CADTH Reimbursement Review
Dostarlimab (Jemperli)
Sponsor: GlaxoSmithKline Inc.
Therapeutic area: Endometrial cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ATE
average treatment effect
BICR
blind independent clinical review
BMI
body mass index
BOR
best overall response
CCO
Cancer Care Ontario
CI
confidence interval
CR
complete response
DCR
disease control rate
dMMR
deficient mismatch repair
DOR
duration of response
DoT
duration of treatment
EC
endometrial cancer
ECOG
Eastern Cooperative Oncology Group
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EOT
end of treatment
EQ-5D-5L
EQ-5D 5-Levels
EQ-VAS
EQ-5D-5L visual analogue scale
ESS
effective sample size
FIGO
International Federation of Gynecology and Obstetrics
GOC
Society of Gynecologic Oncology of Canada
HR
hazard ratio
HRQoL
health-related quality of life
IA-1
first interim analysis
IA-2
second interim analysis
IA-3
third interim analysis
IHC
immunohistochemistry
IPD
individual patient data
IPTW
inverse probability treatment weighting
irAE
immune-related adverse event
irDCR
immune-related disease control rate
irDOR
immune-related duration of response
irORR
immune-related objective response rate
irPFS
immune-related progression-free survival
irRECIST
immune-related Response Evaluation Criteria in Solid Tumours
ITC
indirect treatment comparison
KM
Kaplan-Meier
MAIC
matching-adjusted indirect comparisons
MID
minimally important difference
MMR
mismatch repair
MMR-unk
unknown mismatch repair tumour status
MSI
microsatellite instability
MSI-H
microsatellite instability-high
MSS
microsatellite stable
MUHC
McGill University Health Centre
NCRAS
National Cancer Registration and Analysis Service
NGS
next-generation sequencing
NICE
National Institute of Health Care Excellence
NOC/c
Notice of Compliance with conditions
OH
Ontario Health
ORR
objective response rate
OS
overall survival
PD-1
programmed cell death protein-1
PD-L1
programmed death ligand-1
PD-L2
programmed death ligand-2
PFS
progression-free survival
PHA
proportional hazards assumption
PMCC
Princess Margaret Cancer Centre
PR
partial response
PSM
propensity score matching
RCT
randomized controlled trial
RECIST
Response Evaluation Criteria in Solid Tumours
RWE
real-world evidence
SAE
serious adverse event
SBHSC
Sunnybrook Health Sciences Centre
SCA
Saskatchewan Cancer Agency
SD
standard deviation
TEAE
treatment emergency adverse effect
TTD
time to treatment discontinuation
TTNT
time to next treatment
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Dostarlimab (Jemperli), solution for infusion, 500 mg/10 mL vial (50 mg/mL) |
Indication | Monotherapy for the treatment of adults with dMMR or MSI-H recurrent or advanced endometrial cancer that has progressed on or after prior treatment with a platinum-containing regimen |
Reimbursement request | Per indication |
Health Canada approval status | NOC/c |
Health Canada review pathway | Advance consideration under NOC/c |
NOC date | December 23, 2021 |
Sponsor | GlaxoSmithKline Inc. |
dMMR = deficient mismatch repair; MSI-H = microsatellite instability-high; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions.
Introduction
Endometrial cancer (EC) is the most common gynecological cancer.1 An estimated 7,000 to 8,000 new cases of EC are diagnosed in Canada every year, with approximately 1,400 deaths annually.2,3 Approximately 80% of ECs are diagnosed at an early stage and are curable with surgery.1 Recurrence occurs in 13% to 20% of patients.4,5 The 5-year survival rate of patients diagnosed with metastatic or advanced disease is less than 20%.6 Patients with advanced, metastatic, or recurrent EC have limited effective therapeutic options after front-line standard treatment with a platinum-containing chemotherapeutic.
Mismatch repair-deficient (dMMR) and microsatellite instability-high (MSI-H) tumour status is a predictive biomarker of clinical benefit from checkpoint inhibitors, and represent approximately 25% of primary ECs and 13% to 30% of recurrent ECs.7-9 At first recurrence or primary advanced disease, response rates with platinum-based combination regimens in the first-line setting ranges from 40% to 62%.10-13 However, for patients with advanced or recurrent EC who have progressed on or after platinum-based chemotherapy, there is currently no standard second-line therapy. Single-drug chemotherapies or hormonal therapy may be administered, but these have low response rates and no clear survival benefit.14
Dostarlimab is an anti-programmed cell death protein-1 (PD-1) monoclonal antibody. It targets the cellular pathway between the PD-1 receptor and 2 ligands, PD-L1 and PD-L2, found on immune cells. Dostarlimab binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, which allows the PD-1 pathway-mediated immune response and antitumour immune response to occur.15 On December 23, 2021, dostarlimab was approved by Health Canada for the treatment of adults with dMMR or MSI-H recurrent or advanced EC that has progressed on or after prior treatment with a platinum-containing regimen. The sponsor’s requested reimbursement criteria for dostarlimab align with the Health Canada–approved indication. Dostarlimab underwent review by Health Canada, which used an expedited review process (advance consideration under Notice of Compliance with conditions [NOC/c]). Dostarlimab has no other Health Canada–approved indication and has not previously been reviewed by CADTH. Dostarlimab was approved by the FDA and European Union, and is currently under review by the National Institute of Health Care Excellence (NICE) and the Scottish Medicines Consortium.16,17
Dostarlimab is available as a 500 mg IV infusion and is administered as an IV infusion over 30 minutes. The recommended dosage is 500 mg every 3 weeks for dose 1 through 4, and 1,000 mg every 6 weeks for dose 5 onward. Treatment may continue until disease progression or unacceptable toxicity. Dose reductions are not recommended, but dosing delays and discontinuation may be required based on safety and tolerability. Patients should be selected for treatment based on dMMR or MSI-H tumour status, determined by an accredited laboratory using validated testing methods.
The objective was to perform a systematic review of the beneficial and harmful effects of dostarlimab for the treatment of adults with dMMR or MSI-H recurrent or advanced EC that has progressed on or after prior treatment with a platinum-containing regimen.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Patient and caregiver input used for this review was collected by the Canadian Cancer Society. The input was based on an online survey and patient and caregiver testimonials. A total of 6 testimonials and 22 survey responses were received (20 patients with current or previous EC and 2 caregivers).
Respondents indicated that EC symptoms affected their daily activities, causing detrimental effects on their health-related quality of life (HRQoL). Respondents reported that the most significant side effects related to their current cancer treatment were issues with libido, sexual function, and fatigue. Loss of income due to absence from work and travel costs for cancer treatment were important financial barriers.
Respondents reported that they expect the following key outcomes for any treatment: better HRQoL, longer periods of remission, better affordability, better access, and fewer side effects. Eight of 22 respondents indicated that they had received direct experience with dostarlimab, either receiving it or assisting a patient who received it. All respondents indicated that dostarlimab was easier to use than other therapies because it had few to no side effects, longer intervals between doses, and a shorter infusion time.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of EC.
The clinical experts consulted by CADTH indicated that currently there are no standard, funded second-line treatment options for advanced or recurrent EC. The clinical experts agreed that patients who would most benefit from dostarlimab include those with identified dMMR or MSI-H recurrent or advanced EC. One of the clinical experts noted that dostarlimab could be used as monotherapy in first-line settings or later in the absence of effective treatments. The clinical experts noted that treatment with dostarlimab would not be suitable in patients with the following characteristics: very poor performance status; history of severe autoimmune disease; prior immunotherapy use; known uncontrolled central nervous system metastases and/or carcinomatous meningitis; poor medical risk due to a serious uncontrolled medical disorder; nonmalignant systemic disease or active infection requiring systemic therapy; or microsatellite stable (MSS) EC.
In the opinion of the clinical experts consulted by CADTH, treatment with dostarlimab should be discontinued in the case of disease progression, severe toxicity, or intolerability. The clinical experts indicated that the following outcomes would best assess response to treatment: overall survival (OS); response rate based on clinical and radiological investigation; progression-free survival (PFS); reduction of cancer burden and symptom improvement in activities of daily living; HRQoL; durability of response; and response to subsequent therapies.
In terms of clinically meaningful responses, the clinical experts recommended that in addition to clinical assessment of disease symptoms and duration of disease control, the use of standard immune-related Response Evaluation Criteria in Solid Tumours (irRECIST) for the assessment of response to immunotherapeutic treatments may be useful.
Clinician Group Input
A total of 7 clinician group inputs were submitted from the following groups: British Columbia Cancer Provincial Gynecological Oncology Tumour Group; McGill University Health Centre (MUHC), Division of Gynecologic Oncology; Ontario Health-Cancer Care Ontario (OH-CCO) Gynecological Drug Advisory Committee; Princess Margaret Cancer Centre (PMCC), Gynecologic Cancers Disease Site Group, Medical Oncology Group; Saskatchewan Cancer Agency (SCA); the Society of Gynecologic Oncology of Canada (GOC); and Sunnybrook Health Sciences Centre (SBHSC).
The views of the clinician groups were overall consistent with those of the clinical experts consulted by CADTH. The clinician groups indicated that the most important treatment goals are achieving disease control, delaying worsening of symptoms, prolonging survival, maintaining HRQoL, delaying disease progression, and an acceptable safety profile. All the clinician groups indicated that all patients with recurrent EC would benefit from an effective immunotherapy, but patients with dMMR or MSI-H subtypes would be the most likely to benefit from immune checkpoint inhibitor therapy. All groups recommended that patients diagnosed with metastatic EC should be offered platinum-based chemotherapy as first-line therapy. However, the British Columbia Cancer Provincial Gynecological Oncology Tumour Group did acknowledge that treatment with an immune checkpoint inhibitor may be an appropriate first-line therapy when chemotherapy is contraindicated or not desirable.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The drug plans identified implementation issues related to considerations for the initiation of therapy, continuation or renewal of therapy, and generalizability. The clinical experts consulted by CADTH weighed evidence from the GARNET trial (Study 4010-01-001)18 and other clinical considerations to provide responses to implementation questions posed by drug programs. Refer to Table 4 for more details.
Clinical Evidence
Pivotal Study
Description of Study
The GARNET trial (Study 4010-01-001)18 is an ongoing nonrandomized, noncomparative, multi-centre, open-label, phase I dose-escalation and cohort-expansion study in patients with recurrent or advanced solid tumours. The objective of part 2B of the GARNET trial was to evaluate the safety and antitumour activity of dostarlimab in patients with advanced solid tumours. Cohort A1 included patients with advanced or recurrent dMMR or MSI-H EC that had progressed on or after prior treatment with a platinum-containing regimen. Patients were enrolled from 123 sites in 8 countries (including 8 Canadian sites). Enrolment started on April 10, 2017, and is ongoing.
To be eligible, patients had to be at least 18 years of age, diagnosed with recurrent or advanced dMMR or MSI-H EC, and had to have progressed on or after no more than 2 lines of prior systemic therapy, with at least 1 of these being platinum-based doublet therapy. In addition, patients had to have adequate organ function and an Eastern Cooperative Oncology Group (ECOG) performance status of less than 1. The study consisted of 3 phases: the screening phase (up to 35 days before treatment), the treatment phase, and the follow-up phase. A total of 129 patients were enrolled in cohort A1. All patients received dostarlimab by IV injection (500 mg every 3 weeks for cycles 1 to 4, and 1,000 mg every 6 weeks from cycle 5 onward) for up to 2 years or until disease progression, treatment discontinuation, or withdrawal.
The co-primary outcomes of the GARNET trial were objective response rate (ORR) and duration of response (DOR). The secondary outcomes were OS, disease control rate (DCR), immune-related DCR (irDCR), PFS, immune-related PFS (irPFS), immune-related ORR (irORR), and immune-related DOR (irDOR). HRQoL was an exploratory outcome assessed by the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) and the EQ-5D 5-Levels (EQ-5D-5L). The safety outcomes assessed included treatment-emergent adverse events (TEAEs), serious adverse events (SAEs), immune-related adverse events (irAEs), ECOG Performance Status, clinical laboratory measures, vital signs, electrocardiogram, physical examination, serum pregnancy testing, and concomitant medications.
The GARNET trial has 3 planned interim analyses that are contingent on a combined enrolment of 100, 200, and 300 patients, respectively, in cohort A1 and cohort F and 24 weeks of follow-up. Only data from the second interim analysis (IA-2) was reviewed by CADTH; it included a subset of patients from the first interim analysis (IA-1) (N = 72). Data for this subset of patients is available in Appendix 4. The data cut-off dates were July 8, 2019, March 1, 2020, and November 1, 2021, for IA-1, IA-2 (N = 105), and the third interim analysis (IA-3) (N = 143), respectively. The GARNET trial was sponsored by GlaxoSmithKline Inc.
Because only 2 (< 2%) enrolled patients had mismatch repair (MMR)-unknown but MSI-H tumours (MMR-unk/MSI-H), these patients were included with patients with dMMR tumours. Median age was 64 years (range = 39 to 80 years), median weight was 71 kg (range = 34.0 to 141.4), and median body mass index (BMI) was 27.97 kg/m2 (range = 13.6 to 53.9 kg/m2). Most patients were White (> 75%). The most common histology type of EC was type I endometrioid carcinoma (67.6%), with grade 2 being the most common histology grade at diagnosis (39%). More than two-thirds (67.6%) of patients had stage IV EC. ECOG Performance Status of 1 was the most common (60%), followed by ECOG 0 (40%). All patients received prior anti-cancer treatment. Most patients (88.6%) had 2 or fewer lines of prior anti-cancer regimens, and a smaller proportion had 2 or more lines (11.5%). More than half (56.2%) of patients had received prior regimens for metastatic disease. The patient subgroups of interest, as identified in the CADTH systematic review protocol, included the following: International Federation of Gynecology and Obstetrics (FIGO) stage, histology of tumour type (e.g., type I, type II) and subtype (e.g., clear cell carcinoma), number and type of prior systemic therapies (e.g., chemotherapy, hormonal therapy), prior radiation, and progression-free interval after the most recent platinum-containing anti-cancer therapy.
Outcomes
The key outcomes from cohort A1 of the GARNET trial are summarized in Table 2. At the time of IA-2, the median duration of follow-up was 16.3 months, and the median duration of treatment (DoT) was 26 weeks.
The proportion of patients who achieved an ORR (complete response [CR] or partial response [PR]) was 44.8% (95% confidence interval [CI], 35.0 to 54.8). The Best overall response (BOR) was CR in 11 patients (10.5%), PR in 36 patients (34.3%), and stable disease in 13 patients (37.1%). The DCR was 57.1% (95% CI, 35.0 to 54.8). Of those who responded, 89.5% had an ongoing response. The median DOR was not reached, but 79% of patients who achieved an objective response had a DOR of at least 6 months.
At the time of IA-2, a large proportion of patients (66.7% and 45.7%, respectively) had no OS or PFS events. The median OS was not reached, but Kaplan-Meier (KM) estimates for the probability of survival at 6, 9, and 12 months were 80.9% (95% CI, 71.7 to 87.4), 75.1% (95% CI, 65.2 to 82.6), and 68.9% (95% CI, 58.3 to 77.4), respectively. The median PFS was 5.5 months (95% CI, 3.2 to not reached), with KM estimates of PFS by RECIST 1.1 of 48.6% (95% CI, 38.6 to 57.9) at month 6 and 47.5% (95% CI, 37.4 to 56.8) at month 9 and at month 12. In terms of HRQoL, the EQ-5D-5L visual analogue scale (EQ-VAS) and EORTC QLQ-C30 scores appeared stable over time. Summary data for the EQ-5D-5L descriptive system were not provided.
As part of the sponsor’s feedback on this CADTH reimbursement review report, the sponsor provided CADTH with a summary of the updated analysis (data cut-off November 1, 2021) for certain baseline characteristics, efficacy, and safety outcomes in the GARNET trial. The results of the updated analysis were, overall, consistent with those reported in the previous analyses performed as of March 1, 2020, data cut-off date. The additional results from the November 1, 2021, data cut-off date are available in Appendix 5.
Harms Results
A summary of the key harms reported in cohort A1 of GARNET are summarized in Table 2. Almost all patients (95.3%) experienced at least 1 TEAE. The most common serious TEAEs were abdominal pain, acute kidney injury, sepsis, pulmonary embolism, pyrexia, and urinary tract infection. Grade 3 or higher TEAEs occurred in 48.1% of patients, with the most common being anemia (14.7%), abdominal pain (5.4%), and hyponatremia (3.9%). No patient withdrew due to an adverse event (AE) as a primary reason. Study treatment discontinuation due to AEs occurred in 11.6% of patients, whereas AEs that led to study treatment interruption occurred in 24% of patients. The most common AEs leading to study interruption were anemia (3.1%) and diarrhea (2.3%).
One patient died due to a TEAE (aspiration) during the treatment period, and 4 patients died due to TEAEs (i.e., pleural effusion, pneumonia, sepsis, and shock) during the 90-day safety follow-up. None of the TEAEs leading to death were considered treatment-related, and no TEAEs were the primary cause of death during the long-term follow-up period.
The notable harms associated with dostarlimab included immune-related toxicity. The incidence of irAEs was 34.9% in cohort A1. The most frequently reported irAEs (≥ 5%) were diarrhea and hypothyroidism. A total of 7.9% of patients had a serious irAE, 12.7% had an irAE of grade 3 or higher, and 4.8% had an irAE that led to study treatment discontinuation. Most of the irAEs were considered related to the study treatment.
Table 2: Summary of Key Results From the Pivotal Study (IA-2)
Characteristic | GARNET trial Cohort A1a |
|---|---|
Data cut-off date | March 1, 2020 |
Follow-up time (months), median (range) | 16.3 (NR to NR) |
Primary efficacy analysis population | 105 |
OS | |
n | 105 |
OS (months), median (95% CIb) | NE |
Death, n (%) | 35 (33.3) |
Censored, n (%) | 70 (66.7) |
KM estimates of OS (95% CI) | |
6 months | 80.9 (71.7 to 87.4) |
9 months | 75.1 (65.2 to 82.6) |
12 months | 68.9 (58.3 to 77.4) |
DCR by BICR assessment | |
n | 105 |
DCR, n (%) | 60 (57.1) |
95% CIc | 35.0 to 54.8 |
Best response, n (%) | |
Confirmed CR | 11 (10.5) |
Confirmed PR | 36 (34.3) |
Stable disease | 13 (12.4) |
HRQoL, EORTC QLQ-C30, and EQ-5Dd | |
EORTC QLQ-C30 | |
n | 94 |
Mean change from baseline (SD) | |
Cycle 2 | 2.9 (18.06) |
Cycle 3 | 4.7 (20.96) |
Cycle 4 | 6.5 (24.43) |
Cycle 5 | 6.4 (23.51) |
Cycle 6 | 9.7 (24.14) |
Cycle 7 | 10.2 (23.77) |
EQ-5D-5L | NR |
EQ-VAS | |
n | 89 |
Mean score (SD) | |
Baseline | 69.3 (19.2) |
Week 12 | 77.1 (18) |
Week 18 | 77.4 (17.4) |
Week 42 | 77.4 (NR) |
Mean change from baseline (SD) | |
Week 12 | 5.0 (12.6) |
Week 18 | 4.0 (15.2) |
Week 42 | 4.0 (16.2) |
PFS by BICR assessment | |
PFS (months) median (95% CIb) | 5.5 (3.2 to NR) |
Events (progressive disease or death), n (%) | 57 (54.3) |
Censored, n (%) | 48 (45.7) |
KM estimates of PFS (95% CI) | |
6 months | 48.6 (38.6 to 57.9) |
9 months | 47.5 (37.4 to 56.8) |
12 months | 47.5 (37.4 to 56.8) |
ORR by BICR assessment | |
Objective response, n (%) | 47 (44.8) |
95% CIc | (35.0 to 54.8) |
BOR, n (%) | |
Confirmed CR | 11 (10.5) |
Confirmed PR | 36 (34.3) |
Stable disease | 13 (12.4) |
PD | 39 (37.1) |
NE | 3 (2.9) |
Response ongoinge | 42 (89.4) |
DOR by BICR assessment | |
DOR status, n (%) | |
Events observed | 5 (10.6) |
Censored | 42 (89.4) |
DOR (months), range | 2.63 to ≥ 28.09 |
Duration ≥ 6 months, n (%)e | 37 (78.7) |
DOR distribution function (95% CI) | |
Month 6 | 97.9 (85.8 to 99.7) |
Month 12 | 90.9 (73.7 to 97.1) |
Month 18 | 80.1 (56.8 to 91.7) |
Harms — safety analysis population, n (%) | |
n | 129 |
Any TEAEsf | 123 (95.3) |
Any SAEs | 44 (34.1) |
Any AE leading to study treatment interruption | 31 (24.0) |
Any TEAE leading to discontinuation of study treatment | 15 (11.6) |
Death during the treatment period | 6 (4.7) |
Death during the 90-day safety follow-up periodh | 13 (10.1) |
Death during the long-term follow-up periodi | 17 (13.2) |
Notable harms, n (%) | |
Immune-related reactions | 45 (34.9) |
Diarrhea | 11 (8.5) |
Hypothyroidism | 9 (7.0) |
Pruritus | 4 (3.1) |
Alanine aminotransferase increased | 4 (3.1) |
Blood creatinine increased | 4 (3.1) |
Hyperthyroidism | 4 (3.1) |
Lipase increased | 4 (3.1) |
Amylase increased | 3 (2.3) |
Aspartate aminotransferase increased | 3 (2.3) |
Colitis | 3 (2.3) |
Transaminases increased | 3 (2.3) |
AE = adverse event; BICR = blind independent clinical review; BOR = best overall response; CI = confidence interval; CR = complete response; DCR = disease control rate; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels; EQ-VAS = EQ-5D-5L visual analogue scale; HRQol = health-related quality of life; IA-2 = second interim analysis; KM = Kaplan-Meier; max = maximum; min = minimum; NE = not evaluable; NR = not reported; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PR = partial response; SAEs = serious adverse events; SD = standard deviation; TEAEs = treatment-emergent adverse events.
Note: A “≥” sign indicates that patients’ response is ongoing.
aData from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients with such tumours (3 in the safety analysis dataset and 2 in the primary efficacy analysis dataset); the sponsor noted that data remained similar when these patient groups were pooled together.
b95% CIs were generated using the method of Brookmeyer and Crowley. Biometrics, 1982;38:29-41.
cExact 2-sided 95% CI for the binomial proportion.
dNo summary results were provided for the EORTC QLQ-C30 and EQ-5D-5L scores.
eAll responders who have not yet died or progressed (including clinical progression); the denominator for the percentage is the number of responders.
fTEAEs experienced by ≥ 5% of patients with dMMR or MSI-H EC by preferred term (safety analysis dataset).
gTEAEs were experienced by ≥ 15% of patients with dMMR or MSI-H EC by preferred term (safety analysis dataset).
hWithin 90 days after the EOT visit or until the first follow-up anti-cancer therapy, whichever occurred first.
i90 days after the EOT visit or until the first follow-up anti-cancer therapy, whichever occurred first.
Source: Clinical Study Report.18
Critical Appraisal
Internal Validity
The main limitation of the GARNET trial is the single-arm design, which makes it challenging to interpret the data and determine whether the efficacy and safety events observed were attributable to dostarlimab. Formal hypothesis and statistical significance testing were not performed, limiting the ability to draw conclusions. Given that results were based on an interim analysis, some time-to-event outcomes, including median OS and DOR, were not reached due to data immaturity; therefore, the treatment effect observed with dostarlimab may be overestimated. The risk of overestimating HRQoL benefit and known subjective harms is also high, given the open-label trial design in which treatment was not blinded. To mitigate bias, the sponsor used a blind independent clinical review (BICR) to evaluate treatment response, with standardized criteria for certain efficacy outcomes (i.e., ORR, DOR, PFS, and DCR). Therefore, bias is less of a concern for these end points and OS, and more of a concern for subjective end points, including HRQoL and safety. It is also acknowledged that mature OS data will be confounded by the use of subsequent anti-cancer therapy received by some patients after progressive disease. No analyses were undertaken to account for the potential of confounding. Overall, the magnitude and direction of bias is unclear. The clinical experts agreed that in the absence of robust comparative data on PFS and OS, no firm conclusions could be drawn on how dostarlimab compares with other relevant treatment options, as causal inferences cannot be made from the results of a single-arm trial design.
HRQoL was identified as an important outcome by the patient and clinician groups providing input for this review. However, no conclusions could be drawn from the HRQoL data from the GARNET trial due to several limitations. Given the wide and overlapping CIs, the reduced number of patient responses over time, and the lack of statistical testing and a definition of what constituted a clinically meaningful response, it is not possible to draw conclusions with precision from the available data.
External Validity
Overall, the clinical experts consulted by CADTH agreed that the inclusion and exclusion criteria, baseline patient characteristics, concomitant medications, and prohibited medications present in cohort A1 of the GARNET trial were reflective of patients they see in clinical practice for the indication under review. There were no barriers to identifying patients who would most benefit from the treatment, given that testing for MMR and microsatellite instability (MSI) status is standard practice in Canada. The clinical experts indicated that no difference in treatment effect would be expected based on variation in disease-management practices across participating countries. In the opinion of the clinical experts, as long as patients have dMMR or MSI-H tumour status, dostarlimab would be appropriate to administer after any of the prior therapies received by patients in the trial. However, they noted that clinical benefit may be diminished in patients with more prior lines of systemic therapy. There were a limited number of patients included in the primary efficacy analysis dataset (n = 105) and very few patients from various ethnic backgrounds, which may reduce the generalizability of the results to a real-world practice setting. Furthermore, the subgroup analyses had no statistical comparisons and even smaller sample sizes, which limits generalizability to a broader population.
Indirect Comparisons
Description of Studies
The sponsor submitted 6 reports of indirect treatment comparisons (ITCs) — 3 reports of matching-adjusted indirect comparisons (MAICs), and 3 reports of inverse probability of treatment weighting (IPTW) analyses19 — which aimed to compare survival between dostarlimab from the phase I GARNET trial with the current treatment paradigm in advanced or recurrent EC.
Efficacy Results
The primary end point for all comparisons was OS. Other outcomes included PFS, ORR, DOR, time to treatment discontinuation (TTD), DoT, time to next treatment (TTNT), time to deterioration in HRQoL, and AEs; however, these were less frequently investigated, and outcomes specifically important to patients, including HRQoL, were not assessed. The results of the MAIC and IPTW analyses generally suggest that dostarlimab is favoured for OS over all the included comparators.
Harms Results
The sponsor-submitted MAIC and IPTW reports did not assess safety outcomes.
Critical Appraisal
Although the results of the MAIC and IPTW analyses generally suggest that dostarlimab is favoured for OS over all the included comparators, there was significant uncertainty in the results based on the clinical heterogeneity of the included populations, resulting in reduced sample sizes and wide CIs. There were important differences in the design of the comparator studies that limit the ability to draw strong conclusions about the effectiveness for dostarlimab compared with other treatments. An important limitation of all analyses was the fact that MMR and MSI-H status was unknown for all or most patients in the comparator trial, and it is therefore uncertain whether the comparator population in the ITC analyses would be eligible for treatment with dostarlimab, providing further uncertainty about the comparative effectiveness.
Conclusions
One phase I, singe-arm, open-label trial (GARNET)18 provided evidence of the efficacy and safety of dostarlimab in adults with dMMR or MSI-H recurrent or advanced EC (cohort A1) that had progressed on or after prior treatment with a platinum-containing regimen. The clinical experts consulted by CADTH felt that the response outcomes (ORR and DOR co-primary outcomes) observed in the trial were clinically meaningful and durable for this patient population and, in their opinion, were higher than what is observed with currently used second-line therapies in this setting. The trial results were based on an interim analysis; therefore, there is the possibility of overestimating clinical benefit and underestimating harms. There was uncertainty around the magnitude of the clinical benefit, given the limitations inherent in the single-arm trial design. The trial data on important long-term outcomes were immature, and interpretation of OS will be confounded by the use of subsequent anti-cancer therapies. The clinical experts noted that a randomized controlled trial (RCT) would be needed to directly compare dostarlimab with currently available therapies in the second-line setting to accurately evaluate its efficacy in this patient population. In the absence of a direct comparison of dostarlimab with relevant treatment options, the sponsor submitted multiple ITCs. However, the CADTH critical appraisal of these analyses identified significant limitations with the submitted MAICs and IPTWs, which restricted the ability to interpret the relative treatment-effect estimates obtained. Limitations of the ITCs included heterogeneity across study designs, high risk of confounding and effect modifiers, and uncertainty regarding the inclusion of dMMR or MSI-H status in the comparator groups. The results for HRQoL, an outcome important to patients and clinicians, remained inconclusive due to the lack of statistical analysis, the substantial decline in patients completing questionnaires over time, and the lack of a definition of what constituted a clinical meaningful change from baseline. The notable harms observed with dostarlimab, such as diarrhea and peripheral nephropathy, were considered manageable and consistent with other immunotherapies by the clinical experts and, in their opinion, appeared favourable when naively compared with currently available chemotherapy options. However, interpreting the safety events attributable to dostarlimab was challenging because all patients in cohort A1 received the same treatment. Overall, limitations of GARNET’s single-arm design prohibited the drawing of causal conclusions between the intervention and outcomes.
Introduction
Disease Background
EC is the most common gynecological malignancy among women in Canada.1 EC malignant tumours arise from the cells of the uterine lining. More than 95% of all uterine cancers are endometrial.20 Uterine cancer is the 17th leading cause of cancer death in Canada.3,20 The Canadian Cancer Society estimated that 8,000 women would be diagnosed with uterine cancer in 2021 and that 1,400 women would die of the disease.2,3 EC most often occurs in patients older than 50 years, with an average age of diagnosis at 60 years.21 Diagnosis of EC occurs at an early stage for approximately 80% of patients because of the early presenting symptom of uterine bleeding.1,20 The most common route of diagnosis of EC is endometrial biopsy, followed by endometrial curettage and hysterectomy specimen.1 EC uses the FIGO criteria to determine disease stage, which depends on the size of the tumour and the extent to which the tumour has spread to lymph nodes or distant sites (metastasis).22 Generally, the higher the stage number, the more the cancer has spread.23 Tumour stage is fixed, regardless of tumour type.1
The prognosis of EC is primarily based on stage of cancer, histology of the tumour, and grade. Five-year survival by FIGO stage is 80% to 90% for stage I, 70% to 80% for stage II, and 20% to 60% for stage III and IV.1 In terms of histology, there are 2 subtypes: type I ECs represent 80% of patients and are low-grade (1 or 2) endometrioid tumours,1,20,24 and type II accounts for 10% to 20% of ECs and includes grade 3 endometrioid tumours and tumours of nonendometrioid histology, such as serous clear cell, mucinous, squamous, transitional cell, mesonephric carcinosarcoma, and undifferentiated.1,20 The 5-year survival of type I is around 80% to 90%, while 5-year survival of type II EC is as low as 20%. Other notable prognostic factors for EC include race, age, uterine tumour location, peritoneal cytology results, and lymphovascular space invasion.1
Molecular testing of biomarkers during endometrial biopsy assists in the identification of treatment options and in risk stratification.1 Standard testing includes immunohistochemistry (IHC) and polymerase chain reaction. IHC is used to test for dMMR, in which the cells’ ability to repair mistakes during the division process is impaired. The tumour is immunohistochemically assessed for the loss of at least 1 of the following MMR proteins: MLH1, MSH2, MSH6, and/or PMS2.25 The dMMR proteins cause cellular hypermutations and high levels of microsatellite instability (MSI-H) in sections of DNA. If MMR status cannot be determined from tumour samples, the sample may undergo genetic testing that uses next-generation sequencing (NGS) to identify MSI status. dMMR and MSI-H tumour statuses are predictive of clinical benefit from PD-1 inhibitors and represent approximately 25% of primary ECs and 13% to 30% of recurrent ECs.7-9 Recurrence occurs in approximately 13% to 20% of patients with EC, with rates varying greatly by FIGO stage at diagnosis; rates are highest among patients with stage IV EC (> 65%).4,5,26 The prognosis of patients with recurrent EC is poor, with a median survival of approximately 12 months.4
Standards of Therapy
Treatment options for EC are dependent on stage and pathologic factors identified after initial surgery and are based on estimated risk of disease recurrence. Early-stage EC and/or type I tumour ECs can be cured with surgery alone.5,27,28 Individuals diagnosed with advanced or recurrent EC may require adjuvant radiotherapy and/or chemotherapy, depending on the extent and location of spread and/or pathologic risk factors. CCO and Alberta Health Services recommendations favour combination chemotherapy over single-drug chemotherapy for individuals with advanced or recurrent EC, as combination therapy elicits a higher response rate while maintaining acceptable toxicity levels.5,29-32 The current standard of care for patients with advanced or recurrent disease is platinum-based chemotherapy as a doublet or single drug.29 A standard echoed by the European Society of Gynaecological Oncology, the European Society of Radiotherapy and Oncology, and the European Society of Pathology.33 The most common platinum-based therapy is carboplatin plus paclitaxel.5,29,33 For a subset of patients with low-grade recurrent or metastatic EC who are estrogen- or progesterone-receptor positive or for patients with poor tolerance to systemic therapy, hormonal therapy, such as megestrol, letrozole, and medroxyprogesterone, may be used.29
For patients with newly diagnosed advanced or recurrent EC, response rates for standard first-line treatment range from 40% to 62%.10-13 However, for patients with advanced or recurrent EC who have progressed on or after platinum-based chemotherapy, there is currently no standard effective or curative second-line therapy.5,33 Patients with recurrent EC are typically re-treated with platinum-based chemotherapy with poor outcomes; response rates range from 10% to 15% for all available treatment options.33 The clinical experts consulted by CADTH noted that median survival ranges from 12 to 15 months after re-treatment. Various single-drug chemotherapies may be administered to patients who are resistant or refractory to platinum-based chemotherapy, with response rates typically below 15% and no known clear survival benefit.14 Hormonal treatments may also be used for disease control but are not considered curative.
The clinical experts consulted by CADTH for this review indicated that there is great unmet need for effective therapies with acceptable toxicity profiles that achieve disease control, reduce disease-related symptoms, improve HRQoL, prevent disease progression, and prolong survival among patients with recurrent or advanced EC that has progressed on or after prior treatment with a platinum-containing regimen. There is currently no standard effective second-line therapy for recurrent or refractory disease, and commonly used therapies are noncurative. The clinical experts anticipated more promising benefit with biomarker-driven treatments for patients with dMMR or MSI-H cancers.
Drug
Dostarlimab is an anti-PD-1 monoclonal antibody that targets the cellular pathway between the PD-1 receptor and 2 programmed death ligands, PD-L1 and PD-L2, found on immune cells. Dostarlimab binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2. This suppresses the PD-1-pathway-mediated immune response in the tumour microenvironment, enhancing a patient’s antitumour immune response.15
On December 23, 2021, dostarlimab was issued a Notice of Compliance with conditions (NOC/c) by Health Canada for the treatment of adults with dMMR or MSI-H recurrent or advanced EC that has progressed on or after prior treatment with a platinum-containing regimen. The sponsor’s requested reimbursement criteria for dostarlimab align with the Health Canada indication. Dostarlimab underwent an expedited Health Canada review process (advance consideration under NOC/c); the market authorization comes with conditions, pending the results of trials that confirm its clinical benefit. Dostarlimab has no other Health Canada–approved indications and has not previously been reviewed by CADTH.
Dostarlimab received accelerated approval from the FDA in August 2021 for the treatment of adults with dMMR or MSI-H recurrent or advanced EC that has progressed on or after prior treatment with a platinum-containing regimen.34 Dostarlimab received conditional authorization in the European Union in April 2021 for the same indication.35 Currently, dostarlimab is being reviewed by NICE and the Scottish Medicines Consortium.16,17
Dostarlimab is available as a 500 mg IV infusion and is administered with an IV infusion pump over 30 minutes.15 The recommended dosage of dostarlimab as monotherapy in adults is 500 mg once every 3 weeks for dose 1 through 4, and 1,000 mg once every 6 weeks for dose 5 onward (dose 5 occurs 3 weeks after dose 4). The product monograph states that treatment may continue until disease progression or unacceptable toxicity. Dose reductions of dostarlimab are not recommended, but dosing delays and discontinuation are permitted based on safety and the patient’s tolerability of the treatment. Patients should be selected for treatment based on MSI-H or dMMR tumour status, determined by an accredited laboratory using validated testing methods.
Key characteristics commonly used in the treatment of advanced or recurrent dMMR or MSI-H EC are presented in Table 3.
Table 3: Key Characteristics of Dostarlimab, Platinum-Based Therapy, and Hormonal Therapy Regimens
Detail | Dostarlimab | Platinum-based chemotherapies | Hormonal therapy | |
|---|---|---|---|---|
Carboplatin36 | Paclitaxel37 | Medroxyprogesterone acetate38 | ||
Mechanism of action | Anti-PD-1 IgG4 humanized monoclonal antibody | Antineoplastic drug (synthetic analogue of cisplatin) | Antineoplastic drug | Antineoplastic nonsteroidal drug with potent antiestrogenic properties |
Indicationa | Monotherapy for the treatment of adults with dMMR or MSI-H recurrent or advanced EC that has progressed on or after prior treatment with a platinum-containing regimen | Treatment of ovarian cancer of epithelial origin in first-line therapy, and in second-line therapy after other treatments have failed | Alone or in combination, for the treatment of ovarian, breast, or lung cancer | Adjunctive and/or palliative treatment of recurrent and/or metastatic EC or breast cancer in post-menopausal women. Also indicated for HRT. |
Route of administration | IV | IV | IV | Oral |
Recommended dose | 500 mg once every 3 weeks for the first 4 cycles (1 cycle = 3 weeks), then 1,000 mg once every 6 weeks for subsequent cycles (1 cycle = 6 weeks) | 400 mg/m2 given as a single IV infusion over 15 to 60 minutes in previously untreated adults with normal renal function. Therapy should not be repeated until 4 weeks after the previous carboplatin course. | 175 mg/m2 IV injection over 3 hours in combination with cisplatin 75 mg/m2 every 3 weeks is recommended for as first-line therapy for ovarian cancer. In patients previously treated with chemotherapy, paclitaxel should be administered as a monotherapy. | For EC, 200 mg/day to 400 mg/day is the usual dose. If neither subjective nor objective improvement is noted in 2 to 3 months, therapy should be discontinued. Where improvement is noted and the disease process appears to be stabilized, it may be possible to maintain this improvement with a 200 mg/day dose. |
Serious adverse effects or safety issues | Immune-related adverse reactions | Highly toxic drug with a narrow therapeutic index. Therapeutic effect is unlikely to occur without some evidence of toxicity, such as:
| Should be administered as diluted infusion. Patients should be pre-treated with corticosteroids, antihistamines, and H2 antagonists. Should not be administered to patients with baseline neutrophil counts of less than 1,500 cells/mm3. | Increased risk of myocardial infarction, stroke, invasive breast cancer, pulmonary emboli, and deep vein thrombosis in post-menopausal women. |
dMMR = mismatch repair-deficient; EC = endometrial cancer; PD-1 = programmed cell death protein-1; IgG4 = immunoglobulin G4; HRT = hormone replacement therapy; MSI-H = microsatellite instability-high.
aHealth Canada–approved indication
Source: Application overview, product monograph for dostarlimab,15 carboplatin,36 paclitaxel37 and medroxyprogesterone acetate.38
Stakeholder Perspectives
Patient Group Input
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input for the purpose of this CADTH review. The full patient input received is available in the Stakeholder Input section.
The patient and caregiver input received for this review was collected by the Canadian Cancer Society. The input was sourced from on an online survey and from patient and caregiver testimonials gathered from October 22 to November 3, 2021. Six testimonials and 22 survey responses were received (20 patients with current or previous EC, and 2 caregivers of someone with current or previous EC). Of the 22 survey respondents, 8 (6 patients and 2 caregivers) had experience with dostarlimab. Six of these respondents resided in Quebec and 2 resided in British Columbia. All patients and caregivers who had experience with dostarlimab reported receiving it through a clinical trial.
Respondents indicated a range of EC symptoms that affected their daily activities. The daily activities that were most commonly moderately or severely affected included the ability to preform household chores (46%), travel (41%), exercise (41%), work (36%), fulfill family obligations (32%) and spend time with family and friends (27%). Forty-four percent of patients were not currently being treated for EC and 39% were undergoing immunotherapy. The treatment side effects most commonly reported as having a moderate or severe impact on daily life were issues with libido and sexual function (45%) and fatigue (41%). Fifty-nine percent of patients reported a financial barrier related to their treatment; among these patients, a loss of income due to absence from work (31%) and travel costs for cancer treatment (31%) were the most common barriers.
According to the patient input received, respondents expect the following key outcomes to be improved with any new drug or treatment: quality of life, periods of remission, drug affordability, access across jurisdictions, and fewer side effects (such as skin issues, fatigue, bladder control, stamina, hair loss, pain, arthritis, vaginal dryness, vaginal bleeding after intercourse, and concentration problems). All survey respondents indicated that, compared with other therapies, dostarlimab was easier to use either because it had little to no side effects (75%), longer intervals between doses (13%), or a shorter infusion time (13%).
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of EC.
Unmet Needs
Currently, there is no standard second-line therapy for individuals with recurrent EC who have progressed on or after prior treatment with a platinum-containing regimen. This represents a critical unmet need in this patient population. Both clinical experts agreed that biomarker-driven treatments are needed to guide selection of the most effective and durable treatment option.
Place in Therapy
Currently, patients with advanced or recurrent EC receive carboplatin plus paclitaxel (another platinum chemotherapy doublet) or a single drug as first-line therapy. For a subset of patients with estrogen- or progesterone-receptor-positive indolent low-grade metastatic or recurrent EC, hormonal treatments, such as megestrol, medroxyprogesterone, letrozole, and tamoxifen, are considered. For those who have progressed on platinum chemotherapy and are considered to have platinum-resistant or refractory disease, second-line therapy with single-drug chemotherapy is considered, but it has a low expected response rate and a short DOR with no known survival benefit. Hormonal treatments may also be used for disease control but are not considered curative.
Dostarlimab will likely cause a shift in the current treatment paradigm for patients with dMMR or MSI-H metastatic or recurrent EC. The mechanism of action of dostarlimab would address the underlying disease-specific process and biomarkers for patients with dMMR or MSI-H EC. The clinical experts felt it would be preferable to initiate treatment with dostarlimab before other therapies.
Patient Population
The clinical experts agreed that patients with dMMR or MSI-H EC tumours would most benefit from an immune checkpoint inhibitor. One clinical expert added that patients with dMMR or MSI-H metastatic or recurrent EC of any histology would benefit from dostarlimab, regardless of symptoms or prior treatment; however, the other clinical expert noted that a smaller magnitude of benefit may be observed with increased previous lines of systemic therapy.
The clinical experts mentioned that the diagnosis of EC typically relies on biopsy, which may be conducted in clinic. If technical issues make it difficult to obtain samples from the endometrial lining during an office biopsy, the procedure can be performed in the operating room using dilatation and curettage. IHC testing for MMR status is relatively inexpensive and considered standard practice. If the MMR status of a sample is unknown, it may be further analyzed with genomic testing to determine MSI status. A valid test would involve NGS.
The clinical experts noted that treatment with dostarlimab would not be suitable for patients any of the following characteristics:
very poor performance status
history of severe autoimmune disease
prior therapy with anti-PD-1, anti-PD-L1, or anti-PD-L2 drugs
known uncontrolled nervous system metastases and/or carcinomatous meningitis
poor medical risk due to serious uncontrolled medical disorder
nonmalignant systemic disease or active infection requiring systemic therapy
history of immunosuppression
MSS EC.
Assessing Response to Treatment
According to the clinical experts, the most important goals of treatment for recurrent or advanced EC would be to improve OS and PFS; reduce symptoms of cancer; improve functional status (i.e., ability to perform activities of daily living); improve HRQoL; and reduce the burden of disease on patients and caregivers. One clinical expert noted that many patients with advanced or recurrent EC suffer from pelvic symptoms (e.g., unresectable disease in the pelvis causing bleeding or pain), lung symptoms (e.g., dyspnea from metastases), and neurologic symptoms (e.g., brain metastases) or bone symptoms (e.g., painful bony metastases), and that alleviation of these symptoms would be a benefit of treatment.
In terms of a clinically meaningful response, the clinical experts recommended that in addition to clinical assessment of disease symptoms and duration of disease control, the use of standard irRECIST39 for the assessment of response to immunotherapeutic treatments is useful. One clinical expert noted that the Common Terminology Criteria for Adverse Events40 tool for CT imaging can be used to assess response to treatment. The same clinical expert also suggested that cancer antigen 125 (nonspecific) may be used to assess treatment response in addition to other methods, although it is not commonly used in EC. In terms of the timing of assessments, the clinical experts recommend that response to treatment should be assessed radiologically every 3 months, with blood work every month and clinical assessments every 2 to 3 months.
Discontinuing Treatment
According to the clinical experts, treatment with dostarlimab should be discontinued when there are radiological and clinical signs and symptoms of disease progression, treatment toxicities (e.g., grade 3 or higher adverse reactions), or intolerability to treatment.
Prescribing Conditions
According to the clinical experts consulted by CADTH, the diagnosis, treatment, and monitoring of patients with EC should be undertaken by a specialist, namely a gynecologist oncologist, medical oncologist, and/or surgeon. Biomarker testing to identify dMMR or MSI-H status is also recommended.
Clinical experts recommend that dostarlimab be administered in a hospital clinic that has multidisciplinary medical supports to manage potential immune-related side effects. The clinical experts noted that treatment may also be administered in outpatient clinics.
Additional Considerations
The clinical experts noted that re-treatment with dostarlimab is possible if patients experience recurrence, as long as they had no signs of toxicity or intolerability while using the drug. If a patient completed their treatment and achieved a durable response for up to 2 years, and a significant time period elapsed before they progressed, then re-treatment with dostarlimab could be considered. The clinical experts noted that 50% of patients will have disease recurrence within 1 year of completing treatment for recurrent EC and will need to restart treatment.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
The information in this section is a summary of 7 inputs provided by the registered-clinician groups that responded to CADTH’s call for clinician input for the purpose of this CADTH review. The full clinician group inputs received are available in the Stakeholder Input section. Input was received from the following clinical groups:
British Columbia Cancer Provincial Gynecological Oncology Tumour Group
MUHC, Division of Gynecologic Oncology
OH-CCO Gynecological Drug Advisory Committee
PMCC, Gynecologic Cancers Disease Site Group, Medical Oncology Group
SCA
GOC
SBHSC, Division of Gynecologic Oncology.
Unmet Needs
The views of the clinician groups were overall consistent with the clinical experts consulted by CADTH, indicating that the most important treatment goals for advanced or recurrent dMMR or MSI-H EC are disease control, prolonged survival, delayed worsening of symptoms, maintenance of HRQoL, delayed disease progression, and an acceptable safety profile. Treatment for individuals diagnosed with recurrent EC is a critical unmet need. All clinician groups noted that although the prognosis for patients diagnosed with early-stage disease is good, for those with recurrent or metastatic EC, median OS is short. In fact, there is no effective second-line treatment for Canadians with EC. Response rates to second-line cytotoxic drugs are low (< 20%), with a median PFS of 3 to 4 months.
All clinician groups also noted that although Health Canada recently issued a NOC/c approval for pembrolizumab immunotherapy for dMMR or MSI-H EC, there is no funded access to this treatment. The lack of funding has created disparity in access to immunotherapy, as only those with insurance coverage or the capacity to self-pay can access pembrolizumab. Moreover, PMCC added that there is currently no patient-supported or compassionate-access program for immune checkpoint inhibitor therapies. The MUHC and SBHSC noted that the side-effect profile of combination therapy with pembrolizumab and lenvatinib was significant, with 66.9% of patients experiencing a grade 3 or 4 toxicity. The MUHC added that prolonging poor HRQoL with interventions associated with significant toxicity serves very little purpose.
Place in Therapy
All clinician groups indicated that all patients with advanced or recurrent EC would benefit from effective immunotherapy, but patients with MSI-H or dMMR subtypes would most benefit from treatment with an immune checkpoint inhibitor. All groups recommended that patients diagnosed with advanced or recurrent EC should be offered platinum-based chemotherapy as first-line therapy. However, the British Columbia Cancer Provincial Gynecological Oncology Tumour Group did acknowledge that treatment with immune checkpoint inhibitors may be an appropriate first-line therapy for patients who either refuse chemotherapy or for whom chemotherapy may be contraindicated or known to be poorly tolerated, provided dMMR or MSI-H status is confirmed. The MUHC further stated, “it is false economy to delay starting a highly effective treatment [dostarlimab] with minimal toxicity in this niche population.” The MUHC stated that trying second-line chemotherapy alone in this patient population is not advisable due to toxicity and lack of benefit.
MUHC indicated that when EC recurs in patients who initially responded well and dostarlimab is interrupted for any other reason, restarting treatment with dostarlimab would be appropriate. If patients progress on dostarlimab or other immunotherapies, palliative and supportive care options are considered.
Patient Population
Currently, the standard of care in Canada includes universal molecular characterization of all endometrial carcinomas for dMMR. Thus, identifying EC subtypes should not be a barrier to treatment. The British Columbia Cancer Provincial Gynecological Oncology Tumour Group and the GOC noted that patients whose EC is related to Lynch syndrome would benefit from treatment with dostarlimab, and PMCC added that those with polymerase-epsilon-mutation would benefit as well. The SCA noted that patients with EC suitable for treatment with dostarlimab should be PD-L1-naive and should not have undergone more than 2 prior treatments with a platinum-based therapy.
All clinician groups noted that patients who do not have a dMMR or MSI-H profile and/or have contraindications to immune checkpoint inhibitors are not suitable for treatment with dostarlimab. PMCC and the GOC also indicated that patients with MMR-proficient EC would not be suitable to receive dostarlimab as monotherapy treatment.
SBHSC noted that the EC patient groups less suitable for treatment with dostarlimab would be those that met all the following criteria:
no previous treatment with platinum-based chemotherapy
no dMMR seen on IHC
poor medical risk due to a serious uncontrolled medical disorder
poor performance status (ECOG Performance Status score of at least 3)41
inadequate organ function
a known immunodeficiency or current use of systemic steroids or other immunosuppressant medications.
Assessing Response to Treatment
The British Columbia Cancer Provincial Gynecological Oncology Tumour Group, COG, OH-CCO, and SBHSC noted that improvement in symptoms and physical findings related to advanced or recurrent EC (e.g., pain, bleeding, shortness of breath) would be considered a clinically meaningful treatment outcome. OH-CCO and PMCC stated that durable disease control with no adverse effects on HRQoL is an ideal treatment goal in this population. The SBHSC specified that PFS of at least 6 months would be a clinically meaningful outcome in this patient population. In addition, OH-CCO listed reduced caregiver burden as a meaningful treatment outcome.
The British Columbia Cancer Provincial Gynecological Oncology Tumour Group and the GOC also listed the following outcomes as clinically meaningful responses to treatment:
maintenance or improvement of performance status and ability to perform activities of daily living
evidence of disease regression from imaging studies
disease stabilization (in patients with good baseline performance status and few disease-related symptoms).
OH-CCO and the GOC reported that standard clinical monitoring of therapy, including a physical examination, symptom review, and intermittent CT imaging, should be used to evaluate response to treatment. The MUHC recommended that after the first 3 cycles of dostarlimab, patients should be assessed for response to treatment with a CT scan. If there is no indication of progression and patients are well, the CT-monitoring interval may be spaced out to every 12 weeks. PMCC recommended that tumour assessment by CT scan or MRI be completed every 2 to 3 cycles (i.e., every 6 to 9 weeks). The British Columbia Cancer Provincial Gynecological Oncology Tumour Group and SBHSC stated that CT imaging should occur every 12 weeks, whereas the SCA suggested imaging every 3 to 6 months. The British Columbia Cancer Provincial Gynecological Oncology Tumour Group and SCA also added that laboratory values, as well as respiratory and pulmonary status, should be assessed to ensure that treatment response is occurring without toxicity.
Discontinuing Treatment
All clinician groups indicated that treatment with dostarlimab should be discontinued if patients experience disease progression or serious toxicities. SBHSC indicated that the following side effects warrant consideration of pausing or discontinuing dostarlimab:
grade 2 anemia
pneumonitis
grade 3 colitis
grade 2 asthenia
grade 3 myalgia
pemphigoid
grade 3 increase in transaminases.
PMCC and SBHSC noted that due to the nature of immunotherapies and the possibility of pseudo-progression, patients with progression of disease on the first CT scan after initiation of treatment, but with no other symptoms, should continue treatment until further imaging demonstrates progression of disease. Similarly, the MUHC adds that in cases where disease progression occurs after 3 cycles with dostarlimab, patients should receive follow-up in 6 weeks to rule out pseudo-progression and discontinue treatment if progression is confirmed.
Prescribing Conditions
The British Columbia Cancer Provincial Gynecological Oncology Tumour Group, MUHC, OH-CCO, GOC, and SCA explained that dostarlimab is suitable to be delivered in the community and in outpatient and specialty clinics. Conversely, PMCC and SBHSC suggested that dostarlimab be delivered at cancer centres by gynecologic or medical oncologists. Both groups recommend that dostarlimab be administered in a chemotherapy suite with appropriate supervision by an oncologist familiar with gynecologic cancers and the management of immune-related adverse effects. The British Columbia Cancer Provincial Gynecological Oncology Tumour Group and the GOC noted that patients should be under the care of a treating physician or nursing staff with experience monitoring and evaluating patients for possible toxicities that may be caused by immune checkpoint inhibitor therapy. The GOC stated that this should not be a barrier to treatment because such therapies are now routinely used by most oncologists. The MUHC notes that for patients who do not live close to a treatment centre (e.g., patients in rural, remote, or Indigenous communities), it is appropriate to work closely with local physicians to share the responsibility of patient care.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. Implementation questions from the drug programs and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Consideration for initiation of therapy | |
Which patients with recurrent or advanced EC should have MMR/MSI testing? When during the treatment course should MMR/MSI testing occur? | Testing should occur for all patients with EC of any stage at the time of diagnosis. |
Are there standard definitions of dMMR and MSI-H to help define eligible patients? | IHC testing is the standard screening method to identify patients with dMMR. Screening includes identifying mutations in the following genes: MLH, MSH, and PMS. If MMR status cannot be determined, MSI status is screened using NGS. |
Are patients who received platinum-based therapy only in early-stage disease eligible to receive dostarlimab as first-line therapy for recurrent dMMR or MSI-H disease? | The clinical experts noted that it is reasonable to receive dostarlimab in this case because there is no comparative evidence yet to suggest which therapy is superior. Patient preference is taken into consideration and, at this time, treatment is usually based on availability of treatment and cost. If a patient is keen to pursue immunotherapy as first-line therapy due to its high response rate, then the clinical experts felt it would be acceptable to use dostarlimab or any other immunotherapy as a first-line treatment for recurrent EC because the disease itself has a poor prognosis. |
Are patients with dMMR or MSI-H recurrent EC with a contraindication to or no prior exposure to platinum-containing regimens eligible for treatment with dostarlimab? | In the absence of evidence, this question cannot be answered. If patients have a contraindication to platinum-containing therapy, then there are limited evidence-based treatment options available. Other chemotherapies may be considered (e.g., doxorubicin, hormone therapy), but they are not considered as effective as a platinum doublet regimen. Single-drug chemotherapy and hormone therapy historically have lower response rates than dostarlimab. |
Consideration for continuation or renewal of therapy | |
In the GARNET study, the primary end point was response rate. In clinical practice, what would determine therapeutic response or benefit from dostarlimab for patients with dMMR or MSI-H recurrent EC? | Response rate is important, but the most important outcome is OS, followed by PFS and DOR. |
Considerations for prescribing of therapy | |
Treatment is continued until disease progression or unacceptable toxicity, per the GARNET study. Is there evidence to support weight-based dosing to a maximum capped dose? | Therapies are typically given as either a flat-rate dose or a weighted dose based on a maximum capped dose. Administration of treatment in the GARNET trial seems reasonable if tolerated and effective. |
Generalizability | |
Patients with an ECOG > 1 were excluded from part 2 of the trial (ECOG > 2 for Part 1). Should patients with ECOG > 1 be eligible for treatment with dostarlimab? | The clinical experts noted that the answer to this question is not clear. In other tumour types (e.g., lung cancer), single-drug chemotherapy in combination with immunotherapy is considered for patients with ECOG > 2. If patients in these clinical trials derive benefit from immunotherapy and they performed well, it would be reasonable to apply evidence from the GARNET trial to patients with an ECOG > 1. |
Should patients who have received more than 2 lines of therapy for advanced or recurrent disease, but who would have otherwise fit the trial criteria, be eligible for dostarlimab on a time-limited basis? | The clinical experts noted that these patients should be eligible; however, the magnitude of benefit may be less in patients with more prior lines of therapy. |
For patients currently receiving systemic therapy for recurrent dMMR or MSI-H EC (with prior platinum-based therapy), is a switch to dostarlimab appropriate? | Systemic therapies are continued until disease progression or toxicity. If systemic therapy stops working, then switching to dostarlimab is appropriate, as long as the patient has not received another PD-L1 or PD-1 inhibitor. |
dMMR = mismatch repair-deficient; DOR = duration of response; EC = endometrial cancer; ECOG = Eastern Cooperative Oncology Group performance status; IHC = immunohistochemistry; MMR = mismatch repair; MSI = microsatellite instability; MSI-H = microsatellite instability-high; MSS = microsatellite stable; NGS = next-generation sequencing; OS = overall survival; PD-1 = programmed cell death protein-1; PD-L1 = programmed death ligand-1; PFS = progression-free survival.
