CADTH Reimbursement Review
Tafasitamab (Minjuvi)
Sponsor: Incyte Biosciences Canada Corporation
Therapeutic area: Diffuse large B-cell lymphoma (DLBCL)
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
BR
bendamustine plus rituximab
CAR T-cell
chimeric antigen receptor T-cell
CI
confidence interval
CNS
central nervous system
CR
complete response
CRR
complete response rate
CT
computed tomography
DLBCL
diffuse large B-cell lymphoma
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group performance status
EFS
event-free survival
ePS
estimated propensity score
EOT
end of treatment
ESMO
European Society for Medical Oncology
ESS
effective sample size
GCB
germinal centre B-cell like
HBV
hepatitis B virus
HDC
high-dose chemotherapy
HR
hazard ratio
HRQoL
health-related quality of life
IPI
International Prognostic Index
IRC
independent review committee
ITC
indirect treatment comparison
IV
intravenous
IWG
International Working Group
LC
Lymphoma Canada
LDH
lactate dehydrogenase
MAIC
matching-adjusted indirect comparison
MAS
matched analysis set
NCCN
National Comprehensive Cancer Network
NHL
non-Hodgkin lymphoma
NN
nearest neighbour
NR
not reached
OH-CCO
Ontario Health – Cancer Care Ontario)
ORR
objective response rate
OS
overall survival
PD
progressive disease
pERC
pCODR Expert Review Committee
PET
positron emission tomography
PFS
progression-free survival
PMBL
primary mediastinal large B-cell lymphoma
Pola-BR
polatuzumab vedotin plus bendamustine plus rituximab
PML
progressive multifocal leukoencephalopathy
PR
partial response
PRR
partial response rate
R-CHOP
rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
R-GemOx
rituximab plus gemcitabine plus oxaliplatin
R/R
relapsed or refractory
SAE
serious adverse event
SD
standard deviation
SMD
standardized mean difference
TTNT
time to next treatment
TTP
time to progression
TTR
time to response
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Tafasitamab (Minjuvi), 200 mg single-use vial, 12 mg/kg body weight, IV infusion |
Indication | Indicated in combination with lenalidomide for the treatment of adult patients with relapsed or refractory DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT |
Reimbursement request | As per indication |
Health Canada Approval status | NOC/c |
Health Canada Review pathway | Advance consideration under NOC/c |
NOC/c date | August 19, 2021 |
Sponsor | Incyte Biosciences Canada Corporation |
ASCT = autologous stem cell transplant; DLBCL = diffuse large B-cell lymphoma; IV = IV; NOC/c = Notice of Compliance with Conditions.
Introduction
Non-Hodgkin lymphoma (NHL) is a cancer of the immune system that encompasses more than 60 types of cancer affecting the lymphocytes.1 In 2021, it was estimated that 11,100 of people living in Canada would be diagnosed with NHL and 2,900 of those in Canada would die from NHL that year.2 Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of NHL, constituting 30% to 40% of cases in Canada.3,4 DLBCL represents a heterogeneous group of aggressive B-cell malignancies.4 Some types of indolent B-cell lymphomas can transform into DLBCL (e.g., follicular lymphoma).5 Although the cure rate of DLBCL is high, approximately 30% to 50% of patients in Canada experience relapsed or refractory (R/R) disease after treatment with standard first-line chemotherapy with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) or a similar regimen.4,6
Patients with R/R DLBCL have limited treatment options, ranging from supportive care to conventional salvage therapy and autologous stem cell transplant (ASCT). Eligibility for this salvage approach depends on performance status, age, and comorbidities, and eligibility for ASCT also depends on the response to salvage chemotherapy.4 The prognosis for patients with relapsed DLBCL who do not undergo high-dose therapy and ASCT is poor.4 Even for those patients who respond to salvage chemotherapy and undergo ASCT, 50% are likely to relapse following ASCT.4 In patients with R/R DLBCL who are not eligible for intensive therapies, there is no standard treatment approach. There are numerous chemotherapy options, but response rates are generally low and remission duration is short.4 Polatuzumab vedotin plus bendamustine plus rituximab (pola-BR) is an option for those living in Canada in this setting, if it is funded, according to the clinical experts consulted by CADTH.
Tafasitamab is an Fc-enhanced monoclonal antibody that targets the CD19 antigen expressed on the surface of pre-B and mature B lymphocytes and on several B-cell malignancies, including DLBCL.7 Tafasitamab is indicated in combination with lenalidomide for the treatment of adult patients with R/R DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT. The recommended dosage of tafasitamab is 12 mg/kg body weight administered as an IV (IV) infusion in 28-day cycles.7 According to the product monograph, tafasitamab should be administered with lenalidomide for up to 12 cycles. After a maximum of 12 cycles of combination therapy, patients receive tafasitamab infusions as monotherapy until disease progression or unacceptable toxicity. Tafasitamab has serious warnings and precautions in the Health Canada product monograph for infection, myelosuppression, progressive multifocal leukoencephalopathy (PML), and hepatitis B reactivation.
The objective of this review is to perform a systematic review of the beneficial and harmful effects of tafasitamab (200 mg single-use vial) in combination with lenalidomide for the treatment of adult patients with R/R DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT.
After the draft CADTH pan-Canadian Oncology Drug Review Expert Review Committee (pERC) recommendation for tafasitamab was issued in May 2022, the sponsor submitted additional post hoc analyses from the RE-MIND2 study. The results of these post hoc analyses are presented in Appendix 4. These data were not included in the initial submission to CADTH. After the CADTH recommendation was issued, the sponsor reported that the data became available only after its submission to CADTH.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
One patient advocacy group provided input on tafasitamab for the treatment of DLBCL in adult patients. Lymphoma Canada (LC) conducted 4 anonymous online surveys. Overall, 150 DLBCL patients responded to the surveys, of which 2 (1%) indicated they had received tafasitamab therapy. Commonly reported symptoms affecting patients’ health-related quality of life (HRQoL) at diagnosis included fatigue or lack of energy, enlarged lymph nodes, drenching night sweats, unexplained weight loss, loss of appetite, influenza-like symptoms, and persistent cough. Patients also described mental and emotional problems associated with their disease and treatment that negatively affected their quality of life. Patients rated longer survival and remission than current therapies and controlling disease symptoms as the most important outcomes for a new therapy. Better HRQoL and fewer side effects compared to current therapies were also important considerations.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts reported that the goal of treatment in patients with R/R DLBCL who are not eligible for intensive therapies (i.e., ASCT and or chimeric antigen receptor T-cell [CAR T-cell] therapy) is to control symptoms with minimal toxicity to improve HRQoL, delay disease progression, and prolong life. The clinical experts noted that ASCT and CAR T-cell therapy both have toxicity and feasibility issues that limit broad application. Of the available options for patients who are not eligible for intensive therapies, or who have a relapse after these therapies, there is no standard of care treatment and there is no treatment that is curative (i.e., patients are treated with palliative intent). According to the clinical experts, most currently used treatment options have short durations of response, if patients respond.
The clinical experts indicated that tafasitamab in combination with lenalidomide (tafasitamab plus lenalidomide) would be an option at relapse for second-line therapy in patients who are not eligible for intensive therapy. Tafasitamab plus lenalidomide treatment could also be used in the third-line or later setting for patients who have a relapse after ASCT.
The clinical experts thought that patients who would most likely to benefit from tafasitamab plus lenalidomide are those with relapsed DLBCL, including those with underlying indolent lymphomas. The clinical experts thought that tafasitamab plus lenalidomide may be considered in patients who are not eligible for ASCT or CAR T-cell therapy, or who decline either of these treatments. The clinical experts indicated that it is not possible to identify patients who are most likely to exhibit a response to tafasitamab plus lenalidomide before treatment because there are no data on which patient or tumour characteristics are optimal for this treatment compared to other options. The clinical experts thought that patients with primary refractory DLBCL would be least suitable for treatment with tafasitamab plus lenalidomide because these patients have been excluded from the pivotal L-MIND trial. In addition, the clinical experts noted that patients who cannot come in for frequent IV infusions would not be suitable for this treatment.
The clinical experts consulted by CADTH reported that standard of care for assessing treatment response is imaging with CT (CT) or PET-CT (PET-CT) every 3 to 4 months (or sooner if there is a change in patient’s clinical status) and clinical examination and bloodwork before each treatment. The clinical experts indicated that a clinically meaningful response to treatment would include improvement in survival as well as duration of response (DOR), which would usually correlate with improvement in symptom burden. According to the clinical experts, meaningful response would include complete response (CR), partial response (PR), or stable disease with a tolerable toxicity profile.
The clinical experts noted that any disease progression should be an indication for treatment discontinuation. The clinical experts thought that recurrent infections, serious infection due to B-cell depletion, and hypogammaglobulinemia may also be considerations for discontinuation.
Clinician Group Input
Clinician input on the review of tafasitamab for the treatment of adult patients with R/R DLBCL was received from 2 groups: the Ontario Health – Cancer Care Ontario (OH-CCO) Hematology Drug Advisory Committee and a group of 4 clinicians whose submission was coordinated by LC. The clinician groups agreed that tafasitamab plus lenalidomide would be recommended in patients with DLBCL who do not respond to or relapse after first-line therapies. There were differing opinions on which patients are unsuitable for tafasitamab. The clinicians from OH-COO stated that DLBCL patients who have progressed on CAR T therapies would be least suitable for this therapy, while the LC-coordinated group maintained that there are no specific parameters that make a patient unsuitable.
Drug Program Input
The drug plans noted that primary refractory disease was an exclusion criterion in the L-MIND study and sought clarification on the definition of primary refractory disease, and whether these patients would be eligible for treatment, if the drug were reimbursed. In response, the clinical experts consulted by CADTH noted that the L-MIND study definition of primary refractory disease changed mid-study, thus complicating analysis of benefits in this high-need patient population. In the original study protocol, only patients whose disease relapsed within 3 months of a previous anti-CD20–containing regimen were defined as having primary refractory disease and excluded. After the protocol amendment, primary refractory disease was defined as disease progressing during the course of the first-line treatment as per International Working Group (IWG) response criteria (Cheson et al., [2007])8 and/or showing a response of less than a PR to first-line treatment or disease recurrence or progression within less than 6 months from the completion of first-line therapy. The clinical experts indicated that patients with primary refractory DLBCL are unlikely to have chemosensitive disease to subsequent therapies; these patients have an extremely poor outcome. Due to the changing definition and exclusion criterion, the clinical experts thought that it is difficult to determine whether patients with primary refractory DLBCL should be treated with tafasitamab plus lenalidomide as second-line or later therapy. The clinical experts reported that the pivotal study of pola-BR9 did not specifically exclude this patient population, and, therefore, pola-BR may be a better treatment option for patients with primary refractory DLBCL. However, the clinical experts also noted that there are no standard of care treatment options for these patients with high unmet need; thus, they would consider offering tafasitamab plus lenalidomide to patients with primary refractory disease.
The drug plans also asked whether the following patients would be eligible for tafasitamab: patients with a history of double- or triple-hit genetics DLBCL, patients with central nervous system (CNS) lymphoma, and patients with other histological types of lymphoma (e.g., primary mediastinal large B-cell lymphoma [PMBL] or Burkitt lymphoma). The clinical experts noted that patients with known double- or triple-hit genetics lymphoma were excluded from the L-MIND trial; thus, they indicated they would favour treatment with pola-BR in this population. The clinical experts noted that patients with a history of double- or triple-hit genetics may respond to tafasitamab plus lenalidomide, but data are not currently available to support this. The clinical experts thought that patients with CNS involvement of lymphoma, PMBL, or Burkitt lymphoma should not be treated with tafasitamab plus lenalidomide because there is no evidence to support the use of this treatment in these patients.
The drug plans inquired about whether patients who have received more than 3 prior lines of treatment, but who would otherwise fit the L-MIND trial eligibility criteria, should be eligible for tafasitamab plus lenalidomide on a time-limited basis if reimbursed. The clinical experts consulted by CADTH thought that these patients should be eligible for tafasitamab plus lenalidomide, particularly if they had no prior access to a novel therapy (i.e., CAR T-cell therapy, pola-BR). The clinical experts thought that the number of prior lines of therapy should not affect a patient’s eligibility for treatment with tafasitamab plus lenalidomide.
The drug plans also asked about the sequencing of tafasitamab plus lenalidomide therapy with pola-BR and CAR T-cell therapy. In addition, the drug plans asked how clinicians would decide when to use tafasitamab plus lenalidomide therapy versus pola-BR. The clinical experts consulted by CADTH reported that there is no evidence from prospective clinical studies to guide sequencing of tafasitamab plus lenalidomide with pola-BR and CAR T-cell therapy. The clinical experts indicated they would use tafasitamab plus lenalidomide in patients previously treated with bendamustine or polatuzumab,however they noted that there is no evidence that re-treatment with these drugs is ineffective, and, currently, they do not use these drugs routinely. The clinical experts also thought that tafasitamab plus lenalidomide may be preferred over pola-BR in patients with existing peripheral neuropathy. In addition, the clinical experts noted that the duration of pola-BR treatment is limited, whereas treatment with tafasitamab plus lenalidomide is followed by tafasitamab monotherapy until disease progression or intolerable toxicity. The clinical experts thought that some patients may prefer a treatment option with limited duration. The clinical experts noted that, in Ontario, they do not have the option to proceed with funded CAR T-cell therapy after tafasitamab is given to a patient. The clinical experts reported that pola-BR can be used for bridging for CAR T-cell therapy.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
One single-arm, multi-centre, open-label, phase II study (L-MIND, N = 81) of tafasitamab plus lenalidomide in adult patients with DLBCL who had a relapse after or were refractory to 1 to 3 previous systemic regimens (with at least 1 anti-CD20 therapy), who were not candidates for high-dose chemotherapy (HDC) and subsequent ASCT, was included.10,11 The primary objective of the L-MIND study was to determine the activity of tafasitamab plus lenalidomide in terms of objective response rate (ORR) (CR + PR) in adult patients with R/R DLBCL. Patients received IV tafasitamab (12 mg/kg) and oral lenalidomide (25 mg/day) for up to 12 cycles (28 days each), followed by tafasitamab monotherapy in patients with stable disease or better until disease progression. The primary end point was ORR by independent review committee (IRC). Other efficacy outcomes assessed included ORR by investigator assessment, overall survival (OS), progression-free survival (PFS), time to progression (TTP), event-free survival (EFS), complete response rate (CRR), DOR, time to response (TTR), and time to next treatment (TTNT). Harms outcomes were also examined. HRQoL outcomes were not reported.
In the L-MIND study, the mean age of patients was 69.3 years. Most patients were White (88.9%), had Ann Arbor stage III or IV disease (75.3%), and did not have a prior ASCT (88.9%). Overall, 54.3% of the enrolled patients were male, 55.6% had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 1, 50.6% had an International Prognostic Index (IPI) score of 3 to 5, and 46.9% had disease of germinal centre B-cell like (GCB) cell origin by immunohistochemistry. Mean time since first DLBCL diagnosis was 39.6 months (standard deviation [SD] 34.8). All (100%) patients had at least 1 prior anticancer medication; 50.6% of patients had received 2 or more prior therapy lines; and 44.4% were refractory to their most recent previous therapy. The most common reasons for ASCT ineligibility were older age (46.3%) and chemorefractory status (22.5%).
Efficacy Results
Results for the key efficacy outcomes in the L-MIND study are summarized in Table 2. Three analyses were conducted based on 3 data cuts. The primary analysis had a data cut-off date of November 30, 2018.10,11 Two additional interim analyses, which were not pre-specified in the study protocol, were conducted, with data cut-off dates of November 30, 2019, and October 30, 2020.12-14 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||15
Table 2: Summary of Key Results From L-MIND
Outcome | FAS (N = 80) | ||
|---|---|---|---|
November 30, 2018, DCO (primary analysis) | November 30, 2019, DCO | October 30, 2020, DCO | |
OS (secondary) | |||
Patients who died, n (%) | 29 (36.3) | 37 (46.3) | 41 (51.3) |
Censored, n (%) | 51 (63.8) | 43 (53.8) | |||||||||||| |
Median OSa (95% CIb), months | NR (18.3 to NR) | 31.6 (18.3 to NR) | 33.5 (18.3 to NR) |
Median follow-up timec (95% CIb), months | 19.6 (15.3 to 21.9) | 31.8 (27.2 to 35.9) | 42.7 (||||||||||||) |
PFS by IRC (secondary) | |||
Experienced an event, n (%) | 39 (48.8) | 39 (48.8) | |||||||||||| |
Progression | 32 (40.0) | 32 (40.0) | |||||||||||| |
Death | 7 (8.8) | 7 (8.8) | |||||||||||| |
Censored, n (%) | 41 (51.3) | 41 (51.3) | |||||||||||| |
Median PFSa (95% CIb), months | 12.1 (5.7 to NR) | 16.2 (6.3 to NR) | 11.6 (6.3 to 45.7) |
Median follow-up timec (95% CIb), months | 17.3 (11.5 to 21.2) | 22.6 (22.2 to 27.4) | 33.9 (||||||||||||) |
TTP (secondary) | |||
Experienced an event, n (%) | 35 (43.8) | Not reported | Not reported |
Progression | 32 (40.0) | Not reported | Not reported |
Death due to lymphoma | 3 (3.8) | Not reported | Not reported |
Censored, n (%) | 45 (56.3) | Not reported | Not reported |
Median TTPa (95% CIb), months | 16.2 (7.4 to NR) | Not reported | Not reported |
EFS (exploratory) | |||
Experienced an event, n (%) | 46 (57.5) | Not reported | Not reported |
Progression | 32 (40.0) | Not reported | Not reported |
Death | 7 (8.8) | Not reported | Not reported |
New non-study antineoplastic treatment | 7 (8.8) | Not reported | Not reported |
Censored, n (%) | 34 (42.5) | Not reported | Not reported |
Median EFSa (95% CIb), months | 9.1 (5.3 to 21.0) | Not reported | Not reported |
Median follow-up timec (95% CIb), months | 19.7 (14.3 to 22.0) | Not reported | Not reported |
ORR by IRC (primary) | |||
ORR, n (%) [95% CId] | 48 (60.0) [48.4 to 70.8] | 47 (58.8) [47.2 to 69.6] | 46 (57.5) [45.9 to 68.5] |
Best objective response, n (%) | |||
CR | 34 (42.5) | 33 (41.3) | 32 (40.0) |
PR | 14 (17.5) | 14 (17.5) | 14 (17.5) |
DOR by IRC (secondary) | |||
Patients with response by IRC, n | 48 | 47 | |||||||||||| |
Patients with event, n (%) | 13 (27.1) | 13 (27.1) | |||||||||||| |
Progression | 12 (25.0) | 12 (25.5) | |||||||||||| |
Death | 1 (2.1) | 1 (2.1) | |||||||||||| |
Censored | 35 (72.9) | 34 (72.3) | |||||||||||| |
Median DORa (95% CIb), months | 21.7 (21.7 to NR) | 34.6 (26.1 to 34.6) | 43.9 (26.1 to NR) |
TTR (secondary) | |||
Median TTR (CR or PR), months (minimum, maximum) | 2.0 (1.7 to 16.8) | 2.0 (1.7 to 16.8) | Not reported |
Median time to CR, months (minimum, maximum) | 7.05 (1.7 to 17.0) | 4.00 (1.7 to 17.0) | Not reported |
TTNT (secondary) | |||
Experienced an event, n (%) | 43 (53.8) | 49 (61.3) | Not reported |
Next treatment | 27 (33.8) | 32 (40.0) | Not reported |
Death | 16 (20.0) | 17 (21.3) | Not reported |
Censored | 37 (46.3) | 31 (38.8) | Not reported |
Median TTNTa (95% CIb), months | 15.4 (7.6 to NR) | 12.5 (7.6 to 24.7) | Not reported |
Harms, n (%) – safety analysis set (N = 81) | |||
Aes | — | — | 81 (100) |
SAEs | — | — | 43 (53.1) |
WDAE (discontinuation of 1 or both study drugs) | — | — | |||||||||||| |
Deaths | — | — | 42 (51.9) |
Notable harms, n (%) – safety analysis set (N = 81) | |||
Infection | — | — | 59 (72.8) |
Bronchitis | — | — | 13 (16.0) |
Pneumonia | — | — | 10 (12.3) |
Urinary tract infection | — | — | 10 (12.3) |
Respiratory tract infection | — | — | 9 (11.1) |
Myelosuppression | — | — | |
Neutropenia | — | — | 41 (50.6) |
Anemia | — | — | 30 (37.0) |
Thrombocytopenia | — | — | 25 (30.9) |
Leukopenia | — | — | 12 (14.8) |
Febrile neutropenia | — | — | 10 (12.3) |
Lymphopenia | — | — | 6 (7.4) |
PML | — | — | 1 (1.2) |
Hepatitis B reactivation | — | — | |||||||||||| |
Infusion-related reactions | — | — | |||||||||||| |
Cytokine release syndrome | — | — | Not reportede |
Tumour lysis syndrome | — | — | Not reportede |
AE = adverse event; CI = confidence interval; CR = complete response; DCO = data cut-off; DOR = duration of response; EFS = event-free survival; FAS = full analysis set; IRC = independent review committee; NR = not reached; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PML = progressive multifocal leukoencephalopathy; PR = partial response; SAE = serious adverse event; TTNT = time to next treatment; TTP = time to progression; TTR = time to response; WDAE = withdrawal due to adverse event.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
cCalculated using the reverse Kaplan–Meier method, considering the censored patients as events and patients with events as censored.
d95% CI calculated using 2-sided 95% Clopper-Pearson exact method based on binomial distribution.
eNo patients experienced grade 3 or higher tumour lysis syndrome or cytokine release syndrome. Tumour lysis syndrome or cytokine release syndrome events of any grade were not reported.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Overall Survival
At the primary analysis, median OS was not reached (NR) (95% confidence interval [CI], 18.3 to NR) with a median follow-up time of 19.6 (95% CI, 15.3 to 21.9) months. As of the most recent analysis, the median OS was 33.5 months (95% CI, 18.3 to NR), with a median follow-up time of 42.7 (||||||||||||||||||||||||) months.
Progression-Free Survival
At the primary analysis, median PFS by IRC was 12.1 months (95% CI, 5.7 to NR), with a median follow-up time of 17.3 (95% CI, 11.5 to 21.2) months. As of the most recent analysis, median PFS by IRC was 11.6 months (95% CI, 6.3 to 45.7), with a median follow-up time of 33.9 (||||||||||||||||||||||||) months.
Time to Progression
At the primary analysis, median TTP was 16.2 months (95% CI, 7.4 to NR). TTP was not analyzed at the subsequent interim analyses.
Event-Free Survival
At the primary analysis, median EFS was 9.1 (95% CI, 5.3 to 21.0) months, with a median follow-up time of 19.7 (95% CI, 14.3 to 22.0) months. EFS was not reported at the subsequent interim analyses.
Objective Response Rate
ORR by IRC was the primary end point in L-MIND. At the primary analysis, the ORR by IRC was 60.0% (95% CI, 48.4 to 70.8). The best objective response for patients was CR for 34/80 (42.5%) patients and PR for 14/80 (17.5%) patients. As of the most recent interim analysis, ORR by IRC was 57.5% (95% CI, 45.9 to 68.5). Thirty-two (40.0%) patients had CR and 14 (17.5%) patients had PR.
Duration of Response
At the primary analysis, median DOR by IRC was 21.7 (95% CI, 21.7 to NR) months. Median DOR by IRC in patients with PR was 4.4 months (95% CI, 2.0 to 9.1) and NR (95% CI, 21.7 to NR) in patients with CR. As of the most recent interim analysis, median DOR by IRC was 43.9 (95% CI, 26.1 to NR) months. Median DOR by IRC in patients with PR was 5.6 (95% CI, 2.2 to NR) months compared to NR (95% CI, 43.9 to NR) in patients with CR.
Time to Response
At the primary analysis, median TTR (CR or PR) based on IRC evaluation was 2.0 months (range 1.7 to 16.8 months). At the second analysis, median TTR based on IRC evaluation was 2.0 months (range 1.7 to 16.8 months). TTR was not reported at the most recent interim analysis.
Time to Next Treatment
At the primary analysis, median TTNT was 15.4 (95% CI, 7.6 to NR) months. At the second analysis, median TTNT was 12.5 (95% CI, 7.6 to 24.7) months. TTNT was not reported at the most recent interim analysis.
Health-Related Quality of Life
HRQoL outcomes were not reported in L-MIND.
Harms Results
Harms data from the L-MIND study safety analysis set (N = 81) as of the most recent analysis (October 30, 2020, data cut-off) are summarized in Table 2. As of both the primary analysis and most recent analysis, the median duration of exposure to the study treatment (tafasitamab plus lenalidomide) was 9.2 months.
Adverse Events
All 81 (100%) patients enrolled in L-MIND experienced at least 1 treatment-emergent adverse event (AE). The most common AEs were neutropenia (50.6%), anemia (37.0%), diarrhea (35.8%), thrombocytopenia (30.9%), and cough (27.2%).
Serious Adverse Events
Overall, 53.1% of patients enrolled in L-MIND experienced at least 1 serious adverse event (SAE). The most common SAEs were pneumonia (n = 7, 8.6%), febrile neutropenia (n = 5, 6.2%), and pulmonary embolism (n = 3, 3.7%). Other SAEs reported in more than 1 patient included bronchitis, lower respiratory tract infection, atrial fibrillation, and congestive cardiac failure (n = 2, 2.5% each).
Withdrawals Due to Adverse Events
Overall, 20 (24.7%) patients permanently discontinued treatment with 1 or both study drugs due to AEs: 8 (9.9%) patients discontinued lenalidomide only, 2 (2.5%) discontinued tafasitamab only, and 10 (12.3%) discontinued both study drugs. The only AE that led to permanent discontinuation of study drug in more than 1 patient was neutropenia (n = 3, 3.7%).
Mortality
In total, 42 (51.9%) patients enrolled in L-MIND had died as of the October 30, 2020, data cut-off date. The cause of death was reported to be related to disease progression for 31 (38.3%) patients and unrelated to disease progression in 10 (12.3%) patients.
Notable Harms
Overall, 72.8% of patients enrolled in L-MIND experienced an infection. The most common types of infections were bronchitis (16.0%), pneumonia (12.3%), urinary tract infection (12.3%), and respiratory tract infection (11.1%).
Regarding myelosuppression, 50.6% of patients experienced neutropenia, 37.0% experienced anemia, 30.9% experienced thrombocytopenia, 14.8% experienced leukopenia, 12.3% experienced febrile neutropenia, and 7.4% experienced lymphopenia.
One (1.2%) patient developed worsening PML. Two (2.5%) patients experienced hepatitis B virus (HBV) reactivation. Five (6.2%) patients experienced an infusion-related reaction. No patients experienced grade 3 or higher tumour lysis syndrome or cytokine release syndrome. Tumour lysis syndrome or cytokine release syndrome events of any grade were not reported.
Critical Appraisal
For the primary end point and multiple secondary end points (i.e., PFS, EFS, DOR, TTR), an IRC was appropriately used. Furthermore, there was generally good agreement between the IRC and investigator-assessed outcomes. The CADTH review team and clinical experts evaluated the eligibility criteria and analysis populations as appropriate.
L-MIND is an open-label, single-arm study. There is no direct evidence comparing tafasitamab plus lenalidomide to a control arm. Furthermore, no statistical testing was performed because the L-MIND study was not designed to test hypotheses. Data were analyzed descriptively. Due to these limitations of the study design, the CADTH review team could draw no definitive conclusions from the L-MIND study regarding the efficacy and safety of tafasitamab plus lenalidomide relative to relevant comparators. The open-label design can increase the risk of performance and detection bias, particularly for outcomes that are subjective in measurement and interpretation (e.g., response, AEs). Objective outcomes, such as OS time and mortality, are unlikely to be affected by performance or detection bias. The potential for detection bias was minimized by using IRC assessment for key study outcomes, such as ORR, DOR, and PFS. The time-to-event analyses were appropriate, but causality cannot be inferred in a single-arm trial without a comparator.
Multiple protocol amendments were implemented while the L-MIND study was being conducted, which included changes to the study eligibility criteria. There was a high rate of protocol deviations, which creates uncertainty in the data because protocol deviations could have affected the internal validity of the study. In addition, protocol deviations related to eligibility criteria may have caused selection bias, although the direction of this potential bias is unknown. The L-MIND study consisted of patients with R/R DLBCL diagnosed as per local pathologic analysis. However, central pathologic analysis concluded that approximately 10% of these patients had non-DLBCL histology or alternative diagnoses. This causes selection bias. The clinical experts consulted by CADTH indicated that inclusion of these patients may have confounded results, particularly for OS and PFS, although sensitivity analyses conducted using patients with a DLBCL diagnosis confirmed by central pathologic analysis were generally consistent with the main analysis. Overall, the L-MIND study was a phase II trial that enrolled 80 patients in the full analysis set (FAS). The clinical experts consulted by CADTH indicated that it may not be possible to extrapolate efficacy results from this small sample of patients to the general population of patients with R/R DLBCL in Canada.
The L-MIND trial was an international, multi-centre study, but there were no sites in Canada. The treatment regimen used in the L-MIND trial aligns with Health Canada–recommended dosage of tafasitamab plus lenalidomide. The clinical experts indicated that the baseline characteristics of patients enrolled in L-MIND were generally representative of the R/R DLBCL patient population in Canada, although they noted that the L-MIND study patients would represent the most fit patients in this population. The L-MIND study excluded some groups of patients in the R/R DLBCL patient population, specifically, patients with known double- or triple-hit genetics DLBCL at study entry, which limits the generalizability of results to this patient population. In addition, primary refractory DLBCL was an exclusion criterion in L-MIND. However, the definition changed during the study, complicating the interpretation of the generalizability of study results to this group of patients. Multiple protocol amendments related to the eligibility criteria were implemented during the L-MIND study, which increased the generalizability of results because the trial population was more representative of the general patient population of those living in Canada, according to the clinical experts consulted by CADTH. HRQoL outcomes, which are important to patients, were not reported, which is a key gap in the evidence.
Indirect Comparisons
Description of Studies
Three sponsor-submitted indirect treatment comparisons (ITCs) were included in this review: 2 retrospective observational studies (RE-MIND16,17 and RE-MIND218) that were used as external cohorts for indirect comparison with patients enrolled in the L-MIND trial, using estimated propensity score (ePS)-based nearest neighbour (NN) 1:1 matching methodology; and 1 ITC that used unanchored matching-adjusted indirect comparisons (MAICs). These ITCs were used to inform the pharmacoeconomic models.
RE-MIND16,17 was designed to characterize the effectiveness of lenalidomide monotherapy in the treatment of R/R DLBCL patients not eligible for HDC followed by ASCT. The primary end point was ORR. Other end points assessed included OS, CRR, DOR, PFS, TTNT, and EFS. Data from the L-MIND study used in RE-MIND were from the November 30, 2018, data cut-off (primary analysis).
RE-MIND218 was designed to characterize the effectiveness of systemically administered therapies in the treatment of R/R DLBCL patients (second, third, or fourth line). Eligible systemic therapies included regimens administered in routine clinical care according to National Comprehensive Cancer Network (NCCN) or European Society for Medical Oncology (ESMO) guidelines19,20 for patients who were not eligible for ASCT. This study included the following treatment cohorts: systemic therapies pooled, bendamustine plus rituximab (BR), rituximab plus gemcitabine plus oxaliplatin (R-GemOx), CAR T-cell therapy, and pola-BR. The primary end point was OS. Other end points assessed included ORR, CRR, DOR, PFS, TTNT, EFS, treatment discontinuation due to AEs, and duration of treatment exposure. Data from the L-MIND study used in RE-MIND2 were from the November 30, 2019, data cut-off. The pre-specified main analysis was conducted for systemic therapies pooled, BR, and R-GemOx. Pre-specified analyses could not be conducted for pola-BR and CAR T-cell therapy due to insufficient patient numbers. Thus, only post hoc, exploratory analyses were conducted.
Unanchored MAICs21 of tafasitamab plus lenalidomide in the L-MIND study versus comparator therapies using prospective studies were conducted. In total, |||||||| prospective studies reporting data for ||||||||||||||||||||||||||||||||||||, pola-BR, BR, and R-GemOx were selected for the MAICs against tafasitamab plus lenalidomide. End points assessed included OS, PFS, DOR, ORR, and CRR. Data were used from the L-MIND study analysis with the October 30, 2020, data cut-off.
Efficacy Results
In RE-MIND, ORR was 67.1% (95% CI, 55.4 to 77.5) in the tafasitamab plus lenalidomide cohort compared to 34.2% (95% CI, 23.7 to 46.0) in the lenalidomide-monotherapy cohort (odds ratio [OR] = 3.885; 95% CI, 1.900 to 8.142; P < 0.0001). Median OS was NR (95% CI, 15.5 to NR) in the tafasitamab plus lenalidomide cohort and 9.4 (95% CI, 5.1 to 20.0) months in the lenalidomide-monotherapy cohort (hazard ratio [HR] = 0.499; 95% CI, 0.317 to 0.785; P = 0.0026). Median PFS was 12.1 (95% CI, 5.9 to NR) months in the tafasitamab plus lenalidomide cohort and 4.0 (95% CI, 3.1 to 7.4) months in the lenalidomide-monotherapy cohort (HR = 0.463; 95% CI, 0.307 to 0.698; P = 0.0002). Median DOR was 20.5 (95% CI, 12.3 to NR) months in the tafasitamab plus lenalidomide cohort and 6.6 (95% CI, 4.1 to 17.2) months in the lenalidomide-monotherapy cohort (P < 0.0001).
In RE-MIND2, patients in the tafasitamab plus lenalidomide cohort showed an improvement in OS compared to the cohorts of systemic therapies pooled (HR = 0.553; 95% CI, 0.358 to 0.855; P = 0.0076), BR (HR = 0.418; 95% CI, 0.272 to 0.644; P < 0.0001), and R-GemOx (HR = 0.467; 95% CI, 0.305 to 0.714; P = 0.0004). An improvement was also observed for PFS in the tafasitamab plus lenalidomide cohort compared with the cohorts of systemic therapies pooled (HR = 0.424; 95% CI, 0.278 to 0.647; P < 0.0001), BR (HR = 0.527; 95% CI, 0.344 to 0.809; P = 0.0033), and R-GemOx (HR = 0.433; 95% CI, 0.288 to 0.653; P < 0.0001). The ORR was higher in the tafasitamab plus lenalidomide cohort compared to the cohorts of systemic therapies pooled (|||||||||||||||||||||||||||||||||||||||||||||||| P = 0.0323) and R-GemOx (||||||||||||||||||||||||||||| P = 0.0076). There was no difference in ORR in the tafasitamab plus lenalidomide cohort compared to the BR cohort (|||||||||||||||||||||||||||||||||||||||||||||||| P = 0.1810).
In the MAICs, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. For the comparisons of tafasitamab plus lenalidomide to pola-BR, no differences were observed for OS, PFS by IRC, ORR, and CRR. Overall, the results of some of the comparisons to BR favoured tafasitamab plus lenalidomide, whereas others indicated no difference. Last, in the MAIC of tafasitamab plus lenalidomide versus R-GemOx, results indicated no difference between tafasitamab plus lenalidomide and R-GemOx for all outcomes assessed.
Harms Results
In RE-MIND2, 8 patients (14.5%, 14.5%, and 15.1% in the analysis sets for comparison to systemic therapies pooled, BR, and R-GemOx, respectively) discontinued due to AEs in the tafasitamab plus lenalidomide cohort. In the cohorts of systemic therapies pooled, BR, and R-GemOx, 5 (6.8%), 2 (2.8%), and 4 (5.4%) patients, respectively, had AEs leading to permanent discontinuation of treatment. The types of AEs leading to treatment discontinuation were not reported. The median duration of exposure in the tafasitamab plus lenalidomide cohort was longer (approximately 10 months) than in the cohorts of systemic therapies pooled (2.4 months), BR (3.2 months), and R-GemOx (2.9 months). Harms outcomes were not reported in RE-MIND or the MAICs.
Critical Appraisal
The RE-MIND and RE-MIND2 studies implemented multiple measures to minimize bias. However, important sources of heterogeneity between the L-MIND cohort and observational cohorts could not be accounted for with the methods used. Although the eligibility criteria for enrolment in RE-MIND and RE-MIND2 were based on the eligibility criteria used in the L-MIND trial, differences related to the retrospective nature of the RE-MIND and RE-MIND2 studies studies were noted. Comparison of data from a prospective, interventional trial to retrospective, observational studies using real-world data may be problematic, as a number of notable differences in data collection, outcomes, and assessments were noted (e.g., tumour assessment frequency, imaging modalities, and criteria used to assess response). Most important, unmeasured confounding factors not accounted for in the matching may have had an effect on results. The RE-MIND and RE-MIND2 studies used 9 covariates for matching in their main analyses (age, Ann Arbor stage, refractoriness to last therapy line, number of previous lines of therapy, history of primary refractoriness, prior ASCT, neutropenia, anemia, and elevated LDH). Other known confounders were not accounted for in the matching (e.g., ECOG PS, IPI score, cell of origin) in the main analyses. As a result of these limitations, there is substantial risk of bias in the RE-MIND and RE-MIND2 study results.
There are also limitations to the external validity of the RE-MIND and RE-MIND2 studies. Lenalidomide monotherapy is not used as a treatment for R/R DLBCL in Canada, according to the clinical experts. RE-MIND-2 included relevant comparators, but the clinical experts consulted by CADTH also indicated that R-GemOx and BR are not very commonly used to treat patients with R/R DLBCL in Canada. The clinical experts indicated that pola-BR would be the most relevant comparator, although it is not yet funded. The clinical experts noted that the relevance of CAR T-cell therapy as a comparator for tafasitamab plus lenalidomide in patients who are not eligible for ASCT was debatable. The clinical experts considered CAR T-cell therapy an intensive therapy and thus more comparable to ASCT. The clinical experts indicated that they would not consider using tafasitamab plus lenalidomide in patients who were eligible for CAR T-cell therapy. There are also concerns about whether the systemic therapies pooled cohort adequately reflects current contemporary practice and therapies in Canada.
Although the methods used to conduct the unanchored MAICs followed technical guidance,22 the analyses have limitations that affect internal and external validity. Most important, not all known effect modifiers and prognostic factors identified by the authors could be adjusted for in the analyses, due to the lack of available data. The quality of most of the comparator studies was low. Furthermore, multiple sources of heterogeneity (e.g., study design, eligibility criteria, study end point definitions, timing of tumour assessments) were identified and could not be accounted for in the analyses conducted. Given these issues, there is substantial concern about the risk of bias in the MAIC results. There are also limitations to the external validity of some of the comparators (i.e., lenalidomide monotherapy, BR, and R-GemOx), as previously described. In addition, results may be generalizable only to patients similar to those enrolled in the comparator studies, which may not represent patients typically seen in practice in Canada.
Other Relevant Evidence
No long-term extension studies or additional relevant studies were included in the sponsor’s submission to CADTH.
Conclusions
One phase II, single-arm, open-label trial (L-MIND) of tafasitamab plus lenalidomide in patients with R/R DLBCL was included in the systematic review conducted by CADTH. The L-MIND trial data were analyzed descriptively; no statistical hypotheses were tested. According to the clinical experts consulted by CADTH, the results suggested that tafasitamab plus lenalidomide therapy is clinically effective in this patient population and that there may be a beneficial effect of tafasitamab plus lenalidomide on OS, PFS, ORR, DOR, and other efficacy outcomes. However, there is significant uncertainty because it is a phase II trial and because of its open-label single-arm design and small sample size. Due to the absence of a comparator arm and statistical testing, the CADTH review team could draw no definitive conclusions regarding the efficacy of tafasitamab plus lenalidomide based on the L-MIND trial. HRQoL outcomes were not reported in the L-MIND trial, which represents an important gap in the evidence. All study patients reported treatment-emergent AEs, the most common which was neutropenia, and more than half reported SAEs. The most frequently reported SAEs were pneumonia, febrile neutropenia, and pulmonary embolism. The most common cause of death was disease progression.
No direct evidence on the relative efficacy and safety of tafasitamab plus lenalidomide versus other therapies was identified. Results from the ITCs submitted by the sponsor suggested that tafasitamab plus lenalidomide therapy may be associated with an improvement in clinical outcomes (e.g., ORR, CRR, OS, PFS, EFS, DOR, and TTNT) compared to lenalidomide monotherapy, systemic therapies pooled, BR, R-GemOx, pola-BR, and CAR T-cell therapies. However, the ITCs were associated with substantial risk of bias due to important limitations, including methodological limitations, heterogeneity, matching based on a limited number of variables, and small sample sizes. In view of the uncertainty in the ITC results, the CADTH review team could draw no conclusions on the efficacy of tafasitamab plus lenalidomide compared to other therapies used to treat patients with R/R DLBCL who are ineligible for ASCT. Harms outcomes were assessed in 1 ITC (RE-MIND2). The ITC results showed that a numerically greater proportion of patients treated with tafasitamab plus lenalidomide may discontinue treatment due to AEs than patients treated with systemic therapies pooled, BR, and R-GemOx. However, there were limitations associated with the data (i.e., differences in study design, data collection methods, and duration of exposure to treatment). The potential benefits and safety of tafasitamab plus lenalidomide compared with other therapies remain uncertain.
Introduction
Disease Background
NHL is a cancer of the immune system that encompasses more than 60 types of cancer affecting the lymphocytes.1 In 2021, it was estimated that 11,100 of those living in Canada would be diagnosed with NHL and 2,900 of those in Canada would die from NHL that year.2 The signs and symptoms of NHL vary depending on the type of NHL, where it starts in the body, and how advanced it is.23 Common symptoms of NHL include swollen or enlarged lymph nodes in the neck, armpit, or groin; rash or itchy skin on the chest, stomach, and back; and unexplained fatigue.23 Other systemic symptoms can include unexplained persistent fever, drenching night sweats, and unexplained weight loss.23 DLBCL is the most common subtype of NHL, constituting 30% to 40% of cases in Canada.3,4 DLBCL represents a heterogeneous group of aggressive B-cell malignancies.4 Some types of indolent B-cell lymphomas (e.g., follicular lymphoma) can transform into DLBCL.5
Although the cure rate of DLBCL is high, approximately 30% to 50% of patients in Canada experience R/R disease after treatment with standard first-line chemotherapy with R-CHOP or a similar regimen.4,6 According to the clinical experts, patients with R/R DLBCL typically have disease confirmed on repeat biopsy. There are multiple factors associated with a worse prognosis in this patient population (e.g., GCB cell of origin, double- or triple-hit genetics, older age, greater ECOG PS score).21,24,25
Standards of Therapy
Patients with R/R DLBCL have limited treatment options, ranging from supportive care to conventional salvage therapy and ASCT, with the choice of therapy depending on age and comorbidities. According to the clinical experts consulted by CADTH, patients with R/R disease after first-line therapy are assessed for eligibility for intensive therapy. Intensive therapies include HDC followed by ASCT and CAR T-cell treatment. According to the clinical experts consulted by CADTH, approximately half of R/R DLBCL patients are not eligible for intensive therapy due to age or comorbidities, and these patients are treated with palliative intent.
For eligible patients, the standard treatment approach for patients R/R DLBCL is an intensive salvage chemotherapy regimen followed by ASCT.4,6 However, eligibility for this salvage approach largely depends on performance status, age, and comorbidities, and eligibility for ASCT also depends on the response to salvage chemotherapy.4 Among patients who progress following frontline treatment, only 30% to 40% respond to salvage chemotherapy and proceed with ASCT.4 The prognosis for patients with relapsed DLBCL who do not undergo HDC followed by ASCT is poor.4 Even among those patients who respond to salvage chemotherapy and undergo ASCT, 50% are likely to relapse following ASCT.4 If patients do not show chemosensitivity to salvage therapy (and thus are not transplant-eligible), or relapse following ASCT, they may be eligible for CAR T-cell therapy.4 Patients who are not candidates for intensive therapies due to comorbidities, age, or functional status are usually treated with palliative intent. According to the clinical experts consulted by CADTH, the goals of treatment for patients with R/R DLBCL who are not eligible for intensive therapy are to control symptoms with minimal toxicity, improve quality of life, delay disease progression, and prolong life.
In patients with R/R DLBCL who are not eligible for intensive therapies, there is no standard treatment approach. There are numerous chemotherapy options, but response rates are generally low and remission duration is short.4 Pola-BR would be an option for those living in Canada, in this setting, if it were funded. Pola-BR may be used as a stand-alone treatment or may provide a bridge to future consolidative therapies, including ASCT or CAR T-cell therapy.4 Chemotherapy options for fit patients can include rituximab, ifosfamide, carboplatin, and etoposide; rituximab, oxaliplatin, cytosine arabinoside, and dexamethasone; R-GemOx; and rituximab, dexamethasone, cisplatin, and cytarabine. Palliative strategies can also be employed.
Drug
Tafasitamab is an Fc-enhanced monoclonal antibody that targets the CD19 antigen expressed on the surface of pre-B and mature B lymphocytes and on several B-cell malignancies, including DLBCL.7 Upon binding to CD19, tafasitamab mediates B-cell lysis through apoptosis and immune effector mechanisms. In in vitro laboratory studies conducted in DLBCL tumour cell lines, tafasitamab plus lenalidomide was associated with greater cytotoxicity than when cells were treated with either drug alone.
Tafasitamab is indicated in combination with lenalidomide for the treatment of adult patients with R/R DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT. Key characteristics of tafasitamab and lenalidomide are summarized in Table 3. The sponsor’s reimbursement request is per the Health Canada indication. Tafasitamab has not been previously reviewed by CADTH.
Tafasitamab received a Notice of Compliance with Conditions on August 19, 2021, pending the results of trials to verify its clinical benefit. According to the Health Canada product monograph, authorization was based on ORR, CRR, and durability of response from a single-arm clinical study; an improvement in PFS or OS has not been established.7 According to the Letter of Undertaking,24 the planned confirmatory study is a phase III, randomized, double-blind, placebo-controlled trial comparing the efficacy and safety of tafasitamab plus lenalidomide in addition to R-CHOP versus R-CHOP in previously untreated, high-intermediate, and patients at high risk patients with newly diagnosed DLBCL (the frontMIND study).15,24 ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
The recommended dosage of tafasitamab is 12 mg/kg body weight administered as an IV infusion in 28-day cycles according to the following schedule: during cycle 1, infusions are administered on days 1, 4, 8, 15, and 22; in cycles 2 and 3, infusions are administered on days 1, 8, 15, and 22 of each cycle; and, in cycle 4 and subsequent cycles, infusions are administered on days 1 and 15 of each cycle.7 According to the product monograph, tafasitamab should be administered with lenalidomide for up to 12 cycles. Patients take lenalidomide capsules orally at the recommended starting dosage of 25 mg daily on days 1 to 21 of each cycle. The starting and subsequent dosages of lenalidomide may be adjusted as needed. After a maximum of 12 cycles of combination therapy, lenalidomide treatment is stopped, and patients receive tafasitamab infusions as monotherapy until disease progression or unacceptable toxicity.
Table 3: Key Characteristics of Tafasitamab and Lenalidomide
Tafasitamab | Lenalidomide | |
|---|---|---|
Mechanism of action | Fc-enhanced monoclonal antibody that targets the CD19 antigen expressed on the surface of pre-B and mature B lymphocytes and on several B-cell malignancies, including DLBCL. Upon binding to CD19, tafasitamab mediates B-cell lysis through apoptosis and immune effector mechanisms, including antibody-dependent cellular cytotoxicity and antibody-dependent cellular phagocytosis. | Remains to be fully characterized. Lenalidomide increases hemoglobin expression by erythroid cells; inhibits proliferation of certain hematopoietic tumour cells (including tumour cells with or without deletions of chromosome 5 and MM tumour cells); enhances T-cell and natural killer cell number and activity; inhibits angiogenesis by blocking the migration and adhesion of endothelial cells and the formation of microvessels; and inhibits production of pro-inflammatory cytokines (e.g., TNF-alpha and IL-6) by monocytes. |
Indication(s)a | Indicated in combination with lenalidomide for the treatment of adult patients with relapsed or refractory DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT. | Indicated for the treatment of patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality, with or without additional cytogenetic abnormalities. Indicated in combination with dexamethasone for the treatment of MM patients who are not eligible for stem cell transplant. |
Route of administration | IV infusion | Oral |
Recommended dosage | 12 mg/kg body weight in 28-day cycles:
| For patients with R/R DLBCL, the recommended starting dosage is 25 mg daily on days 1 to 21 of each cycle. The starting and subsequent dosages should be adjusted, as necessary, according to the lenalidomide product monograph. |
Serious adverse effects or safety issues | Infection, myelosuppression, PML, hepatitis B virus reactivation | Potential for human birth defects, stillbirth, and spontaneous abortion; neutropenia and thrombocytopenia; venous and arterial thromboembolism; hepatotoxicity; anaphylaxis |
Other | In in vitro laboratory studies conducted in DLBCL tumour cell lines, tafasitamab, in combination with lenalidomide, was associated with greater cytotoxicity than when cells were treated with either drug alone. | Lenalidomide is only available through a controlled distribution program called RevAid®. Under this program, only prescribers and pharmacists registered with the program can prescribe and dispense the product. In addition, lenalidomide can be dispensed only to patients who are registered and meet all the conditions of the program. |
ASCT = autologous stem cell transplant; CD = cluster of differentiation; DLBCL = diffuse large B-cell lymphoma; IL = interleukin; IV = IV; MM = multiple myeloma; PML = progressive multifocal leukoencephalopathy; R/R = relapsed or refractory; TNF = tumour necrosis factor.
aHealth Canada–approved indication.
Source: Tafasitamab Product Monograph,7 Lenalidomide Product Monograph.26
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient input(s) received by CADTH have been included in the stakeholder section at the end of this report.
One patient advocacy group provided input on tafasitamab for the treatment of R/R DLBCL in adult patients. LC conducted 4 anonymous online surveys for DLBCL patients from April 2018 to December 2021. The most recent survey, conducted from October 2021 to December 2021, included patients receiving tafasitamab. Overall, 150 DLBCL patients responded to the surveys, of whom 2 patients (1%) indicated they had received tafasitamab therapy. The following is a summary of key input from the perspective of the patient group.
Patients described the negative impact of DLBCL on their day-to-day life. Commonly reported symptoms affecting patients’ HRQoL at diagnosis included fatigue or lack of energy, enlarged lymph nodes, drenching night sweats, unexplained weight loss, loss of appetite, influenza-like symptoms, and persistent cough. Patients also described mental and emotional problems associated with their disease and treatment that negatively affected their HRQoL, including fear of disease recurrence, memory loss, anxiety/worry, problems concentrating, difficulty sleeping, loss of sexual desire, stress of diagnosis, and depression.
Patients had received at least 1 line of treatment or were undergoing first-line treatment (with the most commonly reported first-line treatment being R-CHOP). Patients reported that treatment had a significant impact on their ability to work, travel, and participate in daily activities, as nearly all patients reported at least 1 side effect. Fatigue, nausea and/or vomiting, “chemo-brain,” and hair loss were reported as the most difficult side effects to tolerate. Treatment-related side effects reported by 2 patients who received tafasitamab, including neutropenia, rash or itching, diarrhea, and nausea, were short-term and did not affect the patients’ HRQoL. The majority of DLBCL symptoms resolved after patients received tafasitamab, including enlarged lymph nodes, abnormal blood cell counts (platelets, red blood cells, white blood cells), and weight loss and poor appetite.
Patients rated longer survival and remission than current therapies and control of disease symptoms as the most important outcomes for a new therapy. Better HRQoL and fewer side effects compared to current therapies were also important considerations. Some patients (n = 48/114, 42%) indicated that they would tolerate potential side effects of a new treatment if symptoms were short-term and would choose a new treatment with known side effects (potentially serious) if their doctors recommended it as the best option. For other patients (n = 54/114, 47%), choosing a new treatment would depend on the type of side effects, exact length of time they would experience side effects, and whether side effects would outweigh treatment benefit and result in a long-term outcome or cure.
The patient group also stated that patients who are ineligible for ASCT have limited treatment options and indicated a significant unmet need in this population.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of DLBCL.
Unmet Needs
The clinical experts reported that the goal of treatment in patients with R/R DLBCL who are not eligible for intensive therapies (i.e., ASCT and CAR T-cell therapy) is to control symptoms with minimal toxicity to improve HRQoL, delay disease progression, and prolong life. The clinical experts noted that ASCT and CAR T-cell therapy both have toxicity and feasibility issues that limit broad application. Of the available options for patients who are not eligible for ASCT or CAR T-cell therapy, or relapse after these therapies, there is no treatment that is curative. According to the clinical experts, most currently used treatment options have short durations of response, if patients respond at all. The clinical experts indicated that, at relapse of DLBCL, non-intensive treatments are usually prescribed to older adult patients (i.e., ≥ 70 years old), and this population often has comorbidities and may be frail. Thus, tolerability of treatment is important.
Place in Therapy
According to the clinical experts consulted by CADTH, initial therapy for patients with DLBCL who do not have double-hit genetics disease is R-CHOP for 3 to 6 cycles, with or without radiation. Patients with double-hit genetics DLBCL are often treated with more aggressive regimens (e.g., dose-adjusted rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and hydroxydaunorubicin). The clinical experts reported that approximately half of patients with R/R DLBCL are not eligible for intensive therapy (i.e., multi-drug chemotherapy as second-line followed by ASCT or CAR T-cell therapy) due to their age or comorbidities. If patients are not eligible for intensive therapy, there is no standard of care treatment. According to the clinical experts, if ASCT or CAR T-cell treatment are not options, or if the patient relapses following these treatments, the patient would be treated with palliative intent. These treatments aim to improve symptoms, control disease, and prolong life in some cases. Treatment options include steroids, radiotherapy, BR, and non-curative chemotherapy regimens (e.g., rituximab, gemcitabine, cisplatin, and dexamethasone; or rituximab, dexamethasone, cisplatin, and cytarabine). The clinical experts indicated that pola-BR could also be used in this patient population, if it were funded. The clinical experts reported that there is no current standard of care regimen in this population, and oncologists tend to use a regimen that they feel the patient can tolerate based on their prior experience with chemotherapy, the pace of disease progression, and the comorbidities they have.
The clinical experts indicated that tafasitamab plus lenalidomide would be an option at relapse for second-line therapy in patients who are not eligible for intensive therapy. Tafasitamab plus lenalidomide treatment could also be used in the third-line or later setting for people who relapse after ASCT. If both this treatment and pola-BR were available, the clinical experts indicated that they were uncertain how to sequence them, based on the available evidence. The clinical experts thought that patients with peripheral neuropathy or those at higher risk of myelosuppression might do better with tafasitamab plus lenalidomide than pola-BR. The clinical experts also noted that patients with underlying indolent lymphoma were excluded from the pivotal randomized trial evaluating pola-BR,9 so this may be another reason to consider tafasitamab plus lenalidomide. The clinical experts indicated that, if tafasitamab were reimbursed, it would add a potential option for older patients with comorbidities, rather than shifting the treatment paradigm.
In addition, the clinical experts noted that exposure to tafasitamab or any other CD19 antibody would make a patient ineligible for CD19 CAR T-cell therapy in Ontario. Therefore, they would not use tafasitamab plus lenalidomide as a bridging option to CAR T-cell therapy or ASCT. As a result, the clinical experts thought that tafasitamab plus lenalidomide would be an option only for those patients who fail all intensive treatment regimens or for whom an intensive regimen is not an option.
Patient Population
The clinical experts indicated that patients with R/R DLBCL should be diagnosed using biopsy whenever feasible. At times, patients have PR to initial therapy and then progression at this same site. In these cases, repeat biopsy is not feasible, and diagnosis can be made based on clinical reasoning that an alternative diagnosis is unlikely and treatment should be started for probable relapse, according to the clinical experts consulted by CADTH. The clinical experts agreed that both symptomatic and asymptomatic patients with DLBCL require treatment, because it is an aggressive type of lymphoma. The clinical experts indicated that a hematologist or medical oncologist assesses eligibility for treatments at relapse, and, if the patient is not eligible for intensive therapy, then tafasitamab plus lenalidomide would be considered.
The clinical experts consulted by CADTH thought that patients most likely to benefit from tafasitamab plus lenalidomide are those with a relapse of DLBCL, including those with underlying indolent lymphomas. The clinical experts thought that this treatment would be considered in patients who are not eligible for ASCT or CAR T-cell therapy or decline either of these treatments. The clinical experts indicated that it is impossible to identify patients who are most likely to exhibit a response to tafasitamab plus lenalidomide before treatment because there are no data on which patient or tumour characteristics are optimal for this treatment compared to other options.
The clinical experts thought that patients with primary refractory DLBCL would be least suitable for treatment with tafasitamab plus lenalidomide, since these patients have not been studied in trials. In addition, the clinical experts noted that patients who cannot come in for frequent IV infusions, want a time-limited treatment, or for whom intensive therapy might be considered in the future would not be suitable for this treatment.
Assessing Response to Treatment
The clinical experts consulted by CADTH reported that standard of care for assessing treatment response is imaging with CT or PET-CT every 3 to 4 months, with clinical examination and bloodwork before each treatment. Treatment response may be assessed with imaging sooner than every 3 to 4 months if there is a change in clinical status. The clinical experts indicated that a clinically meaningful response to treatment would include improvement in survival as well as DOR, which would usually correlate with improvement in symptom burden. According to the clinical experts, meaningful response would include CR, PR, or stable disease with a tolerable toxicity profile.
Discontinuing Treatment
The clinical experts noted that any disease progression should be an indication for treatment discontinuation. The clinical experts thought that recurrent infections, serious infection due to B-cell depletion, and hypogammaglobulinemia may also be considerations for discontinuation. One clinical expert also thought that inability to tolerate lenalidomide or tafasitamab may be a consideration for discontinuation because there are unclear benefits of monotherapy for either. By contrast, the second clinical expert indicated that they would not stop single-drug treatment if it provided a clinical benefit for the patient.
Prescribing Conditions
The clinical experts thought that treatment with tafasitamab could be carried out in any setting that can monitor for infusion-related reactions and has protocols and processes to deal with a hypersensitivity reaction. The clinical experts noted that this is an outpatient regimen, and standard supportive measures, such as those available for rituximab or other monoclonal antibodies, would be required. The patients would typically have standard pre-medications and management of hypersensitivity with monitoring.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input(s) received by CADTH have been included in the stakeholder section at the end of this report.
Clinician input on the review of tafasitamab for the treatment of adult patients with R/R DLBCL was received from 2 groups: the OH-CCO Hematology Drug Advisory Committee and a group of 4 clinicians whose submission was coordinated by LC.
Both groups agreed that there are poor and limited treatment options for patients with R/R DLBCL. The clinician groups indicated that CAR T-cell therapies and other combinations of palliative drug regimens (e.g., pola-BR) are options for these patients. Patients who are unfit or ineligible for intensive therapies (i.e., ASCT or CAR T-cell therapy) or who have failed prior cellular therapy have the greatest unmet need for treatment, according to the clinician groups. Both groups agreed that the most important goals in treatment are prolonging survival and reducing disease symptoms. The LC-coordinated group added that ensuring a reasonable safety profile, manageable toxicities, ease of administration, and improvement in HRQoL, as well as helping the patients gain their independence and reducing burden on caregivers, are also important.
Both groups agreed that tafasitamab plus lenalidomide would be recommended after first-line therapies. The clinician groups indicated that patients with R/R DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT would be most suitable for this therapy. The LC-coordinated group added that tafasitamab plus lenalidomide would be routinely offered in patients ineligible for CAR T-cell therapy. The OH-CCO clinicians suggested that tafasitamab plus lenalidomide would be an additional option for second-line treatment, while the LC-coordinated group stated that this therapy would be used in the third-line setting or beyond. The LC-coordinated group also mentioned that most clinicians would proceed with intensive salvage therapy first and reserve tafasitamab plus lenalidomide for after failure of salvage therapy.
The LC-coordinated group added that, for patients who fail tafasitamab plus lenalidomide treatment, pola-BR is an option. Additionally, oral prednisone, etoposide, procarbazine, and cyclophosphamide are a palliative oral chemotherapy, and participation in clinical trials is an option as well. Both groups reported that receiving tafasitamab plus lenalidomide could affect patients’ eligibility for subsequent CAR T-cell therapy.
Both groups agreed that patients would be identified by their primary treating physician. They also agreed that tafasitamab plus lenalidomide can be administered on an outpatient basis in certified centres. There were 2 differing opinions on which patients are unsuitable for tafasitamab. The clinicians from OH-COO stated that patients with DLBCL who have progressed on CAR T-cell therapies would be least suitable for this therapy, while the LC-coordinated group maintained that there are no specific parameters that would make a patient unsuitable. The LC-coordinated group thought that the patient should be well enough to tolerate the frequent outpatient visits required for this therapy. Both groups agreed that there are no predictors to identify which patients will exhibit response.
Both groups provided input on how response to treatment is assessed. The group from OH-COO suggested that treatment response should be assessed every 3 months in the first year of treatment and then every 6 months after the first year of treatment. The LC-coordinated group added that clinical assessment before each cycle of treatment is the standard of practice, which may include a review of symptoms, a physical examination, and assessment of lymphadenopathies, organomegaly, and extranodal involvement. The LC-coordinated group also provided insights on additional practices related to assessing response. Imaging studies with CT and/or PET scan, if clinically indicated, are part of the assessment after cycle 4 and after cycle 12 of treatment (i.e., 4 months and 12 months after starting the treatment).
