProcytox: cyclophosphamide tablets USP: 25 mg, 50 mg and cyclophosphamide for injection: 200 mg, 500 mg, 1000 mg, 2000 mg (powder for injection) per vial,, antineoplastic agent [product monograph]. Mississauga, ON: Baxter Corporation; 2012 September 7.
24.PrDoxorubicin: doxorubicin hydrochloride injection, preservative-free solution, 2 mg/mL, 10 mg (5 mL) and 50mg (25 mL), antineoplastic agent [product monograph]. Kirkland, QC: Pfizer Canada; 2021 September 30.
25.PrVincristine sulfate injection USP, 1 mg/mL, antineoplastic agent [product monograph]. Kirkland, QC: Pfizer Canada; 2021 July 26.
26.PrIrinotecan hydrochloride injection USP (irinotecan hydrochloride trihydrate), 20 mg/mL (40 mg/2 mL, 100 mg/5 mL, 500 mg/25 mL), antineoplastic agent [product monograph]. Kirkland, QC: Pfizer Canada; 2019 March 8.
27.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Mar 12.
28.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
29.Fernandez-Teruel C, Lubomirov R, Fudio S. Population Pharmacokinetic-Pharmacodynamic Modeling and Covariate Analyses of Neutropenia and Thrombocytopenia in Patients With Solid Tumors Treated With Lurbinectedin. J Clin Pharmacol. 2021;61(9):1206-1219. PubMed
30.Subbiah V, Paz-Ares L, Besse B, et al. Antitumor activity of lurbinectedin in second-line small cell lung cancer patients who are candidates for re-challenge with the first-line treatment. Lung Cancer. 2020;150:90-96. PubMed
31.Schwartz LH, Litiere S, de Vries E, et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur J Cancer. 2016;62:132-137. PubMed
32.Medical Dictionary for Regulatory Activities (MedDRA) version 21.1. Geneva (CH): International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 2018: https://www.meddra.org. Accessed 2022 Jan 1.
33.Common Terminology Criteria for Adverse Events (CTCAE v4.0: CTCAE v4.03. Bethesda (MD): National Cancer Institute; 2010: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm. Accessed 2022 Jan 1.
34.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for Population-Adjusted Indirect Comparisons in Health Technology Appraisal. Med Decis Making. 2018;38(2):200-211. PubMed
35.US FDA. Clinical trial endpoints for the approval of cancer drugs and biologics: Guidance for industry 2018; https://www.fda.gov/media/71195/download.
36.Farago AF, Drapkin BJ, Lopez-Vilarino de Ramos JA, et al. ATLANTIS: a Phase III study of lurbinectedin/doxorubicin versus topotecan or cyclophosphamide/doxorubicin/vincristine in patients with small-cell lung cancer who have failed one prior platinum-containing line. Fut Oncol. 2019;15(3):231-239. PubMed
37.ClinicalTrials.gov. Clinical trial of lurbinectedin as single-agent or in combination with irinotecan versus topotecan or irinotecan in patients with relapsed small-cell lung cancer (LAGOON). 2021; https:/www.clinicaltrials.gov/ct2/show/NCT05153239.
38.ClinicalTrials.gov. To assess the effectiveness and safety of Zepzelca in adult patients with extensive stage small cell lung cancer (SCLC). 2021; https:/www.clinicaltrials.gov/ct2/show/NCT04894591.
Appendix 1: Literature Search Strategy
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: March 24, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type
Limits: Conference abstracts excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Zepzelca* or lurbinectedin or PM01183 or “PM 01183” or pm1183 or pm 1183 or WHO9397 or WHO 9397 or ly01017 or “ly 01017” or tryptamicidin* or zepsyre* or 2CN60TN6ZS).ti,ab,ot,kf,hw,nm,rn.
1 use medall
*lurbinectedin/ or (Zepzelca* or lurbinectedin or PM01183 or “PM 01183” or pm1183 or pm 1183 or WHO9397 or WHO 9397 or ly01017 or “ly 01017” or tryptamicidin* or zepsyre*).ti,ab,kf,dq.
3 use oemezd
4 not (conference review or conference abstract).pt.
2 or 5
remove duplicates from 6
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search -- Studies with results: Zepzelca/lurbinectedin AND SCLC
WHO ICTRP
International Clinical Trials Registry Platform, produced by WHO. Targeted search used to capture registered clinical trials.
Search terms -- Zepzelca/lurbinectedin AND SCLC
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms -- Zepzelca/lurbinectedin AND SCLC
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms -- Zepzelca/lurbinectedin AND SCLC
Grey Literature
Search dates: March 14 to 17, 2022
Keywords: Zepzelca, lurbinectedin, SCLC
Limits: Publication years: 1996-present
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Appendix 2: Excluded Studies
Reference | Reason for exclusion |
|---|---|
Farago et al. (2019)36 | Intervention (lurbinectedin plus doxorubicin; lurbinectedin dosed at 2 mg/m2) |
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CAV
cyclophosphamide plus doxorubicin plus vincristine
CUA
cost-utility analysis
ICER
incremental cost-effectiveness ratio
KM
Kaplan-Meier
LY
life-year
OS
overall survival
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
RWE
real-world evidence
SCLC
small cell lung cancer
SYNTH
synthetic basket comparator
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Lurbinectedin (Zepzelca), powder for IV infusion |
Submitted price | Lurbinectedin, 4 mg vial: $6,470.00 per vial |
Indication | For the treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-based chemotherapy |
Health Canada approval status | NOC |
Health Canada review pathway | Advance consideration under NOC/c |
NOC date | September 29, 2021 |
Reimbursement request | As per indication |
Sponsor | Jazz Pharmaceuticals, Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions; SCLC = small cell lung cancer.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partition survival mode |
Target population |
|
Treatment | Lurbinectedin |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (25 years) |
Key data source | Single-arm, phase II, basket trial (PM1183-B-005 to 14); naive comparisons with comparators |
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
CAV = cyclophosphamide + doxorubicin + vincristine; ICER = incremental cost-effectiveness ratio; LY = life-year; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; SCLC = small cell lung cancer; SYNTH = synthetic basket comparator derived from real-world evidence.
Conclusions
The sponsor’s economic model is informed by results from the PM1183-B-005-14 trial (hereafter referred to as the B-005 study), a single-arm, phase II trial that aimed to assess the efficacy and safety of lurbinectedin as a second-line treatment for patients with small cell lung cancer (SCLC). Quality of life (QoL) was not assessed in the B-005 trial. The CADTH clinical review concluded that data from the B-005 study were inadequate to interpret the overall survival (OS) and progression-free survival (PFS) findings because of the lack of a comparator group. Owing to the sponsor’s use of naive comparisons of lurbinectedin to all modelled comparators, it is not possible to determine if any observed differences in PFS, OS, or quality-adjusted life-years (QALYs) between treatments are due to the effect of treatment or are instead due to bias or confounding. As a result, the comparative effectiveness of lurbinectedin and other currently available treatments is highly uncertain.
Given the lack comparative data and critical limitations within the sponsor’s model, CADTH was unable to derive a reliable base-case estimation of the cost-effectiveness of lurbinectedin. Notably, the sponsor’s model predicts a survival benefit with lurbinectedin that has not been shown in clinical trials; further, the model predicts that the majority of the benefits of lurbinectedin are realized after patients discontinue treatment, which is not supported by data from clinical trials. In their analysis, the sponsor assumes that all patients receive lurbinectedin for four 21-day treatment cycles, which is not aligned with the monograph-recommended dosing and may underestimate drug acquisition costs. CADTH conducted an exploratory reanalysis to assess the effect of alternative extrapolation curves on PFS and OS; however, CADTH was unable to address critical limitations related to the absence of comparative clinical data in the pharmacoeconomic model or related to the structure of the sponsor’s model. Further, drug costs associated with lurbinectedin acquisition are likely underestimated, owing to the structure of the sponsor’s model.
The results of the CADTH exploratory analyses were consistent with those submitted by the sponsor: lurbinectedin is not cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY. Based on the CADTH exploratory analysis, a price reduction of at least 83% would be needed for lurbinectedin to be considered cost-effective at a WTP threshold of $50,000 per QALY, compared with the synthetic basket comparator derived by the sponsor from real-world data (SYNTH) (i.e., intended to reflect usual care for patients in Canada whose disease has progressed on or after platinum-based chemotherapy). However, this estimate is subject to the limitations discussed, including a lack of comparative clinical data and an underestimation of lurbinectedin acquisition costs. Additional uncertainty is introduced by the use of data from real-world clinical practice and from structured clinical trials. As such, a higher price reduction may be warranted.
CADTH notes that, among patients with platinum-resistant SCLC, lurbinectedin was less effective than the combination of cyclophosphamide, doxorubicin, and vincristine (CAV), and there is no price reduction that would make lurbinectedin cost-effective in this patient subgroup based on data submitted by the sponsor. In patients who have progressed on or after platinum-based chemotherapy, lurbinectedin is more costly than treatment with currently available comparators. There is no reliable information on the comparative clinical effects of lurbinectedin relative to any alternative treatment. As such, there is insufficient evidence to support a price premium over currently available treatments.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission).
Patient input was received from the Lung Health Foundation and Lung Cancer Canada. The Lung Health Foundation collected perspectives through an online survey (2 patients) and interviews (3 patients) with patients with lung cancer. Lung Cancer Canada collected perspectives through interviews (2 patients) and environmental scans. Patients had experience with surgery, radiation, chemotherapy, targeted therapy, and immunotherapy, and noted that the side effects related to currently available treatments affect their QoL and their ability to work and perform activities of daily living; such side effects included fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy. Some respondents noted the financial burden related to the high cost of treatment and travel-related expenses (i.e., to receive treatment), and the need for caregivers to take time off work to provide care. Patients expressed a desire for a treatment that would stop or slow disease, be effective in managing symptoms, have manageable side effects, and allow them to maintain their independence and QoL. Patients who have used lurbinectedin reported side effects such as fatigue, headache, nausea, shortness of breath, and gastrointestinal issues.
Clinician input received from Lung Cancer Canada and the Ontario Health (Cancer Care Ontario) Lung Cancer Drug Advisory Committee indicated that currently available treatments for patients with relapsed SCLC include re-treatment with platinum plus etoposide (for those with platinum-sensitive disease), CAV, topotecan, palliative radiation, supportive care, or clinical trial drugs. Clinicians noted that existing treatments for patients with relapsed SCLC are associated with short PFS and OS. Adverse events (AEs) associated with currently available treatments include nausea and vomiting, neutropenia, thrombocytopenia, infection, mucositis, and fatigue, and clinicians noted that treatments with improved AE profiles are needed. Clinicians noted that tumour shrinkage, PFS, OS, and improved patient-reported symptoms and QoL are key goals of treatment. Clinicians indicated that a clinically meaningful response to treatment would include a reduction in tumour size and an improvement in disease-related symptoms. Clinicians noted that lurbinectedin may be used as a second-line or third-line treatment.
Participating drug plans noted considerations related to clinical evidence, relevant comparators, and potential implementation factors (e.g., drug wastage). The plans noted that the relevant comparators depend on whether relapsed disease is platinum-sensitive or platinum-resistant or -refractory. Plans noted that comparators for platinum-sensitive disease include cisplatin plus etoposide and carboplatin plus etoposide. For platinum-refractory disease, plans noted that the relevant comparators are CAV, topotecan, cisplatin plus irinotecan, and carboplatin plus irinotecan. The plans highlighted that the pivotal trial, B-005, is a single-arm trial, so no comparators were included. The plans noted that, based on the recommended dosage of lurbinectedin, drug wastage is anticipated, and additional pharmacy resources will be required to assess potential drug interactions with lurbinectedin. Plans noted that all chemotherapy comparators have existing confidential negotiated prices in place.
Several of these concerns were addressed in the sponsor’s model:
Subgroup analyses were provided to assess the cost-effectiveness of lurbinectedin among patients with platinum-sensitive and platinum-resistant disease.
The use of a cost-utility approach accounts for some issues related to QoL; however, it is unclear if all QoL concerns noted to be important to patients were captured in the health state utility values adopted by the sponsor. QoL was not assessed in the B-005 trial.
Wastage was considered in the sponsor’s analysis.
CADTH was unable to address the following concerns raised from stakeholder input:
Some comparators identified as being relevant by the drug plans (carboplatin or cisplatin plus irinotecan) could not be included, owing to a lack of clinical information and the structure of the sponsor’s model.
Patients were assumed to discontinue lurbinectedin after 4 treatment cycles regardless of disease progression. This may underestimate drug costs.
Economic Review
The current review is for lurbinectedin (Zepzelca) for adults with SCLC who have received 1 prior chemotherapy-containing line of therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Lurbinectedin is indicated for the treatment of stage III or metastatic SCLC that has progressed on or after platinum-based chemotherapy in adult patients.1 The sponsor submitted a cost-utility analysis (CUA) to assess the cost-effectiveness of lurbinectedin in this population and in 2 subgroups: patients without platinum-sensitive disease and patients with platinum-refractory disease.2 The modelled population is consistent with the reimbursement request. In the full Health Canada–indicated population, lurbinectedin was compared to topotecan, CAV, and to 2 basket comparators: real-world evidence (RWE) comprised of topotecan, CAV, carboplatin plus etoposide, cisplatin plus etoposide, other platinum regimens, and other regimens; and SYNTH comprised of CAV, carboplatin plus etoposide, cisplatin plus etoposide, and other regimens. Among the platinum-sensitive patient subgroup, lurbinectedin was compared to topotecan, carboplatin plus etoposide, cisplatin plus etoposide, and carboplatin plus irinotecan. Among the platinum-refractory patient subgroup, lurbinectedin was compared to topotecan and CAV.
Lurbinectedin is available as a 4 mg vial, with a recommended dosage of 3.2 mg/m2 by IV infusion once every 21 days, until disease progression or unacceptable toxicity.1 At a submitted price of $6,470.00 per 4 mg vial, the 21-day drug cost of lurbinectedin was calculated by the sponsor to be $12,940. The drug costs of comparator regimens were as follows: CAV = $1,457; topotecan = $2,835; carboplatin plus etoposide = $1,360; cisplatin plus etoposide = $1,341; and carboplatin plus irinotecan = $1,326.
The clinical outcomes of interest were QALYs and life-years (LYs). The economic analysis was undertaken from the perspective of the publicly funded health care payer over a 25-year horizon. Discounting (1.5% per annum) was applied to both costs and outcomes. The sponsor assumed an inflation rate of 2.57%.
Model Structure
The sponsor submitted a partitioned survival model (PSM) that included 3 health states: Progression Free, Progressed Disease, and Death. The modelled time cycle was 1 month. The proportion of patients in the Progression-Free state, who moved to Progressed Disease or Death states at any time over the model’s time horizon was derived from nonmutually exclusive survival curves. All patients entered the model in the Progression-Free state. The proportion of patients in the Progressed Disease state was calculated as the proportion of patients alive (based on the OS curve) minus the proportion of patients alive and progression-free (based on the PFS curve). In the model, PFS was capped by OS. Patients in the Progression-Free state were assumed to receive treatment for a defined number of treatment cycles. After disease progression, 24% of patients were assumed to receive subsequent (i.e., third-line) treatment for 3 months. Disutility associated with AEs were assumed to last for 1 month.
Model Inputs
The modelled cohort’s characteristics were based on the B-005 trial (mean age = 60 years; 1.80 m2 body surface area; 60% male). For lurbinectedin, PFS and OS data were obtained from the B-005 study, a single-arm, basket trial that enrolled 110 patients with SCLC. PFS and OS estimates for CAV, topotecan, carboplatin plus etoposide, cisplatin plus etoposide, and carboplatin plus irinotecan were obtained from the literature when available; otherwise, the sponsor assumed equivalence between treatments. For RWE and SYNTH, time to next treatment (as a proxy for PFS) and OS were derived from a retrospective cohort of SCLC patients in Alberta, from 2004 to 2019. The effectiveness of RWE was derived from 577 patients who received a systemic post–platinum-based treatment, and effectiveness of SYNTH was derived from a subset of 224 patients who additionally met the inclusion criteria of the B-005 study. The treatments included in the RWE and SYNTH baskets by the sponsor, and their relative frequencies, are shown in Appendix 3.
For lurbinectedin, Kaplan-Meier (KM) estimates of PFS and OS from the B-005 trial period were used to fit parametric survival curves to extrapolate the observed data beyond the trial period (piecewise model) (OS data were extrapolated from 42 months, PFS data were extrapolated from 19 months). Log-logistic distributions were adopted by the sponsor for lurbinectedin OS and PFS, whereas gamma distributions were adopted for OS and PFS for topotecan and CAV, with the choice between curves based on Akaike information criteria, Bayesian information criteria, and visual inspection. For RWE and SYNTH, KM estimates were used for OS and PFS without extrapolation.
Health state utility values were obtained for the progression-free state (on or off systemic treatment) and the Progressed Disease state from the literature,3 based on 5-Level EQ-5D data from a Canadian cohort of patients with advanced SCLC, valued using Canadian preference weights. The sponsor’s model included grade 3 and 4 AEs, with the prevalence of AEs and associated disutility values obtained from the literature.
The model included costs related to drug acquisition and administration, antiemetic treatments, febrile neutropenia prophylaxis, subsequent treatment after disease progression, AEs, health care resource use, and mortality costs. Drug acquisition costs for lurbinectedin were based on the sponsor’s submitted price, whereas the drug acquisition costs for comparators were obtained from a previous CADTH review.4 Each drug was assigned a risk of emesis (low, moderate, high) and febrile neutropenia (low, intermediate, high), with each risk level associated with a per-cycle treatment cost. Administration costs were assumed to be $223 per hour for drugs administered by IV infusion,5 with the duration of administration for each treatment obtained from Cancer Care Ontario and the literature. Total monthly treatment costs (includes drug acquisition costs, antiemetic and prophylactic febrile neutropenia costs, administration costs) included by the sponsor are as follows: lurbinectedin = $19,203; CAV = $3,701; carboplatin plus etoposide = $4,101; cisplatin plus etoposide = $4,719; carboplatin plus irinotecan = $4,214; topotecan = $6,392; RWE = $3,835; and SYNTH = $3,822. Subsequent treatment was assumed to consist of etoposide for 3 months. The cost of treating AEs was obtained from the Ontario Case Costing Initiative for hematologic and nonhematologic AEs. Health care resource use was assumed to include oncologist visits, imaging (CT, MRI, X-ray), and laboratory diagnostics, with the frequency of use assumed to vary depending on whether the patient was on treatment or under surveillance (stratified by limited-stage or extensive-stage disease) according to National Comprehensive Cancer Network guidelines.6 Mortality costs were assumed to include acute care and hospice care applied to the final 3 months of life.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented here. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
The sponsor-provided base-case analyses were intended to reflect the B-005 trial population (including both platinum-sensitive and platinum-refractory patients), and subgroup analyses were intended to reflect platinum-sensitive and non–platinum-sensitive patients (hereafter referred to as resistant, which includes patients with platinum-resistant and platinum-refractory disease). As noted in the CADTH clinical review, the B-005 trial population includes platinum-sensitive (57.1%) and platinum-resistant patients (42.9%).
