CADTH Reimbursement Review
Zanubrutinib (Brukinsa)
Sponsor: BeiGene Canada ULC
Therapeutic area: Mantle cell lymphoma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
BTK
Bruton tyrosine kinase
CI
confidence interval
CR
complete response
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
ESS
effective sample size
FDG
fluorodeoxyglucose
GI
gastrointestinal
HR
hazard ratio
IRC
independent review committee
ITC
indirect treatment comparison
ITT
intention to treat
MAIC
matching-adjusted indirect comparison
MCL
mantle cell lymphoma
MIPI
Mantle Cell Lymphoma International Prognostic Index
MIPI-b
Combined Biologic Mantle Cell Lymphoma International Prognostic Index
NHL
non-Hodgkin lymphoma
ORR
overall response rate
OS
overall survival
PAG
Provincial Advisory Group
PD
progressive disease
PFS
progression-free survival
QTc
corrected QT interval
RCT
randomized controlled trial
SAE
serious adverse event
sIMPI
simplified Mantle Cell Lymphoma International Prognostic Index
WDAE
withdrawal due to adverse event
WM
Waldenström macroglobulinemia
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Zanubrutinib (Brukinsa) 80 mg capsules; oral |
Indication | For the treatment of adult patients with mantle cell lymphoma who have received at least 1 prior therapy |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 22, 2021 |
Sponsor | BeiGene Canada ULC |
NOC = Notice of Compliance.
Introduction
Mantle cell lymphoma (MCL) is an aggressive B-cell lymphoma arising from cells in the mantle zone of the lymph node. It is a relatively rare cancer and accounts for 5% to 10% of all cases of non-Hodgkin lymphoma (NHL). According to Canadian Cancer Society estimates, in 2021, 11,100 Canadians would have been diagnosed with NHL.1 MCL can begin with an indolent phase, and a small percentage of patients will remain in this indolent phase. In most patients, MCL can become aggressive. It is often diagnosed at a late stage and often present in the gastrointestinal (GI) tract, bone marrow, blood, and other non-lymph node sites. The median survival is between 4 and 5 years. A definitive diagnosis of MCL is achieved through biopsy, which is also used to distinguish it from other NHL subtypes. Imaging is often used to determine the areas of involvement, using CT and/or PET.
Approximately 10% to 15% of patients with MCL do not require treatment, at least initially, and are instead managed with watchful waiting. Most patients with MCL require treatment right away, and the first decision is whether patients are eligible for an autologous stem cell transplant (ASCT). Those eligible for ASCT undergo intensive multi-drug regimens followed by transplant.2 Rituximab maintenance is used post-ASCT for 3 years. Those who are transplant-ineligible (medically unfit or, in most centres, > 65 years of age) receive bendamustine plus rituximab followed by rituximab maintenance until progressive disease (PD) or for 2 years, whichever occurs sooner. At relapse, most patients would receive a Bruton tyrosine kinase (BTK) inhibitor, namely, ibrutinib. In patients who have had a very long remission following initial therapy and wished to avoid indefinite BTK inhibitor therapy, other options that would be considered include bortezomib combination regimens. Therapy for relapsed or refractory MCL is considered palliative, with the goal of improving the quality and quantity of remaining life.
Zanubrutinib is available as 80 mg oral capsules and administered at a dose of 320 mg once daily or 160 mg twice daily. It is indicated for the treatment of adults with MCL who have received at least 1 prior therapy. It is also indicated for the treatment of adults with Waldenström macroglobulinemia (WM) and it is currently under review at CADTH for this indication. Zanubrutinib is a BTK inhibitor. The sponsor’s reimbursement request is for adult patients with MCL who have received at least 1 prior therapy, which is the same as the Health Canada indication.
The objective of this report is to perform a systematic review of the beneficial and harmful effects of zanubrutinib 80 mg for treatment of adult patients with MCL who have received at least 1 prior therapy.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Lymphoma Canada submitted patient input for this review, based on 2 online surveys of patients with MCL conducted between October 19, 2020, and January 11, 2021, and between September 20, 2021, and October 20, 2021, with a total of 85 respondents.
Respondents reported MCL symptoms such as fatigue and symptoms caused by low red blood cell count, which affected their ability to travel, work, exercise, and complete household chores, causing detrimental effects on their quality of life. According to respondents, the most difficult MCL treatment side effects included fatigue, nausea and vomiting, neurocognitive effects such as brain fog or headaches, and hair loss.
Respondents reported that they expect the following key outcomes from any new drug or treatment: faster remission, delay in disease progression, control of disease and symptoms, improved quality of life, and fewer side effects. Most respondents indicated a desire to have a choice in their treatment selection and most would prefer a pill option rather than an IV treatment.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH for this review noted that, until the emergence of BTK inhibitors, treatments for relapsed MCL had not been very effective at generating prolonged remission. The currently funded BTK inhibitor (ibrutinib) improved many of the treatment goals; however, there are side effects in some patients.
The clinical experts believe that zanubrutinib would be an alternative for patients who are unable to tolerate ibrutinib or 1 of its alternatives. Patients with relapsed or refractory MCL who have not progressed on another BTK inhibitor would be candidates for zanubrutinib. The clinical experts believed that zanubrutinib could carry a marginally higher risk of neutropenia than ibrutinib and, therefore, patients who are having issues with neutropenia may not be good candidates for a switch.
The clinical experts believe that the most effective methods for assessing response to treatment are clinical and radiological assessment of lymph node size, and response to therapy would be indicated by a reduction in lymph node size, although preventing the progression of lymphadenopathy and/or symptoms would also be considered valuable. Response to treatment would likely be assessed every month early on and then perhaps every 3 months, and treatment should be discontinued when there is clinical or radiological evidence of disease progression or intolerable side effects.
Clinician Group Input
Experts assembled by Lymphoma Canada and the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee provided input.
There were no major disagreements between the input provided by the clinical experts consulted by CADTH for this review and the clinician groups.
Drug Program Input
The Provincial Advisory Group (PAG) inquired about factors influencing the choice of treatment between zanubrutinib and ibrutinib, and the clinical experts noted that many physicians would be comfortable with ibrutinib because it has been around longer; however, zanubrutinib may have a better safety profile with respect to adverse events (AEs), such as atrial fibrillation. The clinical experts noted that neither of the BTK inhibitors are considered superior to the other in clinical practice.
PAG asked whether patients who progressed on ibrutinib would be candidates for zanubrutinib, and the clinical experts did not see this as a viable clinical pathway. PAG also asked whether zanubrutinib might be appropriate on a case-by-case basis for patients who are unable to receive chemo-immunotherapy due to age or comorbidities. PAG also asked whether reimbursement criteria should align with ibrutinib; the clinical experts believed it should, and that zanubrutinib should also be reimbursed for patients intolerant to ibrutinib.
PAG asked what the most appropriate frequency or modality is to assess treatment response, and the clinical experts stated that, in this population, imaging would be performed if the patient were feeling unwell. PAG asked about the preferred dosing schedule for zanubrutinib, and the clinical experts believed it to be twice daily. PAG asked whether the clinical experts would recommend switching patients who are currently on ibrutinib and not experiencing PD to zanubrutinib, and the clinical experts did not believe there would be a reason to switch patients who are tolerating ibrutinib.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
Two single-arm, multi-centre, sponsor-funded trials, Study 2063,4 (N = 86) and Study 0035,6 (N = 32), were included in this review. The objective of Study 206 was to evaluate the efficacy of zanubrutinib in patients with relapsed or refractory MCL as measured by the overall response rate (ORR) assessed by an independent review committee (IRC) using the Lugano criteria. This single-arm study was conducted entirely in China and enrolled 86 patients after an initial screening phase of up to 28 days, followed by a single-arm treatment phase where patients received zanubrutinib 320 mg daily orally, and a follow-up phase. The treatment phase could last up to 3 years until PD, unacceptable toxicity, death, withdrawal of consent, or until it was terminated by the sponsor for the final analysis. The primary outcome was ORR, while secondary outcomes included progression-free survival (PFS), and duration of response (DOR) while overall survival (OS) was an exploratory outcome. The data cut-off for the final Clinical Study Report was September 8, 2020. Study 003 was divided into 2 parts. The primary objectives of part 1 were to determine the safety and tolerability of zanubrutinib in patients with B-cell lymphoid malignancies, and to determine the recommended phase II dose regimen for oral zanubrutinib. The primary objective of part 2 was to further assess the safety and tolerability of zanubrutinib administered orally either once or twice daily. There were sites in North America, Europe, Australia, New Zealand, and South Korea, although no specific Canadian sites were identified. The total daily dosage for zanubrutinib was 320 mg, administered as either a single daily dose or split into 2 daily doses. Study 003 was not designed to assess efficacy outcomes, but did report outcomes such as ORR, PFS, and OS. The data cut-off for the Clinical Study Report was March 31, 2021.
Patients were a median of 60.5 years of age in Study 206 and 70.5 years of age in Study 003. The majority of patients were male in both Study 206 (78%) and Study 003 (69%). In Study 206, all patients were Chinese, while in Study 003, the majority of patients were White (78%). The majority of patients (70%) in Study 206 had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0, while in Study 003, there were a similar number of patients with an ECOG PS of 0 (47%) or 1 (44%), and the majority of patients in Study 206 (74%) and Study 003 (88%) had stage IV disease. The majority of patients (71%) in Study 206 had 2 or more prior therapies, while the majority of patients in Study 003 had 1 prior therapy.
Efficacy Results
In Study 206, by the time of the final Clinical Study Report,3 and with a median follow-up of 36.8 months (range = 0.3 to 41.6), the median OS was still not estimable (NE). At 30 months, 77.6% of patients were alive (95% confidence interval [CI], 66.8 to 85.3) and at 36 months, 74.8% (95% CI, 63.7 to 83.0). In Study 003, with a median follow-up of 45.8 months (95% CI, 42.0 to 48.6) at the final analysis, the median OS was also NE.
In Study 206, in the final analysis, after a median follow-up of 33.3 months (range = 0.0 to 38.9), the median PFS was 33.0 months (95% CI, 19.4 to NE). In Study 003, in the final analysis5 and after a median follow-up time for PFS of 40.0 months (95% CI, 28.3 to 45.1), the median PFS was 21.1 months (95% CI, 13.2 to NE).
In Study 206, the ORR was 83.7% (95% CI, 74.2 to 90.8), which ruled out the pre-specified null hypothesis of 40%, with a 1-sided P value of less than 0.0001. The complete response (CR) rate was 77.9% (95% CI, 67.7 to 86.1). In Study 003, the ORR at the final analysis was 90.6% (95% CI, 75.0 to 98.0) and the CR rate was 31.3% (95% CI, 16.1 to 50.0). No statistical analysis was planned.
In Study 206, the median DOR in the 72 patients who achieved an ORR was 24.9 months (95% CI, 23.1 months to not reached). The sponsor noted that because the median was reached with the last event occurring when only 3 patients were at risk, the median DOR estimate was “unstable.” In Study 003, the median DOR at the final analysis was 25.2 months after a median follow-up of 36.9 months (95% CI, 32.3 to 42.3).
Health-related quality of life and time to next treatment were not assessed in the included studies.
Harms Results
AEs were reported in 97% of patients in both Study 206 and Study 003; 50% of patients in Study 206 and 69% of patients in Study 003 reported a grade 3 or higher AE, respectively.3,5 The most common AEs in Study 206 were decreased neutrophil count (47% of patients) and upper respiratory tract infections (38%), and the most common grade 3 or higher AEs were decreased neutrophil count (19%) and lung infection (9%). The most common AEs in Study 003 were diarrhea (47%), constipation (41%), and rash (34% of patients), and the most common grade 3 or higher AEs were anemia (12.5%) and pneumonia (12.5%).
Serious adverse events (SAEs) occurred in 29% of patients in Study 206 and 59% of patients in Study 003, with the most common SAE being pneumonia (12% in Study 206, 12.5% in Study 003).
In Study 206, 9% of patients had at least 1 AE leading to the discontinuation of the study drug; pneumonia was the most common event, occurring in 2% of patients. In Study 003, 28% of patients had at least 1 AE leading to the discontinuation of the study drug and, in 6% of patients, this was pneumonia.
In Study 206, 24% of patients died, 9% within 30 days of their last dose of the study drug and 15% more than 30 days after their last dose of the study drug. Among the patients who died within 30 days of their last dose, most (7% overall) died due to an AE, while the remaining deaths were due to PD. For those deaths that occurred more than 30 days after the last dose of the study drug, most (12% overall) were due to PD, while the remaining 3 deaths were due to AE and “other.” In Study 003, 38% of patients died: 16% died within 30 days of their last dose of the study drug (9% due to an AE) and 22% died more than 30 days after their last dose of the study drug (16% due to PD).
Notable harms in Study 206 included infections (65% of patients; 19% grade ≥ 3), platelet count decrease (33% of patients; 7% grade ≥ 3), hemorrhage (36% of patients; 1% grade ≥ 3), and anemia (17% of patients; 6% grade ≥ 3). In Study 003, hemorrhage occurred in 62.5% of patients and infections occurred in 72% of patients.
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies (ITT Population)
Parameters | Study 206 Zanubrutinib N = 86 | Study 003 Zanubrutinib N = 32 |
|---|---|---|
OS | ||
Events, n (%) | 21 (24.4) | — |
Death | 21 (24.4) | 14 (43.8) |
Censored, n (%) | 65 (75.6) | Alive = 18 (56.3) |
Median OS, months (95% CI), final analysis Median follow-up, months (95% CI) | NE (NE to NE) 36.8 (35.4 to 37.2) | NE (26.1 to NE) 45.8 (42.0 to 48.6) |
PFS | ||
Events, n (%) | 42 (48.8) | 18 (56.3) |
PD | 37 (43.0) | 15 (46.9) |
Death | 5 (5.8) | 3 (9.4) |
Censored | 44 (51.2) | 14 (43.8) |
Median PFS, months (95% CI), final analysis Median follow-up, months (95% CI) | 33.0 (19.4 to NE) 33.3 (33.1 to 34.3) | 21.1 (13.2 to NE) 40.0 (28.3 to 45.1) |
ORR | ||
Best overall response, n (%) | Empty cell | Empty cell |
CR | 67 (77.9) | 10 (31.3) |
PR | 5 (5.8) | 19 (59.4) |
SD | 1 (1.2) | 1 (3.1) |
PD | 8 (9.3) | 2 (6.3) |
Discontinued before first assessment | 5 (5.8) | — |
ORR (95% CI) | 83.7 (74.2 to 90.8) | 90.6 (75.0 to 98.0) |
P value, 1-sideda | P < 0.0001 | NR |
Patients with a CR, n (%) | 67 (77.9) | 10 (31.3) |
DOR | ||
Responders, n (%) | 72 (83.7) | 29 (90.6) |
Events, n (%) | 32 (44.4) | 15 (51.7) |
PD | 29 (40.3) | 13 (44.8) |
Death | 3 (4.2) | 2 (6.9) |
Censored, n (%) | 40 (55.6) | 14 (48.3) |
Median DOR, months (95% CI), final analysis Median follow-up, months (95% CI) | NE (24.9 to NE) 30.6 (4 to 31.5) | 25.2 (12.6 to NE) 36.9 (32.3 to 42.3) |
Harms | ||
Patients with ≥ 1 AE, n (%) | 83 (97) | 31 (97) |
Patients with ≥ 1 SAE, n (%) | 25 (29) | 19 (59) |
Patients who stopped treatment due to AE, n (%) | 8 (9) | 15 (47) |
AE = adverse event; CI = confidence interval; CR = complete response; CRR = complete response rate; DOR = duration of response; ITT = intention to treat; NE = not estimable; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; SAE = serious adverse event; SD = stable disease.
aP value (1-sided) was based on the binomial exact test against the null hypothesis (H0): ORR = 0.40.
Source: Clinical Study Report for Study 206 and Study 003.3-6
Critical Appraisal
Both of the included studies lacked a control group. This limits the interpretation of both efficacy and harms, both because there is no control group available as a basis for comparison and because all patients were aware of the treatment they were receiving. The outcomes most at risk of bias from patient unblinding are typically patient-reported outcomes such as health-related quality of life; however, these outcomes were not assessed in the included studies.
ORR was the only outcome that was formally assessed using a statistical comparison, and this was done only in Study 206. Data for key clinical outcomes like OS and PFS were reported; however, the lack of statistical comparisons and lack of a control group makes it challenging to interpret this data. Median OS was NE and there is uncertainty around those outcomes that were estimable due to the lack of comparison. Study 003 was a phase I and II study and was not designed to evaluate efficacy, as only toxicities were important in the dose-finding stage and for the initial phase II outcomes.
Indirect Comparisons
Description of Studies
No indirect comparisons of zanubrutinib were noted in the peer-reviewed literature. One study, a matching-adjusted indirect comparison (MAIC) that was provided by the sponsor, compared zanubrutinib with ibrutinib in patients with relapsed or refractory MCL. Data to inform this analysis were taken from 2 studies from the sponsor, Study 2063,4 and Study 003,5,6 both of which had data available at the individual patient level and were matched to a pooled analysis population from 3 ibrutinib trials7 (PCYC-1104-CA,8 RAY,9 and SPARK10) using entropy balancing. This indirect treatment comparison (ITC) evaluated differences in OS, PFS, response, and safety between the weighted zanubrutinib population relative to the pooled ibrutinib population.
Efficacy Results
Following an entropy weighting adjustment, the zanubrutinib analysis population was reduced to an effective sample size (ESS) of 37 from an available total population of 117. The ORR did not demonstrate statistically significant differences between the weighted zanubrutinib (ORR = 77.7%; 95% CI, 63 to 92.4) and ibrutinib (ORR = 65.7%; 95% CI, 60.6 to 70.5) treatment groups. Similarly, the CR rate did not demonstrate statistically significant differences between the weighted zanubrutinib (CR = 25.5%; 95% CI, 12.5 to 38.5) and ibrutinib (CR = 20%; 95% CI, 16 to 24.4) treatment groups. PFS did not demonstrate statistically significant differences between the weighted zanubrutinib (PFS restricted-mean survival time [RMST] = 13.9 months) and ibrutinib (PFS RMST = 12.6) treatment arms (zanubrutinib versus ibrutinib hazard ratio [HR] = 0.92; 95% CI, 0.63 to 1.33). Similarly, OS did not demonstrate statistically significant differences between the weighted zanubrutinib (RMST = 21.2) and ibrutinib (18.4) treatment arms (zanubrutinib versus ibrutinib HR = 0.74; 95% CI, 0.43 to 1.26).
Upon reconsideration, the sponsor shared the results of an additional ITC analysis that was completed as part of its submission to Australia’s Pharmaceutical Benefits Advisory Committee (PBAC) to support its conclusion that the efficacy and safety of zanubrutinib suggests there is little uncertainty regarding the class effect and overall clinical benefit of BTK. After adjustment, the ITC HRs for both PFS and OS indicate comparable efficacy between zanubrutinib and ibrutinib (PFS = 0.94; 95% CI, 0.65 to 1.36; OS = 0.77; 95% CI, 0.47 to 1.28). While acknowledging the results of the naive comparison and MAIC from the PBAC submission are aligned with the other analytical approaches presented in the original submission and support the claim of noninferiority, the sponsor confirmed that the slight difference between the Canadian and Australian MAIC results was driven by a combination of a new data cut and different type of analysis.11
The clinical experts agreed there is a class effect for BTK inhibitors and expressed that this class effect supports funding BTK inhibitors equally for all relevant diseases and not specifically zanubrutinib in MCL. Nonetheless, the clinical experts agreed there does not appear to be an important risk related to the approval of zanubrutinib in MCL. The clinical experts further explained that approval could provide important alternative access to BTK inhibitors for a subset of patients experiencing significant toxicities with ibrutinib. The clinical experts felt that the MCL data provided, although relatively small, were convincing and supportive, in addition to the data presented from other diseases. The clinical experts also highlighted that the cost to the system should be relatively neutral, given that patients will likely be switched from 1 BTK inhibitor to another and will get funded for BTK inhibitor treatment only until disease progression.
Harms Results
No formal statistical comparison was made of the differences in the safety events between the 2 analysis populations.
Critical Appraisal
The analytical approach that was used resulted in a low ESS, making estimates of comparative efficacy subject to substantial uncertainty. The low ESS is indicative of large differences between the unadjusted patient populations, which demonstrated large between-population differences pre-adjustment. The post-adjustment balance of patient characteristics was assessed using an approach that still allows for differences between patient populations and, therefore, residual confounding due to specified and unspecified patient characteristics may influence the results presented. No conclusion can be made regarding the ITC owing to the statistical approaches used alongside the large differences in the patient populations of the trials within the comparison.
No formal comparisons of patient safety or patient quality of life were made, meaning that comparisons with ibrutinib are not possible from the evidence presented.
Other Relevant Evidence
Upon reconsideration, the sponsor provided evidence supporting the need for other BTK inhibitor alternatives, which came from a new publication that was not available at the time of submission. Shah et al. was a retrospective observational study. The objectives of the study were: to examine the clinical and sociodemographic characteristics of patients receiving a BTK inhibitor, to describe the treatment patterns and compliance for each BTK inhibitor, and to assess the costs and hospitalizations associated with each BTK inhibitor in the real-world setting in the US.12
Shah et al. de-identified data from Integrated Dataverse, an open-source claims database that captures and aggregates data from different claims vendors. Shah et al. included adult patients with MCL who had at least 1 BTK inhibitor prescription claim for 12-month period Patients were stratified into 1 of 3 cohorts based on their index BTK inhibitor, and outcomes included length of hospital stay and inpatient hospital charges. There were 1,242 patients in the ibrutinib cohort, 485 in the acalabrutinib cohort, and 67 in the zanubrutinib cohort.12
Approximately 1-fifth of patients on zanubrutinib or acalabrutinib had been switched from ibrutinib. Ibrutinib was more often used in the front-line setting (68.4%), while use of zanubrutinib (80.6%) and acalabrutinib (68.9%) tended to be in the relapsed or refractory setting. Shah et al. noted that results should be interpreted with caution due to the limited sample size and follow-up period. CADTH would also highlight that the retrospective observational study in poster form included a small sample size in the zanubrutinib cohort compared with the other cohorts.12
In its feedback, the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee agreed that patients who are intolerant to ibrutinib but otherwise responding should be offered a switch to an alternative BTK inhibitor, such as zanubrutinib. The clinician group highlighted that if these patients cannot tolerate any BTK inhibitor, then there are no additional treatment options and the outcomes will be poor.
The clinical experts consulted by CADTH agreed there is a clear need for alternative treatment options, specifically for patients who may be at higher risk for AEs with ibrutinib or who are experiencing AEs with current ibrutinib therapy. The clinical experts explained that data from various sources (in chronic lymphocytic leukemia, WM, and MCL) suggest that the second-generation BTK inhibitors will be less toxic than ibrutinib and they accept these data. The clinical experts noted that AEs of special interest (hypertension, bleeding and, in particular, atrial fibrillation) appear to be less frequent with zanubrutinib. Lastly, the clinical experts acknowledged that although the data for MCL are from a relatively smaller patient subset, the composite data would suggest that some patients with toxicities to ibrutinib may be able to tolerate zanubrutinib, which is consistent with the observation by Shah et al.
Upon reconsideration, the sponsor submitted evidence from a poster of a phase II study by Shadman et al. The study included only patients who were intolerant to ibrutinib and/or acalabrutinib. The primary objective of the study was to evaluate the safety of zanubrutinib compared with ibrutinib and/or acalabrutinib intolerance, as assessed by recurrence and change in severity of AEs. The secondary objective was to evaluate the efficacy of zanubrutinib with respect to investigator-assessed ORR, disease control rate, and PFS, as well as patient-reported outcomes.13
Patients were divided into 2 cohorts: cohort 1 comprised patients intolerant to ibrutinib (N = 50), and cohort 2 comprised patients intolerant to acalabrutinib with or without ibrutinib (N = 10); overall, 4.5% of patients had MCL. All patients were switched to zanubrutinib 160 mg twice daily or 320 mg once daily and were treated until PD, unacceptable toxicity, consent was withdrawn, or the study was terminated. A limitation of this study is that it was only reported as a poster and only 3 patients in this analysis had MCL, which reflects both the small sample size in this study and the fact that only a small proportion of the patients had MCL.13
While the clinical experts acknowledged the concerns related to the dataset for the requested indication (MCL) being relatively small, they agree the data are relevant to the safety and even efficacy comparative data for other diseases (chronic lymphocytic leukemia and WM) that support the safety profile and clinical activity of zanubrutinib, and this provides a relatively high level of confidence that the similar results reported in MCL are valid.
Conclusions
Two pivotal, sponsor-funded, multi-centre, single-arm studies that enrolled a total of 118 patients with relapsed or refractory MCL were included in this review. In the 1 study that included a historical control, ORR was improved for zanubrutinib versus the control, although the control used was not a BTK inhibitor. No conclusions can be drawn about efficacy with respect to other outcomes, such as OS, PFS, and DOR, as no statistical analysis was planned. Common AEs were consistent with those described in the product monograph and included various cytopenias, infections, and hemorrhage. There were no other studies to inform the long-term safety of this second-generation BTK inhibitor; therefore, the long-term safety of zanubrutinib is unknown. The ITC submitted by the sponsor was of limited value for drawing any conclusions about the relative efficacy and safety of zanubrutinib compared with other BTK inhibitors due to significant methodological issues with the approach taken.
Introduction
Disease Background
MCL is an aggressive B-cell lymphoma arising from cells in the mantle zone of the lymph node. It is a relatively rare cancer and accounts for 5% to 10% of all cases of NHL. According to Canadian Cancer Society estimates, in 2021, 11,100 Canadians would have been diagnosed with NHL.1 MCL can begin with an indolent phase and a small percentage of patients will remain in this indolent phase. In most patients, MCL can become aggressive. It is often diagnosed at a late stage and often present in the GI tract, bone marrow, blood, and other non-lymph node sites. The median survival is between 4 and 5 years.