Both groups agreed that improved symptoms, prolonged remission, and improved survival would be considered a clinically meaningful response to treatment. The LC-coordinated group added that clinically meaningful responses would include resolution of all lymphoma-related symptoms, improvement in functional status and HRQoL indicators, and return to normal activities. Both groups agreed that confirmation of disease progression would constitute treatment failure and would prompt treatment discontinuation. The LC-coordinated group suggested that severe toxicities (e.g., grade 3 or higher) should result in temporary discontinuation until improved, and that therapy should be discontinued if the toxicity is unacceptable to either the patient or the physician provider.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Initiation | |
Patients with primary refractory DLBCL were excluded from the L-MIND trial. Can pERC clarify the definition of primary refractory disease, as there was a protocol amendment? | For consideration by pERC. The clinical experts noted that primary refractory DLBCL was an exclusion criterion in the L-MIND study and that the study definition of primary refractory disease changed mid-study, thus complicating analysis of benefits in this high-need patient population. In the original study protocol, only patients whose disease relapsed within 3 months of a previous anti-CD20–containing regimen were defined as having primary refractory disease and excluded. After the protocol amendment, primary refractory disease was defined as disease progressing in the course of the first-line treatment as per IWG response criteria (Cheson et al. [2007]8) and/or showing a response of less than a PR to first-line treatment or disease recurrence or progression < 6 months after the completion of first-line therapy. |
Prior anti-CD20 was a requirement for eligibility. | For consideration by pERC. The clinical experts noted that the vast majority of patients with DLBCL in Canada would have been offered a CD20 antibody. They thought that the requirement for prior CD20 therapy in the L-MIND trial was likely to ensure the patient was fit for multi-drug treatment. The clinical experts thought that treatment with a standard first-line therapy is required before treatment with tafasitamab plus lenalidomide. They noted that there may be rare CD19-positive DLBCL patients who are treated initially with CHOP only and these patients would be eligible for tafasitamab plus lendalidomide. The clinical experts thought that the eligibility criteria should include prior treatment with curative intent initial therapies (i.e., R-CHOP/R-CEOP or CHOP/CEOP if patients are CD20-negative). |
Discontinuation | |
In the L-MIND trial, the investigator could decide whether the patient should continue further tafasitamab in the case of disease progression. Also, if both drugs need to be interrupted for more than 28 days for the same persistent toxicity, then the treatment was discontinued in the trial. Can pERC provide guidance on the discontinuation criteria for tafasitamab plus lenalidomide? | The clinical experts noted that any disease progression should be an indication for treatment discontinuation. The clinical experts thought that recurrent infections, serious infection due to B-cell depletion, and hypogammaglobulinemia may also be considerations for discontinuation. One clinical expert also thought that inability to tolerate lenalidomide or tafasitamab may be a consideration for discontinuation as well, because there are unclear benefits as monotherapies for either, whereas the second clinical expert indicated they would not stop single-drug treatment if it was resulting in a clinical benefit. |
If a patient receives 12 cycles of combination tafasitamab plus lenalidomide therapy, then tafasitamab monotherapy, and the patient stops tafasitamab monotherapy but later progresses on while off therapy, should treatment be re-initiated or should the patient move on to an alternative treatment option? If restarting, would patients receive tafasitamab monotherapy or tafasitamab plus lenalidomide followed by tafasitamab monotherapy? | The clinical experts indicated that this would depend on how long the patient was off treatment and how much their disease had progressed. The clinical experts indicated that, if tafasitamab was briefly held due to a comorbid issue, restarting would be reasonable if the disease were stable. |
Prescribing | |
Tafasitamab dosage is 12 mg/kg IV on days 1, 8, 15, and 22 of 28-day cycle for cycles 1 to 3. From cycle 4, tafasitamab is 12 mg/kg IV on days 1 and 15 every 28 days. In the L-MIND trial, dosage reductions were not permitted. Lenalidomide starting dosage is 25 mg orally daily on days 1 to 21 of a 28-day cycle. The L-MIND trial outlined dosage reductions for lenalidomide in 5 mg increments, only once per cycle. Lenalidomide dosage also requires close monitoring, including for potential dosage adjustments for renal dysfunction. | For consideration by pERC. |
Implementation | |
Can pERC clarify the eligible patient population and whether the following would be eligible for tafasitamab:
| Primary refractory DLBCL: The clinical experts noted that primary refractory DLBCL was an exclusion criterion in the L-MIND study and that the study definition of primary refractory disease changed mid-study, thus complicating analysis of benefits in this high-need patient population. They indicated that patients with primary refractory DLBCL are unlikely to have chemosensitive disease to subsequent therapies; these patients have an extremely poor outcome. Because of the changing definition and exclusion criterion, the clinical experts thought that it is difficult to determine whether patients with primary refractory DLBCL should be treated with tafasitamab plus lenalidomide as second-line or later therapy. The clinical experts reported that the pivotal study of pola-BR9 did not specifically exclude this patient population, and therefore pola-BR may be a better treatment option for patients with primary refractory DLBCL. However, the clinical experts also noted that there are no standard of care treatment options for these patients with high unmet need. Thus, they would consider offering tafasitamab plus lenalidomide. History of double- or triple-hit genetics DLBCL: The clinical experts noted that patients with known double- or triple-hit genetics lymphoma were excluded from the L-MIND trial. Thus, they would favour treatment with pola-BR in this population. The clinical experts noted that patients with a history of double- or triple-hit genetics may respond to tafasitamab plus lenalidomide, but data are not currently available to support this. CNS lymphoma involvement: The clinical experts thought that patients with CNS involvement of lymphoma should not be treated with tafasitamab plus lenalidomide because there is no evidence to support the use of this treatment in these patients. Other histological types of lymphoma: The clinical experts thought that patients with PMBL or Burkitt lymphoma should not be treated with tafasitamab plus lenalidomide because there is no evidence to support the use of tafasitamab plus lenalidomide in these patient populations. In addition, patients with PMBL have the option of treatment with an immune checkpoint inhibitor. |
On a time-limited basis, should patients who have received more than 3 prior lines of treatment, but who would otherwise fit the trial criteria, be eligible for tafasitamab plus lenalinomide? | The clinical experts thought that these patients should be eligible for tafasitamab plus lenalidomide, particularly if they had no prior access to a novel therapy (i.e., CAR T-cell therapy, pola-BR). The clinical experts thought that the number of lines of therapy should not affect a patient’s eligibility for treatment with tafasitamab plus lenalidomide. |
Under what clinical circumstances would tafasitamab plus lenalinomide be preferred over pola-BR? | The clinical experts indicated they would use tafasitamab plus lenalidomide for patients previously treated with bendamustine or polatuzumab; however, they noted there is no evidence that re-treatment with these drugs is ineffective and, currently, they do not use these drugs routinely. The clinical experts also thought that tafasitamab plus lenalidomide may be preferred over pola-BR in patients with existing peripheral neuropathy. The clinical experts also noted that the duration of pola-BR treatment is limited, whereas treatment with tafasitamab plus lenalidomide is followed by tafasitamab monotherapy until disease progression or intolerable toxicity. The clinical experts thought that some patients may prefer a treatment option with limited duration. If the patient has CD19-negative DLBCL, the clinical experts would consider pola-BR. However, the clinical experts acknowledged that not all centres conduct CD19 testing routinely. |
What evidence is available to support the sequencing of tafasitamab plus lenalidomide with pola-BR and with CAR T-cell therapy? | The clinical experts reported that there is no evidence from prospective clinical studies to guide sequencing of tafasitamab plus lenalinomide with pola-BR and with CAR T-cell therapy. The clinical experts noted that, in murine models, treatment with tafasitamab did not impair subsequent CAR T-cell therapy response.27 In relapse following CAR T-cell therapy, there is retrospective evidence28 that looked at 400 patients treated with anti-CD19 CAR T-cell therapy. Of the patients given treatment at relapse, the median PFS was stated to be best for pola-BR (4.5 months). However, this was based on a total of 14 patients. Only 5 patients had tafasitamab plus lenalidomide with median PFS of 1.2 months. The clinical experts thought that there is insufficient evidence to support sequencing of these drugs with CAR T-cell therapy at the time of this review. The clinical experts noted that, in Ontario, they do not have option to proceed with funded CAR T-cell therapy after tafasitamab is given to a patient. The clinical experts reported that pola-BR can be used for bridging for CAR T-cell therapy. |
BL = Burkitt lymphoma; CAR T-cell therapy = chimeric antigen receptor T-cell therapy; CD = cluster of differentiation; CEOP = cyclophosphamide, epirubicin, vincristine, and prednisone; CHOP = cyclophosphamide, doxorubicin, vincristine, and prednisone; CNS = central nervous system; DLBCL = diffuse large B-cell lymphoma; IV = IV; IWG = International Working Group; pERC = pCODR Expert Review Committee; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine plus rituximab; PMBL = primary mediastinal (thymic) large B-cell; PR = partial response; R-CEOP = rituximab, cyclophosphamide, epirubicin, vincristine, and prednisone; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Clinical Evidence
The clinical evidence included in the review of tafasitamab is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. No additional relevant studies were included in the sponsor’s submission to CADTH.
After the draft CADTH pCODR Expert Review Committee (pERC) recommendation for tafasitamab was issued in May 2022, the sponsor submitted additional post hoc analyses from the RE-MIND2 study, which provided indirect evidence on tafasitamab. The results of these post hoc analyses are presented in Appendix 4. These data were not included in the initial submission to CADTH. After the CADTH recommendation was issued, the sponsor reported that the data became available only after it had made its submission to CADTH.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of tafasitamab (200 mg single-use vial) in combination with lenalidomide for the treatment of adult patients with R/R DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered important to patients, clinicians, and drug plans.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist (https://www.cadth.ca/resources/finding-evidence/press).29
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Minjuvi/tafasitamab. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on December 15, 2021. Regular alerts updated the search until the meeting of the CADTH pERC on April 13, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool for Searching Health-Related Grey Literature checklist (https://www.cadth.ca/grey-matters).30 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
In addition, a focused literature search for NMAs dealing with DLBCL was run in MEDLINE All (1946–) on December 15, 2021. No date or language limits were applied.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with R/R DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT Subgroups:
|
Intervention | Tafasitamab (12 mg/kg, IV infusion) in combination with lenalidomide for up to 12 cycles, then as monotherapy |
Comparator | Pola-BR CAR T-cell therapy R-GemOx GDP with or without rituximab PEP-C MEP BR |
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, mortality, notable harms/harms of special interest (e.g., infection, myelosuppression, PML, hepatitis B reactivation, infusion-related reactions, cytokine release syndrome, tumour lysis syndrome) |
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse events; ASCT = autologous stem cell transplantation; BR = bendamustine plus rituximab; CAR T-cell therapy = chimeric antigen receptor T-cell therapy; CRR = complete response rate; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group performance status; EFS = event-free survival; EORTC-QLQ-C30 = European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire; GDP = gemcitabine plus dexamethasone plus cisplatin; HRQoL = health-related quality of life; IPI = International Prognostic Index; IV = IV; MEP = methotrexate plus etoposide plus cisplatin; ORR = objective response rate; OS = overall survival; PEP-C = prednisone plus etoposide plus procarbazine plus cyclophosphamide; PFS = progression-free survival; PML = progressive multifocal leukoencephalopathy; pola-BR = polatuzumab vedotin plus bendamustine plus rituximab; PRR = partial response rate; RCT = randomized controlled trial; R/R = relapsed and/or refractory; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; SAE = serious adverse events; TTNT = time to next treatment; TTR = time to response; WDAE = withdrawal due to adverse events.
aDouble-hit status is defined as genetic lesions in BCL2 and MYC; triple-hit status is defined as genetic lesions in BCL2, MYC, and BCL6.
Findings From the Literature
Eight reports10-14,31,32 of 1 unique study (L-MIND) were identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6. A list of excluded studies is presented in Appendix 2.
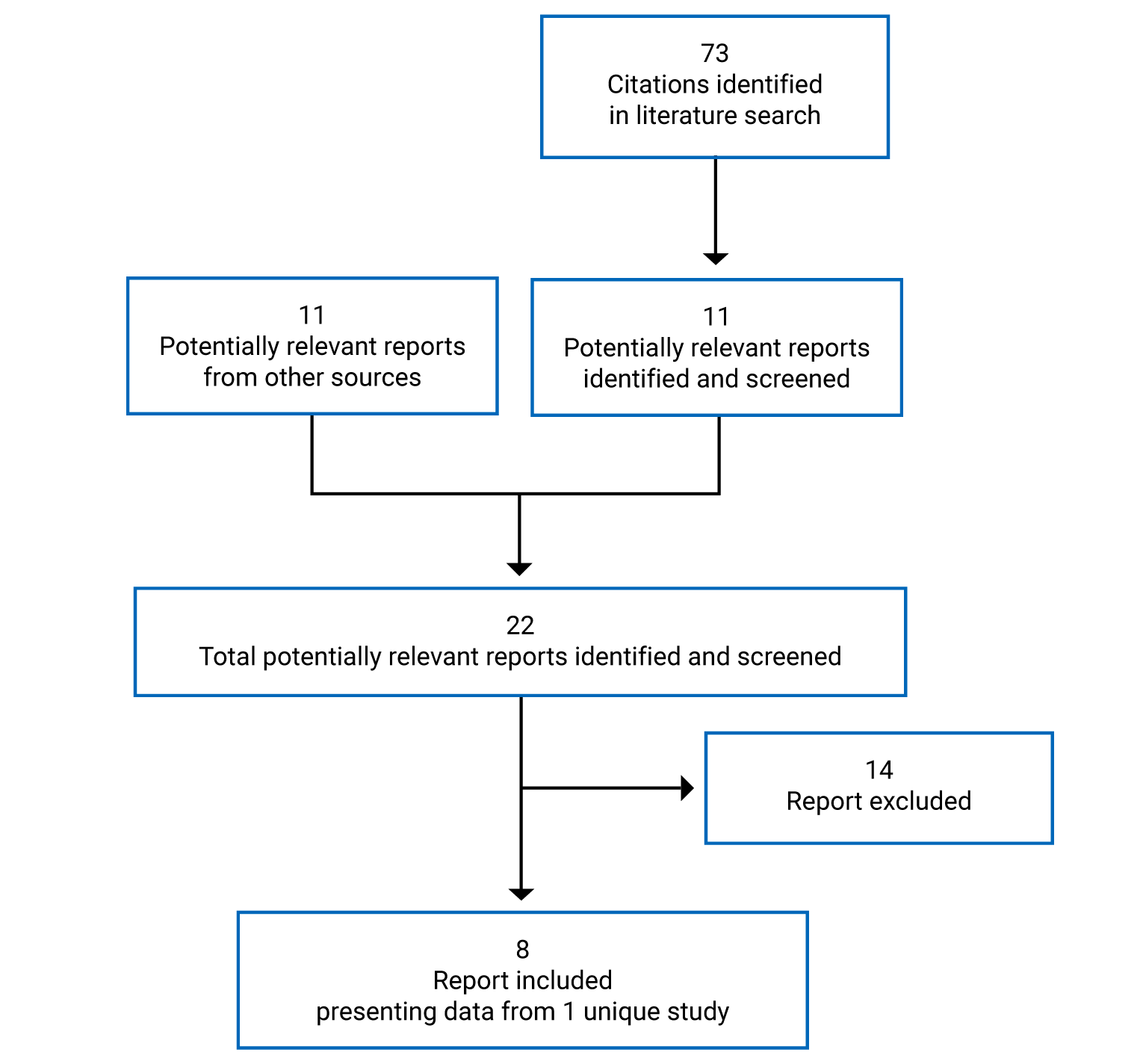
Table 6: Details of Included Study
Detail | L-MIND |
|---|---|
Designs and populations | |
Study design | Single-arm OL, phase II trial |
Locations | 35 sites in 10 countries (Europe and US) |
Enrolment dates | January 18, 2016, to November 15, 2017 |
Enrolled (N) | 81 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Combination of tafasitamab and lenalidomide for up to 12 cycles (28 days each), followed by tafasitamab monotherapy (in patients with stable disease or better) until disease progression
|
Comparator(s) | None |
Duration | |
Phase | |
Screening | 28 days |
Safety run-ind | 22 days |
Treatment | Maximum 12 cycles for tafasitamab plus lenalidomide followed by tafasitamab monotherapy thereafter, until disease progression, unacceptable toxicity, or discontinuation for any other reason |
Survival follow-up | Up to 5 years from cycle 1 day 1 or until withdrawal of consent or death, whichever came first |
Outcomes | |
Primary end point | Best ORR (CR + PR) by IRC according to the 2007 IWG response criteria for malignant lymphoma |
Secondary and exploratory end points | Secondary:
|
Notes | |
Publications | Salles et al. (2020)11 Duell et al. (2021)13 – long-term outcomes Duell et al. (2021)32 – post hoc analysis of CD19 expression |
AE = adverse event; ALT = alanine transaminase; AP = alkaline phosphatase; ASCT = autologous stem cell transplant; AST = aspartate aminotransferase; CNS = central nervous system; CR = complete response; DCR = disease control rate; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; EBV = Epstein-Barr virus; ECOG PS = Eastern Cooperative Oncology Group performance status; EFS = event-free survival; IRC = independent review committee; IV = IV; IWG = International Working Group; NK = natural killer; NOS = not otherwise specified; OL = open-label; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PK = pharmacokinetic; PMBL = primary mediastinal (thymic) large B-cell; PR = partial response; THRLBCL = T-cell or histiocyte-rich large B-cell lymphoma; TTNT = time to next treatment; TTP = time to progression; TTR = time to response; ULN = upper limit of normal.
Note: Four additional reports were included from the sponsor’s submission to CADTH.10,12,14,31
aAdditionally, patients with evidence of histological transformation to DLBCL from an earlier diagnosis of low-grade lymphoma (e.g., an indolent pathology such as follicular lymphoma, marginal zone lymphoma, chronic lymphocytic leukemia) into DLBCL with a subsequent DLBCL relapse were also eligible.
bRelapsed disease was defined as the appearance of any new lesions or an increase in size by 50% or more of previously involved sites, according to the 2007 IWG response criteria, after the most recent systemic therapy. Refractory disease was defined as disease progression per IWG response criteria, showing less than a PR or disease recurrence or progression in less than 6 months from the completion of first-line therapy, or showing less than a PR to the most recently administered systemic therapy.
cThe lesion must have a greatest transverse diameter of 1.5 cm or more and greatest perpendicular diameter of 1.0 cm or more at baseline. The lesion must be positive on PET scan.
dAs the combination of tafasitamab plus lenalidomide had not previously been evaluated in a clinical study, an evaluation of safety data was conducted after the first 6 patients had been accrued. These patients were enrolled sequentially with a 48-hour lag period between enrolments. Patient accrual was held until all 6 patients had been followed for 1 treatment cycle. Once the sixth patient in the cohort had completed the cycle 1 day 22 visit, the safety review panel performed an evaluation based on the number and type of AEs during the first cycle as well as laboratory values (biochemistry and hematology).
Source: Salles et al. (2020),11 L-MIND Clinical Study Report,10 L-MIND Statistical Analysis Plan.24
Description of Studies
L-MIND (N = 81) was a single-arm, multi-centre, open-label, phase II study of tafasitamab plus lenalidomide in adult patients with DLBCL who had relapsed after, or were refractory to, 1 to 3 previous systemic regimens (with at least 1 anti-CD20 therapy), who were not candidates for HDC and subsequent ASCT.10,11 The primary objective of the L-MIND study was to determine the efficacy of tafasitamab plus lenalidomide in terms of ORR (CR + PR) in adult patients with R/R DLBCL. Patients received IV tafasitamab (12 mg/kg) and oral lenalidomide (25 mg/day) for up to 12 cycles (28 days each), followed by tafasitamab monotherapy in patients with stable disease or better until disease progression. Patients were enrolled from 35 sites in 10 countries (Europe and US).
The design of the L-MIND study is summarized in Figure 2. The L-MIND study consisted of 2 parts, which were performed sequentially. The first part was the safety run-in, during which 6 patients were enrolled in the study and completed the first cycle of study treatment. Once the sixth patient in the cohort completed the visit in cycle 1 day 22, a safety review panel performed a clinical safety review. The sponsor opened the second part of the study for enrolment based on the outcome of this interim safety evaluation. The study duration per patient could be up to approximately 5 years, including periods of screening (up to 28 days from the time the patient signed the informed consent form), the treatment period (maximum 12 cycles for tafasitamab plus lenalidomide followed by tafasitamab monotherapy thereafter), and the survival follow-up phase. The survival follow-up phase began with the end-of-treatment (EOT) visit. Patients had an onsite visit for a safety evaluation 30-days after the last administration of study treatment. All patients who discontinued for any reason (other than withdrawal of consent or death) were contacted every 90 days by telephone from the date of the 30-day safety follow-up visit. The survival follow-up covered a duration of up to 5 years from cycle 1 day 1 or until withdrawal of consent or death, whichever came first.
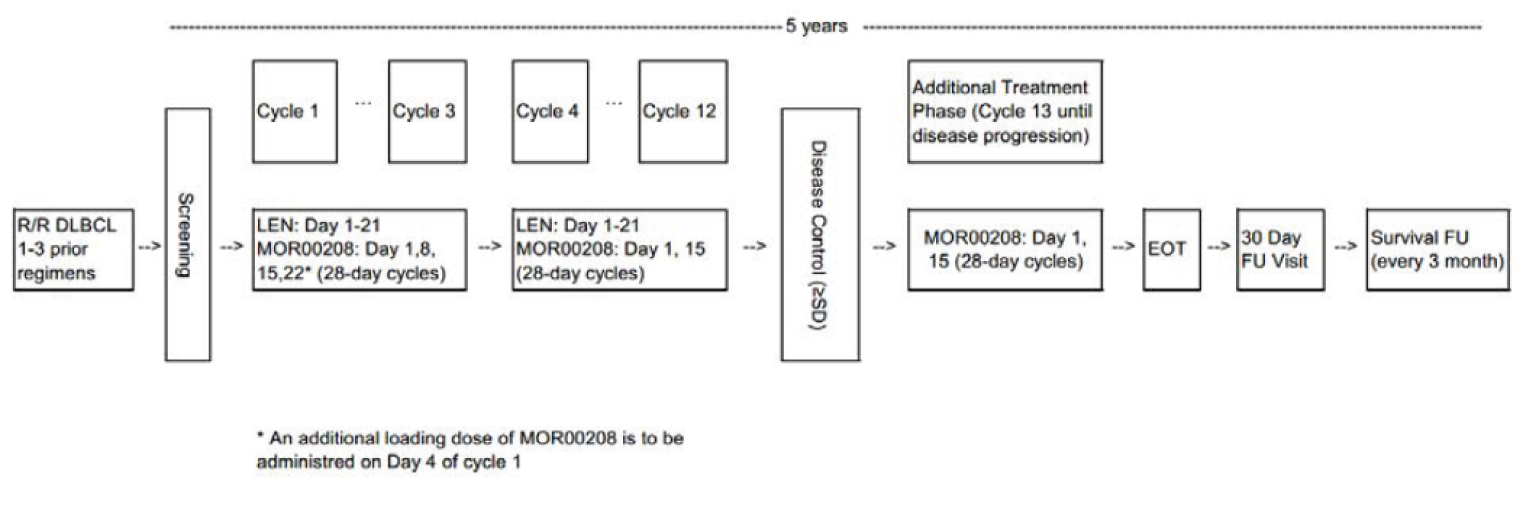
EOT = end of treatment; FU = follow-up; LEN = lenalidomide; MOR00208 = tafasitamab; R/R DLBCL = relapsed or refractory diffuse large B-cell lymphoma; SD = stable disease.
Source: L-MIND Clinical Study Protocol.24
Three analyses based on 3 data cuts were conducted for L-MIND. The primary analysis for L-MIND had a data cut-off date of November 30, 2018, and outcomes were reported after a median follow-up of 13.2 months.10,11 A second efficacy analysis of key outcomes was conducted based on a data cut-off date of November 30, 2019; this analysis was used in the Health Canada approval for tafasitamab plus lenalidomide.12 A third analysis of key outcomes was undertaken with a data cut-off of October 30, 2020, to report long-term outcomes after 35 months or more of follow-up for ORR.13,14 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The analyses with the November 30, 2019, and October 30, 2020, data cut-off dates were not pre-specified in the study protocol. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Populations
Inclusion and Exclusion Criteria
Adult patients (> 18 years of age) were eligible for the L-MIND study if they had histologically confirmed DLBCL (including transformed indolent lymphoma with a subsequent DLBCL relapse); had disease that relapsed after, or was refractory to, at least 1, but no more than 3, systemic regimens (with at least 1 anti-CD20 therapy); and were not candidates for HDC and subsequent ASCT. Patients were also required to have adequate organ function, an ECOG PS of 0 to 2, and measurable disease at baseline. Patients ineligible for ASCT included patients who were older than 70 years, who had organ dysfunction or comorbidities, who had failed previous ASCT, who did not respond to salvage therapy, who had refused ASCT, or who were unable to receive ASCT because of an inability to successfully collect peripheral blood stem cells. Patients were excluded if they had primary refractory DLBCL, double-hit or triple-hit genetics DLBCL (i.e., simultaneous detection of MYC with BCL2 and/or BCL6 translocation), ASCT within the previous 3 months, previous allogenic stem cell transplantation, or CNS lymphoma involvement. Additional details regarding inclusion and exclusion criteria in the L-MIND study are provided in Table 7.
Baseline Characteristics
Baseline characteristics of all patients enrolled in the L-MIND study are presented in Table 7. The mean age of enrolled patients was 69.3 years (SD 9.5 years). Most patients were White (88.9%), had Ann Arbor stage III or IV disease (75.3%), and did not have a prior ASCT (88.9%). Overall, 54.3% of enrolled patients were male, 55.6% had an ECOG PS of 1, 50.6% had an IPI score of 3 to 5, and 46.9% had disease of GCB cell of origin by immunohistochemistry, whereas 8.6% had disease of GCB cell of origin by gene-expression profiling. Mean time since first DLBCL diagnosis was 39.6 months (SD 34.8 months). All (100%) patients had 1 or more prior anticancer medication, 50.6% of patients had received 2 or more prior therapy lines, and 44.4% were refractory to their most recent previous therapy. The most common reasons for ASCT ineligibility were older age (46.3%) and chemorefractory status (22.5%).
Table 7: Summary of Baseline Characteristics in L-MIND – All Enrolled Patients
Characteristic | L-MIND (N = 81) |
|---|---|
Age, mean (SD), years | 69.3 (9.5) |
Sex, n (%) | |
Male | 44 (54.3) |
Female | 37 (45.7) |
Race | |
Asian | 2 (2.5) |
White | 72 (88.9) |
Other | 1 (1.2) |
Missing | 6 (7.4) |
ECOG PS, n (%) | |
0 | 29 (35.8) |
1 | 45 (55.6) |
2 | 7 (8.6) |
Prior systemic treatment lines (DLBCL medications), n (%) | |
1 | 40 (49.4) |
2 | 35 (43.2) |
3 | 5 (6.2) |
4 | 1 (1.2) |
≥ 2 | 41 (50.6) |
Ann Arbor disease stage | |
Stage I and II | 20 (24.7) |
Stage III and IV | 61 (75.3) |
IPI category (score) | |
Low and low-intermediate risk (0 to 2) | 40 (49.4) |
Higher and intermediate-high risk (3 to 5) | 41 (50.6) |
Bulky disease at screening, n (%) | |
Present | 15 (18.5) |
Absent | 65 (80.2) |
Missing | 1 (1.2) |
Cell of origin by immunohistochemistry, n (%) | |
GCB | 38 (46.9) |
Non-GCB | 21 (25.9) |
Missing | 22 (27.2) |
Cell of origin by gene-expression profiling, n (%) | |
GCB | 7 (8.6) |
ABC | 19 (23.5) |
Unclassified | 6 (7.4) |
No evaluable | 5 (6.2) |
Missing | 44 (54.3) |
Reason for ASCT ineligibility, n (%) | |
Chemorefractorya | 18 (22.5) |
Comorbiditiesb | 11 (13.8) |
Older agec | 37 (46.3) |
Refusal of HDT or ASCT | 13 (16.3) |
Other | 1 (1.3) |
Time since first DLBCL diagnosis, mean (SD), months | 39.6 (34.8) |
Time since first DLBCL progression or relapse, mean (SD), months | 9.8 (14.8) |
Relapse after initial diagnosis of DLBCL, n (%) | |
Early (≤ 12 months) | 19 (23.5) |
Late (> 12 months) | 61 (75.3) |
Unknown | 1 (1.2) |
Time since discontinuation of last prior anti-DLBCL medication or ASCT, mean (SD), months | 17.0 (21.8) |
Refractoriness to last prior therapy n (%) | |
Yes | 36 (44.4) |
No | 45 (55.6) |
Primary refractoriness, n (%) | |
Yes | 15 (18.5) |
No | 66 (81.5) |
Prior ASCT, n (%) | |
Yes | 9 (11.1) |
No | 72 (88.9) |
ABC = activated B-cell; ASCT = autologous stem cell transplantation; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group performance status; GCB = germinal centre B-cell like; HDT = high-dose chemotherapy; IPI = International Prognostic Index; SD = standard deviation.
aChemorefractory patients included patients who failed to achieve PR or CR with salvage therapy or who underwent stem cell transplant before enrolment.
bAll patients who were not chemorefractory and who had comorbidities, defined as (1) diffusion lung capacity for carbon monoxide less than 50% by pulmonary function test, (2) left ventricular ejection fraction less than 50% by multiple gated acquisition echocardiogram, or (3) other organ dysfunction or comorbidities precluding the use of HDT or ASCT on the basis of unacceptable risk of treatment.
cAll patients who were not chemorefractory, had no comorbidities as defined in [b], but were too old (age > 70 years).
Source: L-MIND Clinical Study Report.10
Interventions
All patients had received R-CHOP or equivalent chemoimmunotherapy before study entry.24 Study treatment in L-MIND consisted of tafasitamab plus lenalidomide for up to 12 cycles (28 days each), followed by tafasitamab monotherapy in patients with stable disease or better until disease progression.
Tafasitamab (12 mg/kg, IV) was administered over approximately 2 hours. During cycles 1 to 3, tafasitamab infusions were administered weekly on days 1, 8, 15, and 22, with an additional loading dose administered on day 4 of cycle 1. From cycle 4 forward, tafasitamab infusions were administered every 14 days, on days 1 and 15 of each cycle.
Pre-medications as prophylaxis for infusion-related reactions were administered 0.5 to 2 hours before tafasitamab. Pre-medications included antipyretics, histamine (H1 and H2) receptor blockers, glucocorticoids, and meperidine. Pre-medication for patients who did not experience any infusion-related reactions to tafasitamab during the first 3 infusions was optional for subsequent infusions at the discretion of the investigator. Otherwise, pre-medication was continued for all subsequent tafasitamab administrations.
Tafasitamab was interrupted for any grade 2 to 4 infusion-related reactions or other toxicities, and patients discontinued tafasitamab if they experienced a grade 4 infusion-related reaction. Tafasitamab was administered until disease progression, unacceptable toxicity, or discontinuation for any other reason. Continuing tafasitamab despite disease progression was at the discretion of the investigator.
Lenalidomide was self-administered on days 1 to 21 of each 28-day cycle for a maximum of 12 cycles. The starting dosage was 25 mg orally daily. A stepwise dosage reduction of lenalidomide was done in cases of toxicities, with a decrease by 5 mg per day in each step, only once per cycle, without re-escalation. If the lenalidomide dosage was reduced during the previous cycle, then that reduced dosage level was continued in the new cycle. There was no more than 1 dosage reduction from 1 cycle to the next. Once a patient’s lenalidomide dose had been reduced, no dosage re-escalation was permitted.
Concomitant Medications
Patients could receive concomitant medications for the treatment of symptoms, AEs, and intercurrent illnesses. Medications to treat concomitant diseases (e.g., diabetes, hypertension, bronchial asthma, chronic obstructive pulmonary disease) were allowed. Patients could also receive therapy to mitigate side effects of the study medication, as clinically indicated, as well as best supportive care as per institutional guidelines. This included antiemetics, antidiarrheals, anticholinergics, antispasmodics, antipyretics, antihistamines, analgesics, antibiotics, and other medications intended to treat symptoms. Growth factors could be prescribed during the treatment and follow-up periods at the investigator’s discretion.
Patients were not permitted to receive a CD20-targeted therapy, chemotherapy, radiotherapy, investigational anticancer therapy, or other lymphoma-specific therapy within 14 days before cycle 1 day 1 of the study. In addition, no radiotherapy (including limited field radiotherapy) was permitted after the baseline PET-CT scan for initial disease assessment. Other than the study drugs, patients did not receive any other DLBCL-specific therapy during the study treatment period. After disease progression had been recorded, additional antineoplastic therapies were permitted at the discretion of the investigator and in accordance with the local guidelines.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trial included in this review is provided in Table 8. These end points are further summarized in this section.
Efficacy and Tumour Imaging Assessments
Disease response was assessed according to the revised response criteria based on the guidelines of the IWG reported by Cheson et al. (2007).8 In the L-MIND study, disease and response assessments as well as tumour measurements were performed. All response assessments were evaluated locally and centrally. Response assessment made by the central radiology and clinical review at cycle 12 day 28 and/or at the EOT visit, as applicable, were considered for the main efficacy analysis.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | L-MIND |
|---|---|
OS | Secondary |
PFS | Secondary |
TTP | Secondary |
EFS | Exploratory |
ORR | Primary (ORR by IRC), secondary (ORR by investigator) |
CRR | Secondary |
DOR | Secondary |
TTR | Exploratory |
TTNT | Secondary |
HRQoL | Not reported |
Harms | Secondary |
CRR = complete response rate; DOR = duration of response; EFS = event-free survival; HRQoL = health-related quality of life; IRC = independent review committee; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; TTNT = time to next treatment; TTP = time to progression; TTR = time to response.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3,14 and sponsor’s submission to CADTH.24
A local assessment of efficacy or disease response was performed and recorded at cycle 3 day 1, cycle 5 day 1, cycle 7 day 1, and cycle 10 day 1, in addition to central review at the end of cycle 12 (for decisions concerning the additional cycle 13 through cycle 24 tafasitamab treatment) and at the EOT visit (to determine whether disease had progressed). During the additional treatment phase, disease was assessed as follows:
Cycle 13 until cycle 24: The first assessment during this phase was carried out approximately 3 months and 2 days after the cycle 12 day 28 PET-CT assessment, if required. Thus, this assessment was not needed at cycle 13 day 1, and an interval longer than 3 months was acceptable for the cycle 16 day 1 assessment. Thereafter, the assessment was repeated approximately every 3 months plus or minus 2 days from the previous scan.
Cycle 25 onward: appoximately once every year after the previous scan.
Initial disease and disease response assessments for the primary end point were made centrally by PET-CT (CT with IV contrast) at screening, at day 28 (plus or minus 4 days) of cycle 12, and at the EOT visit, if a patient was withdrawn from treatment before the end of cycle 12 for reasons other than progressive disease (PD). PET-CT scanning during the additional treatment phase (cycle 13 onward) with tafasitamab was performed at the discretion of the investigator and was not performed more than approximately once per year.
MRI was permitted in lieu of CT, and PET-MRI in lieu of PET-CT, for patients with contraindications to the administration of contrast drugs, or due to other medical reasons, at the same time points as CT, or in addition to CT, at the discretion of the investigator.
End Point Definitions
OS was defined as the time from the first administration of any study drug until the date of death from any cause. Patients who were alive as of the data cut-off date or dropped out early were censored at the date of last contact.
PFS was defined as the time elapsed between the first administration of any study drug and lymphoma progression or death from any cause, whichever occurred first. The date of progression was the first date for which PD was assessed as the objective response. The tumour assessments were derived according to the IWG treatment response criteria for malignant lymphoma by an IRC. If a patient was alive and progression-free at the data cut-off date, the patient was censored.
TTP was defined as the time from the first administration of any study drug until documented DLBCL progression or death due to lymphoma.
EFS was defined as the time from the date of the first administration of any study drug to the date of tumour progression, first initiation of a new non-study antineoplastic therapy, or death from any cause, whichever comes first.
ORR was defined as the proportion of patients with a CR or PR up until disease progression, based on central radiological/clinical evaluations by the IRC. Objective response was defined as CR or PR after DLBCL evaluation using the IWG treatment response criteria for malignant lymphoma. The best ORR was the primary end point, considering all response assessments recorded until the cut-off for the primary analysis.
CRR was defined as the proportion of patients with a CR.
DOR was defined as the time interval between the initial time point of tumour response (CR or PR, whichever status was recorded first) and the first date that recurrence of PD or death was documented. If a patient was alive and progression-free at the data cut-off date, the patient was censored.
TTR was defined as the time from the first administration of any study drug to the first documented response (CR or PR).
TTNT was defined as the time from the first administration of any study drug to the institution of next antineoplastic therapy (for any reason, including disease progression, treatment toxicity, and patient preference) or death from any cause, whichever came first.
The incidence and severity of AEs were a secondary outcome in the L-MIND study. MedDRA-coded AEs were used to show the incidence of all AEs by system organ class, preferred term, relationship to treatment, severity, and seriousness.
Statistical Analysis
For the L-MIND study, data were analyzed descriptively. No formal statistical hypothesis testing was performed. For continuous variables, the number of non-missing observations, mean, SD, minimum and maximum values, and quartiles (first quartile, median, third quartile) were presented. For categorical variables, the number of non-missing observations, the number of missing observations, the relevant percentage of the analysis population, and frequencies were reported. If not defined otherwise, the percentage denominator was the number of patients with non-missing information. For subcategories, the relative frequencies were calculated on the basis of the patients in the respective category.
Sample Size Determination and Power Calculation
The L-MIND study planned to enrol approximately 80 patients with R/R DLBCL. The sample size was determined using various possible monotherapy and combination effect ORR rates and power assumptions. To determine the sample size, it was assumed that the combination treatment could improve the ORR from a value of 20% (with monotherapy) to 35%. Applying an exact binomial test with a 2-sided significance level of 5% and a power of 85%, the estimated sample size was 73 patients. According to this scenario, an observed ORR of 32% would lead to a statistically significant study outcome. Assuming a dropout rate of 10%, a total sample size of approximately 80 patients was estimated.
Primary End Point
ORR was defined as the proportion of responders whose best response at any time throughout treatment or follow-up was CR or PR. The number and percentage of patients classified as having a best overall response of CR or PR as well as 95% CIs calculated using the Clopper-Pearson exact method were presented.
For the main analysis, the denominator for calculating the ORR was the total number of patients in the FAS population. The tumour assessments were performed by the IRC. Patients with no post-baseline assessment of response were included as nonresponders. Patients with a best response of “not evaluable” were summarized by the reason that their status was unknown. Concordance between IRC assessment and investigator assessment was also analyzed.
Subgroup Analyses
ORR was reported for the following subgroups identified in the CADTH systematic review protocol: refractory to last therapy (yes versus no), primary refractoriness (yes versus no), prior ASCT (yes versus no), number of prior treatment lines, reason for ASCT ineligibility (chemorefractory versus older age or comorbidities versus other), cell of origin, and IPI score. Subgroup analyses were exploratory. All subgroups except for IPI score were pre-specified in the study protocol. All subgroup analyses were conducted using the FAS, based on both IRC and investigator assessment.
Sensitivity Analyses
Sensitivity analyses were conducted based on local investigator assessment, using a per-protocol set (i.e., patients without any post-baseline assessment of DLBCL response or patients who had major protocol deviations were excluded) and excluding the patients with no post-baseline assessment of response or with all post-baseline assessments categorized as “unknown.” The number (and percentage) of patients was descriptively tabulated by categories of individual best outcome in tumour response assessments (CR, PR, stable disease, PD, not evaluable).
Secondary End Points
For time-to-event end points, Kaplan–Meier estimates of the first quartile, median, and third quartile, along with their 95% CIs, were reported. The corresponding Kaplan–Meier survival curves were presented. The OS and PFS probabilities at specific time points (1 month, 3 months, 6 months, 12 months, 18 months, 24 months, 36 months, 48 months) and the associated 95% CIs (Greenwood formula) were summarized.
An AE summary table was used to present the number of events, number of patients, and percentage of patients with treatment-emergent AEs, SAEs, and treatment-emergent AEs that led to study discontinuation.
Censoring
For OS, patients who were alive or dropped out early were censored at the date of last contact. For PFS and DOR, if a patient was alive and progression-free at the data cut-off date, the patient was censored. Patients who had not experienced a lymphoma progression or death from any cause were censored at the last available tumour assessment. The same held true for patients who were lost to follow-up.
Subgroup Analyses
Subgroup analyses were exploratory. PFS, DOR, and OS were reported for the following subgroups identified in the CADTH systematic review protocol: refractory to last therapy (yes versus no), primary refractoriness (yes versus no), prior ASCT (yes versus no), number of prior treatment lines, reason for ASCT ineligibility (chemorefractory versus older age or comorbidities versus other), cell of origin, and IPI score. All subgroup analyses were conducted using the FAS based on both IRC and investigator assessment. Kaplan–Meier curves were provided for PFS, DOR, and OS.
Sensitivity Analyses
Sensitivity analyses for the secondary end points were generally performed in the same manner as for the primary end point. For PFS, an additional sensitivity analysis with patients having more than 1 missed visit, but having an available death date, were included in the time-to-event analysis and considered to have a PFS event (FAS for both IRC and investigator response assessment). For DOR, an additional sensitivity analysis was performed—with DOR stratified by best overall response—and duration of CR was analyzed.
Planned Analyses
A safety run-in analysis was planned for after the first 6 patients had been accrued. At this time, a safety review panel consisting of the sponsor, a representative of the participating investigators, and 2 independent expert clinical hematologists performed a clinical safety review based on the number and type of AEs during the first cycle and on laboratory values (biochemistry and hematology). After discussion of these data, the panel would consider the lenalidomide dosage tolerated or in need of reduction. The sponsor opened the second part of the study for enrolment following the outcome of this discussion.
The primary analysis was conducted after all patients had undergone at least 1 post-baseline response assessment at the earliest. The sponsor decided when to perform the primary analysis (approximately 12 months after the last patient has been enrolled). The primary analysis corresponds to the analysis with a data cut-off date of November 30, 2018.
The final analysis is planned when all patients have completed their EOT visit after database lock.
Protocol Amendments and Deviations
In the original L-MIND study protocol, patients were required to have a histologically confirmed diagnosis of DLBCL (not otherwise specified) according to the Revised European American Lymphoma or WHO classification. Patients with NHL other than DLBCL with classical histology (e.g., including patients with DLBCL transformed from indolent lymphomas) were excluded. In addition, patients who had relapsed within 3 months of prior CD20-targeted therapy were excluded. For the first 6 months (6 cycles) of the study, each cycle consisted of a tafasitamab infusion on day 1, day 8, day 15, and day 22 of the cycle. Additionally, a loading dose was administered on day 4 of cycle 1. Thereafter, tafasitamab was administered biweekly (every 14 days), with infusions on days 1 and 15 of each 28-day cycle. Tafasitamab could be given for up to 24 months in total. A summary of key protocol amendments implemented in the L-MIND study is presented in Table 9. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 9: Summary of Key Protocol Amendments in L-MIND
Amendment (date) | Key changes |
|---|---|
Protocol Amendment 1 (May 27, 2015) |
|
Protocol Amendment 2, (June 27, 2016) |
|
Protocol Amendment 3 |||||||||||||||| |
|
Anti-HBc = hepatitis B core antibody; DLBCL = diffuse large B-cell lymphoma; HBV = hepatitis B virus; NHL = non-Hodgkin lymphoma; PR = partial response; ULN = upper limit of normal.
Source: L-MIND Clinical Study Report,10 L-MIND Clinical Study Protocol.24
A summary of key protocol deviations during the L-MIND study is presented in Table 10. As of the primary analysis (November 30, 2018, data cut), diagnosis of DLBCL was not confirmed by the central pathology lab in 9 patients (11.1%). ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The following subtypes of NHL were classified as non-DLBCL cases: follicular lymphoma (grade 2 + 3A), follicular lymphoma grade 2, mantle-cell lymphoma, classic type, and marginal zone lymphoma. Diagnosis remained unknown because there was insufficient tissue and because no biopsy material was available for a further 2 patients (2.5%). ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Two patients with double- or triple-hit genetics were enrolled in L-MIND.
Table 10: Summary of Key Protocol Deviations – Safety Analysis Set, November 30, 2018, Data Cut-Off (Primary Analysis)
Protocol deviation | L-MIND (N = 81) |
|---|---|
Procedure or test, n (%) | |||||||||||| |
Study drug and treatment, n (%) | |||||||||||| |
Laboratory assessment, n (%) | |||||||||||| |
Informed consent form, n (%) | |||||||||||| |
Eligibility criteria, n (%) | |||||||||||| |
Prohibited concomitant medication, n (%) | |||||||||||| |
Visit schedule and assessment, n (%) | |||||||||||| |
Other, n (%) | |||||||||||| |
Source: L-MIND Clinical Study Report.10
Analysis Populations
The FAS included all patients who received at least 1 dose of tafasitamab and 1 dose of lenalidomide (i.e., both study drugs must have been received at least once).
The safety analysis set included all patients who received at least 1 dose of either study drug (tafasitamab or lenalidomide).
Results
Patient Disposition
Patient disposition in the L-MIND study (summarized in Table 11) was the same at each of the analyses (November 2018, November 2019, and October 2020 data cut-off dates). In total, 156 patients were screened, of whom 75 (48.1%) were screen failures. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| A total of 81 patients were enrolled in L-MIND, and 80 (98.8%) patients received both tafasitamab and lenalidomide (i.e., were included in the FAS). One enrolled patient received tafasitamab only and was excluded from the FAS. Overall, 30 (37.5%) patients successfully completed the combination treatment phase on both study drugs (12 cycles); 50 (62.5%) patients did not successfully complete the combination treatment phase. In total, 45 (56.3%) discontinued both study drugs before completing 12 cycles; the primary reasons were disease relapse (39.5%), AE (9.9%), withdrawal by patient (2.5%), death (2.5%), and other (2.5%). Four (4.9%) patients discontinued lenalidomide only due to AEs before completing combination treatment. One (1.2%) patient discontinued tafasitamab only due to an AE before completing combination treatment. Additional details on the reasons for study treatment discontinuation are presented in Table 12.
As of the October 30, 2020, data cut-off, 34 (42.0%) patients had reached cycle 13 day 1 (beginning of treatment with tafasitamab monotherapy). Fifteen (44.1%) of these patients treated with tafasitamab monotherapy had discontinued tafasitamab treatment as of the data cut-off date due to disease progression (n = 8 patients), withdrawal by patient (n = 4), other reasons (n = 2), or an AE (n = 1). Thus, 19 patients were continuing tafasitamab monotherapy, and 62 patients had discontinued study treatment at the data cut-off. Of the patients who had discontinued study treatment, 42 had died, 13 were in survival follow-up, and 7 were lost to follow-up at the data cut-off.
Table 11: L-MIND Patient Disposition
Disposition | L-MIND |
|---|---|
Screened, N | 156 |
Enrolled, N | 81 |
Treated with tafasitamab plus lenalidomide, N | 80 |
Discontinued from study treatment (cycle 1 to 12), n (%) | 50 (62.5) |
Tafasitamab only | 1 (1.3) |
Lenalidomide only | 4 (5.0) |
Both tafasitamab plus lenalidomide | 45 (56.3) |
Finished combination treatment on both study drugs, n (%) | 30 (37.5) |
Treated with tafasitamab monotherapy (cycle 13 onward), n (%) | 34 (42.5) |
FASa, N | 80 |
Safety, N | 81 |
FAS = full analysis set.
aOne enrolled patient received tafasitamab only and was excluded from the FAS.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Exposure to Study Treatments
Exposure to study treatment in the L-MIND study as of the primary analysis (November 30, 2018. data cut-off date) and most recent interim analysis (October 30, 2020, data cut-off) is summarized in Table 12. Exposure to study treatment was not reported at the interim analysis with the November 30, 2019, data cut-off date. All 81 (100%) patients enrolled in the L-MIND study received at least 1 dose of a study drug. As of the primary analysis and most recent analysis, the median duration of exposure to the study treatment (tafasitamab plus lenalidomide) was 9.2 months.
Concomitant medications were defined as medications with a start date after the start of the treatment period but before study completion or discontinuation. As of the primary analysis, all patients in the FAS reported the use of concomitant medications. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Dosage Interruptions and Reductions
As of the primary analysis (November 30, 2018, data cut-off), 61/81 (75.3%) patients had temporary interruptions of tafasitamab in total, including skipped visits and infusion interruptions. The most common reason for temporary interruption of tafasitamab was AEs (n = 47/61, 77.0%). As of the October 30, 2020, data cut-off, 64/81 (79.0%) patients had temporary interruptions of tafasitamab. The most common reason was AEs (n = 47/64, 73.4%). Dosage reductions of tafasitamab were not permitted per the study protocol.
As of both analyses, 28/80 (35.0%) patients who received lenalidomide had temporary interruptions. The most common reason was AEs, in 25 (89.3%) patients. Thirty-seven (45.7%) patients had at least 1 dosage reduction for lenalidomide.
Table 12: Summary of Exposure to Study Treatment in L-MIND – Safety Analysis Set (N = 81)
Characteristic/study drug | November 30, 2018, DCO (primary analysis) | October 30, 2020, DCO |
|---|---|---|
All study treatment | ||
Duration of exposure to study treatment | ||
Number of patients, n | 81 | 81 |
Mean (SD), months | 11.3 (9.7) | |||||||||||| |
Median (Q1 to Q3), months | 9.2 (2.2 to 18.8) | 9.2 (2.2 to 36.0) |
Duration of exposure to combination therapy or lenalidomide onlya | ||
Number of patients, n | 80 | 80 |
Mean (SD), months | 6.6 (4.3) | |||||||||||| |
Median (Q1 to Q3), months | 6.2 (2.1 to 10.9) | |||||||||||| |
Duration of exposure to tafasitamab monotherapy after lenalidomide discontinuation | ||
Number of patients, n | |||||||||||| | 52 |
Mean (SD), months | |||||||||||| | |||||||||||| |
Median (Q1 to Q3), months | |||||||||||| | 13.9 (0.5 to 31.7) |
Exposure to tafasitamab | ||
Number of patients exposed to tafasitamab, n | 81 | 81 |
Total number of tafasitamab infusions, n | ||
Mean (SD) | |||||||||||| | |||||||||||| |
Median (Q1 to Q3) | |||||||||||| | |||||||||||| |
Patients with ≥ 1 temporary interruption, n (%) | 61 | |||||||||||| |
Infusion-related reaction | 3 (4.9) | |||||||||||| |
AE | 47 (77.0) | |||||||||||| |
Unacceptable toxicity | 1 (1.6) | |||||||||||| |
Other | 33 (54.1) | |||||||||||| |
Patients who discontinued tafasitamab permanently, n (%) | 53 (65.4) | |||||||||||| |
Withdrawal by patient | 3 (3.7) | |||||||||||| |
AE | 10 (12.3) | |||||||||||| |
Disease relapse | 36 (44.4) | |||||||||||| |
Death | 2 (2.5) | |||||||||||| |
Other | 2 (2.5) | |||||||||||| |
Exposure to lenalidomide | ||
Number of patients exposed to lenalidomide, n | 80 | 80 |
Duration of exposure to lenalidomide, weeks | ||
Mean (SD) | |||||||||||| | |||||||||||| |
Median (Q1 to Q3) | |||||||||||| | |||||||||||| |
Patients with temporary interruptions, n (%) | 28 | |||||||||||| |
AE | 25 (89.3) | |||||||||||| |
Unacceptable toxicity | 1 (3.6) | |||||||||||| |
Other | 4 (14.3) | |||||||||||| |
Patients with any dose reductions of lenalidomide, n (%) | 37 (45.7) | |||||||||||| |
Patients who discontinued lenalidomide permanently at any time, n (%) | 80 (98.8) | |||||||||||| |
Withdrawal by patient | 2 (2.5) | |||||||||||| |
AE | 15 (18.5) | |||||||||||| |
Disease relapse | 24 (29.6) | |||||||||||| |
Death | 2 (2.5) | |||||||||||| |
Completion per protocol (after cycle 12) | 30 (37.0) | |||||||||||| |
Other | 1 (1.2) | |||||||||||| |
AE = adverse event; DCO = data cut-off; SD = standard deviation; Q1 = first quartile; Q3 = third quartile.
aDuration of exposure to combination therapy or lenalidomide only was the time from first administration of study drug to the last date of administration of lenalidomide or the last date of exposure to tafasitamab during combination treatment.
Source: L-MIND Clinical Study Report10 and Addendum 3.14
Compliance
Compliance with tafasitamab was calculated by taking the actual tafasitamab infusion doses divided by the planned tafasitamab infusion doses. Compliance was summarized per single infusion and per cycle. A boxplot depicting compliance with tafasitamab by treatment cycle as of the primary analysis (November 30, 2018, data cut-off) is presented in Appendix 3 (Figure 9). As of the primary analysis, the median compliance for tafasitamab for each cycle was 100% until cycle 34.
Compliance with lenalidomide was calculated by taking the total use of lenalidomide doses divided by the total planned lenalidomide doses. If these values were 80% or more and 120% or less of the planned dosage for the cycle, then the patient was considered compliant for that cycle. A boxplot of compliance with lenalidomide by cycle as of the primary analysis is presented in Appendix 3 (Figure 10). As of the primary analysis, the median compliance for lenalidomide was 100% for each cycle.
Subsequent Anticancer Treatments
As of the primary analysis (November 30, 2018, data cut-off date), 25 (31.3%) patients received non-study anticancer treatment after discontinuing study treatment. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in this section. See Appendix 3 for detailed efficacy data, including sensitivity analyses of patients with central pathology-confirmed DLBCL (Table 50).
Time-to-Event Outcomes
Overall Survival
A summary of OS results from the L-MIND as of the 3 data cut-off dates is presented in Table 13. As of the primary analysis (November 30, 2018, data cut-off), median follow-up time for OS was 19.6 months (95% CI, 15.3 to 21.9) and 29 (36.3%) deaths were observed in the FAS. The Kaplan–Meier plot of OS as of the primary analysis is depicted in Figure 3. The Kaplan–Meier estimate for the median OS was NR (95% CI, 18.3 to NR).
As of the October 30, 2020, data cut-off, median follow-up time for OS was 42.7 months (||||||||||||||||||||||||||||||||||||) and 41 (51.3%) deaths had occurred. Thirty-nine patients were censored in the OS analysis ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The Kaplan–Meier plot of OS as of the October 30, 2020, data cut-off is depicted in Figure 4. The Kaplan–Meier estimate for the median OS was 33.5 months (95% CI, 18.3 to NR).
The results of subgroup analyses of OS are presented in Table 14.
Table 13: Summary of OS Results in L-MIND – FAS
Outcome | L-MIND (N = 80) | ||
|---|---|---|---|
November 30, 2018, DCO (primary analysis) | November 30, 2019, DCO | October 30, 2020, DCO | |
Patients who died, n (%) | 29 (36.3) | 37 (46.3) | 41 (51.3) |
Censored, n (%) | |||||||||||| | 43 (53.8) | 39 (48.8) |
Median OSa (95% CIb), months | NR (18.3 to NR) | 31.6 (18.3 to NR) | 33.5 (18.3 to NR) |
Median follow-up time for OSc (95% CIb), months | 19.6 (15.3 to 21.9) | 31.8 (27.2 to 35.9) | 42.7 (||||||||||||) |
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; NR = not reached; OS = overall survival.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
cCalculated using the reverse Kaplan–Meier method, considering the censored patients as events and patients with events as censored.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Figure 3: Kaplan–Meier Plot of OS in L-MIND – FAS; November 30, 2018, Data Cut-Off (Primary Analysis)
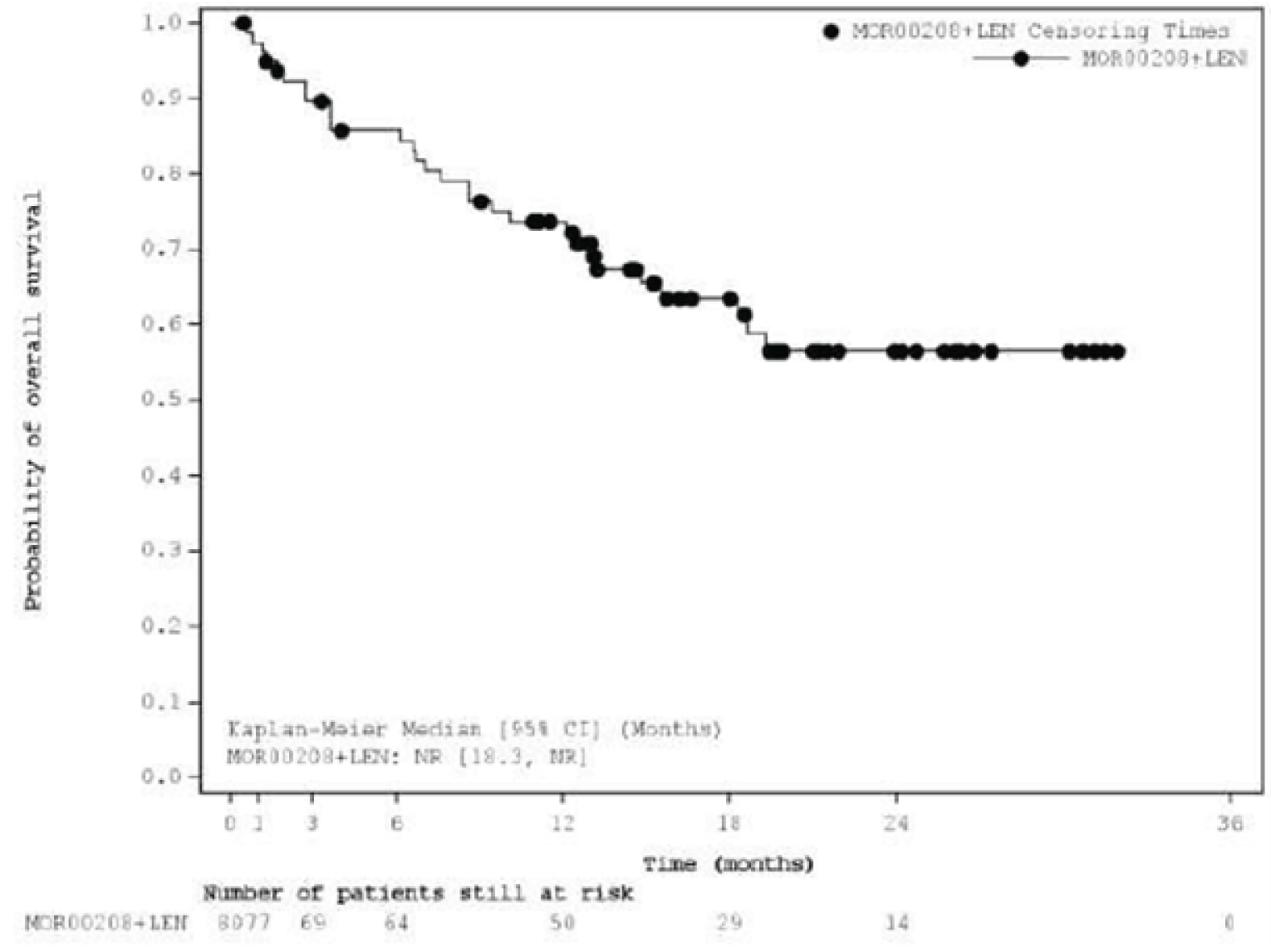
CI = confidence interval; FAS = full analysis set; LEN = lenalidomide; MOR00208 = tafasitamab; NR = not reached.
Source: L-MIND Clinical Study Report.10
Figure 4: Kaplan–Meier Plot of OS in L-MIND – FAS; October 30, 2020, Data Cut-Off
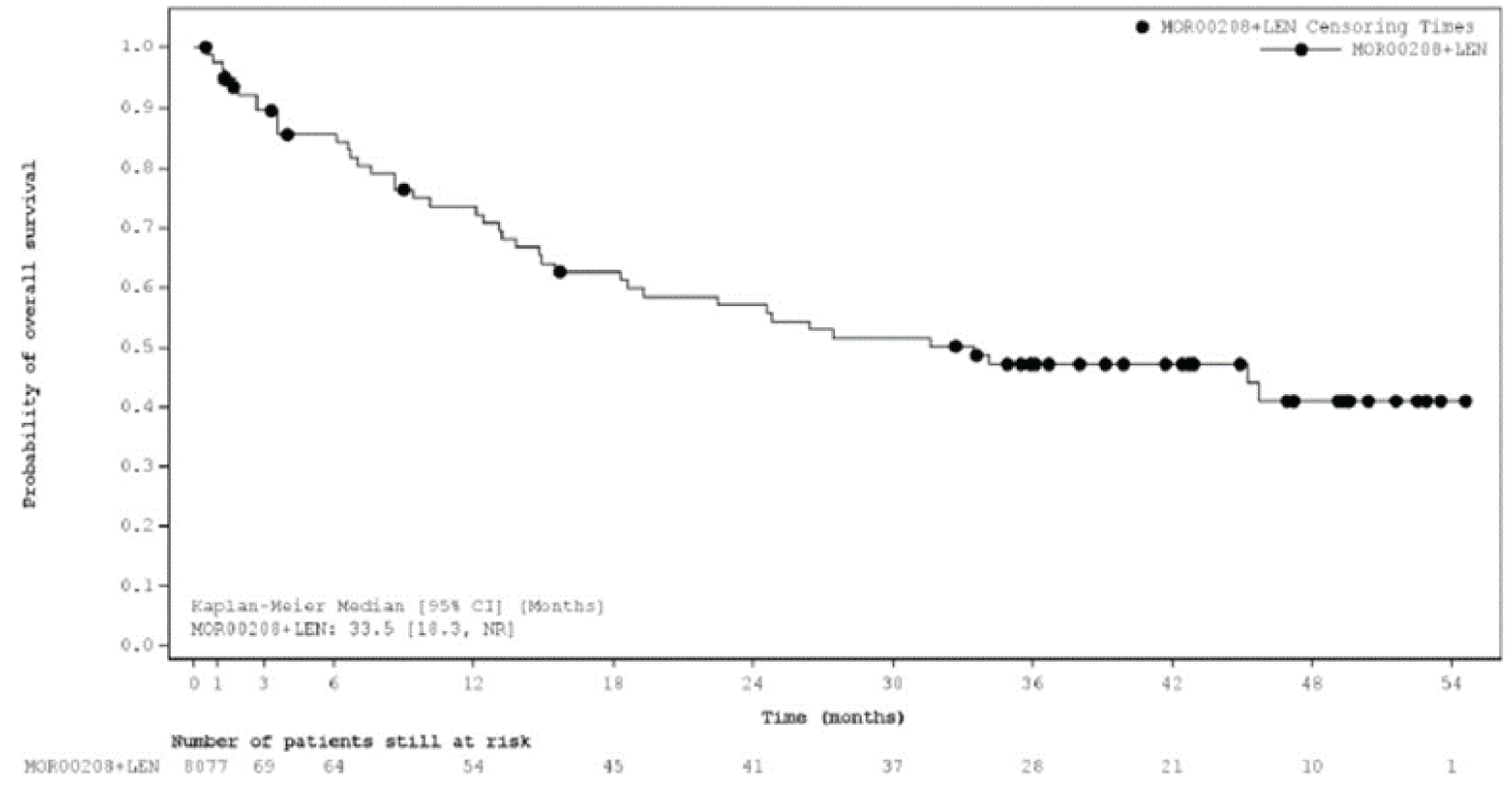
CI = confidence interval; FAS = full analysis set; LEN = lenalidomide; MOR00208 = tafasitamab; NR = not reached.