Base-Case Results
In the B-005 trial population, lurbinectedin was associated with estimated costs of $107,726 and QALYs gains of 0.67 over a 25-year time horizon (Table 3). Treatment with lurbinectedin was more costly and produced more QALYs than all comparators. Based on a sequential analysis, the incremental cost-effectiveness ratio (ICER) for lurbinectedin is $248,709 per QALY compared with CAV. At a WTP of $50,000 per QALY, the probability of lurbinectedin being considered the most likely cost-effective intervention was 0%.
Results were driven by the predicted differences in total LYs between lurbinectedin and comparators (incremental LYs versus CAV = 0.39 years) and increased drug acquisition costs associated with lurbinectedin (incremental costs versus CAV = $50,782). All QALYs were accrued during the B-005 trial period (i.e., none were accrued through extrapolation), and the sponsor’s model predicts that the majority (63%) of QALYs gained with lurbinectedin are accrued in the postprogression health state (i.e., after discontinuation of lurbinectedin treatment). Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results — B-005 Trial Population
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
RWEa | 53,408 | 0.44 | Reference |
SYNTHb | 53,734 | 0.45 | 39,348 vs. RWE |
CAV | 56,944 | 0.47 | 206,020 vs. SYNTH |
Topotecan | 74,778 | 0.46 | Dominated by CAV |
Lurbinectedin | 107,726 | 0.67 | 248,709 vs. CAV |
CAV = cyclophosphamide + doxorubicin + vincristine; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; RWE = basket comparator derived from real-world evidence; SYNTH = synthetic basket comparator derived from real-world evidence.
aBasket containing topotecan, CAV, carboplatin + etoposide, cisplatin + etoposide, other platinum regimens, and other regimens.
bBasket containing CAV, etoposide, carboplatin + etoposide, cisplatin + etoposide, and other regimens.
Source: Sponsor’s pharmacoeconomic submission.2
Among patients with platinum-sensitive disease, lurbinectedin was associated with estimated costs of $118,436 and QALY gains of 0.91 over a 25-year time horizon. In sequential analyses, lurbinectedin was associated with an incremental cost of $61,348 and 0.48 additional QALYs compared with carboplatin plus etoposide over a 25-year horizon, resulting in an ICER of $126,544 per QALY (Table 4). The majority (66%) of QALYs gained with lurbinectedin are predicted by the sponsor’s model to be accrued in the postprogression health state (i.e., after lurbinectedin discontinuation).
Table 4: Summary of the Sponsor’s Economic Evaluation Results — Platinum-Sensitive Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Cisplatin + etoposide | 57,066 | 0.38 | Reference |
Carboplatin + etoposide | 57,088 | 0.43 | 505 vs. cisplatin + etoposide |
Carboplatin + irinotecan | 63,262 | 0.41 | Dominated |
Topotecan | 88,226 | 0.55 | Extended dominance |
Lurbinectedin | 118,436 | 0.91 | 126,544 vs. carboplatin + etoposide |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
Among patients with platinum-refractory disease, lurbinectedin was associated with estimated costs of $92,218 and QALY gains of 0.37 over a 25-year time horizon. In this subgroup, lurbinectedin was dominated by CAV; that is, lurbinectedin was less effective (incremental QALYs = –0.03) and more costly (incremental cost = $41,272) than CAV over a 25-year horizon (Table 5). The majority (57%) of QALYs gained with lurbinectedin are predicted by the sponsor’s model to be accrued in the postprogression health state (i.e., after lurbinectedin discontinuation).
Table 5: Summary of the Sponsor’s Economic Evaluation Results — Platinum-Resistant Subgroup
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
CAV | 51,946 | 0.40 | Reference |
Topotecan | 73,584 | 0.36 | Dominated |
Lurbinectedin | 93,218 | 0.37 | Dominated |
CAV = cyclophosphamide + doxorubicin + vincristine; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analyses Results
The sponsor provided several pairwise scenario and sensitivity analyses for the B-005 trial population, including scenarios including adopting shorter time horizons (i.e., 5 years, 10 years), alternative distributions for OS and PFS, and including irinotecan as a comparator, as well as using a population-adjusted comparison for lurbinectedin PFS. The majority of scenarios included by the sponsor had little impact on the ICER, with the exception of a reduced time horizon and the inclusion of irinotecan.
When irinotecan was included as a comparator, lurbinectedin was dominated in the B-005 trial population (i.e., less effective and more costly) by irinotecan.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
Comparative clinical efficacy of lurbinectedin is unknown. There have been no completed head-to-head trials comparing lurbinectedin (as monotherapy) to any of the comparators included in the model, and a key limitation of the clinical efficacy (i.e., OS and PFS) data informing lurbinectedin in the economic model is that it is based on the B-005 study. Given the nonrandomized, single-arm design of the B-005 trial, the interpretation of all outcomes is hampered by the lack of a control group, which makes the relative magnitude of any benefits highly uncertain. The sponsor provided several indirect treatment comparisons as part of their submission to CADTH; however, these were not used in the pharmacoeconomic submission, and treatment comparators in the pharmacoeconomic submission were based on naive comparison of treatments. That is, in the pharmacoeconomic model, the sponsor incorporated efficacy (i.e., OS, PFS) and safety (i.e., AEs) data directly from clinical trials that involved lurbinectedin or comparators without adjustment or without accounting for differences in patient characteristics. CADTH notes that, owing to the direct use of clinical trial data from various sources, it is not possible to determine whether any observed differences in PFS, OS, or AEs between therapies are solely due to the treatment or, rather, due to bias or confounding (e.g., differences in study populations, definitions of outcomes, study designs). Together, this brings great uncertainty to the naive estimates taken to inform the clinical inputs. As such, the incremental gains in QALYs and LYs predicted by the sponsor’s model for lurbinectedin relative to comparators should be interpreted with a higher degree of uncertainty than is reflected in the sponsor’s probabilistic analysis.
CADTH additionally notes that the sponsor’s base case predicts that, in the B-005 trial population, lurbinectedin is less effective in prolonging PFS than standard care (reflected by the RWE and SYNTH comparators), such that patients remain progression-free for a shorter duration with lurbinectedin than with RWE or SYNTH. Whether this is an artifact of the sponsor’s use of naive comparisons or a true effect of lurbinectedin treatment cannot be determined based on the evidence provided by the sponsor.
CADTH was unable to address the lack of comparative data for lurbinectedin and all model comparators. Given the lack of direct evidence and the use of naive comparisons to inform the model, the cost-effectiveness of lurbinectedin is unknown.
Full Health Canada–indicated population not modelled: The Health Canada indication and the sponsor’s reimbursement request are for the treatment of adult patients with stage III or metastatic SCLC who have progressed on or after platinum-based chemotherapy, without restriction by line of therapy. In contrast, the data used by the sponsor to inform lurbinectedin OS and PFS were obtained from the B-005 trial, which assessed the efficacy and safety of lurbinectedin as a second-line treatment after first-line platinum plus etoposide doublet therapy. As such, the modelled population explores the cost-effectiveness of lurbinectedin in only a subset of the Health Canada–indicated population. The sponsor’s model was not sufficiently flexible to report the cost-effectiveness of lurbinectedin in the full population, and no clinical data were provided to support the use of lurbinectedin in the third-line setting. Thus, the cost-effectiveness of lurbinectedin as a third-line treatment is unknown. Based on the submitted budget impact analysis (BIA), the sponsor estimates that approximately 20% of patients eligible for lurbinectedin would receive it as third-line therapy.
CADTH was unable to address this limitation within the model, owing to a lack of clinical data. The cost-effectiveness of lurbinectedin as a third-line therapy is unknown.
Limitations associated with the sponsor’s chosen modelling approach: The sponsor submitted a PSM, in which treatment efficacy is represented by PFS and OS curves, based on observations from 105 patients with SCLC in the single-arm B-005 study. The sponsor’s model predicts a gain of 1.11 LYs with lurbinectedin, with 0.35 LYs (4.2 months) accrued in the preprogression state. This exceeds the median PFS observed in the B-005 trial (3.5 months), suggesting that the sponsor’s model overestimates the benefits associated with lurbinectedin. Further, the total LYs predicted by the sponsor’s model (1.1 LYs) exceeds the median OS observed in the B-005 trial (9.3 months), which further suggests that the sponsor’s model overestimates the benefits associated with lurbinectedin. CADTH notes that the Health Canada monograph for lurbinectedin indicates that an OS benefit has not been demonstrated with lurbinectedin.7
The sponsor’s model predicts a median OS of approximately 9 months with lurbinectedin, but notes that a small proportion of patients will remain alive until approximately 14 years after initiating second-line treatment. The clinical experts consulted by CADTH for this review indicated that, in the second-line setting, survival in this patient population does not typically exceed 1 year. It is thus unlikely that a proportion of patients will remain alive 14 years after initiating lurbinectedin treatment.
Results from the sponsor’s model suggests that the majority (63%) of the benefits with lurbinectedin treatment are accrued in the postprogression health state (Table 10). This finding implies that the majority of the incremental benefit would be realized after patients have discontinued lurbinectedin. The clinical experts consulted by CADTH for this review noted that there is no clear mechanism by which lurbinectedin would continue to provide clinical benefit after relapse. CADTH asked the sponsor to provide additional evidence to support the implied postprogression benefit of lurbinectedin treatment (0.76 LYs; 0.42 QALYs). The sponsor indicated that they believe this to be “grounded in data from the clinical trials” and “may be expected in SCLC patients with high tumour burden, minimal response to chemotherapy, and high frequency of chemotherapy-induced toxicity.” Based on the evidence provided by the sponsor, CADTH is unable to ascertain whether the postprogression survival benefit predicted in the sponsor’s model is a true effect or an artifact of the use of a B-005 model.
In exploratory reanalysis, CADTH adopted alternative parametric extrapolations for PFS and OS. In all reanalyses, the structural features of the sponsor’s model persist (i.e., the postprogression survival benefit). CADTH was unable to determine the extent to which the implied postprogression benefit was due to the effect of treatment or was due to structural bias within the PSM.
Costs associated with lurbinectedin may be underestimated. In the sponsor’s pharmacoeconomic model, all patients are assumed to receive lurbinectedin for 4 21-day treatment cycles, regardless of disease progression, based on the median duration of treatment in the B-005 trial. This is in contrast with the product monograph, which indicates that lurbinectedin should be administered until disease progression or unacceptable toxicity. The clinical experts consulted by CADTH for this review indicated that they would expect patients who remain disease-free and without AEs to continue to receive lurbinectedin until disease progression. In the B-005 trial, the majority (91%) of patients who discontinued lurbinectedin did so because of disease progression or disease-related death. CADTH notes that, in the sponsor’s pharmacoeconomic model, after the first 3 months of treatment (4 treatment cycles), 53% of patients remain progression-free.
CADTH requested that the sponsor revise its pharmacoeconomic model to reflect the monograph dosing (i.e., until disease progression or unacceptable toxicity7); however, the sponsor declined this request. CADTH notes that, although the median number of cycles per patient in the lurbinectedin clinical trial (B-005) was 4, 47.6% of patients received 5 or more cycles (43.8% received 6 or more cycles). In the B-005 trial, the mean treatment duration with lurbinectedin was 19.7 weeks, and patients received a mean of 5.9 treatment cycles. CADTH additionally notes that the mean number of cycles was higher among patients with platinum-sensitive disease (7.0 cycles; range, 1 to 24 cycles) compared with those with platinum-resistant disease (4.4 cycles; range, 1 to 18 cycles), which was not considered in the sponsor’s analysis.
Drug costs associated with lurbinectedin acquisition are likely higher than quoted by the sponsor. In exploratory scenario analyses, CADTH adopted a treatment duration of 5.9 cycles for lurbinectedin, based on the mean number of treatment cycles in the B-005 trial.
Additional limitations were identified as part of the review process. Because of the lack of robust comparative efficacy evidence, these limitations were considered unlikely to meaningfully impact the assessment of overall cost-effectiveness and could not be addressed in reanalysis.
Generalizability of the model comparators.
The clinical experts consulted by CADTH noted that some comparators included in the sponsor’s model (e.g., topotecan) are not commonly used in clinical practice.
The impact of subsequent treatment after disease progression is uncertain.
The sponsor assumed that 24% of patients would receive subsequent treatment after disease progression, based on “key opinion leaders” consulted by the sponsor. In the B-005 trial, 45% of patients received subsequent treatment after lurbinectedin treatment.
The sponsor assumed that subsequent treatment would consist of etoposide for 4 months; this assumption was not justified by the sponsor, and the clinical experts consulted by CADTH indicated this is not consistent with Canadian clinical practice.
The sponsor assumed that subsequent treatment would affect treatment costs but would have no impact on OS.
The model lacked flexibility to assess relevant subgroups.
The sponsor’s pharmacoeconomic analysis did not adequately consider the cost-effectiveness of lurbinectedin by disease stage (i.e., limited versus extensive). Clinical experts consulted by CADTH indicated that disease stage affects the natural history and treatment paradigm. The sponsor’s model assumes that disease stage will affect only health care resource use.
The sponsor’s pharmacoeconomic model lacked transparency.
The sponsor’s submitted model included more than 93,000 IFERROR statements. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, and it is unclear whether the model is running inappropriately by overriding errors.
Additionally, the key assumptions outlined in Table 6 were made by the sponsor and have been appraised by CADTH.
Table 6: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The sponsor assumed that 31% of patients would have limited-stage disease, based on data from the B-005 study, with the remainder assumed to have extensive-stage disease. | Uncertain. Clinical experts consulted by CADTH for this review noted that, in the second-line setting, a lower proportion of patients would be expected to have limited-stage disease. The sponsor’s pharmacoeconomic model considered disease stage only in the context of health care resource use. CADTH additionally notes that the sponsor adopted different estimates of limited and extensive disease stages in their pharmacoeconomic and budget impact analyses. |
Effectiveness estimates were obtained from clinical trials and observational (real-world) studies. | Uncertain. The clinical effectiveness of lurbinectedin was based on data from the prospective B-005 clinical study, whereas data for some comparators (i.e., RWE, SYNTH) were obtained from a retrospective real-world cohort of SCLC patients in Alberta. Although observational studies may provide information about the real-world effectiveness of treatments, patients who receive treatment outside of a clinical practice may differ from those in clinical trials, and the assessment of outcomes (i.e., PFS) may differ. CADTH notes that there were differences between patients in the retrospective cohort and those in the B-005 study, including for age, disease stage, and chemotherapy-free interval. |
Health care resource use was based on National Comprehensive Cancer Network guidelines.6 | Uncertain. The frequency of health care resource use (including oncologist visits and imaging) in the Canadian context is uncertain. Clinician input received by CADTH for this review indicated that there may be a longer duration between imaging than adopted by the sponsor. |
Drug wastage was assumed. | Uncertain but likely appropriate. Based on the recommended dosage (3.2 mg/m2), each dose will require more than 1 vial. Drug plan input received for this review indicated that vial sharing would only be possible if multiple patients were scheduled to be treated close together at centres near a hazardous sterile compounding pharmacy facility. Per the product monograph, the reconstituted or diluted solution can be stored for up to 24 hours. |
In the calculation of drug costs, the largest vial size was used. | Inappropriate. The sponsor assumed that only the largest vial would be used (despite of the availability of smaller and less costly vials for some drugs) to achieve the required mg per dose (e.g., for carboplatin, vincristine). As a result, the drug acquisition costs of some comparators are likely overestimated. |
RWE = basket comparator derived from real-world evidence; SCLC = small cell lung cancer; SYNTH = synthetic basket comparator derived from real-world evidence.
CADTH Reanalyses of the Economic Evaluation
As noted previously and in Appendix 2, there are key limitations associated with the available clinical data for lurbinectedin and the sponsor’s model. Several limitations of the sponsor’s submission could not be adequately addressed because of data or structural limitations, including notable limitations associated with the lack of comparative clinical data and limitations associated with the sponsor’s modelling approach (i.e., PSM) and practices (i.e., lack of transparency). Further, the sponsor has assumed that lurbinectedin will be received by all patients for 4 treatment cycles, regardless of disease progression or toxicity, which is not aligned with the Health Canada–recommended dosing strategy.7 Although the sponsor has provided a sequential analysis to evaluate the cost-effectiveness of lurbinectedin relative to comparators, such analyses are inappropriate in the context of naive comparisons of treatment effectiveness (i.e., it is unknown whether the model results are a reflection of a true difference in effectiveness or a result of, for example, heterogeneity between study populations). The use of both randomized controlled trials and RWE further compounds uncertainty in the modelled results. CADTH reanalyses cannot address these critical sources of uncertainty in the clinical evidence. CADTH was further unable to assess the cost-effectiveness of lurbinectedin as a third-line treatment, owing to a lack of clinical data.
CADTH was unable to address these fundamental limitations of the sponsor’s model, which represent fundamental problems for interpreting the results of the sponsor’s economic evaluation — the costs and QALYs used to calculate the ICER are derived from highly uncertain evidence and do not reflect the intended dosing strategy — and for conducting any reanalysis using the sponsor’s model. As a result, CADTH was unable to conduct any base-case reanalysis of the sponsor’s model, given that any estimates of the incremental costs and incremental effectiveness would be misleading.