A definitive diagnosis of MCL is achieved through biopsy, which is also used to distinguish it from other NHL subtypes. Imaging is often used to determine the areas of involvement, using CT or PET plus CT. The presence of cyclin D1 and the t[11;14][q13;q32] translocation can be used not only for diagnosis but also for prognosis, as can Ki67, which indicates the proportion of cells that are actively dividing, and TP53 mutations, which may indicate increased risk of treatment failure.
Standards of Therapy
Approximately 10% to 15% of patients with MCL do not require treatment, at least initially, and are instead managed with watchful waiting.2 According to the clinical experts consulted by CADTH for this review, most patients with MCL require treatment right away, and the first decision is whether patients are eligible for an ASCT. Those eligible for ASCT undergo intensive multi-drug regimens such as rituximab plus cyclophosphamide plus doxorubicin plus vincristine plus prednisone (R-CHOP) alternating with rituximab plus dexamethasone plus cytarabine plus cisplatin (R-DHAP) or bendamustine plus rituximab alternating with R-DHAP or rituximab plus cytarabine, followed by transplant.1 According to the clinical experts consulted by CADTH, rituximab maintenance is used post ASCT for 3 years. Those who are transplant-ineligible (medically unfit or, in most centres, > 65 years of age) receive bendamustine plus rituximab followed by rituximab maintenance until PD or for 2 years, whichever occurs sooner. Some centres may use less rituximab maintenance in this group, given the lack of randomized controlled trial (RCT) evidence specifically for rituximab maintenance following bendamustine plus rituximab.
According to the clinical experts, patients are treated similarly upon relapse and most would receive a BTK inhibitor, namely, ibrutinib. In patients who have had a very long remission following initial therapy and wished to avoid indefinite BTK inhibitor therapy, other options that would be considered would include bortezomib combination regimens although, according to the clinical experts, this would be an infrequent occurrence. Therapy for relapsed or refractory MCL is considered palliative, with the goal of improving the quality and quantity of remaining life.
According to the clinical experts consulted by CADTH for this review, treatment goals include establishing disease control (i.e., no or reduced disease-related symptoms) for as long as possible, balanced with acceptable side effects. An improvement in OS would also be optimal.
Drug
Zanubrutinib is available as 80 mg oral capsules and administered at a dose of 320 mg once daily or 160 mg twice daily. It is indicated for the treatment of adults with MCL who have received at least 1 prior therapy. It is also indicated for the treatment of adults with WM. Zanubrutinib is a BTK inhibitor. BTK is a signalling molecule that activates pathways involved in proliferation, trafficking, chemotaxis, and adhesion of B-cells. The sponsor’s reimbursement request is as per indication. Zanubrutinib underwent the standard review pathway at Health Canada, receiving a Notice of Compliance on July 22, 2021.
Table 3: Key Characteristics of BTK Inhibitors
Characteristics | Zanubrutinib | Ibrutinib | Acalabrutinib |
|---|---|---|---|
Mechanism of action | BTK is a signalling molecule in the BCR pathway. BCR may be important in pathogenesis of B-cell malignancies | BTK is a signalling molecule in the BCR pathway. BCR may be important in pathogenesis of B-cell malignancies | BTK is a signalling molecule in the BCR pathway. BCR may be important in pathogenesis of B-cell malignancies |
Indicationa |
| For relapsed or refractory MCL Others:
| For MCL in patients who have received at least 1 prior therapy Others:
|
Route of administration | Oral | Oral | Oral |
Recommended dose | 320 mg once daily or 160 mg twice daily until disease progression or unacceptable toxicity | Ibrutinib (MCL or MZL): 560 mg once daily until disease progression or no longer tolerated by the patient | 100 mg twice daily until disease progression or unacceptable toxicity |
Serious adverse effects or safety issues |
|
|
|
BCR = B-cell antigen receptor; BTK = Bruton tyrosine kinase; cGVHD = chronic graft-versus-host disease; CLL = chronic lymphocytic leukemia; MCL = mantle cell lymphoma; MZL = marginal zone lymphoma; NMSC = non-melanoma skin cancer; TLS = tumour lysis syndrome; WM = Waldenström macroglobulinemia.
aHealth Canada–approved indication.
Source: Product monographs from e-CPS (electronic version of the Compendium of Pharmaceuticals and Specialties) for zanubrutinib, ibrutinib, and acalbrutinib.14
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
One patient group, Lymphoma Canada, submitted patient input for this review. Lymphoma Canada is a national Canadian registered charity that empowers the lymphoma community through education, support, advocacy, and research. The input was based on 2 online surveys of MCL patients; 1 survey was conducted between October 19, 2020, and January 11, 2021, and the other was conducted between September 20, 2021, and October 20, 2021. A total of 85 respondents (33 patients from the first survey and 52 from the second) were included in the patient input. Among the respondents who provided demographic information, 59% live in Canada, 58% are female, and 40% are over the age of 65. Two respondents who live in Canada had experience with zanubrutinib.
Respondents reported that MCL symptoms such as fatigue affect their ability to travel, work, exercise, and complete household chores, causing detrimental effects on their quality of life. According to respondents, the most difficult MCL treatment side effects included fatigue, nausea and vomiting, neurocognitive effects (such as brain fog or headaches), and hair loss. Two respondents with experience with zanubrutinib reported that the treatment was able to manage their MCL symptoms (including fatigue, indigestion, abdominal pain, and bloating), resolve their blood cell counts, reduce weight loss, and improve appetite. Both patients indicated that the side effects they experienced on zanubrutinib did not negatively impact their quality of life.
In the patient input received, respondents reported they expect the following key outcomes from any new drug or treatment: faster remission, delay in disease progression, control of disease and symptoms, improved quality of life, and fewer side effects. Most respondents indicated a desire to have a choice in their treatment selection and most would prefer a pill option rather than an IV treatment.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of MCL.
Unmet Needs
Traditionally, second-line therapies for MCL have not been very effective at generating prolonged remission; however, BTK inhibitors have improved this. The currently funded BTK inhibitor (ibrutinib) meets many of the treatment goals; however, there are side effects in some patients, the most concerning among them being cardiac-related (hypertension, atrial fibrillation), hemorrhage, and/or bruising.
Place in Therapy
Zanubrutinib could be an alternative to ibrutinib, including for patients who are intolerant to ibrutinib. It is unclear whether zanubrutinib has better effectiveness than ibrutinib, but there is a possibility that it may have better overall tolerability, based on results from other studies in other lymphoproliferative disorders. This may make zanubrutinib a suitable choice in case of severe intolerance. The clinical expert noted that acalabrutinib, which is also a second-generation BTK inhibitor, also has Health Canada’s approval for MCL.
While the data are clear that BTK inhibitors are the preferred treatment for relapsed or refractory MCL, it is not clear whether use of 1 BTK inhibitor should be encouraged over another.
Patient Population
Any patient with relapsed or refractory MCL who has not experienced progression with another BTK inhibitor would be expected to experience clear benefits with zanubrutinib. Based on the clinical experts’ overall impression, the only side effect that appears relatively more frequent with zanubrutinib compared with ibrutinib is neutropenia; therefore, in the opinion of the clinical experts, the only patient group where ibrutinib may be favoured over zanubrutinib would be patients who have problems with neutropenia.
MCL is expected to relapse after first-line therapy so any evidence of recurrent adenopathy (clinical or radiological) would be sufficient to establish recurrence of MCL and justify re-treatment. Given the aggressive nature of MCL, relapse treatment is typically indicated when there is evidence of disease recurrence, with or without symptoms. Pathological confirmation of disease recurrence would not likely be routinely sought, as the misdiagnosis of relapse is rare.
Patients with ongoing or recurrent neutropenia would be least suited for zanubrutinib; in these patients ibrutinib would be favoured. With respect to how patients who are best suited for zanubrutinib can be identified, the clinical expert did not identify a subgroup that was likely to experience better efficacy with zanubrutinib, although they did note with respect to BTK inhibitors in general the patients with blastoid histology tend to do better and those with TP53 mutations may do worse.
Assessing Response to Treatment
The most common mechanism for assessing response is clinical and radiological assessment of lymph node size. Meaningful response to therapy would be indicated by a reduction in lymph node size and improvement in lymphoma symptoms (such as night sweats, weight loss, and fatigue). Maintaining disease stability (i.e., preventing progression of lymphadenopathy and/or symptoms) would also be considered valuable. Disease control is expected to correlate with improved survival, as disease progression would lead to death or requirement for other therapy. These other therapies tend to be costly and/or toxic (bispecific antibodies, chimeric antigen receptor [CAR] T-cell therapy or, rarely, allogeneic stem cell transplant. Response to therapy would typically be assessed every month early on and then perhaps every 3 months in patients experiencing good disease control (as durable responses beyond several years are observed in patients whose disease responds to treatment).
Discontinuing Treatment
Treatment should be discontinued upon clinical or radiological evidence of disease progression or intolerable side effects.
Prescribing Conditions
Zanubrutinib can be given in any setting in which MCL is treated, including in community and academic settings, as long as laboratory testing and imaging is available. A specialist is required for optimal management, as knowledge of disease characteristics and the features of the drug, including its toxicity profile, are critical for best management.
Additional Considerations
One clinical expert noted there is a definite role for second-generation BTK inhibitors with improved toxicity profiles due to the increasing importance of BTK inhibitors in the management of MCL and other lymphoproliferative disorders.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
A cross-Canada group of experts in lymphoma was assembled with the help of Lymphoma Canada. Lymphoma Canada was not involved in the development of any content.
The Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee also provided input in the form of evidence-based guidance on drug-related issues in support of Cancer Care Ontario’s mandate.
Unmet Needs
There are limited options for patients with relapsed or refractory MCL. The available options only benefit a fraction of patients (35% to 75%) and typically do not provide durable responses (6 to 18 months). Many of these treatments need to be administered indefinitely and toxicity may impact quality of life. The toxicities associated with the first BTK inhibitor, ibrutinib, and the second publicly available BTK inhibitor, acalabrutinib, are well known and there is room for improvement. Some BTK inhibitors have drug interactions that interfere with their use; therefore, newer BTK inhibitors with fewer interactions would also be an improvement.
Older, more frail patients and younger patients without comorbidities and with good ECOG PS have the greatest unmet need that could potentially be addressed by a drug like zanubrutinib. Choice of BTK inhibitor may be guided by its AE profile and/or contraindications in given patients.
Place in Therapy
Zanubrutinib would likely be used in a manner similar to other BTK inhibitors and could be used in patients with specific contraindications to other BTK inhibitors.
Patients could either try zanubrutinib as their initial BTK inhibitor, with the choice guided by contraindications that a given patient may have to the other BTK inhibitors, or patients who develop specific toxicities to other BTK inhibitors could end up switching to zanubrutinib.
Patient Population
Any patient with MCL could be considered for BTK inhibitor therapy after primary therapy. Among the BTK inhibitors, zanubrutinib could be considered in patients who have experienced cardiovascular toxicities with other BTK inhibitors, as the risk of these events may be lower with zanubrutinib.
Candidates for zanubrutinib would be identified by their hematologist or oncologist. Progression after primary treatment may be identified clinically but is confirmed with laboratory and/or imaging findings. Due to the aggressive nature of MCL at relapse, second-line therapy is generally initiated promptly upon first detection of relapse, even if the patient remains asymptomatic. There are currently no biomarkers that can be used to predict which patients are most likely to exhibit a response.
Patients least suitable for zanubrutinib would be those who have comorbid illnesses that represent contraindications to treatment with zanubrutinib, including disorders associated with serious bleeding and/or cardiovascular disease and obvious uncontrolled infections. Patients with poor ECOG PS and low life expectancy (particularly for other reasons) may not be good candidates either.
Assessing Response to Treatment
Treatment response would be assessed by CT scans and possibly PET scans. Assessments of organ function and bloodwork (including routine blood counts) would also be important. A clinically meaningful response would include an objective response or, at a minimum, disease stabilization (lack of progression). Typically, this would be associated with improvement in disease-related symptoms. Treatment success would be expected to improve quality of life and independence in activities of daily living. Response should be assessed radiologically post-treatment and again several months later. Ongoing imaging may be dependent on symptoms and the results of previous imaging scans, clinical findings, and laboratory results.
Discontinuing Treatment
PD (based on imaging or laboratory findings) would indicate treatment failure. In that event, it would be appropriate to consider initiating a new treatment, and this could include CAR T-cell therapy in eligible patients, where funded.
Prescribing Conditions
As an oral therapy that is well tolerated and third in its class, zanubrutinib could be administered in any setting where patients with cancer may be seen.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Issues with the choice of comparator in the submitted trial The BGB-3111-206 study was a non-comparative trial. Relevant comparators for zanubrutinib include ibrutinib and various chemotherapy (or chemo-immunotherapy Rituximab-based chemotherapy] options. What factors determine the choice of treatment between ibrutinib and zanubrutinib? | Many physicians would be comfortable with ibrutinib, as it has a longer track record. Zanubrutinib may have a better safety profile when it comes to AEs such as atrial fibrillation. With respect to effectiveness, they are likely comparable. |
Other implementation issues regarding relevant comparators (e.g., access or funding, covered population) Acalabrutinib was approved by Health Canada for the treatment of relapsed or refractory MCL; however, it is not reimbursed by jurisdictions and it is no longer available under a compassionate access program. Rituximab-based chemotherapies may be used in the first line. Bortezomib is also indicated for relapsed or refractory MCL in Canada and may be approved in some jurisdictions. Tecartus (brexucabtagene autoleucel) was recently recommended by CADTH for relapsed or refractory MCL after 2 or more lines of systemic therapy that included a BTK inhibitor. | NA |
Other patient characteristics for eligibility (e.g., age restrictions, comorbidities) Should patients who have received treatment with ibrutinib and whose disease has progressed be eligible for treatment with zanubrutinib? | The clinical experts would not select zanubrutinib for patients whose disease has relapsed or is refractory to ibrutinib or vice versa. |
Prior therapies required for eligibility Should zanubrutinib be used as a first-line therapy on a case-by-case basis in patients with MCL who are unable to receive chemo-immunotherapy due to age or comorbidities? | The clinical experts consulted by CADTH are not aware of evidence to support this type of approach. |
Consistency with initiation criteria associated with other drugs reviewed by CADTH in the same therapeutic space Should the reimbursement criteria align with that of ibrutinib? | The reimbursement criteria should align with ibrutinib. Zanubrutinib could also be reimbursed for those who are intolerant to ibrutinib. |
Challenges related to assessment and monitoring of therapeutic response Patients in the BGB-3111-206 trial were assessed via a combined PET and CT scan every 12 weeks for 96 weeks and then every 24 weeks thereafter until disease progression or withdrawal. In clinical practice, what is the most appropriate frequency and modality to determine treatment response? | The clinical experts indicated that, in practice, imaging would be conducted only during follow-up if there were symptoms or signs of deterioration. |
Dosing, schedule or frequency, dose intensity Zanubrutinib dosing in the BGB-3111-206 study was 160 mg (two 80 mg capsules) orally twice daily. Is the alternate dosing schedule of 320 mg once daily also clinically appropriate for this patient population? Is there a preferred dosing schedule? | The clinical experts are of the opinion that a 320 mg once-daily dosing can be an option. However, there are more clinical data to support twice-daily 160 mg dosing. |
Patients on active treatment with a time-limited opportunity to switch to the drug under review Should patients who are currently receiving ibrutinib and have not experienced disease progression be eligible on a time-limited basis? | The clinical experts do not advise switching in patients who are not experiencing toxicities or tolerability issues. |
AE = adverse event; BTK = Bruton tyrosine kinase; MCL = mantle cell lymphoma; NA = not applicable.
Clinical Evidence
The clinical evidence included in the review of zanubrutinib is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section normally includes sponsor-submitted long-term extension studies and additional relevant studies that can be considered to address important gaps in the evidence included in the systematic review; however, no such studies are available for this report.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of zanubrutinib 80 mg, taken as either 320 mg (four 80 mg capsules) once daily or 160 mg (two 80 mg capsules) twice daily, for the treatment of adult patients with MCL who have received at least 1 prior therapy.
Methods
The studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect the outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Adult patients with MCL who have received at least 1 prior therapy Subgroups:
|
Intervention | Zanubrutinib 320 mg daily, taken orally |
Comparators |
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published and unpublished phase III and IV RCTs |
AE = adverse event; CR = complete response; ECOG = Eastern Cooperative Oncology Group; MCL = mantle cell lymphoma; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.15
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Brukinsa (zanubrutinib). Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on November 3, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on March 9, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.16 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 2 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
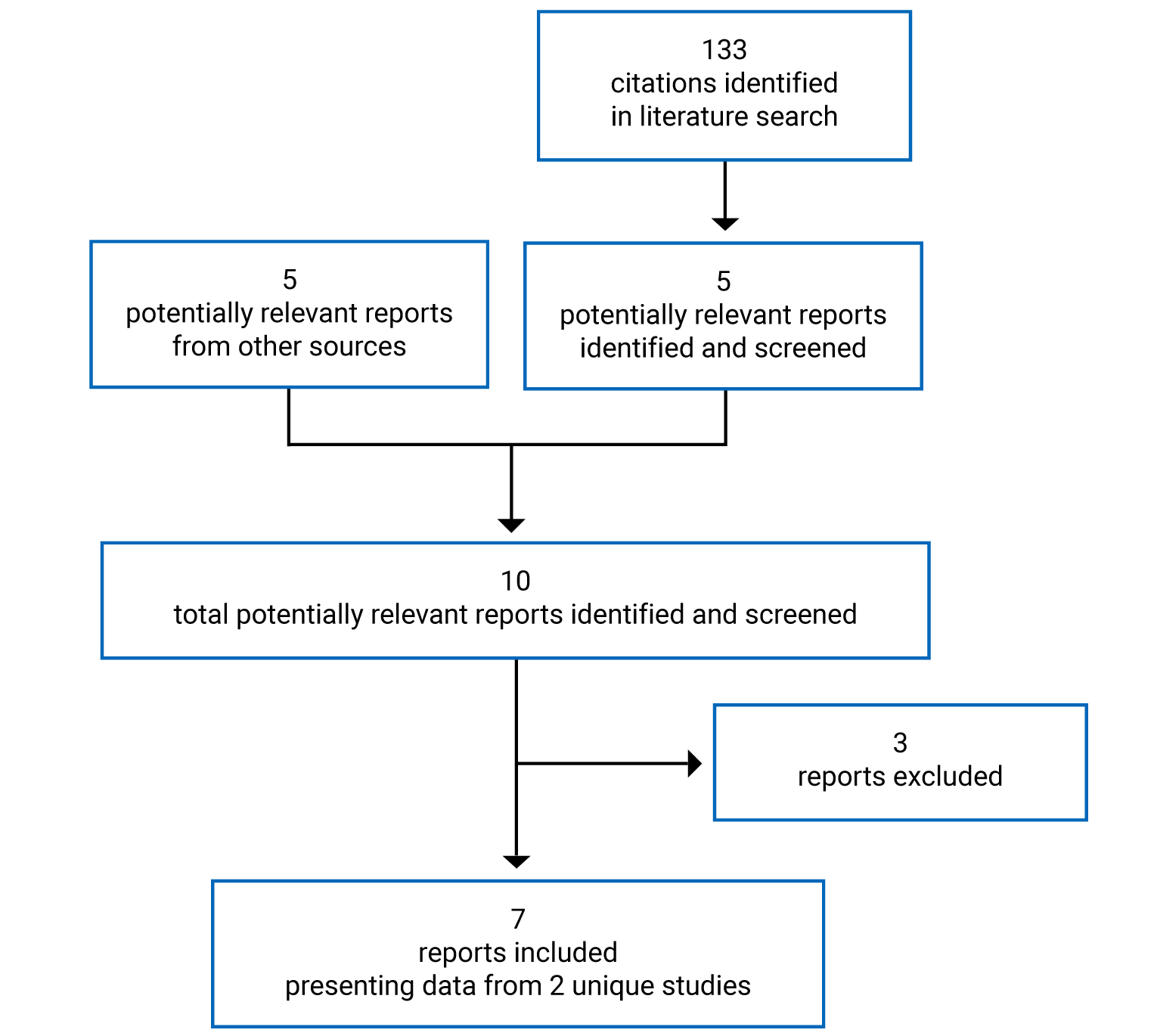
Table 6: Details of Included Studies
Detail | Study 206 | Study 003 |
|---|---|---|
Designs and populations | ||
Study design | Non-RCT (single arm) | Non-RCT (single arm) |
Locations | 14 sites: China | North America, Europe, Australia, New Zealand, South Korea, UK |
Patient enrolment dates |
|
|
Enrolled (N) | 86 | 32 (R/R MCL 320 mg dose) |
Inclusion criteria |
|
Dose-escalation phase:
Safety, schedule, and efficacy expansion:
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | 160 mg zanubrutinib twice daily by mouth | Dose-escalation phase: Zanubrutinib 40 mg/day orally, escalating to 160 mg twice daily or 320 mg once daily |
Comparator(s) | None | None |
Duration | ||
Phase | — | — |
Screening | Up to 28 days | Up to 28 days |
Treatment | Until disease progression or intolerable toxicity | Until disease progression or intolerable toxicity |
Follow-up | Survival follow-up every 3 months | Every 3 months |
Outcomes | ||
Primary end point | ORR (IRC-assessed) | AE, SAE, physical exam, laboratory measurements |
Secondary and exploratory end points | Secondary:
Exploratory:
| Secondary:
Exploratory: Correlation of clinical response with prognostic factors or biomarkers |
Notes | ||
Publications | Song (2020) | Tam (2019) |
ALT = alanine aminotransferase; aPTT = activated partial thromboplastin time; ASCT = autologous stem cell transplant; AST = aspartate transaminase; AV = atrioventricular; BTK = Bruton tyrosine kinase inhibitor; CNS = central nervous system; CR = complete response; DOR = duration of response; ECG = electrocardiogram; ECOG PS = Eastern Cooperative Oncology Group Performance Status; eGFR = estimated glomerular filtration rate; FISH = fluorescence in situ hybridization; GVHD = graft-versus-host disease; INR = international normalized ratio; IRC = independent review committee; MDRD = modified diet in renal disease; MRD = minimal residual disease; NYHA = New York Heart Association; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; QTcF = QT interval corrected for heart rate using the Fridericia formula; RCT = randomized controlled trial; R/R = relapsed or refractory; SD = stable disease; TTR = time to response; ULN = upper limit of normal.
Note: 5 additional reports were included (Clinical Study Reports for studies 206 and 003 and the sponsor’s submission).3-6,17
Source: Clinical Study Report for Study 206 and Study 003.3-6
Description of Studies
Study 206 is a single-arm, sponsor-funded study whose objective was to evaluate the efficacy of zanubrutinib in patients with relapsed or refractory MCL as measured by ORR assessed by IRC using the Lugano criteria. This single-arm study was conducted entirely at 14 sites in China and enrolled 86 patients after an initial screening phase of up to 28 days, followed by a single-arm treatment phase where patients received zanubrutinib 320 mg per day orally, and a follow-up phase. Tumour assessments were performed during the screening phase and consisted of a CT with contrast or MRI scan of the neck, chest, abdomen, and pelvis; a fluorodeoxyglucose PET [FDG-PET]; and a bone marrow biopsy. An endoscopy or GI biopsy was performed for patients with suspected GI involvement. The treatment phase could last up to 3 years, until PD, unacceptable toxicity, death, withdrawal of consent, or until it was terminated by the sponsor for the final analysis. The data cut-off for the Clinical Study Report submitted to CADTH was August 31, 2019; after a subsequent request, the final Clinical Study Report, with a data cut-off of September 8, 2020, was provided. See Figure 2 for study design.
Study 003 is a sponsor-funded, multinational, phase I and II, single-arm, multi-dose, dose-escalation trial. It was divided into 2 parts; the primary objectives of part 1 were to determine the safety and tolerability of zanubrutinib in patients with B-cell lymphoid malignancies and to determine the recommended phase II dose regimen for oral zanubrutinib. The primary objective of part 2 was to further assess the safety and tolerability of zanubrutinib administered orally either once or twice daily. There were sites in North America, Europe, Australia, New Zealand, and South Korea, although no specific Canadian sites were identified. Part 1 was a dose-escalation phase, where patients with a relapsed or refractory B-cell malignancy were enrolled and followed a pre-planned dose-escalation scheme, starting at zanubrutinib 40 mg once daily. The purpose of part 1 was to establish a dose of zanubrutinib that would be recommended for phase II development, while the purpose of part 2 was to establish the optimal dosing regimen (once versus twice daily). Part 2 divided 380 patients by type of B-cell malignancy (subdivided as subparts 2a through 2m). These subparts generally ran parallel to each other, with the exception of part 2a, which contained patients with relapsed or refractory MCL as well as other diffuse large B-cell lymphoma subtypes (N = 40), and part 2g, which contained 20 patients with relapsed or refractory MCL. The total daily dosage for zanubrutinib was 320 mg; initially, however, 2 different regimens were evaluated, 320 mg once daily and 160 mg twice daily, until a protocol amendment recommended the 160 mg twice-daily regimen. The data cut-off for the Clinical Study Report provided to CADTH was December 13, 2018, and, after a subsequent request, the final Clinical Study Report was provided with a data cut-off of March 31, 2021. For this report, data are reported only for the 32 patients with relapsed or refractory MCL who received 320 mg of zanubrutinib daily.