Clinical Evidence
The clinical evidence included in the review of dostarlimab is presented in 2 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to the a priori CADTH protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the CADTH review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of dostarlimab in 500 mg to 1,000 mg doses administered intravenously for the treatment of adults with dMMR or MSI-H advanced or recurrent EC that has progressed on or after prior treatment with a platinum-containing regimen.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. The systematic review protocol was established before the granting of the NOC/c from Health Canada. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults with recurrent or advanced dMMR or MSI-H EC that has progressed on or after prior treatment with a platinum-containing regimen. Subgroups:
|
Intervention | Dostarlimab for IV injection:
|
Comparator |
|
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, mortality, notable harms (immune-related reactions [e.g., infusion reactions, colitis, pneumonitis], neurotoxicity, anemia, nausea, diarrhea, vomiting, pruritus, rash, fever, hypothyroidism) |
Study designs | Published and unpublished phase II, III, and IV RCTs |
AEs = adverse events; BOR = best overall response; DCR = disease control rate; dMMR = mismatch repair-deficient; DOR = duration of response; EC = endometrial cancer; FIGO = International Federation of Gynecology and Obstetrics; HRQoL = health-related quality of life; ICU = intensive care unit; MSI-H = microsatellite instability-high; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PLD = pegylated liposomal doxorubicin; RCT = randomized control trial; SAEs = serious adverse events; WDAEs = withdrawal due to adverse events.
aOnly indicated for patients with hormone-therapy-positive and low-grade endometrioid cancer.
bApproved by Health Canada but not approved for reimbursement.
cThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy, according to the PRESS Peer Review of Electronic Search Strategies checklist.42
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was dostarlimab. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on November 3, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC) on March 9, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.43 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented with a review of bibliographies of key papers and contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies. Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for network meta-analyses dealing with EC was run in MEDLINE All (1946–) on November 3, 2021. No limits were applied to the search.
Findings From the Literature
Two reports of 1 study18,44 were identified from the literature for inclusion in the systematic review (Figure 1); the study is summarized in Table 6. A list of excluded studies is presented in Appendix 2.
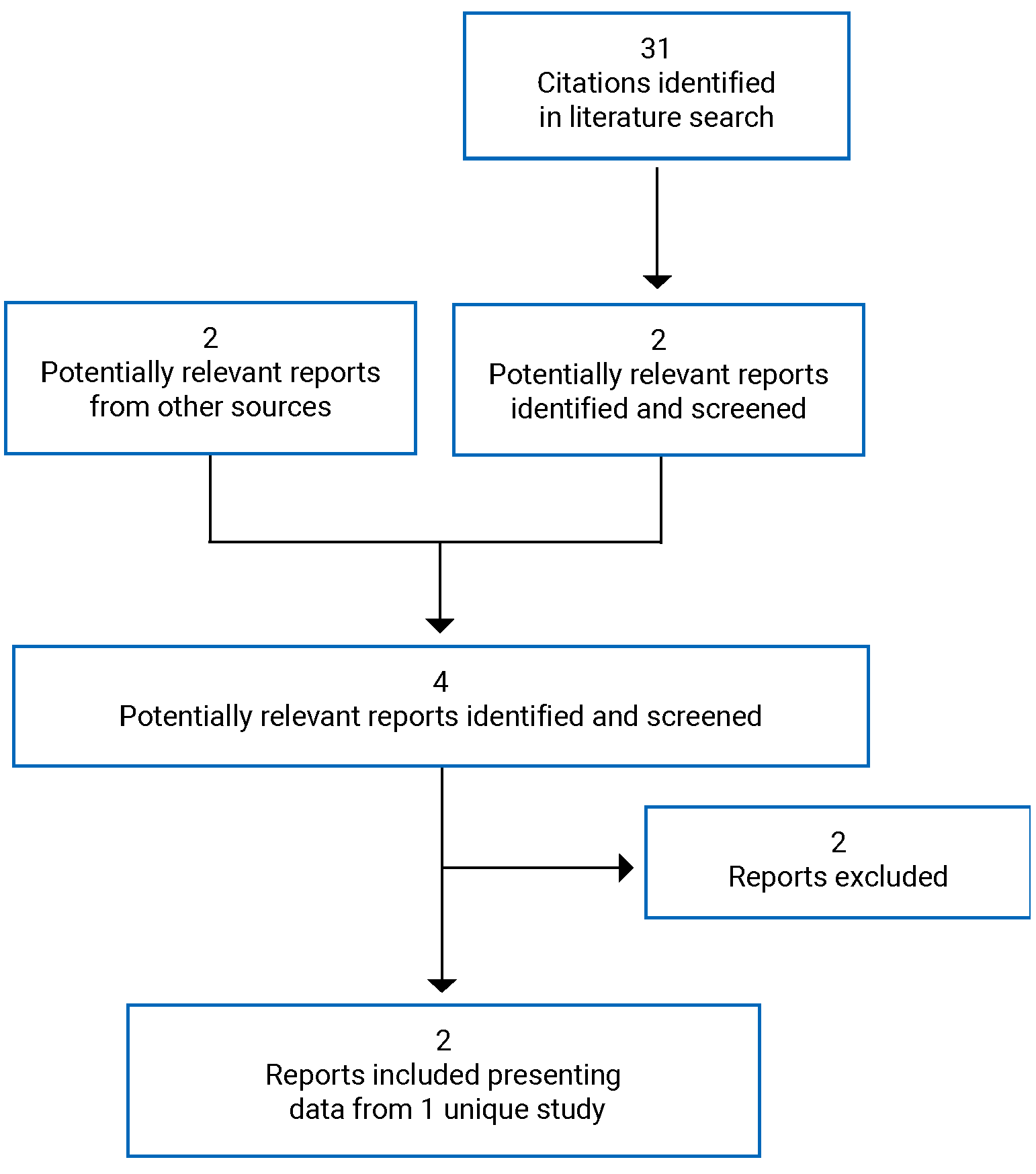
Table 6: Details of Included Study
Detail | GARNET trial, part 2B |
|---|---|
Study design | Nonrandomized, multi-centre, single-arm, open-label, phase I dose-escalation and cohort-expansion study |
Locations | Patients enrolled across 123 sites in 8 countries (Canada, European countries, UK, US) |
Patient enrolment dates | First patient enrolled on April 10, 2017 |
Data cut-off date | March 1, 2020 |
Enrolment | Cohort A1, n = 129
Cohort A2, n = 160
|
Inclusion criteria |
|
Exclusion criteria |
|
Intervention | 500 mg dostarlimab administered by 30-minute IV infusion every 3 weeks for doses 1 to 4 and 1,000 mg dostarlimab administered by 30-minute IV infusion every 6 weeks for dose 5 onward for up to 2 years. |
Comparator(s) | None |
Phase | |
Screening | In the 35 days before treatment with dostarlimab |
Treatment | Up to 24 months |
Follow-up | Safety follow-up: 90 ± 7 days after treatment HRQoL and OS: every 90 days until death or withdrawal from the study Follow-up is ongoing |
Primary end point | ORR and DOR based on BICR using RECIST 1.1 |
Secondary and exploratory end points and safety outcomes | Secondary:
Safety:
Exploratory:
|
Publications | Oaknin et al. (2020)44 |
BICR = blind independent clinical review; CD3+ = cluster of differentiation 3; CNS = central nervous system; cRO = conversion rate optimization; CTCAE = Common Terminology Criteria for Adverse Events; DCR = disease control rate; dMMR = mismatch repair-deficient; DOR = duration of response; EC = endometrial cancer; ECG = electrocardiogram; ECOG = Eastern Cooperative Oncology Group performance status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels; HRQoL = health-related quality of life; IgG4 = immunoglobulin G4; IHC = immunohistochemistry; irAEs = immune-related adverse events; irDCR = immune-related disease control rate; irDOR = immune-related duration of response; irORR = immune-related objective response rate; irPFS = immune-related progression-free survival; irRECIST = immune-related Response Evaluation Criteria in Solid Tumours; MMR = mismatch repair; MSI-H = microsatellite instability-high; MSS = microsatellite stable; NCI = National Cancer Institute; NGS = next-generation sequencing; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PD-1 = programmed cell death protein-1; PD-L1 = programmed death ligand-1; PD-L2 = programmed death ligand-2; POLE = polymerase epsilon; RECIST = Response Evaluation Criteria in Solid Tumours; SAEs = serious adverse events; TEAEs = treatment-emergent adverse events; TILs = tumour infiltrating lymphocytes.
Source: Clinical Study Report.18
Description of Study
The GARNET trial (Study 4010-01-001)18 is an ongoing multi-centre, single-arm, open-label, phase I dose-escalation and cohort-expansion study of patients with recurrent or advanced solid tumours. The primary objective of part 2B of the GARNET trial was to evaluate the safety and antitumour activity of dostarlimab in patients with advanced solid tumours. The secondary objective was to characterize the pharmacokinetic profile and to evaluate immunogenicity and additional measures of clinical benefit of dostarlimab. Exploratory objectives were to characterize the pharmacodynamics of dostarlimab and patient-reported outcomes assessed with EQ-5D-5L and the EORTC QLQ-C30. Patients were assigned to different cohorts based on variety of factors, including cancer type, response to previous therapies, and/or clinical biomarkers. Cohort A1 included patients with recurrent or advanced dMMR or MSI-H EC who had progressed on or after prior treatment with a platinum-containing regimen. Cohort A2 included patients with recurrent or advanced MMR-proficient or MSS EC that had progressed on or after prior treatment with a platinum-containing regimen. Cohort F included patients with nonendometrial dMMR, MSI-H, or polymerase-epsilon-mutation cancers who had progressed after 2 prior lines of systematic therapy for recurrent or advanced disease and had no alternative treatment options. For the purpose of this review, only the results of cohort A1, which aligns with the Health Canada indication, are presented. Refer to Figure 2 for a schematic of the GARNET trial.
A total of 129 patients were enrolled into cohort A1. Enrolment started on April 10, 2017 and is ongoing. Patients were enrolled across 123 sites in 8 countries (refer to Table 6). Of the 123 sites, 8 were located in Canada. Refer to Figure 2 for a schematic of the GARNET trial. All patients received dostarlimab by IV injection (500 mg every 3 weeks for cycles 1 to 4, and 1,000 mg every 6 weeks from cycle 5 onward) for up to 2 years or until disease progression, treatment discontinuation, or study withdrawal. Patients were permitted to continue treatment with dostarlimab beyond 2 years if the treating physician and the sponsor agreed that the treatment continued to provide clinical benefit to the patient.
To be enrolled, patients had to have archival or newly obtained tumour tissue so the tumour microenvironment could be assessed for biomarkers; in cohort A1, this included IHC results from a certified local laboratory that could be used to determine dMMR status. The definition of dMMR was the loss of expression of 1 of the following proteins, determined with IHC testing: MLH1, MSH2, MSH6, and PMS2. In the absence of known MMR status, MSI-H was tested using NGS in a central laboratory. Assay results assessed with local polymerase chain reaction were also acceptable for the determination of MSI status.
The study consisted of 3 phases: the screening phase (up to 35 days before beginning treatment), the treatment phase, and the follow-up phase. During the screening phase, 479 patients were screened for enrolment in either cohort A1 or cohort A2, based on MMR/MSI status. There were 184 screening failures, and 5 patients were screened but not treated. During the follow-up phase, patients were followed for safety (final follow-up visit was 90 ± 7 days after the end of treatment [EOT]), as well as for OS and HRQoL. The date of the final analysis for OS had not been determined at the time of the data cut-off (March 1, 2020).
The GARNET trial has 3 planned interim analyses that are contingent on a combined enrolment of 100, 200, and 300 patients, respectively, in cohort A1 and cohort F. For IA-1, the data cut-off date was July 8, 2019, after a 100 patients with measurable disease at baseline and at least 24 weeks of follow-up had been enrolled in either cohort A1 or cohort F. For IA-2, the data cut-off date was March 1, 2020, after the combined patient enrolment of cohort A1 and cohort F reached approximately 200 patients with measurable disease at baseline and at least 24 weeks of follow-up. For IA-3, the data cut-off date was November 1, 2021, after the combined patient enrolment in cohort A1 and cohort F reached approximately 300 patients with measurable disease at baseline and at least 24 weeks of follow-up. Only data from the IA-2 data cut-off date were reviewed by CADTH, which included a subset of 72 patients from IA-1 (refer to Appendix 4 for further details). The GARNET trial was sponsored by GlaxoSmithKline Inc.
Amendments and Protocol Deviations
The study protocol of the GARNET trial was amended 6 times, although versions 4.0 and 5.0 were not implemented. Amendments to the protocol did not substantially affect the results of the study.
Amendment 2 (October 31, 2016) included updates to the inclusion criteria, tumour assessment criteria, definitions for safety data, and sample size justification of cohort A1 and cohort A2.
Amendment 3 (October 9, 2017) included the addition of HRQoL assessments and interim analyses for patients with dMMR or MSI-H cancer in cohort A1 and cohort F combined.
Amendment 4 (July 3, 2018) included revisions to allow clinically stable patients without major safety issues to continue with dostarlimab treatment after confirmation of progressive disease. Changes were also made to blood sample collection and to increase the number of patients in cohort A2.
Amendment 5 (May 10, 2019) implemented changes to the definition of cohort A1 and cohort A2, based on feedback from a regulatory agency. A recommendation was made to select patients based on the MMR IHC result (local or central test) instead of NGS; hence, the central testing vendor was changed. The sample size for cohort A1 was increased and the assessment of HRQoL was revised from a secondary to an exploratory objective.
Amendment 6 (January 7, 2020) updates to the primary objective included evaluation of the safety and tolerability of dostarlimab, as well as the addition of 2 interim analyses for the combined enrolment of cohort A1 and cohort F to reach 200 and 300 patients, respectively, and for all patients to have measurable disease at baseline and at least 24 weeks of follow-up.
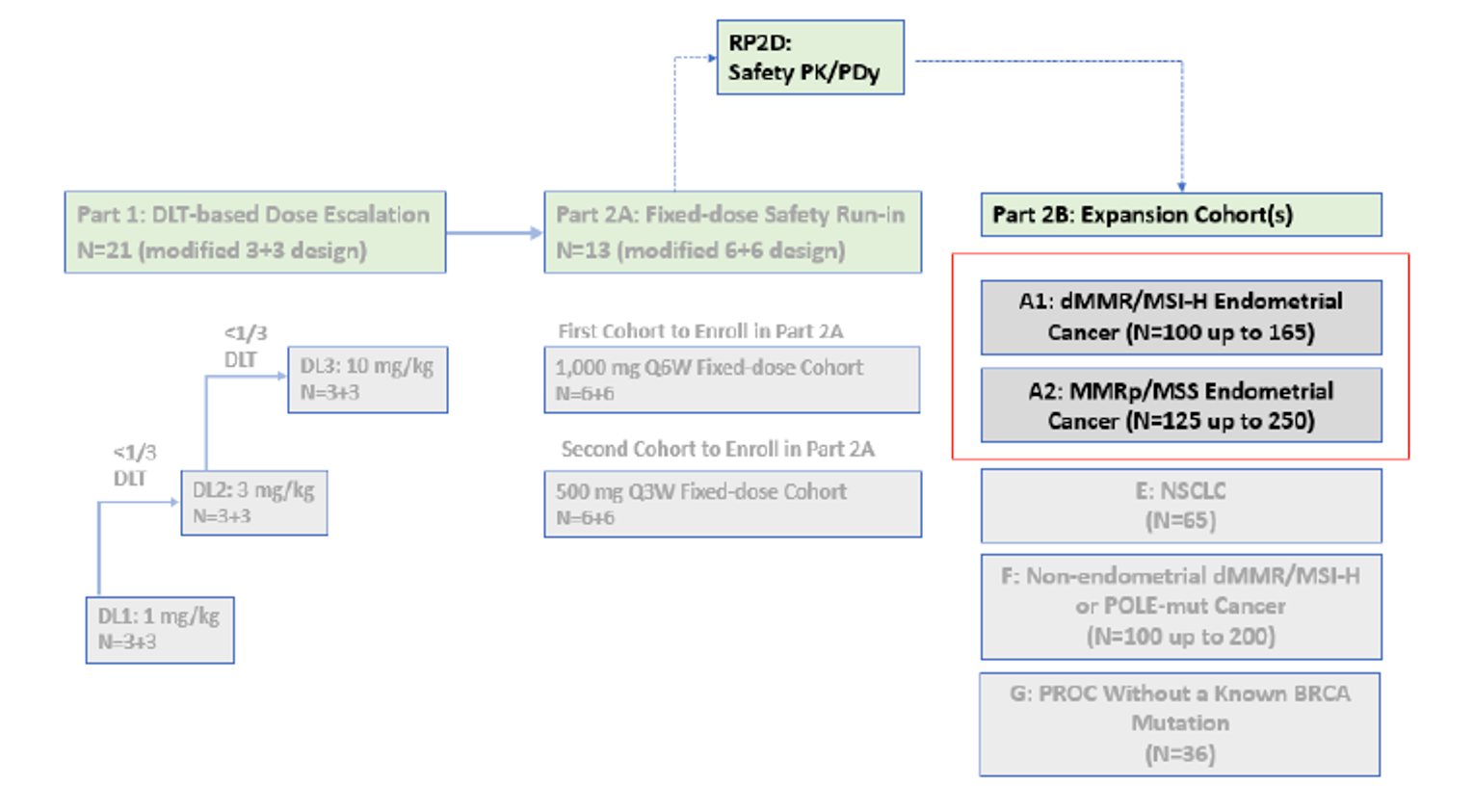
DLT = dose-limiting toxicity; dMMR = mismatch repair-deficient; MMRp = mismatch repair proficient; MSI-H = microsatellite instability-high; MSS = microsatellite instability stable; mut = mutation; NSCLC = non–small cell lung cancer; PDy = pharmacodynamics; PK = pharmacokinetics; POLE = polymerase epsilon; PROC = platinum-resistant ovarian cancer; ; Q3W = every 3 weeks; Q6W = every 6 weeks; RP2D = recommended phase II dose.
Source: Clinical Study Report.18
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria used in cohort A1 of the GARNET trial are summarized in Table 4. Briefly, the trial enrolled adults aged 18 years and older who had been diagnosed with recurrent or advanced EC and who progressed on or after no more than 2 lines of prior systemic therapy (not including hormone therapy), with at least 1 of these being platinum-based doublet therapy. Patients had to have documentation of dMMR or MSI-H status and radiologically measurable disease that met Response Evaluation Criteria in Solid Tumors 1.1 (RECIST 1.1).45 All EC histologies were permitted, except for endometrial sarcoma (including carcinosarcoma). At screening, patients had to have an ECOG Performance Status of 0 or 1, adequate organ function, a negative serum pregnancy test (if of childbearing potential), and 1 highly effective form of contraception throughout the study period. Prior therapy with an anti-PD-1, anti-PD-L1, or anti-PD-L2 drug was not permitted.
Baseline Characteristics
The baseline characteristics of patients who comprised the primary efficacy population of the GARNET trial are summarized in Table 7. At baseline, 105 patients in cohort A1 were identified as having dMMR or MSI-H EC (103 dMMR and 2 MMR-unk/MSI-H EC patients) for the primary efficacy analysis at IA-2. The demographic and baseline characteristics of patients with dMMR or MMR-unk/MSI-H tumours are presented as a total because of the small proportion of patients (n = 2) with MMR-unk/MSI-H tumours; the sponsor noted that the data remained similar when these patient groups were pooled. The median age was 64 years (range = 39 to 80 years), with 50.5% of patients between 39 and 65 years. Median weight was 71 kg (range = 34.0 to 141.4), and median BMI was 27.97 kg/m2 (range = 13.6 to 53.9 kg/m2). Most patients were White (78.1%). The most common histology type was type I endometrioid carcinoma (67.6%); the most common histology grade at diagnosis was grade 2 (39.0%). At the time of study enrolment, more than two-thirds (67.6%) of patients had FIGO stage IV EC. An ECOG Performance Status of 1 was the most common (60%), followed by an ECOG Performance Status of 0 (40%). All patients received prior anti-cancer treatment that included surgery (90.5%), radiotherapy (70.5%), adjuvant or neoadjuvant treatment (53.3%), and/or bevacizumab use (4.8%). Most patients (88.6%) had previously received 2 or fewer lines of anti-cancer regimens; a small portion had previously received 2 or more lines (11.5%). More than half (56.2%) of patients had received prior regimens for metastatic disease.
Table 7: Summary of Baseline Characteristics of Patients in the GARNET Trial (IA-2, Primary Efficacy Analysis Dataset)
Characteristic | Cohort A1a N = 105 |
|---|---|
Race, n (%) | |
White | 82 (78.1) |
Black | 2 (1.9) |
Asian | 4 (3.8) |
American Indian or Alaska Native | 3 (2.9) |
Not reported | 14 (13.3) |
Age (years), median (range) | 64.0 (39 to 80) |
Age group, n (%) | |
< 65 years | 53 (50.5) |
≥ 65 years to < 75 years | 41 (39.0) |
≥ 75 years | 11 (10.5) |
Weight (kg), median (range) | 71.0 (34.0 to 141.4) |
BMI (kg/m2) | |
n | 102 |
Median (range) | 27.97 (13.6 to 53.9) |
ECOG Performance Status, n (%) | |
0 | 42 (40.0) |
1 | 63 (60.0) |
Histology at diagnosis, n (%) | |
Endometrioid carcinoma type I | 71 (67.6) |
Endometrioid carcinoma type II | 33 (31.4) |
Serous carcinoma | 4 (3.8) |
Clear cell carcinoma | 1 (1.0) |
Squamous carcinoma | 1 (1.0) |
Undifferentiated carcinoma | 4 (3.8) |
Mixed carcinoma | 4 (3.8) |
Unspecified | 14 (13.3) |
Otherb | 5 (4.8) |
Unknown | 1 (1.0) |
Grade of disease at diagnosis, n (%) | |
Grade 1 | 31 (29.5) |
Grade 2 | 41 (39.0) |
Grade 3 | 28 (26.7) |
Not assessable | 5 (4.8) |
Most recent FIGO stage, n (%) | |
Stage I | 12 (11.4) |
Stage II | 4 (3.8) |
Stage III | 16 (15.2) |
Stage IV | 71 (67.6) |
Unknown | 2 (1.9) |
Any prior anti-cancer treatment, n (%)c | 105 (100) |
Prior surgery for study indication, n (%) | 95 (90.5) |
Any prior anti-cancer radiotherapy, n (%) | 74 (70.5) |
Prior bevacizumab use, n (%) | 5 (4.8) |
Any prior adjuvant or neoadjuvant anti-cancer treatment, n (%) | 56 (53.3) |
Prior anti-cancer regimens, n (%) | |
1 | 66 (62.9) |
2 | 27 (25.7) |
3 | 9 (8.6) |
≥ 4 | 3 (2.9) |
Prior regimens for metastatic disease, n (%)d | |
0 | 46 (43.8) |
1 | 48 (45.7) |
2 | 10 (9.5) |
3 | 1 (1.0) |
BMI = body mass index; ECOG = Eastern Cooperative Oncology Group; FIGO = International Federation of Gynecology and Obstetrics; IA-2 = second interim analysis.
aPopulation total (n = 105) includes 103 dMMR and 2 MMR-unk/MSI-H patients. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 2) with MMR-unk/MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
bOther may include adenocarcinoma; adenocarcinoma with ambiguous differentiation; biopsies that showed a high-grade adenocarcinoma, which can be seen in tumours of Mullerian origin; carcinoma epidermoid; endometrial adenocarcinoma; endometrial adenocarcinoma not otherwise specified; endometrial neuroendocrine carcinoma; endometrioid adenocarcinoma; high-grade uterine carcinoma; moderately differentiated adenocarcinoma; papillary serous carcinoma; and undifferentiated clear cell carcinoma.
cIncludes surgery, radiotherapy, or drug therapy, excluding hormonal drugs.
dDoes not include neoadjuvant regimens, adjuvant regimens, or hormonal drugs.
Source: Clinical Study Report.18
Interventions
All patients in cohort A1 received dostarlimab as a 30-minute IV infusion (with a permitted window of –5 minutes and + 15 minutes). Doses were administered in hospitals, infusion centres, and outpatient clinics. Dosing schedules were as follows:
cycles 1 to 4: 500 mg every 3 weeks (day 1 of each 21-day cycle)
cycle 5 and onward: 1,000 mg every 6 weeks (day 1 of each 42-day cycle).
Patients continued with the study treatment for up to 2 years or until specific withdrawal criteria were met. Patients discontinued study treatment for the following reasons: AEs, including dose-limiting toxicities; progressive disease based on clinical criteria used by the investigator or outlined in the protocol; patient request or patient pregnancy; risk to patients, determined by the investigator and/or sponsor; and severe noncompliance with the protocol, determined by the investigator and/or sponsor. In some instances, study treatment continued beyond 2 years if the treating physician and the sponsor agreed that treatment with dostarlimab continued to provide clinical benefit to the patient.
Dose Modifications (Delay, Interruption)
Dosing delays were permitted in the case of medical or surgical events or for logistical reasons not related to study treatment (e.g., surgery, unrelated medical events, participant vacation). Continuation of the study treatment should have occurred within 28 days of the scheduled dostarlimab infusion. If a delay was more than 28 days, a patient may have been permitted to continue treatment after discussion with the sponsor.
Dosing interruptions were permitted in the event of toxicities. In general, treatment withheld for drug-related grade 3 toxicities could be resumed if toxicity resolved to grade 1 or lower. Similarly, for all irAEs, dostarlimab was held until the patient was considered clinically and metabolically stable and AEs resolved to grade 1 or lower. Dostarlimab was permanently discontinued after any drug-related grade 4 events and after some grade 3 immunologic-mediated AEs.
Dose reductions were not permitted.
Concomitant Medications
Concomitant medications were allowed to treat AEs and comorbidities. Concomitant medications included any medications other than study treatments taken on or after the initial study treatment dosing date. If systemic steroids were used as a part of irAE management, the total dose of daily steroids allowed was 10 mg or less of prednisone when dostarlimab treatment was resumed. Patients were also allowed to receive rescue medications and appropriate supportive care deemed necessary by the treating investigator.
To ensure accurate assessment of the safety and efficacy of dostarlimab, patients were prohibited from receiving the following therapies during the screening and treatment periods of the study:
systemic anti-cancer or biological therapy
immunotherapy not specified in the protocol
chemotherapy not specified in the protocol
investigational drugs other than dostarlimab
radiation therapy within 3 weeks before study day 1 and during study treatment
note: palliative radiation therapy to a small field more than 1 week before day 1 of study treatment may have been allowed
any surgery that involved tumour lesions
note: radiation therapy or surgery that involved tumour lesions was considered to be progressive disease at the time the procedure was performed.
Subsequent Therapies
Data were collected on whether patients received subsequent anti-cancer therapy after treatment discontinuation or progressive disease. However, these were not defined in the protocol or statistical analysis plan.
Outcomes
A list of the efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized below. A more detailed discussion of the HRQoL and symptom severity outcome measures assessed in the trial is provided in Appendix 3.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | GARNET trial |
|---|---|
OS | Secondary |
DCR and irDCR | Secondary |
HRQoL
| Exploratory |
PFS and irPFS | Secondary |
ORR | Co-primary |
irORR | Secondary |
DOR | Co-primary |
irDOR | Secondary |
BOR | Secondary |
Health care resource use | Not measured |
BOR = best overall response; DCR = disease control rate; DOR = duration of response; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-C30; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels; HRQoL = health-related quality of life; irDCR = immune-related disease control rate; irDOR = immune-related duration of response; irORR = immune-related objective response rate; irPFS = immune-related progression-free survival; ORR = objective response rate; OS = overall survival; PFS = progression-free survival;
Note: In the GARNET trial, DCR, PFS, DOR, and BOR outcomes were assessed based on BICR, per RECIST 1.1 criteria. irDCR, irPFS, irORR, and irDOR outcomes were assessed based on investigator assessment, per irRECIST 1.1.
Response assessment, according to RECIST 1.1,45 was based on radiologic imaging and performed by a BICR at pre-specified time points for the outcomes of OS, DCR, PFS, DOR, and BOR. The first radiographic evaluation occurred 12 weeks (84 ± 10 days) after the first dostarlimab dose and every 6 weeks (42 ± 10 days) thereafter, independent of cycle delays and/or dose interruptions. After 48 weeks of radiographic assessments, imaging and assessment of serum-based tumour markers were performed every 12 weeks (84 ± 10 days) until progressive disease. If a patient discontinued treatment for any reason other than progressive disease, death, withdrawal of consent, or loss to follow-up, radiographic scans and appropriate testing of serum-based tumour markers continued at the specified intervals until progressive disease was confirmed or an alternate anti-cancer therapy was started, whichever occurred first.
Tumour response was also evaluated by the investigator based on irRECIST 1.146,47 for the outcomes of immune-related DCR (irDCR), immune-related PFS (irPFS), immune-related ORR (irORR), and immune-related (irDOR). Investigators used irRECIST-based assessment to make clinical decisions. Per irRECIST, immune-related complete response (irCR) or immune-related partial response (irPR) was confirmed with repeat radiographic evaluation, at the earliest, 4 weeks after the first indication of response or at the next scheduled scan, whichever was clinically indicated. Progressive disease was confirmed with radiographic evaluation a minimum of 4 weeks and up to 6 weeks after the first progressive disease assessment.
Overall Survival
OS was a secondary outcome of the GARNET trial and was defined as the time from the first dose of study treatment to death by any cause. Patients last known to be alive were censored at the date of the last known contact, as follows: OS in days = date of death or censoring – date of first dose + 1.
OS was assessed at EOT, at the safety follow-up visit, and every 3 months after treatment.
Disease Control Rate and Immune-Related Disease Control Rate
DCR and irDCR were secondary outcomes of the GARNET trial. DCR, per RECIST 1.1., was defined as the proportion of patients achieving BOR of confirmed CR, PR, or stable disease. irDCR, per irRECIST 1.1., was defined as the proportion of patients achieving irBOR or irCR. irPR or immune-related stable disease as assessed by the investigator.
BOR was programmatically derived, based on reported time point response assessments determined by a central reader at different evaluation time points, from the first dose until the first documented progressive disease. The irBOR, according to irRECIST, was programmatically derived, based on reported overall time point responses determined by investigators after assessment of radiological scans, at different evaluation time points from the first dose until documented disease progression.
BOR was determined according to the following rules:
CR — at least 2 consecutive determinations of CR more than 4 weeks apart, with no other assessment between the 2 determinations other than not evaluable, CR, or missing.
PR — at least 2 consecutive determinations of PR or better more than 4 weeks apart, with no other assessment between the 2 determinations other than not evaluable, CR, PR, or missing before disease progression (and not qualifying for a CR).
Stable disease — at least 1 stable disease or non-CR or nonprogressive disease assessment (or better) at least 12 weeks to 10 days (i.e., ≥ 74 days) after baseline and before progressive disease (and not qualifying for a CR or PR). For an unconfirmed PR or CR to qualify, it must still meet the requirement of at least 12 weeks to 10 days.
Progressive disease — disease progression after baseline. Note that a determination of CR followed at least 4 weeks later by stable disease resulted in a BOR of progressive disease.
No disease — for central imaging data, when the independent radiologist cannot identify any disease at baseline, all subsequent assessments were documented as no disease if not declared progressive disease or not evaluable.
irBOR followed similar rules, except outcomes were immune-related (e.g., irCR, irPR). Clinical deterioration was not considered to be progressive disease. Only tumour assessments performed before the start of any new anti-cancer treatment were considered in the assessment of BOR and irBOR.
Progression-Free Survival and Immune-Related Progression-Free Survival
PFS and irPFS were secondary outcomes of the GARNET trial. PFS was defined as the time from the first dose to the earlier assessment of progressive disease or to death by any cause in the absence of progressive disease. irPFS time was defined, per irRECIST 1.1, as the time from the first dose to the earlier date of assessment of immune-related disease progression (irPD) event or to death by any cause in the absence of disease progression.
irPD events were considered when:
2 consecutive irPDs were observed; the date of the first was considered to be the date of the irPD event.
the only or most recent tumour assessment before treatment discontinuation was irPD; the date was considered to be the date of the irPD event.
PFS and irPFS times were defined as follows: PFS or irPFS in days = date of PD or irPD event or death ÷ censoring – date of first dose + 1 (refer to Table 9 for details on censoring rules for PFS and irPFS).
Table 9: Censoring Rules for DOR, irDOR, PFS, irPFS
Scenario | Date of event | Censoring outcome |
|---|---|---|
Prior to 48-week tumour assessment: Progression or death ≤ 12 weeks ( + 10 days) after prior post-baseline tumour assessment or ≤ 18 weeks after first dose After 48-week tumour assessment: Progression or death ≤ 24 weeks ( + 10 days) after prior post-baseline tumour assessment | Date of progressive disease or death | Event |
No baseline assessment | First dose date | Censored |
No post-baseline evaluable radiologic tumour assessment | First dose date | Censored |
Prior to 48-week tumour assessment: Progression or death > 12 weeks ( + 10 days) after the prior post-baseline tumour assessment After 48-week tumour assessment: Progression or death > 24 weeks ( + 10 days) after the prior post-baseline tumour assessment | Date of the last evaluable radiologic tumour assessment before progressive disease or death | Censoreda |
No progression | Date of the last evaluable radiologic tumour assessment | Censored |
New anti-cancer therapy given | Date of the last evaluable radiologic tumour assessment before anti-cancer therapy given | Censored |
DOR = duration of response; irDOR = immune-related duration of response; irPFS = immune-related progression-free survival; PFS = progression-free survival.
aTumour assessment was based on a CT or MRI scan. If progression or death was determined and 2 previous scans were missing, the date of progression was not known.
Objective Response Rate and Immune-Related Objective Response Rate
ORR was the primary outcome of the GARNET trial and was defined, per RECIST 1.1 based on BICR, as the proportion of patients that achieved a BOR of CR or PR. irORR was a secondary outcome of the GARNET trial and was defined, per irRECIST 1.1, as the proportion of patients that achieved irBOR of irCR or irPR. Nonresponders were patients who did not have a post-baseline radiographic tumour assessment, who received post-baseline antitumour treatments (including surgery or radiation to tumour lesions) other than the study treatments before reaching a CR or irCR or a PR or irPR, or who died, progressed, or dropped out for any reason before reaching a CR or irCR or a PR or irPR.
Duration of Response and Immune-Related Duration of Response
DOR was a co-primary outcome of the GARNET trial and was defined as the time from first documentation of overall response leading to a confirmed CR or PR until the time of first documentation of disease progression or death. Censoring rules for DOR were similar to those for PFS (refer to Table 9 for details on censoring rules). irDOR was a secondary outcome of the GARNET trial and was defined, per irRECIST 1.1, as the time from first documentation of response leading to a confirmed irCR or irPR until the time of the irPD event or death. As with BOR and irBOR, clinical deterioration was not considered to be documented progressive disease.
Health-Related Quality of Life
The HRQoL outcomes measured in the trial included EORTC QLQ-C30 and EQ-5D-5L scores. Data from both instruments were exploratory outcomes in the GARNET trial and assessed at IA-2. HRQoL assessments were collected at scheduled visits (at each cycle) every 3 weeks (± 7 days) for the first 12 weeks, beginning on cycle 1, day 1, and every 6 weeks (± 7 days) thereafter while the patient was receiving study treatment. After treatment discontinuation, HRQoL assessments were collected from the remaining patients at the EOT visit, at the safety follow-up visit (90 days after EOT), and every 90 days (± 14 days) during the post-treatment follow-up period. A detailed discussion and critical appraisal of the HRQoL measures are provided in Appendix 3.
The EORTC QLQ-C30 is a questionnaire developed specifically to assess HRQoL in cancer patients. The questionnaire consists of 30 questions, 5 function scales (physical, role, cognitive, emotional, and social), 1 global health status/global quality-of-life scale, 3 symptom scales (fatigue, pain, and nausea and vomiting), and 6 single items that assess additional symptoms (dyspnea, appetite loss, sleep disturbance, constipation, and diarrhea) and financial impact.48 Scales and single items range in score from 0 to 100, with higher scores on the functional and global health status/quality-of-life scales indicating higher levels of functioning and health status/quality of life, respectively. Higher scores on symptom scales or items represent a greater presence of symptoms.48
The EQ-5D-5L version 2.0 is a generic HRQoL questionnaire for assessing a patient’s health status in terms of a single index value or utility score. There are 2 components of the questionnaire: a descriptive system that allows patients to rate their level of problems (none, slight, moderate, severe, extreme/unable) in 5 areas (mobility, self-care, usual activities, pain/discomfort, and anxiety/depression), and a visual analogue scale that allows patients to rate their overall health status from 0 (worst imaginable) to 100 (best imaginable).49
Safety
Assessing the safety of dostarlimab was a primary objective of the GARNET trial. The primary safety variables included the following:
TEAEs — all, SAEs, their relationship to the study treatment, and their intensity (based on National Cancer Institute Common Terminology Criteria for Adverse Events v4.03 severity grades)
TEAEs leading to discontinuation
TEAEs leading to death
irAEs
clinical laboratory measures (including hematology, chemistry, coagulation, thyroid function, and urinalysis) and vital signs
ECG
physical examination
ECOG status
serum pregnancy testing
concomitant medications.
Statistical Analysis
Sample Size Determination
For the final analysis of the co-primary end point (ORR) in cohort A1, the sample size of approximately 100 patients with dMMR or MSI-H EC (with potential enrolment of up to 165 patients) was planned after protocol amendment 5. The null hypothesis that the true response rate was 20% or less (expected ORR for conventional therapy) was tested against a 1-sided alternative hypothesis of a response rate of at least 40%. With 65 patients in cohort A1, there was 92% power to rule out an ORR of 20% or less when the true ORR was 40% at the 2.5% type I error rate (1-sided). The increase in sample size allowed the lower-limit boundary of the exact 95% CI to exclude a response rate of 25% or less, assuming the observed ORR was 35%.
Interim Analyses
The 3 planned interim analyses were based on the combined enrolment in cohort A1 and cohort F (nonendometrial cancer). IA-1, IA-2, and IA-3 were conducted when the combined enrolment reached 100, 200, and 300 patients, respectively. All enrolled patients in cohort A1 and cohort F had dMMR tumours with measurable disease at baseline and at least 24 weeks of follow-up. IA-1 had a data cut-off date of July 8, 2019; IA-2 had a data cut-off date of March 1, 2020; and IA-3 had a data cut-off date of November 1, 2021. IA-2 and IA-3 were added after amendment 6.
All interim analyses evaluated the outcomes listed in Table 8, with the exception of IA-1, which included irORR as a primary end point and patient-reported outcomes as a secondary end point.
Primary Outcomes
The co-primary outcomes in the GARNET trial were ORR and DOR. No formal hypothesis-testing or inferential analyses were performed in cohort A1, and no statistical comparisons were planned between cohorts. The analysis of ORR included summary statistics, including the number of patients and percentage for categorical variables and, for continuous variables, the number of patients, mean, standard deviation (SD), median, minimum, and maximum. Two-sided exact 95% CIs based on the Clopper-Pearson method were reported for the ORR.
DOR analyses were performed using KM methods and summarized by minimum, maximum, 25th, 50th (median), and 75th percentiles with associated 95% CIs, the number and percentage of events, the number and percentage of censored observations, and the median duration of follow-up. Censoring rules for DOR are presented in Table 7.
Secondary Outcomes
The analyses for OS were based on the safety analysis and primary efficacy analysis datasets. OS analyses were performed using KM methods and summarized by the 25th, 50th (median), and 75th percentiles with associated 95% CIs, as well as the number and percentage of events and censored observations. Censoring for OS was set at the last known date of contact.
The analyses of DCR and irDCR involved summary statistics, including the number of patients and percentage for categorical variables and, for continuous variables, the number of patients, mean, SD, median, minimum, and maximum. Two-sided exact 95% CIs based on the Clopper-Pearson method were provided for DCR and irDCR.
The analyses of PFS and irPFS were performed using KM methods and summarized by the 25th, 50th (median), and 75th percentiles with associated 95% CIs, as well as the number and percentage of events and censored observations. Censoring rules for PFS and irPFS are presented in Table 9.
The analyses of irORR and irDOR were similar to the those of the co-primary outcomes of ORR and DOR.
Exploratory Outcomes
No analysis plan, objectives, or minimally important difference (MID) for the EORTC QLQ-C30 or the EQ-5D-5L instruments were specified a priori in the statistical analysis plan; however, according to the study protocol, changes from baseline to each pre-specified study time point were to be measured and summarized using descriptive statistics. HRQoL assessments (EQ-5D-5L and EORTC QLQ-C30) were collected during scheduled visits for all patients in cohorts A1 and F enrolled under amendment 3 and subsequent amendments (i.e., every 3 weeks ± 7 days for the first 12 weeks, in alignment with study drug administration, and every 6 weeks ± 7 days thereafter, in alignment with tumour imaging assessments) while the patient was receiving study treatment. Once a patient discontinued treatment, patient-reported outcome assessments were performed during the EOT visit, the safety follow-up visit, and the post-treatment follow-up period every 90 days (± 14 days).
For the EORTC QLQ-C30, scores and changes in scores from baseline for each domain were summarized by the number of patients with values for the mean, SD, median, range, first and third quartiles, and 95% CI. Statistical graphs were produced for each of the 15 domains to show mean scores over time, with 95% CIs. Similar graphic displays were produced for the mean change from baseline over time. In terms of missing data, if at least half of the items from a particular scale were answered, it was assumed that the missing items had values equal to the average of those items that were present. With this method of imputation, none of the single-item measures could be imputed.
Safety Outcomes
Safety and tolerability were evaluated by monitoring the frequency, duration, and severity of AEs and irAEs. In general, all by-visit summaries of safety parameters were only summarized up to and including month 6 and at the treatment discontinuation visit. AEs were organized based on the Medical Dictionary for Regularity Activities version 23.0. Only TEAEs were analyzed, but all AEs occurring during the study were listed. TEAEs were tabulated by preferred term and system organ class. Patients with the same TEAE more than once had that event counted only once for each system organ class and once for each preferred term. By-patient listings of irAEs were presented, if appropriate.
Sensitivity Analyses
No sensitivity analyses were conducted for any of the outcomes outlined in the protocol of the GARNET trial.
Subgroup Analyses
Subgroup analyses were performed for ORR, DOR, and TEAEs. The following subgroup analyses for DOR and ORR were planned a priori in the statistical analysis plan:
MSI status (MSI-H versus MSS versus unknown/missing)
histology (patients with dMMR EC only)
number of prior anti-cancer therapy regimens (1 versus ≥ 2)
prior radiation therapy (yes or no)
prior bevacizumab use (yes or no)
BOR from last platinum-containing prior anti-cancer therapy (CR or PR, SD, progressive disease, or missing)
progression-free interval from last platinum-containing prior anti-cancer therapy (< 6 months versus ≥ 6 months versus missing).
The following subgroups aligned with those pre-specified in the protocol for this CADTH review: histology, number of prior anti-cancer therapy regimens, prior radiation therapy, and progression-free interval from last platinum-containing prior anti-cancer therapy. Only the subgroups identified in the CADTH review protocol are reported in the Outcomes section.
Analysis Populations
The primary efficacy analysis dataset, by RECIST 1.1 per BICR, was defined as all patients in the safety analysis dataset with measurable disease at baseline (defined as the existence of at least 1 target lesion at baseline tumour assessment identified by BICR) who had the opportunity for at least 24 weeks of tumour assessment at the time of analysis (i.e., patients whose first dose of dostarlimab administration was on or before September 15, 2019).
The secondary efficacy analysis dataset, by irRECIST per investigators’ assessment, was defined as all patients in the safety analysis dataset with measurable disease at baseline (defined as the existence of at least 1 target lesion at baseline tumour assessment identified by investigator assessment) who had the opportunity for at least 24 weeks of tumour assessment at the time of analysis (i.e., patients whose first dose of dostarlimab administration was on or before September 15, 2019).
The safety analysis dataset included all patients who received any amount of study drug. In addition, all patient-reported outcome analyses were conducted on patients using the safety analysis dataset enrolled under amendment 3 or subsequent amendments.
Results
Patient Disposition
Details of patient disposition in the cohort A1 safety analysis dataset of the GARNET trial are summarized in Table 10. A total of 479 patients were screened and, of those, 129 were enrolled into cohort A1. In cohort A1, there were 126 patients with dMMR EC and 3 with MMR-unk/MSI-H identified through NGS. All patients enrolled received dostarlimab. Of enrolled patients, 100.0%, 81.4%, and 87.6% were included in the safety, primary, and secondary efficacy analysis datasets, respectively. In IA-2, a subset of 72 patients from IA-1 were included. Refer to Appendix 4 for detailed outcomes data for this subset of patients.
At the time of IA-2, the median duration of follow-up was 16.3 months. A total of 47 (36.4%) patients had withdrawn from the study. The most common reason for study discontinuation was death, which occurred in 36 patients (27.9%); progressive disease was the main cause of death. Two patients were lost to follow-up (1.6%) and 71 (55.0%) discontinued treatment. The primary reasons for treatment discontinuation were confirmed progressive disease (38.0%) followed by AEs (10.9%). Twenty-eight (21.7%) patients continued treatment with dostarlimab beyond initial confirmation of progressive disease. Thirty-six patients (27.9%) died during the study, 31 (24.0%) due to progressive disease and 5 (3.9%) due to an AE.
Table 10: Patient Disposition in Cohort A1 of the GARNET Trial (IA-2, Safety Analysis Dataset)
Characteristic | Cohort A1a (N = 129) |
|---|---|
Data cut-off date | March 1, 2020 |
Discontinued from study, n (%) | 47 (36.4) |
Reason for discontinuation, n (%) | |
Death | 36 (27.9) |
Withdrawal of consent | 8 (6.2) |
Lost to follow-up | 2 (1.6)b |
Other | 1 (0.8)c |
Discontinued from treatment, n (%) | 71 (55.0) |
Reason for discontinuation, n (%) | |
Confirmed progressive disease | 49 (38.0) |
AEs | 14 (10.9) |
Based on investigator’s clinical criteria | 6 (4.7)b |
Patient request | 1 (0.8) |
Other | 1 (0.8)d |
Patients treated beyond initial progressive disease | 28 (21.7) |
Died during study | 36 (27.9) |
Reasons for death, n (%) | |
Progressive disease | 31 (24.0) |
AEs | 5 (3.9) |
Safety analysis dataset, n (%) | 129 (100) |
dMMR | 126 (100) |
MMR-unk/MSI-H | 3 (100) |
Primary efficacy analysis dataset (RECIST 1.1), n (%) | 105 (81.4) |
dMMR | 103 (81.7) |
MMR-unk/MSI-H | 2 (66.7) |
Secondary efficacy analysis dataset (irRECIST 1.1), n (%) | 113 (87.6) |
dMMR | 110 (87.3) |
MMR-unk/MSI-H | 3 (100) |
HRQoL population, n | |
EORTC QLQ-C30 | 94 |
EQ-5D-5L | 89 |
AEs = adverse events; dMMR = mismatch repair-deficient; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D-5L = EuroQol 5-Dimensions 5-Levels; HRQoL = health-related quality of life; IA-2 = second interim analysis; irRECIST = immune-related Response Evaluation Criteria in Solid Tumours; MMR-unk = unknown mismatch repair tumour status; MSI-H = microsatellite instability-high; RECIST = Response Evaluation Criteria in Solid Tumours.
aThe population total (N = 129) included 126 dMMR patients and 3 MMR-unk/MSI-H EC patients. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 3) with MMR-unk/ MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
bIncludes 1 patient with MMR-unk/MSI-H EC.
cThis patient returned to their country of origin.
dPatient died due to progressive disease before the decision to stop treatment; no EOT visit or safety follow-up were done.
Source: Clinical Study Report.18
Protocol Deviations
Protocol deviations in cohort A1 of the GARNET trial are summarized in Table 11. There were 168 important protocol deviations in 75 (58.1%) patients in the safety analysis dataset. Most of these deviations were related to study visits or procedures (n = 61; 42.1%), There were 17 significant protocol deviations in 11 (8.5%) patients, the majority of which were related to eligibility criteria (n = 8; 6.2%). The sponsor noted that “none of these significant protocol deviations were considered to affect the patients’ safety or well-being or the overall integrity of the study.”
Table 11: Summary of Protocol Deviations in Cohort A1 of the GARNET Trial (IA-2)
Deviation categories | Cohort A1 (N = 129)a |
|---|---|
Important protocol deviations, n (%) | 75 (58.1) |
Study visit or procedures | 61 (42.1) |
Investigational product administration or study treatment | 8 (5.5) |
AE and SAE | 5 (3.9) |
Documentation | 0 |
Informed consent | 0 |
Significant protocol deviations, n (%) | 11 (8.5) |
Inclusion or exclusion criteria | 8 (6.2) |
Disallowed medication | 3 (2.8) |
AE or SAE | 1 (0.8) |
Study visit/procedures | 1 (0.8) |
AE = adverse event; IA-2 = second interim analysis; SAE = serious adverse event.
aPopulation total (N = 129) included 126 dMMR EC patients and 3 MMR-unk/MSI-H EC patients. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 3) with MMR-unk/MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
Source: Clinical Study Report.18
Exposure to Study Treatments
Duration and Dose Intensity
Data on exposure to dostarlimab in cohort A1 at IA-2 are summarized in Table 12. Data were available for 129 patients with dMMR or MMR-unk/MSI-H. The median DoT with dostarlimab was 25.6 weeks. The maximum DoT with dostarlimab was 138.9 weeks. Approximately half of patients (53.5%) received dostarlimab treatment for up to 19 to 24 weeks. The median relative dose intensity was 100% throughout the study treatment period because dose reductions were not permitted. Twenty-eight patients continued study treatment despite initial progressive disease.
Treatment Compliance
Treatment compliance was based on number of infusions. At any time during the study, less than 6% of patients with dMMR EC had a missed infusion, and less than 20% had an infusion delay. From cycle 1 to cycle 4, there were 3 missed infusions (2.4%) and 11 infusion delays (8.7%) reported in patients with dMMR EC. From cycle 5 through the EOT, 7 missed infusions (5.6%) and 25 infusion delays (19.8%) were reported in patients with dMMR EC. One infusion interruption (0.8%) was reported between cycle 5 and EOT.
Table 12: Patients With dMMR or MSI-H EC on Treatment by Week Intervals in Cohort A1 of the GARNET Trial (IA-2, Safety Analysis Dataset)
Characteristic | Cohort A1a (N = 129) |
|---|---|
DoT interval,b n (%) | |
Week 1 to ≤ week 3 | 129 (100.0) |
Week 4 to ≤ week 6 | 110 (85.3) |
Week 7 to ≤ week 9 | 97 (75.2) |
Week 10 to ≤ week 12 | 96 (74.4) |
Week 13 to ≤ week 18 | 89 (69.0) |
Week 19 to ≤ week 24 | 69 (53.5) |
Week 25 to ≤ week 30 | 58 (45.0) |
Week 31 to ≤ week 36 | 51 (39.5) |
Week 37 to ≤ week 42 | 52 (40.3) |
Week 43 to ≤ week 48 | 41 (31.8) |
Week 49 to ≤ week 54 | 37 (28.7) |
> Week 54 | 35 (27.1) |
Overall DoT (weeks)c | |
Patients, n | 129 |
Mean (SD) | 40.8 (36.68) |
Median (IQR) | 26.0 (12.0 to 60.9) |
Range | 3.0 to 138.9 |
dMMR = mismatch repair-deficient; DoT = duration of treatment; EC = endometrial cancer; IA-2 = second interim analysis; MSI-H = microsatellite instability-high; SD = standard deviation.
aThe population total (N = 129) included 126 dMMR EC patients and 3 MMR-unk/MSI-H EC patients. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 3) with MMR-unk/MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
bIntervals are inclusive of the upper week number (i.e., week 1 to ≤ week 3 is equivalent to day 1 to day 21, inclusive).
cIf the last cycle of treatment was ≤ 4 cycles, the DoT was calculated as follows: the last dose date − start date + 21 or death date – start date + 1 if death occurred < 21 days after last dose. If the last cycle of treatment was ≥ 5 cycles, the DoT was calculated as follows: last dose − start date + 42 or death date – start date + 1 if death occurred < 42 days after last dose.
Source: Clinical Study Report.18
Concomitant Medication
Nearly all patients in cohort A1 reported taking concomitant medications (99.2% of dMMR EC patients). The most common types of concomitant medications (used by ≥ 40% of patients) were in the Anatomical Therapeutic Chemical (ATC) classes of the following:
other analgesics and antipyretics (76.2%)
opioids (58.7%)
antithrombotic drugs (49.2%)
drugs for peptic ulcer and gastro-esophageal reflux disease (47.6%)
anti-inflammatory and antirheumatic products, nonsteroids (42.1%)
drugs for constipation (41.3%).
Subsequent Treatments
Thirty-three patients received subsequent anti-cancer therapy after treatment with dostarlimab. Of these patients, 27 had progressive disease. These therapies, from most to least frequent, included the following:
radiotherapy (7.8%)
single-drug chemotherapy, such as doxorubicin (4.7%), carboplatin (4%), paclitaxel (4%), and pegylated liposomal doxorubicin (4%)
immunotherapy, such as pembrolizumab (4.0%) and bevacizumab (2.4%)
hormone therapy, such as letrozole (3.1%), megestrol acetate (0.8%) medroxyprogesterone (0.8%), tamoxifen (0.8%), temsirolimus (0.8%)
combination chemotherapy, such as paclitaxel plus carboplatin (2.4%) and carboplatin plus gemcitabine (1.6%)
surgery (0.8%).