Scenario Analysis Results
Although CADTH did not conduct any formal reanalyses of the sponsor’s model, an exploratory analysis was undertaken to explore the impact that changes to model assumptions would have on the ICER (Appendix 4). CADTH selected SYNTH as the main comparator for the B-005 trial population, as it represents an approximation of the standard of care in Canada, the data were derived from patients in Canada, and the cohort was restricted to individuals who met the eligibility criteria of the B-005 clinical trial. CADTH notes that the key limitations of the sponsor’s base-case analysis — noted in the CADTH Appraisal of the Sponsor’s Economic Evaluation and Appendix 2 — apply to this exploratory analysis, including the fundamental limitation that there is no direct evidence to support the comparative efficacy of lurbinectedin relative to any comparator, including SYNTH. As such, this exploratory analysis should not be interpretated as a CADTH base case because there remains uncertainty regarding the true effect of lurbinectedin.
In CADTH exploratory reanalyses, lurbinectedin was associated with an ICER of $307,262, compared with SYNTH. Based on the CADTH exploratory analysis, which is subject to the key limitations of the sponsor’s model, as previously noted, a price reduction of 83% would be required for lurbinectedin to be considered cost-effective compared to SYNTH at a WTP threshold of $50,000 per QALY. Given that the estimates of incremental LYs (and hence QALYs) are highly uncertain and may not be representative of the true incremental effect of lurbinectedin, the true price reduction required for lurbinectedin to be cost-effective is unknown. Details of this exploratory analysis are provided in Appendix 4.
Additional exploratory scenario analyses are provided in Appendix 4.
Issues for Consideration
Two phase III randomized controlled trials have been undertaken in patients with relapsed SCLC. One is comparing lurbinectedin monotherapy to lurbinectedin plus irinotecan and to physicians’ choice of irinotecan or topotecan (NCT05153239); as the other compared lurbinectedin plus doxorubicin to topotecan or CAV (NCT02566993). Direct comparative evidence may reduce the uncertainty associated with the cost-effectiveness estimate. Lurbinectedin in combination with other drugs was not included by the sponsor in its model and the cost-effectiveness of lurbinectedin used in combination with other drugs is unknown.
Overall Conclusions
The pharmacoeconomic results are informed by results from the B-005 study; a single-arm, phase II trial that aimed to assess the efficacy and safety of lurbinectedin as a second-line treatment among patients with SCLC. QoL was not assessed in the B-005 trial. The CADTH clinical review concluded that in the absence of a control group, OS and PFS data from the B-005 study could not be interpreted. Owing to the sponsor’s use of naive comparisons of lurbinectedin to all modelled comparators, it is not possible to determine if any observed differences in PFS, OS, or QALYs between treatments are due to the effect of treatment or are instead due to bias or confounding. As a result, the comparative effectiveness of lurbinectedin relative to other currently available treatments is highly uncertain.
Given the lack comparative data and critical limitations of the sponsor’s model, CADTH was unable to derive a reliable base-case estimation of the cost-effectiveness of lurbinectedin. Notably, the sponsor’s model predicts a survival benefit with lurbinectedin that has not been shown in clinical trials. Further, the model predicts that the majority of the benefits of lurbinectedin are realized after patients discontinue treatment, which is not supported by data from clinical trials. In its analysis, the sponsor assumes that all patients receive lurbinectedin for four 21-day treatment cycles, which is not aligned with the monograph-recommended dosing and may underestimate drug acquisition costs based on the mean treatment duration in the B-005 trial. CADTH conducted exploratory reanalysis to assess the impact of alternative extrapolation curves for PFS and OS; however, CADTH was unable to address critical limitations related to the quality of the comparative clinical data and the structure of the sponsor’s model, and drug costs associated with lurbinectedin acquisition are likely underestimated.
The treatment of patients with SCLC who have progressed on or after platinum-based chemotherapy with lurbinectedin is more costly than treatment with currently available comparators. There is no reliable information on the comparative clinical effects lurbinectedin and any alternative treatment. As such, based on the available evidence, the cost-effectiveness of lurbinectedin is unknown.
Based on the CADTH exploratory analysis, a price reduction of at least 83% would be needed for lurbinectedin to be considered cost-effective at a WTP threshold of $50,000 per QALY, compared with SYNTH, the synthetic basket comparator derived by the sponsor from real-world data (i.e., intended to reflect the usual care of patients in Canada whose disease has progressed on or after platinum-based chemotherapy). However, this estimate is subject to the limitations discussed, including a lack of comparative clinical data and an underestimation of lurbinectedin acquisition costs. Additional uncertainty is introduced by the combination of data from real-world clinical practice and from clinical trials. As such, a higher price reduction may be warranted. CADTH notes that, among patients with platinum-resistant SCLC, lurbinectedin was less effective than CAV, and there is no price reduction that would make lurbinectedin cost-effective in this patient subgroup.
Treatment with lurbinectedin increases costs to the health care system. Because of a lack of comparative efficacy evidence for currently available comparator treatments, the magnitude of benefit associated with lurbinectedin is unknown in terms of OS, PFS, health-related QoL, and other outcomes identified as important to patients and clinicians. Because of limitations in the structure of the sponsor’s model, the level of additional health care system costs was also highly uncertain. There is insufficient evidence to justify a price premium for lurbinectedin over other currently available treatments for SCLC.
References
1.pERC Final Recommendation: Daratumumab (Darzalex) plus lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy. 2017: https://www.cadth.ca/sites/default/files/pcodr/pcodr_daratumumab_darzalex_mm_2ndln_fn_rec.pdf. Accessed 2022 Feb 10.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Zepzelca (lurbinectedin): lyophilized powder, 4 mg/vial, intravenous infusion. Mississauga (ON): Jazz Pharmaceuticals; 2022 Feb 24.
3.Vedadi A, Shakik S, Brown MC, et al. The impact of symptoms and comorbidity on health utility scores and health-related quality of life in small cell lung cancer using real world data. Qual Life Res. 2021;30(2):445-454. PubMed
4.CADTH Reimbursement Review: larotrectinib (Vitrakvi). Can J Health Technol. 2021;1(11).
5.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
6.Kalemkerian GP, Loo BW, Akerley W, et al. NCCN Guidelines Insights: Small Cell Lung Cancer, Version 2.2018. J Natl Compr Canc Netw. 2018;16(10):1171-1182. PubMed
7.Zepzelca (lurbinectedin): lyophilized powder, 4 mg / vial, intravenous infusion [product monograph]. Dublin (IE): Jazz Pharmaceuticals; 2021 Sep 29.
8.Government of Alberta. Interactive Health Data Application. 2022; http://www.ahw.gov.ab.ca/IHDA_Retrieval/selectCategory.do. Accessed 2022 Feb 10.
9.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Zepzelca (lurbinectedin): lyophilized powder, 4 mg/vial, intravenous infusion. Mississauga (ON): Jazz Pharmaceuticals; 2022 Feb 24.
10.Brenner DR, Weir HK, Demers AA, et al. Projected estimates of cancer in Canada in 2020. CMAJ. 2020;192(9):E199-E205. PubMed
11.Canadian cancer statistics: a 2020 special report on lung cancer. Toronto (ON): Canadian Cancer Society, Statistics Canada, Public Health Agency of Canada; 2020: https://cdn.cancer.ca/-/media/files/cancer-information/resources/publications/2020-canadian-cancer-statistics-special-report/2020-canadian-cancer-statistics-special-report-en.pdf?rev=15c66a0b05f5479e935b48035c70dca3&hash=3D51B0D0FB5C3F7E659F896D66495CE8. Accessed 2022 Apr 18.
Appendix 1: Cost Comparison Table
Note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost Comparison Table for Relapsed SCLC
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | 21-day costa ($) |
|---|---|---|---|---|---|---|
Lurbinectedin (Zepzelca) | 4 mg | Vial | 6,470.0000a | 3.2 mg/m2 every 21 days | 616 | 12,940 |
Cyclophosphamide plus doxorubicin plus vincristine (CAV) | ||||||
Cyclophosphamide (Procytox) | 500 mg 1,000 mg | Powder for IV infusion | 93.1400 168.8300 | 800 mg/m2 on day 1 (21-day cycle) | 12 | 262 |
Doxorubicin (generic) | 2 mg/mL | 5 mL 25 mL 100 mL IV infusion | 50.4500 252.2500 973.0000 | 50 mg/m2 on day 1 (21-day cycle) | 24 | 505 |
Vincristine (generic) | 1 mg/mL | 1 mL 2 mL 5 mL IV infusion | 30.6000 61.2000 153.0000 | 1.4 mg/m2 on day 1 (21-day cycle) | 4 | 92 |
CAV | 41 | 858 | ||||
Carboplatin plus etoposide | ||||||
Carboplatin (generics) | 10 mg/mL | 5 mL 15 mL 45 mL 60 mL IV infusion | 70.0000 210.0000 599.9985 775.0020 | Area under the curve (AUC) 5 on day 1 (21-day cycle)b | 29 | 600 |
Etoposide (generic) | 20 mg/mL | 5 mL 10 mL 25 mL 50 mL Injection | 75.0000 150.0000 375.0000 750.0000 | 100 mg/m2 on days 1 to 3 (21-day cycle) | 21 | 450 |
Carboplatin plus etoposide | 50 | 1,050 | ||||
Cisplatin plus etoposide | ||||||
Cisplatin (generics) | 1 mg/mL | 50 mL 100 mL Vial | 135.0000 270.0000 | 75 mg/m2 on day 1 (21-day cycle) | 19 | 405 |
Etoposide (generic) | 20 mg/mL | 5 mL 10 mL 25 mL 50 mL Injection | 75.0000 150.0000 375.0000 750.0000 | 100 mg/m2 on days 1 to 3 of every 21-day cycle | 21 | 450 |
Cisplatin plus etoposide | 41 | 855 | ||||
Cisplatin plus irinotecan | ||||||
Cisplatin (generics) | 1 mg/mL | 50 mL 100 mL Vial | 135.0000 270.0000 | 75 mg/m2 on day 1 (21-day cycle) | 19 | 405 |
Irinotecan (generics) | 20 mg/mL | 2 mL 5 mL 25 mL | 218.0000 535.0000 2,675.0000 | 50 mg/m2 once per 21 days | 25 | 535 |
Cisplatin plus irinotecan | 45 | 940 | ||||
Carboplatin plus irinotecan | ||||||
Carboplatin (generics) | 10 mg/mL | 5 mL 15 mL 45 mL 60 mL Vial for IV infusion | 70.0000 210.0000 599.9985 775.0020 | Area under the curve (AUC) 5 on day 1 (21-day cycle)b | 29 | 600 |
Irinotecan (generics) | 20 mg/mL | 2 mL 5 mL 25 mL | 218.0000 535.0000 2,675.0000 | 50 mg/m2 once per 21 days | 25 | 535 |
Carboplatin plus irinotecan | 54 | 1,135 | ||||
Single drug treatments | ||||||
Topotecan (generic) | 4 mg | Powder | 567.0000 | 1.5 mg/m2 on days 1 to 5 (21-day cycle) | 135 | 2,835 |
Note: All prices are from the Delta IQVIA database (accessed April 2022), unless otherwise indicated, and do not include dispensing fees. Recommended dosage is based on Cancer Care Ontario monographs, unless otherwise indicated. For dosing that depends on weight or body surface area, CADTH assumed 71.0 kg or 1.8 m2 based on the B-005 trial. Total cost estimates per regimen are based on the cheapest combination of the component drugs, with wastage considered for single-use vials.
aLurbinectedin price based on the sponsor’s submission;8 dosage based on the lurbinectedin product monograph.1
bDose assumed by the sponsor: 450 mg.
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The sponsor’s pharmacoeconomic submission assumes that lurbinectedin will be used as a second-line treatment. This is in contrast with the Health Canada indication,7 which does not restrict use to the second-line. Clinicians consulted by CADTH for this review indicated that lurbinectedin may be used in the second- or third-line setting. CADTH additionally notes that the sponsor’s budget impact analysis assumes usage in the second or third-line. Owing to a lack of clinical information and the structure of the sponsor’s model, the cost-effectiveness of lurbinectedin in the third-line is unknown. |
Model has been adequately programmed and has sufficient face validity | No | The duration of lurbinectedin treatment in the model is not aligned with the Health Canada monograph.7 The sponsor was asked to provide a revised model in which the treatment duration reflects the monograph-recommended dosing (i.e., treatment received until disease progression or unacceptable toxicity) but declined. |
Model structure is adequate for decision problem | No | A partitioned-free survival model was used which introduced structural constraints. A Markov model would have been more appropriate. As noted above, the model structure as related to treatment duration is not reflective of expected clinical practice. Relevant subgroups (e.g., limited vs extensive-stage disease) could not be considered. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | For some model parameters, the sponsor arbitrarily incorporated uncertainty using a standard deviation equal to ± 20% of the mean value (e.g., percentage of patients experiencing an adverse event, relative dose intensity, health care costs, adverse event costs), which does not reflect the true uncertainty around the model’s parameters possible. values. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Discrepancies were noted between the PE report and model (e.g., BSA, inflation). |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
Table 9: Composition and Drug Acquisition Costs of RWE and SYNTH Basket Comparators Included in the Sponsor’s Pharmacoeconomic Model
Treatment | RWE | SYNTH |
|---|---|---|
Percentage of patients assumed to receive each treatment (%) | ||
Carboplatin + etoposide | 52% | 54% |
Cisplatin + etoposide | 18% | 21% |
Etoposide | 10% | 9% |
Cyclophosphamide + doxorubicin + vincristine (CAV) | 9% | 9% |
Topotecan | 3% | NA |
Other platinum regimens | 5% | NA |
Other regimens | 4% | 8% |
Drug acquisition costs ($) | ||
21-day cost | 2,078a | 2,097b |
NA = not applicable; RWE = basket comparator derived from real-world evidence; SYNTH = synthetic basket comparator derived from real-world evidence.
aThe sponsor applied an additional $260 per cycle to reflect the cost of antiemetic treatments, as well as a 1-time cost of $1,433 to reflect the overall cost of febrile neutropenia.
bThe sponsor applied an additional $275 per cycle to reflect the cost of antiemetic treatments, as well as a 1-time cost of $1,238 to reflect the overall cost of febrile neutropenia.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — B-005 Trial Population
Parameter | Lurbinectedin | Topotecan | RWE | SYNTH | CAV |
|---|---|---|---|---|---|
Discounted LYs | |||||
Total | 1.11 | 0.72 | 0.64 | 0.65 | 0.72 |
Progression-free | 0.35 | 0.32 | 0.51 | 0.51 | 0.32 |
Progressed disease | 0.76 | 0.41 | 0.13 | 0.14 | 0.41 |
Discounted QALYs | |||||
Total | 0.67 | 0.46 | 0.44 | 0.45 | 0.47 |
Progression-free | 0.25 | 0.21 | 0.36 | 0.37 | 0.22 |
Postprogression | 0.42 | 0.24 | 0.08 | 0.09 | 0.24 |
Discounted costs ($) | |||||
Drug costs | 53,131 | 21,904 | 13,152 | 13,308 | 10,136 |
Initial therapy | 52,820 | 21,593 | 12,876 | 13,013 | 9,825 |
Subsequent therapy | 311 | 311 | 276 | 295 | 311 |
Adverse events | 3,608 | 9,725 | 3,198 | 3,134 | 3,945 |
Progression-free | 1,392 | 7,508 | 1,231 | 1,033 | 1,728 |
Postprogression | 2,216 | 2,217 | 1,967 | 2,101 | 2,217 |
Medical costs | 6,517 | 4,263 | 2,701 | 2,786 | 4,044 |
Progression-free | 1,178 | 1,389 | 1,763 | 1,776 | 1,170 |
On subsequent therapy | 876 | 1,277 | 1,349 | 1,362 | 928 |
Not on subsequent therapy | 302 | 112 | 414 | 414 | 242 |
Postprogression | 5,339 | 2,874 | 938 | 1,010 | 2,874 |
Palliative care costs | 12,187 | 6,408 | 1,907 | 2,056 | 6,408 |
Mortality | 32,140 | 32,369 | 32,409 | 32,403 | 32,369 |
Total | 107,582 | 74,669 | 53,369 | 53,688 | 56,903 |
CAV = cyclophosphamide + doxorubicin + vincristine; LY = life-year; QALY = quality-adjusted life-year; RWE = basket comparator derived from real-world evidence; SYNTH = synthetic comparator (basket comparator derived from real-world evidence).
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Platinum-Sensitive Subgroup
Parameter | Lurbinectedin | Carboplatin | Cisplatin | Carboplatin | Topotecan |
|---|---|---|---|---|---|
Discounted LYs | |||||
Total | 1.52 | 0.65 | 0.59 | 0.63 | 0.89 |
Progression-free | 0.44 | 0.34 | 0.34 | 0.34 | 0.34 |
Postprogression | 1.08 | 0.30 | 0.24 | 0.29 | 0.54 |
Discounted QALYs | |||||
Total | 0.91 | 0.43 | 0.38 | 0.41 | 0.55 |
Progression-free | 0.31 | 0.24 | 0.24 | 0.23 | 0.23 |
Postprogression | 0.60 | 0.18 | 0.14 | 0.18 | 0.33 |
Discounted costs ($) | |||||
Drug costs | 56,142 | 13,236 | 15,411 | 15,151 | 30,807 |
Initial therapy | 55,824 | 12,912 | 15,189 | 14,852 | 30,483 |
Subsequent therapy | 318 | 324 | 222 | 299 | 324 |
Adverse events | 4,010 | 3,373 | 2,494 | 7,898 | 11,285 |
Progression-free | 1,683 | 1,003 | 872 | 5,711 | 8,915 |
Postprogression | 2,327 | 2,370 | 1,622 | 2,187 | 2,370 |
Medical costs | 9,030 | 3,611 | 3,178 | 3,510 | 5,419 |
Progression-free | 1,411 | 1,458 | 1,454 | 1,441 | 1,581 |
On subsequent therapy | 1,005 | 1,304 | 1,301 | 1,287 | 1,499 |
Not on subsequent therapy | 406 | 154 | 153 | 154 | 82 |
Postprogression | 7,619 | 2,153 | 1,724 | 2,069 | 3,838 |
Palliative care costs | 17,597 | 4,734 | 3,829 | 4,563 | 8,699 |
Mortality | 31,656 | 32,132 | 32,155 | 32,140 | 32,017 |
Total | 118,436 | 57,088 | 57,066 | 63,262 | 88,226 |
LY = life-year; QALY = quality-adjusted life-year.