Figure 2: Study Schematic for Study 206
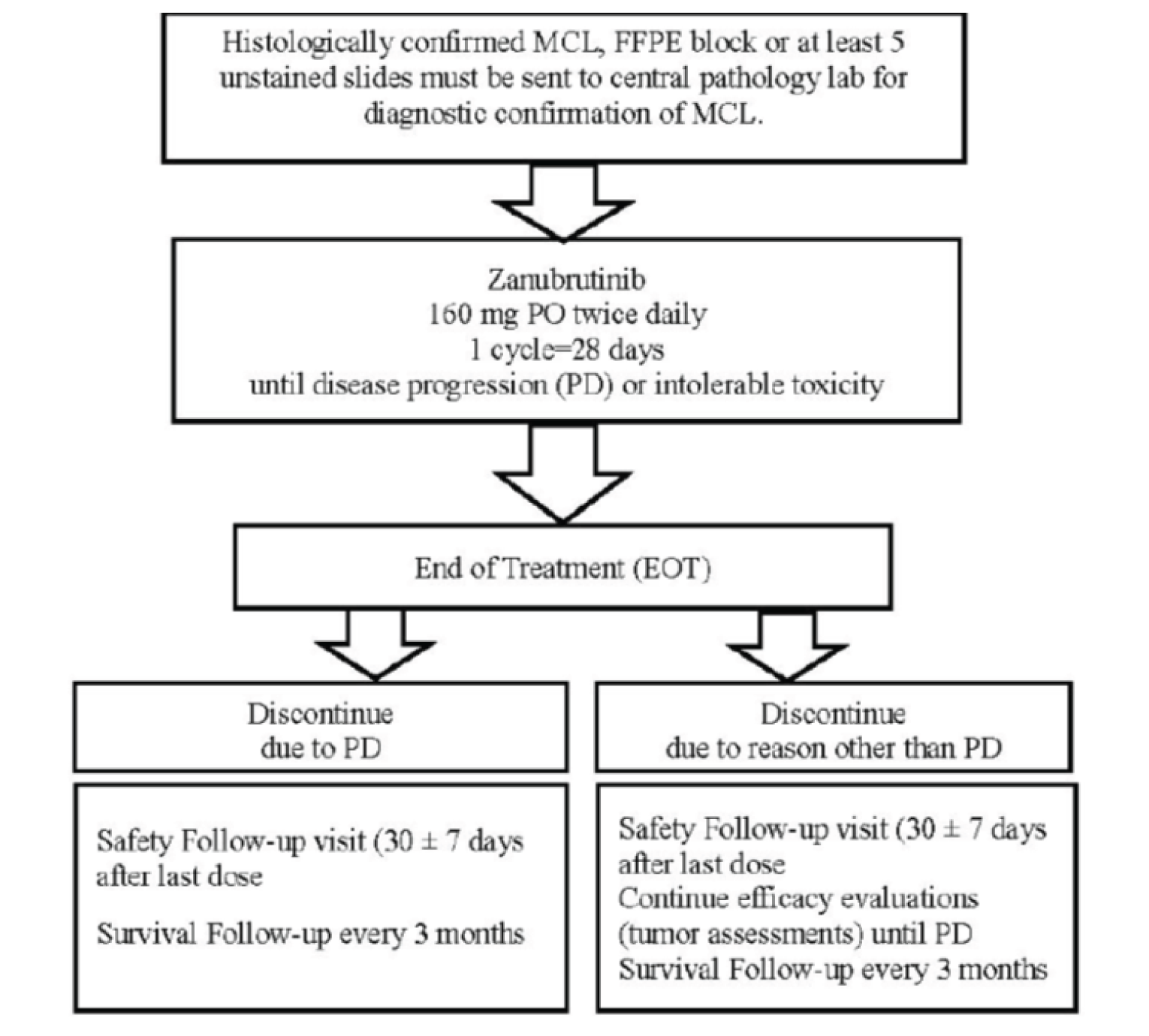
FFPE = formalin-fixed paraffin embedded; MCL = mantle cell lymphoma; PD = progressive disease; PO = orally.
Important protocol amendments to Study 206 included amendment 1 (January 5, 2017), which included the following:
added that the primary objective and end point would be evaluated using Lugano classification
modified the inclusion criteria for measurable disease by defining it as having at least 1 lymph node with a longest diameter greater than 1.5 cm
added electrocardiogram assessment and results requirements to the exclusion criteria
added the requirement for FDG-PET scans for efficacy assessments for patients with FDG-avid disease and IRC assessment for efficacy analysis.
Amendment 2 (October 25, 2017) including the following:
clarified that the primary efficacy objectives or end points would be assessed by an IRC
added ORR assessments by study site investigators as a secondary objective or end point
added exclusion criteria 16 (patient has received allogeneic hematopoietic stem cell transplant before enrolment)
clarified that patients with documented GI tumour involvement at screening should have CR confirmed with endoscopy
clarified that cytology and immunohistochemistry must have been performed for bone marrow aspirates and biopsies.
Amendment 3 (September 6, 2018) clarified the response assessment schedule:
Patients with FDG-avid disease at screening would have a PET and contrast-enhanced CT repeated every 12 weeks for the first 96 weeks and every 24 weeks thereafter until PD or end of study, whichever comes first.
Patients with non–FDG avid disease at screening would have only contrast-enhanced CT performed every 12 weeks for the first 96 weeks and every 24 weeks thereafter until PD or end of study, whichever came first.
Important protocol amendments to Study 003 included the following:
amendment 2, which expanded cohort 2a from 20 to 40 patients
amendment 3, which added an exclusion criterion for patients with corrected QT (QTc) prolongation and added follow-up assessments for progression and survival
amendment 4, which added cohort 2g (20 patients with relapsed or refractory MCL)
amendment 6, which extended the time patients could receive zanubrutinib from 1 year to until PD, added response assessments beyond week 52 to be every 6 months instead of only as clinically indicated, and added a survival follow-up assessment
amendment 7, which clarified the process for evaluation of response and progression by IRC for MCL and added other clarifications for response criteria (imaging for response was to be conducted every 12 weeks from week 64 and every 24 weeks thereafter from week 100 or when a significant change in response was suspected).
Populations
Inclusion and Exclusion Criteria
Men and women aged 18 to 75 years with an ECOG PS of 0 to 2 were enrolled into Study 206. Patients were to have MCL confirmed by immunohistochemistry performed at a central laboratory and have measurable disease as assessed by CT or MRI, defined as having at least 1 lymph node with a longest diameter greater than 1.5 cm and measurable in 2 perpendicular dimensions. Patients were to have received between 1 and 4 prior treatment regimens for MCL and had documented failure to achieve any response (stable disease or PD during treatment) or documented PD after response to the most recent treatment regimen. Patients with current or a history of central nervous system lymphoma were excluded, as were those with prior exposure to a BTK inhibitor and prior treatment with corticosteroids in excess of prednisone 10 mg per day (or equivalent) with neoplastic intent. Study 003 enrolled patients 18 years of age or older with relapsed or refractory disease with a B-lymphoid malignancy following at least 1 previous line of therapy and an ECOG PS similar to Study 206 criteria.
Baseline Characteristics
Patients in Study 206 were slightly younger than patients in Study 003 (mean age of 59.0 ± a standard deviation of 8.18 years versus age 69.7 ± 10.33 years), and there was a higher proportion of males in Study 206 compared with Study 003 (78% versus 69%) (Table 7). All patients in Study 206 were Chinese, while the majority of patients in Study 003 were White (78%). The majority of patients in Study 206 had an ECOG PS of 0 (70%) while, in Study 003, a similar number of patients had an ECOG PS of 0 (47%) or 1 (44%). The majority of patients had stage IV disease in Study 206 (74%) and Study 003 (88%). In Study 206, a similar number of patients had relapsed disease (48%) versus refractory (52%) while, in Study 003, more patients had relapsed disease (70%) than refractory disease (25%). The majority of patients in Study 206 (71%) had had 2 or more prior lines of therapy while, in Study 003, 41% had 2 or more prior lines.
Table 7: Summary of Baseline Characteristics (ITT Population)
Characteristic | Study 206 Zanubrutinib (N = 86) | Study 003 Zanubrutinib (N = 32) |
|---|---|---|
Mean (SD) age | 59.0 (8.18) | 69.7 (10.33) |
Median (range) age | 60.5 (34 to 75) | 70.5 (42 to 86) |
< 65 years | 64 (74.4) | 8 (25.0) |
≥ 65 years | 22 (25.6) | 24 (75.0) |
Male, n (%) | 67 (77.9) | 22 (68.8) |
Race, n (%) | ||
Chinese | 86 (100) | 3 (9.4) |
White | 0 | 25 (78.1) |
Black or African American | 0 | 1 (3.1) |
Other or not reported | 0 | 3 (9.4) |
ECOG PS, n (%) | ||
0 | 60 (69.8) | 15 (46.9) |
1 | 22 (25.6) | 14 (43.8) |
2 | 4 (4.7) | 3 (9.4) |
Time since first diagnosis of MCL, months, mean (SD) | 35.96 (24.264) | 59.80 (44.20) |
Bulky disease, n (%) | ||
Yes (any target lesion LDi > 10 cm) | 7 (8.1) | 3 (9.4) |
No (all target lesion LDi ≤ 10 cm) | 79 (91.9) | 29 (90.6) |
Stage at study entry for MCL, n (%) | ||
I | 1 (1.2) | 2 (6.3) |
II | 7 (8.1) | 1 (3.1) |
III | 14 (16.3) | 1 (3.1) |
IV | 64 (74.4) | 28 (87.5) |
Blastoid form, n (%) | ||
Yes | 12 (14.0) | 2 (6.3) |
No | 68 (79.1) | 28 (87.5) |
Unknown/missing | 6 (7.0) | 2 (6.3) |
Ki67-positive cell percentage, mean (SD) | 35.4 (18.22) | NR |
≤ 30% | 50 (58.1) | NR |
> 30% | 34 (39.5) | NR |
Missing | 2 (2.3) | NR |
MIPI-ba or MIPI, n (%) | ||
Low risk | 12 (14.0) | 9 (28.1) |
Medium risk | 39 (45.3) | 13 (40.6) |
High risk | 33 (38.4) | 10 (31.3) |
Missing | 2 (2.3) | 0 |
Disease status,b n (%) | ||
Relapsed | 41 (47.7) | 22 (68.8) |
Refractory | 45 (52.3) | 8 (25.0) |
Not evaluable | 0 | 2 (6.3) |
Patients with any prior anti-cancer therapy, n (%) | 86 (100) | 32 (100) |
Number of prior systemic therapies | Mean = 2.2 (SD = 0.98) | Median = 1.0 (Range = 1 to 4) |
Number of prior systemic therapies or anti-cancer therapies, n (%) | ||
1 | 25 (29.1) | 19 (59.4) |
2 | 32 (37.2) | 4 (12.5) |
3 | 19 (22.1) | 7 (21.9) |
4 | 10 (11.6) | 2 (6.3) |
Prior radiotherapy, n (%) | ||
Yes | 8 (9.3) | 9 (28.1) |
No | 78 (90.7) | 23 (71.9) |
Time from end of last therapy to first dose of the study drug, months, mean (SD) | 13.13 (17.992) | NR |
ASCT = autologous stem cell transplant; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITT = intention to treat; LDi = longest transverse diameter of a lesion; MCL = mantle cell lymphoma; MIPI = Mantle Cell Lymphoma International Prognostic Index; MIPI-b = Combined Biologic Mantle Cell Lymphoma International Prognostic Index; NR = not reported; SD = standard deviation.
aMIPI-b score was calculated with the following risk cut-offs: low (< 5.7), medium (≥ 5.7 and < 6.5), and high (≥ 6.5).
bDisease status defined by investigator.
Source: Clinical Study Report for Study 206 and Study 003.3-6
Interventions
In Study 206, patients self-administered zanubrutinib at a dose of 160 mg twice daily. If a dose was not taken within the time window (every 12 hours ± 2 hours) it was taken as soon as possible but not less than 4 hours before the next scheduled dose. Dose modifications of zanubrutinib were allowed, following specific protocols. The study drug could have been held for a maximum of 28 consecutive days.
Study 003 began with a dose-escalation phase, based on observations of dose-limiting toxicities within cohorts. The period for the dose-limiting toxicity assessment was 21 days from the first dose of zanubrutinib. A cohort of 3 to 6 patients had to be evaluated before determining the dose and regimen for the subsequent cohort of patients. If more than 1 patient in a cohort experienced a dose-limiting toxicity, then no additional patients were enrolled in that cohort and the maximum tolerated dose was considered to have been exceeded. If the maximum tolerated dose was exceeded, the next-lower dose was to be used in part 2. If 1 patient or no patients in a cohort experienced a dose-limiting toxicity, then the dose in the next cohort was to be increased by up to 100%, as determined by the safety monitoring committee. A dose-limiting toxicity was defined as any event that occurred during the first 21 days after the first dose of zanubrutinib that was not due to disease progression, underlying or concurrent illness, or concomitant medication, and that met any of the following criteria:
grade 4 neutropenia lasting for more than 7 days (while receiving growth factor support), or grade 3 or higher neutropenia with fever
grade 4 thrombocytopenia or grade 3 or higher thrombocytopenia associated with bleeding
any grade 2 or higher toxicity requiring either dose modification of zanubrutinib, or a delay of treatment for 1 week
any other non-hematologic grade 3 or higher event (excluding asymptomatic biochemical abnormalities that were not clinically significant and resolved to grade 2 or less in less than 7 days)
toxicity of any grade that, in the judgment of the investigator or sponsor, required removal of the patient from the study.
Zanubrutinib could be re-initiated in patients experiencing a dose-limiting toxicity if the toxicity improved to grade 1 or lower within 14 days and interruption or the delay of treatment was 21 days or less. When treatment resumed, it was to be at the next-lower dose level tested (or 50% lower if the dose-limiting toxicity occurred at the first dose level).
In Study 206, prohibited concomitant therapies included any anti-cancer therapy, including but not restricted to chemotherapy, immunotherapy, corticosteroids (> 10 mg of prednisone or equivalent daily), experimental therapy, radiotherapy, and Chinese herbal medications. Other anti-cancer therapies were also prohibited in Study 003. In Study 206, concurrent use of drugs that prolong the QTc interval was also prohibited; if patients were required to take a QTc-prolonging drug, their zanubrutinib was held until completion of the QTc-prolonging drug(s), plus 5 half-lives. In Study 003, use of drugs that prolong the QT interval or QTc were to be avoided unless no alternative was available. Zanubrutinib is a cytochrome P (CYP) 3A substrate; therefore, strong inducers or inhibitors of CYP 3A were avoided in both studies.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized subsequently. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 3.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study 206 | Study 003 |
|---|---|---|
Overall survival | Exploratory | Secondary |
Progression-free survival | Secondary | Secondary |
Health-related quality of life | Not investigated | Not investigated |
Objective response | Primary | Secondary |
Duration of response | Secondary | Secondary |
Time to next treatment | Not investigated | Not investigated |
AE | Safety | Safety |
SAE | Safety | Safety |
WDAE | Safety | Safety |
Notable harms | Safety | Safety |
AE = adverse event; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Source: Clinical Study Report for Study 206 and Study 003.3-6
Overall Survival
OS was an exploratory outcome in Study 206, defined as the time from the first dose of the study medication to death due to any cause. Patients who were alive before the data cut-off date or before discontinuation from the study (i.e., those who discontinued the study for reasons other than death) were censored at the time of data cut-off or the last date the patient was known to be alive. Efficacy was not an objective of Study 003; however, OS was assessed. The definition of OS was not described.
Progression-Free Survival
In Study 206, PFS was a secondary outcome and was defined as the time from the date of the first dose of the study drug until documented PD or death from any cause, whichever occurred first, using IRC assessment. Investigator-assessed PFS was performed as a sensitivity analysis. Censoring rules for PFS were as follows:
Patients with no baseline or post-baseline disease assessments were censored at the date of first dose. Patients with progression documented on scheduled visit or between scheduled visits were defined as progressed on the date of first disease assessment showing documented disease progression.
Patients who were alive without documented disease progression at the time of data cut-off or withdrawal from study (including lost to follow-up without disease progression) were censored at the date of last radiographic disease assessment.
Patients with new anti-cancer treatment started before documented disease progression or death were censored at the date of last radiographic disease assessment before or on date of new anti-cancer treatment.
Patients who died before first disease assessment were defined as progressed at the date of death.
Patients who died or progressed after more than 1 missed scheduled disease assessment were censored at the date of last disease assessment without documented disease progression before missed tumour assessments
Efficacy was not an objective of Study 003; however, PFS was assessed. The definition of PFS was defined as the time from the first dose of the study drug until death or PD.
Overall Response Rate
The primary outcome of Study 206 was ORR, which was defined as patients achieving a partial response or CR as determined by IRC, in accordance with the Lugano classification. The best overall response was defined as the best response recorded from the start of study drug until the data cut-off or start of new anti-cancer treatment. Patients with no post-baseline disease assessment (for any reason) were considered nonresponders for ORR.
In Study 206, radiographic tumour assessments were performed at screening, every 12 weeks for the first 96 weeks of the study, and every 24 weeks thereafter and for confirmation of CR. If PD was suspected based on clinical grounds, prompt imaging studies and physical exams were conducted for confirmation. Assessments included contrast-enhanced CT or MRI scans of the neck, chest, abdomen, and pelvis as well as other disease sites in all patients, as well as FDG-PET scans. Imaging of the brain was only indicated if clinical signs or symptoms suggested central nervous system involvement. A contrast-enhanced CT scan of diagnostic quality performed as part of a combined PET and CT was acceptable as long as bi-dimensional nodal and liver and spleen measurements could have been made and the study adhered to specified slice thicknesses and scan parameters. An MRI could be used in place of CT only for anatomic lesions that could not be adequately visualized by CT or when the patient could not undergo a CT scan. Unilateral bone marrow aspiration and biopsy were required during the screening evaluation or within 60 days of the first dose of the study drug as long as there had been no intervening therapy between the time of the procedure and the start of the study drug. A bone marrow biopsy was required for confirmation of CR in patients with bone marrow tumour involvement at baseline, per the Lugano classification. An endoscopy was required to confirm CR for any patient with a documented history of GI involvement.
In Study 003, ORR was a secondary outcome, although no analysis was planned. Imaging was again used for assessment, using CT or MRI and/or PET, and was assessed by 2 independent reviewers. For periodic tumour assessment, CT with contrast was performed at baseline, every 12 weeks during treatment, and at disease progression. Starting with protocol version 6, CT scans were performed every 6 months after 52 weeks of treatment with zanubrutinib, where previously they were performed only as clinically indicated.
Duration of Response
In Study 206, DOR was defined as the interval between the date of the earliest qualifying response (CR or partial response) and the date of PD or death from any cause (whichever occurred earlier). Censoring rules were the same as those for PFS. The censored date for responders without PD or death was based on the last adequate disease assessment that included imaging studies (unless censoring occurred due to other reasons). Efficacy was not an objective of Study 003; however, DOR was assessed. The definition of DOR was not described.
Harms
In Study 206, AEs and SAEs were reported from initiation of therapy until 30 days after the last dose of zanubrutinib. After this period, only SAEs that were believed to be related to the study drug were reported. Safety was a primary end point of Study 003 and was assessed by monitoring AEs using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03, SAEs, a physical exam, and laboratory values.
Statistical Analysis
Primary Outcome(s) of the Studies
Power Calculation
In Study 206, the calculation for sample size was based primarily on the desired level of precision for the ORR estimate. The sponsor assumed an ORR of 70%, compared with a historical control of 40%, and this resulted in approximately 80 patients being needed to demonstrate statistical significance at a 1-sided alpha of 0.025 with power greater than 0.99 using a binomial exact test. For Study 003, the sample sizes for part 1 were initially estimated to be 12 patients to establish the dose and regimen for zanubrutinib, but this was expanded to 25. In part 2, sample sizes for individual disease cohorts were “based on obtaining rigorous descriptions of the safety profile and estimates of the response rates for zanubrutinib in specific B-cell malignancies that have sufficient precision.”
For Study 003, sample sizes for part 2 were based on estimates of response rates for zanubrutinib as well as the safety profile and were performed for each cohort. Examples of how sample sizes were calculated for some cohorts were provided but not for the relapsed or refractory MCL cohort.
Statistical Test or Model
In Study 206, for the primary outcome of ORR, the sponsor performed a binomial exact test to test the null hypothesis (H0) (ORR = 0.40) using a 1-sided significance level of 0.025. The best ORR was defined as the best response recorded from the start of the study drug until the data cut-off or the start of a new anti-cancer treatment. The safety analysis set was the basis for all efficacy and safety analyses provided in the Study 206 Clinical Study Report.
There was no formal hypothesis testing in Study 003.
Data Imputation Methods
In Study 206, patients with no post-baseline response assessments for ORR were considered nonresponders. No data imputation methods were described for Study 003.
Subgroup Analyses
Subgroup data for ORR were presented; however, no statistical analysis was planned in Study 206. The subgroups that were reported that were relevant to our protocol included ECOG PS (0 versus ≥ 1), number of lines of prior therapy for MCL (< 3 versus 3), blastoid histology (yes versus no), and prior anti-cancer drug use. No a priori subgroup analyses were planned for the relapsed refractory subgroup in Study 003.
Sensitivity Analyses
In Study 206, sensitivity analyses consisted of performing an investigator assessment of PFS to support the IRC assessment. There was no formal hypothesis testing in Study 003, and therefore no sensitivity analyses.
Secondary Outcomes of the Studies
In Study 206, median and interquartile ranges for PFS, DOR, and OS were estimated using Kaplan–Meier methodology and 2-sided 95% CIs constructed using the Brookmeyer and Crowley method with log-log transformation. PFS rates at selected landmark time points (6 months, for example) were determined with corresponding 95% CIs using the Greenwood formula, with log-log transformation.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
Study 206 | |||
ORR | Binomial exact test | None described | Investigator assessment of PFS to support the IRC assessment |
Study 003 | |||
No formal hypothesis testing | NA | NA | NA |
IRC = independent review committee; NA = not applicable; ORR = overall response rate; PFS = progression-free survival.
Analysis Populations
The safety analysis set in both studies comprised all patients who received any dose of the study drug, In Study 206, the revised safety analysis set included all patients with pathologically confirmed MCL among the safety analysis population. The per-protocol analysis set defined in Study 206 included all patients who received any dose of the study drug and had no major protocol deviations. Study 003 also defined a dose-limiting toxicity analysis set, which included patients who received treatment with zanubrutinib for 21 days or more in part 1. Patients who had a dose-limiting toxicity event during the dose-limiting toxicity assessment window despite receiving less than 21 days of zanubrutinib were also considered evaluable for dose-limiting toxicity. Study 003 also defined an efficacy evaluable analysis set, which included all MCL patients first dosed 12 weeks or more before the data cut-off date.
Results
Patient Disposition
A summary of patient disposition is provided in Table 10. In Study 206 at the final analysis, 24.4% of patients had died, 15.1% had withdrawn from the study, a further 3.5% were lost to follow-up, and the remainder were in the study when it was terminated by the sponsor. Disposition data were not reported in the final analysis for patients with relapsed or refractory MCL receiving 320 mg daily; therefore, data are reported for the interim analysis in Table 10.
In Study 206, there were 7 patients with at least 1 major protocol deviation and 4 patients had received prohibited medications. There was 1 instance each of accidental overdose, treatment interruption not carried out per protocol, and non-compliance with good clinical practice. In Study 003, protocol deviations were not reported for the specific MCL population of interest for this review. For the wider MCL population (N = 57), protocol deviations occurred due to AEs or SAEs, prohibited medications, and informed consent.
Detail | Study 206 Zanubrutinib N = 86 | Study 003 Zanubrutinib N = 32 |
|---|---|---|
Screened, N | NR | NR |
Enrolled, N (%) | 86 (100) | 32 (100) |
Discontinued from the study drug, N (%) | 86 (100) | 19 (59.4) |
Reason for discontinuation, N (%) | ||
Study terminated by the sponsor | 39 (45.3) | — |
Progressive disease | 37 (43.0) | 10 (31.3) |
Adverse event | 8 (9.3) | 9 (28.1) |
Investigator’s discretion | 1 (1.2) | 0 |
Withdrawal by patient | 1 (1.2) | 0 |
Patients remaining on study treatment, n (%) | 0 | 14 (43.8) |
Patients discontinued from the study, n (%) | 86 (100) | 15 (46.9) |
Reason for discontinuation from the study N (%) | ||
Study terminated by sponsor | 49 (57.0) | — |
Death | 21 (24.4) | 10 (31.3) |
Withdrawal by patient | 13 (15.1) | 0 |
Lost to follow-up | 3 (3.5) | 0 |
Adverse event | 0 | 3 (9.4) |
Other | 0 | 2 (6.3) |
Patients remaining in study, n (%) | 0 | 17 (53.1) |
Patients requiring a dose reduction, n (%) | 2 (2.3) | 6 (18.8) |
Reason: Adverse event | 2 (2.3) | 4 (12.5) |
Patients requiring a dose interruption, n (%) | 24 (27.9) | NR |
Reason | ||
Adverse event | 16 (18.6) | 20 (62.5) |
Other | 10 (11.6) | NR |
Number of dose interruptions per patient, n (%) | ||
1 | 7 (8.1) | NR |
2 | 9 (10.5) | NR |
3 | 5 (5.8) | NR |
4 | 2 (2.3) | NR |
6 | 1 (1.2) | NR |
Patients with missed doses, n (%) | 42 (48.8) | NR |
Reason: Other | 42 (48.8) | NR |
Safety analysis set | 86 (100) | 32 (100) |
Revised safety analysis set | 85 (98.8) | NR |
Per-protocol analysis set | 79 (91.9) | 32 (100) |
NR = not reported.
Source: Clinical Study Reports for Study 206 and Study 003.3-6
Exposure to Study Treatments
The median duration of exposure to zanubrutinib was 27.61 months (range, 0.2 to 41.6 months) in the final analysis for Study 206, with a mean relative dose intensity of 98.34% (standard deviation of 6.829) achieving 320 mg a day. For the final analysis in Study 003, the median duration of exposure was 18.35 months (range, 0.4 to 56.3), and the mean relative dose intensity was 91.04% (standard deviation of 14.661) achieving 320 mg a day.
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported subsequently. See Appendix 3 for detailed efficacy data.
Overall Survival
In Study 206, by the time of the cut-off date for the interim analysis and a median follow-up of 24.9 months (range, 0.3 to 30.0 months), 17 (19.8%) of patients had died, and the median OS was not yet estimable (Table 11). A total of 81.7% (95% CI, 71.5 to 88.6) were alive at 18 months, and 80.4% (95% CI, 69.9 to 87.5) were alive at 21 and 24 months, respectively. By the time of the final Clinical Study Report, with a median follow-up of 36.8 months (range, 0.3 to 41.6), the median OS was still NE. At 30 months, 77.6% of patients were alive (95% CI, 66.8 to 85.3) and, at 36 months, 74.8% of patients were alive (95% CI, 63.7 to 83.0). The Kaplan–Meier analysis of OS is provided in Figure 3.
