CADTH Reimbursement Review
Ripretinib (Qinlock)
Sponsor: Medison Pharma Canada Inc.
Therapeutic area: Gastrointestinal stromal tumours
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
AESI
adverse event of special interest
ANCOVA
analysis of covariance
BSC
best supportive care
CI
confidence interval
CR
complete response
CYP
cytochrome P450
DB
double-blind
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire for Cancer 30 item
GIST
gastrointestinal stromal tumour
HR
hazard ratio
HRQoL
health-related quality of life
IRR
independent radiological review
ITT
intention to treat
KM
Kaplan-Meier
LRGC
Life Raft Group Canada
MedDRA
Medical Dictionary for Regulatory Activities
MID
minimal important difference
mRECIST
modified Response Evaluation Criteria in Solid Tumours
MRI
magnetic resonance imaging
NCI-CTCAE
National Cancer Institute Common Terminology Criteria for Adverse Events
OL
open label
ORR
objective response rate
OS
overall survival
PAG
Provincial Advisory Group
PD
progressive disease
PDGFRA
platelet-derived growth factor alpha
PFS
progression-free survival
PR
partial response
PS
performance status
RCT
randomized controlled trial
SD
standard deviation
SE
standard error
TKI
tyrosine kinase inhibitor
TTR
time to response
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Ripretinib (Qinlock), tablets, 50 mg, oral |
Indication | For the treatment of adult patients with advanced gastrointestinal stromal tumour (GIST) who have received prior treatment with imatinib, sunitinib, and regorafenib |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | June 19, 2020 |
Sponsor | Medison Pharma Canada Inc. |
NOC = Notice of Compliance.
Introduction
Gastrointestinal stromal tumours (GISTs) are rare soft-tissue sarcomas of the gastrointestinal tract.1 GISTs are typically characterized by primary gain-of-function mutations in CD117/c-KIT (80% to 85% of GISTs)2 and platelet-derived growth factor alpha (PDGFRA).3 Small and/or slow-growing GISTs may be clinically more benign, while tumours that grow significantly outward from the bowel wall can cause dysphagia, bleeding, abdominal pain/discomfort, fatigue, vomiting, loss of appetite, and other gastrointestinal issues.4 Both disease symptoms and the side effects of therapy severely impact health-related quality of life (HRQoL). At diagnosis, approximately half of patients with GISTs are eligible for potentially curative surgical resection.3 Among patients undergoing resection, disease will recur in about half within 5 years.5 Patients with advanced disease are transferred to the care of a medical oncologist for systemic therapy with palliative intent. According to the clinical experts consulted by CADTH for this review, in these patients, chemotherapy and radiotherapy are ineffective,1,6 and the mainstay of therapy is sequential treatment with the tyrosine kinase inhibitors (TKIs) imatinib, sunitinib, and regorafenib. Responses to each line of therapy after imatinib are generally short-lived, and resistance and progression develop in most patients within months. Following progression on third-line regorafenib, there are no standard therapy options beyond best supportive care (BSC). Patient outcomes are unfavourable: further progression and death can be expected within a few months.
Published data on the prevalence, incidence, and survival of advanced GIST in Canada are unavailable. Based on an estimated 500 GIST cases diagnosed per year in Canada,7 with 75% representing advanced GIST, and assuming 80%, 70%, and 60% failure/progression rates on imatinib, sunitinib, and regorafenib, respectively, the sponsor estimated a target population of 62 to 86 patients with advanced GIST per year from 2023 to 2025 in Canada (outside of Quebec) who have received prior treatment with imatinib, sunitinib, and regorafenib and would be eligible for fourth-line ripretinib.8
Ripretinib is a TKI administered at a dosage of 150 mg orally (three 50 mg tablets) once daily. Ripretinib is indicated “for the treatment of adult patients with advanced gastrointestinal stromal tumour (GIST) who have received prior treatment with imatinib, sunitinib, and regorafenib.” The objective of this report was to perform a systematic review of the beneficial and harmful effects of ripretinib for the treatment of adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib, and regorafenib.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups submitted patient input for this review: the CanCertainty Coalition and the GIST Sarcoma Life Raft Group Canada (LRGC). CanCertainty raised concerns about the financial and administrative barriers to accessing cancer treatments such as ripretinib in some Canadian jurisdictions (Ontario and the Atlantic provinces). Young patients (younger than 65 years) who require take-home cancer treatment such as ripretinib and do not have private or automatic public prescription drug coverage may incur significant deductibles or co-payments from their personal savings. These costs can become a financial burden and may lead to distress and hardship for Canadian patients with GIST younger than 65 years without private drug coverage (estimated at approximately 5 patients per year).
In September and October 2021, LRGC conducted telephone interviews with 11 patients with advanced GIST (5 Canadian and 6 from the US) who had experience with ripretinib. All respondents were either initially or eventually diagnosed with metastatic GIST, and many experienced delays in diagnosis due to nonspecific symptoms. Patients highlighted the negative impacts of advanced GIST on HRQoL, including symptoms of vomiting, abdominal pain/discomfort, and bowel issues, including diarrhea, severe fatigue, black stools, and loss of appetite. Patients had received 1 line to 4 lines of therapy before ripretinib, and several recounted their rapid progression and sometimes severe side effects during treatment with prior TKIs. Most patients conveyed that ripretinib was generally more tolerable than other TKIs, with milder and acceptable side effects that included hair loss, cramping in body extremities, nausea, fatigue, hand-and-foot syndrome, foot calluses, and curly/kinky hair regrowth. More than half of patients reported improved HRQoL during ripretinib treatment compared with prior TKIs.
Patients with advanced GIST identified an unmet need for novel therapies that can stabilize or enhance HRQoL while effectively reducing disease progression for several years. In addition to improved survival, patients desired access to new drugs with that have improved toxicity profiles and longer-term effectiveness, and that can target specific GIST mutations.
Clinician Input
Input From Clinical Experts Consulted by CADTH
Two clinical experts with expertise in the diagnosis and management of patients with advanced GIST were consulted for this review. According to the clinical experts, not all patients with metastatic GIST respond to available treatments (TKIs), and responses are generally short-lived, especially in later lines of therapy. Following exhaustion of available TKIs (imatinib, sunitinib, and regorafenib), there are no standard treatment options available in Canada, and additional lines of therapy are required to fulfill the unmet needs of these patients. According to the clinical experts, the goals of fourth-line treatment of advanced GIST following progression/intolerance to imatinib, sunitinib, and regorafenib are prolonging survival, delaying disease progression, palliating symptoms, and preventing new symptom development. The clinical experts stated that, based on currently available evidence, ripretinib would be used for fourth-line monotherapy after progression on or intolerance to imatinib, sunitinib, and regorafenib and would not result in a treatment paradigm shift but rather would provide an additional option for later-line therapy in patients with no other good options. According to the clinical experts, there are no established biomarkers of response to ripretinib, and all patients with advanced GIST who experienced progression or intolerance on imatinib, sunitinib, and regorafenib with adequate performance status (PS), organ function, and hematological function would be candidates for ripretinib, irrespective of tumour mutational status. Patients with poor PS, limited organ/hematological function, significant comorbidities (especially cardiac problems), central nervous system metastases, and problems taking or absorbing oral medications would be least suitable for ripretinib treatment.
The clinical experts stated that treatment with ripretinib would be initiated either immediately following progression on third-line treatment (regorafenib) or after symptoms worsen follow discontinuation of third-line treatment. According to the clinical experts, response to ripretinib treatment would be assessed by clinical evaluation, in conjunction with imaging scans, every 2 months to 4 months. Clinically meaningful responses to therapy would be reflected by restricted tumour growth, prolongation of overall survival (OS) and progression-free survival (PFS), maintained or improved HRQoL, and stabilization or reduction of symptom severity. Treatment would be discontinued because of disease progression, significant adverse events (AEs), persistent treatment intolerance despite dosage reductions, or patient preference. The clinical experts also noted the convenience of ripretinib as an oral drug that can be self-administered at home in this advanced disease setting.
Clinician Group Input
One group of 7 Canadian medical oncologists who treat patients with advanced GIST provided input for this review; some of the oncologists are medical advisors to LRGC. No major contrary views were presented. The clinicians echoed the absence of fourth-line treatment options for patients after available TKIs (imatinib, sunitinib, and regorafenib) have been exhausted and the poor outcomes in these patients. Minor discrepancies were noted between the clinical experts and the clinician group input in the frequency of response assessment by imaging scans (2 to 3 months versus 3 to 4 months), possibly due to jurisdictional variation.
Drug Program Input
The Provincial Advisory Group (PAG) identified the following jurisdictional implementation issues: relevant comparators, considerations for continuation or renewal of therapy, considerations for discontinuation of therapy, considerations for prescribing of therapy, and care provision issues. The clinical experts consulted by CADTH for this review weighed evidence from the included study and other clinical considerations to provide responses to PAG’s drug program implementation questions (Table 4).
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
INVICTUS was a phase III, double-blind (DB), placebo-controlled multi-centre randomized controlled trial (RCT; N = 129) with an open-label (OL) period of active treatment.9-11 The primary objective of the study was to assess the efficacy of ripretinib in prolonging PFS per independent radiologic review (IRR) in patients with advanced GIST who had received prior anticancer therapies, including imatinib, sunitinib, and regorafenib. Secondary objectives included comparing objective response rate (ORR) per IRR (hierarchically tested), OS (hierarchically tested), patient-reported changes in disease symptoms and HRQoL from baseline to the start of cycle 2 (hierarchically tested), and other efficacy outcomes, including duration of response (DOR) between the ripretinib and placebo arms. Patients were enrolled at 29 sites in 12 countries (1 site in Toronto, Canada) and randomized to DB treatment with either ripretinib 150 mg orally once daily plus BSC or placebo orally once daily plus BSC. Following initial objective progression per IRR, patients and investigators were unblinded to treatment allocation, and patients could choose either to receive OL ripretinib at the same dosage (150 mg once daily) or to escalate the dosage to OL ripretinib 150 mg twice daily. Following treatment discontinuation, patients entered survival follow-up.
Adult patients with unresectable advanced GIST who had progressed on or developed intolerance to imatinib, sunitinib, and regorafenib were eligible for the study if they had Eastern Ontario Oncology Group (ECOG) PS 0, 1, or 2 and did not have active central nervous system metastases, clinically significant cardiac conditions or other comorbidities, or gastrointestinal problems preventing their taking or absorbing oral medication. The mean age of participants was approximately 60 years, approximately 57% were men, approximately 75% were White, and approximately 47% were enrolled at sites in the US. The most common tumour site was gastric (45.0%), and the most common location of primary tumour mutations was KIT exon 11 (58.1%). Approximately 60% of patients had received 3 prior lines of therapy, while approximately 40% had received 4 or more prior lines of therapy. Baseline demographic and disease characteristics were generally well balanced between study arms, apart from minor imbalances of potential prognostic relevance in age, ECOG PS, and gastric tumour site. The clinical experts consulted by CADTH for this review did not feel that any of these imbalances would be likely to affect the study results.
Efficacy Results
Key efficacy results of the INVICTUS study are summarized in Table 2. Importantly, the OS analysis did not account for crossover from placebo to ripretinib following initial objective progression, post-progression ripretinib treatment, or post-progression dosage escalation to the non–Health Canada–approved dosage of 150 mg twice daily. At the time of the primary analysis (database lock May 31, 2019), median OS was 28.6 weeks (95% confidence interval [CI], 17.9 to 50.4 weeks) in patients originally randomized to the placebo arm and 65.6 weeks (95% CI, 53.6 to 65.6 weeks) in patients originally randomized to the ripretinib arm. The hazard ratio (HR) for OS comparing ripretinib to placebo was 0.36 (95% CI, 0.21 to 0.62). A post hoc subgroup analysis of OS by combined treatment assignment in both the DB and OL periods showed the following results: DB placebo, no crossover, median OS 7.9 weeks (95% CI, 3.7 to 19.6 weeks); DB placebo with crossover to OL ripretinib 150 mg once daily, median OS 30.1 weeks (95% CI, 12.4 weeks to not calculable); and ||| |||||||||| ||| || || |||| || || |||||||||| |||||| || |||| ||||| |||| ||| |||| ||||| || ||| |||||||||||| ||| |||| ||| |||||||||| ||| || || |||| || |||||||||| ||| || ||| |||||| || ||||| ||||| |||| ||| |||| ||||| || ||||||||||||||||. During DB treatment, no objective tumour responses occurred in the placebo arm, while the ORR at the time of the primary analysis was 9.4% (95% CI, 4.2% to 17.7%) in the ripretinib arm (P = 0.0504). Among patients responding to ripretinib, median DOR was not estimable at the time of the primary analysis (May 31, 2019, data cut) but was 14.5 months (95% CI, 3.7 weeks to not estimable) at the more recent data cut of January 15, 2021. At the time of the primary analysis, median PFS during DB treatment was 4.1 weeks (95% CI, 4.0 to 7.3 weeks) in the placebo arm and 27.6 weeks (95% CI, 20.0 to 29.9 weeks) in the ripretinib arm (P < 0.0001). The HR for PFS comparing ripretinib to placebo was 0.15 (95% CI, 0.09 to 0.25). OS, ORR, and PFS results for the most recent cut (January 15, 2021) were similar. The results of the primary PFS analysis were statistically and clinically significant, according to the clinical experts consulted by CADTH for this review. Comparisons of OS and ORR (despite their descriptive nature and non-statistical significance, respectively, and despite the complexities of the OS analysis) were viewed by the clinical experts as supportive of the PFS findings and were judged to be potentially clinically important, given that the population is affected by advanced disease and has no other available treatment options.
HRQoL indicators (European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire for Cancer 30 item [EORTC QLQ-C30] role and physical function, EQ-5D-5L usual activities and pain/discomfort, EQ-5D-5L utility index scores, and EQ-visual analogue scale [VAS]) were similar at baseline in the ripretinib and placebo arms. Interpretation of changes from baseline to cycle 2 day 1 was limited by several factors (refer to Critical Appraisal).
Harms Results
Key harms results of the INVICTUS study are summarized in Table 2. AEs occurred in most patients treated with placebo (97.7%) and ripretinib (98.8%). Serious AEs occurred in larger proportions of patients receiving placebo (44.2%) than ripretinib (30.6%). Withdrawals due to AEs occurred in 11.6% of placebo-treated patients and 8.2% of ripretinib-treated patients. Deaths were more frequent among placebo-treated patients (23.3%) than among ripretinib-treated patients (5.9%), primarily due to disease progression.
Among protocol-specified AEs of special interest, squamous cell carcinoma of the skin occurred in no patients who received placebo and 2 patients (2.4%) who received ripretinib. Actinic keratosis occurred in 1 patient (2.3%) who received placebo and 5 patients (5.9%) who received ripretinib. All notable harms specified in the CADTH review protocol occurred more frequently in patients who received ripretinib than in those who received placebo: cardiac dysfunction, cardiac ischemic events, hypertension, cutaneous malignancies, palmar-plantar erythrodysesthesia syndrome, arthralgia, myalgia, and increased bilirubin. The most common AEs by preferred term were peripheral edema (ripretinib versus placebo: 16.5% versus 7.0%), hypertension (14.1% versus 4.7%), palmar-plantar erythrodysesthesia syndrome (21.2% versus 0%), arthralgia (17.6% versus 4.7%), myalgia (31.8% versus 11.6%), and increased bilirubin (16.5% versus 0%).
Table 2: Summary of Key Results From the INVICTUS Study
Outcome | Placebo | Ripretinib |
|---|---|---|
ITT population | ||
n | 44 | 85 |
OS (weeks) | ||
Events, n (%) | 26 (59.1)a 36 (81.8)b | 26 (30.6)a 46 (54.1)b |
Median OS (95% CI)c | 28.6 (17.9 to 50.4)a 27.4 (17.9 to 43.4)b | 65.6 (53.6 to 65.6)a 79.1 (57.1 to 133.6)b |
HR (95% CI)d | 0.36 (0.21 to 0.62)a 0.41 (0.26 to 0.65)b | |
ORR (%) | ||
ORR (95% CI) | 0% (0.0% to 8.0%)a 0% (0.0% to 8.0%)b | 9.4% (4.2% to 17.7%)a 11.8% (5.8% to 20.6%)b |
ORR difference (95% CI)e | 9.4% (0.2% to 17.5%)a | |
P valuef | 0.0504a | |
DOR (months), median (95% CI)c | NE (NE to NE)a NE (NE to NE)b | NE (16.0 to NE)a 14.5 (3.7 to NE)b |
PFS (weeks) | ||
Events, n (%) | 37 (84.1)a 37 (84.1)b | 51 (60.0)a 71 (83.5)b |
Median PFS (95% CI)c | 4.1 (4.0 to 7.3)a 4.1 (4.0 to 7.3)b | 27.6 (20.0 to 29.9)a 27.6 (20.0 to 35.3)b |
HR (95% CI)d | 0.15 (0.09 to 0.25)a 0.16 (0.10 to 0.27)b | |
P valueg | < 0.0001a | |
HRQoL, change from baseline to cycle 2 day 1 | ||
EORTC QLQ-C30 role functioning, adjusted mean (SE)h | –17.1 (5.0)a | 3.5 (3.5)a |
EORTC QLQ-C30 physical functioning, adjusted mean (SE)h | –8.9 (3.0)a | 1.6 (2.1)a |
EQ-5D-5L usual activities, % of patients reporting improvement or no change | 56.8%a | 70.6%a |
EQ-5D-5L pain/discomfort, % of patients reporting improvement or no change | 52.3%a | 60.1%a |
EQ-5D-5L utility (index) score, adjusted mean (SE)h | –0.0606 (0.02796)a | –0.0094 (0.01957)a |
EQ-VAS, mean (SD) | –8.9 (19.31)a | 3.7 (20.36)a |
Safety population | ||
n | 43 | 85 |
Harms, n (%) | ||
AEs | 42 (97.7) | 84 (98.8) |
SAEs | 19 (44.2) | 26 (30.6) |
WDAEs | 5 (11.6) | 7 (8.2) |
Deaths | 10 (23.3) | 5 (5.9) |
Notable harms, n (%) | ||
AESIs | ||
SCC of the skin | 0 | 2 (2.4) |
Actinic keratosis | 1 (2.3) | 5 (5.9) |
Keratoacanthoma | 0 | 0 |
Cardiac dysfunction and cardiac ischemic events | ||
Peripheral edema | 3 (7.0) | 14 (16.5) |
Sinus bradycardia | 0 | 4 (4.7) |
Chest pain | 1 (2.3) | 3 (3.5) |
Pleural effusion | 0 | 3 (3.5) |
Tachycardia | 0 | 2 (2.4) |
Bradycardia | 0 | 1 (1.2) |
Cardiac failure | 0 | 1 (1.2) |
Chest discomfort | 0 | 1 (1.2) |
Embolism | 0 | 1 (1.2) |
Orthopnea | 0 | 1 (1.2) |
Palpitations | 0 | 1 (1.2) |
Pericardial effusion | 0 | 1 (1.2) |
Ventricular extrasystoles | 0 | 1 (1.2) |
Cardiac murmur | 1 (2.3) | 0 |
Atrial fibrillation | 0 | 0 |
Generalized edema | 0 | 0 |
Mitral valve disease | 0 | 0 |
Peripheral swelling | 0 | 0 |
Hypertension | 2 (4.7) | 12 (14.1) |
Cutaneous malignancies | ||
Fibrous histiocytoma | 0 | 2 (2.4) |
Malignant melanoma in situ | 0 | 2 (2.4) |
SCC of head and neck | 0 | 2 (2.4) |
SCC of skin | 0 | 2 (2.4) |
Neoplasm skin | 0 | 1 (1.2) |
Basal cell carcinoma | 0 | 0 |
SCC | 0 | 0 |
Palmar-plantar erythrodysesthesia | 0 | 18 (21.2) |
Arthralgia | 2 (4.7) | 15 (17.6) |
Myalgia | 5 (11.6) | 27 (31.8) |
Increased bilirubin | 0 | 14 (16.5) |
AE = adverse event; AESI = adverse event of special interest; CI = confidence interval; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire for Cancer 30 item; EQ-VAS = EuroQol visual analogue scale; HR = hazard ratio; HRQoL = health-related quality of life; ITT = intention to treat; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event; SCC = squamous cell carcinoma; SD = standard deviation; SE = standard error; WDAE = withdrawal due to adverse event.
aData are from the May 31, 2019, database lock.
bData are from the January 15, 2021, database lock.
cFrom Kaplan-Meier analysis.
dHR from Cox proportional regression model with treatment and randomization stratification factors as fixed factors.
eFrom exact binomial CI.
fP value from Fisher exact test.
gP value from 2-sided stratified log-rank test. Stratification factors were the same as those applied to randomization (number of prior anticancer treatment and ECOG status at baseline).
hEstimated from an analysis of covariance (ANCOVA) model that included factors for study treatment, number of prior anticancer treatment, and ECOG PS at baseline as fixed effects.
Source: INVICTUS Clinical Study Report,12 CADTH review submission,8 and sponsor’s additional information.13
Critical Appraisal
The major limitation of the INVICTUS trial was its small size and associated uncertainty, although this was expected for a study of a rare disease. Minor baseline differences between arms (in favour of ripretinib) in age, ECOG PS, and tumour site were not considered likely to affect the study results by clinical experts consulted by CADTH for this review. Because of elective crossover of patients from DB placebo to OL ripretinib 150 mg once daily, as well as elective intra-patient post-progression dosage escalation to 150 mg twice daily (a dosage not approved by Health Canada), the relative impacts of ripretinib versus placebo treatment, pre- versus post-progression treatment, and ripretinib dose on OS could not be ascertained from the study data. Early failure of the statistical hierarchy at the time of the primary analysis precluded testing of OS and HRQoL differences between arms. Analyses of HRQoL outcomes were further limited by missing data and uncertainty regarding the measurement properties or minimal important differences (MIDs) of the instruments used in GIST patients. Changes in GIST symptoms were not directly assessed in the study.
The demographic and disease characteristics of the INVICTUS study population were considered broadly reflective of the Canadian population with advanced GIST who would be eligible for ripretinib, according to the clinical experts consulted by CADTH for this review. There were no major generalizability concerns to smaller subgroups (e.g., tumour mutational status). However, the descriptive OS results should not be generalized to clinical practice, due to inability to account for patient crossover from placebo to ripretinib, post-progression OL treatment, and dosage escalation during OL treatment to 150 mg twice daily. The impact of crossover from placebo to OL ripretinib would be expected to bias OS comparisons against ripretinib, while the impacts of post-progression treatment and intra-patient dosage escalation were uncertain.
Indirect Comparisons
No indirect evidence was identified for this review.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Conclusions
Evidence from the INVICTUS study suggested that administration of ripretinib in patients with advanced GIST who had previously received imatinib, sunitinib, and regorafenib was associated with statistically significant and clinically meaningful prolongation of PFS compared with placebo. Administration of ripretinib also resulted in numerically higher ORRs compared with placebo, although this difference was not statistically significant at the 0.05 level at the time of the primary analysis. OS was numerically longer in patients randomized to receive ripretinib compared with those randomized to receive placebo. However, differences in OS between patients randomized to receive ripretinib versus placebo were not tested statistically due to early failure of the statistical hierarchy, precluding definitive conclusions. Changes in patient-reported HRQoL (EORTC QLQ-C30, EQ-5D-5L) following ripretinib administration were difficult to interpret due to absence of formal statistical testing, missing data, wide variation in estimates, and uncertainty regarding HRQoL measurement properties in GIST patients. Ripretinib was generally well tolerated in most patients, and its notable harms were considered expected and acceptable by patients and clinicians. The observed PFS benefits, consistent numeric improvements in other efficacy outcomes, and acceptable toxicity profile in the study were aligned with outcomes identified as important to patients with advanced GIST who currently have no treatment options available.
Introduction
Disease Background
Gastrointestinal stromal tumours (GISTs) are the most common soft-tissue sarcomas (rare cancers of mesenchymal cells) of the gastrointestinal tract.1 GISTs occur more often in older individuals; they are found equally in both genders.14 Tumours arise in interstitial cells of Cajal of the stomach, or less commonly, the small intestine, esophagus, or other locations in the gastrointestinal tract (e.g., the rectum, colon, or mesentery without bowel wall primary).15 GISTs are typically characterized by primary gain-of-function mutations in CD117/c-KIT (80% to 85% of GISTs), especially exons 9 and 112; mutations in PDGFRA are less frequent (5% to 10% of GISTs).3 Small and/or slow-growing GISTs may be benign, while tumours that grow significantly outward from the bowel wall can cause dysphagia, bleeding, abdominal pain/discomfort, fatigue, vomiting, loss of appetite, and other gastrointestinal issues.4 Both disease symptoms and the side effects of therapy severely affect HRQoL.
Data on the prevalence, incidence, and survival of advanced GIST in Canada are unavailable. At diagnosis, approximately half of patients with GISTs are eligible for potentially curative surgical resection.3 Among patients undergoing resection, 5-year survival is approximately 54% and disease-free survival is approximately 45%.5 Based on an estimated 500 GIST cases diagnosed per year in Canada,7 with 75% representing advanced GIST (50% at diagnosis and 25% recurrent disease following resection) and assuming 80%, 70%, and 60% failure/progression rates on imatinib, sunitinib, and regorafenib, respectively, the sponsor estimated a target population of 62 to 86 patients with advanced GIST per year from 2023 to 2025 in Canada (outside of Quebec) who have received prior treatment with imatinib, sunitinib, and regorafenib and would be eligible for fourth-line ripretinib.8 Following progression or failure on third-line regorafenib, patient outcomes are dismal: further progression and death can be expected within several months.
Diagnosis of GIST is typically made by a gastroenterologist or surgeon based on endoscopic biopsy, pathology, and imaging findings. Few tumours are identified incidentally, and screening programs are rare in North America.16 Patients with advanced disease are transferred to the care of a medical oncologist for systemic therapy with palliative intent. Tumour mutational testing may be conducted at diagnosis or following progression after earlier lines of therapy (depending on the centre), which may influence treatment sequencing (e.g., avapritinib first line for patients with PDGFRA D842V).
Standards of Therapy
Clinical experts consulted by CADTH for this review stated that localized GIST can be treated by resection followed by adjuvant imatinib in high-risk patients,17 while locally advanced disease may require a neo-adjuvant approach. In the approximately 50% of patients with metastatic/unresectable GIST at diagnosis, as well as the 25% of GIST patients who undergo resection and subsequently experience recurrence, radiotherapy and chemotherapy are ineffective.1,6 Four TKIs are indicated for the treatment of patients with advanced/unresectable GIST: imatinib, sunitinib, regorafenib, and ripretinib.1,6 Another TKI, avapritinib, is not approved in Canada but may be obtained through special access programs for early-line (typically first) therapy of patients with a rare PDGFRA D842V mutation that is found in approximately 5% of GISTs. Imatinib (approved in 2003) is not generally curative but is associated with high ORRs3,18; progression typically occurs following secondary mutations in the KIT kinase domain.19 Sunitinib (approved in 2006) is used in patients with GIST who progressed on or were intolerant to imatinib; although the drug offers clinical benefit for the subset of patients with imatinib-induced secondary KIT mutations that do not confer sunitinib resistance, most patients will relapse within 6 months to 1 year following additional KIT mutations.20 Regorafenib (approved in 2013) is used as third-line therapy for patients who progressed on or were intolerant to imatinib and sunitinib.21,22 Despite providing clinical benefit for patients with certain secondary KIT mutations, responses are generally brief, and relapse generally occurs within 6 months. Additional KIT secondary mutations occur following third-line regorafenib treatment (particularly in exons 13 and 17) and contribute to resistance.21 The clinical experts consulted by CADTH stated that, in most patients, metastatic disease would be treated sequentially with imatinib, sunitinib, and regorafenib; disease control following TKI administration ameliorates symptoms, including pain, obstruction, and bleeding.
Following development of resistance to imatinib, sunitinib, and regorafenib, rechallenge with previously failed TKIs may offer minor survival benefits,23 but rechallenge is not currently a fourth-line treatment option in Canada. Ripretinib (approved in 2020) is not currently funded in Canadian jurisdictions except through special access programs. Thus, experimental drugs and/or BSC (e.g., analgesics, laxatives, antidiarrheals, antiemetics, and antibiotics) are the only options remaining to patients. According to the clinical experts, the goals of fourth-line treatment of advanced GIST following progression/intolerance to imatinib, sunitinib, and regorafenib are prolonging survival, delaying disease progression, palliating symptoms, or preventing new symptom development.
Drug
Ripretinib is a switch-control TKI that broadly inhibits signalling of KIT, PDGFRA, and other kinases (wild-type and multiple primary and secondary mutations) by binding to both the switch pocket and the activation loop, locking the kinase in an inactive state. Key characteristics of ripretinib are listed in Table 3. The drug is administered at a dosage of 150 mg orally (three 50 mg tablets) once daily. Ripretinib is indicated “for the treatment of adult patients with advanced gastrointestinal stromal tumour (GIST) who have received prior treatment with imatinib, sunitinib, and regorafenib.”24 The drug has not been previously reviewed by CADTH. The sponsor’s reimbursement request is aligned with the Health Canada–approved indication. The drug underwent expedited review by Health Canada under Project Orbis.
Table 3: Key Characteristics of Ripretinib and BSC for Fourth-Line Treatment of Advanced GIST
Characteristic | Ripretinib | BSCa |
|---|---|---|
Mechanism of action | Inhibition of kinase (e.g., KIT, PDGFRA) signalling by binding to the switch pocket and the activation loop | Symptom palliation |
Indicationb | For the treatment of adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib, and regorafenib | NA |
Route of administration | Orally | Generally orally |
Recommended dose | 150 mg once daily | Various |
Serious adverse effects or safety issues | Cardiac dysfunction, cardiac ischemic events, hypertension, cutaneous malignancies, palmar-plantar erythrodysesthesia syndrome, arthralgia, myalgia, increased bilirubin | NA |
BSC = best supportive care; GIST = gastrointestinal stromal tumour; NA = not applicable; PDGFRA = platelet-derived growth factor alpha.
aBSC: analgesics, laxatives, antidiarrheals, antiemetics, antibiotics, and others.
bHealth Canada–approved indication.
Source: CADTH review submission8 and product monograph for ripretinib.24
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The original patient group submission can be found in Appendix 1.
Two patient groups submitted patient input for this review: the CanCertainty Coalition and the GIST Sarcoma Life Raft Group Canada (LRGC). CanCertainty raised concerns about the financial and administrative barriers to accessing cancer treatments such as ripretinib in some Canadian jurisdictions (Ontario and the Atlantic provinces). Young patients (under the age of 65) who require take-home cancer treatment such as ripretinib and do not have private or automatic public prescription drug coverage may incur significant deductibles or co-payments from their personal savings. These costs can become a financial burden and may lead to distress and hardship for Canadian GIST patients under 65 without private drug coverage (estimated at approximately 5 patients per year).
In September and October 2021, LRGC conducted telephone interviews of 11 patients with advanced GIST (5 Canadian and 6 from the US) who had experience with ripretinib. All respondents were either initially or eventually diagnosed with metastatic GIST and many had experienced delays in diagnosis due to nonspecific symptoms. Patients highlighted the negative impacts of advanced GIST on HRQoL, including symptoms of vomiting, abdominal pain/discomfort, and bowel issues, including diarrhea, severe fatigue, black stools, and loss of appetite. Patients had received 1 to 4 lines of therapy before ripretinib, and several recounted their rapid progression and sometimes severe side effects during treatment with prior TKIs. Most patients conveyed that ripretinib was generally more tolerable than other TKIs, with milder and acceptable side effects that included hair loss, cramping in body extremities, nausea, fatigue, hand-and-foot syndrome, foot calluses, and curly/kinky hair regrowth. More than half of patients reported improved HRQoL during ripretinib treatment compared with prior TKIs.
Patients with advanced GIST identified an unmet need for novel therapies that can stabilize or enhance HRQoL while effectively reducing disease progression for several years. In addition to improved survival, patients desired access to new drugs that have improved toxicity profiles and longer-term effectiveness, and that can target specific GIST mutations.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib, and regorafenib.
Unmet Needs
According to the clinical experts consulted by CADTH, not all patients with metastatic GIST respond to available treatments (TKIs). The clinical experts noted that, among responding patients, responses are generally short-lived and become shorter with advancing lines of therapy (e.g., 7 months in second line, 5 months in third line). Eventually, almost all patients will develop refractory disease. Following exhaustion of available TKIs (imatinib, sunitinib, and regorafenib), there are no standard treatment options available in Canada. An additional line of therapy is required to fulfill the unmet needs of these patients.
Place in Therapy
The clinical experts consulted by CADTH for this review stated that, based on currently available evidence, ripretinib would be used as per the Health Canada indication for fourth-line monotherapy after progression on or intolerance to imatinib, sunitinib, and regorafenib. The clinical experts noted that, although use of avapritinib might, in theory, be an option for later-line treatment of patients with PDGFRA D842V mutations, it would be much more common for these patients to receive avapritinib in earlier lines of therapy. According to the clinical experts, ripretinib might theoretically be efficacious in earlier lines of therapy, but this has yet to be determined; trials evaluating ripretinib for second-line therapy are under way. The clinical experts noted that the mechanism of kinase inhibition by ripretinib is unique compared with that of other TKIs (binding to the switch conformation area rather than the adenosine triphosphate binding pocket). Based on the currently available data, clinical experts did not expect that funding of ripretinib would result in a treatment paradigm shift but rather would provide an additional option for later-line therapy that could be offered to patients who currently have no efficacious treatment available to them.
Patient Population
According to the clinical experts consulted by CADTH for this review, there are no established biomarkers of response to ripretinib. Thus, all patients with advanced GIST who experienced progression or intolerance on imatinib, sunitinib, and regorafenib with ECOG PS 0 to 2 and adequate organ and hematological function would be candidates for ripretinib. The clinical experts noted that most patients evaluated in the pivotal trial for ripretinib approval had KIT mutations, while only a few had PDGFRA mutations or wild-type tumours. However, patients would be candidates for ripretinib regardless of mutational status, and mutational analysis would not be a requirement for treatment. Similarly, the clinical experts stated that patients would be candidates for ripretinib regardless of tumour location. Patients with poor PS (e.g., ECOG PS > 2) or inadequate organ/hematological function, patients with significant comorbidities (e.g., class 2 to class 4 heart failure, cerebrovascular accident within 6 months, or venous thromboembolism within 3 months); patients who cannot take or absorb oral medications (e.g., due to bowel obstruction); and patients with central nervous system metastases would be least suitable for treatment with ripretinib treatment. The clinical experts stated that diagnosis of GIST through biopsy and pathologic examination is straightforward, and that misdiagnosis or diagnostic delays are not major issues. The clinical experts noted that GIST patients with progressive disease (PD) who are candidates for later-line therapy would generally be symptomatic. However, if patients remain asymptomatic following failure of prior TKIs, either immediate ripretinib treatment or a break in treatment until symptoms recur would be reasonable options.
Assessing Response to Treatment
According to clinical experts consulted by CADTH for this review, response to ripretinib treatment would be assessed via clinical evaluation (every 2 weeks to 4 weeks) and by CT or MRI scans (every 2 weeks to 4 months). Clinically meaningful responses to therapy would be reflected by prolonged OS and PFS, maintained or improved HRQoL and PS, and stabilized or reduced symptom severity (e.g., pain, bowel transit problems, liver/biliary obstruction, and bleeding).
Discontinuing Treatment
According to the clinical experts consulted by CADTH for this review, ripretinib treatment would be discontinued because of disease progression (either clinical symptomatic progression or radiographic progression), significant AEs (e.g., Grade ≥ 3 elevated liver enzymes), persistent treatment intolerance despite dosage reductions, or patient preference.
Prescribing Conditions
According to the clinical experts consulted by CADTH for this review, GIST diagnosis is typically made by a gastroenterologist or surgeon, but treatment with ripretinib in the fourth-line setting would be handled by a medical oncologist. Appropriate treatment setting would include outpatient oncology clinics (community or academic setting).
Additional Considerations
The clinical experts consulted by CADTH for this review noted the convenience of ripretinib as an oral drug that can be self-administered at home, minimizing clinic visits and potential hospitalizations in this advanced disease setting, and emphasized the absence of alternative viable options for fourth-line GIST therapy.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The original clinician group submission can be found in Appendix 2.
One group of 7 Canadian medical oncologists who treat patients with advanced GIST provided input for this review; some of the oncologists are medical advisors to LRGC. No major contrary views were presented. The clinicians echoed the absence of fourth-line treatment options for patients after available TKIs (imatinib, sunitinib, and regorafenib) have been exhausted and the poor outcomes in these patients. Minor discrepancies were noted between the clinical experts and the clinician group input in the frequency of response assessment by imaging scans (2 to 3 months versus 3 to 4 months), possibly due to jurisdictional variation.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Implementation issues | Clinical experts’ response |
|---|---|
Relevant comparators | |
PAG noted that BSC is a relevant comparator in patients with GIST who have progressed on imatinib, sunitinib, and regorafenib. | For pERC consideration. |
Considerations for continuation or renewal of therapy | |
Patients in the INVICTUS study received tumour assessments (via CT or MRI) every cycle for the first 3 cycles, then every other cycle starting at cycle 4. In clinical practice, what is the most appropriate frequency/modality to determine treatment response? | In Canadian clinical practice, imaging scans would be performed every 2 to 3 months rather than at every cycle. CT would be used more commonly than MRI. |
Considerations for discontinuation of therapy | |
At the time of disease progression in the INVICTUS trial, patients could either escalate the dosage to 150 mg ripretinib twice daily, continue 150 mg daily if there is continued clinical benefit, or discontinue therapy. What would be appropriate discontinuation criteria for ripretinib? | Discontinuation would be based on a combination of factors, including clinical/radiological progression, significant adverse events that may be related to ripretinib, impact on HRQoL, and patient preference. If possible, some clinicians would prefer to continue treating patients with ripretinib after progression, if the patients continued to tolerate the drug, until near the end of life, as there are no other treatment options in these patients and discontinuation may hasten progression or contribute to deterioration of symptoms and HRQoL. The INVICTUS study data suggest that post-progression treatment may offer some degree of benefit, but this is far from certain. Other clinicians may discontinue treatment immediately following or soon after progression they deem that the patient is unlikely to derive continued benefit. |
Considerations for prescribing of therapy | |
PAG noted that the usual dose of ripretinib is 150 mg (three 50 mg tablets) orally once daily. Lexicomp drug information database cautions not to use ripretinib 1 week before elective surgery and not to administer it for at least 2 weeks following surgery or until wound healing is adequate. | For pERC consideration. |
Care provision issues | |
PAG noted that, per the product monograph, ripretinib tablets are 50 mg and supplied in a bottle of 90 tablets. Recommended storage is to “Store in the original container at room temperature.” If the dosage is reduced, this storage restriction (original packaging) could lead to dispensing issues. The original container must continue to retain the desiccant provided. In the US, there is a restricted dispensing program that does not appear in the Health Canada monograph. | For pERC consideration. |
PAG noted that ripretinib has multiple potential drug-drug, drug-food (e.g., grapefruit), and drug-herb interactions, requiring assessment and potential intervention. | For pERC consideration. |
BSC = best supportive care; GIST = gastrointestinal stromal tumour; HRQoL = health-related quality of life; PAG = Provincial Advisory Group; pERC = CADTH pan-Canadian Oncology Drug Review Expert Committee.