Source: L-MIND Clinical Study Report Addendum 3.14
Table 14: Subgroup Analyses of OS – FAS
Characteristic | N | Median OSa (95% CIb), months | ||
|---|---|---|---|---|
November 2018 DCO (primary analysis) | November 2019 DCO | October 2020 DCO | ||
IPI score | ||||
0 to 2 | 40 | NR (NR to NR) | NR (31.6 to NR) | |||||||||||| |
3 to 5 | 40 | 14.8 (8.6 to NR) | 14.8 (8.6 to 24.6) | |||||||||||| |
Refractory to last therapy | ||||
Yes | 35 | NR (8.6 to NR) | 15.5 (8.6 to NR) | |||||||||||| |
No | 45 | NR (18.6 to NR) | NR (19.3 to NR) | |||||||||||| |
Primary refractoriness | ||||
Yes | 15 | NR (1.3 to NR) | 13.8 (1.3 to NR) | |||||||||||| |
No | 65 | NR (18.3 to NR) | 34.1 (18.6 to NR) | |||||||||||| |
Prior ASCT | ||||
Yes | 9 | NR (12.4 to NR) | 31.6 (12.4 to 31.6) | |||||||||||| |
No | 71 | NR (15.5 to NR) | 34.1 (14.9 to NR) | |||||||||||| |
Number of prior treatment lines | ||||
1 | 40 | NR (19.3 to NR) | NR (24.6 to NR) | |||||||||||| |
≥ 2 | 40 | 15.5 (8.6 to NR) | 15.5 (8.6 to 34.1) | |||||||||||| |
Reason for ASCT ineligibility | ||||
Chemorefractory | 18 | 13.2 (7.6 to NR) | 22.5 (7.6 to 31.6) | |||||||||||| |
Comorbidities | 11 | NR (0.6 to NR) | NR (0.6 to NR) | |||||||||||| |
Older age | 37 | NR (15.5 to NR) | 24.8 (13.8 to NR) | |||||||||||| |
Refusal of HDT or ASCT | 13 | NR (14.8 to NR) | NR (26.4 to NR) | |||||||||||| |
Cell of origin by IHC | ||||
GCB | 37 | 19.3 (8.6 to NR) | 24.6 (8.6 to NR) | |||||||||||| |
Non-GCB | 21 | NR (12.4 to NR) | NR (12.4 to NR) | |||||||||||| |
ASCT = autologous stem cell transplantation; CI = confidence interval; DCO = data cut-off; FAS = full analysis set; GCB = germinal centre B-cell; HDT = high-dose chemotherapy; IHC = immunohistochemistry; IPI = International Prognostic Index; NR = not reached; OS = overall survival.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Progression-Free Survival
Results for PFS by IRC and PFS by investigator assessment in the L-MIND trial are summarized in Table 15.
As of the primary analysis (November 30, 2018, data cut-off), the median follow-up time for PFS by IRC in the FAS was 17.3 months (95% CI, 11.5 to 21.2) in the FAS. By IRC assessment, PFS events were observed in 39 (48.8%) patients. The Kaplan–Meier curve of PFS by IRC is depicted in Figure 5. The Kaplan–Meier estimate for the median PFS by IRC was 12.1 months (95% CI, 5.7 to NR). By investigator assessment, 44 (55.0%) patients had experienced an event and median PFS by investigator was 9.1 months (95% CI, 5.5 to 22.3).
As of the October 30, 2020, data cut-off date, the median follow-up time for PFS by IRC evaluation was 33.9 months (95% CI, 26.5 to 35.4) in the FAS. By IRC assessment, PFS events were observed in 42 (52.5%) patients. The Kaplan–Meier curve of PFS by IRC is depicted in Figure 6. The Kaplan–Meier estimate for median PFS by IRC was 11.6 months (95% CI, 6.3 to 45.7). By investigator assessment, 46 (57.5%) patients experienced a PFS event and median PFS by investigator was 9.1 (95% CI, 5.5 to 28.0) months.
Results of the subgroup analyses of PFS by IRC are reported in Table 16.
Table 15: Summary of PFS Results in L-MIND – FAS
Outcome | L-MIND (N = 80) | ||
|---|---|---|---|
November 30, 2018, DCO (primary analysis) | November 30, 2019, DCO | October 30, 2020, DCO | |
PFS by IRC | |||
Patients who experienced an event, n (%) | 39 (48.8) | 39 (48.8) | |||||||||||| |
Progression | 32 (40.0) | 32 (40.0) | |||||||||||| |
Death | 7 (8.8) | 7 (8.8) | |||||||||||| |
Censored, n (%) | 41 (51.3) | 41 (51.3) | |||||||||||| |
Median PFSa (95% CIb), months | 12.1 (5.7 to NR) | 16.2 (6.3 to NR) | |||||||||||| |
Median follow-up time for PFSc (95% CIb), months | 17.3 (11.5 to 21.2) | 22.6 (22.2 to 27.4) | |||||||||||| |
PFS by investigator | |||
Patients who experienced an event, n (%) | 44 (55.0) | 45 (56.3) | |||||||||||| |
Progression | 37 (46.3) | 38 (47.5) | |||||||||||| |
Death | 7 (8.8) | 7 (8.8) | |||||||||||| |
Censored, n (%) | 36 (45.0) | 35 (43.8) | |||||||||||| |
Median PFSa (95% CIb), months | 9.1 (5.5 to 22.3) | 9.1 (5.5 to 28.0) | |||||||||||| |
Median follow-up time for PFSc (95% CIb), months | 19.7 (14.3 to 22.1) | 23.6 (22.4 to 30.4) | |||||||||||| |
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; IRC = independent review committee; NR = not reached; PFS = progression-free survival.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
cCalculated using the reverse Kaplan–Meier method, considering the censored patients as events and patients with events as censored.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Figure 5: Kaplan–Meier Plot of PFS by IRC in L-MIND – FAS; November 30, 2018, Data Cut-Off (Primary Analysis)
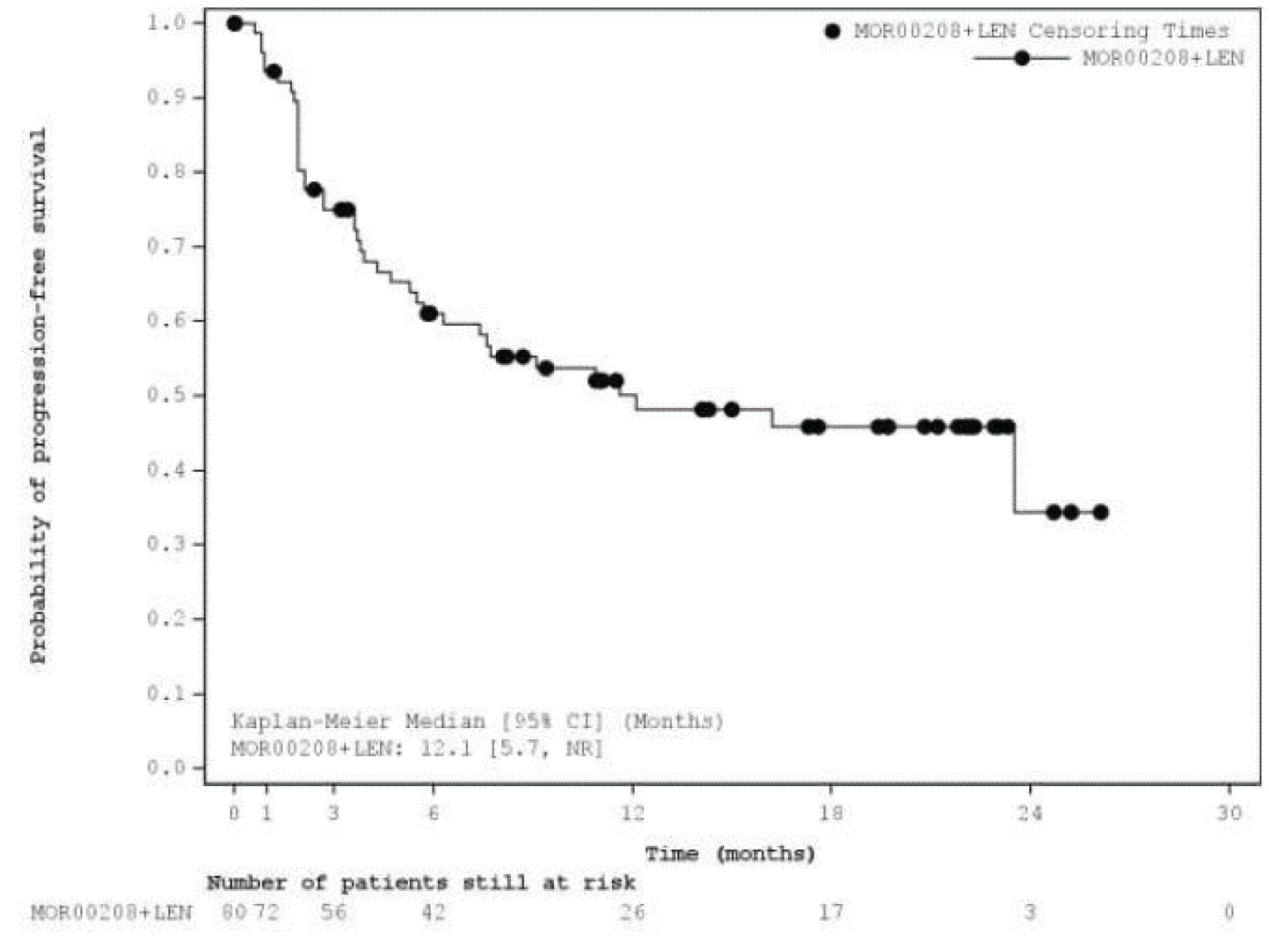
CI = confidence interval; FAS = full analysis set; IRC = independent review committee; LEN = lenalidomide; MOR00208 = tafasitamab; NR = not reached; PFS = progression-free survival.
Source: L-MIND Clinical Study Report.10

Table 16: Subgroup Analyses of PFS by IRC – FAS
Characteristic | N | Median PFSa (95% CIb), months | ||
|---|---|---|---|---|
November 2018 DCO (primary analysis) | November 2019 DCO | October 2020 DCO | ||
IPI score | ||||
0 to 2 | 40 | NR (12.1 to NR) | 36.4 (36.4 to NR) | |||||||||||| |
3 to 5 | 40 | 5.3 (3.6 to 7.7) | 5.7 (3.6 to 11.6) | |||||||||||| |
Refractory to last therapy | ||||
Yes | 35 | 7.6 (2.7 to NR) | 7.6 (2.7 to NR) | |||||||||||| |
No | 45 | 16.2 (7.4 to NR) | 23.5 (7.4 to NR) | |||||||||||| |
Primary refractoriness | ||||
Yes | 15 | 4.3 (0.9 to NR) | 5.3 (0.9 to NR) | |||||||||||| |
No | 65 | 16.2 (7.6 to NR) | 23.5 (7.6 to NR) | |||||||||||| |
Prior ASCT | ||||
Yes | 9 | NR (1.9 to NR) | NR (1.9 to NR) | |||||||||||| |
No | 71 | 12.1 (5.3 to NR) | 16.2 (5.5 to NR) | |||||||||||| |
Number of prior treatment lines | ||||
1 | 40 | 23.5 (7.4 to NR) | 36.4 (7.4 to NR) | |||||||||||| |
≥ 2 | 40 | 6.3 (2.7 to NR) | 7.6 (2.7 to NR) | |||||||||||| |
Reason for ASCT ineligibility | ||||
Chemorefractory | 18 | 7.6 (1.9 to NR) | 7.6 (1.9 to NR) | |||||||||||| |
Comorbidities | |||||||||||| | |||||||||||| | 5.3 (0.6 to NR) | |||||||||||| |
Older age | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Refusal of HDT or ASCT | 13 | 23.5 (1.9 to NR) | NR (1.9 to NR) | |||||||||||| |
Cell of origin by IHC | ||||
GCB | 37 | 7.4 (3.6 to 12.1) | 7.6 (3.6 to 36.4) | |||||||||||| |
Non-GCB | 21 | 23.5 (3.9 to NR) | 28.0 (6.3 to NR) | |||||||||||| |
ASCT = autologous stem cell transplantation; CI = confidence interval; DCO = data cut-off; FAS = full analysis set; GCB = germinal centre B-cell; HDT = high-dose therapy; IHC = immunohistochemistry; IPI = International Prognostic Index; NR = not reached; PFS = progression-free survival.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Time to Progression
TTP based on IRC evaluation was analyzed at the primary analysis and results are presented in Table 17. As of the November 30, 2018, data cut-off, 35 (43.8%) patients had progressed or died and the median TTP was 16.2 months (95% CI, 7.4 to NR). Median follow-up time for TTP was not reported. TTP was not analyzed at the subsequent interim analyses.
Event-Free Survival
EFS was not a protocol-defined end point in L-MIND and thus is considered exploratory. EFS was analyzed at the primary analysis, and results are presented in Table 18. As of the November 30, 2018, data cut-off, 46 (57.5%) patients had experienced progression, death, or received a new non-study antineoplastic treatment. Median EFS was 9.1 (95% CI, 5.3 to 21.0) months. The median follow-up time for EFS was 19.7 months (95% CI, 14.3 to 22.0). EFS was not analyzed at the subsequent interim analyses.
Table 17: Summary of TTP by IRC Results in L-MIND – FAS; November 30, 2018, Data Cut-Off (Primary Analysis)
Outcome | L-MIND (N = 80) |
|---|---|
Patients who experienced an event, n (%) | 35 (43.8) |
Progression | 32 (40.0) |
Death due to lymphoma | 3 (3.8) |
Censored, n (%) | 45 (56.3) |
Median TTPa (95% CIb), months | 16.2 (7.4 to NR) |
CI = confidence interval; FAS = full analysis set; IRC = independent review committee; NR = not reached; TTP = time to progression.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
Source: L-MIND Clinical Study Report.10
|||||||||||| | |||||||||||| |
|---|---|
|||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Response/Remission Rate
Objective Response Rate
The ORR by IRC was the primary end point of the L-MIND study. The ORR by investigator assessment was a secondary end point. Results for ORR by IRC and ORR by investigator, including the proportion of patients who achieved CR (i.e., CRR) and PR, are presented in Table 19.
As of the primary analysis (November 30, 2018, data cut-off), the ORR by IRC was 60.0% (95% CI, 48.4 to 70.8). The best objective response for patients was CR for 34/80 (42.5%) patients and PR for 14/80 (17.5%) patients. Based on investigator assessment, the ORR was 63.8% (95% CI, 52.2 to 74.2).
As of the most recent efficacy analysis (October 30, 2020, data cut-off date), ORR by IRC was 57.5% (95% CI, 45.9 to 68.5). Thirty-two (40.0%) patients had CR and 14 (17.5%) patients had PR. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
At each analysis, 8 patients by IRC and 7 patients by investigator were considered not evaluable because they had no valid post-baseline assessment.
The results of the subgroup analyses of ORR by IRC are presented in Table 20.
Table 19: Summary of ORR Results in L-MIND – FAS
Outcome | L-MIND (N = 80) | ||
|---|---|---|---|
November 30, 2018, DCO (primary analysis) | November 30, 2019, DCO | October 30, 2020, DCO | |
ORR by IRC (primary end point) | |||
ORR, n (%) [95% CIa] | 48 (60.0) [48.4 to 70.8] | 47 (58.8) [47.2 to 69.6] | 46 (57.5) [45.9 to 68.5] |
Best objective response, n (%) | |||
CR | 34 (42.5) | 33 (41.3) | 32 (40.0) |
PR | 14 (17.5) | 14 (17.5) | 14 (17.5) |
SD | 11 (13.8) | 12 (15.0) | 13 (16.3) |
PD | 13 (16.3) | 13 (16.3) | 13 (16.3) |
Not evaluable | 8 (10.0) | 8 (10.0) | 8 (10.0) |
ORR by investigator (secondary end point) | |||
ORR, n (%) [95% CIa] | 51 (63.8) [52.2 to 74.2] | 51 (63.8) [52.2 to 74.2] | ||||||||||||||||||||||||||| |
Best objective response, n (%) | |||
CR | 29 (36.3) | 29 (36.3) | |||||||||||| |
PR | 22 (27.5) | 22 (27.5) | |||||||||||| |
SD | 9 (11.3) | 9 (11.3) | |||||||||||| |
PD | 13 (16.3) | 13 (16.3) | |||||||||||| |
Not evaluable | 7 (8.8) | 7 (8.8) | |||||||||||| |
CI = confidence interval; CR = complete response; DCO = data cut-off; FAS = full analysis set; IRC = independent review committee; ORR = objective response rate; PD = progressive disease; PR = partial response; SD = stable disease.
a95% CI calculated using 2-sided 95% Clopper-Pearson exact method based on binomial distribution.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Table 20: Subgroup Analyses of ORR by IRC (Primary End Point) – FAS
Characteristic | N | November 2018 DCO (primary analysis) | November 2019 DCO | October 2020 DCO | |||
|---|---|---|---|---|---|---|---|
N | ORR (95% CIa), % | n | ORR (95% CIa), % | n | ORR (95% CIa), % | ||
IPI score | |||||||
0 to 2 | 40 | 28 | 70.0 (53.5 to 83.4) | 28 | 70.0 (53.5 to 83.4) | |||||||| | |||||||||||| |
3 to 5 | 40 | 20 | 50.0 (33.8 to 66.2) | 19 | 47.5 (31.5 to 63.9) | |||||||| | |||||||||||| |
Refractory to last therapy | |||||||
Yes | 35 | 21 | 60.0 (42.1 to 76.1) | 21 | 60.0 (42.1 to 76.1) | |||||||| | |||||||||||| |
No | 45 | 27 | 60.0 (44.3 to 74.3) | 26 | 57.6 (42.2 to 72.3) | |||||||| | |||||||||||| |
Primary refractoriness | |||||||
Yes | 15 | 9 | 60.0 (32.3 to 83.7) | 8 | 53.3 (26.6 to 78.7) | |||||||| | |||||||||||| |
No | 65 | 39 | 60.0 (47.1 to 72.0) | 39 | 60.0 (47.1 to 72.0) | |||||||| | |||||||||||| |
Prior ASCT | |||||||
Yes | 9 | 7 | 77.8 (40.0 to 97.2) | Not reported | Not reported | |||||||| | |||||||||||| |
No | 71 | 41 | 57.7 (45.4 to 69.4) | Not reported | Not reported | |||||||| | |||||||||||| |
Number of prior treatment lines | |||||||
1 | 40 | 28 | 70.7 (53.5 to 83.4) | 28 | 70.0 (53.5 to 83.4) | |||||||| | |||||||||||| |
≥ 2 | 40 | 20 | 50.0 (33.8 to 66.2) | 19 | 47.5 (31.5 to 63.9) | |||||||| | |||||||||||| |
Reason for ASCT ineligibility | |||||||
Chemorefractory | 18 | 10 | 55.6 (30.8 to 78.5) | 10 | 55.6 (30.8 to 78.5) | |||||||| | |||||||||||| |
Comorbidities | 11 | 6 | 54.5 (23.4 to 83.3) | 6 | 54.5 (23.4 to 83.3) | |||||||| | |||||||||||| |
Older age | 37 | 24 | 64.9 (47.5 to 79.8) | |||||||| | |||||||||||| | |||||||| | |||||||||||| |
Refusal of HDT or ASCT | 13 | 8 | 61.5 (31.6 to 86.1) | 7 | 53.8 (25.1 to 80.8) | |||||||| | |||||||||||| |
Cell of origin by IHC | |||||||
GCB | 37 | 18 | 48.6 (31.9 to 65.6) | 18 | 47.4 (31.0 to 64.2) | |||||||| | |||||||||||| |
Non-GCB | 21 | 15 | 71.4 (47.8 to 88.7) | 16 | 72.7 (49.8 to 89.3) | |||||||| | |||||||||||| |
ASCT = autologous stem cell transplantation; CI = confidence interval; DCO = data cut-off; FAS = full analysis set; GCB = germinal centre B-cell; HDT = high-dose therapy; IHC = immunohistochemistry; IPI = International Prognostic Index; IRC = independent review committee; n = patients with response; ORR = objective response rate.
a95% CI calculated using 2-sided 95% Clopper-Pearson exact method based on binomial distribution.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Duration of Response
A summary of DOR by IRC and DOR by investigator assessment is presented in Table 21. Kaplan–Meier plots of DOR by IRC assessment by best objective response achieved (CR or PR) as of the primary analysis and most recent analysis are depicted in Figure 7 and Figure 8, respectively.
As of the primary analysis (November 30, 2018, data cut-off), 48 patients had a response (CR or PR) by IRC assessment. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The Kaplan–Meier estimate for the median DOR by IRC was 21.7 (95% CI, 21.7 to NR) months. Median DOR by IRC in patients with PR was 4.4 months (95% CI, 2.0 to 9.1) and NR (95% CI, 21.7 to NR) in patients with CR.
As of the most recent analysis (October 30, 2020, data cut-off), 46 patients had a response by IRC assessment. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The Kaplan–Meier estimate for the median DOR by IRC was 43.9 (95% CI, 26.1 to NR) months. Median DOR by IRC in patients with PR was 5.6 (95% CI, 2.2 to NR) months compared to NR (95% CI, 43.9 to NR) in patients with CR.
Results of the subgroup analyses for DOR by IRC are presented in Table 22.
Table 21: Summary of DOR Results in L-MIND – FAS
Outcome | L-MIND (N = 80) | ||
|---|---|---|---|
November 30, 2018, DCO (primary analysis) | November 30, 2019, DCO | October 30, 2020, DCO | |
DOR by IRC | |||
Patients with response by IRC, n | 48 | 47 | 46 |
Patients with event, n (%) | 13 (27.1) | 13 (27.1) | |||||||||||| |
Progression | 12 (25.0) | 12 (25.5) | |||||||||||| |
Death | 1 (2.1) | 1 (2.1) | |||||||||||| |
Censored, n (%) | 35 (72.9) | 34 (72.3) | |||||||||||| |
Median DORa (95% CIb), months | 21.7 (21.7 to NR) | 34.6 (26.1 to 34.6) | 43.9 (26.1 to NR) |
DOR by investigator | |||
Patients with response by investigator, n | 51 | 51 | |||||||||||| |
Patients with event, n (%) | 20 (39.2) | 21 (41.2) | |||||||||||| |
Progression | 19 (37.3) | 20 (39.2) | |||||||||||| |
Death | 1 (2.0) | 1 (2.0) | |||||||||||| |
Censored, n (%) | 31 (60.8) | 30 (58.8) | |||||||||||| |
Median DORa (95% CIb), months | 20.5 (12.3 to NR) | NR (13.9 to NR) | |||||||||||| |
CI = confidence interval; DCO = data cut-off; DOR = duration of response; FAS = full analysis set; IRC = independent review committee; NR = not reached.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Figure 7: Kaplan–Meier Plot of DOR by IRC by Best Objective Response in L-MIND – FAS; November 30, 2018, Data Cut-Off (Primary Analysis)
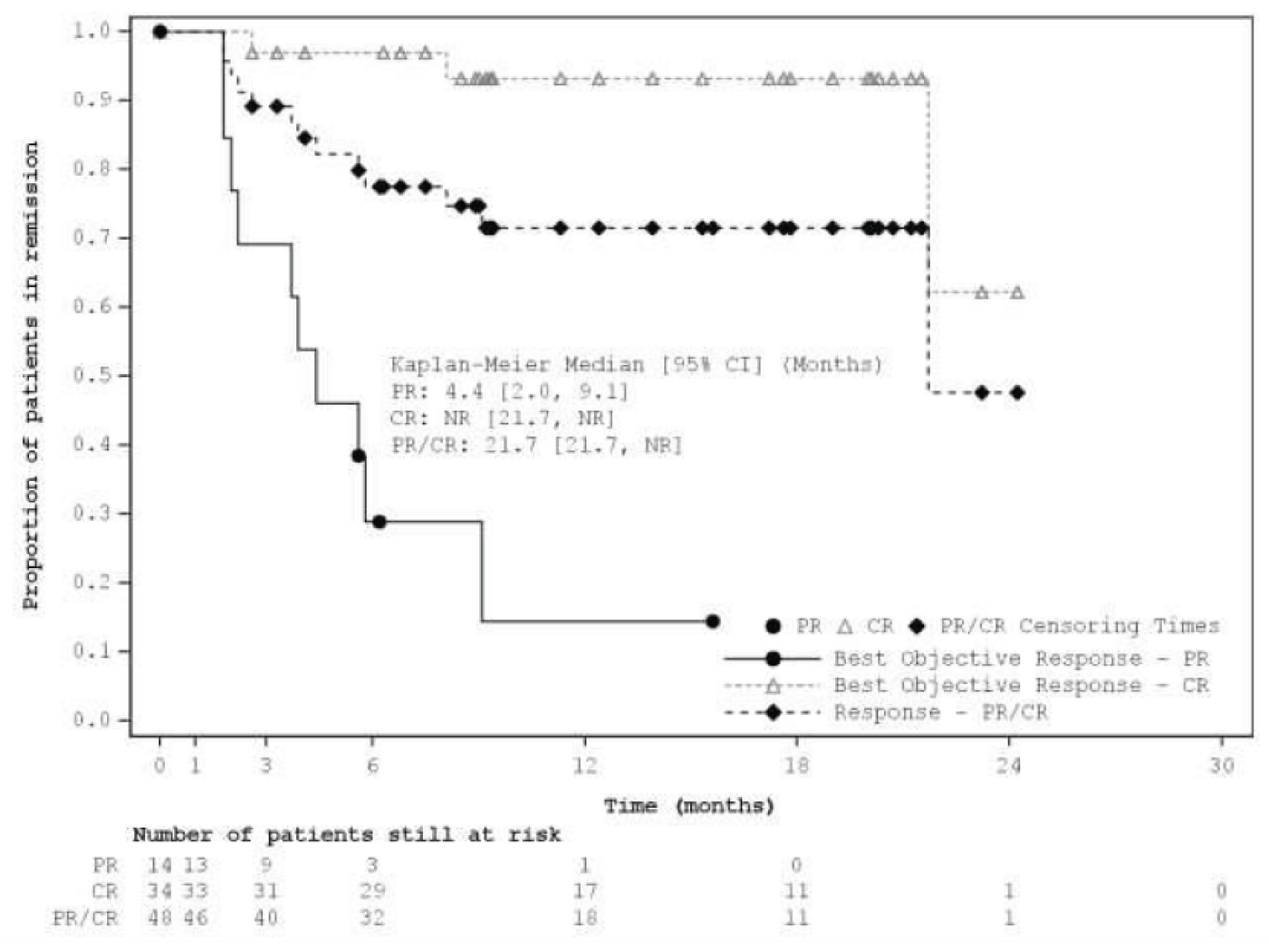
CR = complete response; DOR = duration of response; FAS = full analysis set; IRC = independent review committee; LEN = lenalidomide; NR = not reached; PR = partial response.
Source: L-MIND Clinical Study Report.10
Table 22: Subgroup Analyses of DOR by IRC – FAS
Characteristic | N | Median DORa (95% CIb), months | ||
|---|---|---|---|---|
November 30, 2018, DCO (primary analysis) | November 30, 2019, DCO | October 30, 2020, DCO | ||
IPI score | ||||
0 to 2 | 40 | NR (NR to NR) | 34.6 (NR to NR) | |||||||||||| |
3 to 5 | 40 | 21.7 (3.9 to NR) | 21.7 (4.4 to NR) | |||||||||||| |
Refractory to last therapy | ||||
Yes | 35 | NR (5.8 to NR) | NR (5.8 to NR) | |||||||||||| |
No | 45 | 21.7 (9.1 to NR) | 34.6 (21.7 to 34.6) | |||||||||||| |
Primary refractoriness | ||||
Yes | 15 | 3.7 (1.8 to NR) | NR (1.8 to NR) | |||||||||||| |
No | 65 | NR (21.7 to NR) | 34.6 (26.1 to 34.6) | |||||||||||| |
Prior ASCT | ||||
Yes | 9 | NR (4.4 to NR) | NR (4.4 to NR) | |||||||||||| |
No | 71 | 21.7 (21.7 to NR) | 34.6 (26.1 to 34.6) | |||||||||||| |
Number of prior treatment lines | ||||
1 | 40 | 21.7 (9.1 to NR) | 34.6 (21.7 to 34.6) | |||||||||||| |
≥ 2 | 40 | NR (5.8 to NR) | NR (26.1 to NR) | |||||||||||| |
Reason for ASCT ineligibility | ||||
Chemorefractory | 18 | NR (4.4 to NR) | NR (4.4 to NR) | |||||||||||| |
Comorbidities | ||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Older age | ||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Refusal of HDT or ASCT | 13 | 21.7 (2.0 to NR) | NR (21.7 to NR) | |||||||||||| |
Cell of origin by IHC | ||||
GCB | 37 | NR (2.6 to NR) | |||||||||||| | |||||||||||| |
Non-GCB | 21 | 21.7 (8.1 to 21.7) | |||||||||||| | |||||||||||| |
ASCT = autologous stem cell transplantation; CI = confidence interval; DCO = data cut-off; DOR = duration of response; FAS = full analysis set; GCB = germinal centre B-cell; HDT = high-dose therapy; IHC = immunohistochemistry; IPI = International Prognostic Index; IRC = independent review committee; NR = not reached.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
Source L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Time to Response
Results for TTR as of the primary analysis and November 30, 2019, data cut-off analysis are summarized in Table 23. TTR data were not reported at the most recent interim analysis (October 30, 2020, data cut-off).
As of the primary analysis (November 30, 2018, data cut-off), median TTR (CR or PR) based on IRC evaluation was 2.0 months (range 1.7 to 16.8 months). Median time to PR was 1.90 months (range 1.7 to 16.8 months). Median time to CR was 7.05 months (range 1.7 to 17.0 months). Median time to best response (either CR or PR) was 3.6 months (range 1.7 to 17.0 months).
As of the November 30, 2019, data cut-off, median TTR based on IRC evaluation was 2.0 months (range 1.7 to 16.8). Median time to CR was 4.0 months (range 1.7 to 17.0). Median time to PR and median time to best response were not reported.
Figure 8: Kaplan–Meier Plot of DOR by IRC by Best Objective Response in L-MIND – FAS; October 30, 2020, Data Cut-Off
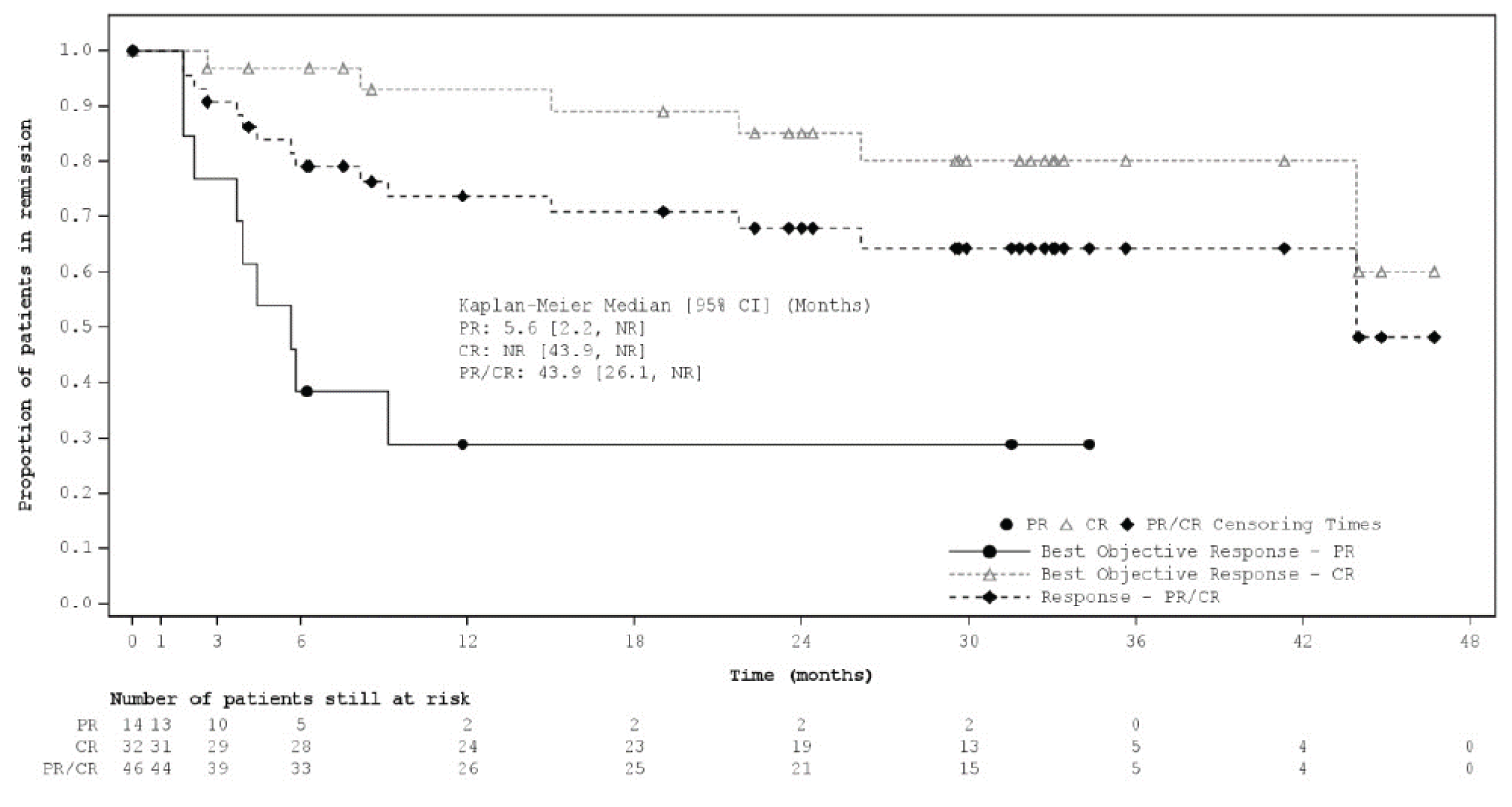
CR = complete response; DOR = duration of response; FAS = full analysis set; IRC = independent review committee; LEN = lenalidomide; NR = not reached; PR = partial response.
Source: L-MIND Clinical Study Report Addendum 3.14
Time to Next Treatment
Results for TTNT as of the primary analysis and November 30, 2019, data cut-off analysis are summarized in Table 24. TTNT was not reported at the most recent interim analysis (October 30, 2020, data cut-off). Median TTNT was 15.4 (95% CI, 7.6 to NR) months at the primary analysis compared to 12.5 (95% CI, 7.6 to 24.7) at the November 2019 interim analysis. Median follow-up time for TTNT was not reported.
Results from a subgroup analysis of TTNT by IPI score conducted at the primary analysis are presented in Table 25. No other subgroup analyses for TTNT were reported.
Table 23: Summary of TTR by IRC Results in L-MIND – FAS
Outcome | L-MIND (N = 80) | |
|---|---|---|
November 30, 2018, DCO (primary analysis) | November 30, 2019, DCO | |
Median TTR (CR or PR), months (minimum, maximum) | 2.0 (1.7, 16.8) | 2.0 (1.7, 16.8) |
Median time to PR, months (minimum, maximum) | 1.90 (1.7, 16.8) | Not reported |
Median time to CR, months (minimum, maximum) | 7.05 (1.7, 17.0) | 4.00 (1.7, 17.0) |
Median time to best response, months (minimum, maximum) | 3.60 (1.7, 17.0) | Not reported |
CR = complete response; DCO = data cut-off; FAS = full analysis set; IRC = independent review committee; PR = partial response; TTR = time to response.
Source: L-MIND Clinical Study Report,10 Addendum 1,12 and Addendum 3.14
Table 24: Summary of TTNT Results in L-MIND – FAS, November 30, 2018, Data Cut-Off (Primary Analysis)
Outcome | L-MIND (N = 80) | |
|---|---|---|
November 30, 2018, DCO (primary analysis) | November 30, 2019, DCO | |
Patients who experienced an event, n (%) | 43 (53.8) | 49 (61.3) |
Next treatment | 27 (33.8) | 32 (40.0) |
Death | 16 (20.0) | 17 (21.3) |
Censored, n (%) | 37 (46.3) | 31 (38.8) |
Median TTNTa (95% CIb), months | 15.4 (7.6 to NR) | 12.5 (7.6 to 24.7) |
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; NR = not reached; TTNT = time to next treatment.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
Source: L-MIND Clinical Study Report10 and Addendum 1.12
Table 25: Subgroup Analysis of TTNT by IPI Score – FAS, November 30, 2018, Data Cut-Off (Primary Analysis)
Characteristic | N | Median TTNTa (95% CIb), months |
|---|---|---|
IPI score | ||
0 to 2 | 40 | NR (15.4 to NR) |
3 to 5 | 40 | 7.0 (3.6 to 8.6) |
CI = confidence interval; FAS = full analysis set; IPI = International Prognostic Index; NR = not reached; TTNT = time to next treatment.
aKaplan–Meier estimate.
b95% CI calculated using the Brookmeyer and Crowley (1982) method.
Source: L-MIND Clinical Study Report.10
Health-Related Quality of Life
HRQoL outcomes were not reported in the L-MIND study.
Harms
Only those harms identified in the review protocol are reported below. See Table 26 for detailed harms data for the safety analysis set as of the most recent analysis of the L-MIND trial (data cut-off date October 30, 2020). Safety data after the longer follow-up was similar to safety data presented at the primary analysis (November 30, 2018, data cut-off). All 81 (100%) enrolled patients received at least 1 dose of the study drug and were included in the safety analysis set.
Adverse Events
As of the October 30, 2020, data cut-off, all 81 (100%) patients enrolled in L-MIND experienced 1 or more treatment-emergent AE. The most common AEs were neutropenia (50.6%), anemia (37.0%), diarrhea (35.8%), thrombocytopenia (30.9%), and cough (27.2%).
Serious Adverse Events
Overall, 53.1% of patients enrolled in L-MIND experienced 1 or more SAEs as of the October 30, 2020, data cut-off. The most common SAEs were pneumonia (n = 7, 8.6%), febrile neutropenia (n = 5, 6.2%), and pulmonary embolism (n = 3, 3.7%). Other SAEs reported in more than 1 patient included bronchitis, lower respiratory tract infection, atrial fibrillation, and congestive cardiac failure (n = 2, 2.5% each).
Withdrawal Due to Adverse Events
Overall, 20 (24.7%) patients permanently discontinued treatment with 1 or both study drugs due to AEs: 8 (9.9%) patients discontinued lenalidomide only, 2 (2.5%) discontinued tafasitamab only, and 10 (12.3%) discontinued both study drugs. The only AE that led to permanent discontinuation of study drug in more than 1 patient was neutropenia (n = 3, 3.7%).
Mortality
In total, 42 (51.9%) patients enrolled in L-MIND had died as of the October 30, 2020, data cut-off date. The cause of death was reported to be related to disease progression for 31 (38.3%) patients and unrelated to disease progression in 10 (12.3%) patients.
Notable Harms
Overall, 72.8% of patients enrolled in L-MIND experienced an infection. The most common types of infections were bronchitis (16.0%), pneumonia (12.3%), urinary tract infection (12.3%), and respiratory tract infection (11.1%).
Regarding myelosuppression, 50.6% of patients experienced neutropenia, 37.0% experienced anemia, 30.9% experienced thrombocytopenia, 14.8% experienced leukopenia, 12.3% experienced febrile neutropenia, and 7.4% experienced lymphopenia.
One (1.2%) patient developed worsening PML. |||||||||||||||||||||||| experienced hepatitis B reactivation. Five (6.2%) patients experienced an infusion-related reaction. No patients experienced a grade 3 or higher tumour lysis syndrome or cytokine release syndrome. Tumour lysis syndrome or cytokine release syndrome events of any grade were not reported.
Table 26: Summary of Harms – Safety Analysis Set; October 30, 2020, Data Cut-Off Date
Harms | L-MIND (N = 81) |
|---|---|
Patients with ≥ 1 AE | |
n (%) | 81 (100) |
Most common events,a n (%) | |
Neutropenia | 41 (50.6) |
Anemia | 30 (37.0) |
Diarrhea | 29 (35.8) |
Thrombocytopenia | 25 (30.9) |
Cough | 22 (27.2) |
Asthenia | 20 (24.7) |
Edema, peripheral | 19 (23.5) |
Pyrexia | 19 (23.5) |
Decreased appetite | 18 (22.2) |
Back pain | 16 (19.8) |
Hypokalemia | 15 (18.5) |
Fatigue | 14 (17.3) |
Constipation | 14 (17.3) |
Bronchitis | 13 (16.0) |
Patients with ≥ 1 SAE | |
n (%) | 43 (53.1) |
Most common events,b n (%) | |
Pneumonia | 7 (8.6) |
Febrile neutropenia | 5 (6.2) |
Pulmonary embolism | 3 (3.7) |
Bronchitis | 2 (2.5) |
Lower respiratory tract infection | 2 (2.5) |
Atrial fibrillation | 2 (2.5) |
Cardiac failure congestive | 2 (2.5) |
Patients who permanently stopped treatment due to AEs | |
One or both study drugs, n (%) | 20 (24.7) |
Most common events,b n (%) | |
Neutropenia | 3 (3.7) |
Lenalidomide only, n (%) | 8 (9.9) |
Tafasitamab only, n (%) | 2 (2.5) |
Both study drugs, n (%) | 10 (12.3) |
Deaths | |
n (%) | 42 (51.9) |
Related to disease progression | 31 (38.3) |
Unrelated to disease progression | 10 (12.3) |
Unknown | 1 (1.2) |
Notable harms | |
Infection,c n (%) | 59 (72.8) |
Bronchitis | 13 (16.0) |
Pneumonia | 10 (12.3) |
Urinary tract infection | 10 (12.3) |
Respiratory tract infection | 9 (11.1) |
Nasopharyngitis | 8 (9.9) |
Upper respiratory tract infection | 8 (9.9) |
Sinusitis | 6 (7.4) |
Gastroenteritis | 5 (6.2) |
Rhinitis | 5 (6.2) |
Myelosuppression, n (%) | |
Neutropenia | 41 (50.6) |
Anemia | 30 (37.0) |
Thrombocytopenia | 25 (30.9) |
Leukopenia | 12 (14.8) |
Febrile neutropenia | 10 (12.3) |
Lymphopenia | 6 (7.4) |
PML, n (%) | 1 (1.2) |
Hepatitis B reactivation, n (%) | |||||||||||| |
Infusion-related reactions, n (%) | |||||||||||| |
Cytokine release syndrome, n (%) | Not reportedd |
Tumour lysis syndrome, n (%) | Not reportedd |
AE = adverse event; PML = progressive multifocal leukoencephalopathy; SAE = serious adverse event.
aFrequency more than 15%.
bOccurred in more than 1 patient.
cFrequency more than 5%.
dNo patients experienced a grade 3 or higher tumour lysis syndrome or cytokine release syndrome. Tumour lysis syndrome or cytokine release syndrome events of any grade were not reported.
Source: L-MIND Clinical Study Report Addendum 3.14
Critical Appraisal
Internal Validity
For the primary end point and multiple secondary end points (i.e., PFS, EFS, DOR, TTR), an IRC was appropriately used to reduce the risk of detection bias. The IRC consisted of independent radiology and hemato-oncology physicians. Furthermore, there was generally good agreement between the IRC and investigator-assessed outcomes. The criteria used to assess response (IWG response criteria reported by Cheson et al. [2007])8 were appropriate, although the clinical experts consulted by CADTH noted that there are more recent guidelines available (i.e., Lugano criteria)33 that include PET scanning criteria more extensively. It is possible that the results could have differed if other criteria had been used, although the extent is uncertain.
The study population in L-MIND was adequately defined, and the clinical experts consulted by CADTH indicated that the eligibility criteria were appropriate. Approximately half of the patients screened were screen failures. Screen failures were driven predominantly by laboratory parameters exclusion criteria, which were considered necessary by the sponsor due to the starting dosage of lenalidomide. Study treatment discontinuation rates were as expected by the clinical experts. It was noted that most discontinuations of study drug(s) were due to disease relapse, which was expected by the clinical experts.
The analysis populations used in the L-MIND trial were appropriate. The efficacy outcomes were analyzed descriptively in the FAS population, which excluded 1 enrolled patient. Exclusion of 1 patient is unlikely to affect outcomes. Safety outcomes were assessed in all patients who were treated with a study drug.
The primary analysis (November 30, 2018, data cut-off date) was pre-specified in the L-MIND study protocol. The sample size of 80 patients was adequate to estimate the primary end point—ORR by IRC—with high statistical precision. The L-MIND trial met its primary end point (i.e., improvement of ORR as per IRC from 20% to 35% with single-drug therapy) at the primary analysis, as there was an ORR by IRC of 60.0% (95% CI, 48.4 to 70.8). The interim analyses with November 30, 2019, and October 30, 2020, data cut-off dates were not pre-specified. The November 30, 2019, data cut-off analysis was used in the Health Canada; the October 30, 2019, data cut-off analysis was conducted to report long-term outcomes after greater follow-up time. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
L-MIND is an open-label, single-arm study. There is no direct evidence comparing tafasitamab plus lenalidomide to a control arm. Furthermore, no statistical testing was performed because the L-MIND study was not designed to test hypotheses. Data were analyzed descriptively. Due to these limitations of the study design, no definitive conclusions can be drawn by the CADTH review team from the L-MIND study regarding the efficacy and safety of tafasitamab plus lenalidomide relative to a relevant comparator. The results were not adjusted for potential confounders (i.e., effect modifiers or prognostic factors) that could affect outcomes. The open-label design can increase the risk of performance and detection bias, particularly for outcomes that are subjective in measurement and interpretation (e.g., response, AEs). There is a high risk of performance and detection bias for known subjective harms, which could be overestimated, since both patients and their treating clinicians knew the treatment received and knew that they were participating in a trial. Objective outcomes such as OS time and mortality are unlikely to be affected by performance or detection bias. The potential for detection bias was minimized by using IRC assessment for key study outcomes such as ORR, DOR, and PFS.
The time-to-event analyses were appropriate, but causality cannot be inferred from a single-arm trial without a comparator. Survival times (median OS and median PFS) were estimated from the Kaplan–Meier models, and patients who did not have an event were censored, which is appropriate. The L-MIND trial is ongoing. Although the planned primary analysis has been conducted (data cut-off date of November 30, 2018), the final analysis has not yet been conducted. As of the October 30, 2020, data cut-off date, 19 (23.5%) patients were ongoing with tafasitamab monotherapy treatment. Per the Clinical Study Report, PFS data were considered mature as of the primary analysis, although numerical changes in median PFS at the subsequent analyses were noted. The median duration of follow-up at the time of the analyses was sufficient for other time-to-event end points (e.g., OS, DOR).
Most of the subgroup analyses of OS, PFS, and DOR were pre-specified, except for the subgroup analyses of IPI score. Sample sizes for the subgroup analyses were small, and the CIs reflected imprecision. In addition, the subgroup analyses were exploratory, and there were no statistical comparisons. As a result, the CADTH review team can draw no conclusions based on the subgroup analyses.
Multiple protocol amendments were implemented while the L-MIND study was being conducted, which included changes to the study eligibility criteria. In the original trial protocol, patients were required to have a histologically confirmed diagnosis of DLBCL not otherwise specified. Patients with NHL other than classical histology DLBCL (e.g., including patients with DLBCL transformed from indolent lymphomas) were excluded. In addition, patients who had relapsed within 3 months of prior CD20-targeted therapy were excluded. The first protocol amendment, which was implemented before patient enrolment began, expanded the study eligibility criteria to include patients with evidence of histological transformation to DLBCL from indolent NHL. Since this change was implemented before study patients were enrolled, it is less likely to have introduced bias. In subsequent protocol amendments, which were implemented after participant enrolment commenced, the eligibility criteria were changed to allow up to 3 lines of prior therapy for DLBCL treatment (previously, 2 prior lines were allowed), to remove the upper age limit of 80 years for study entry, and to allow patients with Gilbert’s syndrome or liver involvement by lymphoma. Changes in study eligibility after enrolment had begun may introduce selection bias, although the direction of bias is unknown. The clinical experts consulted by CADTH noted that the changes in eligibility criteria generally made the trial population more closely resemble the patient population in Canada with R/R DLBCL who are not eligible for ASCT.
In the original trial protocol, HBV serology was required monthly for all patients. An amendment implemented after participant enrolment had begun changed the requirement to HBV serology monthly only for those patients who were anti-HBc antibody–positive and HBV-DNA–negative at screening. This may have biased the detection of HBV reactivation, leading to underestimation of this notable harm.
In the original trial protocol, treatment with tafasitamab was continued until a maximum of 24 cycles (i.e., approximately 2 years). In the last protocol amendment, treatment with tafasitamab was extended beyond cycle 24 until disease progression. The reason for extending the treatment duration was unclear.
A high rate of protocol deviations occurred in L-MIND, which creates uncertainty in the data. Key protocol deviations were related to procedures or tests, study drug and treatment, laboratory assessment, informed consent form, eligibility criteria, and prohibited concomitant mediations. Deviations in study drug and treatment could have biased the efficacy and safety results, thus affecting internal validity. Deviations in eligibility criteria may also have resulted in selection bias. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Of note, the L-MIND study consisted of patients with R/R DLBCL diagnosed as per local pathology. However, central pathologic analysis concluded that approximately 10% of these patients had non-DLBCL histology or alternative diagnoses. The following subtypes of NHL were classified as non-DLBCL cases: follicular lymphoma (grade 2 + 3A), follicular lymphoma grade 2, mantle-cell lymphoma, classic type, and marginal zone lymphoma. The clinical experts consulted by CADTH indicated that patients could be misdiagnosed due to numerous factors (e.g., the biopsy, expertise in lymphoma). However, enrolment of these patients introduced bias because these patients are not representative of the intended patient population. Patients with alternative diagnoses may have had a better prognosis than patients with R/R DLBCL, thus overestimating the treatment effects of tafasitamab plus lenalidomide. Sensitivity analyses were performed on patients who had DLBCL confirmed by central pathologic analysis versus those who had not. Efficacy results for the patients with central pathology-confirmed DLBCL were generally consistent with the FAS.
In L-MIND, flattening of the OS and PFS Kaplan–Meier curves was observed. The clinical experts reported that they do not see flattening of OS and PFS curves with other available treatments in patients with R/R DLBCL who are not eligible for ASCT. The clinical experts indicated that subsequent anticancer treatments may have confounded the PFS and OS results. Furthermore, i a small number of patients were at risk at the tail end of the Kaplan–Meier curves, and these results may have been confounded by the patients with histologies other than DLBCL.
Overall, the L-MIND study was a phase II trial that enrolled 80 patients in the FAS. The clinical experts consulted by CADTH indicated that it may not be possible to extrapolate efficacy results from this small sample of patients to the general population of patients with R/R DLBCL in Canada. The clinical experts indicated that results should be confirmed in a phase III trial with a comparator arm.
External Validity
The L-MIND trial was an international, multi-centre study that included sites in Europe and the US. There were no study sites in Canada. The treatment regimen used in the L-MIND trial aligns with the Health Canada–recommended dose of tafasitamab plus lenalidomide. In the L-MIND study, pre-medications as prophylaxis for infusion-related reactions were administered before tafasitamab infusions, which was appropriate according to the clinical experts. The clinical experts indicated that pre-medications are commonly used in standard practice to prevent infusion-related reactions with other monoclonal antibody treatments.
The clinical experts consulted by CADTH reported that standard of care for assessing treatment response would be imaging with CT-PET every 3 to 4 months with clinical examination and bloodwork before each treatment. In the L-MIND study, imaging to assess disease response was conducted every 2 cycles (i.e., approximately every 2 months) during the 12 months of combination treatment. While the patients received tafasitamab monotherapy, imaging was conducted every 3 months from cycle 13 to 24, then approximately once every year from cycle 25 onward.
The clinical experts indicated that the baseline characteristics of patients enrolled in L-MIND were generally representative of the R/R DLBCL patient population in Canada, although they noted that the L-MIND study patients represent the most fit patients in this population, which is common in clinical trials. Some differences between the L-MIND study population and population of patients in Canada
with R/R DLBCL who are ineligible for ASCT were noted. The clinical experts indicated that the study population was younger than the general population of patients with R/R DLBCL who are eligible for ASCT, as many patients are ineligible due to older age (i.e., > 70 or > 75 years, depending on the site). According to the clinical experts, the population of patients in Canada with R/R DLBCL has a greater proportion of patients with an ECOG PS of 2, more patients have a relapse within 12 months of initial therapy, more patients have primary refractory disease, and more patients have had a prior ASCT that failed compared to the L-MIND study population. Furthermore, the large rate of screen failure indicates that many patients with R/R DLBCL were not eligible for participation in L-MIND, thus limiting generalizability if a large proportion of patients normally seen in clinical practice in Canada would not have been eligible.
Overall, the clinical experts thought that the eligibility criteria used in the L-MIND study were appropriate and allowed enrolment of patients with R/R DLBCL who were generally representative of the patient population in Canada. However, it noted that some groups of patients in the R/R DLBCL patient population were excluded. The L-MIND study excluded patients with known double- or triple-hit genetics DLBCL at study entry, which limits the generalizability of results to this patient population. In addition, primary refractory DLBCL was an exclusion criterion in the L-MIND study. The initial definition led to exclusion of relapses within 3 months of a prior anti-CD20 therapy. In a later amendment of the protocol, the definition of primary refractory DLBCL was revised to less than a PR to first-line therapy or progression within 6 months after completion of first-line therapy, and removed the need to have DLBCL relapse or progression after at least 3 months from completion of prior anti-CD20–containing therapy. As a result of this change in definition, 15 (18%) of patients enrolled in the L-MIND trial were considered to have primary refractory disease. This complicates the interpretation of the generalizability of study results to the population of patients who have primary refractory disease. Because of the change in definition and because primary refractory disease is an exclusion criterion in the pivotal study, the clinical experts consulted by CADTH indicated that it was unclear whether results were generalizable to patients with primary refractory disease.
Multiple protocol amendments related to the eligibility criteria were implemented during the L-MIND study and affect the generalizability of results. The first protocol amendment, implemented before patient enrolment began, expanded the study eligibility criteria to include patients with evidence of histological transformation to DLBCL from indolent NHL. In subsequent protocol amendments, implemented after participant enrolment commenced, the eligibility criteria were changed to allow up to 3 lines of prior therapy for DLBCL treatment (previously, 2 prior lines were allowed), to remove the upper age limit of 80 years for study entry, and to allow patients with Gilbert’s syndrome or liver involvement by lymphoma. According to the clinical experts, these changes to the study eligibility criteria increased the generalizability of results, as they made the trial population more representative of the general patient population in Canada.The clinical experts indicated that the criteria used to defined ASCT ineligibility in the L-MIND study were reasonable. The L-MIND study defined older age as > 70 years old as part of the ineligibility for ASCT criteria. The clinical experts thought that this was reasonable, although they noted that some centres in Canada perform ASCTs in patients up to 75 years of age. The clinical experts noted that organ function and predicted toxicities related to the transplant are important in determining a patient’s eligibility for ASCT.
The outcomes OS, PFS, ORR, and DOR are commonly used in clinical trials of anticancer therapy and are relevant to clinical practice. The patient groups that provided input on this review indicated that longer survival, remission, and controlling disease symptoms were most important outcomes for a new therapy. Better HRQoL and fewer side effects compared to current therapies were also important considerations for patients. Similarly, the clinical experts indicated that a clinically meaningful response to treatment would include improvement in survival and DOR, which they would expect to correlate with improvement in symptom burden. According to the clinical experts, meaningful response would include CR, PR, or stable disease with a tolerable toxicity profile. The L-MIND trial assessed some outcomes that were important to patients (e.g., OS, PFS, EFS, and treatment-emergent AEs). Disease symptoms and HRQoL were not reported in L-MIND, which represents a gap in the evidence.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Because there was no direct evidence comparing tafasitamab plus lenalidomide to other therapies for the treatment of R/R DLBCL, a review of the indirect evidence was undertaken. In addition to reviewing the sponsor’s submission, CADTH conducted a literature search to identify potentially relevant ITCs in patients with DLBCL. A focused literature search for ITCs dealing with DLBCL was run in MEDLINE All (1946–) on December 15, 2021. No date or language limits were applied. Titles, abstracts, and full-text articles were screened for inclusion by 1 reviewer based on the population, intervention, comparator, and outcome criteria outlined in Table 5.
No relevant ITCs were identified in the literature search. Three sponsor-submitted indirect comparisons were included in this review: 2 studies in which retrospective observational cohorts were compared to patients enrolled in L-MIND using ePS-based NN 1:1 matching methodology and unanchored MAICs. These ITCs were used to inform the pharmacoeconomic models.
Description of Indirect Comparison(s)
The RE-MIND16,17 and RE-MIND218 studies were retrospective observational studies conducted to generate an external control for indirect comparison with the L-MIND cohort. A summary of the key study design features and analysis methods of RE-MIND and RE-MIND2 is provided in Table 27.
Table 27: RE-MIND and RE-MIND2 Study Characteristics and Analysis Methods
Detail | RE-MIND | RE-MIND2 |
|---|---|---|
Study design | Retrospective, observational cohort study | Retrospective, observational cohort study |
Sites | 42 sites in 4 countries (Europe, US) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |
Number of patients |
|
|
Population | R/R DLBCL patients not eligible for HDC followed by ASCT who were treated with lenalidomide monotherapy | Patients who received systemic therapies for R/R DLBCL |
L-MIND data cut-off used | November 30, 2018 (primary analysis) | November 30, 2019 |
Treatments (observational cohorts) | Lenalidomide monotherapy (25 mg/day starting dosage for the primary analysis, and other starting dosages for additional analyses) | |
Outcomes | Primary end point:
Other end points:
| Primary end point:
Secondary end points:
|
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Data sources | Patient records from sites | |
Data extraction process | Data were collected using electronic data capture (Medidata RAVE electronic case report form, Cardinal Health survey tool) and electronic health record data extraction Data were entered by trained investigators or trained delegated staff | |
Assessments of tumour response | IWG response criteria reported by Cheson et al. (2014),33 Cheson et al. (2007),8 Cheson et al. (1999),34 or other criteria could be used for physician assessments of tumour response for the observational cohort The following imaging modalities could be used: no radiological assessment done, PET-CT, PET-MRI, PET only, MRI only, CT only, and other | |
ITC methods | ePS-based NN 1:1 matching methodology | |
Covariates used for matching |
| |
AE = adverse event; ANC = absolute neutrophil count; ASCT = autologous stem cell transplant; BR = bendamustine plus rituximab; CAR T-cell = chimeric antigen receptor T-cell; CNS = central nervous system; CRR = complete response rate; CT = CT; DCR = disease control rate; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; EBV = Epstein-Barr virus; EFS = event-free survival; ePS = estimated propensity score; ESMO = European Society for Medical Oncology; FAS = full analysis set; Hb = hemoglobin; HDC = high-dose chemotherapy; ITC = indirect treatment comparison; IWG = International Working Group; LDH = lactate dehydrogenase; MAS 25 = matched analysis set with starting dosage 25 mg per day; MAS_BR = matched analysis set for tafasitamab plus lenalidomide and bendamustine plus rituximab; MAS_Pool = matched analysis set for tafasitamab plus lenalidomide and pooled systemic therapies; MAS_R-GemOx = matched analysis set for tafasitamab plus lenalidomide and rituximab plus gemcitabine plus oxaliplatin; MRI = MRI; NCCN = National Comprehensive Care Network; NN = nearest neighbour; NOS = not otherwise specified; ORR = objective response rate; PET = PET; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine plus rituximab; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; R/R = relapsed or refractory; THRLBCL = T-cell or histiocyte-rich large B-cell lymphoma; TTNT = time to next treatment; ULN = upper limit of normal.
Source: RE-MIND Clinical Study Report,16 RE-MIND2 Clinical Study Report.18
RE-MIND
RE-MIND16,17 was an international, retrospective, observational cohort study designed to characterize the effectiveness of lenalidomide monotherapy in the treatment of R/R DLBCL patients who were not eligible for HDC followed by ASCT. The effectiveness of tafasitamab plus lenalidomide in L-MIND was compared to lenalidomide monotherapy in an observational retrospective cohort study. In total, data were collected from 490 patients treated with lenalidomide and 140 qualified for matching with the L-MIND cohort. Overall, 76 eligible RE-MIND patients were identified and matched 1:1 to 76 L-MIND patients based on baseline characteristics. Data from the L-MIND study were from the November 30, 2018, data cut-off (i.e., the primary analysis).