In Study 003, in the final analysis, after a median follow-up of 45.8 months (95% CI, 42.0 to 48.6), the median OS was NE.
Figure 3: Kaplan–Meier Plot of Overall Survival
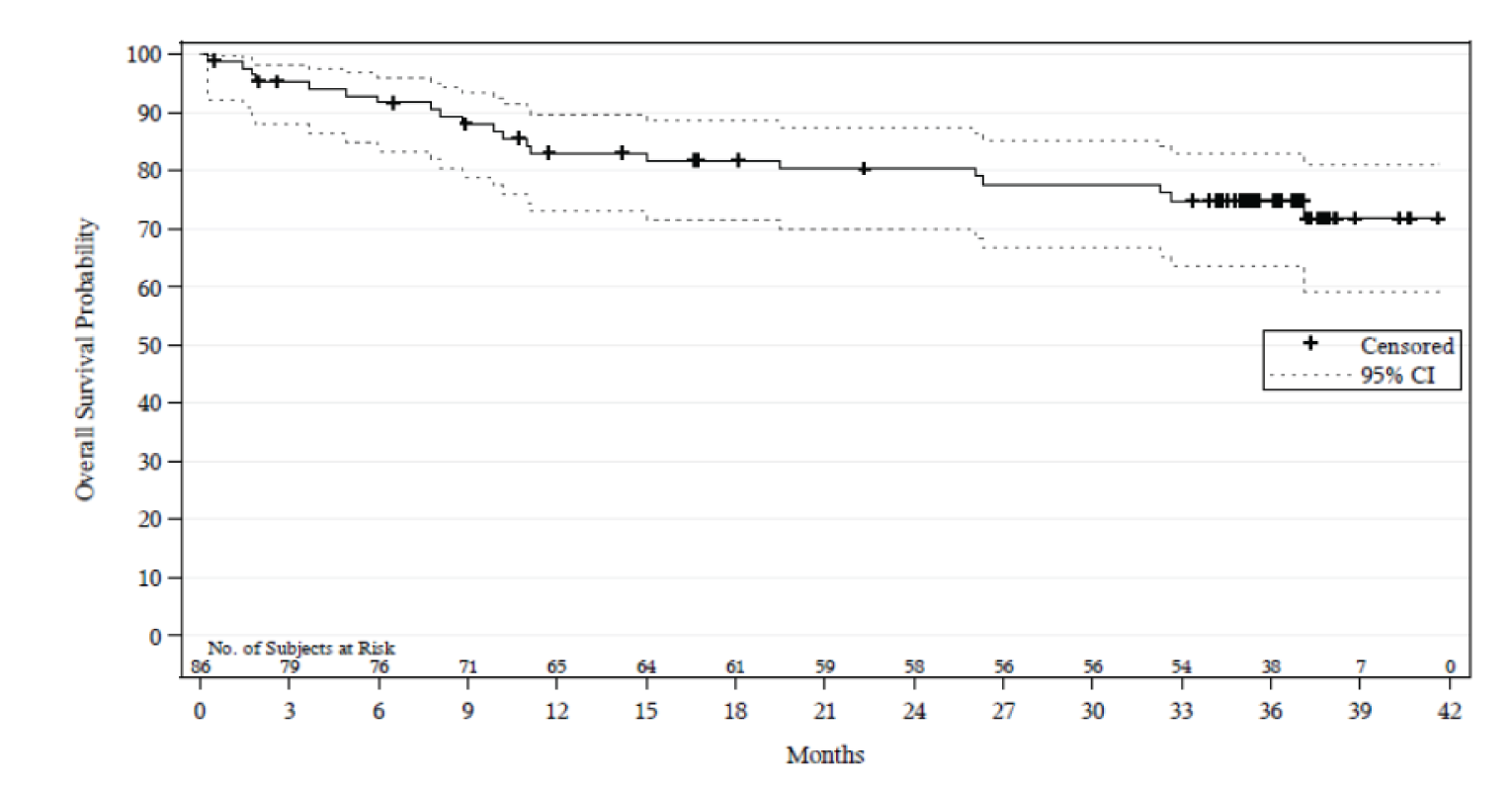
CI = confidence interval.
Source: Clinical Study Report for Study 206.3
As of the final database lock date of May 03, 2021, there have been 14 deaths (43.8% of the patients).
Progression-Free Survival
In Study 206, after a median follow-up of 22 months (range: 0 to 27.6 months) at the interim analysis the median PFS was 27.5 months (95% CI, 19.4 to NE) (Table 11). The sponsor noted that due to the small number of patients at risk and the resulting wide confidence interval the median PFS estimate was unstable. In the final analysis, after a median follow-up of 33.3 months (range, 0.0 to 38.9 months), the median PFS was 33.0 months (95% CI, 19.4 to NE). The Kaplan–Meier analysis of PFS is provided in Figure 4.
In Study 003, after a median follow-up time for PFS of 40.0 months (95% CI, 28.3 to 45.1), the median PFS was reported as 21.1 months (95% CI, 13.2 to NE) in the final Clinical Study Report (Table 12).
Figure 4: Kaplan–Meier Analysis of PFS in Study 206 (Final Analysis)
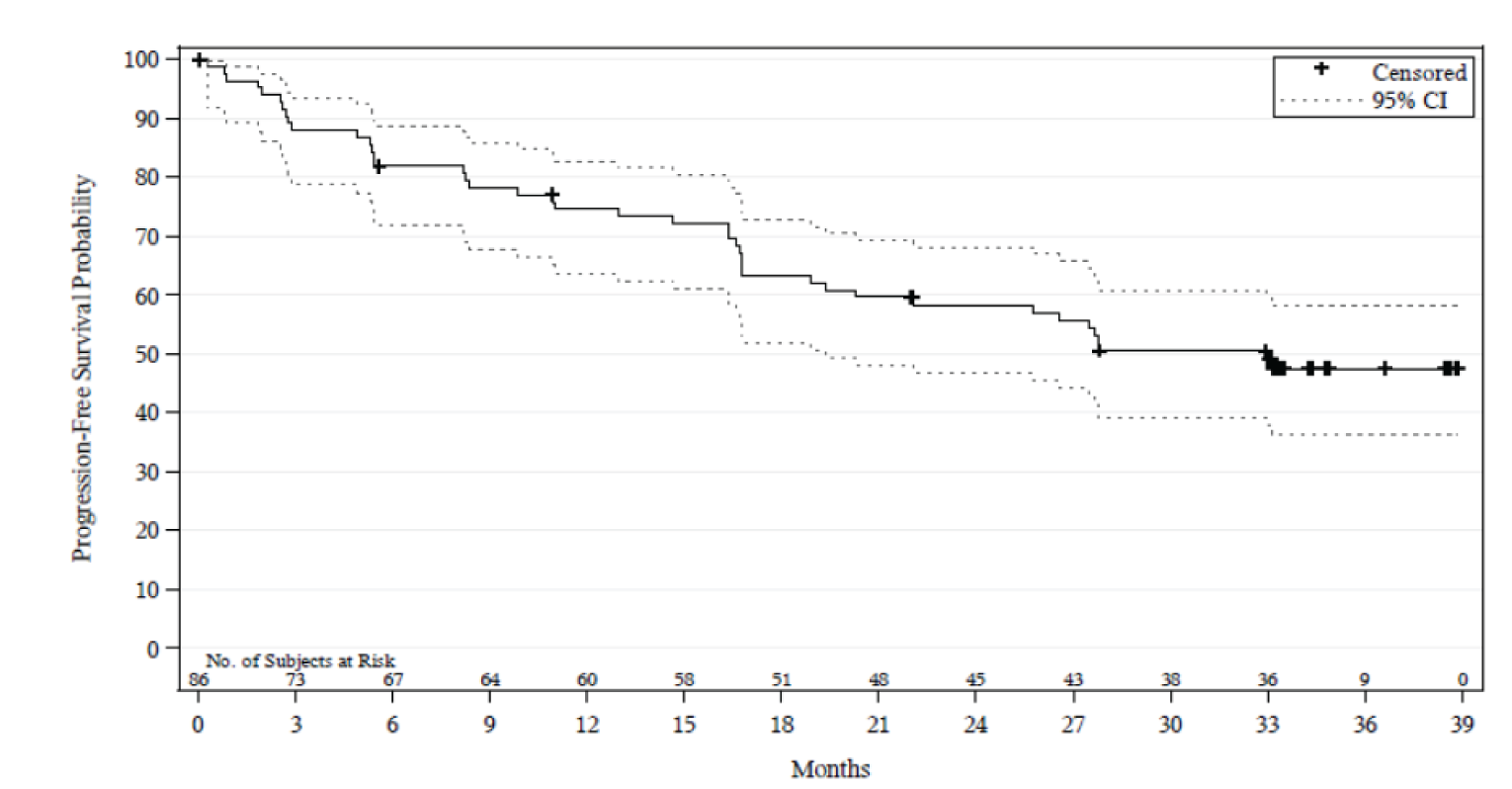
CI = confidence interval; PFS = progression-free survival.
Source: Clinical Study Report for Study 206.3
Health-Related Quality of Life
This outcome was not investigated.
Objective Response Rate
In Study 206, the ORR was 83.7% (95% CI, 74.2 to 90.8), which ruled out the pre-specified null hypothesis of 40% with a 1-sided P value of less than 0.0001. The CR rate was 77.9% (95% CI, 67.7 to 86.1) (Table 11). Subgroup analyses for ORR based on various parameters can be found in Table 23 (Appendix 3). There were no planned analyses of this data, and the results were presented descriptively. There were no clear differences in ORR based on subgroups such as ECOG PS, prior lines of therapy, or prior ASCT, although many of the subgroups contained fewer than 10 patients, thus limiting the ability to make valid and reliable comparisons.
In Study 003, the ORR at the final analysis was 90.6% (95% CI, 75.0 to 98.0) and the CR rate was 31.3% (95% CI, 16.1 to 50.0). No statistical analysis was planned (Table 11).
Detail | Study 206 Zanubrutinib N = 86 | Study 003 Zanubrutinib N = 32 |
|---|---|---|
OS | ||
Events, n (%) | 21 (24.4) | 14 (43.8) |
Censored, n (%) | 65 (75.6) | Alive: 18 (56.3) |
Not known to have died | 65 (75.6) | NR |
Median OS, months (range) | NE (NE to NE) | 27.20 (95% CI, 18.27 to 38.18)b |
Median OS, months (95% CI), final analysis Median follow-up, months (95% CI) | NE (95% CI, NE to NE) 36.8 (35.4 to 37.2) | NE (26.1 to NE) 45.8 (42.0 to 48.6) |
PFS | ||
Events, n (%) | 42 (48.8) | 18 (56.3) |
PD | 37 (43.0) | 15 (46.9) |
Death | 5 (5.8) | 3 (9.4) |
Censored | 44 (51.2) | 14 (43.8) |
No documented disease progression or death | 38 (44.2) | 13 (40.6) |
No baseline or post-baseline assessment | 3 (3.5) | 0 |
Withdrew consent or lost to follow-up | 2 (2.3) | 0 |
Non-protocol anti-cancer therapy | 1 (1.2) | NR |
Median PFS, months (95% CI) | 27.5 (19.4 to NE) | 21.1 (13.2 to NE) |
Median PFS, months, range | 0.0 to 38.9 | 0.7 to 56.3 |
Median PFS, months, (95% CI) final analysis Median follow-up, months (95% CI) | 33.0 (19.4 to NE) 33.3 (33.1 to 34.3) | 21.1 (13.2 to NE) 40.0 (28.3 to 45.1) |
ORR | ||
Best overall response, n (%) | ||
CR | 67 (77.9) | 10 (31.3) |
PR | 5 (5.8) | 19 (59.4) |
SD | 1 (1.2) | 1 (3.1) |
PD | 8 (9.3) | 2 (6.3) |
Discontinued before first assessment | 5 (5.8) | — |
Patients with an OR, n (%) | 72 (83.7) | 29 (90.6) |
ORR (95% CI) | 83.7 (74.2 to 90.8) | 90.6 (75.0 to 98.0) |
P value, 1-sideda | P < 0.0001 | NR |
Patients with a CR, n (%) | 67 (77.9) | 10 (31.3) |
CRR (95% CI) | 77.9 (67.7 to 86.1) | (16.1 to 50.0) |
DOR | ||
Responders, n (%) | 72 (83.7) | 29 (90.6) |
Events, n (%) | 32 (44.4) | 15 (51.7) |
PD | 29 (40.3) | 13 (44.8) |
Death | 3 (4.2) | 2 (6.9) |
Censored, n (%) | 40 (55.6) | 14 (48.3) |
No documented PD or death | 37 (51.4) | 13 (44.8) |
Withdrew consent or lost to follow-up PD or death after > 1 missed assessment | 2 (2.8) — | — 1 (3.4) |
Median DOR, months (95% CI) | 24.9 (23.1 to NE) | 18.53 (12.58 to NE) |
Median DOR, months (95% CI), final analysis Median follow-up | NE (24.9 to NE) 30.6 (95% CI, 30.4 to 31.5) | 25.2 (12.6 to NE) 36.9 (32.3 to 42.3) |
CI = confidence interval; CR = complete response; CRR = complete response rate; DOR = duration of response; NE = not estimable; NR = not reported; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; SD = stable disease.
aP value (1-sided) was based on the binomial exact test against the null hypothesis (H0): ORR = 0.40.
Source: Clinical Study Report for Study 206 and Study 003.3-6
Duration of Response
In Study 206, the median DOR in the 72 patients who achieved an ORR was 24.9 months (95% CI, 23.1 months to NE) (Table 11). The sponsor noted that because the median was reached with the last event occurring while only 3 patients were at risk, the median DOR estimate was “unstable.” At the final analysis, the median DOR was not reached. The Kaplan–Meier analysis for DOR is provided in Figure 5.
In Study 003, the median DOR at the final analysis was 25.2 months after a median follow-up of 36.9 months (95% CI, 32.3 to 42.3).
Figure 5: Kaplan–Meier Analysis of DOR in Study 206 (Final Analysis)
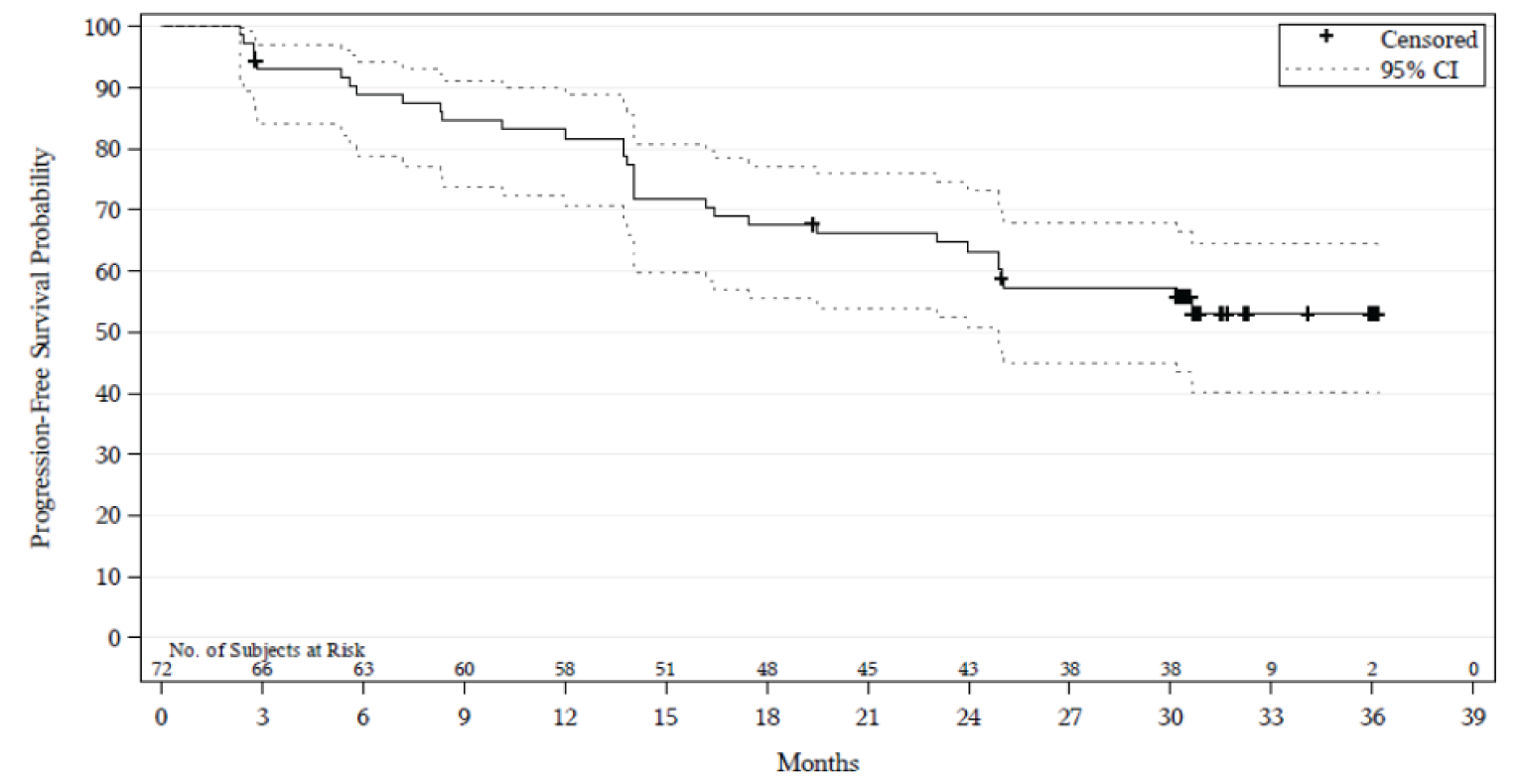
CI = confidence interval; DOR = duration of response.
Source: Clinical Study Report for Study 206.3
Harms
Only those harms identified in the review protocol are reported subsequently. Refer to Table 12 for detailed harms data.
Adverse Events
AEs were reported in 97% of patients in both Study 206 and Study 003; 50% of patients in Study 206 reported an AE of grade 3 or higher compared with 69% of patients in Study 003 (Table 12). The most common AEs in Study 206 were decreased neutrophil count (47% of patients), upper respiratory tract infections (38%), rash (35%), decreased white blood cell count (34%), and decreased platelet count (33%), and the most common grade 3 or higher AEs were decreased neutrophil count (19%), lung infection (9%), and decreased platelet count and white blood cell count (7.0% each). The most common AEs in Study 003 were diarrhea (47%), constipation (41%), and rash (34% of patients), and the most common grade 3 or higher AEs were anemia (12.5%) and pneumonia (12.5%).
Serious Adverse Events
SAEs occurred in 29% of patients in Study 206 and 59% of patients in Study 003, with the most common SAE being pneumonia (12% in Study 206, 12.5% in Study 003) (Table 12).
Withdrawals Due to Adverse Events
In Study 206, 9% of patients had at least 1 AE leading to discontinuation of the study drug, and pneumonia was the most common event, occurring in 2% of patients (Table 12). In Study 003, 28% of patients had at least 1 AE leading to discontinuation of the study drug and, in 6% of patients, this was pneumonia.
Mortality
In Study 206, 24% of patients died, 9% within 30 days of their last dose of the study drug and 15% more than 30 days after the last dose of the study drug (Table 12). Among the patients who died within 30 days of their last dose, most (7% overall) died due to an AE, while the remaining deaths were due to PD. For those deaths that occurred more than 30 days after the last dose of the study drug, most (12% overall) were due to PD, while the remaining 3 deaths were due to AE and “other.” In Study 003, 38% of patients died, 16% within 30 days of their last dose of the study drug ((9% due to an AE) and 22% more than 30 days after their last dose of the study drug (16% due to PD).
In Study 206, 24% of patients died: 9% died within 30 days of their last dose of the study drug and 15% died more than 30 days after their last dose of the study drug (Table 12). Among the patients who died within 30 days of their last dose, most (7% overall) died due to an AE, while the remaining deaths were due to PD. For those deaths that occurred more than 30 days after the patient’s last dose of the study drug, most (12% overall) were due to PD, while the remaining 3 deaths were due to AE and “other.” In Study 003, 38% of patients died, 16% within 30 days of their last dose of the study drug (9% due to an AE), while 22% died more than 30 days after their last dose of the study drug (16% due to PD).
Notable Harms
Notable harms in Study 206 included infections (64% of patients; 16% grade ≥ 3), thrombocytopenia (37%; 7% grade ≥ 3), hemorrhage (30%; 1% grade ≥ 3), and anemia (16%; 6% grade ≥ 3) (Table 12). The most common infections were upper respiratory tract infection (36%; 0 grade ≥ 3), lung infection (12%; 9% grade ≥ 3), and urinary tract infection (12%; 1% grade ≥ 3). The most common hemorrhage events were blood urine present (8%; 0 grade ≥ 3) and hematuria (6%; 0 grade ≥ 3). In Study 003, hemorrhage occurred in 62.5% of patients, the most common events being contusion (44% of patients) and hematuria (16%), and infections occurred in 72% of patients.
Table 12: Summary of Harms (From Final Analysis Unless Otherwise Indicated)
Detail | Study 206 | Study 003 | ||
|---|---|---|---|---|
Zanubrutinib N = 86 all AEs, n (%) | Grade ≥ 3 AEs, n (%) | Zanubrutinib N = 32 all AEs, n (%) | Grade ≥ 3 AE, n (%) | |
Patients with ≥ 1 adverse event | ||||
n (%) | 83 (97) | — | 31 (97) | — |
Grade 3 or higher, n (%) | 43 (50) | Grade ≥ 3 | 22 (69) | Grade ≥ 3 |
Most common events,a n (%) | — | — | — | — |
Investigations | ||||
Neutrophil count decreased | 40 (47) | 16 (19) | 2 (6) | 2 (6) |
White blood cell count decreased | 29 (34) | 6 (7) | 0 | 0 |
Platelet count decreased | 28 (33) | 6 (7) | 4 (12.5) | 2 (6) |
ALT increased | 16 (19) | 1 (1) | 0 | 0 |
AST increased | 9 (11) | 0 | 0 | 0 |
Blood urine present | 11 (13) | 0 | 0 | 0 |
Weight increased | 7 (8) | 1 (1) | 0 | 0 |
Blood creatinine increased | 8 (9) | 0 | 2 (6) | 0 |
Lymphocyte count decreased | 5 (6) | 2 (2) | 0 | 0 |
Skin and subcutaneous tissue | ||||
Rash | 30 (35) | 0 | 11 (34) | 0 |
Metabolism and nutrition | ||||
Hypokalemia | 15 (17) | 1 (1) | 3 (9) | 1 (3) |
Hyperuricemia | 12 (14) | 3 (4) | NR | NR |
Hyperglycemia | 12 (14) | 1 (1) | NR | NR |
Tumour lysis syndrome | NR | NR | 2 (6) | 2 (6) |
GI disorders | — | — | — | — |
Diarrhea | 14 (16) | 0 | 15 (47) | 1 (3) |
Constipation | 6 (7) | 0 | 13 (41) | 1 (3) |
Toothache | 6 (7) | 0 | 0 | 0 |
Nausea | NR | NR | 5 (16) | 1 (3) |
Blood and lymphatic | ||||
Anemia | 15 (17) | 5 (6) | 4 (12.5) | 4 (12.5) |
Neutropenia | 7 (8) | 1 (1) | 3 (9) | 2 (6) |
Thrombocytopenia | 8 (9) | 0 | 1 (3) | 1 (3) |
Leukopenia | 7 (8) | 1 (1) | 0 | 0 |
General disorders | ||||
Pyrexia | 7 (8) | 0 | 1 (3) | 0 |
Peripheral swelling or edema | 3 (3) | 2 (2) | 7 (22) | 2 (6) |
Fatigue | 1 (1) | 0 | 8 (25) | 2 (6) |
Respiratory, thoracic, mediastinal | ||||
Cough | 10 (12) | 0 | 6 (19) | 0 |
Dyspnea | NR | NR | 8 (25) | 1 (3) |
Pleural effusion | NR | NR | 3 (9) | 1 (3) |
Vascular | — | — | — | — |
Hypertension | 13 (15) | 3 (4) | 1 (3) | 1 (3) |
Psychiatric | — | — | — | — |
Insomnia | 5 (6) | 0 | 0 | 0 |
Agitation | NR | NR | 2 (6) | 2 (6) |
Renal and urinary | — | — | — | — |
Hematuria | 6 (7) | 0 | 5 (16) | 0 |
Acute kidney injury | 1 (1) | 0 | 2 (6) | 2 (6) |
Injury, poisoning, procedural complication | ||||
Contusion | 2 (2) | 0 | 12 (37.5) | 0 |
MSK and connective tissue disorders | — | — | — | — |
Back pain | 1 (1) | 0 | 7 (22) | 1 (3) |
Arthralgia | 2 (2) | 0 | 6 (19) | 0 |
Muscle spasms | NR | NR | 5 (16) | 0 |
Myalgia | 1 (1) | 0 | 3 (9) | 3 (9) |
Nervous system disorders | — | — | — | — |
Headache | 4 (5) | 0 | 3 (9) | 0 |
Dizziness | 1 (1) | 0 | 5 (16) | 0 |
Neoplasms, benign, malignant, unspecified | ||||
Basal cell carcinoma | NR | NR | 4 (12.5) | 0 |
Patients with ≥ 1 SAE | ||||
n (%) | 25 (29) | NA | 19 (59) | NA |
Most common events (> 1 patient), n (%) | ||||
Pneumonia | 10 (12) | NA | 4 (12.5) | NA |
Platelet count decreased | 2 (2) | NA | 0 | NA |
Upper GI hemorrhage | 2 (2) | NA | 0 | NA |
Death | 2 (2) | NA | 0 | NA |
Anemia | 1 (1) | NA | 2 (6) | NA |
Patients who stopped treatment due to AEs | ||||
n (%) | 8 (9) | NR | 9 (28) | NR |
Most common events (> 1 patient), n (%) | ||||
Pneumonia | 2 (2) | NR | 2 (6)a | NR |
Deaths | ||||
n (%) | 21 (24) | NA | 12 (37.5)a | NA |
Within 30 days of last dose | 8 (9) | NA | 5 (16)a | NA |
Due to AE | 6 (7) | NA | 3 (9)a | NA |
Due to PD | 2 (2) | NA | 1 (3)a | NA |
Unknown | 0 | NA | 0a | NA |
Other | 0 | NA | 1 (3)a | NA |
Deaths > 30 days of last dose | 13 (15) | NA | 7 (22)a | NA |
Due to AE | 1 (1) | NA | 0a | NA |
Due to PD | 10 (12) | NA | 5 (16)a | NA |
Other reason | 2 (2) | NA | 1 (3)a | NA |
Unknown | 0 | NA | 1 (3)a | NA |
Notable harms | ||||
Infections or infestations, n (%) | 56 (65) | 16 (19) | 23 (72) | 8 (25.0) |
Upper respiratory tract infection | 33 (38) | 0 | 13 (41) | 0 |
Urinary tract infection | 10 (12) | 1 (1) | 5 (16) | 0 |
Nasopharyngitis | 5 (6) | 0 | 4 (12.5) | 0 |
Pneumonia | 14 (16) | 11 (13) | 5 (16) | 4 (12.5) |
Localized infection | NR | NR | 5 (16) | NR |
Cellulitis | NR | NR | 3 (9) | 2 (6) |
Hemorrhage | 31 (36) | 1 (1) | 20 (62.5) | 0 |
Most common (> 1 patient), n (%) | ||||
Blood urine present | 11 (13) | 0 | 0 | 0 |
Hematuria | 6 (7) | 0 | 5 (16) | 0 |
Epistaxis | 3 (4) | 0 | 3 (9) | 0 |
Hemorrhage subcutaneous | 3 (4) | 0 | 0 | 0 |
Upper GI hemorrhage | 3 (4) | 0 | 0 | 0 |
Contusion | 2 (2) | 0 | 14 (44) | 0 |
Ecchymosis | 2 (2) | 0 | 0 | 0 |
Hemoptysis | 2 (2) | 0 | 0 | 0 |
Purpura | 2 (2) | 0 | 0 | 0 |
Increased tendency to bruise | NR | NR | 2 (6) | 0 |
Major hemorrhagea | 3 (4) | 1 (1) | 3 (9) | 0 |
Atrial fibrillation or flutter | 0 | 0 | 2 (6) | 1 (3) |
Second primary malignancy | 0 | 0 | 7 (22) | 0 |
Squamous cell carcinoma, skin | 0 | 0 | 2 (6) | 0 |
Squamous cell carcinoma, head or neck | 0 | 0 | 1 (3) | 0 |
Skin cancer | 0 | 0 | 1 (3) | 0 |
Malignant melanoma | 0 | 0 | 1 (3) | 0 |
Second primary malignancy, skin cancers | 0 | 0 | 7 (22) | 0 |
Basal cell carcinoma | 0 | 0 | 4 (12.5) | 0 |
Squamous cell carcinoma, skin | 0 | 0 | 1 (3) | 0 |
Skin cancer | 0 | 0 | 1 (3) | 0 |
Malignant melanoma | 0 | 0 | 1 (3) | 0 |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate transaminase; GI = gastrointestinal; MSK = musculoskeletal; NA = not applicable; NR = not reported; PD = progressive disease; SAE = serious adverse event.
aMajor hemorrhage was defined as serious or ≥ grade 3 bleeding at any site or central nervous system bleeding of any grade. Events of major hemorrhage are also included in the counts for the other hemorrhage terms.
aInterim analysis; final analysis data not provided.