Outcomes
Only outcomes and analyses of the subgroups identified in the review protocol are reported below. Data tables for the subset of patients from IA-1 included in IA-2 (n = 72) can be found in Appendix 4. In addition, data tables for all immune-related outcomes (except irPFS) are available in Appendix 4. As part of the sponsor’s feedback on this CADTH reimbursement review report, the sponsor provided CADTH with an updated analysis (data cut-off of November 1, 2021) for certain baseline characteristics, efficacy, and safety outcomes in the GARNET trial. The results of the updated analysis were, overall, consistent with those reported in the previous analyses of data from the March 1, 2020, cut-off date. The additional results from the November 1, 2021, data cut-off date are available in Appendix 5.
Overall Survival
The results for OS from cohort A1 of the GARNET trial are summarized in Table 13. The median OS was not reached at the time of IA-2. The median follow-up time was 16.3 months. In total, there were 35 deaths (33.3%), and 70 patients (66.7%) were censored. The probability of patients in cohort A1 surviving to 6, 9, and 12 months was 80.9% (95% CI, 71.7% to 87.4%), 75.1% (95% CI, 65.2% to 86.6%), and 68.9% (95% CI, 58.3% to 77.4%), respectively.
Table 13: KM Analysis of OS for Patients With dMMR or MSI-H EC in Cohort A1 of the GARNET Trial (IA-2, Primary Efficacy Analysis Dataset)
Outcome measure | Cohort A1a (N = 105) |
|---|---|
OS status, n (%) | |
Deaths | 35 (33.3) |
Censored | 70 (66.7) |
OS (months) | |
Quartile (95% CI)b | |
25% | 9.3 (5.2 to 15.4) |
50% | NR (17.1 to NR) |
75% | NR (NR to NR) |
OS distribution function (95% CI) | |
Month 6 | 80.9 (71.7 to 87.4) |
Month 9 | 75.1 (65.2 to 82.6) |
Month 12 | 68.9 (58.3 to 77.4) |
CI = confidence interval; dMMR = mismatch repair-deficient; EC = endometrial cancer; KM = Kaplan-Meier; MSI-H = microsatellite instability-high; NR = not reported; OS = overall survival.
aThe population total (N = 129) included 126 dMMR EC patients and 3 MMR-unk/MSI-H EC patients. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 3) with MMR-unk/MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
b95% CIs were generated using the method of Brookmeyer and Crowley. Biometrics, 1982;38:29-41.
Source: Clinical Study Report.18
Figure 3: KM Plot of OS for Patients With dMMR or MSI-H EC per RECIST 1.1 (IA-2, Secondary Efficacy Analysis Dataset)
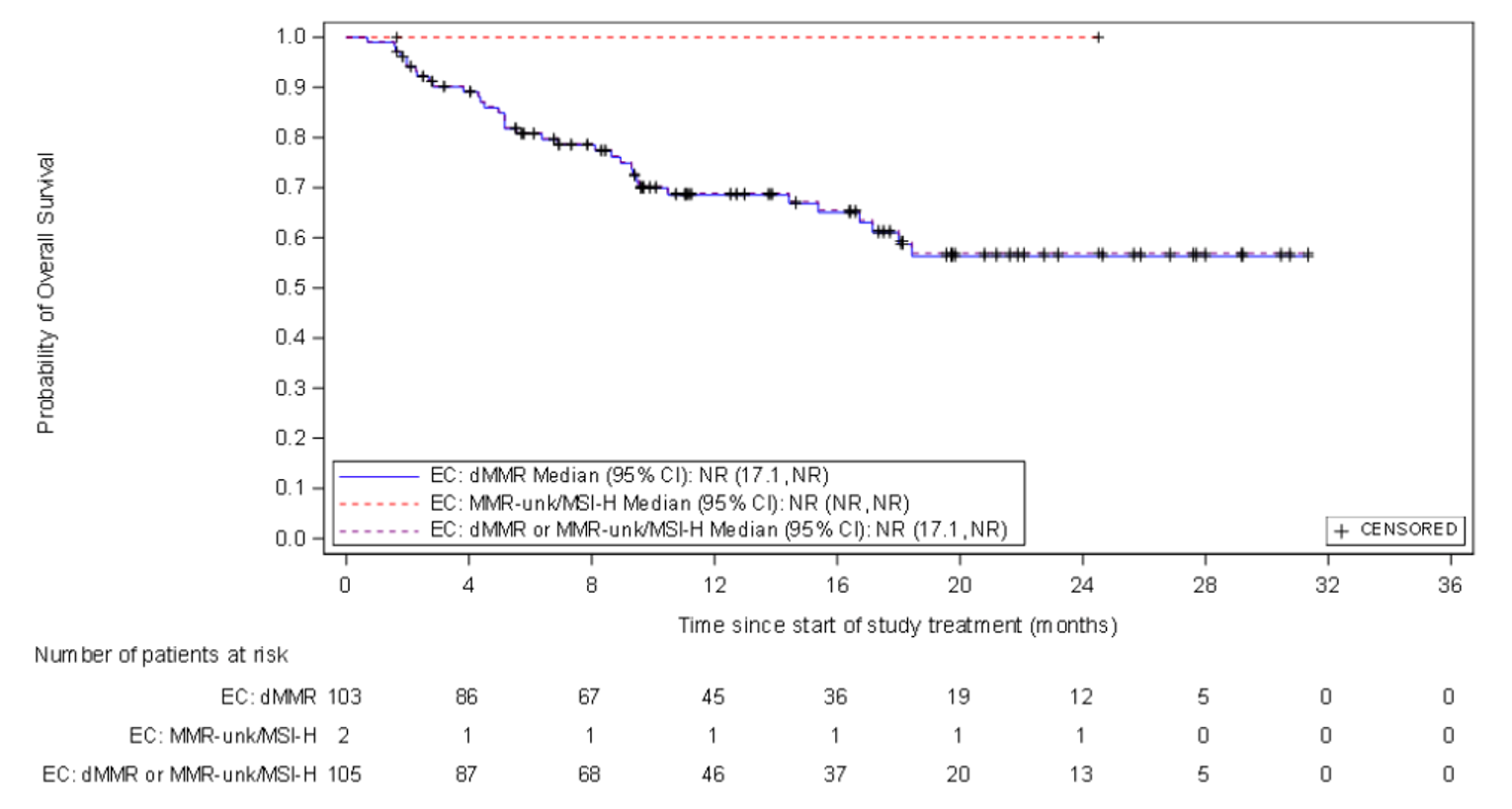
CI = confidence interval; MMR = mismatch repair-deficient; EC = endometrial cancer; IA-2 = second interim analysis; KM = Kaplan-Meier; MMR-unk = unknown mismatch repair tumour status; MSI-H = microsatellite instability-high; OS = overall survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; NR = not reported.
Source: Clinical Study Report18
Disease Control Rate and Immune-Related Disease Control Rate
The DCR results (based on investigator assessment) of the GARNET trial for cohort A1 are summarized in Table 14. The DCR was 57.1%, with 11 patients (10.5%) having a BOR of CR, 36 patients (34.3%) having a BOR of PR, and 13 patients (12.4%) having a BOR of stable disease. Forty-two (89.4%) patients at the time of IA-2 had an ongoing response.
irDCR by investigator assessment per irRECIST 1.1 (n = 113) was 63.7% for patients with dMMR or MSI-H EC, which was slightly higher than the DCR. Eight patients (7.1%) had an irBOR of irCR, 44 patients (38.9%) had an irBOR of irPR, and 20 patients (17.7%) had an irBOR of irSD. Forty-three (82.7%) patients at the time of IA-2 had an ongoing response. Detailed irDCR results are available in Appendix 4.
Table 14: Tumour Response in Patients With dMMR or MSI-H EC in Cohort A1 of the GARNET Trial per RECIST 1.1 Assessed by BICR (IA-2, Primary Efficacy Analysis Dataset)
Outcome measure | Cohort A1a (N = 105) |
|---|---|
BOR by RECIST 1.1, n (%) | |
CR | 11 (10.5) |
PR | 36 (34.3) |
Stable disease | 13 (12.4) |
Progressive disease | 39 (37.1) |
NE | 3 (2.9) |
Not done | 3 (2.9) |
Confirmed ORR by RECIST 1.1, n (%) | 47 (44.8) |
95% CIb | 35.0 to 54.8 |
Response ongoing, n (%)c | 42 (89.4) |
DCR by RECIST 1.1, n (%) | 60 (57.1) |
95% CIb | 47.1 to 66.8 |
BICR = blinded independent central review; BOR = best overall response; CI = confidence interval; CR = complete response; DCR = disease control rate; dMMR = mismatch repair-deficient; EC = endometrial cancer; MSI-H = microsatellite instability-high; NE = not evaluable; ORR = objective response rate; PR = partial response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Note: ORR was defined as the percentage of patients with a RECIST 1.1–confirmed CR or PR. DCR was defined as the percentage of patients with a RECIST 1.1–confirmed PR, confirmed CR, or stable disease. Response assessments were based on BICR.
aThe population total (N = 105) included 103 dMMR EC patients and 2 MMR-unk/MSI-H EC patients. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 2) with MMR-unk/MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
bExact 2-sided 95% CI for the binomial proportion.
cAll responders who have not yet died or progressed (including clinical progression); the denominator for the percentage is the number of responders.
Source: Clinical Study Report.18
Health-Related Quality of Life
EORTC QLQ-C30
EORTC QLQ-C30 data were available for 94 of the 129 patients in cohort A1 of the GARNET trial at the time of IA-2. Completion rates for the EORTC QLQ-C30 instrument declined over time. The completion rate at baseline was 100% and 58.5% by cycle 7 (n = 55). The mean scores and mean changes from baseline at each assessment point for EORTC QLQ-C30 (global health status or quality-of-life scale) are summarized in Table 15 and Figure 4. A definition for what constituted a clinically meaningful change from baseline in the study population was not provided. Overall, summary scores increased or remained stable from baseline to cycle 7 among patients who completed questionnaires.
Table 15: Summary of EORTC QLQ-C30 Results — Global Health Status and QoL Function From Baseline to Cycle 7 (IA-2, Safety Analysis Dataset)
Study time point | n | Mean (SD) | Mean change from baseline (SD) |
|---|---|---|---|
Baseline | 94 | 63.3 (21.71) | — |
Cycle 2 | 81 | 65.3 (20.07) | 2.9 (18.06) |
Cycle 3 | 78 | 67.8 (19.42) | 4.7 (20.96) |
Cycle 4 | 74 | 69.2 (18.85) | 6.5 (24.43) |
Cycle 5 | 64 | 71.2 (19.87) | 6.4 (23.51) |
Cycle 6 | 48 | 73.2 (18.57) | 9.7 (24.14) |
Cycle 7 | 40 | 73.1 (18.12) | 10.2 (23.77) |
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; IA-2 = second interim analysis; QoL = quality of life; SD = standard deviation.
A summary of the disease-related symptom subscales can be found in Figure 5. Patients who received dostarlimab and completed the questionnaire reported decreased symptoms or remained stable over time in key disease-related symptom subscales, including pain and fatigue.
Mean change in symptomatic AE response from baseline over time is illustrated in Figure 6. Most patients who experienced symptomatic AEs, including nausea, vomiting, constipation, diarrhea, or tiredness, remained stable or had improvement from baseline in these symptoms over the treatment course. Less than 25% of patients reported single-category worsening in these AE symptoms, and less than 6% reported 2- or 3-category worsening.
Figure 4: Mean Change in Global Health Status and QoL From Baseline (IA-2, Safety Analysis Dataset)
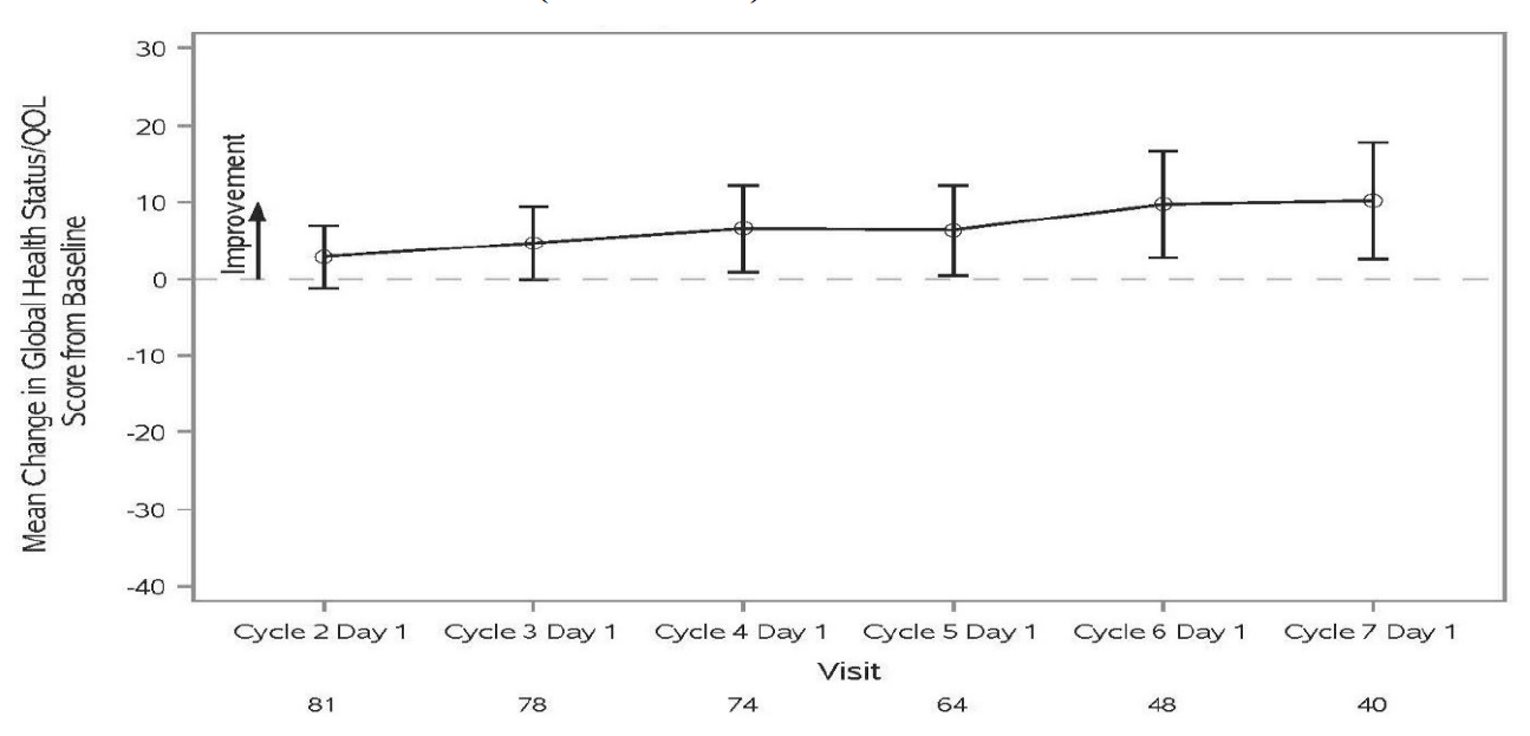
IA-2 = second interim analysis; QoL = quality of life.
Figure 5: Pain, Fatigue, and Physical Functioning Mean Change From Baseline (IA-2, Safety Analysis Dataset)
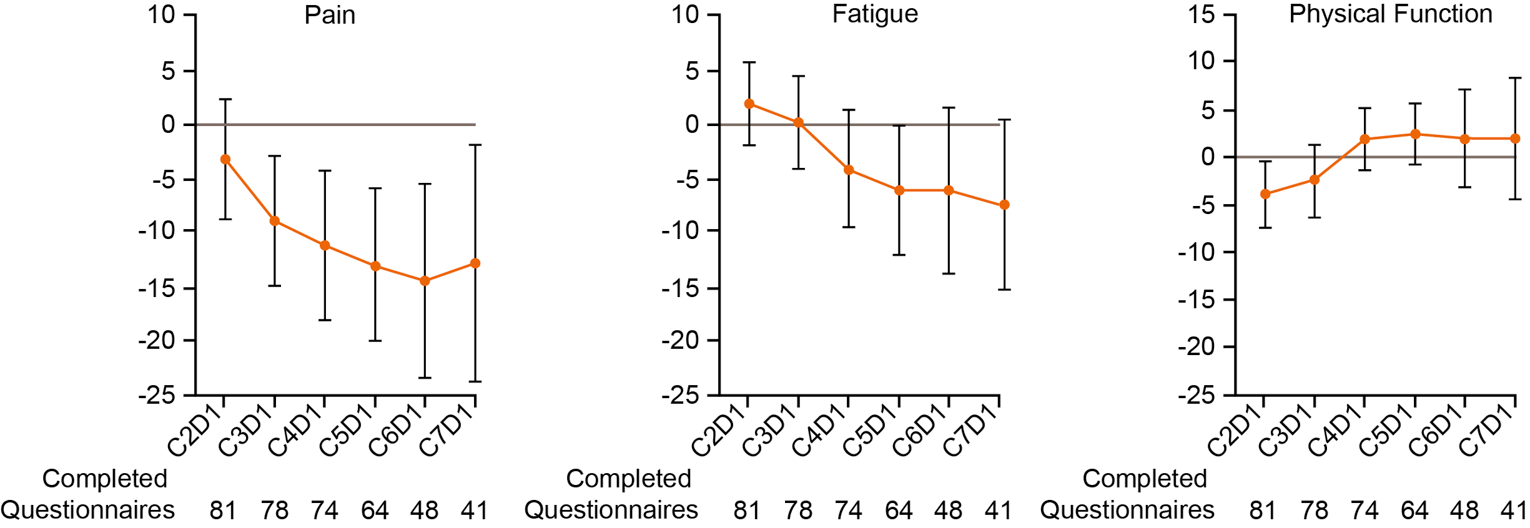
CxDx: cycle X, day X; IA-2 = second interim analysis.
Figure 6: Symptomatic AE Change in Response From Baseline (IA-2, Safety Analysis Dataset)
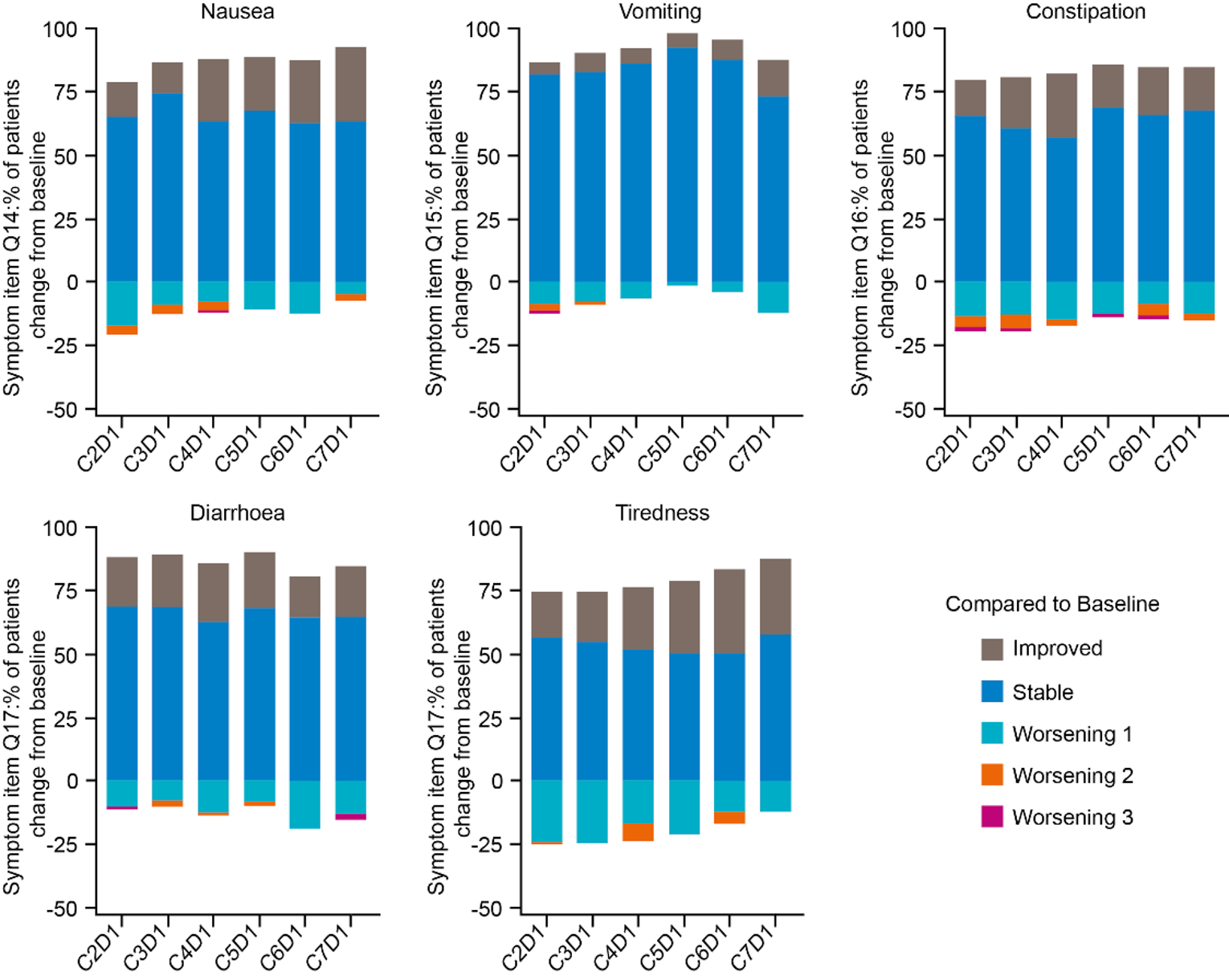
AE = adverse event; CxDx = cycle X, day X; IA-2 = second interim analysis; Q = question.
EQ-5D-5L
EQ-5D-5L data were available for 89 of 129 patients in cohort A1 at the time of IA-2. Summary data for the descriptive system of the EQ-5D-5L were not reported for any of the 5 dimensions. Overall, completion rates for the EQ-5D-5L and corresponding EQ-VAS instrument declined over time; there were 89 patients who completed questionnaires at baseline and 5 or fewer patients at each post-treatment visit.
EQ-VAS
At baseline, the mean EQ-VAS score was 69.3 (SD = 19.2) (refer to Figure 7). The following changes were observed in the EQ-VAS scores over time:
At week 12, the mean score was 77.1 (SD = 18.0) and mean change from baseline was 5.0 (SD = 12.6).
At week 18, mean score was 77.4 (SD = 17.4) and mean change from baseline was 4.0 (SD = 15.2).
At week 42, the change from baseline was 4.0 (SD = 16.2).
Greater change in scores was observed after EOT; however, the number of patients at each post-treatment visit was markedly low (< 5 patients).
Figure 7: Adjusted Mean Change From Baseline in EQ-VAS (IA-2, Safety Analysis Dataset)
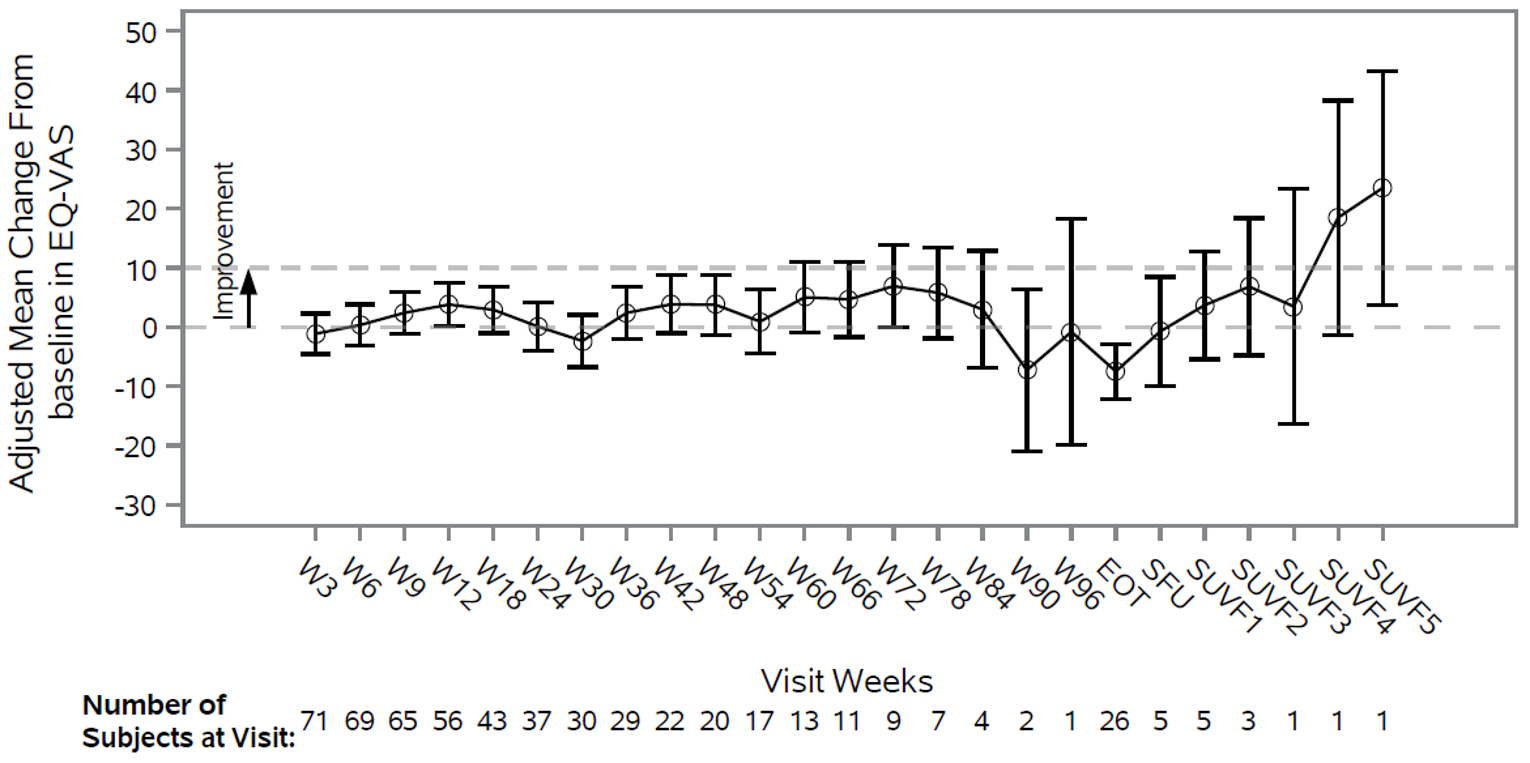
EOT = end of treatment; EQ-VAS = EQ-5D-5L visual analogue scale; IA-2 = second interim analysis; SUVF: survival follow-up; WX = week X.
Note: Data for the adjusted mean and 95% CI are derived from mixed model repeated measures with week of visit and ECOG Performance Status as factors and baseline score as continuous covariate as well as an unstructured covariance structure.
Progression-Free Survival and Immune-Related Progression-Free Survival
The results for PFS in cohort A1 of the GARNET trial, based on BICR, are summarized in Table 16. At IA-2, 57 (54.3%) patients had a PFS event and median PFS was 5.5 (95% CI, 3.2 to NR) months. KM estimates of PFS by RECIST 1.1 were 48.6% (95% CI, 38.6% to 56.8%) at month 6 and 47.5% (95% CI, 37.4% to 56.8%) at month 9 and at month 12. Refer to Figure 8 for the PFS KM curve.
In terms of irPFS, 57.3% of patients had an irPFS event at the time of IA-2. The median irPFS time was 10.3 months (95% CI 5.2 to 18.0). Based on investigator assessment, the probability of having irPFS based on KM estimates were 54.7% at 6 months, 50.7% at 9 months, and 47% at 12 months for patients with dMMR or MSI-H EC (95% CI not reported). KM estimates for irPFS were higher than that of PFS by BICR. Refer to Figure 13 in Appendix 4 or the irPFS KM curve.
Table 16: KM Analysis of PFS in Patients With dMMR or MSI-H EC in Cohort A1 of the GARNET Trial per RECIST 1.1 Assessed by BICR (IA-2, Primary Efficacy Analysis Dataset)
Outcome measure | Cohort A1a (N = 105) |
|---|---|
PFS status, n (%) | |
Events observed | 57 (54.3) |
Censored | 48 (45.7) |
PFS (months) | |
Quartile (95% CI)b in months | |
25% | 2.7 (2.5 to 2.9) |
50% | 5.5 (3.2 to NR) |
75% | NR (NR to NR) |
PFS distribution function (95% CI) | |
Month 6 | 48.6 (38.6 to 57.9) |
Month 9 | 47.5 (37.4 to 56.8) |
Month 12 | 47.5 (37.4 to 56.8) |
BICR = blinded independent central review; CI = confidence interval; dMMR = mismatch repair-deficient; EC = endometrial cancer; IA-2 = second interim analysis; KM = Kaplan-Meier; MSI-H = microsatellite instability-high; NR = not reported; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Note: PFS per RECIST 1.1 was based on BICR.
aThe population total (N = 105) included 103 dMMR EC patients and 2 MMR-unk/MSI-H EC patients. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 2) with MMR-unk/MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
b95% CIs were generated using the method of Brookmeyer and Crowley. Biometrics, 1982;38:29-41.
Source: Clinical Study Report.18
Figure 8: KM Plot for PFS in Patients With dMMR or MSI-H EC per RECIST 1.1 Assessed by BICR (IA-2, Primary Efficacy Analysis Dataset)
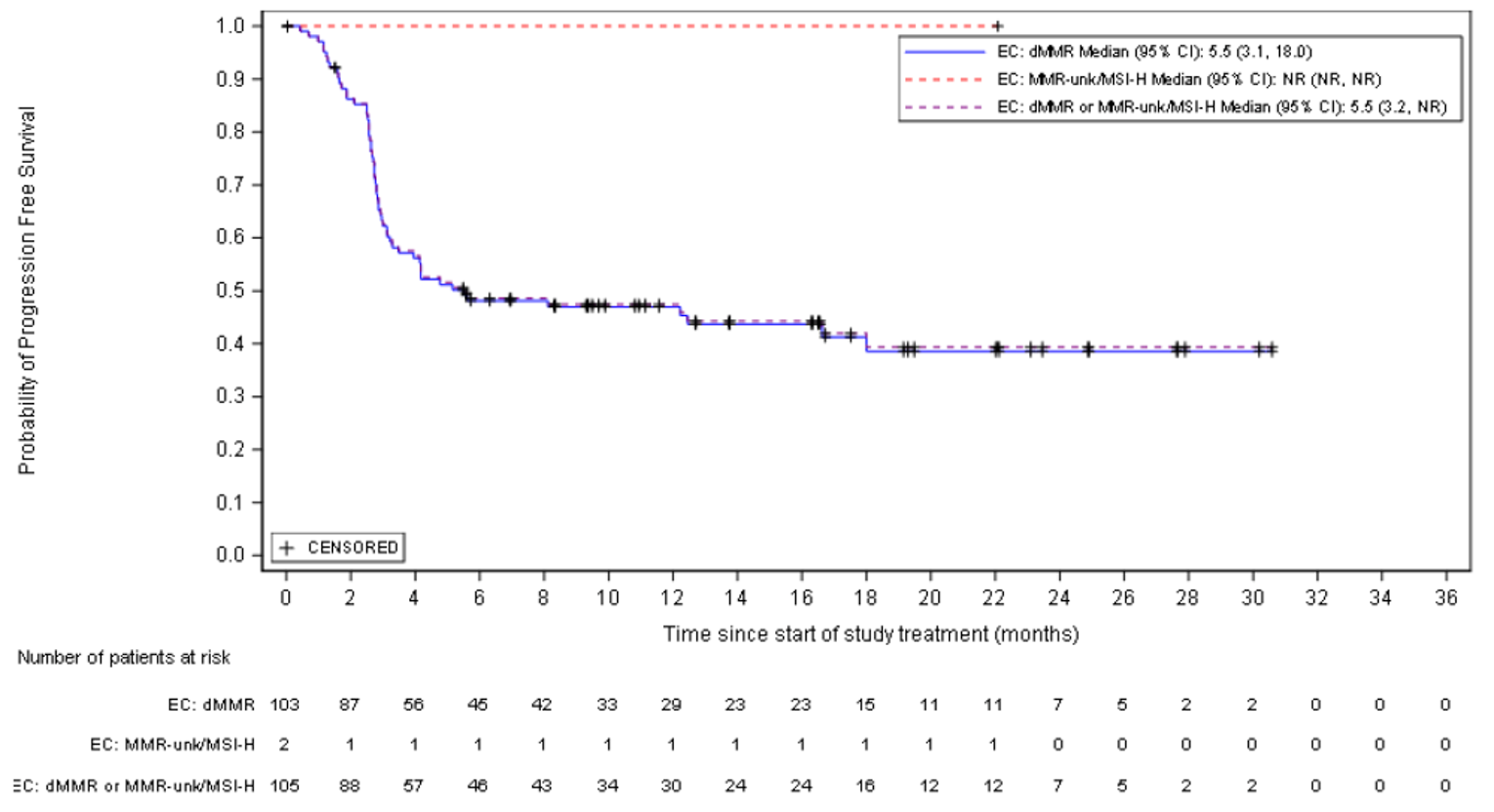
BICR = blinded independent central review; CI = confidence interval; dMMR = mismatch repair-deficient; EC = endometrial cancer; IA-2 = second interim analysis; KM = Kaplan-Meier; MMR-unk = unknown mismatch repair tumour status; NR = not reported; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Source: Clinical Study Report.18
Objective Response Rate and Immune-Related Objective Response Rate
The results for ORR (based on BICR) for cohort A1 of the GARNET trial are summarized in Table 12. The ORR results were generally consistent with the results for irORR.
At the time of IA-2, the ORR, per RECIST 1.1, was 44.8% (95% CI, 35.0% to 54.8%); 47 of 105 patients achieved either a CR (n = 11) or PR (n = 36) (Table 14). The median follow-up time was 16.3 months. The ORR results were generally consistent with the results of irORR; the irORR, per irRECIST 1.1, was 46% (95% CI, 36.6% to 55.6%), with 82.7% of responders having an ongoing immune-related response at the time of IA-2.
The ORR results for pre-specified patient subgroups of interest to the CADTH review are summarized in Table 17. The ORR was 49.2% in patients with 1 line of prior anti-cancer therapy, compared with 36.8% in patients who received 2 or more lines of prior anti-cancer therapy. The ORR was 46.6% in patients with prior radiation, compared with 40.0% in patients who had not received prior radiation. The ORR was 46.0% in patients with a progression-free interval of at least 6 months, compared with 39.5% in patients with a progression-free interval of less than 6 months. The ORR was 56.3% in patients with type II endometrioid carcinoma and 40.0% in patients with type I endometrioid carcinoma. Note that 95% CIs were not provided for any of the ORR subgroup analyses.
Table 17: Subgroup Analysis Results for ORR in Cohort A1 of the GARNET Trial (IA-2, Primary Efficacy Analysis Dataset)
Subgroups | GARNET cohort A1, n N = 105 | ORR (%)a |
|---|---|---|
Number of prior anti-cancer therapy regimens | ||
1 | 65 | 49.2 |
≥ 2 | 38 | 36.8 |
Prior radiation therapy, n | ||
Yes | 73 | 46.6 |
No | 30 | 39.5 |
Progression-free interval from most recent platinum-containing prior anti-cancer therapy | ||
≥ 6 months | 63 | 46.0 |
< 6 months | 38 | 39.5 |
Histology | ||
Type II endometrioid carcinoma | 32 | 56.3 |
Type I endometrioid carcinoma | 70 | 40.0 |
IA-2 = second interim analysis; ORR = objective response rate.
a95% CIs were not provided for any of the subgroup analyses.
The sponsor noted that there were too few patients of each endometrioid carcinoma type II subtype to make a meaningful comparison based on histologic subtype.
Duration of Response and Immune-Related Duration of Response
The DOR results (based on BICR) for cohort A1 of the GARNET trial are summarized in Table 18.
Of the 47 patients who achieved an objective response, 42 (89.4%) had an ongoing response at the time of the IA-2 data cut-off. The median duration of follow-up was 16.3 months; however, the median DOR was not reached among responders. The DOR in these patients ranged from 2.63 to 28.09 months as at the time of the IA-2data cut-off, with 37 patients (78.7% of responders) achieving a DOR of at least 6 months. Based on these results, the probability of responders maintaining a confirmed objective response, per RECIST 1.1, was estimated to be 97.9% (95% CI, 85.8% to 99.7%), 90.9% (95% CI, 73.7% to 97.1%), and 80.1% (95% CI, 56.8% to 91.7%) at months 6, 12, and 18, respectively.
The sponsor noted that the small sample size of responders hampered the ability to estimate the DOR of patient subgroups, so these data were not reported.
The results of investigator-assessed irDOR were generally consistent with DOR assessed by BICR (refer to Appendix 4 for further details). At the time of the IA-2, 43 patients had an ongoing immune-related response out of the 52 who achieved an irCR or irPR. The median irDOR was not reached among responders after a median follow-up of 16.5 months. Approximately 76.9% of responding patients achieved an irDOR of at least 6 months. Based on KM estimates, the probability of maintaining a response, per irRECIST, at 6 months was 96.1% (95% CI, 85.2 to 99.0) and at 12 months and at 18 months was 79.2% (95% CI, 62.1% to 89.2%).
Table 18: KM Analysis of DOR for Patients With dMMR or MSI-H EC in Cohort A1 of the GARNET Trial per RECIST 1.1 Assessed by BICR (IA-2, Primary Efficacy Analysis Dataset of Patients With Objective Response)
Outcome measure | Cohort A1a (N = 47) |
|---|---|
Follow-up duration (months), median | 16.3 |
DOR status, n (%) | |
Events observed | 5 (10.6) |
Censored | 42 (89.4) |
DOR (months), range | 2.63 to > 28.09 |
Quartile (95% CI)b | |
25% | NR (9.8 to NR) |
50% | NR (NR to NR) |
75% | NR (NR to NR) |
Duration ≥ 6 months, n (%) | 37 (78.7) |
DOR distribution function (95% CI) | |
Month 6 | 97.9 (85.8 to 99.7) |
Month 12 | 90.9 (73.7 to 97.1) |
Month 18 | 80.1 (56.8 to 91.7) |
BICR = blinded independent central review; CI = confidence interval; dMMR = mismatch repair-deficient; DOR = duration of response; EC = endometrial cancer; KM = Kaplan-Meier; MSI-H = microsatellite instability-high; NR = not reported; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Note: DOR, per RECIST 1.1, was based on BICR. A “>” sign indicates that patients’ response is ongoing.
aThe population total (N = 47) included 46 dMMR EC patients and 1 MMR-unk/MSI-H EC patients with an objective response. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 1) with MMR-unk/ MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
b95% CIs were generated using the method of Brookmeyer and Crowley. Biometrics, 1982;38:29-41.
Harms
Only those harms identified in the CADTH review protocol are reported in this section. A summary of harms data is provided in Table 19.
Adverse Events
The most frequently reported TEAEs (≥ 15%) in patients with dMMR or MSI-H EC were nausea, diarrhea, anemia, fatigue, asthenia, constipation, vomiting, abdominal pain, cough, arthralgia, urinary tract infection, and back pain. With the exception of anemia, these TEAEs were reported to be mild or moderate in severity in most patients.
Grade 3 or higher TEAEs occurred in 48.1% of patients in cohort A1. The most commonly reported grade 3 or higher TEAEs were anemia (14.7%), abdominal pain (5.4%), and hyponatremia (3.9%). Grade 4 TEAEs were reported for hyponatremia, pulmonary embolism, pneumonia, and sepsis. Sepsis was reported as a grade 4 TEAE in 3 patients with dMMR EC and 1 patient with MMR-unk/MSI-H. Other grade 3 or higher TEAEs included acute kidney injury (3.1%) and back pain (3.1%).
Serious Adverse Events
The percentage of patients that experienced a serious TEAE was 34.1% in cohort A1. The most common serious TEAEs were abdominal pain, acute kidney injury, and sepsis, each occurring in 3.1% of patients, and pulmonary embolism, pyrexia, and urinary tract infection, each occurring in 2.3% of patients.
Withdrawals Due to AEs
None of the patients in cohort A1 who withdrew from the GARNET trial had an AE as a primary reason. AEs that led to study treatment discontinuation occurred in 11.6% of patients, whereas AEs that led to study treatment interruption occurred in 24.0% of patients. The most common AEs leading to study treatment interruption were anemia (3.1%) and diarrhea (2.3%).
Mortality
TEAEs leading to death occurred relatively rarely in cohort A1 during the treatment period (n = 1; 0.8%) and included aspiration. TEAEs leading to death during the 90-day safety follow-up occurred in 4 patients (3.1%), and included pleural effusion, pneumonia, sepsis, and shock. None of the TEAEs leading to death were considered by the investigators to be treatment-related. No TEAEs were the primary cause of death during the long-term follow-up period.
Notable Harms
Notable harms as specified in the CADTH review protocol included immune-related toxicity.
The incidence of irAEs was 34.9% in cohort A1. The most frequently reported irAEs (≥ 5%) were diarrhea and hypothyroidism. A total of 7.9% of patients had a serious irAE, 12.7% had an irAE that was grade 3 or higher, and 4.8% of patients with dMMR EC had an irAE that led to study treatment discontinuation. Most of the irAEs were considered by the investigators to be related to study treatment.
Table 19: Summary of Harms in Cohort A1 of the GARNET Trial
Category | Cohort A1 (N = 129)a |
|---|---|
TEAEs experienced by ≥ 15% of patients, n (%) | |
Any TEAEsb | 123 (95.3) |
Most common events | |
Nausea | 42 (32.6) |
Diarrhea | 36 (27.9) |
Anemia | 35 (27.1) |
Fatigue | 32 (24.8) |
Asthenia | 28 (21.7) |
Constipation | 25 (19.4) |
Vomiting | 24 (18.6) |
Abdominal pain | 21 (16.3) |
Cough | 21 (16.3) |
Arthralgia | 20 (15.5) |
Urinary tract infection | 20 (15.5) |
Back pain | 19 (14.7) |
Most common grade ≥ 3 TEAEs, n (%) | |
Any | 62 (48.1) |
Anemia | 19 (14.7) |
Abdominal pain | 7 (5.4) |
Hyponatremia | 5 (3.9) |
Acute kidney injury | 4 (3.1) |
Back pain | 4 (3.1) |
Pulmonary embolism | 4 (3.1) |
Sepsis | 4 (3.1) |
Alanine aminotransferase increased | 3 (2.3) |
Diarrhea | 3 (2.3) |
Hypertension | 3 (2.3) |
Lipase increased | 3 (2.3) |
Pneumonia | 3 (2.3) |
Urinary tract infection | 3 (2.3) |
SAEs experienced by ≥ 2 patients, n (%) | |
Any SAEs | 44 (34.1) |
Abdominal pain | 4 (3.1) |
Acute kidney injury | 4 (3.1) |
Sepsis | 4 (3.1) |
Pulmonary embolism | 3 (2.3) |
Pyrexia | 3 (2.3) |
Urinary tract infection | 3 (2.3) |
Bronchitis | 2 (1.6) |
Colitis | 2 (1.6) |
General physical health deterioration | 2 (1.6) |
Intestinal obstruction | 2 (1.6) |
Pain | 2 (1.6) |
Pneumonia | 2 (1.6) |
Pyelonephritis | 2 (1.6) |
Tumour pain | 2 (1.6) |
Treatment-related TEAEs, n (%) | |
Any treatment-related TEAE | 82 (63.6) |
Diarrhea | 21 (16.3) |
Asthenia | 18 (14.0) |
Fatigue | 17 (13.2) |
Nausea | 16 (12.4) |
Arthralgia | 11 (8.5) |
Pruritus | 11 (8.5) |
Anemia | 9 (7.0) |
Hypothyroidism | 9 (7.0) |
Rash | 7 (5.4) |
Discontinued treatment due to TEAEs, n (%) | |
Any TEAE leading to discontinuation of study treatment | 15 (11.6) |
Alanine aminotransferase increased | 2 (1.6) |
Transaminases increased | 2 (1.6) |
Acute kidney injury | 1 (0.8) |
Apraxia | 1 (0.8) |
Aspartate aminotransferase increased | 1 (0.8) |
Aspiration | 1 (0.8) |
Bronchitis | 1 (0.8) |
Gamma-glutamyl transferase increased | 1 (0.8) |
Intestinal obstruction | 1 (0.8) |
Pancreatitis | 1 (0.8) |
Pleural effusion | 1 (0.8) |
Pneumonia | 1 (0.8) |
Pneumonitis | 1 (0.8) |
Sepsis | 1 (0.8) |
Interrupted treatment due to TEAEs, n (%) | |
Any AE leading to study treatment interruption | 31 (24.0) |
Anemia | 4 (3.1) |
Diarrhea | 3 (2.3) |
Colitis | 2 (1.6) |
Gastroenteritis radiation | 2 (1.6) |
Lipase increased | 2 (1.6) |
Pneumonia | 2 (1.6) |
Death, n (%) | |
Death during the treatment period | 6 (4.7) |
Primary reason for death | |
Progressive disease | 5 (3.9) |
AE | 1 (0.8) |
Death during the 90-day safety follow-up periodc | 13 (10.1) |
Primary reason for death | |
Progressive disease | 9 (7.0) |
AE | 4 (3.1) |
Death during the long-term follow-up periodd | 17 (13.2) |
Primary reason for death | |
Progressive disease | 17 (13.2) |
Notable harms, n (%) | |
Immune-related reaction | 45 (34.9) |
Diarrhea | 11 (8.5) |
Hypothyroidism | 9 (7.0) |
Pruritus | 4 (3.1) |
Alanine aminotransferase increased | 4 (3.1) |
Blood creatinine increased | 4 (3.1) |
Hyperthyroidism | 4 (3.1) |
Lipase increased | 4 (3.1) |
Amylase increased | 3 (2.3) |
Aspartate aminotransferase increased | 3 (2.3) |
Colitis | 3 (2.3) |
Transaminases increased | 3 (2.3) |
Hyperglycemia | 2 (1.6) |
Pneumonitis | 2 (1.6) |
Adrenal insufficiency | 1 (0.8) |
Arthritis | 1 (0.8) |
Facial paresis | 1 (0.8) |
Gastritis | 1 (0.8) |
Gastroenteritis | 1 (0.8) |
Iridocyclitis | 1 (0.8) |
Nephritis | 1 (0.8) |
Neuropathy peripheral | 1 (0.8) |
Pancreatitis | 1 (0.8) |
Pancreatitis, acute | 1 (0.8) |
Pemphigoid | 1 (0.8) |
Rash maculopapular | 1 (0.8) |
AE = adverse event; SAEs = serious adverse event; TEAE = treatment-emergent adverse event.
Note: AEs were coded using Medical Dictionary for Regularity Activities version 23.0. For each preferred term, a patient was included only once, even if they experienced multiple events in that preferred term. TEAEs are new AEs that began, or any pre-existing condition that worsened in severity, after at least 1 dose of study treatment was administered and throughout the treatment period until 90 days after the EOT visit (or until the start of alternate anti-cancer therapy, whichever occurred first).
aThe population total (N = 129) included 126 dMMR EC patients and 3 MMR-unk/MSI-H EC patients. Data from patients with dMMR tumours and MMR-unk/MSI-H tumours are presented as a total due to the small proportion of patients (n = 3) with MMR-unk/MSI-H tumours; the sponsor noted that data remained similar when these patient groups were pooled together.
bTEAEs experienced by ≥ 5% of patients with dMMR or MSI-H EC by preferred term (safety analysis dataset).
cIn the 90 days after the EOT visit or until the first follow-up anti-cancer therapy, whichever occurred first.
d90 days after the EOT visit or until the first follow-up anti-cancer therapy, whichever occurred first.
Source: Clinical Study Report.18
Critical Appraisal
Internal Validity
The main limitation of the GARNET trial stems from the single-arm trial design and the lack of a comparator group. Such a design makes it challenging to interpret the efficacy and safety events attributable to dostarlimab, because all patients in cohort A1 received the same treatment. The lack of comparison with an active comparator or with standard of care or placebo precludes the ability to assess the relative therapeutic benefit or safety of dostarlimab. Formal statistical significance and hypothesis-testing were not performed, except for ORR. The risk of selection bias cannot be ruled out, given the lack of transparency regarding the selection and timing of the enrolment of patients in the trial. In the absence of such information, the risk of bias is unclear. GARNET is an open-label trial, and the study investigators and patients were aware of their treatment status, which increases the risk of detection and performance bias; this has the potential to influence results and outcomes in favour of dostarlimab if the assessor (investigator or patient) believes the study drug is likely to provide a benefit. Furthermore, if study personnel and patients knew that the treatment was dostarlimab (which is known to cause irAEs and other neurotoxicity), this also could have influenced the reporting of harms outcomes. To mitigate the impact of such bias, the investigators used a BICR to evaluate responses using standardized criteria for certain efficacy outcomes, such as ORR, DOR, PFS, and DCR. Bias is therefore less a concern for these efficacy end points and OS, and more of a concern for subjective end points such as HRQoL and safety. Overall, the magnitude and direction of this bias remain unclear.
Median OS was not reached at the time of IA-2, so the survival data from the trial were immature. Because results were based on an interim analysis, the treatment benefit may be overestimated and harms may be underestimated. In addition, the potential for confounding cannot be ruled out, given the lack of adjustment in the analysis for known confounders and treatment-effect modifiers that would typically be accounted for in an RCT. Interpretation of time-to-event end points such as OS or PFS is limited in single-arm studies. The clinical experts agreed that in the absence of robust comparative data on PFS and OS, no firm conclusions could be drawn on how dostarlimab compares with other relevant treatment options.
The open-label, single-arm design also limits the ability to interpret HRQoL data from the GARNET trial. Loss of patients completing HRQoL questionnaires over the course of the study led to small sample sizes at many assessment time points (in some cases < 5 patients) and resulted in imprecise measurement, with wide and overlapping CIs. Statistical analyses of changes in scale scores over time were not conducted, and a definition of what was considered a clinically meaningful response or minimally important difference was not reported. As well, summary data were not reported for the EQ-5D-5L descriptive system. For these reasons, no conclusions should be drawn from the HRQoL data. This is considered a significant limitation of the GARNET trial, because HRQoL was identified as an important outcome by the patient and clinician groups providing input for this review.
External Validity
Overall, the clinical experts consulted by CADTH agreed that the inclusion and exclusion criteria, baseline patient characteristics, concomitant medications, and prohibited medications in cohort A1 of the GARNET trial were reflective of patients they see in clinical practice for the indication under review. There were no barriers to the identification of patients who would most benefit from the treatment, given that testing for MMR and MSI status is standard practice in Canada. The population enrolled in the trial was consistent with the population expected to be treated in Canadian clinical practice, but the clinical experts noted that patients in the GARNET trial were slightly younger than those seen in clinical practice. The clinical experts also noted that patients with certain comorbidities (e.g., obesity) and of certain racial groups (e.g., Black) may have poorer prognosis; however, the findings of this study may not necessarily be generalizable to these subgroups, given the small sample size and lower level of representation of ethnic groups in the study (n = 2). Furthermore, the clinical experts indicated that no different treatment effect would be expected based on different disease-management practices in participating countries. In the opinion of the clinical experts, as long as patients have dMMR or MSI-H tumour status, dostarlimab would be appropriate to administer after any of the prior therapies received by patients in the trial. The majority of patients (88%) in cohort A1 of the GARNET trial had received 2 or fewer prior lines of systemic therapy before trial enrolment, and 11% of patients had received 3 or more lines of prior systemic therapy. However, the clinical experts noted that experience from clinical practice suggests that dostarlimab may have a lower magnitude of treatment benefit in patients with more prior lines of systemic therapy. This aligns with the results of the ORR subgroup analysis, in which the ORR was higher in patients with 1 line of prior anti-cancer therapy than in patients with 2 or more lines of prior anti-cancer therapy (49.2% versus 36.8% [95% CI, NR to NR]). However, these results are exploratory, and conclusions should be interpreted with caution. The clinical experts and clinician groups agreed that patients should not have previously been treated with immunotherapies. In cohort A1, 3 patients could not have their MMR tumour status determined, but their tumours were classified as MSI-H. The clinical experts noted that these 2 groups are generally thought to be synonymous, and data could be pooled together, especially given the small number of patients with MSI-H status. Concomitant medications received by patients in the trial appeared to reflect the medications that patients would receive in Canadian clinical practice, according to the clinical experts. It is also acknowledged that mature OS data will be confounded by the use of subsequent anti-cancer therapy received by some patients after progressive disease.
According to the clinical experts consulted by CADTH, OS, PFS, and HRQoL are clinically meaningful end points for patients with dMMR or MSI-H recurrent or advanced EC that has progressed on or after treatment with a platinum-containing regimen. Tumour response outcomes are also important in this patient population because of the accompanying delay in the worsening of symptoms and the slower decline in ECOG performance status.
There were a limited number of patients in the primary efficacy dataset (N = 105). The magnitude of the treatment-effect estimates observed in a small study sample may not be replicable in a larger study sample or generalizable to the target population in real-world clinical practice. Furthermore, the subgroup analyses had no statistical comparisons and even smaller sample sizes, which limits the generalizability to a broader population. These data should be considered exploratory in nature, and no conclusions should be drawn based on the results.
Summary Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The objective of this section is to provide an appraisal and summary of the indirect evidence submitted by the sponsor, which included 6 reports of ITCs (3 reports of MAICs and 3 of IPTW analyses),19 which compare dostarlimab with other treatments used for patients with advanced or recurrent EC that has progressed on or after a platinum-based regimen. Results for the MAIC and IPTW analyses are presented separately.
A focused literature search for network meta-analyses dealing with EC was run in MEDLINE All (1946–) on November 3, 2021. No limits were applied to the search. The literature search identified 31 citations, none of which met the eligibility criteria. Given the lack of comparative evidence, and the phase I, single-arm nature of the GARNET trial, the sponsor-submitted ITCs were used to inform the pharmacoeconomic model. The submitted analyses were appraised and summarized.
Matching-Adjusted Indirect Comparisons
Description of MAIC Analyses
The sponsor submitted 3 MAIC reports to demonstrate the efficacy and safety of dostarlimab compared with relevant treatments for advanced or recurrent EC19:
Two MAIC reports that compared individual patient data (IPD) from the phase I GARNET trial with a GARNET-like real-world evidence (RWE) cohort in the UK that received current treatment paradigms (hereafter called MAIC report 1), and a selection of 5 specific treatments for advanced or recurrent EC (hereafter called MAIC report 2) from the National Cancer Registration and Analysis Service (NCRAS) database.19
One MAIC report that compared IPD from the phase I GARNET trial with relevant comparator data from the published literature (hereafter called MAIC report 3).19
Methods of MAIC Analyses
Objectives
In the absence of direct comparative evidence from trials, the aim of each analysis was to compare the efficacy and safety (e.g., OS, PFS, time on treatment, ORR, and/or SAEs) of dostarlimab with current UK treatment paradigms based on RWE cohorts and published literature, using the MAIC method.19
Study Selection Methods
The index trial in all cases was based on IPD from cohort A1 of part 2B of the phase I, single-arm GARNET trial (dMMR or MSI-H EC cohorts, safety population set; n = 129).19 Study selection methods to identify relevant comparators varied in the 3 sponsor-submitted MAIC reports.