Table 12: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Platinum-Resistant Subgroup
Parameter | Lurbinectedin | CAV | Topotecan |
|---|---|---|---|
Discounted LYs | |||
Total | 0.58 | 0.60 | 0.56 |
Progression-free | 0.25 | 0.34 | 0.34 |
Postprogression | 0.33 | 0.26 | 0.22 |
Discounted QALYs | |||
Total | 0.37 | 0.40 | 0.36 |
Progression-free | 0.18 | 0.24 | 0.23 |
Postprogression | 0.19 | 0.16 | 0.13 |
Discounted costs ($) | |||
Drug costs | 49,094 | 8,553 | 26,030 |
Initial therapy | 48,770 | 8,230 | 25,707 |
Subsequent therapy | 324 | 323 | 323 |
Adverse events | 3,463 | 4,009 | 9,051 |
Progression-free | 1,143 | 1,694 | 6,736 |
Postprogression | 2,320 | 2,315 | 2,315 |
Medical costs | 3,207 | 3,019 | 2,852 |
Progression-free | 890 | 1,160 | 1,307 |
On subsequent therapy | 704 | 909 | 1,145 |
Not on subsequent therapy | 186 | 251 | 162 |
Postprogression | 2,317 | 1,859 | 1,545 |
Palliative care costs | 5,116 | 4,037 | 3,301 |
Mortality | 32,339 | 32,328 | 32,349 |
Total | 93,218 | 51,946 | 73,584 |
CAV = cyclophosphamide + doxorubicin + vincristine; LY = life-year; QALY = quality-adjusted life-year.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Scenario Analyses
While CADTH did not conduct any formal reanalyses of the sponsor’s model, the economic review team performed an exploratory analysis to explore the impact of several key limitations on the ICER, notably, the impact of adopting alternate parametric survival distributions for PFS and OS. All CADTH exploratory reanalyses are deterministic owing to limitations with the programming of the sponsor’s model.
The fundamental limitations in the sponsor’s model persist within this exploratory analysis. There is no direct evidence to support the comparative efficacy of lurbinectedin to any comparator, and the pharmacoeconomic model is informed by naive comparisons. Therefore, this exploratory analysis should not be interpreted as a formal CADTH reanalysis to which credence should be given to the results; in particular, the incremental QALY benefit estimated as part of this exploratory analysis remains unlikely to be representative of the true effect of lurbinectedin, such that the corresponding ICER is unlikely to be reflective of the true cost-effectiveness of lurbinectedin.
Table 13: CADTH Revisions to the Submitted Economic Evaluation — B-005 Trial Population
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH exploratory base case | ||
1. Extrapolation of PFS | Log-logistic | Gompertz |
2. Extrapolation of OS | Log-logistic | Weibull |
CADTH exploratory reanalysis | 1 + 2 | |
OS = overall survival; PFS = progression-free survival.
CADTH undertook a stepped analysis, incorporating each change proposed in Table 14 to sponsor’s base case to highlight the impact of each change. Given the sponsor’s use of naive comparisons, a sequential analysis would be inappropriate. As such, CADTH explored the cost-effectiveness of lurbinectedin relative to SYNTH, as this synthetic control arm was intended by the sponsor to reflect the standard care of patients living in Canada who meet the eligibility criteria of the B-005 clinical trial. As per the sponsor’s analysis, the CADTH exploratory analyses found that lurbinectedin is more costly and more effective than SYNTH and would not be cost-effective at a $50,000 per QALY threshold compared to SYNTH.
Table 14: Summary of the Stepped Analysis of the CADTH Reanalysis Results — B-005 Trial Population (Lurbinectedin vs. SYNTH)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base casea,b,c | SYNTH | 53,734 | 0.45 | Reference |
Lurbinectedin | 107,726 | 0.67 | 247,181 | |
CADTH reanalysis 1: PFS | SYNTH | 53,688 | 0.45 | Reference |
Lurbinectedin | 107,945 | 0.67 | 248,355 | |
CADTH reanalysis 2: OS | SYNTH | 53,688 | 0.45 | Reference |
Lurbinectedin | 104,400 | 0.62 | 302,179 | |
CADTH exploratory analysis (1 + 2)a | SYNTH | 53,688 | 0.45 | Reference |
Lurbinectedin | 104,696 | 0.62 | 307,232 |
ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year; SYNTH = synthetic comparator.
aThe sponsor’s results are probabilistic; all remaining analyses are deterministically presented, including the CADTH exploratory base case.
bReference product is SYNTH, a synthetic comparator arm (i.e., basket of treatments) intended to reflect standard of care among patients who meet the B-005 eligibility criteria, based on real-world data. Includes carboplatin + etoposide, cisplatin + etoposide, CAV (cyclophosphamide + doxorubicin + vincristine), etoposide, and “other regimens” (not defined by sponsor).
cSponsor’s submitted base case: lurbinectedin vs. SYNTH only.
Table 15: Disaggregated Summary of CADTH’s Economic Evaluation Results — B-005 Trial Population
Parametera | Lurbinectedin | SYNTH | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 0.97 | 0.65 | 0.31 |
Progression-free | 0.34 | 0.51 | −0.17 |
Postprogression | 0.63 | 0.14 | 0.49 |
Discounted QALYs | |||
Total | 0.62 | 0.45 | 0.17 |
Progression-free | 0.24 | 0.37 | −0.13 |
Postprogression | 0.38 | 0.09 | 0.29 |
Discounted costs ($) | |||
Drug costs | 53,131 | 13,308 | 39,823 |
Initial therapy | 52,820 | 13,013 | 39,807 |
Subsequent therapy | 311 | 295 | 16 |
Adverse events | 3,609 | 3,134 | 475 |
Progression-free | 1,392 | 1,033 | 359 |
Postprogression | 2,217 | 2,101 | 116 |
Medical costs | 5,613 | 2,786 | 2,827 |
Progression-free | 1,167 | 1,776 | −609 |
On subsequent therapy | 876 | 1,362 | −486 |
Not on subsequent therapy | 291 | 414 | −123 |
Postprogression | 4,446 | 1,010 | 3,436 |
Palliative care costs | 10,094 | 2,056 | 8,038 |
Mortality | 32,251 | 32,403 | −152 |
Total | 104,696 | 53,688 | 51,008 |
ICER ($/QALYs) | $307,232 vs. SYNTH | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SYNTH = synthetic comparator (basket comparator derived from real-world evidence).
aDeterministic analysis.
Several scenario and sensitivity analyses were conducted on the CADTH exploratory reanalysis. These scenario analyses explored the impact of the following model parameters and assumptions:
Lurbinectedin acquisition cost based on mean treatment duration in the B-005 trial (19.7 weeks).
Cost-effectiveness of lurbinectedin in the platinum-sensitive subgroup.
Table 16: CADTH Scenario Analyses
Scenario | CADTH base case | CADTH scenario |
|---|---|---|
Scenario analyses | ||
1. Duration of lurbinectedin treatment | 4 cycles | 5.9 cyclesa |
2. Patient population | B-005 trial population; includes platinum-sensitive (57.1%) and platinum-resistant patients (42.9%) | Platinum-sensitive subgroup:b,c a. lurbinectedin vs. carboplatin + etoposide b. lurbinectedin vs. topotecan |
aBased on the mean number of treatment cycles in the B-005 trial.
bGompertz distribution adopted for PFS; GenGamma distribution adopted for OS.
cNo subgroup analyses were undertaken for the platinum-resistant subgroup, as lurbinectedin was less effective and more costly (dominated) compared to CAV in the sponsor’s submitted analysis.
Table 17: CADTH Scenario Analyses Results
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
CADTH exploratory analysis | |||
SYNTH | 53,688 | 0.45 | Reference |
Lurbinectedin | 104,696 | 0.62 | 307,232 |
Scenario 1: Lurbinectedin treatment duration | |||
SYNTH | 53,688 | 0.45 | Reference |
Lurbinectedin | 134,250 | 0.61 | 496,021 |
Scenario 2a: Platinum-sensitive subgroup (lurbinectedin vs. carboplatin + etoposide) | |||
Carboplatin + etoposide | 57,197 | 0.43 | Reference |
Lurbinectedin | 115,120 | 0.85 | 135,625 |
Scenario 2b: Platinum-sensitive subgroup (lurbinectedin vs. topotecan) | |||
Topotecan | 88,226 | 0.55 | Ref. |
Lurbinectedin | 115,120 | 0.85 | 90,128 |
Price Reduction Analysis
As no formal CADTH reanalysis was performed, price reduction analyses were conducted using only the sponsor’s base-case assumptions and the CADTH exploratory reanalysis. This deterministic analysis was subject to the key limitations of the sponsor’s model as noted in the CADTH Appraisal of the Sponsor’s Economic Evaluation section. Based on the CADTH exploratory analysis, a reduction in the price of lurbinectedin by 83% would be required for lurbinectedin to be cost-effective at a WTP threshold of $50,000 per QALY compared to SYNTH. In the platinum-sensitive subgroup, price reductions of 22% and 68% would be required for lurbinectedin to be cost-effective at a WTP of $50,000 per QALY compared to topotecan and to carboplatin + etoposide, respectively.
It is important to note that this price reduction estimate is based on estimates of incremental LYs (and hence QALYs) that are highly uncertain and may not be representative of the true incremental effect of lurbinectedin. Similarly, this price reduction estimate is based on lurbinectedin being received by all patients for 4 treatment cycles, which does not reflect the Health Canada–recommended dosing strategy. Should lurbinectedin be received for longer, the drug acquisition costs will be higher, and a greater price reduction would be required for lurbinectedin to be considered cost-effective at a WTP of $50,000 per QALY.
Table 18: CADTH Price Reduction Analyses — B-005 Trial Population
Price reduction | ICERs for lurbinectedin vs. SYNTH ($) | |
|---|---|---|
Sponsor’s base case | CADTH exploratory analysis | |
No price reduction | 244,546 | 307,232 |
10% | 221,138 | 276,160 |
20% | 197,730 | 245,087 |
30% | 174,321 | 214,014 |
40% | 150,913 | 182,942 |
50% | 127,505 | 151,869 |
60% | 104,097 | 120,796 |
70% | 80,689 | 89,724 |
80% | 57,280 | 58,651 |
83% | 50,258 | 49,329 |
90% | 33,872 | 27,578 |
100% | 10,464 | LUR dominant |
ICER = incremental cost-effectiveness ratio; NA = not applicable; vs. = versus.
Note: All analyses in this table are deterministic and are subject to limitations within the sponsor’s economic model.
Appendix 5: Submitted BIA and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 19: Summary of Key Takeaways
Key takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA estimating the incremental budget impact of reimbursing lurbinectedin for the treatment of patients with stage III or metastatic SCLC that has progressed on or after prior platinum-containing therapy.9 The BIA was undertaken from the perspective of the Canadian public drug plans over a 3-year time horizon, and the sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec). Key inputs to the BIA are documented in Table 21.
The sponsor estimated the eligible population using an epidemiologic approach (Figure 1), based on an annual lung cancer incidence of 97 of 100,000. Of these, 12.1% of patients were assumed to have SCLC.10 The sponsor assumed that, of patients with SCLC, 25% will have stage III (limited stage) and that 67% will have stage IV (extensive-stage) disease at diagnosis. The incident population at both stage III and IV was further segmented by line of therapy and by platinum-sensitive and platinum-resistant disease status (based on chemotherapy-free interval). In the reference scenario, patients were assumed to receive platinum-based chemotherapy, topotecan, or CAV. In the new drug scenario, lurbinectedin was assumed to be reimbursed and prescribed as second- or third-line therapy and to take market share from these therapies in a manner proportional to their market share in the scenario without lurbinectedin. The market share of lurbinectedin varied by line of therapy, disease stage, and platinum sensitivity (refer to Table 20) based on sponsor’s internal data, market research, and clinician input (reiterated at the CADTH presubmission meeting).
In the sponsor’s base case, costs related to drug acquisition were captured. In the sponsor’s base case, wastage was assumed, such that unused portions of drug vials would be discarded. Lurbinectedin was assumed to be received for 4 21-day cycles based on the median treatment duration in the B-005 trial. Duration of treatment for comparators ranged from 3 to 6 cycles. The cost of lurbinectedin was based on the sponsors submitted price ($6,470.00 per 4 mg vial). Drug costs of other regimens were obtained from a previous CADTH review.4 Costs related to dispensing, markup, administration, or subsequent therapy were not included.
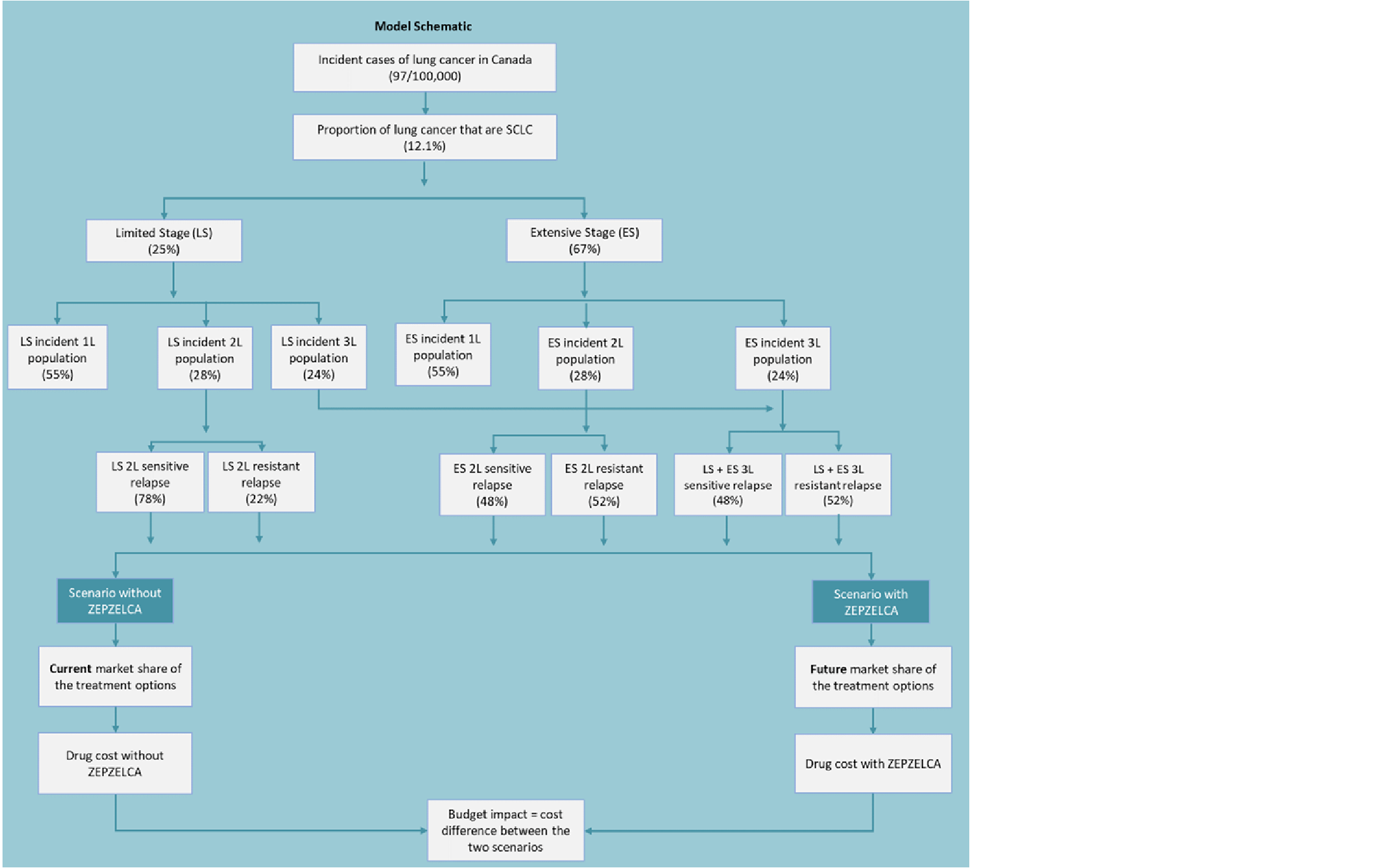
Table 20: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (year 1 / year 2 / year 3) | |
|---|---|---|
Target population | ||
Adult population in Canadaa | 25,326,885 / 25,682,617 / 26,038,350 | |
Annual lung cancer incidence | 97 / 100,000 | |
Proportion of lung cancer that is SCLC | 12.1%10 | |
Proportion of SCLC that is limited stage (stage III) | 25%11 | |
Proportion of SCLC that is extensive stage (stage IV) | 67%11 | |
Receiving 1L therapy | 55% | |
Receiving 2L therapy | 28% | |
Receiving 3L therapy | 24% | |
Number of patients eligible for the drug under review | 529 / 537 / 544 | |
Second-line | 427 / 433 / 439 | |
Third-line | 102 / 104 / 105 | |
Market uptake (3 years) | ||
Uptake (reference scenario) | Platinum-sensitive disease | Platinum-resistant disease |
Second-line | ||
Limited stage | ||
Platinum chemotherapy | 74% / 74% / 74% | 0% / 0% / 0% |
Topotecan | 25% / 25% / 25% | 71% / 71% / 71% |
CAV | 1% / 1% / 1% | 29% / 29% / 29% |
Extensive stage | ||
Platinum chemotherapy | 76% / 76% / 76% | 0% / 0% / 0% |
Topotecan | 21% / 21% / 21% | 65% / 65% / 65% |
CAV | 3% / 3% / 3% | 35% / 35% / 35% |
Third-line | ||
Platinum chemotherapy | 23% / 23% / 23% | 0% / 0% / 0% |
Topotecan | 50% / 50% / 50% | 41% / 41% / 41% |
CAV | 28% / 28% / 28% | 59% / 59% / 59% |
Uptake (new drug scenario) | Platinum-sensitive disease | Platinum-resistant disease |
Second-line | ||
Limited stage | ||
Lurbinectedin | 40% / 50% / 55% | 50% / 60% / 70% |
Platinum chemotherapy | 44% / 37% / 33% | 0% / 0% / 0% |
Topotecan | 15% / 12% / 11% | 35% / 28% / 21% |
CAV | 1% / 1% / 1% | 15% / 12% / 9% |
Extensive-stage | ||
Lurbinectedin | 40% / 50% / 55% | 50% / 60% / 70% |
Platinum chemotherapy | 46% / 38% / 34% | 0% / 0% / 0% |
Topotecan | 12% / 10% / 9% | 33% / 26% / 20% |
CAV | 2% / 2% / 2% | 17% / 14% / 10% |
Third-line | ||
Lurbinectedin | 45% / 35% / 30% | 45% / 35% / 30% |
Platinum chemotherapy | 12% / 15% / 16% | 0% / 0% / 0% |
Topotecan | 28% / 33% / 35% | 22% / 26% / 28% |
CAV | 15% / 18% / 19% | 33% / 39% / 42% |
Cost of treatment (per patient) | Per 21-day treatment cycle | Total drug acquisition cost included in the BIAb |
Lurbinectedin | $12,940 | $51,760 |
Platinum chemotherapy | Carboplatin + etoposide: $1,360 Cisplatin + etoposide: $1,742 | $7,754c |
Topotecan | $2,835 | $17,010 |
CAV | $1,457 | $4,371 |
aSum of adult patients in participating drug plans (i.e., excluding Quebec, including NIHB).
bThe sponsor assumed that each treatment would be administered for a fixed duration, regardless of treatment response. Lurbinectedin: 4 cycles; platinum chemotherapy: 5 cycles; topotecan: 6 cycles; CAV: 3 cycles (all 21-day cycles).
cAssumed to be the average of carboplatin + etoposide and cisplatin + etoposide, over a duration of 5 cycles.