RE-MIND2
RE-MIND218 was an international, retrospective, observational cohort study designed to characterize the effectiveness of systemically administered therapies in the treatment of R/R DLBCL patients (second, third, and fourth line). Eligible systemic therapies included regimens administered in routine clinical care according to NCCN or ESMO guidelines19,20 for patients who were not eligible for ASCT. The effectiveness of systemically administered therapies was then compared using the data collected retrospectively with the efficacy outcomes of tafasitamab plus lenalidomide in the L-MIND study after the cohorts were balanced. This study included the following treatment cohorts: systemic therapies pooled, BR, R-GemOx, rituximab plus lenalidomide, CAR T-cell therapy, pola-BR, or pixantrone monotherapy. Rituximab plus lenalidomide and pixantrone monotherapy are not relevant comparators in Canada, according to the clinical experts consulted by CADTH. Therefore, results for these cohorts will not be presented in the CADTH Clinical Review Report. Data used from L-MIND were from the second analysis (i.e., November 30, 2019, data cut-off). In total, 3,454 patients were enrolled in the observational study. In the pre-specified analysis, systemic therapies pooled, BR, and R-GemOx were compared to tafasitamab plus lenalidomide. Exploratory, post hoc analyses were conducted for the comparisons to pola-BR and CAR T-cell therapy. A propensity score–based, 1:1 matched comparison was conducted between the real-world data collected in the retrospective observational study and clinical trial data from L-MIND.
MAICs
Unanchored MAICs of tafasitamab plus lenalidomide in the L-MIND study versus comparators using prospective studies were conducted. The authors conducted a systematic literature review to identify relevant sources of evidence for comparator treatments for patients with R/R DLBCL who were not eligible for ASCT. In total, |||||||||||| prospective studies reporting data for ||||||||||||||||||||||||, pola-BR, BR, and R-GemOx were selected for the MAICs against tafasitamab plus lenalidomide. Data were used from the most recent interim analysis of the L-MIND study (i.e., October 30, 2020, data cut-off).
Methods of RE-MIND and RE-MIND2
Objectives
The primary objective of the RE-MIND study was to characterize the effectiveness of lenalidomide monotherapy compared with tafasitamab plus lenalidomide combination therapy in the treatment of R/R DLBCL patients.
The primary objective of the RE-MIND2 study was to compare the efficacy outcomes of the L-MIND cohort with the effectiveness in a matched patient population treated with systemic regimens administered in routine clinical care and listed in the NCCN or ESMO guidelines.19,20
Populations
RE-MIND
Eligibility criteria for the RE-MIND observational cohort were aligned with the L-MIND study. Eligible patients were aged 18 years or older with histologically confirmed DLBCL, R/R after 1 to 3 prior systemic therapies (including 1 or more CD20-targeting regimens), and were not candidates for HDC and subsequent ASCT. Exclusion criteria included CNS lymphoma involvement, receiving lenalidomide in combination with another anti-lymphoma therapy (including radiation), prior treatment with anti-CD19 therapy or immunomodulatory imide drugs (e.g., thalidomide or lenalidomide), known double-hit or triple-hit genetics DLBCL, or a prior history of malignancies other than DLBCL (unless disease free for ≥ 5 years). Study sites were selected in Europe and the US (i.e., according to geographic distribution in L-MIND) based on data completeness and number of available patients. Eligible patients were identified from patient health records. The RE-MIND observational cohort did not have eligibility criteria related to ECOG PS or laboratory values.
RE-MIND2
Eligibility criteria for the RE-MIND2 observational cohort were also based on the L-MIND patient population. Eligible patients were aged 18 years or older at the initial DLBCL diagnosis; had a histologically confirmed diagnosis of DLBCL not otherwise specified, T-cell or histiocyte-rich large B-cell lymphoma, Epstein-Barr virus–positive DLBCL of the elderly, grade 3b follicular lymphoma, or composite lymphoma with a DLBCL component and a subsequent DLBCL relapse. Additionally, patients with the evidence of histological transformation to DLBCL from an earlier diagnosis of low-grade lymphoma into DLBCL with a subsequent DLBCL relapse were eligible. Patients were required to have R/R DLBCL and to have received at least 2 systemic regimens for the treatment of DLBCL, including at least 1 anti-CD20–containing therapy. Patients were excluded from the RE-MIND2 observational cohort if they had CNS involvement by lymphoma at initial DLBCL diagnosis, were treated with CD19-targeted therapy or immunomodulatory drugs (e.g., thalidomide, lenalidomide) as frontline DLBCL therapy, had undergone an allogenic stem cell transplant, had a prior history of malignancies other than DLBCL (unless the patient had been free of the disease for ≥ 5 years before inclusion), or had received tafasitamab. The RE-MIND2 observational cohort did not have eligibility criteria related to ECOG PS or laboratory values.
Eligible patients were identified from patient health records from sites selected across Europe, North America, and the Asia Pacific region, based on data completeness and number of available patients. Potential sites were identified from sponsor and contract research organization databases, as well as using input from external medical experts in the DLBCL indication. A Site Identification Questionnaire was designed and rolled out to the potential sites to obtain information on availability of eligible R/R DLBCL patients as well as on the availability and completeness of key variables. A site was selected for study participation if the availability of eligible R/R DLBCL patients and presence of key information were confirmed.
Recommended therapies for R/R DLBCL per NCCN or ESMO guidelines19,20 eligible for inclusion in the RE-MIND2 observational cohort are listed in Appendix 3 (Table 52).
Outcomes
In both RE-MIND and RE-MIND2, the IWG response criteria reported by Cheson et al. (2014),33 Cheson et al. (2007),8 Cheson et al. (1999),34 or other criteria could be used for physician assessments of tumour response for the observational cohort. The following imaging modalities could be used to assess tumours: no radiological assessment done, PET-CT, PET-MRI, PET only, MRI only, CT only, and other. The interval between 2 tumour disease assessments was expected to be approximately 3 months (plus or minus 14 days).
RE-MIND
The primary end point for RE-MIND was ORR. Other end points included CRR, OS, DOR, PFS, TTNT, and EFS.
Best ORR was defined as the proportion of patients for whom CR or PR was the best response achieved at any time within ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
CRR was defined as the proportion of patients for whom CR was the best response achieved at any time within ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
OS was defined as the time from the index date until death from any cause (documented by the time of death). ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
DOR was defined as the elapsed time between the date of first documented response (CR or PR) and the date of an event defined as the first documented progression or death (from any cause).
PFS was defined as the time from the index date to the date of tumour progression or death from any cause. The date of progression was the first date for which PD was assessed as the objective response. PD was used as documented by the treating physician. Disease progression qualifying for a PFS event had to be confirmed by radiology assessment, bone marrow aspiration with confirmed lymphoma involvement, or tissue biopsy with confirmed lymphoma infiltration. Patients who did not experience an event were censored.
TTNT was defined as the time from index date to the start of next anti-DLBCL therapy ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
EFS was defined as the time from the index date to the date of disease progression, initiation of a new non-study anti-DLBCL treatment, or death ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
RE-MIND2
The primary end point for RE-MIND 2 was OS. Secondary end points included ORR, CRR, DOR, EFS, PFS, TTNT, treatment discontinuation rate due to AEs, and duration of treatment exposure.
OS was defined as the time from the index date for a given line until death from any cause (documented by the date of death). ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ORR was defined as the proportion of patients for whom CR or PR was the best response achieved at any time within the analysis window or between index date for a given line and date of initiation of a new anti-DLBCL medication.
CRR was defined as the proportion of patients for whom CR was the best response achieved at any time within the analysis window or between index date for a given line and date of initiation of a new anti-DLBCL medication or death.
DOR was defined as the elapsed time between the date of first documented response (CR or PR) for a given line and the date of event defined as the first documented progression or death (from any cause).
PFS was defined as the time from the index date for a given line to the date of tumour progression or death from any cause. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TTNT was defined as the time from index date for a given line to the start of next anti-DLBCL therapy (therapies included ASCT, a new systemic anti-DLBCL medication, surgery, or radiotherapy for any reason) or death due to any cause, whichever came first. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
EFS was defined as the time from the index date to the date of disease progression, initiation of a new anti-DLBCL treatment, or death from any cause, whichever came first.
ITC Analysis Methods
RE-MIND
Data were collected retrospectively from health records of patients in either real-world, compassionate use, and/or clinical trial settings. The effectiveness of lenalidomide monotherapy was then compared using the data collected retrospectively with the efficacy outcomes of tafasitamab plus lenalidomide combination therapy in the L-MIND study after the non-randomized cohorts were balanced on relevant, observable variables. Safety data were not collected except to document the reason for change in lenalidomide treatment.
Multiple sensitivity analyses were conducted. A sensitivity analysis was performed by applying NN 1:1 matching with caliper to ensure a high degree of balance in a subgroup of patients. The biggest caliper ensuring a standardized mean difference (SMD) of less than 0.2 for all 9 covariates involved in 1:1 matching was chosen. For residual imbalance after NN 1:1 matching for 1 or more covariates, a sensitivity analysis with doubly robust estimation was performed by adding the covariates with SMD of more than 0.2 following NN 1:1 matching in the respective logistic (for ORR and DCR) or Cox proportional hazard models (for PFS, OS, EFS, and TTNT).
A 6-month follow-up rule was implemented to prevent an overestimation of the rate of nonresponders in the RE-MIND observational cohort. A minimum of 6 months’ follow-up time was met if a patient responded (CR or PR) or progressed or died within 6 months from index date (from study day 1 to 183), or a responding patient (CR or PR as best response during analysis window) had a baseline tumour assessment and at least 1 post-baseline response assessment available at 6 months or later (on or after study day 184), or any patient who had at least 1 disease response assessment with SD, “indeterminate,” “not evaluable,” or “other” within 6 months from index date (from study day 1 to 183), with at least 1 assessment or death at 6 months or later (on or after study day 184).
The maximum follow-up time for an individual patient in the L-MIND study in the data cut-off used for the RE-MIND analyses was 32 months (first patient enrolled in March 2016, primary completion cut-off date November 30, 2018). To ensure a comparable distribution of follow-up times of patients in the observational study, an analysis window was applied, defined from index date to 32 months (974 days). This analysis window was applied for all analyses.
Statistical Analysis
Estimated propensity score–based NN 1:1 matching methodology was used to balance cohorts using the following 9 baseline covariates to estimate the propensity score: age (as categorical variable with subgroups < 70 versus ≥ 70 years of age); Ann Arbor stage I-II versus III-IV; refractoriness to last therapy line (yes versus no); number of prior lines of therapy (1 versus 2 or 3); history of primary refractoriness (yes versus no); prior ASCT (yes versus no); elevated lactate dehydrogenase (LDH; LDH > ULN versus LDH ≤ ULN); neutropenia (absolute neutrophil count < 1.5 × 109/L versus absolute neutrophil count ≥ 1.5 × 109/L); anemia (hemoglobin < 10 g/dL versus ≥ 10 g/dL). A sensitivity analysis using imputation of missing data in baseline covariates was also performed.
For all end point models, a doubly robust estimation was conducted, in which 1 or more covariates were added to the end point estimation model to address any residual imbalance. Covariates added for doubly robust estimation were those with SMD of less than 0.20 following balancing, and/or covariates pre-specified as the strongest potential confounders from among the original list of covariates. The suggested order of the strongest potential confounder covariates for inclusion in the end point model was: primary refractory, refractory to last treatment, prior ASCT, number of prior therapies, age, LDH, Ann Arbor stage, neutropenia, and anemia.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Patients with a starting dosage of lenalidomide of 20 or 25 mg/day, available baseline and at least 1 post-baseline (after the index date) scan, 6 months of follow-up time, physician recorded tumour response assessment, and relevant available clinical data qualified for this validation of response assessment.
Determination of Sample Size
As 81 patients were enrolled and treated in the L-MIND study, the ePS-based 1:1 matching could result in a maximum sample size of n = 162. With an assumed difference of 23% in ORR for lenalidomide monotherapy (35%) versus tafasitamab plus lenalidomide therapy (58%), the achieved power was 80% and minimal detectable difference was 17% in ORR under Fisher’s exact test for unpaired data. Based on an assumed difference of 35% in ORR for lenalidomide monotherapy, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Approximately 500 patients needed to be included in the observational cohort to complete successful cohort balancing for 9 dichotomized baseline covariates before conducting comparative efficacy outcome analyses.
Analysis of Outcomes
For the analysis of ORR and CRR, Fisher’s exact tests were used and P values were presented. The primary analysis was based on Fisher’s exact test on the primary analysis population. ORs and 95% CIs were estimated using logistic regression analysis. The cohort status covariate was used in the model. Difference in ORR and CRR between the 2 cohorts was estimated, and exact 95% CI were presented. The number and percentage of patients with CR, PR, SD, PD as best response, and deaths before any post-baseline assessments were presented.
For the analysis of PFS, EFS, OS, and TTNT, the difference in the specific time-to-event measures between the 2 cohorts was compared using the log-rank test. HRs and 95% CI were estimated using Cox proportional hazards model, with cohort status as covariate. For the analysis of DOR, the difference between the 2 cohorts was compared using the log-rank test. Patients without a response were assigned a duration of zero.
Analysis Sets
The primary analysis population was the matched analysis set 25 (MAS 25), which consisted of 1:1 matched patients from the L-MIND study and the observational cohort with a lenalidomide starting dosage of 25 mg per day using 9 baseline covariates.
RE-MIND2
Data such as baseline characteristics, effectiveness outcomes, and treatment termination or dropout due to AEs were retrospectively collected from existing health records, including paper or electronic records of patients treated for R/R DLBCL. Since RE-MIND2 was an observational retrospective study, no patient visits or laboratory tests were required. Only data that had been previously collected were in scope.
Following data collection, the efficacy outcomes of the L-MIND cohort (tafasitamab plus lenalidomide) were compared with the effectiveness in a matched patient population treated with systemic regimens administered in routine clinical care and listed in NCCN or ESMO guidelines19,20 or pre-specified treatments after cohort balancing using matching and weighting applications of the ePS. Patients who received at least 2 therapy lines for DLBCL were assigned an index date (index date second, third, or fourth line) for each eligible therapy line.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Patients were assigned an index date based on the first documented treatment record of the systemically administered therapy for R/R DLBCL under consideration. The pre-index period for each patient was defined as the time between first documented DLBCL diagnosis, or history of cancer other than DLBCL, and the index date (start of R/R DLBCL treatment of second, third, or fourth line). If a patient had received more than 1 treatment regimen (therapy lines) for R/R DLBCL, the patient was assigned an index date for each applicable therapy line. For observational cohorts, the index date for a given line was the start date of any component of R/R DLBCL treatment in that line. Patients who received at least 2 therapy lines for DLBCL were assigned an index date (index date second, third, or fourth line) for the respective therapy line. For the L-MIND study, the index date was the date of first dose of any study drug (lenalidomide or tafasitamab).
The observational period was defined as the time between the index date and end of follow-up. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| A 6-month follow-up rule was implemented in RE-MIND2 as well.
Data were collected for patients who were initially diagnosed with DLBCL between 2010 and 2020. Data from the L-MIND study database, with a data cut-off date of November 30, 2019 (i.e., approximately 2 years after the last patient was enrolled in the study), were used for comparison with the observational cohort of RE-MIND2. For patients in the observational cohort, an analysis window was applied in place of data cut-off date. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The analysis window was applied per therapy line.
Statistical Analysis
To balance the L-MIND cohort with systemically administered therapies, subgroup strata were categorized on the basis of number of lines of therapy (i.e., 2, 3, or 4 therapy lines). Then 1:1 NN matching without replacement was performed using the remaining 8 baseline covariates per stratum to obtain each matched population set. The ratio of the L-MIND cohort to the observational cohort was 1:1, with a maximum ratio of |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The matched population with SMD of 0.2 or less for all baseline characteristics and the highest matching ratio was selected as the primary analysis set for end point calculation. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| The final matched population for analysis was the aggregation of the matched population of each stratum. Additional matched cohorts were created on the basis of the following 2 subgroups of the L-MIND cohort: (1) 1 prior line before tafasitamab plus lenalidomide, and (2) 2 or 3 prior lines before tafasitamab plus lenalidomide.
To balance the L-MIND cohort with pre-specified treatment regimens, 1:1 NN matching for 9 baseline characteristics was performed using the same covariates as in RE-MIND. Comparative analysis with the L-MIND cohort was performed only if a certain balance of baseline characteristics had been achieved (SMD ≤ 0.2 for all covariates). Patients with different treatment regimens were used in matched population sets under different treatment regimens. Matching was performed only if the number of patients eligible for matching in the pre-specified cohort was larger than number of patients in the L-MIND cohort FAS population used for the RE-MIND2 study (N = 76).
Patients who fulfilled eligibility criteria qualified for matching if they had a baseline response assessment, had a sufficient follow-up for a documented response or progression to the respective treatment regimen, had non–double- or triple-hit genetics lymphoma, were non–transplant-eligible, did not receive ASCT for the given therapy line, had no prior CNS involvement, and had data on all baseline covariates available at the start of the respective treatment.
A pre-specified analysis was not feasible for patients treated with pola-BR and CAR T-cell therapies because of the low number of identified patients. To produce comparative efficacy estimates of the tafasitamab and lenalidomide combination against pola-BR, CAR T-cell therapy, and rituximab and lenalidomide (R2), the following alternative matching methods were conducted in post hoc analyses:
inverse probability treatment weighting (IPTW) matching using 9 baseline covariates
IPTW matching using 9 baseline covariates with multiple imputations
1:1 NN matching using 6 baseline covariates
1:1 NN matching using 6 baseline covariates with multiple imputation
1:1 NN matching using 9 baseline covariates with multiple imputation
The 9 covariates used for the post hoc analysis were age, Ann Arbor stage, refractoriness to last therapy line, number of prior lines of therapy, primary refractoriness, prior ASCT, elevated LDH levels, anemia, and neutropenia. For the methods that used only 6 covariates, these were age, refractoriness to last therapy line, number of prior lines of therapy, primary refractoriness, prior ASCT and ECOG status. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Determination of Sample Size
In general, approximately 2,800 patients who had received systemic therapies as per NCCN or ESMO guidelines19,20 were needed to complete successful cohort balancing before conducting comparative efficacy outcome analyses. The L-MIND primary analysis set consisted of 76 patients for comparison in RE-MIND2. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Analysis of Outcomes
Time-to-event end points (i.e., OS, PFS, TTNT, DOR, and EFS) were analyzed using standard Kaplan–Meier methodology; log-rank test and HR along with 95% CI and the associated P values estimated using Cox proportional hazard model were reported. Binary end points, including ORR and CR rate, were compared using Fisher’s exact tests, and P values were reported. Treatment effect in terms of difference in ORR or CR rate between the 2 cohorts was estimated, and exact 95% CI was presented. In addition, ORs, ratio of ORR or CR rate, and the ratio of the proportions were presented.
Analysis Sets
The primary analysis in RE-MIND2 used 3 analysis sets: MAS_Pool, MAS_BR, and MAS_R-GemOx. MAS_Pool included patients who met criteria for matching and included 1:1 matched patients from the L-MIND study and the observational cohort using baseline covariates. MAS_BR and MAS_R-GemOx included patients who met criteria for matching using baseline covariates and included 1:1 matched patients from the L-MIND study and observational cohort for each pre-specified treatments.
For the post hoc exploratory analyses of pola-BR and CAR T-cell therapy, the MAS with 9 covariates for specified treatment after multiple imputation (MASMI_9cov) was used to inform the economic model.24 There was a MASMI_9cov for each pre-specified treatment (i.e., MASMI_Pola-BR_9cov, MASMI_CAR-T_9cov).
Results of RE-MIND and RE-MIND2
Summary of Included Studies
RE-MIND
Data from 524 patients were collected from 42 sites in the US and Europe. Following review, data for 34 patients were excluded due to incomplete data, lack of R/R condition, lack of ASCT ineligibility reason, or double- or triple-hit genetics DLBCL. Of the data collected for the 490 patients included, data for 140 patients indicated they had fulfilled the inclusion criteria for RE-MIND, received a lenalidomide starting dose of 25 mg, fulfilled the 6-month follow-up criteria, and had data on the pre-specified baseline covariates available at baseline. Of the 81 patients enrolled in L-MIND, 5 were excluded from the analysis: 1 did not receive lenalidomide, and 4 did not meet the 6-month follow-up criteria. As a result, 76 patients from the L-MIND study were included in RE-MIND.
Following ePS-based NN 1:1 matching, the primary analysis set (MAS 25) comprised 76 patients from each cohort. Patients in the lenalidomide-monotherapy cohort were treated between January 2007 and April 2019, and patients in the combination-therapy cohort received treatment between March 2016 and November 2017. Most patients (63 patients; 82.9%) in the lenalidomide-monotherapy cohort commenced treatment between 2014 and 2019.
Baseline characteristics for the MAS 25 are shown in Table 28 and were generally balanced between the tafasitamab plus lenalidomide combination therapy and lenalidomide-monotherapy cohorts, with SMDs of less than 0.13 for 7 of the 9 baseline characteristics. Residual imbalance was observed for 2 of the 9 covariates: the number of prior lines of therapy (SMD 0.29) and Ann Arbor stage (SMD 0.23). These residual imbalances were addressed in sensitivity analyses that confirmed the primary analysis. There were baseline imbalances for ECOG PS, IPI score, cell of origin, and race, which all had large amounts of missing data. ECOG PS was not a balancing characteristic in the primary analysis but was included as such in a sensitivity analysis.
In the tafasitamab plus lenalidomide and lenalidomide-monotherapy cohorts, the median lenalidomide dose intensity was 17.6 mg per day (interquartile range [IQR] 14.4 to 19.2) versus 19.0 mg per day (IQR 17.5 to 19.5), median follow-up for OS was 21.5 months (IQR 15.1 to 26.5) versus 20.9 months (IQR 15.5 to 29.6), and median time to first post-baseline assessment was 1.9 versus 3.1 months, respectively. In the tafasitamab plus lenalidomide cohort, 96% of assessments were made by CT only or PET-CT, compared with 82% in the lenalidomide-monotherapy cohort; the median frequency of assessment for response in the combination-therapy cohort was 2.1 months (IQR 1.8 to 2.8) and in the lenalidomide-monotherapy cohort, 3.2 months (IQR 1.9 to 4.5).
Table 28: RE-MIND Demographics and Baseline Characteristics – MAS 25
Characteristic | Tafasitamab plus lenalidomide (N = 76) | Lenalidomide monotherapy (N = 76) |
|---|---|---|
Age at index date, years, mean (SD) | 69.1 (9.71) | 70.0 (8.65) |
Age group, n (%) | ||
< 70 years | 33 (43.4) | 31 (40.8) |
≥ 70 years | 43 (56.6) | 45 (59.2) |
Sex, n (%) | ||
Female | 36 (47.4) | 33 (43.4) |
Male | 40 (52.6) | 43 (56.6) |
Race, n (%) | ||
Black or African American | 0 | 4 (5.3) |
American Indian or Alaska Native | 0 | 1 (1.3) |
White | 70 (98.6) | 50 (65.8) |
Unknown | 0 | 18 (23.7) |
Other | 1 (1.4) | 3 (3.9) |
Ann Arbor stage, n (%) | ||
I or II | 19 (25.0) | 12 (15.8) |
III or IV | 57 (75.0) | 64 (84.2) |
IPI score, n (%) | ||
0 to 2 | 40 (52.6) | 16 (21.1) |
3 to 5 | 36 (47.4) | 32 (42.1) |
Missing | 0 | 28 (33.8) |
ECOG PS, n (%) | ||
0 | 29 (38.2) | 5 (6.6) |
1 | 41 (53.9) | 36 (47.4) |
2 | 6 (7.9) | 19 (25.0) |
3 | 0 | 6 (7.9) |
≥ 2 | 6 (7.9) | 25 (32.9) |
Missing | 0 | 10 (13.2) |
Prior ASCT, n (%) | ||
Yes | 9 (11.8) | 6 (7.9) |
No | 67 (88.2) | 70 (92.1) |
Number of prior systemic treatment lines, n (%) | ||
1 | 39 (51.3) | 28 (36.8) |
2 | 32 (42.1) | 42 (55.3) |
3 | 5 (6.6) | 6 (7.9) |
2 or 3 | 37 (48.7) | 48 (63.2) |
Cell of origin by IHC, n (%) | ||
GCB | 34 (44.7) | 14 (18.4) |
Non-GCB | 20 (26.3) | 16 (21.1) |
Missing | 22 (28.9) | 46 (60.5) |
Primary refractoriness, n (%) | ||
Yes | 14 (18.4) | 16 (21.1) |
No | 62 (81.6) | 60 (78.9) |
Refractoriness to last prior therapy, n (%) | ||
Yes | 34 (44.7) | 34 (44.7) |
No | 42 (55.3) | 42 (55.3) |
Neutropenia, n (%) | ||
Yes | 2 (2.6) | 2 (2.6) |
No | 74 (97.4) | 74 (97.4) |
Anemia, n (%) | ||
Yes | 6 (7.9) | 5 (6.6) |
No | 70 (92.1) | 71 (93.4) |
Elevated LDH, n (%) | ||
Yes | 41 (53.9) | 45 (59.2) |
No | 35 (46.1) | 31 (40.8) |
Creatinine clearance, mL/min, n (%) | ||
≥ 60 | 69 (90.8) | 42 (55.3) |
Missing | 7 (9.2) | 34 (44.7) |
Time since first DLBCL diagnosis, months | ||
Mean (SD) | 39.99 (35.958) | 38.04 (35.466) |
Median (Q1, Q3) | 25.92 (16.77, 54.70) | 24.94 (14.49, 45.34) |
Time since discontinuation of last prior anti-DLBCL medication or ASCT, months | ||
Mean (SD) | 17.39 (22.295) | 13.62 (19.643) |
Median (Q1, Q3) | 9.23 (5.17, 20.67) | 6.46 (1.28, 14.77) |
ASCT = autologous stem cell transplant; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group performance status; GCB = germinal centre B-cell like; IHC = immunohistochemistry; IPI = International Prognostic Index; LDH = lactate dehydrogenase; MAS = matched analysis set; SD = standard deviation; Q1 = first quartile; Q3 = third quartile; ULN = upper limit of normal.
Source: RE-MIND Clinical Study Report.16
RE-MIND2
A total of 3,454 patients were enrolled in the observational cohort for RE-MIND2. Of these, 3,449, 1,837, and 894 patients were treated in second, third, and fourth line of anti-DLBCL therapy, respectively. The number of patients eligible for matching in the cohort of systemic therapies pooled was 961. In the cohorts with the pre-specified treatments BR, R-GemOx, CAR T, and pola-BR, there were 282, 235, 51, and 36 patients, respectively. In the 2 cohorts with the pre-specified treatments BR (N = 282) and R-GemOx (N = 235), the number of patients eligible for matching was larger than the number of patients in the L-MIND study FAS population (N = 76). In the L-MIND cohort, 76 of the 81 patients enrolled were eligible for matching (2 patients were excluded because they did not meet the eligibility criteria and 3 patients were excluded because they did not meet the 6 months follow-up rule). Matching exercises and comparative analyses were not performed in other pre-specified treatment cohorts because of the limited number of patients eligible for matching (i.e., less than 76 patients).
Table 29: RE-MIND2 Demographics and Baseline Characteristics – Primary Analysis Sets (MAS_Pool, MAS_BR, MAS_R-GemOx)
Characteristic | MAS_Pool | MAS_BR | MAS_R-GemOx | |||
|---|---|---|---|---|---|---|
Tafa plus LEN (N = 76) | Systemic therapies pooled (N = 76) | Tafa plus LEN (N = 75) | BR (N = 75) | Tafa plus LEN (N = 74) | R-GemOx (N = 74) | |
Age at index date, mean (SD) | 69.1 (9.71) | 68.7 (11.88) | 69.0 (9.75) | 69.3 (9.23) | 69.5 (9.59) | 71.0 (9.64) |
Age group, n (%) | ||||||
< 70 years | 33 (43.4) | 31 (40.8) | 33 (44.0) | 33 (44.0) | 31 (41.9) | 26 (35.1) |
≥ 70 years | 43 (56.6) | 45 (59.2) | 42 (56.0) | 42 (56.0) | 43 (58.1) | 48 (64.9) |
Sex, n (%) | ||||||
Female | 36 (47.4) | 32 (42.1) | 36 (48.0) | 30 (40.0) | 35 (47.3) | 38 (51.4) |
Male | 40 (52.6) | 44 (57.9) | 39 (52.0) | 45 (60.0) | 39 (52.7) | 36 (48.6) |
Race, n (%) | ||||||
Black or African American | 0 | 6 (7.9) | 0 | 12 (16.0) | 0 | 10 (13.5) |
American Indian or Alaska Native | 0 | 1 (1.3) | 0 | 0 | 0 | 1 (1.4) |
Asian | 0 | 9 (11.8) | 0 | 13 (17.3) | 0 | 2 (2.7) |
Native Hawaiian or other Pacific Islander | 0 | 0 | 0 | 0 | 0 | 1 (1.4) |
White | 70 (92.1) | 49 (64.5) | 69 (92.0) | 36 (48.0) | 68 (91.9) | 49 (66.2) |
Unknown | 0 | 4 (5.3) | 0 | 11 (14.7) | 0 | 3 (4.1) |
Other | 1 (1.3) | 7 (9.2) | 1 (1.3) | 3 (4.0) | 1 (1.4) | 8 (10.8) |
Missing | 5 (6.6) | 0 | 5 (6.7) | 0 | 5 (6.8) | 0 |
Ann Arbor stage, n (%) | ||||||
I or II | 19 (25.0) | 19 (25.0) | 18 (24.0) | 19 (25.3) | 18 (24.3) | 15 (20.3) |
III or IV | 57 (75.0) | 57 (75.0) | 57 (76.0) | 56 (74.7) | 56 (75.7) | 59 (79.7) |
IPI score, n (%) | ||||||
0 to 2 | 40 (52.6) | 21 (27.6) | 39 (52.0) | 25 (33.3) | 38 (51.4) | 20 (27.0) |
3 to 5 | 36 (47.4) | 40 (52.6) | 36 (48.0) | 35 (46.7) | 36 (48.6) | 43 (58.1) |
Missing | 0 | 15 (19.7) | 0 | 15 (20.0) | 0 | 11 (14.9) |
ECOG PS, n (%) | ||||||
0 to 1 | 70 (92.1) | 44 (57.9) | 69 (92.0) | 40 (53.3) | 68 (91.9) | 37 (50.0) |
≥ 2 | 6 (7.9) | 21 (27.6) | 6 (8.0) | 23 (30.7) | 6 (8.1) | 28 (37.8) |
Missing | 0 | 11 (14.5) | 0 | 12 (16.0) | 0 | 9 (12.2) |
Prior ASCT, n (%) | ||||||
Yes | 9 (11.8) | 10 (13.2) | 9 (12.0) | 14 (18.7) | 8 (10.8) | 8 (10.8) |
No | 67 (88.2) | 66 (86.8) | 66 (88.0) | 61 (81.3) | 66 (89.2) | 66 (89.2) |
Number of prior systemic treatment lines, n (%) | ||||||
1 | 39 (51.3) | 39 (51.3) | 39 (52.0) | 39 (52.0) | 39 (52.7) | 41 (55.4) |
2 | 32 (42.1) | 32 (42.1) | 31 (41.3) | 22 (29.3) | 30 (40.5) | 26 (35.1) |
3 | 5 (6.6) | 5 (6.6) | 5 (6.7) | 14 (18.7) | 5 (6.8) | 7 (9.5) |
Primary refractoriness, n (%) | ||||||
Yes | 14 (18.4) | 12 (15.8) | 14 (18.7) | 19 (25.3) | 14 (18.9) | 14 (18.9) |
No | 62 (81.6) | 64 (84.2) | 61 (81.3) | 56 (74.7) | 60 (81.1) | 60 (81.1) |
Refractoriness to last prior therapy, n (%) | ||||||
Yes | 34 (44.7) | 35 (46.1) | 33 (44.0) | 32 (42.7) | 33 (44.6) | 29 (39.2) |
No | 42 (55.3) | 41 (53.9) | 42 (56.0) | 43 (57.3) | 41 (55.4) | 45 (60.8) |
Neutropenia, n (%) | ||||||
Yes | 2 (2.6) | 2 (2.6) | 2 (2.7) | 4 (5.3) | 2 (2.7) | 5 (6.8) |
No | 74 (97.4) | 74 (97.4) | 73 (97.3) | 71 (94.7) | 72 (97.3) | 69 (93.2) |
Anemia, n (%) | ||||||
Yes | 6 (7.9) | 5 (6.6) | 6 (8.0) | 5 (6.7) | 6 (8.1) | 5 (6.8) |
No | 70 (92.1) | 71 (93.4) | 69 (92.0) | 70 (93.3) | 68 (91.9) | 69 (93.2) |
Elevated LDH, n (%) | ||||||
Yes | 41 (53.9) | 44 (57.9) | 41 (54.7) | 37 (49.3) | 41 (55.4) | 48 (64.9) |
No | 35 (46.1) | 32 (42.1) | 34 (45.3) | 38 (50.7) | 33 (44.6) | 26 (35.1) |
ASCT = autologous stem cell transplant; BR = bendamustine plus rituximab; ECOG PS = Eastern Cooperative Oncology Group performance status; IPI = International Prognostic Index; LDH = lactate dehydrogenase; LEN = lenalidomide; MAS = matched analysis set; Pool = systemic therapies pooled; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; SD = standard deviation; Tafa = tafasitamab.
Source: RE-MIND2 Clinical Study Report.18
|||||||||||| | |||||||||||| | |||||||||||| | ||
|---|---|---|---|---|
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | ||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Note: Some redacted cells have been removed
Baseline characteristics of the MAS_Pool, MAS_BR, and MAS_R-GemOx (tafasitamab plus lenalidomide cohort versus pre-specified treatment cohort) used for the main analysis are presented in Table 29. A high degree of covariate balance was achieved for MAS_Pool with absolute standardized difference between 0 and 0.08. The absolute standardized difference for MAS_BR and MAS_R-GemOx was between 0 and 0.19. There were baseline imbalances in race, IPI score, and ECOG PS; these characteristics had between 5% to 20% of missing data. ECOG PS was not a balancing characteristic in the primary analysis but was included as such in a sensitivity analysis.
Baseline characteristics of the MASMI_9cov groups used for the exploratory post hoc analyses of pola-BR and CAR T-cell therapies are presented in Table 30. There were baseline imbalances in Ann Arbor stage, IPI score, and ECOG PS.
Results
RE-MIND
Results of the RE-MIND efficacy outcome analyses are presented in Table 31.
The primary end point of the RE-MIND study was met. The best ORR was 67.1% (95% CI, 55.4 to 77.5) in the tafasitamab plus lenalidomide cohort compared to 34.2% (95% CI, 23.7 to 46.0) in the lenalidomide-monotherapy cohort (OR = 3.885; 95% CI, 1.900 to 8.142; P < 0.0001). The results of the primary analytical approach used for ORR was supported by all sensitivity analyses.
Table 31: RE-MIND Efficacy Outcome Results – MAS 25
Outcome | Tafasitamab plus lenalidomide (N = 76) | Lenalidomide monotherapy (N = 76) |
|---|---|---|
OS, median (95% CI), months | NR (15.5 to NR) | 9.4 (5.1 to 20.0) |
Mediana follow-up time, months | 21.5 | 20.9 |
HRb (95% CI) [P valuec,d] | NA | 0.499 (0.32 to 0.79) [0.0026] |
PFS, median (95% CI), months | 12.1 (5.9 to NR) | 4.0 (3.1 to 7.4) |
Mediana follow-up time, months | 19.7 | 12.6 |
HRb (95% CI) [P valuec,d] | NA | 0.463 (0.307 to 0.698) [0.0002] |
EFS, median (95% CI), months | 12.1 (5.5 to 21.0) | 4.0 (3.1 to 6.2) |
Mediana follow-up time, months | 21.9 | 15.4 |
HRb (95% CI) [P valuec,d] | NA | 0.439 (0.296 to 0.650) [< 0.0001] |
ORR, % (95% CI) | 67.1 (55.4 to 77.5) | 34.2 (23.7 to 46.0) |
ORb (95% CI) [P valued,e] | NA | 3.89 (1.90 to 8.14) [< 0.0001] |
CRR, % (95% CI) | 39.5 (28.4 to 51.4) | 13.2 (6.5 to 22.9) |
P valued,e | NA | < 0.0001 |
DOR, median (95% CI), months | 20.5 (12.3 to NR) | 6.6 (4.1 to 17.2) |
P valued,e | NA | < 0.0001 |
TTNT, mediana (95% CI), months | 16.7 (7.6 to NR) | 5.1 (4.7 to 7.3) |
P valued | Not reported | Not reported |
CI = confidence interval; CRR = complete response rate; DOR = duration of response; EFS = event-free survival; HR = hazard ratio; MAS = ?; NA = not applicable; NR = not reached; OR = odds ratio; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; TTNT = time to next treatment.
aMedian is the Kaplan–Meier estimate.
bA HR less than 1 favours tafasitamab plus lenalidomide. An OR greater than 1 favours tafasitamab plus lenalidomide.
cCox proportional hazard model.
dP value is not adjusted for multiple comparisons.
eFisher’s exact test.
fLog-rank test.
Source: RE-MIND Clinical Study Report,16 Zinzani et al. (2021).17
In the MAS 25, secondary end points were consistent with the primary end point. Furthermore, sensitivity analyses results were consistent and supported the results seen in the MAS 25 primary analysis set for all secondary end points.
Median OS was NR (95% CI, 15.5 to NR) in the tafasitamab plus lenalidomide cohort and 9.4 (95% CI, 5.1 to 20.0) months in the lenalidomide-monotherapy cohort (HR = 0.499; 95% CI, 0.317 to 0.785; P = 0.0026). The median follow-up times for OS in the tafasitamab plus lenalidomide and lenalidomide-monotherapy cohorts were 21.49 and 20.90 months, respectively.
Median PFS was 12.1 (95% CI, 5.9 to NR) months in the tafasitamab plus lenalidomide cohort and 4.0 (95% CI, 3.1 to 7.4) months in the lenalidomide-monotherapy cohort (HR = 0.463; 95% CI, 0.307 to 0.698; P = 0.0002). The median follow-up times for PFS in the tafasitamab plus lenalidomide and lenalidomide-monotherapy cohorts were 19.68 and 12.62 months, respectively.
Median EFS was 12.1 (95% CI, 5.5 to 21.0) months in the tafasitamab plus lenalidomide cohort and 4.0 (95% CI, 3.1 to 6.2) months in the lenalidomide-monotherapy cohort. The HR for EFS was 0.439 (95% CI, 0.296 to 0.650) in favour of the tafasitamab plus lenalidomide cohort (P < 0.0001). The median follow-up times for EFS in the tafasitamab plus lenalidomide and lenalidomide-monotherapy cohorts were 21.9 and 15.4 months, respectively.
The CRR was higher in the tafasitamab plus lenalidomide cohort (39.5%; 95% CI, 28.4 to 51.4) compared with the lenalidomide-monotherapy cohort (13.2%; 95% CI, 6.5 to 22.9; P < 0.0001).
Median DOR excluding nonresponders was 20.5 (95% CI, 12.3 to not estimable) months in the tafasitamab plus lenalidomide cohort and 6.6 (95% CI, 4.1 to 17.2) months in the lenalidomide-monotherapy cohort (P < 0.0001).
Median TTNT was 16.7 (95% CI, 7.6 to NR) months in the tafasitamab plus lenalidomide cohort and 5.1 (95% CI, 4.7 to 7.3) months in the lenalidomide-monotherapy cohort. This difference was not statistically significant (P value not reported).
Physician-reported, real-world tumour response assessments were validated via independent radiology and clinical review assessments in the retrospective, observational cohort of R/R DLBCL patients treated with lenalidomide monotherapy in the RE-MIND study. The combined concordance for the responders (CR + PR) and nonresponders (stable disease + PD) was 79.8%. The concordance was 59.1% for the responders (CR + PR) and 89.3% for the nonresponders (stable disease + PD).
RE-MIND2
Efficacy: Results for the pre-specified efficacy analyses conducted in the RE-MIND2 for outcomes that were identified as outcomes of interest in the CADTH systematic review protocol (Table 5) are summarized in Table 32. The results are discussed for the primary analysis sets: MAS_Pool (N = 76 each for tafasitamab plus lenalidomide and systemic therapies pooled), MAS_BR (N = 75, each for tafasitamab plus lenalidomide and BR), and MAS_R-GemOx (N = 74, each for tafasitamab plus lenalidomide and R-GemOx).
The primary end point of the study was met. Patients in the tafasitamab plus lenalidomide cohort showed an improvement in OS compared to the cohorts of systemic therapies pooled (HR = 0.553; 95% CI, 0.358 to 0.855; P = 0.0076), BR (HR = 0.418; 95% CI, 0.272 to 0.644; P < 0.0001), and R-GemOx (HR = 0.467; 95% CI, 0.305 to 0.714; P = 0.0004). The median duration of follow-up for OS in the tafasitamab plus lenalidomide cohort was 31.8 months for MAS_Pool and 32.9 months each for MAS_BR and MAS_R-GemOx. The median duration of follow-up for OS in the cohorts of systemic therapies pooled, BR, and R-GemOx was 33.3, 25.0, and 33.2 months, respectively. The results of the primary analytical approach used for OS were supported by sensitivity analyses using the 11 baseline covariates for matching.
Table 32: RE-MIND2 Results – Pre-specified Efficacy Analyses, Primary Analysis Sets (Systemic Therapies Pooled, BR, R-GemOx)
Outcome | MAS_Pool | MAS_BR | MAS_R-GemOx | |||
|---|---|---|---|---|---|---|
Tafasitimab plus lenalidomide (N = 76) | Systemic therapies pooled (N = 76) | Tafasitimab plus lenalidomide (N = 75) | BR (N = 75) | Tafasitimab plus lenalidomide (N = 74) | R-GemOx (N = 74) | |
OS | ||||||
Mediana (95% CIb), months | 34.1 (18.3 to NR) | 11.6 (8.8 to 16.1) | 31.6 (18.3 to NR) | 9.9 (5.3 to 13.7) | 31.6 (18.3 to NR) | 11.0 (7.9 to 16.8) |
HR (95% CI) [P value]c,d | 0.553 (0.358 to 0.855) [0.0076] | 0.418 (0.272 to 0.644) [< 0.0001] | 0.467 (0.305 to 0.714) [0.0004] | |||
PFS | ||||||
Mediana (95% CIb), months | 12.1 (5.9 to 22.5) | 5.8 (3.1 to 6.4) | 12.1 (5.5 to 22.5) | 7.9 (4.3 to 11.3) | 14.1 (6.3 to 28.0) | 5.1 (3.5 to 9.5) |
HR (95% CI) [P value]c,d | 0.424 (0.278 to 0.647) [< 0.0001] | 0.527 (0.344 to 0.809) [0.0033] | 0.433 (0.288 to 0.653) [< 0.0001] | |||
EFS | ||||||
Mediana (95% CIb), months | 8.7 (5.5 to 21.0) | 4.1 (3.0 to 5.8) | 8.7 (5.5 to 21.0) | 5.2 (3.0 to 7.9) | 9.1 (5.5 to 22.3) | 4.0 (3.2 to 6.0) |
HR (95% CI) [P value]c,d | 0.380 (0.256 to 0.563) [< 0.0001] | 0.464 (0.313 to 0.688) [0.0001] | 0.397 (0.272 to 0.580) [< 0.0001] | |||
ORR | ||||||
ORR (95% CIe), % | 67.1 (55.4 to 77.5) | 48.7 (37.0 to 60.4) | 66.7 (54.8 to 77.1) | 54.7 (42.7 to 66.2) | 68.9 (57.1 to 79.2) | 45.9 (34.3 to 57.9) |
OR (95% CI) [P value]d,f | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||
CRR | ||||||
CRR (95% CIe), % | 38.2 (27.2 to 50.0) | 21.1 (27.2 to 50.0) | 38.7 (27.2 to 50.0) | 28.0 (27.2 to 50.0) | 39.2 (27.2 to 50.0) | 23.0 (27.2 to 50.0) |
OR (95% CI) [P value]d,f | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||
DOR | ||||||
Mediana (95% CIb), months | 26.1 (13.9 to NR) | 6.6 (4.4 to 11.8) | 26.1 (13.9 to NR) | 9.2 (5.3 to 12.5) | 26.1 (13.9 to NR) | 9.5 (5.5 to 13.2) |
TTNT | ||||||
Mediana (95% CIb), months | 12.5 (7.6 to 24.7) | 6.3 (3.3 to 8.3) | 12.1 (7.3 to 24.7) | 6.9 (4.2 to 10.6) | 12.5 (7.6 to 28.0) | 5.7 (4.0 to 7.2) |
HR (95% CI) [P value]c,d | 0.461 (0.314 to 0.676) [< 0.0001] | 0.527 (0.357 to 0.780) [0.0013] | 0.423 (0.289 to 0.619) [< 0.0001] | |||
BR = bendamustine plus rituximab; CI = confidence interval; CRR = complete response rate; DOR = duration of response; EFS = event-free survival; HR = hazard ratio; MAS = matched analysis set; NR = not reached; OS = overall survival; OR = odds ratio; ORR = objective response rate; PFS = progression-free survival; Pool = systemic therapies pooled; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; TTNT = time to next treatment.
Notes: Analysis window for the tafasitamab plus lenalidomide cohort was defined as the interval between index date and data cut-off date (30 November 2019). Analysis window for observational cohorts was defined as the interval between index date for a given line + 44 months (1,338 days).
For the PFS results, the authors state that caution should be taken in interpretation due to the unmet assumption of the Cox proportional hazard model and other limitations.
aKaplan–Meier estimate.
b95% CIs calculated using Brookmeyer and Crowley method.
cHR was estimated using the observational cohort as reference group in Cox proportional hazard models. HR less than 1 favours tafasitamab plus lenalidomide. P values for HRs calculated with Wald test.
dP value has not been adjusted for multiple comparisons.
e95% CIs calculated using Clopper-Pearson exact method.
fOR and their 95% CI was estimated using logistic regression, with observational cohort as the reference group in the model. OR more than 1 favours tafasitamab plus lenalidomide. P value for ORs from logistic regression model.
Source: RE-MIND2 Clinical Study Report.18
An improvement in PFS, EFS, and TTNT was observed in the tafasitamab plus lenalidomide cohort compared with the cohorts of systemic therapies pooled, BR, and R-GemOx. Regarding PFS, an improvement was also observed for PFS in the tafasitamab plus lenalidomide cohort compared with the cohorts of systemic therapies pooled (HR = 0.424; 95% CI, 0.278 to 0.647; P < 0.0001), BR (HR = 0.527; 95% CI, 0.344 to 0.809; P = 0.0033), and R-GemOx (HR = 0.433; 95% CI, 0.288 to 0.653; P < 0.0001). However, the sponsor reported that the PFS data has limitations because of invalidated progression assessments and higher censoring rates due to missing radiological assessments. Conclusions about this outcome are also limited because the assumption of the Cox proportional hazards model was not met.
The ORR was higher in the tafasitamab plus lenalidomide cohort compared to the cohorts of systemic therapies pooled (OR = 2.139; 95% CI, 1.061 to 4.377; P = 0.0323) and R-GemOx (OR = 2.591; 95% CI, 1.265 to 5.407; P = 0.0076). There was no difference in ORR for the tafasitamab plus lenalidomide cohort compared to the BR cohort (OR = 1.653; 95% CI, 0.814 to 3.393; P = 0.1810).
The CRR was higher in the tafasitamab plus lenalidomide cohort compared to the cohort of systemic therapies pooled (||||||||||||||||||||||||||||||||||||||||||||||||). There was no difference in CRR between the tafasitamab plus lenalidomide cohort and the BR (||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||) and R-GemOx cohorts (||||||||||||||||||||||||||||||||||||).
Median DOR appeared greater in the tafasitamab plus lenalidomide cohort compared with the cohorts of systemic therapies pooled, BR, and R-GemOx. However, the CADTH review team could draw no conclusions for this outcome because the cohorts were not compared statistically.
Median TTNT was greater in the tafasitamab plus lenalidomide cohort than in the cohorts of systemic therapies pooled (HR = 0.461; 95% CI, 0.314 to 0.676; P < 0.0001), BR (HR = 0.527; 95% CI, 0.357 to 0.780; P = 0.0013), and R-GemOx (HR = 0.423; 95% CI, 0.289 to 0.619; P < 0.0001).
Sensitivity analyses results by applying 11 covariates in the MAS_Pool_11Cov were consistent and supported the results seen in the MAS_Pool primary analysis set across primary and secondary end points.
Results of the post hoc exploratory analyses of tafasitamab plus lenalidomide versus pola-BR and CAR T-cell therapy for the analysis set used to inform the pharmacoeconomic model are presented in Table 33. In all analysis sets used in the post hoc analyses, median OS was longer in the tafasitamab plus lenalidomide cohort than in the pola-BR cohort. There was no difference in median OS for the tafasitamab plus lenalidomide cohort compared to the CAR T-cell therapy cohort in all analysis sets assessed in the post hoc analyses.
|||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | ||
|---|---|---|---|---|
||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |
|||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Note: Some redacted cells have been removed
Safety
Eight patients discontinued due to AEs in the tafasitamab plus lenalidomide cohort for MAS_Pool (14.5%), MAS_BR (14.5%), and MAS_R-GemOx (15.1%). In the cohorts of systemic therapies pooled, BR, and R-GemOx, 5 (6.8%), 2 (2.8%), and 4 (5.4%) patients, respectively, had AEs leading to permanent discontinuation of treatment. The types of AEs leading to treatment discontinuation were not reported. Results of the sensitivity analysis were consistent with the primary analysis.
The median duration of exposure in the tafasitamab plus lenalidomide cohort was longer (approximately 10 months) than that in the cohorts of systemic therapies pooled (2.4 months), BR (3.2 months), and R-GemOx (2.9 months). The median duration of exposure in the sensitivity analysis was consistent with the primary analysis.
Critical Appraisal of RE-MIND and RE-MIND2
RE-MIND and RE-MIND2 were both retrospective observational studies that were used to generate an external cohort for indirect comparison to the L-MIND cohort. Both studies used an ePS-based NN 1:1 matching methodology. The success of ePS matching depends on the availability of a large pool of patients from which to select a closely matched population. Both studies enrolled an adequate numbers of patients in the observational cohorts. The CADTH review team identified various potential sources of bias associated with ePS-based NN 1:1 matching, including a limited ability to apply similar inclusion and exclusion criteria between cohorts, potential difficulties with the fidelity of available data from patient records, variations in outcome assessments, and differences or changes in treatment strategies across geographic regions or over the time frame of the study parameters.
Although the eligibility criteria for enrolment in RE-MIND and RE-MIND2 were based on the eligibility criteria used in the L-MIND trial, a number of differences were noted. Many of these differences in eligibility criteria were related to retrospective nature of the RE-MIND and RE-MIND2 studies, whereas the L-MIND study was a prospective, interventional study. First, there were no restrictions for patient ECOG PS in RE-MIND and RE-MIND2, whereas patients enrolled in L-MIND were required to have an ECOG PS of 0 to 2. This lack of ECOG PS restrictions was likely because ECOG scores are typically not well-documented in routine clinical care, as they are in prospective clinical studies. However, the RE-MIND and RE-MIND2 studies included patients with an ECOG PS greater than 2, which could bias results in favour of tafasitamab plus lenalidomide. RE-MIND2 did not exclude patients with double- or triple-hit genetics. Both RE-MIND and RE-MIND2 did not exclude patients with ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. It is likely that a proportion of patients enrolled in RE-MIND and RE-MIND2 would have disease history or comorbidities that would have excluded them from participating in L-MIND, thus biasing results in favour of tafasitamab plus lenalidomide, since the clinical experts considered patients in L-MIND to be the most fit subset of patients with R/R DLBCL. Primary refractoriness was an exclusion criterion in L-MIND, but patients considered to have primary refractory disease were enrolled as a result of changing definitions ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. However, a comparable distribution of primary refractory patients between the tafasitamab plus lenalidomide and the observational cohorts was ensured by having primary refractoriness (yes versus no) as a baseline covariate for ePS-based matching. Pre-specified laboratory values for neutrophil count, thrombocyte count, as well as creatinine clearance, liver enzymes, and bilirubin were part of the L-MIND study’s eligibility criteria, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. However, neutropenia (yes or no) and anemia (yes or no) were applied as baseline covariates for ePS-based matching. Although the eligibility criteria used in RE-MIND and RE-MIND2 were based on the L-MIND trial eligibility criteria, patients enrolled in RE-MIND and RE-MIND2 still may not have been eligible for participation in L-MIND, thus limiting the comparability of the study populations.
Comparison of data from a prospective, interventional trial to retrospective, observational studies using real-world data may be problematic, since there were a number of notable differences in data collection, outcomes, and assessments. Observational studies are prone to unique biases that cannot be controlled for using matching methodology. Differences in response assessment frequency or failure to capture response to therapy in daily clinical practice are sources of bias in the RE-MIND and RE-MIND2 studies. The studies attempted to minimize this bias by applying the 6-month follow-up rule to prevent overestimation of nonresponse in the main analysis. However, there were differences in frequency of imaging and assessments to determine when response or disease progression occurred. Patients in the tafasitamab plus lenalidomide cohort used for RE-MIND and RE-MIND2 were treated under the L-MIND clinical trial protocol, and therefore prospectively followed with a defined tumour assessment scheme and defined response criteria. Patients in the observational cohorts of RE-MIND and RE-MIND2 were treated and followed in real-world clinical practice without an overarching protocol, leading to heterogeneity with regard to tumour assessment frequency and criteria. Furthermore, definitions of response used for patients in the observational cohorts did not always match the criteria used in the L-MIND trial (i.e., IWG response criteria by Cheson et al. [2007]8). This creates significant uncertainty in the results of the outcomes based on tumour assessments (i.e., ORR, CRR, DOR, PFS, EFS). Also, data for the IRC-assessed outcomes from the L-MIND trial were generally used, whereas data in the observational cohorts would be investigator-assessed.
Additionally, differences in outcome definitions were noted between the L-MIND study and the RE-MIND and RE-MIND2 studies. In RE-MIND and RE-MIND2, best ORR was defined as the proportion of patients for whom CR or PR was the best response achieved at any time within the analysis window or between index date and date of initiation of a new anti-DLBCL treatment or death. Similarly, CRR was defined as the proportion of patients for whom CR was the best response achieved at any time within the analysis window or between index date and date of initiation of a new anti-DLBCL treatment or death. In L-MIND, ORR was defined as the proportion of patients with a CR or PR up until disease progression by the IRC, and CRR was the defined as the proportion of patients with a CR by the same criteria. These differences in definitions make the comparisons between the L-MIND cohort and observational cohorts problematic. The definitions of other outcomes assessed in RE-MIND and RE-MIND2 were similar to those in L-MIND, although differences in the assessments (e.g., criteria used, imaging modalities, timing of assessments) and prospective data collection versus retrospective data collection remain sources of heterogeneity.
Furthermore, unmeasured confounding factors not accounted for in the matching may have affected the results. The RE-MIND and RE-MIND2 studies used 9 clinically important covariates for matching in their main analyses: age, Ann Arbor stage, refractoriness to last therapy line, number of previous lines of therapy, history of primary refractoriness, prior ASCT, neutropenia, anemia, and elevated LDH. However, these covariates do not represent all important confounders. Most notably, ECOG PS was not used for matching. Other important effect modifiers and prognostic factors include IPI score, cell of origin, extranodal involvement, cytogenetic factors (e.g., double- or triple-hit genetics DLBCL), DOR to prior therapy, and presence of bulky disease. In addition, it is unclear how these 9 covariates were selected.
RE-MIND included several other measures to reduce bias and ensure that the identified lenalidomide-monotherapy cohort would provide a legitimate comparator for the L-MIND cohort. Study sites were selected from the same geographic regions as those in L-MIND. A feasibility questionnaire was used to identify sites that could provide patient data that satisfied key requirements for inclusion and exclusion criteria, patient disposition, and outcomes. The RE-MIND cohort included only patients who started lenalidomide at a dosage of 25 mg per day, which was the starting dosage of lenalidomide in L-MIND. In addition, patients were treated at similar times as L-MIND. The start date of lenalidomide treatment was comparable between the 2 cohorts (main distribution in the MAS 25 between 2015 and 2019).
In RE-MIND, patient disposition and demographic factors in the MAS 25 were generally well balanced between the cohorts, based on assessment of the measured variables. Overall, the 2 cohorts in RE-MIND were well matched, with an SMD of less than 0.20 for 7 of the 9 covariates. Residual imbalance for the other 2 covariates (Ann Arbor stage and number of prior systemic treatments) were addressed in sensitivity analyses. Slight differences were noted for Ann Arbor stage, ECOG, IPI score, number of prior systemic treatments, and time since discontinuation of last prior anti-DLBCL medication. The difference in the IPI score between the 2 cohorts may be attributable to missing data for its components (e.g., ECOG, information on extranodal sites) in the retrospective, observational study setting. All sensitivity analyses were consistent with the primary analysis.
In RE-MIND, duration of exposure to lenalidomide monotherapy was higher in the tafasitamab plus lenalidomide cohort than in the lenalidomide-monotherapy cohort by approximately 4 months. However, mean dose intensity was comparable between the 2 cohorts. The difference in duration of exposure to study treatment may have biased results; the direction of the potential bias is unknown.
The primary end point of the RE-MIND study was met. For the primary end point ORR, a subset of the physician-reported, real-world tumour response assessments were validated via independent radiology and clinical reviewer assessments in the retrospective, observational cohort of R/R DLBCL patients treated with lenalidomide monotherapy. The results of the validation assessment demonstrated a combined concordance for the responders and nonresponders. Secondary end points analyses were consistent with those of the primary end point. The Kaplan–Meier estimates of median OS, PFS, TTNT, and EFS were greater in the tafasitamab plus lenalidomide cohort than in the lenalidomide-monotherapy cohort. However, median PFS follow-up time was shorter in the lenalidomide-monotherapy cohort, which may have biased results and creates uncertainty in the data. Sensitivity analyses performed to assess an alternative cohort balancing approach—with overlap weights, the impact of missing data in baseline covariates via multiple imputation, frequency of response assessment, and potential unmeasured confounding—confirmed the results obtained in the primary analysis. Although the results of the RE-MIND study suggest that tafasitamab adds benefit to treatment with lenalidomide alone, the differences in study designs of the L-MIND cohort and the observational cohort, along with likely differences between cohorts (both observed and unobserved confounders) create significant uncertainty in the data. As a result, the CADTH review team can draw no conclusions regarding whether there is additional benefit of combination treatment compared to lenalidomide monotherapy.
Regarding external validity, lenalidomide monotherapy is not commonly used as treatment for patients with R/R DLBCL in Canada, according to the clinical experts consulted by CADTH. As a consequence, lenalidomide monotherapy is not a truly relevant comparator for tafasitamab plus lenalidomide.
Similar to RE-MIND, the RE-MIND2 study implemented multiple measures to minimize bias. Eligibility criteria for the observational cohort were aligned with those from the L-MIND study. Only those patients who fulfilled the eligibility criteria, had sufficient follow-up, and had complete data on the baseline covariates were considered eligible for 1:1 matching. Matching was performed only if the number of patients eligible for matching in the pre-specified therapy cohort was larger than the number of patients in the L-MIND cohort. In the 2 cohorts with the pre-specified treatments BR (N = 282) and R-GemOx (N = 235), as well as in the cohort of systemic therapies pooled (N = 961), the number of patients eligible for matching was larger than the number of patients in the L-MIND study (N = 76). Hence, the 3 comparisons with patient cohorts of systemic therapies pooled, BR, and R-GemOx were presented in the main analysis. Matching and comparative analyses were not performed in other pre-specified treatment cohorts (i.e., pola-BR and CAR T-cell therapy) because an insufficient number of patients were enrolled and were eligible for matching. For the primary analysis, the tafasitamab plus lenalidomide cohort and the 3 comparator cohorts of systemic therapies pooled, BR, and R-GemOx were generally well matched for the 9 baseline covariates. For the comparison with systemic therapies pooled, the largest SMD was for the covariate of elevated LDH. For the comparison with BR and R-GemOx, the largest SMDs among all covariates were for prior ASCT and elevated LDH. A residual imbalance was noted in the main analysis in the distribution of ECOG performance score at baseline, which was investigated in a sensitivity analysis. Multiple sensitivity analyses were performed to demonstrate the robustness of the matched comparison from the main analysis.
The primary end point of the RE-MIND2 study was met. Patients in the tafasitamab plus lenalidomide cohort showed an improvement in OS compared to the cohorts of systemic therapies pooled, BR, and R-GemOx. Results of all sensitivity analyses were consistent with the main analysis. However, when assessing the treatment effect on OS, duration of OS follow-up should be taken into consideration. Patients in the cohorts of systemic therapies pooled and R-GemOx had a similar median duration of follow-up for OS (approximately 33 months) compared to the tafasitamab plus lenalidomide cohort (approximately 32 months). In contrast, the median duration of follow-up for OS in the BR cohort was shorter (approximately 25 months). This may have biased the results. Consistent with the results for the primary outcome of OS, results favouring tafasitamab plus lenalidomide were reported across most of the secondary end points (i.e., ORR, CRR, PFS, |||||||| TTNT, and DOR) compared with the other observational cohorts. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In RE-MIND2, the median duration of exposure in the tafasitamab plus lenalidomide cohort was longer than that in the cohorts of systemic therapies pooled, BR, and R-GemOx. This difference may be attributed to the respective treatment regimens. In the L-MIND study, tafasitamab plus lenalidomide was administered for 12 cycles (i.e., approximately 12 months), followed by tafasitamab monotherapy until disease progression, death, or withdrawal. In comparison, the majority of therapies administered in the cohorts of systemic therapies pooled, as well as the BR and R-GemOx regimens, were immunochemotherapies, which are typically administered over a fixed, limited treatment duration.