Clinical Evidence
The clinical evidence included in the review of ripretinib is presented in a single section (the Systematic Review) that includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. No indirect evidence met the inclusion criteria for this review. No additional relevant studies were identified that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of ripretinib (150 mg orally once daily) for the treatment of adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib, and regorafenib.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults (age ≥ 18 years) with GIST who have received prior treatment with imatinib, sunitinib, and regorafenib Subgroups:
|
Intervention | Ripretinib (150 mg orally once daily) |
Comparator |
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; BSC = best supportive care; DOR = duration of response; GIST = gastrointestinal stromal tumour; HRQoL = health-related quality of life; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; RCT = randomized controlled trial; SAE = serious adverse event; TTR = time to response; WDAE = withdrawal due to adverse event.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.25
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974—) via Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Qinlock/ripretinib. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 3 for the detailed search strategies. The initial search was completed on November 12, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on March 9, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.26 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 3 for more information on the grey literature search strategy.
A focused literature search for network meta-analyses dealing with Qinlock/ripretinib was run in MEDLINE All (1946–) on November 12, 2021. No limits were applied.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
Four reports of a single study9-12 were identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6.
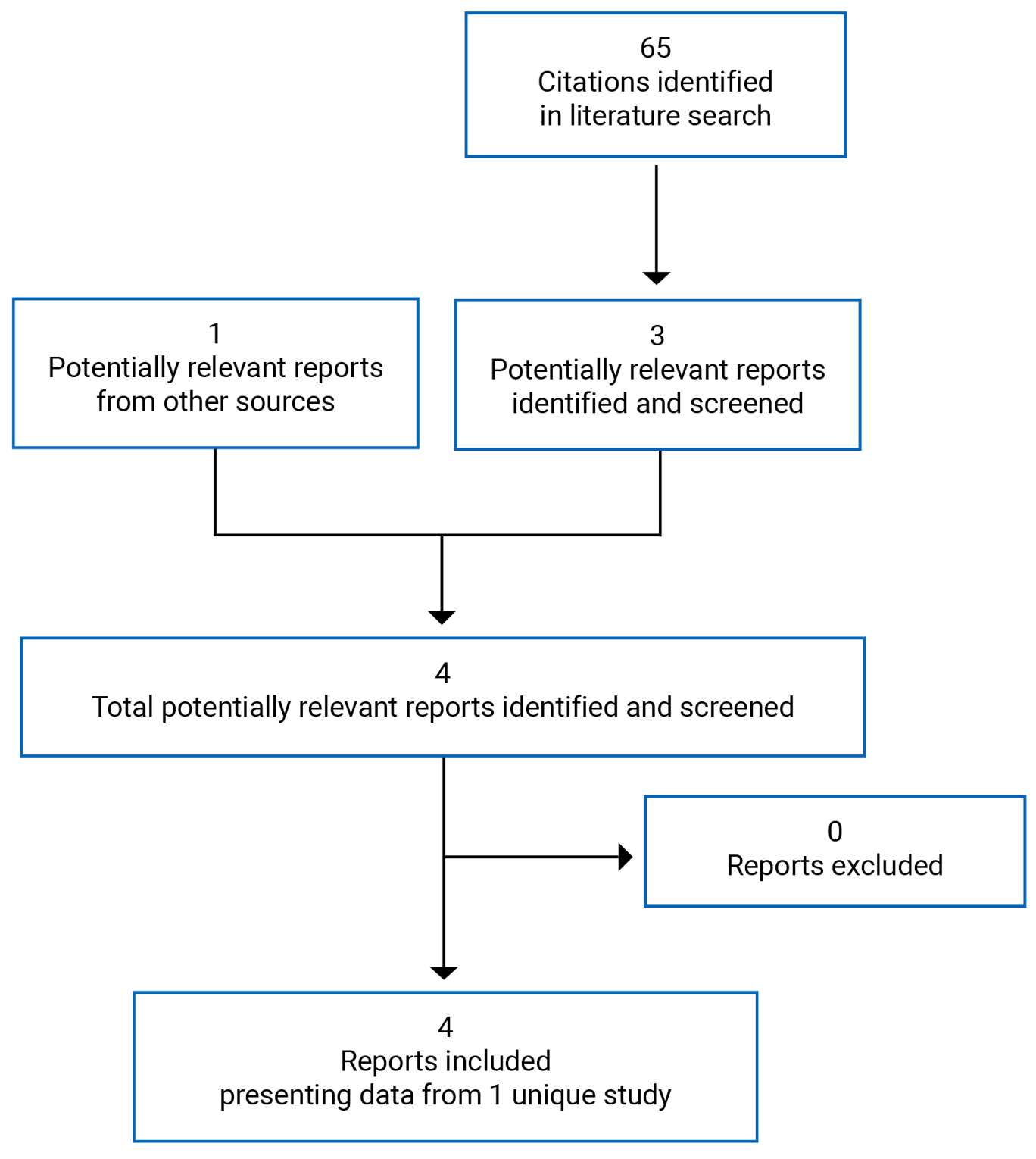
Table 6: Details of the Included Study
Item | INVICTUS |
|---|---|
Design and population | |
Study design | Phase III DB placebo-controlled multi-centre RCT |
Locations | 29 sites in 12 countries (US, Canada, Australia, Belgium, UK, France, Germany, Italy, Netherlands, Poland, Singapore, and Spain) |
Patient enrolment dates | February 27, 2018 to NRa |
Data cut-off | May 31, 2019 (first database lock); January 15, 2020 (second database lock) |
Randomized (N) | 129 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention |
|
Comparator |
|
Duration | |
Phase | |
Screening | 4 weeks |
DB treatment | Until first PD, unacceptable toxicity, withdrawal of IC, or death, whichever came first |
OL treatment | Until second PD, unacceptable toxicity, withdrawal of IC, or death, whichever came first |
Follow-up | Every 3 months until withdrawal of IC, death, or data cut-off, whichever came first |
Outcomes | |
Primary end point |
|
Secondary and exploratory end points | Key secondary:
Secondary:
Exploratory objectives:
|
Notes | |
Publicationsc | Blay et al. (2020)10 Bauer et al. (2021)9 Zalcberg et al. (2021)11 |
ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; BCRP = breast cancer resistance protein; b.i.d. = twice daily; BSC = best supportive care; CNS = central nervous system; CYP3A4 = cytochrome P450 3A4; DB = double-blind period; DOR = duration of response; ECG = electrocardiogram; ECOG PS = Eastern Cooperative Oncology Group performance status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire for Cancer 30 item; GIST = gastrointestinal stromal tumour; HBV = hepatitis B virus; HCV = hepatitis C virus; HRQoL = health-related quality of life; IC = informed consent; INR = international normalized ratio; IRR = independent radiological review; LVEF = left ventricular ejection fraction; mRECIST = modified Response Evaluation Criteria in Solid Tumours; NR = not reported; OL = open label; ORR = objective response rate; OS = overall survival; PD = progressive disease; PDGFRA = platelet-derived growth factor alpha; PFS = progression-free survival; p.o. = orally; PPI = proton pump inhibitor; PrT = prothrombin time; q.d. = once daily; QTc = QT interval corrected; QTcF = QT interval corrected by Fridericia’s formula; RCT = randomized controlled trial; TKI = tyrosine kinase inhibitor; TTP = time to progression; TTR = time to response; ULN = upper limit of normal; VAS = visual analogue scale; WHO = WHO.
aEnrolment was not stated in the clinical study report, but completion of enrolment was announced in a press release dated November 15, 2018.27
bThis dosage (150 mg twice daily orally) was allowed following disease progression by mRECIST per IRR and is outside the Health Canada–approved indication. Efficacy outcomes for this dose are not presented in this report.
cOne additional report was included (INVICTUS Clinical Study Report).
Source: INVICTUS Clinical Study Report.12
Description of Studies
INVICTUS was a phase III, DB, placebo-controlled multi-centre RCT (N = 129) with an OL period of active treatment.9-11 The study was funded by Deciphera Pharmaceuticals; an exclusive agreement was subsequently reached between Deciphera and the sponsor, Medison Biopharma Canada Inc., to commercialize and distribute ripretinib in multiple regions, including Canada. The primary objective of the study was to assess the efficacy of ripretinib in prolonging PFS per IRR in patients with advanced GIST who had received prior anticancer therapies, including imatinib, sunitinib, and regorafenib. Secondary objectives included comparing ORR per IRR (hierarchically tested to control for type I error), OS (hierarchically tested), patient-reported changes in disease symptoms and HRQoL from baseline to the start of cycle 2 (hierarchically tested), time to response (TTR) per IRR, DOR per IRR, and time to progression per IRR between the ripretinib and placebo arms. A summary of the design of the INVICTUS study is shown in Figure 2. Patients aged ≥ 18 years with inoperable advanced GIST who had progressed or become intolerant to all 3 prior TKIs and ECOG PS 0 to 2 were enrolled from 27 February 2018 until 15 November 2019 at 29 sites in 12 countries (including 1 site in Toronto, Canada); patients with active central nervous system metastases, clinically significant cardiac conditions or other comorbidities, and gastrointestinal problems preventing absorption of medication were excluded. Patients were screened for eligibility within 4 weeks of starting protocol therapy.
Patients were randomized 2:1 using an interactive response technology system to receive either: (i) ripretinib 150 mg orally once daily plus BSC or (ii) placebo orally once daily plus BSC, both on 28-day cycles. The randomization algorithm/procedure was not explicitly stated. Randomization was stratified by number of prior lines of therapy (3 versus ≥ 4; enrolment of patients who had received ≥ 4 prior lines of therapy was capped at 40%) and ECOG PS (0 versus 1 or 2). During the DB period, patients were treated until the first designation of PD by investigator assessment and/or IRR, unacceptable toxicity, withdrawal by patient or physician, or death, whichever came first. Following initial objective PD per IRR, patients and investigators were unblinded to treatment allocation, patients randomized to receive ripretinib 150 mg once daily during the DB period could choose from the following OL treatment options: continue ripretinib 150 mg once daily, escalate the dosage to ripretinib 150 mg twice daily, or discontinue ripretinib. Following initial progression, patients randomized to receive placebo during the DB period could choose from the following OL treatment options: (i) crossover to ripretinib 150 mg once daily or (ii) discontinue the study. Patients initially randomized to the placebo arm who chose to cross over to ripretinib 150 mg following initial progression and thereafter were designated with a second objective PD per IRR could choose from the following OL treatment options: continue ripretinib 150 mg once daily, escalate the dosage to ripretinib 150 mg twice daily, or discontinue ripretinib. The 150 mg twice daily dosage is not aligned with the Health Canada–approved dosage, and data for patients receiving this dosage are not presented in this report.
Following treatment discontinuation, patients entered survival follow-up (every 3 months until study withdrawal, death, or data cut-off, whichever came first). The database was closed on May 31, 2019, and these data were used for regulatory approval. Data for a second database lock of January 15, 2021, were made available in the CADTH review submission.
Figure 2: Design of the INVICTUS Study
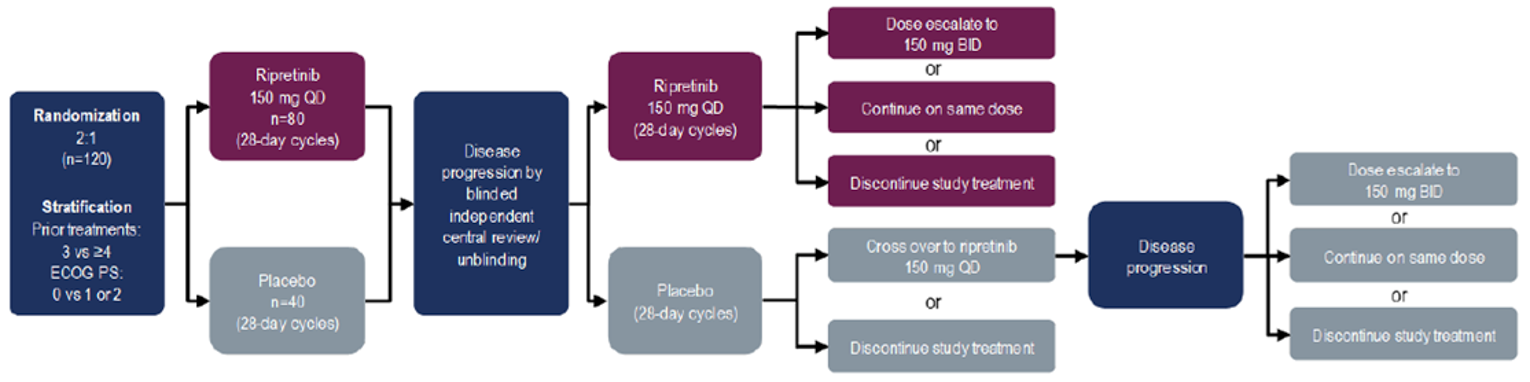
BID = twice daily; ECOG PS = Eastern Cooperative Oncology Group performance status; QD = once daily; vs = versus.
Source: INVICTUS Clinical Study Report.12
Populations
Inclusion and Exclusion Criteria
Key inclusion and exclusion criteria for the INVICTUS study are summarized in Table 6. Adult patients (age ≥ 18 years) with inoperable, advanced, histologically confirmed GIST and ECOG PS 0 to 2 were eligible if they had experienced PD on imatinib, sunitinib, and regorafenib or had documented intolerance to these TKIs. Patients had to have ≥ 1 measurable lesion by modified Response Evaluation Criteria in Solid Tumours (mRECIST) version 1.1 – GIST-specific, adequate hematological and organ function, and have recovered from any toxicity (to Grade ≤ 1) from prior therapies. Patients previously treated with ripretinib and patients with active central nervous system metastases, cardiac conditions (e.g., class II to IV heart disease or active ischemia), recent arterial thrombotic or embolic events, recent venous thrombotic events or pulmonary arterial events, electrocardiogram abnormalities, reduced ejection fraction, clinically significant comorbidities, or active bleeding were excluded. Use of proton pump inhibitors or other drugs that increase gastric pH was prohibited, as was use of drugs or substances with known drug-drug, drug-herb, or drug-food interactions with TKIs. Patients with gastrointestinal abnormalities that would impair taking or absorbing oral medications were excluded.
Baseline Characteristics
The baseline demographic characteristics of patients in the INVICTUS study are shown in Table 7. In the placebo and ripretinib arms, 59.1% and 55.3% of patients were men, respectively, and the mean ages were 62.0 and 59.1 years, respectively. A higher proportion of patients in the ripretinib arm were younger than 65 years (67.1%) compared with those in the placebo arm (50.0%). Approximately 75% of patients were White, approximately 90% were non-Hispanic, and approximately half were from the US. The proportions of patients with ECOG PS 0, 1, or 2 were 38.6%, 54.5%, 6.8%, respectively, in the placebo arm, and 43.5%, 47.1%, or 9.4%, respectively, in the ripretinib arm. Approximately 60% of patients had received 3 prior lines of therapy, while approximately 40% had received 4 or more prior lines of therapy. Baseline demographic characteristics were generally well balanced between study arms during the DB period, apart from minor imbalances in age and ECOG PS.
Table 7: Summary of Baseline Demographic Characteristics in the INVICTUS Study (ITT Population)
Characteristic | DB period | OL period | ||
|---|---|---|---|---|
Placebo N = 44 | Ripretinib N = 85 | Ripretinib 150 mg q.d. (DB: placebo) N = 29 | Ripretinib 150 mg q.d. (DB: ripretinib) N = 11 | |
Gender, n (%) | ||||
Female | 18 (40.9) | 38 (44.7) | 13 (44.8) | 1 (9.1) |
Male | 26 (59.1) | 47 (55.3) | 16 (55.2) | 10 (90.9) |
Age at informed consent (years) | ||||
Mean (SD) | 62.0 (13.50) | 59.1 (10.84) | 62.4 (14.08) | 58.5 (11.29) |
Median (range) | 64.5 (33 to 83) | 59.0 (29 to 82) | 68.0 (33 to 81) | 55.0 (47 to 82) |
Age category, n (%) | ||||
18 to 64 years | 22 (50.0) | 57 (67.1) | 12 (41.4) | 9 (81.8) |
65 to 74 years | 12 (27.3) | 20 (23.5) | 10 (34.5) | 0 |
75 years or older | 10 (22.7) | 8 (9.4) | 7 (24.1) | 2 (18.2) |
Race, n (%) | ||||
Asian | 5 (11.4) | 4 (4.7) | 2 (6.9) | 1 (9.1) |
Black or African American | 2 (4.5) | 8 (9.4) | 2 (6.9) | 1 (9.1) |
White | 33 (75.0) | 64 (75.3) | 21 (72.4) | 7 (63.6) |
Not reported | 4 (9.1) | 8 (9.4) | 4 (13.8) | 1 (9.1) |
Other | 0 | 1 (1.2) | 0 | 1 (9.1) |
Ethnicity, n (%) | ||||
Hispanic or Latino | 0 | 1 (1.2) | 0 | 0 |
Not Hispanic or Latino | 38 (86.4) | 76 (89.4) | 23 (79.3) | 10 (90.9) |
Not reported | 5 (11.4) | 5 (5.9) | 5 (17.2) | 0 |
Unknown | 1 (2.3) | 3 (3.5) | 1 (3.4) | 1 (9.1) |
Region | ||||
US | 20 (45.5) | 40 (47.1) | 14 (48.3) | 5 (45.5) |
Non-US | 24 (54.5) | 45 (52.9) | 15 (51.7) | 6 (54.5) |
Height (cm) | ||||
Mean (SD) | 169.7 (11.72) | 169.7 (10.38) | 169.1 (11.65) | 172.4 (9.75) |
Median (range) | 170.0 (151 to 190) | 169.3 (147 to 192) | 170.0 (151 to 189) | 173.4 (158 to 184) |
Weight (kg) | ||||
Mean (SD) | 71.4 (18.04) | 73.9 (19.02) | 69.3 (19.24) | 76.4 (22.94) |
Median (range) | 67.5 (44 to 110) | 73.0 (39 to 133) | 67.0 (44 to 110) | 72.2 (49 to 133) |
BMI (kg/m2) | ||||
Mean (SD) | 24.5 (5.08) | 25.6 (6.22) | 23.9 (4.65) | 25.6 (8.19) |
Median (range) | 22.9 (16 to 39) | 24.4 (13 to 47) | 22.3 (16 to 34) | 23.0 (19 to 47) |
ECOG PS at screening, n (%) | ||||
0 | 17 (38.6) | 37 (43.5) | 11 (37.9) | 2 (18.2) |
1 | 24 (54.5) | 40 (47.1) | 18 (62.1) | 7 (63.6) |
2 | 3 (6.8) | 8 (9.4) | 0 | 2 (18.2) |
ECOG PS stratum at screening, n (%) | ||||
0 | 19 (43.2) | 38 (44.7) | 11 (37.9) | 2 (18.2) |
1 or 2 | 25 (56.8) | 47 (55.3) | 18 (62.1) | 9 (81.8) |
Number of prior systemic anticancer treatments, n (%) | ||||
3 | 27 (61.4) | 54 (63.5) | 20 (69.0) | 4 (36.4) |
4 or more | 17 (38.6) | 31 (36.5) | 9 (31.0) | 7 (63.6) |
BMI = body mass index; DB = double-blind; ECOG PS = Eastern Cooperative Oncology Group performance status; ITT = intention to treat; OL = open label; q.d. = once daily; SD = standard deviation.
Source: INVICTUS Clinical Study Report.12
The baseline disease characteristics of patients in the INVICTUS study are shown in Table 8. The most common site of the primary tumour was gastric (40.9% of the placebo arm and 47.1% of the ripretinib arm). The most common location of primary tumour mutations was KIT exon 11 (63.6% of the placebo arm and 55.3% of the ripretinib arm). Few patients had PDGFRA mutations (none in the placebo arm and 3.5% of the ripretinib arm), and a small subset had wild-type KIT and PDGFRA genes (6.8% of the placebo arm and 8.2% of the ripretinib arm). Approximately two-thirds of patients had stage IV disease at diagnosis. The most common histologic type at diagnosis was spindle cell (70.5% of the placebo arm and 43.5% of the ripretinib arm). The mean time elapsed since diagnosis was approximately 7 years. Approximately 80% of patients had undergone prior surgery for GIST, while approximately 20% had received radiotherapy. Among patients who had received 4 or more prior lines of therapy, the most common drugs (other than imatinib, sunitinib, and regorafenib) were sorafenib, pazopanib, and nilotinib. Baseline disease characteristics were generally well balanced between study arms, apart from minor imbalances in tumour site and histology.
Table 8: Summary of Baseline Disease Characteristics in the INVICTUS Study (ITT Population, DB Period)
Characteristic | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
Site of primary tumour, n (%) | ||
Gastric | 18 (40.9) | 40 (47.1) |
Duodenum | 8 (18.2) | 2 (2.4) |
Jejunum/ileum | 8 (18.2) | 20 (23.5) |
Colon/rectum | 0 | 9 (10.6) |
Mesenteric/omental | 6 (13.6) | 6 (7.1) |
Other | 4 (9.1) | 7 (8.2) |
Unknown | 0 | 1 (1.2) |
Tumour mutation gene, n (%) | ||
KIT exon 9 | 6 (13.6) | 14 (16.5) |
KIT exon 11 | 28 (63.6) | 47 (55.3) |
KIT other exons | 2 (4.5) | 2 (2.4) |
PDGFRA | 0 | 3 (3.5) |
KIT wt/PDGFRA wt | 3 (6.8) | 7 (8.2) |
Not available | 5 (11.4) | 11 (12.9) |
Not done | 0 | 1 (1.2) |
Stage at initial diagnosis, n (%) | ||
Stage I | 0 | 2 (2.4) |
Stage IA | 1 (2.3) | 1 (1.2) |
Stage IB | 0 | 2 (2.4) |
Stage II | 1 (2.3) | 1 (1.2) |
Stage IIIA | 0 | 7 (8.2) |
Stage IIIB | 6 (13.6) | 7 (8.2) |
Stage IV | 30 (68.2) | 56 (65.9) |
Unknown | 6 (13.6) | 9 (10.6) |
Histology at initial diagnosis, n (%) | ||
Epithelioid | 3 (6.8) | 17 (20.0) |
Mixed spindle cell and epithelioid | 4 (9.1) | 16 (18.8) |
Spindle cell | 31 (70.5) | 37 (43.5) |
Other | 4 (9.1) | 10 (11.8) |
Unknown | 2 (4.5) | 5 (5.9) |
Time since initial diagnosis (years) | ||
Mean (SD) | 7.16 (4.328) | 7.11 (4.129) |
Median (range) | 5.42 (1.4 to 17.5) | 5.87 (1.5 to 16.4) |
Prior anti-GIST therapy, n (%) | ||
Systemic therapya | 44 (100.0) | 85 (100.0) |
Imatinib | 44 (100.0) | 85 (100.0) |
Regorafenib | 44 (100.0) | 85 (100.0) |
Sunitib | 44 (100.0) | 85 (100.0) |
Sorafenib | 3 (6.8) | 9 (10.6) |
Pazopanib | 5 (11.4) | 8 (9.4) |
Nilotinib | 6 (13.6) | 7 (8.2) |
Masitinib | 1 (2.3) | 4 (4.7) |
Avapritinib | 3 (6.8) | 3 (3.5) |
Cabozantinib | 1 (2.3) | 2 (2.4) |
Nivolumab | 0 | 2 (2.4) |
Ponatinib | 2 (4.5) | 1 (1.2) |
Total number of prior systemic therapies | ||
3 | 26 (59.1) | 54 (63.5) |
4 | 12 (27.3) | 21 (24.7) |
5 | 4 (9.1) | 6 (7.1) |
6 | 0 | 1 (1.2) |
7 | 2 (4.5) | 3 (3.5) |
Surgery | 36 (81.8) | 71 (83.5) |
Radiotherapy | 10 (22.7) | 18 (21.2) |
DB = double-blind; GIST = gastrointestinal stromal tumour; ITT = intention to treat; PDGFRA = platelet-derived growth factor alpha; SD = standard deviation; wt = wild-type.
aPrior systemic therapies administered in ≥ 1 patient in either arm are reported.
Source: INVICTUS Clinical Study Report.12
Interventions
During the DB period, patients were randomized 2:1 to receive either ripretinib 150 mg orally once daily plus BSC or placebo orally once daily plus BSC, both on repeated 28-day cycles. Both ripretinib and placebo were supplied as identically sized, shaped, and coloured tablets (ripretinib: 50 mg per tablet). Concomitant use of medications for symptomatic relief (e.g., analgesics, laxatives, antiemetics) was permitted. Medications that increased gastric pH (e.g., antacids) other than proton pump inhibitors were permitted provided they were not administered within 2 hours before or after administration of study drug. Proton pump inhibitors, strong or moderate inhibitors or inducers of cytochrome P450 (CYP)3A4, grapefruit or grapefruit juice, substrates or inhibitors of breast cancer resistance protein, and other systemic anticancer therapies (including investigational therapies) were prohibited. Patients were cautioned against taking strong or moderate inhibitors or inducers of CYP2D6, CYP2C8, or CYP2E1; substrates or inhibitors of P-glycoprotein 1; substrates of organic anion transporter polypeptides 1B1 and 1B3; and medications dependent on CYP2C8, CYP2C9, CYP2C19, or CYP2D6 for their metabolism. Patients taking any of these drugs were closely monitored for drug-drug interactions.
Treatment was continued until initial PD (either investigator assessed and/or per IRR), unacceptable toxicity, withdrawal by patient or physician, death, pregnancy, loss to follow-up, or nonadherence to study drug. Treatment was discontinued for the following toxicities: Grade 4 dermatologic toxicities or arthralgia/myalgia (unless not life threatening), Stevens-Johnson syndrome of any grade, Grade 4 hypertension, and clinically significant Grade 3 or higher laboratory AEs, including creatine phosphokinase and lipase elevation. In addition, treatment could be interrupted for no more than 1 cycle (28 days) and/or the dosage could be reduced stepwise (first reduction: 100 mg once daily; second reduction: 50 mg once daily) for the following toxicities: any Grade ≥ 3 toxicity, Grade 2/3 dermatologic toxicities or arthralgia/myalgia, symptomatic Grade 3 hypertension, and asymptomatic/not clinically significant Grade 3 or higher laboratory AEs that persist for more than 10 days. Per protocol, attempts were made to re-escalate to the dosage level at which toxicity occurred. If the AE returned to Grade 1 or baseline after dosage reduction, the patient could restart at the next higher step and remain at this dosage level for 1 cycle without interruption before escalating to the starting dosage level (if required). If treatment was delayed longer than 28 days or patients required dosages lower than 50 mg once daily, treatment was discontinued.
Following initial PD, patients randomized to receive placebo during the DB period could choose to cross over and receive ripretinib 150 mg once daily plus BSC. Patients randomized to receive ripretinib 150 mg once daily during the DB period, as well as the subset of patients initially randomized to receive DB placebo who crossed over to OL ripretinib 150 mg once daily and developed subsequent PD per IRR, were eligible to escalate the dosage to 150 mg twice daily plus BSC (at the first and second designation of PD, respectively).
Outcomes
A list of efficacy and safety end points identified in the CADTH review protocol that were assessed in INVICTUS study is provided in Table 9. These end points are further summarized in this section.
Table 9: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | INVICTUS |
|---|---|
PFSa per IRR in the DB period | Primary |
ORRb per IRR | Key secondary |
OSc | Secondary |
HRQoL: EORTC QLQ-C30d (change from baseline to cycle 2 day 1 in role function and physical function) | Secondary |
HRQoL: EQ-5D-5Le (change from baseline to cycle 2 day 1 in pain/discomfort, usual activities, and index utility score) | Secondary |
HRQoL: EQ-VASf (change from baseline to cycle 2 day 1) | Secondary |
DOR per IRRg | Secondary |
TTR per IRRh | Secondary |
AEs, SAEs, WDAEs, deaths | Safety |
AESIs (SCC of the skin, actinic keratosis, keratoacanthoma) | Safety |
Notable harms: cardiac dysfunction, cardiac ischemic events, hypertension, cutaneous malignancies, palmar-plantar erythrodysesthesia syndrome, arthralgia, myalgia, increased bilirubin | Safety |
AE = adverse event; AESI = adverse event of special interest; DB = double-blind; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire for Cancer 30 item; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels questionnaire; HRQoL = health-related quality of life; IRR = independent radiological review; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; SAE = serious adverse event; SCC = squamous cell carcinoma; TTR = time to response; VAS = visual analogue scale; WDAE = withdrawal due to adverse event.
Note: For HRQoL outcomes, all components of each patient-reported instrument were presented. Pre-specified hypotheses regarding changes in HRQoL outcomes are listed in parentheses.
aDefined as the interval between the date of randomization and the earliest documented evidence of first disease progression based on IRR or death due to any cause. PFS was censored at randomization for patients without evaluable radiological assessment unless they died within 2 cycles of treatment. For patients who only had nonmeasurable lesions, PFS was censored at the date of latest evaluable progression-free radiologic assessment. PFS was censored for patients who underwent surgery or palliative radiotherapy and patients who received other anticancer therapy before progression at the last evaluable progression-free radiologic assessment. For patients who progressed or died after 2 or more missed/nonevaluable assessments, PFS was censored at the last evaluable radiologic assessment. For patients who did not progress or die, PFS was censored at last progression-free radiologic assessment.
bDefined as the proportion of patients with confirmed complete or partial response per IRR (2 repeat measurements at least 4 weeks apart) during the DB phase before PFS events or censoring. Patients with unknown or missing response were classified as nonresponders.
cDefined as the interval between the date of randomization and death from any cause. Patients were censored at the last date known alive.
dScores for role function and physical function range from 0 to 100 with higher scores indicating better HRQoL.
eScores for pain/discomfort and usual activities range from 1 (no problems) to 5 (extreme problems). The index (utility) score ranges from less than 0 (worse than dead) to 1.00 (perfect health) and is calculated using a scoring function to assign a value to self-reported health states from a set of country-specific (or if unavailable, the set from the nearest neighbouring country with available set) population-based preference weights.
fScores range from 0 (worst imaginable health) to 100 (best imaginable health).
gDefined as the interval between the first assessment of confirmed complete response or partial response until the first disease progression or death, whichever came first. If PFS was censored, DOR was censored at the last evaluable progress-free radiologic assessment.
hDefined as the interval between date of randomization and the earliest date of first documented confirmed complete response or partial response.
Source: INVICTUS Clinical Study Report.12
OS, ORR, DOR, TTR, and PFS are standard and accepted outcome measures in oncology trials. A detailed discussion and critical appraisal of the HRQoL measures used in the INVICTUS study (EORTC QLQ-C3028 and EQ-5D-5L29) is provided in Appendix 2. Neither the measurement properties nor the MID of either instrument have been specifically evaluated in patients with GIST. Among all cancer patients, the measurement properties of these instruments have been investigated. Differences of approximately 10 points in EORTC QLQ-C30 individual items and scale scores (range: 0 to 100, with higher scores on function scales reflecting better function and higher scores on symptom scales reflecting increased symptoms), approximately 7 to 12 points in EQ-VAS scores (range: 0 to 100, with 0 and 100 representing “worst imaginable health” and ”best imaginable health,” respectively), and approximately 0.07 to 0.12 (using the UK algorithm) in EQ-5D-5L utility index scores (range: < 0 to 1, with 0 and 1 representing the health states “dead” and ”perfect health,” respectively) are typically considered significant. MIDs for EQ-5D-5L descriptive system dimension scores (range: 1 to 5, representing no problems, slight problems, moderate problems, severe problems, and extreme problems, respectively) are uncertain.
During treatment, tumour response was assessed by CT or MRI at screening (within 21 days of cycle 1 day 1) and then on day 1 of each cycle until cycle 4. Subsequently, pelvic and abdominal scans were performed every other cycle, and chest scans were performed only for patients with lung metastases at baseline or with lung symptoms. The same imaging modality was used for each patient throughout the study. Copies of all scans were sent for blinded IRR by an independent radiologist.
Tumour response was assessed using mRECIST version 1.1 – GIST-specific. No lymph nodes were chosen as target lesions, and enlarged lymph nodes were followed as nontarget lesions. No bone lesions were chosen as target lesions. Partial response (PR) was defined as at least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters. Complete response (CR) was defined as disappearance of all target lesions with reduction of the short axis of any pathological lymph nodes to less than 10 mm. PD was defined as a predefined increase ( + 20%), taking as reference the smallest sum on study, in the sum of target lesions or the appearance of new nontarget lesions; the sum must also demonstrate an absolute increase of at least 5 mm. A progressively growing new tumour nodule within a pre-existing tumour mass was considered unequivocal evidence of progression if the lesion was at least 2 cm in size and definitively a new active GIST lesion or the lesion was expanding on at least 2 sequential imaging studies. Stable disease was defined as neither sufficient shrinkage (compared to baseline) to qualify for PR nor sufficient increase (taking as reference the smallest sum diameters while on study) to qualify for PD.
An initial indication of PR or CR per investigator assessment was confirmed 4 or more weeks later, following IRR. The investigator decided whether to discontinue protocol therapy due to PD based on local imaging scans and clinical evaluation in conjunction with IRR. If the IRR confirmed no disease progression, the patient continued to receive study drug unless there was a medical need (i.e., rapid progression or clinical deterioration requiring treatment discontinuation). For investigator-determined progression based on clinical deterioration, a scan was performed and sent for IRR to determine PD. Following confirmation of PD by IRR, the patient’s treatment assignment was unblinded, and the patient’s dosage could be escalated or the patient could cross over.
During treatment, patient-reported HRQoL instruments (EORTC QLQ-C30 and EQ-5D-5L) were completed on days 1 and 15 of cycle 1, on day 1 of each cycle thereafter, and at the end-of-treatment visit. Following treatment discontinuation, an end-of-treatment visit within 7 days of the final dose included clinical evaluation, imaging scans, and completion of HRQoL instruments. Thereafter, patients entered survival follow-up every 3 months (by phone).
All efficacy outcomes, except OS and PFS, were evaluated during the DB treatment period. OS was censored at the date patients were last known alive but not for patients who crossed over from placebo to OL ripretinib and/or who had a dosage escalation to 150 mg twice daily during the OL treatment period. For patients randomized to receive placebo during the DB period who had objective PD per IRR and chose to cross over to ripretinib, second PFS was defined as the interval between the date of the first ripretinib dose and the first subsequent objective PD per IRR or death, whichever came first. See the footnotes to Table 9 for PFS censoring rules.
Harms outcomes included treatment-emergent AEs, serious AEs, AEs requiring dosage interruption or reduction, withdrawals due to AE, and AEs of special interest (AESIs). AESIs were squamous cell carcinoma, actinic keratosis, and keratoacanthoma. AEs that began or worsened on or after the start of protocol therapy until 30 days after the last dose of study drug were captured. AEs were defined as any untoward medical occurrence and were coded according to Medical Dictionary for Regulatory Activities (MedDRA) version 21.130 and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.03.31
Statistical Analysis
Statistical analysis of efficacy outcomes in the INVICTUS study is summarized in Table 10. No interim analyses were planned or conducted. The trigger for the final primary analysis (May 31, 2019, database lock) was the occurrence of 90 PFS events, which was expected to occur approximately 6 months after the last patient was enrolled in the trial. An unplanned analysis of a more recent data cut (January 15, 2021) was also made available for CADTH review.
The planned sample size of 120 patients (80 ripretinib and 40 placebo) was based on the primary efficacy analysis of PFS per IRR during the DB treatment period, as well as the key secondary outcome of ORR per IRR and safety assessment, assuming 15% patient dropout. In the resulting final sample of 105 patients (n = 70 ripretinib, n = 35 placebo), it was assumed that 90 PFS events would occur (55 ripretinib, 35 placebo) in the final analysis. This would yield at least 90% power to detect a difference in PFS (by log-rank test, refer to following discussion), assuming median PFS of 4.5 months for ripretinib and 1 month for placebo, 9 months of uniform recruitment, and 6 months of additional follow-up (maximum patient follow-up 15 months). This sample size was expected to yield approximately 80% power to detect a 20% difference in ORR (by Fisher’s exact test, refer to following discussion), assuming an ORR of 22% for ripretinib and 2% for placebo.
Type I error was controlled using a hierarchical testing strategy. Hypothesis tests for treatment differences were performed at a 2-sided alpha significance level of 0.05 sequentially, in the following order: (1) primary analysis of PFS per IRR, (2) key secondary analysis of ORR per IRR, (3) analysis of OS, and (4) analysis of HRQoL reflected in changes from baseline to cycle 2 day 1 in EORTC QLQ-C30 role function and physical function (each at a 0.025 significance level). Once a hypothesis test was nonsignificant at the 0.05 level, the remaining analyses were viewed as descriptive.
For the primary PFS analysis, PFS per IRR was compared in the intention-to-treat (ITT) population during the DB period between the ripretinib and placebo arms using a 2-sided stratified log-rank test. The HR comparing treatment arms was calculated using stratified Cox proportional hazard models with treatment and randomization stratification factors as fixed factors. Stratification factors for these analyses were those used for randomization (3 versus ≥ 4 prior systemic therapies and ECOG PS 0 versus 1 or 2). 95% CIs for the HR were calculated using the Wald method. The PFS function was estimated using the Kaplan-Meier (KM) product limit method. PFS time (25th percentile, median, 75th percentile) and PFS rates at pre-specified time points (26, 39, and 52 weeks), each with 2-sided 95% CIs, were derived from KM analysis. Sensitivity analyses for the primary PFS analysis included analysis using randomization stratification factor values collected on the electronic case record form instead of the interactive response technology, analysis in the per-protocol and safety populations instead of the ITT population, and analysis of PFS per investigator assessment.
For the key secondary ORR analysis, ORR per IRR was calculated in the ITT population during the DB period in each treatment arm along with the exact binomial 95% CI. ORRs were compared between treatment arms using 2-sided Fisher’s exact test. The ORR difference between treatment arms was calculated by simple subtraction and a 95% Newcombe score CI was constructed. Sensitivity analyses of the key secondary ORR analysis included analysis in the per-protocol population instead of the ITT population and analysis of ORR per investigator assessment.
For the secondary analysis of changes from baseline to cycle 2 day 1 of the DB period in EORTC QLQ-C30 role and physical function in the ITT population (included in the statistical hierarchy), a stratified analysis of covariance (ANCOVA) model was used to calculate adjusted mean changes from baseline and to compare changes from baseline between treatment arms. The ANCOVA model included randomization stratification factors as factors (3 versus ≥ 4 prior systemic therapies and ECOG PS 0 versus 1 or 2). Analysis of changes from baseline to cycle 2 day 1 of the DB period in other HRQoL outcomes outlined in the remainder of this paragraph were not adjusted for multiplicity. For changes from baseline in EQ-5D-5L pain/discomfort and usual activities, a Cochran-Mantel-Haenszel test was used to assess differences in response distribution between treatment arms. For index utility scores, a stratified ANCOVA model was used to calculated adjusted mean changes from baseline and to compare changes from baseline between treatment arms, as previously described. For changes from baseline in EQ-VAS scales, a t-test was used to assess differences in mean change from baseline between treatment arms.