For the 2 reports that used RWE as a comparator (MAIC reports 1 and 2), no systematic literature review (SLR) was conducted. Instead, the investigators created GARNET-like cohorts based on a subgroup of patients from the NCRAS in the UK, including a base-case analysis of current treatment paradigms, an analysis of 5 treatment-specific cohorts, and a sensitivity analysis of patients with an ECOG Performance Status of 0 or 1 at index. Notably, information on dMMR or MSI-H status (an inclusion criterion in GARNET and the indication under review) was not available in the RWE cohorts. In MAIC report 3, a SLR of clinical and observational studies was conducted in accordance with the highest standard for evidence synthesis (date last searched, April 5, 2020).51 Although there were no limitations to study eligibility by risk of bias, the authors noted that the included studies were generally at low risk of bias per the NICE tool,52 and bias was either high or unclear per the CASP tool.19,53
The selection characteristics used to develop the GARNET-like cohorts and the population, intervention, comparison, outcomes, and study (PICOS) criteria used to identify published literature are summarized in Table 20.
Table 20: Selection Criteria for GARNET-Like Subgroups and for Relevant Comparator Studies for Sponsor-Submitted MAICs
Variable | Selection criteria for GARNET-like subgroups for MAICs using NCRAS RWE | Selection criteria for studies for MAIC report 3 (published literature) |
|---|---|---|
Population | Patients with recurrent (for stage I and II at diagnosis, probable recurrence is captured by a gap of > 90 days between treatment and surgery or systemic anti-cancer therapy, or radiation therapy followed by treatment with any treatment) or advanced (stage III or IV) EC who meet all the eligibility criteria for GARNET As the data were unavailable in NCRAS, patients were eligible regardless of MMR or MSI status |
|
Interventions and comparators |
|
|
Outcomes |
|
|
Study design | Routine, linked patient-level health data available through NCRAS |
|
Publication characteristics | NA | For any study to be included in the analysis, a sample size of ≥ 30 will be required for each treatment arm |
AEs = adverse events; CR = complete response; DCR = disease control rate; DFS = disease-free survival; DOR = duration of response; EC = endometrial cancer; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-5D = EuroQol 5-Dimensions; HRQoL = health-related quality of life; MAIC = matching-adjusted indirect comparison; MMR = mismatch repair; MSI = microsatellite instability; NA = not applicable; NCRAS = National Cancer Registry Analysis System; ORR = objective response rate; OS = overall survival; PD-L1 = programmed death (ligand) 1; PFS = progression-free survival; PLD = pegylated liposomal doxorubicin; PR = partial response; RCT = randomized controlled trial; RWE = real-world evidence.
Source: Sponsor-Submitted ITC Reports19
The outcomes evaluated in the MAICs are summarized in Table 21. OS was the pre-specified primary outcome in all MAICs. In MAIC report 3, PFS was also included as a pre-specified primary outcome. PFS and TTD were the pre-specified secondary outcomes of MAIC reports 1 and 2. These were summarized descriptively, as the measurement definitions for PFS and the time period evaluations associated with the RWE cohort were too dissimilar to those in GARNET. As PFS was not recorded in the NCRAS database, TTNT and TTD were used as proxies. Pre-specified secondary end points in MAIC report 3 included ORR, DOR, HRQoL, and SAEs; however, no definitions were provided for these outcomes in the report.19
Table 21: End Point Definitions in Included Studies
End point | GARNET trial | MAIC reports 1 and 2 (NCRAS RWE cohorts) | MAIC report 3 (published literature) |
|---|---|---|---|
OS | Time from first dose of study treatment to death by any cause. Patients last known to be alive were censored at date of last known contact. | OS from second-line treatment defined as the time from the initiation of the second-line current treatment paradigm therapies (i.e., index date equal to start date of second-line therapy) until failure (all-cause death). Patients lost to follow-up or still alive at the end of the study period were censored. | NR |
PFS | Defined as the time from first dose to the previous date of assessment of progression or death by any cause in the absence of progression, based on the time of first documentation of progressive disease, per RECIST 1.1. | TTNT and TTD were used as proxies for PFS in the RWE cohorts. TTNT was defined as time from the start of therapy until failure (the earliest of all-cause death or the start of a new line of treatment). Patients lost to follow-up or still in same line of treatment at the end of the study period were censored. TTD was defined as time to discontinuation from second-line current treatment paradigms was defined as time at risk (in years) calculated from the initiation of second-line therapy to the earliest of: the end date of the first second-line drug administration that is followed by a gap of > 90 days before the resumption of drug administration; the end date of the first second-line drug administration, where a gap in treatment of > 90 days occurred without any resumption of drug administration; or death. | NR |
TTNT | NR | Refer to PFS | NR |
TTD | NR | Refer to PFS | NR |
ORR | NA | NA | NR |
SAE | NA | NA | NR |
MAIC = matching-adjusted indirect comparison; NA = not applicable; NR = not reported; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; RWE = real-world evidence; SAE = serious adverse event; TTD = time to treatment discontinuation; TTNT = time to next treatment.
Source: Sponsor-submitted ITC reports.19
MAIC Analysis Methods
Given that the GARNET trial was a single-arm trial, unanchored MAICs were necessary to conduct the ITCs because the single-arm nature precluded the use of anchored ITC methods. A summary of the analysis methods in each MAIC is provided in Table 23.
GARNET Versus NCRAS RWE Cohort MAIC Methods (MAIC Reports 1 and 2)
Analysis methods for the MAICs conducted using RWE data (MAIC reports 1 and 2) were identical, only differing in the RWE cohorts used. The GARNET trial was a phase I, open-label, dose-escalation study, whereas the RWE study was a descriptive, noninterventional study of patient-level data available through the NCRAS to describe characteristics, treatments, and outcomes of patients diagnosed with advanced or recurrent EC. The dataset for analysis consisted of IPD for all patients in the GARNET safety cohort (n = 129), and aggregated summary information from the initial RWE cohort of incident patients at second-line treatment (n = 999; n = 501 for patients with an ECOG Performance Status of 0 or 1 at index). Treatment-specific RWE cohorts included patients who received paclitaxel monotherapy (n = 116), carboplatin plus paclitaxel (n = 279), carboplatin plus liposomal doxorubicin (n = 141), liposomal doxorubicin monotherapy (n = 130), and carboplatin monotherapy (n = 93).19
Key differences were noted by the sponsor between the GARNET trial and the NCRAS RWE cohort’s inclusion criteria. One of the main differences between GARNET and the RWE cohort was the MMR or MSI status of patients. All patients in GARNET had dMMR or MSI-H EC, whereas this information was not available in the RWE cohort, so patients were included regardless of MMR or MSI status. Another key difference between GARNET and the RWE cohort’s inclusion criteria was that the RWE cohort used platinum doublet therapy as the key milestone regarding lines of therapy. The RWE cohort did not analyze the lines of therapy before the diagnosis of advanced or recurrent EC. The first-line therapy received for advanced or recurrent diagnosis was platinum doublet therapy for nearly all (99.3%) patients. Thus, the incident population at second-line therapy gave rise to the base case sample size (n = 999). Last, although all patients in GARNET had an ECOG Performance Status of 0 or 1, the inclusion criteria on ECOG Performance Status were partially met in the RWE cohort. In GARNET, ECOG Performance Status was ascertained at the index date of the trial (i.e., the start of second-line therapy), whereas in the RWE cohort, ECOG Performance Status was recorded at diagnosis. Also, there were many patients with ECOG Performance Status documented as “not recorded” in the RWE cohort. A sensitivity analysis conducted with the RWE cohort was restricted to patients with a known ECOG Performance Status of 0 or 1. This was referred to as the RWE cohort (ECOG ≤ 1).19
A total of 8 analyses (4 base-case and 4 scenario analyses) were conducted in the initial RWE cohort of MAIC report 1. Hazard ratios (HRs), 95% CIs, and corresponding P values were calculated only for the OS outcome (comparisons were descriptive for other outcomes), as follows19:
OS in GARNET (N = 129) versus OS in RWE cohort (base case, n = 999; ECOG ≤ 1, n = 501)
PFS in GARNET (N = 129) versus TTNT in RWE cohort (base case, n = 999; ECOG ≤ 1, n = 501)
PFS in GARNET (N = 129) versus TTD in RWE cohort (base case, n = 999; ECOG ≤ 1, n = 501)
TTD in GARNET (N = 129) versus TTD in RWE cohort (base case, n = 999; ECOG ≤ 1, n = 501).
A total of 4 analyses were performed for each treatment-specific RWE cohort in MAIC report 2. HRs, 95% CIs, and P values were calculated only for the OS outcome (comparisons were descriptive for other outcomes), as follows19:
OS in GARNET (N = 129) versus OS in treatment-specific RWE cohorts
PFS in GARNET (N = 129) versus TTNT in treatment-specific RWE cohorts
PFS in GARNET (N = 129) versus TTD in treatment-specific RWE cohorts
TTD in GARNET (N = 129) versus TTD in treatment-specific RWE cohorts.
A range of prognostic variables typically associated with survival in EC were identified in a targeted literature review (conducted May 2020), and validated through a panel of oncologists in Canada, Germany, and the UK, as follows19:
race and/or ethnicity (Black, other, unknown versus White)
age category (≥ 65 years versus < 65 years)
ECOG Performance Status at treatment initiation (1 versus 0)
histology at initial diagnosis (nonendometrioid, unknown versus endometrioid)
FIGO stage (stages III and IV versus stages I and II)
grade of disease at diagnosis (grades 3 and 4, unknown versus grades 1 and 2)
number of prior platinum-based therapies (0 or 1 versus ≥ 2)
prior surgery for study indication (yes versus no).
In MAIC report 1, assessment of the effect modification and prognostic value of each potential matching variable was done using a Cox proportional hazard model for each outcome (OS, PFS, and TTD). Cox regression models were fit separately for the GARNET data and the RWE data. Patient characteristics that exhibited an association at a level of significance of at least 0.1 in at least 1 of the 2 datasets were considered prognostic. P values evaluated in a multivariable model were obtained from a full model (including all variables listed above), and then backward stepwise selection, with a P ≤ 0.1 threshold, was applied. Effect-modifying status was assessed semi-empirically by comparing regression coefficients (or HRs) for the variable of interest from the 2 regression models fitted to the GARNET and the RWE dataset, where large differences between coefficients were considered to be potentially effect-modifying. The following variables were considered prognostic for each outcome19:
OS: ECOG, race and/or ethnicity, grade, histology, stage at diagnosis, and prior surgery
PFS: ECOG, and histology
TTNT: Stage at diagnosis, grade, and prior surgery
TTD: ECOG, race and/or ethnicity, grade, histology, and prior surgery.
The IPD from the GARNET trial were matched to the RWE cohort using the selected matching variables to estimate the above comparisons. A method of moments, as introduced by Signorovitch et al. (2010),54 was used to allow a propensity score logistic regression model to be estimated without IPD for the RWE cohort.19 After matching, re-weighted outcome data from the GARNET trial and RWE cohort were compared. This included calculating KM survival estimates for OS, PFS, TTD, and TTNT at given time points after index treatment initiation, as well as creating KM curves. The primary end point analysis used a Cox proportional hazard model, with weights obtained using the MAIC method, to re-estimate HRs for dostarlimab in the GARNET trial versus the RWE cohort. In the treatment-specific RWE cohort, weighted Cox proportional hazard models were fitted, and OS HRs for dostarlimab versus the 5 comparator treatments of interest were re-estimated, together with 95% CIs and P values. For use in these regression models, OS data from the RWE cohort were digitized, and pseudo-IPD were constructed from KM curves using the method described by Guyot et al. (2012).55 Log-cumulative hazards, as well as Schoenfeld residuals, were plotted for OS to examine the proportional hazards assumption (PHA) for the treatment group variable before and after matching.19
To assess the impact of each potential covariate on effective sample size (ESS) and estimated treatment effect, a “leave 1 out” approach was applied. This approach started with the calculation of ESS and a measure of treatment effect (HR for OS in the case of this study) for a full model, and then the calculation of the same 2 measures after removal of the covariate from the model; all remaining matching variables in the model were kept.19
The following matching scenarios were considered in both MAICs of the RWE cohorts19:
Scenario 1 included all matching variables considered most relevant by expert opinion (grade, histology [1 patient from GARNET with unknown histology was removed], number of prior platinum-based therapies in the advanced or recurrent setting [patients from GARNET with 0 or > 2 therapies were removed]).
Scenario 2 included all matching variables from scenario 1 except grade, because of poor reporting of grade in the RWE cohort (histology, number of prior platinum-based therapies in the advanced or recurrent setting). Scenario 2 was considered to be a sensitivity analysis of scenario 1 that was supposed to maintain a larger ESS.
Scenario 3 included the matching variables that were considered prognostic, based on the empirical regression analyses, with the exception of grade. Moreover, ECOG Performance Status was only used as a matching variable in the sensitivity analysis for patients with an ECOG Performance Status of 0 or 1 from the RWE cohort.
GARNET Versus Published Literature MAIC Methods (MAIC Report 3)
Three studies were eligible for the MAIC (MAIC report 3) based on the SLR. GARNET was a phase I single-arm trial of dostarlimab. McMeekin et al. (2015)56 was a global, open-label, phase III RCT comparing ixabepilone with paclitaxel or doxorubicin, Julius et al. (2013)57 was a retrospective review of medical records in the US focusing on pegylated liposomal doxorubicin, and Mazgani et al. (2008)58 was a retrospective analysis of carboplatin plus paclitaxel in patients with relapsed EC at 1 centre in Vancouver, British Columbia. The individual comparator studies were assessed for eligibility based on reporting of outcomes and appropriateness of the MAIC based on sample size and population (had to be adequately comparable to GARNET).19 Differences in eligibility criteria across studies were not discussed; however, MMR and MSI-H status was not accounted for in the comparator studies.
The specific (regression-type) modelling approach used in the analyses depended on the outcome data type. All P values presented were exploratory in nature and not conclusive. The tests performed were conducted post hoc, were not formally powered, and were not adjusted for multiplicity, so the reported P values are not confirmatory. When available, KM curves were digitized (using GetData Graph Digitizer 2.26) and the algorithm of Guyot et al. (2012)55 was used to produce reconstructed IPD for the aggregate trial data. As the GARNET trial had less than 36 months of follow-up for OS, KM data from each digitized comparator study were restricted (censored) to 36 months or less to improve the comparability of results.19
For all studies, each KM curve was digitized separately and the reconstructed IPD were synthesized. Curves created from these reconstructed IPD were plotted against curves displaying the raw digitized survival probability time points, and visual comparisons were made against the published curves. These pseudo-IPD were considered alongside the IPD from the GARNET trial. A weighted Cox regression was then applied to the dataset, combining the reconstructed IPD with the GARNET data (no covariates other than treatment). To assess the assumption of proportional hazards in the Cox survival modelling between the treatment groups compared, a (MAIC-weighted) log-cumulative hazard plotted against log time was visually assessed for parallelism, and Schoenfeld residuals plots were produced. If the PHA was violated, an accelerated-failure-time model with a Weibull distribution was used.19
A method of moments analytical technique was used to produce individual patient-specific weights for patients from the GARNET trial to produce weighted prognostic means. Comparison of means between the aggregate trial data before and after adjustment of the GARNET trial, as well as the ESS were presented.19
In MAIC report 3, pre-specified prognostic factors were identified in the same manner as for the RWE cohort MAICs. Additionally, BMI, number of prior anti-cancer regimens, and MMR or MSI molecular profile were considered to be prognostic factors but were not included in weighting. Because of limitations of data availability, the GARNET data could only be matched on a small number of prognostic factors for McMeekin et al. (2015)56 (age, ECOG, histology, race), Julius et al. (2013)57 (age, race, number of prior chemotherapies), and Mazgani et al. (2008)58 (histology).19 The distribution of baseline characteristics by treatment group were summarized before and after matching.
To minimize heterogeneity between separate study populations before statistical adjustments, the comparator study inclusion and exclusion criteria were applied to the GARNET IPD. The definition of outcomes in the comparator studies were also compared. Disease expertise was sought to identify the most likely criteria to have been used in each study.19 The inclusion criteria of GARNET that were modified to match the comparator studies and the resulting modified sample sizes are summarized in Table 22.
Table 22: Criteria and Sample Size Used in Each MAIC Published in the Literature (MAIC Report 3)
Comparator study (OS base case) | GARNET inclusion criteria modified to match named comparator study | GARNET ITT, n | Modified base, n |
|---|---|---|---|
McMeekin et al. (2015)56 | Hemoglobin ≥ 9 g/dL Total bilirubin ≤ 1.5 × ULN Aspartate aminotransferase ≤ 2.5 × ULN Serum creatinine ≤ 1.5 × ULN | 129 | 122 |
Julius et al. (2013)57 | No information of inclusion and exclusion criteria | 129 | 129 |
Mazgani et al. (2008)58 | Histology (endometrioid or serous) | 129 | 90 |
ITT = intention to treat; OS = overall survival; ULN = upper limit of normal.
Source: Sponsor-submitted ITC reports.19
No sensitivity or subgroup analyses were conducted for MAIC report 3.
Table 23: Summary of MAIC Analysis Methods
Category | RWE NCRAS datasets | MAIC report 3 (published literature) | |
|---|---|---|---|
MAIC report 1 (initial RWE cohort) | MAIC report 2 (treatment-specific RWE cohort) | ||
MAIC Methods | Unanchored MAIC | Unanchored MAIC | |
Justification | No common comparator | No common comparator | |
Differences in inclusion and exclusion criteria |
|
| |
Covariates used for weightinga |
|
| |
Outcomes | OS, PFS, TTNT, and TTD (as proxies for PFS) | OS, PFS, ORR, SAEs | |
Population | GARNET trial: IPD from cohort A1 of part 2B (dMMR or MSI-H EC). RWE cohort: Patients in the UK with recurrent (stage I or II at diagnosis) or advanced (stages III or IV) EC | GARNET: IPD from cohort A1 (dMMR or MSI-H EC) McMeekin et al. (2015)56: multi-national patients with advanced EC Julius et al. (2013)57: patients who had received PLD as treatment for recurrent EC at 1 centre in Texas Mazgani et al. (2008)58: patients with relapsed EC at 1 centre in Vancouver, British Columbia; endometrioid histology only | |
Analysis datasets | GARNET IPD, safety analysis dataset (n = 129) RWE cohort (base case; n = 999) | GARNET IPD, safety analysis dataset (n = 129) RWE cohorts:
| |
Analyses |
|
| |
Sensitivity analyses |
| Unweighted raw GARNET data for survival outcomes | None |
dMMR = mismatch repair-deficient; EC = endometrial cancer; ECOG = Eastern Cooperative Oncology Group performance status; FIGO = International Federation of Gynecology and Obstetrics; IPD = individual patient data; MAIC = matching-adjusted indirect comparison; MSI-H = microsatellite instability-high; NCRAS = National Cancer Registration and Analysis Service; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PLD = pegylated liposomal doxorubicin; RWE = real-world evidence; SAE = serious adverse events; TTD = time to treatment discontinuation; TTNT = time to next treatment; vs. = versus.
aFor MAIC reports 1 and 2, matching variables are histology, grade, and number of prior platinum-based therapies for scenario 1; histology and number of prior platinum-based therapies for scenario 2; and race/ethnicity, histology, stage at initial diagnosis and surgery for scenario 3. For MAIC report 3, matching variables included age, ECOG, histology, and race for comparisons with McMeekin et al. (2015)56; age, race, and number of prior chemotherapies for comparisons with Julius et al. (2013)57; and histology for comparisons with Mazgani et al. (2008).58
Source: Sponsor-submitted ITC reports.19
Results of MAIC Analyses
Summary of Included Studies
Key design characteristics and sources of heterogeneity of the studies included in the RWE cohort MAICs and the published literature MAICs are summarized in Table 24.
GARNET Versus NCRAS RWE Cohort (MAIC Reports 1 and 2)
In total, the GARNET cohort included 129 patients and the base-case RWE cohort included 999 patients. The RWE cohort used for the sensitivity analysis (ECOG ≤ 1) included 501 patients.19
There was some variation in the baseline characteristics of patients in GARNET and MAIC reports 1 and 2 (initial and treatment-specific RWE cohorts, respectively). Age range in the study cohorts varied, with 31.2%% to 51.2% of patients younger than 65 years and with 48.8% to 68.8% of patients older than 65 years. The majority of patients were White (GARNET = 76.0%; RWE cohorts = 75.0% to 92.5%); however, the GARNET trial had a greater proportion of unknown race (15.5% versus 0.7% to 5.4%). The proportion of patients with endometrioid histology was greater in GARNET (69.8%) than in the initial RWE cohort and treatment-specific cohorts (37.6% to 44.6%). In general, the GARNET trial had a notably higher proportion of patients with an ECOG Performance Status of 0 or 1 (ECOG status for many patients in the RWE cohorts was unknown), FIGO stage I/II (44.2% versus 15.8% to 35.4%), and grade 1/2 disease (67.4% versus 22.4% to 29.0%). The number of prior platinum-based therapies in the advanced or recurrent setting was 1 for all patients in the RWE cohort, whereas in the GARNET ITT cohort, it was 0 or 2 or more for some patients, based on the method of recording prior treatment for the RWE cohort. All patients in the GARNET trial were dMMR or MSI-H. There was no information on MMR or MSI-H status in the initial RWE cohort population.19
GARNET Versus Published Literature (MAIC Report 3)
In MAIC report 3, comparing GARNET with published literature, 23 records representing 14 trial publications and 13 unique studies were included in the SLR. Three unique studies — McMeekin et al. (2015),56 Julius et al. (2013),57 and Mazgani et al. (2008)58 — were eligible for the MAIC. It was noted that several identified studies were not deemed appropriate for a MAIC, primarily due to either a low sample size or the study population being systemically different than the GARNET trial.19
The GARNET, Julius et al. (2013),57 and Mazgani et al. (2008)58 studies were nonrandomized studies of dostarlimab, pegylated liposomal doxorubicin, and carboplatin plus paclitaxel, respectively, whereas the McMeekin et al. (2015)56 study was an open-label, RCT comparing ixabepilone with paclitaxel or doxorubicin. Only the treatment arm of paclitaxel or doxorubicin was included in the MAIC with McMeekin et al. (2015).56 The sample size of included studies ranged from 31 to 248 patients.19
An assessment of the baseline characteristics before weighting was not provided in MAIC report 3. The majority of baseline characteristics that were considered important, including age range, FIGO stage, disease grade, prior platinum-based therapy, and surgery, were not reported in the published literature. When reported, race was relatively similar between GARNET and the published literature trials. When reported, the proportion of patients with endometrioid histology and the proportion of patients with an ECOG Performance Status of 0 and 1 was greater in GARNET than in the studies included in the published literature MAIC.19
Results
GARNET Versus NCRAS Initial RWE Cohort (MAIC Report 1)
OS, PFS, and TTD in the GARNET ITT and initial RWE cohort are summarized in Table 25. For the initial RWE cohort, matching scenario 1, where studies were matched on histology, grade, and number of prior platinum-based therapies, resulted in the smallest ESS, with just 34 patients. The ESS was higher for scenario 2 and scenario 3, which did not include grade for 74 and 75 patients, respectively.19
Under all matching scenarios, dostarlimab was favoured over the current treatment paradigm for OS (scenario 1 HR = 0.52 [95% CI, 0.29 to 0.92]; scenario 2 HR = 0.35 [95% CI, 0.22 to 0.55]; scenario 3 HR = 0.31 [95% CI, 0.20 to 0.49]). Results for the sensitivity analysis of patients with an ECOG Performance Status of 0 or 1 were consistent with the base case. For PFS, 2 analyses were conducted: PFS from GARNET compared with TTNT from the RWE cohort; and PFS from GARNET compared with TTD from the RWE cohort. In general, the median PFS and PFS rate at 6, 12, and 18 months in all scenarios was higher than the comparator of current treatment paradigm, particularly for the analysis comparing GARNET PFS with RWE TTD. The median TTD and TTD rates at 6, 12, and 18 months were higher in all scenarios for dostarlimab than in the RWE cohort.19
The analysis of the leave-1-out approach to assess the impact of each potential covariate on ESS and estimated treatment effect is summarized for OS in Table 26. Omission of disease grade had the largest impact on ESS and estimated treatment effect, as a substantial proportion of patients in the RWE cohort had unknown grade.19
Table 24: Key Study and Baseline Characteristics in the GARNET and Comparator Studies
Characteristic | GARNET | RWE NCRAS datasets | MAIC report 3 (published literature) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
MAIC report 1 (initial RWE cohort) | MAIC report 2 (treatment-specific cohorts) | McMeekin et al. (2015)56 | Julius et al. (2013)57 | Mazgani et al. (2008)58 | |||||||
Base case | ECOG ≤ 1 | Paclitaxel | Carboplatin + paclitaxel | Carboplatin + liposomal doxorubicin | Liposomal doxorubicin | Carboplatin | |||||
Study design characteristics | |||||||||||
Study design | Nonrandomized, prospective, interventional | Descriptive, noninterventional, patient-level data from NCRAS | Open-label, RCT | Retrospective review of medical records | Retrospective study | ||||||
Region | Global | UK | Global | US | Canada | ||||||
Treatments | Dostarlimab | Current treatment paradigm | Paclitaxel | Carboplatin + paclitaxel | Carboplatin + liposomal doxorubicin | Liposomal doxorubicin | Carboplatin | Paclitaxel or doxorubicin | PLD | Carboplatin + paclitaxel | |
Outcomes | OS, PFS, ORR, TTD, SAEs | OS, PFS, TTNT, TTD | OS, ORR, SAE | OS | OS, PFS, ORR | ||||||
Sample size | 129 | 999 | 501 | 116 | 279 | 141 | 130 | 93 | 248 | 41 | 31 |
Included population | Adults with advanced, recurrent, or metastatic EC who have dMMR or MSI-H | Adults with recurrent (stage I/II at diagnosis) or advanced (stage III or IV) EC who meet all of the applied inclusion criteria and none of the applied exclusion criteria for the GARNET trial | Women with advanced EC | Recurrent EC between January 1, 1996, and June 30, 2006 | Patients with relapsed EC | ||||||
ECOG | 0 or 1 | 0, 1, or unknown | NR (KPS) | NR | NR | ||||||
Prior therapy | No more than 2 lines of anti-cancer therapy for recurrent or advanced (≥ stage IIIB) disease, including platinum doublet therapy | Prior platinum doublet therapy in the advanced/recurrent setting; all patients were considered second line | NR | NR | NR | ||||||
Baseline characteristics | |||||||||||
Age category, n (%) | |||||||||||
< 65 years | 66 (51.2) | 428 (42.8) | 202 (40.3) | 52 (44.8) | 110 (39.4) | 59 (41.8) | 43 (33.1) | 29 (31.2) | NR | NR | NR |
≥ 65 years | 63 (48.8) | 571 (57.2) | 299 (59.7) | 64 (55.2) | 169 (60.6) | 82 (58.2) | 87 (66.9) | 64 (68.8) | NR | NR | NR |
Race, n (%) | |||||||||||
White | 98 (76.0) | 841 (84.2) | 439 (87.6) | 87 (75.0) | 242 (86.7) | 119 (84.4) | 112 (86.2) | 86 (92.5) | 213 (86) | 44 (73.3) | NR |
Black | 3 (2.3) | 57 (5.7) | 21 (4.2) | 10 (8.6) | 18 (6.5) | 6 (4.3) | 4 (3.1) | 3 (3.2) | 18 (7.3) | 10 (16.7) | NR |
Other | 8 (6.2) | 78 (7.8) | 33 (6.6) | 18 (15.5) | 17 (6.1) | 12 (8.5) | 7 (5.4) | 3 (3.2) | 17 (7) | NR | NR |
Unknown | 20 (15.5) | 23 (2.3) | 8 (1.6) | 1 (0.9) | 2 (0.7) | 4 (2.8) | 7 (5.4) | 1 (1.1) | NR | NR | NR |
Histology, n (%) | |||||||||||
Endometrioid | 90 (69.8) | 424 (42.4) | 213 (42.5) | 47 (40.5) | 117 (41.9) | 55 (39.0) | 58 (44.6) | 35 (37.6) | 138 (56.0) | NR | 62 (58.86) |
Nonendometrioid | 38 (29.5) | 575 (57.6) | 288 (57.5) | 69 (59.5) | 162 (58.1) | 86 (61.0) | 72 (55.4) | 58 (62.4) | 109 (44.0) | NR | 49 (44.14) |
Unknown | 1 (0.8) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | NR | NR | NR |
ECOG, n (%) | |||||||||||
0 | 55 (42.6) | 320 (32.0) | 320 (63.9) | 41 (35.3) | 97 (34.8) | 45 (31.9) | 37 (28.5) | 30 (32.3) | 86 (35.0) | NR | NR |
1 | 74 (57.4) | 181 (18.1) | 181 (36.1) | 21 (18.1) | 44 (15.8) | 35 (24.8) | 23 (17.7) | 14 (15.1) | 79 (32) | NR | NR |
Unknown | 0 (0) | 498 (49.8) | 0 (0) | 54 (46.6) | 138 (49.5) | 61 (43.3) | 70 (53.8) | 49 (52.7) | NR | NR | NR |
FIGO Stage, n (%) | |||||||||||
Stages I and II | 57 (44.2) | 221 (22.1) | 121 (24.2) | 33 (28.4) | 44 (15.8) | 31 (22.0) | 46 (35.4) | 19 (20.4) | NR | NR | NR |
Stages III and IV | 72 (55.8) | 778 (77.9) | 380 (75.8) | 83 (71.6) | 235 (84.2) | 110 (78.0) | 84 (64.6) | 74 (79.6) | NR | NR | NR |
Disease grade, n (%) | |||||||||||
Grades 1 and 2 | 87 (67.4) | 274 (27.4) | 141 (28.1) | 26 (22.4) | 79 (28.3) | 32 (22.7) | 34 (26.2) | 27 (29.0) | NR | NR | NR |
Grades 3 and 4 | 36 (27.9) | 389 (38.9) | 206 (41.1) | 48 (41.4) | 112 (40.1) | 54 (38.3) | 54 (41.5) | 30 (32.3) | NR | NR | NR |
Unknown | 6 (4.7) | 336 (33.6) | 154 (30.7) | 42 (36.2) | 88 (31.5) | 55 (39.0) | 42 (32.3) | 36 (38.7) | NR | NR | NR |
Number of prior platinum therapies, n (%) | |||||||||||
0 | 2 (1.6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | NR | NR | NR |
1 | 110 (85.2) | 999 (100) | 501 (100) | 116 (100) | 279 (100) | 141 (100) | 130 (100) | 93 (100) | NR | NR | NR |
≥ 2 | 17 (13.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | NR | NR | NR |
Prior surgery, n (%) | |||||||||||
Yes | 116 (89.9) | 815 (81.6) | 413 (82.4) | 97 (83.6) | 244 (87.5) | 117 (83.0) | 98 (75.4) | 77 (82.8) | NR | NR | NR |
No | 13 (10.1) | 184 (18.4) | 88 (17.6) | 19 (16.4) | 35 (12.5) | 24 (17.0) | 32 (24.6) | 16 (17.2) | NR | NR | NR |
dMMR = mismatch repair-deficient; EC = endometrial cancer; ECOG = Eastern Cooperative Oncology Group performance status; KPS = Karnofsky Performance Status; MAIC = matching-adjusted indirect comparison; MSI-H = microsatellite instability-high; NCRAS = National Cancer Registration and Analysis Service; NR = not reported; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PLD = pegylated liposomal doxorubicin; RCT = randomized controlled trial; RWE = real-world evidence; SAEs = serious adverse events; SD = standard deviation; TTD = time to treatment discontinuation; TTNT = time to next treatment.
Note: The sample size of the study by Julius et al. 2013 was 60 patients; however, only 41 patients were included in the MAIC.
Source: Sponsor-submitted ITC reports.19
Table 25: Survival Outcomes Before and After Matching — GARNET Versus Initial RWE Cohort (MAIC Report 1)
Outcome | Initial RWE base case | Scenario analysis (ECOG ≤ 1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
Base case | GARNET ITT before matching | Scenario 1a | Scenario 2b | Scenario 3c | ECOG ≤ 1 | Scenario 1a | Scenario 2b | Scenario 3c | |
ESS, n | 999 | 129 | 34 | 74 | 75 | 501 | 38 | 75 | 58 |
OS | |||||||||
OS (months), median (95% CI) | 10.3 (9.2 to 11.1) | NE (18.4 to NE) | 18.0 (6.4 to NE) | NE (18.0 to NE) | NE (21.6 to NE) | 10.3 (9.0 to 11.1) | 18.0 (9.3 to NE) | NE (18.0 to NE) | NE (21.6 to NE) |
OS rate (95% CI) | |||||||||
6 months | 0.70 (0.67 to 0.73) | 0.83 (0.75 to 0.89) | 0.87 (0.74 to 0.94) | 0.86 (0.75 to 0.92) | 0.88 (0.80 to 0.93) | 0.72 (0.68 to 0.76) | 0.87 (0.74 to 0.94) | 0.86 (0.75 to 0.92) | 0.90 (0.80 to 0.95) |
12 months | 0.44 (0.40 to 0.47) | 0.72 (0.62 to 0.80) | 0.71 (0.46 to 0.86) | 0.75 (0.63 to 0.84) | 0.78 (0.66 to 0.86) | 0.43 (0.38 to 0.47) | 0.71 (0.48 to 0.86) | 0.75 (0.63 to 0.84) | 0.83 (0.71 to 0.90) |
18 months | 0.29 (0.26 to 0.32) | 0.63 (0.51 to 0.72) | 0.46 (0.21 to 0.68) | 0.62 (0.45 to 0.75) | 0.69 (0.55 to 0.80) | 0.27 (0.23 to 0.32) | 0.48 (0.23 to 0.69) | 0.62 (0.45 to 0.75) | 0.73 (0.56 to 0.84) |
HR (95% CI) | — | 0.39 (0.28 to 0.54) | 0.52 (0.29 to 0.92) | 0.35 (0.22 to 0.55) | 0.31 (0.20 to 0.49) | — | 0.49 (0.27 to 0.87) | 0.34 (0.22 to 0.55) | 0.24 (0.14 to 0.42) |
P valued | — | < 0.0001 | 0.0250 | < 0.0001 | < 0.0001 | — | 0.0155 | < 0.0001 | < 0.0001 |
PFS from GARNET compared with TTNT from RWE cohort | |||||||||
PFS (months), median (95% CI) | 7.7 (7.1 to 8.2) | 8.1 (3.3 to NE) | 5.5 (2.7 to NE) | 12.5 (4.2 to NE) | 13.8 (4.2 to NE) | 7.6 (6.8 to 8.2) | 5.6 (2.7 to NE) | 12.5 (4.2 to NE) | 18.0 (5.5 to NE) |
PFS rate (95% CI) | |||||||||
6 months | 0.61 (0.58 to 0.64) | 0.50 (0.40 to 0.59) | 0.48 (0.29 to 0.65) | 0.54 (0.41 to 0.65) | 0.56 (0.44 to 0.67) | 0.63 (0.58 to 0.67) | 0.49 (0.31 to 0.65) | 0.54 (0.41 to 0.65) | 0.63 (0.49 to 0.74) |
12 months | 0.28 (0.25 to 0.31) | 0.49 (0.39 to 0.58) | 0.46 (0.27 to 0.62) | 0.51 (0.38 to 0.63) | 0.54 (0.41 to 0.66) | 0.27 (0.23 to 0.31) | 0.47 (0.29 to 0.63) | 0.51 (0.38 to 0.63) | 0.62 (0.47 to 0.73) |
18 months | 0.16 (0.14 to 0.19) | 0.39 (0.29 to 0.50) | 0.35 (0.18 to 0.53) | 0.38 (0.23 to 0.53) | 0.42 (0.27 to 0.56) | 0.15 (0.12 to 0.19) | 0.36 (0.19 to 0.53) | 0.38 (0.23 to 0.53) | 0.49 (0.31 to 0.64) |
PFS from GARNET compared with TTD from RWE cohort | |||||||||
PFS (months), median (95% CI) | 3.4 (3.2 to 3.4) | 8.1 (3.3 to NE) | 5.5 (2.7 to NE) | 12.5 (4.2 to NE) | 13.8 (4.2 to NE) | 3.4 (3.1 to 3.5) | 5.6 (2.7 to NE) | 12.5 (4.2 to NE) | 18.0 (5.5 to NE) |
PFS rate (95% CI) | |||||||||
6 months | 0.12 (0.10 to 0.15) | 0.50 (0.40 to 0.59) | 0.48 (0.29 to 0.65) | 0.54 (0.41 to 0.65) | 0.56 (0.44 to 0.67) | 0.11 (0.09 to 0.14) | 0.49 (0.31 to 0.65) | 0.54 (0.41 to 0.65) | 0.63 (0.49 to 0.74) |
12 months | 0.02 (0.01 to 0.03) | 0.49 (0.39 to 0.58) | 0.46 (0.27 to 0.62) | 0.51 (0.38 to 0.63) | 0.54 (0.41 to 0.66) | — | 0.47 (0.29 to 0.63) | 0.51 (0.38 to 0.63) | 0.62 (0.47 to 0.73) |
18 months | — | 0.39 (0.29 to 0.50) | 0.35 (0.18 to 0.53) | 0.38 (0.23 to 0.53) | 0.42 (0.27 to 0.56) | — | 0.36 (0.19 to 0.53) | 0.38 (0.23 to 0.53) | 0.49 (0.31 to 0.64) |
TTD | |||||||||
TTD (months), median (95% CI) | 3.4 (3.2 to 3.4) | 9.1 (5.5 to 17.0) | 7.8 (4.1 to NE) | 9.7 (5.5 to NE) | 14.0 (6.4 to 17.9) | 3.4 (3.1 to 3.5) | 8.2 (5.1 to NE) | 9.7 (5.5 to NE) | 20.7 (9.7 to NE) |
TTD rate (95% CI) | |||||||||
6 months | 0.12 (0.10 to 0.15) | 0.56 (0.47 to 0.64) | 0.62 (0.43 to 0.77) | 0.61 (0.48 to 0.71) | 0.63 (0.51 to 0.72) | 0.11 (0.09 to 0.14) | 0.62 (0.44 to 0.76) | 0.61 (0.48 to 0.71) | 0.68 (0.55 to 0.78) |
12 months | 0.02 (0.01 to 0.03) | 0.48 (0.38 to 0.56) | 0.45 (0.28 to 0.61) | 0.49 (0.37 to 0.60) | 0.53 (0.41 to 0.64) | — | 0.46 (0.30 to 0.62) | 0.49 (0.37 to 0.60) | 0.62 (0.49 to 0.73) |
18 months | — | 0.36 (0.26 to 0.46) | 0.39 (0.23 to 0.55) | 0.41 (0.28 to 0.54) | 0.37 (0.24 to 0.50) | — | 0.40 (0.24 to 0.56) | 0.41 (0.28 to 0.54) | 0.52 (0.36 to 0.65) |
CI = confidence interval; ECOG = Eastern Cooperative Oncology Group performance status; ESS = effective sample size; HR = hazard ratio; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; NE = not estimable; OS = overall survival; PFS = progression-free survival; RWE = real-world evidence; vs. = versus; TTD = time to treatment discontinuation.
aScenario 1: Matching variables are histology, grade, and number of prior platinum-based therapies.
bScenario 2: Matching variables are histology and number of prior platinum-based therapies.
cScenario 3: Matching variables are race/ethnicity, histology, stage at initial diagnosis, and surgery.
dP values are exploratory and not adjusted for multiplicity, thus at an increased risk of type I error.
Source: Sponsor-submitted ITC reports19
Table 26: Summary of Leave-1-Out Approach — Initial RWE Cohort (Base-Case and Scenario Analyses, MAIC Report 1)
Covariates omitted | Base case | Scenario analysis (ECOG ≤ 1) | ||
|---|---|---|---|---|
ESS | HR (95% CI) for OS | ESS | HR (95% CI) for OS | |
Full model | 26 | 0.483 (0.249 to 0.937) | 27 | 0.406 (0.208 to 0.795) |
Race and ethnicity | 32 | 0.486 (0.276 to 0.856) | 32 | 0.418 (0.230 to 0.758) |
Age | 26 | 0.484 (0.250 to 0.938) | 27 | 0.403 (0.205 to 0.794) |
ECOGa | — | — | 30 | 0.460 (0.237 to 0.891) |
Histology | 27 | 0.480 (0.246 to 0.934) | 29 | 0.396 (0.201 to 0.781) |
FIGO stage | 27 | 0.530 (0.275 to 1.021) | 28 | 0.414 (0.213 to 0.806) |
Disease grade | 53 | 0.307 (0.174 to 0.543) | 46 | 0.267 (0.143 to 0.499) |
Number of prior platinum therapies | 28 | 0.480 (0.252 to 0.912) | 29 | 0.396 (0.206 to 0.761) |
Surgery for advanced or recurrent EC | 26 | 0.480 (0.250 to 0.921) | 26 | 0.410 (0.215 to 0.784) |
CI = confidence interval; EC = endometrial cancer; ECOG = Eastern Cooperative Oncology Group performance status; ESS = effective sample size; FIGO = Federation of Gynecology and Obstetrics; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; OS = overall survival; RWE = real-world evidence.
aFor the base-case RWE cohort, ECOG Performance Status is not used as a matching variable because 49.8% of patients have an unknown ECOG status and adjusting for this imbalance would correspond to performing the analysis in the RWE ECOG ≤ 1 cohort (where patients with unknown ECOG have been removed).Source: Sponsor-submitted ITC reports.19
GARNET Versus NCRAS Treatment-Specific Cohorts (MAIC Report 2)
For the treatment-specific MAICs, Table 27 summarizes the OS for dostarlimab compared with the 5 specific treatments in the RWE cohort. All results for scenario analyses using unweighted GARNET data were consistent with the primary analyses for OS. For paclitaxel monotherapy, the ESS in scenarios 1, 2, and 3 was 30, 72, and 63, respectively. Under all matching scenarios, the median OS (and HRs) favoured dostarlimab over paclitaxel monotherapy (scenario 1: HR = 0.36; 95% CI, 0.19 to 0.65; scenario 2: HR = 0.24; 95% CI, 0.15 to 0.40; scenario 3: HR = 0.18; 95% CI, 0.11 to 0.30), indicating that patients taking dostarlimab had a lower risk of death than patients receiving paclitaxel. The OS rates at 6, 12, and 18 months were greater for dostarlimab than for paclitaxel in the RWE cohort.19
For carboplatin plus paclitaxel, the ESS in scenarios 1, 2, and 3 was 36, 74, and 69, respectively. Dostarlimab was favoured over carboplatin plus paclitaxel only in scenarios 2 and 3 (scenario 2: HR = 0.48; 95% CI, 0.29 to 0.78; scenario 3: HR = 0.42; 95% CI, 0.25 to 0.68), but not scenario 1 (HR = 0.70; 95% CI, 0.39 to 1.28). The OS rates at 6, 12, and 18 months were greater for dostarlimab than for carboplatin plus paclitaxel in the RWE cohort.19
For carboplatin plus liposomal doxorubicin, the ESS in scenarios 1, 2, and 3 was 26, 69, and 74, respectively. Dostarlimab was favoured over carboplatin plus liposomal doxorubicin only in scenarios 2 and 3 (scenario 2: HR = 0.45; 95% CI, 0.27 to 0.76; scenario 3: HR = 0.40; 95% CI, 0.24 to 0.67), but not scenario 1 (HR = 0.74; 95% CI, 0.39 to 1.41). After matching, the 6-, 12-, and 18-month OS rates were greater for dostarlimab.19
For liposomal doxorubicin monotherapy, the ESS in scenarios 1, 2, and 3 was 37, 78, and 76, respectively. Under all matching scenarios, the median OS (and HRs) favoured dostarlimab over liposomal doxorubicin monotherapy (scenario 1: HR = 0.20; 95% CI, 0.09 to 0.44; scenario 2: HR = 0.17; 95% CI, 0.10 to 0.29; scenario 3: HR = 0.16; 95% CI, 0.09 to 0.27), indicating that patients taking dostarlimab had a lower risk of death than patients taking liposomal doxorubicin. Under all matching scenarios, the OS rates at 6 and 12 months were greater in the GARNET ITT cohort than in the RWE cohort.19
For carboplatin monotherapy, the ESS in scenarios 1, 2, and 3 was 23, 67, and 69, respectively. Dostarlimab was favoured over carboplatin in all matching scenarios (scenario 1: HR = 0.53; 95% CI, 0.29 to 0.98; scenario 2: HR = 0.32; 95% CI, 0.19 to 0.55; scenario 3: HR = 0.28; 95% CI, 0.16 to 0.48), indicating that patients taking dostarlimab had a lower risk of death than patients taking carboplatin monotherapy. Under all matching scenarios, the OS rates at 6, 12, and 18 months were greater in the GARNET ITT cohort compared with in the RWE cohort.19
Table 27: OS Before and After Matching — GARNET Versus Treatment-Specific RWE Cohort (MAIC Report 2)
Outcome | Treatment-specific cohort | GARNET ITT before matching | Scenario 1a | Scenario 2b | Scenario 3c |
|---|---|---|---|---|---|
Paclitaxel | |||||
ESS | 116 | 129 | 30 | 72 | 63 |
OS (months), median (95% CI) | 6.6 (5.7 to 8.0) | NE (18.4 to NE) | 17.1 (6.4 to NE) | NE (18.0 to NE) | NE (21.6 to NE) |
OS rate (95% CI) | |||||
6 months | 0.54 (0.44 to 0.62) | 0.83 (0.75 to 0.89) | 0.88 (0.73 to 0.95) | 0.86 (0.75 to 0.92) | 0.90 (0.82 to 0.94) |
12 months | 0.28 (0.20 to 0.37) | 0.72 (0.62 to 0.80) | 0.71 (0.44 to 0.86) | 0.76 (0.63 to 0.85) | 0.80 (0.68 to 0.88) |
18 months | 0.15 (0.09 to 0.23) | 0.63 (0.51 to 0.72) | 0.45 (0.19 to 0.68) | 0.62 (0.45 to 0.76) | 0.72 (0.58 to 0.82) |
HR (95% CI) | — | 0.26 (0.18 to 0.39) | 0.36 (0.19 to 0.65) | 0.24 (0.15 to 0.40) | 0.18 (0.11 to 0.30) |
P valued | — | < 0.0001 | 0.0008 | < 0.0001 | < 0.0001 |
Carboplatin + paclitaxel | |||||
ESS | 279 | 129 | 36 | 74 | 69 |
OS (months), median (95% CI) | 14.2 (12.4 to 16.1) | NE (18.4 to NE) | 18.0 (6.9 to NE) | NE (18.0 to NE) | NE (21.6 to NE) |
OS rate (95% CI) | |||||
6 months | 0.83 (0.78 to 0.87) | 0.83 (0.75 to 0.89) | 0.87 (0.74 to 0.94) | 0.86 (0.75 to 0.92) | 0.88 (0.79 to 0.94) |
12 months | 0.58 (0.51 to 0.64) | 0.72 (0.62 to 0.80) | 0.71 (0.47 to 0.86) | 0.75 (0.63 to 0.84) | 0.79 (0.67 to 0.87) |
18 months | 0.37 (0.31 to 0.44) | 0.63 (0.51 to 0.72) | 0.47 (0.22 to 0.68) | 0.62 (0.45 to 0.75) | 0.70 (0.54 to 0.81) |
HR (95% CI) | — | 0.53 (0.37 to 0.76) | 0.70 (0.39 to 1.28) | 0.48 (0.29 to 0.78) | 0.42 (0.25 to 0.68) |
P valued | — | 0.0006 | 0.2480 | 0.0032 | 0.0006 |
Carboplatin + liposomal doxorubicin | |||||
ESS | 141 | 129 | 26 | 69 | 74 |
OS (months), median (95% CI) | 13.9 (11.2 to 15.7) | NE (18.4 to NE) | 15.4 (6.4 to NE) | NE (18.0 to NE) | NE (21.6 to NE) |
OS rate (95% CI) | |||||
6 months | 0.86 (0.79 to 0.91) | 0.83 (0.75 to 0.89) | 0.88 (0.73 to 0.95) | 0.86 (0.75 to 0.92) | 0.88 (0.80 to 0.94) |
12 months | 0.57 (0.48 to 0.65) | 0.72 (0.62 to 0.80) | 0.69 (0.40 to 0.86) | 0.76 (0.63 to 0.85) | 0.78 (0.66 to 0.87) |
18 months | 0.39 (0.30 to 0.47) | 0.63 (0.51 to 0.72) | 0.42 (0.16 to 0.66) | 0.62 (0.45 to 0.76) | 0.69 (0.55 to 0.80) |
HR (95% CI) | — | 0.51 (0.35 to 0.74) | 0.74 (0.39 to 1.41) | 0.45 (0.27 to 0.76) | 0.40 (0.24 to 0.67) |
P value d | — | 0.0005 | 0.3660 | 0.0027 | 0.0004 |
Liposomal doxorubicin | |||||
ESS | 130 | 129 | 37 | 78 | 76 |
OS (months), median (95% CI) | 4.9 (4.2 to 6.1) | NE (18.4 to NE) | 18.0 (9.3 to NE) | NE (18.0 to NE) | NE (21.6 to NE) |
OS rate (95% CI) | |||||
6 months | 0.41 (0.33 to 0.50) | 0.83 (0.75 to 0.89) | 0.87 (0.73 to 0.94) | 0.85 (0.75 to 0.92) | 0.87 (0.79 to 0.92) |
12 months | 0.16 (0.10 to 0.23) | 0.72 (0.62 to 0.80) | 0.72 (0.49 to 0.86) | 0.75 (0.63 to 0.84) | 0.76 (0.64 to 0.85) |
18 months | — | 0.63 (0.51 to 0.72) | 0.48 (0.23 to 0.69) | 0.62 (0.46 to 0.75) | 0.68 (0.53 to 0.79) |
HR (95% CI) | — | 0.20 (0.13 to 0.30) | 0.20 (0.09 to 0.44) | 0.17 (0.10 to 0.29) | 0.16 (0.09 to 0.27) |
P valued | — | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 |
Carboplatin | |||||
ESS | 93 | 129 | 23 | 67 | 69 |
OS (months), median (95% CI) | 8.3 (6.7 to 11.4) | NE (18.4 to NE) | 15.4 (6.4 to NE) | NE (18.0 to NE) | NE (21.6 to NE) |
OS rate (95% CI) | |||||
6 months | 0.68 (0.57 to 0.77) | 0.83 (0.75 to 0.89) | 0.88 (0.75 to 0.95) | 0.86 (0.75 to 0.92) | 0.88 (0.78 to 0.93) |
12 months | 0.35 (0.25 to 0.45) | 0.72 (0.62 to 0.80) | 0.68 (0.37 to 0.86) | 0.76 (0.63 to 0.85) | 0.78 (0.65 to 0.87) |
18 months | 0.24 (0.15 to 0.34) | 0.63 (0.51 to 0.72) | 0.39 (0.14 to 0.63) | 0.62 (0.45 to 0.76) | 0.69 (0.53 to 0.80) |
HR (95% CI) | — | 0.35 (0.23 to 0.54) | 0.53 (0.29 to 0.98) | 0.32 (0.19 to 0.55) | 0.28 (0.16 to 0.48) |
P valued | — | < 0.0001 | 0.0427 | < 0.0001 | < 0.0001 |
CI = confidence interval; ESS = effective sample size; HR = hazard ratio; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; NE = not estimable; OS = overall survival; RWE = real-world evidence.
aScenario 1: Matching variables are histology, grade, and number of prior platinum-based therapies.
bScenario 2: Matching variables are histology and number of prior platinum-based therapies.
cScenario 3: Matching variables are race/ethnicity, histology, stage at initial diagnosis, and surgery.
dP values are exploratory and not adjusted for multiplicity, so at increased risk of type I error.