Regarding external validity of RE-MIND2, the clinical experts consulted by CADTH indicated that R-GemOx and BR are not commonly used to treat patients with R/R DLBCL in Canada. There are also concerns about whether the systemic therapies pooled cohort adequately reflects current contemporary practice or therapies. The clinical experts indicated that pola-BR would be the most relevant comparator. The clinical experts noted that the relevance of CAR T-cell therapy as a comparator for tafasitamab + lenalidomide in patients who are not eligible for ASCT was debatable. The clinical experts considered CAR T-cell therapy an intensive therapy, more similar to ASCT. The clinical experts indicated that they would not consider using tafasitamab plus lenalidomide in patients who were eligible for CAR T-cell therapy.
The RE-MIND study did not assess safety. The RE-MIND2 study did assess safety, using the outcomes of duration of exposure to study treatment and treatment discontinuations due to AEs for systemic therapies pooled, BR, and R-GemOx. However, there are limitations to comparing safety data recorded during routine clinical care with data stringently collected during the prospective L-MIND study. In addition, the longer exposure to treatment in the tafasitamab plus lenalidomide cohort also needs to be considered in the context of these data. The CADTH review team could draw no conclusions regarding the safety of tafasitamab plus lenalidomide relative to the comparator therapies.
The RE-MIND and RE-MIND2 studies did not report HRQoL because this outcome was not reported in L-MIND. This is an important gap in the evidence, as HRQoL is an outcome identified as important to patients.
The RE-MIND study used the November 30, 2018, data cut-off (primary analysis) of the L-MIND study, whereas the RE-MIND2 study used the L-MIND November 30, 2019, data cut-off. According to the L-MIND Clinical Study Report, PFS data were mature at the primary analysis. However, CADTH noted that median PFS changed numerically at the subsequent analyses with longer follow-up time. Furthermore, OS and DOR data were likely not fully mature at either of these analyses. Results of the RE-MIND and RE-MIND2 comparisons could differ if more mature data from the L-MIND study were used, although this is currently unknown.
The issues highlighted above as limitations of the RE-MIND and RE-MIND2 studies may have affected the comparisons of tafasitamab plus lenalidomide with other active therapies in the analyses. Although multiple methods were employed in the RE-MIND and RE-MIND2 studies to minimize bias in the comparisons, the propensity matching method is not a replacement for a randomized controlled study. There is likely significant bias affecting the results due to heterogeneity that could not be accounted for in the methods implemented, which makes the results of the RE-MIND and RE-MIND2 studies uncertain.
Methods of the MAICs
Objectives
The objective of the sponsor-submitted unanchored MAIC21 was to conduct an MAIC of tafasitamab plus lenalidomide in the FAS population of the L-MIND study versus appropriate comparators on OS, PFS, DOR, ORR, and CRR.
Study Selection Methods
Details on the study selection methods used for the MAICs are provided in Table 34. The sponsor conducted a systematic literature review to identify relevant sources of evidence for comparator treatments for patients with R/R DLBCL who were not eligible for ASCT.21,25 Key comparators identified by the sponsor as relevant to Canada included R-GemOx, pola-BR, and CAR T-cell therapies. Additional treatments not currently used in clinical practice in Canada were included because the scope of the MAIC was international. These were rituximab plus lenalidomide, lenalidomide monotherapy, BR, and pixantrone monotherapy.
The databases searched are listed in Table 34. Multiple databases were searched to identify relevant clinical studies.25 The original searches were conducted in February 2021, and ||||||||||||||||||||||||||||||||||||||||||||||||||||. The electronic database and grey literature searches were supplemented with a review of relevant published systematic literature reviews. Relevant publications identified in the bibliographies were cross-referenced with the database search results to identify any additional published studies. Studies published after 2011 and abstracts published after 2016 were considered. The search strategy removed non-human studies, in vitro studies, case studies, letters, commentaries, and editorials.
Study selection criteria were developed ||||||||||||||||||||||||||||. These criteria were used to assess whether the L-MIND and comparator studies identified by the authors’ systematic literature review were comparable. Studies that enrolled patients eligible for stem cell transplant, reported large proportions of patients with untransformed follicular lymphoma or mantle-cell lymphoma, a large proportion of patients with double- or triple-hit genetics lymphoma (i.e., high-grade B-cell lymphoma, with MYC and BCL2 and/or BCL6 rearrangements), or enrolled a majority of patients treated in the fifth-line setting or beyond were considered not comparable to the L-MIND study by the sponsor’s clinical experts and thus excluded from the analyses. Two independent researchers screened titles and abstracts to determine potential relevance. Full-text screening was conducted for articles that were not definitively categorized via title/abstract. Discrepancies were addressed through discussion, and detailed reasons for study inclusion and exclusion were documented.
Table 34: Study Selection Criteria and Methods for MAICs
Criteria | MAICs |
|---|---|
Population | Adult patients with transplant ineligible R/R DLBCLa |
Intervention | Tafasitamab plus lenalidomide, as in the L-MIND study |
Comparators in study selection search criteria (broad search)b |
|
Outcomes | Efficacy:
|
Study design | Prospective studies, including:
|
Exclusion criteria |
|
Databases searched | Embase, MEDLINE (PubMed), the Cochrane Library (including CENTRAL and the Cochrane Database of Systematic Reviews), the University of York Centre for Reviews and Dissemination Database, EconLit via the American Economics Association, and PsycInfo using the American Psychological Association PsycNet platform A search of the grey literature, including a search for conference abstracts on Embase, as well as select regulatory and health technology assessment websites, including NICE, SMC, AWMSG, CADTH, IQWiG, HAS, the Institute for Clinical and Economic Review, and PBAC |
Date range | 2011 to 2021 for papers and 2016 to 2021 for conference abstracts |
Selection process | Two independent researchers examined all titles and abstracts to determine potential relevance; full-text screening was conducted for articles that were not definitively categorized via title/abstract; discrepancies were addressed through discussion |
Data extraction process | Data were extracted by a single investigator and validated by a second; any disagreements were resolved by a third investigator |
Quality assessment | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |
ACVBP = doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone; ASHAP = doxorubicin, solumedrol, cytarabine, platinum; AWMSG = All Wales Medicines Strategy Group; BEAM = carmustine, etoposide, cytarabine, melphalan; BR = bendamustine plus rituximab; CEOP = cyclophosphamide, etoposide, vincristine, prednisone; CEPP = cyclophosphamide, etoposide, prednisone, procarbazine; CHOP = cyclophosphamide, doxorubicin, vincristine, prednisone; DA-EPOCH = dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin; DHAOx = dexamethasone, cisplatin, oxaliplatin; DHAP = dexamethasone, cisplatin, cytarabine; DHL = double-hit genetics lymphoma; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; EPOCH = etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin; ESHAP = etoposide, methylprednisolone, cytarabine, cisplatin; GDP = gemcitabine plus dexamethasone plus cisplatin; GemOx = gemcitabine and oxaliplatin; HAS = Haute Autorité de Santé; ICE = ifosfamide, carboplatin, and etoposide; IEV = ifosfamide, etoposide, epirubicin; IGEV = ifosfamide, gemcitabine, vinorelbine, prednisone; IQWiG = Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen; MAIC = matching-adjusted indirect comparison; MINE = mesna, ifosfamide, mitoxantrone, etoposide; NICE = UK National Institute for Health and Care Excellence; ORR = objective response rate; OS = overall survival; PBAC = Pharmaceutical Benefits Advisory Committee; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine plus rituximab; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone; RCT = randomized controlled trial; R/R = relapsed or refractory; SCT = stem cell transplant; SMC = Scottish Medicines Consortium; THL = triple-hit genetics lymphoma.
aRefractory is defined as disease that does not respond to initial treatment or that gets worse or stays the same within 6 months after the end of initial treatment. Relapsed is disease that responds to treatment but then returns, and patients must be on at least second-line treatment. Studies that contained only transplant-eligible patients or salvage therapy including ASCT-eligible patients were excluded. Studies that contained a mix of transplant-eligible and -ineligible patients and did not report their results separately were excluded.
Source: Sponsor-submitted MAIC,21 MAIC feasibility assessment.25
In total, 36 studies reporting data for the comparators of interest were identified by the sponsor’s systematic literature review. Comparator study designs were assessed for comparability to the L-MIND study design. Following this initial selection process, inclusion and exclusion criteria of each of the studies were applied to the L-MIND population to estimate the retained sample size of the L-MIND population for further population adjustment. A final decision on the inclusion of the evidence in the MAIC was made after assessing this sample size against the extent of the remaining imbalances in population characteristics between the L-MIND and comparator studies. Reasons for excluding studies were reported (e.g., study design, patient population, concerns about survivor bias, small number of patients retained after prefiltering of the L-MIND population to match the eligibility criteria of the studies).
In total, 5 prospective studies reporting data for lenalidomide monotherapy, pola-BR, BR, and R-GemOx were selected for the MAIC against tafasitamab plus lenalidomide.
ITC Analysis Methods
MAIC weights were estimated through propensity score-like regressions. Since the L-MIND study was a single-arm study, an unanchored MAIC was performed. The authors’ matching strategy was intended to preserve at least 20% (i.e., effective sample size [ESS] ≥ 16) of the original L-MIND FAS (n = 80), while adjusting for as many effect modifiers and prognostic factors as possible. Three matching scenarios were investigated: (1) adjust for all mutually available baseline characteristics among the L-MIND and comparator studies; 2i) adjust for all mutually available prognostic factors and effect modifiers, as identified from the systematic literature review and the sponsor’s clinical experts, as listed in Table 35; and (3) prioritize matching for age, ECOG PS score, IPI score, refractoriness of patients (primary refractoriness or refractoriness to prior lines of therapy), number of prior treatment lines, prior ASCT, and cell type of origin of the disease, per the advice of the sponsor’s clinical experts.
Table 35: Prognostic Factors and Effect Modifiers in DLBCL Identified by the Sponsor’s Clinical Experts
Factors | Prognostic factor | Effect modifier |
|---|---|---|
General health | ||
Age | X | X |
Sex | — | X |
ECOG PS | X | — |
Creatinine clearance | X | X |
Disease characteristics | ||
Primary refractory disease | X | X |
IPI score | X | X |
LDH levels | X | X |
Cell of origin (ABC and GCB) | X | X |
Ann Arbor disease stage | X | — |
Extra nodal involvement | X | — |
Cytogenetics factors (MYC, BCL2, BCL6, and double- or triple-hit genetics DLBCL) | X | X |
P53 positivity staining, anti-CD10, CD20, CD30 staining, MUM1 | X | X |
High Ki67 index (> 40%) | X | X |
Prior therapies | ||
Refractoriness to last line of therapy or to rituximab | X | X |
Duration of response to prior therapy | X | — |
Prior ASCT | X | — |
Number of prior therapies | X | — |
ABC = activated B-cell; ASCT = autologous stem cell transplant; BCL2 = B-cell lymphoma 2; BCL6 = B-cell lymphoma 6; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group performance status; GCB = germinal centre B-cell; IPI = International Prognostic Index; LDH = lactate dehydrogenase.
Source: Sponsor-submitted MAIC.21
The comparability of the comparator studies relative to the L-MIND study was assessed in a feasibility assessment25 before conducting the MAIC. Potential sources of heterogeneity were identified and reported by the authors.
The MAIC analyses included assessment of ORR, CRR, DOR, PFS, and OS. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For time-to-event outcomes, the relative efficacy estimates were quantified as an HR with a 95% CI. The HRs were obtained using a Cox regression analysis fitted on the L-MIND data and the reconstructed individual patient data of the comparator study used in the matching. Reconstructed individual-patient data were generated from digitized coordinates using the algorithm presented by Guyot et al.{Guyot, 2012 #209} The assumption of proportional hazards was evaluated by plotting the log-log survival versus log time after applying the weights.
For binary outcomes, the relative efficacy estimates were quantified as an OR with a 95% CI. The OR was obtained using logistic regression analysis fitted on the L-MIND data and the reconstructed individual-patient data of the comparator study used in the matching.
Robust sandwich estimators were used for the calculation of the standard errors. The regression models were fitted using the weighted L-MIND population and the unweighted L-MIND data against comparator data to estimate the reduction in the bias induced by the population adjustment.
Since multiple studies were identified with BR as a comparator, a pooling of the HRs obtained was performed using direct meta-analyses. Several estimates of relative efficacy of tafasitamab plus lenalidomide against BR could be estimated for PFS, DOR, ORR, and CRR using the different sources of evidence. Direct meta-analyses were conducted by pooling the results from multiple MAICs (i.e., estimates of mean treatment effects on the ln scale between tafasitamab plus lenalidomide and BR on PFS, ORR, DOR, and CRR to obtain the direct estimate for the comparison and its standard error using frequentist meta-analysis). Random-effects models were used in the primary analysis to account for any unexplained heterogeneity in the MAIC estimates. Fixed-effects meta-analyses were also run. Results of the direct meta-analyses were presented as a central estimate of the relative effect of interest (HR and OR) along with 95% CI.
Results of the MAICs
Summary of Included Studies
Five studies were included in the sponsor-submitted MAICs: |||||||||||||||||||||||,35 the GO29365 trial (Sehn et al. [2020]),9 the Ohmachi et al. (2013) study,36 Vacirca et al. (2014) study,37 and the Mounier et al. (2013) study.38 A summary of key differences in study characteristics compared to the L-MIND trial is provided in Table 36. A summary of baseline characteristics of the study populations is provided in Table 37.
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The ||||||||||||||||||||||| was a phase II/III, 2-stage, multi-centre, open-label randomized trial of lenalidomide monotherapy against investigator choice of treatment in patients with R/R DLBCL who had received at least 2 prior lines of therapy (including an anti-CD20 drug) and who were considered ineligible for stem cell transplant. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Patients enrolled in the |||||||||||||| study could be treated with lenalidomide until disease progression, and there was no maximum number of treatment cycles. In contrast, lenalidomide intake was limited to a maximum of 12 cycles in the L-MIND study. The |||||||||||||| was conducted among patients who had received at least 2 prior lines of therapy, including a prior line containing rituximab and an anthracycline equivalent in addition to either a combination therapy or a conditioning regimen following ASCT. The L-MIND study, however, was conducted in patients with 1 to 3 prior lines of therapy. Since no maximum of prior lines of therapy seemed to have been set for inclusion of patients in the ||||||||||||||, patients from the |||||||||||||| were expected to be more heavily pre-treated than patients enrolled in the L-MIND study. In addition, the |||||||||||||| only enrolled patients with a life expectancy of 3 months or more, whereas the L-MIND study could enrol patients irrespective of their life expectancy. The |||||||||||||| excluded patients with history of transformed indolent lymphoma or with follicular lymphoma, whereas the L-MIND trial included these patients. Furthermore, the L-MIND study and |||||||||||||| used different criteria to evaluate response of patient receiving therapy in DLBCL.
The GO29365 trial (Sehn et al. [2020]) provided data on pola-BR and BR for the sponsor-submitted MAICs. The GO29365 trial was a phase Ib/II, multi-centre, open-label study of pola-BR or obinutuzumab. As part of the MAIC, only the phase II component of the study was considered. The phase II component of the study was a randomized trial of pola-BR against BR in patients with R/R DLBCL who were ineligible for stem cell transplant and who had received at least 1 prior line of therapy, including at least 1 anti-CD20 drug. The GO29365 trial was high quality, according to the quality assessment performed by the authors of the ITC. Inclusion and exclusion criteria of patients based on DLBCL diagnosis differed from those in the L-MIND trial. Patients with transformed DLBCL and grade IIIb follicular lymphoma were specifically excluded from the GO29365 trial but could have been enrolled in the L-MIND study. Patients with a known history of double- or triple-hit genetics DLBCL were excluded from the L-MIND study, but not from the GO29365 trial. Patients enrolled in the L-MIND study were capped to a maximum of 3 prior lines of therapies, while this was not the case in the GO29365 trial. As a result, patients more heavily pre-treated could have been enrolled in the GO29365 trial. The L-MIND study and the GO29365 trial used different criteria to evaluate patients’ response to treatment in DLBCL. Furthermore, the schedule of the initial disease assessment differed between the L-MIND study and GO29365 trial. The index time for OS in the GO29365 trial was not the date of treatment initiation but the date of randomization. Similarly, although no explicit definition for PFS is provided, it is expected that the index time for PFS would also be the date of randomization.
Data on BR were also obtained from the Ohmachi et al. (2013) and Vacirca et al. (2014) studies. The Ohmachi et al. (2013) study was a phase II multi-centre, open-label, single-arm study of BR in patients with CD20-positive R/R DLBCL (excluding transformed lymphoma from low-grade B-NHL), who had received 1 to 3 prior anticancer therapy lines and who were not eligible for stem cell transplant. The primary outcome of this study was ORR; CRR and PFS were also investigated. The Vacirca et al. (2014) study was a phase II multi-centre, single-arm study of BR in patients with CD20-positive R/R DLBCL who received at least 1 prior line of therapy and who were not eligible for stem cell transplant. Both the Ohmachi et al. (2013) and the Vacirca et al. (2014) studies were low quality, according to the quality assessment performed by the authors of the MAIC. In both the Ohmachi and Vacirca studies, patients could be treated with BR for a maximum of 6 cycles, while patients from the L-MIND study could be treated with tafasitamab until progression, and with lenalidomide for up to 12 cycles. The maximum follow-up duration in the L-MIND study was longer than in the Vacirca et al. (2014) study (5 years versus 3 years). Median follow-up could not be retrieved for the Vacirca et al. (2014) study. Median follow-up for the Ohmachi et al. (2013) study was short (4.7 months). Patients could not be enrolled in the Ohmachi et al. (2013) study if they were older than 75 years, whereas 31 patients from the L-MIND study were 75 years or older. Only patients with ECOG PS 0 or 1 could be enrolled in the Ohmachi et al. (2013) study, whereas patients with ECOG PS 2 could be enrolled in the L-MIND study. To be eligible for inclusion in the Ohmachi et al. (2013) study, patients were required to have achieved at least PR to any of their prior therapies, which was not required for patients enrolled in L-MIND. Patients with transformed indolent lymphoma were excluded from the Ohmachi et al. (2013) study. The Ohmachi et al. (2013) and Vacirca et al. (2014) studies only enrolled patients with a life expectancy of 3 months or more. The inclusion criteria for the number of prior lines of therapy received by patients differed between the L-MIND and the Vacirca et al. (2014) study. OS was not reported in the Vacirca et al. (2014) study, although it was listed as an end point of interest. It is unclear whether patients with double-hit or triple-hit genetics DLBCL were enrolled in the Ohmachi et al. (2013) and Vacirca et al. (2014) studies.
The Mounier et al. (2013) study provided data on R-GemOx for the sponsor-submitted MAIC. This study was a phase II, multi-centre, open-label, single-arm study of R-GemOx in patients with CD20-positive R/R DLBCL, in first or second relapse, who were not eligible for high-dose therapy. The primary end point of the study was ORR, assessed after 4 treatment cycles. The study also investigated ORR at completion of the therapy, CRR after 4 cycles and at therapy completion, plus OS, PFS, and DOR. The Mounier et al. study was low quality, according to the quality assessment performed by the authors of the ITC. Disease monitoring was performed in the Mounier et al. study at the end of the 4 cycles of induction R-GemOx therapy, and then at the EOT, as opposed to at regular cycles in the L-MIND study. Patients could not be enrolled in the Mounier et al. study if they were older than 75 years, and patients were required to be in their first or second relapse. No patient enrolled in the Mounier et al. study appeared to have been treated in the fourth-line setting or beyond. The Mounier et al. study enrolled only patients with a life expectancy of 3 months or more. The L-MIND study and Mounier et al. study used different criteria to evaluate response of patients receiving therapy in DLBCL. ORR and CRR were evaluated at the end of the 4-cycle induction therapy planned in the Mounier et al. study, and not as best ORRs, which differs from the L-MIND study. The exact definitions for time-to-event end points (OS, PFS, DOR) were not reported for the Mounier et al. study. No information was provided on the proportion of patients who would have had double-hit or triple-hit genetics DLBCL in the Mounier et al. study; however, it is unclear whether such patients were enrolled in that study.
Table 36: Summary of Key Characteristics and Differences Between L-MIND and Comparator Studies
Characteristic | L-MIND | DLC-001 | GO29365 (Sehn et al. [2020]) | Ohmachi et al. (2013) | Vacirca et al. (2014) | Mounier et al. (2013) |
|---|---|---|---|---|---|---|
Population size | Enrolled N = 81 FAS N = 80 | Enrolled N = 54 Treated N = 51 | Enrolled N = 40 Treated N = 40 | Enrolled N = 63 Treated N = 59 | Enrolled N = 61 Treated N = 59 | Enrolled N = 49 Treated N = 48 |
Study design | Phase II, SA, MC, OL | Phase II/III, randomized, MC, OL | Phase II, randomized, MC, OL | Phase II, SA, MC, OL | Phase II, SA, MC, OL | Phase II, SA, MC, OL |
Primary end point | ORR | ORR | ORR | ORR | ORR | ORR after 4 cycles of induction |
Other end points | OS, PFS, CRR, DOR | OS, PFS, CRR | OS, PFS, CRR, DOR | PFS, CRR | OS, PFS, CRR, DOR | ORR at EOT, PFS, CRR after 4 cycles of induction, CRR at EOT, OS, DOR |
Countries | US and Europe | US, Australia, and Europe | Europe, UK, US, Canada, Australia, Turkey | Japan and South Korea | US | France |
Enrolment years | 2016 to 2020 | Not reported | 2014 to 2016 | 2010 to 2011 | 2008 to 2011 | 2003 to 2009 |
Intervention(s) | Tafasitamab plus lenalidomide | Lenalidomide monotherapy | Pola-BR BR | BR | BR | R-GemOx |
Prior treatments | Received 1 to 3 previous systemic regimens for treatment of DLBCL | Received 2 or more prior lines of therapy | Received at least 1 prior line of therapy | Received 1 to 3 previous systemic regimens for treatment of DLBCL | Received at least 1 prior line of therapy | Patients were required to be in first or second relapse |
Clinical trial eligibility criteria | ECOG PS 0 to 2 ≥ 18 years Included grade 3b follicular lymphoma, and patients with the evidence of histological transformation an earlier diagnosis of low-grade lymphoma with a subsequent DLBCL relapse were also eligible Excluded patients with history of DHL or THL genetics | ECOG PS 0 to 2 ≥ 18 years Life expectancy of at least 3 months Excluded patients with history of low-grade B-cell NHL; evidence of concurrent follicular lymphoma, or history of known transformed large-cell NHL | Excluded patients with history of transformation of indolent disease to DLBCL, or grade 3b follicular lymphoma Patients with DHL or THL genetics were not excluded | ECOG PS 0 to 1 20 to 75 years old Life expectancy of at least 3 months Unclear whether patients with DHL or THL enrolled Excluded transformed lymphoma from low-grade B-NHL Excluded patients with failure to achieve CR, CR unconfirmed, or PR in any prior treatment | ECOG PS 0 to 2 ≥ 18 years Life expectancy of at least 3 months Unclear whether patients with DHL or THL genetics enrolled | ECOG PS 0 to 2 18 to 75 years old Life expectancy of at least 3 months Unclear whether patients with DHL or THL genetics enrolled |
Treatment duration | Treatment with lenalidomide for a maximum of 12 cycles Treatment with tafasitamab until PD, death, intolerable toxicity, or withdrawal | Treatment until PD, unacceptable toxicity, or voluntary withdrawal | Treatment with pola-BR for up to 6 cycles Treatment with BR for up to 6 cycles | Up to 6 treatment cycles | Up to 6 treatment cycles | Induction therapy of 4 cycles, followed by an additional 4 cycles for patients who achieved at least a partial response following induction therapy |
Treatment doses | Tafasitamab 12 mg/kg Lenalidomide 25 mg | Lenalidomide dose based on creatinine clearance: either 25 mg (CrCl ≥ 60 mL/min) or 10 mg (CrCl ≥ 30 mL/min but < 60 mL/min) | Polatuzumab 1.8 mg/kg Rituximab 375 mg/m2 Bendamustine 90 mg/m2 | Rituximab 375 mg/m2 Bendamustine 120 mg/m2 | Rituximab 375 mg/m2 Bendamustine 120 mg/m2 | Rituximab 375 mg/m2 Gemcitabine 1,000 mg/m2 Oxaliplatin 100 mg/m2 |
On treatment tumour assessment schedule | Day 1 of cycles 3, 5, 7, and 10, and then every 3 months thereafter | Not reported | Response was assessed after 3 cycles (interim) and at EOT (primary) | In cycles 2, 4, and 6, as well as at study discontinuation or termination | Every 2 cycles | Not reported |
Response criteria | IWG response criteria reported by Cheson et al. (2007)8 | IWG response reported in Cheson et al. (1999)34 | Lugano modified criterion reported in Cheson et al. (2014)33 | IWG response reported by Cheson et al. (2007)8 | IWG response reported by Cheson et al. (2007)8 | IWG response in Cheson et al. (1999)34 |
EOT visit / follow-up monitoring | Safety visit 30 days after treatment discontinuation. 90 days follow-up following this visit, for living patients who did not withdraw consent, for up to 5 years | Not reported | PFS: 6 to 8 weeks after cycle 6 day 1 (cycle length 21 or 28 days) or last dose of study drug (up to 28 weeks overall) Follow-up CT scans every 6 months for 2 years or until PD or patient withdrawal | Not reported | Maximum follow-up duration was set, with patients censored after 3 years of follow-up | Disease monitoring seems to have been performed at end of 4 cycle of induction R-GemOx therapy, and then at the EOT |
Definitions of end points | OS: time elapsed between treatment initiation and death PFS: time between first study drug administration and tumour progression or death from any cause, whichever occurs first DOR: elapsed time between the date of first documented response (CR or PR) | OS: time from randomization until death of any cause PFS: time from randomization to the first documented disease progression or death due to any cause DOR: length of time of overall response (CR + CR unconfirmed + PR) | OS: time between randomization and death from any cause PFS: not defined explicitly DOR: not reported | OS: not investigated PFS: time from day 1 of the first cycle of study treatment to either disease progression, commencement of another treatment, death from any cause, or discontinuation of assessment | OS definition not reported PFS: time from the start of treatment to the date of disease progression or death as a result of any cause, up to 3 years DOR: time from the first documented response to the date of disease progression | Not reported |
Median follow-up duration | 42.7 months | Not reported | 27.0 months | 4.7 months | Not reported | 65 months |
BR = bendamustine and rixtuximab; CR = complete response; CrCl = creatinine clearance; CRR = complete response rate; CT = CT; DHL = double-hit genetic lymphoma; DLBCL = diffuse large B-cell lymphoma; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group performance status; EOT = end of treatment; FAS = full analysis set; IWG = International Working Group; MC = multi-centre; NHL = non-Hodgkin lymphoma; OL = open-label; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine plus rituximab; PR = partial response; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; SA = single-arm; THL = triple-hit genetic lymphoma.
Source: MAIC Feasibility Assessment Report.25
Table 37: Comparison of Baseline Characteristics Between Included Trials
Characteristic | L-MIND | GO29365 pola-BR (Sehn et al. [2020]) | GO29365 BR (Sehn et al. [2020]) | Ohmachi et al. (2013) | Vacirca et al. (2014) | Mounier et al. (2013) |
|---|---|---|---|---|---|---|
N | 68 | 40 | 40 | 59 | 61 | 49 |
Age | ||||||
Median, years | 71.5 | 67 | 71 | 67 | 74 | 69 |
< 65 years, n (%) | 20 (29.4) | 17 (42.5) | 14 (35.0) | 22 (37.3) | NR | NR |
≥ 65 years, n (%) | 48 (70.6) | 23 (57.5) | 26 (65.0) | 37 (62.7) | NR | NR |
Sex, n (%) | ||||||
Male | 37 (54.4) | 28 (70) | 25 (62.5) | 25 (42.4) | 30 (49.2) | 27 (55) |
Female | 31 (45.6) | 12 (30.0) | 15 (37.5) | 34 (57.6) | 31 (50.8) | 22 (45) |
ECOG PS, n (%) | ||||||
0 | 26 (38.2) | NR | NR | 39 (66.1) | 26 (42.6) | NR |
1 | 36 (52.9) | NR | NR | 20 (33.9) | 31 (50.8) | NR |
0 or 1 | 62 (91.2) | 33 (82.5) | 31 (77.5) | 59 (100) | 57 (93.4) | 38 (78) |
2 | 6 (8.8) | 6 (15.0) | 8 (20.0) | NA | 4 (6.6) | NR |
≥ 2 | 6 (8.8) | 6 (15.0) | 8 (20.0) | NA | 4 (6.6) | 11 (22) |
Ann Arbor disease stage | ||||||
I | 4 (5.9) | NR | NR | 5 (8.5) | NR | NR |
II | 11 (16.2) | NR | NR | 18 (30.5) | NR | NR |
III | 15 (22.1) | NR | NR | 21 (35.6) | NR | NR |
IV | 38 (55.9) | NR | NR | 15 (25.4) | NR | NR |
I or II | 15 (22.1) | 6 (15.0) | 4 (10.0) | NR | 6 (9.8) | 6 (12) |
III or IV | 53 (77.9) | 34 (85) | 36 (90) | NR | 54 (88.5) | 43 (88) |
IPI score | ||||||
0 to 2 | 26 (57.8) | NR | NR | 41 (69.5) | NR | 15 (31) |
3 to 5 | 19 (42.2) | NR | NR | 18 (30.5) | NR | 34 (69) |
0 | 4 (5.9) | 0 | 0 | NR | NR | NR |
1 | 9 (13.2) | 9 (22.5) | 3 (7.5) | NR | NR | NR |
2 | 20 (29.4) | 9 (22.5) | 8 (20.0) | NR | NR | NR |
≥ 3 | 35 (51.5) | 22 (55.0*) | 29 (72.5) | NR | NR | NR |
Revised IPI | ||||||
Very good | 5 (6.2) | NR | NR | NR | 0 | NR |
Good | 35 (43.8) | NR | NR | NR | 22 (36.1) | NR |
Poor | 40 (50.0) | NR | NR | NR | 38 (62.3) | NR |
Unknown | 0 | NR | NR | NR | 1 (1.6) | NR |
Lines of previous systemic treatment, n (%) | ||||||
1 | 35 (51.5) | 11 (27.5) | 12 (30.0) | 38 (64.4) | 31 (50.8) | 42 (86) |
2 | 29 (42.6) | 11 (27.5) | 9 (22.5) | 13 (22.0) | 13 (21.3) | 7 (14) |
3 | 3 (4.4) | NR | NR | 8 (13.6) | 8 (13.1) | NR |
4 | 1 (1.5) | NR | NR | NR | NR | NR |
≥ 3 | 4 (5.9) | 18 (45.0) | 19 (47.5) | NR | 9 (14.8) | NR |
Cell of origin based on gene-expression profiling, n (%) | ||||||
GCB | 7 (10.3) | 15 (37.5) | 17 (42.5) | NR | NR | NR |
ABC | 19 (27.9) | 19 (47.5) | 19 (47.5) | NR | NR | NR |
Unclassified | 5 (7.4) | NR | NR | NR | NR | NR |
Not evaluable | 5 (7.4) | NR | NR | NR | NR | NR |
Missing | 32 (47.1) | 6 (15.0) | 4 (10.0) | NR | NR | NR |
NHL subtype, central pathology | ||||||
DLBCL NOS | 68 (100) | 38 (95.0) | 40 (100.0) | NR | NR | NR |
Burkitt lymphoma | 0 | 1 (2.5) | NR | NR | NR | NR |
Follicular lymphoma | 0 | 1 (2.5) | NR | NR | NR | NR |
Bulky disease present, n (%) | 12 (17.6) | 10 (25.0) | 15 (37.5) | NR | NR | NR |
Refractoriness to last prior therapy, n (%) | ||||||
Yes | 30 (44.1) | 30 (75.0) | 34 (85.0) | 51 (86.4) | NR | NR |
No | 38 (55.9) | 10 (25.0) | 6 (15.0) | 8 (13.6) | NR | NR |
Prior ASCT, n (%) | ||||||
Yes | 9 (13.2) | 10 (25) | 6 (15) | NR | 5 (8.2) | 17 (34.7) |
No | 59 (86.8) | 30 (75) | 34 (85) | NR | 56 (91.8) | NR |
Primary reason for transplantation ineligibility, n (%) | ||||||
Age | 32 (47.1) | 13 (32.5) | 19 (47.5) | NR | NR | NR |
Comorbidities | 9 (13.2) | 1 (2.5) | 1 (2.5) | NR | NR | NR |
Age + comorbidities | 41 (60.3) | NR | NR | NR | NR | NR |
Performance status | NA | 0 | 2 (5.0) | NR | NR | NR |
Insufficient response to salvage therapy | NA | 12 (30.0) | 9 (22.5) | NR | NR | NR |
Chemorefractory patients | 18 (26.5) | NR | NR | NR | NR | NR |
Failed prior transplantation | NR | 10 (25.0) | 6 (15.0) | NR | NR | NR |
Patient refused | 8 (11.8) | 2 (5.0) | 2 (5.0) | NR | NR | NR |
Other | 1 (1.5) | 2 (5.0) | 1 (2.5) | NR | NR | NR |
ABC = activated B-cell; ASCT = autologous stem cell transplant; BR = bendamustine plus rituximab; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group performance status; GCB = germinal centre B-cell; IPI = International Prognostic Index; NA = not applicable; NHL = non-Hodgkin lymphoma; NOS = not otherwise specified; NR = not reported; pola-BR = polatuzumab vedotin plus bedamustine plus rituximab.
Source: MAIC Feasibility Assessment Report.25
Results
Comparison to Lenalidomide Monotherapy
Data for lenalidomide monotherapy were obtained from the DLC-001 trial, which assessed OS, PFS by IRC, DOR by IRC, ORR by IRC, and CRR by IRC. Only median DOR by IRC was available for lenalidomide monotherapy, with the upper bound of the 95% CI not estimable. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Following the population adjustment, the population characteristics of the studies at the treatment arm level were matched successfully.
Table 38 presents the baseline characteristics of the original L-MIND population and the weighted L-MIND population against characteristics of the lenalidomide-monotherapy cohort of the DLC-001 trial. Due to population-matching on the other factors included in the population adjustment, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Compared to the original L-MIND population, the weighted L-MIND population were observed to be ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Patients with history of transformed indolent lymphoma were not included in the weighted L-MIND population.
No concerns were identified by the authors concerning the distribution of the MAIC weights within the L-MIND population.
|||||||||||||| | |||||||||||||| | |||||||||||||| | |
|---|---|---|---|
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
|||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Results of the comparisons using data for tafasitamab plus lenalidomide before and after the population adjustment against the reported data for lenalidomide are provided in Table 39. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| |
|||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| |
|||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| |
|||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| |
|||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Comparison to Pola-BR
Data for pola-BR were obtained from the GO29365 trial, which assessed OS, PFS by IRC, ORR, CRR, and DOR. For PFS by IRC, the authors used an analysis of the GO29365 trial that explicitly censored PFS records of patients who received a subsequent anticancer treatment without a recorded progression event at the time of the last progression assessment available because similar censoring rules were used in the L-MIND study.
|||||||| matching models were investigated. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The base-case model matched populations on all available characteristics that were considered factors that should be prioritized for matching by the sponsor’s clinical experts. Therefore, population adjustment was not carried out for sex, Ann Arbor stage, DOR, or bulky disease at baseline. Despite being an important factor, cell of origin of the disease was not included in the adjustment ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Adjustment on prior lines of therapy was based on the proportion of patients with 1 versus 2 or more prior lines combined.
Two sensitivity analyses were conducted. The first sensitivity analysis used a model that matched populations on all available characteristics that were identified to be relevant by the sponsor’s clinical experts, excluding the cells of origin of DLBCL. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. The resulting ESS (n = 24.8) was satisfactory to the authors. The second was based on the first sensitivity analysis, excluding DOR to prior therapy from the list of matching factors. The ESS (n = 25.49) obtained by this model was satisfactory to the authors.
The authors did not identify any concerns about the distribution of the MAIC weights in the L-MIND population.
Table 40 presents the baseline characteristics of the original L-MIND population and the weighted L-MIND population against reported characteristics for the pola-BR cohort from the GO29365 trial.
Table 40: Baseline Characteristics of the L-MIND Study, the Weighted L-MIND Population, and the GO29365 Trial (Pola-BR)
Detail | L-MIND unweighted | L-MIND weighted | GO29365 (pola-BR) |
|---|---|---|---|
Sample size | 80 | 29.149 | 40.000 |
Age ≥ 65 | 0.712 | 0.575 | 0.575 |
DLBCL histologya | 0.886 | 0.950 | 0.950 |
History of transformed indolent lymphomaa | 0.100 | 0.000 | 0.000 |
ECOG PS 0 or 1a | 0.925 | 0.825 | 0.825 |
IPI score 3 to 5 | 0.500 | 0.550 | 0.550 |
1 prior line of therapya | 0.500 | 0.275 | 0.275 |
Refractory to last prior line of therapya | 0.438 | 0.750 | 0.750 |
Prior ASCTa | 0.112 | 0.250 | 0.250 |
ASCT = autologous stem cell transplant; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group performance status; IPI = International Prognostic Index; pola-BR = polatuzumab plus bendamustine plus rituximab.
aFactors included in the matching.
Source: Sponsor-submitted MAIC Technical Report.21
Indirect comparisons of tafasitamab plus lenalidomide versus pola-BR before and after the MAIC base case are provided in Table 41. No differences were found for OS and PFS by IRC. Relative estimates for DOR by IRC after population adjustment showed a significant treatment effect of tafasitamab plus lenalidomide over pola-BR, but the authors of the MAIC indicated that these results should be interpreted with caution due to the small ESS. No differences were observed for ORR and CRR. The sensitivity analysis produced similar results to the base-case model.
The sponsor performed a visual assessment of the proportional hazard assessment for OS, PFS by IRC, and DOR by IRC. There were some concerns about the proportional hazard assumption in the analyses of OS and PFS by IRC. In both cases, the proportional hazards assumption appeared not to hold, as the tafasitamab plus lenalidomide and pola-BR curves were observed to cross around 4 months. As a result, time-varying HRs were investigated using a splitting time point at 4 months, when the trends in the OS and PFS logarithm of the cumulative hazards were observed to change. Results using a splitting time point at 4 months are presented in Appendix 3 (Table 51).
Table 41: Relative Efficacy Estimates for Tafasitamab plus Lenalidomide Versus Pola-BR (GO29365 Trial)
Outcome | Unadjusted comparison | Population-adjusted comparison |
|---|---|---|
OS, HR (95% CI) [P value] | |||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| |
PFS by IRC, HR (95% CI) [P value] | |||||||||||||||||||||||||||| | |||||||||||||||||||||||||||| |
DOR by IRC, HR (95% CI) [P value] | 0.49 (0.23 to 1.04) [0.062] | 0.34 (0.12 to 0.98) [0.045] |
ORR by IRC, OR (95% CI) [P value] | 0.81 (0.37 to 1.80) [0.607] | 0.68 (0.25 to 1.86) [0.450] |
CRR by IRC, OR (95% CI) [P value] | 0.67 (0.31 to 1.46) [0.309] | 0.74 (0.27 to 2.07) [0.571] |
CI = confidence interval; CRR = complete response rate; DOR = duration of response; IRC = independent review committee; HR = hazard ratio; NE = not estimable; OR = odds ratio; ORR = objective response rate; PFS = progression-free survival.
Note: HR less than 1 favours tafasitamab plus lenalidomide. OR more than 1 favours tafasitamab plus lenalidomide.
Source: Sponsor-submitted MAIC Technical Report.21
Comparison to BR
Three studies reporting data for BR were included in the MAIC analyses: the GO29365 trial of pola-BR versus BR, the Vacirca et al. (2014) study, and the Ohmachi et al. (2013) study. Results of comparisons against the individual studies and pooled results are summarized in Table 45.
MAIC Using GO29365 Trial Data
For the MAIC of tafasitamab plus lenalidomide versus BR using data from the GO29365 trial, ||||||| matching models were investigated. Of the ||||||| matching models investigated, 3 had sufficient ESS above the threshold of 20% of the L-MIND population. In ||||||| of the models, a small number of patients were given extreme weights and were, therefore, very influential on the results. As a result, ||||||| investigated further. The base-case model matched populations on all available characteristics that were considered by the sponsor’s clinical experts as factors that should be prioritized for matching. Therefore, population adjustment was not carried out for sex, Ann Arbor stage, DOR, and bulky disease at baseline. Despite being an important factor, cell of origin of the disease was not included in the adjustment |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Adjustment on prior lines of therapy was based on the proportion of patients with 1 prior line versus 2 or more prior lines combined. No matching models were selected for sensitivity analyses.
No concerns were identified by authors concerning the distribution of the MAIC weights in the L-MIND population for the base case.
Baseline characteristics of the original L-MIND population and the weighted L-MIND population against reported characteristics for the BR arm of the GO29365 trial are presented in Table 42.
Table 42: Baseline Characteristics of the L-MIND Study, the Weighted L-MIND Population, and the GO29365 Trial (BR)
Detail | L-MIND unweighted | L-MIND weighted | GO29365 (BR) |
|---|---|---|---|
Sample size | 80 | 20.866 | 40.000 |
Age ≥ 65a | 0.712 | 0.650 | 0.650 |
DLBCL histologya | 0.886 | 1.000 | 1.000 |
History of transformed indolent lymphomaa | 0.100 | 0.000 | 0.000 |
ECOG PS 0 or 1a | 0.925 | 0.775 | 0.775 |
IPI score 3 to 5 | 0.500 | 0.725 | 0.725 |
1 prior line of therapya | 0.500 | 0.300 | 0.300 |
Refractory to last prior line of therapya | 0.438 | 0.850 | 0.850 |
Prior ASCTa | 0.112 | 0.150 | 0.150 |
ASCT = autologous stem cell transplant; BR = bendamustine plus rituximab; DLBCL = diffuse large B-cell lymphoma; ECOG PS = Eastern Cooperative Oncology Group performance status; IPI = International Prognostic Index.
aFactors included in the matching.
Source: Sponsor-submitted MAIC Technical Report.21
Results of the comparisons are provided in Table 45. Results indicated a statistically significant advantage in favour of tafasitamab plus lenalidomide in OS, PFS by IRC, DOR by IRC, and ORR by IRC against BR, before and after the population adjustment. The authors of the MAIC indicated the DOR by IRC results may be unreliable due to the small ESS supporting the comparison. There was no difference for CRR by IRC.
There were no major concerns about the proportional hazard assumption in the analyses of OS, PFS by IRC, or DOR by IRC. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
MAIC Using Vacirca et al. (2014) Study Data
For the MAIC using data from the Vacirca et al. (2014) study, | matching models were investigated. For the base-case model, populations were matched on all available characteristics that the sponsor’s clinical experts identified as relevant, which included age ≥ 60, proportion of women, ECOG PS of 0 or 1, revised IPI score (very good and good), Ann Arbor stage I to II, 1 prior line of treatment, and prior ASCT. Another matching model, similar to the base case but with increased the granularity of the matching on prior therapy lines and ECOG PS, was used as a sensitivity analysis. The ESS obtained from the sensitivity analysis model was satisfactory according to the authors of the MAIC, and the model converged. Overall, the results of the base case and sensitivity models were aligned.
No concerns were identified concerning the distribution of the MAIC weights in the L-MIND population.
Baseline characteristics in Vacirca et al. (2014) study were reported for 61 patients, and efficacy results were presented for 59 patients. Baseline characteristics of the original L-MIND population and the weighted L-MIND population against reported characteristics for the Vacirca et al. (2014) study are presented in Table 43. Following the population adjustment, the population characteristics of the 2 studies at the treatment arm level were matched successfully.
Table 43: Baseline Characteristics of the L-MIND Study, the Weighted L-MIND Population, and the Vacirca et al. (2014) Study (BR)
Detail | L-MIND unweighted | L-MIND weighted | Vacirca et al. (BR) (2014) |
|---|---|---|---|
Sample size | 80.000 | 67.376 | 61.000 |
Age ≥ 65a | 0.712 | 0.852 | 0.852 |
Proportion of womena | 0.462 | 0.508 | 0.508 |
ECOG PS 0 to 1a | 0.925 | 0.934 | 0.934 |
Revised IPI: very gooda | 0.062 | 0.000 | 0.000 |
Revised IPI: gooda | 0.438 | 0.361 | 0.361 |
Ann Arbor stage I to IIa | 0.250 | 0.098 | 0.098 |
1 prior line of therapya | 0.500 | 0.508 | 0.508 |
Prior ASCTa | 0.112 | 0.082 | 0.082 |
ASCT = autologous stem cell transplant; BR = bendamustine plus rituximab; ECOG PS = Eastern Cooperative Oncology Group performance status; IPI = International Prognostic Index.
aFactors included in the matching.
Source: Sponsor-submitted MAIC Technical Report.21
Results of the comparison analyses are provided in Table 45. OS data were not reported in the Vacirca et al. (2014) study; thus, no comparisons could be made for the end point. Tafasitamab plus lenalidomide significantly prolonged PFS by IRC after the population adjustment. There was no difference for DOR by IRC and ORR. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Patients receiving tafasitamab plus lenalidomide were significantly more likely to achieve CR.
No major concerns were identified with respect to the proportionality of hazards assumption in the analyses of PFS by IRC or DOR by IRC.
MAIC Using Ohmachi et al. (2013) Study Data
For the MAIC using data from the Ohmachi et al. (2013) study, ||||||| matching models were investigated. Three of the models investigated achieved satisfactory ESSs. In the base case, patients were matched on history of transformed indolent lymphoma, 1 prior treatment line, prior ASCT, and refractoriness to prior line of therapy. The other 2 models were used for sensitivity analyses: a model with matched populations on all available characteristics that clinical experts identified as relevant and excluding L-MIND patients with an ECOG PS of 2, and a model based on the base case but not excluding patients aged 75 or older. Overall, the results of the base case and sensitivity models were aligned.
After weighting, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Baseline characteristics of the original L-MIND population and the weighted L-MIND population against reported characteristics for the Ohmachi et al. (2013) study are presented in Table 44. Patients with a history of indolent lymphoma were not included in the analyses.
Table 44: Baseline Characteristics of the L-MIND Study, the Weighted L-MIND Population, and the Ohmachi et al. (2013) Study (BR)
Detail | L-MIND unweighted | L-MIND weighted | Ohmachi et al. (BR) (2013) |
|---|---|---|---|
Sample size | 80.000 | 20.249 | 59.000 |
Age ≥ 65a | 0.712 | 0.627 | 0.627 |
Aged ≥ 75a | 0.388 | 0.000 | 0.000 |
ECOG PS 1a | 0.562 | 0.339 | 0.339 |
ECOG PS 2a | 0.075 | 0.000 | 0.000 |
IPI score 3 to 5 | 0.500 | 0.305 | 0.305 |
History of transformed indolent lymphomaa | 0.100 | 0.000 | 0.000 |
1 prior line of therapya | 0.500 | 0.644 | 0.644 |
Prior ASCTa | 0.112 | 0.136 | 0.136 |
Refractory to last prior line of therapya | 0.438 | 0.136 | 0.136 |
ASCT = autologous stem cell transplant; BR = bendamustine plus rituximab; ECOG PS = Eastern Cooperative Oncology Group performance status; IPI = International Prognostic Index.
aFactors included in the matching.
Source: Sponsor-submitted MAIC Technical Report.21
Results of the comparison analyses are provided in Table 45. There was no difference between groups for PFS by IRC, CRR, and ORR.
No concerns were raised with respect to the proportional hazard assumption on the analyses of PFS by IRC.
Pooled Results
Pooling of the relative efficacy estimates was conducted:
A pooled HR for PFS by IRC using relative efficacy estimates obtained using evidence from the 3 studies was estimated.
A pooled HR for DOR by IRC using relative efficacy estimates obtained using evidence from the GO29365 trial and the Vacirca et al. (2014) study was estimated.
A pooled OR for ORR by IRC using relative efficacy estimates obtained using evidence from all 3 studies was estimated.
A pooled OR for CRR by IRC using relative efficacy estimates obtained using evidence from all 3 studies was estimated.
During the pooling of the population-adjusted estimates of HRs for PFS by IRC, ORR by IRC, and CRR by IRC, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. It is likely that ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Results are presented in Table 45. Using the pooled estimates, tafasitamab plus lenalidomide significantly prolonged PFS by IRC and DOR by IRC compared to BR, and patients receiving tafasitamab plus lenalidomide were significantly more likely to achieve a CR. There was no statistically significant difference between groups for ORR by IRC.
Table 45: Relative Efficacy Estimates for Tafasitamab Plus Lenalidomide Versus BR
Outcome | Unadjusted comparison | Population-adjusted comparison |
|---|---|---|
GO29365 trial | ||
OS, HR (95% CI) [P value] | 0.27 (0.16 to 0.44) [< 0.001] | 0.39 (0.18 to 0.82) [0.014] |
PFS by IRC, HR (95% CI) [P value] | 0.28 (0.18 to 0.44) [< 0.001] | 0.35 (0.18 to 0.71) [0.003] |
DOR by IRC, HR (95% CI) [P value] | 0.20 (0.09 to 0.43) [< 0.001] | 0.15 (0.05 to 0.51) [0.002] |
ORR by IRC, OR (95% CI) [P value] | 4.06 (1.72 to 9.60) [0.001] | 3.40 (1.05 to 11.02) [0.041] |
CRR by IRC, OR (95% CI) [P value] | 2.30 (0.95 to 5.57) [0.066] | 2.36 (0.68 to 8.21) [0.177] |
Vacirca et al. (2014) study | ||
PFS, HR (95% CI) [P value] | 0.32 (0.22 to 0.47) [< 0.001] | 0.35 (0.24 to 0.52) [< 0.001] |
DOR by IRC, HR (95% CI) [P value] | 0.37 (0.19 to 0.73) [0.004] | 0.44 (0.22 to 0.88) [0.019] |
ORR by IRC, OR (95% CI) [P value] | 1.60 (0.81 to 3.19) [0.178] | 1.48 (0.72 to 3.03) [0.281] |
CRR by IRC, OR (95% CI) [P value] | 3.70 (1.58 to 8.68) [0.003] | 3.36 (1.40 to 8.07) [0.007] |
Ohmachi et al. (2013) study | ||
PFS, HR (95% CI) [P value] | 0.72 (0.46 to 1.11) [0.139] | 0.59 (0.31 to 1.15) [0.122] |
ORR by IRC, OR (95% CI) [P value] | 0.80 (0.40 to 1.62) [0.542] | 1.00 (0.35 to 2.85) [0.995] |
CRR by IRC, OR (95% CI) [P value] | 1.12 (0.56 to 2.26) [0.750] | 1.51 (0.51 to 4.46) [0.459] |
Pooled resultsa | ||
PFS, HR (95% CI) [P value] | 0.40 (0.23 to 0.71) [0.002] | 0.39 (0.29 to 0.53) [< 0.001] |
DOR by IRC, HR (95% CI) [P value] | 0.30 (0.23 to 0.41) [< 0.001] | 0.35 (0.25 to 0.50) [< 0.001] |
ORR by IRC, OR (95% CI) [P value] | 1.69 (0.69 to 4.14) [0.252] | 1.59 (0.94 to 2.69) [0.086] |
CRR by IRC, OR (95% CI) [P value] | 2.05 (1.00 to 4.17) [0.049] | 2.43 (1.33 to 4.41) [0.004] |
CI = confidence interval; CRR = complete response rate; DOR = duration of response; IRC = independent review committee; HR = hazard ratio; OR = odds ratio; ORR = objective response rate; OS = overall survival; PFS = progression-free survival.
HR less than 1 favours tafasitamab plus lenalidomide. OR more than 1 favours tafasitamab plus lenalidomide.
aA pooled HR for PFS by IRC using relative efficacy estimates obtained using evidence from the 3 studies was estimated. A pooled HR for DOR by IRC using relative efficacy estimates obtained using evidence from the GO29365 trial and the Vacirca et al. (2014) study was estimated. A pooled OR for ORR by IRC using relative efficacy estimates obtained using evidence from all 3 studies was estimated. A pooled OR for CRR by IRC using relative efficacy estimates obtained using evidence from all 3 studies was estimated.
Source: Sponsor’s MAIC Technical Report.21
Comparison to R-GemOx
The Mounier et al. (2013) study provided data on R-GemOx for the MAIC analyses, which assessed OS, PFS by investigator, DOR by investigator (only median, without 95% CI reported), ORR by investigator, and CRR by investigator.
||||||| matching models were investigated, of which ||||||| did not converge and the ESS was too small. The base-case model achieved an ESS above the pre-specified threshold and included the variables age older than 60 years, ECOG PS 0 or 1, IPI score 3 to 5, GCB immunohistochemistry, 3 or more prior lines of therapy, and prior ASCT. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Table 46 presents the baseline characteristics of the original L-MIND population and the weighted L-MIND population against reported characteristics for the Mounier et al. (2013) study. Baseline characteristics in Mounier et al. (2013) study were reported for 49 patients, and efficacy results were presented for 48 patients. Although a population adjustment was made to exclude patients who received at least 3 prior lines of therapy from the weighted cohort, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. As a result, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In addition, 34.6% of the patients for which outcomes were evaluated were rituximab-naive in the Mounier et al. (2013) study. As no such patients were enrolled in the L-MIND study, no adjustment was possible on this factor. As part of the eligibility criteria of Mounier et al. (2013), no patients older than 75 should have been included in this study. However, as some patients older than 75 were enrolled in Mounier et al. (2013) (the maximum age for enrolled patients was reported to be 77) and |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Results of the comparisons are provided in Table 47. There was no difference between the tafasitamab plus lenalidomide and R-GemOx groups. No concern was raised with respect to the proportional hazards assumption.
Table 46: Baseline Characteristics of the L-MIND Study, the Weighted L-MIND Population, and the Mounier et al. (2013) Study (R-GemOx)
Characteristic | L-MIND unweighted | L-MIND weighted | Mounier et al. (R-GemOx) (2013) |
|---|---|---|---|
Sample size | 80 | 19.604 | 49.000 |
Age ≥ 60a | 0.788 | 0.694 | 0.694 |
ECOG PS 0 or 1a | 0.925 | 0.776 | 0.776 |
IPI score 3 to 5a | 0.500 | 0.694 | 0.694 |
GCB immunohistochemistrya | 0.475 | 0.265 | 0.265 |
Refractory to first line of prior therapy | 0.075 | 0.048 | 0.122 |
Relapsed after first line of prior therapy | 0.425 | 0.331 | 0.735 |
Refractory to second line of prior therapy | 0.312 | 0.365 | 0.000 |
Relapsed after second line of prior therapy | 0.112 | 0.256 | 0.143 |
3 or more lines of prior therapya | 0.075 | 0.000 | 0.000 |
Prior ASCTa | 0.112 | 0.347 | 0.347 |
ASCT = autologous stem cell transplant; ECOG PS = Eastern Cooperative Oncology Group performance status; GCB = germinal centre B-cell; IPI = International Prognostic Index.
aFactors included in the matching.
Source: Sponsor’s MAIC Technical Report.21
Table 47: Relative Efficacy Estimates for Tafasitamab Plus Lenalidomide Versus R-GemOx
Outcome | Unadjusted comparison | Population-adjusted comparison |
|---|---|---|
OS, HR (95% CI) [P value] | 0.54 (0.35 to 0.83) [0.006] | 0.55 (0.28 to 1.06) [0.073] |
PFS by INV, HR (95% CI) [P value] | 0.58 (0.39 to 0.88) [0.010] | 0.59 (0.30 to 1.17) [0.133] |
DOR by INV, ratio of medians | 4.39 | 4.39 |
ORR by INV, OR (95% CI) [P value] | 1.22 (0.57 to 2.58) [0.609] | 1.42 (0.46 to 4.38) [0.543] |
CRR by INV, OR (95% CI) [P value] | 0.73 (0.35 to 1.54) [0.409] | 1.09 (0.34 to 3.54) [0.882] |
CI = confidence interval; CRR = complete response rate; DOR = duration of response; HR = hazard ratio; INV = investigator-assessed; OS = overall survival; OR = odds ratio; ORR = objective response rate; PFS = progression-free survival; R-GemOx = rituximab plus gemcitabine plus oxaliplatin.
Note: HR less than 1 favours tafasitamab plus lenalidomide. OR more than 1 favours tafasitamab plus lenalidomide.
Source: Sponsor’s MAIC Technical Report.21
Critical Appraisal of the MAICs
Since the pivotal L-MIND study was single-arm, an unanchored MAIC was an appropriate method for indirect comparison. In general, the methods used to conduct the MAICs followed technical guidance.22 The literature search conducted by the authors of the MAIC to identify relevant studies was appropriate. A feasibility assessment was conducted before performing the MAICs, which is appropriate. In this feasibility assessment, the comparator studies were thoroughly assessed for comparability to the L-MIND study, and potential sources of heterogeneity were adequately detailed. Study selection criteria , which is a potential source of selection bias. Otherwise, study selection methods were appropriate. Appropriate databases were sea|||||||||||||||||||||||||||||||||||rched, and duplicate reviewers were used to select relevant studies for inclusion in the MAICs. Similarities between the L-MIND study and MAIC comparator studies were noted. All included studies were phase II trials, and the primary end point was ORR. In addition, all studies were open-label. It was noted that the sample size of all comparator studies (ranging from N = 40 to N = 63) was smaller than that of L-MIND (N = 80 in the FAS).
The quality assessments used to assess the included studies were appropriate, ||||||||||||||||||||||||||||||||||||||||||. The GO29365 study was high quality, but the quality of all other studies assessed was considered low, which is a limitation of the available evidence, although the direction of any bias is unknown. In the comparison using the data reported in Vacirca et al. (2014), the authors reported |||||||||||||||||||||||||||||||||||||||||||||||||. The authors indicated that the |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
For comparisons in unanchored MAICs to be unbiased, matching on all known and available effect modifiers and prognostic factors is required. The list of effect modifiers and confounders provided by the sponsor was comprehensive, although the clinical experts consulted by CADTH identified 2 additional variables that could have been used for matching (presence of bulky disease and prior use of radiation). The methods used by the sponsor to identify effect modifiers and prognostic factors were credible and appropriate. Although the sponsor likely identified the majority of known effect modifiers and prognostic factors, matching of the baseline patient characteristics between trials was limited by lack of data availability in L-MIND and the published comparator studies. Multiple matching models were investigated for each comparison. Due to lack of data availability and overlap in population characteristics, a relatively small number of variables were used in the base-case matching models for each comparison to have an adequate ESS. Sensitivity analyses with an increased number of variables used for matching were performed for some MAICs to increase the internal validity of the results, but these models also did not include all important effect modifiers and prognostic factors. Exclusion of important confounders in the matching introduces bias and creates substantial uncertainty in the results because residual bias could have affected the relative efficacy estimates. Unanchored forms of population-adjusted indirect comparisons make the much stronger assumption of “conditional constancy of absolute effects.”22 This means that the absolute treatment effects are assumed to be constant at any given level of the effect modifiers and prognostic variables, and all effect modifiers and prognostic variables must be known. This assumption is unlikely to have been met in the sponsor-submitted unanchored MAICs, especially since not all known effect modifiers and prognostic factors could be included in the models. Therefore, no conclusions can be made from the data.
Specifically, in the MAIC of tafasitamab plus lenalidomide versus BR based on the Vacirca et al. (2014) study, some important factors, such as the cell of origin of the disease and patient refractoriness, were not reported and, therefore, could not be matched. Regarding the MAIC of tafasitamab plus lenalidomide versus lenalidomide monotherapy, few baseline characteristics were reported for the DLC-001 trial. As a result, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In addition, cell of origin for DLBCL could not be included in the population adjustment due to the extent of the missing data. In the MAIC of tafasitamab plus lenalidomide versus pola-BR, no adjustment could be made on patients’ cell of origin due to missing data in the L-MIND study, despite cell of origin being an important prognostic factor and effect modifier of DLBCL. The population adjustment for the comparison of tafasitamab plus lenalidomide and R-GemOx was incomplete. Due to the poor overlap of L-MIND and Mounier et al. (2013), ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In addition, 34.6% of the patients for whom outcomes were evaluated were rituximab-naive in Mounier et al. (2013) As no such patients were enrolled in the L-MIND study, no adjustment on this factor was possible. Populations were still unbalanced with respect to their number of prior therapy lines and refractory status after matching. As a result of these limitations in the matching that could be conducted, the results of the MAICs might be confounded by unobserved imbalances in these important prognostic factors or treatment effect modifiers. The direction of the residual bias is unknown.
In addition, the definition of some key baseline characteristics, such as patient refractoriness, could not be found in the Ohmachi et al. (2013) and in the Mounier et al. (2013) studies. It was assumed that these definitions were comparable to the definition used in the L-MIND study when performing the population adjustment. As this assumption may not be correct, this represents a source of potential bias. The direction of bias is unknown.
Furthermore, an MAIC can adjust only for heterogeneity that is directly related to differences in baseline patient characteristics. Other sources of heterogeneity, such as those related to differences in study design, definitions of study outcomes, or changes in the management of patients over time, cannot be adjusted for. Many of the sources of heterogeneity identified by the authors of MAIC feasibility assessment could not be accounted for in the MAIC analyses conducted, particularly differences in study design, definitions of end points, and timing of outcome assessments.
With respect to definitions of end points, different versions of the IWG response criteria were used across studies to assess disease response. The L-MIND study, the Vacirca et al. (2014) study, and the Ohmachi et al. (2013) study used the version of the IWG response criteria defined in Cheson et al. (2007).8 As a result, the surrogate outcomes investigated in these 3 studies are expected to be comparable in that respect. However, the DLC-001 trial and the Mounier et al. (2013) study used the earlier version of the IWG response criteria presented in Cheson et al. (1999),34 and the GO29365 trial used revised Lugano 2014 criteria.33 As the updates in the IWG response criteria were motivated by technological advances in imaging and monitoring of DLBCL, the later version of the criteria may better capture the disease response to treatment. This implies that the surrogate outcomes may not be entirely comparable in the analyses of L-MIND against the GO29365 trial, DLC-001 trials, and the Mounier et al. (2013) study. The extent and direction of the potential bias to the MAIC results caused by differences in response criteria could not be determined.