Analysis of OS and time to progression as secondary outcomes during the DB period was performed in the same manner as the primary PFS analysis, as was analysis of second PFS during the OL treatment period. TTR was assessed using descriptive and summary statistics. DORs and their 95% CIs in each treatment arm were calculated using KM methodology. Of the analyses described in this paragraph, only the OS analysis was adjusted for multiplicity.
Subgroup analyses of PFS and ORR per IRR during the DB period were conducted for pre-specified subgroups (by age, gender, race, region, ECOG PS, and number of prior therapies), as per the primary and key secondary efficacy analysis, but in exploratory fashion. A post hoc subgroup analysis of ORR per IRR by tumour mutational status was also conducted. Data for a post hoc subgroup analysis of OS by treatment assignment in both the DB and OL periods was made available for CADTH review. The study was not specifically powered to evaluate outcomes in individual strata. Safety data were tabulated and presented using descriptive statistics (frequencies).
For time-to-event analyses (PFS, OS, DOR), missing data were accounted for by censoring. TTR was evaluated among responders and, thus, missing data were not possible. For analysis of ORR, patients without tumour response assessment were classified as nonresponders. For analysis of EORTC QLQ-C30 role and physical function, missing data for cycle 2 day 1 were replaced with data from the end of study treatment during the DB period.
Table 10: Statistical Analysis of Efficacy End Points in the INVICTUS Study
End point | Position in statistical hierarchy | Statistical model | Adjustment factors | Sensitivity analyses | Censoring rules |
|---|---|---|---|---|---|
Double-blind period | |||||
PFS per IRR | 1 (2-sided alpha = 0.05) |
|
|
|
|
ORR per IRR | 2 (2-sided alpha = 0.05) |
| None |
| NA |
OS | 3 (2-sided alpha = 0.05) | As per primary PFS analysis | As per primary PFS analysis | None | OS was censored at the date patients were last known alive |
HRQoL: Change from baseline to cycle 2 day 1 in EORTC QLQ-C30 role function and physical function) | 4 (2-sided alpha = 0.025 each) |
| ANCOVA model: randomization stratification factors as factors (3 vs. ≥ 4 prior systemic therapies and ECOG PS 0 vs. 1 or 2) | None | NA |
HRQoL: Change from baseline to cycle 2 day 1 in EQ-5D-5L dimensions | Not included |
| ANCOVA model: randomization stratification factors as factors (3 vs. ≥ 4 prior systemic therapies and ECOG PS 0 vs. 1 or 2) | None | NA |
HRQoL: Change from baseline to cycle 2 day 1 in EQ-VAS | Not included | t-test for difference in mean change from baseline between treatment arms | None | None | NA |
TTR | Not included | Descriptive and summary statistics | None | None | NA |
DOR | Not included | DORs and 2-sided 95% CIs from KM methodology | None | None | If PFS was censored, DOR was censored at the last evaluable progress-free radiologic assessment |
TTP | Not included | As per primary PFS analysis | As per primary PFS analysis | None | At date of death for patients who died without disease progression |
Safety (AEs, SAEs, WDAEs, mortality, notable harms) | Not included | Descriptive and summary statistics | None | None | NA |
Open-label period | |||||
Second PFS in the OL period (for patients who crossed over from placebo to ripretinib 150 mg once daily) | Not included | As per primary PFS analysis | As per primary PFS analysis | None | As per primary PFS analysis, using the date of the first OL ripretinib dose |
Safety (AEs, SAEs, WDAEs, mortality, notable harms) | Not included | Descriptive and summary statistics | None | None | NA |
AE = adverse event; ANCOVA = analysis of covariance; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group performance status; eCRF = electronic case record form; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire for Cancer 30 item; HR = hazard ratio; HRQoL = health-related quality of life; IRR = independent radiological review; IRT = interactive response technology; KM = Kaplan-Meier; NA = not applicable; OL = open label; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PP = per protocol; SAE = serious adverse event; TTP = time to progression; TTR = time to response; VAS = visual analogue scale; vs. = versus; WDAE = withdrawal due to adverse event.
Source: INVICTUS Clinical Study Report.12
Analysis Periods
The DB treatment analysis period was defined as 1 of the following intervals:
from the randomization date to the last follow-up date if a patient did not have disease progression based on IRR, if a placebo patient did not cross over to ripretinib treatment, or if a patient was initially treated with ripretinib and did not continue to receive ripretinib at 150 mg once daily or escalate to 150 mg twice daily after disease progression based on IRR
from the randomization date to the first disease progression date if a patient was initially treated with ripretinib and continued to receive ripretinib at 150 mg once daily after disease progression based on IRR
from the randomization date to the day immediately before the first dosage of ripretinib 150 mg twice daily if a patient’s dosage was escalated to ripretinib 150 mg twice daily after disease progression based on IRR
from the randomization date to the day immediately before the first dose of ripretinib 150 mg once daily if a patient crossed over from placebo to receive ripretinib at 150 mg once daily after disease progression based on IRR.
The OL treatment analysis period was defined as 1 of the following intervals:
from the day immediately after the first disease progression based on IRR to the last follow-up date if a patient was initially treated with ripretinib and continued to receive ripretinib 150 mg once daily after disease progression based on IRR
from the first dose date of ripretinib 150 mg twice daily to the last follow-up date if a patient was initially treated with ripretinib and the patient’s dosage was escalated to ripretinib 150 mg twice daily after disease progression based on IRR
from the first dose date of ripretinib 150 mg once daily to the last follow-up date if a patient crossed over from placebo to receive ripretinib 150 mg once daily after disease progression based on IRR.
The OL treatment analysis period was further subdivided into 2 subperiods: before intra-patient dosage escalation and following intra-patient dosage escalation.
All efficacy outcomes were evaluated in the DB treatment period, with the following exceptions. OS was evaluated during the entire study period. Among patients who were randomized to receive placebo in the DB period and subsequently crossed over to ripretinib in the OL period following initial progression per IRR, second PFS was evaluated in the period before intra-patient dosage escalation.
Safety outcomes were evaluated separately during each analysis period (DB period and OL period). For the OL period, safety analyses were conducted separately for the 2 subperiods (before and following intra-patient dosage escalation).
Analysis Populations
The ITT population was defined as all patients who provided informed consent and were randomized. The per-protocol population was defined as randomized patients who did not have important protocol deviations that were expected to compromise efficacy and/or safety assessments, as follows: (i) patients with pre-specified inclusion/exclusion criteria deviations, (ii) patients who received the wrong treatment, (iii) patients who received an incorrect dose, and (iv) patients who received pre-specified prohibited medications. The safety population was defined as all patients who received at least 1 dose of study drug, and data were analyzed according to the treatment the patient actually received.
Results
Unless otherwise noted, data presented are for the primary analysis (May 31, 2019, database lock).
Patient Disposition
Patient disposition during the DB period of the INVICTUS study is summarized in Table 11. For disposition during the OL period, refer to Appendix 3. A total of 154 patients were screened, of whom 25 (16.2%) were screen failures. The remaining 129 patients were randomized to the ripretinib (n = 85) and placebo arms (n = 44) for DB treatment. As of the May 31, 2019, data cut, approximately 30% and 20% of patients in the placebo and ripretinib arms, respectively, had discontinued treatment. The most common reasons for treatment discontinuation were PD and death. Few patients (n = 2, 4.7% in the placebo arm and n = 3, 3.5% in the ripretinib arm) discontinued protocol therapy due to AEs. Approximately 32% and 18% of patients in the placebo and ripretinib arms, respectively, had discontinued the study. The most common reason for study discontinuation was death. Attrition due to losses to follow-up or withdrawal of consent were minimal.
As of the May 31, 2019, data cut, a total of 71 patients (55.0%), including 29 (67.4%) patients randomized to the placebo arm and 42 (49.4%) of patients randomized to the ripretinib 150 mg once daily arm for DB treatment, had entered the OL treatment period. Among the 29 patients initially randomized to the placebo arm for DB treatment who crossed over to ripretinib 150 mg once daily, 10 (22.7%) subsequently had an escalation in dosage to 150 mg twice daily, while 19 (43.2%) continued to receive the 150 mg once daily dosage. Among the 42 patients initially randomized to the ripretinib 150 mg once daily arm for DB treatment who chose to receive OL ripretinib, 31 (36.5%) had an escalation in dosage to 150 mg twice daily, while 11 (12.9%) continued to receive the 150 mg once daily dosage.
Table 11: Patient Disposition in the INVICTUS Study (DB Period)
Disposition | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
Screened, N | 154 | |
Screen failure, N | 25 | |
Randomized, N (%) | 44 (100.0) | 85 (100.0) |
Discontinued treatment,a n (%) | 13 (30.2) | 17 (20.0) |
Primary reason for treatment discontinuation, n (%)a | ||
Adverse event | 2 (4.7) | 3 (3.5) |
Clinical progression | 3 (7.0) | 4 (4.7) |
Death | 4 (9.3) | 3 (3.5) |
Physician decision | 1 (2.3) | 1 (1.2) |
Confirmed PD by investigator assessment | 0 | 3 (3.5) |
Confirmed PD by IRR | 2 (4.7) | 1 (1.2) |
Withdrawal of IC from study | 0 | 2 (2.4) |
Withdrawal of IC from treatment | 1 (2.3) | 0 |
Discontinued study,b n (%) | 14 (31.8) | 15 (17.6) |
Primary reason for study discontinuation, n (%)b | ||
Death | 13 (29.5) | 12 (14.1) |
Withdrawal of IC from study | 1 (2.3) | 3 (3.5) |
Analysis populations, n (%)b,c | ||
ITT population | 44 (100.0) | 85 (100.0) |
Safety population | 43 (97.7) | 85 (100.0) |
PP population | 42 (95.5) | 81 (95.3) |
Entered OL, n (%) | 29 (67.4) | 42 (49.4) |
Treatment ongoing at data cut-off, n (%) | 1 (2.3) | 26 (30.6) |
DB = double-blind; IC = informed consent; IRR = independent radiological review; ITT = intention to treat; OL = open label; PD = progressive disease; PP = per protocol.
Note: Data are from the May 31, 2019, database lock.
aDenominator is the safety population.
bDenominator is the ITT population.
cFor the OL period, all patients received assigned treatments per protocol.
Source: INVICTUS Clinical Study Report.12
Important protocol deviations during the DB treatment period of the INVICTUS study as of the May 31, 2019, data cut are summarized in Table 12. Important protocol deviations occurred in 2 (4.5%) patients randomized to the placebo arm and 4 (4.7%) patients randomized to the ripretinib 150 mg once daily arm.
Table 12: Important Protocol Deviations in the INVICTUS Study (ITT Population, DB Period)
Deviation | Placebo N = 43 | Ripretinib N = 85 |
|---|---|---|
Any important deviations, n (%) | 2 (4.5) | 4 (4.7) |
Type of important deviation, n (%) | ||
Patient did not satisfy entry criteria | 1 (2.3) | 2 (2.4) |
Patient received incorrect dose | 1 (2.3) | 1 (1.2) |
Patient received prohibited medication | 1 (2.3) | 1 (1.2) |
DB = double-blind; ITT = intention to treat.
Note: Data are from the May 31, 2019, database lock.
Source: INVICTUS Clinical Study Report.12
Exposure to Study Treatments
Treatment exposure in the INVICTUS study is summarized in Table 13. Treatment adherence was assessed by site personnel at each site visit, both verbally and by ongoing study drug count. Patients were instructed to return all unused, partially used, and used study drug bottles to the site at each visit. As of the May 31, 2019, data cut, the mean treatment duration was shorter in patients who received placebo compared with those who received ripretinib 150 mg once daily during the DB period (8.25 and 24.44 weeks, respectively). On mean, patients who received ripretinib 150 mg once daily during the DB period had completed 6.11 cycles and achieved 96.5% dose intensity with 97.74% treatment adherence. Dose was modified in 21 (24.7%) of patients treated with ripretinib in the DB period, most commonly dose was interrupted (n = 18, 21.2%).
Table 13: Treatment Exposure in the INVICTUS Study (Safety Population)
Treatment exposure | DB period | OL period | ||
|---|---|---|---|---|
Placebo N = 43 | Ripretinib N = 85 | Ripretinib 150 mg q.d. (DB: placebo) N = 29 | Ripretinib 150 mg q.d. (DB: ripretinib) N = 11 | |
Treatment duration (weeks)a | ||||
Mean (SD) | 8.25 (6.757) | 24.44 (13.941) | 16.87 (12.418) | 5.23 (6.169) |
Median (range) | 6.00 (0.4 to 38.4) | 23.86 (1.3 to 59.4) | 12.00 (1.0 to 44.1) | 3.86 (0.3 to 20.0) |
Treatment duration category, n (%)a | ||||
Less than 1 month | 8 (18.6) | 4 (4.7) | 3 (10.3) | 6 (54.5) |
1 to < 3 months | 29 (67.4) | 19 (22.4) | 12 (41.4) | 4 (36.4) |
3 to < 6 months | 5 (11.6) | 23 (27.1) | 7 (24.1) | 1 (9.1) |
6 to < 12 months | 1 (2.3) | 36 (42.4) | 7 (24.1) | 0 |
12 months or longer | 0 | 3 (3.5) | 0 | 0 |
Number of cyclesb | ||||
Mean (SD) | 2.06 (1.689) | 6.11 (3.485) | 4.22 (3.105) | 1.31 (1.542) |
Median (range) | 1.50 (0.1 to 9.6) | 5.96 (0.3 to 14.9) | 3.00 (0.3 to 11.0) | 0.96 (0.1 to 5.0) |
Relative dose intensity (%)c | ||||
Mean (SD) | 91.6 (11.96) | 96.5 (7.62) | 92.5 (12.56) | 86.7 (19.23) |
Median (range) | 97.0 (56 to 100) | 100.0 (64 to 100) | 100.0 (50 to 100) | 100.0 (42 to 100) |
Adherence (%)d | ||||
Mean (SD) | 92.00 (10.945) | 97.74 (5.178) | 93.71 (11.289) | 91.04 (14.713) |
Median (range) | 96.97 (60.3 to 100.0) | 100.0 (71.0 to 100.0) | 100.00 (50.0 to 100.0) | 100.00 (56.3 to 100.0) |
Adherence category, n (%)d | ||||
Less than 75% | 5 (11.6) | 1 (1.2) | 2 (6.9) | 2 (18.2) |
75% to < 80% | 2 (4.7) | 0 | 1 (3.4) | 0 |
80% or greater | 36 (83.7) | 84 (98.8) | 26 (89.7) | 3 (27.3) |
Dose modifications, n (%) | 9 (20.9) | 21 (24.7) | 10 (34.5) | 3 (27.3) |
Type of dose modification, n (%) | ||||
Dose increase | 0 | 3 (3.5) | 0 | 0 |
Dose reduction | 1 (2.3) | 7 (8.2) | 2 (6.9) | 0 |
Dose interruption | 8 (18.6) | 18 (21.2) | 9 (31.0) | 3 (27.3) |
DB = double-blind; OL = open label; q.d. = once daily; SD = standard deviation.
Note: Data are from the May 31, 2019, database lock.
aCalculated as (date of last treatment – date of first treatment + 1)/7. For patients who entered the OL period, the end date of the DB treatment period is used as the date of last treatment for calculation.
bCalculated as (date of last treatment – date of first treatment + 1)/28. Date of last treatment is defined as in footnote a.
cCalculated as total dose (mg) / total planned dose (mg) × 100.
dCalculated as total number of days dosed / treatment duration in days × 100. Treatment duration in days calculated as (date of last treatment – date of first treatment + 1) with date of last treatment defined as in footnote a.
Source: INVICTUS Clinical Study Report.12
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in this section. Refer to Appendix 3 for detailed efficacy data (sensitivity and subgroup analyses; second PFS among patients randomized to receive DB placebo who crossed over to ripretinib 150 mg once daily following initial progression per IRR).
Overall Survival
OS results are shown in Table 14, Figure 3, and Figure 4. This outcome was positioned in the statistical hierarchy after a nonsignificant result for a prior outcome (ORR) at the time of the primary analysis; the analysis was viewed as descriptive. OS was analyzed for the study as a whole and reflects both the DB and OL treatment periods, including patients who crossed over from placebo to ripretinib and patients who had an escalation in dosage to 150 mg twice daily for OL treatment. As of the May 31, 2019, database lock and in the ITT population, OS events had occurred in 26 (59.1%) of patients originally randomized to the placebo arm and 26 (30.6%) of patients originally randomized to the ripretinib arm. Median OS was 28.6 weeks (95% CI, 17.9 to 50.4 weeks) for patients originally randomized to the placebo arm and 65.6 weeks (95% CI, 53.6 to 65.6 weeks) for patients originally randomized to the ripretinib arm. The HR for OS comparing ripretinib with placebo was 0.36 (95% CI, 0.21 to 0.62). Results were similar for the January 15, 2021, database lock.
Table 14: OS in the INVICTUS Study (ITT Population)
OS | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
Patients with events, n (%) | 26 (59.1)a 36 (81.8)b | 26 (30.6)a 46 (54.1)b |
Patients censored, n (%) | 18 (40.9)a 8 (18.2)b | 59 (69.4)a 46 (54.1)b |
Median OS (95% CI), weeksc | 28.6 (17.9 to 50.4)a 27.4 (17.9 to 43.4)b | 65.6 (53.6 to 65.6)a 79.1 (57.1 to 133.6)b |
P valued | 0.0004a | |
HR (95% CI)e | 0.36 (0.21 to 0.62)a 0.41 (0.26 to 0.65)b | |
OS rates (95% CI)a | ||
26 weeks | 55.9 (39.9 to 69.2) | 84.3 (74.5 to 90.6) |
39 weeks | 43.1 (27.9 to 57.5) | 71.2 (59.3 to 80.1) |
52 weeks | 25.9 (7.2 to 49.9) | 65.4 (51.6 to 76.1) |
CI = confidence interval; HR = hazard ratio; ITT = intention to treat; OS = overall survival.
aData are from the May 31, 2019, database lock.
bData are from the January 15, 2021, database lock.
cFrom KM analysis.
dP value from 2-sided stratified log-rank test. Stratification factors were the same as those applied to randomization (number of prior anticancer treatment and ECOG status at baseline). Testing occurred after failure of the statistical hierarchy; therefore, the P value should be considered descriptive.
eHR from Cox proportional regression model with treatment and randomization stratification factors as fixed factors.
Source: INVICTUS Clinical Study Report,12 CADTH review submission,8 and sponsor’s additional information.13
Figure 3: Kaplan-Meier Plot of OS in the INVICTUS Study (ITT Population; Data Cut: May 31, 2019)
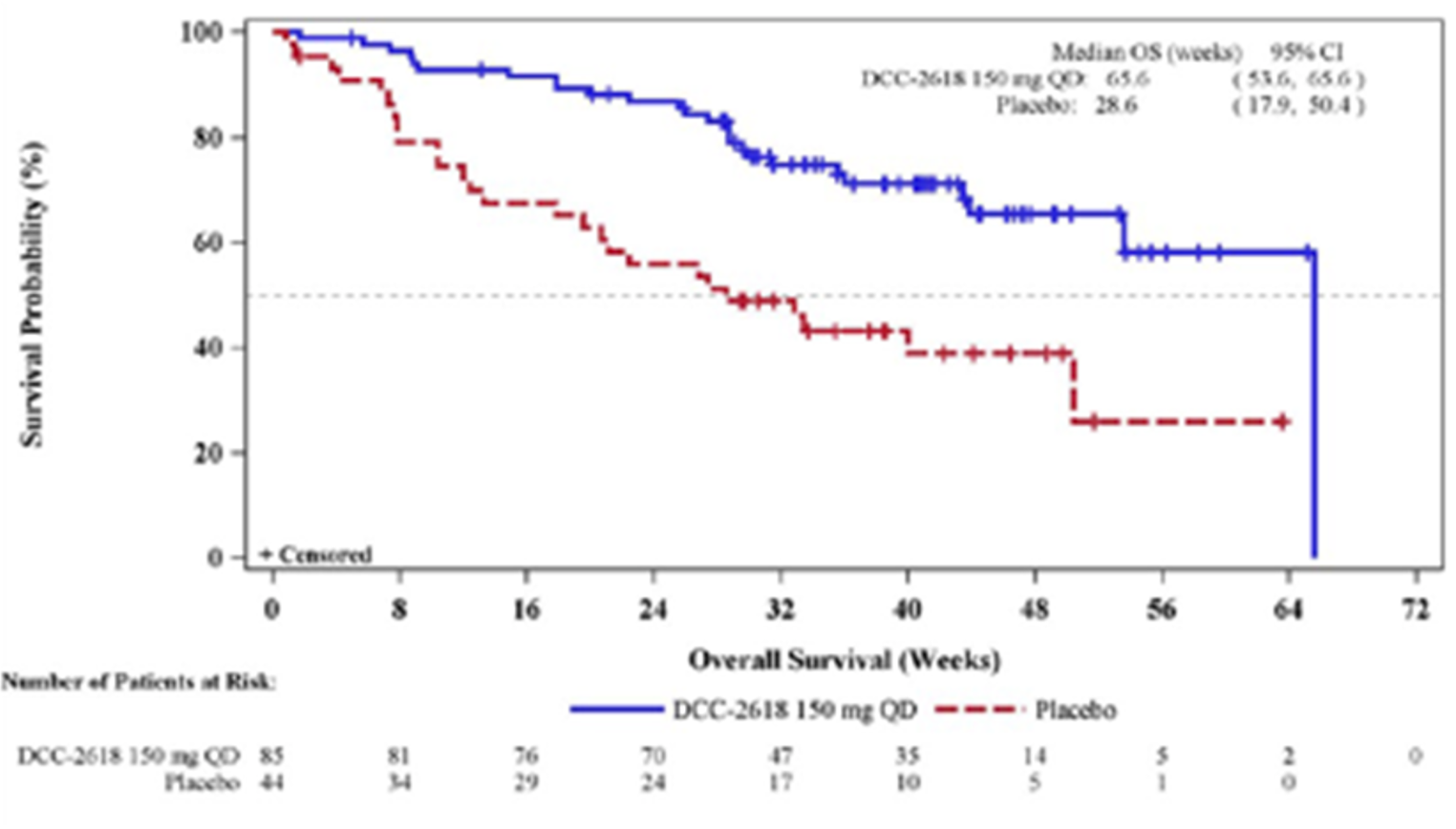
CI = confidence interval; DCC-2618 = ripretinib; ITT = intention to treat; OS = overall survival; QD = once daily.
Note: Plus symbols represent censored observations.
Source: INVICTUS Clinical Study Report.12
Figure 4: Kaplan-Meier Plot of OS in the INVICTUS Study (ITT Population; Data Cut: January 15, 2021)
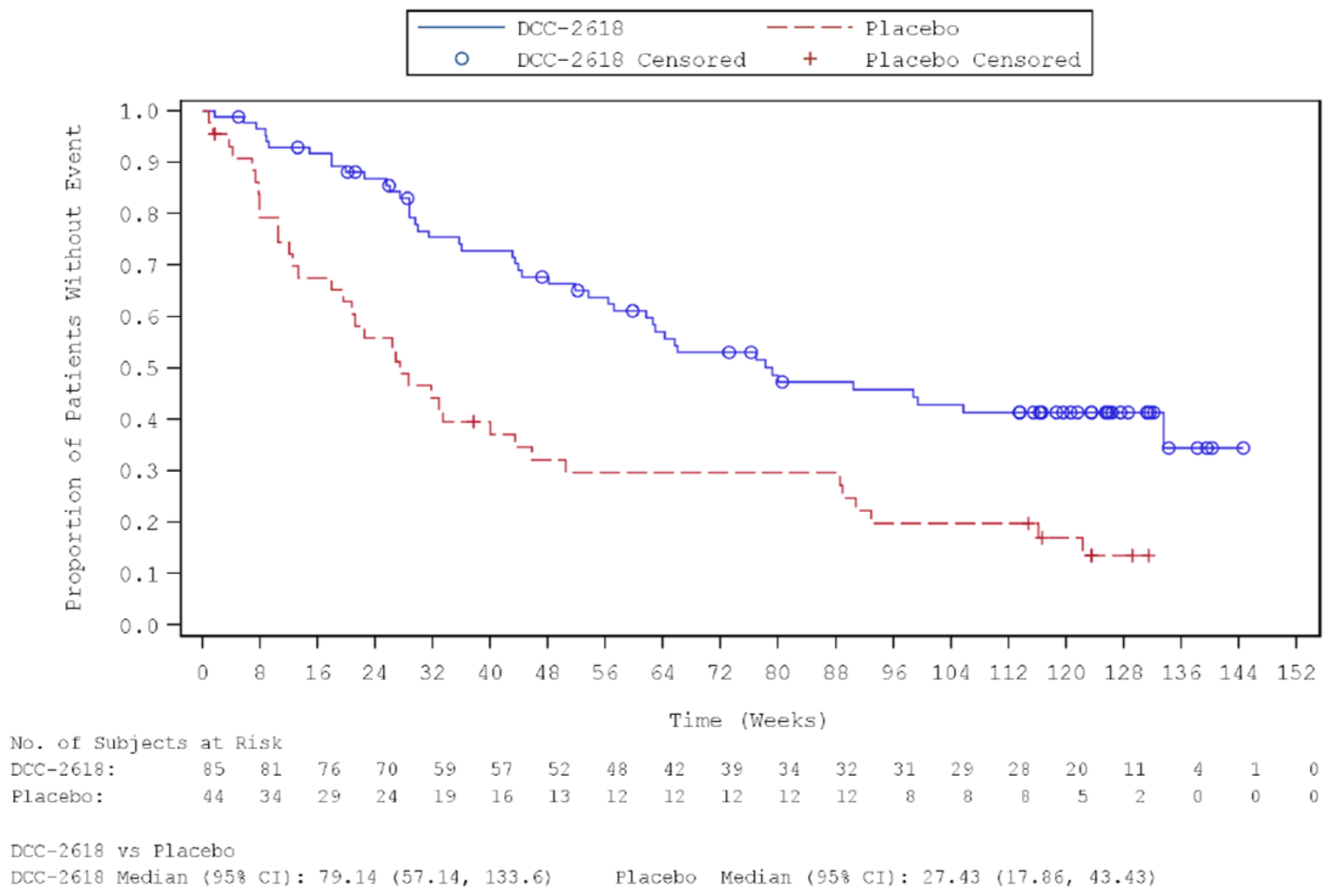
CI = confidence interval; DCC-2618 = ripretinib; ITT = intention to treat; OS = overall survival.
Note: Circle and plus symbols represent censored observations.
Source: Sponsor’s additional information.13
Refer to Appendix 3 for subgroup analyses of OS. A post hoc subgroup analysis of OS by combined treatment assignment in both the DB and OL periods showed the following results: DB placebo, no crossover (n = 14), median OS 7.9 weeks (95% CI, 3.7 to 19.6 weeks); DB placebo with crossover to OL ripretinib 150 mg once daily (n = 14), median OS 30.1 weeks (95% CI, 12.4 weeks to not calculable); and ||||| ||| |||||||||| ||| || || |||| || || ||||||||| ||||||| |||||| || |||| ||||| |||| ||| |||| ||||| || ||| |||||||||||| ||| |||| ||| |||||||||| ||| || || |||| || |||||||||| ||| || || ||||||| |||||| || ||||| ||||| |||| ||| |||| ||||| || ||| ||||||||||||
Health-Related Quality of Life
Changes in EORTC QLQ-C30 role and physical functioning scores from baseline to cycle 2 day 1 by treatment arm in the ITT are shown in Table 15. This outcome was positioned in the statistical hierarchy after a nonsignificant result for a prior outcome (ORR) at the time of the primary analysis; the analysis was viewed as descriptive. Baseline role functioning (placebo: mean = 73.8; ripretinib: mean = 69.4) and physical functioning (placebo: mean = 76.0; ripretinib: mean = 75.7) were similar in both treatment arms. The adjusted mean changes from baseline in role functioning were –17.1 (standard error [SE] = 5.0) in the placebo arm and 3.5 (SE = 3.5) in the ripretinib arm; the difference in adjusted mean changes from baseline comparing ripretinib with placebo was 20.61 (95% CI, 8.58 to 32.63). The adjusted mean changes from baseline in physical functioning were –8.9 (SE = 3.0) in the placebo arm and 1.6 (SE = 2.1) in the ripretinib arm; the difference in adjusted mean changes from baseline comparing ripretinib with placebo was 10.48 (95% CI, 3.37 to 17.59).
Table 15: EORTC QLQ-C30 Scores in the INVICTUS Study (ITT Population, DB Period)
EORTC QLQ-C30 domain | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
Role functioning | ||
Baseline | ||
Patients, n | 42 | 74 |
Score, mean (SD) | 73.8 (30.39) | 69.4 (30.10) |
Cycle 2 day 1 | ||
Patients, n | 33 | 79 |
Score, mean (SD) | 65.2 (27.75) | 75.1 (26.13) |
Change from baseline | ||
Patients, n | 32 | 70 |
Score, mean (SD) | –17.2 (30.38) | 3.3 (27.31) |
Score, adjusted mean (SE)a | –17.1 (5.0) | 3.5 (3.5) |
Difference in adjusted means (95% CI)a | 20.61 (8.58 to 32.63) | |
P valuea | 0.001 | |
Physical functioning | ||
Baseline | ||
Patients, n | 42 | 74 |
Score, mean (SD) | 76.0 (26.47) | 75.7 (21.58) |
Cycle 2 day 1 | ||
Patients, n | 33 | 80 |
Score, mean (SD) | 75.2 (20.23) | 79.4 (17.34) |
Change from baseline | ||
Patients, n | 32 | 71 |
Score, mean (SD) | –9.0 (19.28) | 1.5 (16.03) |
Score, adjusted mean (SE) | –8.9 (3.0) | 1.6 (1) |
Difference in adjusted means (95% CI)a | 10.48 (3.37 to 17.59) | |
P valuea | 0.004 | |
CI = confidence interval; DB = double-blind; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire for Cancer 30 item; ITT = intention to treat; NA = not applicable; SD = standard deviation; SE = standard error.
Note: EORTC QLQ-C30 role and physical functioning are scored from 0 (worse HRQoL) to 100 (better HRQoL). Data are for the May 31, 2019, database lock.
aEstimated from an ANCOVA model that included factors for study treatment, number of prior anticancer treatment, and ECOG status at baseline as fixed effects. Testing occurred after failure of the statistical hierarchy; therefore, the P value should be considered descriptive.
Source: INVICTUS Clinical Study Report.12
Changes in EQ-5D-5L usual activities and pain/discomfort domains, and index utility scores, from baseline to cycle 2 day 1 by treatment arm in the ITT are shown in Table 16. These outcomes were outside the statistical hierarchy and not adjusted for multiplicity. Baseline scores were generally similar between study arms, although the proportion of patients in the placebo arm who responded 1 (“no problems”) was higher in the placebo arm (47.7%) compared with the ripretinib arm (35.3%). The proportions of patients reporting improvements in usual activities were 6.8% in the placebo arm and 23.5% in the ripretinib arm; the proportions of patients reporting no change were 50.0% in the placebo arm and 47.1% in the ripretinib arm. The proportions of patients reporting improvements in pain/discomfort were 15.9% in the placebo arm and 17.7% in the ripretinib arm; the proportions of patients reporting no change were 36.4% in the placebo arm and 42.4% in the ripretinib arm. The adjusted mean changes from baseline in utility index scores were –0.0606 (SE = 0.02796) in the placebo arm and –0.0094 (SE = 0.01957) in the ripretinib arm; the difference in adjusted mean changes from baseline comparing ripretinib with placebo was 0.05 (95% CI, –0.02 to 0.12).
Table 16: EQ-5D-5L Scores in the INVICTUS Study (ITT Population, DB Period)
EQ-5D-5L domain | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
Usual activities score | ||
Baseline, n (%) | ||
1 | 21 (47.7) | 30 (35.3) |
2 | 10 (22.7) | 25 (29.4) |
3 | 8 (18.2) | 14 (16.5) |
4 | 2 (4.5) | 5 (5.9) |
5 | 1 (2.3) | 0 |
Missing | 2 (4.5) | 11 (12.9) |
Cycle 2 day 1, n (%) | ||
1 | 14 (31.8) | 42 (49.4) |
2 | 13 (29.5) | 25 (29.4) |
3 | 4 (9.1) | 10 (11.8) |
4 | 2 (4.5) | 1 (1.2) |
5 | 0 | 1 (1.2) |
Missing | 11 (25.0) | 6 (7.1) |
Change from baseline | ||
–2 | 1 (2.3) | 3 (3.5) |
–1 | 2 (4.5) | 17 (20.0) |
0 | 22 (50.0) | 40 (47.1) |
1 | 3 (6.8) | 9 (10.6) |
2 | 2 (4.5) | 1 (1.2) |
3 | 2 (4.5) | 0 |
Missing | 12 (27.3) | 15 (17.6) |
P valuea | 0.066 | |
Pain/discomfort score | ||
Baseline, n (%) | ||
1 | 10 (22.7) | 18 (21.2) |
2 | 18 (40.9) | 36 (42.4) |
3 | 9 (20.5) | 14 (16.5) |
4 | 4 (9.1) | 6 (7.1) |
5 | 1 (2.3) | 0 |
Missing | 2 (4.5) | 11 (12.9) |
Cycle 2 day 1, n (%) | ||
1 | 9 (20.5) | 18 (21.2) |
2 | 11 (25.0) | 36 (42.4) |
3 | 9 (20.5) | 20 (23.5) |
4 | 4 (9.1) | 3 (3.5) |
5 | 0 | 1 (1.2) |
Missing | 11 (25.0) | 7 (8.2) |
Change from baseline | ||
–2 | 2 (4.5) | 1 (1.2) |
–1 | 5 (11.4) | 14 (16.5) |
0 | 16 (36.4) | 36 (42.4) |
1 | 7 (15.9) | 14 (16.5) |
2 | 2 (4.5) | 5 (5.9) |
Missing | 12 (27.3) | 15 (17.6) |
P valuea | 0.617 | |
Index (utility) score | ||
Baseline | ||
n | 42 | 74 |
Mean (SD) | 0.7547 (0.25210) | 0.7606 (0.20846) |
Cycle 2 day 1 | ||
n | 33 | 78 |
Mean (SD) | 0.7545 (0.22010) | 0.7762 (0.16802) |
Change from baseline | ||
n | 32 | 70 |
Mean (SD) | –0.0596 (0.19535) | –0.0058 (0.13986) |
Adjusted mean (SE)a | –0.0606 (0.02796) | –0.0094 (0.01957) |
Difference in adjusted means (95% CI)b | 0.05 (–0.02 to 0.12) | |
P valuec | 0.133 | |
CI = confidence interval; DB = double-blind; ITT = intention to treat; SD = standard deviation; SE = standard error.
Note: Usual activities and pain/discomfort are scored on a 1 to 5 scale representing, no problems, slight problems, moderate problems, severe problems, and unable to/extreme problems, respectively. The index utility score ranges from < 0 (worse than dead) to 1 (perfect health). Data are for the May 31, 2019, database lock.
aP value from Cochran-Mantel-Haenszel test.
bDifference of placebo arm – ripretinib arm.
cEstimated from an ANCOVA model that included factors for study treatment, number of prior anticancer treatment, and ECOG status at baseline as fixed effects. Analysis not adjusted for multiplicity.
Source: INVICTUS Clinical Study Report.12
Changes in EQ-VAS scores from baseline to cycle 2 day 1 by treatment arm in the ITT are shown in Table 17. This outcome was outside the statistical hierarchy, and the analysis was not adjusted for multiplicity. Baseline EQ-VAS scores were similar between treatment arms. The mean change from baseline in EQ-VAS score was –8.9 (standard deviation [SD] = 19.31) in the placebo arm and 3.7 (SD = 20.36) in the ripretinib arm. The difference in change from baseline between treatment groups was not reported.
Table 17: EQ-VAS Scores in the INVICTUS Study (ITT Population, DB Period)
Testing time | Placebo N = 43a | Ripretinib N = 85 | ||
|---|---|---|---|---|
n | EQ-VAS score, mean (SD) | n | EQ-VAS score, mean (SD) | |
Baseline | 42 | 65.6 (22.91) | 74 | 63.9 (22.05) |
Cycle 2 day 1 | 33 | 64.1 (23.25) | 78 | 69.5 (20.47) |
Change from baseline | 32 | –8.9 (19.31) | 70 | 3.7 (20.36) |
P valueb | 0.004 | |||
DB = double-blind; EQ = EuroQol; ITT = intention to treat; NA = not applicable; SD = standard deviation; VAS = visual analogue scale.
Note: EQ-VAS scores range from 0 (worst imaginable health) to 100 (best imaginable health). Data are for the May 31, 2019, database lock.
aLabelled as N = 43 in Table 14.2.10 of the INVICTUS Clinical Study Report. The ITT population consisted of N = 44 patients randomized to receive placebo.
bP value from t-test.
Source: INVICTUS Clinical Study Report.12
Objective Response Rate
ORR (best overall response of CR or PR) per IRR in the ITT population is shown in Table 18. At the time of the primary analysis (as of the May 31, 2019, database lock) and in the ITT population, no patients in the placebo arm and 8 patients (9.4%) in the ripretinib arm had achieved objective responses (P = 0.0504). The ORR difference between arms was 9.4% (95% CI, 0.2% to 17.5%). The proportions of patients in the placebo and ripretinib arms with stable disease were 20.5% and 65.9%, respectively. Results were similar for the January 15, 2021, database lock.
Table 18: ORR per IRR in the INVICTUS Study (ITT Population, DB Period)
Response | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
ORR | ||
Responders, n (%) | 0 | 8 (9.4)a 10 (11.8)b |
ORR 95% CI, %c | 0.0 to 8.0a 0.0 to 8.0b | 4.2 to 17.7a 5.8 to 20.6b |
ORR difference (95% CI)e | 9.4 (0.2 to 17.5)a | |
P value | 0.0504d | |
BOR, n (%) | ||
Complete response | 0 | 0 |
Partial response | 0 | 8 (9.4) |
Stable diseasef | 9 (20.5) | 56 (65.9) |
Progressive disease | 28 (63.6) | 16 (18.8) |
Not evaluable | 3 (6.8) | 4 (4.7) |
No response assessment | 4 (9.1) | 1 (1.2) |
BOR = best overall response; CI = confidence interval; DB = double-blind; IRR = independent radiological review; ITT = intention to treat; NA = not applicable; ORR = objective response rate.
aData are from the May 31, 2019, database lock.
bData are from the January 15, 2021, database lock.
cFrom exact binomial CI.
dP value from Fisher’s exact test.
eNewcombe Score 95% CI of the difference in ORR between treatment arms.
fDefined as absence of PD for at least 6 weeks.