Summary of the Sponsor’s BIA Results
The sponsor estimated the 3-year budget impact of reimbursing lurbinectedin for the treatment of adult patients with stage III or IV SCLC who have progressed on or after prior platinum-containing therapy to be $32,910,109 (Year 1: $9,589,408; Year 2: $11,058,221; Year 3: $12,262,481).
The 3-year budget impact of reimbursing lurbinectedin as a second-line therapy was estimated to be $28,197,746 (Year 1: $7,684,727; Year 2: $9,555,995; Year 3: $10,957,024), and the impact of reimbursing lurbinectedin as third-line therapy was estimated to be $4,712,363 (Year 1: $1,904,680; Year 2: $1,502,225; Year 3: $1,305,457).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The number of patients eligible for lurbinectedin is uncertain. There is uncertainty in the number of eligible patients for several reasons. First, the sponsor used an epidemiological approach to estimate the target population size, and there is uncertainty in several inputs. The sponsor assumed that, in each year, all patients would be newly diagnosed (i.e., incident cases), noting the short OS of this population. The sponsor applied the same incidence rate (97 per 100,000 population) in each year of the BIA to determine the number of newly diagnosed SCLC cases. It is uncertain whether the incidence of SCLC will remain constant over the budget impact horizon, as the Canadian Cancer Society notes that the age-standardized incidence rates for SCLC are decreasing over time. The sponsor further assumed that 25% of newly diagnosed patients will have limited-stage disease and 67% would have extensive-stage disease, based on estimates from the Canadian Cancer Society reflecting data from 1992 to 2016. It is uncertain whether this distribution is consistent across Canadian jurisdictions, as clinical experts consulted by CADTH noted that, in their practices, about 75% of patients have extensive-stage disease at presentation, and data from an Alberta cohort suggest that 58% have extensive-stage disease at presentation. Clinical experts further noted that there is considerable uncertainty associated with how the sponsor has grouped patients by disease stage and line of therapy and that, in practice, patients can move between groups within a given year. For example, a patient at first line (i.e., not eligible for lurbinectedin) in the first year of the BIA would become eligible for lurbinectedin should their disease progress, which could occur within the same year or in the subsequent year. Such patients were not accounted for in the sponsor’s estimate of the eligible population. Additionally, the introduction of lurbinectedin may be associated with an increased number of patients undergoing treatment, as noted by a clinical expert consulted by the sponsor.9
CADTH explored the impact of uncertainty in input parameters related to the number of eligible patients in scenario analyses.
Unclear market shares for comparators: The sponsor assumed that between 59% and 91% of patients (depending on line of therapy, disease stage, platinum sensitivity) would receive a treatment deemed to be a comparator to lurbinectedin, and the sponsor reweighted the market share of the remaining treatments (platinum chemotherapy, CAV, topotecan). Noncomparator regimens were assumed to included irinotecan + platinum, radiation, best supportive care, and clinical trial regimens. Clinical experts consulted by CADTH for this review indicated the irinotecan-based regimens would be relevant in this patient population, and CADTH notes that carboplatin + irinotecan was included as a comparator in the sponsor’s pharmacoeconomic model for the platinum-sensitive subgroup).
CADTH was unable to validate the proportion of patients who receive “noncomparator regimens.” In CADTH reanalyses, irinotecan + platinum was included as a comparator, based on a sponsor-provided option in the model.
Uncertainty regarding the uptake of lurbinectedin. The sponsor assumed that the uptake of lurbinectedin will vary by disease stage, line of therapy, and platinum sensitivity (refer to Table 20), and that lurbinectedin will take market share from platinum-based chemotherapy (among platinum-sensitive patients), topotecan, and CAV. Clinical experts consulted by CADTH for this review indicated that, among platinum-sensitive patients, lurbinectedin is likely to take market share from CAV and topotecan but not platinum-based regimens, although this may vary by line of therapy and by patient characteristics (e.g., age, performance status). Clinical experts indicated that the sponsor’s assumption of higher uptake among patients with platinum-resistant disease is reasonable.
CADTH explored the impact of alternative uptake assumptions in scenario analyses.
Lurbinectedin acquisition costs may be underestimated. In the calculation of drug costs, the sponsor assumed that patients would receive lurbinectedin for 5 cycles. This is in contrast with the product monograph, which recommends that lurbinectedin be administered until disease progression or unacceptable toxicity. CADTH requested that the sponsor revise their BIA to reflect the monograph dosing; however, the sponsor declined this request, stating that “the appropriate number of treatment cycles for ZEPZELCA based on the relevant clinical data is 4.” CADTH notes that, while the median number of cycles per patient in the lurbinectedin B-005 clinical trial was 4, 15.2% of patients received exactly 4 cycles, and 47.6% of patients received 5 or more cycles (43.8% received 6 or more cycles). In the B-005 trial, the mean number of treatment cycles was 5.9 cycles. CADTH additionally notes that the median number of cycles was higher among patients with platinum-sensitive disease (6 cycles; range 1 to 24 cycles) compared to those with platinum-resistant disease (2 cycles; range: 1 to 18 cycles). This variation in dosing by platinum-sensitivity status was not accounted for in the sponsor’s budget analysis.
Drug costs associated with lurbinectedin acquisition are likely higher than quoted by the sponsor. If patients receive lurbinectedin for a mean duration of 19.7 weeks at a cost of $12,940 per 21-days, lurbinectedin treatment cost will be $84,973 over the mean duration of therapy. This estimate does not include the cost of subsequent treatment after lurbinectedin discontinuation. CADTH explored the impact of treatment duration in scenario analyses.
Costs related to subsequent treatment after lurbinectedin discontinuation were not considered. Such costs are relevant under the drug plan perspective and were included in the sponsor’s CUA. In the CUA, the sponsor assumed that 24% of patients would receive third-line treatment after lurbinectedin, while in the B-005 trial, 47% of patients received subsequent treatment after discontinuation of lurbinectedin. Clinical experts consulted by CADTH for this review noted that, in practice, the proportion of patients who receive subsequent therapy varies by line of therapy.
CADTH was unable to consider costs associated with subsequent treatment owing to the structure of the sponsor’s model and a lack of clinical data. The inclusion of costs associated with subsequent treatments after lurbinectedin would increase the costs associated with its reimbursement.
Additional limitations were identified but were not considered to be key limitations.
In the calculation of drug costs, the sponsor assumed that only the largest vial size would be used (regardless of the availability of smaller and less costly vials for some drugs) to achieve the required mg per dose (e.g., for carboplatin, vincristine). As a result, the drug acquisition costs of some comparators are overestimated.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by including irinotecan-based regimens as a relevant comparator.
Table 21: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Irinotecan + platinum | Excluded | Included, based on a sponsor-provided option in the model (i.e., market share was provided by the sponsor)a |
CADTH base case | Reanalysis 1 | |
aMarket share for irinotecan + platinum was provided by the sponsor and was assumed to vary by disease stage, platinum sensitivity, and line of therapy (range: 5% to 13% in the baseline year).
The results of the CADTH step-wise reanalysis are presented in summary format in Table 22 and a more detailed breakdown is presented in Table 23. Based on the CADTH reanalysis, the budget impact of the reimbursement of lurbinectedin for the treatment of patients with stage III or metastatic SCLC that has progressed on or after prior platinum-containing therapy is expected to be $9,582,252 in year 1, $11,052,096 in year 2, and $12,257,895 in year 3, with a 3-year total of $32,892,244.
The estimated budget impact of reimbursing lurbinectedin is sensitive to the duration for which lurbinectedin is received. When the duration of lurbinectedin treatment was aligned with the mean treatment duration from the B-005 trial, the estimated budget impact increased by 74% increase (Table 23).
Table 22: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 32,910,109 |
CADTH reanalysis 1 (CADTH base case) | 32,892,244 |
BIA = budget impact analysis.
CADTH also conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 23.
Assuming a 10% higher number of eligible patients.
Assuming 75% of patients have extensive-stage disease at presentation.
Assuming that all patients will receive lurbinectedin for 5.9 cycles, based on the mean number of treatment cycles in the B-005 trial.
Assuming 15% higher uptake of lurbinectedin.
Price of lurbinectedin is reduced by 83%.
Table 23: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 5,692,558 | 5,773,653 | 5,854,747 | 5,935,842 | 17,564,241 |
New drug | 5,692,558 | 15,363,060 | 16,912,968 | 18,198,322 | 50,474,350 | |
Budget impact | 0 | 9,589,408 | 11,058,221 | 12,262,481 | 32,910,109 | |
CADTH base case | Reference | 5,713,251 | 5,794,640 | 5,876,029 | 5,957,419 | 17,628,088 |
New drug | 5,713,251 | 15,376,892 | 16,928,126 | 18,215,314 | 50,520,332 | |
Budget impact | 0 | 9,582,252 | 11,052,096 | 12,257,895 | 32,892,244 | |
CADTH sensitivity analysis: 10% more eligible patients | Reference | 6,284,576 | 6,374,104 | 6,463,632 | 6,553,161 | 19,390,897 |
New drug | 6,284,576 | 16,914,581 | 18,620,938 | 20,036,845 | 55,572,365 | |
Budget impact | 0 | 10,540,477 | 12,157,306 | 13,483,685 | 36,181,469 | |
CADTH sensitivity analysis: 75% of patients with extensive-stage disease | Reference | 6,198,481 | 6,286,783 | 6,375,085 | 6,463,387 | 19,125,255 |
New drug | 6,198,481 | 16,686,358 | 18,368,047 | 19,769,708 | 54,824,112 | |
Budget impact | 0 | 10,399,574 | 11,992,962 | 13,306,321 | 35,698,857 | |
CADTH scenario analysis: Lurbinectedin received for 5.9 cycles | Reference | 6,693,558 | 6,788,912 | 6,884,267 | 6,979,621 | 20,652,800 |
New drug | 6,693,558 | 21,714,315 | 24,102,150 | 26,083,366 | 71,899,832 | |
Budget impact | 0 | 14,925,403 | 17,217,884 | 19,103,745 | 51,247,032 | |
CADTH scenario analysis: 15% higher lurbinectedin uptake | Reference | 5,713,251 | 5,794,640 | 5,876,029 | 5,957,419 | 17,628,088 |
New drug | 5,713,251 | 16,814,230 | 18,585,940 | 20,053,998 | 55,454,169 | |
Budget impact | 0 | 11,019,590 | 12,709,911 | 14,096,580 | 37,826,081 | |
CADTH scenario analysis: 83% price reduction for lurbinectedin | Reference | 5,713,251 | 5,794,640 | 5,876,029 | 5,957,419 | 17,628,088 |
New drug | 5,713,251 | 5,258,851 | 5,254,768 | 5,251,235 | 15,764,854 | |
Budget impact | 0 | −535,789 | −621,261 | −706,184 | −1,863,234 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Lung Health Foundation / The Ontario Lung Association
About the Lung Health Foundation / The Ontario Lung Association
The Ontario Lung Association (now named Lung Health Foundation) is registered with the CADTH and pCODR (www.lunghealth.ca). The Lung Health Foundation (Ontario Lung Association) is a registered charity that assists and empowers people living with or caring for others with lung disease. It is a recognized leader, voice and primary resource in the prevention and control of respiratory illness, tobacco cessation and prevention, and its effects on lung health. The Foundation provides programs and services to patients and health-care providers, invests in lung research and advocates for improved policies in lung health. It is run by a board of directors and has approximately 46 employees, supported by thousands of dedicated volunteers.
Information Gathering
The information provided from the Lung Health Foundation in this submission was obtained from an online survey and three phone interviews that were conducted between September and December 2021. The interviews were with two female patients and one male patient living with lung cancer. All the patients interviewed were over the age of 50. One of the female patients is based in Ontario and the other patient is based in Manitoba. The male patient was from Quebec. There were 2 survey respondents and demographic data was not collected. Input from a Registered Nurse is also included based on information gathered from monthly support groups attended by patients and their caregivers. Input from a certified respiratory educator was also obtained for this submission. The individual reviewed sections related to disease experience, experiences with available treatments and outcomes.
Disease Experience
Patients interviewed expressed that they found it difficult to cope with a lung cancer diagnosis. Lung cancer is associated with a poor prognosis and is the leading cause of cancer related deaths (Canadian Cancer Statistics, 2021). The patients interviewed report that the symptoms they experience with lung cancer were, in most cases, mild and are often associated with other conditions which led to a late diagnosis. The symptoms reported were shortness of breath, fatigue and pain. One patient interviewed, reported that she had a lingering cough for over six months before she was screened for lung cancer. She had been considered low risk because she did not have a smoking history. Another patient interviewed reported that she received her diagnosis during the peak of the COVID pandemic. Delays in getting diagnostic tests and starting treatment was a great source of distress for her. Patients found the psychosocial effects of having a disease with a poor prognosis challenging and they also struggled with side effects associated with some treatments.
Some of the psychosocial effects reported were anxiety (66%), distress (100%) and depression (66%). One patients reported that having lung cancer was particularly isolating because of the stigma associated with lung cancer. She withdrew from all activities because she did not want people to know that she was diagnosed with lung cancer. Another patient interviewed described having a challenging time maintaining relationships with families and friends. They felt short tempered and impatient. Physical and emotional intimacy were also reported to be a challenge.
The side effects related to some treatments severely impact day to day and quality of life. One of the patients interviewed reported that he struggled with the side effects of chemotherapy. Prior to starting treatment he was active and played sports, but once he started chemotherapy, he was unable to participate in his usual activities. He reported having hair loss, loss of appetite, weight loss, poor sleep, difficulty breathing and this severely impacted his quality of life. This was very challenging for him. He also reported that the hair loss impacted his self-esteem because he looked visibly ill.
Another patient interviewed, reported that she experienced neuropathy, difficulty swallowing, fatigue and scarring in her lungs resulting in breathing difficulties. This negatively impacted her quality of life and ability to work and care for her family.
Family members and caregivers of those living with lung cancer share the same psychosocial burdens as the patients. They also have the added responsibility of providing care. Being a caregiver affects their ability to work, their relationships with family and friends and their emotional well-being. Their independence and ability to travel and socialize are often impacted as well. Having to take time off work to drive those they are caring for to get groceries, run errands or attend medical appointments can be problematic for caregivers. Feelings of fatigue and emotional exhaustion are not uncommon.
Experiences With Currently Available Treatments
The treatments tried by the respondents included surgery, radiation, chemotherapy, targeted therapy and immunotherapy. The medications tried included Cisplatin, Docetaxel, Gefitinib, Entrectnib, Alectinib, Brigatinib, Opdivo+Yervoy and Tagrisso.
The benefits experienced with the treatments were prolonged life, delayed disease progression and a reduction in the severity of disease-related symptoms. Although these benefits were noted, most patients struggled with lingering side effects. Respondents who received surgery, reported deconditioning and chronic fatigue. Some of the side effects reported from radiation were fatigue, skin changes, hair loss and tissue scarring.
With oral and subcutaneous medications, the side effects reported included fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy. Side effects from chemotherapy severely impacted the patients’ quality of life, ability to work and in some cases, the ability to perform activities of daily living.
When asked about challenges with access to treatment, the respondents reported that they struggled with the cost of treatments and navigating the healthcare system. Some respondents reported travelling several hours to access treatments and sometimes they needed to stay overnight in hotels. This added a financial burden to the treatment process.
Patients also found delays in treatment and diagnostic testing to be a great source of distress because lung cancer progresses quickly and advanced disease is associated with poorer outcomes.
Improved Outcomes
Key treatment outcomes for this group of lung cancer patients include stopping or slowing the progression of the disease with minimal side effects and medications that are effective for advanced disease. Due to the poor outcomes associated with advanced disease, patients describe feeling very anxious about any sign or prospect of disease progression. Patients state that if treatments were more effective in treating lung cancer at any stage, then a diagnosis would not feel like a “death sentence”.
Patients would also like treatments with minimal side effects so that they can carry on with regular activities while on treatment. The importance of maintaining some quality of life cannot be overstated.
When choosing therapy, patients are most interested in the efficacy of the medication. One respondent commented that they would be more receptive to side effects if there was a guarantee that the medication would stop or slow down the progression of lung cancer.
Experience With Drug Under Review
No patients within this evidence group submission had experience with the medication under review.
The majority of small cell lung cancer patients are diagnosed at advanced stages (65%) with an extremely poor prognosis. There is an urgent need for treatment options for this population as if a patient progresses on chemotherapy, the options are limited. (Yang, S., Zhang, Z., & Wang, Q. (2019). Emerging therapies for small cell lung cancer. Journal of Hematology & Oncology, 12(1),1-11.)
Companion Diagnostic Test
Not applicable
Anything Else?
Not applicable
Reference
Canadian Cancer Statistics Advisory Committee in collaboration with the Canadian Cancer Society, Statistics Canada and the Public Health Agency of Canada. Canadian Cancer Statistics 2021. Toronto, ON: Canadian Cancer Society; 2021
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Conflict of Interest Declaration for Lung Health Foundation/Ontario Lung Association
Company | Check Appropriate Dollar Range | |||||
|---|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In excess of $50,000 | |||
Jazz Pharmaceuticals | — | — | — | X | ||
Lung Cancer Canada
About Lung Cancer Canada
Lung Cancer Canada is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research, and advocacy. Lung Cancer Canada is a member of the Global Lung Cancer Coalition and is the only organization in Canada focused exclusively on lung cancer.
https://www.lungcancercanada.ca/
Lung Cancer Canada is registered with CADTH.