Source: Clinical Study Report for Study 206 and Study 003.3-6
Critical Appraisal
Internal Validity
Both study 206 and 003 are open-label, single-arm studies that did not include a control group. The lack of a control group increases the potential for bias in the estimate of treatment effect. Without a comparator, natural fluctuations in a patient’s disease and other unidentified prognostic factors cannot be accounted for; thus, there is a risk of overestimation of the impact of treatment. Assessment of patient-reported outcomes, such as AEs, also increases the risk of bias, as patients are aware of their assigned treatment; however, hard clinical outcomes, such as those assessed in the included studies, are likely to be impacted less (e.g., ORR).
The time-to-event analyses were appropriate, but the data are difficult to interpret in a single-arm trial without a comparator. Moreover, in both studies, the survival data would be considered immature, as the median survival was not achieved, and it is therefore unclear whether the survival curves would have maintained their current trends if there had been additional follow-up and events. Moreover, due to the small number of events and patients at risk, the 95% CI could not be estimated around many of the end points, further adding to the uncertainty of the data. Due to these limitations of the study design, no definitive conclusions can be drawn from the data regarding the efficacy of zanubrutinib relative to a comparator.
Formal statistical analysis was performed for only 1 outcome, ORR, in Study 206, and was not performed for any outcomes in Study 003. The pre-planned analysis was performed on interim data, although final analysis data were also provided in the final Clinical Study Report, and the ORR remained essentially unchanged. For this analysis, the sponsor used a historical control of 40% as a reference. The sponsor based this estimate on a study of bortezomib (where there was an ORR of 31%), an ORR of 28% for lenalidomide, and an ORR of 22% with temsirolimus, none of which are BTK inhibitors. It is not clear why the sponsor did not instead use data from studies of other BTK inhibitors for their historical control, as these would be the most appropriate comparators for zanubrutinib. The clinical experts agreed that other BTK inhibitors would be the most appropriate comparators for estimating a historical control, which would raise the ORR estimate to 65% or 70%. If the historical control of 40% used by the sponsor is indeed an underestimate, then this would potentially bias in favour of finding a statistically significant improvement for zanubrutinib where none existed. That said, the actual ORR from Study 206 was 84%, which was higher than the estimate used for the power calculation of 70%. Additionally, the population in Study 206 is younger than would typically be seen for this indication, and it is not clear whether the populations in the studies used in the historical controls were as young.
Another limitation of using a historical control is that there is no opportunity to ensure key baseline characteristics are balanced between the groups being compared, an important consideration that would normally be achieved through randomization. Aside from differences in populations, there may have been differences in the study design, including in the concomitant therapies patients received in the historical controls compared with the study patients. The studies for the non–BTK inhibitors cited by the sponsor were published between 2007 and 2013 and, given the rapid progress made in the management of relapsed or refractory MCL, it is possible that the background therapies that patients may have received in these earlier trials would differ from those received in Study 206. Thus, the magnitude of the benefit may not be as large with zanubrutinib if a more contemporary control group had been chosen as the baseline ORR.
Data for pre-specified subgroups were reported; however, no statistical analyses were planned. Furthermore, small sample sizes in these subgroups limited any conclusions that could be drawn from these data.
No definitions for OS, PFS, or DOR were provided in Study 003, making interpretation of the outcomes difficult. Study 003 was a phase I and II study and was never designed to assess clinical outcomes beyond toxicities. There were only 32 patients who met the Health Canada–approved indication and dosing; this was a small, difficult-to-assess subsample of all patients enrolled in this phase I and II study. In the final Clinical Study Report, there were a number of efficacy and harms outcomes that did not report data specifically for this cohort and, after a subsequent data request to the sponsor, there remains some harms outcomes for which we lack data for the final analysis.
For the primary and most secondary end points, an IRC was appropriately used in Study 206. However, in some secondary assessments, only investigator judgment was used to assess occurrence of the end point, which could lead to observer bias due to the open-label nature of the study. In general, the investigator assessments were in line with the IRC when both were completed on the same end point (e.g., PFS in Study 206).
A large proportion of patients (43%) discontinued from Study 206 and, although 24% of overall withdrawals were due to deaths, 15% of patients withdrew for unknown reasons. The clinical experts consulted by CADTH for this review believed this to be a relatively large number of withdrawals and may bias the study results in favour of zanubrutinib; however, it is also acknowledged that it is difficult to determine the significance of the withdrawals without a comparator group.
Important outcomes such as PFS and OS were not formally assessed in the included studies. PFS data were reported; however, with no control group, it is difficult to put these findings into context. OS was NE in Study 206 by the time of the final analysis, and while this may be considered a positive development because it suggests that zanubrutinib might have significantly improved survival, the lack of an estimate for OS is a limitation of the findings emanating from Study 206.
Health-related quality of life was not assessed in either of the included studies. This is a limitation of the data, as MCL can have a significant impact on health-related quality of life, as evidenced by the patient input provided to CADTH. However, due to the open-label design, had health-related quality of life been assessed in either of the included studies, these data would be prone to bias due to patient knowledge of their assigned treatment.
External Validity
The clinical experts consulted by CADTH for this review indicated that patients in Study 206 appeared younger than one would expect to see in clinical practice. The median age of patients in a real-world study was 73 and the median age of patients in Study 206 was 61.5 years; thus, there is a considerable difference between the 2 studies. The enrolment of younger and potentially healthier patients in clinical trials is not an uncommon phenomenon; however, without a formal comparison group, these prognostic differences would be expected to bias the study results in favour of zanubrutinib. Otherwise, the populations in the included studies appeared to reflect patients in Canada who would be expected to be treated with zanubrutinib, according to the clinical experts consulted by CADTH for this review.
The fact that Study 206 was conducted entirely in China and all patients enrolled were Chinese may affect the generalizability of the study results; however, the majority of patients in Study 003 were White. It is not clear whether there are genetic characteristics that might suggest Chinese patients would respond differently to zanubrutinib than non-Chinese patients; therefore, the impact of having a completely Chinese population is unknown.
Indirect Evidence
Routine care of patients with MCL who have received at least 1 prior therapy in Canada is typically treatment with ibrutinib. Evidence for zanubrutinib is limited to single-arm evidence, and no direct comparisons have been identified which compare zanubrutinib to ibrutinib. This comparison is considered for a pharmacoeconomic model of zanubrutinib relative to ibrutinib. Accordingly, understanding the available evidence for the comparative efficacy of zanubrutinib relative to ibrutinib is important in contextualizing the relative economic, efficacy, and safety implications of these 2 therapies relative to one another.
For this submission, a focused literature search for network meta-analyses dealing with MCL was run in MEDLINE All (1946–) on November 2, 2021. No limits were applied. No relevant studies were identified from this process.
Description of Indirect Treatment Comparison
In total, 1 ITC provided by the sponsor was reviewed.18
Methods of Sponsor-Provided ITC
Objectives
The purpose of the sponsor-provided ITC is to indirectly compare zanubrutinib with ibrutinib for the treatment of relapsed or refractory MCL.
Study Selection Methods
A systematic literature review was undertaken in June 2020 to identify relevant trials for inclusion in the ITC. Studies were assessed against PICOS (population, intervention, comparison, outcomes, and study) criteria, as demonstrated in Table 13.
Table 13: Population, Intervention, Comparator, Outcome, and Study Design Criteria
Item | Description | Exclusions |
|---|---|---|
Population | Adults with R/R MCL who have had at least 1 prior therapy |
|
Interventions | Zanubrutinib, Ibrutinib | — |
Outcomes | ORR, CRR, PFS, OS, safety (AEs) | — |
Study design | Prospectively planned, interventional studies including single-arm trials |
|
AE = adverse event; BSC = best supportive care; CRR = complete response rate; MA = meta-analysis; MCL = mantle cell lymphoma; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; R/R = relapsed or refractory; SLR = systematic literature review.
Table 14: Sponsor ITC Analysis Methods
Detail | Sponsor ITC |
|---|---|
ITC method | Unanchored comparison of weighted sample relative to a pooled comparator trial |
Outcomes | Overall survival, PFS, overall response rate, complete response rate |
Covariates included | Age at least 65 years, sex, ECOG PS of at least 2, sMIPI (low, medium, high), bulky disease (at least 5 cm), lactate dehydrogenase > ULN, extranodal disease, bone marrow involvement, at least 3 prior lines of therapy (lenalidomide, bortezomib, stem cell transplant, rituximab, high-intensity therapy) |
Follow-up time points | Not reported |
Sensitivity analyses | None |
Subgroup analysis | None |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITC = indirect treatment comparison; PFS = progression-free survival; sMIPI = simplified Mantle Cell Lymphoma International Prognostic Index; ULN = upper limit of normal.
Source: Sponsor-provided ITC report.18
EMBASE, MEDLINE, the Cochrane Library, and the US Trials Registry were searched in June 2020. A single reviewer screened titles and abstracts, followed by a single reviewer assessment of full-text articles. Where uncertainty existed for an article, a secondary reviewer provided input on article relevance. Data extraction was conducted by a single reviewer, with quality control being provided by a secondary reviewer. No specification was noted on the number of articles assessed by the secondary reviewer. No assessment of study quality was recorded.
In total, 5 studies were identified. Two single-arm trials of zanubrutinib (referred to as BGB-3111-2063,4 and BGB-3111-AU0035,6), and 3 trials of ibrutinib (referred to as PCYC-1104-CA,8 RAY,9 and SPARK10). Additionally, a pooled analysis of the 3 ibrutinib trials was identified (referred to as the pooled study).7 The pooled study used individual-level patient data from each of the 3 contributory studies to construct the referenced baseline demographics and clinical outcomes.
For time-to-event outcomes (OS, PFS), data were extracted using a manual graphical digitization process. First, WebPlotDigitizer (https://automeris.io/WebPlotDigitizer/) was used to manually reconstruct the reported Kaplan–Meier (KM) curves. Second, pseudo individual-level patient data were generated from the digitized KM using a methodology referred to as IPDfromKM. For response, the authors noted that 1 included study, BGB-3111-AU003,5,6 used a PET-based response as opposed to a CT-based assessment for its’ primary analysis. To facilitate comparison to ibrutinib-based studies, the CT-based response thresholds were based on investigator assessment as opposed to a PET-based response.
ITC Analysis Methods
As no direct evidence was available, the sponsor attempted an unanchored MAIC analysis utili4zing method of moments to match patients. The sponsor noted that this method was ineffective under the data being assessed, resulting in an ESS of 4.643 patients, with issues in convergence owing to an insufficient overlap of observations. As a result, the sponsor conducted a 3-step process to finalize their analysis population:
Determined the weights assigned for a given sample size based on an entropy-balancing approach.
For a calculated weight, assessed the difference in baseline variables using univariate regression (logistic regression for binary variables and ordinal regression for ordinal variables) to construct P values of differences between baseline variables.
Repeated steps 1 and 2 for a grid of ESSs and determined the largest ESS where P values for each baseline covariate were greater than 0.05.
As such, the metric for model fit was based on ESS with retention of non-significant differences between baseline characteristics. The choice of baseline characteristics for the analysis was not described within the report. Results are only provided for the pre-weighting and post-weighting analysis population cohorts of the pooled trial,7 without a breakdown of the individual trials that constitute the pooled trial7 population. The sponsor used a Chi squared test to determine the significance of differences in both the pre- and post-weighted analysis populations. Comparative efficacy of time-to-event outcomes were recorded as differences in restricted-mean survival times.
No sensitivity analyses were reported, and no subpopulation-specific analyses were performed. Owing to the lack of direct evidence, no assessment of consistency was possible. All therapeutics considered within the analysis were reported to be of the same dose, formulation, and frequency.
Results of Sponsor-Provided ITC
Summary of Included Studies
A summary of the studies included within the sponsor’s ITC are provided in Table 15. The sponsor evaluated characteristics of the included studies, comparing study phase, geography of the trial, dosage of therapies, patient population criteria, sample size and which version of the international myeloma working group (IWG) were used within the trial.
Table 15: Overview of Included Studies
Detail | BGB-3111-206 | BGB-3111-AU003 | Pooled study | ||
|---|---|---|---|---|---|
PCYC-1104-CA | RAY | SPARK | |||
Study design | Phase II, single-arm open-label trial | Phase I, multi-centre, open-label trial | Phase II, open-label trial | Phase III, open-label, multi-centre trial | Phase II, multi-centre, single-arm trial |
Country, sites | China, 13 sites | International, 24 sites | International, 18 sites | International, 21 countries | International, multi-centre |
Intervention | Zanubrutinib 160 mg twice daily | Zanubrutinib 160 mg twice dailya | Ibrutinib 560 mg once daily | Ibrutinib 560 mg once daily | Ibrutinib 560 mg once daily |
Patient population | Adult patients with relapsed or refractory MCL | Adult patients with relapsed or refractory MCLb | Adult patients with relapsed or refractory MCL | Adults with relapsed or refractory MCL | Adults with MCL who had received a rituximab-containing regimen and had progressed after at least 2 cycles of bortezomib therapy |
Sample size, N | 85c | 32 | 111 | 139 | 120 |
IWG criteria version | 2014 | 2014 | 2007 | 2007 | NR |
ITC = indirect treatment comparison; IWG = International Working Group; MCL = mantle cell lymphoma; NR = not reported.
aThe intervention dose for patients included within the analysis. Patients provided with zanubrutinib 320 mg once daily were not included within the analysis.
bPatients in BGB-3111-AU003 included patients with B-cell malignancies; only patients with relapsed or refractory MCL were included in the analysis.
cOne patient with unconfirmed MCL was not included within the analysis.
Source: Sponsor-provided ITC report.18
Studies were varied with regard to study phase (ranging from phase I for BGB-3111-AU003 through to phase III for RAY). All studies were multi-site, and all but 1 study (BGB-3111-206) were conducted internationally. BGB-3111-206 was exclusively conducted within China. Studies demonstrated notable variation with regard to sample size, ranging from 32 through to 139. No variations were noted in the dose of the comparator, formulation or frequency in the ibrutinib trials being treated with 560 mg once daily. One study, BGB-3111-AU003, enrolled patients with B-cell malignancies and provided 2 dosages for zanubrutinib: 320 mg once daily and 160 mg twice daily. Patients included within the pooled zanubrutinib population from BGB-3111-AU003 were restricted to those with relapsed or refractory MCL who received zanubrutinib at the Health Canada–approved dosage. One patient from BGB-3111-206 was excluded from analysis, as their MCL status was not confirmed. Both zanubrutinib studies reported the use of 2014 response criteria, in contrast with 2 ibrutinib studies (RAY and PCYC-1104-CA) that used 2007 response criteria; the SPARK trial did not report any response criteria data. No details were provided with regard to the medical dictionary used for AE reporting. Data from 2 zanubrutinib studies (Study 206 and AU003) were taken from an interim analysis of the clinical trials. The data for Study 206 were from February 15, 2019; for AU003, the data were taken from an interim analysis on December 13, 2018.
The inclusion of patients was restricted to those with relapsed or refractory MCL with no further specification, although 1 study within the pooled analysis, SPARK, had a more specific treatment history requirement; patients had to have received a rituximab-containing regimen and shown evidence of progression following at least 2 cycles of bortezomib therapy. No details were provided on the individual patient demographics of the included trials, although a comparison of the pooled patient demographics from the zanubrutinib and ibrutinib trials is provided in Table 16.
Table 16: Patient Demographics Between Pooled Cohorts, Before Matching
Parameter | Zanubrutinib (%) (Study 206 + AU003) (N = 117) | Ibrutinib (%) (RAY, SPARK, PCYC-1104-CA) (N = 370) |
|---|---|---|
Age at least 65 | 38.5 | 62.4 |
Sex: Male | 75.2 | 78.1 |
ECOG PS at least 2 | 5.1 | 6.5 |
sMIPI: Low | 49.6 | 23.8 |
sMIPI: Medium | 36.8 | 44.3 |
sMIPI: High | 13.7 | 31.9 |
Bulky disease at least 5 cm | 36.8 | 48.9 |
Lactate dehydrogenase > ULN | 35.0 | 53.8 |
Extranodal disease | 59.8 | 58.1 |
Bone marrow involvement | 48.7 | 45.7 |
Number of prior lines of therapy | ||
At least 3 | 31.6 | 43.8 |
1 | 37.6 | 26.8 |
2 | 30.8 | 29.5 |
Prior lenalidomide | 9.4 | 15.7 |
Prior bortezomib | 6.8 | 53.5 |
Prior stem cell transplant | 6.8 | 23.0 |
Prior rituximab | 79.5 | 96.8 |
Prior high-intensity therapy | 17.1 | 33.5 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; sMIPI = simplified Mantle Cell Lymphoma International Prognostic Index; ULN = upper limit of normal.
Source: Sponsor-submitted indirect treatment comparison, Table 6.18
Table 17: Assessment of Homogeneity for ITC 1
Detail | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Imbalanced before statistical weighting, efficacy analyses are presented post-adjustment |
Clinical trial eligibility criteria | Variable in 1 out of 3 studies with regard to treatment history |
Definitions of end points | Variable across pooled studies, unadjusted in analysis |
Timing of end point evaluation or trial duration | Not reported |
Withdrawal frequency | Not reported |
Clinical trial setting | A mixture of single-country and international trials, variable study sample size |
Study design | Variable from phase I through to phase III |
ITC = indirect treatment comparison.
Results: Efficacy and Safety
The sponsor provided an assessment of the distribution of patient baseline characteristics following their weighting algorithm. The results of this adjusted population analysis are provided in Table 18.
Table 18: Patient Characteristics of the Adjusted Zanubrutinib Population
Parameter | Zanubrutinib (Study 206 + AU003) ESS = 37 % | Ibrutinib (RAY, SPARK, PCYC-1104-CA) N = 370 % |
|---|---|---|
Age at least 65 | 59.7 | 62.4 |
Sex: male | 77.3 | 78.1 |
ECOG PS of at least 2 | 8.6 | 6.5 |
sMIPI: Low | 26.2 | 23.8 |
sMIPI: Medium | 47.9 | 44.3 |
sMIPI: High | 25.9 | 31.9 |
Bulky disease at least 5 cm | 44.5 | 48.9 |
Lactate dehydrogenase > ULN | 52.8 | 53.8 |
Extranodal disease | 56.4 | 58.1 |
Bone marrow involvement | 49.5 | 45.7 |
Prior lines of therapy | ||
At least three | 41.8% | 43.8% |
1 | 27.3 | 26.8 |
2 | 30.9 | 29.5 |
Prior lenalidomide | 15.2 | 15.7 |
Prior bortezomib | 33.9 | 53.5 |
Prior stem cell transplant | 15.7 | 23.0 |
Prior rituximab | 96.6 | 96.8 |
Prior high-intensity therapy | 29.0 | 33.5 |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; ESS = effective sample size; sMIPI = simplified Mantle Cell Lymphoma International Prognostic Index; ULN = upper limit of normal.
Source: Sponsor-submitted indirect treatment comparison, Table 6.18
Indirect treatment comparisons were presented for zanubrutinib relative to ibrutinib for the pooled zanubrutinib patient-level data and the aggregate-level ibrutinib data. Comparative estimates of treatment efficacy were provided for pre- and post-weighting for response (ORR, CR rate), PFS, and OS. A summary of these results is provided in Table 19. ORR did not demonstrate statistically significant differences between the weighted zanubrutinib (ORR = 77.7%; 95% CI, 63 to 92.4) and ibrutinib (ORR = 65.7%; 95% CI, 60.6 to 70.5) treatment groups. Similarly, the CR rate did not demonstrate statistically significant differences between the weighted zanubrutinib (CR = 25.5%; 95% CI, 12.5, 38.5) and ibrutinib (CR = 20%; 95% CI, 16 to 24.4) treatment groups. PFS did not demonstrate statistically significant differences between the weighted zanubrutinib (PFS RMST = 13.9 months) and ibrutinib (PFS RMST = 12.6) treatment arms (HR of zanubrutinib versus ibrutinib = 0.92; 95% CI, 0.63 to 1.33). Similarly, OS did not demonstrate statistically significant differences between the weighted zanubrutinib (OS RMST = 21.2) and ibrutinib (OS RMST = 18.4) treatment arms (HR of zanubrutinib versus ibrutinib = 0.74; 95% CI, 0.43 to 1.26).
For safety, the sponsor provided numerical comparisons of treatment-emergent AEs with a prevalence rate of 10% or greater in either the weighted zanubrutinib population or pooled ibrutinib comparator population. No formal statistical comparison of event rates was presented. A summary of the AEs of special interest is provided in Table 20. Data were not presented for the following AEs of special interest owing to a lack of reported data from the associated ibrutinib studies: serious infections, secondary malignancies, atrial fibrillation or flutter, interstitial lung disease, hemorrhage, and increased blood pressure.
No alternative subgroup or sensitivity analyses were presented.
Table 19: Efficacy Results of the Sponsor’s ITC
Detail | Zanubrutinib (Weighted Study 206 + AU003) ESS = 37 | Ibrutinib (RAY, SPARK, PCYC-1104-CA) N = 370 |
|---|---|---|
Overall response | ||
Overall response rate (95% CI) | 77.7% (63.0% to 92.4%) | 65.7% (60.6% to 70.5%) |
Odds ratio (95% CI) | 1.82 (0.85 to 4.30) | |
P value | 0.142 | |
Complete response | ||
Complete response rate (95% CI) | 25.5% (12.5% to 38.5%) | 20.0% (16.0% to 24.4%) |
Odds ratio (95% CI) | 1.37 (0.60 to 2.89) | |
P value | 0.431 | |
Progression-free survival | ||
Restricted-mean progression-free survival, months | 13.9 | 12.6 |
Difference in restricted-mean progression-free survival, months (95% CI) | 1.25 (−1.31 to 3.80) | |
Hazard ratio (95% CI) | 0.92 (0.63 to 1.33) | |
Overall survival | ||
Restricted-mean overall survival, months | 21.2 | 18.4 |
Difference in restricted-mean overall survival, months (95% CI) | 2.77 (−0.22 to 5.76) | |
Hazard ratio (95% CI) | 0.74 (0.43 to 1.26) | |
CI = confidence interval; ESS = effective sample size; ITC = indirect treatment comparison.