Source: INVICTUS Clinical Study Report12 and sponsor’s additional information.13
Refer to Appendix 3 for sensitivity and subgroup analyses of ORR. Sensitivity analyses of ORR were consistent with the primary analysis. A planned subgroup analysis of ORR by number of lines of prior therapy was suggestive of similar response rates among patients in the ripretinib arm who had received 3 (5/54, 9.3%) and 4 or more (3/31, 9.7%) lines of prior therapy. A post hoc subgroup analysis of ORR by tumour mutational status was uninformative as all responses occurred in patients with mutations in KIT exon 11, the most common primary mutational type among patients in the study.
Duration of Response
DOR among patients responding to ripretinib is shown in Table 19. As of the 31 May 2019 database lock, only 1 responding patient in the ripretinib arm had progressed per IRR and the median DOR was not estimable. As of the January 15, 2021, database lock, median DOR in the ripretinib arm was 14.5 months (95% CI, 3.7 months to not estimable).
Table 19: DOR in the INVICTUS Study (ITT Population, DB Period)
Response | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
Responders, n (%) | 0a 0b | 8 (9.4)a 10 (11.8)b |
Patients with events, n (%) | 0 | 1 (1.2) |
Patients censored, n (%) | 0 | 7 (8.2) |
DOR (months), median (95% CI) c | NE (NE to NE)a NE (NE to NE)b | NE (16.0 to NE)a 14.5 (3.7 to NE)b |
CI = confidence interval; DB = double-blind; DOR = duration of response; ITT = intention to treat; NE = not estimable.
aData are from the May 31, 2019, database lock.
bData are from the January 15, 2021, database lock.
cFrom KM analysis.
Source: INVICTUS Clinical Study Report,12 CADTH review submission,8 and sponsor’s additional information.13
Time to Response
TTR among patients responding to ripretinib is shown in Table 20. As of the May 2019 database lock, the mean TTR in the ripretinib arm was 9.1 (SD = 5.83) weeks.
Table 20: TTR in the INVICTUS Study (ITT Population, DB Period)
Response | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
Responders, n (%) | 0 | 8 (9.4) |
TTR (weeks), mean (SD) | NA | 9.1 (5.83) |
Median (range) | NA | 8.1 (4.0 to 20.1) |
ITT = intention to treat; NA = not applicable; SD = standard deviation; TTR = time to response.
Note: Data are from the May 31, 2019, database lock.
Source: INVICTUS Clinical Study Report.12
Progression-Free Survival
The results of the primary PFS analysis are shown in Table 21, Figure 5, and Figure 6. As of the May 31, 2019 database lock and in the ITT population, PFS events had occurred in 37 (84.1%) of patients randomized to the placebo arm and 51 (60.0%) of patients randomized to the ripretinib arm. Median PFS was 4.1 weeks (95% CI, 4.0 to 7.3 weeks) for patients randomized to the placebo arm and 27.6 weeks (95% CI, 20.0 to 29.9 weeks) for patients randomized to the ripretinib arm (P < 0.0001). The HR for PFS comparing ripretinib with placebo was 0.15 (95% CI, 0.09 to 0.25). Results were similar for the January 15, 2021, database lock.
Table 21: PFS per IRR in the INVICTUS Study (ITT Population, DB Period)
Outcome | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
Patients with events, n (%) | 37 (84.1)a 37 (84.1)b | 51 (60.0)a 71 (83.5)b |
Patients censored, n (%) | 7 (15.9)a 7 (15.9)b | 34 (40.0)a 14 (16.5)b |
Median PFS (95% CI)c | 4.1 (4.0 to 7.3)a 4.1 (4.0 to 7.3)b | 27.6 (20.0 to 29.9)a 27.6 (20.0 to 35.3)b |
P valued | < 0.0001a | |
HR (95% CI)e | 0.15 (0.09 to 0.25)a 0.16 (0.10 to 0.27)b | |
PFS rate (95% CI)c | ||
26 weeks | 3.2 (0.2 to 13.8) | 51.0 (39.4 to 61.4) |
39 weeks | NE (NE to NE) | 34.4 (22.9 to 46.2) |
52 weeks | NE (NE to NE) | 21.0 (9.0 to 36.3) |
CI = confidence interval; DB = double-blind; HR = hazard ratio; IRR = independent radiological review; ITT = intention to treat; NE = not estimable; PFS = progression-free survival.
aData are from the May 31, 2019, database lock.
bData are from the January 15, 2021, database lock.
cFrom Kaplan-Meier analysis.
dP value from 2-sided stratified log-rank test. Stratification factors were the same as those applied to randomization (number of prior anticancer treatment and ECOG status at baseline).
eHR from Cox proportional regression model with treatment and randomization stratification factors as fixed factors.
Source: INVICTUS Clinical Study Report,12 CADTH review submission,8 and sponsor’s additional information.13
Analysis of second PFS in the OL period (for patients who crossed over from placebo to ripretinib 150 mg once daily) is presented in Appendix 3. Among patients who crossed over from placebo to ripretinib 150 mg once daily following initial designation of objective PD per IRR (before any dosage escalation; n = 29), subsequent PFS events (second PFS) occurred in 13 (44.8%) patients. Median second PFS was 20.0 weeks (95% CI, 8.0 weeks to not estimable) in this group of patients.
Figure 5: Kaplan-Meier Plot of PFS in the INVICTUS Study (ITT Population; Data Cut: May 31, 2019)
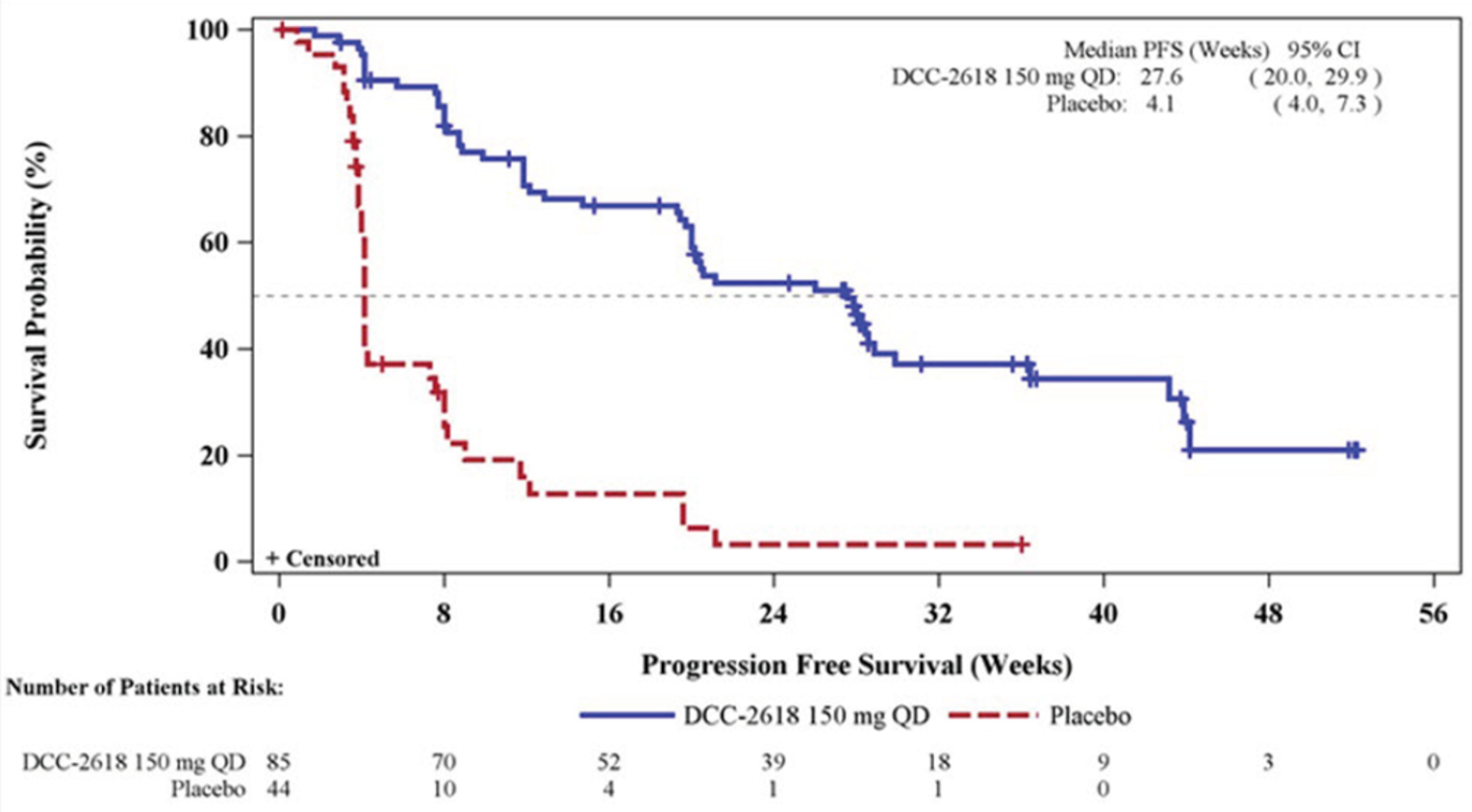
CI = confidence interval; DCC-2618 = ripretinib; ITT = intention to treat; PFS = progression-free survival; QD = once daily.
Note: Symbols represent censored observations.
Source: INVICTUS Clinical Study Report.12
Figure 6: Kaplan-Meier Plot of PFS in the INVICTUS Study (ITT Population; Data Cut: January 15, 2021)
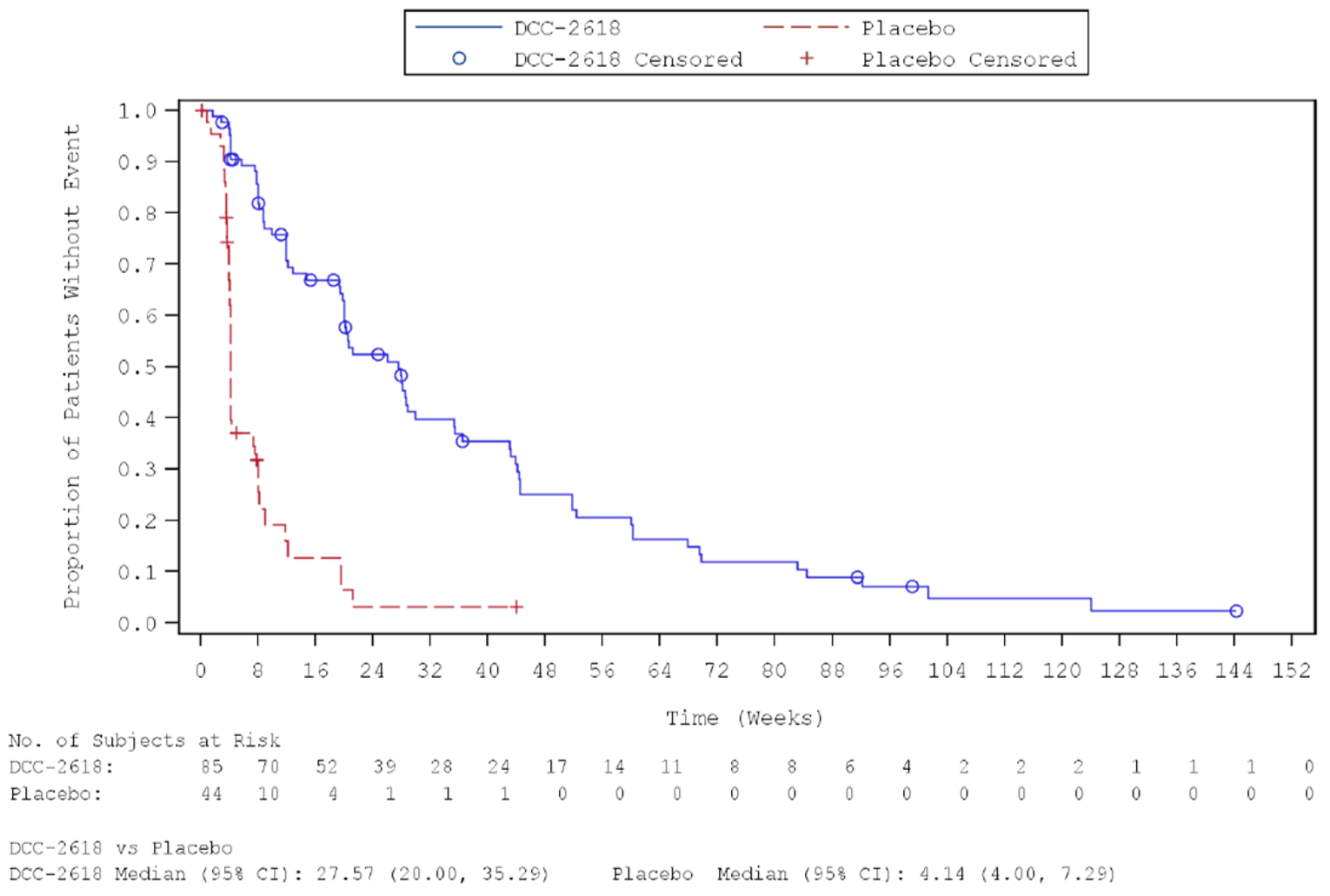
CI = confidence interval; DCC-2618 = ripretinib; ITT = intention to treat.
Note: Symbols represent censored observations.
Source: Sponsor’s additional information.13
Refer to Appendix 3 for sensitivity and subgroup analyses of PFS. Sensitivity analyses of PFS were consistent with the primary analysis. A planned subgroup analysis of PFS by number of lines of prior therapy was suggestive of similar effect sizes of ripretinib on PFS among patients who had received 3 (HR 0.15; 95% CI, 0.08 to 0.25) and 4 or more (HR 0.24; 95% CI, 0.12 to 0.51) lines of prior therapy. No subgroup analysis of PFS by tumour mutational status was conducted.
Symptom Severity
Symptom severity was not specifically assessed in the INVICTUS study, other than via the symptom scales of generic patient-reported HRQoL instruments (EORTC QLQ-C30, EQ-5D-5L).
Harms
Only those harms identified in the review protocol are reported in this section. See Table 22 for detailed harms data.
Adverse Events
Almost all patients who received placebo (97.7%) and ripretinib (98.8%) experienced at least 1 AE during the DB treatment period. Common AEs that occurred more frequently in ripretinib-treated patients than in placebo-treated patients included alopecia (51.8% versus 4.7%), fatigue (42.4% versus 23.3%), nausea (38.8% versus 11.6%), constipation (34.1% versus 18.6%), myalgia (31.8% versus 11.6%), diarrhea (28.2% versus 14.0%), and vomiting (21.2% versus 7.0%).
Serious Adverse Events
Serious adverse events occurred in 44.2% of patients who received placebo and in 30.6% of patients who received ripretinib during the DB treatment period.
Adverse Events Leading to Dosage Modification
AEs leading to dosage reduction occurred in 1 (2.3%) patient who received placebo and 6 (7.1%) patients who received ripretinib during the DB treatment period. AEs leading to dosage interruption occurred in 9 (20.9%) patients who received placebo and 20 (23.5%) patients who received ripretinib during the DB treatment period.
Withdrawals Due to Adverse Events
Five patients (11.6%) who received placebo and 7 patients (8.2%) who received ripretinib withdrew from the protocol therapy due to AEs during the DB treatment period.
Mortality
Ten patients (23.3%) who received placebo and 5 patients (5.9%) who received ripretinib died within 30 days of the last dose of study drug during the DB treatment period. The primary cause of death in most patients was PD.
Notable Harms
Among protocol-specified AESIs, squamous cell carcinoma of the skin occurred in no patients who received placebo and 2 patients (2.4%) who received ripretinib during the DB treatment period. Actinic keratosis occurred in 1 patient (2.3%) who received placebo and 5 patients (5.9%) who received ripretinib during the DB treatment period. No patients experienced keratoacanthoma during the study.
All notable harms specified in the CADTH review protocol occurred more frequently in patients who received ripretinib than in those who received placebo during the DB treatment period: cardiac dysfunction, cardiac ischemic events, hypertension, cutaneous malignancies, palmar-plantar erythrodysesthesia syndrome, arthralgia, myalgia, and increased bilirubin. The most common AEs by preferred term were peripheral edema (ripretinib versus placebo: 16.5% versus 7.0%), hypertension (14.1% versus 4.7%), palmar-plantar erythrodysesthesia syndrome (21.2% versus 0%), arthralgia (17.6% versus 4.7%), myalgia (31.8% versus 11.6%), and increased bilirubin (16.5% versus 0%).
Table 22: Summary of Harms in the INVICTUS Study (Safety Population)
Harms | DB period | OL period | ||
|---|---|---|---|---|
Placebo N = 43 | Ripretinib N = 85 | Ripretinib 150 mg q.d. (DB: placebo) N = 29 | Ripretinib 150 mg q.d. (DB: ripretinib) N = 11 | |
Patients with ≥ 1 AE | ||||
n (%) | 42 (97.7) | 84 (98.8) | 28 (96.6) | 11 (100.0) |
Common AEs, n (%)a | ||||
Alopecia | 2 (4.7) | 44 (51.8) | 8 (27.6) | 2 (18.2) |
Fatigue | 10 (23.3) | 36 (42.4) | 10 (34.5) | 2 (18.2) |
Nausea | 5 (11.6) | 33 (38.8) | 4 (13.8) | 1 (9.1) |
Abdominal pain | 13 (30.2) | 31 (36.5) | 8 (27.6) | 3 (27.3) |
Constipation | 8 (18.6) | 29 (34.1) | 9 (31.0) | 0 |
Myalgia | 5 (11.6) | 27 (31.8) | 10 (34.5) | 1 (9.1) |
Diarrhea | 6 (14.0) | 24 (28.2) | 3 (10.3) | 2 (18.2) |
Decreased appetite | 9 (20.9) | 23 (27.1) | 6 (20.7) | 5 (45.5) |
Vomiting | 3 (7.0) | 18 (21.2) | 3 (10.3) | 2 (18.2) |
Patients with ≥ 1 SAE | ||||
n (%) | 19 (44.2) | 26 (30.6) | 12 (41.4) | 7 (63.6) |
Common SAEs, n (%)b | ||||
Abdominal pain | 2 (4.7) | 4 (4.7) | 2 (6.9) | 1 (9.1) |
Anemia | 1 (2.3) | 3 (3.5) | 0 | 0 |
Death | 4 (9.3) | 3 (3.5) | 4 (13.8) | 3 (27.3) |
Nausea | 0 | 2 (2.4) | 0 | 1 (9.1) |
Vomiting | 0 | 2 (2.4) | 0 | 2 (18.2) |
Acute kidney injury | 2 (4.7) | 1 (1.2) | 1 (3.4) | 1 (9.1) |
Sepsis | 2 (4.7) | 1 (1.2) | 0 | 0 |
Asthenia | 2 (4.7) | 0 | 0 | 0 |
Patients with ≥ 1 AE leading to dosage reduction | ||||
n (%) | 1 (2.3) | 6 (7.1) | 2 (6.9) | 0 |
Patients with ≥ 1 AE leading to dosage interruption | ||||
n (%) | 9 (20.9) | 20 (23.5) | 10 (34.5) | 5 (45.5) |
WDAEs | ||||
n (%) | 5 (11.6) | 7 (8.2) | 4 (13.8) | 0 |
Deaths | ||||
n (%) | 10 (23.3) | 5 (5.9) | 5 (17.2) | 4 (36.4) |
Primary cause of death, n (%) | ||||
Disease progression | 8 (18.6) | 4 (4.7) | 5 (17.2) | 4 (36.4) |
AESIs, n (%) | ||||
SCC of the skin | 0 | 2 (2.4) | 0 | 0 |
Actinic keratosis | 1 (2.3) | 5 (5.9) | 1 (3.4) | 0 |
Keratoacanthoma | 0 | 0 | 0 | 0 |
Notable harms, n (%) | ||||
Cardiac dysfunction and cardiac ischemic events | ||||
Peripheral edema | 3 (7.0) | 14 (16.5) | 3 (10.3) | 0 |
Sinus bradycardia | 0 | 4 (4.7) | 0 | 0 |
Chest pain | 1 (2.3) | 3 (3.5) | 0 | 0 |
Pleural effusion | 0 | 3 (3.5) | 2 (6.9) | 0 |
Tachycardia | 0 | 2 (2.4) | 0 | 1 (9.1) |
Bradycardia | 0 | 1 (1.2) | 0 | 0 |
Cardiac failure | 0 | 1 (1.2) | 0 | 0 |
Chest discomfort | 0 | 1 (1.2) | 0 | 0 |
Embolism | 0 | 1 (1.2) | 1 (3.4) | 0 |
Orthopnea | 0 | 1 (1.2) | 0 | 0 |
Palpitations | 0 | 1 (1.2) | 0 | 0 |
Pericardial effusion | 0 | 1 (1.2) | 0 | 0 |
Ventricular extrasystoles | 0 | 1 (1.2) | 0 | 0 |
Cardiac murmur | 1 (2.3) | 0 | 0 | 0 |
Atrial fibrillation | 0 | 0 | 0 | 0 |
Generalized edema | 0 | 0 | 0 | 1 (9.1) |
Mitral valve disease | 0 | 0 | 1 (3.4) | 0 |
Peripheral swelling | 0 | 0 | 1 (3.4) | 0 |
Hypertension | 2 (4.7) | 12 (14.1) | 4 (13.8) | 0 |
Cutaneous malignancies | ||||
Fibrous histiocytoma | 0 | 2 (2.4) | 0 | 0 |
Malignant melanoma in situ | 0 | 2 (2.4) | 0 | 0 |
SCC of head and neck | 0 | 2 (2.4) | 0 | 0 |
SCC of skin | 0 | 2 (2.4) | 0 | 0 |
Neoplasm skin | 0 | 1 (1.2) | 0 | 0 |
Basal cell carcinoma | 0 | 0 | 1 (3.4) | 0 |
SCC | 0 | 0 | 0 | 0 |
Palmar-plantar erythrodysesthesia | 0 | 18 (21.2) | 5 (17.2) | 0 |
Arthralgia | 2 (4.7) | 15 (17.6) | 4 (13.8) | 0 |
Myalgia | 5 (11.6) | 27 (31.8) | 10 (34.5) | 1 (9.1) |
Increased bilirubin | 0 | 14 (16.5) | 0 | 0 |
AE = adverse event; AESI = adverse event of special interest; DB = double-blind; OL = open label; q.d. = once daily; SAE = severe adverse event; SCC = squamous cell carcinoma; WDAE = withdrawal due to adverse event.
Note: Treatment-emergent AEs reported in this table were defined as any untoward medical occurrence occurring after administration of the first dose of study drug and within 30 days of the last dose of study drug. AEs were coded using MedDRA version 21.1 and graded according to NCI-CTCAE version 4.03.
aAEs with frequency ≥ 20% in either study arm during the DB period are reported (excluding notable harms reported separately).
bSAEs affecting ≥ 2 patients in either study arm during the DB period are reported.
Source: INVICTUS Clinical Study Report.12
Critical Appraisal
Internal Validity
INVICTUS was a relatively small, phase III DB multi-centre RCT with an OL period of active treatment (N = 129) conducted in patients with advanced GIST who had prior progression or intolerance on imatinib, sunitinib, and regorafenib.9-11 Randomization appeared generally successful in balancing baseline demographic and disease characteristics between study arms. However, as expected for a smaller study of a rare condition, small baseline imbalances were present between study arms, several of which were of potential prognostic significance. The younger age, higher proportion of patients with ECOG PS 0, and higher proportion of patients with gastric tumours in the ripretinib arm would be expected to potentially favour ripretinib; however, according to the clinical experts consulted by CADTH for this review, these slight imbalances would not limit the interpretation of study results.
Many of the outcomes used in the INVICTUS study (OS, ORR, DOR, TTR, PFS) are standard in oncology trials, and tumour responses were objectively evaluated using mRECIST version 1.1 – GIST-specific per IRR. According to the clinical experts consulted by CADTH for this review, the definition of stable disease used in the study (6 weeks) was arbitrary. Although there is no standardized definition of stable disease in GIST patients, the clinical experts stated that this interval is shorter than what most clinicians would consider stable, especially as scans would typically be conducted every 2 to 4 months in clinical practice. Few patients in the study had important deviations (< 5%) and there was very good adherence and dose intensity (> 95%) for patients in the ripretinib arm. Withdrawal of consent and losses to follow-up were both very low. However, there were several limitations of the basic design of the study. First, only 44 patients were randomized to receive placebo, many of whom progressed very rapidly, resulting in unblinding of patient and investigator. Development of characteristic AEs, including AESIs, may also have led to partial unblinding. Thus, the extent to which investigators remained blinded over the course of the study was unclear, although the clinical experts consulted for this review did not expect that any resulting bias (direction uncertain) would limit interpretation of the study results. Second, and more importantly, interpretation of OS results was limited by elective crossover of patients from DB placebo to OL ripretinib 150 mg once daily and by intra-patient post-progression dosage escalation to 150 mg twice daily, a dosage not approved by Health Canada. Crossover, post-progression treatment, and dosage escalation were not accounted for by censoring or other techniques. Thus, the relative effects of ripretinib versus placebo treatment, pre- versus post-progression treatment, and ripretinib dosage on OS could not be easily ascertained from the available data. The lack of adjustment for crossover would be expected to bias the analysis of OS differences toward the null (against ripretinib). The magnitudes and directions of biases in the OS analysis attributable to post-progression treatment and dosage escalation were uncertain. However, based on the similar proportions of patients in each arm who received OL post-progression treatment and escalated dosage, these biases may have been nondirectional, although this interpretation was associated with substantial uncertainty.
Several statistical issues should be considered when interpreting the results of the INVICTUS study. Overall statistical tests were appropriate, and the results of the primary PFS analysis and key secondary ORR analysis were robust to several sensitivity analyses. Although not explicitly tested, the proportional hazards assumption appeared likely to be satisfied for Cox models based on KM survival curves. Multiplicity was controlled using an appropriate hierarchical testing strategy. However, the second step in the hierarchy was the hypothesis test of ORR, which had only 80% power to detect a difference in ORR between the ripretinib and placebo arms of 20%. According to the clinical experts consulted by CADTH for this review, ORRs of this magnitude were unlikely to occur in this patient population. Because of the modest power to detect differences in ORR of magnitudes that, according to the clinical experts, would be somewhat optimistic in this advanced disease setting, statistical testing of ORR returned a nonsignificant result at the time of the primary analysis, precluding subsequent testing of important outcomes to clinicians and patients (HRQoL and OS). Only 1 of the subgroup analyses of interest for this review was specified a priori and was based on a stratification factor (number of prior lines of therapy). Subgroup analysis by tumour mutational status was conducted only for ORR, in an exploratory fashion. The study was not specifically powered to evaluate strata among subgroups; there were no tests for differences among subgroups; and subgroup analyses were not controlled for multiplicity.
Analyses of HRQoL data (EORTC QLQ-C30 role and physical function; EQ-5D-5L usual activities and pain/discomfort dimensions; EQ-5D-5L utility index scores; EQ-VAS) were limited by several factors, including absence of formal statistical testing (due to failure of the statistical hierarchy at the time of the primary analysis at an earlier stage or hypothesis tests being outside the hierarchy). Several HRQoL outcomes (e.g., EQ-5D-5L dimensions) were affected by high rates of missing data for cycle 2 day 1. The strategy of replacing missing data with those from the end of treatment (e.g., for EORTC QLQ-C30 functional scales), which may have been significantly earlier or later than cycle 2 day 1, was not clearly justified. Importantly, the measurement properties of both HRQoL instruments have not been investigated, and MIDs have not been identified specifically for GIST patients. The degree to which these generic HRQoL instruments capture the symptoms of GIST patients, which were considered highly important outcomes by patients and clinicians, was unclear. Moreover, limited evidence is available documenting the responsiveness to change of the EORTC QLQ-C30 and the reliability and responsiveness to change of the EQ-5D-5L among all cancer patients. In addition, the clinical experts consulted by CADTH for this review expressed uncertainty as to whether it is plausible that HRQoL would improve owing to tumour response during the first cycle of ripretinib. Analysis of longer-term trends in HRQoL data were not conducted in the study. Safety outcomes were generally well captured, although the small size of the study may have reduced the likelihood of detecting very rare harms.
External Validity
According to the clinical experts consulted by CADTH for this review, the demographic and disease characteristics of patients enrolled in the INVICTUS study9-11 reflect the Canadian population of advanced GIST patients they would treat in their clinical practice. Similarly, the clinical experts felt that the study eligibility criteria would be expected to result in recruitment of a patient population that reflects Canadian practice. As with most oncology trials, however, the study likely enrolled a healthier cross-section of patients with better PS who were more likely to tolerate and respond to therapy than the general population of GIST patients. The clinical experts confirmed that exclusion criteria related to cardiac problems would eliminate, because of safety concerns, a small percentage of patients who would not be good candidates for ripretinib in clinical practice. The clinical experts noted that few patients with ECOG PS 2 were enrolled in the study and that generalizability to patients with ECOG PS greater than 2 was even less certain. The clinical experts stated that, although most patients in the INVICTUS study were White and had gastric tumours with primary KIT exon 11 mutations, this would not limit generalizability to other patients (e.g., the smaller numbers of patients with other tumour sites and mutations).
The clinical experts consulted by CADTH for this review confirmed that placebo plus BSC was an appropriate comparator for ripretinib, as there are currently no options for fourth-line therapy following failure of imatinib, sunitinib, and regorafenib. Dosages of ripretinib administered during the DB period of the INVICTUS study (150 mg once daily) were aligned with the Health Canada—approved dosage and with clinical practice. However, dosage during the OL period (patients could choose to receive ripretinib beyond initial PD and to escalate the dosage to 150 mg twice daily) was not aligned with Health Canada–approved dosage or an accepted standard of clinical practice. Thus, generalizability of the OS results of the INVICTUS study to Canadian patients is uncertain.
Several of the outcomes assessed in the INVICTUS study, including OS, HRQoL, and PFS, were identified as clinically important by both patients and clinical experts. Patients and clinicians alike indicated that improved survival is the most important outcome of treatment but that stabilization or improvement of HRQoL is also critical. According to the clinical experts, the generic HRQoL instruments use in the INVICTUS study are research tools that are not used in clinical practice. Moreover, improvement in GIST symptoms was identified as a critical outcome of treatment but was not directly assessed in the study; instead, it was assessed indirectly using generic HRQoL instruments. The duration of follow-up was adequate for assessment of the primary and secondary efficacy outcomes and safety outcomes, especially for the unplanned analysis from the recent database lock of January 15, 2021.
Patients in the INVICTUS study underwent imaging scans more frequently (every cycle; 28 days) and may have had better access to the treating clinician and team via participation in the trial compared with the expected treatment setting in Canada. However, the clinical experts consulted by CADTH for this review did not feel this would limit generalizability of the study findings.
Indirect Evidence
No indirect evidence was identified for this review.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Discussion
Summary of Available Evidence
One phase III, DB multi-centre RCT with an OL period of active treatment (INVICTUS, N = 129)9-11 contributed evidence to this report. The study enrolled patients with advanced GIST who had previously progressed on or developed intolerance to imatinib, sunitinib, and regorafenib. Patients were randomized 2:1 to receive either ripretinib 150 mg once daily or placebo during the DB period until the first objective PD per IRR or unacceptable toxicity. The primary outcome was PFS per IRR, and hierarchically tested secondary outcomes were ORR per IRR, change from baseline to cycle 2 day 1 in EORTC QLQ-C30 role function and physical function, and OS. Other secondary outcomes assessed outside of the statistical hierarchy included TTR, DOR, and change from cycle 2 day 1 in EQ-5D-5L usual activities and pain/discomfort dimensions, EQ-5D-5L utility index score, and EQ-VAS.
According to the clinical experts consulted by CADTH for this review, the baseline characteristics of the INVICTUS study population were broadly representative of Canadian GIST patients who have progressed on or developed intolerance to imatinib, sunitinib, and regorafenib and would be candidates for fourth-line ripretinib. Most patients were White and from the US, with a mean age of approximately 60 years, and most had ECOG PS 0/1. The most common tumour site was gastric; the most common location of primary tumour mutations was KIT exon 11; and patients were roughly evenly split between having received 3 versus 4 or more prior lines of therapy. A major limitation of the included study was uncertain interpretation of changes due to ripretinib treatment in 2 outcomes identified as critically important by both patients and clinicians (OS and HRQoL). The OS analysis did not account for crossover, post-progression treatment, and dosage escalation to a not approved by Health Canada. Analyses of HRQoL data were limited by absence of formal statistical testing, missing data (up to 25% of the randomized population), and lack of instrument validation specifically in patients with GIST. Changes in patient symptoms, which were noted as highly important by both patients and clinicians, were not directly measured in the study.
Interpretation of Results
Efficacy
Patients originally randomized to the ripretinib arm in the INVICTUS study had numerically improved OS (median 65.6 weeks) compared with those originally randomized to the placebo arm (median = 28.6 weeks). However, the statistical hierarchy failed at the time of the primary analysis before testing of this outcome, precluding definitive conclusions. Interpretation of the OS data was additionally limited by complexities associated with patient crossover, post-progression treatment, and intra-patient dosage escalation, which were not adjusted for in the analysis. The impact of crossover from placebo to OL ripretinib would be expected to bias OS comparisons toward the null (i.e., bias against ripretinib), while the impacts of post-progression treatment and intra-patient dosage escalation were uncertain but possibly nondirectional. Nonetheless, the clinical experts consulted by CADTH judged that the difference in OS between patients originally randomized to DB ripretinib or placebo was potentially clinically relevant in this patient population that is affected by advanced disease and has no other treatment options. Numerical differences in OS were also observed for the subgroup of patients who received only DB treatment with ripretinib versus placebo before initial objective PD (without any OL treatment), as well as for patients who crossed over from placebo to OL ripretinib 150 mg once daily versus patients randomized to the placebo arm who did not cross over.
The clinical experts consulted by CADTH for this review felt that the difference in ORR per IRR during the DB treatment period of the INVICTUS study was encouraging and potentially clinically relevant for fourth-line treatment of patients with advanced GIST. However, the comparison of ORR between the ripretinib and placebo arms did not reach statistical significance at the time of the primary analysis, precluding definitive conclusions. The clinical experts stated that the relatively low ORR in the ripretinib arm was expected and that, in tandem with the higher proportion of patients in the ripretinib arm with stable disease (65.9% versus 20.5%), the ORR findings supported and were consistent with the OS and PFS results.
Administration of ripretinib during the DB treatment period of the INVICTUS study resulted in statistically significant prolongation of PFS (median = 27.6 weeks) compared with placebo (median = 4.1 weeks; P < 0.0001), which the clinical experts consulted by CADTH for this review judged as highly clinically meaningful. An unplanned analysis of mature PFS data from a more recent data cut (January 15, 2021) were consistent with the primary PFS analysis.
Patient input identified HRQoL and symptom relief as very important outcomes for patients with GIST. Unfortunately, assessment of changes in HRQoL from baseline was limited by absence of formal statistical testing, missing data, large variability in estimates, and lack of HRQoL instrument validation specifically in patients with GIST. Consistent numeric differences in the INVICTUS study provided potential signals of HRQoL stabilization or improvement in the ripretinib arm versus placebo arm across all outcomes assessed. However, these differences in HRQoL outcomes were not easily interpretable, and conclusions were uncertain. Validated tools for studying HRQoL and symptoms in GIST patients are needed.
Harms
According to input from the clinical experts consulted by CADTH for this review, as well as clinician and patient groups, the safety profile of ripretinib reflected in the INVICTUS study was acceptable and manageable, especially considering the seriousness of the underlying disease and the lack of other treatment options. Clinicians and patients both noted that the tolerability and toxicity profile of ripretinib appeared to be improved compared with prior lines of therapy for GIST, including other TKIs. Similar and low proportions of patients treated with ripretinib and placebo arms withdrew from the protocol therapy due to AEs, and ripretinib dosage reductions were required in relatively few (approximately 7%) patients receiving the drug. SAEs occurred less often in patients receiving ripretinib compared to placebo, potentially due to delayed disease progression. Although the notable harms of ripretinib are varied and, in many cases, serious, the clinical experts consulted by CADTH for this review emphasized that most can be managed through careful patient selection and close monitoring, without posing undue risk to patients.
Conclusions
Evidence from the INVICTUS study suggested that administration of ripretinib in patients with advanced GIST who had previously received imatinib, sunitinib, and regorafenib was associated with statistically significant and clinically meaningful prolongation of PFS compared with placebo. Administration of ripretinib also resulted in numerically higher ORR compared with placebo, although this difference was not statistically significant at a P value of less than 0.05 at the time of the primary analysis. OS was numerically longer in patients randomized to receive ripretinib compared with those randomized to receive placebo. However, differences in OS between patients randomized to receive ripretinib versus placebo were not tested statistically due to early failure of the statistical hierarchy, precluding definitive conclusions. Changes in patient-reported HRQoL (EORTC QLQ-C30, EQ-5D-5L) following ripretinib administration were difficult to interpret due to absence of formal statistical testing, missing data, wide variation in estimates, and uncertainty regarding HRQoL measurement properties in GIST patients. Ripretinib was generally well tolerated in most patients, and its notable harms were considered expected and acceptable by patients and clinicians. The observed PFS benefits, consistent numeric improvements in other efficacy outcomes, and acceptable toxicity profile in the study were aligned with outcomes identified as important to patients with advanced GIST who currently have no treatment options available.
References
1.Aubin F, Blanke CD. Metastatic gastrointestinal stromal tumors. Cancer Chemother Pharmacol. 2011;67(Suppl 1):S9-14. PubMed
2.Emile JF, Brahimi S, Coindre JM, et al. Frequencies of KIT and PDGFRA mutations in the MolecGIST prospective population-based study differ from those of advanced GISTs. Med Oncol. 2012;29(3):1765-1772. PubMed
3.Kee D, Zalcberg JR. Current and emerging strategies for the management of imatinib-refractory advanced gastrointestinal stromal tumors. Ther Adv Med Oncol. 2012;4(5):255-270. PubMed
4.Caterino S, Lorenzon L, Petrucciani N, et al. Gastrointestinal stromal tumors: correlation between symptoms at presentation, tumor location and prognostic factors in 47 consecutive patients. World J Surg Oncol. 2011;9:13. PubMed
5.Maleddu A, Pantaleo MA, Nannini M, Biasco G. The role of mutational analysis of KIT and PDGFRA in gastrointestinal stromal tumors in a clinical setting. J Transl Med. 2011;9:75. PubMed
6.Eisenberg BL, Smith KD. Adjuvant and neoadjuvant therapy for primary GIST. Cancer Chemother Pharmacol. 2011;67(Suppl 1):S3-8. PubMed
7.About GIST. Cobourg (ON): GIST Sarcoma Life Raft Group Canada; 2020: https://liferaftgroup.ca/about-gist/. Accessed 2022 Jan 1.
8.Drug Reimbursement Review sponsor submission: Qinlock (ripretinib) 50 mg oral tablets [internal sponsor's package]. Toronto (ON): Medison; 2021 Oct 15.