Differences in the definitions of study end points were identified as well. The definitions of PFS and of censoring rules differed between the L-MIND study and the GO29365 trial, and the definitions of PFS and of the censoring rules might have differed as well between the L-MIND study and other comparator studies. In L-MIND, PFS was defined as the time between first study drug administration and tumour progression or death from any cause, whichever occurs first. PFS was defined as time from randomization to the first documented disease progression or death due to any cause in the DLC-001 trial, and a similar definition seems to have been used in the GO29365 trial. In the absence of an accurate definition for these outcomes in comparator trials, the authors of the MAIC assumed that the definition used in the L-MIND and remaining comparator studies were similar, which may not be true. As a result, the comparison of PFS is associated with significant limitations. Definitions of OS also differed. In L-MIND, OS was defined as time elapsed between treatment initiation and death. In the DLC-001 and GO29365 trials, OS was defined as time from randomization until death of any cause. This could have an impact on the comparability of the time-to-event outcomes if there was a large period between randomization and initiation of therapy. Further adding to the uncertainty of the OS results, median duration of follow-up differed across studies.
In addition, ORR and CRR were evaluated at the end of the 4-cycle induction therapy planned in the Mounier et al. (2013) study, rather than as best ORRs. As a result, the comparison against best overall responses obtained from the L-MIND study could be biased, as response rates could include patients who would have lost response if responses were assessed after 4 cycles of treatment.
Heterogeneity in the timing of outcome assessments was noted. As the schedule of the initial disease assessment differed between the L-MIND study and the GO29365 trial, assessment-time bias may have affected the results. Assessment-time bias can arise in comparing surrogate outcomes, such as PFS, when data from 2 treatment arms investigated using different schedules of assessment for tumour progression are compared. Due to the differences in study designs, disease progression could be detected earlier in the treatment arm for which an earlier disease assessment schedule was planned. In the case of the comparison of the L-MIND study and the GO29365 trial, such assessment-time bias could exist, since the initial disease assessment was performed after 3 cycles in L-MIND but after 4 cycles in the GO29365 trial. This bias may have affected the results of the MAICs comparing tafasitamab plus lenalidomide to pola-BR and BR. Similarly, assessment-time bias could favour L-MIND in the comparison against the Ohmachi et al. (2013) study, since the initial assessment was performed in cycle 2 in the latter study but in cycle 3 in the L-MIND study. In the Mounier et al. (2013) study, disease monitoring seems to have been performed at the end of the 4 cycles of induction R-GemOx therapy, and then at the EOT, rather than at regular cycles, as in the L-MIND study. This could lead to assessment-time bias.
There were also important differences in study eligibility criteria that could not be accounted for in the matching. All comparator studies, except for the GO29365 trial, enrolled only patients with a life expectancy of 3 months or more, whereas the L-MIND study could enrol patients irrespective of their life expectancy. As a result, frailer patients could have been enrolled to the L-MIND study, which would bias results against tafasitamab plus lenalidomide. No adjustment could be made to correct this potential source of bias. In addition, the Mounier et al. (2013) study specifically excluded patients older than 75 years or who had relapsed following more than 2 prior therapy lines, which could bias results against tafasitamab plus lenalidomide. In the Mounier et al. (2013) study, matching was based on age 60 years or older and 3 or more lines of prior therapy. The Ohmachi et al. (2013) study also excluded patients with failure to achieve CR, CR unconfirmed, or PR in any prior treatment, which was not an exclusion criterion in L-MIND. These differences in eligibility criteria indicate heterogeneity between the study patient populations that could not be fully accounted for with the methods used in the MAIC analyses.
Another source of heterogeneity was that treatment duration differed across studies, which could not be accounted for in the MAICs. In the L-MIND trial, combination treatment was given for 12 cycles, followed by tafasitamab monotherapy until progression, death, or withdrawal. Treatment duration of lenalidomide monotherapy was similar in the DLC-001 trial. In contrast, treatment duration was 6 cycles in the GO29365, Ohmachi et al. (2013), and Vacirca et al. (2014) studies, and 4 cycles in the Mounier et al. (2013) study.
The ESS achieved after the population adjustment was substantially smaller than the L-MIND original sample size in the comparisons versus BR (using the GO29365 trial and the Ohmachi et al. [2013] study) as well as versus R-GemOx (using the Mounier et al. [2013] study). This suggests there was likely significant heterogeneity between the L-MIND study and comparator studies. The results of comparisons with major reductions of ESS indicate that the weights are highly variable due to a lack of population overlap, and that the resulting estimate may not be reliable. Furthermore, the small ESS reduced the statistical power with which inference could be made by increasing the overall uncertainty of the results, because small ESSs lead to substantial losses in precision. Regarding the MAIC of tafasitamab plus lenalidomide versus pola-BR, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
In some comparisons, large balancing weights were attributed to only a small number of patients. Therefore, these patients would have a disproportionate influence on the MAIC results. In the MAIC of tafasitamab plus lenalidomide to R-GemOx, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. It is expected that ||||||||||||||||||||||||||||||||||| influenced the results of the MAIC and creates uncertainty in the results. In the MAIC to BR using the Ohmachi et al. (2013) study data, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. When the distribution of balancing weights is skewed |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| results are uncertain.
The authors of the MAICs noted limitations regarding the DOR analyses. Regarding the MAIC of tafasitamab plus lenalidomide versus lenalidomide monotherapy, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Consequently, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Another limitation of the MAIC of tafasitamab plus lenalidomide versus pola-BR was that there was evidence of violation of the proportional hazards assumption for the analyses of OS and PFS; therefore, the results are unreliable. Similarly, there was potential violation of the proportional hazard assumption for the BR comparison using the GO29365 trial; therefore, results may not be reliable.
The limitations of the unanchored MAICs highlighted previously and multiple sources of heterogeneity that could not be accounted for in the analyses all may have affected the comparisons of tafasitamab plus lenalidomide to other active therapies in the analyses and introduced bias in the results. Given these issues, there is substantial concern for risk of bias in the MAIC results. The CADTH review team can draw no definitive conclusions on the relative efficacy of tafasitamab plus lenalidomide.
No HRQoL or safety outcomes were assessed in the MAICs, which represent gaps in the evidence.
Regarding external validity, the clinical experts consulted by CADTH indicated that lenalidomide monotherapy is not used to treat patients with R/R DLBCL in Canada. According to the clinical experts, although BR and R-GemOx were considered relevant comparators for this review because there is no standard treatment approach for patients with R/R DLBCL who are not eligible for intensive therapies, these treatment regimens are not commonly used in Canada. Other chemotherapy regimens are used more frequently. Although there is no standard of care treatment for patients with R/R DLBCL ineligible for ASCT in Canada, the clinical experts indicated that pola-BR would be the most relevant comparator for this review. Only the GO29365 trial included study sites in Canada. In the other studies, sites were located in the US, Europe, Australia, and Asia. In addition, the Ohmachi et al. (2013), Vacirca et al. (2014), and Mounier et al. (2013) studies enrolled patients more than a decade ago. Enrolment years were not reported for the DLC-001 trial. Differences in the patient populations and clinical practice related to the temporal differences between studies may also limit generalizability. Furthermore, results of the MAICs can be generalized only to patients similar to those enrolled in the comparator trials. In some of the comparator trials, there were eligibility criteria that could limit generalizability. For example, the Ohmachi et al. (2013) study limited enrolment to patients with an ECOG PS of 0 or 1, which is likely healthier than many patients who may be treated in clinical practice. Many of the comparator trials also excluded patients with history of transformation from indolent lymphoma or follicular lymphoma.
Other Relevant Evidence
No additional relevant studies were included in the sponsor’s submission to CADTH.
Discussion
Summary of Available Evidence
One open-label, single-arm phase II trial (L-MIND, N = 81) was included in the CADTH systematic review. The primary objective of the L-MIND study was to determine the activity of tafasitamab plus lenalidomide in terms of ORR. The trial included adult patients with DLBCL who had relapsed after or were refractory to 1 to 3 previous systemic regimens (with at least 1 anti-CD20 therapy), who were not candidates for HDC and subsequent ASCT. Patients received IV tafasitamab (12 mg/kg) and oral lenalidomide (25 mg per day for days 1 to 21) for up to 12 cycles (28 days each), followed by tafasitamab monotherapy in patients with stable disease or better until disease progression. The primary end point was ORR. Other efficacy outcomes assessed in the L-MIND study included OS, PFS, TTP, EFS, CRR, DOR, TTR, and TTNT. Harms outcomes were also examined.
In the L-MIND study, the mean age of patients was 69.3 years. Most patients were White (88.9%), had Ann Arbor stage III or IV disease (75.3%), and did not have a prior ASCT (88.9%). Overall, 54.3% of enrolled patients were male, 55.6% had an ECOG PS of 1, 50.6% had an IPI score of 3 to 5, and 46.9% had disease of GCB cell origin by immunohistochemistry. Mean time since first DLBCL diagnosis was 39.6 months. All (100%) patients had 1 or more prior anticancer medication, 50.6% of patients had received 2 prior or more therapy lines, and 44.4% were refractory to their most recent previous therapy. The most common reasons for ASCT ineligibility were older age (46.3%) and chemorefractory status (22.5%).
Since the primary clinical review of tafasitamab plus lenalidomide consisted of a single-arm trial, a review of the available indirect evidence was conducted. Three sponsor-submitted ITCs were summarized and critically appraised in this review: 2 retrospective observational studies conducted to generate external controls for indirect comparison to the L-MIND study using ePS-based NN 1:1 matching methodology (RE-MIND and RE-MIND2), and 1 ITC that was performed using unanchored MAICs. RE-MIND was designed to characterize the effectiveness of lenalidomide monotherapy in the treatment of R/R DLBCL patients not eligible for HDC followed by ASCT compared to tafasitamab plus lenalidomide. RE-MIND2 was designed to characterize the effectiveness of systemically administered therapies in the treatment of R/R DLBCL (second, third, and fourth line), which included pooled systemic therapies (i.e., any systemic therapy listed in the NCCN or ESMO guidelines for patients ineligible for ASCT), BR, R-GemOx, CAR T-cell therapy, and pola-BR. The unanchored MAICs compared tafasitamab plus lenalidomide to lenalidomide monotherapy, pola-BR, BR, and R-GemOx.
No long-term extension studies or additional relevant studies were included in the sponsor’s submission to CADTH.
Interpretation of Results
Efficacy
Patients with R/R DLBCL who are not eligible for ASCT have limited treatment options and poor outcomes. Both the clinical experts and patient groups indicated that there is significant unmet need in this patient population. According to the clinical experts consulted by CADTH, there is no standard of care for in patients with R/R DLBCL who are ineligible for ASCT.
The L-MIND trial investigated tafasitamab plus lenalidomide in adult patients with R/R DLBCL who are not candidates for HDC and subsequent ASCT. For the primary end point, the L-MIND trial aimed to show an improvement in ORR by IRC assessment from 20% to 35%. The primary end point of the study was met at the primary analysis, since the ORR observed was 60.0% (95% CI, 48.4 to 70.8). In the updated efficacy analyses with longer follow-up, the results were generally consistent with the primary analysis of the study. The DOR results indicated that response was durable. The reported median DOR was longer than the clinical experts expected. Notably, the long DOR in responders was driven by patients who had achieved a CR, according to the clinical experts consulted by CADTH and the sponsor’s assessment. Median DOR in patients who achieved PR was more modest. Other time-to-event end points, such as PFS, TTP, TTNT, and OS, were consistent with the ORR and DOR results in suggesting a benefit from tafasitamab plus lenalidomide treatment.
Multiple subgroup analyses that aligned with subgroups of interest in the CADTH review protocol were conducted in the L-MIND study. Most of the subgroups were pre-specified in the L-MIND study protocol. Results were broadly numerically consistent across various subgroups of interest. However, because of the small sample sizes and a lack of statistical testing, the subgroup analysis results were considered exploratory, and the CADTH review team could draw no definitive conclusions.
Patients were enrolled in the L-MIND study with the diagnosis of DLBCL by local investigator assessment. Approximately 10% of patients in the L-MIND FAS did not have DLBCL by central pathologic assessment. Inclusion of patients with alternative diagnoses resulted in selection bias and creates uncertainty in the results. However, sensitivity analyses were conducted on patients with DLBCL confirmed by central pathology and, in general, estimates for efficacy end points were numerically comparable between patients with DLBCL by investigator diagnosis (i.e., the FAS) and patients with DLBCL by central pathologic analysis.
The clinical experts consulted by CADTH indicated that the median OS and median PFS reported in the L-MIND trial were longer than expected in patients with R/R DLBCL who are ineligible for ASCT. The clinical experts also noted a large numerical difference between median PFS and median OS, which is not expected in patients with R/R DLBCL. However, they also noted that TTNT and EFS results were consistent with the PFS results. Furthermore, the clinical experts indicated that the flattening of the Kaplan–Meier curves for OS and PFS observed in L-MIND is not expected for treatments in patients with R/R DLBCL who are ineligible for ASCT. The clinical experts thought that the results, especially for OS and PFS, may have been confounded by factors such as subsequent anticancer therapies and the fact that some patients enrolled in L-MIND were found to not have a diagnosis of DLBCL by central pathologic analysis. The clinical experts indicated that the DOR data suggested that patients who achieve CR may experience more benefit, whereas those who achieve PR may experience less benefit. However, it was noted that it is not possible to identify which patients will achieve CR before initiating treatment with tafasitamab plus lenalidomide. The clinical experts indicated that the median time to CR was greater than expected.
The primary limitations of the L-MIND trial that affect the interpretation of results were the absence of a comparator arm and statistical hypothesis testing. Due to these limitations of the study design, the CADTH review team could draw no definitive conclusions from the L-MIND study regarding the efficacy and safety of tafasitamab plus lenalidomide relative to a comparator. In addition, the L-MIND trial is open-label. The open-label design can increase the risk of performance and detection bias, particularly outcomes that are subjective in measurement and interpretation (e.g., response or AEs). Objective outcomes such as OS time are unlikely to be affected by performance or detection bias. The potential for detection bias was minimized by using IRC assessment for key study outcomes such as ORR, DOR, and PFS. Furthermore, the IRC and investigator-assessed outcome results were generally consistent.
Another limitation of the L-MIND study is that it is a phase II trial with a relatively small sample size. In general, the clinical experts thought that it was difficult to make reliable extrapolations to the larger R/R DLBCL population in Canada based on 80 patients, especially when there are the confounding factors of subsequent anticancer therapies and 10% of patients enrolled in L-MIND were found not to have DLBCL by central pathologic analysis. The clinical experts indicated that the results demonstrated that treatment with tafasitamab plus lenalidomide appears to confer a clinical benefit. However, the magnitude of the benefit is unknown and the relationship between the treatment and the outcomes is uncertain, given the lack of direct comparative evidence. The clinical experts thought that results of the L-MIND study would need to be reproduced in a phase III trial in order for them to have confidence in the results.
Tafasitamab received a Notice of Compliance with Conditions, pending the results of trials to verify its clinical benefit. Authorization was based on ORR, CRR, and durability of response from the L-MIND study.7 Per the Health Canada product monograph, an improvement in PFS or OS has not been established.7 Per the Letter of Undertaking included in the sponsor’s submission to CADTH, the planned confirmatory study is a phase III, randomized, double-blind, placebo-controlled trial comparing the efficacy and safety of tafasitamab plus lenalidomide in addition to R-CHOP versus R-CHOP alone in previously untreated, high-intermediate, and patients at high risk with newly diagnosed DLBCL (the frontMIND study), which is different than the indication under review.15,24 According to the sponsor’s response to CADTH’s request for additional information, there are no other current or planned trials for the submitted regimen besides L-MIND in the R/R DLBCL population (i.e., in line with the submitted indication).15
In the RE-MIND and RE-MIND2 studies, ePS-based NN 1:1 matching methodology was used to compare patients treated with tafasitamab plus lenalidomide in L-MIND to patients retrospectively enrolled in observational cohorts. Overall, matching was successful using the selected covariates. Both the RE-MIND and RE-MIND2 studies met their primary end points. In RE-MIND, ORR was greater in the tafasitamab plus lenalidomide cohort than in the lenalidomide-monotherapy cohort. Regarding the primary end point of RE-MIND2, patients in the tafasitamab plus lenalidomide cohort showed an improvement in OS compared to the cohorts of systemic therapies pooled, BR, and R-GemOx. In RE-MIND2, the pre-specified analysis could not be conducted for patients treated with pola-BR and CAR T-cell therapies because of the small number of identified patients in the observational cohort.
In RE-MIND, the results of the secondary end point analyses were consistent with the primary end point in suggesting a benefit of tafasitamab plus lenalidomide compared to lenalidomide monotherapy. Results showed a statistically significant benefit in favour of tafasitamab plus lenalidomide for CRR, OS, PFS, and EFS; there was no difference in median DOR and TTNT. Furthermore, sensitivity analyses were consistent with the primary analysis. In RE-MIND2, analyses of the secondary end points similarly supported the results of the primary end point analysis, suggesting a benefit of tafasitamab plus lenalidomide compared to systemic therapies pooled, BR, and R-GemOx. A statistically significant improvement in PFS, EFS, and TTNT was observed in the tafasitamab plus lenalidomide cohort compared with the cohorts of systemic therapies pooled, BR, and R-GemOx. However, the sponsor reported that the ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. ORR was higher in the tafasitamab plus lenalidomide cohort than in the cohorts of systemic therapies pooled and R-GemOx; no significant differences were observed for ORR in the tafasitamab plus lenalidomide cohort compared to the BR cohort. CRR was higher in the tafasitamab plus lenalidomide cohort than in the cohort of systemic therapies pooled; no significant differences were observed for CRR in the tafasitamab plus lenalidomide cohort compared to the BR and R-GemOx cohorts.
Only post hoc, exploratory analyses were conducted to compare tafasitamab plus lenalidomide to pola-BR and CAR T-cell therapy in RE-MIND2. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.There were no significant differences in median OS in the tafasitamab plus lenalidomide cohort compared to the pola-BR cohort. Similarly, there were no significant differences in median OS in the tafasitamab plus lenalidomide cohort compared to the CAR T-cell therapy cohort. Since only exploratory post hoc analyses were conducted on a small number of patients, the CADTH review team could draw no definitive conclusions regarding whether there is a benefit of tafasitamab plus lenalidomide compared to pola-BR or CAR T-cell therapy based on the RE-MIND2 data.
Although multiple measures were implemented to minimize bias in RE-MIND and RE-MIND2, there were important sources of heterogeneity between the L-MIND cohort and observational cohorts that could not be accounted for in the methods used. Although the eligibility criteria for enrolment in RE-MIND and RE-MIND2 were based on the eligibility criteria used in the L-MIND trial, differences were noted, many of which were related to the retrospective nature of RE-MIND and RE-MIND2 studies compared to the L-MIND study’s prospective, interventional nature. As a result, patients enrolled in RE-MIND and RE-MIND2 may not have been eligible for participation in L-MIND, thus limiting the comparability of the study populations. Comparison of data from a prospective, interventional trial to retrospective, observational studies using real-world data may be problematic, as a number of notable differences in data collection, outcomes, and assessments were noted. Differences in response assessment frequency or failure to capture response to therapy in daily clinical practice are sources of bias in the RE-MIND and RE-MIND2 studies. Patients in the tafasitamab plus lenalidomide cohort used for RE-MIND and RE-MIND2 were treated under the L-MIND clinical trial protocol, and therefore prospectively followed with a defined tumour assessment scheme and defined response criteria. Patients in the observational cohorts of RE-MIND and RE-MIND2 were treated and followed in real-world clinical practice without an overarching protocol, thus leading to heterogeneity with regard to tumour assessment frequency and criteria. In addition, imaging modalities used to assess response and the definitions of response used for patients in the observational cohorts did not always match the criteria used in the L-MIND trial. Last, and most important, unmeasured confounding factors not accounted for in the matching may have affected the results. The RE-MIND and RE-MIND2 studies used 9 clinically important covariates for matching in their main analyses: age, Ann Arbor stage, refractoriness to last therapy line, number of previous lines of therapy, history of primary refractoriness, prior ASCT, neutropenia, anemia, and elevated LDH. Although these variables are clinically important, other known confounders were not accounted for in the matching in the main analyses (e.g., ECOG PS, IPI score, cell of origin), and there are likely other unknown confounders that could not be accounted for. As a result of these sources of heterogeneity that could not be accounted for, there is substantial risk of bias in the RE-MIND and RE-MIND2 study results.
There are also limitations to the external validity of the RE-MIND and RE-MIND2 studies. The RE-MIND study has limited external validity because lenalidomide monotherapy is not used as a treatment for R/R DLBCL in Canada. The clinical experts consulted by CADTH reported that lenalidomide monotherapy is not commonly used because it is not considered effective, in their clinical opinion. As a result, the results of RE-MIND may not be relevant for decision-making. Regarding external validity of RE-MIND2, the clinical experts consulted by CADTH indicated that R-GemOx and BR are not very commonly used to treat patients with R/R DLBCL in Canada, although they are considered relevant comparators in the CADTH systematic review protocol. There is no standard of care for patients with R/R DLBCL who are not eligible for intensive therapies; therefore, many different treatment options are used. The clinical experts indicated that pola-BR would be the most relevant comparator in terms of evidence for decision-making. Although pola-BR is not funded at the time of review, it has received a recommendation for reimbursement from CADTH.39 The clinical experts noted that the relevance of CAR T-cell therapy as a comparator for tafasitamab plus lenalidomide in patients who are not eligible for ASCT was debatable. The clinical experts considered CAR T-cell therapy to be an intensive therapy and thus more similar to ASCT. The clinical experts indicated that they would not consider using tafasitamab plus lenalidomide in patients who were eligible for CAR T-cell therapy. There are also concerns about whether the systemic therapies pooled cohort adequately reflects current contemporary practice and therapies.
In addition to the RE-MIND and RE-MIND2 studies, the sponsor submitted unanchored MAICs of tafasitamab plus lenalidomide versus lenalidomide monotherapy, pola-BR, BR, and R-GemOx. The outcomes OS, PFS, ORR, DOR, and CRR were assessed. Results favouring tafasitamab plus lenalidomide compared to lenalidomide monotherapy were observed for OS, PFS, and CRR, and no differences were observed for DOR and ORR. For the comparisons of tafasitamab plus lenalidomide to pola-BR, the MAIC for OS, PFS by IRC, ORR, and CRR showed no differences. Overall, the comparisons to BR favoured tafasitamab plus lenalidomide, although some results were not statistically significant and thus indicated no differences. Last, in the MAIC of tafasitamab plus lenalidomide versus R-GemOx, a numerical advantage in favour of tafasitamab plus lenalidomide was observed for all outcomes assessed. Although the methods used to conduct the MAICs followed technical guidance, the analyses have a number of limitations that affect the internal and external validity. Most important, not all known effect modifiers and prognostic factors identified by the authors could be used for matching due to the lack of availability of data in L-MIND and the comparator studies. Furthermore, the quality of most of the comparator studies was low. Last, multiple other sources of heterogeneity (e.g., study design, eligibility criteria, study end point definitions, timing of tumour assessments, response criteria used) were identified that could not be accounted for in the analyses conducted. Given these issues, there is substantial concern about the risk of bias in the MAIC results. There are also limitations to the external validity of some of the comparators (i.e., lenalidomide monotherapy, BR, and R-GemOx), as described earlier. In addition, the results can be generalized only to patients who are similar to those enrolled in the comparator studies. Some of the comparator studies had eligibility criteria (e.g., ECOG PS 0 or 1) that may limit generalizability to patients with R/R DLBCL typically seen in practice in Canada.
Patients indicated that they want treatments that result in longer survival, remission, control of disease symptoms, and better quality of life compared to current therapies. The L-MIND study and indirect comparative evidence assessed the outcomes OS, PFS, ORR, and CRR, which align with some of the outcomes that are important to patients. Due to the limitations of the L-MIND study design and indirect comparative evidence, it is unknown whether treatment with tafasitamab plus lenalidomide confers a benefit in OS and PFS. ORR and CRR results from the L-MIND study suggest that tafasitamab plus lenalidomide is effective in patients with R/R DLBCL who are ineligible for ASCT, although the relative magnitude of the treatment benefit compared to other therapies is unknown. HRQoL and symptoms were not assessed in the L-MIND trial or sponsor-submitted ITCs, which is an important gap in the evidence.
Harms
The patient groups that provided input for this review indicated that they want treatments that have fewer side effects than current therapies. Similarly, the clinical experts consulted by CADTH reported that the goal of treatment in patients with R/R DLBCL who are not eligible for intensive therapies (i.e., ASCT and CAR T-cell therapy) is to control symptoms with minimal toxicity to improve quality of life, and that the tolerability of treatment is important in this patient population.
The L-MIND trial assessed the safety of tafasitamab plus lenalidomide. All study patients experienced at least 1 treatment-emergent AE. The most frequently reported AE was neutropenia. Other frequently reported AEs included anemia, diarrhea, thrombocytopenia, and cough. Overall, 24.7% of patients permanently discontinued 1 or both study drugs because of AEs. The incidence of SAEs in L-MIND was 53.1%. The most frequently reported SAEs were pneumonia, febrile neutropenia, and pulmonary embolism. According to the clinical experts consulted by CADTH, the safety data from the L-MIND trial were what they expected, based on the known safety profiles of tafasitamab and lenalidomide. Overall, 51.9% of patients had died as of the most recent analysis |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Tafasitamab has serious warning and precautions in the Health Canada product monograph for infection, myelosuppression, PML, and hepatitis B reactivation, which were included as notable harms in the CADTH systematic review protocol. Most (72.8%) patients enrolled in L-MIND experienced an infection. The most common types of infection were bronchitis, pneumonia, and urinary tract infection. Regarding myelosuppression, the most frequently reported AEs were neutropenia, anemia, and thrombocytopenia, which the clinical experts expected. One patient experienced worsening PML, and 2 patients experienced hepatitis B reactivation. Other notable harms specified in the CADTH review protocol included cytokine release syndrome and tumour lysis syndrome, and no patients in L-MIND experienced grade 3 or higher cytokine release syndrome or tumour lysis syndrome (although it is unclear whether any patients experienced cytokine release syndrome or tumour lysis syndrome lesser than grade 3). Five patients experienced infusion-related reactions, which is a notable harm that the clinical experts indicated is important to treating clinicians.
The L-MIND trial is open-label, which may have affected the reporting of AEs. There is a high risk of performance and detection bias for known subjective harms, which could be overestimated, since both patients and their treating clinicians knew the treatment received and that they were participating in a trial. Objective outcomes such as mortality are unlikely to be affected by performance or detection bias.
Regarding the available comparative evidence, the RE-MIND2 study assessed safety using duration of exposure to study treatment and treatment discontinuations due to AEs. Tafasitamab plus lenalidomide were compared to systemic therapies pooled, BR, and R-GemOx. Median duration of exposure was numerically longer in the tafasitamab plus lenalidomide cohorts compared to systemic therapies pooled, BR, and R-GemOx. A numerically greater proportion of patients in the tafasitamab plus lenalidomide cohort discontinued treatment due to AEs compared to those in the systemic therapies pooled, BR, and R-GemOx cohorts. However, there are limitations to comparing safety data recorded during routine clinical care with data that were collected during the prospective L-MIND study. Safety was not assessed in the RE-MIND study or in the MAICs. Due to the limited comparative safety data available, the CADTH review team can draw no conclusions regarding the relative safety of tafasitamab plus lenalidomide to relevant comparators.
Conclusions
One phase II, single-arm, open-label trial (L-MIND) of tafasitamab plus lenalidomide in patients with R/R DLBCL was included in the systematic review conducted by CADTH. The L-MIND trial data were analyzed descriptively; no statistical hypotheses were tested. According to the clinical experts consulted by CADTH, the results suggested that tafasitamab plus lenalidomide therapy is clinically effective in this patient population and there may be a beneficial effect of tafasitamab plus lenalidomide on OS, PFS, ORR, DOR, and other efficacy outcomes. However, there is significant uncertainty because the trial is phase II, has an open-label single-arm design, and has a small sample size. Due to the absence of a comparator arm and statistical testing, the CADTH review team can draw no definitive conclusions regarding the efficacy of tafasitamab plus lenalidomide based on the L-MIND trial. HRQoL was not assessed in the L-MIND trial, which represents an important gap in the evidence. All study patients reported treatment-emergent AEs, the most common of which was neutropenia, and more than half reported SAEs. The most frequently reported SAEs were pneumonia, febrile neutropenia, and pulmonary embolism. The most common cause of death was disease progression.
No direct evidence on the relative efficacy and safety of tafasitamab plus lenalidomide versus other therapies was identified. Results of the ITCs submitted by the sponsor suggested that tafasitamab plus lenalidomide therapy may be associated with an improvement in clinical outcomes (e.g., ORR, CRR, OS, PFS, EFS, DOR, and TTNT) compared to lenalidomide monotherapy, systemic therapies pooled, BR, R-GemOx, pola-BR, and CAR T-cell therapies. However, the ITCs were associated with substantial risk of bias due to important limitations, including methodological limitations, heterogeneity, matching based on a limited number of variables, and small sample sizes. In view of the uncertainty in the ITC results, the CADTH review team can draw no conclusions on the efficacy of tafasitamab plus lenalidomide compared to other therapies used to treat patients with R/R DLBCL who are ineligible for ASCT. Harms outcomes were assessed in 1 ITC (RE-MIND2). The ITC results showed that a numerically greater proportion of patients treated with tafasitamab plus lenalidomide may discontinue treatment due to AEs compared to systemic therapies pooled, BR, and R-GemOx; however, there were limitations associated with the data (i.e., differences in study design, data collection methods, and duration of exposure to treatment). The potential benefits and safety of tafasitamab plus lenalidomide compared with other therapies remain uncertain.
Critical Appraisal
RE-MIND2 was a retrospective observational study that was used to generate an external cohort for indirect comparison to the L-MIND cohort using ePS-based NN 1:1 matching methodology. The success of ePS matching depends on the availability of a large pool of patients from which to select a closely matched population. The RE-MIND2 study enrolled adequate numbers of patients into the observational cohorts. The CADTH review team identified various potential sources of bias associated with ePS-based NN 1:1 matching, including a limited ability to apply similar inclusion and exclusion criteria between cohorts, potential difficulties with the fidelity of available patient record data, variations in outcome assessments, and differences or changes in treatment strategies across geographic regions or over the time frame of the study parameters. Furthermore, the analyses presented in this appendix were conducted post hoc and thus considered exploratory. No conclusions can be drawn based on these post hoc analyses.
References
1.Non-Hodgkin lymphoma. 2022; https://www.lymphoma.ca/lymphoma/non-hodgkin-%20lymphoma/. Accessed 2022 Feb 3.
2.Non-Hodgkin lymphoma statistics. https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/statistics. Accessed 2022 Feb 3.
3.DLBCL – Diffuse large B cell lymphoma. Mississauga (ON): Lymphoma Canada; 2022: https://www.lymphoma.ca/resources/healthcare-professionals/dlbcl-diffuse-large-b-cell-lymphoma/. Accessed 2022 Feb 3.
4.Shafey M, Savage KJ, Skrabek P, Elsawy M, Bosch M, Kuruvilla J. Canadian evidence-based guideline for the treatment of relapsed/refractory diffuse large B-cell lymphoma. Mississauga (ON): Lymphoma Canada; 2021: https://www.lymphoma.ca/wp-content/uploads/2021/09/LymphomaCanada_Guideline_Relapsed_Refractory_DLBCL_VF_Digital.pdf. Accessed 2022 Feb 3.
5.Diffuse large B-cell lymphoma. Toronto (ON): Canadian Cancer Society: https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/what-is-non-hodgkin-lymphoma/diffuse-large-b-cell-lymphoma. Accessed 2022 Feb 3.
6.Sarkozy C, Sehn LH. Management of relapsed/refractory DLBCL. Best Pract Res Clin Haematol. 2018;31(3):209-216. PubMed
7.PrMinjuviTM (tafasitamab): lyophilized powder for solution for injection (200 mg single-use vial) [product monograph]. Wilmington (DE): Incyte; 2021 Aug 19.
8.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579-586. PubMed
9.Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol. 2020;38(2):155-165. PubMed
10.Clinical Study Report: MOR208C203. A phase II, single-arm, open-label, multicentre study to evaluate the safety and efficacy of lenalidomide combined with MOR00208 in patients with relapsed or refractory diffuse large B-cell lymphoma (R-R DLBCL) [internal sponsor's report]. Pointe Claire (QC): Incyte Biosciences Canada; 2019.
11.Salles G, Duell J, Gonzalez Barca E, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020;21(7):978-988. PubMed
12.Clinical Study Report Addendum 1: MOR208C203. A phase II, single-arm, open-label, multicentre study to evaluate the safety and efficacy of lenalidomide combined with MOR00208 in patients with relapsed or refractory diffuse large B-cell lymphoma (R-R DLBCL) [internal sponsor's report]. Pointe Claire (QC): Incyte Biosciences Canada; 2020.
13.Duell J, Maddocks KJ, Gonzalez-Barca E, et al. Long-term outcomes from the phase II L-MIND study of tafasitamab (MOR208) plus lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Haematologica. 2021;106(9):2417-2426. PubMed
14.Clinical Study Report Addendum 3.0: MOR208C203. A phase II, single-arm, open-label, multicentre study to evaluate the safety and efficacy of lenalidomide combined with MOR00208 in patients with relapsed or refractory diffuse large B-cell lymphoma (R-R DLBCL) [internal sponsor's report]. Pointe Claire (QC): Incyte Biosciences Canada; 2021.
15.Incyte Biosciences Canada response to February 2, 2022 DRR request for additional information regarding tafasitamab DRR review [internal sponsor's additional information]. Pointe Claire (QC): Incyte Biosciences Canada; 2022.
16.Clinical Study Report: MOR208C206. An observational retrospective cohort study of lenalidomide monotherapy in relapsed or refractory diffuse large B-cell lymphoma (R/R DLBCL) to generate a historical control for clinical trial MOR208C203 (RE-MIND) [internal sponsor's report]. Pointe Claire (QC): Incyte Biosciences Canada; 2019.
17.Zinzani PL, Rodgers T, Marino D, et al. RE-MIND: comparing tafasitamab + lenalidomide (L-MIND) with a real-world lenalidomide monotherapy cohort in relapsed or refractory diffuse large B-cell lymphoma. Clin Cancer Res. 2021;27(22):6124-6134. PubMed
18.Clinical Study Report: MOR208C213. An observational retrospective cohort study of systemic therapies for relapsed or refractory diffuse large B-cell lymphoma (R/R DLBCL), to compare outcomes to those from tafasitamab + lenalidomide in the L-MIND study (RE-MIND2) [internal sponsor's report]. Pointe Claire (QC): Incyte Biosciences Canada; 2021.
19.B-cell lymphomas. NCCN guideline. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2021: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1480. Accessed 2022 Feb 16.
20.Tilly H, Gomes da Silva M, Vitolo U, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v116-125. PubMed
21.Updated MAIC of tafasitamab + lenalidomide against comparators in R/R DLBCL using the L-MIND October 2020 Datacut: technical report [internal sponsor's report]. Pointe Claire (QC): Incyte Biosciences Canada; 2021.
22.Population-adjusted indirect comparisons (MAIC and STC). NICE Decision Support Unit (DSU). Sheffield (UK): University of Scheffield: https://nicedsu.sites.sheffield.ac.uk/tsds/population-adjusted-indirect-comparisons-maic-and-stc. Accessed 2022 Feb 17.
23.Symptoms of non-Hodgkin lymphoma. Toronto (ON): Canadian Cancer Society: https://cancer.ca/en/cancer-information/cancer-types/non-hodgkin-lymphoma/signs-and-symptoms. Accessed 2022 Feb 3.
24.Drug Reimbursement Review sponsor submission: Minjuvi (tafasitamab): lyophilized powder for solution for infusion (200 mg / vial) [internal sponsor's package]. Pointe Claire (QC): Incyte Biosciences Canada; 2021 Nov 19.
25.MAIC of tafasitamab + lenalidomide against comparators in R/R DLBCL feasibility assessment report [sponsor's internal report]. 2020.
26.PrRevlimid® (lenalidomide): 2.5 mg, 5 mg, 10 mg, 20 mg, 25 mg capsules [product monograph]. Mississauga (ON): Celgene; 2019 Aug 20: https://media.celgene.com/content/uploads/sites/23/Revlimid-Product_Monograph_-_English_Version.pdf. Accessed 2022 Feb 2.
27.Sakemura R, Horvei P, Manriquez-Roman C, et al. 2412 The impact of prior treatment with a CD19 targeting monoclonal antibody on subsequent treatment with CD19 targeting CART cell therapy in preclinical models [abstract]. Presented at: 63rd ASH Annual Meeting & Exposition; Dec 2021; Atlanta (GA). https://ash.confex.com/ash/2021/webprogram/Paper145649.html. Accessed 2022 Feb 7.
28.Zurko JC, Epperla N, Nizamuddin I, et al. 884 Outcomes and treatment patterns in patients with aggressive B-Cell lymphoma after failure of anti-CD19 CAR T-cell therapy [abstract]. Presented at: 63rd ASH Annual Meeting & Exposition; Dec 2021; Atlanta (GA). https://ash.confex.com/ash/2021/webprogram/Paper147433.html. Accessed 2022 Feb 7.
29.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
30.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Dec 6.
31.Health Canada reviewer's report: Minjuvi (tafasitamab) [internal sponsor's additional information]. In: Drug Reimbursement Review sponsor submission: Minjuvi (tafasitamab): lyophilized powder for solution for infusion (200 mg / vial). Pointe Claire (QC): Incyte Biosciences Canada; 2021 Nov 19.
32.Duell J, Obr A, Augustin M, et al. CD19 expression is maintained in DLBCL patients after treatment with tafasitamab plus lenalidomide in the L-MIND study. Leuk Lymphoma. 2022;63(2):468-472. PubMed
33.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-3067. PubMed
34.Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17(4):1244. PubMed
35.Czuczman MS, Trněný M, Davies A, et al. A phase 2/3 multicenter, randomized, open-label study to compare the efficacy and safety of lenalidomide versus investigator's choice in patients with relapsed or refractory diffuse large B-cell lymphoma. Clin Cancer Res. 2017;23(15):4127-4137. PubMed
36.Ohmachi K, Niitsu N, Uchida T, et al. Multicenter phase II study of bendamustine plus rituximab in patients with relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol. 2013;31(17):2103-2109. PubMed
37.Vacirca JL, Acs PI, Tabbara IA, Rosen PJ, Lee P, Lynam E. Bendamustine combined with rituximab for patients with relapsed or refractory diffuse large B cell lymphoma. Ann Hematol. 2014;93(3):403-409. PubMed
38.Mounier N, El Gnaoui T, Tilly H, et al. Rituximab plus gemcitabine and oxaliplatin in patients with refractory/relapsed diffuse large B-cell lymphoma who are not candidates for high-dose therapy. A phase II Lymphoma Study Association trial. Haematologica. 2013;98(11):1726-1731. PubMed
39.Polatuzumab vedotin (Polivy) for DLBCL. pCODR expert review committee (pERC) final recommendation. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/pcodr/Reviews2021/10227PolatuzumabDLBCL_fnRec_EC21Apr2021_final.pdf. Accessed 2022 Feb 8.
40.Corporation IBC. Incyte Biosciences Canada Corporation additional information regarding the Minjuvi CRR provided May 19, 2022 [internal additional sponsor's information]. 2022.
Appendix 1: Literature Search Strategy
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: December 15, 2021
Alerts: Biweekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type
Limits: Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Monjuvi* or Minjuvi* or tafasitamab* or "MOR 00208" or MOR 208 or MOR00208 or MOR208 or WHO 10835 or WHO10835 or XmAb 5574 or XmAb5574 or xenp 5574 or xenp5574 or QQA9MLH692).ti,ab,ot,kf,hw,nm,rn.
1 use medall
*tafasitamab/
(Monjuvi* or Minjuvi* or tafasitamab* or "MOR 00208" or MOR 208 or MOR00208 or MOR208 or WHO 10835 or WHO10835 or XmAb 5574 or XmAb5574 or xenp 5574 or xenp5574).ti,ab,kf,dq.
3 or 4
5 use oemezd
6 not (conference abstract or conference review).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search terms – Minjuvi/tafasitamab OR (MOR208 and topic: DLBCL)
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search terms – Minjuvi/tafasitamab
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms – Minjuvi/tafasitamab
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms – Minjuvi/tafasitamab
Grey Literature
Search dates: December 8 – 13, 2021
Keywords: Minjuvi, Monjuvi, tafasitamab, DLBCL
Limits: None
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free).
Appendix 2: Excluded Studies
Reference | Reason for exclusion |
|---|---|
Duell J, Maddocks KJ, González-Barca E, et al. Long-term outcomes from the phase II L-MIND study of tafasitamab (MOR208) plus lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Haematologica. 2021 Aug 19;08:19 [Online ahead of print] | Duplicate |
Salles G, Duell J, González Barca E, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020;21(7):978-988. | Duplicate |
Düll J, Maddocks KJ, González-Barca E, et al. Long-term analyses from L-MIND, a phase II study of tafasitamab (MOR208) combined with lenalidomide (LEN) in patients with relapsed or refractory diffuse large B-cell lymphoma (R/R DLBCL). J Clin Oncol. 2021. 39(Suppl_15):7513. doi: 10.1200/JCO.2021.39.15_suppl.7513 | Abstract |
Maddocks KJ, Duell J, González-Barca E, et al. 021 Long-term subgroup analyses from L-MIND, a Phase II Study of Tafasitamab (MOR208) combined with lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Presented at: 62nd American Society of Hematology Annual Meeting and Exposition. 2020; Virtual. https://ash.confex.com/ash/2020/webprogram/Paper 140314.html | Abstract |
Salles G, Duell J, González-Barca E, et al. EP1201: Long-term outcomes from the phase II L-MIND study of tafasitamab (MOR208) plus lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Presented at 25th European Hematology Association Annual Congress (Virtual Edition). 2020. | Abstract |
Duell J, Maddocks KJ, González-Barca E, et al. Subgroup analyses from L-MIND, a phase II study of tafasitamab (MOR208) combined with lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Blood. 2019; 134(Suppl_1):1582. doi: 10.1182/blood-2019-122573 | Abstract |
González-Barca E, Duell J, Cavallo F et al. Efficacy of tafasitamab (MOR208) combined with lenalidomide in patients with high-risk relapsed or refractory diffuse large B-cell lymphoma in the L-MIND study. Presented at: American Society of Hematology Annual Meeting and Exposition. 2020 | Abstract |
Duell J, Maddocks KJ, Gonzalez-Barca E, et al. Long-term L-MIND study outcomes of tafasitamab from the (MOR208) phase II plus lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma. Haematologica. 2021;106(9):2417-2426. | Duplicate |
Dull J, Topp M, Salles G. The use of tafasitamab in diffuse large B-cell lymphoma. Ther Adv Hematol. 2021;12:20406207211027458. | Review article |
Klisovic RB, Leung WH, Brugger W, et al. A phase 2a, single-arm, open-label study of tafasitamab, a humanized, Fc-modified, anti-CD19 antibody, in patients with relapsed/refractory B-precursor cell acute lymphoblastic leukemia. Cancer. 2021;127(22):4190-4197. | Study population |
Staber PB, Jurczak W, Greil R, et al. Tafasitamab combined with idelalisib or venetoclax in patients with CLL previously treated with a BTK inhibitor. Leuk Lymphoma. 2021;62(14):3440-3451. | Study population |
Tilch MK, Robak T, Ghiggi C, et al. Safety of the anti-CD19 antibody tafasitamab in long term responders from a phase II trial for relapsed lymphoma. Clin Lymphoma Myeloma Leuk. 2021;16:16. | Study population |
Anonymous. Diffuse large B-cell lymphoma responds to tafasitamab plus lenalidomide. Cancer Discov. 2020;10(8):1091. | Study design |
Anonymous. Tafasitamab-cxix. Am J Health Syst Pharm. 2020;77(24):2029-2031. | Study design |
Appendix 3: Detailed Outcome Data
Figure 9: Boxplot of Compliance With Tafasitamab by Cycle in L-MIND – FAS, November 30, 2018, Data Cut-off (Primary Analysis)
Source: L-MIND Clinical Study Report10
Figure 10: Boxplot of Compliance With Lenalidomide by Cycle in L-MIND – FAS, November 30, 2018, Data Cut-off (Primary Analysis)
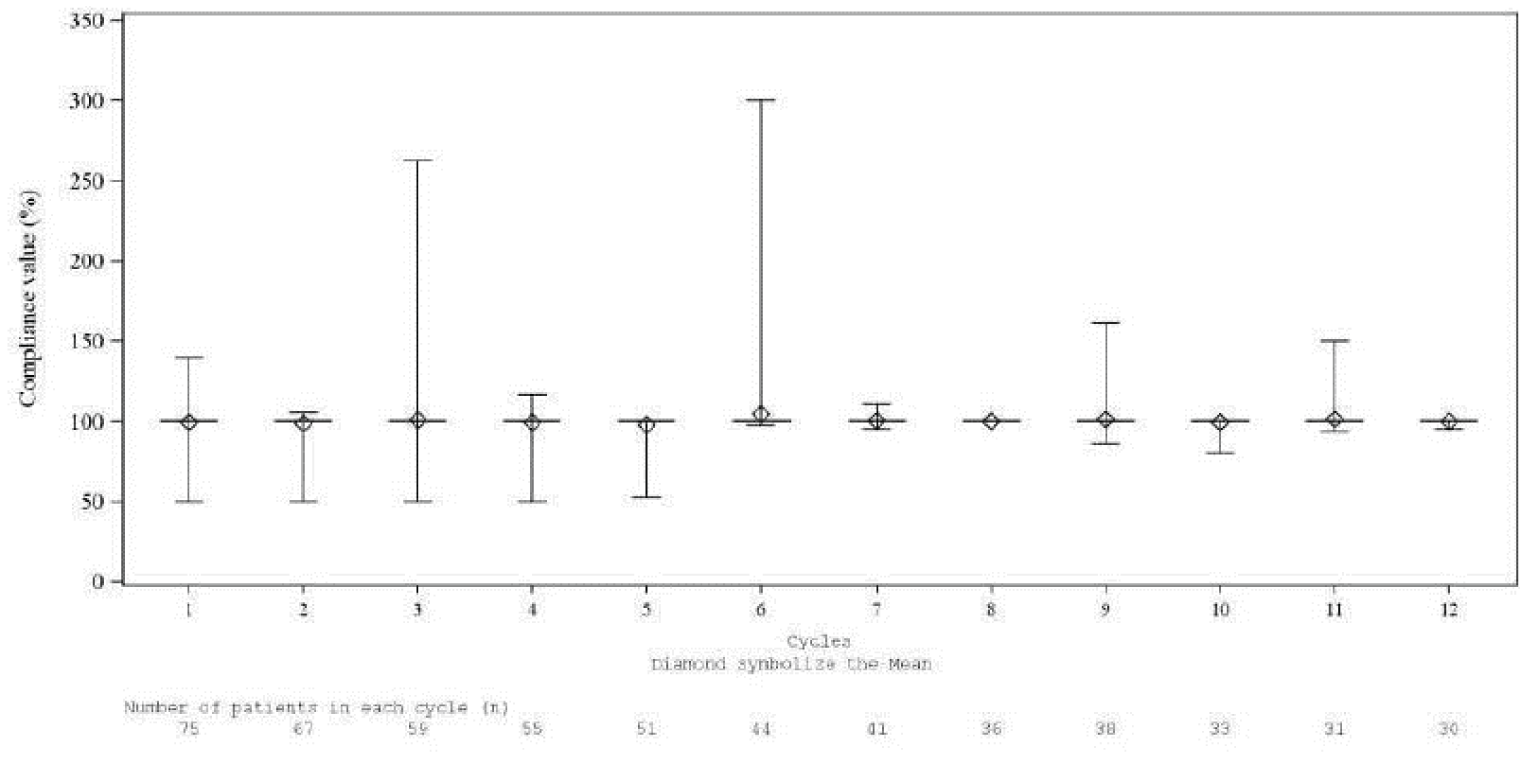
Source: L-MIND Clinical Study Report10
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||
|---|---|---|---|---|---|---|
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |
|||||||||||||||||| | ||||||
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | ||||||
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | ||||||
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | ||||||
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 51: Time-varying HRs at 4 Months of Tafasitamab plus Lenalidomide versus Pola-BR – MAIC Analyses
Outcome | Time Frame | Unadjusted Comparison | Population-Adjusted Comparison |
|---|---|---|---|
OS, HR (95% CI) [P value] | 0 to 4 months | 1.08 (0.38, 3.09) [0.886] | 1.82 (0.58, 5.65) [0.302] |
4 months to end of follow-up | 0.48 (0.27, 0.86) [0.013] | 0.41 (0.19, 0.90) [0.026] | |
PFS by IRC, HR (95% CI) [P value] | 0 to 4 months | 0.98 (0.50, 1.95) [0.961] | 1.42 (0.65, 3.09) [0.376] |
4 months to end of follow-up | 0.61 (0.30, 1.27) [0.186] | 0.39 (0.14, 1.06) [0.065] |
CI = confidence interval; HR = hazard ratio; OS = overall survival; PFS = progression-free survival; pola-BR = polatuzumab vedotin plus bendamustine plus rituximab.
HR < 1 favours tafasitamab plus lenalidomide.
Source: Sponsor-submitted MAIC Technical Report21
Table 52: Recommended Therapies for R/R DLBCL per NCCN/SMO Guidelines Eligible for Inclusion in RE-MIND2 Observational Cohort
Recommended therapies for R/R DLBCL per NCCN/ESMO guidelines19,20 |
|---|
|
DHAP = dexamethasone, cisplatin, cytarabine; DHAX = dexamethasone, cytarabine, oxaliplatin; ESHAP = etoposide, methylprednisolone, cytarabine, cisplatin; GDP = gemcitabine, dexamethasone, cisplatin, or gemcitabine, dexamethasone, carboplatin; GemOx = gemcitabine plus oxaliplatin; ICE = ifosfamide, cisplatin, etoposide; MINE = mesna, ifosfamide, mitoxantrone, etoposide; CEPP = cyclophosphamide, etoposide, prednisone, procarbazine; CEOP = cyclophosphamide, etoposide, vincristine, procarbazine; DA-EPOCH = cyclophosphamide, doxorubicin, etoposide, vincristine.
Source: RE-MIND2 Clinical Study Report18
Appendix 4: New Data Submitted by Sponsor for Reconsideration
Note that this appendix has not been copy-edited.
Following the issuance of the draft CADTH pERC recommendation for tafasitamab in May 2022, the sponsor-submitted post hoc analyses from the RE-MIND2 study. The results of these post hoc analyses are presented in Table 53, Table 54, and Table 55 below. These data were not included in the initial submission to CADTH. After the CADTH recommendation was issued, the sponsor reported that the data only became available after their submission to CADTH.
The sponsor provided the data presented in Table 53 and Table 54 to show the proportion of patients with 3 types of comorbidities in the BR and R-GemOx groups in the RE-MIND2 study.
In the sponsor’s Request for Reconsideration, it was acknowledged that cell of origin is a contributing factor in disease prognosis. The sponsor presented the data reported in Table 55 and reported that cell of origin data are incomplete for a proportion of patients in RE-MIND2 BR and R-GemOx arms.
Table 53: Analysis of Comorbidities By L-MIND and RE-MIND 2 (Match BR Analysis Set)
Variable | L-MIND | RE-MIND2 | P valuea |
|---|---|---|---|
Comorbidities, n (%) | |||
(i) Diffusion lung capacity for carbon monoxide < 50% by pulmonary function test | 2 (18.2) | 1 (5.3) | 0.0541 |
(ii) Left ventricular ejection fraction < 50% by multiple gated acquisition echocardiogram | 2 (18.2) | 12 (63.2) | Reference |
(iii) Other organ dysfunction or comorbidities precluding the use of HDT/ASCT on the basis of unacceptable risk of treatment | 7 (63.6) | 6 (31.6) | Reference |
ASCT = allogenic stem cell transplant; BR = bendamustine plus rituximab; HDT = high-dose chemotherapy.
aP value was calculated by chi-square method.
Source: Additional data submitted by sponsor.40
Table 54: Analysis of Comorbidities By L-MIND and RE-MIND 2 (Match R-GemOx Analysis Set)
Variable | L-MIND | RE-MIND2 | P valuea |
|---|---|---|---|
Comorbidities, n (%) | |||
(i) Diffusion lung capacity for carbon monoxide < 50% by pulmonary function test | 2 (18.2) | 3 (15.0) | 0.1192 |
(ii) Left ventricular ejection fraction < 50% by multiple gated acquisition echocardiogram | 2 (18.2) | 11 (55.0) | Reference |
(iii) Other organ dysfunction or comorbidities precluding the use of HDT/ASCT on the basis of unacceptable risk of treatment | 7 (63.6) | 6 (30.0) | Reference |
ASCT = allogenic stem cell transplant; BR = bendamustine plus rituximab; HDT = high-dose chemotherapy.
aP value was calculated by chi-square method.
Source: Additional data submitted by sponsor.40
Table 55: Analysis of initial diagnosis of DLBCL (Match Analysis Set)
Characteristic | BR (N = 75) | R-GemOX (N = 74) |
|---|---|---|
Cell of origin (Immunohistochemistry, Hans’ algorithm) – n (%) | ||
GCB | 21 (28.0) | 21 (28.4) |
Non-GCB | 25 (33.3) | 13 (17.6) |
Missing | 29 (38.7) | 40 (54.1) |
Cell of origin (Gene-expression profiling) – n (%) | ||
GCB | 1 (1.3) | 2 (2.7) |
ABC | 0 (0.0) | 1 (1.4) |
Missing | 74 (98.7) | 71 (95.9) |
ABC = activated B cell; BR = bendamustine plus rituximab; GCB = germinal centre B cell like.
Source: Additional data submitted by Sponsor.40
Pharmacoeconomic Review
Abbreviations
AE
adverse event
ASCT
autologous stem cell transplant
axi-cel
axicabtagene ciloleucel
CAR T-cell
chimeric antigen receptor T-cell
DLBCL
diffuse large B-cell lymphoma
GDP
gemcitabine plus dexamethasone plus cisplatin
ICER
incremental cost-effectiveness ratio
MAIC
matching-adjusted indirect comparison
NICE
National Institute for Health and Care Excellence
NOC/c
Notice of Compliance with Conditions
OS
overall survival
PFS
progression-free survival
pola-BR
polatuzumab plus bendamustine plus rituximab
QALY
quality-adjusted life-year
R/R
relapsed or refractory
R-CHOP
rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
R-GDP
rituximab plus gemcitabine plus dexamethasone plus cisplatin
R-GemOx
rituximab plus gemcitabine plus oxaliplatin
tisa-cel
tisagenlecleucel
WTP
willingness to pay
Executive Summary
The Executive Summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Tafasitamab (Minjuvi), powder for solution for IV injection 12 mg/kg body weight |
Submitted price | Tafasitamab: $1,167.86 per 200 mg single-use vial |
Indication | In combination with lenalidomide for the treatment of adult patients with relapsed or refractory DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT |
Health Canada Approval Status | NOC/c |
Health Canada Review Pathway | Advance consideration under NOC/c |
NOC date | August 19, 2021 |
Reimbursement request | As per indication |
Sponsor | Incyte Biosciences Canada Corporation |
Submission history | Previously reviewed: No |
ASCT = autologous stem cell transplant; DLBCL = diffuse large B-cell lymphoma; NOC/c = Notice of Compliance with Conditions.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation |
|
Target population | Patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) who are not eligible for autologous stem cell transplant |
Treatment | Tafasitamab in combination with lenalidomide |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 20 years |
Key data sources |
|
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
axi-cel = axicabtagene ciloleucel; CAR T-cell = chimeric antigen receptor T-cell; DLBCL = diffuse large B-cell lymphoma; GDP = gemcitabine plus dexamethasone plus cisplatin; ICER = incremental cost-effectiveness ratio; LY = life-year; MAIC = matching-adjusted indirect comparison; NICE = National Institute for Health and Care Excellence; NOC/c = Notice of Compliance with Conditions; pola-BR = polatuzumab plus bendamustine plus rituximab; PSM = partitioned survival model; QALY = quality-adjusted life-year; R-GDP = rituximab plus gemcitabine plus dexamethasone plus cisplatin; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; R/R = relapsed or refractory; tisa-cel = tisagenlecleucel.
Conclusions
The CADTH clinical review noted that there are significant limitations with the L-MIND trial, including the phase II, open-label single-arm design; the small sample size; and the descriptive analysis of the data (i.e., no statistical hypotheses were tested). Due to the absence of a comparator arm and statistical testing, no definitive conclusions can be drawn regarding the efficacy of tafasitamab plus lenalidomide based on the L-MIND trial. CADTH also noted the commentary from Health Canada, that the Notice of Compliance with Conditions (NOC/c) for tafasitamab (when used in combination with lenalidomide) was given pending the results of additional trials to verify its clinical benefit. The NOC/c was based on overall response rate, complete response rate, and durability of response. An improvement in progression-free survival (PFS) or overall survival (OS) has not been established based on the evidence reviewed. As the sponsor’s economic evaluation was based on a partitioned survival model that considered pre-progression (based on PFS), post-progression (based on the difference between PFS and OS), and death, the clinical benefit predicted for tafasitamab plus lenalidomide based on this approach is highly uncertain.
Results from the indirect comparative evidence submitted by the sponsor suggested that tafasitamab plus lenalidomide may be associated with an improvement in clinical outcomes compared with relevant comparators (e.g., rituximab plus gemcitabine plus oxaliplatin [R-GemOx], polatuzumab plus bendamustine plus rituximab [pola-BR]). However, the CADTH clinical review noted that the indirect evidence is associated with substantial risk of bias due to important limitations (including methodological limitations, heterogeneity, matching based on a limited number of variables, and small sample sizes). In view of the uncertainty in the indirect evidence results, no conclusions can be drawn concerning the efficacy and safety of tafasitamab plus lenalidomide compared to other therapies used to treat patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) who are ineligible for autologous stem cell transplant (ASCT).
While CADTH could correct for biases in the model, the lack of comparative evidence for tafasitamab plus lenalidomide remains, and the evidence for PFS and OS are evolving. CADTH estimated an incremental cost-effectiveness ratio (ICER) for tafasitamab (in combination with lenalidomide) of $228,224 per quality-adjusted life-year (QALY) compared with gemcitabine plus dexamethasone plus cisplatin (GDP; the least costly reference treatment). This estimate is based on the sponsor’s clinical assumptions that predict an additional 3.46 life-years and 2.52 QALYs for those receiving tafasitamab plus lenalidomide. Based on the CADTH-corrected sponsor’s analysis, a price reduction of 90% is required to achieve an ICER of $50,000 per QALY. When this benefit in survival is not realized, as reported in exploratory analyses, a greater price reduction would be required for tafasitamab (in combination with lenalidomide) to be considered cost-effective at a willingness to pay threshold of $50,000 per QALY.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission).
Patient group input was received from Lymphoma Canada. The input was based on a series of anonymous online surveys conducted in 2021 to capture the perspectives of patients with DLBCL. Survey links were also made available via social media and health care professionals across Canada. Feedback was received from 150 patients with DLBCL, of whom 2 patients had experience with tafasitamab. Of total respondents, 73% were from Canada, although no Canadian patients had experience with tafasitamab. No caregivers participated in the survey. Key aspects of DLBCL that affect quality of life were reported to be fatigue or lack of energy, night sweats, weight loss, loss of appetite, influenza-like symptoms, persistent cough, memory loss, stress, depression, impacts on personal relationships, limitations on attendance at school or work, and fear of disease recurrence. Respondents had received at least 1 therapy, with the majority of respondents having received chemoimmunotherapy with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). Twenty-three percent of patients had undergone ASCT, while 4% had undergone allogenic stem cell transplant. Current treatments were noted to have the important side effects, including hair loss, fatigue, neutropenia, memory issues, nausea, neuropathy, constipation, diarrhea, anemia, infection, pain, skin rashes, pain, irregular heartbeat, viral reactivation, and bowel obstruction. Current treatments also have a negative impact on quality of life, due to fatigue, treatment-emergent adverse events, infusion reactions, infections, time spent at clinic visits and drug administration, and missed activities because of travel for health care; and have an impact on personal relationships. Patients are seeking new treatments with longer remission and survival, better disease control, improved quality of life, and fewer adverse events.
Clinician input was received from 2 groups: the OH-CCO Hematology Drug Advisory Committee and a group of 4 clinicians whose submission was coordinated by Lymphoma Canada. Clinicians stated that current treatment options for patients with R/R DLBCL are limited. The population with the greatest unmet needs is patients not currently eligible for more intensive treatments (i.e., ASCT or chimeric antigen receptor T-cell [CAR T-cell] therapy). Tafasitamab is most likely to be used in patients who are not eligible for ASCT, although clinician groups noted that use of tafasitamab may affect eligibility for CAR T-cell therapy use in the future. Tafasitamab plus lenalidomide is likely to be used as another treatment option, alongside pola-BR, in patients with R/R DLBCL. The clinical goal of treatment is to improve symptoms, prolong remission, and improve survival. Benefits associated with tafasitamab (in combination with lenalidomide) were reported to be the ease of administration and lower risks of important adverse events compared with current chemoimmunotherapy regimens (e.g., bone marrow suppression, long-term immune suppression, neurotoxicity).