Source: Sponsor-submitted ITC reports.19
Results for PFS for all comparators are summarized in Table 28. When compared with the RWE cohort TTNT and TTD, the median PFS was longer for dostarlimab than for all relevant comparators in all scenarios except scenario 1 of the TTNT comparison for paclitaxel monotherapy, carboplatin plus paclitaxel, carboplatin plus liposomal doxorubicin, and carboplatin monotherapy. For the TTD comparison, dostarlimab was favoured over all comparators in all scenarios except carboplatin monotherapy in scenario 1. The PFS rate for dostarlimab was greater in all scenarios and at all time points for the TTD comparison for all relevant comparators. In contrast with TTNT from the RWE cohort, the PFS rate for dostarlimab was greater than both paclitaxel and liposomal doxorubicin monotherapy at all time points and in all scenarios. The PFS rate was not greater at the 6-month assessment in any scenario for carboplatin plus paclitaxel, carboplatin plus liposomal doxorubicin, or carboplatin monotherapy. Otherwise, dostarlimab had a greater PFS rate at the 12- and 18-month time points in all scenarios.19
Table 28: PFS Before and After Matching — GARNET Versus Treatment-Specific RWE Cohort (MAIC Report 2)
Outcome | Treatment-specific cohort | GARNET ITT before matching | Scenario 1a | Scenario 2b | Scenario 3c |
|---|---|---|---|---|---|
Paclitaxel | |||||
ESS | 116 | 129 | 30 | 72 | 63 |
PFSd (compared with TTNT) | |||||
PFS (months), median (95% CI) | 5.8 (3.9 to 6.5) | 8.1 (3.3 to NE) | 5.5 (2.7 to NE) | 12.5 (4.2 to NE) | 16.6 (5.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.45 (0.36 to 0.54) | 0.50 (0.40 to 0.59) | 0.48 (0.28 to 0.66) | 0.54 (0.41 to 0.66) | 0.60 (0.47 to 0.71) |
12 months | 0.20 (0.13 to 0.28) | 0.49 (0.39 to 0.58) | 0.46 (0.27 to 0.64) | 0.51 (0.38 to 0.63) | 0.59 (0.45 to 0.70) |
18 months | — | 0.39 (0.29 to 0.50) | 0.35 (0.17 to 0.54) | 0.38 (0.23 to 0.53) | 0.46 (0.30 to 0.61) |
PFSe (compared with TTD) | |||||
PFS (months), median (95% CI) | 3.2 (2.5 to 3.7) | 8.1 (3.3 to NE) | 5.5 (2.7 to NE) | 12.5 (4.2 to NE) | 16.6 (5.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.11 (0.06 to 0.18) | 0.50 (0.40 to 0.59) | 0.48 (0.28 to 0.66) | 0.54 (0.41 to 0.66) | 0.60 (0.47 to 0.71) |
12 months | — | 0.49 (0.39 to 0.58) | 0.46 (0.27 to 0.64) | 0.51 (0.38 to 0.63) | 0.59 (0.45 to 0.70) |
18 months | — | 0.39 (0.29 to 0.50) | 0.35 (0.17 to 0.54) | 0.38 (0.23 to 0.53) | 0.46 (0.30 to 0.61) |
Carboplatin + paclitaxel | |||||
ESS | 279 | 129 | 36 | 74 | 69 |
PFSd (compared with TTNT) | |||||
PFS (months) median (95% CI) | 10.0 (9.0 to 11.3) | 8.1 (3.3 to NE) | 5.5 (2.7 to NE) | 12.5 (4.2 to NE) | 13.8 (4.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.74 (0.68 to 0.79) | 0.50 (0.40 to 0.59) | 0.48 (0.30 to 0.65) | 0.54 (0.41 to 0.65) | 0.55 (0.42 to 0.67) |
12 months | 0.37 (0.31 to 0.43) | 0.49 (0.39 to 0.58) | 0.46 (0.28 to 0.62) | 0.51 (0.38 to 0.63) | 0.53 (0.39 to 0.65) |
18 months | 0.19 (0.14 to 0.25) | 0.39 (0.29 to 0.50) | 0.35 (0.18 to 0.52) | 0.38 (0.23 to 0.53) | 0.40 (0.25 to 0.54) |
PFSe (compared with TTD) | |||||
PFS (months), median (95% CI) | 3.4 (3.4 to 3.5) | 8.1 (3.3 to NE) | 5.5 (2.7 to NE) | 12.5 (4.2 to NE) | 13.8 (4.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.13 (0.09 to 0.18) | 0.50 (0.40 to 0.59) | 0.48 (0.30 to 0.65) | 0.54 (0.41 to 0.65) | 0.55 (0.42 to 0.67) |
12 months | — | 0.49 (0.39 to 0.58) | 0.46 (0.28 to 0.62) | 0.51 (0.38 to 0.63) | 0.53 (0.39 to 0.65) |
18 months | — | 0.39 (0.29 to 0.50) | 0.35 (0.18 to 0.52) | 0.38 (0.23 to 0.53) | 0.40 (0.25 to 0.54) |
Carboplatin + liposomal doxorubicin | |||||
ESS | 141 | 129 | 26 | 69 | 74 |
PFSd (compared with TTNT) | |||||
PFS (months) median (95% CI) | 9.9 (8.3 to 11.2) | 8.1 (3.3 to NE) | 5.2 (2.7 to NE) | 12.5 (4.2 to NE) | 13.8 (4.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.78 (0.70 to 0.84) | 0.50 (0.40 to 0.59) | 0.46 (0.25 to 0.64) | 0.54 (0.41 to 0.66) | 0.56 (0.43 to 0.67) |
12 months | 0.38 (0.30 to 0.46) | 0.49 (0.39 to 0.58) | 0.44 (0.24 to 0.62) | 0.51 (0.38 to 0.63) | 0.54 (0.41 to 0.65) |
18 months | 0.20 (0.13 to 0.27) | 0.39 (0.29 to 0.50) | 0.33 (0.15 to 0.52) | 0.38 (0.22 to 0.54) | 0.41 (0.26 to 0.55) |
PFSe (compared with TTD) | |||||
PFS (months) median (95% CI) | 4.6 (4.0 to 4.6) | 8.1 (3.3 to NE) | 5.2 (2.7 to NE) | 12.5 (4.2 to NE) | 13.8 (4.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.14 (0.09 to 0.21) | 0.50 (0.40 to 0.59) | 0.46 (0.25 to 0.64) | 0.54 (0.41 to 0.66) | 0.56 (0.43 to 0.67) |
12 months | — | 0.49 (0.39 to 0.58) | 0.44 (0.24 to 0.62) | 0.51 (0.38 to 0.63) | 0.54 (0.41 to 0.65) |
18 months | — | 0.39 (0.29 to 0.50) | 0.33 (0.15 to 0.52) | 0.38 (0.22 to 0.54) | 0.41 (0.26 to 0.55) |
Liposomal doxorubicin | |||||
ESS | 130 | 129 | 37 | 78 | 76 |
PFSd (compared with TTNT) | |||||
PFS (months), median (95% CI) | 4.1 (3.4 to 4.6) | 8.1 (3.3 to NE) | 8.1 (2.7 to NE) | 12.2 (4.2 to NE) | 13.8 (4.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.30 (0.22 to 0.38) | 0.50 (0.40 to 0.59) | 0.50 (0.31 to 0.66) | 0.54 (0.41 to 0.65) | 0.56 (0.43 to 0.67) |
12 months | — | 0.49 (0.39 to 0.58) | 0.48 (0.30 to 0.64) | 0.51 (0.38 to 0.63) | 0.54 (0.41 to 0.66) |
18 months | — | 0.39 (0.29 to 0.50) | 0.36 (0.19 to 0.54) | 0.38 (0.24 to 0.53) | 0.41 (0.25 to 0.56) |
PFSe (compared with TTD) | |||||
PFS (months), median (95% CI) | 2.8 (2.1 to 2.9) | 8.1 (3.3 to NE) | 8.1 (2.7 to NE) | 12.2 (4.2 to NE) | 13.8 (4.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.12 (0.07 to 0.19) | 0.50 (0.40 to 0.59) | 0.50 (0.31 to 0.66) | 0.54 (0.41 to 0.65) | 0.56 (0.43 to 0.67) |
12 months | — | 0.49 (0.39 to 0.58) | 0.48 (0.30 to 0.64) | 0.51 (0.38 to 0.63) | 0.54 (0.41 to 0.66) |
18 months | — | 0.39 (0.29 to 0.50) | 0.36 (0.19 to 0.54) | 0.38 (0.24 to 0.53) | 0.41 (0.25 to 0.56) |
Carboplatin | |||||
ESS | 93 | 129 | 23 | 67 | 69 |
PFSd (compared with TTNT) | |||||
PFS (months), median (95% CI) | 6.9 (5.8 to 8.3) | 8.1 (3.3 to NE) | 3.3 (2.7 to 18.0) | 12.5 (4.2 to NE) | 12.5 (4.2 to NE) |
PFS rate (95% CI) | |||||
6 months | 0.59 (0.48 to 0.69) | 0.50 (0.40 to 0.59) | 0.42 (0.22 to 0.61) | 0.54 (0.41 to 0.66) | 0.55 (0.41 to 0.66) |
12 months | 0.18 (0.10 to 0.27) | 0.49 (0.39 to 0.58) | 0.40 (0.21 to 0.58) | 0.51 (0.38 to 0.64) | 0.52 (0.38 to 0.64) |
18 months | — | 0.39 (0.29 to 0.50) | 0.30 (0.14 to 0.49) | 0.38 (0.22 to 0.54) | 0.38 (0.23 to 0.53) |
PFSe (compared with TTD) | |||||
PFS (months), median (95% CI) | 3.4 (2.8 to 3.5) | 8.1 (3.3 to NE) | 3.3 (2.7 to 18.0) | 12.5 (4.2 to NE) | 12.5 (4.2 to NE) |
PFS rate (95% CI) | |||||
6 months | — | 0.50 (0.40 to 0.59) | 0.42 (0.22 to 0.61) | 0.54 (0.41 to 0.66) | 0.55 (0.41 to 0.66) |
12 months | — | 0.49 (0.39 to 0.58) | 0.40 (0.21 to 0.58) | 0.51 (0.38 to 0.64) | 0.52 (0.38 to 0.64) |
18 months | — | 0.39 (0.29 to 0.50) | 0.30 (0.14 to 0.49) | 0.38 (0.22 to 0.54) | 0.38 (0.23 to 0.53) |
CI = confidence interval; ESS = effective sample size; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; NE = not estimable; PFS = progression-free survival; RWE = real-world evidence; TTD = time to treatment discontinuation; vs. = versus.
aScenario 1: Matching variables are histology, grade, and number of prior platinum-based therapies.
bScenario 2: Matching variables are histology and number of prior platinum-based therapies.
cScenario 3: Matching variables are race/ethnicity, histology, stage at initial diagnosis, and surgery.
dPFS from GARNET is compared with TTNT from RWE cohort.
ePFS from GARNET is compared with TTD from RWE cohort.
Source: Sponsor-submitted ITC reports.19
For the outcome of TTD (Table 29), the median TTD and TTD rates were longer for dostarlimab than for all relevant comparators in the RWE cohort at all time points and across all 3 matching scenarios.19
Table 29: TTD Before and After Matching — GARNET Versus Treatment-Specific RWE Cohort (MAIC Report 2)
Outcome | Treatment-specific cohort | GARNET ITT before matching | Scenario 1a | Scenario 2b | Scenario 3c |
|---|---|---|---|---|---|
Paclitaxel | |||||
ESS | 116 | 129 | 30 | 72 | 63 |
TTD (months), median (95% CI) | 3.2 (2.5 to 3.7) | 9.1 (5.5 to 17.0) | 8.2 (4.1 to NE) | 9.7 (5.5 to NE) | 14.0 (6.4 to NE) |
TTD rate (95% CI) | |||||
6 months | 0.11 (0.06 to 0.18) | 0.56 (0.47 to 0.64) | 0.63 (0.42 to 0.78) | 0.61 (0.48 to 0.71) | 0.65 (0.52 to 0.75) |
12 months | — | 0.48 (0.38 to 0.56) | 0.46 (0.27 to 0.63) | 0.49 (0.36 to 0.61) | 0.56 (0.42 to 0.67) |
18 months | — | 0.36 (0.26 to 0.46) | 0.40 (0.22 to 0.57) | 0.41 (0.28 to 0.54) | 0.39 (0.25 to 0.53) |
Carboplatin + paclitaxel | |||||
ESS | 279 | 129 | 36 | 74 | 69 |
TTD (months) median (95% CI) | 3.4 (3.4 to 3.5) | 9.1 (5.5 to 17.0) | 8.2 (4.2 to NE) | 9.7 (5.5 to NE) | 12.6 (5.5 to 17.9) |
TTD rate (95% CI) | |||||
6 months | 0.13 (0.09 to 0.18) | 0.56 (0.47 to 0.64) | 0.62 (0.43 to 0.76) | 0.61 (0.48 to 0.71) | 0.61 (0.48 to 0.72) |
12 months | — | 0.48 (0.38 to 0.56) | 0.46 (0.29 to 0.61) | 0.49 (0.37 to 0.60) | 0.51 (0.39 to 0.63) |
18 months | — | 0.36 (0.26 to 0.46) | 0.39 (0.23 to 0.55) | 0.41 (0.28 to 0.54) | 0.37 (0.24 to 0.50) |
Carboplatin + liposomal doxorubicin | |||||
ESS | 141 | 129 | 26 | 69 | 74 |
TTD (months), median (95% CI) | 4.6 (4.0 to 4.6) | 9.1 (5.5 to 17.0) | 6.4 (4.1 to NE) | 9.7 (5.5 to NE) | 13.8 (5.8 to 17.9) |
TTD rate (95% CI) | |||||
6 months | 0.14 (0.09 to 0.21) | 0.56 (0.47 to 0.64) | 0.62 (0.40 to 0.78) | 0.61 (0.48 to 0.72) | 0.62 (0.50 to 0.72) |
12 months | — | 0.48 (0.38 to 0.56) | 0.44 (0.24 to 0.61) | 0.49 (0.36 to 0.61) | 0.52 (0.39 to 0.63) |
18 months | — | 0.36 (0.26 to 0.46) | 0.38 (0.20 to 0.56) | 0.41 (0.28 to 0.54) | 0.36 (0.23 to 0.49) |
Liposomal doxorubicin | |||||
ESS | 130 | 129 | 37 | 78 | 76 |
TTD (months), median (95% CI) | 2.8 (2.1 to 2.9) | 9.1 (5.5 to 17.0) | 9.1 (5.1 to NE) | 9.7 (5.5 to NE) | 14.0 (6.4 to 17.9) |
TTD rate (95% CI) | |||||
6 months | 0.12 (0.07 to 0.19) | 0.56 (0.47 to 0.64) | 0.63 (0.45 to 0.77) | 0.61 (0.48 to 0.71) | 0.64 (0.52 to 0.73) |
12 months | — | 0.48 (0.38 to 0.56) | 0.48 (0.30 to 0.63) | 0.49 (0.37 to 0.60) | 0.53 (0.40 to 0.64) |
18 months | — | 0.36 (0.26 to 0.46) | 0.41 (0.24 to 0.57) | 0.41 (0.28 to 0.53) | 0.35 (0.22 to 0.49) |
Carboplatin | |||||
ESS | 93 | 129 | 23 | 67 | 69 |
TTD (months), median (95% CI) | 3.4 (2.8 to 3.5) | 9.1 (5.5 to 17.0) | 6.4 (4.1 to NE) | 9.7 (5.5 to NE) | 12.6 (5.5 to 17.1) |
TTD rate (95% CI) | |||||
6 months | — | 0.56 (0.47 to 0.64) | 0.60 (0.36 to 0.78) | 0.61 (0.48 to 0.72) | 0.61 (0.48 to 0.72) |
12 months | — | 0.48 (0.38 to 0.56) | 0.40 (0.21 to 0.58) | 0.49 (0.36 to 0.61) | 0.50 (0.37 to 0.62) |
18 months | — | 0.36 (0.26 to 0.46) | 0.34 (0.18 to 0.52) | 0.41 (0.28 to 0.54) | 0.35 (0.22 to 0.48) |
CI = confidence interval; ESS = effective sample size; ITT = intention to treat; MAIC = matching-adjusted indirect comparison; NE = not estimable; PFS = progression-free survival; RWE = real-world evidence; TTD = time to treatment discontinuation; vs. = versus.
aScenario 1: Matching variables are histology, grade, and number of prior platinum-based therapies.
bScenario 2: Matching variables are histology and number of prior platinum-based therapies.
cScenario 3: Matching variables are race/ethnicity, histology, stage at initial diagnosis, and surgery.
Source: Sponsor-submitted ITC reports.19
GARNET Versus Published Literature (MAIC Report 3)
Table 30 presents the ESS and weighted baseline characteristics for the MAICs versus published literature sources. A full list of baseline characteristics in GARNET after matching is summarized in Table 52. Seven patients were removed from GARNET who did not meet inclusion or exclusion criteria for McMeekin et al. (2015),56 resulting in a MAIC base of 122 patients. These 122 patients from GARNET were then weighted for each prognostic variable available, resulting in an ESS of 87.274. Because of a lack of information on inclusion and exclusion criteria in Julius et al. (2013),57 no patients were removed from the GARNET trial before weighting. After matching on available prognostic variables, the resulting ESS compared with Julius et al. (2013)57 was 81.992. After the removal of 39 GARNET patients who did not meet the inclusion or exclusion criteria for Mazgani et al. (2008),58 the resulting MAIC base was 90 patients, with an ESS after matching of 29.08.19
The covariate values used to derive the weights in the MAICs included age, ECOG, race, histology, and number of prior chemotherapies. Covariates not used in weighting varied across studies. Individual MAICs were conducted for each study identified in the literature.19
Table 30: Sample Size and Baseline Characteristics After Matching (MAIC Report 3)
Comparison | Modified n, or ESS | Mean age, years | ECOG (1, not 0), % | Race (White), % | Endometrioid type I, % | Prior chemotherapies, n |
|---|---|---|---|---|---|---|
McMeekin et al. (2015)56 | ||||||
Dostarlimab, unweighted | n = 122 | 63.11 | 55.7 | 75.4 | 65.6 | NR |
Paclitaxel or doxorubicin | n = 248 | 64.00a | 33.2b | 85.9 | 55.6 | NR |
Dostarlimab, weighted | ESS = 87.274 | 64.00 | 33.2b | 85.9 | 55.6 | NR |
Julius et al. (2013)57 | ||||||
Dostarlimab unweighted | n = 129 | 63.095 | NR | 75.97 | NR | 1.516 |
PLD | n = 41 | 66.800 | NR | 73.3 | NR | 2 |
Dostarlimab, weighted | ESS = 81.992 | 66.800 | NR | 73.3 | NR | 2 |
Mazgani et al. (2008)58 | ||||||
Dostarlimab, unweighted | n = 90 | NR | NR | NR | 94.44 | NR |
Paclitaxel and carboplatin | n = 31 | NR | NR | NR | 61.29 | NR |
Dostarlimab, weighted | ESS = 29.08 | NR | NR | NR | 61.29 | NR |
ECOG = Eastern Cooperative Oncology Group performance status; ESS = effective sample size; MAIC = matching-adjusted indirect comparison; NR = not reported; PLD = pegylated liposomal doxorubicin.
aMedian age is used for McMeekin et al. (2015).56
bECOG converted from Karnofsky performance scores in McMeekin et al. (2015).56
Source: Sponsor-submitted ITC Reports.19
The median OS and PFS for dostarlimab (GARNET) before and after matching compared with data from each individual study are summarized in Table 31. In all cases, the median OS was not reached before or after adjustment for the dostarlimab group. After adjustment, OS favoured dostarlimab over paclitaxel and over doxorubicin using data from the McMeekin et al. (2015)56 study (HR = 0.407; 95% CI, 0.252 to 0.657) and over pegylated liposomal doxorubicin using data from the Julius et al. (2013)57 study (HR = 0.287; 95% CI, 0.170 to 0.486). After adjustment, there was no difference in OS for dostarlimab compared with carboplatin plus paclitaxel using data from the Mazgani et al. (2008)58 study (HR = 0.559; 95% CI, 0.256 to 1.220). Data for PFS were only available from the Mazgani et al. (2008)58 study. After adjustment, there was no difference in PFS between dostarlimab and carboplatin plus paclitaxel.19
For OS, the test of the PHA did not show a violation for the comparisons with data from the McMeekin et al. (2015),56 Julius et al. (2013),57 or Mazgani et al. (2008)58 studies (P = 0.28, 0.17, and 0.62, respectively). A Cox proportional hazards model with MAIC was fitted to the data. The HRs (0.407; 95% CI, 0.252 to 0.657 and 0.287; 95% CI, 0.170 to 0.486) indicated a difference between the 2 treatments for OS, with a 59.3%, and 71.3% lower risk of death for dostarlimab than for paclitaxel plus doxorubicin and for pegylated liposomal doxorubicin, respectively; median survival was not reported. For PFS, the PHA was violated (P = 0.014). As such, the accelerated-failure-time model with Weibull distribution was fitted to the data. Despite the accelerated-failure-time model, no difference was noted between dostarlimab and carboplatin plus paclitaxel in PFS.19
Table 31: Summary of OS and PFS Before and After Matching (MAIC Report 3)
Outcome | Dostarlimab, unadjusted | Dostarlimab, adjusted | Comparator |
|---|---|---|---|
McMeekin et al. (2015)56 | |||
OS | |||
OS (months), median (95% CI) | NR (21.6 to NR) | NR (NR to NR) | 12.6 (11.0 to 16.7) |
GARNET vs. McMeekin et al. (2015)56 HR (95% CI) | 0.407 (0.252 to 0.657) | ||
P valuea | 0.0002 | ||
Julius et al. (2013)57 | |||
OS | |||
OS (months), median (95% CI) | NR (21.62 to NR) | NR (18.43 to NR) | 7.1 (5.83 to 11.0) |
GARNET vs. Julius et al. (2013)57 HR (95% CI) | 0.287 (0.170 to 0.486) | ||
P valuea | < 0.0001 | ||
Mazgani et al. (2008)58 | |||
OS | |||
OS (months), median (95% CI) | NR (17.1 to NR) | NR (15.4 to NR) | 19.4 (10.5 to NR) |
GARNET vs. Mazgani et al. (2008)58 HR (95% CI) | 0.559 (0.256 to 1.220) | ||
P valuea | 0.144 | ||
PFS | |||
PFS (months), median (95% CI) | 4.17 (2.96 to NR) | 4.17 (2.96 to 16.6) | 8.56 (6.52 to 16.5) |
GARNET vs. Mazgani et al. (2008)58 HR (95% CI) | 1.278 (0.767 to 2.128) | ||
P value | 0.346 | ||
AFT model (Weibull) HR (95% CI) | 1.492 (0.879 to 2.534) | ||
P valuea | 0.136 | ||
AFT = accelerated-failure time; CI = confidence interval; HR = hazard ratio; MAIC = matching-adjusted indirect comparison; NR = not reached; OS = overall survival; PFS = progression-free survival; vs. = versus.
aP values are exploratory and not adjusted for multiplicity, so at increased risk of type I error.
Source: Sponsor-submitted ITC reports.19
The weighted ORR results for dostarlimab compared with paclitaxel or doxorubicin using the data from the McMeekin et al. (2015)56 study and carboplatin plus paclitaxel using data from the Mazgani et al. (2008)58 study are summarized in Table 32. For the comparison of dostarlimab with paclitaxel or doxorubicin in McMeekin et al. (2015),56 dostarlimab was favoured over paclitaxel and over doxorubicin using data from the McMeekin et al. (2015)56 study (OR = 0.202; 95% CI, 0.116 to 0.352). There was no difference between dostarlimab and paclitaxel plus carboplatin using data from the Mazgani et al. (2008)58 study.19
Table 32: Summary of ORR Before and After Matching (MAIC Report 3)
Outcome | Odds ratio, comparator vs. dostarlimab | Relative risk, comparator vs. dostarlimab | Risk difference, comparator – dostarlimab |
|---|---|---|---|
McMeekin et al. (2015),56 paclitaxel or doxorubicin | |||
Estimate (95% CI) | 0.202 (0.116 to 0.352) | 0.328 (0.225 to 0.477) | –0.322 (–0.423 to –0.222) |
P valuea | < 0.0001 | < 0.0001 | < 0.0001 |
Mazgani et al. (2008),58 carboplatin + paclitaxel | |||
Estimate (95% CI) | 0.919 (0.327 to 2.583) | 0.951 (0.510 to 1.772) | –0.020 (–0.219 to 0.179) |
P valuea | 0.8733 | 0.8733 | 0.8432 |
CI = confidence interval; MAIC = matching-adjusted indirect comparison; ORR = overall response rate.
aP values are exploratory and not adjusted for multiplicity, so at increased risk of type I error.
Source: Sponsor-submitted ITC reports.19
SAEs were only compared for the GARNET and McMeekin et al. (2015)56 studies and are summarized in Table 33. The results demonstrate no difference in SAEs between dostarlimab and paclitaxel or doxorubicin.
Table 33: Summary of SAEs Before and After Matching (MAIC Report 3)
Outcome | Odds ratio, comparator vs. dostarlimab | Relative risk, comparator vs. dostarlimab | Risk difference, comparator – dostarlimab |
|---|---|---|---|
McMeekin et al. (2015),56 paclitaxel or doxorubicin | |||
Estimate (95% CI) | 0.802 (0.475 to 1.353) | 0.858 (0.600 to 1.227) | –0.047 (–0.147 to 0.054) |
P valuea | 0.4082 | 0.4012 | 0.3619 |
CI = confidence interval; MAIC = matching-adjusted indirect comparison; SAEs = serious adverse events.
aP values are exploratory and not adjusted for multiplicity, so at increased risk of type I error.
Source: Sponsor-submitted ITC reports.19
Inverse Probability Treatment Weighting
Description of IPTW Analyses
In addition to the 3 reports of MAIC analyses, the sponsor submitted 3 IPTW and propensity score matching (PSM) analyses to compare clinical outcomes in patients who received dostarlimab in GARNET with patients who received other treatments for advanced or recurrent EC, as follows19:
1 analysis comparing IPD from GARNET with real-world patients receiving current treatment paradigms in a US real-world data cohort, using data from the Flatiron Health database (hereafter called IPTW report 1).
1 analysis comparing IPD from GARNET with a GARNET-like RWE cohort in the UK from the NCRAS database (the base-case RWE cohort described in the MAIC section, hereafter called IPTW report 2).
1 analysis comparing GARNET IPD with IPD from the doxorubicin arm of the ZoptEC trial, identified using the same SLR that was used to inform comparators for the previously described MAICs (hereafter called IPTW report 3).
Methods of ITPW Analyses
Objectives
The aim of each analysis was to compare clinical outcomes of survival (OS) in patients treated with dostarlimab from GARNET with OS in patients treated with current treatment paradigms for advanced or recurrent EC, based on external control cohorts from RWE databases and 1 clinical trial using IPTW methods. Additional objectives across the analyses included comparisons of outcomes of PFS, response (ORR, DOR), time on treatment (DoT, TTNT), HRQoL, and AEs, although these were not conducted for each comparison.19
Study Selection Methods
Study selection methods varied in the 3 sponsor-submitted IPTW reports. The index trial in all cases was based on IPD from IA-2 for cohort A1 of part 2B of the phase I, single-arm GARNET trial (dMMR or MSI-H EC cohort; n = 129). The selection characteristics used to develop the GARNET-like cohorts for the IPTW analyses for IPTW reports 1 and 3 are summarized in Table 34. The selection criteria for IPTW report 2 (using data from the NCRAS database) are summarized in Table 20.19
In IPTW report 1, no SLR was conducted to identify relevant comparator studies. Instead, an external real-world control cohort was created, leveraging data from the Flatiron Health database, a US nationwide, longitudinal, demographically and geographically diverse database derived from electronic health record data from more than 280 cancer clinics. Patients with recurrent or advanced EC who had progressed on or after no more than 2 prior lines of systemic chemotherapy for advanced or recurrent disease, with at least 1 being platinum-based therapy, were identified from the Flatiron Health Endometrial Cancer Analytic Cohort. The inclusion and exclusion criteria for the real-world retrospective cohort study were divided into 2 categories: criteria that were used to create the Flatiron Health Endometrial Cancer Analytic Cohort (n = 4,264); and additional criteria (adult age; no more than 2 prior lines of therapy, including at least 1 line of platinum-based chemotherapy [prior hormone therapy was allowed but did not count toward line of therapy]; additional line of therapy after platinum therapy for advanced or recurrent EC, and an ECOG Performance Status of 0 or 1 at index) (n = 185 after applying all criteria).19
In IPTW report 2, no SLR was conducted to identify relevant comparator studies. Instead, data from NCRAS were used, as previously described, for the MAIC analyses (refer to Table 20). The initial RWE cohort base case (N = 999) was used for the IPTW analysis, with patients with an ECOG Performance Status of 0 or 1 included as a scenario analysis (N = 501). There were a few minor differences between patient-level data for the RWE cohort and aggregated data for the same cohort used previously to conduct the MAICs, including differences in age and race/ethnicity, although these changes would not likely affect the results.19
In IPTW report 3, in consultation with clinicians and payer advisors, doxorubicin was identified as 1 of the most used chemotherapies in EC patients who progressed on platinum-based treatments. A clinical trial investigating doxorubicin (ZoptEC) was identified from the same SLR that was used to identify relevant comparator studies for the MAIC comparing data from GARNET with that from published literature. ZoptEC was a phase III, open-label, randomized, active-controlled, international study evaluating the efficacy and safety of AEZS-108 and doxorubicin in advanced or recurrent EC patients who received 1 platinum plus taxane combination regimen as a first-line treatment and progressed. The trial was discontinued in 2017 as it did not achieve its primary or secondary end points. The transfer of the IPD for the doxorubicin arm (N = 255) of this trial was negotiated by the sponsor. Key differences in clinical criteria between the GARNET and ZoptEC trials included prior anti-cancer regimens (up to 2 prior lines in GARNET versus 1 in ZoptEC), biomarker status (dMMR or MSI-H in GARNET versus not evaluated in ZoptEC), and ECOG Performance Status (≤ 1 in GARNET versus ≤ 2 in ZoptEC). Although dMMR or MSI-H status was not known in ZoptEC, as previously noted, its influence as a prognostic or treatment-effect modifier in advanced or recurrent EC is unknown. Thus, it was assumed that the GARNET and ZoptEC patients had comparable molecular profiles.19
Table 34: Selection Criteria for GARNET-Like Subgroups and for Relevant Comparator Studies for Sponsor-Submitted IPTW Analyses
Outcome | Selection criteria for GARNET-like subgroups for IPTWs using Flatiron RWE | Selection criteria for GARNET-like subgroups for IPTWs using the ZoptEC trial |
|---|---|---|
Population | Patients with recurrent or advanced (≥ stage III) EC who have progressed on or after no more than 2 prior lines systemic chemotherapy for advanced (≥ stage III) or recurrent disease, with at least 1 of them being platinum-based therapy | Advanced and/or recurrent EC patients who received 1 platinum + taxane combination regimen as a first-line treatment and progressed |
Interventions and comparators | Current treatment paradigm | Doxorubicin |
Outcomes |
|
|
Study design | Routine, linked patient-level health data available through the Flatiron Health Endometrial Cancer Analytic Cohort | Synthetic control arm consisting of patient-level data from the doxorubicin arm of the ZoptEC RCT |
Publication characteristics | N/A | ZoptEC was a phase III, open-label RCT |
AE = adverse event; DOR = duration of response; DoT = duration of treatment; EC = endometrial cancer; IPTW = inverse probability of treatment weights; N/A = not applicable; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; QoL = quality of life; RCT = randomized controlled trial; RWE = real-world evidence; TTD = time to treatment discontinuation; TTNT = time to next treatment.
Source: Sponsor-submitted ITC reports.19
The primary outcome of each analysis was OS, which was previously defined for the GARNET trial and the NCRAS RWE cohort (Table 21). In the Flatiron database cohort (IPTW report 1), OS was defined as the interval between the index date and the date of death, with the index date defined by the initiation date of therapy after post-platinum therapy (could consist of multiple lines of platinum therapy). OS was the only outcome for the IPTW analysis comparing GARNET with the NCRAS cohort (IPTW report 2). In the ZoptEC trial (IPTW report 3), OS was defined as time from date of the first dose of study treatment to the date of death by any cause.19
Other outcomes in the analyses using the comparison with the Flatiron database included TTD, previously defined for GARNET and icalculated as the time between the start date and end date of the index treatment, or censoring if the patient died, was lost to follow-up, or still on-therapy in the Flatiron database, as well as descriptive analyses of DoT and TTNT. In the comparison with the ZoptEC trial (IPTW report 3), additional outcomes included19:
PFS, calculated as time from the first dose to the previous date of assessment of progression or death by any cause in the absence of progression based on the time of first documentation of disease progression
ORR, defined as the proportion of patients that achieved BOR of CR or PR, per RECIST 1.1
DOR, defined as time from the first documentation of overall response leading to a confirmed CR or PR, when confirmation is required by RECIST 1.1, until time of the first documentation of overall response that included disease progression or death
TTD in HRQoL, defined as time from the first cycle to the earliest time with a decrease of at least 10 points (deterioration) from baseline in the global HRQoL; however, no rationale for this threshold was provided
AEs, including TEAEs, SAEs.
IPTW Analysis Methods
The method of each IPTW analysis varied. A summary of the analysis methods in each MAIC is provided below.
GARNET Versus Flatiron Cohort Methods (IPTW Report 1)
Five patients in GARNET who received anti-PD-L1/2 therapies after dostarlimab were excluded because of differences in availability across countries. As such, the analysis set from the GARNET trial was the safety cohort (n = 124), as was the efficacy analysis dataset (n = 103). After the application of inclusion and exclusion criteria to the 4,564 patients from the Flatiron Health Endometrial Cancer Analytic Cohort, the final study population of the real-world retrospective cohort study was 185 patients.19
A feasibility assessment was conducted to determine whether the retrospective real-world cohort was suitable for use as an external control arm to the GARNET dMMR or MSI-H EC cohort. Feasibility was based on assessments of data quality and information availability, consideration of sample size, availability of key prognostic factors and outcomes, and comparability with the GARNET trial. Overall, the feasibility assessment revealed that the definitions of all prognostic variables were comparable between cohorts; however, there were noted differences in reporting of ECOG Performance Status, BMI, number of prior anti-cancer regimens, prior surgery, and MMR or MSI-H status. Notably, MMR or MSI status was not known for most patients, and there were concerns about the under-reporting of prior surgeries in the Flatiron cohort. Based on the results of the feasibility assessment, a decision was made to proceed with the analysis.19
Prognostic variables in EC were identified and validated in accordance with the methods described previously for the initial RWE cohort MAIC. The prognostic factors used as covariates in the models included race, age, ECOG Performance Status, histology, FIGO stage, BMI, disease grade, number of prior platinum-based regimens, and prior surgery.19
The KM method was used to describe the distribution of OS and TTD by cohort. Survival curves were presented graphically. Median OS (95% CI) and estimated survival probabilities at specific time points were reported by cohort. The HR for OS and its 95% CI for the cohort variable (GARNET versus real-world cohort) were calculated from a Cox proportional hazard model in which cohort was included as a single covariate. The corresponding P value was also reported. The PHA was checked graphically by log-cumulative hazard plots for the covariate and by Schoenfeld residual plots. To assess whether the PHA was met, an interaction between time and the study variable was added to the Cox models as a time-dependent covariate.19
Covariate-adjusted comparisons of OS and TTD between cohorts were performed using regression modelling (only for OS), IPTW, and PSM. Propensity scores for each patient in the real-world control cohort were estimated as a patient’s predicted probability of being assigned to the GARNET cohort, which was estimated from a logistic regression model. The propensity score model included the key prognostic variables as covariates.19
Propensity scores for each patient were estimated from a logistic regression model as a patient’s predicted probability of being assigned to the GARNET cohort. Variable selection in the propensity score model considered clinical relevance (determined by key opinion leaders and medical experts), frequency of reporting in the literature, and strength of association with OS. Two propensity score models were built19:
A “lean” propensity score model that only included a small number of the most relevant of the variables; namely, histology, grade of disease at initial EC diagnosis, ECOG Performance Status, and number of prior platinum-based therapies in the advanced or recurrent setting.
A propensity score model that included all variables identified except prior surgery for EC.
The overlap of the distribution of the propensity scores across the GARNET cohort and real-world control cohort was assessed with a histogram of the estimated propensity scores in the 2 cohorts. PSM formed pairs of subjects from the GARNET and the real-world cohorts, such that matched subjects had similar propensity score values. To account for the imbalance in sample size between the 2 cohorts, a 1:2 matching ratio was used, where 2 patients from the real-world cohort were matched to each patient from the GARNET cohort. The matching was based on the greedy nearest neighbourhood matching without replacement, using a caliper width of 0.2. After matching, OS and TTD were compared between cohorts in the matched sample. This included the calculation of KM curves and estimation of median survival and corresponding 95% CIs.19
Adjusted HRs were obtained from multivariable Cox regression models, and included the prognostic variables as covariates in addition to the cohort variable. The same 2 sets of covariates as in the propensity score models were considered in the regression models.19
IPTW was performed using weights from estimated propensity scores. Weights were calculated from propensity scores so that resulting estimates referred to the average treatment effect (ATE). To assess balance in baseline characteristics between cohorts before and after weighting and matching, absolute standardized differences were calculated for each covariate, in which the standardized difference was defined as the difference in means or proportions divided by the pooled SD. Using the IPTW method, OS and TTD were compared between the cohorts in the weighted sample. This included the creation of weighted KM curves and the estimation of weighted median survival and corresponding CIs. A weighted Cox regression model for OS was also fitted to estimate adjusted HRs for the cohort variable.19
GARNET Versus NCRAS RWE Cohort Methods (IPTW Report 2)
In the analyses comparing GARNET with the RWE cohort from NCRAS conducted to assess balance in baseline characteristics between cohorts before weighting, absolute standardized differences were calculated for each covariate, in which the standardized difference is defined as the difference in means or proportions divided by the pooled SD. Statistical differences in baseline characteristics between the 2 treatment cohorts were evaluated using t-tests for continuous variables and chi-square tests for categorical outcomes.19
As above, the KM method was used to describe median (95% CI) OS and estimated survival probabilities by cohort. The HR for OS and its 95% CI for the cohort variable (GARNET versus real-world cohort) was calculated from a Cox proportional hazard model that included cohort as a single covariate. The PHA was checked graphically with log-cumulative hazard plots for the covariate and plots of Schoenfeld residuals for the treatment variable over time. In addition, Schoenfeld global tests that evaluated independence between Schoenfeld residuals and time were also performed.19
Covariate-adjusted comparisons of OS between cohorts were performed using IPTW based on the propensity score. Propensity scores for each patient were estimated as a patient’s predicted probability of being assigned to the GARNET cohort, which was estimated from a logistic regression model. The propensity score model included the key prognostic variables, identified following the method previously described for the RWE cohort MAIC, as covariates.19
Three different propensity score models were fitted. The sets of covariates used in the 3 propensity score models were identical to the sets of matching variables that defined the 3 MAIC scenarios described previously. The overlap of the distribution of propensity scores across the GARNET trial cohort and the RWE cohort was assessed with a histogram distribution graph of propensity scores across cohorts.19
Adjustment for potential imbalances in relevant prognostic variables between cohorts was done with the IPTW method. In the principal analysis, weights were calculated from propensity scores so that resulting estimates referred to the ATE. As a secondary analysis, weights were calculated from propensity scores so that resulting estimates referred to the ATE for the controls. The use of the ATE for controls perspective was motivated by the fact that this approach was similar to the weighting approach previously used in the MAIC, where each patient in the control cohort was assigned a fixed weight of 1.19
Using the IPTW method, OS was compared between the cohorts in the weighted sample. This included the creation of weighted KM curves and the estimation of weighted median survival and corresponding CIs. In the weighted KM curves, numbers at risk represent the sums of individual weights of patients at risk, rather than the number of patients at risk. A weighted Cox regression model was also fitted to estimate adjusted HRs (and 95% CIs, P values) for the cohort variable.19
GARNET Versus ZoptEC Methods (IPTW Report 3)
To facilitate a comparison between the single-arm GARNET trial and control patients using doxorubicin in the ZoptEC RCT, IPD from GARNET and ZoptEC were merged to create a comparator control arm.19
To make the baseline characteristics between the 2 trials comparable before statistical analysis, exclusion criteria were applied to each trial. Patients in the ZoptEC comparator trial who would have been ineligible in GARNET were removed before analysis if such criteria were easily determined from the dataset. Patients were excluded from the ZoptEC trial if they had a follow-up longer than 36 months, and patients were excluded if they did not have an ECOG Performance Status score of 0 or 1. Patients were excluded from the GARNET trial if they had previously received more than 1 prior platinum-based therapy. The main analysis dataset included 92 patients from GARNET and 233 patients from ZoptEC, and was used for OS, PFS, and ORR.19 A summary of datasets in the analysis is provided in Table 35.
A sensitivity analysis that included all patients from the GARNET and ZoptEC trials was conducted to ensure that the removal of patients did not violate the positivity or exchangeability assumption for the IPTW method. The positivity assumption can be empirically verified by showing there is a positive probability of each treatment for each set of covariates, whereas the exchangeability assumption assesses the potential impact of unmeasured confounding on the results using the methods of Mortimer et al. (2005)59 and Brumback et al. (2004),60 respectively. Additional sensitivity analyses included 1 that used the safety analysis dataset to assess whether this would affect the OS results; another of TTD in HRQoL was conducted using a 15-point decrease as the threshold.19
Table 35: Summary of Included Patients in Each Analysis Dataset
Cohort | GARNET, N | ZoptEC, N | Comment |
|---|---|---|---|
ITT | 129 | 255 | — |
Safety analysis dataset Received at least 1 dose of treatment | 129 | 249 | 6 patients in ZoptEC did not receive a dose of doxorubicin |
Main analysis dataset Reduced sample size for main analysis after application of exclusion criteria to make the trial populations similar | 92 | 233 | Main dataset for OS, PFS, and ORR end points |
DOR dataset DOR is contingent on response | 40 | 32 | 40 of 92 patients in GARNET and 32 of 233 patients in ZoptEC achieved a response |
TTD in HRQoL dataset Dataset is contingent on having a HRQoL score to assess deterioration | 62 | 188 | 62 of 92 patients in GARNET and 188 of 233 patients in ZoptEC had a baseline and a subsequent HRQoL score |
DOR = duration of response; HRQoL = health-related quality of life; ITT = intention to treat; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; TTD = time to treatment discontinuation.
Source: Sponsor-submitted ITC reports.19
The main analysis dataset sample size — 233 for doxorubicin and 92 for dostarlimab — contained all available patients after the exclusion criteria were applied; thus, a sample size calculation was not applicable. However, a power analysis for OS was performed. If the HR for OS was greater than 0.65, the analysis would be underpowered; however, if the HR was less than or equal to 0.65, the analysis would be sufficiently powered. As with any statistical model using IPTW, power is decreased because IPTW inflates type I errors. Therefore, a stabilized IPTW was used, which is better suited in handling the inflation of type I errors and can also minimize any issues with statistical power.19
In the comparison between GARNET and the ZoptEC study, prognostic variables were identified and ranked with methods previously described for the RWE cohort published literature MAICs.19
A multivariable method with interaction terms for the assessment of effect modification was used for this analysis. For OS, Cox proportional hazards were used with covariates: treatment, candidate for effect modification (e.g., age or ECOG), and the interaction term (treatment × candidate of effect modification). If the P value for interaction was less than 0.05, the variable was considered an effect modifier. If effect modification was present, a subgroup analysis for each level of the effect modifier, in addition to the main analysis, was conducted.19
Because of the lack of randomization, there was a need to control for confounding bias at baseline, so an IPTW method was chosen. PSM was not chosen because there was a restricted number of patients in each arm coming from the clinical trial. A stabilized weight was estimated for each patient to reduce inflation of the type I error rate or to unintentionally increase statistical power. Once weighted, a Cox proportional hazards regression model with IPTW was used for the comparison of OS between dostarlimab and doxorubicin. KM analysis was used for PFS, DOR, and TTD in HRQoL. For binary outcomes such as an ORR, descriptive analyses were used. Specifically, the Clopper-Pearson method was used to calculate the 95% CI of the proportion. All assumptions, such as unmeasured confounding, positivity, and consistency, were checked in the validation of the IPTW method.19
Results of IPTW Analyses
Summary of Included Studies
Key study design characteristics and sources of heterogeneity of the studies included in the IPTW analyses are summarized in Table 36. There was considerable variation in the design of the 4 studies that were used in the IPTW analyses. The GARNET trial evaluated patients with dMMR or MSI-H EC, whereas the Flatiron, NCRAS, and ZoptEC studies included patients with advanced or recurrent EC, regardless of biomarker status. A total of 128 (69.2%) patients in the Flatiron database had unknown MMR or MSI-H status at index. There was also variation in the treatments included, with GARNET evaluating dostarlimab, the RWE cohorts evaluating the current treatment paradigms in advanced or recurrent EC, and the ZoptEC trial evaluating doxorubicin. Eligible patients typically had an ECOG Performance Status of 0 or 1; however, the ZoptEC trial enrolled patients with an ECOG Performance Status of 2, and ECOG status was unknown in most of the NCRAS RWE cohort. The line of therapy also varied across studies, with GARNET and the Flatiron RWE cohorts including patients with no more than 2 lines of therapy, 1 of which was a platinum doublet. The NCRAS real-world cohort used platinum doublet therapy as the key milestone regarding lines of therapy and did not analyze the lines of therapy before advanced or recurrent diagnosis. Moreover, the ZoptEC study only required 1 prior line of therapy.19 No consideration was noted regarding the differences in dosage, treatment or follow-up duration, or supportive care or cointerventions in the included studies.
Table 36: Key Study Characteristics in the GARNET and Comparator Studies
Variable | GARNET | IPTW report 1, Flatiron RWE cohort | IPTW report 2, NCRAS RWE cohort | IPTW report 3, ZoptEC trial |
|---|---|---|---|---|
Study design | Nonrandomized, single-arm, prospective, interventional study | Retrospective real-world study of electronic health records | Descriptive, noninterventional, patient-level data from NCRAS | Phase III, open-label, randomized, active-controlled, international study |
Region | Global | US | UK | Global |
Treatments | Dostarlimab | Current treatment paradigm | Current treatment paradigm | Doxorubicin |
Outcomes | OS, TTD | OS, TTD, DoT, TTNT | OS | OS, PFS, ORR, DOR, AEs, TTD in QoL |
Sample size | 129 (or 124)a | 185 | 999 | 255 |
Included population | Adults with advanced, recurrent, or metastatic EC who have dMMR or MSI-H | Recurrent or advanced (≥ stage III) EC | Patients with recurrent or advanced EC | Adults with advanced, recurrent, or metastatic EC |
ECOG | 0 or 1 | 0 or 1 | 0 or 1 | ≤ 2 |
Prior therapy | No more than 2 lines of anti-cancer therapy for recurrent or advanced (≥ stage IIIB) disease, including platinum doublet therapy | Received no more than 2 prior lines of systemic chemotherapy for advanced or recurrent disease, including ≥ 1 line of platinum-based chemotherapy | Prior platinum doublet therapy in the first-line setting; all patients were considered second line | One prior platinum plus taxane combination therapy |
AEs = adverse events; dMMR = mismatch repair-deficient; DOR = duration of response; DoT = duration of treatment; EC = endometrial cancer; ECOG = Eastern Cooperative Oncology Group performance status; IPTW = inverse probability of treatment weights; MSI-H = microsatellite instability-high; NCRAS = National Cancer Registration and Analysis Service; OS = overall survival; PFS = progression-free survival; RWE = real-world evidence; TTD = time to treatment discontinuation; TTNT = time to next treatment; QoL = quality of life.
aFive patients from GARNET were excluded in the comparison with the Flatiron database, as anti-PD-L1/2 treatments were not approved for this indication in many countries.
Source: Sponsor-submitted ITC reports.19
GARNET Versus Flatiron Cohort (IPTW Report 1)
The baseline characteristics before weighting in the IPTW analysis of GARNET and the Flatiron RWE cohort are summarized in Table 37. In total, the GARNET safety population included 124 patients and the real-world cohort included 185 patients. There were 5 patients in GARNET (n = 129) not eligible for the comparison and excluded who received anti-PD-L1/2 after dostarlimab. In general, the populations of the GARNET trial and the Flatiron RWE cohort were relatively similar, except the GARNET trial had a higher proportion of White patients (75.0% versus 61.1%), endometrioid histology (66.1% versus 57.3%), FIGO stage I/II (43.5% versus 35.7%), and prior surgery (89.54% versus 56.2%). There was a substantial difference in disease grade between the GARNET and Flatiron cohorts because of the large amount of missing data for grade at diagnosis in the Flatiron cohort (38.4%). Comparability of the following variables between the 2 cohorts was limited because of discrepancies in variable definitions or data under-reporting of surgery and radiation in the Flatiron database: prior surgery for EC, history of radiation therapy before second-line therapy, and duration of the platinum- and progression-free interval at second line (because of the unavailability of response data in Flatiron, the platinum-free interval was measured differently in Flatiron and GARNET). The most frequent first-line regimen in the Flatiron cohort was carboplatin plus paclitaxel, received by the majority of patients (72.4%). During second-line treatment, the top 3 regimens were pegylated liposomal doxorubicin (17.8%), carboplatin plus paclitaxel (15.1%), and carboplatin plus docetaxel (9.7%). Only 105 patients received third-line therapy, with the most common being pegylated liposomal doxorubicin (15.2%), bevacizumab (9.5%), and carboplatin plus paclitaxel (7.6%).19
GARNET Versus NCRAS RWE Cohort (IPTW Report 2)
The baseline characteristics before weighting of the IPTW analysis of GARNET and the NCRAS RWE cohort are summarized in Table 37. In total, the GARNET cohort included 129 patients and the RWE base-case cohort included 999 patients. Patients in the GARNET trial were younger (51.2% versus 44.5% < 65 years). Overall, there were key differences in most characteristics of the patient populations; compared with the NCRAS cohort, GARNET had fewer White patients (76.0% versus 84.2%), and a greater proportion of patients with endometrioid histology (69.8% versus 42.4%), FIGO stage I and II disease (44.2% versus 22.1%), an ECOG Performance Status of 0 (42.6% versus 32.0%) or 1 (57.4% versus 18.1%), and a disease of grade 1 and 2 (67.4% versus 27.4%). The number of prior platinum-based therapies in the advanced or recurrent setting was 1 for all patients in the NCRAS RWE cohort, whereas it was 0 or 2 or more for 1.6% and 13.2% of patients in the GARNET cohort, respectively.19
GARNET Versus ZoptEC (IPTW Report 3)
The baseline characteristics before weighting of the IPTW analysis of GARNET and the ZoptEC trial are summarized in Table 37. The sponsor noted that the baseline characteristics were similar enough to warrant comparison. There were fewer White patients in GARNET than in the ZoptEC trial (76.0% versus 94.1%), and more patients with FIGO stage III and IV disease (85.3% versus 36.9%). The proportion of patients with endometrioid histology was similar. Comparability of some key variables, including BMI, disease grade, number of prior platinum therapies, and prior surgery, was limited because of a lack of reported data.19
Table 37: Baseline Characteristics of Patients by Cohort Before Weighting
Characteristic | IPTW Report 1, GARNET vs. Flatiron RWE cohort | IPTW report 2, GARNET vs. NCRAS RWE cohort | IPTW report 3, GARNET vs. ZoptEC | |||
|---|---|---|---|---|---|---|
GARNET population (N = 124) | Flatiron RWE cohort (N = 185) | GARNET population (N = 129) | NCRAS RWE cohort (N = 999) | GARNET population (N = 129) | ZoptEC doxorubicin arm (N = 255) | |
Age, n (%) | ||||||
< 65 years | 63 (50.8) | 83 (44.9) | 66 (51.2) | 445 (44.5) | 66 (51.2) | 136 (53.3) |
≥ 65 years | 61 (49.2) | 102 (55.1) | 63 (48.8) | 554 (55.5) | 63 (48.8) | 119 (46.7) |
Race, n (%) | ||||||
White | 93 (75.0) | 113 (61.1) | 98 (76.0) | 841 (84.2) | 98 (76.0) | 240 (94.1) |
Asian | 5 (4.0) | 4 (2.2) | 20 (15.5) | NR | 5 (3.9) | 5 (2.0) |
Black | 3 (2.4) | 41 (22.2) | 3 (2.3) | 55 (5.5) | 3 (2.3) | 7 (2.7) |
Other | 3 (2.4) | 22 (11.9) | 8 (6.2) | 78 (7.8) | 3 (2.3) | 3 (1.2) |
Unknown or NR | 20 (16.1) | 5 (2.7) | 0 (0) | 25 (2.5) | 20 (15.5) | 0 (0) |
BMI (kg/m2) | ||||||
Mean (SD) | 29.62 (8.07) | 32.07 (8.24) | NR | NR | 29.7 (8.1) | NR |
Median (range) | 28.83 (13.6 to 53.9) | 30.45 (17.9 to 66.5) | NR | NR | 29.0 (23.1 to 34.9) | NR |
MMR or MSI-H status, n (%) | ||||||
dMMR or MSI-H | 121 (97.6) | 10 (5.4) | NR | NR | NR | NR |
Unknown | 3 (2.4) | 128 (69.2) | NR | NR | NR | NR |
Histology, n (%) | ||||||
Endometrioid | 82 (66.1) | 106 (57.3) | 90 (69.8) | 424 (42.4) | 85 (65.9) | 164 (64.3) |
Nonendometrioid | 41 (33.1) | 79 (42.7) | 38 (29.5) | 575 (57.6) | 43 (33.4) | 91 (35.7) |
Unknown | 1 (0.8) | 0 (0) | 1 (0.8) | 0 (0) | 1 (0.8) | NR |
ECOG, n (%) | ||||||
0 | 54 (43.5) | 86 (46.5) | 55 (42.6) | 320 (32.0) | 55 (42.6) | 125 (49.0) |
1 | 70 (56.5) | 99 (53.5) | 74 (57.4) | 181 (18.1) | 74 (57.4) | 118 (46.3) |
Unknown | 0 (0) | 0 (0) | NR | 498 (49.8) | NR | NR |
FIGO stage, n (%) | ||||||
Stage I and II | 54 (43.5) | 66 (35.7) | 57 (44.2) | 221 (22.1) | 17 (13.2) | NR |
Stage III and IV | 70 (56.6) | 105 (56.7) | 72 (55.8) | 778 (77.9) | 110 (85.3) | 94 (36.9) |
Unknown | 0 (0) | 14 (7.6) | 0 (0) | 0 (0) | 2 (1.4) | NR |
Disease grade, n (%) | ||||||
Grade 1 and 2 | 83 (66.9) | 70 (37.8) | 87 (67.4) | 274 (27.4) | NR | NR |
Grade 3 and 4 | 35 (28.2) | 44 (23.8) | 36 (27.9) | 389 (38.9) | NR | NR |
Unknown | 6 (4.8) | 71 (38.4) | 6 (4.7) | 336 (33.6) | NR | NR |
Prior anti-cancer therapy for adjuvant/neoadjuvant disease, n (%)a | ||||||
Yes | 69 (55.6) | 25 (13.5) | NR | NR | 129 (100) | 92 (36.9) |
No | 55 (44.4) | 160 (86.5) | NR | NR | 0 (0) | NR |
Number of prior platinum therapies, n (%) | ||||||
0 | 2 (1.6) | 0 (0) | 2 (1.6) | 0 (0) | NR | NR |
1 | 105 (84.7) | 166 (89.7) | 110 (85.3) | 999 (100.0) | NR | NR |
≥ 2 | 17 (13.7) | 19 (10.3) | 17 (13.2) | 0 (0) | NR | NR |
Prior radiotherapy, n (%) | ||||||
Yes | 92 (74.2) | 50 (27.0) | NR | NR | 94 (72.9) | 138 (55.4) |
No or unknown | 32 (25.8) | 135 (73.0) | NR | NR | NR | NR |
Number of lines of therapy post-index, n (%) | ||||||
0 | 104 (83.9) | 108 (58.4) | NR | NR | NR | NR |
1 | 13 (10.5) | 43 (23.2) | NR | NR | NR | NR |
≥ 2 | 7 (5.6) | 34 (18.4) | NR | NR | NR | NR |
Prior surgery, n (%) | ||||||
Yes | 111 (89.5) | 104 (56.2) | 116 (89.9) | 815 (81.6) | 116 (89.9) | 222 (89.2) |
No | 13 (10.5) | 81 (43.8) | 13 (10.1) | 184 (18.4) | NR | NR |
BMI = body mass index; ECOG = Eastern Cooperative Oncology Group performance status; FIGO = International Federation of Gynecology and Obstetrics; IPTW = inverse probability of treatment weights; MMR = mismatch repair; MSI-H = microsatellite instability-high; NCRAS = National Cancer Registration and Analysis Service; NR = not reported; RWE = real-world evidence; SD = standard deviation; vs. = versus.
aAdjuvant or neoadjuvant therapy refers to pharmacological treatment, including chemotherapy and hormone therapy.