9.Bauer S, Heinrich MC, George S, et al. Clinical activity of ripretinib in patients with advanced gastrointestinal stromal tumor harboring heterogeneous KIT/PDGFRA mutations in the phase III INVICTUS study. Clin Cancer Res. 2021;27(23):6333-6342. PubMed
10.Blay JY, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923-934. PubMed
11.Zalcberg JR, Heinrich MC, George S, et al. Clinical benefit of ripretinib dose escalation after disease progression in advanced gastrointestinal stromal tumor: an analysis of the INVICTUS study. Oncologist. 2021;26(11):e2053-e2060. PubMed
12.Clinical Study Report: DCC-2618-03-001. A phase 3, interventional, double-blind, placebo-controlled study to assess the safety and efficacy of DCC-2618 in patients with advanced gastrointestinal stromal tumors who have received treatment with prior anticancer therapies (INVICTUS) [internal sponsor's report]. Waltham (MA): Deciphera Pharmaceuticals; 2019.
13.Medison Pharma Canada Inc. response to 16 December 2021 DRR request for additional information regarding ripretinib DRR review [internal additional sponsor's information]. Toronto (ON): Medison Pharma Canada; 2021.
14.Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod Pathol. 2014;27(Suppl 1):S1-16. PubMed
15.Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23(2):70-83. PubMed
16.Nishida T, Blay JY, Hirota S, Kitagawa Y, Kang YK. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer. 2016;19(1):3-14. PubMed
17.Laurent M, Brahmi M, Dufresne A, et al. Adjuvant therapy with imatinib in gastrointestinal stromal tumors (GISTs)-review and perspectives. Transl Gastroenterol Hepatol. 2019;4:24. PubMed
18.Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632. PubMed
19.Care RS, Valk PJ, Goodeve AC, et al. Incidence and prognosis of c-KIT and FLT3 mutations in core binding factor (CBF) acute myeloid leukaemias. Br J Haematol. 2003;121(5):775-777. PubMed
20.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878. PubMed
21.Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302. PubMed
22.George S, Wang Q, Heinrich MC, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30(19):2401-2407. PubMed
23.Kang YK, Ryu MH, Yoo C, et al. Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2013;14(12):1175-1182. PubMed
24.PrQinlockTM (ripretinib): 50mg oral tablets [product monograph]. Waltham (MA): Deciphera Pharmaceuticals; 2020 Jun 19.
25.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
26.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Nov 9.
27.Deciphera Pharmaceuticals completes enrollment in the INVICTUS pivotal phase 3 clincial study of DCC-2618 in patients with advanced gastrointestinal stromal tumours [press release]. San Francisco (CA): Business Wire; 2018: https://www.businesswire.com/news/home/20181115006037/en/Deciphera-Pharmaceuticals-Completes-Enrollment-in-the-INVICTUS-Pivotal-Phase-3-Clinical-Study-of-DCC-2618-in-Patients-with-Advanced-Gastrointestinal-Stromal-Tumors. Accessed 2022 Jan 1.
28.EORTC Questionnaires: CORE. 2021: https://qol.eortc.org/core/. Accessed 2021 Nov 24.
29.EuroQol Group. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
30.Medical Dictionary for Regulatory Activities (MedDRA) version 21.1. Geneva (CH): International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 2018: https://www.meddra.org. Accessed 2022 Jan 1.
31.Common Terminology Criteria for Adverse Events (CTCAE v4.0: CTCAE v4.03. Bethesda (MD): National Cancer Institute; 2010: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm. Accessed 2022 Jan 1.
32.Luo N, Fones CS, Lim SE, Xie F, Thumboo J, Li SC. The European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-c30): validation of English version in Singapore. Qual Life Res. 2005;14(4):1181-1186. PubMed
33.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. J Patient Rep Outcomes. 2021;5(1):4. PubMed
34.Naamala A, Eriksson LE, Orem J, Nalwadda GK, Kabir ZN, Wettergren L. Psychometric properties of the EORTC QLQ-C30 in Uganda. Health Qual Life Outcomes. 2021;19(1):131. PubMed
35.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
36.Snyder CF, Blackford AL, Sussman J, et al. Identifying changes in scores on the EORTC-QLQ-C30 representing a change in patients' supportive care needs. Qual Life Res. 2015;24(5):1207-1216. PubMed
37.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-117. PubMed
38.Richardson J, Iezzi A, Khan MA, Chen G, Maxwell A. Measuring the sensitivity and construct validity of 6 utility instruments in 7 disease areas. Med Decis Making. 2016;36(2):147-159. PubMed
39.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. PubMed
40.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
41.EORTC Quality of life: FAQ. Brussels (BE): EORTC; 2021: https://qol.eortc.org/faq/. Accessed 2021 Nov 24.
42.EORTC QLC-C30 scoring manual. Brussels (BE): EORTC; 2001: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2021 Nov 24.
43.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
44.EQ-5D-5L user guide: basic information on how to use the EQ-5D-5L instrument. Rotterdam (NL): EuroQol Research Foundation; 2019: https://euroqol.org/wp-content/uploads/2021/01/EQ-5D-5LUserguide-08-0421.pdf. Accessed 2021 Nov 24.
45.Ameri H, Yousefi M, Yaseri M, Nahvijou A, Arab M, Akbari Sari A. Mapping the cancer-specific QLQ-C30 onto the generic EQ-5D-5L and SF-6D in colorectal cancer patients. Expert Rev Pharmacoecon Outcomes Res. 2019;19(1):89-96. PubMed
46.Sinnott PL, Joyce VR, Barnett PG. Preference measurement in economic analysis. Guidebook. Menlo Park (CA): Health Economics Research Center; 2007: https://www.herc.research.va.gov/files/BOOK_419.pdf. Accessed 2021 Nov 24.
Appendix 1: Literature Search Strategy
Note this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: November 12, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits: Conference abstracts excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
cctr | Ovid database code; Cochrane Central Register of Controlled Trials |
Multi-Database Strategy
(Qinlock* or ripretinib* or DCC2618 or DCC-2618 or 9XW757O13D).ti,ab,ot,kf,hw,nm,rn.
1 use medall
*ripretinib/ or (Qinlock* or ripretinib* or DCC2618 or DCC-2618).ti,ab,kf,dq.
3 use oemezd
4 not (conference abstract or conference review).pt.
2 or 5
remove duplicates from 6
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search terms – Qinlock/ripretinib
WHO ICTRP
International Clinical Trials Registry Platform, produced by WHO. Targeted search used to capture registered clinical trials.
Search terms – Qinlock/ripretinib
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms – Qinlock/ripretinib
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms – Qinlock/ripretinib
Grey Literature
Search dates: November 2 to 8, 2021
Keywords: Qinlock, ripretinib
Limits: None
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Appendix 2: Description and Appraisal of Outcome Measures
Note this appendix has not been copy-edited.
Aim
To describe the following outcome measures and their measurement properties (validity, reliability, responsiveness to change, and MID):
EORTC QLQ-C30
EQ-5D-5L and EQ-VAS
Findings
A focused literature search was conducted to evaluate the psychometric properties and the MID of each of these outcome measures. The findings on reliability, validity, responsiveness, and the MID of each outcome measure are summarized in Table 24.
Table 24: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 version 3.012 | Cancer-specific self-reported measure of HRQoL. 30-item questionnaire, consisting of 5 functional scales (physical, role, emotional, social, and cognitive), 9 symptom scales (fatigue, nausea/vomiting, pain, dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties), and a global health status scale. A higher score for functional scales and for global health status represents better functioning ability or HRQoL. A higher score for symptom scales represents a higher level of symptoms. | Not assessed in GIST patients. In cancer patients: Validity Construct validity: moderate to strong correlations between EORTC QLQ-C30 and SF-36 scales, ranging from 0.35 to 0.67.32 Convergent validity: items showed at least moderate correlation with their own scales (r ≥ 0.4).33 Known-groups comparison: except for emotional functioning, all functioning scales and the global quality of life scale showed better scores in patients with mild symptoms than those with severe symptoms.32 Reliability Internal consistency reliability measured using Cronbach alpha: ≥ 0.70 for 6 of the 9 assessed QLQ-30 scales in a Singaporean study.32 Cronbach alpha ≥ 0.70 in all scales expect for cognitive function in a Kenyan study.33 Cronbach alpha ranged from 0.50 (cognitive function scale) to 0.96 (global quality of life scale) in a Ugandan study.34 Responsiveness No relevant studies identified. | Not assessed in GIST patients. In cancer patients: 10 points change for the individual items and scale scores.35,36 Point change for improvement (deterioration).37 Physical: 10.1 (7.2) Role: 15.8 (13.5) Emotional: 14.7 (12.2) Cognitive: 9.1 (0.3) Social: 5.3 (11.1) |
EQ-5D-5L and EQ-VAS12 | Generic preference based HRQoL scale consisting of a descriptive system evaluating 5 dimensions as well as a VAS with anchors of 100 (best imaginable health) and 0 (worst imaginable health) as judged by the patient. A health state profile can be derived by assessing the 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/ depression. Five response levels for each dimension ranging from experiencing no problems to extreme problems. Societal preference weights (utilities) can be used to assign a summary index score to each health state. Index scores range from < 0 (worse than death) to 1 (full health), with 0 representing death and higher scores indicating higher health utility. | Not assessed in GIST patients. In cancer patients: Validity Discriminant validity: mean utility scores differed (0.88 in healthy, 0.18 in patients with cancer).38 Convergent validity: strongly correlated with the physical component of the SF-36 (r = 0.66) and moderately correlated with preference measures of VAS and time trade-off on own health state (r = 0.43).38 Reliability No relevant studies identified. Responsiveness No relevant studies identified. | Not assessed in GIST patients. In advanced cancer patients: 7 to 12 for the EQ-VAS.39 0.10 to 0.12 for UK utility scores and 0.07 to 0.09 for US utility scores anchored by ECOG PS.39 |
ECOG PS = Eastern Cooperative Oncology Group performance status; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-Core 30; GIST = gastrointestinal stromal tumour; HRQoL = health-related quality of life; MID = minimal important difference; SF-36 = 36 item short-form survey; VAS = visual analogue scale.
No literature was identified that assessed validity, responsiveness, or reliability of either of these HRQoL instruments in patients with GIST. No MID information for either HRQoL instrument was identified in patients with GIST.
Studies assessing the psychometric properties of the instruments were only summarized if the assessment was done for the English version and in a sample of mixed (more than 1 type) cancer patients. No literature was identified regarding responsiveness to change of the instruments based on these criteria.
European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-Core 30 (EORTC QLQ-C30)
Description
The EORTC Quality of Life Questionnaire-Core 30, (EORTC QLQ-C30), is 1 of the most commonly used patient-reported outcome measures in oncology clinical trials.28,40 It is a multi-dimensional, cancer-specific, evaluative measure of HRQoL. It was designed specifically for the purpose of assessing changes in participants’ HRQoL in clinical trials in response to treatment.41 The core questionnaire of the EORTC QLQ-C30 consists of 30 questions across 5 multi-item functional scales, 3 multi-item symptom scales, 6 single-item symptom scales, and a 2-item global HRQoL scale (Table 25). Version 3.0 of the questionnaire, used in the INVICTUS study, is the most current version and has been in use since December of 1997.42 The instrument is available more than 110 languages28 and is intended for use in adult populations only.33 Notably, the global HRQoL scale is also known as global health status which was an outcome in the INVICTUS study.33 Patients in the INVICTUS study self-reported their responses to the EORTC QLQ-C30 on a tablet computer.
Table 25: Scales of the EORTC QLQ-C30
Functional Scales (15 Questions) | Symptom Scales (7 Questions) | Single-Item Symptom Scales (6 Questions) | Global Quality of Life (2 Questions) |
|---|---|---|---|
Physical function (5) | Fatigue (3) | Dyspnea (1) | Global quality of life (2) |
Role function (2) | Pain (2) | Insomnia (1) | — |
Cognitive function (2) | Nausea and vomiting (2) | Appetite loss (1) | — |
Emotional function (4) | — | Constipation (1) | — |
Social function (2) | — | Diarrhea (1) | — |
— | — | Financial impact (1) | — |
Scoring
The EORTC QLQ-C30 uses a 1-week recall period in assessing function and symptoms. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 (not at all) to 4 (very much).42 For the 2 items that form the global quality of life scale, however, responses are on a 7-point Likert-type scale, with anchors of 1 (very poor) and 7 (excellent).42
Raw scores for each scale are computed as the average of the items that contribute to a particular scale. This scaling approach is based upon the assumption that it is appropriate to provide equal weighting to each item that comprises a scale. There is also an assumption that, for each item, the interval between response options is equal (for example, the difference in score between “not at all” and “a little” is the same as “a little” and “quite a bit”). Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, higher symptoms on the symptom scales, and better HRQoL on the global quality of life scale. Thus, a decline in scores on symptom scales would reflect an improvement, whereas an increase in scores on function and quality of life scales would reflect an improvement.42
Psychometric Properties
Validity
Three studies with a sample of mixed (more than 1 type) cancer patients were identified that evaluated the psychometric properties of the English version of the EORTC QLQ-30.
One cross-sectional study aimed to validate the EORTC QLQ-30 in a convenience sample of 57 cancer patients (mean 43 [range 15 to 79] years, 44% male) receiving chemotherapy in Singapore.32 Most patients had breast and colorectal cancer (n = 32, 56%), but leukemia, lung cancer, lymphoma, germ cell tumour, and other cancers were also reported. Construct validity was assessed by cross-sectional correlational evidence and discriminative evidence. First, convergent validity was assessed using Spearman’s correlations between 8 pairs of QLQ-30 and Short Form-36 (SF-36) scales (which measures HRQoL within the past 4 weeks), hypothesizing moderate to strong correlation (defined as correlation coefficients of 0.35 to 0.5, and > 0.5, respectively) between scales of these 2 instruments measuring similar dimensions of HRQoL. Moderate to strong correlations were observed between the QLC-30 and SF-36 scales, ranging from 0.35 (comparison of QLQ-C30 role functioning and SF-36 role-emotional scales) to 0.67 (comparison of QLQ-C30 pain and SF-36 bodily pain scales). Next, the known-groups approach was used to compare 6 QLQ-30 scale scores between patients reporting mild and severe symptoms, as well as by stage of disease and presence of comorbid conditions. With the exception of emotional functioning, the remaining 5 scales showed better scores in patients with mild symptoms than those with severe symptoms. There were no significant differences found in QLQ-30 scores between patients in early stages of cancer (or with no comorbid conditions) compared to those in advanced disease stages (or with comorbid conditions).32 This may be attributable to the small sample size of the study which may not have been sufficiently powered to detect a significant difference.
A recent cross-sectional study in Kenya was conducted to evaluate the psychometric properties of the EORTC QLQ-C30, using the English or Kiswahili version in 100 patients with cancer (median 53.5 [range 18 to 83] years, 41% male).33 Most (40%) patients had breast cancer, followed by prostate cancer, Kaposi sarcoma, lung cancer, and other cancers. Convergent validity was assessed by examining Pearson’s intra-scale correlations among the subscales of the EORTC QLQ-C30. Regarding convergent validity, items showed at least moderate correlation with their own scales (r ≥ 0.4) with higher correlations with their own scales than with other scales. This included strong correlations between physical and role functioning (r = 0.66), emotional and cognitive functioning (r = 0.58), pain and fatigue (r = 0.73), and sleep disorders and fatigue (r = 0.68). Furthermore, with the exception of cognitive functioning, emotional functioning, nausea and vomiting, dyspnea, appetite loss, constipation, and diarrhea, global health status correlated moderately with the remaining subscales (r ≥ 0.30).33
Another recent cross-sectional study in Uganda evaluated the English version of EORTC QLQ-C30 among 168 adult patients with cancer (37% male).34 A total of 168 patients with cancers (cervical, breast, Kaposi sarcoma, leukemia, and other cancers) completed the English version of the EORTC QLQ-C30. The study assessed the presence of floor and ceiling effects that were defined as acceptable if they did not exceed 15%. Subscales that exceeded the acceptable floor effect included role and social functioning as well as nausea/vomiting. Subscales exceeding the acceptable ceiling effect included role, emotional, and cognitive functioning, as well as pain. This may be explained by the high number of patients with advanced stages of cancer with reduced role and social functioning due to fatigue and pain. Confirmatory factor analysis using the maximum likelihood method was conducted to assess construct validity of the EORTC QLQ-C30, with coefficients of ≥ 0.4 considered acceptable. Satisfactory correlations were observed ranging from 0.46 to 0.97, suggesting strong construct validity. Further evidence of construct validity of the instrument was demonstrated using the known-groups comparisons approach by examining the capacity to discriminate between patients differing in disease stage (stages I/II versus stages III/IV). Effect sizes (ES) were calculated and 0.2, 0.5 to 0.8, and > 0.8 were classified as small, moderate, and large differences, respectively. There were statistically significant differences for all subscales except for cognitive function (ES = 0.23) and emotional function (ES = 0.21). Small effect sizes (ranging from 0.14 for social functioning to 0.46 for fatigue) were identified for all scales except for physical function which displayed a moderate ES (0.70). These results demonstrated that the instrument was able to discriminate between early and late stages of cancer among the surveyed patients.34 Criterion validity was assessed by examining associations between 2 EORTC QLQ-C30 subscales (global quality of life and physical function) and the Karnofsky Performance Scale (KPS). Positive correlations were observed between KPS and the 2 subscales, with r = 0.72 and r = 0.76 for each of global quality of life and physical function, respectively.
Reliability
The Singaporean cross-sectional study also assessed internal consistency reliability by calculating Cronbach alpha for all EORTC QLQ-C30 scales. Cronbach alpha was ≥ 0.70 for 6 of the 9 assessed QLQ-30 scales; cognitive functioning, physical functioning, and nausea and vomiting had Cronbach alpha ranging from 0.19 to 0.68.32
Both the Kenyan and Ugandan studies previously described assessed the internal consistency of each scale of the questionnaire using Cronbach alpha-coefficients. In the Kenyan study, with the exception of the cognitive function scale, all of the scales had Cronbach alpha ≥ 0.70.33 In the Ugandan study, Cronbach alpha ranged from 0.50 (cognitive function scale) to 0.96 (global quality of life scale).34
Studies evaluating the responsiveness to change of the English version of the EORTC QLQ-C30 among mixed cancer patients were not identified.
MID
For use in clinical trials, scores on the EORTC QLQ-C30 can be compared between different groups of patients or within a group of patients over time. One study of patients in 2 randomized trials of chemotherapy (1 for breast cancer and 1 for small-cell lung cancer) conducted in 1998 estimated a clinically relevant change in score on any scale of the EORTC QLQ-C30 to be 10 points.35 The study used an anchor-based approach to estimate the MID in which patients who reported “a little” change (for better or worse) on the subjective significance questionnaire had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately 5 to 10 points. Participants who reported a “moderate” change had corresponding changes in the EORTC QLQ-C30 of about 10 to 20 points, and those who reported having “very much” changed HRQoL had corresponding changes of more than 20 points.35
More recently in 2015, a Canadian study estimated the MIDs of EORTC QLQ C-30 scales using data from 193 newly diagnosed breast and colorectal cancer patients who had recently undergone surgery (mean = 60 [range = 22 to 88] years, 20% male).36 The Supportive Care Needs Survey-Short Form-34 (SCNS-SF34) was used as an anchor; mean changes in EORTC QLQ C-30 scales associated with improvement, worsening, and no change in supportive care needs based on the SCNS-SF34 were then calculated. MIDs were assessed for the following scales: physical function, role function, emotional function, global health status, pain, and fatigue. For improvement, MIDs associated with improved supportive care needs ranged from 10 to 32 points. For worsening, MIDs associated with a worsening of supportive care needs ranged from 9 to 21 points. The range for unchanged supportive care needs was from 1-point worsening to 16-point improvement in EORTC QLQ-C30 score.36 Based on this, the authors suggested that a 10-point change in EORTC QLQ C-30 score represented changes in supportive care needs, and therefore should be considered for clinical use.36
In 2014, another Canadian study estimated the MID for EORTC QLQ-C30 among 369 patients with advanced cancer (mean = 57.7 [SD = 12.8] years, 22% male) who completed the questionnaire at baseline and 1-month post-radiation.37 The most common cancer types were breast cancer (n = 270, 70%), followed by lung, prostate, gastrointestinal, renal cell, and other cancers. MID was estimated using both anchor- and distribution-based methods for improvement and deterioration. Two anchors of overall health and overall HRQoL were used, both taken directly from the EORTC QLQ-C30 (questions 29 and 30) where patients rated their overall health and HRQoL themselves. The MIDs were determined by calculating the difference in mean change in scores between patients with improved versus unchanged overall health and between patients with deteriorated versus unchanged overall health. Improvement and deterioration were categorized as an increase or decrease of 2 units to account for the natural fluctuation of patient scoring (i.e., assuming that 1 unit may not be sensitive). With these 2 anchors, the estimated MIDs (95% CI) across all EORTC QLQ-C30 scales ranged from 9.1 (1.4 to 16.7) to 23.5 (31.9 to 15.2) units for improvement, and from 7.2 (0.2 to 14.2) to 13.5 (3.7 to 23.3) units for deterioration. For the overall health anchor, none of the symptom scales showed a significant MID at the 1-month follow-up which may be due to differing symptoms among the patient groups examined. Distribution-based estimates were closest to 0.5 SD.
EQ-5D-5L
Scoring
The EQ-5D is a generic HRQoL instrument that may be applied to a wide range of health conditions and treatments.29,43 The first of 2 parts of the EQ-5D is a descriptive system that classifies respondents (aged ≥ 12 years) based on the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. In 2005, updates were made to the EQ-5D-3L to create the EQ-5D-5L which included creating 5 response levels of severity (no problems, slight problems, moderate problems, severe problems, unable to/extreme problems) in each of the 5 existing dimensions.44 Respondents are asked to choose the level that reflects their health state for each of the 5 domains resulting in 3,125 possible health states.45 A scoring function can be used to assign a value to self-reported health states from a set of population-based preference weights.29,43 The second part is a 20 cm visual analogue scale (EQ-VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to rate their health by drawing a line from an anchor box to the point on the EQ-VAS which best represents their health on that day.
The EQ-5D index score is generated by applying societal preference weights for the various aforementioned health states.46 Health state index scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states ‘dead’ and ‘perfect health,’ respectively.
Psychometric Properties
Validity
Richardson et al. (2016)38 examined various instruments, including the EQ-5D-5L, among respondents who were healthy and who had a chronic disease (i.e., arthritis, asthma, cancer, depression, diabetes, hearing loss, and heart disease) through an online survey in Australia, Canada, Germany, Norway, the UK, and the US (total N = 7,933; cancer N = 772). For discriminant validity, the mean EQ-5D-5L utility score of healthy respondents was compared to patients with cancer (and other chronic diseases). The mean utility scores differed in these subgroups (0.88 in healthy individuals, 0.18 in patients with cancer). Regarding convergent validity, the EQ-5D-5L was compared to other related scales using Pearson’s correlations. The EQ-5D-5L was strongly correlated with the physical component of the SF-36 in cancer patients (r = 0.66), moderately correlated with the psychosocial content of the mental component of the SF-36, the Capabilities Instrument, and the Subjective Well-Being Instrument of the UK Office of National Statistics (r = 0.50), and moderately correlated with preference measures of VAS and time trade-off on own health state (r = 0.43).38
Studies evaluating the reliability and responsiveness of the English version of instrument among mixed cancer patients were not identified.
MID
Pickard et al. (2007)39 estimated the MID of the EQ-5D VAS based on cross-sectional data collected from 534 patients (mean 59 [SD = 12] years, 52% male) in the US with advanced (stage III or IV) cancer of the bladder, brain, breast, colon or rectum, head or neck, liver or pancreas, kidney, lung, lymphoma, ovary, or prostate.39 EQ-5D index-based utility (UK and US) scores were estimated using both anchor and distribution-based approaches. Groups were anchored by ECOG PS and then distribution approaches including the standard error of the mean and 0.5 SD were applied to each anchor-based category. MID estimates were similar across all cancers. For utility scores, MID estimates for all cancers and the lung cancer subgroup ranged from 0.10 to 0.12 for UK scores and 0.07 to 0.09 for US scores. MIDs for the EQ-5D VAS ranged from 8 to 12 based on the ECOG PS, and from 7 to 10 based on Functional Assessment of Cancer Therapy HRQoL questionnaire quintiles.39
Appendix 3: Detailed Outcome Data
Note this appendix has not been copy-edited.
Table 26: Patient Disposition in the INVICTUS Study (OL Period)
Disposition | Ripretinib 150 mg q.d. (DB: placebo) N = 29 | Ripretinib 150 mg q.d. (DB: ripretinib) N = 11 |
|---|---|---|
Screened, N | NA | NA |
Screen failure, N | NA | NA |
Randomized, N (%) | NA | NA |
Discontinued treatmenta | ||
n (%) | 9 (31.0) | 10 (90.9) |
Primary reason for treatment discontinuation, n (%)a | ||
Adverse event | 0 | 0 |
Clinical progression | 4 (13.8) | 2 (18.2) |
Death | 0 | 1 (9.1) |
Physician decision | 0 | 1 (9.1) |
Confirmed PD by investigator assessment | 3 (10.3) | 1 (9.1) |
Confirmed PD by IRR | 0 | 2 (18.2) |
Withdrawal of IC from study | 1 (3.4) | 1 (9.1) |
Withdrawal of IC from treatment | 1 (3.4) | 1 (9.1) |
Discontinued studyb | ||
N (%) | 7 (24.1) | 7 (63.6) |
Primary reason for study discontinuation, n (%)b | ||
Death | 6 (20.7) | 4 (36.4) |
Withdrawal of IC from study | 1 (3.4) | 3 (27.3) |
Analysis populations, n (%)b,c | ||
ITT population | 29 (100.0) | 11 (100.0) |
Safety population | 29 (100.0) | 11 (100.0) |
PP population | 29 (100.0) | 11 (100.0) |
Entered OL | ||
n (%) | NA | NA |
Treatment ongoing at data cut-off | ||
n (%) | 10 (34.5) | 1 (9.1) |
IC = informed consent; IRR = independent radiological review; ITT = intention to treat; NA = not applicable; OL = open label; PD = progressive disease; PP = per protocol; q.d. = once daily.
Note: Data are from the May 31, 2019, database lock.
aDenominator is the safety population.
bDenominator is the ITT population.
cFor the OL period, all patients received assigned treatments per protocol.
Source: INVICTUS Clinical Study Report.12
Figure 7: Kaplan-Meier Plot of OS in the INVICTUS Study by Subgroups of DB and OL Treatment (ITT Population; Data Cut: January 15, 2021)
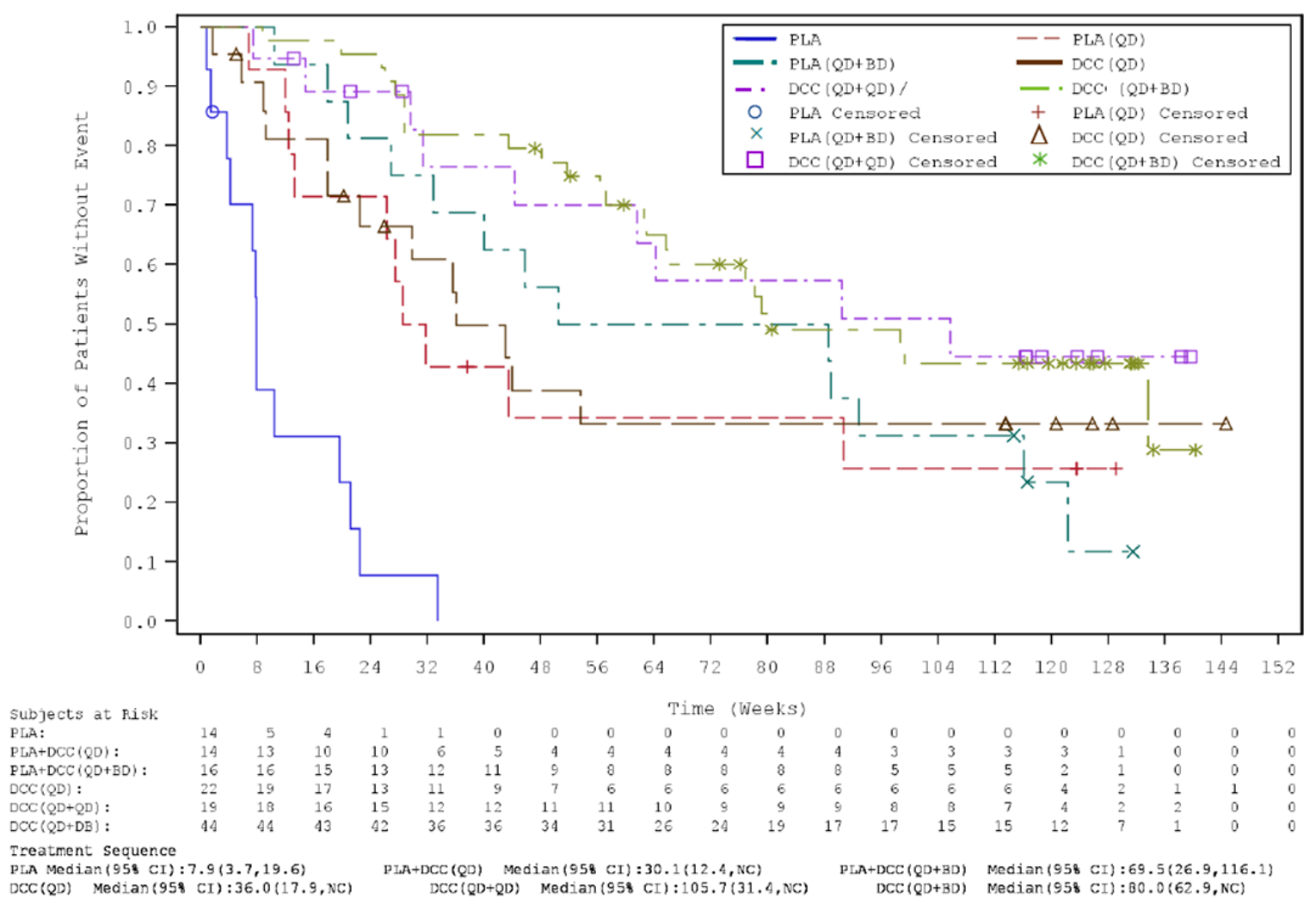
BD = twice daily; CI = confidence interval; DB = double-blind; DCC = ripretinib; ITT = intention to treat; NC = not calculable; OL = open label; OS = overall survival; PLA = placebo; QD = once daily.
Note: symbols represent censored observations.
Source: Sponsor’s additional information.13
Table 27: Sensitivity Analyses of ORR in the INVICTUS Study
Outcome | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
ORR | ||
Responders, n (%) | 0 | 8 (9.9) |
95% CIa | NR | NR |
ORR difference (95% CI)c | 9.9 (0.2 to 18.3) | |
P value b | 0.0500 | |
BOR, n (%) | ||
Complete response | 0 | 0 |
Partial response | 0 | 8 (9.4) |
Stable diseased | 7 (16.7) | 53 (65.4) |
Progressive disease | 28 (66.7) | 16 (19.8) |
Not evaluable | 3 (7.1) | 3 (3.7) |
No response assessment | 4 (9.5) | 1 (1.2) |
ORR | ||
Responders, n (%) | 0 | 9 (10.6) |
95% CIa | 0.0, 8.0 | 5.0, 19.2 |
ORR difference (95% CI)c | 10.6 (1.2, 18.9) | |
P valueb | 0.0275 | |
BOR, n (%) | ||
Complete response | 0 | 0 |
Partial response | 0 | 9 (10.6) |
Stable diseased | 8 (18.2) | 48 (56.5) |
Progressive disease | 28 (63.6) | 21 (24.7) |
Not evaluable | 4 (9.1) | 6 (7.1) |
No response assessment | 4 (9.1) | 1 (1.2) |
BOR = best overall response; CI = confidence interval; IRR = independent radiological review; ITT = intention to treat; NA = not applicable; ORR = objective response rate.
aFrom exact binomial CI.
bP value from Fisher’s exact test.
cNewcombe Score 95% CI of the difference in ORR between treatment arms.
dDefined as absence of PD for at least 6 weeks.
Source: INVICTUS Clinical Study Report.12
Table 28: Subgroup Analysis of ORR per IRR in the INVICTUS Study
Item | Placebo | Ripretinib | ORR difference (95% CI)a | ||
|---|---|---|---|---|---|
N | Responders, n (%) | N | Responders, n (%) | ||
Number of prior systemic anticancer treatments | |||||
3 or more (no subgroups) | 44 | 0 | 85 | 8 (9.4) | 9.4 (0.2 to 17.5) |
3 | 27 | 0 | 54 | 5 (9.3) | 9.3 (–4.3 to 19.9) |
4 or more | 17 | 0 | 31 | 3 (9.7) | 9.7 (–9.8 to 24.9) |
Tumour mutational status | |||||
Any (no subgroups) | 44 | 0 | 85 | 8 (9.4) | 9.4 (0.2 to 17.5) |
KIT exon 9 | 6 | 0 | 14 | 0 | NE (NE to NE) |
KIT exon 11 | 28 | 0 | 47 | 8 (17.0) | 17.0 (2.5 to 30.1) |
KIT other exons | 2 | 0 | 2 | 0 | NE (NE to NE) |
PDGFRA | 0 | 0 | 3 | 0 | NE (NE to NE) |
KIT wt/PDGFRA wt | 3 | 0 | 7 | 0 | NE (NE to NE) |
Not available | 5 | 0 | 11 | 0 | NE (NE to NE) |
Not done | 0 | 0 | 1 | 0 | NE (NE to NE) |
CI = confidence interval; IRR = independent radiological review; NE = not estimable; ORR = objective response rate.
aNewcombe Score 95% CI of the difference in ORR between treatment arms.
Source: INVICTUS Clinical Study Report.12
Table 29: Sensitivity Analyses of PFS in the INVICTUS Study
PFS analysis | Placebo N = 44 | Ripretinib N = 85 |
|---|---|---|
PFS per IRR in ITT using randomization stratification factor values collected on the eCRF instead of in the IRT | ||
Patients with events, n (%) | 37 (84.1) | 51 (60.0) |
Patients censored, n (%) | 7 (15.9) | 34 (40.0) |
Median PFS (95% CI)a | 4.1 (4.0, 7.3) | 27.6 (20.0, 29.9) |
P valueb | < 0.0001 | |
HR (95% CI)c | 0.15 (0.09, 0.25) | |
PFS rate (95% CI)a | ||
26 weeks | 3.2 (0.2, 13.8) | 51.0 (39.4, 61.4) |
39 weeks | NE (NE, NE) | 34.4 (22.9, 46.2) |
52 weeks | NE (NE, NE) | 21.0 (9.0, 36.3) |
PFS per IRR in PP population | ||
Patients with events, n (%) | 36 (85.7) | 49 (60.5) |
Patients censored, n (%) | 6 (14.3) | 32 (39.5) |
Median PFS (95% CI)a | 4.1 (3.9, 4.3) | 27.6 (200, 28.9) |
P valueb | < 0.0001 | |
HR (95% CI)c | 0.13 (0.08, 0.23) | |
PFS rate (95% CI)a | ||
26 weeks | 0.0 (0.0, 0.0) | 50.3 (38.6, 60.9) |
39 weeks | 0.0 (0.0, 0.0) | 34.8 (22.9, 46.9) |
52 weeks | 0.0 (0.0, 0.0) | 21.2 (9.1, 36.7) |
PFS per IRR in safety population | ||
Patients with events, n (%) | 37 (86.0) | 51 (60.0) |
Patients censored, n (%) | 6 (14.0) | 34 (40.0) |
Median PFS (95% CI)a | 4.1 (4.0, 7.3) | 27.6 (20.0, 29.9) |
P valueb | < 0.0001 | |
HR (95% CI)c | 0.15 (0.09, 0.25) | |
PFS rate (95% CI)a | ||
26 weeks | 3.2 (0.2, 13.8) | 51.0 (39.4, 61.4) |
39 weeks | NE (NE, NE) | 34.4 (22.9, 46.2) |
52 weeks | NE (NE, NE) | 21.0 (9.0, 36.3) |
PFS by investigator assessment in ITT | ||
Patients with events, n (%) | 36 (81.8) | 45 (52.9) |
Patients censored, n (%) | 8 (18.2) | 40 (47.1) |
Median PFS (95% CI)a | 4.1 (3.9, 6.0) | 20.4 (18.4, 35.6) |
P valueb | < 0.0001 | |
HR (95% CI)c | 0.19 (0.12, 0.32) | |
PFS rate (95% CI)a | ||
26 weeks | 3.9 (0.3, 16.2) | 47.5 (35.9, 58.1) |
39 weeks | NE (NE, NE) | 37.9 (25.7, 50.0) |
52 weeks | NE (NE, NE) | 37.9 (25.7, 50.0) |
CI = confidence interval; eCRF = electronic case record ford; HR = hazard ratio; IRR = independent radiological review; IRT = interactive response technology; ITT = intention to treat; NE = not estimable; NR = not reported; PFS = progression-free survival; PP = per protocol.
aFrom KM analysis.
bP value from 2-sided stratified log-rank test. Stratification factors were the same as those applied to randomization (number of prior anticancer treatment and ECOG status at baseline).
cHR from Cox proportional regression model with treatment and randomization stratification factors as fixed factors.
Source: INVICTUS Clinical Study Report.12
Table 30: Subgroup Analysis of PFS in the INVICTUS Study
Number of prior systemic anticancer treatments | Placebo N | Ripretinib N | HR (95% CI)a |
|---|---|---|---|
3 or more (no subgroups) | 44 | 85 | 0.15 (0.09, 0.25) |
3 | 27 | 54 | 0.15 (0.08, 0.29) |
4 or more | 17 | 31 | 0.24 (0.12, 0.51) |
CI = confidence interval; HR = hazard ratio; PFS = progression-free survival.
aHR from Cox proportional regression model with treatment and randomization stratification factors as fixed factors.
Source: INVICTUS Clinical Study Report.12
Table 31: PFS per IRR in the INVICTUS Study (ITT Population, OL Period)
PFS analysis | Ripretinib 150 mg q.d. (DB: placebo) N = 29 | Ripretinib 150 mg q.d. (DB: ripretinib) N = 11 |
|---|---|---|
Patients with events, n (%) | 13 (44.8) | NR |
Patients censored, n (%) | 16 (55.2) | NR |
Median PFS (95% CI)a | 20.0 (8.0, NE) | NR |
P valueb | NR | NR |
HR (95% CI)c | NR | NR |
PFS rate (95% CI)a | ||
26 weeks | 44.4 (21.7, 65.0) | NR |
39 weeks | 22.2 (1.8, 57.0) | NR |
52 weeks | NE (NE, NE) | NR |
CI = confidence interval; HR = hazard ratio; IRR = independent radiological review; ITT = intention to treat; NE = not estimable; NR = not reported; OL = open label; PFS = progression-free survival.
Note: Data are from the May 31, 2019, database lock.
aFrom KM analysis.
bP value from 2-sided stratified log-rank test. Stratification factors were the same as those applied to randomization (number of prior anticancer treatment and ECOG status at baseline).
cHR from Cox proportional regression model with treatment and randomization stratification factors as fixed factors.