CADTH-participating drug plans highlighted several implementation and economic considerations for tafasitamab plus lenalidomide. Drug plans indicated that, due to administration requirements, there was likely to be no vial sharing; therefore, drug wastage was of concern. Pharmacy preparation time associated with tafasitamab plus lenalidomide was considered high and complex, while infusion time was between 1.5 and 2.5 hours. Overall, resource use (i.e., chair time, patient visits, pharmacy preparation time) for tafasitamab plus lenalidomide is likely to be more intensive than for other relevant comparator regimens for R/R DLBCL patients, which were considered to be gemcitabine plus oxaliplatin, GDP, and other palliative regimens, with or without rituximab. The input noted that rituximab is not funded in some jurisdictions, while newer regimens, such as pola-BR and CAR T-cell therapy, may also be available in some jurisdictions. Drug plans also noted that lenalidomide is not currently available to this patient population in most jurisdictions, and additional prescriber steps would be required to dispense via Health Canada mandated Restricted Drug Distribution Programs. While multiple brands of lenalidomide are available, these are not easily interchangeable, and capsule strengths differ by manufacturer, which may be have an impact if the dose is reduced and may increase costs. Additional growth factor support would be needed, given the incidence of neutropenia associated with tafasitamab plus lenalidomide. Finally, drug plans noted concerns that tafasitamab plus lenalidomide would be associated with a significant budget impact, depending on the extent of uptake and due to the indefinite treatment duration.
Several of these concerns were addressed in the sponsor’s model:
The sponsor included chemotherapy and chemoimmunotherapies in the submitted base-case analysis. Pola-BR was included, although only in a scenario analysis.
The sponsor included relevant adverse events, and their impact on costs and utilities, in the submitted base-case analysis.
The sponsor assumed no vial sharing for any treatments in the submitted base-case analysis.
The sponsor’s economic evaluation included survival outcomes and quality of life (via utility values), but did not consider response or remission.
CADTH was unable to address the following concerns raised from stakeholder input:
Several issues identified by the drug plan could not be addressed by CADTH, but were noted as issues for consideration. CADTH noted that there was a difference of opinion between drug plans and clinicians regarding complexity of treatment administration.
Economic Review
The current review is for tafasitamab (Minjuvi) for patients with R/R DLBCL who are not eligible for subsequent ASCT.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing tafasitamab plus lenalidomide with various chemotherapies with or without rituximab (i.e., R-GemOx; rituximab, gemcitabine, dexamethasone, and cisplatin [R-GDP]; and GDP). The sponsor also included scenario analyses with the following comparators: pola-BR, CAR T-cell therapies (axicabtagene ciloleucel [axi-cel] and tisagenlecleucel [tisa-cel]), and a pooled analysis including R-GemOx (10%), R-GDP (30%), GDP (40%), pola-BR (15%), and CAR T-cell therapies (5%). The reimbursement population aligns with the Health Canada–indicated population. The sponsor explored the R/R patient group (i.e., patients with at least 1 prior line of treatment) in the base case, as well as a second-line only subgroup (i.e., patients with 1 prior line of treatment) in a scenario analysis.1
Tafasitamab is available as a 200 mg single-use vial of powder for solution for IV injection. The recommended dose is 12 mg tafasitamab per kilogram; for the first 28-day cycle, it is infused on days 1, 4, 8, 15, and 22; for the second and third cycles, on days 1, 8, 15, and 22; and for the cycle 4 and beyond, on days 1 and 15. Patients take lenalidomide capsules at a starting dosage of 25 mg daily on days 1 through 21 of each 28-day cycle for up to 12 cycles. Dosage may be adjusted as necessary per the lenalidomide product monograph. Tafasitamab was administered until disease progression or unacceptable toxicity; lenalidomide was administered for a maximum of 12 cycles, or until disease progression or unacceptable toxicity.2 The dose intensity was assumed to be 93% for lenalidomide and 100% for all other treatments.1 At the submitted price of $1,167.86 per 200 mg single-use vial, tafasitamab costs $29,196 in the first cycle, $23,357 in the second and third cycles, and $11,679 in the fourth cycle and beyond. Lenalidomide costs $2,078 per 28-day cycle for up to 12 cycles.
The outcomes of interest were QALYs and life-years. The analysis takes the public payer perspective. The sponsor specified the time horizon in the base case as 20 years. The discount rate for costs and outcomes was 1.5% annually.
Model Structure
The sponsor submitted a partitioned survival model with the following health states: PFS, progressive disease, and death. All patients entered the model in the PFS state. The progressive disease state captures patients who have progressed but have not died. The death state is an absorbing state, i.e., all patients transitioning to this health state are assumed to occupy it indefinitely (Figure 1, Appendix 3). A cycle length of 4 weeks (28 days) was used in the model.1
The model also captures the proportion of patients on and off treatment in each health state using the same partition approach: patients falling under the time-to-treatment discontinuation curve are on treatment, while the patients between the time-to-treatment discontinuation and PFS curves must be in the pre-progression health state and are off treatment.1
Due to the nature of partitioned survival models, the model does not incorporate the transition of patients between the health states. Rather, the proportion of patients who are progression-free and the proportion who are alive at each time point are estimated independently.
Model Inputs
The baseline population characteristics used to inform the model were based on the L-MIND trial (October 30, 2020, data cut-off), a single-arm, phase II, open-label trial of tafasitamab plus lenalidomide. The mean age of patients in the trial was 69 years (standard deviation ||||), and mean height and weight values resulted in a mean body surface area of 1.91 m2 (standard deviation ||||).1,3
The clinical efficacy and safety of tafasitamab plus lenalidomide were derived from the L-MIND trial (n = 80, October 30, 2020, data cut-off). Due to the lack of head-to-head evidence comparing tafasitamab plus lenalidomide with the included comparators, the sponsor employed 2 approaches to generate comparative effectiveness estimates: the RE-MIND2 study and matching-adjusted indirect comparisons (MAICs).4,5 RE-MIND2 was a study in which a real-world dataset of patients who received R-GemOx, pola-BR, and CAR T-cell therapy was statistically matched to patients from the L-MIND study. While using the MAIC approach, individual patient data from the L-MIND trial were matched to cohort data from a phase II study of R-GemOx6 and to cohort data from the GO29365 trial of pola-BR.7 As R-GDP and GDP were not included in the RE-MIND2 study, the sponsor assumed these regimens would have efficacy and safety equal to the R-GemOx regimen.
Long-term efficacy for tafasitamab plus lenalidomide was estimated by fitting parametric survival models to patient-level OS and PFS data. Model selection was based on clinical validity and statistical fit via Akaike information criterion and Bayesian information criterion, visual assessment, and clinical plausibility. In the base case, long-term OS for tafasitamab plus lenalidomide was predicted using the lognormal function; long-term PFS was predicted using the generalized gamma function; and time-to-treatment discontinuation was predicted using the lognormal function.1 Long-term OS for R-GemOx, based on the RE-MIND2 data, was predicted using the lognormal function; long-term PFS was predicted using the exponential function; and time to discontinuation was based on Kaplan–Meier curves. Data for R-GDP and GDP were assumed to be equivalent to R-GemOx, taking on the same shape and scale. Efficacy information for pola-BR and CAR T-cell therapy were incorporated from the RE-MIND2 study and MAICs; PFS and OS data for pola-BR were incorporated with time-varying hazard ratios. The sponsor indicated that this approach was taken because there was evidence of a violation of the proportionality of hazard assumptions between the MAIC-adjusted estimates of OS and PFS and the estimates for pola-BR.1
Treatment-emergent adverse events (AEs) of grade 3 or higher occurring in 5% or more of study subjects in the L-MIND population were included in the model. AEs for the comparator treatments were also included, although source information for the inputs were not reported.1
Health state utility values were derived from the UK National Institute for Health and Care Excellence (NICE) review of pola-BR for patients with R/R DLBCL for the PFS (0.72) and post-progression health states (0.65).8 If durable remission is considered, the sponsor assumed the same utility as PFS. Disutilities related to AEs were applied for a varied duration as a 1-off occurrence for each treatment; values were based on a wide variety of published sources or assumptions.
The sponsor incorporated a variety of costs in the model. Drug acquisition cost of tafasitamab was based on the sponsor’s submitted price, while the treatment acquisition price for lenalidomide and the comparator treatments was derived from public drug formularies, IQVIA’s Delta PA database, prior pCODR reports, and published literature.1 Dosing regimens, dose intensities, and number of administrations were based on the provincial treatment guidelines, NICE reviews, the L-MIND trial, published literature, or assumptions. It was assumed that there was no re-treatment with CAR T-cell therapy products. Administration costs were applied to IV or subcutaneous treatments based on published literature,9 while monitoring costs were based predominantly on Ontario Schedule of Benefits sources.10,11 Monitoring costs were informed by the NICE review of tisa-cel12 and the Ontario Schedule of Benefits.10 Additionally, disease management costs were incorporated for both the PFS and post-progression health states based on provincial sources and published literature. Resource use for monitoring and disease management items were based on information from the L-MIND trial and the sponsor’s survey of physicians.1,10,13 A 1-off terminal care cost was also applied.14 Use of subsequent treatments were informed by the RE-MIND2 study and the sponsor’s survey of physicians; a proportion of patients were considered eligible for ASCT post-treatment with the comparator treatments, although not patients who received tafasitamab plus lenalidomide.1 Costs for concomitant medications and costs to treat AEs were also incorporated.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically using 5,000 iterations for the base-case and scenario analyses. The sponsor reported both probabilistic and deterministic results. The ICERs differed somewhat, driven by higher life-years accrued in the deterministic analysis. The probabilistic findings are reported below.
Base-Case Results
In the sponsor’s base-case analysis, the results indicated that treatment with tafasitamab plus lenalidomide was associated with higher incremental costs and greater incremental QALYs than the chemotherapy regimens (GDP, R-GDP, and R-GemOx). Over the 20-year time horizon, tafasitamab plus lenalidomide had an ICER of $199,353 per QALY gained compared with GDP. Disaggregated results are provided in Table 8, Appendix 3. The sponsor’s cost-effectiveness acceptability curve indicated that tafasitamab plus lenalidomide had a 0% probability of being cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY. The sponsor’s analysis predicted that tafasitamab plus lenalidomide was associated with a longer duration of life (i.e., life-years) than the current treatment regimens (5.37 for tafasitamab plus lenalidomide and 1.90 to 1.91 for R-GemOx, R-GDP, and GDP).1
Importantly, given the duration of the clinical trial observation period in contrast to the model time horizon (20 years), most of the QALYs realized by patients receiving tafasitamab plus lenalidomide in the model were gained outside of what was observed in the clinical trial (i.e., extrapolated period). However, the extent of this could not be examined, given the sponsor’s model structure and programming. Reducing the time horizon to 4 years (maximum duration of patients on study), 56% of the total QALYs, and 73% of the incremental QALYs, were derived beyond 4 years. When considering the median follow-up (approximately 3 years), 63% of the total QALYs and 81% of the incremental QALYs were derived beyond 3 years.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug regimens | Total costs ($) | Total LYs | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
GDP | 163,948 | 1.91 | 1.29 | Reference |
Tafasitamab plus lenalidomide | 667,021 | 5.37 | 3.81 | 199,353 |
Dominated treatments | ||||
R-GemOx | 196,303 | 1.90 | 1.29 | Extendedly dominated through GDP and tafasitamab plus lenalidomide |
R-GDP | 200,545 | 1.90 | 1.29 | Dominated by GDP |
GDP = gemcitabine plus dexamethasone plus cisplatin; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab plus gemcitabine plus oxaliplatin.
Note: As noted in the summary of the sponsor’s submission, GDP and R-GDP were assumed to have the same efficacy as R-GemOx. Differences in life-years and QALYs were attributed to different AE and subsequent treatment assumptions.
Source: Adapted from the sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor submitted a series of scenario analyses, the results of which are of limited use, as tafasitamab plus lenalidomide has been reported as the reference product throughout. When looking at the results compared with GDP (the reference regimen in the sequential analysis), the largest impact on the results was related to a change in time horizon (10 years; ICER = $234,044 per QALY), the assumption of durable remission (included; ICER = $173,407 per QALY), the assumption that patients with durable remission do not continue treatment (ICER = $138,726 per QALY); and alternative OS distributions for R-GemOx, R-GDP, and GDP (Weibull; ICER = $251,941 per QALY).
The sponsor also presented a scenario analysis that compared tafasitamab plus lenalidomide with pooled CAR T-cell therapies (axi-cel and tisa-cel), pola-BR, and pooled chemotherapy regimens (R-GemOx, R-GDP, and GDP). The results were presented pairwise only and suggested ICERs for tafasitamab plus lenalidomide of $5,216 per QALY versus CAR T-cell therapy, $162,718 per QALY versus pola-BR, and $227,851 per QALY versus pooled comparator.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The clinical data for tafasitamab plus lenalidomide are based on a single-arm study with evidence of clinical response: The clinical efficacy of tafasitamab plus lenalidomide was assessed in a single-arm, open-label, phase II trial. Although the sponsor attempted to minimize the risk of bias by using independent review committee assessment for key study outcomes, the open-label, single-arm design can increase the risk of bias in reporting outcomes that are subjective in measurement and in interpretation, such as response. Furthermore, CADTH identified concerns relating to the small number of enrolled patients and multiple protocol amendments, and it noted that between 10% and 11% of patients enrolled in the L-MIND trial were misdiagnosed with DLBCL, according to a central pathology analysis. While the clinical experts noted that these patients were likely healthier than most Canadians eligible for tafasitamab plus lenalidomide, they did not consider these differences would affect generalizability of the evidence to the Canadian setting. The data cuts submitted to CADTH were interim analyses, and final analysis is not expected until 2023.15 Whether the final efficacy results will conform to the interim results is unknown.
There were 3 data cuts provided to CADTH, of which the dataset used in the economic evaluation was from October 2020. Median PFS at the October 2020 data cut-off was 11.6 months, which had numerically reduced from prior data cuts; the sponsor considered the PFS data mature at the October 2020 data cut-off. Median OS was reached at the November 2019 and October 2020 data cuts, although no upper bound was reported at either the 2019 or 2020 data cut-off. As the data were analyzed descriptively—i.e., no statistical hypotheses were tested—there may be confounders, such as subsequent treatment, which may have affected PFS and OS in patients receiving tafasitamab plus lenalidomide.
At the October 2020 data cut-off, median follow-up ranged from just under 34 months (PFS) to 43 months (OS). The sponsor’s PFS Kaplan–Meier curve in the model suggested that |||||||||% of patients were progression-free at ||||||||| (approximately |||||||||), which is longer than duration of follow-up reported at the October 2020 data cut-off. At 12 months, |||||||||% of patients were in PFS, based on the sponsor’s Kaplan–Meier curve. The Kaplan–Meier curve appears to flatten around 12 months, although there was a large reduction in the N at each time point (n = 30 at 12 months, n = 11 at 36 months, and n = 1 at 48 months).
CADTH was unable to address the limitations associated with the submitted clinical data. The clinical uncertainty directly affects confidence in the results of the economic model.
The comparative clinical efficacy of tafasitamab plus lenalidomide is highly uncertain: Due to the lack of head-to-head evidence comparing tafasitamab plus lenalidomide with relevant comparators in a randomized controlled trial, the sponsor took alternative measures to derive comparative efficacy information. The sponsor compared tafasitamab plus lenalidomide with various comparators in the REMIND2 study, in which patients from L-MIND were matched with observation data from a retrospective review. Additional sources of data via MAICs comparing tafasitamab plus lenalidomide with pola-BR, CAR T-cell therapy, and R-GemOx were also presented.
The CADTH clinical review identified several concerns with the REMIND2 study, noting key differences in inclusion and exclusion criteria between cohorts, difficulties with the fidelity of available patient record data, variations in outcome assessments, and differences or changes in treatment strategies across geographic regions or over the time frame of the study parameters. Moreover, CADTH noted that matching data from difference sources is associated with bias and that it is impossible to assess the impact of unmeasured confounding factors. Additionally, there were several limitations associated with the MAICs, including important effect modifiers and prognostic factors that could not be adjusted for, low quality of evidence, and multiple sources of heterogeneity. These limitations are all underpinned by the limitations of the data on tafasitamab plus lenalidomide from the L-MIND trial, which limit confidence in the assumed clinical efficacy. Due to the substantial methodological limitations of the comparative clinical evidence, CADTH could not draw any conclusions from the comparative evidence identified in the review.
Although there are limitations with the comparative evidence, CADTH observed that statistical significance was not met in some effect measures when tafasitamab plus lenalidomide was compared with chemotherapy and chemoimmunotherapy (i.e., confidence intervals were extremely wide and crossed 1) based on the MAIC, suggesting that tafasitamab plus lenalidomide may be no more effective than chemotherapy or chemoimmunotherapy. While the REMIND2 study suggested a statistically significant difference in PFS, the sponsor reported that the PFS data have limitations because of invalidated progression assessments and higher censoring rates due to missing radiological assessments. Furthermore, the assumption of the Cox proportional hazards model was not met.
Although CADTH could not address the methodological limitations with the evidence, CADTH undertook several exploratory analyses incorporating alternative assumptions for comparative efficacy.
The sponsor’s model structure is not appropriate: Health Canada gave tafasitamab plus lenalidomide for patients with R/R DLBCL an NOC/c in August 2021.16 Health Canada stated that “authorization was based on overall response rate, complete response rate, and durability of response from a single-arm clinical study. An improvement in PFS or OS has not been established.” Further information on the NOC/c is provided as an issue for consideration (later in this report). Given that PFS and OS have not been established in this patient population, and there is no robust evidence to confirm that response measures are a prognostic marker of PFS or OS, the sponsor’s partitioned survival model (which incorporated PFS and OS data based on the progression-free and post-progression health states) was not appropriate or supported by the available evidence. A model based on response rates may have been more appropriate, based on the available data, although the output of such a model would still be constrained by the quality of the data used to inform it, which may have limitations similar to those described in the appraisal of the clinical evidence.
CADTH could not address this limitation due to the submitted model structure.
Key comparator excluded from the base case: Feedback from the clinical experts consulted for this review confirmed that there is no standard of care for patients with R/R DLBCL who are not eligible for intensive therapy (i.e., CAR T-cell therapy, ASCT). Treatment options include radiation and noncurative chemotherapy regimens. Feedback from the same experts indicated that, in jurisdictions that fund pola-BR, the most relevant comparator for tafasitamab plus lenalidomide is likely to be pola-BR. Pola-BR was not included in the sponsor’s base case, although the sponsor considered it in a scenario analysis.
CADTH included pola-BR in CADTH exploratory reanalyses.
Clinical information was not incorporated appropriately: The sponsor allowed for the incorporation of data from MAICs and REMIND2 in the economic evaluation. Regardless of the data source selected, the clinical data for tafasitamab plus lenalidomide remained the same, while clinical data for the comparator changed to the alternative source. MAICs are pairwise analyses that use the base population from the comparator trial and adjust the population from the L-MIND trial to match characteristics of the comparator trial population. This issue extends to the REMIND2 study, in which patients receiving tafasitamab plus lenalidomide were matched to observed cohorts, which changed the sample size of tafasitamab plus lenalidomide. The submitted analysis does not appropriately consider the different baseline characteristics for the patient populations and different treatment efficacy for tafasitamab plus lenalidomide for the specific populations analyzed in each data source used to inform the economic evaluation. Furthermore, when incorporating the MAIC information in the broader analysis set, this was still considered as part of a sequential analysis, which is also not an appropriate approach.
For the comparison of tafasitamab plus lenalidomide and pola-BR, CADTH noted that the data used to inform the analyses appear to overpredict survival for tafasitamab plus lenalidomide (Figure 2) and substantially underpredict survival for pola-BR. The sponsor acknowledged the latter in its pharmacoeconomic submission, as it noted that data from the GO29365 trial17 indicated 2-year OS of approximately 38% for pola-BR, which was considerably higher than those produced by the sponsor’s adjusted and unadjusted parametric models. This suggested the parametric fits may have substantially underpredicted OS in relation to the GO29365 trial data. The sponsor hypothesized that differences between the OS predictions and the GO29365 2020 trial data may have been related to underlying differences in the study populations.
CADTH also noted differences in the modelled treatment discontinuation relative to the L-MIND trial. The model predicted that approximately 55% of patients remained on tafasitamab (20% in combination with lenalidomide) after 12 cycles (i.e., 48 weeks), while, in the L-MIND trial, approximately ||||% of patients received tafasitamab plus lenalidomide for the 12-cycle period, and approximately ||||% received tafasitamab for the 12 cycles and continued receiving it as monotherapy beyond 12 cycles. Notably, these numbers do not align with the proportion of patients in PFS. For context, the product monograph indicates that patients should be treated until progression or intolerance. The difference may be due to differences between investigator assessments and independent review committee assessments.
o CADTH was unable to appropriately address this limitation, given the available information and model setup.
Model lacks transparency and lacks face validity: The submitted economic model includes many hidden sheets and hidden columns and rows, making it difficult to track inputs and outputs throughout the model. For example, there was duplication of key parameters across multiple sheets, making it unclear which parameter needed to be edited to implement a change. Likewise, the sponsor’s submitted model also included numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors. The lack of transparency and lack of user guide for the sponsor’s macros further complicated the validation process.
CADTH was unable to address these deficiencies and cautions that results from the submitted economic model could not be fully validated.
Resource use and costs for health states, subsequent treatments, monitoring, and administration of tafasitamab plus lenalidomide were underestimated: The sponsor assumed different subsequent treatment use based on each comparator, while assuming the same efficacy regardless of subsequent treatment. The sponsor assumed patients receiving tafasitamab plus lenalidomide would incur much less of costly treatments (e.g., CAR T-cell therapy, ASCT) and a greater proportion of lower-cost treatments, while accruing the same relative benefit regardless of subsequent treatment. Feedback from the clinical experts consulted by CADTH suggested that this is an unrealistic assumption and biases the results in favour of tafasitamab plus lenalidomide. Furthermore, the sponsor assumed that health state costs would be lower for patients receiving tafasitamab plus lenalidomide than for those receiving other treatments. Health state costs should be exclusive to the health state and should not differ based on treatment. Feedback from the clinical experts consulted by CADTH concurred that there should be no differences between treatments for these disease management costs based on health state.
Additionally, the sponsor assumed that tafasitamab plus lenalidomide would be associated with lower monitoring costs and administration costs than all comparator treatments. Feedback from the CADTH-participating drug plans indicated that administration costs and resource use associated with tafasitamab plus lenalidomide were underestimated. The clinical experts consulted by CADTH agreed and also indicated that monitoring costs are likely to be similar across the treatments.
CADTH undertook reanalyses with alternative cost and resource use assumptions.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
R-GDP and GDP were assumed to have the same efficacy as R-GemOx | While there are likely numerical differences between these treatments, the clinical experts consulted for this review agreed that these treatments would be considered to have similar efficacy in Canadian practice. |
Dose intensity for tafasitamab (and most other included treatments) is 100% | Appropriate |
Lifetime time horizon is assumed to be 20 years | Reasonable |
Health state utilities were obtained from NICE TA649 | Reasonable |
Assumption of potential long-term remission in the model for patients who continued to respond for 5 years based on PFS data that the sponsor considered to be mature (scenario analysis) | Not appropriate. Feedback from the clinical experts consulted for this review indicated that this was not an appropriate interpretation of this consideration. This assumption would only be reasonable if a patient did not progress after 5 years off treatment. The clinical experts also did not agree with the sponsor’s suggestion that the plateau observed with the PFS data could be interpreted as the potential that tafasitamab plus lenalidomide may be considered curative. Given the small number of patients observed at the time points and other limitations associated with the L-MIND trial, the clinical experts indicated that the current evidence is insufficient to suggest that tafasitamab plus lenalidomide may be considered curative or associated with long-term remission. |
Assumption that 28-day monitoring costs would be lower for tafasitamab plus lenalidomide (less than half) compared with all other comparators | Feedback from the clinical experts consulted by CADTH indicated that monitoring costs for tafasitamab plus lenalidomide should be similar to and, in some cases, greater than current standard therapies. This input does not have a notable impact on the results. |
Assumption that administration costs would be lower for tafasitamab plus lenalidomide compared with all other comparators | Feedback from public drug plans noted that tafasitamab plus lenalidomide is a complex product to prepare, therefore the assumption that administration costs would be lower for tafasitamab plus lenalidomide is unlikely to be accurate. Furthermore, the source of chemotherapy costs may not reflect the same administration costs, as these costs were applied to each component of a regimen. This input does not have a notable impact on the results. |
GDP = gemcitabine plus dexamethasone plus cisplatin; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab plus gemcitabine plus oxaliplatin.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH could not adequately assess the efficacy of tafasitamab plus lenalidomide compared with any of the relevant comparators (e.g., R-GemOx, R-GDP, GDP, pola-BR) due to the limitations associated with the comparative clinical evidence. In view of the uncertainty associated with the comparative efficacy data, the CADTH clinical review could not draw any conclusions regarding the efficacy of tafasitamab plus lenalidomide compared to other therapies used to treat patients with R/R DLBCL who are ineligible for ASCT. The clinical experts consulted for this review also expressed concerns regarding the clinical implausibility of the results derived from the model. As a result, the cost-effectiveness of tafasitamab plus lenalidomide in patients with R/R DLBCL who are ineligible for ASCT is unknown.
CADTH identified basic input errors in the sponsor’s economic evaluation results, which are reported in Table 9, Appendix 4. These changes increase the sequential ICER for tafasitamab plus lenalidomide from $199,353 per QALY to $228,224 per QALY when compared to GDP (the other treatment on the cost-effectiveness frontier). These estimates should not be construed as the CADTH base case.
Exploratory Analysis Results
CADTH undertook exploratory analyses of some of the inputs associated with substantial uncertainty to highlight the impact of these components on the cost-effectiveness of tafasitamab plus lenalidomide. Descriptions of the exploratory analyses are provided in Appendix 4 and included alternative efficacy assumptions as well as pola-BR.
The results of these analyses suggested that the ICER for tafasitamab plus lenalidomide ranged from $225,000 to $490,000, if tafasitamab plus lenalidomide was considered to have an incremental benefit compared with all comparators. When an incremental benefit associated with tafasitamab plus lenalidomide was removed, tafasitamab plus lenalidomide was dominated (i.e., resulted in greater costs but no additional QALYs) by comparator treatments.
CADTH conducted price-reduction scenarios on the corrected sponsor’s base case, which suggested that a price reduction of 90% is required for tafasitamab plus lenalidomide to be considered cost-effective at a $50,000 per QALY WTP threshold (Table 5). Even a price reduction of this magnitude may underestimate the price reduction needed to achieve an ICER of $50,000 per QALY, given the limitations associated with the clinical evidence, which appear to overpredict the QALYs associated with tafasitamab plus lenalidomide that could not be appropriately addressed by CADTH. CADTH explored price reduction in exploratory analyses, which found that, even at a 99% price reduction, tafasitamab plus lenalidomide was not cost-effective at a $50,000 per QALY WTP threshold.
Table 5: CADTH Price-Reduction Analyses
Analysis | ICERs for tafasitamab plus lenalidomide vs. chemotherapy/chemoimmunotherapy (GDP) ($/QALY) |
|---|---|
Price reduction | Corrected sponsor’s base case |
No price reduction | 228,224 |
20% | 189,680 |
40% | 150,583 |
60% | 109,090 |
80% | 69,356 |
90% | 49,132 |
GDP = gemcitabine plus dexamethasone plus cisplatin; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Price-reduction analysis is vs. GDP, as it was the only other treatment on the cost-effectiveness frontier.
Note: Price reduction is applied only to tafasitamab, as tafasitamab is the base product submitted for review, and lenalidomide is available as a generic.
Issues for Consideration
Health Canada gave tafasitamab (in combination with lenalidomide) an NOC/c, pending the results of trials to verify its clinical benefit.2,16 The sponsor’s Letter of Undertaking indicated that the planned study to verify the clinical benefit of tafasitamab was in a different population than the original NOC/c (previously untreated, high-intermediate and high-risk patients with newly diagnosed DLBCL) and that the drug would be used in combination with both lenalidomide and R-CHOP.18 This would represent a different population than the current population being assessed; therefore, validation of the efficacy of the submitted regimen in the population under review is not currently forthcoming.
The clinical experts consulted for this review were concerned that use of tafasitamab plus lenalidomide may limit or be limited by the availability and use of other treatments that act on the CD19 pathway (i.e., CAR T-cell therapy). Specifically, the clinical experts consulted by CADTH noted that exposure to tafasitamab or any other CD19 antibody would make a patient ineligible for CD19 CAR T-cell therapy in Ontario. This would also likely preclude use of tafasitamab after prior CAR T-cell therapy, narrowing the eligible patient population who would likely receive this treatment and potentially limiting the treatment options for patients.
The product monograph states that tafasitamab should be used until treatment progression.2 Other relevant comparator treatments for patients with R/R DLBCL not eligible for ASCT are typically given on a time-limited basis. As time to progression is unclear, duration of treatment is also unclear and longer than currently available options, resulting in greater uncertainty in estimates of total cost of treatment for tafasitamab plus lenalidomide.
Feedback from the CADTH-participating drug plans indicated that rituximab and lenalidomide are not currently funded in this population in most jurisdictions. If tafasitamab were to become available for use in this population, this may have additional implementation considerations due to changes to listing requirements for lenalidomide. Drug plans also noted that lenalidomide is not currently available for this patient population in most jurisdictions, that additional prescriber steps would be required to dispense the product, and that issues relating to interchangeability may arise.
Overall Conclusions
The CADTH clinical review noted that there are significant limitations with the L-MIND trial, as a phase II, open-label single-arm design, with a small sample size and descriptive analysis of data—i.e., no statistical hypotheses were tested. Due to the absence of a comparator arm and statistical testing, CADTH can draw no definitive conclusions regarding the efficacy of tafasitamab plus lenalidomide based on the L-MIND trial. CADTH also noted the commentary from Health Canada that the NOC/c for tafasitamab plus lenalidomide was given pending the results of additional trials to verify its clinical benefit. The authorization was based on overall response rate, complete response rate, and durability of response. An improvement in PFS or OS has not been established based on the evidence reviewed. As the sponsor’s economic evaluation was based on a partitioned survival model that considered pre-progression (based on PFS), post-progression (based on the difference between PFS and OS), and death, the clinical benefit predicted for tafasitamab plus lenalidomide based on this approach is highly uncertain.
The CADTH clinical review noted that, although results from the indirect comparative evidence submitted by the sponsor suggested that tafasitamab plus lenalidomide therapy may be associated with an improvement in clinical outcomes compared with relevant comparators (e.g., R-GemOx, pola-BR), the indirect evidence is associated with substantial risk of bias due to important limitations (including methodological limitations, heterogeneity, matching based on a limited number of variables, and small sample sizes). In view of the uncertainty in the indirect evidence results, CADTH can draw no conclusions on the efficacy and safety of tafasitamab plus lenalidomide compared to other therapies used to treat patients with R/R DLBCL who are ineligible for ASCT.
In addition to the aforementioned limitations regarding the clinical effectiveness, comparative effectiveness, and economic model structure, CADTH identified several additional limitations with the sponsor’s model, including inappropriate cost and resource use assumptions, a lack of transparency with the model (approximately 25,000 IFERROR statements and a lack of transparent trace to derive the deterministic results), exclusion of a key comparator (pola-BR) from the base case, and incorporation of clinical evidence. While CADTH could correct for obvious biases in the model, the lack of comparative evidence for tafasitamab plus lenalidomide remains, and the evidence for PFS and OS are evolving. CADTH estimated an ICER for tafasitamab plus lenalidomide of $228,224 per QALY compared with GDP (the least costly, reference treatment). This estimate is based on the sponsor’s clinical assumptions that predict an additional 3.46 life-years and 2.52 QALYs. These estimates are highly uncertain, given the limitations of the sponsor’s model. Based on the CADTH-corrected sponsor’s analysis, a price reduction of 90% is required to achieve an ICER of $50,000 per QALY. When this benefit in survival is not realized, a greater price reduction would be required for tafasitamab plus lenalidomide to be considered cost-effective at a $50,000 per QALY threshold. Additional price reductions based on exploratory analyses indicated that, with a 99% price reduction, tafasitamab plus lenalidomide was still not on the cost-effectiveness frontier. As tafasitamab is the only relevant treatment included in the model that is not given for a limited duration, the total relative cost of treatment is highly dependent upon the duration of treatment, which is currently uncertain.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Minjuvi (tafasitamab): lyophilized powder for solution for infusion (200 mg / vial). Pointe Claire (QC): Incyte Biosciences Canada; 2021 Nov 19. Evidera; 2021.
2.PrMinjuviTM (tafasitamab): lyophilized powder for solution for injection (200 mg single-use vial) [product monograph]. Wilmington (DE): Incyte; 2021 Aug 19.
3.Clinical Study Report: MOR208C203. A phase II, single-arm, open-label, multicentre study to evaluate the safety and efficacy of lenalidomide combined with MOR00208 in patients with relapsed or refractory diffuse large B-cell lymphoma (R-R DLBCL) [internal sponsor's report]. Pointe Claire (QC): Incyte Biosciences Canada; 2019.
4.Updated MAIC of tafasitamab + lenalidomide against comparators in R/R DLBCL using the L-MIND October 2020 datacut: technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Minjuvi (tafasitamab): lyophilized powder for solution for infusion (200 mg / vial). Pointe Claire (QC): Incyte Biosciences Canada; 2021 Nov 19.
5.Clinical Study Report: MOR208C213. An observational retrospective cohort study of systemic therapies for relapsed or refractory diffuse large B-cell lymphoma (R/R DLBCL), to compare outcomes to those from tafasitamab + lenalidomide in the L-MIND study (RE-MIND2) [internal sponsor's report]. Pointe Claire (QC): Incyte Biosciences Canada; 2021.
6.Mounier N, El Gnaoui T, Tilly H, et al. Rituximab plus gemcitabine and oxaliplatin in patients with refractory/relapsed diffuse large B-cell lymphoma who are not candidates for high-dose therapy. A phase II Lymphoma Study Association trial. Haematologica. 2013;98(11):1726-1731. PubMed
7.Sehn LH, Salles G. Diffuse large B-cell lymphoma. N Engl J Med. 2021;384(9):842-858. PubMed
8.Polatuzumab vedotin with rituximab and bendamustine for treating relapsed or refractory diffuse large B-cell lymphoma [ID1576]: committee papers. London (UK): National Institute for Health and Care Excellence (NICE); 2020: https://www.nice.org.uk/guidance/ta649/evidence/committee-papers-pdf-8840199997. Accessed 2021 Dec 28.
9.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
10.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2021 Dec 3.
11.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2021 Dec 3.
12.Tisagenlecleucel for treating relapsed or refractory diffuse large B-cell lymphoma after 2 or more systemic therapies. NICE technology appraisal guidance [TA567]. London (UK): National Institute of Health and Care Excellence (NICE); 2019: https://www.nice.org.uk/guidance/ta567. Accessed 2021 Dec 28.
13.Primary market research in diffuse large B cell lymphoma (DLBCL) to support economic evaluation of tafasitamab (Minjuvi™): technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Minjuvi (tafasitamab): lyophilized powder for solution for infusion (200 mg / vial). Pointe Claire (QC): Incyte Biosciences Canada; 2021 Nov 19.
14.de Olivera C, Pataky R, Bremner KE, al. e. Phase-specific and lifetime costs of cancer care in Ontario, Canada. BMC Cancer. 2016;16(1):1-12. PubMed
15.Incyte Biosciences Canada response to February 2, 2022 DRR request for additional information regarding tafasitamab DRR review [internal additional sponsor's information]. Pointe Claire (Quebec): Incyte Biosciences Canada; 2022.
16.Notice of Compliance with conditions: Minjuvi (tafasitamab) 200 mg / vial. Ottawa (ON): Health Canada; 2021 Aug 19.
17.Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol. 2020;38(2):155-165. PubMed
18.Letter of Undertaking: tafasitamab (Minjuvi) 200 mg / vial. Wilmington (DE): Incyte; 2021.
19.DeltaPA. [Ottawa (ON)]: IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Dec 28.
20.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Dec 28.
21.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Dec 28.
22.Funded evidence-informed regimens. 2021; https://www.cancercareontario.ca/en/drugformulary/regimens. Accessed 2021 Dec 28.
23.Polatuzumab vedotin (Polivy) for DLBCL. pCODR expert review committee (pERC) final recommendation. Ottawa (ON): CADTH; 2021: https://cadth.ca/sites/default/files/pcodr/Reviews2021/10227PolatuzumabDLBCL_fnRec_EC21Apr2021_final.pdf. Accessed 2021 Dec 28.
24.Chemotherapy protocol: lymphoma cyclophosphamide-etoposide oral. Southampton (UK): NHS University Hospital Southampton; 2015.
25.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Minjuvi (tafasitamab): lyophilized powder for solution for infusion (200 mg / vial). Pointe Claire (QC): Incyte Biosciences Canada; 2021 Nov 19.
26.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics 2019. Toronto (ON): Canadian Cancer Society; 2019: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2019-statistics/canadian-cancer-statistics-2019-en.pdf?rev=11e32d8aa5b344a09184fc6d463f1e63&hash=3FAAF41B5247B50C59C6E4AA827B656E&_ga=2.195584236.1305238684.1643896630-911802007.1632235443. Accessed 2022 Jan 17.
27.Raut LS, Chakrabarti PP. Management of relapsed-refractory diffuse large B cell lymphoma. South Asian J Cancer. 2014;3(1):66-70. PubMed
28.Diffuse large B cell lymphoma (DLBCL). Mississauga (ON): Lymphoma Canada; 2020: https://www.lymphoma.ca/diffuse-large-b-cell-lymphoma/. Accessed 2022 Jan 17.
29.INESSS. Tisagenlecleucel pour le traitement du lymphome diffus à grandes cellules B récidivant ou réfractaire. Québec, QC: Gouvernement du Québec,; 2019: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Rapports/Therapies_cellulaires/INESSS_Avis_Kymriah_LDGCB.pdf. Accessed 2022 Jan 18.
30.Chaganti S, Illidge T, Barrington S, et al. Guidelines for the management of diffuse large B-cell lymphoma. Br J Haematol. 2016;174(1):43-56. PubMed
31.Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood. 2010;116(12):2040-2045. PubMed
32.Tilly H, Gomes da Silva M, Vitolo U, et al. Diffuse large B-cell lymphoma (DLBCL): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v116-125. PubMed
33.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics 2021. Toronto (ON): Canadian Cancer Society; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf?rev=2b9d2be7a2d34c1dab6a01c6b0a6a32d&hash=01DE85401DBF0217F8B64F2B7DF43986&_ga=2.24767994.1305238684.1643896630-911802007.1632235443. Accessed 2022 Jan 17.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and CADTH-participating drug plans. Comparators may be recommended practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CADTH Cost Comparison Table for R/R DLBCL
Treatment | Strength / concentration | Form | Price | Recommended dosage | Cost per cycle or 1-time use | Cost per 28 days | |||
|---|---|---|---|---|---|---|---|---|---|
Tafasitamab plus lenalidomide | |||||||||
Tafasitamab (Minjuvi) | 200 mg vial | Powder for IV infusion | 1,167.8600a | 28-day cycles: 12 mg/kg on days 1, 4, 8, 15 & 22 for the first cycle, then on days 1, 8, 15 & 22 for the second and third cycles, then on days 1 & 15 for the fourth cycle and beyond | first cycle: 29,196 second & third cycles: 23,357 fourth+ cycle: 11,679 | first cycle: 29,196 second & third cycles: 23,357 fourth+ cycle: 11,679 | |||
Lenalidomide (Generics) | 2.5 mg 5 mg 10 mg 15 mg 20 mg 25 mg | Tablet | 82.3752 85.0000 90.2500 95.5000 100.7500 106.0000 | 28-day cycles: 25 mg on days 1 to 21 for up to 12 cycles | 2,226 | 2,226 | |||
Tafasitamab plus lenalidomide regimen cost (28-day cycle) | first: 31,422 second-third: 25,583 fourth-12th: 13,905 13th+: 11,679 | first: 31,422 second-third: 25,583 fourth-12th: 13,905 13th+: 11,679 | |||||||
Pola-BR | |||||||||
Bendamustine (generics) | 25 mg vial 100 mg vial | Powder for IV infusion | 250.0000 1,000.0000 | 21- or 28- day cycles: 90 mg/m2 on days 1 and 2c | 3,500 | 3,500 to 4,667 | |||
Rituximab (biosimilars) | 100 mg vial 500 mg vial | IV infusion | 297.0000b 1,485.0000b | 21- or 28- day cycles: 375 mg/m2 on day 1c | 2,079 | 2,079 to 2,772 | |||
Polatuzumab (Polivy) | 140 mg vial | Lyophilized powder for solution | 14,750.0000d | Per 21-day cycle: 1.8 mg/kg on day 1c | 14,750 | 19,667 | |||
BR regimen cost (28-day cycle) | 5,579 | 5,579 | |||||||
Pola-BR regimen cost (21-day cycle) | 20,329 | 27,105 | |||||||
Cyclophosphamide plus etoposide | |||||||||
Cyclophosphamide (Procytox) | 25 mg 50 mg | Tablet | 0.3545b 0.4773b | 21-day cycles: 100 mg on days 1 through 5e | 5 | 6 | |||
Etoposide (Vepesid) | 50 mg | Capsule | 41.5875b | 21-day cycles: 100 mg on days 1 through 5e | 416 | 555 | |||
CE regimen cost (21-day cycle) | 421 | 561 | |||||||
DHAP(R) | |||||||||
Dexamethasone (generic) | 4 mg | Tablet | 0.3046b | 21- or 28-day cycles: 40 mg days 1 to 4c | 12 | 12 to 16 | |||
Cytarabine (generic) | 500 mg 2,000 mg | 100 mg/mL IV solution | 76.8500 306.5000 | 21- or 28-day cycles: 2000 mg/m2 every 12 hours on Day 2c | 1,228 | 1,228 to 1,637 | |||
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/mL IV solution | 135.0000 270.0000 | 21- or 28-day cycles: 100 mg/m2 on Day 1c | 540 | 540 to 720 | |||
Rituximab (biosimilars) | 100 mg vial 500 mg vial | IV infusion | 297.0000b 1,485.0000b | 21- or 28-day cycles: 375 mg/m2 on day 1c | 2,079 | 2,079 to 2,772 | |||
DHAP regimen cost (21- or 28-day cycle) | 1,780 | 1,780 to 2,373 | |||||||
R-DHAP regimen cost (21- or 28-day cycle) | 3,859 | 3,859 to 5,145 | |||||||
GDP(R) | |||||||||
Gemcitabine (generic) | 1,000 mg 2,000 mg | Lyophilized powder | 270.0000 540.0000 | 21-day cycles: 1,000 mg/m2 days 1 and 8c | 1,080 | 1,440 | |||
Dexamethasone (generics) | 4 mg tab | Tablet | 0.3046b | 21-day cycles: 40 mg days 1 to 4c | 12 | 16 | |||
Cisplatin (generic) | 50 mg vial 100 mg vial | 1 mg/mL solution for injection | 135.0000 270.0000 | 21-day cycles: 75 mg/m2 on Day 1c | 405 | 540 | |||
Rituximab (biosimilars) | 100 mg vial 500 mg vial | IV infusion | 297.0000b 1,485.0000b | 21-day cycles: 375 mg/m2 on day 1c | 2,079 | 2,772 | |||
GDP regimen cost (21-day cycle) | 1,497 | 1,996 | |||||||
R-GDP regimen cost (21-day cycle) | 3,576 | 4,768 | |||||||
Gemcitabine monotherapy | |||||||||
Gemcitabine (generic) | 1,000 mg 2,000 mg | Lyophilized powder | 270.0000 540.0000 | 21- or 28-day cycles: 1,200 mg/m2 days 1 and 8c | 1,080 | 1,080 to 1,440 | |||
Gemcitabine regimen cost (21- or 28- day cycle) | 1,080 | 1,080 to 1,440 | |||||||
GemOx | |||||||||
Gemcitabine (generic) | 1,000 mg 2,000 mg | Lyophilized powder | 270.0000 540.0000 | 21-day cycles: 1,200 mg/m2 on Day 1c | 810 | 1,080 | |||
Oxaliplatin (generic) | 100 mg 200 mg | 5 mg/mL solution for injection | 72.54 145.08 | 21-day cycles: 120 mg/m2 on Day 1c | 145 | 290 | |||
GemOx regimen cost (21-day cycle) | 1,028 | 1,370 | |||||||
R-GemOx | |||||||||
Gemcitabine (generic) | 1,000 mg 2,000 mg | Lyophilized powder | 270.0000 540.0000 | 14-day cycles: 1,000 mg/m2 on Day 1c | 540 | 1,080 | |||
Oxaliplatin (generic) | 100 mg 200 mg | 5 mg/mL Solution for injection | 72.54 145.08 | 14-day cycles: 100 mg/m2 on Day 1c | 145 | 290 | |||
Rituximab (biosimilars) | 100 mg vial 500 mg vial | IV infusion | 297.0000b 1,485.0000b | 14-day cycles: 375 mg/m2 on day 1c | 2,079 | 4,158 | |||
R-GemOx regimen cost (14-day cycle) | 2,764 | 5,528 | |||||||
ICE(R) | |||||||||
Ifosfamide (Ifex) | 1,000 mg vial 3,000 mg vial | Powder for solution | 131.7500 403.4700 | 21- or 28-day cycles:1,667 mg/m2 on days 1 to 3c | 1,186 | 1,186 to 1,581 | |||
Carboplatin (generic) | 50 mg 150 mg 450 mg 600 mg | 10 mg/mL vial for injection | 70.0000 210.0000 600.0000 775.0020 | 21- or 28- day cycles: AUC 5 on day 1; maximum dose for AUC 5 is 750 mgc | Max: 985 | Max: 985 to 1,313 | |||
Etoposide (generic) | 100 mg | 20 mg/mL injection | 75.0000 | 21- or 28-day cycles: 100 mg/m2 on days 1 to 3c | 450 | 450 to 600 | |||
Rituximab (biosimilars) | 100 mg vial 500 mg vial | IV infusion | 297.0000b 1,485.0000b | 21-day cycles: 375 mg/m2 on day 1c | 2,079 | 2,772 | |||
ICE regimen cost (21- or 28-day cycle) | 2,621 | 2,621 to 3,494 | |||||||
ICE-R regimen cost (21-day cycle) | 4,700 | 6,266 | |||||||
PEP-C | |||||||||
Procarbazine (Matulane) | 50 mg | Capsule | 61.1031b | 28-day cycles: 60 mg/m2 days 1 to 10c | 1,833 | 1,833 | |||
Etoposide (generic) | 100 mg 200 mg 500 mg 1,000 mg | 20 mg/mL injection | 75.0000 150.0000 375.0000 750.0000 | 28-day cycles: 70 mg/m2 day 1c | 150 | 150 | |||
Etoposide (Vepesid) | 50 mg | Capsule | 41.5875b | 28-day cycles: 140 mg/m2 on days 2 and 3c | 416 | 416 | |||
Prednisone (generic) | 5 mg 50 mg | Tablet | 0.0220b 0.1735b | 28-day cycles: 60 mg/m2 days 1 to 10c | 4 | 4 | |||
Cyclophosphamide (Procytox) | 500 mg 1,000 mg 2,000 mg | Powder for injection | 93.1400 168.8300 310.6000 | 28-day cycles: 600 to 750 mg/m2 days 1 and 8c | 524 | 524 | |||
PEP-C regimen cost (28-day cycle) | 2,927 | 2,927 | |||||||
R-CEOP | |||||||||
Cyclophosphamide (Procytox) | 500 mg 1,000 mg 2,000 mg | Powder for injection | 93.1400 168.8300 310.6000 | 21-day cycles: 750 mg/m2 day 1c | 262 | 349 | |||
Etoposide (generics) | 100 mg 200 mg 500 mg 1,000 mg | 20 mg/mL injection | 75.0000 150.0000 375.0000 750.0000 | 21-day cycles: 50 mg/m2 on day 1c | 75 | 100 | |||
Etoposide (Vepesid) | 50 mg | Capsule | 41.5875b | 21-day cycles: 100 mg/m2 daily days 2 & 3c | 333 | 444 | |||
Vincristine (generic) | 1 mg 2 mg 5 mg | 1 mg/mL injection | 30.6000 62.0000 153.0000 | 21-day cycles: 1.4 mg/m2 on day 1, max 2 mgc | 62 | 83 | |||
Prednisone (generic) | 5 mg 50 mg | Tablet | 0.0220b 0.1735b | 21-day cycles: 100 mg days 1 to 5c | 2 | 2 | |||
Rituximab (biosimilars) | 100 mg vial 500 mg vial | IV infusion | 297.0000b 1,485.0000b | 21-day cycles: 375 mg/m2 on day 1c | 2,079 | 2,772 | |||
R-CEOP regimen cost (21-day cycle) | 2,812 | 3,750 | |||||||
CAR T-cell therapiesf | |||||||||
Axicabtagene ciloleucel (Yescarta) | Refer to dosage | Suspension for IV infusion | No public price available | Target of 2 x 106 anti-CD19 CAR T-cells/kg body weight (range: 1 x 106 to 2.4 x 106 cells/kg) to a maximum of 2 x 108 anti-CD19 CAR T-cells | No public price available | NA | |||
Tisagenlecleucel (Kymriah) | Refer to dosage | Suspension for IV infusion | No public price available | 0.6 to 6.0 x 108 CAR-positive viable T-cells (non-weight based) | No public price available | NA | |||
CAR T-cell = chimeric antigen receptor T-cell; NA = not applicable.
Note: All prices are wholesale from IQVIA Delta PA (accessed December 2021),19 unless otherwise indicated, and do not include dispensing fees. Calculations assume a patient body weight of 75kg and a body surface area of 1.8 m2. Recommended dosing is from the respective product monographs unless otherwise indicated.
aSponsor’s submitted price.1
bList price from the Ontario Drug Benefit formulary or the Ontario Drug Benefit Exceptional Access Program (accessed December 2021).20,21
cCancer Care Ontario Regimens Database.22
dPrice submitted during CADTH’s review of Polivy.23
eNational Health Service (UK) regimen.24
fLisocabtagene maraleucel (Breyanzi) is also currently under review by CADTH for the treatment of R/R DLBCL at the time of this review.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CADTH Appraisal |
Model has been adequately programmed and has sufficient face validity | No | Refer to CADTH Appraisal |
Model structure is adequate for decision problem | No | Refer to CADTH Appraisal |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CADTH Appraisal |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | None |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The submitted technical reports and models were convoluted and overly complex. The data inputs included in the models did not appear to align with the clinical evidence identified. |
HR = hazard ratio; MAIC = matching-adjusted indirect comparison; pola-BR = polatuzumab plus bendamustine plus rituximab.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
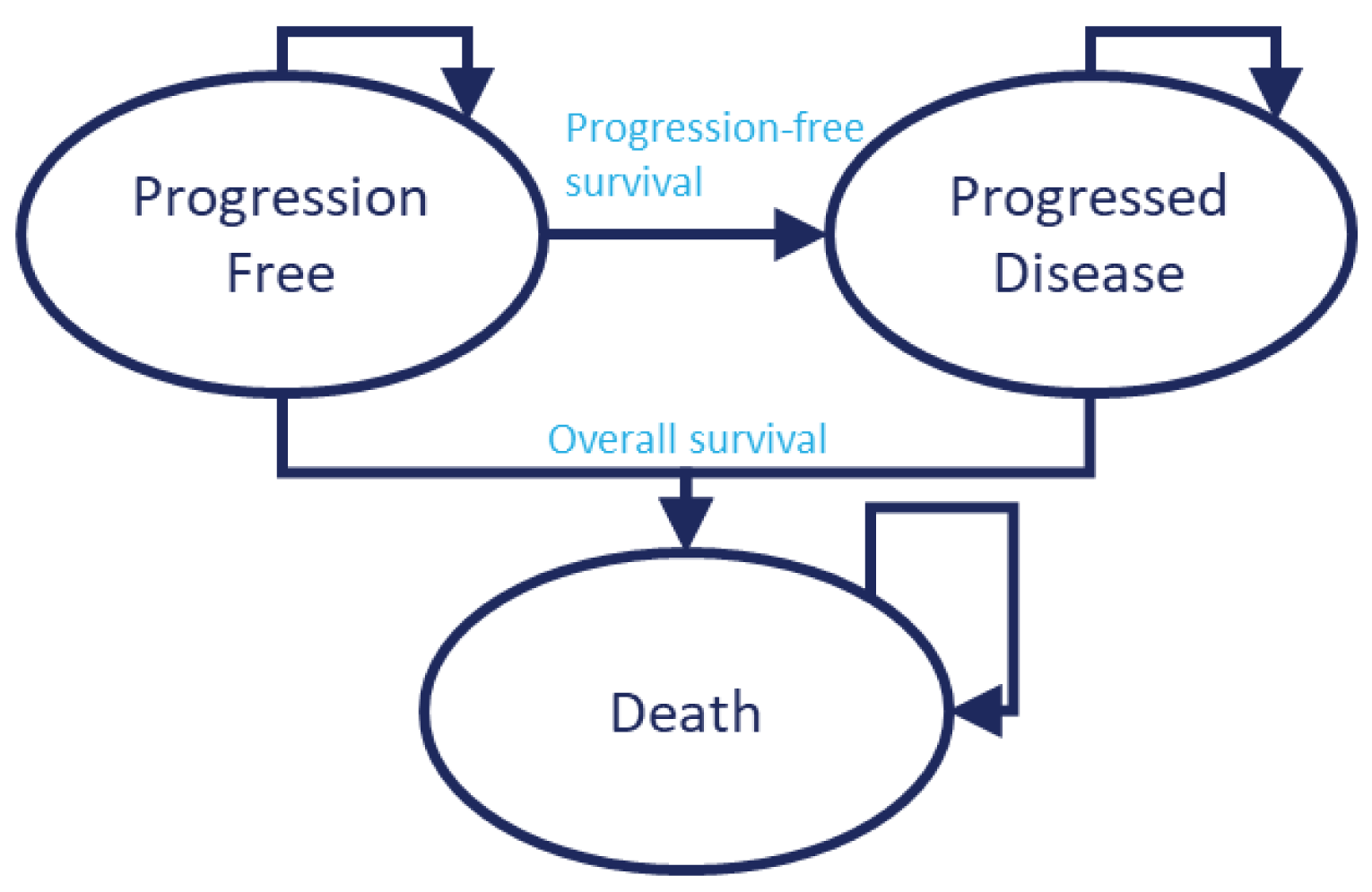
Detailed Results of the Sponsor’s Base Case
Table 8: Disaggregated Summary of Sponsor’s Economic Evaluation Results
Treatment | Component | Value | Incremental (versus reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
GDP | Progression-free | 0.73 | NA | NA |
Post-progression | 1.17 | NA | NA | |
Total | 1.90 | NA | NA | |
R-GemOx | Progression-free | 0.73 | 0 | NA |
Post-progression | 1.18 | 0.01 | NA | |
Total | 1.91 | 0.01 | NA | |
R-GDP | Progression-free | 0.73 | 0 | 0 |
Post-progression | 1.17 | 0 | –0.01 | |
Total | 1.90 | 0 | –0.01 | |
tafasitamab plus lenalidomide | Progression-free | 4.59 | 3.86 | 3.86 |
Post-progression | 0.77 | –0.40 | –0.40 | |
Total | 5.37 | 3.47 | 3.47 | |
Discounted QALYs | ||||
GDP | Progression-free | 0.53 | NA | NA |
Post-progression | 0.76 | NA | NA | |
Total | 1.29 | NA | NA | |
R-GemOx | Progression-free | 0.53 | 0 | NA |
Post-progression | 0.77 | 0.01 | NA | |
Total | 1.29 | 0 | NA | |
R-GDP | Progression-free | 0.53 | 0 | 0 |
Post-progression | 0.76 | 0 | –0.01 | |
Total | 1.29 | 0 | 0 | |
tafasitamab plus lenalidomide | Progression-free | 3.31 | 2.78 | 2.78 |
Post-progression | 0.50 | –0.26 | –0.26 | |
Total | 3.81 | 2.52 | 2.52 | |
Discounted costs ($) | ||||
GDP | Acquisition – induction | 1,248 | NA | NA |
Acquisition – maintenance | 0 | NA | NA | |
Administration – induction | 2,893 | NA | NA | |
Administration – maintenance | 0 | NA | NA | |
Co-medication – induction | 1,353 | NA | NA | |
Co-medication – maintenance | 0 | NA | NA | |
Monitoring | 1,536 | NA | NA | |
AEs | 146 | NA | NA | |
Disease management – pre-progression | 6,787 | NA | NA | |
Disease management – post-progression | 26,753 | NA | NA | |
Disease management – death | 71,851 | NA | NA | |
Subsequent treatment | 51,380 | NA | NA | |
Total | 163,948 | NA | NA | |
R-GemOx | Acquisition – induction | 15,496 | 14,248 | NA |
Acquisition – maintenance | 8,047 | 8,047 | NA | |
Administration – induction | 2,429 | –464 | NA | |
Administration – maintenance | 1,262 | 1,262 | NA | |
Co-medication – induction | 809 | –544 | NA | |
Co-medication – maintenance | 0 | 0 | NA | |
Monitoring | 1,536 | 0 | NA | |
AEs | 146 | 0 | NA | |
Disease management – pre-progression | 6,870 | –7 | NA | |
Disease management – post-progression | 27,008 | 255 | NA | |
Disease management – death | 71,832 | 0 | NA | |
Subsequent treatment | 60,958 | –19 | NA | |
Total | 196,303 | 32,355 | ||
R-GDP | Acquisition – induction | 15,105 | 13,587 | –391 |
Acquisition – maintenance | 0 | 0 | –8,047 | |
Administration – induction | 3,903 | 1,010 | 1,474 | |
Administration – maintenance | 0 | 0 | –1,262 | |
Co-medication – induction | 1,351 | –2 | 542 | |
Co-medication – maintenance | 0 | 0 | 0 | |
Monitoring | 1,529 | –7 | –7 | |
AEs | 146 | 0 | 0 | |
Disease management – pre-progression | 6,756 | –31 | –24 | |
Disease management – post-progression | 26,846 | 93 | –162 | |
Disease management – death | 71,851 | 0 | 19 | |
Subsequent treatment | 73,057 | 0 | 12,099 | |
Total | 200,545 | 36,597 | 4,242 | |
Tafasitamab plus lenalidomide | Acquisition – induction | 149,279 | 148,031 | 134,174 |
Acquisition – maintenance | 370,179 | 370,179 | 370,179 | |
Administration – induction | 4,813 | 1,920 | 910 | |
Administration – maintenance | 13,301 | 13,301 | 13,301 | |
Co-medication – induction | 9,644 | 8,291 | 8,293 | |
Co-medication – maintenance | 18,662 | 18,662 | 18,662 | |
Monitoring | 5,871 | 4,335 | 4,342 | |
AEs | 1,821 | 1,675 | 1,675 | |
Disease management – pre-progression | 7,728 | 941 | 972 | |
Disease management – post-progression | 16,490 | –10,263 | –10,356 | |
Disease management – death | 62,929 | –8,922 | –8,922 | |
Subsequent treatment | 6,302 | –45,078 | –66,755 | |
Total | 667,021 | 503,073 | 466,476 | |
Treatment | ICER versus reference ($) | Sequential ICER ($) | ||
GDP | Ref. | Ref. | ||
R-GemOx | Not estimablea | Extendedly dominated through GDP and tafasitamab plus lenalidomide | ||
R-GDP | Not estimablea | Dominated by GDP | ||
Tafasitamab plus lenalidomide | 199,353 | 199,353 | ||
GDP = gemcitabine, dexamethasone, and cisplatin; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; Ref. = reference.
aThe sponsor’s report assumed tafasitamab plus lenalidomide as the reference product which is an inappropriate assumption. CADTH reanalyzed the results to derive the majority of results presented in this table, with the exception of the sequential ICER.
Note: the results may not tally up. These differences are likely due to rounding.
Source: adapted from the Sponsor’s Pharmacoeconomic Report.1
Figure 2: PFS Curves from Sponsor’s Economic Evaluation Base Case

Note: This figure was redacted at the request of the sponsor.
Source: Sponsor’s Pharmacoeconomic Submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Corrected Sponsor’s Base-Case Analyses
CADTH identified several errors in drug prices. CADTH undertook revisions to the sponsor’s drug prices to align with the CADTH Cost Table (Table 6).
Additionally, the sponsor assumed lower health care/disease management costs for tafasitamab plus lenalidomide for patients in the PFS and post-progression health states compared with all other comparators. Discussion with the clinical experts consulted by CADTH noted that as these are health state costs, there should be no differences in between treatments for these disease management costs. As such, the health state costs were aligned with those for R-GemOx (and other chemotherapies).
Finally, the sponsor assumed a different set of subsequent treatments based on the treatment being assessed. While this makes sense in some manner (e.g., tafasitamab plus lenalidomide patients are unlikely to receive CAR T-cell therapy due to both treatments impact on CD-19), in other areas, these assumptions have not been appropriately justified (e.g., no subsequent ASCT for tafasitamab plus lenalidomide, while 10% to 22% of patients on other therapies receive ASCT). Furthermore, these differences in subsequent treatments were assumed to result in the same clinical effects. Feedback from the clinical experts consulted for this review indicated that these were not accurate assumptions. As such, an assumption of equivalent subsequent treatments (in the absence of appropriate clinical evidence) has been incorporated.