Source: Sponsor-provided ITC report.18
Table 20: Safety Events of Special Interest From the Sponsor’s ITC
Adverse events | Zanubrutinib (Weighted Study 206 + AU003) ESS = 37 % | Ibrutinib (RAY, SPARK, PCYC-1104-CA) N = 370 % |
|---|---|---|
Adverse events, any grade | ||
Anemia | 31.4 | 17 |
Neutropenia | 15.3 | 18.9 |
Thrombocytopenia | 9.9 | 19.7 |
Adverse events, grade ≥ 3 | ||
Anemia | 22.5 | 8.1 |
Neutropenia | 5.9 | 16.5 |
Thrombocytopenia | 5.0 | 11.1 |
ESS = effective sample size; ITC = indirect treatment comparison.
Source: Sponsor-provided ITC report.18
Critical Appraisal of Sponsor-Provided ITC
In this ITC, there was no connected evidence and, therefore, indirect comparison methods necessitating connectivity of trials (i.e., a shared treatment arm) were not available. A critical challenge with the sponsor-submitted ITC is the algorithm used to generate the weighted sample. The sponsor noted that a method of moment matching did not converge and had an ESS of 4.643. The sponsor did not, however, specify which weighting methodologies were tested as an alternative to their original approach outside of the reported algorithm. Further, no justification is provided for the use of the parameters included within the provided adjustment analysis. While the proposed algorithm does provide a larger ESS, estimates from this analysis are associated with wide CIs and substantial uncertainty. With the substantial reduction in ESS observed, there was likely significant heterogeneity between the zanubrutinib studies and comparator studies. The results for comparisons with major reductions in ESS indicate that the weights are highly variable due to a lack of population overlap, and that the resulting estimate may not be reliable. A potential issue with the sponsor’s approach is that their algorithm successively imbalances patient demographics up to a set threshold. An unanchored indirect comparison using MAIC methods will only provide an unbiased comparison if all prognostic and effect-modifying factors are included in the weighting process. While between-cohort comparisons do not reach statistical significance, they remain less balanced than is otherwise achievable by their proposed methods, and this may be influential with regard to treatment outcomes. The use of a P value–based assessment for covariate balance is confounded by the relationship between ESS and the power to detect significant P value thresholds. Additionally, details were not provided on the statistical method used to assess for the generation of a P value. Beyond the potential statistical differences, qualitatively, a number of large differences in characteristics were evident even after the sponsor’s algorithm approach was used to match the data (ECOG PS, sMIPI, prior stem cell therapy). Moreover, unanchored forms of population-adjusted indirect comparisons make the much stronger assumption of conditional constancy of absolute effects. This means that the absolute treatment effects are assumed constant at any given level of the effect modifiers and prognostic variables, and all effect modifiers and prognostic variables are required to be known. This assumption is unlikely to have been met in this unanchored MAIC; therefore, no conclusions can be made from these data.
Further, the nature of the fact that the originally proposed treatment balancing did not converge owing to substantial between-group heterogeneity (see Table 16 for pre-balancing details) is critical to consider. Of the 18 baseline covariates assessed, the sponsor assessment of baseline characteristics suggests that only 5 (sex, ECOG PS of at least 2, extranodal disease, bone marrow involvement, and prior lenalidomide) were not statistically significant different before adjustment. The substantial reduction in ESS from 117 to 37 (and 4.3 in the original weighting method) is a representation of the dissimilarity between the trial populations being compared. Minimization of these differences is partially achieved through the proposed treatment weighting method used, although it is important to consider the potential influence of unmeasured and unadjusted confounders. Additionally, it is of note that within the unadjusted population cohort, the direction of worse prognostic factors heavily favours the zanubrutinib population. For example, age of at least 65 years, proportions of patients with high or medium sMIPI scores, bulky disease, and greater than 3 prior lines of therapy are all higher in the unadjusted ibrutinib population to a statistically significant degree when compared with the zanubrutinib population, which would be expected to bias the ITC results in favour of zanubrutinib.
By virtue of the cohorts being significantly different with respect to almost all demographic features before weighting, it may be reasonably expected that other potentially significant demographic details which were not weighted, such as race, refractory status, and TP-54 mutation status, would follow a similar trend. Accordingly, despite the statistical adjustments undertaken to minimize between-group differences, it is critical to note that heterogeneity between the base population cohorts being compared still likely exists. Further, aspects of study design and temporality are unaccounted for in this analytical framework and may contribute to further confounding in a way that cannot be explored analytically.
With regard to AEs, many of those noted to be of special significance were not available for review. Accordingly, there is substantial uncertainty with the comparative safety profile of zanubrutinib relative to ibrutinib. No data were available for review for the other comparators of interest noted in the protocol (acalabrutinib, bendamustine plus rituximab plus cytarabine, bendamustine plus rituximab plus bortezomib, and bortezomib with or without rituximab).
The sponsor had indicated that its search for evidence was performed in June 2020. Accordingly, it is not possible to assess whether evidence generated between June 2020 up to the submission time may have an influence on the findings of this ITC.
No formal assessment of study quality was conducted by the sponsor for the ITC and, as such, the influence of potential risk of bias cannot be appraised.
The data from the zanubrutinib-containing trials were taken from an interim analysis. Accordingly, the outputs of the reported ITC may be subject to change in the associated final trial analysis, as it would include more events and longer follow-up times.
Summary of Indirect Evidence
Overall, 1 study, a sponsor-performed ITC, was available to assess the relative efficacy and safety of zanubrutinib versus ibrutinib. Using an unanchored MAIC approach, the sponsor did not note any statistically significant differences between zanubrutinib and ibrutinib with regard to response, PFS, or OS. Numerical differences were noted with respect to the proportion of patients experiencing AEs, although no formal comparison was made between zanubrutinib and ibrutinib.
The 1 ITC evaluable for this submission has significant limitations owing to the lack of connected evidence, the substantial pre-weighting differences in patient demographics, and the associated low ESS of the zanubrutinib treatment population. Accordingly, substantial uncertainty exists as to the relative efficacy and safety profile of zanubrutinib relative to ibrutinib for adult patients with relapsed or refractory MCL and no conclusions can be made from the data.
Discussion
Summary of Available Evidence
Two pivotal single-arm, multinational, sponsor-funded studies were included in this review. Study 206 enrolled 86 adult patients with MCL who had received between 1 and 4 prior regimens and whose disease failed to achieve any response. Patients were treated with zanubrutinib 160 mg twice daily until disease progression or unacceptable toxicity. Study 003 enrolled patients with a variety of B-cell malignancies, following at least 1 prior line of therapy, and 32 of these patients had relapsed or refractory MCL. Part 1 of Study 003 was devoted to finding an optimal dose that could then be used in part 2, known as the expansion phase. The primary outcome of Study 206 was IRC-assessed ORR, and secondary outcomes included PFS, DOR, and investigator-assessed ORR, while OS was an exploratory outcome. The primary outcome of Study 003 was related to harms, while secondary outcomes included ORR, CR rate, partial response rate, minimal residual disease clearance rate, PFS, OS, and DOR.
Patients were a median age of 60.5 years in Study 206 and 70.5 years in Study 003. The majority of patients were male in both Study 206 (78%) and Study 003 (69%). Study 206 was entirely conducted in China, and all patients were Chinese while in Study 003, the majority of patients were White (78%). The majority of patients (70%) in Study 206 had a ECOG PS of 0, while in Study 003, patients had a similar ECOG PS of 0 (47%) or 1 (44%), and the majority of patients in Study 206 (74%) and Study 003 (88%) had stage IV disease. The majority of patients (71%) in Study 206 had 2 or more prior therapies, while the majority of patients in Study 003 had 1 prior therapy.
Interpretation of Results
Efficacy
Other BTK inhibitors are the most appropriate comparator for zanubrutinib, and the lack of a comparison group in either of the included studies is a limitation of the evidence from this review. Compared with a historical control, zanubrutinib appeared to improve ORR in Study 206; however, there is some question regarding the estimate of 40% used for the historical control, as it was not based on studies of other BTK inhibitors. If studies of other BTK inhibitors had been used as a historical control, the ORR would likely be around 65% to 70%. The CR rate in Study 206 seemed exceptionally high (78%), according to the clinical experts consulted by CADTH for this review, and was much higher than the CR rate (31%) reported in Study 003. The sponsor acknowledged the difference in CR rates between studies and hypothesized that the significant difference in age between the 2 studies and perhaps use of different imaging techniques may have contributed to these differences.
No other statistical comparisons were performed in either study, although data for PFS, OS, and DOR were reported in each. Without a control group, it is challenging to place this data into context. The median OS was NE by the time of the final Clinical Study Report, and the median PFS was 33.0 months in Study 206.3,4
Health-related quality of life is clearly an important outcome to patients, as evidenced in the input they provided for this review; however, this outcome was not assessed in either included study. Patients with MCL report fatigue as a significant symptom that impacts their ability to carry out normal activities, and the side effects associated with their treatments, including nausea and vomiting, neurocognitive effects, headaches, and alopecia, also have an impact on their health-related quality of life. Without an assessment of the impact of zanubrutinib on health-related quality of life, no conclusions can be drawn about the impact of zanubrutinib on this important outcome.
Without a control group in the included studies, indirect evidence becomes the only source of comparative data for zanubrutinib. The only available ITC was submitted by the sponsor and was reviewed in detail earlier in this report. Due to significant methodological issues with this unanchored MAIC, there is substantial uncertainty as to the relative efficacy and safety of zanubrutinib compared with ibrutinib. There were no statistically significant differences noted between zanubrutinib and ibrutinib with respect to ORR, PFS, or OS; however, given the limitations of the analysis, no conclusions can be made from the data.
Harms
The assessment of harms is complicated by the lack of a control group. The most common AE included various cytopenias (reduced neutrophil count, platelet count, and blood count), and upper respiratory tract infections, and these are consistent with the warnings and safety issues identified in the product monograph for zanubrutinib.
The first-generation BTK inhibitor, ibrutinib, is known for causing cardiac toxicities such as arrhythmia and hypertension. It is believed that interactions with phosphoinositide-3 kinase (PI3K) and other pathways that play a cardioprotective role may explain, or at least partially explain, these cardiotoxic effects. Specifically, the arrhythmias include atrial fibrillation, ventricular tachycardia or fibrillation, premature ventricular contractions, and prolongation of the QT interval, with atrial fibrillation being the most common. Risk factors for ibrutinib-associated atrial fibrillation include being treated for chronic lymphocytic leukemia (as opposed to MCL), prior history of atrial fibrillation, age older than 65 years, pre-existing hypertension and hyperlipidemia, and high Shanafelt risk score in chronic lymphocytic leukemia.19 The hope is that second-generation BTK inhibitors like zanubrutinib, with their enhanced selectivity, will have a reduced risk of cardiotoxicity. At this time, however, the mechanism of cardiotoxicity has not been established; therefore, it is not known whether this is an off-target effect or part of the primary target for BTK inhibitors, in which case the more selective second-generation inhibitors may not prove to be advantageous with respect to this safety issue. There is no head-to-head comparison of zanubrutinib with ibrutinib in MCL; however, in the ASPEN study, which compared these 2 drugs in WM, there was a lower number of zanubrutinib-treated patients with atrial fibrillation or flutter, hypertension, and major hemorrhage. Nevertheless, these findings must be demonstrated in a trial that includes patients with MCL before 1 can conclude a safety advantage of 1 drug over the other for the indication under review. In the sponsor-submitted ITC, there was some evidence of a difference in the risk of some AEs between zanubrutinib and ibrutinib; however, methodological limitations preclude drawing any conclusions about these data.
Conclusions
Two pivotal, sponsor-funded, multi-centre, single-arm studies that enrolled a total of 118 patients with relapsed or refractory MCL were included in this review. In the 1 study that included a historical control, ORR was improved for zanubrutinib versus the control, although the control used was not a BTK inhibitor. No conclusions can be drawn about efficacy with respect to other outcomes such as OS, PFS, and DOR, as no statistical analysis was planned. The common AEs were consistent with those described in the product monograph and included various cytopenias, infections, and hemorrhage. There were no other studies to inform the long-term safety of this second-generation BTK inhibitor; therefore, the long-term safety of zanubrutinib is unknown. The ITC submitted by the sponsor was of limited value for drawing any conclusions about the relative efficacy and safety of zanubrutinib compared with other BTK inhibitors due to significant methodological issues with the approach taken.
References
1.Canadian Cancer Society. Mantle cell lymphoma. 2021; https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/what-is-non-hodgkin-lymphoma/mantle-cell-lymphoma. Accessed 2021 Dec 15.
2.Leukemia & Lymphoma Society (LLS). Mantle cell lymphoma (MCL). 2021; https://www.lls.org/research/mantle-cell-lymphoma-mcl. Accessed 2021 Dec 15.
3.Clinical Study Report: BGB-3111-206. A single-arm, open-label, multicenter phase 2 study to evaluate efficacy and safety of BGB-3111, a Bruton’s tyrosine kinase (BTK) inhibitor, in subjects with relapsed or refractory mantle cell lymphoma (MCL) [internal sponsor's report]. Beijing (CN): BeiGene (Beijing) Co., Ltd.; 2021 Apr 09.
4.Clinical Study Report: BGB-3111-206. A single-arm, open-label, multicenter phase 2 study to evaluate efficacy and safety of BGB-3111, a Bruton’s tyrosine kinase (BTK) inhibitor, in subjects with relapsed or refractory mantle cell lymphoma (MCL) [internal sponsor's report]. Beijing (CN): BeiGene (Beijing) Co., Ltd.; 2020 Jun 05.
5.Clinical Study Report: BGB-3111-AU-003. A phase 1/2, open-label, multiple-dose, dose escalation and expansion study to investigate the safety and pharmacokinetics of the BTK inhibitor BGB-3111 in patients with B-cell lymphoid malignancies [internal sponsor's report]. San Mateo (CA): BeiGene, Ltd.; 2021 Sept 21.
6.Clinical Study Report: BGB-3111-AU-003. A phase 1/2, open-label, multiple-dose, dose escalation and expansion study to investigate the safety and pharmacokinetics of the BTK inhibitor BGB-3111 in patients with B-cell lymphoid malignancies [internal sponsor's report]. San Mateo (CA): BeiGene, Ltd.; 2020 Jun 15.
7.Rule S, Dreyling M, Goy A, et al. Outcomes in 370 patients with mantle cell lymphoma treated with ibrutinib: a pooled analysis from three open‐label studies. Br J Haematol. 2017;179(3):430-438. PubMed
8.Pharmacyclics LLC. NCT01236391: Safety and efficacy of PCI-32765 in participants with relapsed/refractory mantle cell lymphoma (MCL). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2015: https://ClinicalTrials.gov/show/NCT01236391. Accessed 2022 Feb 11.
9.Janssen Research & Development LLC. Study of ibrutinib (a Bruton's tyrosine kinase inhibitor), versus temsirolimus in patients with relapsed or refractory mantle cell lymphoma who have received at least one prior therapy. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2018: https://ClinicalTrials.gov/show/NCT01646021. Accessed 2022 Feb 11.
10.Janssen Research & Development LLC. NCT01599949: A study to evaluate the efficacy and safety of ibrutinib, in patients with mantle cell lymphoma who progress after bortezomib therapy. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2016: https://ClinicalTrials.gov/show/NCT01599949. Accessed 2022 Feb 11.
11.BeiGene (Canada) ULC provided additional information regarding Brukinsa (zanubrutinib) DRR review: new ITC analysis [internal additional sponsor's information]. Mississauga (ON): BeiGene (Canada) ULC; 2022 Apr 22.
12.Shah BD, Yang K, Liu S, et al. Abstract #3046: Real-world Bruton tyrosine kinase inhibitor treatment patterns, compliance, costs, and hospitalizations in patients with mantel cell lymphoma in the United States. Poster presented at the 63rd American Society of Hematology Annual Meeting and Exposition, Atlanta, GA [internal additional sponsor's information]. Mississauga (ON): BeiGene (Canada) ULC; 2021 Dec 11-14.
13.Shadman M, Flinn I, Levy MY, et al. Abstract #148544: Phase 2 study of zanubrutinib in BTK inhibitor-intolerant patients with relapsed/refractory B-cell malignancies. Poster presented at the 63rd American Society of Hematology Annual Meeting and Exposition, Atlanta, GA [internal additional sponsor's information] Mississauga (ON): BeiGene (Canada) ULC; 2021 Dec 11-14.
14.e-CPS. Ottawa (ON): Canadian Pharmacists Association; 2021: https://www.e-therapeutics.ca. Accessed 2021 Dec 15.
15.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
16.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Oct 21.
17.BeiGene (Canada) ULC response to December 10, 2021 DRR request for additional information regarding Brukinsa DRR review: harms data in BGB-3111-AU-003 [internal additional sponsor's information]. Mississauga (ON): BeiGene (Canada) ULC; 2021 Dec 17.
18.BRUKINSA® (zanubrutinib) for the treatment of adult patients with mantle cell lymphoma who have received at least one prior therapy. Indirect Treatment Comparison Report November 2021 [internal sponsor's report]. In: Drug Reimbursement Review sponsor provided submission: Brukinsa (zanubrutinib) 80 mg oral capsules. Mississauga (ON): BeiGene (Canada) ULC; 2021 Nov 12.
19.Giudice V, Vecchione C, Selleri C. Cardiotoxicity of novel targeted hematological therapies. Life (Basel). 2020;10(12):344. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: November 3, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Brukinsa* or zanubrutinib* or BGB 3111* or BGB3111* or AG9MHG098Z*).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*zanubrutinib/
(Brukinsa* or zanubrutinib* or BGB 3111* or BGB3111*).ti,ab,kf,dq.
or/3-4
use oemezd
not conference abstract.pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | brukinsa OR zanubrutinib OR BGB-3111 OR BGB3111]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- brukinsa OR zanubrutinib OR BGB-3111 OR BGB3111]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- brukinsa OR zanubrutinib OR BGB-3111 OR BGB3111]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- brukinsa OR zanubrutinib OR BGB-3111 OR BGB3111]
Grey Literature
Search dates: October 21, 2021, to November 3, 2021
Keywords: [brukinsa OR zanubrutinib OR BGB 3111 OR BGB3111 OR AG9MHG098Z OR mantle cell OR lymphoma OR MCL]
Limits: Publication years: no limit
Updated: Search updated prior to the meeting of CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC)
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Das 2019 | Review |
Tam 2019 | Review |
Xu 2020 | Indication |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 23: Subgroups (Study 206)
Detail | Study 206 Zanubrutinib (N = 86) | |
|---|---|---|
ORR (investigator-assessed) by | ||
ORR, n/N, by | — | — |
ECOG PS | Response/Patients | Overall response rate (95% CI) |
0 | 53/60 | 88.3 (77.4, 95.2) |
1 or more | 19/26 | 73.1 (52.2, 88.4) |
Prior lines | — | — |
< 3 | 51/57 | 89.5 (78.5, 96.0) |
3 or more | 21/29 | 72.4 (52.8, 87.3) |
Prior ASCT | — | — |
Yes | 3/3 | 100.0 (29.2, 100.0) |
No or unknown | 69/83 | 83.1 (73.3, 90.5) |
Prior rituximab or rituximab-containing regimen | — | — |
Yes | 52/64 | 81.3 (69.5, 89.9) |
No/unknown | 20/22 | 90.9 (70.8, 98.9) |
Prior Hyper-CVAD or hyper-CVAD-like regimen | — | — |
Yes | 11/13 | 84.6 (54.6, 98.1) |
No/unknown | 61/73 | 83.6 (73.0, 91.2) |
Prior lenalidomide | — | — |
Yes | 9/12 | 75.0 (42.8, 94.5) |
No/unknown | 63/74 | 85.1 (75.0, 92.3) |
Prior bortezomib | — | — |
Yes | 4/7 | 57.1 (18.4, 90.1) |
No/unknown | 68/79 | 86.1 (76.5, 92.8) |
Prior bendamustine | — | — |
Yes | 0/2 | 0.0 (0.0, 84.2) |
No/unknown | 72/84 | 85.7 (76.4, 92.4) |
Blastoid histology | — | — |
Yes | 8/12 | 66.7 (34.9, 90.1) |
No | 59/68 | 86.8 (76.4, 93.8) |
Unknown | 5/6 | 83.3 (35.9, 99.6) |
ASCT = autologous stem cell transplant; CVAD = cyclophosphamide, vincristine, doxorubicin, and dexamethasone; ECOG PS = Eastern Cooperative Oncology Group Performance Status; ORR = overall response rate.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
BTK
Bruton tyrosine kinase
ITC
indirect treatment comparison
MCL
mantle cell lymphoma
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Zanubrutinib (Brukinsa), oral capsules |
Submitted price | Zanubrutinib: $67.98 per 80 mg capsule |
Indication | For the treatment of adult patients with mantle cell lymphoma who have received at least 1 prior therapy |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 22, 2021 |
Reimbursement request | As per indication |
Sponsor | BeiGene Canada ULC |
Submission history |
|
NOC = Notice of Compliance.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis |
Target population | Adult patients with MCL who have received at least 1 prior therapy |
Treatment | Zanubrutinib |
Comparator | Ibrutinib |
Perspective | Canadian publicly funded health care payer |
Time horizon | One year |
Key data source | A sponsor-submitted indirect treatment comparison of zanubrutinib compared with ibrutinib based on Study AU003 and Study 206 (zanubrutinib) and PCYC-1104-CA, SPARK, and RAY trials (ibrutinib). The studies for each treatment were pooled for the analysis, and individual patient data for the zanubrutinib studies were matched to the pooled ibrutinib cohort based on sponsor-defined criteria. |
Costs considered | Drug acquisition costs. |
Submitted results | Zanubrutinib is associated with an incremental cost savings of $46,503 per patient annually. |
Key limitations |
|
CADTH reanalysis results |
|
BTK = Bruton tyrosine kinase; MCL = mantle cell lymphoma.
Conclusions
The sponsor’s cost-minimization analysis is based on the assumption of similar clinical efficacy and safety for zanubrutinib and ibrutinib. Based on the CADTH Clinical Review, there was no direct head-to-head evidence comparing zanubrutinib with ibrutinib or any other relevant comparators. Evidence from a sponsor-commissioned indirect treatment comparison (ITC), which included 4 single-arm trials and 1 comparative trial, was of limited value for drawing any conclusions about the relative efficacy and safety of zanubrutinib compared with other Bruton tyrosine kinase (BTK) inhibitors, due to significant methodological issues with the approach taken.
If zanubrutinib is considered to have similar clinical efficacy and safety compared with ibrutinib, treatment with zanubrutinib may result in cost savings of $46,503 per patient per year, as estimated by the sponsor. It should be noted that the CADTH pan-Canadian Oncology Drug Review Expert Review Committee (pERC) previously recommended that ibrutinib be funded with a condition of a substantial price reduction for patients with mantle cell lymphoma (MCL). The estimated incremental savings are based on publicly available list prices and may not reflect actual prices paid by Canadian public drug plans. Furthermore, the estimated cost savings are based on the assumption that zanubrutinib will be used for the same duration as ibrutinib and that these treatments will not be used sequentially in patients who experience toxicity; the lack of data on duration of treatment and the potential for treatment switching suggests that the projected cost savings are overestimated, though the magnitude is uncertain.
Economic Review
The current review is for zanubrutinib (Brukinsa) for the treatment of adult patients in Canada with MCL who have received at least 1 prior therapy.
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a cost-minimization analysis1 for zanubrutinib compared with ibrutinib for the treatment of adult patients with MCL who have received at least 1 prior therapy. The reimbursement population aligns with the Health Canada–indicated population. The sponsor’s key assumption was that the only relevant comparator for zanubrutinib is ibrutinib. Ibrutinib is the only other BTK inhibitor currently reimbursed in Canada and the patient population intended to be treated with zanubrutinib and ibrutinib is functionally the same.
Zanubrutinib is available as 80 mg capsules for oral consumption. The recommended dosage of zanubrutinib is 320 mg once daily or 160 mg twice daily. At the submitted price of $67.98 per 80 mg capsule, the cost of zanubrutinib is $271.93 per day. Ibrutinib was considered at a cost of $99.83 per 140 mg capsule. At the recommended dosage of 560 mg once daily, the sponsor estimated a daily per-patient treatment cost of $399.34.
The sponsor assumed zanubrutinib was associated with similar health benefits to ibrutinib, based on a sponsor-commissioned ITC.2 The sponsor adopted dosing as per product monographs3,4 and assumed 100% adherence in estimating treatment costs. As a result, all clinical benefits and resource use beyond drug acquisition costs were assumed to be equivalent, and the sponsor’s base case considered only drug acquisition costs. The analysis was conducted from the perspective of the publicly funded health payer over a time horizon of 1 year. As such, discounting was not applied.
The sponsor’s submitted base case estimated an annual treatment cost of $99,256 per patient with zanubrutinib, while the annual cost of ibrutinib was estimated to be $145,759 per patient. Based on the sponsor’s submission, treatment with zanubrutinib resulted in estimated cost savings of $46,503 per patient per year compared with ibrutinib.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($) | Incremental costs ($) |
|---|---|---|---|---|
Ibrutinib | 145,759 | Reference | 145,759 | Reference |
Zanubrutinib | 99,256 | −46,503 | 99,256 | −46,503 |
Source: Sponsor’s economic submission.1
The sponsor did not present any scenario analyses.