Source: INVICTUS Clinical Study Report.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
GIST
gastrointestinal stromal tumour
ICER
incremental cost-effectiveness ratio
LY
life-year
OS
overall survival
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
RDI
relative dose intensity
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ripretinib (Qinlock), oral tablets |
Submitted price | Ripretinib, 50 mg tablet: $216.32 |
Indication | For the treatment of adult patients with advanced gastrointestinal stromal tumour (GIST) who have received prior treatment with imatinib, sunitinib, and regorafenib |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review (Project Orbis) |
NOC date | June 19, 2020 |
Reimbursement request | As per indication |
Sponsor | Medison Pharma Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Partitioned survival model (PSM) |
Target population | Adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib, and regorafenib |
Treatment | Ripretinib |
Comparator | Best supportive care (BSC; basket of medications for managing symptoms of GIST related to pain, GI support, anemia, nutritional support, sleep, emotional support, and infections) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (16 years) |
Key data source | Clinical efficacy was modelled using overall survival (OS) and progression-free survival (PFS) observed in the INVICTUS trial. Health state utility values were estimated using EQ-5D-5L data collected from the same trial |
Submitted results | ICER = $103,743 per QALY (including QALYs: 2.107; including costs: $218,621) |
Key limitations |
|
CADTH reanalysis results |
|
BSC = best supportive care; GI = gastrointestinal; GIST = gastrointestinal stromal tumour; ICER = incremental cost-effectiveness ratio; LY = life-year; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; RDI = relative dose intensity; WTP = willingness to pay.
Conclusions
The CADTH clinical review found that treatment with ripretinib resulted in a statistically significant and clinically meaningful survival advantage for progression-free survival (PFS) compared to placebo in adult patients with advanced gastrointestinal stromal tumour (GIST) who have received prior treatment with imatinib, sunitinib, and regorafenib. However, the overall survival (OS) benefit for ripretinib compared to placebo is highly uncertain, due to the absence of formal statistical testing and issues relating to post-progression and dose escalation in the INVICTUS trial data.
CADTH identified several limitations with the sponsor’s economic evaluation: OS was overestimated in extrapolation; the model structure created unrealistic results; treatment beyond progression in the ripretinib arm was inappropriately characterized; utilities lacked face validity; and relative dose intensity (RDI) was applied inappropriately in calculating drug costs. In the CADTH base-case reanalysis, CADTH used a Weibull parametric function to extrapolate OS, applied a 2-stage complex adjustment with recensoring for treatment beyond progression in the ripretinib arm to estimate OS, substituted the sponsor’s utilities with those determined to be more clinically relevant, and revised the RDI to 100%. CADTH was unable to evaluate the impact of treatment beyond progression due to structural limitations in the sponsor’s pharmacoeconomic model. Furthermore, CADTH’s estimates of cost-effectiveness are likely biased in favour of ripretinib, as reanalysis was unable to address the constraints introduced by the submitted model structure and its effect on post-progression survival benefit.
In the CADTH reanalysis, ripretinib was associated with an incremental cost-effectiveness ratio (ICER) of $242,365 per quality-adjusted life-year (QALY), and the probability of cost-effectiveness at a willingness-to-pay (WTP) threshold of $50,000 per QALY was 0%. A price reduction of 83% is necessary to achieve cost-effectiveness at this threshold. The cost-effectiveness of ripretinib is driven by assumptions concerning the extrapolation of OS, adjustment for treatment beyond progression, and utility benefit associated with treatment.
Ripretinib is more costly than best supportive care (BSC) in treating adult patients with advanced GIST. CADTH’s reanalysis was unable to address the uncertainty concerning OS evidence from the INVICTUS trial. As a consequence of this and other limitations, CADTH’s cost-effectiveness and price-reduction estimates are highly uncertain.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received patient input from the CanCertainty Coalition and GIST Sarcoma Life Raft Group Canada, 2 national-level advocacy organizations dedicated to supporting Canadians living with cancer. The CanCertainty Coalition primarily highlighted issues with navigating access to drugs for GIST, with administrative barriers to access, and with financial burden in Ontario and the Atlantic provinces, potentially affecting an estimated 5.4 patients with GIST younger than 65 years who are uninsured out of a total approximated 101 yearly GIST cases across Canada. Concerns were also raised surrounding safety issues regarding community dispensing of take-home cancer drugs, such as incorrect dosing and handling, limited monitoring, nonadherence, serious toxicity, morbidity, and mortality. The GIST Sarcoma Life Raft Group Canada commissioned Filomena Servido-Italiano from the Blue Ribbon Project Inc. to oversee, analyze, and conduct telephone interviews of 11 patients with metastatic GIST, of whom 5 resided in Canada. All 11 patients accessed first-line imatinib, 6 accessed sunitinib in the second-line setting, and 3 accessed regorafenib in the third-line setting. Side effects of imatinib, sunitinib, and regorafenib include reduced kidney function, abdominal cramping, fatigue, anemia, poor wound healing, hand and foot syndrome, high fevers, body aches, elevated blood pressure, diarrhea, and gastrointestinal bleeding. Patients noted that they would like ripretinib to improve survival, increase patient quality of life, and induce fewer side effects in the long term, which they felt was observed in ripretinib treatment. Patients noted that side effects of ripretinib were fewer and less toxic than previously accessed therapies and included hair loss, fatigue, nausea, hand and foot syndrome, skin lesions, and elevated blood counts. Two patients out of 11 experienced early disease progression on ripretinib, but the remaining 9 had experienced durable responses, ranging from 10 months to 5.5 years. Patients provided input regarding the need for integration of KIT and platelet-derived growth factor alpha mutational analysis testing into guidelines for treatment selection. Ripretinib would address the lack of available, effective treatments for GIST in patients beyond the third-line setting who remain resistant to existing therapies.
CADTH received registered clinician input from 7 Canadian clinicians who treat patients with GIST. The clinicians stated that current pathway of care is first-line imatinib, followed by second-line sunitinib and third-line regorafenib. Selected patients with advanced GIST who are intolerant to or have progressed on imatinib, sunitinib, and regorafenib have had access to ripretinib in the fourth-line setting through the Special Access Program. Clinicians expect that ripretinib would be prescribed as monotherapy in the fourth-line setting in Canada, with no expected shift in the current treatment paradigm.
CADTH received drug plan input noting challenges in assessing and monitoring therapeutic response, given the higher frequency of CT or MRI tumour assessments in the INVICTUS trial compared to clinical practice. They also expressed concerns about the consistency of discontinuation criteria compared to other drugs in the same therapeutic space, given the option for patients receiving ripretinib in the INVICTUS trial to dose escalate, continue treatment, or discontinue therapy. In the event of dose reduction, drug plans commented that storage restrictions regarding original packaging may be restricting. Finally, drug plans noted multiple potential drug-drug, drug-food, and drug-herb interactions that require assessment and potential intervention.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s model compared ripretinib to BSC following previous treatment with imatinib, sunitinib, and regorafenib.
CADTH also addressed some of these concerns:
CADTH increased the market share of ripretinib in the budget impact analysis to reflect clinical expert feedback suggesting that ripretinib would become standard of care in the fourth-line setting.
CADTH was unable to address the following concerns raised from stakeholder input:
Dose escalation, discontinuation, and treatment beyond progression were not explicitly modelled for ripretinib and therefore could not be explored.
Economic Review
The current review is for ripretinib (Qinlock) for the treatment of adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib, and regorafenib.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of ripretinib compared with BSC for the treatment of adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib, and regorafenib. The model population consisted of patients from the INVICTUS randomized controlled trial receiving ripretinib and BSC.1,2 The target population aligns with the Health Canada–indicated population and reimbursement request.
Ripretinib is available as 50 mg oral tablets in 90-tablet bottles. The recommended dosage of ripretinib is 150 mg (three 50 mg tablets) taken orally once daily until the drug is not tolerated.3 The recommended reduced dosage in the event of adverse reactions is 100 mg orally once daily.3 Ripretinib is intended to be used as fourth-line therapy for patients with GIST who have received prior treatment with imatinib, sunitinib, and regorafenib. The cost for ripretinib is $216.32 per 50 mg tablet, and the 28-day cost is $18,171 as calculated by CADTH (Table 8).3
The comparator for this economic analysis is BSC, defined as a basket of medications for management of GIST symptoms related to pain, gastrointestinal support, anemia, nutritional support, sleep, emotional support, and infections.1 Medications used for the treatment of comorbidities, such as high blood pressure, high lipid levels, or diabetes, were excluded from the basket of drugs.2 The sponsor’s definition of BSC was based on patients from the INVICTUS trial who had received prior treatment with imatinib, sunitinib, and regorafenib in Canada. These patients received the basket of medications for symptom management and no active therapy in the post-regorafenib setting. BSC was also administered to ripretinib patients as concomitant medications as per the INVICTUS trial. The cost per 28-day cycle is $176.71 and $177.24 for the basket of medications administered to those receiving BSC alone and ripretinib plus BSC, respectively.4 Additional details about the cost, dosage, and usage for the basket of medications used to represent BSC are presented in Appendix 3.
The sponsor calculated a 28-day cycle cost for ripretinib based on an RDI of 96.50%.2 Administration costs were not incorporated into either ripretinib or BSC treatment arms because all treatments incorporated were orally self-administered, except sodium chloride IV infusions, the cost of which was assumed to be negligible.
Outcomes modelled included quality-adjusted life-years (QALYs) and life-years (LYs) over a lifetime time horizon of 16 years. The base-case analysis was conducted from the Canadian public health care system perspective, with costs and outcomes discounted at 1.5%. The cycle length was 4 weeks with a half-cycle correction.
Model Structure
The sponsor submitted a partitioned survival model (PSM) that consists of 3 mutually exclusive health states: progression-free, progressed disease, and death. All patients enter the model in the progression-free health state, where they are randomized to receive ripretinib plus BSC or BSC alone. These patients can then transition directly to the death state or to the progressed disease state, where they remain until they transition to the death state. The proportion of patients in the progression-free state is estimated by the PFS curves of the ripretinib and BSC treatment arms from the INVICTUS trial, where progression is defined as an increase in size of target lesions or appearance of new lesions as per the modified Response Evaluation Criteria in Solid Tumours (mRECIST) version 1.1.2 The proportion of patients in the progressed disease state was equal to the difference between the OS and PFS curves from the INVICTUS trial. Patients transitioning to the death state remained there until the end of the model time horizon.
Model Inputs
The population used for this model was derived from the INVICTUS trial (n = 129 patients with GIST).1,2 Median patient age was 60 years, and 57% of patients were men.1,2 All patients enrolled had failed previous treatment with imatinib, sunitinib, and regorafenib.
The sponsor used parametric modelling to extrapolate the PFS and OS data from the INVICTUS trial. For PFS, a log-logistic distribution was selected for the ripretinib and BSC arms, based on best statistical fit and visual inspection. Log-logistic distributions were also selected for both ripretinib and BSC OS, based on best statistical fit and visual inspection. A 2-stage approach to adjust for crossover with recensoring was assumed to be adequate to account for trial crossover in the BSC arm. The sponsor noted that treatment continuation beyond disease progression was not standard; therefore, treatment discontinuation was capped by PFS. However, patients were offered the option to continue ripretinib treatment in the open-label phase.
The dosage of ripretinib used in the model is consistent with the description in the Overview section, based on the INVICTUS trial and the product monograph.2,3
Health-related quality-of-life data were collected from the INVICTUS trial for the progression-free and disease progressed health states by using the EQ-5D-5L instrument. The progression-free and disease progressed utilities were calculated to be 0.817 and 0.807, respectively.2 Disutilities due to Grade 3 and 4 adverse events (AEs) were incorporated as utility decrements, ranging from 0.069 to 0.085, that were applied based on probability of occurrence in each treatment arm.2,4 Disutilities were sourced from published literature for cancer, nonspecific to GIST. These AEs included abdominal pain, anemia, and hypertension.
All costs used in the model were inflated to 2020 Canadian dollars. Drug-acquisition costs included the cost per ripretinib tablet, with no administrative costs. Cost of medications for the ripretinib and BSC arms were calculated based on usage from the INVICTUS trial for each treatment arm and were $176.71 and $177.24 for BSC and ripretinib, respectively.3 Costs of subsequent treatments were not included due to lack of further options for the GIST patient population. The model included disease management costs related to monitoring resources: $105 per outpatient visit, $108 per CT scan, $4 per blood test, $3 per liver function test, and $51 per kidney function test.5,6 These costs were all from the Ministry of Health of Ontario Schedule of Benefits. Costs for managing Grade 3/4 AEs described in the Overview were from the Ontario Schedule of Benefits, Ontario Drug Benefit Formulary, and published literature. These costs varied from $8.81 to $5,363.17.4-6 Last, a 1-time terminal care cost of $16,033 was applied, based on expenditure per patient for palliative end-of-life care in the final month of life.7
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in this section.
Base-Case Results
Ripretinib was associated with incremental costs of $218,621 and 2.11 QALYs in comparison to BSC, resulting in an ICER of $103,743 per QALY gained (Table 3). Approximately 54% of the incremental QALYs in the sponsor’s base case were accrued after the period of the INVICTUS trial data (33 months). The sponsor’s submission estimated mean LYs of 3.14 for patients treated with ripretinib, with 0.54 LYs estimated for patients treated with BSC. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. BSC ($/QALY) |
|---|---|---|---|---|---|
BSC | 18,941 | Reference | 0.44 | Reference | Reference |
Ripretinib | 237,563 | 218,621 | 2.54 | 2.11 | 103,743 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.4
Sensitivity and Scenario Analysis Results
The sponsor conducted several sensitivity and scenario analyses involving discount rate, time horizon, extrapolation assumptions for PFS and OS, adjustment for treatment beyond progression in the ripretinib arm using the 2-stage complex method with recensoring, and adjustment for crossover using the 2-stage complex method without recensoring. In these analyses, the ICER was most sensitive to adjustment for treatment beyond progression in the ripretinib arm, which resulted in an ICER of $139,903.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Comparative efficacy of ripretinib on OS is uncertain: CADTH’s clinical review of the sponsor’s submitted trial evidence found that no definitive conclusions could be drawn about the effect of ripretinib on OS versus placebo, owing to a lack of formal statistical testing and inability to account for patient crossover, post-progression treatment, and dose escalation in the INVICTUS study. The sponsor’s submitted pharmacoeconomic model relies on the trial data to estimate incremental survival and thereby incremental cost-effectiveness. Given that no definitive conclusions can be drawn regarding the effect of ripretinib on OS, CADTH’s reanalysis is subject to a high degree of uncertainty. This uncertainty is compounded by the extent of clinical benefits predicted by the model beyond the INVICTUS trial period.
CADTH could not address the high degree of uncertainty around OS due to the limitations identified of the available clinical data. As the sponsor’s model depends on OS, there remains a high level of uncertainty in any reanalyses.
Survival extrapolations likely overestimate incremental effectiveness of ripretinib: The sponsor assumed a log-logistic distribution for extrapolation of OS data beyond the observation period of the INVICTUS trial. When using this distribution, the 19% of patients receiving ripretinib were still alive at 5 years, while all patients receiving BSC were dead. Clinical experts consulted by CADTH suggested that the sponsor’s base-case extrapolated survival curves for both ripretinib and BSC were higher than expected and lacked face validity.
In reanalysis, CADTH used a Weibull parametric function to extrapolate OS for both ripretinib and BSC, based on feedback from clinical experts.
Model structure may overestimate comparative efficacy: Results from the sponsor’s model suggested that ripretinib was associated with longer survival after disease progression. While the INVICTUS trial demonstrated a statistically significant impact of ripretinib on PFS, there was no clear mechanism by which ripretinib would continue to provide clinical benefit after disease progression. The sponsor’s use of a PSM introduces structural assumptions about the relationship between PFS and OS that are unlikely to accurately reflect causal relationships in the disease pathway. These assumptions may produce a post-progression survival benefit that favours ripretinib. Because of the structural independence between OS and PFS end points assumed in a PSM, extrapolations for each end point may reflect within-trial trends in the rates of disease progression and death.
CADTH asked the sponsor to provide additional evidence to support the post-progression benefit predicted in those receiving ripretinib (1.88 gain in LYs; 1.52 gain in QALYs). In response to this request, the sponsor stated that a post-progression survival benefit was plausible, given that patients with GIST managed with BSC were likely to progress quickly. The sponsor further noted that post-progression survival data were confounded by the presence of post-progression treatment in the placebo arm of the INVICTUS trial. However, the CADTH clinical review team noted that there is high uncertainty concerning the OS benefit due to lack of statistical testing and failure to adjust for post-progression treatment beyond progression in the submitted clinical data. Therefore, CADTH notes that there remains considerable uncertainty surrounding the extent to which the post-progression benefit was due to treatment with ripretinib versus structural bias in the PSM.
CADTH could not address this limitation in reanalysis. Consequently, incremental QALYs may be overestimated.
Treatment beyond progression is not adequately adjusted for: The sponsor’s pharmacoeconomic model uses cost and survival data from the INVICTUS trial. Patients in the INVICTUS trial could receive ripretinib after disease progression and/or dose escalation. The product monograph for ripretinib does not include treatment beyond progression or dose escalation. In the sponsor’s base case, patients were assumed to discontinue ripretinib at progression but without any adjustment in survival due to treatment discontinuation (i.e., patients in the model continued to receive the treatment benefit, and the potential benefit of dose escalation, but did not incur the corresponding treatment cost). These assumptions underestimate treatment cost and introduce a bias in estimated cost-effectiveness that favours ripretinib.
CADTH addressed this limitation by adjusting for treatment beyond progression in the ripretinib arm using the 2-stage complex method with recensoring through the sponsor-provided option in the sponsor’s model.
Inappropriate utility values for the disease progressed health state: The sponsor uses utility values of 0.817 and 0.807 for progression-free and disease progressed patients, respectively, mapped to the EQ-5D-5L from INVICTUS trial data. However, clinical experts consulted by CADTH suggested that patient quality of life declines steadily after progression. Consequently, the utility value for the disease progressed health state should be meaningfully lower than that of the progression-free health state. The mean age of patients in the sponsor’s pharmacoeconomic model was 60 years. The expected corresponding utility of the general population for people of this age is 0.828.8 The implication of the sponsor’s submitted utility values is that patients with advanced GIST have health-related quality of life similar to that of the general population, both before and after disease progression. Clinical experts consulted by CADTH suggested that this was implausible. The use of the sponsor’s submitted utility values introduces a bias in favour of ripretinib. Based on the CADTH literature search, the A6181004 study was identified as a reasonable proxy for the INVICTUS trial population, with utility values 0.712 and 0.577 for the progression-free and disease progressed health states, respectively.9 This approach remains conservative, as patients receiving previous lines of treatment are expected to have a higher utility than those in fourth-line therapy due to the rapid decline in health utility experienced by patients with GIST.
In reanalysis, CADTH applied the utility values 0.712 and 0.577 for the progression-free and disease-progressed health states, respectively. The sponsor’s original utility values were included in a scenario analysis.
The application of RDI may underestimate drug costs: The sponsor incorporated a RDI of 96.50% for ripretinib, which was multiplied by the drug-acquisition cost. The RDI was calculated from the INVICTUS trial as the mean dose received per patient divided by the planned dose. The sponsor’s approach to estimating drug-acquisition costs does not consider other factors that influence dosage. Dosage may vary based on delays in dosing, dose reductions to manage toxicity (reduction to 100 mg once daily as per product monograph), and dose escalation (150 mg twice daily), as observed in INVICTUS trial. The sponsor’s exclusion of these dose-altering options in its modelling introduces uncertainty in deriving drug-acquisition costs for ripretinib.
In its reanalysis, CADTH assumed an RDI of 100% for ripretinib, while including the sponsor’s RDI assumptions in a scenario analysis.
Additionally, the sponsor made the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Treatment continuation beyond disease progression is not standard practice in Canada, and ripretinib is not expected to be continued beyond disease progression. | Uncertain. Clinical experts consulted by CADTH recommended that treatment with ripretinib would continue well beyond progression until inability to swallow in palliative care or death, since it is the last line of therapy available to patients with GIST. CADTH was unable to explore an extended duration of treatment in the sponsor’s model. Furthermore, the pivotal trial includes a large number of patients in the open-label period who dose escalated, which is not representative of the Health Canada indication. |
Two-stage method used to adjust for treatment beyond progression assumes no unmeasured confounders at the point of secondary baseline. | Uncertain. The complex model included time to progression, ECOG performance status, quality of life, and age as covariates. There are likely to be important prognostic factors that are missing from this complex model, which introduces bias with an unclear direction or magnitude. At the time of crossover, there may also have been unmeasured differences at the secondary baseline between those who crossed over and those who did not. |
A gamma distribution was used to represent probabilistic uncertainty for drug-acquisition costs. | Inappropriate. These costs are unlikely to vary and should not be included in probabilistic sensitivity analyses. |
All patients experiencing Grade 3/4 abdominal pain will be hospitalized. | Inappropriate. This overestimates resource use costs for both treatment arms. The sponsor’s overestimation introduces a bias against ripretinib due to a slightly higher incidence of abdominal pain in the ripretinib arm. Clinical experts consulted by CADTH suggested that 50% or fewer of these patients would likely need to be hospitalized for abdominal pain. CADTH could not address this limitation because of lack of flexibility in the sponsor’s model, but notes that the impact on the ICER is minimal. |
ECOG = Eastern Cooperative Oncology Group; GIST = gastrointestinal stromal tumour; ICER = incremental cost-effectiveness ratio.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. Changes to the sponsor’s analyses are summarized in Table 5 and include alterations to the OS extrapolation, adjustment for treatment beyond progression, utility values, and RDI.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Parametric OS modelling | Log-logistic function | Weibull function |
2. Treatment beyond progression | No adjustment for treatment beyond progression in the ripretinib arm; 2-stage crossover adjustment with recensoring is used to adjust for treatment beyond progression in the BSC arm | Adjustment was made for patients receiving treatment beyond progression in the ripretinib arm and BSC arms using the sponsor-provided option of 2-stage crossover adjustment with recensoring |
3. Utilities | Utilities collected from the INVICTUS trial using the EQ-5D-5L instrument were used for the progression-free and disease progressed health states (0.817 and 0.807, respectively) | Alternate utilities from the A6181004 study were applied for the progression-free and disease progressed health states (0.712 and 0.577, respectively) |
4. Relative dose intensity | Relative dose intensity was 96.50% for the ripretinib arm, which was used to determine treatment costs | Relative dose intensity was set to 100% in the ripretinib arm for calculating treatment costs |
CADTH base case | — | Reanalyses 1 + 2 + 3 + 4 |
BSC = best supportive care; OS = overall survival.
In the CADTH base case, ripretinib was associated with a total cost of $229,317 and 1.15 QALYs, compared to $18,501 and 0.28 QALYs for patients receiving BSC. Approximately 19% of the incremental QALYs in the CADTH reanalysis were accrued after the observation period of the INVICTUS trial (a decrease from 54% in the sponsor’s base case). The ICER for ripretinib compared to BSC was $242,365 per QALY, with a probability of being cost-effective at a WTP of $50,000 of 0%. Detailed information and disaggregated results are presented in Table 11, Appendix 4.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|---|
Sponsor’s base case (deterministic) | BSC | 18,940 | 0.53 | 0.43 | Reference |
Ripretinib | 237,227 | 3.13 | 2.53 | 103,943 | |
CADTH reanalysis 1: OS modelling | BSC | 18,466 | 0.43 | 0.35 | Reference |
Ripretinib | 233,553 | 2.42 | 1.96 | 132,937 | |
CADTH reanalysis 2: Treatment beyond progression | BSC | 18,940 | 0.53 | 0.43 | Reference |
Ripretinib | 234,254 | 2.42 | 1.96 | 140,771 | |
CADTH reanalysis 3: Utilities | BSC | 18,940 | 0.53 | 0.34 | Reference |
Ripretinib | 237,227 | 3.13 | 1.93 | 137,041 | |
CADTH reanalysis 4: Relative dose intensity | BSC | 18,940 | 0.53 | 0.43 | Reference |
Ripretinib | 244,726 | 3.13 | 2.53 | 107,514 | |
CADTH base case (reanalyses 1 + 2 + 3 + 4) | BSC | 18,501 | 0.43 | 0.28 | Reference |
Ripretinib | 229,317 | 1.77 | 1.15 | 242,365 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; OS = overall survival; QALY = quality-adjusted life-year.
Scenario Analysis Results
CADTH performed price-reduction analyses based on the sponsor’s base case and CADTH’s base-case reanalysis. Based on the CADTH base case, a price reduction of approximately 83% would be required to achieve cost-effectiveness at a WTP threshold of $50,000 per QALY (Table 7).
Table 7: CADTH Price-Reduction Analyses
Analysis | ICERs for ripretinib vs. BSC ($/QALY) | |
|---|---|---|
Price reduction | Sponsor’s base case | CADTH reanalysis |
No price reduction | 103,743 | 242,365 |
10% | 93,978 | 219,032 |
20% | 84,212 | 195,698 |
30% | 74,447 | 172,365 |
40% | 64,682 | 149,032 |
50% | 54,917 | 125,699 |
60% | 45,151 | 102,366 |
70% | 35,386 | 79,032 |
80% | 25,621 | 55,699 |
83% | 22,691 | 48,699 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
CADTH performed scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of ripretinib, as follows:
The sponsor’s original utility values for the progression-free and disease progressed health states were applied.
The sponsor’s original RDI assumptions were applied.
The results of these analyses are presented in Table 12, Appendix 4. The scenario analysis using the sponsor’s original utility values resulted in an ICER of $192,632 per QALY, indicating that the reanalysis is highly sensitive to uncertainty concerning the health state. The stepwise analysis results also indicate that the estimated ICER was also sensitive to assumptions concerning treatment beyond progression.
Issues for Consideration
Clinical experts consulted by CADTH noted that ripretinib would likely be administered to patients beyond disease progression until inability to swallow in palliative care or death, since it is the last line of therapy available to patients with GIST. Notably, there would be continuous costs incurred for ripretinib, but clinical efficacy is unknown in this setting. Therefore, the impact of treatment beyond progression on cost-effectiveness of ripretinib is unknown.
Similarly, the cost-effectiveness of ripretinib following dose escalation to 150 mg twice daily is unknown. In the INVICTUS trial, 41 of 71 patients in the open-label period dose escalated, meaning that drug-acquisition costs would have increased.2 The sponsor did not model dose escalation and notes that it is unclear whether treatment beyond progression confers any benefit.
Overall Conclusions
The CADTH clinical review found that treatment with ripretinib resulted in a statistically significant and clinically meaningful survival advantage of in terms of PFS compared to placebo in adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib, and regorafenib. However, definitive conclusions about improvement in OS could not be made due to absence of formal statistical testing. The OS analysis was further limited by the inability to account for patient crossover, post-progression treatment, and dose escalation in the trial data. Consequently, the effect of ripretinib treatment on patient OS is highly uncertain.
CADTH identified several limitations with the sponsor’s economic evaluation: a lack of definitive OS benefit for ripretinib compared to placebo, unrepresentative OS extrapolation, the suggestion of a post-progression benefit, accounting for treatment beyond progression in the ripretinib arm, unrealistic estimates of health state utility, and assumptions about RDI. In the CADTH base-case reanalysis, CADTH used a Weibull parametric function to extrapolate OS, applied a 2-stage complex adjustment with recensoring for treatment beyond progression in the ripretinib arm to estimate overall survival, substituted utilities determined to be more clinically relevant, and adjusted the RDI to 100%. Based on the CADTH base-case reanalysis, ripretinib was associated with an ICER of $242,365 per QALY, and the probability of cost-effectiveness at a WTP threshold of $50,000 per QALY was 0%. A price reduction of 83% is necessary to achieve cost-effectiveness at this threshold.
The cost-effectiveness of ripretinib is driven by assumptions concerning the extrapolation of OS, adjustment for treatment beyond progression, and utility benefit associated with treatment. The model was sensitive to assumptions about health state utility and the choice of extrapolation function for long-term OS. CADTH was unable to evaluate the impact of treatment beyond progression because the sponsor did not model it. As highlighted by the drug plans, there is considerable uncertainty surrounding discontinuation criteria for ripretinib. Clinical experts suggested that the decision to discontinue would depend on the prescribing physician and that some may advise continuation of treatment until inability to swallow, which is not reflected in the INVICTUS trial data. Given the lack of existing evidence to inform the benefits of continuation of treatment after progression, CADTH was unable to explore these benefits in reanalyses, but they remain a key driver of the economic model. Last, the extent to which structural bias in the PSM inflated the post-progression benefit observed in those receiving ripretinib is uncertain and could not be addressed by CADTH.
Treatment with ripretinib is more costly than BSC in adult patients with advanced GIST. CADTH’s reanalysis was unable to address the lack of clear evidence of OS benefit in the INVICTUS trial. As a consequence of this and other limitations, CADTH’s cost-effectiveness and price-reduction estimates are highly uncertain.
References
1.Blay JY, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923-934. PubMed
2.Clinical Study Report: DCC-2618-03-001. A phase 3, interventional, double-blind, placebo-controlled study to assess the safety and efficacy of DCC-2618 in patients with advanced gastrointestinal stromal tumors who have received treatment with prior anticancer therapies (INVICTUS) [internal sponsor’s report]. Waltham (MA): Deciphera Pharmaceuticals; 2019.
3.PrQinlockTM (ripretinib): 50mg oral tablets [product monograph]. Waltham (MA): Deciphera Pharmaceuticals; 2020 Jun 19.
4.Pharmacoeconomic evaluation [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Qinlock (ripretinib) 50 mg oral tablets. Toronto (ON): Medison; 2021 Oct 15.
5.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf. Accessed 2021 Nov 1.
6.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2021 Nov 1.
7.Cheung MC, Earle CC, Rangrej J, et al. Impact of aggressive management and palliative care on cancer costs in the final month of life. Cancer. 2015;121(18):3307-3315. PubMed
8.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-518. PubMed
9.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338. PubMed
10.Table: 17-10-0009-01: Population estimates, quarterly. Ottawa (ON): Statistics Canada; 2020 Dec 17: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000901. Accessed 2022 Jan 5.
11.About GIST. Cobourg (ON): GIST Sarcoma Life Raft Group Canada; 2016: https://liferaftgroup.ca/about-gist/. Accessed 2022 Jan 5.
12.Canadian Clinical Experts interview report. Toronto (ON): Medison Canada; 2021.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Treatment of Patients With Gastrointestinal Stromal Tumours
Treatment | Strength/ Concentration | Form | Pricea ($) | Recommended dosageb | Daily cost ($) | 28-day costa ($) |
|---|---|---|---|---|---|---|
Ripretinib | 50 mg | Tab | 216.3194 | 150 mg daily | 648.96 | 18,171 |
Note: All prices are from the sponsor’s pharmacoeconomic submission, unless otherwise indicated, and do not include dispensing fees.
aSponsor submitted price.
bThe recommended dosages are from the respective product monographs.3
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | CADTH identified inflexibilities in the sponsor’s model where certain calculations did not include possible alterations for frequency of use for resource costs related to hospitalization due to abdominal pain. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | The sponsor included drug-acquisition costs in probabilistic sensitivity analyses, which are typically excluded since they are not associated with uncertainty. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Sponsor’s Base-Case PFS Extrapolation for Ripretinib and BSC
BSC = best supportive care; PFS = progression-free survival.
Source: Sponsor’s pharmacoeconomic submission.4
Figure 2: Sponsor’s Base-Case OS Extrapolation for Ripretinib and BSC
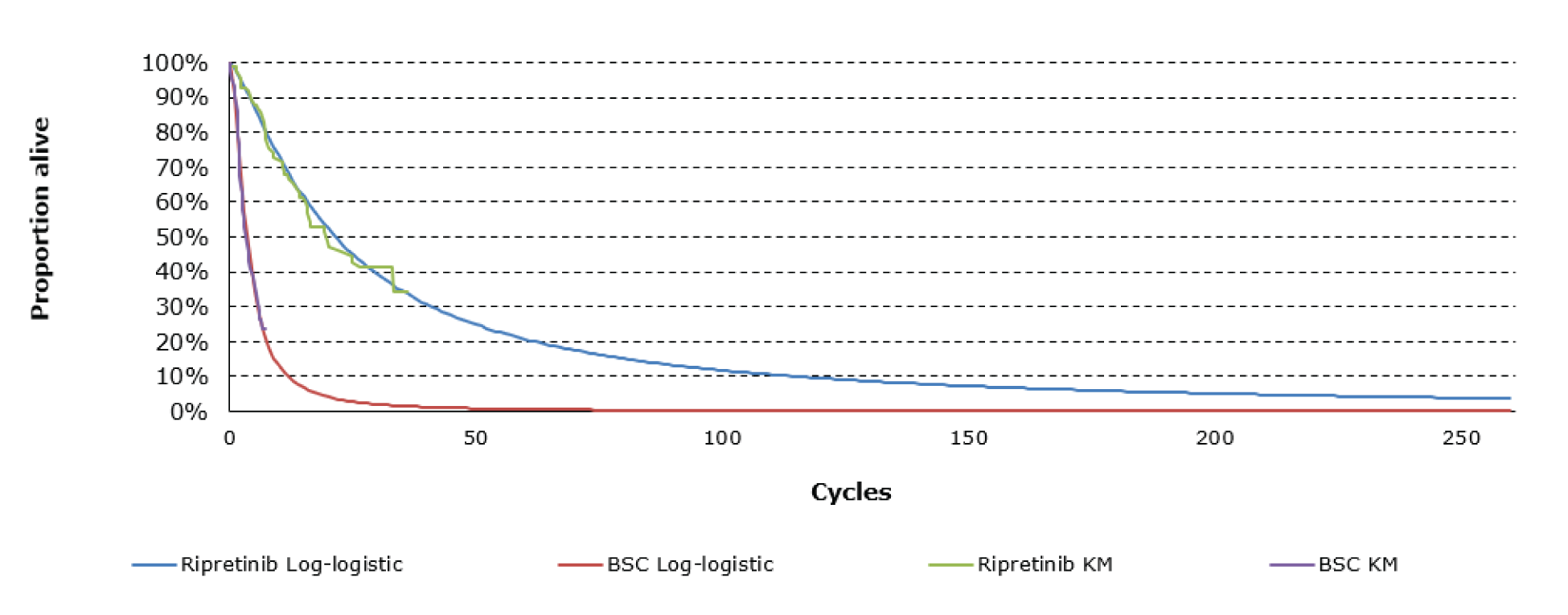
BSC = best supportive care; OS = overall survival.
Source: Sponsor’s pharmacoeconomic submission.4
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Results of the Sponsor’s Base-Case Analysis
Parameter | Ripretinib | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 3.14 | 0.54 | 2.60 |
Pre-progression | 0.93 | 0.21 | 0.72 |
Post-progression | 2.21 | 0.33 | 1.88 |
Discounted QALYs | |||
Total QALYs | 2.54 | 0.44 | 2.11 |
Pre-progression | 0.76 | 0.17 | 0.59 |
Post-progression | 1.79 | 0.27 | 1.52 |
Discounted costs ($) | |||
Total costs | 237,563 | 18,941 | 218,621 |
Treatment cost | 214,318 | 1,241 | 213,077 |
Health state cost | 8,108 | 1,395 | 6,713 |
Adverse event cost | 458 | 355 | 103 |
End-of-life treatment cost | 14,680 | 15,950 | -1,270 |
ICER ($/QALY) | 103,743 | ||
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life year.
Table 11: Concomitant Medications for Best Supportive Care Arm
Drug | % BSC | % Ripretinib | Posology | Unit dose/form | Cost per unit ($) | Units per cycle | BSC cost per 28-day cycle ($) | Ripretinib cost per 28-day cycle ($) |
|---|---|---|---|---|---|---|---|---|
Paracetamol | 60.5% | 42.4% | 500 mg 4 times daily | 500 mg tablet | $0.0285 | 112 | $1.93 | $1.35 |
Oxycodone | 34.9% | 31.8% | 400 mg daily | 20 mg tablet | $0.6001 | 560 | $117.23 | $106.75 |
Fentanyl | 9.3% | 12.9% | 50 mcg every 72 hours | 50 mcg/hr transdermal patch | $6.8838 | 9 | $5.98 | $8.29 |
Morphine sulphate | 18.6% | 11.8% | 40 mg daily | 20 mg capsule | $0.7985 | 56 | $8.32 | $5.26 |
Hydromorphone | 14.0% | 7.0% | 2 mg every 4 hours during daytime | 2 mg tablet | $0.1417 | 112 | $2.22 | $1.11 |
Amoxicillin | 11.6% | 3.5% | 250 mg 3 times daily | 250 mg capsule | $0.0672 | 21 | $0.16 | $0.05 |
Ciprofloxacin | 11.6% | 3.5% | 250 mg twice daily | 250 mg tablet | $0.4454 | 14 | $0.72 | $0.22 |
Loperamide hydrochloride | 16.3% | 8.2% | 8 mg daily | 2 mg caplet | $0.0952 | 28 | $0.43 | $0.22 |
Ondansetron | 23.3% | 34.1% | 8 mg twice daily | 8 mg tablet | $4.9930 | 14 | $16.26 | $23.85 |
Ibuprofen | 14.0% | 20.0% | 400 mg 3 times daily | 400 mg tablet | $0.0936 | 21 | $0.28 | $0.39 |
Sodium Chloride | 11.6% | 16.5% | 1,000 mL once a month | 9 mg/mL injection | $0.1500 | 1,000 | $17.40 | $24.75 |
Ranitidine | 25.6% | 31.8% | 150 mg daily | 150 mg tablet | $0.1197 | 7 | $0.21 | $0.27 |
Macrogol | 34.9% | 32.9% | 240 mL/day | powder for reconstitution | $0.0041 | 1,680 | $2.41 | $2.28 |
Docusate sodium | 11.6% | 12.9% | 100 mg daily | 100 mg capsule | $0.0328 | 7 | $0.03 | $0.03 |
Sennoside A+b | 14.0% | 12.9% | 8.6 mg daily | 8.6 mg tablet | $0.0595 | 7 | $0.06 | $0.05 |
Metoclopramide | 18.6% | 22.4% | 10 mg twice daily | 10 mg tablet | $0.0659 | 56 | $0.69 | $0.82 |
Potassium chloride | 16.3% | 12.9% | 600 mg twice daily | 600 mg tablet | $0.0899 | 56 | $0.82 | $0.65 |
Diazepam | 30.2% | 11.8% | 2 mg twice daily | 2 mg tablet | $0.0532 | 56 | $0.90 | $0.35 |
Levothyroxine | 34.9% | 29.4% | 100 mcg daily | 100 mcg tablet | $0.0416 | 28 | $0.41 | $0.34 |
Vitamins | 16.3% | 12.9% | 1 mL per week | 8,288 IU/mL solution | $0.4011 | 4 | $0.26 | $0.21 |
Total | $176.71 | $177.24 | ||||||
BSC = best supportive care.