Source: Sponsor-submitted ITC reports.19
Results
GARNET Versus Flatiron Cohort (IPTW Report 1)
Propensity scores for each patient were estimated from a logistic regression model as a patient’s predicted probability of being assigned to the GARNET trial. Results from the lean and full propensity score models fitted on the GARNET trial compared with the Flatiron cohort are summarized in Table 53. The overlap of the distribution of the propensity scores was assessed with a histogram distribution graph of propensity scores across cohorts (refer to Figure 12 and Figure 13 in Appendix 4). The lean model demonstrated greater overlap than the full model, so only the lean model was used.19
The distributions of baseline and prognostic factors considered for ITC analyses for GARNET and the Flatiron cohort after lean model IPTW and PSM are summarized in Table 54. Before and after matching, baseline and prognostic variables of race, FIGO, and BMI were different in GARNET and the Flatiron cohort. The matched GARNET cohort had a sample size of 93, and the matched real-world cohort had a sample size of 103.19
Results for OS and TTD before and after IPTW and PSM are summarized in Table 38. Median OS after IPTW was not estimable in GARNET (95% CI, 15.4 to not estimable) and was 13.1 (95% CI, 8.3 to 15.9) in the Flatiron cohort. Median OS after PSM was not estimable in GARNET (95% CI, 17.1 to not estimable) and was 15.9 (95% CI, 10.4 to 33.2) in the Flatiron cohort. Survival rates were higher for dostarlimab than for the Flatiron current treatment paradigm cohort; after IPTW, 24-month OS rates were 0.529 (95% CI, 0.367 to 0.667) in the GARNET cohort and 0.338 (95% CI, 0.262 to 0.415) in the real-world cohort. After PSM, 24-month OS rates were 0.536 (95% CI, 0.387 to 0.664) in the GARNET cohort and 0.410 (95% CI, 0.305 to 0.512) in the real-world cohort. Although the point estimates differ, 95% CIs overlapped, indicating no difference in survival rates.19
Cox proportional hazards regression models were fitted for OS to examine the association between cohort (GARNET versus Flatiron) and the risk of death. The PHA was checked graphically with a log-cumulative hazard plot and with a plot of Schoenfeld residuals for the treatment variable over time. In addition, an interaction between time and study was added to the Cox model as a time-dependent covariate. If the interaction term was not statistically significant, it was reasonable to assume that the PHA was valid.19
Unadjusted Cox proportional hazards regression models for both GARNET and the Flatiron cohort suggested that GARNET patients had a lower risk.(HR = 0.447; 95% CI, 0.305 to 0.653). When interaction for study and time were included, there was no difference in risk of death between studies (HR = 0.989; 95% CI, 0.927 to 1.055). For the model fitted after IPTW, the interaction term was significant when only the study variable was included (HR = 0.559; 95% CI, 0.385 to 0.812) but was not statistically significant when study and time variables were included. After PSM, the interaction terms were not statistically significant (HR for study = 0.653; 95% CI, 0.413 to 1.033; HR for study and time = 1.030; 95% CI, 0.955 to 1.111) (Table 55), so it was reasonable to assume that the PHA holds.19
Results from the multivariable Cox proportional hazards model for OS where 2 sets of covariates were adjusted are summarized in Table 56. The interaction term of study and time was not statistically significant in either adjustment scenario, which indicated that the PHA was valid for study. The results suggested that the study variable favoured the GARNET trial for risk of death after adjustment for the covariates in both adjustment scenarios. Table 57 shows the Cox proportional hazards model results for OS adjusted for covariates and propensity scores. For the study variable, the HRs adjusted for propensity score and for propensity score and covariates were consistent.19
For the exploratory outcome of TTD before and after IPTW and PSM, the results were congruent with the primary analysis of OS, with higher TTD rates at 24 months in the GARNET trial than in the Flatiron database (Table 38). The TTD rate after IPTW was 0.480 (95% CI, 0.319 to 0.625) in GARNET and 0.149 (95% CI, 0.081 to 0.237) in the real-world cohort. After PSM, the TTD rate at 24 months was 0.397 (95% CI, 0.272 to 0.520) in GARNET and 0.173 (95% CI, 0.084 to 0.287) in the real-world cohort; however, the overlap in the 95% CIs indicated that there was no difference.19
Table 38: OS Before and After IPTW and PSM — GARNET Versus Flatiron Real-World Cohort (IPTW Report 1)
Outcome | Before weighting | After IPTW | After PSM | |||
|---|---|---|---|---|---|---|
GARNET, N = 124 | Flatiron cohort, N = 185 | GARNET, N = 121 | Flatiron cohort, (N = 185) | GARNET, N = 93 | Flatiron cohort, N = 103 | |
OS | ||||||
OS (months), median(95% CI) | NE (18.4 to NE) | 11.1 (8.1 to 15.2) | NE (15.4 to NE) | 13.1 (8.3 to 15.9) | NE (17.1 to NE) | 15.9 (10.4 to 33.2) |
OS rate (95% CI) | ||||||
6 months | 0.823 (0.737 to 0.884) | 0.690 (0.617 to 0.752) | 0.831 (0.703 to 0.907) | 0.695 (0.618 to 0.760) | 0.833 (0.733 to 0.898) | 0.722 (0.620 to 0.801) |
12 months | 0.729 (0.628 to 0.807) | 0.497 (0.422 to 0.568) | 0.715 (0.569 to 0.819) | 0.510 (0.430 to 0.585) | 0.715 (0.598 to 0.804) | 0.574 (0.468 to 0.667) |
18 months | 0.638 (0.519 to 0.735) | 0.373 (0.301 to 0.445) | 0.569 (0.405 to 0.703) | 0.399 (0.321 to 0.476) | 0.598 (0.459 to 0.713) | 0.481 (0.375 to 0.579) |
24 months | 0.588 (0.458 to 0.697) | 0.319 (0.250 to 0.391) | 0.529 (0.367 to 0.667) | 0.338 (0.262 to 0.415) | 0.536 (0.387 to 0.664) | 0.410 (0.305 to 0.512) |
TTD | ||||||
TTD (months), median (95% CI) | 14.0 (6.0 to NE) | 5.3 (4.2 to 6.0) | 20.7 (7.3 to NE) | 5.3 (4.1 to 6.0) | 14.0 (5.7 to NE) | 4.9 (3.9 to 6.2) |
TTD rate (95% CI) | ||||||
6 months | 0.596 (0.498 to 0.681) | 0.412 (0.324 to 0.498) | 0.656 (0.510 to 0.768) | 0.402 (0.310 to 0.492) | 0.586 (0.472 to 0.683) | 0.386 (0.269 to 0.501) |
12 months | 0.533 (0.434 to 0.623) | 0.179 (0.108 to 0.265) | 0.615 (0.468 to 0.733) | 0.191 (0.115 to 0.282) | 0.547 (0.433 to 0.647) | 0.219 (0.120 to 0.336) |
18 months | 0.440 (0.334 to 0.540) | 0.140 (0.076 to 0.222) | 0.514 (0.352 to 0.654) | 0.149 (0.081 to 0.237) | 0.431 (0.311 to 0.545) | 0.173 (0.084 to 0.287) |
24 months | 0.414 (0.304 to 0.520) | 0.140 (0.076 to 0.222) | 0.480 (0.319 to 0.625) | 0.149 (0.081 to 0.237) | 0.397 (0.272 to 0.520) | 0.173 (0.084 to 0.287) |
CI = confidence interval; IPTW = inverse probability of treatment weights; NE = not estimable; OS = overall survival; PSM = propensity score matching; TTD = time to treatment discontinuation; vs. = versus.
Note: The propensity score was constructed using the following covariates: histology, grade, ECOG Performance Status, and number of prior platinum-based therapies
Source: Sponsor-submitted ITC reports.19
GARNET Versus NCRAS RWE Cohort (IPTW Report 2)
Baseline characteristics of the GARNET and NCRAS base-case cohorts after IPTW from 3 propensity score models are summarized in Table 58. Propensity score model 1 and 2 removed patients with unknown histology and patients with 0 or at least 2 prior platinum-based therapies from the GARNET cohort, which resulted in a sample size of 109 for the GARNET cohort; propensity score model 3 only removed patients with unknown histology from the GARNET cohort, which resulted in a sample size of 128 for the GARNET cohort. The standardized differences for variables used in the propensity score models were smaller after IPTW, compared with unweighted differences.19
Prior to IPTW, median OS could not be estimated for the GARNET cohort and was 10.3 months (95% CI, 9.2 to 11.1) for the RWE base-case cohort. The survival rates remained higher in the GARNET than in the RWE cohort during the study. At month 24, the survival rate was 0.578 (95% CI, 0.450 to 0.686) in the GARNET cohort and 0.215 (95% CI, 0.186 to 0.244) in the RWE base-case cohort. OS after IPTW that compared ATE weights for GARNET with the NCRAS RWE base-case cohort is summarized in Table 39. After IPTW, results for all propensity score models were consistent, with a greater median OS for dostarlimab than for the current treatment paradigm in the NCRAS RWE cohort (median OS was not evaluable at follow-up in GARNET and was 10.3 for all NCRAS models). Across all models after weighting, survival rates at 24 months ranged from 0.517 (95% CI, 0.326 to 0.678) to 0.606 (95% CI, 0.440 to 0.736) for GARNET and from 0.216 (95% CI, 0.187 to 0.246) to 0.218 (95% CI, 0.189 to 0.248) for the NCRAS RWE cohort. Using ATE for controls weights, results were consistent with ATE. Results of sensitivity analyses in patients with a ECOG Performance Status of 0 or 1 were consistent with the primary analysis.19
The HR for the cohort variable (GARNET versus real-world cohort) were calculated from a Cox proportional hazards model that included cohort as a single covariate. In all models, the HRs ranged from 0.310 (95% CI, 0.271 to 0.355) to 0.438 (95% CI, 0.386 to 0.498) (Table 39).19
The PHA was checked graphically with means of log-cumulative hazard plots for the covariate and plots of Schoenfeld residuals for the treatment variable over time. Both the graphs and the tests suggest that the PHA was valid for the unadjusted Cox models, as well as for the weighted Cox models based on weights calculated from propensity score model 3. The weighted Cox models based on weights calculated from propensity score models 1 and 2 do not seem to meet the PHA.19
Table 39: OS Before and After IPTW — GARNET Versus RWE Base-Case Cohort Propensity Score Models (ATE; IPTW Report 2)
Outcome | Before weighting | Propensity score model 1 | Propensity score model 2 | Propensity score model 3 | ||||
|---|---|---|---|---|---|---|---|---|
GARNET, N = 129 | NCRAS cohort, N = 99 | GARNET, N = 109 | NCRAS cohort, N = 999 | GARNET, N = 109 | NCRAS cohort, N = 999 | GARNET, N = 128 | NCRAS cohort, N = 999 | |
OS (months), median (95% CI) | NE (18.4 to NE) | 10.3 (9.2 to 11.1) | NE (15.4 to NE) | 10.3 (9.3 to 11.1) | NE (18.0 to NE) | 10.3 (9.2 to 11.1) | NE (21.6 to NE) | 10.3 (9.2 to 11.1) |
HR (95% CI), | 0.380 (0.272 to 0.531) | 0.438 (0.386 to 0.498) | 0.346 (0.302 to 0.396) | 0.310 (0.271 to 0.355) | ||||
P valuea | < 0.001 | < 0.001 | < 0.001 | < 0.001 | ||||
OS rate (95% CI) | ||||||||
6 months | 0.830 (0.746 to 0.888) | 0.700 (0.670 to 0.728) | 0.855 (0.707 to 0.931) | 0.702 (0.672 to 0.730) | 0.852 (0.743 to 0.917) | 0.702 (0.672 to 0.730) | 0.880 (0.774 to 0.938) | 0.703 (0.673 to 0.731) |
12 months | 0.716 (0.616 to 0.795) | 0.436 (0.403 to 0.468) | 0.726 (0.554 to 0.840) | 0.440 (0.407 to 0.472) | 0.751 (0.621 to 0.841) | 0.438 (0.405 to 0.470) | 0.778 (0.647 to 0.865) | 0.437 (0.404 to 0.469) |
18 months | 0.647 (0.534 to 0.740) | 0.287 (0.257 to 0.318) | 0.578 (0.379 to 0.733) | 0.292 (0.262 to 0.324) | 0.671 (0.520 to 0.784) | 0.290 (0.260 to 0.321) | 0.707 (0.556 to 0.815) | 0.288 (0.258 to 0.320) |
24 months | 0.578 (0.450 to 0.686) | 0.215 (0.186 to 0.244) | 0.517 (0.326 to 0.678) | 0.218 (0.189 to 0.248) | 0.606 (0.440 to 0.736) | 0.217 (0.188 to 0.247) | 0.597 (0.407 to 0.744) | 0.216 (0.187 to 0.246) |
ATE = average treatment effect; CI = confidence interval; HR = hazard ratio; IPTW = inverse probability of treatment weights; NCRAS = National Cancer Registration and Analysis Service; NE = not estimable; OS = overall survival; RWE = real-world evidence; vs. = versus.
aP values are exploratory and not adjusted for multiplicity, so at increased risk of type I error.
Source: Sponsor-submitted ITC reports.19
GARNET Versus ZoptEC (IPTW Report 3)
The median OS of dostarlimab was not reached (95% CI, 17.150 to not reached), whereas the median OS for doxorubicin was 11.039 months (95% CI, 9.988 to 13.010). A naive HR was calculated using the lower-bound CI for dostarlimab, resulting in an unadjusted HR of 0.644. The main analysis was performed using a Cox proportional hazards model with stabilized IPTW to estimate the HR for OS (dostarlimab versus doxorubicin). A proportionality test was conducted to ensure that the PHA was not violated. As the P value was greater than 0.05, the PHA was supported, and the adjusted analysis used a Cox proportional hazards model with stabilized IPTW. KM curves for OS before and after adjustment are shown in Table 37. After adjusted stabilized IPTW, results of the Cox proportional hazards model suggested that dostarlimab was favoured over doxorubicin for OS (HR, 0.409 [95% CI, 0.277 to 0.605]). Survival time was significantly longer for dostarlimab, with median OS not reached (95% CI, 18.004 to not reached); for doxorubicin, median OS was 11.170 months (95% CI, 9.988 to 13.076).19
Figure 9: KM Curves for OS Without and With Adjusted Stabilized IPTW (IPTW Report 3)
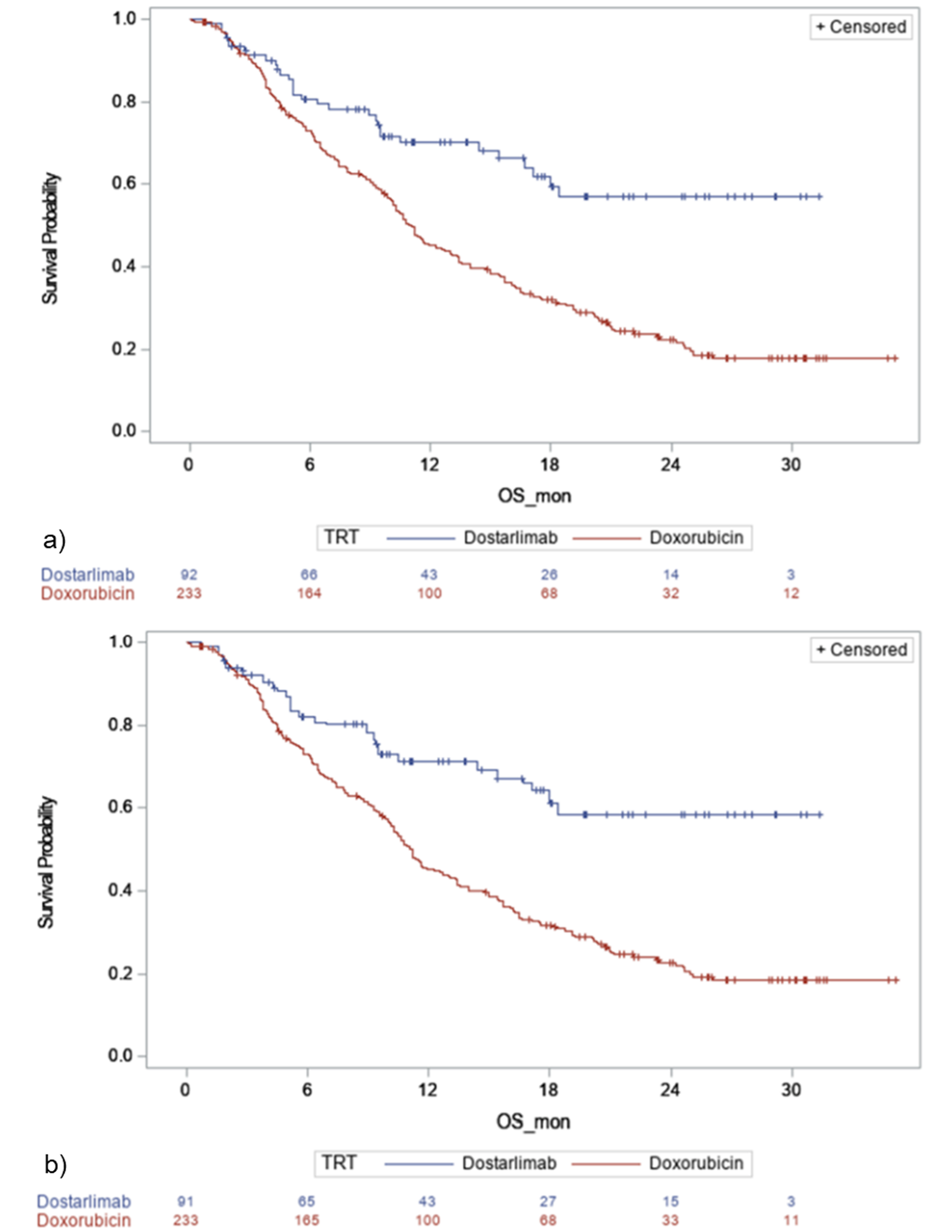
IPTW = inverse probability of treatment weights; KM = Kaplan-Meier; OS = overall survival; OS_mon = overall survival monotherapy; TRT = treatment.
Note: a = KM curves for OS without adjusted stabilized IPTW for the main analysis dataset; b = KM curves for OS with adjusted stabilized IPTW for the main analysis dataset.
The number at risk with IPTW adjustment may differ slightly from the total sample size. This is because the number at risk has been weighted by IPTW.
Source: Sponsor-submitted ITC reports.19
A sensitivity analysis was conducted using the full safety analysis population from the GARNET (n = 129) and ZoptEC (n = 249) trials. Results were consistent with the main analysis, with dostarlimab favoured over doxorubicin for OS (HR, 0.403 [95% CI, 0.280 to 0.581]).19
Critical Appraisal of Indirect Evidence
Six ITC reports were submitted by the sponsor: 3 MAIC reports and 3 IPTW reports. The choice to conduct unanchored MAICs and IPTW analyses was justified by the lack of a common comparator. In all cases, the GARNET trial was used as the index trial. In 1 MAIC and 1 IPTW analysis, the comparator data were identified with a SLR, so there is a low risk of selection bias. For the remaining MAICs and IPTWs, no SLR was conducted to identify comparator studies. Instead, data from the UK NCRAS RWE database and the Flatiron Health database were used. As these were not selected using a systematic approach, there is a high risk of selection bias. It is not possible to know whether the results would have differed if data from a different database had been used.
There were important differences in the design of the comparator studies that limit the ability to draw strong conclusions about the efficacy of dostarlimab compared with other treatments. GARNET was a phase I, single-arm trial, whereas the comparators included real-world cohorts from the Flatiron electronic health records in the US and the NCRAS real-world database in the UK, published literature that consisted of retrospective and prospective studies, and a phase III RCT. Data analyzed retrospectively from databases and medical records are prone to unique biases (e.g., selection bias, confounding), compared with those collected from prospective, interventional studies (like RCTs and single-arm trials) that cannot be controlled for using MAICs or IPTWs.
The data-collection period and setting of the included studies varied, with enrolment for some beginning as far back as 2008 and for GARNET beginning in 2017. Some studies recruited internationally, whereas others recruited from single nations (Canada or US). There may be differences in clinical practice by region at various time points, although the direction of potential bias is unclear. Additionally, line of therapy definitions may not always be reflective of those used in clinical trials. GARNET included patients who had received no more than 2 lines of anti-cancer therapy for recurrent disease, whereas the NCRAS RWE database did not analyze lines of therapy before diagnosis of advanced or recurrent EC (all patients were receiving strictly second-line therapy). In the ZoptEC trial, patients only received 1 prior line of platinum doublet therapy. Follow-up duration also varied. Because the GARNET trial had an OS follow-up of less than 36 months in the published literature MAIC, the sponsor restricted the follow-up of each comparator study to no more than 36 months, and in the GARNET versus ZoptEC IPTW, patients with 36 months or more of follow-up were excluded. The comparative evidence is therefore limited to the longest follow-up for GARNET, for which survival data are still immature. Future comparisons after a longer follow-up may be more meaningful.
An important limitation of the MAICs and IPTWs is that in all comparator studies, MMR and MSI status was unknown for all or most patients. The indicated population for dostarlimab, according to the product monograph, is dMMR or MSI-H patients with advanced or recurrent EC. Further, the clinical experts consulted by CADTH for this review noted that MMR or MSI-H status has potential prognostic implications in these patients. It is therefore uncertain whether the comparator population in the MAIC and IPTW analyses would be eligible for treatment with dostarlimab, precluding strong conclusions about comparative efficacy. Furthermore, the comparators were based on those relevant to the advanced or recurrent EC population, rather than the dMMR or MSI-H subgroup; however, given the lack of advancement in funded treatments for advanced or recurrent EC overall, it may be reasonable that dMMR and MSI-H patients would receive similar options. The comparators included in the MAIC and IPTW were paclitaxel monotherapy, carboplatin plus paclitaxel, carboplatin plus liposomal doxorubicin, liposomal doxorubicin monotherapy, doxorubicin monotherapy, pegylated liposomal doxorubicin monotherapy, carboplatin monotherapy, and, because of limitations of the available data from RWE databases, the current treatment paradigm, which included a basket of possible treatments in advanced or recurrent EC. According to the clinical experts consulted by CADTH, these comparator treatments were relevant to Canadian clinical practice; however, 1 of the experts noted that MSI-H EC may be generally less responsive to platinum chemotherapy, although it is still used as first-line treatment in the absence of an alternative for dMMR or MSI-H patients. There were additional single-drug chemotherapies that could have been considered but were not included, such as gemcitabine and etoposide, because evidence for the efficacy of these treatments is lacking.
All comparisons investigated OS, an important outcome, according to the clinical experts consulted by CADTH for this review. Other outcomes were less frequently investigated, and those that are specifically important to patients (e.g., harms, HRQoL) were notably absent because comparative data were unavailable for those outcomes. There were some notable differences in outcome definitions, time of assessment, and follow-up duration across the comparators. Progression was not recorded in the NCRAS database, and TTNT and TTD were used as proxies, which may both over- and underestimate PFS. As such, strong conclusions cannot be made for this comparison outcome.
In the submitted MAIC analyses, an attempt to match patients based on inclusion and exclusion criteria was only conducted for the analysis versus published literature. In the MAIC of the NCRAS RWE cohort, patients met all of the applied inclusion criteria and none of the applied exclusion criteria for the GARNET trial; however, no information on this process was provided, and no matching of inclusion and exclusion criteria was done before weighting. An attempt to match inclusion and exclusion criteria was illustrated in the IPTW analysis with the Flatiron cohort, as well as in the IPTW and PSM comparisons with ZoptEC. Regardless of the scenarios or treatments, in the majority of cases, the ESS was greatly reduced, representing substantial losses to the precision of estimates. Thus, there was either considerable heterogeneity between GARNET and the RWE studies for the variables included in the weighting process, or the inclusion and exclusion criteria differed greatly in the studies, which is an important limitation of the relative treatment-effect estimates. In the absence of such evidence, the NICE Decision Support Unit considers the amount of bias in an unanchored MAIC likely to be substantial.61 For the primary outcome of OS, all results were associated with moderately to severely wide CIs, highlighting losses in precision and reducing the ability to draw strong conclusions.
In the IPTW analyses for the Flatiron and NCRAS RWE cohorts, propensity scores for each patient were estimated as a patient’s predicted probability of being assigned to the GARNET cohort, which was estimated from a logistic regression model. In the GARNET versus ZoptEC IPTW, exclusion criteria were applied to each trial in the analysis with the ZoptEC trial, followed by propensity scoring and weighting and a Cox proportional hazards model. One limitation of the IPTW technique is that it presumes all biases and confounding have been adjusted for in the model. To address this limitation, multiple PSMs were constructed in the analyses (particularly for Flatiron and NCRAS RWE cohorts) and the sets of covariates were defined based on expert opinion and results from the empirical assessment of effect modification and prognostic value of each covariate. However, it is still likely that there exist other confounders that were not observed and collected in the data. Moreover, in analyses of the Flatiron cohort and ZoptEC trial, the method of calculation was not reported, so the validity of the propensity scores could not be assessed. It was also unclear in all cases if any patients were excluded after propensity scoring.
The sponsors conducted a search of the literature and consulted with clinical experts to identify potential prognostic factors or treatment-effect modifiers in patients with advanced or recurrent EC. The key limitation of the MAICs is inherent in unanchored indirect comparisons, which assume that absolute outcomes can be predicted from the covariates (i.e., all effect modifiers and prognostic factors are accounted for in the model). This assumption is largely considered impossible to meet, according to the NICE Decision Support Unit technical guidance report on methods for population-adjusted indirect comparisons.61 The list of potential prognostic and treatment-effect modifiers was comprehensive and populated using appropriate methods; however, not all variables were available in each trial and, therefore, were not included in the models. Baseline characteristics were presented before and after weighting for all analyses. After the matching of baseline characteristics, the populations in all MAIC analyses were similar; however, in the IPTW analyses, some differences remained after IPTW and PSM, so the comparisons were not balanced for confounders and were not mutually randomizable populations. Aside from the known differences related to the availability of certain characteristics, such as ECOG Performance Status, prior lines of therapy, disease grade, and prior surgery, there were no additional concerns with the baseline characteristics after matching.
Overall, the phase I nature of the GARNET trial limits the ability to make definitive conclusions on comparative efficacy, given the short duration of follow-up resulting in immature data and the limited comparability with other studies because of differences in design. Moreover, because of the heterogeneity of the included populations, the comparisons conducted are not reflective of the Health Canada indication for dostarlimab; all comparator trials were not specific to dMMR or MSI-H patients as there was no comparative evidence identified for this population. The results of the MAICs and IPTWs were generally uncertain because of the clinical differences between populations, which resulted in reduced sample sizes after matching and the wider CIs. Moreover, it is highly unlikely that all prognostic factors and effect modifiers were accounted for, increasing the uncertainty in the results. As such, comparative efficacy in terms of improved survival must consider these limitations and interpretations must be made with caution, as results may not be generalizable to the indicated population. Last, outcomes important to patients, including improved HRQoL and reduced AEs, were not analyzed in the ITCs, so the comparative efficacy of dostarlimab for these outcomes remains uncertain.
Discussion
Summary of Available Evidence
The CADTH systematic review included 1 phase I trial that evaluated the efficacy and safety of dostarlimab in patients with dMMR or MSI-H recurrent or advanced EC that had progressed on or after prior treatment with a platinum-containing regimen. The GARNET trial (N = 129) is an ongoing, multi-centre, open-label, single-arm, phase I dose-escalation and cohort-expansion study of patients with recurrent or advanced solid tumours. This CADTH review focused on cohort A1, which aligned with the proposed Health Canada indication and the reimbursement request. All patients received dostarlimab through IV injection (500 mg every 3 weeks for cycles 1 to 4, and 1,000 mg every 6 weeks from cycle 5 onward) for up to 2 years or until disease progression or unacceptable toxicity, whichever occurred first. The co-primary outcomes were ORR and DOR, and secondary outcomes were OS, PFS, DCR, irPFS, irDCR, irORR, and irDOR. HRQoL was an exploratory outcome. AEs and irAEs were also monitored and reported. The statistical analysis plan of the GARNET trial specified 3 interim analyses. The focus of this CADTH review was IA-2, which was the most recent interim analysis available (median follow-up time = 16.3 months). IA-3 was planned for November 1, 2021, but data from this analysis were not submitted to CADTH until later in the review process so were not included as part of the main analysis. The IA-3 results were, overall, consistent with those reported in IA-2. For further details on IA-3, refer to Appendix 5.
In cohort A1 of the GARNET trial, the majority of enrolled patients with dMMR or MSI-H EC were White, had type II and grade 3 endometrioid tumours, had FIGO stage IV disease, had an ECOG Performance Status of 1, had received 1 or 2 previous lines of systemic therapy for recurrent or advanced EC, and had a median age of 64 (range = 39 to 84) years. Limitations of the GARNET trial included potential biases inherent to its single-arm, noncomparative trial design, which prohibits the ability to draw causal conclusions about the intervention and outcomes.
In the absence of comparative evidence, the sponsor submitted 6 reports of ITCs — 3 MAIC reports and 3 IPTW and PSM reports — that compared dostarlimab with currently available treatment regimens using RWE and data from the published literature. The primary end point for all comparisons was OS. Other outcomes included PFS, ORR, DOR, TTD, DoT, TTNT, TTD in HRQoL, and AEs; however, these were less frequently investigated, and outcomes specifically important to patients, such as HRQoL, were not assessed. There were important differences in the design of the comparator studies that limit the ability to draw strong conclusions about the efficacy of dostarlimab compared with other treatments. An important limitation of all analyses was that MMR and MSI-H status was unknown for all or most patients in the comparator trials, so it is uncertain whether the comparator populations in the ITC analyses would be eligible for treatment with dostarlimab; this creates further uncertainty about comparative efficacy.
Interpretation of Results
Outcomes
The achieved ORR and DOR (co-primary end points in cohort A1) were considered clinically meaningful by the clinical experts consulted by CADTH. The clinical experts felt that the ORR of 44.8% (95% CI, 35.0% to 54.8%) was both clinically meaningful and durable for this patient population. Although the median DOR was not reached, approximately 79% of dostarlimab responders had a DOR of at least 6 months, which the clinical experts noted was impressive compared with currently available treatments. The GARNET trial included no formal hypothesis or statistical significance testing; as such, point estimates with 95% CIs were reported to estimate the magnitude of treatment effect associated with dostarlimab. Given that the trial was not designed to detect differences in treatment effects across subgroups, no conclusions can be drawn from pre-specified subgroup results. OS and PFS were assessed as secondary outcomes in the GARNET trial; however, interpretation of time-to-event end points is limited in a single-arm trial. Because all patients in cohort A1 of the GARNET trial received the same treatment, it remains unclear if the observed survival rates were due to the natural history of the disease or treatment with dostarlimab. Another limitation was that the median OS was not reached at the time of IA-2. Although KM estimates were provided at different time points, these data may overestimate the efficacy of the treatment given the immaturity of the data.
According to the clinical experts consulted by CADTH and the registered-clinician groups that provided input for this submission, the tumour response outcomes, including the CR rate and DOR, achieved with dostarlimab in the GARNET trial were clinically meaningful and much higher than those observed with therapies currently used in this setting. According to the clinical experts, the CR and response rates achieved in the study population were excellent compared with the CR rates they see in clinical practice and with other historical immunotherapies. Although the results of the MAIC and IPTW analyses generally suggest that dostarlimab is favoured for OS over all the included comparators, there was significant uncertainty in the results based on the clinical heterogeneity of the included study populations, resulting in reduced sample sizes and treatment-effect estimates that had wide CIs.
The clinical experts emphasized the clinical relevance and importance of maintaining stable disease to prevent an otherwise fast decline in patients; for many, second-line therapy may be their last line of treatment. This view was echoed in the input provided by the patient-advocacy group, which highlighted tumour response, maintenance of response, delay in disease progression, and HRQoL as important treatment goals for patients. Although the clinical experts agreed that, based on the available evidence, it was not possible to conclude whether the antitumour activity expressed as responses would translate into PFS and OS benefit, they felt that the preliminary survival results from the trial (median PFS was 5.5 months [95% CI, 3.2 to NR] and median OS was not reached) were encouraging, and that the durable responses observed could potentially delay tumour progression and result in prolonged survival in this patient population.
Input received from the patient-advocacy and clinician groups, as well as the clinical experts consulted by CADTH, highlighted HRQoL as an important outcome and treatment goal for patients. The clinical experts consulted by CADTH noted that HRQoL in patients with advanced or recurrent EC is low and unstable, and they did not foresee worsening of HRQoL with dostarlimab, given its acceptable toxicity profile. However, conclusions could not be drawn from the HRQoL evidence from the GARNET trial, given its noncomparative, open-label trial design, the substantial decline over time in patients who completed questionnaires, the lack of statistical analyses, and the lack of a MID or definition of what constituted a clinically meaningful change from baseline in the trial population. The clinical experts and clinician groups anticipated that given the responses and manageable toxicities observed in the GARNET trial, dostarlimab would likely improve or at least maintain patients’ HRQoL; however, these expectations need to be confirmed in a randomized clinical trial.
Although patients in the GARNET trial were considered to be representative of patients in Canadian clinical practice, the clinical experts noted that study patients were slightly younger than the patients typically seen in clinical practice; still, they noted, results would be generalizable. The clinical experts anticipated that because of its acceptable safety profile, they would expect to see a benefit of treatment with dostarlimab, regardless of the number of previous lines of systemic therapy, in patients with dMMR or MSI-H EC. However, patients should not have previously been treated with other anti-PD-1 or anti-PD-L1 immunotherapies.
Harms
The single-arm, nonrandomized design of the GARNET trial made it a challenge to interpret the safety events attributable to dostarlimab, because all patients in cohort A1 received the same treatment. Almost all patients in cohort A1 experienced at least 1 TEAE (n = 123; 95.3%) and almost 50% of TEAEs were grade 3 or higher. The clinical experts consulted by CADTH noted that most TEAEs associated with dostarlimab are manageable with supportive care and are not life-threatening. Further, they noted that treatment discontinuation due to TEAEs was relatively rare. The prevalence of treatment toxicities was in line with what is expected with other immunotherapies. From the review of notable harms, the clinical experts noted that the immune-related toxicities seemed to be associated with dostarlimab treatment. Overall, death from AEs was minimal, and no TEAEs that led to death were considered to be treatment-related. The clinical experts agreed with clinician group input that the TEAEs observed with dostarlimab were, overall, acceptable and could be adequately managed in clinical practice. This was reflective of patients’ experience with dostarlimab reported in the patient input, which stated that, overall, they had few challenges dealing with side effects of dostarlimab. Furthermore, it was emphasized by the clinical experts consulted by CADTH that the toxicity associated with dostarlimab appeared favourable, compared with currently available chemotherapy options. Examples of side effects from chemotherapy that may be weaker with dostarlimab include alopecia, fatigue, peripheral neuropathy, neutropenia, and febrile neutropenia, according to the clinical experts. The sponsor-submitted ITCs reports did not assess safety outcomes.
Conclusions
One phase I, singe-arm, open-label trial (GARNET)18 provided evidence on the efficacy and safety of dostarlimab in adults with dMMR or MSI-H recurrent or advanced EC (cohort A1) that had progressed on or after prior treatment with a platinum-containing regimen. The clinical experts consulted by CADTH felt that the co-primary response outcomes of ORR and DOR observed in the trial were clinically meaningful and durable for this patient population and, in their opinion, were higher than what is observed with currently used second-line therapies in this setting. The trial results were based on an interim analysis, so it is possible that clinical benefit was overestimated and harms were underestimated. There was uncertainty around the magnitude of the clinical benefit, given the limitations inherent to the single-arm trial design. The trial data on important long-term outcomes were immature, and interpretation of OS will be confounded by the use of subsequent anti-cancer therapies. The clinical experts noted that a RCT would be needed to directly compare dostarlimab with currently available therapies in the second-line setting to accurately evaluate its efficacy in this patient population. In the absence of a direct comparison of dostarlimab with relevant treatment options, the sponsor submitted multiple ITCs. However, the CADTH critical appraisal of these analyses identified significant limitations with the submitted MAICs and IPTWs that restricted the ability to interpret the relative treatment-effect estimates obtained. Limitations of the ITCs included heterogeneity across study designs, high risk of confounding and effect modifiers, and uncertainty regarding the inclusion of dMMR or MSI-H in the comparator groups. The results for HRQoL, an outcome important to patients and clinicians, remained inconclusive because of the lack of statistical analysis, substantial decline in patients completing questionnaires over time, and the lack of a definition of what constituted a clinical meaningful change from baseline. The notable harms observed with dostarlimab, such as diarrhea and peripheral nephropathy, were considered manageable and consistent with other immunotherapies by the clinical experts and, in their opinion, appeared favourable when naively compared with currently available chemotherapy options. However, interpretation of the safety events attributable to dostarlimab was a challenge because all patients in cohort A1 received the same treatment. Overall, limitations of the single-arm design of the GARNET trial prohibited the ability to draw causal conclusions between the intervention and outcomes.
References
1.Plaxe S, Mundt A. Overview of endometrial carcinoma. In: Post TW, ed. UptoDate. Waltham (MA): UptoDate; 2021: www.uptodate.com. Accessed 2021 Dec 16.
2.Canadian Cancer Society. Uterine cancer statistics. 2021; https://cancer.ca/en/cancer-information/cancer-types/uterine/statistics. Accessed 2021 Dec 17.
3.Cancer-specific stats 2021. Toronto (ON): Canadian Cancer Society, Government of Canada; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021_cancer-specific-stats.pdf?rev=bbadfd869b66415e97be9655a842858c&hash=3E552CE702E1BD7657E4E91FB7BA624B&_ga=2.72656595.1540445839.1639772303-397212376.1636582496. Accessed 2021 Dec 17.
4.Fung-Kee-Fung M, Dodge J, Elit L, et al. Follow-up after primary therapy for endometrial cancer: a systematic review. Gynecol Oncol. 2006;101(3):520-529. PubMed
5.Endometrial cancer. Edmonton (AB): Alberta Health Services; 2015: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-gyne002-endometrial.pdf. Accessed 2022 Jan 14.
6.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics: a 2018 special report on cancer indicence by stage. Toronto (ON): Canadian Cancer Society, Statistics Canada, the Public Health Agency of Canada; 2018: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2018-statistics/canadian-cancer-statistics-2018-en.pdf. Accessed 2021 Dec 30.
7.Green AK, Feinberg J, Makker V. A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. Am Soc Clin Oncol Educ Book. 2020;40:1-7. PubMed
8.Lorenzi M, Amonkar M, Zhang J, Mehta S, Liaw K-L. Epidemiology of Microsatellite Instability High (MSI-H) and Deficient Mismatch Repair (dMMR) in Solid Tumors: A Structured Literature Review. J Oncol. 2020;2020:1807929.
9.Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12(1):54. PubMed
10.Sovak MA, Dupont J, Hensley ML, et al. Paclitaxel and carboplatin in the treatment of advanced or recurrent endometrial cancer: a large retrospective study. Int J Gynecol Cancer. 2007;17(1):197-203. PubMed
11.Sorbe B, Andersson H, Boman K, Rosenberg P, Kalling M. Treatment of primary advanced and recurrent endometrial carcinoma with a combination of carboplatin and paclitaxel-long-term follow-up. Int J Gynecol Cancer. 2008;18(4):803-808. PubMed
12.Makker V, Hensley ML, Zhou Q, Iasonos A, Aghajanian CA. Treatment of advanced or recurrent endometrial carcinoma with doxorubicin in patients progressing after paclitaxel/carboplatin: Memorial Sloan-Kettering Cancer Center experience from 1995 to 2009. Int J Gynecol Cancer. 2013;23(5):929-934. PubMed
13.Miller DS, Filiaci VL, Mannel RS, et al. Carboplatin and Paclitaxel for Advanced Endometrial Cancer: Final Overall Survival and Adverse Event Analysis of a Phase III Trial (NRG Oncology/GOG0209). J Clin Oncol. 2020;38(33):3841-3850. PubMed
14.Brooks RA, Fleming GF, Lastra RR, et al. Current recommendations and recent progress in endometrial cancer. CA Cancer J Clin. 2019;69(4):258-279. PubMed
15.Jemperli (dostarlimab): solution for infusion, 500 mg/10 mL vial (50 mg/mL) [product monograph]. Mississauga (ON): GlaxoSmithKline Inc.; 2021 Mar 26.
16.National Institute for Health Care and Excellence. Dostarlimab for previously treated advanced or recurrent endometrial cancer with high microsatellite instability or mismatch repair deficiency [ID3802]. 2021; https://www.nice.org.uk/guidance/indevelopment/gid-ta10670. Accessed 2021 Dec 20.
17.Dostarlimab (Jemperli). Glasgow (GB): Scottish Medicines Consortium; 2021: https://www.scottishmedicines.org.uk/medicines-advice/dostarlimab-jemperli-full-smc2404/ Accessed 2021 Dec 20.
18.Clinical Study Report: 4010-01-001. A phase 1 dose escalation and cohort expansion study OF TSR-042, an anti-PD-1 monoclonal antibody, in patients with advanced solid tumors - part 2B, endometrial cancer (cohorts A1 and A2) [internal sponsor's report]. Barcelona (ES): GlaxoSmithKline Research & Development Limited; 2020 Nov 16.
19.Sponsor's ITC reports [internal sponsor's report]. In: Jemperli, solution for infusion, 500 mg/10 mL vial (50 mg/mL) [internal sponsor's package]. Mississauga (ON): GlaxoSmithKline Inc; 2021 Sep 22.
20.Canadian Cancer Society. Cancerous tumours of the uterus. 2021; https://cancer.ca/en/cancer-information/cancer-types/uterine/what-is-uterine-cancer/cancerous-tumours. Accessed 2021 Dec 17.
21.American Society of Clinical Oncology. Uterine cancer: risk factors and prevention. 2020; https://www.cancer.net/cancer-types/uterine-cancer/risk-factors-and-prevention. Accessed 2021 Dec 20.
22.American Cancer Society. Endometrial cancer stages. 2019; https://www.cancer.org/cancer/endometrial-cancer/detection-diagnosis-staging/staging.html Accessed 2021 Dec 22.
23.Canadian Cancer Society. Stages of uterine cancer. 2021; https://cancer.ca/en/cancer-information/cancer-types/uterine/staging. Accessed 2021 Dec 22.
24.American Cancer Society. Endometrial cancer: detection, diagnosis, staging, survival rates. 2021; https://www.cancer.org/cancer/endometrial-cancer/detection-diagnosis-staging/survival-rates.html1. Accessed 2021 Dec 17.
25.McEachron J, Zhou N, Spencer C, et al. Adjuvant chemoradiation associated with improved outcomes in patients with microsatellite instability-high advanced endometrial carcinoma. Int J Gynecol Cancer. 2021;31(2):203-208. PubMed
26.Sorbe B, Juresta C, Ahlin C. Natural history of recurrences in endometrial carcinoma. Oncol Lett. 2014;8(4):1800-1806. PubMed
27.Guidelines & Protocols Advisory Committee. Genital tract cancers in females: endometrial cancer. Victoria (BC): BCGuidelines.ca; 2014: https://www2.gov.bc.ca/gov/content/health/practitioner-professional-resources/bc-guidelines/endometrial-cancer. Accessed 2022 Jan 14.
28.PDQ Adult Treatment Editorial Board. PDQ endometrial cancer treatment. Bethesda (MD): National Cancer Institute; 2021: https://www.cancer.gov/types/uterine/hp/endometrial-treatment-pdq#_36 Accessed 2021 Dec 20.
29.Systemic therapy for advanced or recurrent endometrial cancer and advanced or recurrent uterine papillary serous carcinoma. Toronto (ON): Cancer Care Ontario; 2019: https://www.cancercareontario.ca/en/guidelines-advice/types-of-cancer/501. Accessed 2022 Jan 14.
30.Fleming GF, Brunetto VL, Cella D, et al. Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: a Gynecologic Oncology Group Study. J Clin Oncol. 2004;22(11):2159-2166. PubMed
31.Thigpen JT, Brady MF, Homesley HD, et al. Phase III trial of doxorubicin with or without cisplatin in advanced endometrial carcinoma: a gynecologic oncology group study. J Clin Oncol. 2004;22(19):3902-3908. PubMed
32.van Wijk FH, Aapro MS, Bolis G, et al. Doxorubicin versus doxorubicin and cisplatin in endometrial carcinoma: definitive results of a randomised study (55872) by the EORTC Gynaecological Cancer Group. Ann Oncol. 2003;14(3):441-448. PubMed
33.Concin N, Matias-Guiu X, Vergote I, et al. ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int J Gynecol Cancer. 2021;31(1):12-39. PubMed
34.Center for Drug Evaluation and Research. Multi-discipline review(s). Jemperli (dostarlimab-gxly). Company: GlaxoSmithKline LLC. Application No. 761223. Approval date: 17/08/2021 (FDA drug approval package). Rockville (MD): U.S. Food and Drug Administration (FDA); 2021: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=761223. Accessed 2021 Dec 20.
35.European Public Assessment Report (EPAR): Jemperli (dostarlimab). London (GB): European Medicines Agency; 2021: https://www.ema.europa.eu/en/medicines/human/EPAR/jemperli Accessed 2021 Dec 20.
36.Carboplatin injection BP (carboplatin injection): 10 mg / mL (50 mg, 150 mg, 450 mg, 600 mg of carboplatin per vial) [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2019: https://www.pfizer.ca/sites/default/files/202001/Carboplatin_PM_E_231317_16Dec2019.pdf. Accessed 2022 Jan 20.
37.Paclitaxel injection USP: solution for injection, 6 mg/mL [product monograph]. Kirkland (QC): Accord Healthcare Inc; 2020: https://pdf.hres.ca/dpd_pm/00057982.PDF. Accessed 2022 Jan 20.
38.Provera (medroxyprogesterone acetate tablets USP): 2.5 mg, 5 mg and 10 mg tablets [product monograph]. Kirkland (QC): Pfizer Canada Inc; 2014: https://www.pfizer.ca/sites/default/files/201710/Provera_PM_E_176877_29September2014.pdf. Accessed 2021 Dec 22.
39.Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18(3):e143-e152. PubMed
40.Common terminology criteria for adverse events (CTCAE): version 5.0. Rockville (MD): U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute; 2017: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf. Accessed 2022 Jan 14.
41.Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5(6):649-655. PubMed
42.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
43.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jan 14.
44.Oaknin A, Tinker AV, Gilbert L, et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020;6(11):1766-1772. PubMed
45.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247. PubMed
46.Drug Reimbursement Review sponsor submission: pemigatinib, 4.5 mg, 9 mg, and 13.5 mg, oral tablets [internal sponsor's package]. Pointe Claire (QC): Incyte Biosciences Canada Corporation; 2021 Jun 23.
47.Canadian Cancer Trial Group. irRECIST. 2021; https://recist.eortc.org/irecist/. Accessed 2022 Jan 17.
48.EORTC quality of life. https://qol.eortc.org/. Accessed 2021 Nov 22.
49.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
50.Clinical Study Report: 4010-01-001. A phase 1 dose escalation and cohort expansion study of TSR-042, an anti-PD-1 monoclonal antibody, in patients with advanced solid tumors - part 2B, non-endometrial cancer (cohort F) [internal sponsor's report]. Paris (FR): GlaxoSmithKline Research & Development Limited; 2020 Nov 13.
51.Cochrane Handbook for Systematic Reviews of Interventions. 2nd Edition. In: Higgins JPT, Thomas J, Chandler J, et al., eds. Chichester (UK): John Wiley & Sons; 2019: https://training.cochrane.org/handbook. Accessed 2022 Jan 6.
52.National Institute for Health and Care Excellence. The guidelines manual: process and methods [PMG6]. 2012: https://www.nice.org.uk/process/pmg6/chapter/introduction. Accessed 2022 Jan 6.
53.Programme CAS. CASP Cohort Study Checklist. 2018: https://casp-uk.net/wp-content/uploads/2018/01/CASP-Cohort-Study-Checklist_2018.pdf. Accessed 2022 Jan 6.
54.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28(10):935-945. PubMed
55.Guyot P, Ades AE, Ouwens MJNM, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12(1):9. PubMed
56.McMeekin S, Dizon D, Barter J, et al. Phase III randomized trial of second-line ixabepilone versus paclitaxel or doxorubicin in women with advanced endometrial cancer. Gynecol Oncol. 2015;138(1):18-23. PubMed
57.Julius JM, Tanyi JL, Nogueras-Gonzalez GM, et al. Evaluation of pegylated liposomal doxorubicin dose on the adverse drug event profile and outcomes in treatment of recurrent endometrial cancer. Int J Gynecol Cancer. 2013;23(2):348-354. PubMed
58.Mazgani M, Le N, Hoskins PJ. Reuse of carboplatin and paclitaxel in patients with relapsed endometrial cancer--the British Columbia Cancer Agency experience. Gynecol Oncol. 2008;111(3):474-477. PubMed
59.Mortimer KM, Neugebauer R, van der Laan M, Tager IB. An application of model-fitting procedures for marginal structural models. Am J Epidemiol. 2005;162(4):382-388. PubMed
60.Brumback BA, Hernán MA, Haneuse SJ, Robins JM. Sensitivity analyses for unmeasured confounding assuming a marginal structural model for repeated measures. Stat Med. 2004;23(5):749-767. PubMed
61.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU technical support document 18: methods for population-adjusted indirect comparisons in submissions to NICE. Sheffield (GB): NICE Decision Support Unit; 2016: http://nicedsu.org.uk/wp-content/uploads/2017/05/Population-adjustment-TSD-FINAL.pdf. Accessed 2022 Jan 7.
62.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
63.Davda J, Kibet H, Achieng E, Atundo L, Komen T. Assessing the acceptability, reliability, and validity of the EORTC Quality of Life Questionnaire (QLQ-C30) in Kenyan cancer patients: a cross-sectional study. J Patient Rep Outcomes. 2021;5(1):4. PubMed
64.Osoba D, Zee B, Pater J, Warr D, Kaizer L, Latreille J. Psychometric properties and responsiveness of the EORTC quality of Life Questionnaire (QLQ-C30) in patients with breast, ovarian and lung cancer. Qual Life Res. 1994;3(5):353-364. PubMed
65.Butler L, Bacon M, Carey M, Zee B, Tu D, Bezjak A. Determining the relationship between toxicity and quality of life in an ovarian cancer chemotherapy clinical trial. J Clin Oncol. 2004;22(12):2461-2468. PubMed
66.Zhao H, Kanda K. Translation and validation of the standard Chinese version of the EORTC QLQ-C30. Qual Life Res. 2000;9(2):129-137. PubMed
67.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
68.Bedard G, Zeng L, Zhang L, et al. Minimal important differences in the EORTC QLQ-C30 in patients with advanced cancer. Asia Pac J Clin Oncol. 2014;10(2):109-117. PubMed
69.EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
70.Ameri H, Yousefi M, Yaseri M, Nahvijou A, Arab M, Akbari Sari A. Mapping the cancer-specific QLQ-C30 onto the generic EQ-5D-5L and SF-6D in colorectal cancer patients. Expert Rev Pharmacoecon Outcomes Res. 2019;19(1):89-96. PubMed
71.Sinnott PL, Joyce VR, Barnett PG. Guidebook: preference measurement in economic analysis. Menlo Park (CA): Health Economics Research Center; 2007: https://www.herc.research.va.gov/files/BOOK_419.pdf. Accessed 2021 Nov 23.
72.Lang HC, Chuang L, Shun SC, Hsieh CL, Lan CF. Validation of EQ-5D in patients with cervical cancer in Taiwan. Support Care Cancer. 2010;18(10):1279-1286. PubMed
73.Setiawan D, Dusafitri A, Galistiani GF, van Asselt ADI, Postma MJ. Health-Related Quality of Life of Patients with HPV-Related Cancers in Indonesia. Value Health Reg Issues. 2018;15:63-69. PubMed
74.Hu X, Jing M, Zhang M, Yang P, Yan X. Responsiveness and minimal clinically important difference of the EQ-5D-5L in cervical intraepithelial neoplasia: a longitudinal study. Health Qual Life Outcomes. 2020;18(1):324. PubMed
75.Bjordal K, de Graeff A, Fayers PM, et al. A 12 country field study of the EORTC QLQ-C30 (version 3.0) and the head and neck cancer specific module (EORTC QLQ-H&N35) in head and neck patients. EORTC Quality of Life Group. Eur J Cancer. 2000;36(14):1796-1807. PubMed
76.EORTC QLC-C30 scoring manual. European Organisation for Research and Treatment of Cancer (EORTC) 2001; https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf. Accessed 2021 Nov 23.
77.Bergman B, Sullivan M, Sörenson S. Quality of life during chemotherapy for small cell lung cancer. II. A longitudinal study of the EORTC Core Quality of Life Questionnaire and comparison with the Sickness Impact Profile. Acta Oncol. 1992;31(1):19-28. PubMed
78.Bjordal K, Kaasa S. Psychometric validation of the EORTC Core Quality of Life Questionnaire, 30-item version and a diagnosis-specific module for head and neck cancer patients. Acta Oncol. 1992;31(3):311-321. PubMed
79.Greimel E, Nordin A, Lanceley A, et al. Psychometric validation of the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire-Endometrial Cancer Module (EORTC QLQ-EN24). Eur J Cancer. 2011;47(2):183-190. PubMed
80.Gallardo-Rincon D, Toledo-Leyva A, Bahena-Gonzalez A, et al. Validation of the QLQ-EN24 instrument for the assessment of health-related quality of life for women with endometrial cancer in Mexico. Arch Gynecol Obstet. 2021;304(3):773-782. PubMed
81.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-Defined Estimates of the Minimally Important Difference for EQ-5D-5L Index Scores. Value Health. 2017;20(4):644-650. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: November 3, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(dostarlimab* or jemperli* or P0GVQ9A4S5 or P-0GVQ9A4S5 or UNIIP0GVQ9A4S5 or UNIIP-0GVQ9A4S5 or TSR042 or TSR-042 or WBP285 or WBP-285 or GSK4057190 or GSK-4057190 or ANB011 or ANB-011).ti,ab,kf,ot,hw,nm,rn.
1 use medall
*dostarlimab/
(dostarlimab* or jemperli* or TSR042 or TSR-042 or WBP285 or WBP-285 or GSK4057190 or GSK-4057190 or ANB011 or ANB-011).ti,ab,kf,dq.
3 or 4
5 use oemezd
(conference review or conference abstract).pt.
6 not 7
2 or 8
remove duplicates from 9
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search terms – dostarlimab or Jemperli]
WHO ICTRP
International Clinical Trials Registry Platform, produced by WHO. Targeted search used to capture registered clinical trials.
[Search terms – dostarlimab or Jemperli]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – dostarlimab or Jemperli]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – dostarlimab or Jemperli]
Grey Literature
Search dates: October 30 – November 3, 2021
Keywords: dostarlimab, Jemperli, endometrial cancer
Limits: None
Updated: Search updated before the meeting of CADTH pan-Canadian Oncology Drug Review Expert Committee (pERC).
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Oaknin A, Tinker AV, Gilbert L, et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020;6(11):1766-1772. | Additional report without new data |
Lu S, Bowsher RR, Clancy A, et al. An Integrated Analysis of Dostarlimab Immunogenicity. AAPS J. 2021;23(5):96. | Additional report without new data |
Appendix 3: Description and Appraisal of Outcome Measures
Note this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) version 3.0
EQ-5D-5L version 2.0
Findings
Table 42: Summary of Relevant Secondary Outcomes and Their Measurement Properties
Instrument | Description | Measurement properties and validation | MCID |
|---|---|---|---|
EORTC QLQ-C30 | A self-reported instrument designed to measure the physical, psychological, and social functions of patients with cancers. The EORTC QLQ-C30 incorporates 5 functional scales (Physical, Role, Cognitive, Emotional and Social); 3 symptom scales (Fatigue; Pain and Nausea) and vomiting a global health and quality-of-life scale; and single items that assess additional symptoms commonly reported by patients with cancer (dyspnea, appetite loss, sleep disturbance, constipation, and diarrhea), as well as the perceived financial impact of the disease and treatment.48,62,63 | Validity: Construct, criterion and discriminate validity was demonstrated in patients with ovarian and gestational trophoblastic disease cancers.64 The EORTC QLQ-C30 was found to adequately assess the effect of expected toxicities on patients’ HRQoL during and following treatment.65 No reported validation studies were found for patients with EC. Reliability: Minimum reliability with Cronbach alpha > 0.70 was met in 7 of 9 subscales in patients with gestational trophoblastic disease, ovarian cancer and other types of gynecological cancers.66 Responsiveness: There were no data available for responsiveness. | Among patients with various advanced cancers67,68:
No reported MID was found for patients with EC |
EQ-5D-5L | A generic self-reported HRQoL tool that assesses 5 domains: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each domain has 5 levels: No problems; slight problems; moderate problems; severe problems; and extreme problems. The EQ-5D-5L also includes an EQ-VAS, a vertical visual analogue scale that takes values between 100 (best imaginable health) and 0 (worst imaginable health), on which patients provide a global assessment of their health.49,69-71 | Validity: Among patients with cervical cancer, the EQ-5D-5L was shown to have good convergent and discriminant validity (Cohen kappa range from 0.54 to 0.73).72 Among patients with HPV- related cancer, the EQ-5D-5L was shown have good construct and convergent validity and significantly correlated with mapped subscale of the EORTC QLQ-C30.73 No reported validation studies were found for patients with EC. Reliability: Among patients with HPV-related cancer, the EQ-5D-5L demonstrated good to excellent test-retest reliability across domains (ICCs > 0.80) and good internal consistency (Cronbach alpha = 0.84).72 Responsiveness: In patients with CIN, the instrument’s subscales of mobility, pain/discomfort and anxiety/depression were responsive to changes post-surgery.74 | For patients with CIN, the mean (range) value for the MID using distribution and anchor-based approaches was 0.039 (0.023 to 0.064) for the EQ-5D-5L and 5.35 (3.12 to 6.99) for the EQ-VAS.74 No reported MID was found for patients with EC. |
CIN = cervical intraepithelial neoplasia; EC = endometrial cancer; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EQ-VAS = EQ visual analogue scale; HRQoL = health-related quality of life; ICC = interclass correlation.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30)
Description
The EORTC QLQ-C30 is a self-reported instrument designed to measure the physical, psychological, and social functions of patients with cancers.48 The EORTC QLQ-C30 consists of 30 items that are scored to create 5 multi-item functional scales, 3 multi-item symptom scales, 6 single-item symptom scales, and 2 global quality of life (QoL) scales (Table 43) Version 3.0 of the questionnaire is the most current version and has been in use since December of 1997.75,76 It is intended for use in the adult population only.48
Table 43: EORTC QLQ-C30 Scales
Functional Scales (15 Questions) | Symptom Scales (7 Questions) | Single-Item Symptom Scales (6 Questions) | Global Quality of Life (2 Questions) |
|---|---|---|---|
Physical function (5) Role function (2) Cognitive function (2) Emotional function (4) Social function (2) | Fatigue (3) Pain (2) Nausea and vomiting (2) | Dyspnea (1) Insomnia (1) Appetite loss (1) Constipation (1) Diarrhea (1) Financial impact (1) | Global Quality of Life (2) |
Scoring
The EORTC QLQ-C30 uses a 1-week recall period to assess function and symptoms.76 Twenty-eight questions are scored on a 4-point Likert scale (1: not at all; 2: a little; 3: quite a bit; 4: very much). The 2 questions that make up the global HRQoL scale are scored on a 7-point Likert scale with anchors at 1 (“very poor”) and 7 (“excellent”).