Information Gathering
Data Collection
The information discussed throughout this submission consists of the thoughts and experiences of small cell lung cancer patients and caregivers, conducted through interviews and environmental scans. All data was sourced in March 2022, including interviews.
Demographic Data
Small cell lung cancer (SCLC) represents a minority of the lung cancer patient population, representing only about 10-15% of all lung cancer patients. However, as the indication is for metastatic SCLC, it was very difficult to source patients for this submission, as metastatic SCLC is rather aggressive, and the scope of patients is rather narrow. Thus, we were only able to interview a limited number of patients for this submission. All patients discussed have small cell lung cancer and have experience with lurbinectedin. Specific treatment experience can be found in the section Experience With Drug Under Review.
Table 2: Patient and Caregiver Information
Name | Patient/Caregiver | Type of lung cancer | Stage | Location | Source |
|---|---|---|---|---|---|
AL | Patient | SCLC | Localized | Canada (QC) | Phone interview |
MH | Patient | SCLC | Metastatic | Canada (MB) | Phone interview |
KW | Caregiver | SCLC (initially Exon-18 EGFR NSCLC that had mutated) | Metastatic | USA (CA) | Environmental scan |
GM | Patient | SCLC | Metastatic | USA (IL) | Environmental scan |
FX | Caregiver | SCLC | Metastatic | USA (PA) | Environmental scan |
Disease Experience
AL had been working as a computer programmer for nearly 30 years when in September 2020, he had some symptoms of COVID-19, and his voice was bothering him – he felt something was off and lost it frequently. After doctors brushed it off for months as emphysema and other viral lung infections, tests later confirmed that it was small cell lung cancer by February 2021. Although his disease was very localized and had no spread anywhere in his body, his doctor gave him a prognosis of about a year left. While he started the first chemotherapy treatment, it had weakened his body and its lingering side effects impaired his balance so much that he fell and broke a rib and ended up hospitalized for a few days. Throughout the last year, he underwent 3 different chemotherapy drug treatments, which unfortunately did not work for him. However, when he made the switch to lurbinectedin at the end of December 2021, he has felt a significant difference in his functionality and his doctors saw quite a bit of improvement in his scans. AL’s experience with lurbinectedin and other treatments are further outlined in this document.
KW’s 59-year-old father had been very busy working as an entrepreneur in his business and often travelled back and forth from China to visit family, leading an active lifestyle that did not seem to slow down. Throughout 2018 and into 2019, he had a persistent cough that was not going away, thinking they were allergies and did not think it was worth checking out with a physician. When the cough returned in April 2019 while worsening over the course of the month, he finally agreed to see a doctor. An x-ray showed a 7cm mass in his lung, possibly cancer, and the doctor recommended a CT scan. Living in California, he was frightened and immediately booked a flight to return home to China with KW to follow-up with the CT because he was worried of how expensive healthcare in the United States could be. In China, doctors determined it was determined to very likely be cancer, so further biopsies back in the US confirmed it was EGFR Exon 18 non-small cell lung cancer. After treatment with two targeted therapies starting in June and September of 2019, by February of 2020, KW’s cough had returned while on the second targeted therapy, which was strange as his primary tumour was stable and had not grown, and the other site had only changed slightly. His cough became so severe he had difficulty breathing, and thus another biopsy was ordered. Unfortunately, the results showed the cancer had mutated into small cell lung cancer, which is much more aggressive. These cases are rare but has been documented to occur in about 5% of EGFR cases, particularly after the use of EGFR-targeted therapies due to resistance (Oser et al., 2016). KW started the standard treatment for his SCLC throughout 2020, but after several months, he had progression of metastases to his liver and brain. In December 2020, KW started treatment for lurbinectedin as his third-line treatment for SCLC after chemotherapy and radiation and continued to be on the treatment for about 1 month.
Lung cancer is the most common cancer and by far the leading killer of all cancers in Canada. It accounts for 25% of all cancer deaths and the five-year survival rate is 22%, with even lower rates for cases in advanced stages (Canadian Cancer Statistics, 2021). Though small cell lung cancer (SCLC) accounts for only 10-15% of all lung cancer cases, it is much more aggressive with a high symptom burden and poorer outcomes, yet there are major gaps and few treatment options available for those with SCLC. Compared to non-small cell lung cancer, SCLC spreads much more rapidly, and thus, by the time patients experience symptoms like coughing up blood, persistent cough, weight loss, and shortness of breath, their cancer has likely already metastasized and therefore making recovery less likely. Median survival for those with extensive-stage SCLC is less than a year, 7-11 months at best with treatment, according to the Canadian Cancer Society. While non-small cell lung cancer has seen waves of new developments in research, treatment, and improved patient outcomes in recent years, this is unfortunately not the case for small cell lung cancer. There is a huge unmet need for a wider variety of treatment options for SCLC patients, as there have been no new treatment options approved in this paradigm in the last three decades until recently.
Lurbinectedin received Health Canada approval in September 2021 as a second-line treatment for patients with metastatic small cell lung cancer with disease progression after platinum-based chemotherapy, which is the current baseline treatment for extensive stage SCLC. It has demonstrated impressive outcomes in multiple trials, most notably the NCT02454972 basket study that included 105 SCLC patients (Trigo et al., 2020). From the trial, a median response duration of 5.3 months and progression-free survival of 3.5 months as observed in 86% of participants (Trigo et al., 2020). Median overall survival was 9.3 months, which is noteworthy in the second-line SCLC setting, especially for a population that included patients with resistant and aggressive disease, as is in most of the SCLC population (Trigo et al., 2020). Positive activity was also observed in patients with brain metastases. Improvements in quality of life is one of the most important aspects that a treatment option, such as lurbinectedin, can provide for patients with metastatic disease because of the aggressiveness and rapid progression that typically occurs in SCLC.
Lurbinectedin is a very promising treatment in second-line SCLC, representing the first progress in this setting in more than a decade, and the only new Health Canada-approved treatment for SCLC other than immunotherapy. Though chemotherapy currently represents the standard of care for these patients, it has been seen to be very limited in its potential as a long-term treatment. Receiving a lung cancer diagnosis is already quite devastating, but with recent developments in research and healthcare, new treatments like lurbinectedin are now available and can make all the difference for cancer patients. Patients can slow their disease progression, manage their symptoms, and have a good quality of life. Patients already have a huge burden coping with their lung cancer diagnosis; the battle to survive this disease should be made easier by ensuring the availability of treatments that work beyond what is already the standard in Canada. Being able to broaden the treatment landscape for small-cell lung cancer in Canada with the approval of lurbinectedin is a critical step forward towards the future of patient care.
References
Oser, M. G., Niederst, M. J., Sequist, L. V., & Engelman, J. A. (2015). Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. The Lancet Oncology, 16(4), e165–e172. DOI: 10.1016/S1470-2045(14)71180-5
Trigo, J., Subbiah, V., Besse, B., Moreno, V., López, R., Sala, M. A., ... & Paz-Ares, L. (2020). Lurbinectedin as second-line treatment for patients with small-cell lung cancer: a single-arm, open-label, phase 2 basket trial. The Lancet Oncology, 21(5), 645-654. DOI: 10.1016/S1470-2045(20)30068-1
Experiences With Currently Available Treatments
Systemic chemotherapy currently remains the cornerstone of first and second-line treatment for metastatic SCLC, with little progress and substantial changes in the past three decades until the recent approvals of durvalumab and atezolizumab in first-line treatment in the past 12 months. There are limited treatment options for those with second line disease and beyond aside from chemotherapy; thus, there is a large unmet need for more effective and less toxic treatment options for these patients.
Chemotherapy
For patients with metastatic disease, chemotherapy is typically presented as the first line of treatment and has been a long-standing and well-documented standard of care for cancer patients. It does see some benefits and has been found to work. However, it is limited in its use as a viable long-term treatment option due to its harsh side effects, impact on the individual’s functionality, and increases dependence on caregivers in their daily activities. These have been well documented in previous LCC submissions.
GM was diagnosed with Stage 4 SCLC in February 2019, which to him, was a death sentence. He immediately began chemotherapy and the newly approved immunotherapy drug Tecentriq. About 3 rounds of chemo and atezolizumab later, he was doing pretty well and stabilized his disease for the next two years. However, in January 2021, new scans showed that his disease had spread to his liver and brain, thus concluding his cancer became immune to immunotherapy. Thus, he started second-line treatment with lurbinectedin a few weeks later by February and continued to be on it for the next couple months until the end of June 2021.
MH started carboplatin chemotherapy in October a week after she had gamma knife radiosurgery, undergoing 6 treatments until her final treatment on January 1st. It was successful and seemed to be working to shrink her tumour; however, the side effects from the chemotherapy were hard and took a toll on her physical and mental health. She was tired and nauseated, had a lot of heartburn, lost her hair, and it even affected her eyesight. Overall, she was not feeling well at all, nor did she feel like herself, she lost interest in everything and just wanted to sleep all day. In that moment, MH felt like she wanted to give up, but knew she couldn’t for her family. She went through 2 more treatments of chemotherapy in August, but only completed 1-2 rounds of each before switching as scans revealed the tumour was not responding at all. At that point in November, she was put on palliative care before her physician suggested starting her on lurbinectedin the following January. By early February 2022, she started her first round of treatment with lurbinectedin and has been on it ever since.
Since AL had relatively localized disease with no spread to other areas of the body, his physicians were more optimistic but still started him on continuous treatment due to the aggressive nature of SCLC. Within one year after he was diagnosed, AL completed 3 different chemotherapy treatments, each with different drug combinations, but none of the treatments showed any improvement in his scans yet still made recovery from each one very hard. He did not have many side effects from chemotherapy aside from low red blood cell count, which dropped to 40 g/L at one point compared to the normal level of about 140 g/L. After he terminated his 3rd chemotherapy treatment, he switched over to lurbinectedin, which he has been on since late December 2021.
After it was discovered KW’s EGFR NSCLC had mutated into small cell, he began standard first-line treatment with chemotherapy (afatinib + cisplatin + etoposide) in February 2020. He underwent three rounds of this chemotherapy between February to June, which took a hard toll on his body due to the side effects. He was bedridden during the first week due to extreme nausea that worsened when standing or sitting, nor were they relieved with anti-nausea medications. This also made eating and drinking difficult, and after ending up on the ER due to dehydration, he was scheduled for salt water IV infusions following chemo rounds. During the second round, KW was also prescribed another anti-nausea medication which relieved nausea but also caused drowsiness and he was sleeping all day. The third round of chemotherapy had to be terminated after the second infusion due to harsh and severe side effects, in addition to lingering nausea and weight loss from radiation. KW had lost 28 lbs during his entire chemotherapy experience. However, 5 weeks after the final round of chemotherapy, he had regained some weight and was able to walk around the neighbourhood.
Unfortunately, after scans in June 2020 revealed progression to the liver, KW’s second-line of treatment replaced afatinib was terminated and replaced with doxcatel, which he continued for 6 months until December. He experienced far less side effects than the previous line, though still experienced nausea for the first 2 days after treatment. He was able to do 20-minute walks after 3 days and about 40–50-minute walks after 1 week on the treatment, which was nearly impossible before. Unfortunately, after 2 months there was progression to his brain, and by November, there was even more progression to his liver, growth to the primary tumour, vertebrae, and lymph node. He had to terminate the current treatment and was started on lurbinectedin a few weeks later in December 2020.
Radiation
After the second round of chemotherapy, KW also underwent radiation for the metastases on his vertebrae and brain. The radiation was effective, but the side effects seemed particularly long lasting, particularly nausea. His nausea persisted for the next 6 weeks, and was most uncomfortable when getting up from bed or encountering certain foods. He completed radiation over 5 days and then continued on with his chemotherapy regimens before starting lurbinectedin a few months later.
After being diagnosed on September 2nd, 2020, with stage 4 SCLC, MH’s cancer had already spread to her brain and pelvic bone, though she had no symptoms of cancer at all. MH quickly underwent a session of gamma knife radiosurgery by the end of September to target the lesions on her brain, then started chemotherapy. In March 2021 during chemotherapy, she had to go in for another round of gamma knife radiation, which was also effective in shrinking the lesion in her brain. A few months later at the end of July, she then underwent full brain radiation for 12 days, which was hard on her and her young son to see the aftereffects of. However, it was effective, and she started back on chemotherapy a week later.
Improved Outcomes
Advancements in SCLC research have been slow and limited in the past three decades, and outcomes for patients have remained poor in comparison to the rapid developments that have been made in recent years for NSCLC. Around 13% of all people diagnosed with lung cancer have small cell, and the lack of treatment advances for this population have been disappointing, until now. The first-line treatment options for SCLC patients have been limited to the standard of care, which is generally chemotherapy or radiation as needed, and for metastatic SCLC, there are even more limited options due to the rapid progression that occurs at this stage. The addition of lurbinectedin will bring significant change to the treatment paradigm for SCLC as patients value being able to have additional treatment options in the market when other treatments have failed, or there are comorbidities that prevent them from accessing certain treatments. For new therapies, patients most value:
Delaying disease progression to settle patients into long-term remission for improved survivorship
Improvements in managing their symptoms while having manageable side effects
Being able to maintain their independence and functionality to minimize the burden on their caregivers and loved ones
Being able to have a full and worthwhile quality of life
Experience With Drug Under Review
Lung Cancer Canada was only able to speak to 2 SCLC patients with experience on lurbinectedin, as many other patients were not well enough to speak, or unfortunately had already passed away. Sourcing patient and caregiver experience was difficult as the nature of metastatic SCLC is very aggressive and progresses rapidly, which continues to show the urgent need for effective treatments for this patient population.
Table 3: Patient and Caregiver Experience
Name | Diagnosis | Drug access method | Period on lurbinectedin | Duration on lurbinectedin | Currently on lurbinectedin? |
|---|---|---|---|---|---|
AL | February 2021 | Compassionate Access Program | Dec 2021 - present | 4 months | Yes |
MH | September 2020 | Compassionate Access Program | Feb 2022 - present | 1 month | Yes |
KW | April 2019 (EGFR NSCLC); February 2020 (mutated to SCLC) | Insurance | December 2020 – January 2021 | 2 months | No |
GM | February 2019 | Unknown | February - June 2021 | 4 months | Unknown |
FX | August 2020 | Unknown | Jan 2021 - Unknown | At least 1 month | Unknown |
Lurbinectedin is effective at shrinking tumours while reducing disease symptoms.
For AL, he had a scan prior to his last round of lurbinectedin 3 weeks ago at the end of February 2022, which showed quite significant improvement in his tumour, though his physicians did not specify details on how much it had shrunk. However, he does feel a difference in his symptoms in that they’re much more tolerable than what he used to experience with his previous chemo treatments. Recovery for his first-line chemo treatment specifically was very hard, and with lurbinectedin, he is able to carry on with his daily activities without much help, even driving himself to his lurbinectedin treatments. He notes the recovery and recouperation period after each treatment is only a few days, and the most important aspect is he’s still alive 1.5 years later after his initial diagnosis when his physician gave him a prognosis of only about a year. AL is still doing well to this day and continues to be very optimistic on lurbinectedin.
With her previous treatments, MH was able to “feel” the tumour in her lungs whenever she took a deep breath, where her breathing was vibrated and had a hard cough, but after starting her first treatment with lurbinectedin, this went away and she doesn’t feel this consistent push in her lungs whenever she’s deep breathing, which is likely a good sign of improvement. At the time of writing, she is scheduled for a scan in April 2022 after her 3rd treatment for details on how the treatment is working. However, as discussed below, MH is feeling much more like herself with this new treatment and many of her close friends noticed her “sparkle’ is coming back.
A month into lurbinectedin treatment after Christmas 2020, KW’s dad was doing very well. His symptoms were gone, and he could do light activity on most days. However, his most recent scans just before Christmas showed a new 3mm spot, but KW did not have any new or worsening symptoms. The only major symptoms he had were cough, nausea, and loss of appetite, which all subsided by the third week after each lurbinectedin treatment, similar to AL and MH’s experiences.
After only 2 lurbinectedin treatments, GM’s scans showed stability and even a little shrinkage in his lung and liver lesions, which was very pleasing to his oncologist. The only main symptoms he had on lurbinectedin is gastrointestinal and fatigue issues, which are both very manageable in his sense. He continues to occasionally play golf, which is one of his hobbies that has been a very good distraction and stress reliever throughout his entire journey.
Lurbinectedin allowed patients to return to a level of functionality that wasn’t previously possible.
During chemotherapy, the constant feeling of being unwell and nauseated made it very tough for MH to do much around the house without having to lie down every so often. It was virtually impossible for her to mow the lawn, walk the dogs, and shovel the snow in the driveway because of the harsh toll that chemotherapy had taken on her body. She had to rely on her husband and son for most things around the house, which was odd because typically MH was the caregiver in the family. However, when she started lurbinectedin, she was slowly able to start doing more and more small tasks around the house to help her husband out with chores, even if it takes her longer than before. Being able to return to a level of normalcy and do basic tasks again made MH feel much more confident that things are going well for her. Though vacuuming and cleaning the house may still be tough for her, she’s able to return to cooking family meals, shower herself, help out with raking and mowing the lawn, go grocery shopping, and even go for slow walks around the neighborhood with her two dogs. Although her pace has slowed down quite a bit compared to pre-diagnosis, she has made adjustments and helps out whenever she can and relies much less on her husband for independence. When patients like MH are faced with such a debilitating disease and diagnosis, the thought of returning to some level of normalcy is what drives her to continue to fight.
For FX, a few days after their first infusion treatment with lurbinectedin, they were able to bounce back rather quickly, feeling terrific and even tried to help his mom shovel the driveway. Subsequent treatments got easier as he knew what to expect and how to adjust his life around each cycle and worked his way around each “rough first week” to continue to enjoy his time.