Note: All costs, dosages, and usage information taken from the sponsor’s pharmacoeconomic report.4
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Ripretinib | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total LYs | 1.78 | 0.43 | 1.35 |
Pre-progression | 0.89 | 0.21 | 0.68 |
Post-progression | 0.89 | 0.22 | 0.67 |
Discounted QALYs | |||
Total QALYs | 1.15 | 0.28 | 0.87 |
Pre-progression | 0.63 | 0.15 | 0.49 |
Post-progression | 0.51 | 0.13 | 0.39 |
Discounted costs ($) | |||
Total costs | 229,317 | 18,501 | 210,816 |
Treatment cost | 208,551 | 990 | 207,561 |
Health state cost | 4,613 | 1,111 | 3,502 |
Adverse event cost | 460 | 356 | 104 |
End-of-life treatment cost | 15,694 | 16,045 | –350 |
ICER ($/QALY) | 242,365 | ||
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY= life-year; QALY = quality-adjusted life-year.
Scenario Analyses
CADTH performed a scenario analysis to examine the impact of applying the sponsor’s original utility values on cost-effectiveness. The sponsor’s original utility values of 0.817 for the progression-free health state and 0.807 for the disease progressed health state were applied to the CADTH base case. Lastly, CADTH performed a scenario analysis to examine the impact of including the sponsor’s original RDI assumptions on cost-effectiveness. The sponsor’s RDI of 96.5% was applied in determining costs of treatment to the CADTH base case.
Table 13: Summary of CADTH Scenario Analyses
Scenario | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CADTH base case | BSC | 18,501 | 0.28 | Reference |
Ripretinib | 229,317 | 1.15 | 242,365 | |
1. Utility values | BSC | 18,502 | 0.35 | Reference |
Ripretinib | 229,441 | 1.44 | 192,632 | |
2. RDI | BSC | 18,443 | 0.28 | Reference |
Ripretinib | 222,934 | 1.14 | 235,191 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life year.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis (BIA) estimated the introduction of ripretinib for the treatment of adult patients with advanced gastrointestinal stromal tumour (GIST) who have received prior treatment with imatinib, sunitinib, and regorafenib. The analysis took the perspective of Canadian public drug plans using a top-down epidemiological approach and incorporating drug-acquisition costs. A time horizon of 3 years between 2023 to 2025 was taken, with 2022 being the base year of the model. The target population size was estimated using the incidence of all types of GIST in Canadian adults, followed by further specifications of population size based on patients requiring non-surgical treatment options and proportion of patients progressing or failing imatinib, sunitinib, and regorafenib. A detailed summary of the sponsor’s methodology for calculating eligible target population is presented in Table 15. The reference case scenario included BSC alone (basket of medications for symptom management). The new drug scenario included ripretinib plus BSC and BSC alone. Key inputs to the BIA are documented in Table 15.
The sponsors assume that 100% of patients were assumed to be receiving BSC (basket of drugs) in the reference scenario.
Key inputs to the BIA are documented in Table 15.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
0.00163% | |
Proportion of patients requiring non-surgical treatment12 | 75% |
Proportion of patients progressing or failing on imatinib12 | 80% |
Proportion of patients progressing or failing on sunitinib12 | 70% |
Proportion of patients progressing or failing on regorafenib12 | 60% |
Number of patients eligible for ripretinib | 64 / 76 / 87 |
Market uptake (3 years) | |
Uptake (reference scenario) Ripretinib BSC | 0% / 0% / 0% 100% / 100% / 100% |
Uptake (new drug scenario) Ripretinib BSC | |||||||||| |||||||||| |
Cost of treatment (per patient) | |
Cost of treatment over lifetime Ripretinib plus concomitant BSC (6.3-month duration) Best supportive care alone (1 month duration) | $125,571 $192 |
BSC = best supportive care.
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding ripretinib for the treatment of adult patients with GIST who have received prior treatment with imatinib, sunitinib, and regorafenib was $7,974,106 in year 1, $9,478,655 in year 2, and $10,933,052 in year 3, for a 3-year total of $28,385,813.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Market shares for ripretinib are likely underestimated: The sponsor anticipated a gradual uptake of ripretinib. Given that there is no fourth-line treatment available for patients with GIST, clinical experts noted that the market shares for ripretinib were likely underestimated given clinicians’ anticipated preference for the drug when considering the high failure rates and high toxicity AEs associated with imatinib, sunitinib, and regorafenib. Both clinician and drug plan inputs indicated that ripretinib would replace BSC as the new standard of care in a fourth-line setting. Therefore, rapid uptake of this product is anticipated if it were to be made available. The clinical experts consulted by CADTH estimated the market share of ripretinib to be 90% by year 2.
CADTH increased the market shares of ripretinib plus BSC in each year included in the BIA and proportionately reduced the market shares of BSC alone.
PFS from the INVICTUS trial was used to approximate mean duration of treatment in drug-acquisition cost calculations: The sponsor used median PFS derived from the INVICTUS trial to calculate drug-acquisition costs per patient based on the recommended daily dosing schedule of ripretinib. In the base case, the median PFS of 1 month and 6.3 months for patients receiving BSC and ripretinib, respectively, were assumed to be the average duration of treatment applied in drug cost calculations. The sponsor justified this assumption by stating that all patients were required to discontinue treatment upon progression or intolerance in their respective study arms. However, treatment following disease progression occurred in the INVICTUS trial and clinical experts consulted by CADTH suggested that treatment with ripretinib would continue beyond progression until inability to swallow in palliative care or death since it is the last line of therapy available to patients with GIST. They estimated that mean treatment duration would be extended in clinical practice by approximately 3 months.
CADTH used the mean treatment duration of 8.78 months for ripretinib from Study DCC-2618-01-001 to calculate drug-acquisition costs per patient for the ripretinib arm as a scenario analysis to align with clinical expert feedback reflecting a potentially longer treatment duration that is expected in clinical practice. The mean treatment duration for BSC was changed to 1.90 months, reflecting the available data from the INVICTUS trial.
CADTH Reanalyses of the BIA
Based on the key limitations identified in the sponsor’s analysis, CADTH increased the market shares for ripretinib.
Table 16: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Market shares underestimated for the uptake scenario | Ripretinib = |||||||||| BSC = |||||||||| | Ripretinib = 75% / 90% / 90% BSC = 25% / 10% / 10% |
CADTH base case | Reanalysis 1 | |
BSC = best supportive care.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18. Based on the CADTH base case, the budget impact of the reimbursement of ripretinib for the treatment of adult patients with GIST who have received prior treatment with imatinib, sunitinib, and regorafenib is expected to be $9,967,633 in year 1, $12,186,842 in year 2, and $12,299,683 in year 3. The 3-year total budget impact for ripretinib is $34,454,158. A scenario analysis assessing the budget impact if the price of the drug under review reflected the price in which the ICER would be under the threshold of $50,000 per QALY resulted in a 3-year budget impact of $6,090,087. An additional scenario analysis applying mean treatment duration instead of median PFS led to a 3-year budget impact of $47,966,343. The mean treatment duration used was from the open-label ripretinib study and reflected an anticipated longer duration of treatment in clinical practice as advised by clinical experts consulted by CADTH. The submitted analysis is based on the publicly available prices of the comparator treatments.
Table 17: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $28,385,813 |
CADTH reanalysis 1 – market shares / CADTH base case | $34,454,158 |
BIA = budget impact analysis.
Table 18: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | BSC | $19,965 | $20,349 | $20,732 | $20,924 | $62,006 |
Ripretinib | $19,965 | $7,994,455 | $9,499,387 | $10,953,976 | $28,447,818 | |
Budget impact | $0 | $7,974,106 | $9,478,655 | $10,933,052 | $28,385,813 | |
CADTH base case | BSC | $19,965 | $20,349 | $20,732 | $20,924 | $62,006 |
Ripretinib | $19,965 | $9,987,982 | $12,207,574 | $12,320,607 | $34,516,163 | |
Budget impact | $0 | $9,967,633 | $12,186,842 | $12,299,683 | $34,454,158 | |
CADTH scenario analysis: 83% price reduction | BSC | $19,965 | $20,349 | $20,732 | $20,924 | $62,006 |
Ripretinib | $19,965 | $1,782,219 | $2,174,868 | $2,195,006 | $6,152,093 | |
Budget impact | $0 | $1,761,870 | $2,154,136 | $2,174,081 | $6,090,087 | |
CADTH scenario analysis: mean treatment duration for extended treatment | Reference | $37,908 | $38,637 | $39,365 | $39,730 | $117,732 |
New drug | $37,908 | $13,915,362 | $17,005,626 | $17,163,086 | $48,084,075 | |
Budget impact | $0 | $13,876,726 | $16,966,261 | $17,123,356 | $47,966,343 |
BIA = budget impact analysis; BSC = best supportive care.
Stakeholder Input
Patient Group Input
The CanCertainty Coalition
About CanCertainty
The CanCertainty Coalition is the united voice of more than 30 Canadian patient groups, cancer health charities, and caregiver organizations from across the country, joining together with oncologists and cancer care professionals to significantly improve the affordability and accessibility of cancer treatment.
For more information about the CanCertainty Coalition, please visit: https://www.cancertaintyforall.ca/
Information Gathering
Ripretinib is indicated for patients with gastrointestinal stromal tumours (GIST) who have progressed on previous treatments. As an orally administered oncology drug, ripretinib would not automatically be funded by certain provincial governments. In Ontario and the Atlantic provinces, only individuals over the age of 65 are automatically covered for oral oncology medication. For the small number of patients under 65 (with GIST) living in these provinces, their diagnosis could lead to severe economic hardships. However, if ripretinib were to be fully funded for all age groups, patients would instead be able to focus on their treatment and spending time with their family and friends instead of dealing with the added burden of financial hardship and difficulties in accessing treatment.
Our data collection efforts aimed to estimate the number of patients who are at risk of severe financial burden as a result of their diagnosis. To do this, we calculated the number of GIST cases in Canada each year among the under 65 population who do not have private or automatic public prescription drug coverage.
Ripretinib is a novel, highly selective inhibitor of kinases KIT and PGDFRA that helps keep cancer cells from growing (Smith, Bryan et al. 2019. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug- Resistant KIT and PDGFRA Variants. Cancer Cell, 35(5), 738–751.e9. doi:10.1016/j.ccell.2019.04.006). It is intended to supplant the use of multi-targeted kinase inhibitors that were only marginally affective against these kinases.
GIST is a rare disease. It is estimated that about 500 Canadians are diagnosed with GIST each year (GIST Sarcoma Life Raft Group Canada. About GIST. liferaftgroup.ca. Published August 21, 2016. Accessed February 17, 2021. https://liferaftgroup.ca/about-gist/).
Medison Pharma Canada Inc. used this figure to calculate the number of Canadians that will become eligible for ripretinib each year. Of the 500 diagnoses each year, they estimate that 101 patients will become eligible for ripretinib. Sixty-two of these patients will be under the age of 65; depending on where these individuals live, their oral oncology medication may not be covered by their provincial government. For the 25 patients under 65 living in British Columbia, Alberta, Saskatchewan, and Manitoba, their oral oncology medication is automatically covered. Residents of Ontario and the Atlantic provinces under the age of 65 are not automatically covered for orally administered treatments under public plans. Their route to treatment access is not simple. By our estimations, about five of these Ontario cancer patients will not have private health insurance. Before they can receive their medication these patients will have to navigate a complicated process of funding applications, approval delays, locating a pharmacy, and waiting for their prescription. They will incur out-of-pocket costs and a sizeable portion of their income may go towards their medication. This small number of patients would be unduly impacted by such restrictive treatment funding policies.
GIST is a disease that exemplifies the injustice of not providing oral oncology coverage for Canadians under 65. GIST is present in a higher proportion of under 65 cases than among the over 65 population. These younger patients (and their families) are at risk of financial toxicity if they live in Ontario or the Atlantic provinces. Furthermore, patients who are prescribed ripretinib may have already been prescribed imatinib, sunitinib, and/or regorafenib, three oral oncology medications that are also not automatically covered in Ontario and the Atlantic provinces. Throughout the course of their treatment, these patients and their families may be subject to the financial toxicity of paying for mulitple oral oncology medications.
Data Collection
CanCertainty made a formal request to Medison Pharma Canada Inc. to share incidence and prevalence data for GIST patients that would potentially meet the eligibility criteria for ripretinib. They estimate the proportion of advanced GIST patients requiring non-surgical treatment options to be about 75%. This means that three quarters of GIST patients could require oral oncology medication. Ripretinib is indicated for patients who have progressed on three previous oral oncology treatments. Medison extracted data on treatment progression from key Canadian physicians (Medison Canada. Canadian Clinical Experts Interview Report. Data on file). They estimate that 80% of patients progress or fail on imatinib, 70% of patients progress or fail on sunitinib, and 60% of patients progress or fail on regorafenib.
Therefore, the patients indicated for ripretinib are those who required oral oncology medication and have already progressed on three previous medications. This number is estimated to be 101 patients per year (excluding Quebec).
Incidence data for advanced GIST by age group in Canada is unavailable. According to the INVICTUS trial (Blay J-Y, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double- blind, randomised, placebo-controlled, phase 3 trial. The Lancet Oncology. 2020;21(7):923-934. doi:10.1016/S1470-2045(20)30168-6), of the 129 patients in the intent-to-treat (ITT) population, the mean (SD) age of the study population at informed consent was 60.1 (11.84) years, including 79 (61.2%) patients in the age range of 18 to 64 years, 32 (24.8%) patients in the age range of 65 to 74 years, and 18 (14.0%) patients ≥ 75 years of age. Medison used these percentages to calculate the number of eligible patients aged 18-65.
We measured “potential financial toxicity” using data on lack of private drug coverage. The Canadian Life and Health Insurance Association (Sutherland, Greg, and Thy Dinh. Understanding the Gap: A Pan-Canadian Analysis of Prescription Drug Insurance Coverage. Published in Canada. All rights reserved. Agreement No. 40063028 *Incorporated as AERIC Inc.) provides data on “extended health coverage.” For each province, we extracted the percentage of individuals under the age of 65 without private drug coverage AND without automatic public drug coverage. These province specific percentages were applied to the GIST case rates to arrive at the final estimation: the number of yearly GIST cases among the under 65 population without private or automatic public prescription drug coverage.
Assuming ripretinib is ultimately funded by the provinces and territories, the following chart details the number of patients in each province/territory that would be face financial barriers in accessing this treatment:
Table 1: Estimation of the Yearly Number of Gastrointestinal Stromal Tumour Cancer Patients Without Private Drug Coverage
Province | Canadian populationi | Cases of gastrointestinal stromal tumourii | Patients without private drug coverageiii | |||
|---|---|---|---|---|---|---|
Over 65 | 18 to 65 | Over 65 | 18 to 65 | Over 65 | 18 to 65 | |
Totaliv | 4,766,291 | 20,719,798 | 39 | 62 | 0 | 5.4 |
BC | 912,748 | 3,626,769 | 7 | 11 | 0 | 0 |
AB | 550,944 | 3,197,822 | 5 | 9 | 0 | 0 |
SK | 178,828 | 828,171 | 2 | 2 | 0 | 0 |
MB | 207,999 | 971,496 | 2 | 3 | 0 | 0 |
ON | 2,423,015 | 10,404,301 | 19 | 32 | 0 | 4.8 |
NB | 159,716 | 538,069 | 1 | 2 | 0 | 0.2 |
NS | 195,114 | 674,503 | 1 | 2 | 0 | 0.2 |
PE | 29,833 | 107,963 | 0 | 1 | 0 | 0.2 |
NL | 108,094 | 370,704 | 1 | 1 | 0 | 0.0 |
(i) From Stats Canada for the year 2018 to align with incidence calculations.
(ii) Age-specific incidence rates were into two groups, over 65 years old and 18 to 65 years old.
(iii) Province specific private drug coverage rates provided by The Canadian Life and Health Insurance Association.
(iv) Excluding Quebec (who do not report cancer cases in the same manner) and the territories (for whom we do not have health insurance data).
Limitations
We calculated these estimates to highlight an issue, not to be absolutely precise.
Just because someone younger than 65 does not have private insurance does not mean that they are without financial support for their oral oncology medication. In each province, multiple programs exist to support individuals with high drug costs. Based on our experience as a patient advocacy group, we made the assumption that individuals with private health insurance incur less cost when prescribed oral oncology drugs.
Our calculations are based on estimations that were not analysed for statistical significance. We did not directly measure the incidence of uninsured GIST patients. Instead we used estimated percentages to narrow down our population of interest. These calculations should be considered within the context of this report and may not be appropriate for extraction to other studies.
Disease Experience
The access problems are so difficult that in many hospitals and cancer centres across Canada, such as those in Ontario, a new type of social worker known as a drug access navigator has been established (and funded) to assist patients and clinicians navigate the byzantine treatment access structures. In Ontario, the organization that supports these navigators is known as the Oncology Drug Access Navigators of Ontario (ODANO). They describe the problem that their association works to resolve as follows: Drugs are an important part of cancer treatment, yet patients often have difficulty accessing coverage for the most effective medicines. The complexity of cancer drug coverage in Canada can overwhelm patients and families.
And
For example, although cancer drugs administered in hospitals and clinics are often offered free of charge to patients, half of all new cancer drugs are taken at home and, therefore, many are not covered by the public health system. Unfortunately, many of our patients do not have any private insurance. If a patient is fortunate enough to have private coverage, many drug plans require a 20% co-payment, which can quickly become a financial burden to patients on expensive medications.
British Columbia, Alberta, Saskatchewan, Manitoba, Quebec, NWT, Yukon, and Nunavut cover the reimbursement of oral cancer drugs for all in need. Ontario and the Atlantic provinces do not.
In Ontario and Atlantic provinces, with respect to access to approved cancer treatments, there is institutional discrimination against those who are young, uninsured and who have cancer requiring take-home cancer treatment. With 60% of all new cancer drugs being developed with oral formulations, this issue urgently needs to be resolved through policy change. Traditionally, cancer treatments were administered to patients by an IV in the hospital. Over the past 15 or so years, an increasing number of effective cancer treatments can be taken at home by pill or injection. Take-home cancer medications are now a fundamental part of today’s cancer treatments and should be recognized equally within our health care systems. Patients requiring an intravenous treatment can start that medication as soon as needed and don’t face any financial or administrative burdens provided the drug is included on the provincial formulary.
However, when take-home cancer medications are prescribed, patients in Ontario and the Atlantic provinces, who are under 65, and lack adequate private insurance, have to apply to a variety of funding assistance programs and ultimately pay a significant deductible or co-pay from their personal savings. In some cases, the cost to the patient might be as high as $23,400 annually, based upon Nova Scotia’s Family Pharmacare Program. To qualify for assistance programs, patients and their families have to submit significant amounts of personal and financial information and often face weeks of stressful delay in starting their cancer treatment until the paperwork and approvals are resolved.
Even for patients with private drug insurance, the reality is that many face significant co-pays, deductibles or annual/lifetime caps. For example, some private insurance plans have a cap of $2,000 for prescription drugs for the entire year. The majority of take-home cancer drugs cost more than $20,000 per year. Two-tiered pharmacare in Ontario and the Atlantic Provinces discriminates on the basis of age, income, geography, cancer type, and cancer treatment, and is financially ruining many lives.
A survey (Strategic Directions. Cancertainty & Strategic Directions IVR Report. 2017. Available at: https://d3n8a8pro7vhmx.cloudfront.net/cancertainty/pages/119/attachments/original/1490212245/CanCertaintySurvey_October2016 .pdf) of over 1,600 Nova Scotians, commissioned by the CanCertainty Coalition, demonstrates that drug coverage for cancer patients is a serious and growing problem.
More than half (57 percent) of Nova Scotians expect the provincial health care system will pay for take- home cancer medications. In reality, patients will ultimately pay a significant deductible or co-pay from their personal funds.
Three out of five people in Nova Scotia (60 percent) said they would consider leaving the province if faced with having to pay for their cancer drugs. Only seven percent could afford monthly drug costs of over $200.
Experiences With Currently Available Treatments
Take-home cancer drugs (THCD) are medications used for the active treatment of cancer and are usually dispensed for administration in the home (e.g., oral chemotherapy). These drugs have become a standard treatment for many cancers and present opportunities for patients, providers, and the health system. However, flaws in our current drug coverage system result in some patients not being able to access these treatments.
The term “financial toxicity” describes the distress and hardship arising from the financial burden of cancer treatment. Even in counties with government funded universal healthcare, financial toxicity is an issue for cancer patients and their families. Financial toxicity comes in many forms: out of pocket costs, lost income, travel expenses etc. Patients may deal with their financial burden by delaying or foregoing care. They may take less medication than prescribed, utilize over-the-counter drugs in place of prescribed medications, decline procedures, and skip appointments in an attempt to defray costs. The combination of high drug prices, particularly of oral targeted anticancer drugs, and increased cost sharing has made patients more vulnerable to medication non-adherence. Patients who are younger, have lower income, and are uninsured appear to be at greater risk of medication non-adherence. Although government funded public healthcare exists in many very high development index countries, financial toxicity is still common among cancer patients and caregivers. The evidence suggests that those with a shorter time since diagnosis, not currently working, and with more severe cancers have higher rates of financial toxicity, including stress and strain (Longo, C.J., Fitch, M.I., Banfield, L. et al. Financial toxicity associated with a cancer diagnosis in publicly funded healthcare countries: a systematic review. Support Care Cancer 28, 4645–4665 (2020). https://doi.org/10.1007/s00520-020-05620-9).
An unfunded oral oncology drug is financially toxic compared to a funded IV oncology drug. The disease experience of cancer patients that require oral drugs is a dual track of disease and economic hardships. After receiving their diagnosis, deciding on a medication, and dealing with the side effects, patients in Ontario and the Atlantic provinces have to consider the financial side of their diagnosis. “Hearing that you have cancer is devastating. Finding out that you can’t pay for the medication that will make you well is catastrophic. It doesn’t have to be this way” (Lisa Machado, Ontario).
The financial side of cancer treatment is unnecessarily burdensome. “When you are going through any kind of sickness, whatever the severity of it, the last thing you should have to worry about is your medication cost” (Ed, Ontario). In addition to dealing with cancer, and not being well enough to work, patients in Ontario and the Atlantic provinces spend days on end, sometimes months, wading through paperwork in order to get approval for coverage of the oral chemotherapy that has kept them alive. Because some cancer treatments are not automatically funded, treatment is delayed for many patients. They wait weeks for government approval before dealing with insurance companies and pharmacies to receive their prescription. Patients often pay out of pocket for the first few weeks of their treatment, which they may not be reimbursed for. “My doctor prescribed a new drug that is not covered by the government therefore I had to find insurance to cover it which costs around $5000.00 a month, I came up with insurance to cover it but I had to pay the pharmacy first then the insurance would reimburse me some time later. My problem I do not have the $5000 to pay out let alone wait till they reimburse me” (Sharon, Ontario).
“Cancer isn’t fair, but access to treatment should be!” (Tammy, Ontario).
Experience with Drug Under Review
CanCertainty’s focus for this submission is on issues related the distress and hardship arising from the financial burdens associated with cancer treatment. If ripretinib were to be reimbursed for patients with GIST who have progressed on previous treatments, there would be some patients under 65 in Ontario and Atlantic Canada that would face significant financial and administrative barriers in accessing treatment.
Companion Diagnostic Test
N/A
Anything Else?
Equitable Access
We recommend that pCODR, when assessing and reporting on implementation issues with respect to ripretinib, examine the issues of equitable access across all Canadian jurisdictions.
Safety
With respect to implementation, we believe pCODR should also examine the issue of safety with respect to take-home cancer drugs. From 2006 to 2001, it is estimated that Ontario’s computerized provider entry system, the Oncology Patient Information System (OPIS) prevented 8,500 adverse drug events, 5,000 physician office visits, 750 hospitalizations, 57 deaths, and saved millions in annual healthcare costs. But, this system is only used for only IV Drugs (eHealth Ontario. Cancer Care Ontario and eHealth Ontario Partner to Deliver Safer Chemotherapy Treatment. Toronto, ON: 2011. Available at: https://ehealthontario.on.ca/en/news/view/cancer-care-ontario-ehealth-ontario-partner-to-deliver-safer-chemotherapy). As a result, patients requiring take-home cancer drugs (THCD) in Ontario are (currently) subject to significant safety challenges, and health systems are subject to significant annual costs (physician office visits, hospitalizations etc).
In Ontario, dispensing and delivery models for THCD have been documented to be inconsistent and pose serious safety concerns for patients and their families. Some patients receive their medication from hospital pharmacies, some from specialty pharmacies, and some from community pharmacies that lack specialization and training in the handling of toxic cancer medications. This contrasts with the robust guidelines and clear processes that have been developed for intravenous cancer drugs (IVCD) where delivery is more comprehensive, organized, safer and patient-centred than THCD. There are numerous known safety and quality deficits related to the current method of community dispensing of THCD including incorrect dosing and handling, limited monitoring and non-adherence (which can lead to under or overdosing), serious toxicity, morbidity, and mortality. Patient lives and well-being are at stake. Ontario urgently needs to reform its systems for THCD dispensing that embed high-quality, safe practices that recognize the unique aspects of these drugs.
In April 2017, Cancer Care Ontario organized the Oncology Pharmacy Task Force with the mandate to advise Cancer Care Ontario (CCO) on how to enhance the current system for THCD delivery to optimize quality and safety; and subsequently, to deliver a report to the Ministry of Health and Long-Term Care (MOHLTC) based on the findings of the Task Force. The Task Force included representatives from patient advocacy groups, pharmacy and pharmacist associations, regulatory and standard setting organizations, and subject matter experts. On March 25th, 2019 the report was completed and published on the CCO website, but there has been no follow up or action taken to the many important recommendations. The report Enhancing the Delivery of Take-Home Cancer Drugs in Ontario (March 2019) can be found at: https://www.cancercareontario.ca/sites/ccocancercare/files/guidelines/full/1_CCO_THCD_Report_25Apr2019.pdf
CanCertainty suggests that pCODR examine the issues of safety and dispensing when examining and reporting on issues concerning pan-Canadian implementation of ripretinib.
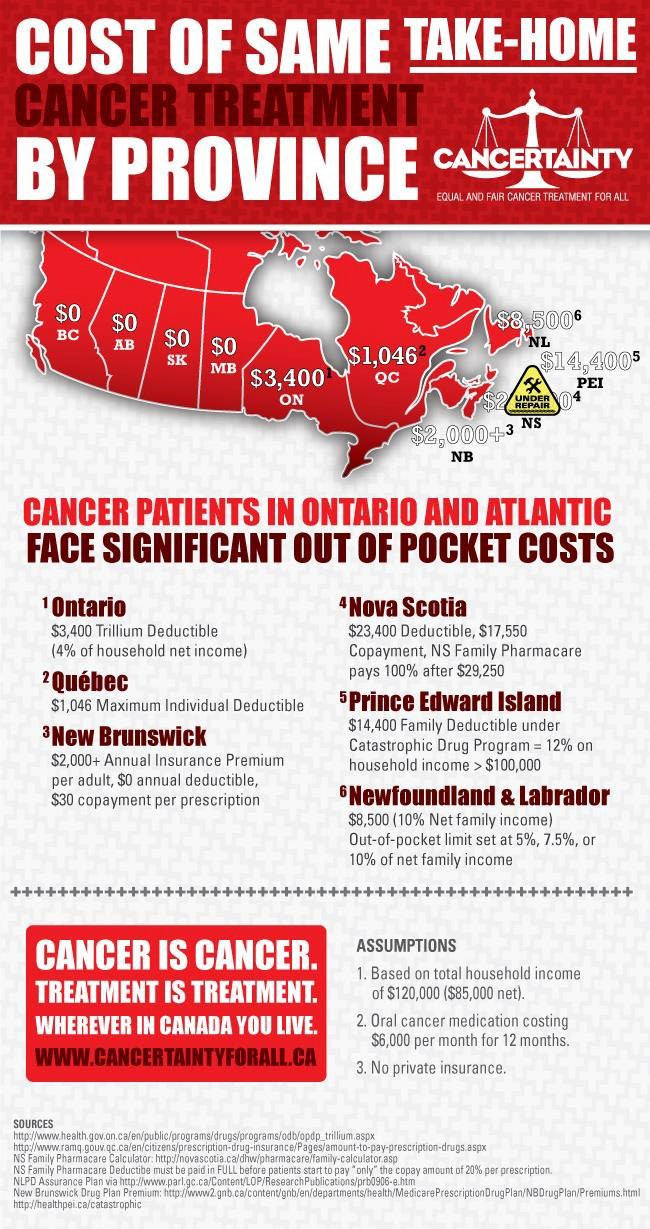
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
This submission was completed exclusively using CanCertainty resources and personnel and contract personnel.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Data was collected and analyzed using CanCertainty personnel/contract personnel.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 2: Conflict of Interest Declaration for The CanCertainty Coalition
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
None declared | — | — | — | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: CanCertainty
Date: Nov 5, 2021
GIST Sarcoma Life Raft Group Canada (LRGC)
About GIST Sarcoma Life Raft Group Canada (LRGC)
GIST Sarcoma Life Raft Group Canada (herein ‘LRGC’) is a registered national patient organization dedicated to supporting Canadians living with gastrointestinal stromal tumour (GIST), a rare life threatening cancer that imposes a number of challenges on the patient and their family members. The organization’s mission is to ensure the survival of patients diagnosed with GIST while optimizing their quality of life.
Information Gathering
To help capture the critically important patient perspective for the therapy under review, ripretinib, LRGC reached out to Filomena Servidio-Italiano from Blue Ribbon Project Inc. to commission its services for the purposes of project managing the ripretinib patient input submission. Acting on behalf of LRGC, Blue Ribbon Project Inc. developed a comprehensive GIST patient telephone interview questionnaire that was employed throughout the patient telephone interviews (please see attached APPENDIX for the qualitative data captured from patients). On September 22nd, 2021, Blue Ribbon Project Inc. respectfully reached out to 5 Canadian GIST clinicians and the Life Raft Group U.S.A, for assistance identifying patients who had/have experience with ripretinib, and who would be kindly willing to participate in a telephone interview to share that experience for the Canadian ripretinib HTA submission. On the same date, a similar outreach was made to advanced GIST patients within Canada, who had been identified by LRGC, to determine if they would be willing to provide their valuable experience with ripretinib for the patient evidence submission via the telephone interview process. Eleven advanced GIST patients were identified through these three outreach methods, all having experience with the therapy under review. Telephone interviews took place between September 29th, 2021 and October 9th, 2021 inclusive, with each patient providing first hand, compelling, relevant and high quality input regarding their:
experience with respect to the diagnosis of their GIST,
experience with respect to their GIST journey,
experience with respect to the drug therapies administered prior to ripretinib,
experience with respect to ripretinib.
Five Canadian patients and six American patients provided input with a mean age of 60.5 years and a median age of 62 years. There was a disproportionate number of males in comparison to females interviewed: 8:3. The qualitative data from the patient interviews is summarized and represented entirely in the attached APPENDIX (TABLE 1 AND TABLE 2) and will serve as the basis for this qualitative submission.
Disease Experience
Gastrointestinal stromal tumours (GISTs) are a rare type of tumour most commonly located in the stomach and small intestine but can develop anywhere throughout the gastrointestinal tract, such as the esophagus, rectum or elsewhere in the abdominal cavity. The majority of the patients who were interviewed were diagnosed with a GIST in the stomach (4) and in the small intestine (5) and the balance were diagnosed with a GIST in the colon (1) and connective tissue (1).
GISTs are aggressive tumours that have historically portended a poor prognosis because in advanced GIST, the primary tumour silently proliferates and disseminates to other organs in the body, most commonly the liver and peritoneum. Our interviewed patients had a wide range of modes of presentation, from asymptomatic to metastatic at diagnosis, depending in part on the anatomic organ in which their GIST originated. Their GIST-induced symptoms varied in extent depending on location of the primary tumour. For example, interviewed patients who were symptomatic reported experiencing the following symptoms prior to their diagnosis:
Vomiting
Abdominal pain/discomfort
Bowel issues, which included diarrhea
Black stools
Horrible fatigue
Loss of appetite/early satiety
Since these symptoms are nonspecific, obtaining a GIST diagnosis became quite the challenge for some of our interviewed patients who experienced a significant delay in the healthcare system when turning to their primary care provider or other entrusted health care professional:
“I did have symptoms of abdominal pain, vomiting, bad fatigue, diarrhea on and off, for what seemed like years before I was diagnosed. And then finally, I landed in the hospital for a week but they never found my GIST. Imagine. ..” Patient G
“I had been experiencing pains in my abdomen so I went to St. Joe’s who thought I might have a twisted bowel syndrome. They sent me home but the pain was really bad so I went to William Osler who did an x ray of the abdomen and found a mass and they kept me in the hospital.” Patient K
Nine of the eleven patients reported experiencing cancer induced symptoms that varied in severity from debilitating and challenging, thereby requiring an emergency room visit, to mild discomfort and concerning, thereby warranting a visit to their primary care physician. Patient B discovered his GIST quite unexpectedly, as an incidental finding from routine bloodwork that led to additional investigations:
“I went for routine bloodwork and when the results came in, I was asked to repeat that bloodwork. When those results came in, the doctor then sent me for a CT scan. And it was in that CT scan that something suspicious was picked up. I was then sent for a PET scan where a cantaloupe size tumour in my abdomen on the left side was sitting on the tail of my pancreas. It had originated in the stomach apparently.”
The same was true of Patient H whose small bowel GIST was quite accidentally discovered through a routine medical exam as a requirement to permit him to continue his service in the Peace Corps.: “…They were looking at my prostate but actually detected the GIST as an incidental finding in my small intestine. They saw this tumour, probably the size of a goose egg, that was picked up by that ultrasound…”
Once patients are officially diagnosed with GIST, the initial treatment for their disease is surgical removal of the primary tumour, provided it is localized and resectable, often followed by adjuvant treatment with imatinib (Gleevec®). Seven of the eleven interviewed patients did have their GIST surgically resected when they received their diagnosis. The balance (Patients B, G, J and K), however, proceeded to first line imatinib because their disease was discovered at an advanced stage (metastatic) which was not amenable to surgical resection. These four advanced GIST patients had little in common other than the diagnosis of their GIST. Patient B experienced no symptoms prior to receiving his diagnosis; Patient G had been experiencing symptoms “of abdominal pain, vomiting, bad fatigue, diarrhea on and off for what seemed like years before I was diagnosed.” Patient J “hadn’t been feeling well for about 4 days and couldn’t eat anything. That’s how long it took for the onset of the symptoms’….prior to his diagnosis. And Patient K had been experiencing pain in his abdomen which necessitated two visits to the hospital ER within 3 days prior to receiving his diagnosis. All four patient cases highlight the sudden onset of symptoms correlating to the aggressive nature of GIST and the need to seek medical attention for the onset of those abrupt symptoms.
The seven GIST patients, who underwent surgical resection, eventually did experience a recurrence which required systemic treatment with first line imatinib therapy to help regress their disease. Interviewed patients whose metastatic GISTs were imatinib resistant/intolerant (n=6) then accessed second line sunitinib to help control their disease. And four patients reported having accessed Regorafenib in the third line setting after having progressed on both imatinib and sunitinib. All eleven interviewed patients were either initially or eventually diagnosed with metastatic GIST and accessed ripretinib in 2nd to 5th line of therapy.
Interviewed patients were asked if they had any GIST-induced symptoms before starting ripretinib with which they struggled. Three of the eleven patients reported some rather troubling cancer induced symptoms which significantly compromised their quality of life. These symptoms included: abdominal pain (Patient A), back pain and bloody stools (Patient B), diarrhea and vomiting/nausea (Patient G), all of which were consistent with GIST symptomology. All patients had experienced disease progression prior to starting ripretinib and were quite thoughtful in providing input regarding the therapies accessed to control their disease prior to ripretinib. Please see Section 4 below.
Experiences With Currently Available Treatments
Tyrosine kinase inhibitors (TKIs) are the drugs (imatinib, sunitinib and regorafenib) used to target very specific proteins (tyrosine kinases) such as KIT and PDGFRA that are responsible for the growth and spread of the disease. TABLE 1 of the attached APPENDIX contains a detailed summary of each patient’s treatment journey. TABLE 2 located within the same APPENDIX (page 26) is a summary of the patient reported side effects organized according to the drug therapy accessed (imatinib, sunitinib, regorafenib and ripretinib) and is reported according to the severity of treatment-induced toxicity (severe, manageable or minor). Thirty nine treatment induced side effects were identified based on the patient interviews performed, and have been captured in TABLE 2.
The eleven patients who were interviewed were each identified with metastatic GIST. Hence, they each accessed first line imatinib for the treatment of their disease, hoping that a sustained and durable response would ensue. While durable responses did ensue for a number of patients (n=9) Patients B, C, D, G, H, J and K (n=7) experienced the most debilitating side effects while undergoing imatinib therapy. Each patient’s respective quality of life was significantly compromised as each endured ghastly treatment induced side effects.
“I had horrible side effects on Gleevec. I had swollen eyes, cramps in my legs, pain in my side , trouble with constipation which wouldn’t resolve for days and days.” Patient B
“With respect to Gleevec, my quality of life wasn’t very good. The diarrhea was disabling. You can never be far from a bathroom.” Patient C
Other patient reported imatinib side effects were: reduced kidney function, abdominal cramping, nasal congestion, leg edema, fatigue, anemia, loss of appetite, poor wound healing, and hypothermia, all of which prevented patients from participating in their life with true engagement, zeal and joy.
Evidence suggests that response to imatinib is not experienced by all patients and most patients with GIST will ultimately develop resistance to imatinib, most commonly due to the development of secondary mutations in KIT. Speaking to the former point, for example, Patient B derived no benefit whatsoever from imatinib therapy. He accessed imatinib in both the first and third line setting hoping his recurrent metastatic disease would respond but there was no benefit observed. In his words: “I had no control over my cancer, my GIST, it did what it wanted to do. I used to get a lot of chills from my cancer and the therapies too. It just kept progressing despite being on those therapies. That was the most difficult part of it.” Patient B learned of the benefits of tumour mutational analysis through LRG USA. After having a sample of his recurrent disease analyzed, he learned that his second GIST contained secondary mutations which could not be targeted through imatinib. The secondary mutations accounted for the lack of response to not only imatinib, but sunitinib and regorafenib as well for this particular patient. He was devastated to learn he had undergone multiple lines of therapy, accompanied by significant toxic side effects which he endured needlessly.
Similarly, Patient K received imatinib as neoadjuvant therapy and did respond which permitted him to proceed to surgical resection. When the patient experienced a recurrence one year later, imatinib was accessed with no response. “When I was on Gleevec the second time (first line therapy), I was only on it for 3 months. We had already started to notice progression which is why we requested the 2nd opinion.”
For patients who develop progressive disease on imatinib, sunitinib is the indicated second line treatment. Seven of the eleven interviewed patients (Patients A, B, C, D, E, H, I) accessed sunitinib in the second line setting, with the exception of one patient who accessed it in the third line setting (Patient E). All patients reported experiencing horrific and debilitating side effects while undergoing therapy with sunitinib. They found it difficult to tolerate this therapy, citing blisters under their feet, high fevers at night, body aches and pain as just some of the toxicities that compromised daily living. According to Patient D:
“As for Sutent, that was extremely toxic. I had really, really bad diarrhea constantly and bad mouth sores. My kidney numbers were down in the 30s. My blood work was not great overall and I had wicked foot blisters that made it so hard to walk. I had hand and foot syndrome. I couldn’t walk. It was so bad.”