Raw scores for each scale are computed as the average of the items that contribute to a particular scale.76 This scaling approach is based on the assumption that it is appropriate to provide equal weighting to each item that comprises a scale. There is also an assumption that, for each item, the interval between response options is equal (for example, the difference in score between “not at all” and “a little” is the same as “a little” and “quite a bit,” at a value of 1 unit). Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, higher symptoms on the symptom scales, and better HRQoL (i.e., higher scores simply reflect higher levels of response on that scale). Thus, a decline in score on the symptom scale would reflect an improvement, whereas an increase in score on the function and HRQoL scales would reflect an improvement. According to the EORTC QLQ-C30 scoring algorithm, if there are missing items for a scale (i.e., the patient did not provide a response), the score for the scale can still be computed if there are responses for at least one-half of the items. In calculating the scale score, the missing items are simply ignored — an approach that assumes that the missing items have values equal to the average of those items for what the respondent completed.
Psychometric Properties
The EORTC QLQ-C30 was originally validated in patients with lung cancer and head and neck cancer from various European and North American countries, as well as from Australia.62,77,78 The scales of the EORTC QLQ-C30 have been found to assess distinct components of HRQoL; distinguishing between patients with different performance status and degrees of weight loss, and responsive to change over time.62,77,78
A literature search was conducted to identify validation information of the EORTC QLQ-C30 in patients with EC and none were identified. Of note, there is a validated version of the EORTC QLQ that was developed specific for endometrial cancer: EORTC QLQ-EN24.79 The EORTC QLQ-EN24 was designed to assess disease and treatment-specific aspects of the HRQoL of patients with EC. A validation study of the Mexican-Spanish version of the EORTC QLQ-EN24 in 189 patients with EC included a brief validation of the QLQ-C30.80 The study confirmed the internal consistency and reliability of the QLQ-C30 and found that its convergent and discriminant validity (Cronbach alpha range = 0.77 to 0.89) was consistent with its original report.80
In a further validation study of the EORTC QLQ-C30, patients with breast cancer (n = 121), ovarian cancer (n = 111) lung cancer (n = 160) and a heterogenous group of other cancers (n = 121) completed the questionnaire before and on day 8 of chemotherapy.64 The item-domain correlations of the EORTC QLQ-C30 were not different across the primary tumour sites (i.e., breast, ovary or lung). The correlations for all items, except for item 5 (whether the responders needed help with eating, dressing, washing, or using the toilet) and the physical function domain (r = −0.3), were highly correlated within their own domain than with any other domains (r = −0.65 to 0.95). At day 8 of chemotherapy, the item-domain for item 5 and the physical function domain was 0.49 for the entire group and ranged from −0.38 for ovarian cancer to −0.55 for breast cancer. These higher values at day 8 suggest that item 5 was more relevant in the week after chemotherapy than before chemotherapy.64 Similarly, items asking about vomiting showed a higher correlation with domains for nausea/vomiting on day 8 after chemotherapy (r = 0.89) than before chemotherapy (r = 0.74). The questionnaire also demonstrated good internal consistency for most domains at baseline and at day 8 (Cronbach alpha > 0.70). However, values were < 0.70 at baseline and day 8 for role function (0.66 and 0.53, respectively) and cognitive function (0.63 and 0.58, respectively). An examination of the discrimination of the domain scores according to primary cancer site found that the mean scores at baseline were not significantly different from each other in all groups for emotional function, cognitive function, or nausea/vomiting. However, the mean scores for each of the other domains differed between the group, with patients with breast cancer tending to have better physical function, role function, social function, less fatigue, and pain and better global HRQoL. Patients with lung and ovarian cancer reported lower scores for all these domains, with patients with ovarian cancer reporting the lowest scores for all domains. After chemotherapy, many of the differences seen at baseline between the groups were no longer evident. At day 8, patients with ovarian cancer has the smallest magnitude of change, with no change in role function, social function and global HRQoL, while being the only cancer group reporting a significant improvement in pain.64
An analysis of data from a Canadian RCT of paclitaxel and cisplatin versus cyclophosphamide and cisplatin in the treatment of 153 patients with epithelial ovarian cancer found the EORTC QLQ-C30 adequately assessed the effect of expected toxicities on patients HRQoL during and after treatment.65 At baseline, before the initiation of treatment, there was close agreement in the “mild or none” category between the symptoms recorded on case report forms (CRFs) and paired EORTC QLQ-C30 questions. The greatest degree of agreement ranged between 0.80 (95% CI, 0.75 to 0.86) to 0.98 (95% CI, 0.92 to 0.99). The pairing of lethargy with Question 18, and mood with Question 22 were slightly weaker in agreement compared to the other pairs at 0.72 and 0.73, respectively. The weakest pairs were constipation with Question 16, and lethargy with Question 18 at 0.44 and 0.44, respectively. During treatment and at the end of cycles 3 and 6, all but 1 symptom and HRQoL pairs demonstrated marked agreement ranging from 0.71 to 0.93. The 1 exception was the pair assessing symptom hair loss and Question 42 with a degree of agreement of 0.50 and 0.37 at cycles 3 and 6, respectively. A regression model predicting global HRQoL scores based on baseline grades of the most frequently observed toxicities and scores corresponding to HRQoL question found that the questions related to motor weakness (question 12), anorexia (question 13), mood (question 24), gastrointestinal pain (question 40) and vomiting (question 15) explained 60% of the variance in baseline global HRQoL on the EROTC QLQ-C30. When patients were off chemotherapy, 78% of symptoms and HRQoL pairs had high level of agreement (> 0.80).
Minimal Important Difference
A literature search was conducted to identify the MID of the EORTC QLQ-C30 in patients with EC and none were identified. Below is a summary of the MID of the EORTC QLQ-C30 in patients with cancer in general.
Change in the EORTC QLQ-C30 may be interpreted in terms of small, moderate or large changes in HRQoL.67 A study of patients with breast cancer and small cell lung cancer estimated that a clinically relevant change in score on any of the EORTC QLQ-C30 scales to be 10 points.67 Using an anchor-based approach to estimate the MID in which patients who reported “a little” change (for better or worse) on the subjective significance questionnaire (SSQ) had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately 5 to 10 points. Patients who reported a “moderate” change had corresponding changes in the EORTC QLQ-C30 of about 10 to 20 points, and those who reported being “very much” changed had corresponding changes of more than 20 points.
A Canadian study estimated the MID for the EORTC QLQ-C30 among 369 patients with advanced cancer, the most common cancer being breast cancer, followed by lung, prostate, gastrointestinal, renal cell, and other cancers.68 Patients completed the questionnaire at baseline and 1-month post-radiation. Using both an anchor- and distribution-based methods for improvement and deterioration, 2 anchors of overall health and overall HRQoL were used, both taken directly from the EORTC QLQ-C30 (questions 29 and 30) where patients rated their overall health and HRQoL themselves. Improvement and deterioration were categorized as an increase or decrease by 2 units to account for the natural fluctuation of patient scoring. With these 2 anchors, the estimated MIDs across all EORTC QLQ-C30 scales ranged from 9.1 units to 23.5 units for improvement, and from 7.2 units to 13.5 units for deterioration. Distribution-based estimates were closest to 0.5 SD.
EQ-5D-5L
Description
The EQ-5D-5L is a generic self-reported HRQoL outcome measure that may be applied to a variety of health conditions and treatments.49,69 The first 2 components of the EQ-5D-5L assesses 5 domains: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. Each domain has 5 levels: no problem; slight problems; moderate problems; severe problems; and extreme problems. A descriptive system that classifies respondents (aged ≥ 12 years) based on the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. The EQ-5D-5L has 5 possible levels for each domain and respondents are asked to choose the level that reflects their health state for each of the 5 domains resulting in 3,125 possible health states.70 The second component of the EQ-5D-5L part is a 20 cm visual analogue scale (EQ-VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to rate their health by drawing a line from an anchor box to the point on the EQ-VAS which best represents their health on that day. Thus, the EQ-5D-5L produces 3 types of data for each respondent:
A profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, e.g., 15121, 33211;
A population preference-weighted health index score based on the descriptive system; and
A self-reported assessment of health status based on the EQ-VAS.
Scoring
The EQ-5D-5L index score is generated by applying a multi-attribute utility function to the descriptive system.71 Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states ‘dead’ and ‘perfect health,’ respectively.
Psychometric Properties
A literature search was conducted to identify validation information of the EQ-5D-5L in patients with EC and none were identified. Below is a summary of the psychometric property of the EQ-5D-5L in patients with gynecological cancers.
Among 530 patients with cervical cancer in Taiwan, the EQ-5D-5L was found to be both a reliable and valid assessment of HRQoL.72 The interclass correlation of the EQ-5D-5L was 0.8, with Cohen kappa values for the different dimension ranging from 0.54 to 0.73. Its convergent and discriminant validities were examined using the EORTC QLQ-30 and the clinical indicators of the functional performance assessment using the Karnofsky Performance Scale (KPF) and disease status. The EQ-5D was strongly correlated with all EORTC QLQ-C30 functioning scales, and its index and VAS scores were higher for patients with higher KPS score and disease-free status.72
Among a group of 300 patients (mean age: 51.5 ± 11.5 years) with HPV-related cancer (i.e., head and neck cancer - 70%; cervical cancer - 13.4%; and nasopharyngeal cancer - 10%) in Indonesia, the EQ-5D-5L demonstrated good to excellent test-retest reliability across domains (ICCs: Mobility = 0.97; Self-care = 0.95; Usual activities = 0.79; Pain/discomfort = 0.84; Anxiety/depression = 0.82; EQ-5D visual analogue scale = 0.73) and good internal consistency (Cronbach alpha = 0.84).73 Construct and convergent validity of the EQ-5D-5L in this population was assessed by mapping its subscales to those of the EORTC QLQ-C30. Significant correlation between almost all the dimension of the Indonesian version of the EQ-5D-5L with mapped subscale of the EORTC QLQ-C30, including physical function, role function, fatigue and pain was observed. Only the mobility dimension of the EQ-5D-5L was not correlated with the social function subscale of the EORTC QLQ-C30.73
The responsiveness of the EQ-5D-5L was evaluated in a longitudinal study of 50 patients with cervical intraepithelial neoplasia in China. The EQ-5D-5L demonstrated only small to moderate responsiveness post-surgery. While scores for self-care and usual activities did not change, scores of mobility, pain/discomfort and anxiety/depression decreased by 0.003, 0.004 and 0.016, indicating improvement of these dimensions at follow-up.
Minimal Important Difference
A literature search was conducted to identify MID of the EQ-5D-5L in patients with EC and none were identified. Below is a summary of MID of the EQ-5D-5L in patients within the general population and in patients with other gynecological cancers.
A simulation-based approach using an instrumental-defined single-level transitions was used to estimate the MID of the EQ-5D-5L in the general population for each country-specific scoring algorithm. An estimated MID between 0.037 and 0.069 was determined for 6 countries (Canada, China, Spain, Japan, England, and Uruguay).81 The country-specific scoring algorithm were as follows Canada, 0.056 ± 0.011; China, 0.069 ± 0.007; Spain, 0.061 ± 0.008; Japan, 0.048 ± 0.004; England, 0.063 ± 0.013; and Uruguay, 0.063 ± 0.019. Differences in MID estimates reflect differences in population preferences, in valuation techniques used, as well as in modelling strategies. After excluding the maximum-valued scoring parameters, the MID estimates (mean ± SD) were as follows: Canada, 0.037 ± 0.001; China, 0.058 ± 0.005; Spain, 0.045 ± 0.009; Japan, 0.044 ± 0.004; England, 0.037 ± 0.008; and Uruguay, 0.040 ± 0.010.81
The MID of the of the EQ-5D-5L was determined in a longitudinal study of 50 patients with cervical intraepithelial neoplasia (CIN) in China using 3 methods: distribution-based, anchor-based, and instrument-defined methods.74 The MIDs, by anchor-based, instrument-defined, and anchor-based methods were 0.041, 0064 and 3.12, respectively. The MIDs estimated in this study; however, only represents truly meaningful change of HRQoL scores at the group level, not the individual level.74
Appendix 4: Detailed Outcome Data
Note that this appendix has not been copy-edited.
The results of OS from the subset of patients from IA-1 (N = 72) included in IA-2 was 26 deaths (36.1%) and 46 (63.9%) patients censored.
Table 44: KM Analysis of OS in Patients With dMMR or MSI-H EC (IA-1 Subset of Patients in IA-2 — Primary Efficacy Analysis)
Outcome measure | GARNET trial Cohort A1a (N = 72) |
|---|---|
OS status, n (%) | |
Events observed | 26 (36.1) |
Censored | 46 (63.9) |
OS (months) | |
Quartile (95% CIb) | |
25% | 9.3 (5.0, 16.7) |
50% | NR (17.1, NR) |
75% | NR (NR, NR) |
OS distribution function (95% CI) | |
Month 6 | 81.0 (69.5, 88.5) |
Month 9 | 76.3 (64.2, 84.8) |
Month 12 | 70.1 (57.5, 79.6) |
CI = confidence interval; KM = Kaplan-Meier; NR = not reported; OS = overall survival.
aThe population total 72 included dMMR EC and MMR-unk/MSI-H EC patients. Results were similar when data from patients with MMR-unknown, but MSI-H tumours were pooled with those of patients with dMMR tumours.
b95% CIs were generated using the method of Brookmeyer and Crowley (1982).
Source: Clinical Study Report50
Figure 10: KM Plot for OS in Patients With dMMR or MSI-H EC (IA-1 Subset of Patients in IA-2)
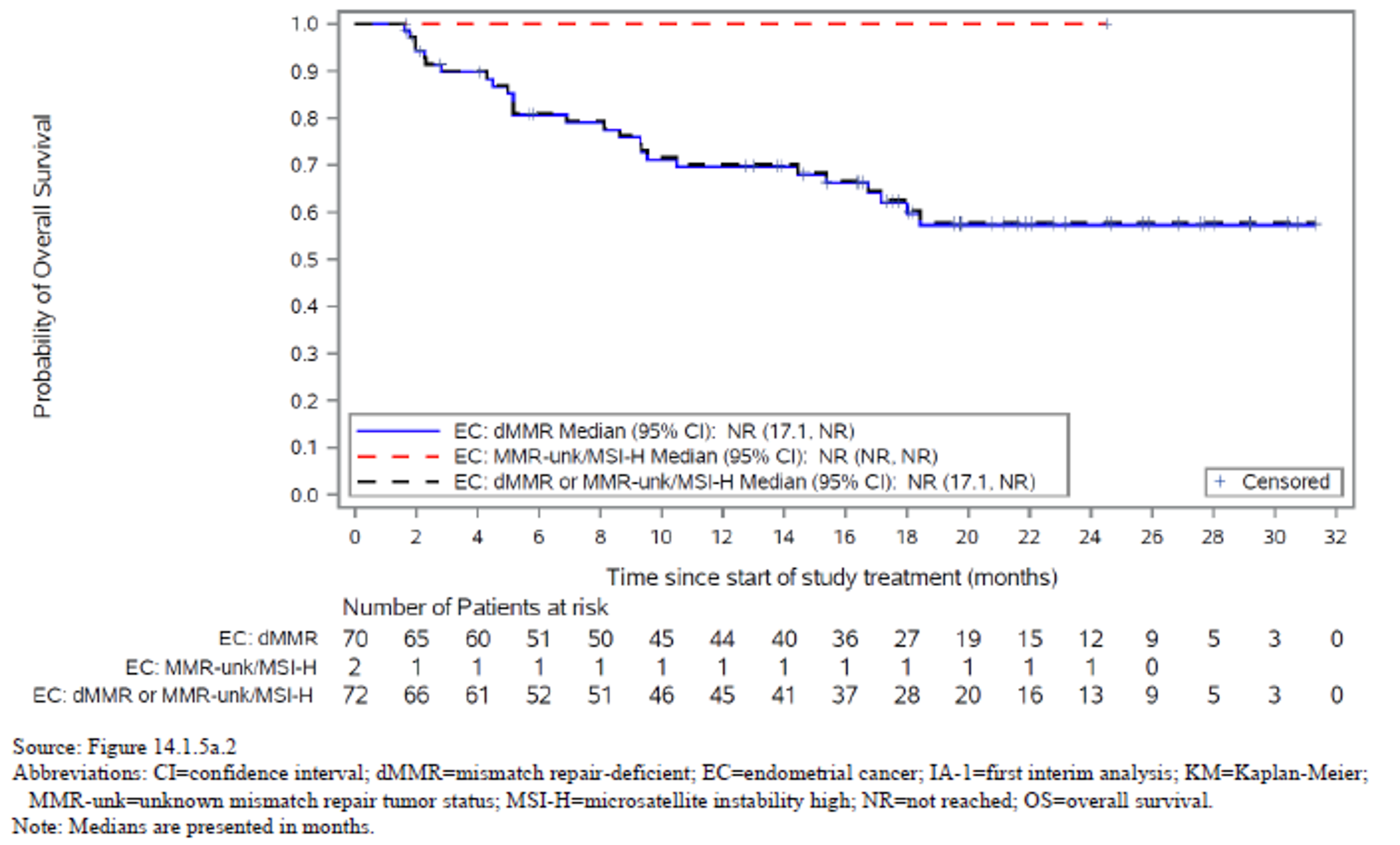
Source: Clinical Study Report50
Disease Control Rate (IA-1 subset of patients in IA-2)
The results of DCR (based on investigator assessment) for the subset of patients from IA-1 included in IA-2 were 58.3%. In total there were 10 patients with CRs (13.9%), 23 patients with PRs (31.9%), and 9 patients with stable disease (12.5%).
Objective Response Rate (IA-1 subset of patients in IA-2)
The ORR of the subset of patients included from IA-1 (median follow-up time 19.2 months) was 45.8%.
Table 45: Tumour Response Summary in Patients With dMMR or MSI-H EC — RECIST 1.1 Assessed by BICR (IA-1 Subset of Patients in IA-2– Primary Efficacy Analysis)
Outcome measure | GARNET trial Cohort A1a (N = 72) |
|---|---|
BOR by RECIST 1.1, n (%) | |
CR | 10 (13.9) |
PR | 23 (31.9) |
SD | 9 (12.5) |
PD | 26 (36.1) |
NE | 3 (4.2) |
No disease | 0 |
Not done | 1 (1.4) |
Confirmed ORR by RECIST 1.1, n (%) | 33 (45.8) |
95% CIb | 34.0 to 58.0 |
Response ongoingc | 28 (84.8) |
DCR by RECIST 1.1, n (%) | 42 (58.3) |
95% CIb | 46.1 to 69.8 |
BICR = blinded independent central review; BOR = best overall response; CI = confidence interval; CR = complete response; DCR = disease control rate; dMMR = mismatch repair-deficient; EC = endometrial cancer; MMR-unk = unknown mismatch repair tumour status; MSI-H = microsatellite instability-high; NE = not evaluable; ORR = objective response rate; PD = progressive disease; PR = partial response; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1; SD = stable disease.
Note: ORR was defined as the percentage of patients with a RECIST 1.1-confirmed CR or PR. DCR was defined as the percentage of patients with a RECIST 1.1-confirmed PR, confirmed CR, or SD. Response assessments were based on BICR.
aThe population total includes dMMR EC and MMR-unk/MSI-H EC patients. Results were similar when data from patients with MMR-unknown, but MSI-H tumours were pooled with those of patients with dMMR tumours.
bExact 2-sided 95% CI for the binomial proportion.
cAll responders who have not yet died or progressed (including clinical progression); the denominator for the percentage is the number of responders.
Source: Clinical Study Report50
PFS (IA-1 Subset of Patients in IA-2)
In the subset of patients from IA-1 (N = 72) included in IA-2 was 52.8% having a PFS event and median PFS of 12.2 months (95% CI, 3.0 to NR).
Table 46: KM Analysis of PFS in Patients With dMMR or MSI-H EC — RECIST 1.1 Based on BICR (IA-1 Subset of Patients in IA-2 – Primary Efficacy Analysis)
Outcome measure | GARNET trial Cohort A1 (N = 72) |
|---|---|
PFS status, n (%) | |
Events observed | 38 (52.8) |
Censored | 34 (47.2) |
PFS (months) | |
Quartile (95% CIa) | |
25% | 2.7 (1.7, 3.0) |
50% | 12.2 (3.0, NR) |
75% | NR (NR, NR) |
PFS distribution function (95% CI) | |
Month 6 | 51.7 (39.3, 62.8) |
Month 9 | 50.1 (37.7, 61.3) |
Month 12 | 50.1 (37.7, 61.3) |
BICR = blinded independent central review; CI = confidence interval; dMMR = mismatch repair-deficient; EC = endometrial cancer; KM = Kaplan-Meier; MMR-unk = unknown mismatch repair tumour status; MSI-H = microsatellite instability-high; NR = not reached; PFS = progression-free survival; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Note: PFS per RECIST 1.1 was based on BICR.
a95% CIs were generated using the method of Brookmeyer and Crowley (1982).
Source: Clinical Study Report50
Duration of Response (IA-1 Subset of Patients With an Objective Response in IA-2)
The results of the DOR for the subset of patients from IA-1 included in IA-2 who had an objective response (N = 33) was similar to that of the full cohort. Median DOR was not reached in this subgroup of the population either. With a median follow-up of 19.2 months, 84.8% of the responders had an ongoing response at the time of IA-2. Approximately 91% of patients with a response had a DOR of ≥ 6 months.
Table 47: KM Analysis of DOR in Patients With dMMR or MSI-H EC — RECIST 1.1 Based on BICR (IA-1 Subset of Patients With an Objective Response at IA-2 – Primary Efficacy Analysis)
Outcome measure | GARNET trial Cohort A1 (N = 33) |
|---|---|
Median duration of follow-up (months) | 19.2 |
DOR status, n (%) | |
Events observed | 5 (15.2) |
Censored | 28 (84.8) |
DOR (months) | |
Range | 2.6 to > 28.1 |
Quartile (95% CI) | |
25% | NR (9.8 to NR) |
50% | NR (NR to NR) |
75% | NR (NR to NR) |
Duration ≥ 6 months, n (%) | 30 (90.9) |
DOR distribution function (95% CI) | |
Month 6 | 97.0 (80.4 to 99.6) |
Month 12 | 90.0 (72.1 to 96.7) |
Month 18 | 79.4 (56.1 to 91.2) |
BICR = blinded independent central review; CI = confidence interval; dMMR = mismatch repair-deficient; DOR = duration of response; EC = endometrial cancer; IA-1 = first interim analysis; KM = Kaplan-Meier; max = maximum; min = minimum; MSI-H = microsatellite instability-high; NR = not reached; RECIST 1.1 = Response Evaluation Criteria in Solid Tumours Version 1.1.
Note: DOR per RECIST 1.1 was based on BICR. A “>” indicates that the patients’ response is ongoing.
a95% CIs were generated using the method of Brookmeyer and Crowley (1982)
Source: Clinical Study Report50
Table 48: Tumour Response Summary in Patients With dMMR or MSI-H EC — irRECIST Based on Investigators’ Assessment (Secondary Efficacy Analysis Set)
Outcome measure | GARNET trial Cohort A1 (N = 113) |
|---|---|
irBOR by irRECIST 1.1, n (%) | |
irCR | 8 (7.1) |
irPR | 44 (38.9) |
irSD | 20 (17.7) |
irPD | 36 (31.9) |
NE | 2 (1.8) |
Not done | 3 (2.7) |
Confirmed irORR by irRECIST 1.1, n (%) | 52 (46.0) |
95% CIb | (36.6 to 55.6) |
Response ongoingc | 43 (82.7) |
irDCR by RECIST 1.1, n (%) | 72 (63.7) |
95% CIb | (54.1 to 72.6) |
CI = confidence interval; dMMR = mismatch repair-deficient; EC = endometrial cancer; irBOR = immune-related best overall response; irCR = immune-related complete response; irDCR = immune-related disease control rate; irORR = immune-related objective response rate; irPD = immune-related progressive disease; irPR = immune-related partial response; irRECIST = immune-related Response Evaluation Criteria in Solid Tumours; irSD = immune-related stable disease; MMR-unk = unknown mismatch repair tumour status; MSI-H = microsatellite instability-high; NE = not evaluable.
Note: irORR was defined as the percentage of patients with an irRECIST-confirmed irCR or irPR. irDCR was defined as the percentage of patients with irPR, irCR, or irSD.
aExact 2-sided 95% CI for the binomial proportion.
bAll responders who have not yet died or progressed (including clinical progression); the denominator for the percentage is the number of responders.
Source: Clinical Study Report50
Table 49: KM Analysis of irDOR in Patients With dMMR or MSI-H EC — irRECIST Based on Investigators’ Assessment (Secondary Efficacy Analysis Set — Patients With Objective Response)
Outcome measure | GARNET trial Cohort A1a (N = 52) |
|---|---|
Median duration of follow-up (months) | 16.5 |
irDOR status, n (%) | |
Events observed | 9 (17.3) |
Censored | 43 (82.7) |
irDOR (months) | |
Range | 1.41 to 28.09 |
Quartile (95% CIb) | |
25% | 20.5 (8.4 to NR) |
50% | NR (20.5 to NR) |
75% | NR (NR to NR) |
Duration ≥ 6 months, n (%) | 40 (76.9) |
irDOR distribution function (95% CI) | |
Month 6 | 96.1 (85.2 to 99.0) |
Month 12 | 79.2 (62.1 to 89.2) |
Month 18 | 79.2 (62.1 to 89.2) |
CI = confidence interval; dMMR = mismatch repair-deficient; EC = endometrial cancer; irDOR = immune-related duration of response; irRECIST = immune-related Response Evaluation Criteria in Solid Tumours; KM = Kaplan-Meier; max = maximum; min = minimum; MMR-unk = unknown mismatch repair tumour status; MSI-H = microsatellite instability-high; NR = not reached.
aPopulation total (N = 52) included n = 50 dMMR EC patients and n = 2 MMR-unk/MSI-H EC patients. Results were similar when data from patients with MMR-unknown, but MSI-H tumours were pooled with those of patients with dMMR tumours
b95% CIs were generated using the method of Brookmeyer and Crowley (1982).
Source: Clinical Study Report50
Figure 11: KM Plot for irPFS in Patients With dMMR or MSI-H EC — irRECIST Based on Investigators’ Assessment (IA-2, Secondary Efficacy Analysis Set)
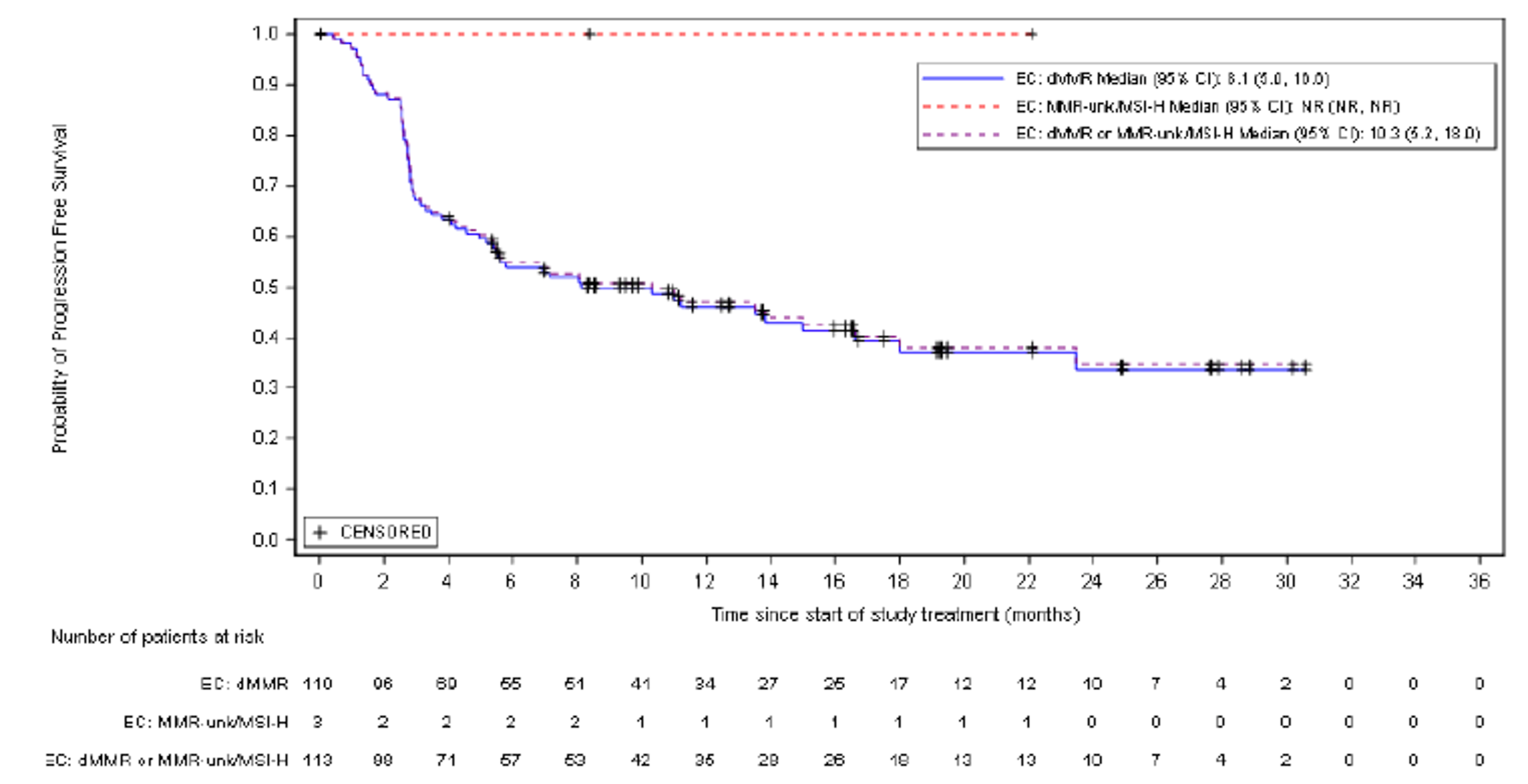
CI = confidence interval; dMMR = mismatch repair-deficient; EC = endometrial cancer; irPFS = immune-related progression-free survival; irRECIST = immune-related Response Evaluation Criteria in Solid Tumours; KM = Kaplan-Meier; MMR-unk = unknown mismatch repair tumour status; MSI-H = microsatellite instability-high; NR = not reached.
Source: Clinical Study Report.50
Table 50: Sample Size and Baseline Characteristics After Matching — Initial RWE Cohort (Base-Case and Scenario Analysis)
Baseline characteristics | Initial RWE cohort (base case) | ECOG ≤ 1 cohort | ||||
|---|---|---|---|---|---|---|
Scenario 1 | Scenario 2 | Scenario 3 | Scenario 1 | Scenario 2 | Scenario 3 | |
ESS | 34 | 74 | 75 | 38 | 75 | 58 |
Age Category | ||||||
< 65 years | 41.4% | 48.9% | 56.7% | 42.4% | 48.9% | 47.4% |
≥ 65 years | 58.6% | 51.1% | 43.3% | 57.6% | 51.1% | 52.6% |
Race | ||||||
White | 76.1% | 77.8% | 84.2% | 75.8% | 77.7% | 87.6% |
Black | 1.9% | 2.5% | 5.7% | 2.0% | 2.5% | 4.2% |
Other Race | 5.7% | 7.4% | 7.8% | 6.0% | 7.4% | 6.6% |
Unknown | 16.2% | 12.4% | 2.3% | 16.2% | 12.4% | 1.6% |
Histology | ||||||
Endometrioid | 42.4% | 42.4% | 42.4% | 42.5% | 42.5% | 42.5% |
Nonendometrioid | 57.6% | 57.6% | 57.6% | 57.5% | 57.5% | 57.5% |
Unknown | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
ECOG | ||||||
0 | 48.7% | 41.0% | 38.9% | 48.4% | 41.0% | 63.9% |
1 | 51.3% | 59.0% | 61.1% | 51.7% | 59.0% | 36.1% |
Unknown | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
FIGO Stage | ||||||
Stage I/II | 30.2% | 40.9% | 22.1% | 30.9% | 40.9% | 24.2% |
Stage III/IV | 69.8% | 59.1% | 77.9% | 69.1% | 59.1% | 75.8% |
Disease Grade | ||||||
Grade 1/2 | 27.5% | 48.5% | 45.8% | 28.2% | 48.6% | 41.9% |
Grade 3/4 | 38.9% | 45.5% | 48.6% | 41.1% | 45.4% | 51.9% |
Unknown | 33.6% | 6.0% | 5.6% | 30.7% | 6.0% | 6.2% |
Number of Prior Platinum Therapies | ||||||
0 | 0.0% | 0.0% | 1.9% | 0.0% | 0.0% | 1.1% |
1 | 100.0% | 100.0% | 79.4% | 100.0% | 100.0% | 83.2% |
≥ 2 | 0.0% | 0.0% | 18.7% | 0.0% | 0.0% | 15.7% |
Prior Surgery | ||||||
Yes | 78.1% | 89.9% | 81.6% | 79.3% | 89.9% | 82.4% |
No | 21.9% | 10.1% | 18.4% | 20.7% | 10.1% | 17.6% |
ECOG = Eastern Cooperative Oncology Group; ESS = effective sample size; FIGO = Federation of Gynecology and Obstetrics; ITT = intent to treat; RWE = real-world evidence.
Note: Percentages for matching variables are highlighted in bold.
Source: Sponsor-submitted ITC reports.19
Table 51: Sample Size and Baseline Characteristics After Matching — Treatment-Specific RWE Cohort
Baseline characteristic | Dostarlimab (N = 129) vs. Paclitaxel (N = 116) | Dostarlimab (N = 129) vs. Carboplatin + Paclitaxel | Dostarlimab (N = 129) vs. Carboplatin + Liposomal Doxorubicin (N = 141) | Dostarlimab (N = 129) vs. Liposomal Doxorubicin | Dostarlimab (N = 129) vs. Carboplatin (N = 93) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Scenario | Scenario | Scenario | Scenario | Scenario | |||||||||||
1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
ESS | 30 | 72 | 63 | 36 | 74 | 69 | 26 | 69 | 74 | 37 | 78 | 76 | 23 | 67 | 69 |
Age Category | |||||||||||||||
< 65 years | 40.9% | 48.8% | 56.8% | 41.9% | 48.9% | 59.0% | 39.4% | 48.7% | 56.0% | 42.7% | 49.0% | 51.9% | 37.9% | 48.6% | 55.9% |
≥ 65 years | 59.1% | 51.2% | 43.2% | 58.1% | 51.1% | 41.0% | 60.7% | 51.3% | 44.0% | 57.3% | 51.0% | 48.1% | 62.1% | 51.4% | 44.1% |
Race | |||||||||||||||
White | 75.5% | 77.8% | 75.0% | 76.2% | 77.8% | 86.7% | 76.6% | 77.9% | 84.4% | 74.8% | 77.6% | 86.1% | 79.2% | 78.0% | 92.5% |
Black | 2.0% | 2.5% | 8.6% | 2.0% | 2.5% | 6.5% | 1.8% | 2.5% | 4.3% | 2.0% | 2.4% | 3.1% | 1.7% | 2.6% | 3.2% |
Other Race | 5.8% | 7.5% | 15.5% | 5.9% | 7.4% | 6.1% | 5.5% | 7.6% | 8.5% | 5.9% | 7.2% | 5.4% | 5.1% | 7.7% | 3.2% |
Unknown | 16.7% | 12.2% | 0.9% | 15.9% | 12.4% | 0.7% | 16.0% | 12.0% | 2.8% | 17.4% | 12.7% | 5.4% | 14.0% | 11.8% | 1.1% |
Histology | |||||||||||||||
Endometrioid | 40.5% | 40.5% | 40.5% | 41.9% | 41.9% | 41.9% | 39.0% | 39.0% | 39.0% | 44.6% | 44.6% | 44.6% | 37.6% | 37.6% | 37.6% |
Nonendometrioid | 59.5% | 59.5% | 59.5% | 58.1% | 58.1% | 58.1% | 61.0% | 61.0% | 61.0% | 55.4% | 55.4% | 55.4% | 62.4% | 62.4% | 62.4% |
Unknown | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
ECOG | |||||||||||||||
0 | 49.8% | 40.8% | 39.4% | 48.2% | 40.9% | 40.0% | 49.4% | 40.6% | 38.4% | 49.7% | 41.2% | 37.2% | 47.3% | 40.5% | 37.6% |
1 | 50.2% | 59.2% | 60.6% | 51.8% | 59.1% | 60.0% | 50.6% | 59.4% | 61.6% | 50.3% | 58.8% | 62.8% | 52.7% | 59.5% | 62.4% |
Unknown | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% |
FIGO Stage | |||||||||||||||
Stage I/II | 28.2% | 40.4% | 28.4% | 30.7% | 40.8% | 15.8% | 27.4% | 40.0% | 22.0% | 30.6% | 41.5% | 35.4% | 28.8% | 39.7% | 20.4% |
Stage III/IV | 71.8% | 59.6% | 71.6% | 69.3% | 59.2% | 84.2% | 72.6% | 60.0% | 78.0% | 69.4% | 58.5% | 64.6% | 71.2% | 60.3% | 79.6% |
Disease Grade | |||||||||||||||
Grade 1/2 | 22.4% | 47.3% | 44.1% | 28.4% | 48.2% | 45.7% | 22.7% | 46.3% | 43.4% | 26.2% | 50.0% | 48.6% | 29.0% | 45.3% | 43.4% |
Grade 3/4 | 41.4% | 46.7% | 51.2% | 40.1% | 45.8% | 49.3% | 38.3% | 47.7% | 51.0% | 41.5% | 44.0% | 44.9% | 32.3% | 48.6% | 50.8% |
Unknown | 36.2% | 6.0% | 4.7% | 31.5% | 6.0% | 5.0% | 39.0% | 6.0% | 5.6% | 32.3% | 6.0% | 6.5% | 38.7% | 6.1% | 5.8% |
Number of Prior Platinum Therapies | |||||||||||||||
0 | 0.0% | 0.0% | 1.7% | 0.0% | 0.0% | 2.2% | 0.0% | 0.0% | 2.0% | 0.0% | 0.0% | 1.7% | 0.0% | 0.0% | 2.3% |
1 | 100.0% | 100.0% | 81.2% | 100.0% | 100.0% | 78.0% | 100.0% | 100.0% | 79.4% | 100.0% | 100.0% | 81.4% | 100.0% | 100.0% | 77.8% |
≥ 2 | 0.0% | 0.0% | 17.1% | 0.0% | 0.0% | 19.9% | 0.0% | 0.0% | 18.6% | 0.0% | 0.0% | 16.9% | 0.0% | 0.0% | 19.9% |
Prior Surgery | |||||||||||||||
Yes | 76.9% | 90.0% | 83.6% | 78.9% | 90.0% | 87.5% | 75.7% | 89.9% | 83.0% | 78.6% | 89.9% | 75.4% | 76.0% | 89.5% | 82.8% |
No | 23.1% | 10.0% | 16.4% | 21.1% | 10.1% | 12.5% | 24.3% | 10.1% | 17.0% | 21.4% | 10.1% | 24.6% | 24.0% | 10.1% | 17.2% |
ECOG = Eastern Cooperative Oncology Group; ESS = effective sample size; FIGO = Federation of Gynecology and Obstetrics; ITT = intention to treat; RWE = real-world evidence.
Percentages for matching variables are highlighted in bold.
Source: Sponsor-submitted ITC reports.19
Table 52: Baseline Characteristics Following Matching for Each MAIC Versus Published Literature
Baseline characteristics | vs. McMeekin (N = 122) | vs. Julius et al. (2013)57 (N = 129) | vs. Mazgani (N = 90) |
|---|---|---|---|
Age Category | |||
Mean (SD) | 63.1 (8.91) | 63.1 (8.72) | 63.7 (8.66) |
Median (Range) | 64.0 (39, 80) | 64.0 (39, 80) | 64.0 (41, 80) |
< 65 years, % | 50.8% | 51.2% | 51.1% |
≥ 65 years, % | 49.2% | 48.8% | 48.9% |
Race, n (%) | |||
White | 92 (75.4) | 98 (76.0) | 66 (73.3) |
Black | 3 (2.5) | 3 (2.3) | 2 (2.2) |
Other Race | 8 (6.6) | 8 (6.2) | 4 (4.4) |
Unknown | 1 (0.8) | 0 | 1 (1.1) |
Not reported | 18 (14.8) | 19 (14.7) | 18 (20.0) |
BMI | |||
Mean (SD) | 29.7 (8.11) | 29.7 (8.08) | 31.1 (7.80) |
Median (Range) | 29.2 (14, 54) | 29.0 (14, 54) | 31.1 (14, 54) |
Histology, n (%) | |||
Endometrioid | 80 (65.6) | 85 (65.9) | 85 (94.4) |
Nonendometrioid | 34 (27.8) | 43 (33.3) | 5 (5.6) |
Unknown | 8 (6.6) | 1 (0.8) | 0 |
ECOG, n (%) | |||
0 | 54 (44.3) | 55 (42.6) | 38 (42.2) |
1 | 68 (55.7) | 74 (57.4) | 52 (57.8) |
FIGO Stage, n (%) | |||
Stage I/II | 52 (42.6) | 57 (44.2) | 48 (53.3) |
Stage III/IV | 70 (57.4) | 72 (55.8) | 42 (46.7) |
Number of Prior Therapies, n (%) | |||
0 | NR | NR | NR |
1 | 79 (64.8) | 82 (63.6) | 57 (63.3) |
≥ 2 | 43 (35.2) | 47 (36.4) | 33 (36.6) |
Prior Surgery, n (%) | |||
Yes | 109 (89.3) | 116 (89.9) | 83 (92.2) |
No | NR | NR | NR |
BMI = body mass index; ECOG = Eastern Cooperative Oncology Group; FIGO = Federation of Gynecology and Obstetrics; SD = standard deviation.
Source: Sponsor-submitted ITC reports.19
Table 53: Propensity Score Model (Logistic Regression) — Lean and Full Models GARNET Versus Flatiron Real-World Cohort
Characteristic | Lean model | Full model | ||
|---|---|---|---|---|
Estimate (SE) | P value | Estimate (SE) | P value | |
Intercept | 0.0109 (0.2240) | 0.9610 | 1.9507 (0.7026) | 0.0055 |
Age | ||||
≥ 65 vs. < 65 years | — | — | –0.4126 (0.3034) | 0.01738 |
BMI | — | — | –0.0543 (0.0193) | 0.0050 |
Race | ||||
Black vs. White | — | — | –2.0273 (0.6771) | 0.0028 |
Other vs. White | — | — | –0.4724 (0.5207) | 0.3642 |
Unknown vs. White | — | — | 2.3482 (0.6614) | 0.0004 |
Histology | ||||
Nonendometrioid vs. Endometrioid | 1.9115 (0.4723) | < 0.0001 | 2.0825 (0.5361) | 0.0001 |
FIGO Stage | ||||
Stage III/IV vs. Stage I/II | — | — | —0.0503 (0.3213) | 0.8756 |
Grade | ||||
Grade 3/4 vs. Grade 1/2 | –1.4630 (0.4213) | 0.0005 | –1.5923 (0.4894) | 0.0011 |
Unknown vs. Grade 1/2 | –4.3394 (0.6425) | < 0.0001 | –4.4005 (0.7199) | < 0.0001 |
ECOG | ||||
1 vs. 0 | 0.0727 (0.2654) | 0.7841 | 0.1988 (0.3028) | 0.5115 |
Number of Prior Platinum Therapies | ||||
2+ vs. 1 | 0.1666 (0.4060) | 0.6816 | 0.2987 (0.4596) | 0.5158 |
BMI = body mass index; ECOG = Eastern Cooperative Oncology Group; FIGO = Federation of Gynecology and Obstetrics.
Source: Sponsor-submitted ITC reports.19
Table 54: Baseline and Prognostic Factors Considered for Analyses — GARNET Versus Flatiron Real-World Cohort (After IPTW/PSM [Lean Model])
Characteristic | After IPTW | After PSM | ||
|---|---|---|---|---|
GARNET | Flatiron Cohort | GARNET | Flatiron Cohort | |
Age, % | ||||
< 65 years | 47.7% | 44.4% | 53.8% | 47.3% |
≥ 65 years | 52.3% | 55.6% | 46.2% | 52.7% |
Race, % | ||||
White | 70.5% | 62.0% | 75.3% | 66.7% |
Black | 1.8% | 22.3% | 3.2% | 16.7% |
Other Race | 5.2% | 13.2% | 4.3% | 14.0% |
Unknown/NR | 22.5% | 2.5% | 17.2% | 2.7% |
ECOG, % | ||||
0 | 51.2% | 47.3% | 36.6% | 49.5% |
1 | 48.8% | 52.7% | 63.4% | 50.5% |
Histology, % | ||||
Endometrioid | 69.1% | 61.9% | 77.4% | 81.7% |
Nonendometrioid | 30.9% | 38.1% | 22.6% | 18.3% |
FIGO Stage, % | ||||
Stage I/II | 39.1% | 39.1% | 44.1% | 49.5% |
Stage III/IV | 60.9% | 53.8% | 55.9% | 42.5% |
Unknown | — | 7.2% | — | 8.1% |
BMI (kg/m2) | ||||
Mean (SD) | 28.56 (7.51) | 32.56 (8.36) | 29.73 (8.19) | 32.99 (8.09) |
Disease Grade, % | ||||
Grade 1/2 | 50.9% | 50.1% | 71.0% | 75.3% |
Grade 3/4 | 22.9% | 24.4% | 22.6% | 18.3% |
Unknown | 26.2% | 25.5% | 6.5% | 6.5% |
Number of Prior Platinum Therapies, % | ||||
1 | 89.5% | 86.7% | 81.7% | 89.2% |
≥ 2 | 10.5% | 13.3% | 18.3% | 10.8% |
Prior Surgery, % | ||||
Yes | 81.3% | 58.3% | 88.2% | 67.7% |
No | 18.7% | 41.7% | 11.8% | 32.3% |
Source: Sponsor-submitted ITC reports19
Figure 12: Distribution of Propensity Scores GARNET Versus Flatiron Real-World Cohort (Lean Model)
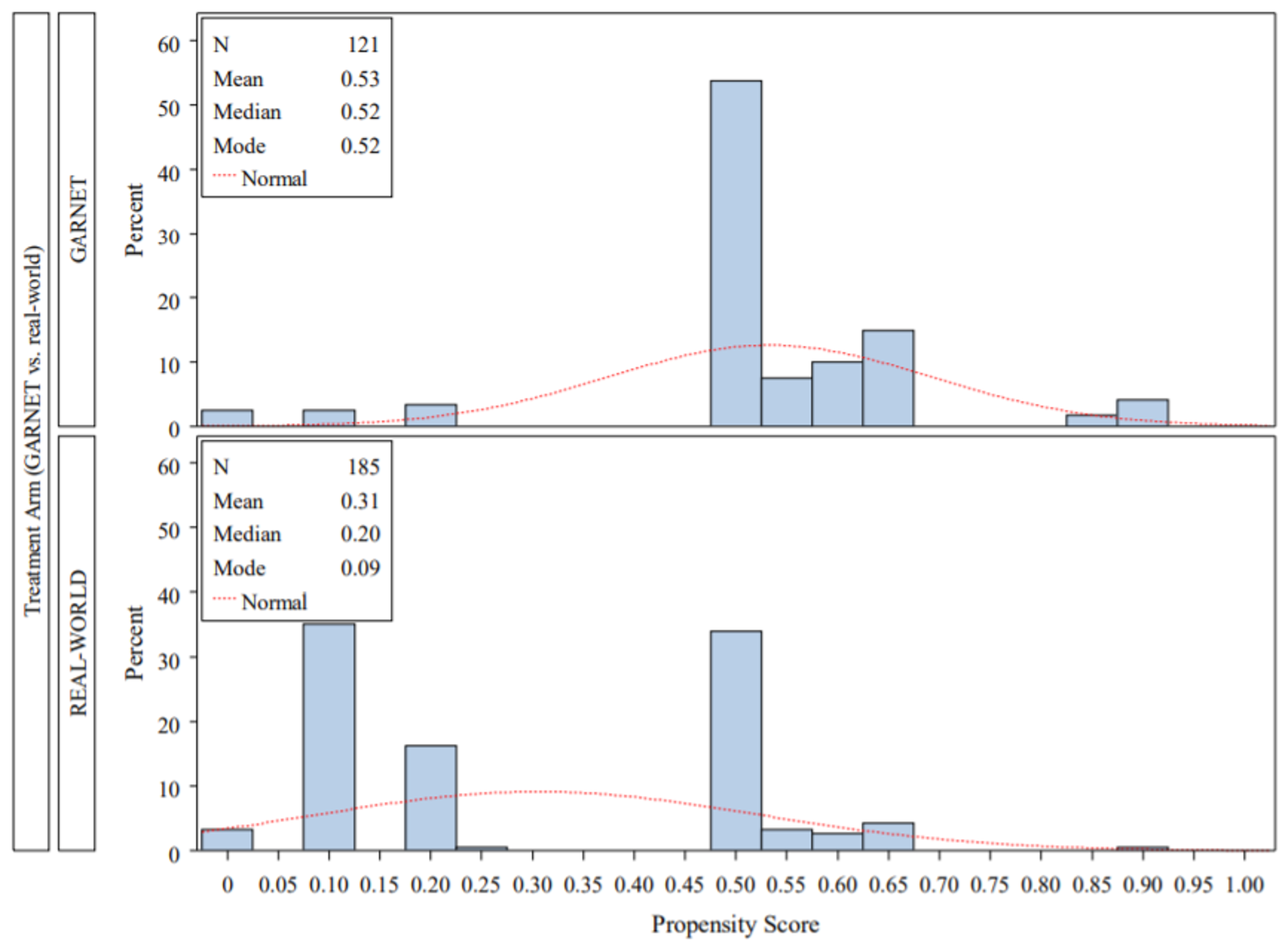
Source: Sponsor-submitted ITC reports19
Figure 13: Distribution of Propensity Scores GARNET Versus Flatiron Real-World Cohort (Full Model)
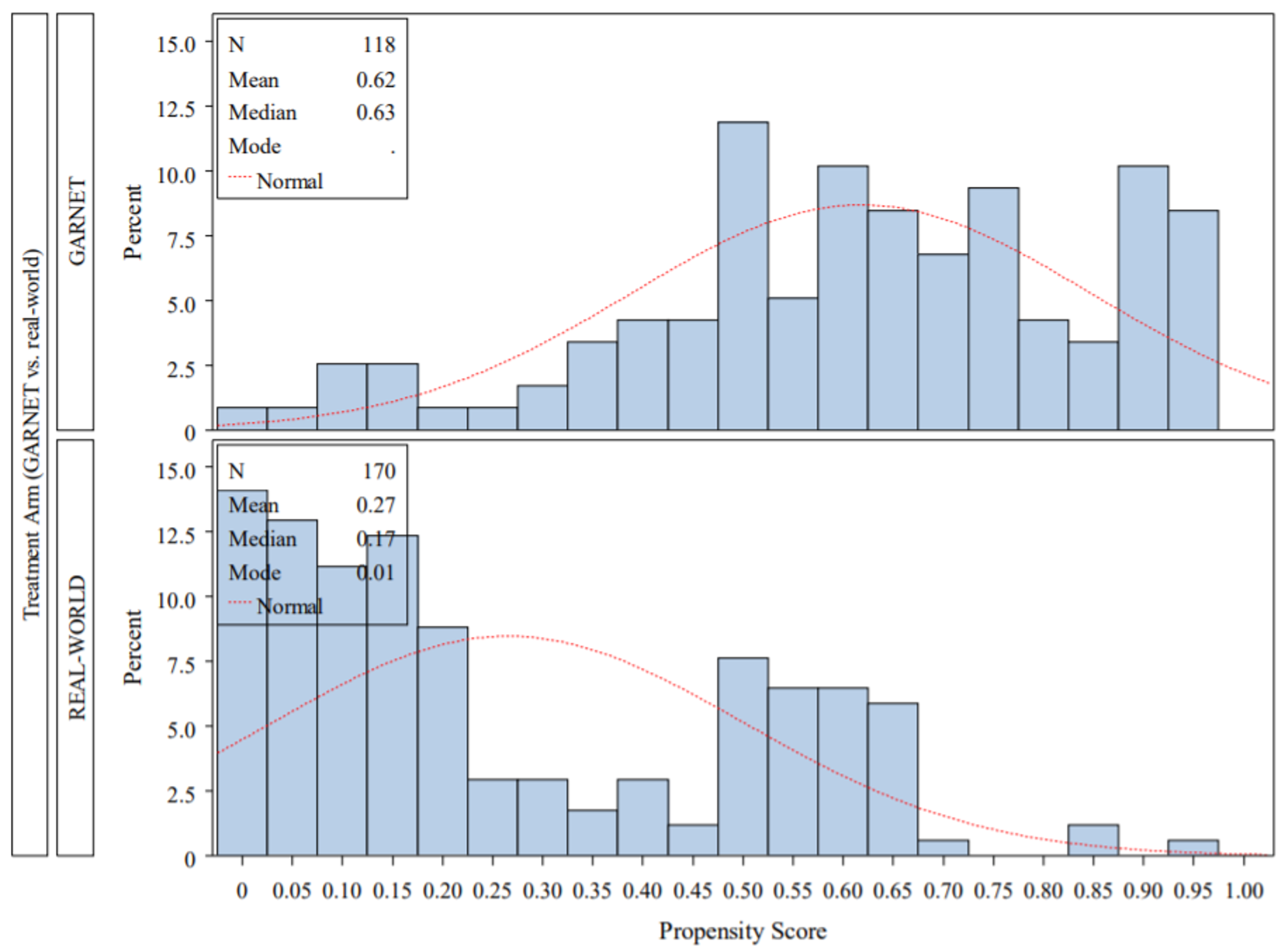
Source: Sponsor-submitted ITC reports19
Table 55: Regression Models for OS Before and After IPTW/PSM — GARNET Versus Flatiron Cohort
Variable | Unadjusted (N = 309) | After IPTW (N = 306) | After PSM (N = 196) | |||
|---|---|---|---|---|---|---|
HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | |
With study (GARNET vs. RWC) | 0.447 (0.305; 0.653) | < 0.001 | 0.559 (0.385; 0.812) | 0.002 | 0.653 (0.413; 1.033) | 0.068 |
With study and time | 0.989 (0.927; 1.055) | 0.738 | 1.001 (0.939; 1.067) | 0.977 | 1.030 (0.955; 1.111) | 0.445 |
CI = confidence interval; HR = hazard ratio; IPTW = inverse probability of treatment weighting; PSM = propensity score matching; RWC = real-world cohort.