CADTH Appraisal of the Sponsor’s Economic Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The assumption of comparable clinical efficacy between zanubrutinib and ibrutinib is uncertain: In the absence of a direct head-to-head comparison between zanubrutinib and ibrutinib, the sponsor commissioned an indirect treatment comparison (ITC) assessing the comparative efficacy and safety of zanubrutinib (BGB-3111 to 206 and BGB-311-AU003) and ibrutinib (PCYC-1104-CA, SPARK, and RAY). The sponsor’s ITC took the form of a matching-adjusted indirect comparison that suggested that patients have higher response rates, longer survival, and fewer adverse events overall if treated with zanubrutinib when compared with those receiving ibrutinib. However, these results were not statistically significant. CADTH’s Clinical Review identified several limitations with the sponsor’s submitted ITC. First, CADTH noted the ITC was based on 4 single-arm open-label studies and 1 randomized open-label comparative study (ibrutinib versus temsirolimus) that did not explicitly test the hypothesis of clinical equivalence or noninferiority between the 2 therapies. Second, there was substantial between-group heterogeneity between the included studies, which reduced the effective sample size substantially (from 117 to 37). Furthermore, the sponsor’s matching approach was not able to correct imbalances in patient demographics at baseline, which may have confounded treatment outcomes, though the influence of unmeasured and unadjusted confounders could not be assessed and heterogeneity still likely exists despite the statistical adjustments undertaken. As such, the results of the ITC are highly susceptible to bias and introduce substantial uncertainty in the sponsor’s assumption of similar long-term clinical efficacy and safety between zanubrutinib and ibrutinib. Should patients receiving zanubrutinib persist on treatment for longer, and/or experience longer survival, greater health care costs may be accrued than for patients receiving ibrutinib, and a cost-utility analysis would be more appropriate than a cost-minimization analysis.
CADTH is unable to address this limitation.
Extended treatment duration on BTK inhibitors may accrue additional costs: The clinical experts consulted by CADTH for this review noted that some patients may discontinue ibrutinib treatment due to toxicity. Currently, patients who experience toxicity may move on to bortezomib, chemotherapy, chemo-immunotherapy regimens, or no treatment. With the availability of zanubrutinib, patients may switch to zanubrutinib and, thus, continue receiving a BTK inhibitor until progression or subsequent toxicity. The additional cost associated with continued treatment has not been captured in the sponsor’s submission because the sponsor does not include treatments other than ibrutinib. If patients switch from ibrutinib to zanubrutinib instead of to 1 of the other current options, additional costs would be incurred. Further, the differences in treatment effectiveness in this setting are unknown, given the limitations with the available clinical information for zanubrutinib.
CADTH is unable to address this limitation.
Time horizon may not accurately capture all relevant costs: In the product monograph, treatment with zanubrutinib is recommended until toxicity or disease progression. The clinical experts consulted for this review noted treatment duration with zanubrutinib would be considerably longer than the sponsor’s 1-year time horizon, particularly if patients tolerate zanubrutinib better than ibrutinib. As such, cost differences may exist between zanubrutinib and ibrutinib, particularly if a longer time horizon is explored. Further, some patients may switch treatment from ibrutinib to zanubrutinib, and the time to treatment switch is heterogenous because some patients may experience toxicity early on and some later during treatment. As such, there may be additional costs and cost differences accrued beyond the first year, and these are not captured within the 1-year time horizon adopted by the sponsor.
CADTH is unable to address this limitation.
CADTH Reanalyses of the Economic Information
CADTH did not undertake a base-case reanalysis, as the limitations related to uncertainty in comparable clinical efficacy, treatment switching, and duration of treatment (short time horizon), could not be addressed by CADTH and thus limit any assessment of a cost comparison.
If the clinical efficacy of zanubrutinib and ibrutinib are considered similar and there is no treatment switching to prolong treatment with a BTK inhibitor, zanubrutinib resulted in an estimated cost savings of $46,503 per patient per year compared with ibrutinib. The estimated incremental savings are based on the drug acquisition price for zanubrutinib, which is approximately 32% less than the published price of ibrutinib and may not reflect actual prices paid by Canadian public drug plans for ibrutinib.
Issues for Consideration
Acalabrutinib received Health Canada approval for use in patients with relapsed or refractory MCL but has not been reviewed by CADTH for this indication. Acalabrutinib was previously available through compassionate access programs but feedback from CADTH-participating drug plans indicated these programs have ended.
Zanubrutinib may be used as a bridging therapy to chimeric antigen receptor (CAR) T-cell therapy. According to the clinical input received for this review by CADTH, zanubrutinib may be used as a bridging therapy to CAR T-cell therapy for patients with relapsing and refractory MCL once it becomes available. If zanubrutinib were to be used as a bridging treatment to CAR T-cell therapy, additional costs might be incurred by the health care system if the currently used bridging therapy is less costly than zanubrutinib. If zanubrutinib replaces ibrutinib as a bridging therapy, it may reduce costs to the drug plans based on the publicly available prices. The relative efficacy of zanubrutinib with treatments in this setting is unknown.
Analysis is based on publicly available list prices. Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. The negotiations for ibrutinib were concluded with a letter of intent,5 suggesting a confidential price has been negotiated, in line with the prior CADTH recommendation for ibrutinib.6 The actual costs paid by public drug plans are unknown.
Conclusions
Based on the CADTH Clinical Review, there was no direct head-to-head evidence comparing zanubrutinib with ibrutinib or any other relevant comparators. Evidence from a sponsor-commissioned ITC, which included 4 single-arm trials and 1 comparative trial, was of limited value for drawing any conclusions about the relative efficacy and safety of zanubrutinib versus other BTK inhibitors due to significant methodological issues with the approach taken. While the results of the sponsor-submitted matching-adjusted indirect comparison did not note any statistically significant differences between zanubrutinib and ibrutinib with regard to response, progression-free survival, or overall survival, numerical differences were noted with respect to the proportion of patients experiencing adverse events, though no formal comparison was made between zanubrutinib and ibrutinib. Substantial limitations were found with the sponsor’s ITC, owing to the lack of connected evidence, the substantial pre-weighting differences in patient demographics, and the associated low effective sample size of the zanubrutinib treatment population. Accordingly, substantial uncertainty exists as to the relative efficacy and safety profile of zanubrutinib relative to ibrutinib for adult patients with relapsed or refractory MCL, and no conclusions can be made from the data.
The sponsor’s cost-minimization analysis is based on the assumption of similar clinical efficacy and safety of zanubrutinib and ibrutinib. However, given the findings of CADTH’s Clinical Review, the validity of the sponsor’s cost-minimization analysis is uncertain. If zanubrutinib is considered to have similar clinical efficacy and safety to ibrutinib, treatment with zanubrutinib may result in estimated cost savings of $46,503 per patient per year, as estimated by the sponsor based on the drug acquisition price of zanubrutinib, which is approximately 32% less than the published price of ibrutinib. It should be noted that pERC previously recommended that ibrutinib be funded with a condition of a substantial price reduction for patients with MCL. The estimated incremental savings are based on publicly available list prices and may not reflect the actual prices paid by Canadian public drug plans. Furthermore, the estimated cost savings are based on the assumption that zanubrutinib will be used for the same duration as ibrutinib and that these treatments will not be used sequentially in patients who experience toxicity; the lack of data on the duration of treatment and the potential for treatment switching suggests that the projected cost savings are overestimated, though the magnitude is uncertain.
Additional Details on the Sponsor’s Submission
No additional information from the sponsor’s submitted pharmacoeconomic evaluation was considered in the review of zanubrutinib.
Additional Details on the CADTH Reanalyses and Additional Analyses
CADTH did not conduct any additional pharmacoeconomic analyses in the review of zanubrutinib.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Brukinsa (zanubrutinib) 80 mg oral capsules. Mississauga (ON): BeiGene (Canada) ULC; 2021 Oct 21.
2.Summary on indirect comparison between zanubrutinib (BGB-3111-206and BGB-3111-AU003) and ibrutinib(1104, SPARK and RAY) for Council of Economic Advisors version 1 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Brukinsa (zanubrutinib) 80 mg oral capsules. Mississauga (ON): BeiGene (Canada) ULC; 2020 Aug 26.
3.Brukinsa (zanubrutinib): 80 mg oral capsules [product monograph]. Basel (CH): BeiGene Switzerland GmbH; 2021 Jul 21.
4.Imbruvica (ibrutinib): 140 mg oral capsules [product monograph]. Toronto (ON): Janssen Inc.; 2018 Jul 24: https://pdf.hres.ca/dpd_pm/00046525.PDF. Accessed 2021 Dec 16.
5.pan-Canadian Pharmaceutical Alliance. Brand Name Drug Negotiations Status. 2021; https://www.pcpacanada.ca/negotiations. Accessed 2021 Dec 20.
6.CADTH pCODR Expert Review Committee (pERC) final recommendation: Imbruvica (ibrutinib). Ottawa (ON): CADTH; 2016 Jul 19: https://www.cadth.ca/sites/default/files/pcodr/pcodr_ibrutinib_imbruvica_mcl_fn_rec.pdf. Accessed 2021 Dec 20.
7.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Oct 21.
8.DeltaPA. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2022 Feb 10.
9.Zilioli VR, Gabutti C, Meli G, et al. Rituximab plus bortezomib still represents an effective treatment option for patients with relapsed or refractory mantle cell lymphoma. Blood. 2017;130(Supplement 1):5162.
10.Cancer Care Ontario (CCO). Cancer Care Ontario: funded evidence-informed regimens. 2021; https://www.cancercareontario.ca/en/drugformulary/regimens. Accessed 2021 Oct 21.
11.Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood. 2011;117(10):2807-2812. PubMed
12.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Jan 10.
13.Calquence (acalabrutinib): 100 mg oral capsules [product monograph]. Mississauga (ON): AstraZeneca Canada Inc.; 2019 Aug 22: https://www.astrazeneca.ca/content/dam/az-ca/downloads/productinformation/calquence-product-monograph-en.pdf. Accessed 2021 Nov 18.
14.Bortezomib (as mannitol boronic ester): 3.5 mg sterile lyophilized powder for intravenous or subcutaneous injection [product monograph]. Toronto (ON): Teva Canada Limited; 2016 May 26: https://pdf.hres.ca/dpd_pm/00035071.PDF. Accessed 2021 Nov 18.
15.Treanda (bendamustine hydrochloride): 25 mg/ vial or 100 mg/ vial of lyophilized powder for intravenous injection [product monograph]. Toronto (ON): Teva Canada Limited; 2018 Jan 10: https://pdf.hres.ca/dpd_pm/00043152.PDF. Accessed 2021 Nov 18.
16.Rituxan (rituximab): 10 mg/mL intravenous infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd.; 2021 Jan 28: https://www.rochecanada.com/PMs/Rituxan/RituxanIV_PM_E.pdf. Accessed 2021 Dec 22.
17.Cytarabine injection (cytarabine): 100 mg/mL solution for intravenous infusion, subcutaneous injection, or intrathecal injection [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2020 Mar 13: https://www.pfizer.ca/sites/default/files/202006/Cytarabine_PM_E_13Mar2020_L3.pdf. Accessed Nov 18 2021.
18.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Brukinsa (zanubrutinib) 80 mg oral capsules. Mississauga (ON): BeiGene (Canada) ULC; 2021 Oct 21.
19.Brenner DR, Weir HK, Demers AA, et al. Projected estimates of cancer in Canada in 2020. CMAJ. 2020;192(9):E199-E205. PubMed
20.Mantle cell lymphoma. Danbury (CT): National Organization for Rare Disorders (NORD); 2021: https://rarediseases.org/rare-diseases/mantle-cell-lymphoma/. Accessed 2021 Nov 08.
21.Table 17-10-0005-01 Population estimates on July 1st, by age and sex. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2021 Aug 19.
Appendix 1: Additional Economic Information
Note that this appendix has not been copy-edited.
Cost Comparison Table
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: CADTH Cost Comparison Table for Refractory or Relapsed Mantle Cell Lymphoma
Treatment | Strength | Form | Price ($) | Recommended dosage | Average daily cost ($) | 28-day cost ($) |
|---|---|---|---|---|---|---|
Zanubrutinib (Brukinsa) | 80 mg | Capsule | 67.9833a | 320 mg once daily or 160 mg twice daily | 271.93 | 7,614 |
BTK inhibitors | ||||||
Ibrutinib | 140 mg | Capsule | 99.8350b | 560 mg once daily | 399.34 | 11,182 |
Acalabrutinib | 100 mg | Capsule | 135.9750c,d | 100 mg twice daily | 271.95 | 7,614 |
Chemotherapy or chemo-immunotherapy regimens | ||||||
Bortezomib | 3.5 mg | Vial IV infusion | 186.9457c,d | 1.3 mg/m2 on days 1, 4, 8, and 11 q.3.w.f | 83.32 | 2,333 |
Bortezomib (monotherapy) | 83.32 | 2,333 | ||||
Bortezomib | 3.5 mg | Vial IV infusion | 186.9457c | 1.6 mg/m2 on days 1, 8, 15, and 22 of | 61.53 | 1,723 |
Rituximab | 10 mg/mL | Vial IV infusion 10 mL 50 mL | 297.0000 1,485.0000 | 375 mg/m2 on days 1, 8, 15, and 22 of | 237.60 | 6,653 |
Bortezomib + rituximab | 290.65 | 8,376 | ||||
Bendamustine | 25 mg 100 mg | Vial IV infusion | 250.000c 1,000.00c | 70 mg/m2 of bendamustine on days 2 and 3 q.4.w.f | 90.00 | 2,520 |
Rituximab | 10 mg/mL | Vial IV infusion 10 mL 50 mL | 297.0000 1,485.0000 | 375 mg/m2 of rituximab on day 1 q.4.w. f | 71.60 | 2,005 |
Cytarabine | 100 mg/mL | 10 mL 20 mL | 153.2500 306.5000 | 500 to 800 mg/m2 of cytarabine on days 2 to 4 q.4.w.f | 14.78 to 23.64 | 414 to 662 |
Bendamustine + rituximab + cytarabine | 176.38 to 185.24 | 4,939 to 5,187 | ||||
Bendamustine | 25 mg 100 mg | Vial IV infusion | 250.000 1,000.000 | 90 mg/m2 bendamustine on days 1 and 4 q.4.w.g | 115.71 | 3,240 |
Rituximab | 10 mg/mL | Vial IV infusion 10 mL 50 mL | 297.0000 1,485.0000 | 375 mg/m2 rituximab on day 1 q.4.w.g | 71.60 | 2,005 |
Bortezomib | 3.5 mg | Vial IV infusion | 186.9457 c | 1.3 mg/m2 bortezomib on days 1, 4, 8, 11 q.4.w.g | 62.49 | 1,750 |
Bendamustine + rituximab + bortezomib | 249.81 | 6,995 | ||||
q.3.w. = every 3 weeks; q.4.w. = every 4 weeks.
Note: All prices are from the Ontario Drug Benefit Formulary12 (accessed November 18 2021), unless otherwise indicated, do not include dispensing fees and do not assume vial sharing. Dosage is based on Health Canada product monographs,13-17 Cancer Care Ontario formulary10 and published literature. CADTH assumed 70 kg or 1.8m2.
Note 2: Treatment with other proteasome inhibitor, lenalidomide, and the mTOR inhibitor, temsirolimus, were identified by clinical input received for this review but are not funded by any provincial cancer agencies. According to clinician input, anti-CD19 chimeric antigen receptor (CAR) T-cell therapy (Tecartus, Gilead) has been Health Canada–approved but currently remains unfunded at a provincial level.
aSponsor’s submitted price.1
bOntario Exceptional Access Program7 (accessed November 18, 2021).
cIQVIA Delta PA database8 (accessed November 9, 2021).
dAcalabrutinib and bortezomib is approved by Health Canada but not currently publicly funded.
eRecommended dosage obtained from published literature9 and verified by clinical experts consulted for this review by CADTH.
fCancer Care Ontario formulary10 (accessed November 18, 2021).
gRecommended dosage obtained from published literature11 and verified by clinical experts consulted for this review by CADTH.
Appendix 2: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 5: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
In the submitted budget impact analysis (BIA),18 the sponsor assessed the expected budget impact of reimbursing zanubrutinib for the treatment of adult patients with MCL who have received at least one prior therapy in Canada. The BIA was undertaken from the perspective of the Canadian public payer, over a 3-year time horizon (2022-2024), and included only drug acquisition costs. Pharmacy markup and dispensing fees were not included.
The analytic framework, which used an epidemiological-based approach, leveraged data from published literature19-21 to estimate the number of patients eligible for zanubrutinib. The sponsor included ibrutinib as the only comparator of interest under the reference scenario. To estimate the annual number of new (incident) patients with non-Hodgkin’s lymphoma among the total Canadian population, the sponsor adopted an incidence rate of 24.4 in 100,000 persons.19 The sponsor assumed an incidence rate of 6% of MCL among patients with non-Hodgkin’s lymphoma based on published literature.20 The sponsor assumed all MCL patients relapse and/or become refractory, and have public coverage. Among this population, the sponsor assumed 70% of refractory/relapsed MCL patients are treated with a BTK inhibitor based on expert opinion,18 and that zanubrutinib would only be used in this manner, and not displace other treatments.
The recommended doses of ibrutinib and zanubrutinib were as per product monographs.3,4 The cost of zanubrutinib was based on the sponsors submitted price,18 while the cost of ibrutinib was based on the public list price.7 Key inputs to the BIA are documented in Table 6.
Table 6: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Total populationa | 30,109,734 / 31,244,773 / 31,618,06721 |
Incidence rate of non-Hodgkin lymphoma (age-standardized) | 24.4 in 100,00019 |
Proportion of MCL cases | 6% |
Proportion of relapsed and refractory cases | 100% |
Proportion covered by payer | 100% |
Proportion treated with a BTK inhibitor | 70% |
Number of patients eligible for drug under review | 311 / 315 / 319 |
Market uptake (3 years) | |
Uptake (reference scenario) Ibrutinib | 100% / 100% / 100% |
Uptake (new drug scenario) Zanubrutinib Ibrutinib | 15% / 30% / 50% 85% / 70% / 50% |
Cost of treatment (per patient) | |
Cost of treatment over 1 year Zanubrutinib Ibrutinib | $99,256 $145,759 |
aIncludes participating programs (Newfoundland and Labrador, Prince Edward Island, Nova Scotia, New Brunswick, Ontario, Manitoba, Saskatchewan, Alberta, and British Columbia).
Summary of the Sponsor’s BIA Results
The sponsor estimated that the introduction of zanubrutinib for adult patients with MCL who have received at least one prior therapy in Canada resulted in a cost saving of $2,166,432 in year 1, $4,389,008 in year 2, and $7,408,585 in year 3, for an overall 3-year budget savings of $13,964,025 to the public payer.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
There is uncertainty in estimated population size and displacement of ibrutinib: According to the clinical experts consulted for this review by CADTH, not all MCL patients become refractory or relapsed because some patients are not treated in the first place. The clinical experts noted some indolent MCL patients are initially watched; some patients may not get treated with first-line therapy and some patients die from comorbidities before relapsing. As such, the clinical experts noted that the proportion of MCL cases that become refractory or relapsed would be less than 100% which may overestimate the cost savings from reimbursing zanubrutinib.
In CADTH scenario analysis, the budget impact associated with zanubrutinib was estimated by assuming arbitrary 90% of RR MCL patients become refractory or relapsed.
Further, the clinical experts noted that the proportion of patients treated with a BTK inhibitor may be in the range of 50% to 70%. Should fewer patients be treated with a BTK inhibitor, the cost savings from reimbursing zanubrutinib may be overestimated.
In scenario analysis, CADTH explored the impact of assuming 50% of patients are treated with a BTK inhibitor.
As the sponsor’s population estimate included the proportion of patients treated with a BTK inhibitor, the sponsor assumed 100% of market share is captured by ibrutinib. The public drug plan and clinical input received for this review identified other treatments that may be used to treat a subset of patients with late relapse. As such, some relevant comparators may have been excluded from the market mix by the sponsor and it is unclear whether zanubrutinib would only displace ibrutinib in all clinical situations across all jurisdictions. Should zanubrutinib displace treatments other than ibrutinib, such as chemotherapy regimens or immuno-chemotherapy regimens, zanubrutinib may no longer be a cost saving, depending on the magnitude of treatment displacement.
• The sponsor also assumed no total market growth (i.e., market share uptake for zanubrutinib only comes from patients on ibrutinib). The clinical experts noted that some clinicians may prefer zanubrutinib over ibrutinib due to difference in the adverse event profile. Should this change the size of the patient population, the estimated cost savings associated with zanubrutinib may not be realized.
CADTH is unable to address this limitation.
There is uncertainty in estimated cost savings in scenarios where patients switch treatment: The clinical experts consulted for this review by CADTH noted that there may be a subset of patients who switch treatment to zanubrutinib after experiencing adverse events while on ibrutinib (although continuing to respond to treatment). Clinical experts consulted by CADTH estimated that this subset of patients may be in the range of 10% to 20% who exhibit intolerance to ibrutinib and discontinue treatment with ibrutinib before disease progression. If zanubrutinib is reimbursed, these patients, who otherwise would have discontinued treatment with a BTK inhibitor, would now persevere on a BTK inhibitor. Should patients switch from ibrutinib to zanubrutinib, these patients would accrue both the costs of ibrutinib (until treatment switch) and zanubrutinib (post-switch), and therefore, zanubrutinib may no longer lead to the estimated cost savings.
CADTH is unable to address this limitation.
The price of drugs paid for by public drug plans is uncertain: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. As ibrutinib has gone through negotiations at pCPA, the prices paid by public drug plans are not known.
This limitation could not be addressed by CADTH. Confidential negotiated prices for ibrutinib, may lead to budgetary savings being limited or eliminated.
CADTH Reanalyses of the BIA
CADTH did not undertake a base case reanalysis. Instead, CADTH conducted several scenario analyses which included:
Assuming 90% of MCL patients become refractory or relapsed.
Assuming 50% of patients are treated with a BTK inhibitor.
Results are presented in Table 7. The reimbursement of zanubrutinib was associated with cost savings in all scenario analyses. Savings decreased as the proportion of MCL patients who become refractory or relapse decreased or the proportion of patients treated with a BTK inhibitor decreased but not substantially.
The reimbursement of zanubrutinib is a cost saving at the publicly available prices. However, if the reimbursement of zanubrutinib allows patients to preserve on a BTK inhibitor or displaces treatments other than ibrutinib, zanubrutinib may not be cost saving. CADTH was not able to address these limitations.
Table 7: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $44,682,803 | $45,269,380 | $45,855,958 | $46,442,535 | $137,567,873 |
New drug | $44,682,803 | $43,102,948 | $41,466,950 | $39,033,950 | $123,603,848 | |
Budget impact | $0 | -$2,166,432 | -$4,389,008 | -$7,408,585 | -$13,964,025 | |
CADTH scenario analysis: 90% become refractory or relapse | Reference | $40,214,523 | $40,742,442 | $41,270,362 | $41,798,282 | $123,811,086 |
New drug | $40,214,523 | $38,792,653 | $37,320,255 | $35,130,555 | $111,243,463 | |
Budget impact | $0 | -$1,949,789 | -$3,950,107 | -$6,667,726 | -$12,567,623 | |
CADTH scenario analysis: 50% treated | Reference | $31,916,288 | $32,335,272 | $32,754,256 | $33,173,239 | $98,262,767 |
New drug | $31,916,288 | $30,787,820 | $29,619,250 | $27,881,393 | $88,288,463 | |
Budget impact | $0 | -$1,547,452 | -$3,135,006 | -$5,291,846 | -$9,974,304 |
BIA = budget impact analysis
Stakeholder Input
Patient Input
Lymphoma Canada
About Lymphoma Canada
Lymphoma Canada is a national Canadian registered charity that empowers the lymphoma community through education, support, advocacy, and research. Based out of Toronto (ON), we collaborate with patients, caregivers, healthcare professionals, and other organizations and stakeholders, to promote early detecting, find new and better treatments for lymphoma patients, help patients access those treatments, learn about the causes of lymphoma, and working together to find a cure. Resources are provided for both English and French Canadians. For more information about our organization, please visit us at www.lymphoma.ca
Information Gathering
Lymphoma Canada (LC) conducted two anonymous online survey of Mantle Cell Lymphoma (MCL) patients between October 19 2020 – January 11, 2021, and between September 20, 2021 – October 20, 2021. Links to the surveys were sent via e-mail to patients registered through the LC database. The links were also made available via LC Twitter and Facebook accounts, Canadian and American Cancer Society message boards, Facebook groups organized for lymphoma patients and survivors, physician specialists across Canada, physicians at leading clinical trial sites across the United States of America (USA), and international lymphoma organizations’ individual contacts. The survey involved a combination of multiple choice, rating and open‐ended questions. Skipping logic was built into surveys so that respondents were asked questions only relevant to them. Open-ended responses to surveys that reflected the sentiment of a majority are included verbatim to provide a deeper understanding of patient perspectives.
There were 33 patients that provided input on their experience with their Mantle Cell Lymphoma from the first survey, and 52 patients that provided their experience with MCL from the second survey; there were 2 patients able to provide their experience with the Zanubrutinib. As there were no clinical trial sites in Canada where patients could gain access to this treatment, patient sampling was extended outside of Canada for therapy experience. Though LC had tremendous difficulty finding patients with MCL that received zanubrutinib, there were two Canadian patients that were able to provide their feedback. There were no caregivers that participated in this survey. Of patients who provided their demographic information (see Tables 1 and 2), the majority lived in Canada (59%), 58% are female, and 40% of patients are over the 65 years.
Table 1: Country of Survey Respondents (85 Respondents)
Respondents | CAN | USA | Australia/New Zealand | Europe | Skipped | Total |
|---|---|---|---|---|---|---|
Patients WITHOUT Zanubrutinib experience | 50 | 7 | 4 | 3 | 19 | 83 |
Patients WITH Zanubrutinib experience | 2 | 0 | 0 | 0 | 0 | 2 |
Table 2: Gender and Age of Survey Respondents (85 Respondents)
Respondents | Age Range | Gender | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
45-54 | 55-64 | 65-74 | 75-84 | Skipped | Female | Male | Skipped | ||
Patients WITHOUT Zanubrutinib experience | 10 | 21 | 26 | 6 | 20 | 29 | 34 | 20 | 83 |
Patients WITH Zanubrutinib experience | 0 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 2 |
Disease Experience
Patients were asked about symptom experience related to their mantle cell lymphoma and the impact on their quality of life at diagnosis and currently. On a scale of 1 – 5 (1- no impact, 5- significant negative impact), patients had the following symptom experience at diagnosis and currently. The most common symptoms negatively impacting patients both at diagnosis and currently include fatigue, enlarged spleen/lymph nodes, and symptoms caused by blood count impacts (Table 3). Therefore, it is important for treatment options for MCL patients to be able to manage and improve these symptoms.