AL has also similarly seen improvements in his functionality where he used to rely on his wife for help with daily activities and constantly had to push back dates when he needed to go in for work. However, after 3 rounds of lurbinectedin, he has felt a significant difference and when he hit the high points of each cycle by the 2nd and 3rd weeks, he’s able to return to a sense of normalcy in his livelihood and daily activities. He’s able to clean the house, go shopping, care for his cat, shovel the snow, and go for long drives out of town. Lurbinectedin has allowed patients like AL, FX, and MH the freedom to continue to have a good quality of life without constant side effects that impede their abilities to enjoy their lives while they can.
Lurbinectedin had less severe side effects than other chemotherapy treatments.
In the 2 weeks in between KW’s previous chemotherapy and before starting lurbinectedin in early December 2020, his symptoms had increased rather quickly. His cough had come back, and he got new aches in his back. Towards the end of the waiting period, talking became difficult, as saying more than two sentences started triggering some coughs, which was very frustrating for KW to communicate. However, when KW started his first treatment with lurbinectedin, his cough was gone the day after, and side effects seemed to be less than those experienced with the previous platinum-based chemotherapy. The day after, he was able to eat small items like noodles, walk around a little, and vomited just once compared to multiple times per day with other treatments. His aches seem to be less and is able to manage pain with over-the-counter Tylenol.
Both AL and MH seemed to have very similar side effect experiences with lurbinectedin. Both noted the first couple of days to a week was the worst for side effects, where MH had headaches, nausea, joint aches, and some minor shortness of breath. AL has low energy and fatigue, changes in his voice becoming raspier, and occasional shortness of breath the first few days after each treatment. However, the side effects subside and eases up after the first week after each round, and they feel better and better throughout the next 2 weeks since both their cycles are every 21 days and are at their highest peaks by the time they go back for their next round of treatment. Both AL and MH agreed the side effects with lurbinectedin have been significantly easier to manage than their previous treatments with chemotherapy because they resolve after a couple days, while chemotherapy’s effects are long-lasting and consistent, making it tough to do much.
FX’s dad was diagnosed August 2020 with SCLC with extensive metastases to the liver and brain. He did very well through his 4 cycles of chemo (carboplatin/etoposide/atezolizumab) and then after the second cycle of solo-atezolizumab, his scans unfortunately showed growth of the primary tumor and some spread to bones. He started lurbinectedin on January 28th, 2021, and the first few days, nausea wasn't too bad, but the fatigue and muscle pain were terrible. Since he has bone mets, he's also very sore and slept about 18-20 hours about 4 days after the treatment. FX had to have a blood transfusion during his initial chemotherapy treatment because of anemia; however, subsequent treatments went much more smoothly, and he had very minimal side effects other than nausea and backaches. Lurbinectedin’s side effects have been noticeably much more manageable for patients compared to other treatments for their SCLC, and this allows them to return to a level of functionality that improves their quality of life, which is what patients with metastatic disease need.
For some patients, the success of lurbinectedin has allowed them to continue working throughout their cancer journeys.
Throughout his 1.5-year cancer journey, AL has continued to work and has never really taken much time off. As a computer programmer who mainly works from home, even when he was diagnosed in September 2020, he never took much time off work. He’s still able to answer client calls and help them out remotely on the computer, so it did not impact his career or financials too much. Although he did have to initially postpone some days where he’d have to travel and go into work physically, AL has continued to work nearly full-time hours during the weeks he’s not as impacted by side effects directly after lurbinectedin treatment, particularly the first week. He loves his job and isn’t planning on retiring anytime soon.
Prior to diagnosis, MH had been working at the public school in her neighborhood as a secretary for several years as well as a grocery store clerk. She was very fortunate to have very supportive co-workers and employers, and when she was diagnosed, she turned to one of her co-workers at the school who also had breast cancer and helped support each other through their cancer experiences. She had to quit her job at the grocery store when she started her initial first-line treatments as it was impossible for her to be on her feet for several hours at a time. However, she has continued working at the school throughout treatments as it was not as demanding and mostly included paperwork. Nowadays, MH says it’s been a very nice distraction from her cancer to be able to connect with others and the kids and helps take her mind off her disease. She currently continues to work at the school and foresees herself doing so as long as she can.
Patients such as MH and AL deserve flexibility in that when they’re faced with an aggressive disease as metastatic SCLC, they have treatment options that are effective with minimal side effects and allow them to continue with their careers and livelihoods and not require them to drop everything in their life because of the cancer. As of March 2022, AL and MH are both still doing very well on lurbinectedin, and both continue to work most days of the week.
Lurbinectedin has allowed patients to foresee a future and make longer-term goals.
With extra time gained thanks to lurbinectedin, MH was able to foresee a future with her grandkids and make longer-term goals to see her kids get married, see her grandkids go to school, and maybe even see her grandson graduate. AL hasn’t travelled much since he was diagnosed with cancer, but he’s staying optimistic and if lurbinectedin continues to go well for him, he wanted to make vacation plans with his wife to take her on a cruise. Unfortunately, KW passed away in January 2021 after also contracting COVID-19, after only 2 treatments with lurbinectedin. However, his son shared he is immensely grateful for the extra time he was able to have with his father, travelling to China to visit family and friends even during his cancer journey. When faced with such an aggressive disease with slim prognosis, patients deserve the ability and freedom to enjoy the time they have. Lurbinectedin has allowed these patients like AL and MH to make longer term goals and wishes for the future.
Companion Diagnostic Test
Lurbinectedin for SCLC does not require any biomarker testing.
Anything Else?
Small Cell Lung Cancer (SCLC) patients have a huge unmet need, particularly as this condition comes with a high symptom burden, spreads and progresses rapidly, and has very few viable treatment options. With the approval of lurbinectedin, this would represent needed progress in the SCLC treatment setting. Patients already have a huge burden coping with their lung cancer diagnosis; the battle to survive this disease should be made easier by ensuring the availability of treatments that work beyond what is already the standard in Canada.
As highlighted throughout this submission, lurbinectedin has seen promising results for patients and has given them an additional treatment option when for most, it seemed like the end of the road. Lurbinectedin was the second line (or beyond) treatment option for the patients interviewed. They had prior treatments with primarily chemotherapy that were not effective, or overtime, their cancer became resistant to these treatments. Some patients were even considering palliative care when these options failed before they had a chance with lurbinectedin. Extensive-stage small cell lung cancer has a median survival of 7-11 months with treatment, according to the Canadian Cancer Society, and many of the aforementioned patients had come very close or already reached this point right before they started lurbinectedin. Because there have been very limited advancements in small cell lung cancer due to the aggressive and rapid progressive nature of the disease, they had virtually no options left. The approval of lurbinectedin allowed these patients to enjoy a second chance at treating their cancer. Lurbinectedin also has much more manageable side effects compared to standard chemotherapy and is less harsh while still being effective at delaying the progression of disease and treating the tumour.
In the NCT02454972 clinical trial, lurbinectedin was effective at improving overall survival and progression-free survival and also treating brain metastases. These extra few months are extremely valuable to such patients who are faced with such an aggressive cancer as metastatic SCLC. Time is the most valuable asset to patients with advanced disease, and it is critical to have additional options in the current treatment paradigm as it can change the lives of patients across the country. Being able to broaden the treatment landscape for small-cell lung cancer in Canada with the approval of lurbinectedin is a critical step forward towards the future of patient care and would also give clinicians the flexibility to determine which treatment options are best for their patients. This group of patients cannot afford to wait and deserve to have access to treatments that can help prolong and maintain their lives now. LCC hopes CADTH provides a positive recommendation for this submission.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 4: Financial Disclosures for Lung Cancer Canada
Company | Check Appropriate Dollar Range | ||||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Jazz Pharmaceuticals | — | — | — | X | |
Clinician Input
Ontario Health (CCO) Lung Cancer Drug Advisory Committee
About Ontario Health (CCO) Lung Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
This input was jointly discussed via Drug Advisory Committee meeting and email.
Current Treatments
Small cell lung cancer represents approximately 15% of lung cancers in Canada. Two thirds of patients present with metastatic (extensive stage) disease with a median overall survival of no more than a year. The majority of patients with stages I-III (limited stage) disease relapse with median overall survival of approximately two years and five years survival of 20-25%. Therefore there is a high need for better treatments for this disease.
Standard systemic therapy for both limited and extensive stage disease is platinum (either cisplatin or carboplatin) in combination with etoposide. Patients with limited stage disease would routinely be offered thoracic radiation and prophylactic cranial irradiation. There are data supporting the addition of atezolizumab or durvalumab to platinum and etoposide in extensive stage disease. Both are Health Canada approved. However, neither agent is currently publicly funded. Atezolizumab was turned down by CADTH, while durvalumab was recommended for funding. This is currently at PCPA awaiting a recommendation on pricing. There is some access through compassionate access programs to durvalumab.
Despite high response rates to initial therapy, relapse is common and often soon after the completion of first-line therapy. Current standards of care for relapsed SCLC include:
Retreatment with platinum and etoposide if there is a treatment free interval greater than 3 months
Chemotherapy with cyclophosphamide, doxorubicin and vincristine on day 1 every 21 days
Chemotherapy with topotecan on days 1-5 every 21 days
Palliative radiation
Supportive care alone
Response rates to second line chemotherapy are generally no better than 20-25% with short progression free survival (4-6 months) and median overall survival around 8-9 months. Based on data comparing oral topotecan to BSC, second line therapy improves survival by a couple of months. Lurbinectidin represents another treatment option to consider in this group of patients with relapsed SCLC and poor overall outlook.
Treatment Goals
The most important goals for patients and clinicians are tumor shrinkage (response rates), progression free survival, overall survival and improvement in patient symptoms and quality of life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Existing treatments for relapsed SCLC have relatively low response rates, short PFS and poor overall survival. The AE profile for CAV is not great. There are high rates of nausea/vomiting, neutropenia and infection, mucositis and fatigue. Topotecan is a 5 day regimen and therefore does not represent a good option for patients with a limited life expectancy. More active treatments associated with improved profile of adverse events, and short treatment administrations are needed.
Which patients have the greatest unmet need for an intervention such as the drug under review?
All patients with relapsed SCLC are incurable and need improved treatment options. However, patients with treatment free intervals of less than three months are considered platinum resistant or refractory. These patients have the worst outcomes and are the least likely to respond to current treatment options for relapsed SCLC with expected response rates around 10%. Lurbinectidin has reasonable response rates for both platinum sensitive (45%) and platinum resistant patients (22%). Therefore it may offer greater benefit in those patients who are platinum resistant.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Lurbinectidin is used as a single agent. The combination of lurbinectidin and doxorubicin was found to be no better than topotecan. However, the dose of lurbinectidin used in combination, is lower than the single agent dose and may have compromised its efficacy. It is a novel cytotoxic which might offer advantages over other agents, but this is a biological argument. Current data comes from a single arm phase II clinical trial. The reported response rate was 35.2% which is higher than the reported response rates of other cytotoxic agents in randomized trials in relapsed SCLC. This agent would be used in SCLC patients who have failed platinum and etoposide chemotherapy. There is no data currently for use in the first line setting. It would likely represent an additional treatment rather than a replacement therapy, although the proportion of patients who would get third line therapy is small.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
There is no randomized data to answer this question. Based on response rates from the single arm trial, and the reported profile of adverse events it is a reasonable consideration for second line therapy. Other options have either unfavourable AE profiles, or scheduling.
How would this drug affect the sequencing of therapies for the target condition?
This agent has an indication in patients who have failed platinum and etoposide. It may be used as the first treatment after failure of platinum, or as an additional option following current second line therapies.
Which patients would be best suited for treatment with the drug under review?
This agent would be suitable for any patients with SCLC who have relapsed after platinum and etoposide
How would patients best suited for treatment with the drug under review be identified?
Patients would be easily identified as they would have an established diagnosis of SCLC and have failed platinum and etoposide already
Which patients would be least suitable for treatment with the drug under review?
As stated above, this agent would be suitable for any patient with relapsed SCLC
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
These patients with relapsed SCLC would have disease that can be assessed on conventional imaging studies. It would be appropriate to do imaging after every two to three cycles of therapy to determine if the patient is benefiting. Most commonly that would be CT imaging
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
The most easily assessed outcome would be the absence of disease progression on radiological imaging, together with an acceptable profile of AEs
What would be considered a clinically meaningful response to treatment?
Tumor shrinkage and improvement in disease related symptoms
How often should treatment response be assessed?
Following every two to three cycles of therapy.
What factors should be considered when deciding to discontinue treatment?
Tumor progression or unacceptable side effects, or patient unwilling to continue
What settings are appropriate for treatment with the drug under review?
This treatment would be administered in an oncology clinic under the supervision of a medical oncologist or pulmonologist with expertise in systemic therapy of thoracic malignancies
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
NA
Additional Information
No.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Stacey Hubay
Position: OH-CCO Lung DAC Member
Date: 16-02-2022
Table 5: Conflict of Interest Declaration for Ontario Health (CCO) Lung Cancer Drug Advisory Committee Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Stephanie Brule
Position: OH-CCO Lung DAC Member
Date: 16-02-2022
Table 6: Conflict of Interest Declaration for Ontario Health (CCO) Lung Cancer Drug Advisory Committee Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Natasha Leighl
Position: OH-CCO Lung DAC Member
Date: 16-02-2022
Table 7: Conflict of Interest Declaration for Ontario Health (CCO) Lung Cancer Drug Advisory Committee Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Mohammad Rassouli
Position: OH-CCO Lung DAC Member
Date: 16-02-2022
Table 8: Conflict of Interest Declaration for Ontario Health (CCO) Lung Cancer Drug Advisory Committee Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr. Sara Kuruvilla
Position: OH-CCO Lung DAC Member
Date: 16-02-2022
Table 9: Conflict of Interest Declaration for Ontario Health (CCO) Lung Cancer Drug Advisory Committee Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Lung Cancer Canada
About Lung Cancer Canada
Lung Cancer Canada (LCC) is a national charity with the purpose of increasing awareness about lung cancer, providing support and education to lung cancer patients and their families, to support research and to advocate for access to the best care for all lung cancer patients in all provinces and territories.
Through the LCC Medical Advisory Committee (MAC), we have been providing clinician input for submissions of new lung cancer drugs to the HTA process for many years. The LCC MAC is made up of clinicians and key opinion leaders in the field of lung cancer across the country.
Information Gathering
The feedback on the questions listed in this Clinical Group Input is based on current clinical data and practice guidelines, which will be referenced in the corresponding sections.
Current Treatments
Small cell lung cancer (SCLC) represents 15-20% of all lung cancer diagnosed per annum in Canada. About 1/3 of the patients are diagnosed with limited-staged disease (LD-SCLC) by which there is no metastatic disease, reasonable lung function as defined by pulmonary function test and the thoracic disease can be encompassed within the radiation field without compromise to the lung and heart functions. These patients will be treated with concurrent platinum/etoposide chemotherapy with thoracic radiation, followed by prophylactic cranial radiation (PCI) for those who respond to chemotherapy. The remaining 2/3 of the patients are considered to have extensive-staged disease (ED-SCLC). Until recently, these patients would receive platinum/etoposide for 4-6 cycles, with optional PCI and thoracic radiation for those who have at least stable disease after chemotherapy. Since Q2 2021, ED-SCLC patients who do not have contraindication to PD(L)1 therapy will receive platinum/etoposide and durvalumab, PDL-1 antibody, for 4 cycles followed by durvalumab maintenance until disease progression, intolerance or patient wishes. No PCI or thoracic radiation will be given [Pas-Ares et al. Lancet Oncology 2019; 394(10212):1929-1939]. The addition of durvalumab to chemotherapy as first line treatment of ED-SCLC received a ‘Reimburse with conditions’ recommendation by CADTH in July 2021 (https://www.cadth.ca/durvalumab).
Unfortunately, first-line systemic therapy will categorically fail in almost all ED-SCLC patients with a median survival of 9-12 months. Even with the addition of durvalumab, only 11.5% will have no progression of disease and 23% of patients will survive for 2 years. In LD-SCLC, the median survival will be 18-24 months with >50% patients recur in the first 2 years.
The treatment options for small-cell lung cancer patients who have disease recurrence after initial combined chemotherapy and radiation for their curative limited-staged disease or chemotherapy +/- durvalumab for their extensive-staged disease are limited and are dependent on (1) time from the last cycle of platinum-based chemotherapy to disease progression or recurrence (2) ECOG performance status, and (3) patient wishes based on toxicity and efficacy.
For patients who have ECOG 0-2, and progression or recurrence of disease at least 3 months from the last dose of platinum-based chemotherapy, they will be considered as platinum-sensitive disease. If patients wish for further treatment, options will include:
Retreatment with platinum/etoposide: The phase III study that compared retreatment with carboplatin/etoposide and topotecan demonstrated an improvement in median progression-free survival (mPFS, 4.7 m versus 2.7 m, p=0.0041, HR=0.57), higher response rate (ORR, 49% versus 25%, p=0.0024) and lower incidences of grade 3 or higher toxicity (14% versus 22%) in the carboplatin/etoposide-treated patients while similar incidence of hematological toxicity, except higher incidences of grade 3-5 neutropenia and febrile neutropenia in the topotecan-arm (14% versus 25% and 6% versus 11%, respectively) and median overall survival (mOS) of 7.5 months. The use of G-CSF use was not reported [Baize et al. Lancet Oncol 2020;21(9):1224-1233].
Treatment with platinum/etoposide and durvalumab: LD-SCLC patients who have received prior curative cisplatin/etoposide and concurrent radiation and relapsed at least 2-3 months post therapy are eligible for platinum/etoposide and durvalumab based on the CAPSIAN trial. ED-SCLC patients treated with platinum/etoposide and durvalumab not only have an improvement in mOS (12.9 months versus 10.5 months, p=0,0003, HR=0.71) but also in the 3-year OS rate (17.6% versus 5.8%) [Paz-Ares et al. ESMO 2021 and Paz-Ares et al. Lancet Oncol 2019;394(10212):1929-1939]. Although the combination has received regulatory and reimbursement approvals with pending PCPA negotiation and provincial reimbursement, durvalumab is currently available through compassionate access program only. All in all, this is a treatment option for a minority of the platinum-sensitive SCLC patients. For the remainder of the Clinician Input, this option will not be discussed.