These same sentiments and feelings of helplessness and distress were echoed by the balance of patients who accessed sunitinib (Patients A, B, C, D, E, H, I). While some of these patients did manage to secure a durable response while on sunitinib, (Patient A=4 years; Patient E=14 months; Patient I=3 years), it is important to consider the effect of the therapy on a patient’s quality of life and without question, according to the patients’ input, their quality of life while undergoing sunitinib therapy was woefully lacking.
“Then I went on Sutent….I had a hard time managing the things I needed to do in life overall such as grocery shopping, housework, cooking, self-bathing and hygiene, the simple things in life that we take for granted but are really necessary.” Patient I
Additionally, of noteworthy importance, is the fact that not all advanced GIST patients respond to second line sunitinib, as was the case for PATIENTS B, D, and H. Due to the extensive heterogeneity of the disease, there exists an unmet need for therapies designed to show activity against a broad spectrum of mutations. Neither imatinib nor sunitinib are designed to inhibit the full spectrum of known mutations in KIT and PDGFRA.
Regorafenib (Stivarga®) was accessed by four of our interviewed patients (Patients A, B, D and H) in the third line setting as a treatment option for people with metastatic GIST whose disease had progressed on prior treatment with imatinib and sunitinib (please note that Patient B accessed regorafenib in the fourth line setting). All four patients reported debilitating side effects while being on regorafenib despite having spent a relatively short time on the drug therapy (3 months, 6 months, 4 months, 1.5 months respectively), and sadly none of the patients reported having benefited from the treatment. Regorafenib-induced side effects included: cramping in extremities, abdominal cramping, hand and foot syndrome, knee pain, elevated blood pressure, skin rash, diarrhea, fatigue, GI bleed, dry mucosal membranes, and eye pain. Patients were disheartened to learn of the lack of response and their quality of life was significantly impacted to the point where they were unable to function because they were physically unwell and debilitated. They were quite emphatic about their experience with regorafenib, despite the fact that it was short lived:
“And Stivarga was the worst of them all but I didn’t respond on that drug so I was immediately taken off that drug after 3 months.” (Patient A)
“Stivarga was the worst of them all. I had pain in my eyes and knees, cramps everywhere, my blood pressure was out of control, blisters on my feet which made it impossible to walk, couldn’t walk at all from the couch to the bathroom, my skin broke out in a horrible rash (brown spots on my body everywhere) and I had to take a biopsy of those terrible spots. This was not an easy procedure for someone my age.” (Patient B)
“And Stivarga…..diarrhea was bad, several times a day, every day. My body was depleted, and I couldn’t take it anymore. I wanted to stay on the full dose of the drug. I did but it didn’t work! I was so sad. Overall, what kind of quality of life is this? What kind of life is this?” (Patient H)
The patient-reported input overall was heart wrenching; but the regorafenib- reported data was particularly gut-wrenching because it highlighted the patients’ overwhelming desire to access a therapy that might provide some small benefit for their aggressive and rare type of cancer, amidst the almost certain diminished quality of life patients were prepared to endure for that small clinical benefit.
As patients were interviewed, they were asked to rate their treatment-induced side effects for each therapeutic according to severity (severe, manageable or minor) and this included the therapy under review as well. The 39 side effects reported by the 11 interviewed patients were captured in TABLE 2 under the attached APPENDIX. Please note that the severity of the treatment induced side effect is represented by shade of the symbol appearing under the respective therapy (O, O, O, respectively) with the darker symbol representing the severe side effect and the lightest symbol representing the minor side effect. The legend is provided at the bottom of the table. The number of patients who reported treatment-induced side effects for each of the four therapies appears at the top of that respective therapeutic column. While this exercise has not been deemed to be statistically validated, we do wish to highlight that ripretinib had no patient reported severe treatment-induced side effects but did have the highest number of patient reported minor side effects (per patient) associated with the therapy under review. No other therapy had any reported minor side effects. Please see TABLE 2 (page 26) of the attached APPENDIX.
Improved Outcomes
All interviewed patients provided their perspective on the improvements they would wish to see associated with a new therapy – improvements that are currently not available with standard of care therapies for the management of advanced GIST. They passionately expressed the following: a desire to access a therapy that would promote good quality of life while effectively reducing their disease for several years, if not forever. Nine of the eleven patients focused heavily on being able to access a treatment that could be free of toxicities, allowing them the freedom to live their life without the constant and painful reminder they are a cancer patient actively undergoing cancer treatments.
According to the data captured in TABLE 1 of the attached APPENDIX, patients would wish to see improvements in:
Extension in survival, if not a cure altogether
The drug’s toxicity profile, inducing no side effects
The drug’s long term effectiveness
The drug’s ability to target the patient’s specific type of GIST (“I guess a therapy that targets my cancer i.e. SDH- D GIST, nothing really helps people like me in a meaningful way…” PATIENT G)
According to our patients, accessing a therapy that can prolong life significantly according to a wide variety of tumour mutations, with minimal to no side effects that promotes quality of life, is what would significantly ameliorate their lives. It would allow them to resume normal activities , be gainfully employed, spend time with their friends and families and would permit them the freedom to “live life well”. Furthermore, nine of the eleven patients maintained that ripretinib currently possesses these desired improvements and were grateful to have been able to access this remarkable therapy. Patients B and D provided the following input respectively when asked if they believed ripretinib had the desired improvements:
“Yes, I do! Compared to other treatments, it sure has. I am so much better on Qinlock and others should benefit from this drug too because of it.”
”I 100% do! Having had experience with other drugs, I don’t even feel I am on a drug right now with Qinlock. In comparison, it is so much better than the other treatments I have been on.”
GIST is a complex disease and the majority of patients who initially respond to traditional tyrosine kinase inhibitors, such as imatinib, sunitinib and regorafenib, eventually develop not only tumour progression due to secondary mutations, but also long term side effects from having accessed those respective therapies. Our interviewed patients certainly spoke to both the former and, more importantly, the latter – long term side effects which continue well beyond treatment cessation: “The Gleevec impacted my kidney function. …and till this day, they really haven’t come back to normal levels.” Patient A. “My kidney function deteriorated (on Gleevec) and I was considered to have chronic kidney disease.” Patient D
Experience With Drug Under Review
After having interviewed the eleven advanced GIST patients, it became copiously clear from their thoughtful input provided that a significant unmet medical need exists for this patient population: Despite currently available therapies, the majority of metastatic KIT-driven GIST patients will experience multiple mutations that cause their disease to progress, causing them to become resistant to existing therapies as these current therapies fail to inhibit all known mutations (Patients A, B, C, D, H, I). Additionally, based on our patient input, some patients will not respond to currently approved targeted therapies, causing their tumours to continue to grow and progress in an uncontrolled manner (Patients B, F, K). Hence, while approved kinase inhibitors control certain initiating and drug resistance- causing mutations in KIT and PDGFRA, the kinases that drive disease progression in most GIST patients, and the complex heterogeneity of KIT mutations within individual tumours and individual patients, are definitely a major cause of resistance to existing drug therapies. This became abundantly evident through the patient interview input as highlighted below.
The attached APPENDIX contains TABLE 1 which includes the demographics and ripretinib-related experiences for 11 patients diagnosed with advanced GIST. Four U.S.-based patients accessed the therapy through the special access program, two U.S.-based patients accessed it through a clinical trial setting, four Canadian patients accessed it through a clinical trial setting and one U.S. patient accessed it through their private insurance. All patients accessed the therapy with great anticipation and hope for they had either exhausted standard of care therapies which included 3 lines of tyrosine kinase inhibitors, or had been recommended to enroll in a clinical trial comparing ripretinib to a standard of care tyrosine kinase inhibitor (sunitinib) and were fortunate enough to receive ripretinib, known to target the broad spectrum of KIT and PDGRFA mutations that drive GIST progression.
One patient experienced no side effects while undergoing ripretinib therapy, except for slightly elevated blood pressure. The balance of patients experienced relatively mild side effects or side effects which were extremely manageable and patients were grateful to be on the therapy “whose side effects were fewer and far less toxic than previously accessed therapies.”
“I experience the occasional cramp like a Charlie horse, in my fingers and legs but not bad at all, and not every day, like previously. It is just occasionally. And these side effects are much better. I have had some hair loss. These side effects are far more tolerable in comparison to the previous side effects I had to endure.” Patient B
“My hair had fallen out. My body hair is less, some of which I have welcomed. My skin is delicate. I have calluses on my feet so I have to keep them moist and soft. My gums bleed. I do have sleep issues and I get tired between 2 and 3 p.m. during the day. These are very manageable and not intrusive at all. I can do everything I wanna do in life. Qinlock has been so good for me. There are days I can forget I have GIST.” Patient C
The balance of patients experienced side effects which included: hair loss - body and head (and hair turning white), mild nausea, mild fatigue, mouth sores, Hand & Foot Syndrome, foot calluses, curly/kinky hair regrowth, skin lesions, elevated blood counts. Patients considered these side effects to be quite tolerable and relatively minor in comparison to previously administered therapies and rated their quality of life with high scores of either: 7 (n=2), 7.5 (n=1), 9 (n=4), 9.5 (n=1) or 10 (n=3), generating an average score of 8.8. Except for one patient, all patients maintain that ripretinib has delivered a clinically meaningful response wherein their disease has regressed significantly or achieved stability while providing them with an excellent quality of life:
“My response has been unbelievable. Qinlock has been wiping out everything in my body, it has literally been melting my cancer. In the first 6 months, it destroyed everything in there! In my most recent scan, the September scan which was just a couple weeks ago, it showed just one liver met that puffed up a bit and another one under the liver that had just slightly increased but all other tumours have been destroyed. So, I am off to surgery one months time because of Qinlock!” Patient C
Six patients (Patients A, B, C, D, E and G) cited how much easier the therapy was to use in so far as it had allowed them to achieve a superior quality of life when compared to previously administered therapies:
“Oh yes, it most certainly was. The side effects were almost nil.” Patient A
“The other therapies were oral too, but they had horrible side effects. This medicine is great with respect to side effects. With the other medicines, you could see the pain in my face and I also had to get up at night because of the cramps and pain. But Qinlock is the best therapy so far when compared to the others so yes, it has definitely been easier to use because of “no collateral damage.” Patient B
“100% yes! I actually feel human and normal on Qinlock. I never felt normal on the other drugs. I am not swollen or in pain and I can do stuff. I can walk and I feel good.” Patient D
“Yes, because it has been easier on me overall. …the drug is giving me a great quality of life and that’s why it’s been easier to use with no additional painstaking efforts to manage the side effects for example.” Patient G
The number of side effects with ripretinib compared with other TKIs is summarized in TABLE 2 in the attached APPENDIX. While it is impossible to make firm conclusions regarding differences between agents because there are likely differences in patient characteristics, disease type and response to each TKI, there are definitely some noteworthy observations regarding the number of reported severe toxicities. There appeared to be differences in the incidence of:
Hand Foot Syndrome (also reported as feet blisters) (highest with sunitinib)
Fatigue (highest with imatinib)
Nausea (highest with imatinib)
Diarrhea (highest with imatinib/sunitinib)
Alopecia (highest with ripretinib)
The majority of reported minor side effects were associated with ripretinib, whereas the majority of reported severe side effects were associated with imatinib.
Three patients (Patients A, B, and G) struggled with GIST induced symptoms prior to starting ripretinib therapy and in each case the therapy provided significant resolution of those symptoms. In Patient A’s case, their abdominal pain resolved despite the fact that there was no radiographic evidence to support clinical efficacy for ripretinib. Patient B was experiencing back pain and bloody stools prior to starting the therapy but these symptoms completely resolved according to the patient input. Patient G had debilitating diarrhea, nausea and vomiting which was ameliorated upon initiation of the therapy and the patient reported feeling immediately better and no longer spending an inordinate “amount of time on the toilet”. Patients J and K provided some rather curious replies to Question 17 when asked if they had been experiencing any cancer symptoms before starting ripretinib therapy. Their thoughtful and provocative replies were as follows:
“Ummm…Other than the progression, which Qinlock was able to stop and regress, no!” Patient K
“It is keeping me alive after my disease progression, is that a cancer symptom? As far as I am concerned, that is a symptom. I was given 2 years to live and if it were not for Qinlock, last Christmas was supposed to have been my last Christmas and I was supposed to have died last January 2021. Qinlock has saved my life! I am here because of Qinlock. My family is very grateful. My enemies, well, not so much.” Patient I
Two patients experienced disease progression while on ripretinib therapy and were therefore required to stop the therapy quite early on (Patients A and E). From the remaining 9 patients, two patients experienced a treatment interruption (Patients F and J) for five and two days respectively due to a flu and liver function test irregularities. The balance of the patients have been undergoing therapy for extended periods of time, reflective of sustained and durable responses: 10 months, 9 months, 14 months, 15 months, 25 months, 14 months, 21 months, 20 months, and 5.5 years. Efficacy was radiographically confirmed in each patient’s case through either CT or PET. Patients repeatedly expressed their appreciation to be accessing an easily administered oral therapy that can be taken in the comfort of their homes that has remarkably improved their quality of life, regressed their disease, and in some cases allowed patients to proceed to surgical resection in the metastatic setting. All interviewed patients expressed profound disappointment with having accessed previous therapies that either failed to successfully treat their cancer and caused indescribable pain, suffering and anguish, or in accessing those therapies that helped to regress their cancer, had to endure the unimaginable and debilitating side effects that ensued from those therapies. Patients believe the therapy has been and continues to be their lifeline, a “miracle” drug without which they would not be alive today. They credit their longevity and ability to function at an almost normal level entirely to ripretinib:
“Well, I can probably say I have been able to see the birth of my grand daughter. How do you put a price tag on that? It is one of those moments in life that most people aspire to and give thanks for. I was able to experience that because of Qinlock. I was also able to travel comfortably because of no diarrhea even though we had covid. I can have lunch with my girlfriends and I had a reunion which was fantastic, all because of Qinlock. Look what I was able to do and more because of this therapy. I have my life back. I am so appreciative and grateful. And I get to go to surgery and aspire for more and more. it’s really a gift.” Patient C
“So, here I was feeling well, and I couldn’t really celebrate much last year but this year, it has been a miracle because of Qinlock for me. Like celebrating milestone birthday. I got to reach and celebrate my 80th birthday because I managed to make it to 80 years of age because of being on Qinlock. What a blessing. We were all together for that. I got to celebrate with my grandchildren who are so near and ear to me. I went on a formal vacation right next to Vancouver island (San Juan island in Washington). So overall, I would say in a year, I managed to do quite a bit at my ripe old age because of this miracle drug.” Patient I
Additional patients (Patients B, D, E, F, G, H, J, K) were not only able to resume what they considered to be a “normal” life while undergoing ripretinib therapy, but were also able to fulfill and accomplish a great deal while on the therapy. They mention being able to travel, oversee businesses, care for young families, see the birth of their grandchildren, perform volunteer work, see their young children reach milestone ages, spend quality time with long lost family and friends, be in attendance for the loss of a parent and settle that parent’s estate, and so much more (Please see TABLE 1, Q23). This life-altering therapy has been repeatedly referred to as a “gift” and a “miracle” because it has offered significant life extending properties while improving the patient’s quality of life. Additionally, it is worth mentioning that adverse reactions resulting in permanent discontinuation of the drug occurred in 0% of patients, dosage interruptions due to an adverse reaction occurred in 0% patients and dose reductions due to an adverse reaction occurred in 0% of our interviewed patients who received ripretinib. Patients were overcome with gratitude and emotion throughout the interviews when speaking of their experience with ripretinib.
Companion Diagnostic Test
Ripretinib currently meets the growing need in Canada for a cutting edge therapy in the treatment of advanced GIST, as it inhibits the full spectrum of primary and secondary mutations, which drive resistance and disease progression in patients who have exhausted 3 or more lines of therapy. There is no currently approved companion diagnostic test required to proceed to ripretinib. Patients who have confirmed radiographic disease progression are identified and offered the therapy by their treating oncologist.
Interviewed patients did, however, provide input regarding the need for upfront mutational analysis testing and the implications the results may have for treatment selection. KIT and PDGFRA mutational analysis may be of great assistance in predicting responses to kinase inhibitors for patients with unresectable, metastatic or recurrent GIST who are undergoing therapy with selective TKIs in earlier settings. However, mutational analysis has not yet been incorporated into treatment guidelines despite the ever present need. Patients expressed the need to access upfront mutational analysis testing to avoid, for example, unnecessary treatment toxicity in the event a therapy is deemed ineffective due to the identification of a mutation, such as the PDGFRA exon 18 (specifically PDGFRA D842V mutation). Tumours that carry this specific mutation rarely respond to first line imatinib. And, in the case of Patient F, whose SDH-deficient GIST did not respond to current drug therapies approved to manage advanced GIST, perhaps molecular classification of GIST would have proven helpful to inform optimal therapeutic selection in the management of this highly heterogeneous pathology.
Anything Else?
Despite the progress that has been made over the past few years in developing treatments for advanced GIST, a subset of the advanced GIST patient population will fail to respond to the TKIs, leaving their cancer in an uncontrolled state of perpetual growth and spread. Also, for the advanced GIST patients who do derive clinical benefit from TKIs, the majority of these patients will experience disease progression due to secondary resistance mutations. Based on our patient input, for some patients, the TKIs administered in the second and third line setting were not very tolerable or were accompanied by significant adverse side effects, resulting in dose reductions, interruptions or discontinuation altogether.
Due to the extensive heterogeneity of advanced GIST, there exists an unmet need for therapies designed to show activity against a broad spectrum of mutations. As appropriately stated by our interviewed patients, ripretinib showed activity in patients who had progressed after three TKIs. Since multiple types of secondary mutations can develop in TKI-resistant GIST, treatments that inhibit as many mutant genes as possible are highly sought after by patients in order to block both the group of mutant genes that are present and those that may arise subsequently. Our interviewed patients maintain that ripretinib has the potential to target primary and secondary mutations which is why their cancers responded as well as they did, including GISTs resistant to imatinib, sunitinib and regorafenib. “My first GIST was identified to have a KIT exon 11 mutation. But my second GIST was identified to have a KIT exon 17 mutation. They were clearly different GISTs.” Patient B
For all these reasons and more, ripretinib will help to address and overcome an unmet medical need for metastatic GIST patients beyond the third line setting. This much needed therapeutic option should be provided for these patients for whom there are existing agents with only limited benefit or no approved treatments.
The eleven interviewed patients provided thoughtful and compelling examples of why ripretinib was worth accessing. Their priorities, values and preferences were consistently echoed throughout TABLE 1 as to how this therapy managed to transform their lives as highlighted by the following accomplishments: Patient B was able to make it to his next birthday because of ripretinib – a birthday he was told he would not live to see. He is alive and well today, surviving but, more importantly, thriving. Patient D has been able to resume travel with his wife leaving the cold winters of Michigan behind them. Ripretinib therapy made this a reality. He and his wife have assumed operation of their businesses because his quality of life has improved considerably. Patient G is able to volunteer his services for a non profit because his physical endurance has improved significantly on ripretinib therapy. According to the patient, one of his greatest accomplishments has been being in attendance for his daughter’s 13th birthday celebration which was a dream come true for him, one he never thought possible – all because of ripretinib. Patient K has been able to take vacations, work – truly work and be a functioning member of society – and contribute meaningfully on so many levels, celebrate birthdays, travel the world, take care of his family, all because of ripretinib to which he has been successfully responding for over 5 years. The balance of patients had similar compelling and moving experiences on ripretinib.
They have all been afforded the luxury of additional time with family, friends and coworkers, time they claim they would not have otherwise been afforded had they not accessed ripretinib. Patients repeatedly stipulated they have been provided with the “gift of hope”:
“It’s a hands down improvement to the medicine I was on previously… Just the quality of life is vastly improved compared to Gleevec. It has definitely given me a hope that it will continue to shrink my tumours and be effective at prolonging my life. .. so there should be no question about making this medicine available to patients.” Patient G
“It’s just about what we all want – more time with our families and when you get the time, there is more of a chance of another drug coming along to give you more time again. That’s what it’s all about – HOPE.” Patient I
“It gives other patients the hope they are looking for. Do not deprive them of that….” Patient J
The patients who received ripretinib reported significant improvements in health status while undergoing ripretinib therapy with respect to physical function and overall quality of life. According to patients, ripretinib has a favorable toxicity profile and yielded improvements in patient-reported quality of life compared to previously administered TKIs. Patients expressed their profound gratitude for being on this therapy because, for the most part, it delivered a robust, durable, safe, and effective response compared to previously accessed therapies with a substantially favorable toxicity profile that was repeatedly stressed throughout the captured data. To deny advanced GIST patients access to this highly effective drug would be a shame. Most of our interviewed patients had exhausted/failed previous treatments for their advanced GIST or had accessed ripretinib in earlier lines of therapy. Ripretinib demonstrated a level of benefit unlike any other previously accessed treatment in these patients because it allowed them to resume a “normal lifestyle”. Additionally, to have observed a benefit in the GIST wild type patients (Patient F) is also quite noteworthy for there are relatively few effective treatment options available for this subset of the patient population. To have observed the magnitude of responses in our interviewed patients overall, who had either progressed following prior early lines of treatment or who had no remaining acceptable alternative treatments, confirms that ripretinib is effective and amenable for long term administration, as is the case with Patient K. Ripretinib can safely achieve antitumor activity after multiple lines of therapy, providing a wonderful and fulfilling quality of life for advanced GIST patients.
If publicly funded, ripretinib would be an extremely important therapeutic option for advanced GIST patients because according to our interviewed patients, it was associated with durable improvements in quality of life and a highly favorable toxicity profile. Funding a targeted therapeutic that inhibits primary and secondary mutations, including mutants resistant to imatinib, sunitinib, and regorafenib, aligns well with the patient perspectives captured within this submission. According to our patient input, ripretinib was effective against disease that had been deemed imatinib-, sunitinib and regorafenib-resistant. And the relative incidence of patient reported severe adverse events was 0% when compared with other TKIs, as summarized in TABLE 2. While the safety and efficacy of a treatment are indeed critically important, the effect of the therapy on the patient’s quality of life is equally important and was deemed to be outstanding according to patient input. We, therefore, strongly support and urge that a positive funding recommendation be issued for ripretinib for the treatment of adult patients with advanced GIST who have received prior treatment with imatinib, sunitinib and regorafenib. We believe ripretinib aligns well with the identified patient need for a new, effective, easily administered treatment option that is capable of maintaining a high quality of life while targeting multiple primary and secondary KIT and PDGFRA mutations in the fourth line setting.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH CDR and pCODR programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Yes, LRGC commissioned the services of Filomena Servidio-Italiano from Blue Ribbon Project Inc. to author this patient input submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Yes, LRGC commissioned the services of Filomena Servidio-Italiano from Blue Ribbon Project Inc. to oversee the planning, coordination, data collection and analysis of this patient input submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 3: Conflict of Interest Declaration for GIST Sarcoma Life Raft Group Canada (LRGC)
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Medison Pharma Canada Inc | — | — | X | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: GIST Sarcoma Life Raft Group Canada
Date: November 8, 2021
Clinician Group Input
Canadian GIST Clinicians
About Canadian GIST Clinicians
Please describe the purpose of your organization. Include a link to your website (if applicable).
This submission represents the collective perspectives of 7 Canadian clinicians who treat patients with Gastrointestinal Stromal Tumours (GIST). They collaborated to produce a thoughtful and compelling submission on the therapy under review, Ripretinib (Qinlock®). The goal is to help inform the expert committee’s deliberative process for a tumour site (GIST) in need of a therapeutic in the fourth line setting for the advanced GIST patient population. The respondents who have contributed to this submission represent an informal group of physicians who provide care for patients diagnosed with advanced GIST, some of whom are medical advisors to Life Raft Group Canada, a registered Canadian Charity dedicated to improving the lives of GIST patients throughout Canada.
The clinicians who collaborated to provide the input are as follows:
Dr. Amirrtha Srikanthan
Dr. Kevin Zbuk
Dr. Yoo-Joung Ko
Dr. Bruce Colwell
Dr. Shantanu Banerji
Dr. Jonathan Noujaim
Dr. Habeeb Majeed
Information Gathering
Please describe how you gathered the information included in the submission.
To ensure the valuable clinician perspective was captured and provided for the therapy under review, Blue Ribbon Project Inc. assisted with the coordination and preparation of the joint clinician input submission on Ripretinib. On September 23, 2021, Blue Ribbon Project Inc. reached out to 13 Canadian clinicians who treat GIST patients to gauge their interest in participating in a joint clinician evidence submission for ripretinib, 8 of whom kindly agreed to participate, half of whom had clinical experience with Ripretinib. An online clinician survey was prepared by Blue Ribbon Project and the lead author of the submission, Dr. Ko. The survey was then sent for the clinicians’ kind completion on September 28th, 2021 – October 12th, 2021. The survey was closed on October 13th, 2021, and the data was generated for review and analysis and incorporated into the submission on October 13 and 14th, 2021. The submission was sent to the clinicians for their review on October 15, 2021, seven of whom agreed to participate in this submission review process (including the lead author). The online clinician survey results are attached (Appendix A).
Current treatments
Describe the current treatment paradigm for the disease
GIST is the most common sarcoma of the GI tract. Over the past two decades, the treatment of GIST has become an example of personalized medicine in the oncology field. Imatinib mesylate remains a key treatment in both the adjuvant and metastatic setting. Many patients with advanced disease experience durable responses to imatinib but the majority of patients eventually develop imatinib-resistant disease, and few are intolerant to the medication. In Canada, sunitinib and regorafenib have been approved by Health Canada and funded in the imatinib refractory setting. Neither surgery nor radiation therapy have a significant role in the advanced disease setting. Since the results of the INVICTUS trial became available, some patients who have experienced disease progression on regorafenib in 3rd line therapy, have had access to Ripretinib in 4th line through the Special Access Program. The development of acquired mutations in the KIT and PDGFRA genes limit the activity of imatinib, sunitinib and regorafenib.
Ripretinib is designed to stabilize KIT and PDGFRA tyrosine kinases in an inactive conformation by binding to switch pocket regions and has been shown to inhibit a wide spectrum of primary and secondary resistant mutations in GIST. The INVICTUS trial demonstrated that the median progression free survival (primary endpoint) was longer in the ripretinib arm than in the placebo arm (6.3 versus 1.0 months) with an improved overall survival (15.1 versus 6.6 months). Ripretinib demonstrated an acceptable safety profile and stabilized patients’ quality of life.
Treatment goals
What are the most important goals that an ideal treatment would address?
An ideal therapy should prolong life, delay disease progression in a disease setting where there are no other treatment options. GIST patients often have significant disease-induced symptoms such as nausea, vomiting and abdominal pain with disease progression in this setting. Hence, an ideal treatment would also reduce the severity of the cancer induced symptoms. According to the clinician survey results, 100% of clinicians maintain that an ideal treatment should prolong life and reduce severity of GIST-induced symptoms.
Treatment gaps (unmet needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Although many advanced GIST patients experience clinical benefit from the existing therapies, almost all will develop refractory disease. Following treatment with regorafenib in the 3rd line setting, there are no standard treatment options in Canada. An additional line of therapy is, therefore, required for the advanced GIST patient population to help address this unmet medical need. Ripretinib, the drug under consideration, would create a 4th line setting for the advanced GIST patient population.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Patients with advanced GIST who have progressed (or are intolerant of, despite dose reductions) on regorafenib, sunitinib and imatinib have the greatest unmet medical need.
Place in therapy
How would the drug under review fit into the current treatment paradigm?
Ripretinib would be prescribed as monotherapy in the 4th line setting. There would be no shift in the current treatment paradigm.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
GIST patients should have received imatinib, sunitinib, and regorafenib prior to being offered ripretinib. They should have experienced disease progression on regorafenib or be intolerant of regorafenib despite dose reductions. Ripretinib can also be considered as 4th line therapy for advanced GIST with PDGFRA mutations.
Also, according to the online clinician survey results (Q10), physicians believe ripretinib is clinically preferable than current available treatments for the defined patient population because there are no Health Canada approved treatments in the 4th line setting. The exception may lie with a PDGFRA D842V mutation where Avapritinib could be considered through the special access program. Once imatinib, sunitinib, and regorafenib have been exhausted, however, ripretinib should be considered a reasonable fourth line treatment for the advanced GIST population.
How would this drug affect the sequencing of therapies for the target condition?
Ripretinib should be considered as 4th line therapy for the advanced GIST patient population. It would be preceded by imatinib, sunitinib and regorafenib, respectively.
Which patients would be best suited for treatment with the drug under review?
Patients with advanced GIST who have progressed on imatinib, sunitinib and regorafenib, or documented intolerance to any of these therapies despite dose modification, who have an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2, with an adequate organ function and bone marrow reserve, would be best suited for ripretinib.
How would patients best suited for treatment with the drug under review be identified?
Mutational analysis of tumor samples is not required to proceed to ripretinib. The Clinician will identify the appropriate patients. Patients who have radiologic disease progression despite lack of symptoms should be offered ripretinib.
Q8 of the clinician survey asked, “How would patients best suited for the treatment under review be identified?” The most frequently selected replies were clinician examination and diagnostic tools (such as CT, PET, MRI). One physician provided the following open-ended reply: “…needs to be physically well, with appropriate labs, and no critical/life-threatening findings on imaging.”
Which patients would be least suitable for treatment with the drug under review?
According to the clinician replies provided in the survey results (Q9), patients with poor performance status (ECOG >3) or those who cannot take or absorb oral medication due to conditions such as bowel obstruction, would be least suitable for treatment. Additionally, inadequate counts, poor renal or hepatic function, central nervous system metastases, class 2-4 heart failure, cerebrovascular accident within 6 months prior to commencing the therapy, and venous thromboembolism within 3 months prior to commencing therapy.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Not at this time. Responses based on mutational analysis from the INVICTUS trial are being analyzed. This is based on Ripretinib targeting multiple molecular alterations present in GIST, including several KIT mutations (exon 9, 11, 13, 14, 17 and 18), PDGFRA mutations in exon 18, including the D842V resistance mutation and the D816V secondary resistance mutation in exon 17. Depending upon the results of the INVICTUS trial, these mutations might be identified through mutational analysis.
100% of survey respondents ordered mutational analysis for the advanced GIST patients at time of diagnosis (Q6). Some of the barriers they experienced in obtaining mutational testing of their patients were delays in generating the patient report, funding and availability of extended mutational analysis testing.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
The outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials for this cancer. These include overall survival, progression free survival, blood work and toxicity assessment, radiologic response (CT or MR), quality of life and clinical assessment of treatment tolerance. Assessing a patient’s clinical response (e.g., improvement in pain) is critical.
What would be considered a clinically meaningful response to treatment?
A clinically meaningful response to treatment includes an improvement in survival, delay in disease progression, stabilization/reduction in disease burden, reduction in disease related symptoms (i.e. pain, nausea, vomiting) ability to perform activities of daily living, improvement or maintenance of quality of life and performance status. These are standard assessments for physicians who treat patients with advanced GIST.
One of the clinicians provided a thoughtful open-ended reply in Q21 of the survey results:
“The overall survival improvement in addition to generally well tolerated drug is very meaningful and a valuable addition for this patient population.”
How often should treatment response be assessed?
According to the survey results of Q18, all clinicians agreed that radiologic disease assessment should be performed at least every 8-12 weeks.
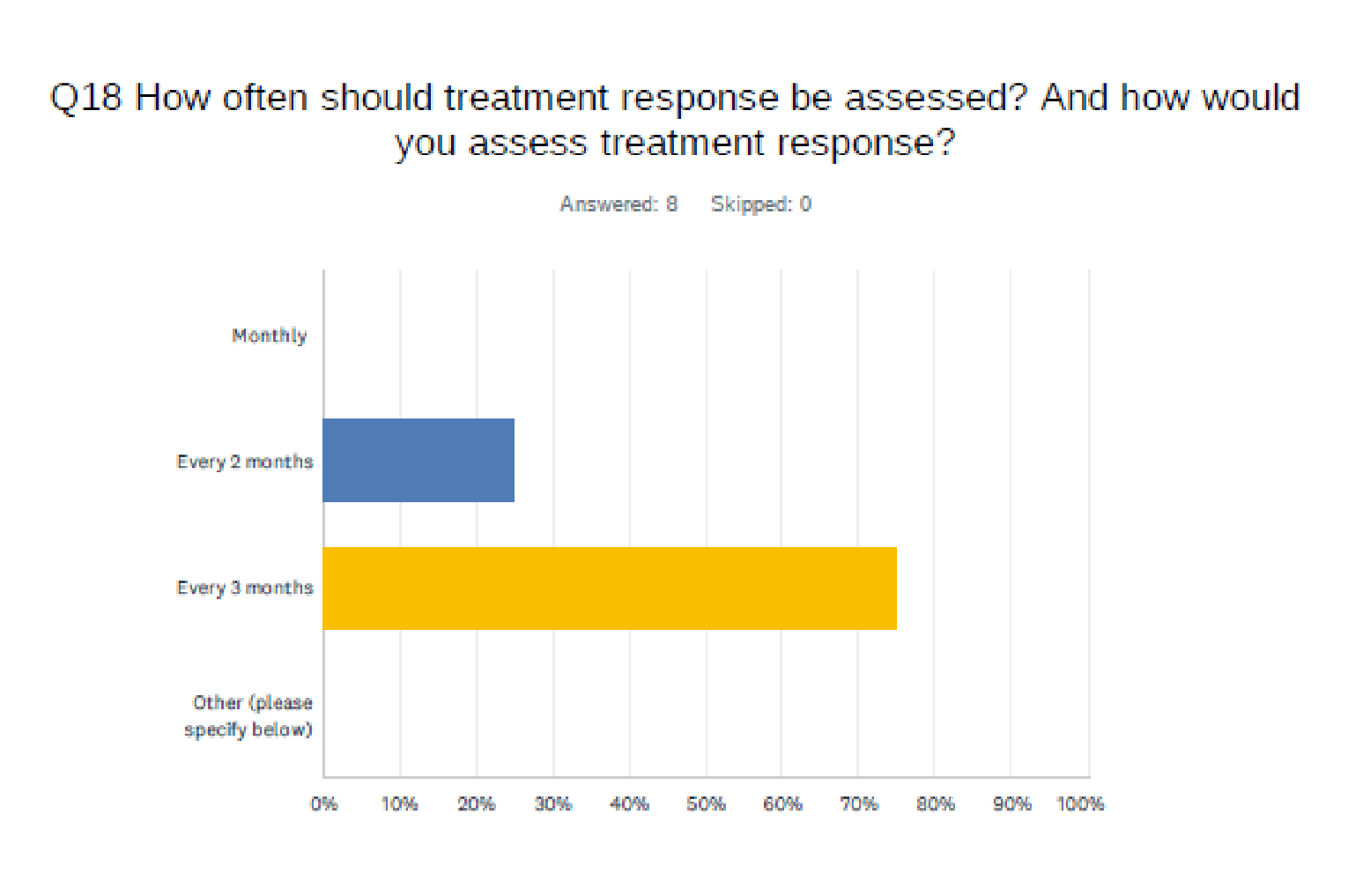
Clinical response assessment should be performed more frequently, at least q2-4 weeks.
What factors should be considered when deciding to discontinue treatment?
Numerous factors are considered when deciding to discontinue treatment and include but are not limited to clinically significant symptomatic disease progression, significant radiographic disease progression and patient specific preferences and adherence (please see Q19 of the survey results). Adverse events may include grade 3 or higher elevated liver enzymes for example. Treatment should also be stopped if there is treatment intolerance despite dose reductions.
What settings are appropriate for treatment with the drug under review?
Ripretinib can be appropriate in the oncology outpatient clinic setting – in either community or academic setting. The therapy has the benefit and convenience of oral administration, thereby providing the patient with ease in administration and a reduction in a burden to the healthcare system.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
N/A
Additional information
Is there any additional information you feel is pertinent to this review?
This therapy is an oral agent which is easily administered in the comfort of a patient’s home, minimizing clinic visits and potential hospitalizations, especially in this advanced disease setting. It is well tolerated, with a favourable side effect profile whose safety data has been regarded acceptable. Patients have the option of taking the therapy with food or fasting which is considered a convenience.
If publicly funded, ripretinib would be an extremely important fourth line therapeutic option for the small population of advanced GIST patients in Canada who have progressed on 3rd line and have no viable therapy available to therm.
Collectively, we strongly support and encourage a positive funding recommendation for ripretinib for the fourth line treatment of adult patients with GIST who have received prior treatment with imatinib, sunitinib, and regorafenib. We maintain it aligns well with the identified need for an additional, effective, quickly and easily administered treatment option that is capable of maintaining a reasonable quality of life and potential improvement in extension in life. It provides a clinically meaningful improvement in quality of life in addition to fewer adverse events and a preferred toxicity profile. As per the open-ended replies provided in the survey results (Q13):
“It has reasonable efficacy for heavily pretreated patients. Side effects profile is very manageable.”
“Well tolerated. Better than regorafenib. Oral administration is preferred by patients.”
“In my experience, Qinlock is far more efficient in controlling disease in the 4th line compared to other TKIs (carbozantinib, pazoparib)”
Respectfully, Ripretinib should become the standard of care for the advanced GIST patient population in the 4th line setting throughout Canada.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes. GIST Sarcoma Life Raft Group Canada commissioned the services of Filomena Servidio-Italiano from Blue Ribbon Project Inc. to assist with the planning, coordination and facilitation of the joint clinician input submission, its data analysis and assistance preparing this submission.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Yes. GIST Sarcoma Life Raft Group Canada commissioned the services of Filomena Servidio-Italiano from Blue Ribbon Project to assist with the planning, coordination and facilitation of the joint clinician input submission, its data analysis and assistance preparing this submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Yoo-Joung Ko
Position: Medical Oncologist/Medical Director, Oncology & Endoscopy Program, Unity Health
Date: (15-10-2021)
Table 4: Declaration for Canadian GIST Clinicians Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Medison | X | — | — | — |
Declaration for Clinician 2
Name: Amirrtha Srikanthan
Position: Medical Oncologist, The Ottawa Hospital Cancer Centre
Date: (15-10-2021)
Table 5: Declaration for Canadian GIST Clinicians Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Kevin Zbuk
Position: Medical Oncologist, Juravinski Cancer Centre
Date: 21-10-2021
Table 6: Declaration for Canadian GIST Clinicians Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Bruce D. O. Colwell
Position: Medical Oncologist, Associate Professor Dalhousie University
Date: 23-10-2021
Table 7: Declaration for Canadian GIST Clinicians Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Shantanu Banerji
Position: Medical Oncologist, Cancer Care Manitoba
Date: 24-10-2021
Table 8: Declaration for Canadian GIST Clinicians Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Medison | X | — | — | — |
Declaration for Clinician 6
Name: Jonathan Noujaim
Position: Medical Oncologist, GIST and Sarcoma Lead, Maisonneuve-Rosemont Hospital
Date: 25-10-2021
Table 9: Declaration for Canadian GIST Clinicians Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 7
Name: Habeeb Majeed
Position: Medical Oncologist, Sunnybrook Health Sciences Centre
Date: 24-10-2021
Table 10: Declaration for Canadian GIST Clinicians Clinician 7
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.