Table 3: Symptom Experience and Impact at Diagnosis
MCL Symptom Experience at Diagnosis (n=85) | MCL Symptom Experience Currently (n=75) | ||||
|---|---|---|---|---|---|
MCL Symptom | No Impact | Significant Negative Impact (4-5) | MCL Symptom | Did not Experience | Significant Negative Impact (4-5) |
Enlarged Lymph Node(s) | 23% | 33% | Aches and Pain | 21% | 14% |
Enlarged Spleen | 20% | 24% | Enlarged Spleen | 32% | 8% |
Fatigue | 8% | 38% | Fatigue | 17% | 20% |
Indigestion, abdominal pain or bloating | 9% | 21% | Low red blood cell count | 32% | 8% |
Low red blood cell count | 20% | 19% | Reduced appetite | 32% | 8% |
Of these MCL symptoms, patients were asked what aspects of their life were impacted by these symptoms. Patients stated that their symptoms most greatly impacted their ability to travel, ability to exercise, ability to concentrate, ability to perform daily activities like household chores, and their ability to perform regular duties like work or volunteer.
Respondents were asked which aspects of their life, including mental and emotional problems associated with their disease, have NEGATIVELY impacted their quality of life at diagnosis compared to currently (at the time of survey completion). All respondents (n=85) rated that their quality of life (QoL) was impacted. The most common impacts to a patients QoL as a result of their MCL include the stress of their diagnosis (even later on into their MCL journey), anxiety and worry, and difficulty sleeping (Table 4).
Table 4: Impacts to QoL at Diagnosis and Currently
Impacts to QoL Diagnosis (n=85) | Impacts to QoL Currently (n=75) | ||
|---|---|---|---|
Impact | Percentage | Impact | Percentage |
Stress of Diagnosis | 82% | Anxiety/Worry | 53% |
Anxiety/Worry | 71% | Stress of Diagnosis | 40% |
Difficulty Sleeping | 34% | Difficulty Sleeping | 32% |
Frequency of Healthcare Appts. | 28% | Problems Concentrating | 26% |
Loss of Sexual Desire | 27% | Memory Loss | 24% |
As reported by MCL patients:
“Was told prognosis very poor with MCL and is an incurable disease. Was on an emotional roller coaster thinking I didn't have much time left. The sense of loss at the time was overwhelming. I'd never see my kids graduate, get married and have grand kids. I'd never grow old with my lovely wife and never accomplish so many things ahead of me.” – Anonymous MCL patient
“I had young children at home to care for. It was a stressful time and I worried about what would happen to them through my treatment and if I did not survive.” – Anonymous MCL patient
Further, with these many negative impacts to QoL, these can be further exacerbated by COVID- 19 and the need for treatment in clinic. As there is no end is site with the ongoing pandemic, it is important to consider the impacts of the pandemic. As one patient indicated:
“COVID response has increased negative impacts to quality of life, in conjunction with the chemo and immunotherapy which increases COVID risk.” – Anonymous MCL patient
Experiences With Currently Available Treatments
67 respondents provided information about their experience with MCL treatments. Following diagnosis 78% of patients required immediate treatment, while 22% remained in Watch & Wait. Of those in Watch and Wait (W&W), the average length of W&W for these patients thus far is 21 months. Though patients in W&W do not require treatment right away, the majority of diagnosed MCL patients often require treatment right away and options in both frontline and relapsed/refractory settings need to be available and accessible. As reported by one patient:
“Watch and wait is stressful because I don't know when I will get treatment. Having other options other that Chemo and SCT would be great.” - Anonymous MCL patient
The most commonly reported first-line treatment was stem-cell transplantation (46%), followed by the chemoimmunotherapy regimen R-CHOP (36%), Bendamustine-Rituximab (30%), radiation (19%), and BEAM therapy (15%) (67 respondents). 27% of patients received rituximab maintenance therapy following first line treatment to extend remission outcome.
Of these patients that received frontline therapy, 21% had relapsed, thus requiring further lines of treatment (63 respondents). Of those who received further therapy, the most commonly reported therapy received were BTK inhibitors, which were received in second-line, third-line, fourth-line and fifth-line for MCL patients (34 respondents). Other treatment options were variable and included stem-cell transplantation, CAR T-cell therapy, or further chemotherapy. This indicates important use of BTK inhibitors in the relapsed/refractory setting for MCL patients.
On a scale of 1 to 10 (1- strongly disagree, 10 – strongly agree), 82% patients agreed (rating of 8-10) that their most recent therapy, including BTK inhibitors, was able to manage their MCL symptoms (67 respondents).
Side effects of current MCL treatments: The most common side effects respondents experienced during their MCL treatments are listed in Table 5. Only 7% of patients did not experience treatment related side effects.
Table 5: Side Effects From Treatment (67 Respondents)
Side effect | % of resp. | Side effect | % of resp. |
|---|---|---|---|
Fatigue | 55% | Confusion/Memory Loss | 36% |
Hair Loss | 52% | Neutropenia | 36% |
Thrombocytopenia | 40% | Anemia | 33% |
Diarrhea | 39% | Mouth Sores | 27% |
Nausea/vomiting | 36% | Constipation | 27% |
When asked which side effects they found most difficult to tolerate, respondents most often reported fatigue, nausea/vomiting, neurocognitive effects such as brain fog or headaches, and hair loss were the most difficult to handle (49 respondents). As reported by patients:
“Don’t have as much energy as before. I need to rest a bit in the afternoon otherwise feel very tired.” - Anonymous MCL patient
“The treatment regimen was aggressive. Although thankfully in a long-term remission, the side effects I still live with today are significant. This includes peripheral neuropathy in fingers and toes, cardiomyopathy, chronic sinusitis, brain fog and fatigue, PTSD (undiagnosed). This has impacted my physical well-being, emotional health, personal relationships, ability to support family and career / financial independence.” - Anonymous MCL patient
“I work full time. Since having MCL and going through chemo, I have a harder time concentrating and focusing.” - Anonymous MCL patient
Impact of treatments on quality of life: When asked about the impact of various aspects of treatment on daily living (on a scale of 1 – 5, where 1= No impact and 5 = significant negative impact), respondents noted that treatment-related fatigue, low activity level, infusion related impacts such as length of infusion and reactions, and other late side effects of treatment had the most significant impact on patients quality of life (Table 6).
Table 6: Impact of Treatment on Quality of Life (67 Respondents)
Treatment aspect | Significant negative impact (rating = 3-5) |
|---|---|
Treatment-related Fatigue | 61% |
Low Activity/Exercise Level | 48% |
Infusion time | 43% |
Side Effects of Treatment | 43% |
Number of Clinic visits | 42% |
As noted from these results, infusion time and the number of clinic visits required have similar negative impacts to patient’s QoL as would treatment-related side effects. Therefore, treatment administration is an important consideration for patients, and ease and simplicity of treatment administration should be noted.
Access and Financial Impacts of treatments on quality of life: 75% of MCL patients could access treatment locally (68 respondents). For the 25% of patients that could not, the main reasons were that the treatment was not available at their local cancer centre and travel was required (12%), or that they live in a community without a cancer centre (4%).
For certain treatments, or if the treatment was not available locally, patients may be required to be away from home for a certain amount of time. 52% of patients did not have to stay away from home, however for the remaining patients that did, the majority (33%) were away for up to a month (66 respondents). Being away from home and support systems can have a significant negative impact to patients as they go through their treatment. For those patients that were unable to access treatment locally, this caused psychosocial impacts such as emotional hardship (15%) and worry over survival/prognosis (17%), as well as negative impacts to relationships and daily activities (10%) (41 respondents).
Patients were asked to select all financial implications that their MCL treatment has caused. Though 32% of patients did not experience any financial implications of their treatment, and 12% of respondents have not received treatment yet, the remaining patient population did experience financial impacts of their MCL treatment (68 respondents). These included absence from work/school (35%), travelling costs (24%), and drug and supplementary costs (18%). It is important to understand the impacts of accessing and affording treatment so that future treatments may address these challenges. As reported by two patients:
“I have taken an unpaid leave of 7 months this year which greatly impacted income.” - Anonymous MCL patient
“Could not work for 13 months owing to hospital commitments.” - Anonymous MCL patient
Improved Outcomes
Patient preferences: Respondents were asked to rate on a scale of 1 (not important) to 5 (extremely important), how important it is for a new MCL drug to be able to control various aspects of the patient’s disease. “Faster remission” and “allowing the patient to live longer” than current therapies were rated as the most important outcomes for a new therapy (Table 7).
Table 7: Treatment Preferences (67 Respondents)
Treatment outcome or factor | Very – Extremely Important (4-5) |
|---|---|
Bring about remission | 97% |
Allow me to live longer | 97% |
Control disease and symptoms | 93% |
Improve Quality of life | 87% |
Fewer Side Effects | 76% |
As reported by one patient:
“I want the most effective treatment that will provide the longest remission with the least side effects.” - Anonymous MCL patient
Respondents were asked if they would be willing to tolerate the side effects of a new treatment if they were short term. 69% of respondents would be willing to tolerate potential side effects, while 29% were not sure; only 2% of patients said no (65 respondents). Respondents were also asked if they would choose a treatment with known side effects, potentially serious, if their doctor recommended it was the best option for them. Of the 65 respondents who answered this question, 60% selected “Yes”, while the remaining were unsure (37%); 3% stated no. This indicates a lot of trust in the patient’s doctor and the importance of coming to an agreement on treatment decisions together to ensure they align with MCL patient’s values and needs.
On average, patients rated the importance of having a choice in their treatment selection as 8.2 on a scale from 1 to 10 (1- not important, 10 being extremely important). Having a number of options are important to patients as this helps them to see that they have more therapies available to receive in case they relapse (65 respondents). The large majority of patient’s (88%) further agree that there is a need for more effective therapy options. As reported by one patient:
“I worry about relapse and about the effectiveness and my eligibility to future treatment options.” - Anonymous MCL patient
Patient’s listed which MCL symptoms would be most important for new treatments to control. This includes abdominal discomfort (nausea/diarrhea/constipation) (73%), enlarged lymph nodes or spleen (71%), changes in vision (66%), aches/body or joint pain (63%), and headaches or cognitive changes (63%) (41 respondents).
There can be a different burden placed on patients for those who receive intravenous therapy compared to those receiving oral therapies. Burden can include different treatment-related side effects, length of treatment administration, impacts due to travel and frequency of infusions, etc. Patients were asked if they would rather receive treatment orally rather than intravenously. 78% of patients would prefer a pill option (41 respondents), while 15% were unsure and wanted more information. As reported by patients related to oral therapy:
“Quicker, less travel requirements.” - Anonymous MCL patient
“It is much easier to be treated in the comfort of my home with oral chemotherapy.” - Anonymous MCL patient
“It would be more convenient, and I could take it myself at home.” - Anonymous MCL patient
Patients further described their expectations for new treatment options:
“I think more research needs to be done on the delivery of personalized cancer care to ensure that I will receive the right therapy at the right time. To me efficacy and quality of life associated with new therapies are both very important to the patient and should be duly recognized in the drug approval process. The patient perspective is crucial.” - Anonymous MCL patient
“It is important that new treatments be accessible to people over 70 as currently there are age cutoffs.” - Anonymous MCL patient
Experience With Drug Under Review
Zanubrutinib experience was provided by two Canadian patients. Both patients were able to access treatment locally and there was no financial impact from receiving treatment. Details related to specific access and treatment history can be found in Table 8.
Table 8: Treatment Experience with Zanubrutinib (2 Respondents)
Patient # | Gender | Age | Date Received | Access | Previous Treatment Experience | Stage of Receipt |
|---|---|---|---|---|---|---|
1 | M | 75-84 | Aug-21 | Compassionate Access Program | BR Zanubrutinib *received as 2nd-line therapy | Still receiving |
2 | M | 75-84 | Aug-21 | Clinical Trial | CHOP Zanubrutinib *received as 2nd-line therapy | Still receiving |
Symptom Experience: Both patients had 100% of their MCL symptoms resolved with taking Zanubrutinib. Specific symptoms that Zanubrutinib was able to manage included fatigue, indigestion/abdominal pain/bloating, resolve blood cell counts (platelets, RBC, WBC), and improve weight loss/appetite. As both of these patients have indicated:
“Before taking Zanubrutinib, I was having trouble breathing. Within a week, my breathing was normal.” - Anonymous MCL patient
“In general, zanubrutinib is well tolerated and I feel stronger, have better appetite.” - Anonymous MCL patient
Side Effect Experience: Patient’s experienced the following side effects from Zanubrutinib treatment: easy bruising/bleeding (n=2), rash/itching (n=1), ache and joint pain (n=1), peripheral neuropathy (n=1), and nausea/vomiting (n=1); both patients indicated that these side effects did not impact their quality of life. As one patient indicated:
“I’ve had a very quick positive response & few side effects. My life is back to normal.” Anonymous MCL patient
Treatment Experience and Impacts to QoL: Overall both patients did not experience any negative impacts to QoL related to treatment administration such as number of clinic visits required, length/frequency of taking the drug, and challenges with swallowing the pill.
Zanubrutinib instead has improved patients’ overall quality of life, by first and foremost improving both patients general fitness/health level as well as their mental health. Other improvements indicated included the ability to maintain family/friend relationships and intimate relationships and increasing the ability to continue daily activities and to travel.
Overall Experience and Comparison with Previous Treatments: Patients were asked whether Zanubrutinib was better or worse than past treatments for their MCL. Patients stated that:
Zanubrutinib managed/controlled their MCL symptoms better than previous treatments (n=1)
Zanubrutinib had fewer side effects compared to previous treatments (n=2)
With zanubrutinib, there was a faster and better response rate compared to previous treatments (n=2)
Needed to take zanubrutinib for a shorter amount of time then the length of past treatments (n=1)
Zanubrutinib negatively impacted my quality of life less then past treatments (n=2)
Overall Experience: Based on patients experience with zanubrutinib, 100% of patients would recommend it to other patients with relapsed/refractory MCL. Patients rated their overall experience with zanubrutinib as very good to excellent. Patients would 100% of the time take it again if their doctor recommended it was the best treatment option for them. As one patient indicated:
“It's the only treatment that works to date.” - Anonymous MCL patient
Companion Diagnostic Test
There is no companion diagnostic testing for use of this therapy.
Anything Else?
N/A
Patient Group Conflict of Interest Declaration — Lymphoma Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 9: Conflict of Interest Declaration for Lymphoma Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Beigene | — | — | — | X |
Janssen | — | — | — | X |
AstraZeneca | — | — | — | X |
Clinician Input
Lymphoma Canada
About Lymphoma Canada
Please describe the purpose of your organization. Include a link to your website (if applicable).
Lymphoma Canada, a national non-for-profit organization for Canadian lymphoma and CLL patients, and assisted in the administrative coordination of the group clinician response. Lymphoma Canada was not involved in the development of the content of the submission. For more information about Lymphoma Canada, please visit www.lymphoma.ca.
Information Gathering
Please describe how you gathered the information included in the submission.
Clinicians provided responses to the questions in the submission based on research results, clinical experience, and understanding of patient needs and challenges
Current Treatments
Describe the current treatment paradigm for the disease.
Mantle cell lymphoma (MCL) is a incurable subtype of non-Hodgkin’s lymphoma although there is some disease heterogeneity with both more indolent or more aggressive presentations possible. Historically, the median survival for patients with MCL was approximately three years but this has improved substantially through treatments defined by randomized controlled trials including immunochemotherapy with rituximab-based regimens, the use of autologous stem cell transplantation (ASCT) in eligible patients, and rituximab maintenance as part of primary therapy. The majority of MCLs behave more like the aggressive B cell lymphoma and requires aggressive treatment. Observation may be considered in asymptomatic patients if they have no other indication for therapy such as cytopenia’s related to lymphoma.
For younger patients often less than age 70 or with favourable comorbidity profiles, aggressive chemotherapy regimens including anthracycline-based chemotherapy combined with cytarabine-based chemotherapy are generally used for induction treatment and followed by consolidative ASCT. Maintenance therapy with rituximab is offered post treatment. For patients who are not eligible for a stem cell transplant or who are felt to have a more indolent variety of MCL, treatment with initial watchful waiting and less intense chemotherapy with Bendamustine and Rituximab (BR) would be offered as first line chemotherapy treatment upon development of symptomatic progressive disease. Rituximab maintenance is also offered to these patients. The median PFS for patients undergoing ASCT as part of primary therapy for MCL approaches 8-10 years while patients receiving R-chemo that are ineligible for transplant will have median PFS in the range of 3-5 years.
Second line therapies for patients with relapsed/refractory (RR) MCL are less defined. Novel drugs in MCL with proven single agent activity include the proteasome inhibitor bortezomib, lenalidomide and the mTOR inhibitor temsirolimus. While bortezomib is only funded in select provinces (Alberta), the other options of lenalidomide or temsirolimus are not funded by any provincial cancer agencies. Median PFS in these trials ranged between 6-12 months. Clinicians have employed these agents in the context of clinical trials and when compassionate access may be available. Chemotherapy (or immunochemotherapy with rituximab) was typically of less benefit historically as typical patients would relapse early after primary therapy and derive less benefit from these traditional approaches. However, with more modern treatment, there remains a smaller subset of patients who may be treated with immunochemotherapy in the setting of late relapse (ie. beyond 5 years) and may be expected to have favourable outcomes with a regimen such as bendamustine-rituximab if they have not been exposed to this as part of primary therapy. More recently, anti-CD19 CAR-T cell therapy (Tecartus, Gilead) has been Health Canada approved but currently remains unfunded at a provincial level.
The mainstay of therapy for RR-MCL is the BTK inhibitor ibrutinib or more recently, the second-generation agent acalabrutinib (Health Canada approved but not currently publicly funded). Ibrutinib is approved and funded for use in relapsed/refractory MCL based on a single arm pivotal trial and a subsequent confirmatory phase III trial against temsirolimus demonstrating significant benefit in (median 15 versus 6 months PFS with 3-year follow-up). Unfortunately, additional novel agents in RR-MCL are typically unavailable if patients experience toxicity or progression. Medical comorbidity (AF, hypertension etc.) that may limit the use of ibrutinib in CLL is typically managed more aggressively in MCL given the lack of alternative agents.
There is no clear standard beyond immunochemotherapy, autologous transplantation and BTK inhibitors in MCL. As the disease is typically incurable, patients will likely require all of these therapies through their lifetime if they maintain acceptable performance status and are medically fit for specific treatments. Clinicians may attempt to access unfunded targeted therapies or enrol patients in clinical trials. Allogeneic stem cell transplantation has been employed for younger patients that typically have disease progression following primary immunochemotherapy and BTK inhibitor therapy. Allogeneic stem cell transplantation may also be considered upfront in ultra-high risk subsets such as those with TP53 mutation, although this is not universally screened for.
Treatment Goals
What are the most important goals that an ideal treatment would address?
The most important goal of therapy for relapsed and refractory MCL is to produce clinical responses and remission that may prolong life. Relief of disease-related symptoms to improve health related quality of life is an important objective. Doing this in a fashion that is non-toxic would be preferable. Treatments that are finite and not continued indefinitely may be preferable to patients. Unfortunately, treatments in RR-MCL are typically given indefinitely until progression, associated with significant costs and toxicity and ultimately are not curative in intent.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
At present there are a limited number of options for patients with relapsed and refractory MCL. In addition, these options benefit only a fraction of patients (35-75%) and typically do not offer durable responses (approximately 6-18 months). Many of these treatments must be administered indefinitely toxicity may adversely affect quality of life. There are important considerations to improve on the toxicity profile of available standard of care agents. The toxicity of BTK inhibitors are well understood with the first-in-class (ibrutinib) product and the second publicly funded agent (acalabrutinib) in Canada and there remains an opportunity to improve the tolerability of BTK inhibitors for patients. Some of the BTK inhibitors have important and common drug-drug interactions which interfere with their use, so newer BTKi with less interactions would be of benefit.
Which patients have the greatest unmet need for an intervention such as the drug under review?
In patients with RR-MCL, there will be two patient populations – an older/frailer population that would not be eligible for more aggressive therapy and a patient population that will typically be younger, without comorbidity and with good performance status. When funding for CAR-T therapy is available, patients will likely require BTK inhibitor exposure and failure in order to be eligible to receive subsequent cell therapy. The majority of patients with RR-MCL would be treated with a BTK inhibitor. Ideally, as the agents appear to have very similar efficacy (ibrutinib, acalabrutinib, zanubrutinib), clinicians may use toxicity differences to select a specific agent for a specific patient. At a minimum, patients with contraindications to a specific agent may be better suited to an alternate agent based on the adverse event profile of the drug.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
BTK inhibitors are currently available in Canada with public funding across provinces. The use of zanubrutinib would be similar to other BTK inhibitors. Zanubrutinib could be used in patients with specific contraindications to acalabrutinib or ibrutinib or could be considered in patients based on a comparison of the toxicity profile of all three agents. The randomized ASPEN study in Waldenstrom’s macroglobulinemia highlights favourable cardiovascular toxicity event rates (in addition to most other BTK-associated toxicities) in patients receiving zanubrutinib when compared prospectively to ibrutinib. However, the incidence of atrial fibrillation in the Phase 1/2 study of zanubrutinib in MCL demonstrated similar rates (6%) as first generation BTKi's.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
There are two potential approaches here – patients could start zanubrutinib as the BTK inhibitor of choice (based on differences in toxicity profile or contraindications to other BTK inhibitors) or in patients that develop specific toxicities on other BTK inhibitors (where the rate of these events may be lower with zanubrutinib) in which switching to another agent to improve tolerance would be considered.
How would this drug affect the sequencing of therapies for the target condition?
The availability of zanubrutinib would not affect sequencing as it would be considered one of the BTK inhibitor options that could be considered when that type of therapy would be indicated
Which patients would be best suited for treatment with the drug under review?
Any MCL patient after primary therapy could be considered. The best suited patients would be those that have higher likelihood of experience toxicities such as cardiovascular events which appear to be lower in patients receiving zanubrutinib.
How would patients best suited for treatment with the drug under review be identified?
Patients who would be candidates for this therapy would be identified by the treating haematologist or oncologist. Progression after primary treatment may be identified clinically but is confirmed with laboratory and/or imaging findings. This is a concept setting for clinicians treating lymphoma and there are no likely issues. Due to the typically aggressive nature of MCL at relapse, 2nd line therapy is generally promptly initiated at first detection of relapse even if patients remain asymptomatic.
Which patients would be least suitable for treatment with the drug under review?
Patients with comorbid illnesses that represent contraindications to zanubrutinib including disorders/treatments associated with significant bleeding and/or cardiovascular disease would be patients that may be less suitable for this class of agent. Patient with obvious uncontrolled infections would not be acceptable candidates. Patients with extremely poor performance and low life expectancy (particularly for other reasons) may not be good candidates.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Currently no biomarkers to identify these patients.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Standard clinical parameters would be used to document clinical response including CT scans and possibly PET scans. Bloodwork and assessments of organ function and routine blood counts would also be important.
What would be considered a clinically meaningful response to treatment?
Clinically meaningful results to therapy include objective response to therapy or at a minimum stabilization of disease/lack of progression. Typically, this would be associated with improvement in disease-related symptoms. Success with this treatment would be expected to improve quality of life and independence in activities of daily living.
How often should treatment response be assessed?
Response to treatment should be assessed radiologically post treatment and several months again post treatment. Ongoing imaging may be dependent upon symptoms and the results of the previous testing, clinical findings as well as laboratory results. This would be no different than for the other BTK inhibitors available for treatment in this disease.
What factors should be considered when deciding to discontinue treatment?
Progression of disease (typically based on imaging or laboratory findings) would indicate treatment failure. Consideration of initiating a new treatment at that time would be appropriate. This could include CAR-T cell therapy in eligible patients when funded.
What settings are appropriate for treatment with the drug under review?
As an oral therapy that is well tolerated and third in class, zanubrutinib could be administered in any setting where cancer patients may be seen allowing for these patients to be treated and followed in their local community.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Not applicable
Additional Information
Is there any additional information you feel is pertinent to this review?
N/A
OH-CCO’s Drug Advisory Committee
About OH-CCO’s Drug Advisory Committee
Please describe the purpose of your organization. Include a link to your website (if applicable).
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
Please describe how you gathered the information included in the submission.
This input was jointly discussed via email.
Current Treatments
Describe the current treatment paradigm for the disease
In R/R MCL, ibrutinib, acalabrutinib (available via manufacturer compassionate program) or retreatment w/ rituximab-chemo or chemo alone (if rituximab refractory) are treatment options.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Increase survival, delay disease progression, symptom improvement, improve health-related quality of life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Ibrutinib is currently available. Zanubrutinib may have a more favourable toxicity profile. Both drugs delay disease progression and can improve survival.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Patient in whom ibrutinib may be associated with higher risk of toxicities (e.g., patients on anticoagulation or patients with cardiac comorbidities).
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
As an alternative to ibrutinib.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
No.
How would this drug affect the sequencing of therapies for the target condition?
It would not alter the sequencing. Zanubrutinib would be an alternative to ibrutinib.
Which patients would be best suited for treatment with the drug under review?
Would potentially be appropriate for all MCL patients. But better suited for patients with ibrutinib-related side effects.
How would patients best suited for treatment with the drug under review be identified?
Within routine practices of hematologists.
Which patients would be least suitable for treatment with the drug under review?
None.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
No.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Reduction in disease burden and symptoms. PFS aligns with outcome typically used in trials.
What would be considered a clinically meaningful response to treatment?
Significant reduction in lymphoma burden and improvement in symptoms
How often should treatment response be assessed?
Clinically every 1-2 months with imaging as required.
What factors should be considered when deciding to discontinue treatment?
Disease progression or treatment-related adverse events
What settings are appropriate for treatment with the drug under review?
Community setting – this is a take-home anticancer drug
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
N/A
Additional Information
Is there any additional information you feel is pertinent to this review?
N/A
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.