IV topotecan: Topotecan was found to have comparable ORR (24.3% versus 18.3%, p=0.285), mPFS (13.3 weeks versus 12.3 weeks, p=0.552) and mOS (25 weeks versus 24.7 weeks (p=0.795) but improvement in patient reported symptoms to cyclophosphamide/adriamycin/vincristine (CAV) in SCLC patients with disease progression at least 60 days from last dose of chemotherapy [von Pawel et al. JCO 1999; 17(2):658-667]. The randomized phase II study comparing IV and oral topotecan in SCLC who had disease progression of at least 6 months from the last dose of chemotherapy in the first-line setting reported an ORR of 15%, mPFS of 13.1 weeks and mOS of 25.1 week, with 16.4% with dose reduction. The most common grade 3 or 4 toxicity were neutropenia, thrombocytopenia and anemia, in decreasing order, with 12.1% patients required G-CSF {von Pawel et al. JCO 2001;19(16):1743-1749]. Comparable ORR (21.9%, mPFS (14.6 weeks) and mOS (35 weeks) was reported in the IV topotecan-arm of the phase III study comparing IV to oral topotecan in SCLC patients with recurrence or progression after first-line chemotherapy regardless of time to failure. Again, the most common grade 3 and 4 toxicity were hematological with 16% patients required G-CSF [Eckhardt et al. JCO 2009;25(25):2086-2092]. In the meta-analysis of both IV or oral topotecan in the platin-sensitive setting, the ORR was 17% with an 1-year OS rate at 27% [Horita et al. Sci Rep 2015;5:15437].
CAV: see above.
Clinical trial.
For patients who have ECOG 0-2 and progression or recurrence of disease while on treatment to less than 3 months from last dose of platinum-based chemotherapy, they will be considered as platinum-resistant or refractory. The treatment option is very limited even for those who wish for further treatment:
IV topotecan: Horita et al reported an ORR of 5% and 1 year OS rate of 9% in the meta-analysis of IV and oral topotecan in the platin-refractory SCLC patients [Horita et al. Sci Rep. 2015;5:15437].
CAV with an ORR of 5% [Tiseo et al. JTO 2007;2(8):764-772}.
Clinical trial.
Any of the standard options in the recurrent or refractory setting provide a short duration of symptom or quality of life improvement and modest gain in both mPFS and mOS at the expanse of toxicity. There is urgent need to have better understanding of the biology of SCLC and to develop more effective therapy with tolerable toxicity profile.
Treatment Goals
The goals of treatment in the recurrent or relapse setting for SCLC are:
Improvement in mOS
Prolonged mPFS and prolonged duration of response
High response rate and high and rapid reduction of tumour burden
Reasonable toxicity with low incidence of dose modification and immature termination of treatment due to toxicity
Improvement of health-related quality-of-life and disease-related symptoms.
Chemotherapy unit utilization.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
In platinum-sensitive disease, although retreatment with platinum/etoposide and IV topotecan are considered as standard of care, the ORR, mPFS and mOS were still modest with a high incidence of grade 3 or higher toxicity, particularly neutropenia, febrile neutropenia and thrombocytopenia. The combination of platinum/etoposide and durvalumab will only be applicable to a small number of platinum-sensitive relapse SCLC with initial presentation as LD-SCLC.
In the platinum-refractory or resistant setting, the currently available therapy of topotecan or CAV offers minimal benefit. The incidences of grade 3 and 4 neutropenia, thrombocytopenia and anemia were 69%, 41% and 24%, respectively after treatment with topotecan, while grade 3-4 non-hematological toxicity was less common.
In addition, IV platinum/etoposide requires 3 treatment days, which can be reduced to 1 day with the use of IV carboplatin/etoposide on day 1 and oral etoposide on days 2-3, and IV topotecan requires 5 consecutive treatment days.
In either setting, there is still significant room to improve the mOS, mPFS, ORR, toxicity, health-related quality of life and administration schedule.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Table 10: Therapy for Platinum-Sensitive and Platinum-Refractory Disease
Relapse | Carboplatin/Etoposide | Topotecan | Lurbinectedin | Lurbinectedin >180 days from last cycle of platinum-combination |
|---|---|---|---|---|
Platinum-Sensitive Relapse | ORR: 49% mDOR: 5.4 months mPFS: 4.7 months mOS: 7.5 months 1-year OS: 20% G-CSF use: Not reported | ORR: 17% mDOR: 4.1 months mPFS: 2.7-3.7 months mOS: 6.25-9.75 months 1-year OS: 27% G-CSF use: 12-16% | ORR: 45% mDOR: 8.2 months mPFS: 4.6 months mOS: 11.9 months 1-year OS: 48.3% G-CSF use: 22% | ORR:60% mDOR: 5.5 months mPFS: 4.6 months mOS: 16.2 months 1-year OS: 60.9% G-CSF use: not reported |
Platinum-refractory Relapse | N/A | ORR: 5% mDOR: NA mPFS: NA mOS: NA i-year OS: 9% G-CSF use: NA | ORR 22% mDOR: 4.7 months mPFS: 2.6 months mOS: 5 months 1-year OS: 15.9% G-CSF use: 22% | N/A |
The incidences of grade 3-4 neutropenia, febrile neutropenia, thrombocytopenia and anemia with lurbinectedin were 46%, 5%, 7% and 9%, respectively.
Based on the current available data, the platinum-refractory SCLC patients have an urgent need to have more tolerable and effective therapy as available therapy is generally considered as ineffective. But availability of novel and effective therapy in the platinum-sensitive SCLC patients is still important.
Trigo et al. Lancet Oncol 2020;21:645-654.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
In the platinum-sensitive setting,
the benefit of lurbinectedin was numerically comparable to retreatment with platinum/etoposide for ORR and mPFS while numerically better mDOR, 1-year OS and mOS. It is still not clear if retreatment with platinum/etoposide will be the preferred option in platinum-sensitive relapsed patients with a treatment-free period greater than 6 months.
the benefit of lurbinectedin was numerically improved in ORR, mDOR, mOS and 1-year OS when compared to topotecan.
Thus, lurbinectedin can be considered as an option for patients with platinum-sensitive relapsed SCLC in addition to retreatment with carboplatin/etoposide and IV topotecan. There are insufficient data to determine the role of lurbinectedin in patients who have rechallenge platinum/etoposide.
In the platinum-refractory setting,
lurbinectedin yielded a favourable ORR, and 1-year OS rate when compared to IV topotecan.
Thus, lurbinectedin may be considered as more efficacious platinum-refractory relapsed SCLC patients.
In addition, the incidence of hematological toxicity seemed to be lower than both IV topotecan and retreatment with carboplatin/etoposide.
All of the above is limited by the fact that the study reported by Trigo et al was a single-arm phase 2 study with a small sample size for both platinum-sensitive and platinum-relapse SCLC patients, it is still uncertain the best setting that lurbinectedin should be used in clinical practice.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
In the platinum-sensitive setting,
the benefit of lurbinectedin was numerically comparable in ORR and mPFS while numerically better in mDOR, 1-year OS and mOS than retreatment with carboplatin/etoposide.
the benefit of lurbinectedin was numerically improved in ORR, mDOR, mOS and 1-year OS when compared to topotecan.
Thus, lurbinectedin can be considered as an option for patients with platinum-sensitive relapse SCLC in addition to retreatment with carboplatin/etoposide and IV topotecan. If lurbinectedin is limited to SCLC patients who have received only 1 prior line of platinum/etoposide, oncologists may choose to use lurbinectedin as second-line therapy instead of standard chemotherapy like retreatment with carboplatin/etoposide or topotecan.
In the platinum-refractory setting,
lurbinectedin yielded a higher ORR, and 1-year OS rate when compared to IV topotecan.
Thus, lurbinectedin may have better efficacy and the possibility of better improvement in disease related symptoms than IV topotecan. In addition, the incidence of hematological toxicity seemed to be lower than both IV topotecan and retreatment with carboplatin/etoposide.
Limited by the fact that the study reported by Trigo et al. was a single-arm phase 2 study with a small sample size for both platinum-sensitive and platinum-relapse SCLC patients, it is hard to be certain the best setting that lurbinectedin is truly better than that of current standard chemotherapy regimens in the relapse setting.
How would this drug affect the sequencing of therapies for the target condition?
For the platinum-sensitive relapsed SCLC patients, lurbinectedin will be considered as a treatment option. The impact treatment algorithm will depend on the line of therapy lurbinectedin will be reimbursed.
If lurbinectedin will be reimbursed only after first relapse of at least 3 months from initial platinum/etoposide in either the LD-SCLC or ED-SCLC setting, lurbinectedin may be used preferentially over retreatment of platinum/etoposide or topotecan. It is still unclear as to whether oncologists will choose lurbinectedin or retreatment platinum/etoposide in those who have progression greater than 6 months.
If lurbinectedin will be reimbursed in platinum-sensitive relapse after initial platinum/etoposide in either the LD-SCLC and ED-SCLC settings with or without intervening therapy, lurbinectedin will be used either as second- or third-line therapy.
For the platinum-refractory relapsed SCLC patients, lurbinectedin is a treatment option given the argument in Section 5.2.
Which patients would be best suited for treatment with the drug under review?
The candidates for lurbinectedin will include
SCLC patients initially presented with LD-SCLC and failed concurrent platinum/etoposide and radiation within or after 3 months from the last dose of platinum/etoposide.
SCLC patients initially presented with ED-SCLC and failed first-line platinum/etoposide +/- durvalumab within or after 3 months from the last dose of platinum/etoposide,
Who have an ECOG 0-2, adequate hematological, renal and hepatic function.
How would patients best suited for treatment with the drug under review be identified?
At this time, aside from the clinical criteria listed in Section 6.4, there is no other biomarker that will help to identify the subpopulation of relapsed SCLC patients who will benefit the most from lurbinectedin.
Which patients would be least suitable for treatment with the drug under review?
Patients with ECOG PS of 3 or worse and those with significant liver and renal dysfunction should not be offered lurbinectedin.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
At this point, there is no predictive biomarker for efficacy with lurbinectedin identified in previously platinum-treated SCLC patients.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
CT scan was done every 6 weeks until cycle 6 and then every 9 weeks thereafter for evaluation of response as reported in the study by Trigo et al., which is in line with current clinical practice.
What would be considered a clinically meaningful response to treatment?
Relapsed SCLC, regardless of being platinum-sensitive or platinum-refractory, presents with disease related symptoms. Improvement in symptoms predates radiological stabilization or improvement, which can be considered as a clinical meaningful response to treatment. But radiological response/stabilization and the duration of response/stabilization will be considered as a more definitive demonstration of clinical benefit to treatment.
How often should treatment response be assessed?
CT scan was done every 6 weeks until cycle 6 and then every 9 weeks thereafter for evaluation of response as reported in the study by Trigo et al., which is in line with current clinical practice.
What factors should be considered when deciding to discontinue treatment?
Patients will terminate treatment with lurbinectedin if one or more of the following occurs:
Disease progression, except those with CNS progression without extracranial disease progression as lurbinectedin is not known to cross the blood-brain barrier. These patients should have radiation to the brain and continue with lurbinectedin.
Clinically important toxicity that jeopardizes patient safety with or without prior dose reduction of lurbinectedin and/or
Patient wishes to stop treatment.
What settings are appropriate for treatment with the drug under review?
Lurbinectedin can be administered in both the academic and community oncology outpatient chemotherapy settings. `
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
N/A
Additional Information
N/A
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Quincy Chu
Position: Medical Oncologist, Cross Cancer Institute
Date: March 18, 2022
Table 11: Conflict of Interest Declaration for Lung Cancer Canada Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Abbvie | X | — | — | — |
Amgen | X | — | — | — |
AnHeart | X | — | — | — |
Astellas | X | — | — | — |
Astra Zeneca | — | X | — | — |
BI | X | — | — | — |
BMS | X | — | — | — |
Eli Lilly | — | X | — | — |
Eisai | X | — | — | — |
J and J | — | X | — | — |
Jazz | X | — | — | — |
Merck | X | — | — | — |
Novartis | X | — | — | — |
Pfizer | X | — | — | — |
Roche | X | — | — | — |
Sanofi | — | X | — | — |
Takeda | X | — | — | — |
Merck KgaA- DSMB | — | — | — | — |
Astra Zeneca research funding | — | — | X | — |
Declaration for Clinician 2
Name: Dr. David Dawe
Position: Medical Oncologist, CancerCare Manitoba
Date: Mar 18, 2022
Table 12: Conflict of Interest Declaration for Lung Cancer Canada Clinician 2
Name of Organization | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
AstraZeneca | Advisory boards | X | — | — | — |
Merck | Advisory boards | X | — | — | — |
AstraZeneca | Research Grant | — | — | X | — |
Boehringer-Ingelheim | Honoraria | X | — | — | — |
Declaration for Clinician 3
Name: Dr. Donna Maziak
Position: Thoracic Surgeon, The Ottawa Hospital
Date: Mar 18, 2022
Table 13: Conflict of Interest Declaration for Lung Cancer Canada Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Paul Wheatley-Price
Position: Medical Oncologist, The Ottawa Hospital. Associate Professor, Department of Medicine, University of Ottawa
Date: Mar 18, 2022
Table 14: Conflict of Interest Declaration for Lung Cancer Canada Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi | X | — | — | — |
Astra Zeneca | X | — | — | — |
Jazz Pharmaceuticals | X | — | — | — |
Amgen | X | — | — | — |
Janssen | X | — | — | — |
Novartis | X | — | — | — |
Merck | X | — | — | — |
BMS | X | — | — | — |
Roche | X | — | — | — |
EMD Serono | X | — | — | — |
Pfizer | X | — | — | — |
Bayer | X | — | — | — |
Novartis | X | — | — | — |
Declaration for Clinician 5
Name: Dr. Silvana Spadafora
Position: Medical Oncologist, Sault Area Hospital
Date: Mar 18, 2022
Table 15: Conflict of Interest Declaration for Lung Cancer Canada Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Dr. Ronald Burkes
Position: Medical oncologist, Mount Sinai Health
Date: Mar 18, 2022
Table 16: Conflict of Interest Declaration for Lung Cancer Canada Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 7
Name: Dr. Geoffrey Liu
Position: Medical Oncologist, Princess Margaret Cancer Centre
Date: Mar 18, 2022
Table 17: Conflict of Interest Declaration for Lung Cancer Canada Clinician 7
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Takeda Canada | Advisory Board, Health Technology Assessment Submission Advice, Speaker’s Bureau, past 10 years | — | — | X | — |
Takeda Canada | (To institution, not individual) Observational Study funding, past 10 years | — | — | — | X |
Hoffman La Roche | Advisory Board, Health Technology Assessment Submission Advice, past 10 years | — | — | X | — |
Pfizer | Advisory Board, Health Technology Assessment Submission Advice, part 10 years | — | — | X | — |
AstraZeneca | Advisory Board, Health Technology Assessment Submission Advice, Speaker’s Bureau, past 10 years, | — | — | X | — |
AstraZeneca | (To institution, not individual) Observational Study funding, past 10 years | — | — | — | X |
Bristol Myers Squibb | Advisory Board | X | — | — | — |
Boehringer Ingerheim | (To institution, not individual) Observational Study funding, past 10 years | — | — | X | — |
Abbvie | Advisory Board, past 10 years | — | X | — | — |
Merck | Advisory Board, Health Technology Assessment Submission Advice, past 10 years | — | X | — | — |
EMD Serono | Speaker’s Bureau, past 10 years | X | — | — | — |
Novartis | Advisory Board,past 10 years | — | — | X | — |
Glaxo Smith Kline | Advisory Board, past 10 years | — | X | — | — |
Declaration for Clinician 8
Name: Dr. Kevin Jao
Position: Medical Oncologist, Hôpital Sacré-Cœur, Montreal
Date: Mar 18, 2022
Table 18: Conflict of Interest Declaration for Lung Cancer Canada Clinician 8
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Bristol-Myers Squibb | Advisory Role | X | — | — | — |
Declaration for Clinician 9
Name: Dr. Rosalyn Juergens
Position: Chair, LCC Medical Advisory Committee; Medical Oncologist, Juravinski Cancer Center
Date: Mar 18, 2022
Table 19: Conflict of Interest Declaration for Lung Cancer Canada Clinician 9
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Bristol Myers Squibb | X | — | — | — |
Astra Zeneca | — | X | — | — |
Merck Sharp and Dohme | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 10
Name: Dr. Shaqil Kassam
Position: Medical Oncologist, Southlake Regional Hospital
Date: Mar 18, 2022
Table 20: Conflict of Interest Declaration for Lung Cancer Canada Clinician 10
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Roche | X | — | — | — |
Merck | X | — | — | — |
BMS | X | — | — | — |
Takeda | X | — | — | — |
Novartis | X | — | — | — |
Ipsen | X | — | — | — |
Sanofi | X | — | — | — |
Pfizer | X | — | — | — |
Declaration for Clinician 11
Name: Dr. Cheryl Ho
Position: Medical Oncologist, BC Cancer
Date: Mar 18, 2022
Table 21: Conflict of Interest Declaration for Lung Cancer Canada Clinician 11
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Bayer | Advisory role | X | — | — | — |
Roche | Advisory role, travel, research grants | — | — | — | X |
Declaration for Clinician 12
Name: Dr. Zhaolin Xu
Position: Pathologist, QEII Health Sciences Centre
Date: Mar 18, 2022
Table 22: Conflict of Interest Declaration for Lung Cancer Canada Clinician 12
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Astra Zeneca | X | — | — | — |
Declaration for Clinician 13
Name: Dr Nicole Bouchard
Position: Respirologist, Sherbrooke University Hospital
Date: Mar 18, 2022
Table 23: Conflict of Interest Declaration for Lung Cancer Canada Clinician 13
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Astra Zeneca | Advisory Role/Conference | X | — | — | — |
Bristol-Myers Squibb | Advisory Role/Research | X | — | — | — |
Merck | Advisory Role /Research/Conference | X | — | — | — |
Bayer | Advisory Role | X | — | — | — |
Pfizer | Conference/Research | X | — | — | — |
Declaration for Clinician 14
Name: Dr Catherine Labbé
Position: Head of Respiratory Medicine Service, Université de Laval
Date: Mar 18, 2022
Table 24: Conflict of Interest Declaration for Lung Cancer Canada Clinician 14
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Amgen | X | — | — | — |
Astra Zeneca | — | X | — | — |
Brystol-Myers Squibb | X | — | — | — |
Jazz Pharmaceuticals | X | — | — | — |
LEO Pharma | X | — | — | — |
Merck | X | — | — | — |
Pfizer | X | — | — | — |
Roche | X | — | — | — |
Sanofi Genzyme | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.