Table 9: CADTH Corrections to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Bendamustine price | $263.35 per 25 mg | $250.00 per 25 mg |
2. Gemcitabine price | $12.40 per 200 mg | $54.00 per 200 mg |
3. Oxaliplatin price | $10.20 per 1 mg $1,865.20 per 5 mg (40 units) | $72.54 per 100 mg $145.08 per 200 mg |
4. Cisplatin price | $19.00 per 100 mg $9.50 per 50 mg | $270.00 per 100 mg $135.00 per 50 mg |
5. Disease Management costs per cycle | tafasitamab plus lenalidomide, PFS: $128.86 tafasitamab plus lenalidomide, post-progression: $1,633.36 | tafasitamab plus lenalidomide, PFS: $707.73 tafasitamab plus lenalidomide, post-progression: $1,760.37 (i.e., Equal to R-GDP, GDP, and R-GemOx) |
6. Subsequent treatment cost assumptions | tafasitamab plus lenalidomide: $7,399.24 R-GDP: $61,383.50 GDP: $51,665.56 R-GemOx: $73,381.19 | All assumptions informing these outputs assumed the same (equivalent to tafasitamab plus lenalidomide). |
Corrected sponsor’s base case | 1+2+3+4+5+6 | |
GDP = gemcitabine, dexamethasone, and cisplatin; PFS = progression-free survival; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab plus gemcitabine plus oxaliplatin.
Results of CADTH-Corrected Sponsor’s Base Case
Corrections to the sponsor’s model result in an increase to the ICER (Table 10).
Table 10: Summary of the Stepped Analysis of the CADTH-Corrected Analysis Results
Stepped analysis | Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|---|
Sponsor’s base case | GDP | 163,948 | 1.91 | 1.29 | Ref. |
tafasitamab plus lenalidomide | 667,021 | 5.37 | 3.81 | 199,353 | |
Corrections 1-4: Drug costs | GDP | 171,973 | 1.89 | 1.29 | Ref. |
tafasitamab plus lenalidomide | 664,823 | 5.36 | 3.80 | 195,499 | |
Correction 5: Health state costs | GDP | 164,260 | 1.91 | 1.30 | Ref. |
tafasitamab plus lenalidomide | 669,854 | 5.31 | 3.77 | 204,082 | |
Correction 6: Subsequent treatment costs | GDP | 119,901 | 1.90 | 1.29 | Ref. |
tafasitamab plus lenalidomide | 667,718 | 5.37 | 3.81 | 217,428 | |
Corrected base case (1+2+3+4+5+6) | GDP | 124,587 | 1.91 | 1.30 | Ref. |
tafasitamab plus lenalidomide | 700,466 | 5.37 | 3.82 | 228,224 |
GDP = gemcitabine, dexamethasone, and cisplatin; ICER = incremental cost-effectiveness ratio; QALY= quality-adjusted life-year; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; Ref. = reference.
Exploratory Analyses
Although CADTH could not address the underlying limitations with the submitted model and clinical data, in addition to the corrections to the sponsor’s base case, CADTH undertook exploratory analyses to highlight the impact of alternate data inputs on key drivers in the model (Table 11).
Table 11: CADTH Revisions to the Submitted Economic Evaluation
Exploratory analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
1. Comparative efficacy of tafasitamab plus lenalidomide with R-GemOx (and thus R-GDP and GDP) | Various parametric distributions | Based on HRs compared to tafasitamab plus lenalidomide per REMIND2 |
2. Comparative efficacy of tafasitamab plus lenalidomide with R-GemOx (and thus R-GDP and GDP) | Various parametric distributions | Based on HRs compared to tafasitamab plus lenalidomide. PFS HR = |||| and OS HR = ||||, per lower CI from the REMIND2 study |
3. Comparative efficacy of tafasitamab plus lenalidomide with R-GemOx (and thus R-GDP and GDP) | Various parametric distributions | Based on HRs compared to tafasitamab plus lenalidomide. PFS and OS HRs = 1 (as lower CI from MAIC is less than 1) |
4. tafasitamab plus lenalidomide duration of treatment | As per trial data, extrapolated | As per product monograph (until progression) |
5. Comparative efficacy of tafasitamab plus lenalidomide with pola-BR | Pola-BR excluded | Pola-BR included, based on REMIND2, parametric curve |
6. Comparative efficacy of tafasitamab plus lenalidomide with pola-BR | Pola-BR excluded (when included, base assumption using HRs based on REMIND2): Pola-BR PFS HR of |||| Pola-BR OS HR of ||||) | Pola-BR included; HRs based on REMIND2; lower CI: Pola-BR PFS HR of |||| Pola-BR OS HR of |||| |
7. Comparative efficacy of tafasitamab plus lenalidomide with pola-BR | Pola-BR excluded (when included, base assumption using HRs based on REMIND2): Pola-BR PFS HR of |||| Pola-BR OS HR of ||||) | Pola-BR included, PFS and OS HRs = 1 based on expert opinion |
Ci = confidence interval; GDP = gemcitabine, dexamethasone, and cisplatin; ICER = incremental cost-effectiveness ratio; QALY= quality-adjusted life-year; R-GDP = rituximab, gemcitabine, dexamethasone, and cisplatin; R-GemOx = rituximab plus gemcitabine plus oxaliplatin; Ref. = reference.
Results of CADTH Exploratory Analyses
The results of all CADTH’s exploratory analyses must be viewed within the context of the base clinical information used to inform the analyses. As the model was set up with tafasitamab plus lenalidomide as the reference treatment, and baseline efficacy data from L-MIND were used as the basis of the model, all analyses overpredict the number of life-years, and thus, QALYs associated with tafasitamab plus lenalidomide. This limitation has spillover effects on to comparator treatments, as changes to hazard ratios – which were used to inform differences with the comparators – will also overpredict life-years and QALYs for the comparators. While feedback from the clinical experts consulted by CADTH indicated that, based on the available data, tafasitamab plus lenalidomide may be associated with greater health benefits compared to chemotherapy or chemoimmunotherapy regimens, and may have similar health benefits to pola-BR; CADTH were limited by the available data to assess this. The closest comparison to address the feedback from the clinical experts appears to be exploratory analysis 7, although as noted earlier, this overpredicts benefits associated with pola-BR.
Based on the REMIND2 study, the hazard ratios suggested that pola-BR was substantially less effective than the chemotherapy regimens (Table 12, Figure 2), which does not align with other available data, clinical expert opinion, and the sponsor’s caveats associated with the comparative efficacy previously noted in the CADTH critical appraisal.
CADTH undertook price reductions on the exploratory analyses, several of which indicated that even at a 99% price reduction, tafasitamab plus lenalidomide was not cost-effective at a $50,000 per QALY WTP threshold.
Table 12: Summary of the CADTH Exploratory Analysis Results
Stepped analysis | Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|---|
Sponsor’s base case | GDP | 163,948 | 1.91 | 1.29 | Ref. |
tafasitamab plus lenalidomide | 667,021 | 5.37 | 3.81 | 199,353 | |
Sponsor’s corrected base case | GDP | 124,587 | 1.91 | 1.30 | Ref. |
tafasitamab plus lenalidomide | 700,466 | 5.37 | 3.82 | 228,224 | |
CADTH EA 1: Comparative efficacy based on HRs per REMIND2 | GDP | 124,320 | 3.55 | 1.54 | Ref. |
tafasitamab plus lenalidomide | 700,439 | 5.41 | 3.82 | 250,684 | |
CADTH EA 2: Comparative efficacy based on HR per REMIND2 (lower CI) | GDP | 139,994 | 3.81 | 2.67 | Ref. |
tafasitamab plus lenalidomide | 703,711 | 5.38 | 3.81 | 490,424 | |
CADTH EA 3: Comparative efficacy based on HR = 1 | GDPa | 150,611 | 5.43 | 3.85 | Ref.a |
CADTH EA 4: Duration of tafasitamab plus lenalidomide until progression | GDP | 124,527 | 1.91 | 1.29 | Ref. |
tafasitamab plus lenalidomide | 994,475 | 5.36 | 3.80 | 346,515 | |
CADTH EA 5: Include pola-BR, REMIND2 HRs | GDP | 235,423 | 1.90 | 1.29 | Ref. |
tafasitamab plus lenalidomide | 699,946 | 5.39 | 3.82 | 227,383 | |
CADTH EA 6: Include pola-BR, REMIND2 HRs at Lower 95% CI for pola-BR compared with tafasitamab plus lenalidomide | GDP | 124,602 | 1.29 | 1.29 | Ref. |
Pola-BR | 258,622 | 4.22 | 3.01 | 72,159 | |
tafasitamab plus lenalidomide | 702,325 | 5.38 | 3.81 | 225,225 | |
CADTH EA 7: Include pola-BR, HR = 1 compared with tafasitamab plus lenalidomide | GDP | 124,174 | 1.91 | 1.29 | Ref. |
Pola-BR | 271,709 | 5.38 | 3.81 | 54,398 |
EA = exploratory analysis; GDP = gemcitabine, dexamethasone, and cisplatin; HR = hazard ratio; ICER = incremental cost-effectiveness ratio; QALY= quality-adjusted life-year; pola-BR = polatuzumab plus bendamustine and rituximab; Ref. = reference.
Note: Only treatments on cost-effectiveness frontier are included in the table. Other treatments are dominated (i.e., more costly, and not more effective than the other treatments), or extendedly dominated (i.e., less cost-effective than the next, more effective, alternative).
aall other included treatments (tafasitamab plus lenalidomide, R-GDP, R-GemOx) were dominated.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the introduction of tafasitamab plus lenalidomide for the treatment of adult patients with R/R DLBCL not otherwise specified, including DLBCL arising from low-grade lymphoma, who are not eligible for ASCT. The BIA was from the perspective of a Canadian public drug payer over a 3-year time horizon using an epidemiological approach (refer to Figure 3), with year 1 effectively starting September 2021. Comparators included GDP, R-GDP, R-GemOx, tisa-cel, axi-cel, and pola-BR regimens with the sponsor including acquisition costs associated with all comparators, including wastage and administration. A scenario analysis incorporating Ontario Drug Benefit markups and dispensing fees for the full Canadian population (excluding Quebec) can also be explored.
Key inputs to the BIA are documented in Table 15.
State the key assumptions:
Previous and subsequent treatments will not affect the budget impact of tafasitamab plus lenalidomide.
The population of eligible patients includes those who were never eligible for ASCT, as well as those who were unresponsive to salvage chemotherapy prior to ASCT and therefore could not receive ASCT, and those who relapsed following ASCT, and this combined population will use comparators under the same market share assumptions.
Tafasitamab in combination with lenalidomide will be used for patients in both a palliative and curative setting, leading to it displacing CAR T-cell therapy.
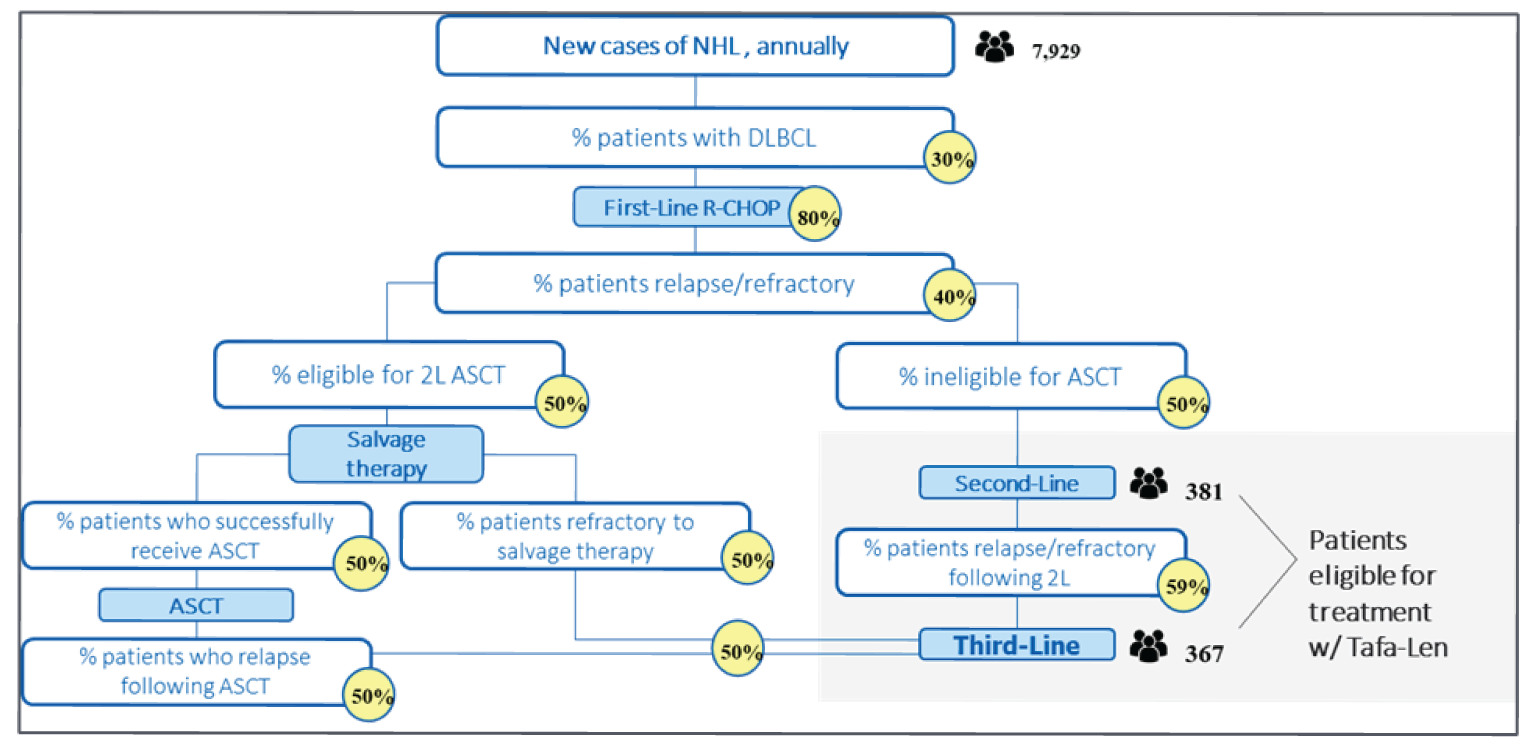
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Incident NHL cases | 7,929a |
Proportion patients with NHL who have DLBCL | 30%b |
Annual change in DLBCL incidence | 1.1%a |
Proportion of patients receiving 1L therapy | 80%c |
Proportion of DLBCL patients who are relapsed/refractory | 40%d |
Proportion of patient ineligible for ASCT | 50%e |
Proportion of ASCT-ineligible patients R/R to 2L therapy | 59%c |
Proportion of ASCT-eligible patients who are refractory to pre-ASCT salvage chemo | 50%e |
Proportion of patients who relapse after ASCT | 50%e |
Proportion of patients who do not continue to 3L chemotherapy | 50%c |
Number of patients eligible for drug under review | 748 / 756 / 764 |
Market Uptake – reference scenario (3 years) | |
GDP | 35.47% / 33.11% / 30.74% |
R-GDP | 26.56% / 24.79% / 23.02% |
R-GemOx | 9.00% / 8.40% / 7.80% |
Tisa-cel | 1.99% / 1.85% / 1.72% |
Axi-cel | 1.99% / 1.85% / 1.72% |
Pola-BR | 25.00% / 30.00% / 35.00% |
Market Uptake – new drug scenario (3 years) | |
tafasitamab plus lenalidomide | 15.00% / 30.00% / 40.00% |
GDP | 30.15% / 23.17% / 18.44% |
R-GDP | 22.58% / 17.35% / 13.81% |
R-GemOx | 7.65% / 5.88% / 4.68% |
Tisa-cel | 1.69% / 1.30% / 1.03% |
Axi-cel | 1.69% / 1.30% / 1.03% |
Pola-BR | 21.25% / 21.00% / 21.00% |
Cost of treatment including administration (per patient, first 52 weeks)f | |
Tafasitamab plus lenalidomide (median duration in months: 9.2 tafasitamab, 6.21 lenalidomide) | $226,589 |
GDP (median duration in months: 1.38) | $5,543 |
R-GDP (median duration in months: 2.76) | $24,703 |
R-GemOx (mean duration in months: 2.76) | $19,717 |
Tisa-cel (1-time administration) | $450,218 |
Axi-cel (1-time administration) | $450,218 |
Pola-BR (mean duration in months: 2.42) | $130,646 |
ASCT = autologous stem cell transplant; axi-cel = axicabtagene ciloleucel; DLBCL = diffuse large B-cell lymphoma; GDP = gemcitabine, dexamethasone, cisplatin; NHL = non-Hodgkin lymphoma; pola-BR = polatuzumab, bendamustine, rituximab; R-GDP = rituximab, gemcitabine, dexamethasone, cisplatin; R-GemOx = rituximab, gemcitabine, oxaliplatin; Tisa-cel = tisagenlecleucel.
aIncident NHL cases derived from Canadian Cancer Statistics 2019,26 Table 1.5, with Quebec removed, with an annual increase of 1.1%, and prorated to yield a base year representing September 2021 to August 2022 (no population inflation occurred between the base year and year 1, thus year 1 effectively starts September 2021). 1.1% appears to be the midpoint of 1.3% and 0.9% in Figure 1.7 for the annual change in age-standardized incidence rates for men and women, respectively.26
bA figure of 30% to 40% from Raut and Chakrabarti (2014)27 and Lymphoma Canada (2020).28
cSponsor-conducted survey cited as Tafasitamab stakeholder research CRF results, October 2021.25
dFrom the INESSS report on tisa-cel,29 citing the literature.30-32
eSehn and Salles, 2021.7
fAdministration costs were estimated at $218 per IV drug per infusion day derived from Tam et al. 2013.9,25
Summary of the Sponsor’s BIA Results
Results of the sponsor’s base case BIA suggest that the incremental expenditure associated with the reimbursement of tafasitamab plus lenalidomide for the indicated population would be $6,702,952 in year 1, $22,448,732 in year 2, and $41,769,216 in year 3, for a 3-year cumulative budget impact of $70,920,899, not including dispensing fees or markups.
The sponsor also conducted sensitivity analyses varying parameters within the model by 20% of their mean value. Of these, varying the cost of tafasitamab, the number of new NHL cases, the percentage of patients with DLBCL, the proportion of patients receiving 1L treatment, proportion of R/R DLBCL, and the mean weight of patients had the largest effect on the budgetary impact of tafasitamab plus lenalidomide.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The model lacked transparency: The submitted BIA model included many, often unlabelled, cell offsets and indexing of parameters, making it difficult to trace inputs to their resulting outputs. Calculations and outputs for the base year of the analysis were not conducted or reported, and the submitted budget impact report was insufficiently detailed in terms of methodology (e.g., when patients entered the model was not described). Furthermore, while inputs were usually referenced, they were often hard-coded, and the calculations used to derive them from their source were not included, decreasing transparency and replicability (e.g., the number of new NHL cases estimated for the base year and the year 1 population being equivalent to the base year).
CADTH was unable to fully compensate for this limitation. Decreased transparency provided to the review increases uncertainty in any resulting analyses.
Updated epidemiological data available: While the method was not reported, CADTH reviewers replicated the sponsor’s estimate of the number of incident cases in their base year (September 2021 to August 2022) by inflating the 2019 estimates of new NHL cases in Canada, excluding Quebec, as reported in 2019 by the Canadian Cancer Society,26 and inflating them by the average of the male and female annual changes in incidence as reported in the same publication. This resulted in an estimated 7,870 new cases projected for 2021, or 7,929 new cases for the modelled base year. However, the Canadian Cancer Society published more recent data after the sponsor submitted its analysis, which projected that the number of new cases of NHL for 2021 to be 8,650, resulting in an estimate of 8,713 patients with newly diagnosed NHL in the base year.33
CADTH increased the number of NHL patients in the model to be consistent with the new 2021 Cancer Statistics from the Canadian Cancer Society.33
Proportion of NHL patients with DLBCL is underestimated: The sponsor assumes that 30% of patients with NHL will have DLBCL, citing Lymphoma Canada and Raut and Chakrabarti 2014.27,28 However, both of these citations estimate that DLBCL makes up 30 to 40% of NHL cases.
CADTH increased the proportion of NHL patients who have DLBCL to 35%.
Proportion of patients receiving first-line therapy is underestimated: The sponsor estimated that of patients diagnosed with DLBCL, 80% would receive a first-line therapy, citing an internal survey of 20 Canadian physicians which was not provided to CADTH. According to the experts consulted by CADTH, the proportion of patients with DLBCL who would receive a 1L therapy would be approximately 90%, with the other 10% being patients who were not healthy enough to begin treatment and/or who died before treatment could begin, and those who instead participated in clinical trials at any included line of therapy.
CADTH revised the proportion of patients receiving a 1L therapy (represented by R-CHOP in the model), to be 90%.
Subsequent therapies not modelled appropriately: The sponsor’s model combines all patients considered eligible for tafasitamab plus lenalidomide, including both 2L and 3L patients, into a single population with identical market share assumptions. As such, the model does not consider that the more patients use any given therapy at 2L, the fewer would use it at 3L and thus combining these populations into one with identical market share assumptions is inappropriate. For example, the way the patient population is counted suggests that out of patients ineligible for ASCT in year 3 (refer to Figure 3, right-most stream), the model implicitly assumes approximately 9.4% would receive tafasitamab plus lenalidomide twice (40% receive it at 2L, 59% relapse, 40% receive it at 3L). Additionally, the potential for subsequent therapies after 3L, for example palliative therapies, are not considered.
CADTH was unable to address this limitation.
CAR T-cell products are unlikely to be displaced: The sponsor’s model underestimates the number of patients likely to receive a CAR T-cell product over the 3-year time horizon and assumes the market of CAR T-cell therapy is shrinking. Rather than considering tafasitamab plus lenalidomide a comparator to one-time potentially curative intensive treatments like ASCT and CAR T-cell therapy, the clinical experts consulted by CADTH indicated that given the current level of evidence, tafasitamab plus lenalidomide is a continuous palliative therapy more appropriately compared to pola-BR and salvage chemotherapy regimens; they did not foresee physicians or patients choosing tafasitamab plus lenalidomide if CAR T-cell therapy was available to them. CADTH therefore reframed the 50% of patients the sponsor assumed would leave the model due to death or palliative options to instead represent an assumed 50% of patients who would leave the model due to death or due to receiving CAR T-cell therapy, while the remaining 50% of patients relapsing after ASCT or refractory to pre-ASCT salvage chemotherapy would not be eligible for CAR T-cell therapy and go on to receive one of tafasitamab plus lenalidomide, pola-BR, R-GemOx, R-GDP, or GDP.
CADTH removed the CAR T-cell products tisa-cel and axi-cel from the analysis, increasing the other comparators’ market shares proportionally.
Market uptake may be overestimated: The sponsor estimated that tafasitamab plus lenalidomide would take up 15%, 30%, and 40% of the overall market share of the eligible population in Years 1, 2, and 3 of its reimbursement, giving it approximately twice the market share of pola-BR by the third year by displacing almost all growth of pola-BR’s market share. The experts consulted by CADTH did not agree that this was a likely scenario, instead estimating that tafasitamab plus lenalidomide and pola-BR would each capture about 35% of the market share for the eligible population by the third year of tafasitamab plus lenalidomide’s reimbursement.
CADTH reduced the market uptake of tafasitamab plus lenalidomide to 15%, 30%, and 35% of the eligible population in Years 1, 2, and 3, respectively. Pola-BR also continued expanding in a similar manner with 25%, 30%, and 35% in Years 1, 2, and 3.
Duration of therapy is uncertain: The sponsor’s model incorporated a lognormal extrapolation for time-to-treatment discontinuation of tafasitamab based on patients treated in the L-MIND trial, while time to discontinuation of lenalidomide was based directly on KM estimates. Durations of therapy for other comparators were based on exponential distributions derived from median time on treatment reported in the literature. Given the limitations with the modelled treatment discontinuation for tafasitamab plus lenalidomide noted in CADTH’s appraisal of the sponsor’s economic evaluation, notably the lack of agreement with the proportion of patients in PFS, and the uncertainty inherent in combining durations of therapy from multiple study populations in varying times and places, the relative duration of treatment between tafasitamab plus lenalidomide and its comparators is highly uncertain.
CADTH was not able to appropriately address this limitation.
Relative administration costs are uncertain: The sponsor’s model predicted tafasitamab plus lenalidomide would be associated with lower administration costs than all comparator treatments except CAR T-cell therapies due an assumption that each IV drug product would incur a $218 administration cost per infusion, regardless of reconstitution complexity, administration complexity, or the number of products infused at the same time. Feedback from the CADTH-participating drug plans indicated that administration costs and resource use associated with tafasitamab plus lenalidomide were underestimated. The clinical experts consulted by CADTH agreed.
CADTH was not able to address this limitation.
An additional limitation was identified but was not considered to be key limitation. Dose intensity was inappropriately modelled, with all patients assumed to receive a reduced dose each administration, rather than an assumption that some administrations would not take place. This resulted in no decrease in the cost of tafasitamab plus lenalidomide despite an inappropriately reduced dose intensity relative to the other comparators due to wastage of excess product in vial. Thus, despite the dose intensity of tafasitamab plus lenalidomide being artificially low relative to all other comparators, there was no effect on model results.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s submitted analysis by: updating comparator costs to publicly available sources, updating the number of new NHL cases in the base year, increasing the proportion of NHL patients who have DLBCL, increasing the proportion of DLBCL patients who receive a 1L therapy, removing CAR-Ts as direct comparators, and reducing market uptake of tafasitamab plus lenalidomide and its displacement of pola-BR (Table 15).
The results of the CADTH step-wise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17. Applying these changes resulted in a 3-year budget impact of $133,373,822.
CADTH also conducted scenarios assuming a 90% reduction in the price of tafasitamab, consistent with that required for the sponsor’s corrected economic base case to be cost-effective at a WTP of $50,000 per QALY, as well as a 99% reduction. Refer to Table 17.
Although baseline inputs were provided, the sponsor’s model was not built to calculate outputs nor report results for the base year, making comparisons between multiple analyses less transparent.
Table 15: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Bendamustine price | $263.35 per 25 mg | $250.00 per 25 mg |
2. Gemcitabine price | $12.40 per 200 mg | $54.00 per 200 mg |
3. Oxaliplatin price | $10.20 per 1 mg $1,865.20 per 5 mg (40 units) | $72.54 per 100 mg $145.08 per 200 mg |
4. Cisplatin price | $19.00 per 100 mg $9.50 per 50 mg | $270.00 per 100 mg $135.00 per 50 mg |
Corrected sponsor’s base case | 1+2+3+4 | |
Changes to derive the CADTH base case | ||
1. Updated number of new NHL cases in base year | 7,929 | 8,713b |
2. Proportion of NHL patients who have DLBCL | 30% | 35%c |
3. Proportion of DLBCL patients receiving 1L therapy | 80% | 90%d |
4. Removal of CAR-Ts as comparators (base year market shares)d | GDP = 40.20% R-GDP = 30.10% R-GemOx = 10.20% Tisa-cel = 2.25% Axi-cel = 2.25% Pola-BR = 15.00% | GDP = 42.09% R-GDP = 31.52% R-GemOx = 10.68% Tisa-cel = 0% Axi-cel = 0% Pola-BR = 15.71% |
5. Market uptake similar to Pola-BRe | tafasitamab plus lenalidomide = 15% / 30% / 40% Pola-BR = 21% / 21% / 21% Other = 64% / 49% / 39% | tafasitamab plus lenalidomide = 15% / 30% / 35% Pola-BR = 25% / 30% / 35% Other = 60% / 40% / 30% |
CADTH base case | 1 through 5 | |
1L = first-line therapy; axi-cel = axicabtagene ciloleucel; CAR T-cell = chimeric antigen receptor T-cell; DLBCL = diffuse large B-cell lymphoma; GDP = gemcitabine, dexamethasone, cisplatin; NHL = non-Hodgkin lymphoma; pola-BR = polatuzumab, bendamustine, rituximab; R-GDP = rituximab, gemcitabine, dexamethasone, cisplatin; R-GemOx = rituximab, gemcitabine, oxaliplatin; Tisa-cel = tisagenlecleucel.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses) that are not identified as limitations.
bDerived from the Canadian Cancer Society Canadian Cancer Statistics 2021.33
cFrom Raut and Chakabarti 2014 and Lymphoma Canada website.27,28
dDerived from feedback of clinical experts consulted by CADTH.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $70,920,899 |
Corrected base case | $68,962,238 |
CADTH reanalysis 1: Increased patients with NHL per year | $75,781,054 |
CADTH reanalysis 2: Increased proportion of NHL is DLBCL | $80,455,944 |
CADTH reanalysis 3: Increased proportion receiving 1L therapy | $77,582,518 |
CADTH reanalysis 4: CAR T-cell therapies removed | $79,677,006 |
CADTH reanalysis 5: tafasitamab plus lenalidomide uptake similar to pola-BR | $74,921,935 |
CADTH base case (1 through 5) | $133,373,822 |
1L = first-line therapy; BIA = budget impact analysis; CAR T-cell = chimeric antigen receptor T-cell; DLBCL = diffuse large B-cell lymphoma; NHL = non-Hodgkin lymphoma.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | NR | $31,995,943 | $36,901,782 | $39,299,392 | $108,197,117 |
New drug | NR | $38,698,895 | $59,350,514 | $81,068,608 | $179,118,016 | |
Budget impact | NR | $6,702,952 | $22,448,732 | $41,769,216 | $70,920,899 | |
Corrected base case | Reference | NR | $34,384,453 | $39,398,771 | $41,597,953 | $115,381,178 |
New drug | NR | $40,729,128 | $61,140,575 | $82,473,712 | $184,343,416 | |
Budget impact | NR | $6,344,675 | $21,741,804 | $40,875,759 | $68,962,238 | |
CADTH base case | Reference | NR | $31,657,798 | $38,100,179 | $41,958,212 | $111,716,189 |
New drug | NR | $46,069195 | $81,126,606 | $117,894,210 | $245,090,011 | |
Budget impact | NR | $14,411,397 | $43,026,427 | $75,935,998 | $133,373,822 | |
CADTH Scenario A: 90% reduction in price of tafasitamab | Reference | NR | $31,657,798 | $38,100,179 | $41,958,212 | $111,716,189 |
New drug | NR | $33,143,831 | $43,596,962 | $52, 182,623 | $128,923,417 | |
Budget impact | NR | $1,486,033 | $5,496,783 | $10,224,411 | $17,207,228 | |
CADTH Scenario B: 99% reduction in price of tafasitamab | Reference | NR | $31,657,798 | $38,100,179 | $41,958,212 | $111,716,189 |
New drug | NR | $31,851,295 | $39,843,998 | $45,611,465 | $117,306,757 | |
Budget impact | NR | $193,497 | $1,743,819 | $3,653,252 | $5,590,568 |
BIA = budget impact analysis; NR = not reported.
Stakeholder Input
Patient Input
Lymphoma Canada
About Lymphoma Canada
Lymphoma Canada is a national Canadian registered charity that empowers the lymphoma community through education, support, advocacy, and research. Based out of Toronto (ON), we collaborate with patients, caregivers, healthcare professionals, and other organizations and stakeholders, to promote early detection, find new and better treatments for lymphoma patients, help patients access those treatments, learn about the causes of lymphoma, and work together to find a cure. Resources are provided in both English and French. For more information about our organization, please visit us at www.lymphoma.ca
Information Gathering
Lymphoma Canada (LC) conducted a number of anonymous online surveys for Diffuse Large B-Cell Lymphoma patients. The most recent survey, released from October 16, 2021 – December 2, 2021, was targeted towards the experience of patients receiving the therapy under review. The previous surveys released (June 21, 2021 – August 25, 2021; August 31, 2020 – October 5, 2020; April 18, 2018 – June 15, 2018) provided general experience data for patients with DLBCL related to their disease experience and quality of life. Links to the surveys were sent via e-mail to patients registered through the LC database. These surveys were also made available via social media outlets, including Twitter, Instagram and Facebook accounts, and were also sent to healthcare professionals across Canada to share with their patients. The survey had a combination of multiple choice, rating and open‐ended questions. Skipping logic was built into the survey so that respondents were asked questions only relevant to them. Open-ended responses to surveys that reflected the sentiment of a majority are included verbatim to provide a deeper understanding of patient perspectives.
Disease Experience
There were 150 Diffuse Large B-Cell Lymphoma (DLBCL) patients that responded to the surveys, of which two patients had direct experience with Tafasitamab therapy. LC had a tremendous difficulty finding patients with Tafasitamab treatment experience as there were no Canadian clinical trial sites nor access to the lead clinical investigators of global trial sites. Despite significant effort by LC staff, two patients were able to provide their experience. There were no caregivers that participated in this survey. Of the DLBCL patients who provided their demographic information for this survey (refer to Table 1 and Table 2), 73% live in Canada, 46% are male, and 45% are ≥ 60 years-old.
Table 1: Country of Survey Respondents (150 respondents)
Respondents | CAN | USA | Other | Skipped | Total |
|---|---|---|---|---|---|
Patients WITHOUT Tafasitamab experience | 110 | 8 | 4 | 26 | 148 |
Patient WITH Tafasitamab Experience | 0 | 2 | 0 | 0 | 2 |
Table 2: Age and Gender of Survey Respondents (150 respondents)
Respondents | Age Range | Gender | ||||||
|---|---|---|---|---|---|---|---|---|
< 20 | 20-39 | 40-59 | ≥ 60 | skipped | Female | Male | Skipped | |
Patients WITHOUT Tafasitamab experience | 2 | 15 | 37 | 65 | 29 | 69 | 50 | 29 |
Patient WITH Tafasitamab Experience | 0 | 0 | 0 | 2 | 0 | 0 | 2 | 0 |
Symptoms of DLBCL that most commonly affected respondents’ quality of life at diagnosis (129 respondents) were fatigue or lack of energy (71%), enlarged lymph nodes (47%), drenching night sweats (41%), unexplained weight loss (33%), loss of appetite (30%), flu-like symptoms (23%), and persistent cough (21%). Other symptoms affecting quality of life for ≥ 10% of respondents included bodily aches and pains, chest pain and trouble breathing.
Respondents were asked which aspects of their life, including mental and emotional problems associated with their disease and treatment, NEGATIVELY impacted their quality of life. The majority of respondents (87%) had one or more symptoms negatively impact their quality of life (Table 3).
Table 3: Impact of DLBCL on Patients’ Mental and Emotional Well Being (129 respondents)
Symptom | Number of respondents | Percentage of respondents |
|---|---|---|
Fear of disease recurrence | 86 | 67% |
Memory loss | 43 | 33% |
Anxiety/worry | 58 | 45% |
Problems concentrating | 52 | 40% |
Difficulty sleeping | 41 | 32% |
Loss of sexual desire | 33 | 26% |
Stress of diagnosis | 36 | 28% |
Depression | 25 | 19% |
None of these | 17 | 13% |
As one patient shared, “It was a huge shock of diagnosis at young age of 33 years old. Quality of life impacted by potentially poor prognosis and change in life plans as we held off on having children. Inability for family to attend in person appointments with medical info due to COVID-19 pandemic, this definitively added to my stress.”
As described by another patient related to psychosocial impacts, “I retired early due to memory loss, lack of concentration and ongoing depression.”
Further, respondents indicated how their mental health, emotions, disease and treatment, have had a NEGATIVE impact on different aspects of their life. Notably, 59% of patients indicated that their DLBCL had a negative impact on their ability to work or attend to family/friend obligations. Additional responses are summarized in Table 4.
Table 4: Effect of DLBCL on Day-to-Day Life of Patients (126 respondents)
Aspect of life NEGATIVELY impacted by DLBCL | Number of respondents | Percentage of respondents |
|---|---|---|
Ability to work/school | 74 | 59% |
Family/friend obligations | 75 | 59% |
Personal image | 39 | 39% |
Intimate relations | 30 | 30% |
Continuing daily activities | 17 | 68%* |
*Only 25 respondents replied to this question, therefore percentage provided was the number of respondents divided by the number that answered this question
As described by two patients:
“It affected our personal lives my husband had to stay home from work to help me. We had no income. Very stressful.”
“It has limited my work options. It has limited my ability to do routine daily chores around the home at times and I can only perform limited exercise due to fatigue.”
Experiences With Currently Available Treatments
124 respondents provided information about their experience with DLBCL treatments. All respondents had received at least one line of treatment or were undergoing first-line treatment for DLBCL. Of these, 63 respondents (51%) had received more than one line of treatment, of which 22% had received 3 or more lines of treatment. The most commonly reported first-line treatment (82% of respondents) was the chemoimmunotherapy regimen R-CHOP. Of those who received more than one line of treatment, 23% had undergone an autologous stem cell transplant and 4% had undergone an allogeneic stem cell transplant. Side effects of current treatments: The most common side effects respondents experienced during their DLBCL treatments are listed in Table 5. Nearly all patients (96%) reported at least one side effect.
Table 5: Side-Effects From Treatment (124 respondents)
Side effect (n) | % of resp. | Side effect (n) | % of resp. | Side effect (n) | % of resp. |
|---|---|---|---|---|---|
Hair loss (108) | 87% | Mouth sores (52) | 42% | Trouble breathing (28) | 23% |
Fatigue (106) | 85% | Thrombocytopenia (48) | 39% | Skin rashes/severe itching (28) | 23% |
Neutropenia (83) | 67% | Anemia (42) | 34% | Cough (27) | 22% |
Memory problems or confusion (77) | 62% | Infections (40) | 32% | Loss of menstruation (18) | 15% |
Nausea (68) | 55% | Diarrhea (35) | 28% | Irregular heartbeat (18) | 15% |
Peripheral neuropathy (60) | 48% | Pain (34) | 27% | Viral reactivation (11) | 9% |
Constipation (58) | 47% | Other (32) | 26% | Bowel obstruction (9) | 7% |
When asked which side effects they found most difficult to tolerate, respondents most often reported fatigue (35/85; 41%), nausea/vomiting (16/85; 19%), chemo-brain (13/85; 15%), and hair loss (8/85; 9%) (85 respondents).
Impact of treatments on quality of life: When asked about the impact of various aspects of treatment on daily living (on a scale of 1 – 5, where 1= No impact and 5 = significant negative impact), respondents noted that treatment-related fatigue and other side effects had the most significant impact on their quality of life (Table 6). This shows that the side effects of treatments are more important and impact patients greater than treatment administration.
Table 6: Impact of Treatment on Quality of Life (111-115 respondents)
Treatment aspect | Weighted average | Significant negative impact (rating = 4-5) | Number of responses |
|---|---|---|---|
Treatment-related Fatigue | 3.7 | 63% | 115 |
Side-effects | 3.5 | 54% | 114 |
Infusion reaction | 2.5 | 27% | 113 |
Number of infections | 2.1 | 23% | 111 |
Number of clinic visits | 2.4 | 23% | 114 |
Infusion time | 2.4 | 21% | 112 |
Treatment also had a very significant impact on many respondents’ ability to work, travel and participate in daily activities. 92% of patients had their daily activities negatively impacted by their treatment or were even unable to continue with their daily activities (96 respondents). Patients’ ability to travel (89%) and continue with work/school (88%) were additionally negatively impacted. Patients additionally experienced challenges to their relationships, with their treatment impacting their intimate relationships (83%) and family and friendships (71%).
Patient’s shared further details as to the psychosocial impacts of receiving treatment for their DLBCL:
“I am afraid of how chemo will affect my quality of life as I receive more treatments.”
“[Fear of disease recurrence] is very high and consumes a lot of my thought process almost every day, even after two years since my Chemo treatments finished and I had a complete response.”
“Learning to not to push myself with physical activity i.e. yard work, house reno etc. Not taking on extra duties at work, and possibly retiring early in age.”
When asked about the financial implications of treatment, a large proportion of patients reported that their absence from work or school (47%) and the costs associated with travelling to their treatment centre (i.e. parking, accommodation, driving, etc.) (44%), had the greatest negative financial impact. Patient’s shared further details as to these impacts:
“I had to travel 1 ½ hours away and stay overnight for treatment days. It is exhausting and expensive to travel to receive treatment.”
“Had to give up a new career and job to have treatment”
“I was unable to continue working so I had to retire early, and therefore I lost my salary and health benefits”
“There is always some stress getting time off work to attend check-ups with oncologist. I am tired after work so I do very little during the work week to make sure I will have enough energy for my job.”
Improved Outcomes
Patient preferences: Respondents were asked to rate, on a scale of 1 -5 (1 = not important; 5 = extremely important), the importance of various factors regarding a new drug or therapy for DLBCL. “Longer survival and remission than current therapies” and “controlling disease symptoms” were rated as the most important outcomes for a new therapy (Table 7). “Fewer side effects” was rated as the least important outcome overall but is still an important consideration for patients.
Table 7: Treatment Preferences (18-114 respondents)
Treatment outcome or factor | Rating = 4-5 (very-extremely important) | Weighted average |
|---|---|---|
Longer remission than current therapies | 96% | 4.8 |
Control disease symptoms | 94% | 4.8 |
Longer survival than current therapies | 94% | 4.8 |
Better quality of life than current therapies | 89% | 4.6 |
Fewer side effects than current therapies | 72% | 4.1 |
Respondents were asked if they would be willing to tolerate the side effects of a new treatment if they were short term. 42% (n=48) of respondents would be willing to tolerate potential side effects, while 6% were not; the remaining were unsure (n=52), as this would depend on the exact length they would experience them for and whether this would outweigh treatment benefit (114 respondents). Respondents were also asked if they would choose a treatment with known side effects, potentially serious, if their doctor recommended it was the best option for them. Of the 114 respondents who answered this question, (n=57) 50% selected “Yes”, while only (n=3) 3% selected “No”. The remaining 47% of respondents selected “I’m not sure”, as it would depend on the type of side effect and its severity, how long they would experience it for, and whether the treatment would result in long-term outcome or cure. As shared by one patient: “Overall well-being and quality of life is equally as important as the treatment/drug being provided.”
Experience With Drug Under Review
Unfortunately, Lymphoma Canada had tremendous difficulty locating DLBCL patients to share their experience with tafasitamab, however two patients were able to participate in this survey and provide their experience. Both patients were able to access treatment locally and there was no financial impact from receiving treatment. Details related to specific access and treatment history can be found in Table 8.
Table 8: Treatment Experience With Tafasitamab (2 respondents)
Patient # | Gender | Age | Date Received | Access | Previous Treatment Experience | Stage of Receipt |
|---|---|---|---|---|---|---|
1 | M | 65-74 | 2020 | Not reported | R-CHOP *received as 2nd line therapy | Received all courses |
2 | M | 75-84 | 2021 | Not reported | CHOP Chemotherapy (unspecified) *received as 3rd line therapy | Received all courses |
Symptom Experience: Both patients had the majority of their DLBCL symptoms resolved after receiving tafasitamab including enlarged lymph nodes, resolved blood cell counts (platelets, RBC, WBC), and improvement in weight loss/appetite. As one patient described:
“In general, I tolerated tafasitamab well. I feel less fatigued and have a better appetite.”
Side Effect Experience: Patient’s experienced the following side effects from tafasitamab treatment, however the majority of them were short term and did not impact quality of life: neutropenia (n=2), rash/itching (n=1), diarrhea (n=1), and nausea (n=1). As one patient indicated:
“I’ve had a positive response to this treatment and the few side effects I experienced were minor.”
Treatment experience and impacts to QoL: Overall both patients did not experience any negative impacts to QoL related to treatment administration such as number of clinic visits required and the length/frequency of taking the drug. Tafasitamab has improved patients’ overall quality of life by improving their general activity level as well as their mental health (n=2).
Overall experience: Based on patients experience with tafasitamab, both patients would recommend this treatment to patients that do not meet the criteria to receive curative therapies in the second or later line settings. As one patient shared:
“I did not have many options available to me. There is no standard approach for patients like me and having options that work and are safe are important.”
The two patients rated their overall experience with Zanubrutinib as very good (n=1) to excellent (n=1). Patients would 100% of the time take it again if their doctor recommended it was the best treatment option for them.
Companion Diagnostic Test
To our knowledge, there is no companion diagnostic testing required to receive this therapy.
Anything Else?
Tafasitamab provides an effective and safe treatment option for DLBCL patients that do not meet the strict eligibility criteria for standard of care approaches including stem-cell transplant. There are limited treatment options in this setting and tafasitamab addresses the need for an effective treatment, while aligning with patient values based on the feedback received.
Conflict of Interest Declaration — Lymphoma Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Patient organizations that work with the DLBCL patient population (such as the Lymphoma and Leukemia Society of Canada) helped to promote this survey to their constituents. Adam Waiser, an independent consultant, helped promote the patient survey in 2018.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 9: Financial Disclosures for Lymphoma Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Incyte | — | X | — | — |
Roche | — | — | X | — |
Gilead | — | — | X | — |
Novartis | — | — | X | — |
BMS | — | — | X | — |
Clinician Input
Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
About the Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
This input was jointly discussed via Drug Advisory Committee meeting and email.
Current Treatments
Describe the current treatment paradigm for the disease.
Response: Patients that are relapsed/refractory DLBCL have CAR-T therapies and other combinations of palliative drug regimens, such as Polatuzumab Vedotin with BR, as options for treatment.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Response: The most important goal would address prolongation of survival, reduction in disease symptoms.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Response: There are poor and limited treatment options for this patient population.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Response: All patients within this population would have the greatest unmet need. Patients who are ineligible for CAR-T therapies would be the target population.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: Tafasitamab in combination with lenalidomide would fit after first line therapies. Tafasitamab would be an option for non-transplant eligible, disease progression post-transplant, or not CAR-T eligible for other reasons.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Tafasitamab would be recommended after first line therapies and progression-after transplant, specifically if patient is not transplant or CAR-T eligible.
How would this drug affect the sequencing of therapies for the target condition?
Response: Patients would get Polatuzumab vedotin with BR and Tafasitamab in limited treatment options. Tafasitamab with lenalidomide would be an additional option for second-line. Tafasitamab with lenalidomide would potentially impact the eligibility for CAR-T therapy, depending on evolving evidence.
Which patients would be best suited for treatment with the drug under review?
Response: Patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, including DLBCL arising from low grade lymphoma, who are not eligible for autologous stem cell transplant (ASCT).
How would patients best suited for treatment with the drug under review be identified?
Response: Clinician examination or judgement.
Which patients would be least suitable for treatment with the drug under review?
Response: DLBCL patients who have progressed on CAR-T therapies would be least suitable.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: No.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Response: Yes. Standard lymphoma response measures.
What would be considered a clinically meaningful response to treatment?
Response: Improvement of symptoms, prolonged remission and improved survival.
How often should treatment response be assessed?
Response: Treatment response should be assessed every 3 months within the first year of treatment and then every 6 month after the first year of treatment.
What factors should be considered when deciding to discontinue treatment?
Response: Progression on treatment or development of unacceptable toxicity.
What settings are appropriate for treatment with the drug under review?
Response: Cancer centre-based treatment.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: N/A
Additional Information
Response: N/A
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Tom Kouroukis
Position: Provincial Head – Complex Malignant Hematology (OH-CCO)
Date: 18/11/2021
Table 10: Conflict of Interest Declaration for Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee — Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Pierre Villeneuve
Position: Hematologist/oncologist
Date: 18/11/2021
Table 11: Conflict of Interest Declaration for Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee — Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Jordan Herst
Position: Hematologist/oncologist
Date: 18-11/2021
Table 12: Conflict of Interest Declaration for Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee — Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Lee Mozessohn
Position: Hematologist/oncologist
Date: 18-11-2021
Table 13: Conflict of Interest Declaration for Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee — Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr. Mark Brown
Position: Clinical pharmacist
Date: 18-11-2021
Table 14: Conflict of Interest Declaration for Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee — Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Dr. Joanna Graczyk
Position: Hematologist
Date: 02-12-2021
Table 15: Conflict of Interest Declaration for Ontario Health (Cancer Care Ontario) Hematology Cancer Drug Advisory Committee — Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Lymphoma Canada
About Lymphoma Canada
Lymphoma Canada, a national non-for-profit organization for Canadian lymphoma and CLL patients, assisted in the coordination of the group clinician response. Lymphoma Canada was not involved in the development of this submission, nor are the clinicians’ involved members of Lymphoma Canada. For more information about Lymphoma Canada, please visit www.lymphoma.ca.
Information Gathering
Responses to the questions in this submission are based on current research results from clinical trials and real word experience, clinical experience with this population, and understanding of patient needs and challenges in practice.
Current Treatments
Response: Diffuse large B cell lymphoma (DLBCL) is the most common type of lymphoma in adult population that has been diagnosed in almost 30% of the adult population with lymphoproliferative diseases. It is estimated that 60% of patients will be cured with either standard frontline therapy, or with subsequent salvage chemotherapy and autologous stem cell transplantation for those who are considered transplant-eligible. Although CAR T-cell therapy is now available in Canada for patients with relapsed/refractory DLBCL who have failed multiple lines of treatment, this treatment modality is not suitable for a significant proportion of these patients (~50%), particularly those who are elderly, unfit medically, or too frail to undergo this intensive treatment. In addition, there are many other exclusions for CAR T therapy based on type of disease; as an example, any DLBCL transformed from any other entity besides follicular lymphoma are not eligible for CAR T therapy.
Patients with relapsed/refractory DLBCL who are not fit for CAR-T therapy generally receive palliative treatments, or enroll on clinical trials, with no expectation for long-term survival, as the estimated median survival in this group is 6 months.
Polatuzumab Vedotin in combination with Bendamustine-Rituximab is an option for the category of relapsed/refractory DLBCL patients. It was granted market authorization by Health Canada in September 2020 but not publicly funded through provincial cancer care. The indication does not include relapsed/refractory lymphoma arising from low grade lymphoproliferative disease. As preclinical studies in Canada show, the objective response rate is 41.5% with a median progression free and overall survival of 6.6 and 12.5 months, respectively.
Tafasitamab in combination with Lenalidomide is Health Canada approved and currently available through a compassionate access program. This combination was well tolerated. Besides, a high proportion of DLBCL patients who relapsed or had refractory disease after previous treatment with one to three systemic regimens (with at least one anti-CD20 therapy) and ineligible for intensive therapy (autologous stem cell transplant) achieving complete responses, which has translated into significantly improved progression free and overall survival (11.6 months and 33 months, respectively). This is a dramatic difference compared to what is expected in this population, particularly when compared to other palliative therapies.
From symptom management and quality of life standpoint, most of the symptoms in patients with relapsed/refractory lymphoma are either resulted directly from the disease extension in the patients, including massive lymphadenopathies, pleural effusion, gastrointestinal involvement and bone lesions or caused by inflammatory/cytokine associated symptoms including weight loss, cachexia, fever, night sweats and fatigue. Response to therapy is often associated with symptom control and improvement in quality of life, particularly in patients who achieve complete remissions that are durable.
Treatment Goals
Response: The most important goal in treatment of an aggressive lymphoma is achieving a complete response and a durable remission. Prolonged disease-free survival and overall survival are the important statistical values in addressing the durable response. Parallel to the survival benefit, a reasonable safety profile, manageable toxicities and ease of administration positively impacts patients’ compliance and tolerance to treatment. Improvement in quality of life, helping the patients in gaining their independence and reducing burden on caregivers are considered clinical goals. An objective response and progression free interval has a positive impact on this aspect of patients’ care.
Treatment Gaps (Unmet Needs)
Response: Salvage chemotherapy and autologous stem cell transplant is feasible in about 50% of patients in relapsed/refractory DLBCL. Among those who are able to complete this treatment modality, long term remission is generally achieved in 30-40% of cases. This will leave about 70% of relapsed/refractory DLCBL with the unanswered clinical question on the available treatment options in third line setting.
CAR T-cell therapy will be a reasonable option for selected cases among these challenging DLBCL cases, but this too only leads to durable remissions in 35-45% of patients. Those who are either ineligible for cellular therapy, or who have failed it, have a very poor prognosis and represent an unmet need given the lack of other effective therapies.
Availability of third line chemo-immunotherapy with manageable toxicity and reasonable safety profile that provides the chance of durable response is required to meet the needs of this population. Tafasitamab in combination with lenalidomide is considered an attractive option not only for the complete response, progression free and overall survival benefits as per clinical studies, but for the ease of administration with one IV monoclonal antibody in combination with the oral lenalidomide. Unlike other chemo-immunotherapy combinations with potential risk of bone marrow suppression and long term immune suppression, such as bendamustine combination chemoimmunotherapy, this combination is not empirically affect lymphocyte activity and function. Apart from hematologic toxicity, that is common among all the treatment options in this clinical setting, grade 3 toxicity of tafasitiamab/lenalidomide combination is overall reported in less than 5% of patients and unlike polatuzumab/bendamustine combination treatment, no neurotoxocity has been reported.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Response: Patients who are unfit or ineligible for intensive therapies (i.e. autologous SCT or CAR-T therapy) or who have failed prior cellular therapy have the greatest unmet need that would be met by the use of tafasitamab in combination with lenalidomide. As mentioned previously, a significant proportion of patients with relapsed/refractory disease will be ineligible for intensive treatments, including those not included in the CAR-T therapy program (e.g. aggressive lymphoma arising from any indolent lymphoma other than follicular lymphoma).
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: Tafasitamab is an Fc-enhanced humanised anti-CD19 monoclonal antibody, which mediates antibody-dependent cellular cytotoxicity and antibody-dependent cellular phagocytosis, and exerts direct cytotoxicity. As a single agent it was well tolerated and showed encouraging activity in patients with relapsed/refractory B-cell malignancies with some achieving durable responses. Lenalidomide has direct antineoplastic activity, stimulates proliferation and activation of natural killer cells, and enhances natural killer cell-mediated antibody-dependent cellular cytotoxicity with tafasitamab in vitro. The combination of tafasitamab and lenalidomide has shown to be synergistic and well tolerated in clinical studies. Lenalidomide is now available as a generic compound and as such is readily available for use in combination with tafasitamab.
The landmark study addressing the combination of tafasitamab and lenalidomide was performed in patient’s ineligible for autologous transplantation (including a small proportion of patients who had failed a prior transplant). It was given as second line therapy in about 50% of patients on this study. It is reasonable to consider this therapy in second line for patients who are unfit for standard salvage chemotherapy (e.g. GDP), or for a patient that is not eligible to receive ASCT or CAR-T.
The most likely setting tafasitamab/lenalidomide would be used is in the third line or beyond setting. This would be routinely offered in patients ineligible for autologous stem cell transplant and/or CAR-T therapy. Most clinicians would proceed with intensive salvage therapy first if this was an option for their patient, and reserve tafasitamab/lenalidomide if failure of this therapy occurs.
Given the superior outcomes and general tolerability of tafasitamab/lenalidomide we would anticipate that this therapy would be chosen ahead of polatuzumab-BR in patients with relapsed/refractory DLBCL unfit for more intensive treatments. Although primary FDA approval for Tafasitamb/ lenalidomide combination has been based on a phase 2 clinical trial, the long-term outcome as per Duell et. al publication shows sustained durable response with remarkable complete response rate (40%). Even in tha absence of a randomised phase 3 clinical trial, these results are superior to the available data in phase 3 clinical trials in similar clinical setting and relapsed/refractory DLBCL patient population.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: In relapsed/refractory DLBCL, selected patients with a good performance status and limited comorbidities fit for intensive cellular therapies should be offered standard salvage chemotherapy and offered high-dose therapy and autologous stem cell transplant if chemosensitive, or CAR-T therapy if chemo-refractory or failure after high-dose therapy. A curative treatment approach is preferred.
For those who are considered relapsed/refractory to rituximab-based chemotherapy and ineligible for intensive cellular therapies, Tafasitamab/lenalidomide combination is a non-cytotoxic, immunomodulatory combination treatment with favorable safety profile and durable response in the non-curative setting.
How would this drug affect the sequencing of therapies for the target condition?
Response: In patients who fail tafasitamab/lenalidomide treatment, polatuzumab plus bendamustine and rituximab is an option. Oral PEP-C is also a palliative oral chemotherapy that is usually considered to control symptoms rather than elicit disease response. Participation in clinical trials is generally recommended for all patients with relapsed/refractory disease who have failed standard lines of therapy.
There are case reports of CAR T-cell therapy being feasible after disease progression on Tafasitamab/lenalidomide combination treatment. As per case reports and available data, this combination treatment is not compromising T cell harvest and processing. However, more clinical information and data is needed in this specific clinical setting, particularly impact on efficacy after CD19-directed therapy. In these cases, repeat biopsy would be suggested to confirm persistent CD19+ disease prior to proceeding with CAR T therapy (as we would with other anti-CD19 directed therapies, the most studied being blinatumomab in B-cell ALL for which CAR-T therapy is approved).
As tafasitamab is given indefinitely until disease progression, we would not anticipate re-treatment in this setting. Re-treatment would only be considered if discontinued due to toxicity reasons in responding patients.
Which patients would be best suited for treatment with the drug under review?
Response: Patients with one prior line of therapy had a trend for better outcomes than those with ≥2 prior lines: objective response rate, 70.0% vs 50.0%; and 12-month overall survival rate, 86.9% vs 60.1%. However, the 12-month duration of response rate was similar regardless of the number of prior lines (one prior line: 70.5% [95% CI: 47.2-85.0] vs ≥2 prior lines: 72.7% [95% CI: 46.3-87.6]).
For patients who were refractory to primary therapy or their last line of therapy, similar objective response rates were observed to non-refractory patients (60.0% vs 60.0%); 12-month duration of response rate was similar regardless of refractory status to last therapy; and 12-month OS rates were higher in non-refractory patients.
As expected, patients with a low/low-intermediate International Prognostic Index (IPI) score had better outcomes than those with an intermediate-high/high score: objective response rate 70.0% vs 50.0%; 12-month duration of response rate, 86.5% vs 50.4%; and 12-month overall survival rate, 87.0% vs 59.9%.
Of note, as per primary inclusion criteria, patients with primary refractory disease (ie. no response or progressive disease (PD) within <3 months of frontline therapy) were excluded. However, a protocol amendment in June 2016, adjusted the definition of primary refractoriness as per “B-cell lymphoma National Comprehensive Cancer Network guidelines” to those patients with no response to or PD following a rituximab-containing regimen within <6 months of completion of therapy. In light of this adjustment, one can conclude that the primary refractory cases with no response or progression within six months of treatment may not show the best outcome, but more studies may shed more light into this specific category of patients with dismal prognosis.
How would patients best suited for treatment with the drug under review be identified?
Response: The patients are identified by their primary treating physician. There are standard guidelines that address the approach to relapsed patients, including when to refer for intensive cellular therapies ahead of using tafasitamab/lenalidomide. Diagnosis and staging are standard and widely available. Patients without symptoms should be considered for therapy given the aggressive nature of this lymphoma. For patients with a history of indolent lymphoma with biopsy-proven transformation that has been previously treated, it is important to establish that recurrent disease is also due to transformation and not recurrence of the indolent disease, thus biopsy is required in this setting. Unlike most of the targeted therapies that dictate presence of the targeting marker in the biopsy sample, transformation into aggressive lymphoma is enough to consider a potential therapeutic role for tafasitamab/lenalidomide combination in this clinical setting.
Which patients would be least suitable for treatment with the drug under review?
Response: There are no specific parameters that deem a patient unsuitable for this specific treatment. The patient should be well enough to tolerate the frequent outpatient visits required for this therapy. Those with limited life span or competing life-threatening conditions should be not offered this therapy.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: Not at this time. There are no predictors to accurately identify which patients will exhibit response and which will not.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Response: Standard Lugano criteria for lymphoma is used to confirm remission status (via CT +/- PET scans). Clinical improvement in patients’ performance status, resolution of fever, night sweats, weight loss and cachexia and regression of lymphadenopathies and splenomegaly are the clinical findings suggestive for clinical response to treatment.
What would be considered a clinically meaningful response to treatment?
Achievement of remission would be considered clinically meaningful, with complete remission (CR) associated with long-term outcomes. Clinically meaningful responses would include resolution of all lymphoma-related symptoms, improvement in functional status and quality of life indicators, and return to normal activities.
How often should treatment response be assessed?
Response: Clinical assessment prior to each cycle of treatment, including review of symptoms, and physical examination and assessing lymphadenopathies and organomegaly and extra nodal involvement is the standard of practice.
Laboratory findings including LDH, CBC, Liver and kidney function tests, albumin are not only suggested response to treatment, but evaluate potential safety signals and toxicity profile.
Imaging study with CT scan and/or PET Ct scan if clinically indicated are part of the assessment after cycle 4 and after cycle 12 of treatment (4 months and 12 months after starting the treatment).
What factors should be considered when deciding to discontinue treatment?
Response: Treatment failure occurs with confirmation of disease progression and this would prompt treatment discontinuation. If there is a significant change in the patient’s status this could result in treatment discontinuation. Severe toxicities (e.g. grade 3 or higher) should result in temporary discontinuation until improved, with discontinuation of therapy if unacceptable toxicity to either patient or physician provider
What settings are appropriate for treatment with the drug under review?
Response: This therapy can be administered in any centre that is certified to administer chemotherapy in the outpatient setting.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: N/A
Additional information
Is there any additional information you feel is pertinent to this review?
Response: Tafasitamab in combination with lenalidomide is an important addition to the treatment landscape for a particularly poor prognosis group of patients with relapsed/refractory DLBCL. It provides a well tolerated effective therapy in patient’s ineligible for other curative-intent treatments, providing them with superior survival outcomes compared to all other currently available therapies, filling an unmet need for this population.
Conflict of Interest Declarations — Lymphoma Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Lymphoma Canada was solely involved in assisting to coordinate the group clinician responses for this submission, and were not involved in analyzing or including feedback to any of the responses within this submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Mona Shafey
Position: Clinical Associate Professor
Date: 25-Nov-2021
Table 16: Conflict of Interest Declaration for Lymphoma Canada — Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Incyte | X | — | — | — |
Novartis | X | — | — | — |
Kite/Gilead | X | — | — | — |
BMS | X | — | — | — |
Declaration for Clinician 2
Name: Ghazaleh Shoja E Razavi
Position: Clinical assistant professor
Date: 25-11-2021
Table 17: Conflict of Interest Declaration for Lymphoma Canada — Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Incyte | X | — | — | — |
Declaration for Clinician 3
Name: Dr. John Kuruvilla
Position: Hematologist, Princess Margaret Cancer Centre
Date: 06-12-2021
Table 18: Conflict of Interest Declaration for Lymphoma Canada — Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Laurie Sehn
Position: Medical Oncologist, BC Cancer Agency
Date: 06-12-2021
Table 19: Conflict of Interest Declaration for Lymphoma Canada — Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Incyte | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.