Source: Sponsor-submitted ITC reports19
Table 56: Regression Models for OS Adjusting for Covariates — GARNET Versus Flatiron Real-World Cohort
Variable | Without interaction time x study (proportional hazards) | With interaction time x study (proportional hazards) | ||
|---|---|---|---|---|
HR (95% CI) | P value | HR (95% CI) | P value | |
Adjusted for histology, grade, ECOG and number of prior platinum-based therapies in advanced/recurrent setting (n = 309) | ||||
Study (GARNET vs. RWC) | 0.514 (0.335 to 0.789) | 0.002 | 0.571 (0.296 to 1.102) | 0.095 |
Study and time | N/A | N/A | 0.987 (0.925 to 1.053) | 0.681 |
Histology | ||||
Nonendometrioid vs. Endometrioid | 0.832 (0.512 to 1.353) | 0.458 | 0.834 (0.513 to 1.355) | 0.463 |
Unknown vs. Endometrioid | 13.743 (1.816 to 104.021) | 0.011 | 13.042 (1.700 to 100.055) | 0.013 |
Grade | ||||
Grade 3/4 vs. Grade 1/2 | 2.069 (1.341 to 3.191) | 0.001 | 2.079 (1.346 to 3.210) | < 0.001 |
Unknown vs. Grade 1/2 | 2.080 (1.155 to 3.744) | 0.015 | 2.080 (1.155 to 3.745) | 0.015 |
ECOG Performance Status | ||||
ECOG status: 1 vs. 0 | 1.461 (1.064 to 2.007) | 0.019 | 1.462 (1.064 to 2.008) | 0.019 |
Number of Prior Platinum Therapies | ||||
0 vs. 1 | 0.000 (0.000 to NE) | 0.979 | 0.000 (0.000 to NE) | 0.979 |
2+ vs. 1 | 0.995 (0.598 to 1.655) | 0.984 | 0.997 (0.599 to 1.658) | 0.989 |
Adjusted for age group, BMI, race, ECOG status, histology, FIGO stage, grade, and number of prior platinum-based therapies in advanced/recurrent setting (n = 305) | ||||
Study (GARNET vs. RWC) | 0.462 (0.290 to 0.737) | 0.001 | 0.496* (0.247 to 0.995) | 0.049 |
Study and Time | N/A | N/A | 0.991 (0.929 to 1.058) | 0.793 |
Age | ||||
≥ 65 years vs. < 65 years | 1.053 (0.759 to 1.462) | 0.757 | 1.056 (0.760 to 1.467) | 0.746 |
BMI | 0.993 (0.972 to 1.015) | 0.556 | 0.993 (0.972 to 1.015) | 0.554 |
Race | ||||
Black vs. White | 1.096 (0.722 to 1.664) | 0.665 | 1.098 (0.723 to 1.667) | 0.661 |
Other vs. White | 0.802 (0.472 to 1.363) | 0.415 | 0.803 (0.473 to 1.365) | 0.418 |
Unknown vs. White | 1.297 (0.613 to 2.745) | 0.497 | 1.284 (0.604 to 2.728) | 0.516 |
ECOG Performance Status | ||||
1 vs. 0 | 1.533 (1.102 to 2.132) | 0.011 | 1.533 (1.102 to 2.133) | 0.011 |
Histology | ||||
Nonendometrioid vs. Endometrioid | 0.876 (0.533 to 1.440) | 0.602 | 0.879 (0.535 to 1.445) | 0.611 |
Unknown vs. Endometrioid | 15.905 (1.914 to 132.155) | 0.010 | 15.310 (1.810 to 129.507) | 0.012 |
FIGO Stage | ||||
Stage III/IV vs. Stage I/II | 1.357 (0.944 to 1.950) | 0.099 | 1.357 (0.944 to 1.950) | 0.099 |
Unknown vs. Stage I/II | 1.688 (0.788 to 3.615) | 0.178 | 1.696 (0.791 to 3.637) | 0.175 |
Grade | ||||
Grade 3/4 vs. Grade 1/2 | 1.723 (1.085 to 2.736) | 0.021 | 1.725 (1.086 to 2.741) | 0.021 |
Unknown vs. Grade 1/2 | 1.589 (0.839 to 3.008) | 0.155 | 1.584 (0.836 to 3.001) | 0.158 |
Number of Prior Platinum Therapies | ||||
0 vs. 1 | 0.000 (0.000 to NE) | 0.979 | 0.000 (0.000 to NE) | 0.979 |
2+ vs. 1 | 1.000 (0.592 to 1.689) | 0.999 | 1.000 (0.592 to 1.689) | 0.999 |
CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; FIGO = Federation of Gynecology and Obstetrics; HR = hazard ratio; N/A = Not applicable; RWC = real-world cohort.
Source: Sponsor-submitted ITC reports19
Table 57: Regression Models for OS Adjusting for Covariates and Propensity Scores — GARNET Versus Real-World Cohort
Variable | Without interaction time x study (proportional hazards) | With interaction time x study (proportional hazards) | ||
|---|---|---|---|---|
HR (95% CI) | P value | HR (95% CI) | P value | |
Before adjusting for covariates (n = 306) | ||||
Study (GARNET vs. RWC) | 0.575 (0.375; 0.880) | 0.011 | 0.620* (0.321; 1.197) | 0.154 |
Study and time | N/A | N/A | 0.990 (0.928; 1.057) | 0.770 |
Propensity scorea | 0.319 (0.148; 0.688) | 0.004 | 0.319 (0.148; 0.687) | 0.004 |
Adjusted for histology, grade, ECOG and number of prior platinum-based therapies in advanced/recurrent setting (n = 306) | ||||
Study (GARNET vs. RWC) | 0.562 (0.359; 0.880) | 0.012 | 0.637* (0.325; 1.246) | 0.188 |
Study and time | N/A | N/A | 0.984 (0.922; 1.050) | 0.632 |
Histology | ||||
Nonendometrioid vs. endometrioid | 1.540 (0.507; 4.678) | 0.446 | 1.549 (0.510; 4.710) | 0.440 |
Grade | ||||
Grade 3/4 vs. Grade 1/2 | 1.182 (0.445; 3.137) | 0.737 | 1.185 (0.446; 3.147) | 0.734 |
Unknown vs. Grade 1/2 | 0.489 (0.047; 5.116) | 0.550 | 0.485 (0.046; 5.082) | 0.546 |
ECOG Performance Status | ||||
ECOG status: 1 vs. 0 | 1.517 (1.099; 2.094) | 0.011 | 1.518 (1.100; 2.096) | 0.011 |
Number of prior platinum therapies | ||||
2+ vs. 1 | 1.068 (0.635; 1.795) | 0.804 | 1.071 (0.637; 1.800) | 0.796 |
Propensity Scorea | 0.123 (0.005; 3.004) | 0.198 | 0.121 (0.005; 2.973) | 0.196 |
CI = confidence interval; ECOG = Eastern Cooperative Oncology Group; FIGO = Federation of Gynecology and Obstetrics; HR = hazard ratio; N/A = Not applicable; RWC = real-world cohort.
aThe propensity score was constructed using the following covariates: histology, grade, ECOG and number of prior platinum-based therapies in advanced/recurrent setting
Source: Sponsor-submitted ITC reports19
Table 58: Baseline Characteristics After IPTW — GARNET Versus NCRAS Base-Case Propensity Score Models (ATE)
Baseline characteristics | Propensity score model 1 | Propensity score model 2 | Propensity score model 3 | |||
|---|---|---|---|---|---|---|
GARNET (N = 109) | NCRAS Cohort (N = 999) | GARNET (N = 109) | NCRAS Cohort (N = 999) | GARNET (N = 128) | NCRAS Cohort (N = 999) | |
Age, (%) | ||||||
< 65 years | 45.4% | 45.5% | 49.0% | 45.2% | 55.9% | 45.4% |
≥ 65 years | 54.6% | 54.5% | 51.0% | 54.8% | 44.1% | 54.6% |
Race, (%) | ||||||
White | 72.7% | 84.3% | 77.6% | 84.2% | 83.7% | 83.3% |
Black | 1.8% | 5.4% | 2.4% | 5.4% | 4.6% | 5.1% |
Other Race | 5.5% | 7.8% | 7.2% | 7.9% | 8.0% | 7.5% |
Unknown/NR | 19.9% | 2.6% | 12.8% | 2.6% | 3.8% | 4.1% |
Histology, (%) | ||||||
Endometrioid | 55.7% | 45.4% | 45.4% | 45.4% | 42.4% | 45.6% |
Nonendometrioid | 44.3% | 54.6% | 54.6% | 54.6% | 57.6% | 54.4% |
ECOG, (%) | ||||||
0 | 51.3% | 32.2% | 41.3% | 32.1% | 38.9% | 32.3% |
1 | 48.7% | 18.1% | 58.7% | 18.0% | 61.1% | 18.0% |
Unknown | — | 49.7% | — | 49.8% | — | 49.8% |
FIGO Stage, (%) | ||||||
Stage I/II | 34.3% | 22.9% | 41.7% | 22.6% | 23.4% | 24.8% |
Stage III/IV | 65.7% | 77.1% | 58.3% | 77.4% | 76.6% | 75.2% |
Disease Grade, (%) | ||||||
Grade 1/2 | 31.7% | 31.5% | 50.6% | 28.9% | 45.8% | 29.3% |
Grade 3/4 | 38.8% | 37.6% | 43.5% | 38.9% | 48.6% | 38.9% |
Unknown | 29.5% | 30.9% | 5.9% | 32.2% | 5.6% | 31.9% |
Number of Prior Platinum Therapies, (%) | ||||||
0 | — | — | — | — | 1.9% | — |
1 | 100.0% | 100.0% | 100.0% | 100.0% | 79.7% | 100.0% |
≥ 2 | — | — | — | — | 18.4% | — |
Prior Surgery, (%) | ||||||
Yes | 79.9% | 81.8% | 89.9% | 81.7% | 82.2% | 82.6% |
No | 20.1% | 18.2% | 10.1% | 18.3% | 17.8% | 17.4% |
Appendix 5: Data From Sponsor (IA-3)
Note that this appendix has not been copy-edited.
Table 59: Patient Population EC (A1) Data Cut-Off November 1, 2021
Cohort | MMR status | Safety population (n) | Efficacy populationa (n) |
|---|---|---|---|
A1 | dMMR | 150 | 141 |
MSI-H/MMR-unknown | 3 | 2 | |
Total (dMMR/MSI-H) | 153 | 143 |
Table 60: Patient Disposition Safety Analysis Set, Data Cut-Off November 1, 2021
Variable, reason [n (%)] | dMMR (N = 150) | dMMR/MSI-H (N = 153) |
|---|---|---|
Discontinued treatment | 107 (71.3) | 108 (70.6) |
Adverse event | 24 (16.0) | 24 (15.7) |
Confirmed disease progression | 66 (44.0) | 66 (43.1) |
Patient request | 6 (4.0) | 6 (3.9) |
Based on clinical criteria by investigator | 8 (5.3) | 9 (5.9) |
Other | 3 (2.0) | 3 (2.0) |
Discontinued study | 72 (48.0) | 73 (47.7) |
Withdrawal of consent | 12 (8.0) | 12 (7.8) |
Lost to follow-up | 2 (1.3) | 3 (2.0) |
Death | 57 (38.0) | 57 (37.3) |
Other | 1 (0.7) | 1 (0.7) |
Subjects treated beyond initial disease progression | 43 (28.7) | 43 (28.1) |
Died while on study | 57 (38.0) | 57 (37.3) |
Disease progression | 46 (30.7) | 46 (30.1) |
Adverse event | 6 (4.0) | 6 (3.9) |
Unknown | 2 (1.3) | 2 (1.3) |
Other | 3 (2.0) | 3 (2.0) |
Table 61: Demographics and Baseline Characteristics EC Safety Analysis Set, Data Cut-Off November 1, 2021
Characteristic | dMMR (N = 141) | dMMR/MSI-H (N = 143) |
|---|---|---|
Race [n (%)] | ||
White | 108 (76.6) | 110 (76.9) |
Black | 4 (2.8) | 4 (2.8) |
Asian | 5 (3.5) | 5 (3.5) |
American Indian or Alaska Native | 3 (2.1) | 3 (2.1) |
Unknown | 1 (0.7) | 1 (0.7) |
Not Reported | 20 (14.2) | 20 (14.0) |
Ethnicity [n (%)] | ||
Hispanic or Latino | 6 (4.3) | 6 (4.2) |
Not Hispanic or Latino | 109 (77.3) | 111 (77.6) |
Unknown | 3 (2.1) | 3 (2.1) |
Not Reported | 23 (16.3) | 23 (16.1) |
Age (years) n | 141 | 143 |
Mean (std) | 63.7 (8.80) | 63.6 (8.81) |
Median | 65.0 | 65.0 |
Min, Max | 39, 85 | 39, 85 |
< 65 years | 66 (46.8) | 68 (47.6) |
≥ 65 years - < 75 years | 61 (43.3) | 61 (42.7) |
≥ 75 years | 14 (9.9) | 14 (9.8) |
ECOG Performance Status [n (%)] | ||
0 | 54 (38.3) | 56 (39.2) |
1 | 87 (61.7) | 87 (60.8) |
FIGO Cancer stage at diagnosis | ||
Stage I | 49 (34.8) | 49 (34.3) |
Stage II | 12 (8.5) | 13 (9.1) |
Stage III | 50 (35.5) | 51 (35.7) |
Stage IV | 30 (21.3) | 30 (21.0) |
Histology at diagnosis | ||
Endometrioid Carcinoma type I | 91 (64.5) | 92 (64.3) |
Endometrial Carcinoma type II | 48 (34.0) | 49 (34.3) |
Serous Carcinoma | 7 (5.0) | 7 (4.9) |
Grade 3 Endometrioid | 21 (14.9) | 21 (14.7) |
Mixed Carcinoma | 7 (5.0) | 7 (4.9) |
Unspecified | 4 (2.8) | 4 (2.8) |
Clear Cell Carcinoma | 1 (0.7) | 1 (0.7) |
Undifferentiated Carcinoma | 4 (2.8) | 4 (2.8) |
Squamous Carcinoma | 1 (0.7) | 1 (0.7) |
Othera | 3 (2.1) | 4 (2.8) |
Any prior anti-cancer treatment [n (%)] | 141 (100) | 143 (100) |
Prior surgery for study indication [n (%)] | 125 (88.7) | 127 (88.8) |
Any prior anti-cancer radiotherapy [n (%)] | 100 (70.9) | 101 (70.6) |
Any prior anti-cancer treatment for Adjuvant/Neoadjuvant disease [n (%)] | 73 (51.8) | 75 (52.4) |
Number of prior anti-cancer regimen [n (%)] | ||
0 | 0 | 0 |
1 | 89 (63.1) | 90 (62.9) |
2 | 35 (24.8) | 35 (24.5) |
3 | 14 (9.9) | 15 (10.5) |
≥ 4 | 3 (2.1) | 3 (2.1) |
Number of prior regimen for metastatic disease [n (%)] | 60 (42.6) | 61 (42.7) |
1 | 68 (48.2) | 68 (47.6) |
2 | 12 (8.5) | 13 (9.1) |
3 | 1 (0.7) | 1 (0.7) |
≥ 4 | 0 | 0 |
aIncludes dedifferentiated, endometrial adenocarcinoma, endometrial adenocarcinoma nos, endometrial neuroendocrine carcinoma, high-grade uterine carcinoma, undifferentiated clear cell carcinoma.
Table 62: Efficacy — ORR (RECIST 1.1 by BICR) Primary Efficacy Analysis Set, Data Cut-Off November 1, 2021
Response summary [n (%)] | dMMR (N = 141) | dMMR/MSI-H (N = 143) | IA-2 (01 March 2020) dMMR/MSI-H N = 105 |
|---|---|---|---|
Confirmed objective response rate (%) | 64 (45.4) | 65 (45.5) | 47 (44.8) |
95% CIa | (37.0, 54.0) | (37.1, 54.0) | (35.0, 54.8) |
Best overall response by RECIST 1.1 | |||
CR | 22 (15.6) | 23 (16.1) | 11 (10.5) |
PR | 42 (29.8) | 42 (29.4) | 36 (34.3) |
Stable disease | 21 (14.9) | 21 (14.7) | 13 (12.4) |
PD | 51 (36.2) | 51 (35.7) | 39 (37.1) |
Not Evaluable | 3 (2.1) | 3 (2.1) | 3 (2.9) |
Not Done | 2 (1.4) | 3 (2.1) | 3 (2.9) |
Response ongoingb | 53 (82.8) | 54 (83.1) | 42 (89.4) |
Disease control rate (DCR) | 85 (60.3) | 86 (60.1) | 60 (57.1) |
95% CIa | (51.7, 68.4) | (51.6, 68.2) | (47.1 66.8) |
CR = complete response, PD = progressive disease; PR = partial response.
aExact 2 sided 95% CI for the binomial proportion
bAll responders who have not yet died or progressed (including clinical progression), denominator for percentage is number of responders.
Table 63: Efficacy — Duration of Response (RECIST 1.1 by BICR) Primary Efficacy Analysis Set, Data Cut-Off November 1, 2021
Variable | dMMR (N = 64) | dMMR/MSI-H (N = 65) |
|---|---|---|
Median duration of follow-up (months) | 27.6 | 27.6 |
Median DOR (months) | NR | NR |
Min, Max DOR (months) | 1.18+, 47.21+ | 1.18+, 47.21+ |
Duration > = 6 months [n (%)] | 58 (90.6) | 59 (90.8) |
Duration ≥ 12 months [n (%)] | 51 (79.7) | 52 (80.0) |
Probability of maintaining response (95% CI)a [Kaplan-Meier estimate] | ||
Month 6 | 96.7 (87.5, 99.2) | 96.8 (87.7, 99.2) |
Month 12 | 93.1 (82.7, 97.4) | 93.3 (83.0, 97.4) |
Month 18 | 85.7 (73.3, 92.6) | 85.9 (73.8, 92.7) |
NR = Not reached.
Note: + indicates patients’ response is ongoing.
a95% Confidence intervals generated using the method of Brookmeyer and Crowley (1982).
Table 64: Overall Summary of TEAEs — Data Cut-Off November 1, 2021(Safety Analysis Set)
Adverse event category [n (%)] | dMMR (N = 150) | dMMR/MSI-H (N = 153) |
|---|---|---|
Adverse events | 149 (99.3) | 152 (99.3) |
Grade 3 or Greater AEs | 84 (56.0) | 87 (56.9) |
Treatment-related AEs | 106 (70.7) | 108 (70.6) |
Grade ≥ 3 Treatment-Related AEs | 27 (18.0) | 27 (17.6) |
AE with outcome of death | 6 (4.0) | 6 (3.9) |
Treatment-Related AE Leading to Death | 0 | 0 |
Serious AEs | 57 (38.0) | 58 (37.9) |
Treatment-Related Serious AEs | 18 (12.0) | 18 (11.8) |
AEs leading to withdrawal of study treatment | 24 (16.0) | 24 (15.7) |
TRAEs Leading to Withdrawal of Study Treatment | 13 (8.7) | 13 (8.5) |
AE leading to study treatment infusion interruption | 2 (1.3) | 2 (1.3) |
AE leading to study drug treatment interruption | 42 (28.0) | 43 (28.1) |
Immune-related AE | 58 (38.7) | 59 (38.6) |
Treatment-related immune-related AE | 41 (27.3) | 42 (27.5) |
TEAE = treatment-emergent adverse event(s), SAE = serious adverse event(s).
Note: Immune-related Adverse Events are identified as any > = Grade 2 AEs based on a pre-specified preferred terms list. Related, Possibly Related and Missing relationship Adverse Events are considered as Related Adverse Events.
For each category, patients are included only once, even if they experienced multiple events in that category.
Table 65: TEAEs Regardless of Causality — Safety Analysis Set, at Least 15% Patients, Data Cut-Off November 1, 2021
AE preferred term | dMMR (N = 150) | dMMR/MSI-H (N = 153) |
|---|---|---|
Patients with at least 1 TEAE | 149 (99.3) | 152 (99.3) |
Anemia | 51 (34.0) | 51 (33.3) |
Nausea | 48 (32.0) | 50 (32.7) |
Diarrhea | 44 (29.3) | 45 (29.4) |
Fatigue | 40 (26.7) | 41 (26.8) |
Asthenia | 36 (24.0) | 36 (23.5) |
Constipation | 35 (23.3) | 35 (22.9) |
Vomiting | 34 (22.7) | 34 (22.2) |
Arthralgia | 31 (20.7) | 33 (21.6) |
Urinary tract infection | 28 (18.7) | 30 (19.6) |
Pruritus | 29 (19.3) | 29 (19.0) |
Abdominal pain | 27 (18.0) | 27 (17.6) |
Rash | 23 (15.3) | 24 (15.7) |
Cough | 22 (14.7) | 24 (15.7) |
Decreased appetite | 23 (15.3) | 23 (15.0) |
Table 66: Related TEAEs EC (A1) Safety Analysis Set, at Least 5% Patients, Data Cut-Off November 1, 2021
AE Preferred Term | dMMR (N = 150) | dMMR/MSI-H (N = 153) |
|---|---|---|
Patients with at least 1 drug-related TEAEa | 106 (70.7) | 108 (70.6) |
Diarrhea | 24 (16.0) | 25 (16.3) |
Asthenia | 24 (16.0) | 24 (15.7) |
Fatigue | 21 (14.0) | 21 (13.7) |
Nausea | 19 (12.7) | 19 (12.4) |
Pruritus | 19 (12.7) | 19 (12.4) |
Hypothyroidism | 16 (10.7) | 17 (11.1) |
Arthralgia | 16 (10.7) | 16 (10.5) |
Rash | 14 (9.3) | 14 (9.2) |
Anemia | 11 (7.3) | 11 (7.2) |
Alanine aminotransferase increased | 9 (6.0) | 9 (5.9) |
Decreased appetite | 8 (5.3) | 8 (5.2) |
aRelated, Possibly Related and Missing relationship Adverse Events are considered as Related Adverse Events.
Table 67: Grade 3 or Higher TEAEs Regardless of Causality — Safety Analysis Set, at Least 2% Patients, Data Cut-Off November 1, 2021,
AE Preferred Term | dMMR (N = 150) | dMMR/MSI-H (N = 153) |
|---|---|---|
Patients with at least 1 grade 3 and above TEAE | 84 (56.0) | 87 (56.9) |
Anemia | 26 (17.3) | 26 (17.0) |
Abdominal pain | 7 (4.7) | 7 (4.6) |
Hyponatremia | 6 (4.0) | 7 (4.6) |
Urinary tract infection | 6 (4.0) | 6 (3.9) |
Back pain | 5 (3.3) | 5 (3.3) |
Pulmonary embolism | 5 (3.3) | 5 (3.3) |
Sepsis | 5 (3.3) | 5 (3.3) |
Hypertension | 4 (2.7) | 5 (3.3) |
Acute kidney injury | 4 (2.7) | 4 (2.6) |
Alanine aminotransferase increased | 4 (2.7) | 4 (2.6) |
Diarrhea | 4 (2.7) | 4 (2.6) |
Dyspnea | 3 (2.0) | 3 (2.0) |
Fatigue | 3 (2.0) | 3 (2.0) |
Arthralgia | 3 (2.0) | 3 (2.0) |
Lipase increased | 3 (2.0) | 3 (2.0) |
Neuralgia | 3 (2.0) | 3 (2.0) |
Pneumonia | 3 (2.0) | 3 (2.0) |
Table 68: Serious TEAEs Regardless of Causality — Safety Analysis Set, at Least 2% Patients, Data Cut-Off November 1, 2021
AE preferred term | dMMR (N = 150) | dMMR/MSI-H (N = 153) |
|---|---|---|
Patients with at least 1 serious TEAE | 57 (38.0) | 58 (37.9) |
Urinary tract infection | 6 (4.0) | 6 (3.9) |
Sepsis | 5 (3.3) | 5 (3.3) |
Abdominal pain | 4 (2.7) | 4 (2.6) |
Acute kidney injury | 4 (2.7) | 4 (2.6) |
COVID-19 | 3 (2.0) | 3 (2.0) |
Pneumonia | 3 (2.0) | 3 (2.0) |
Pneumonitis | 3 (2.0) | 3 (2.0) |
Pulmonary embolism | 3 (2.0) | 3 (2.0) |
Pyrexia | 3 (2.0) | 3 (2.0) |
Table 69: Summary of Overall irAEs Regardless of Causality — Safety Analysis Set, Over 2 Patients, Data Cut-Off November 1, 2021
AE preferred term | dMMR (N = 150) | dMMR/MSI-H (N = 153) |
|---|---|---|
Patients with at least 1 treatment-emergent irAEa | 58 (38.7) | 59 (38.6) |
Hypothyroidism | 13 (8.7) | 14 (9.2) |
Arthralgia | 10 (6.7) | 10 (6.5) |
Pruritus | 7 (4.7) | 7 (4.6) |
Alanine aminotransferase increased | 6 (4.0) | 6 (3.9) |
Hyperthyroidism | 6 (4.0) | 6 (3.9) |
Pneumonitis | 5 (3.3) | 5 (3.3) |
Aspartate aminotransferase increased | 4 (2.7) | 4 (2.6) |
Rash | 4 (2.7) | 4 (2.6) |
Colitis | 3 (2.0) | 3 (2.0) |
Gastritis | 3 (2.0) | 3 (2.0) |
Transaminases increased | 3 (2.0) | 3 (2.0) |
airAE = Immune-related Adverse Events. irAE events are TEAE with grade ≥ 2 from a pre-specified list of preferred terms.
Table 70: Summary of AEs Leading to Death — Safety Analysis Set Data, Cut-Off November 1, 2021
Adverse event category [n (%)] | dMMR/MSI-H (N = 153) |
|---|---|
AE with outcome of deatha | 6 (4.0) |
Shock | 1 |
Urinary tract infection | 1 |
Sepsis | 1 |
Pleural effusion | 1 |
Aspiration | 1 |
Pneumonia | 1 |
aNone are considered to be related to treatment by the Investigator.
Table 71: Progression-Free Survival (RECIST 1.1 by BICR) Primary Efficacy Analysis Set EC (A1 and A2), Data Cut-Off November 1, 2021
Variable | dMMR or (MMR-unk and MSI-H) EC | MMRp or (MMR-unk and MSS) EC | All EC Overall (N = 299) | ||||
|---|---|---|---|---|---|---|---|
dMMR (N = 141) | MMR-unk/MSI-H (N = 2) | Total (N = 143) | MMRp (N = 142) | MMR-unk/MSS (N = 14) | Total (N = 156) | ||
PFS Status [n (%)] | |||||||
Events observed | 83 (58.9) | 0 (0.0) | 83 (58.0) | 126 (88.7) | 10 (71.4) | 136 (87.2) | 219 (73.2) |
Censored | 58 (41.1) | 2 (100.0) | 60 (42.0) | 16 (11.3) | 4 (28.6) | 20 (12.8) | 80 (26.8) |
PFS (months) Quartile (95% CI)a | |||||||
25% | 2.7 (2.6, 2.8) | NR (NR, NR) | 2.7 (2.6, 2.8) | 2.1 (1.4, 2.5) | 2.7 (1.2, 2.7) | 2.1 (1.7, 2.5) | 2.6 (2.3, 2.6) |
50% | 5.6 (4.1, 16.6) | NR (NR, NR) | 6.0 (4.1, 18.0) | 2.7 (2.6, 2.8) | 2.8 (2.6, 6.8) | 2.7 (2.6, 2.8) | 3.1 (2.8, 4.1) |
75% | NR (41.6, NR) | NR (NR, NR) | NR (41.6, NR) | 5.5 (4.0, 7.0) | 5.4 (2.7, NR) | 5.5 (4.0, 7.0) | 19.3 (10.0, 41.6) |
PFS distribution function (95% CI) | |||||||
Month 6 | 49.2 (40.6,57.2) | 100.0 (100.0,100.0) | 49.5 (41.0,57.5) | 22.7 (16.0,30.1) | 25.0 (6.0,50.5) | 22.9 (16.5,30.0) | 35.8 (30.3,41.4) |
Month 9 | 47.6 (39.0,55.7) | 100.0 (100.0,100.0) | 48.0 (39.4,56.0) | 15.4 (9.8,22.1) | 16.7 (2.7,41.3) | 15.5 (10.1,22.0) | 31.3 (26.0,36.8) |
Month 12 | 46.0 (37.4,54.1) | 100.0 (100.0,100.0) | 46.4 (37.8,54.5) | 13.0 (7.9,19.4) | 16.7 (2.7,41.3) | 13.3 (8.3,19.5) | 29.4 (24.1,34.8) |
NR = Not reachable.
Note: PFS: Progression-Free Survival per RECIST 1.1 based on blind independent central review (BICR).
a95% Confidence intervals generated using the method of Brookmeyer and Crowley (1982).
Figure 14: KM Plot for Progression-Free Survival per RECIST 1.1 by BICR) — Primary Efficacy Analysis Set, Data Cut-Off November 1, 2021
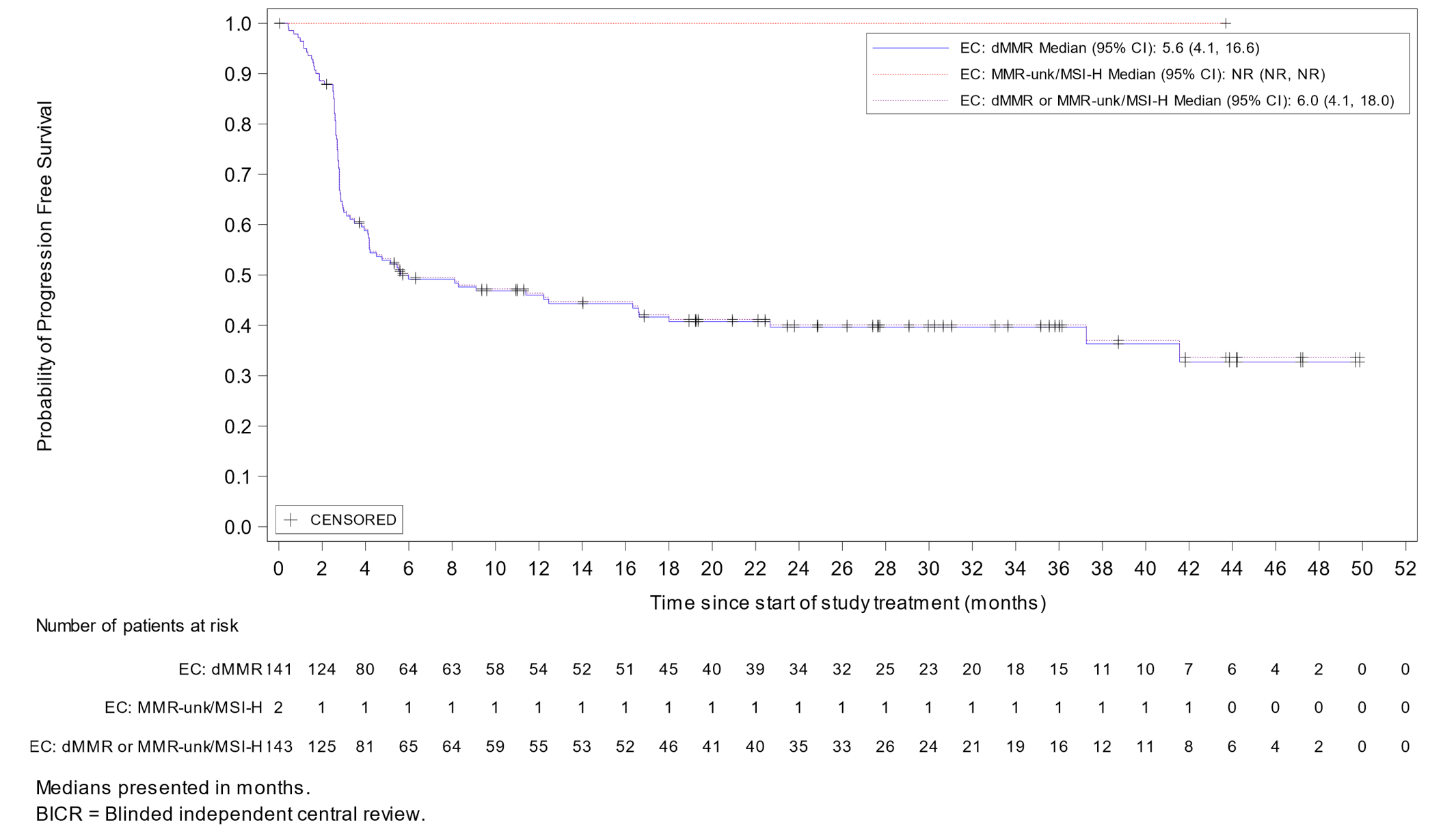
Table 72: OS (Cohort A1 and A2) — Primary Efficacy Analysis Set, Data Cut-Off November 1, 2021
Variable | dMMR or (MMR-unk and MSI-H) EC | MMRp or (MMR-unk and MSS) EC | All EC Overall (N = 299) | ||||
|---|---|---|---|---|---|---|---|
dMMR (N = 141) | MMR-unk/MSI-H (N = 2) | Total (N = 143) | MMRp (N = 142) | MMR-unk/MSS (N = 14) | Total (N = 156) | ||
OS Status [n (%)] | |||||||
Events observed | 55 (39.0) | 0 (0.0) | 55 (38.5) | 101 (71.1) | 8 (57.1) | 109 (69.9) | 164 (54.8) |
Censored | 86 (61.0) | 2 (100.0) | 88 (61.5) | 41 (28.9) | 6 (42.9) | 47 (30.1) | 135 (45.2) |
OS (months) Quartile (95% CI)a | |||||||
25% | 9.3 (6.2, 14.5) | NR (NR, NR) | 9.3 (6.2, 14.5) | 6.0 (4.3, 8.5) | 6.4 (2.6, 15.3) | 6.0 (4.3, 7.8) | 6.9 (5.7, 9.5) |
50% | NR (25.7, NR) | NR (NR, NR) | NR (25.7, NR) | 18.0 (13.0, 22.2) | 15.3 (4.5, NR) | 16.9 (13.0, 21.8) | 22.5 (18.0, 28.3) |
75% | NR (NR, NR) | NR (NR, NR) | NR (NR, NR) | 32.8 (28.3, 42.3) | NR (11.8, NR) | 32.8 (29.1, NR) | NR (42.3, NR) |
OS Distribution Function (95% CI) | |||||||
Month 6 | 83.7 (76.3, 89.0) | 100.0 (100.0, 100.0) | 83.9 (76.5, 89.1) | 74.9 (66.6, 81.3) | 76.9 (44.2, 91.9) | 75.1 (67.2, 81.3) | 79.3 (74.1, 83.5) |
Month 9 | 76.0 (67.7, 82.4) | 100.0 (100.0, 100.0) | 76.2 (68.0, 82.5) | 67.2 (58.5, 74.4) | 61.5 (30.8, 81.8) | 66.7 (58.4, 73.7) | 71.2 (65.5, 76.1) |
Month 12 | 71.2 (62.6, 78.2) | 100.0 (100.0, 100.0) | 71.4 (62.9, 78.3) | 61.8 (52.9, 69.4) | 53.8 (24.8, 76.0) | 61.1 (52.6, 68.4) | 66.0 (60.1, 71.2) |
NR = Not Reached; OS = Overall Survival.
a95% Confidence intervals generated using the method of Brookmeyer and Crowley (1982).
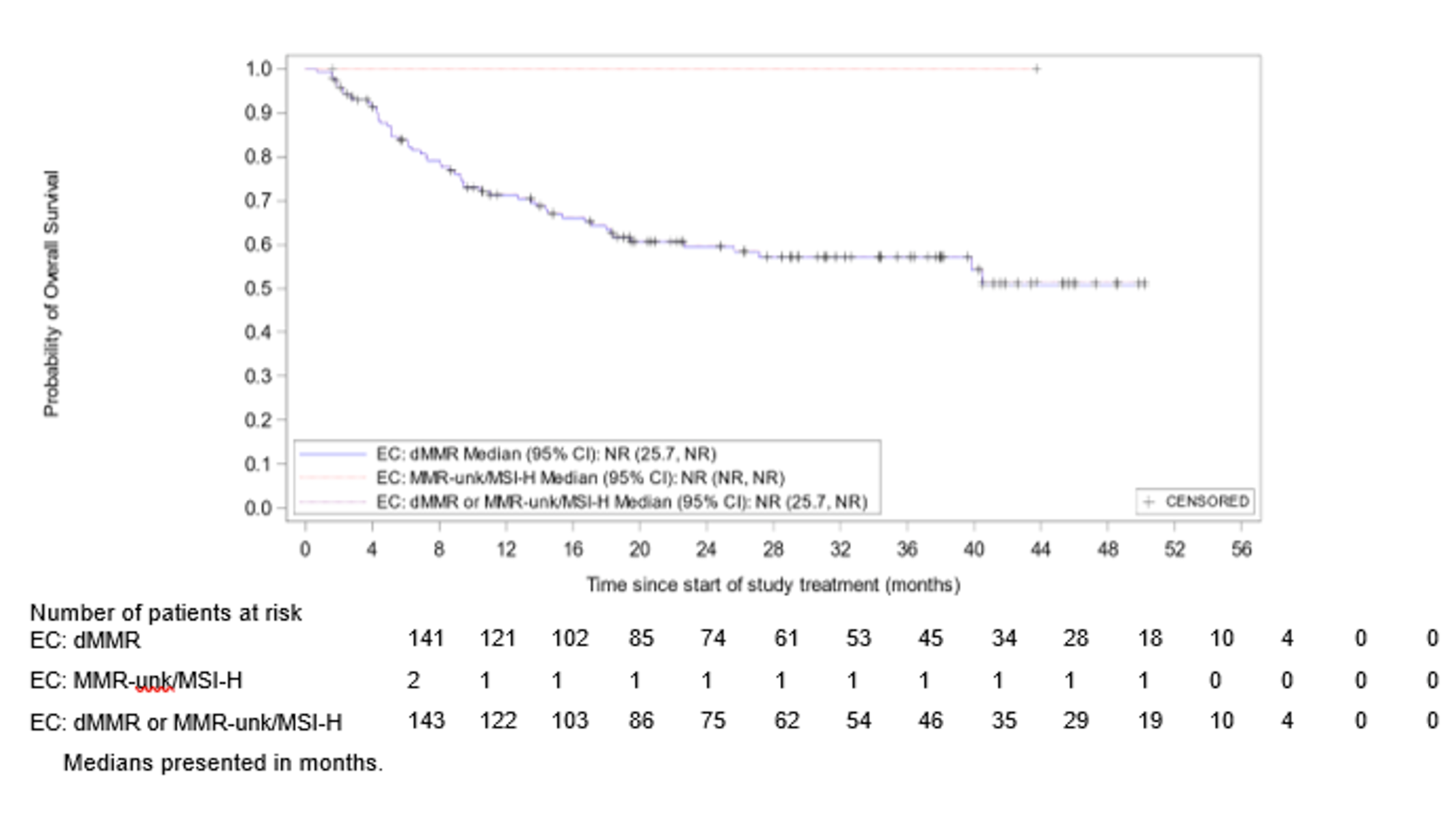
Pharmacoeconomic Review
Abbreviations
CMT
current mix of treatments
dMMR
deficient mismatch repair
EC
endometrial cancer
EQ-5D-5L
EQ-5D-5-Level
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IPTW
inverse probability treatment weighting
KM
Kaplan-Meier
MAIC
matching-adjusted indirect comparison
MSI-H
microsatellite instability-high
NCRAS
National Cancer Registration and Analysis Service
OS
overall survival
PLD
pegylated liposomal doxorubicin
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
RWE
real-world evidence
TTD
time to treatment discontinuation
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Dostarlimab (Jemperli), 500 mg/10 mL vial (50 mg/mL) |
Submitted price | Dostarlimab, 50 mg/mL, single-use vial: $10,270.00 per 500 mg vial |
Indication | Monotherapy for the treatment of adults with dMMR or MSI-H recurrent or advanced endometrial cancer that has progressed on or after prior treatment with a platinum-containing regimen |
Health Canada approval status | NOC/c |
Health Canada review pathway | Advance consideration under NOC/c |
NOC date | December 23, 2021 |
Reimbursement request | As per indication |
Sponsor | GlaxoSmithKline Inc. |
Submission history | Previously reviewed: No |
dMMR = deficient mismatch repair; MSI-H = microsatellite instability-high; NOC = Notice of Compliance; NOC/c = Notice of Compliance with conditions.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target population | Adults with dMMR or MSI-H advanced or recurrent EC who have been previously treated with PBCT (cohort A1 of the GARNET study) |
Treatment | Dostarlimab |
Comparators | CMT, a weighted distribution of chemotherapies and hormone therapies, which included cisplatin plus doxorubicin, carboplatin plus gemcitabine, cisplatin, cisplatin in combination with cyclophosphamide plus doxorubicin, and gemcitabine The following single-agent and combination therapies were also included as individual comparators:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (40 years) |
Key data source | Treatment efficacy of dostarlimab (i.e., OS and PFS) was informed by the GARNET study, a nonrandomized, single-arm, multi-centre, open-label study Comparative efficacy of comparator treatments was estimated using a combination of approaches based on unanchored ITCs, including the use of hazard ratios estimated from MAICs, IPTW methods, and/or parametric survival distributions, and was sourced from a UK RWE cohort (NCRAS database) |
Submitted results | The sponsor conducted a series of pairwise comparisons and sequential analyses. Sequential analyses were not presented due to the lack of comparability among patient populations.a
|
Key limitations |
|
CADTH reanalysis results |
|
CMT = current mix of treatments; dMMR = deficient mismatch repair; EC = endometrial cancer ; ICER = incremental cost-effectiveness ratio; IPTW = inverse probability treatment weighting; ITCs = indirect treatment comparisons; LYs = life-years; MAICs = matching-adjusted indirect comparisons; MSI-H = microsatellite instability-high; NCRAS = National Cancer Registration and Analysis Service; OS = overall survival; PBCT = platinum-based chemotherapy; PFS = progression-free survival; PLD = pegylated liposomal doxorubicin; PMS = partitioned survival model; QALYs = quality-adjusted life-years; RWE = real-world evidence; vs. = versus.
aIncremental cost and incremental QALY results are not presented in this table for reader ease. These and additional results are presented in Appendix 3.
Conclusions
The pharmacoeconomic results are informed by results from the GARNET study, a single-arm, phase I trial that aimed to assess the efficacy and safety of dostarlimab among patients with deficient mismatch repair (dMMR) or microsatellite instability-high (MSI-H) recurrent or advanced endometrial cancer (EC). According to the CADTH clinical review, the lack of a comparator group precludes the ability to assess the relative therapeutic benefit or safety of dostarlimab to relevant comparators (i.e., active treatment, standard of care). According to the CADTH clinical review, it is not clear from the GARNET study whether dostarlimab may improve progression-free survival (PFS). There is indirect evidence to suggest that dostarlimab may improve PFS, compared with the standard of care (chemotherapeutic regimens); however, no strong conclusions can be drawn because of many limitations in the indirect comparisons. The relative effects of dostarlimab on patient-relevant outcomes, such as overall survival (OS) and quality of life, are uncertain.
CADTH was not able to conduct a CADTH base-case analysis because of foundational limitations with the sponsor’s model and submitted clinical evidence. Notably, the comparator population was not matched by dMMR or MSI-H status and, thus, the cost-effectiveness of dostarlimab compared with second-line treatments in the indicated population is unknown. The sponsor’s choice of a partitioned survival model (PSM) may overestimate incremental quality-adjusted life-years (QALYs), and the long-term extrapolations of OS and PFS are highly uncertain. CADTH conducted an exploratory analysis, the results of which suggest that the model is highly sensitive to several model assumptions, including long-term survival extrapolation, stopping rules, and treatment-effectiveness waning after 24 months.
The sponsor submitted a model comparing the cost-effectiveness of dostarlimab with second-line treatments (pegylated liposomal doxorubicin [PLD]; doxorubicin; paclitaxel; current mix of treatments [CMT]; carboplatin plus PLD; carboplatin plus paclitaxel; and carboplatin). According to the comparative evidence in the submitted model, carboplatin plus paclitaxel was the treatment with the smallest amount of uncertainty related to OS and PFS. Based on the sponsor’s submitted base case, the probability that dostarlimab is cost-effective was 0% at a willingness-to-pay threshold of $50,000 per QALY. The sponsor’s base-case results indicate that 89% of the incremental benefit of dostarlimab was obtained beyond the single-arm trial period, when cost-effectiveness results vary widely, depending on the chosen extrapolation function. Based on the CADTH exploratory analysis, a price reduction of 83% would be needed for dostarlimab to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY; however, this estimate is subject to limitations in the sponsor’s submission, most crucially the inappropriateness of the estimated efficacy of comparator treatments in the dMMR or MSI-H population. An additional price reduction may be warranted.
Treatment of dMMR or MSI-H EC with dostarlimab increases costs, compared with other currently available treatments. CADTH could not estimate the amount of health improvement that may result from treatment with dostarlimab because of highly uncertain clinical efficacy data and a lack of information about the efficacy of comparator treatments. Because of methodological limitations identified in the model and clinical evidence, the cost-effectiveness of dostarlimab is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Two patient groups — the Canadian Cancer Society and the Division of Gynecologic Oncology at McGill University Health Centre — provided patient input for this review. Patient input raised concerns about the inconsistent availability of treatment options in different provinces (e.g., carboplatin plus paclitaxel is the only available option in Manitoba). Patients shared their interest and desire to have access to treatment options that are available outside of Canada. Patients noted that important goals of treatment include longer remission, improved quality of life, and the ability to enjoy life and loved ones outside of clinic visits. Patients reported that they wanted new treatments, like immunotherapy, to have fewer nondebilitating side effects. Patients reported that commonly experienced side effects of existing treatments include skin issues, fatigue, bladder control, stamina, vaginal bleeding after intercourse, vaginal dryness, hair loss, pain, concentration problems (i.e., chemotherapy “fog”) and arthritis. Patient input raised concerns about the affordability of treatment, especially considering the way it can interrupt daily activities.
Two registered clinicians and 4 clinician-input groups — the Society for Gynecologic Oncology of Canada; the British Columbia Cancer Provincial Gynecology Oncology Tumour Group; clinicians from the Divisions of Gynecologic Oncology at McGill and Sunnybrook Hospitals; and Ontario Health-Cancer Care Ontario — reported that the standard of care in the first-line treatment setting for advanced or metastatic disease is platinum-based chemotherapy, typically with carboplatin and paclitaxel. Registered clinicians indicated that dostarlimab fills an unmet need for an entire biomarker-defined population with recurrent disease. The current place in therapy for dostarlimab is in the second-line treatment setting, given that pembrolizumab is currently not funded, and there are currently no available nor accessible second-line treatments for Canadian patients. Dostarlimab is unlikely to be used again in a later line of therapy, whereas endocrine therapy and radiation therapy may be used in some circumstances. Among patients who respond to treatment, dostarlimab is expected to prolong disease progression, reduce severity of symptoms, improve quality of life, compared with available standard chemotherapy or hormone-based treatments.
Feedback from the drug plans identified several items for CADTH to take into consideration during the review. First, drug plans noted that there is no current standard-of-care treatment for patients who progress on platinum-containing regimens. Drug plans noted that relevant comparators include chemotherapy (combination and monotherapy) and hormonal therapy, and that the funding of relevant comparators varies across jurisdictions. Drug plans highlighted that although pembrolizumab is approved by Health Canada for this indication, it is not publicly funded in any jurisdiction. Drug plans anticipated the potential for indication creep, given patient eligibility for dostarlimab in the following scenarios: as a first-line therapy for those who received platinum-based therapy only in early-stage disease; for patients with dMMR or MSI-H with a contraindication to or no prior exposure to platinum-containing regimens; for patients who have received more than 2 lines of therapy for advanced or recurrent disease; and for those who wish to switch to dostarlimab from current systemic therapy for recurrent dMMR or MSI-H EC (with prior platinum-based therapy). Drug plans also require clarity about whether patients with an Eastern Cooperative Oncology Group Performance Status of 2 or greater are eligible for dostarlimab. Drug plans anticipate that dostarlimab will likely be less resource-intensive than other IV chemotherapy comparators with regard to chair time, blood-work frequency, and pharmacy sterile compounding time. Importantly, drug plans indicated that mismatch repair and microsatellite instability testing in EC is not standard in all jurisdictions. Drug plans further noted that additional resources (e.g., specialists) will be required to monitor and manage potential immune-mediated adverse effects. Drug plans raised concerns about the applicability of weight-based dosing of dostarlimab in Canadian clinical practice, as it is anticipated that drug wastage is likely. However, drug wastage is not expected if a flat dose schedule is applied, as outlined in the product monograph for dostarlimab. Drug plans shared concerns about the anticipated budget impact, given that treatment duration is likely to be significantly longer with dostarlimab than with currently funded chemotherapy comparators.
Several of these concerns were addressed in the sponsor’s model:
The sponsor incorporated several relevant comparators, including single-agent (i.e., carboplatin, paclitaxel, PLD) and combination (carboplatin plus paclitaxel, carboplatin plus PLD) chemotherapies and a mix of treatments that comprise various chemotherapies and hormonal therapies.
PFS and OS were modelled for the overall population. Health-related quality of life (HRQoL) was incorporated into the model by progression status.
The costs of dMMR and MSI-H testing were included.
CADTH was unable to address the following concerns raised from stakeholder input:
In the economic model, the sponsor incorporated a fixed-dosing approach (i.e., 500 mg every 3 weeks, 1,000 mg every 6 weeks) for dostarlimab, as in the GARNET study. The sponsor did not include an option to assess the effects of weight-based dosing for dostarlimab and, as such, CADTH was unable to explore this option further. However, drug wastage of comparator treatments was incorporated, as appropriate. The cost-effectiveness of weight-based dosing for dostarlimab remains unknown.
Economic Review
The current review is for dostarlimab (Jemperli) in adults with dMMR or MSI-H advanced or recurrent EC who have been previously treated with platinum-based chemotherapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Dostarlimab is indicated as monotherapy for the treatment of adults with recurrent or advanced dMMR or MSI-H EC that has progressed on or after prior treatment with a platinum-containing regimen.
The sponsor submitted a cost-utility analysis to assess the cost-effectiveness of dostarlimab as monotherapy, compared with multiple comparator treatments, including doxorubicin monotherapy, carboplatin monotherapy; PLD, paclitaxel monotherapy, carboplatin plus paclitaxel, and carboplatin plus PLD. The sponsor further included a basket comparator representing CMT (i.e., chemotherapies, hormone therapies, and radiation therapy).1 Chemotherapies considered as part of the CMT included cisplatin plus doxorubicin, carboplatin plus gemcitabine, cisplatin, cisplatin in combination with cyclophosphamide plus doxorubicin, and gemcitabine. Hormone therapies considered part of the mix of treatments included medroxyprogesterone, megestrol, tamoxifen, anastrozole, and exemestane. The modelled population is consistent with the reimbursement request and is aligned with the GARNET trial population, a multi-centre, open-label, phase I dose-escalation study involving patients with dMMR or MSI-H recurrent or advanced EC who have progressed on or after prior treatment with a platinum-containing regimen.1
Dostarlimab is supplied in single-use vials at a submitted price of $10,270.00 per injectable 10 mL vial (50 mg/mL). The recommended dosage for dostarlimab is 500 mg every 3 weeks for doses 1 to 4, followed by 1,000 mg every 6 weeks from dose 5 until disease progression or unacceptable toxicity. The dosages of all comparator treatments, including those in the CMT, were based on Cancer Care Ontario regimen monographs for doxorubicin (60 mg/m2 on day 1 and then every 21 days), for PLD (50 mg/m2 on day 1 and then every 28 days), for carboplatin plus paclitaxel (400 mg/m2 carboplatin on day 1 and then every 28 days, and 175 mg/m2 paclitaxel on day 1 and then every 21 days), for carboplatin (400 mg/m2 on day 1 and then every 28 days), and for paclitaxel (175 mg/m2 on day 1 and then every 28 days).1 The dosage regimen for carboplatin plus PLD was 400 mg/m2 carboplatin and 50 mg/m2 PLD on day 1 and then every 28 days based on Julius et al. (2013).2 The sponsor’s calculated cost (including administration costs and wastage) of dostarlimab is $10,270 for cycles 1 to 4 and $10,270 for subsequent cycles. Using similar assumptions, the sponsor estimated per-cycle costs for each comparator to be doxorubicin = $554.50, PLD = $2,495.5, carboplatin = $738.75, paclitaxel = $4,040.00, carboplatin and paclitaxel = $4,778.75, carboplatin and PLD = $3,234.26, CMT (chemotherapies) = $2,668.67, and CMT (hormone therapies) = $14.55.1
The clinical outcomes of interest were QALYs and LYs. The economic analysis was undertaken over a lifetime horizon (40 years) from the perspective of a publicly funded health care payer. Costs and outcomes were discounted at a rate of 1.5% annually.
Model Structure
A PSM was submitted to capture the long-term costs and effects associated with the natural history of recurrent or advanced dMMR or MSI-H EC over the model time horizon. The model consisted of 3 primary health states: PFS, progressive disease, and death.1 The proportion of patients in each health state at any time over the model’s time horizon was derived from nonmutually exclusive Kaplan-Meier (KM) survival curves. The modelled time cycle was 3 weeks. OS and PFS curves were derived from the GARNET trial for dostarlimab, and were used to determine the proportion of patients in each health state (Appendix 3, Figure 1).1 Specifically, all patients entered the model in the progression-free state. The proportion of progression-free patients was derived from the area under the PFS curves, whereas the proportion of patients with progressed disease was derived from the difference in the area under the curve between the OS and PFS curves.1 Progression in the GARNET trial was defined as time from the first dose to the earliest date of assessment of disease progression or death by any cause, according to the assessment of response by a blind independent central review that used Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1).1,3 OS was defined as the time from the first dose of study treatment to the date of death by any cause. Time to treatment discontinuation (TTD) was calculated using the GARNET trial to identify the proportion of patients who were alive and who remained on treatment for the full duration of the treatment cycle after the last recorded exposure. TTD accounted for treatment discontinuation due to death or cessation of treatment.1
Model Inputs
The patient cohort comprised patients with dMMR or MSI-H advanced (stage ≥ IIIB) or recurrent ECs from cohort A1 of the GARNET study who had been previously treated with platinum-based chemotherapy.3 The modelled population was comprised mainly of adult women, with an average age of 63 years, with a mean weight of 75.8 kg, and body surface area of 1.8 m2, based on the intention-to-treat population from the GARNET trial.1
Key clinical efficacy inputs (i.e., OS and PFS) and treatment duration (i.e., TTD) for dostarlimab were based on the results of the GARNET trial (i.e., data cut-off March 11, 2020). KM estimates of PFS, OS, and TTD from the trial period (median follow-up = 11.2 months) were used to fit parametric survival curves to extrapolate the treatment effect beyond the observed trial data (median follow-up = 16.3 months) for dostarlimab at the data cut-off (i.e., date of final analysis) over the model’s time horizon (40 years).1,3 Several parametric survival functions were fitted to the PFS, OS, and TTD data to determine the best fitting distribution based on diagnostic plots, goodness-of-fit statistics, visual inspection, and clinical expectations regarding long-term progression risk and survival. The chosen parametric survival distribution of PFS and of OS for dostarlimab was the generalized gamma distribution. TTD data for dostarlimab were obtained from the GARNET trial, and the parametric survival distribution chosen to extrapolate TTD for dostarlimab over the lifetime was the 1-knot spline distribution.1
Estimates of OS for the CMT and all other individual chemotherapies were based on individual patient data from the full real-world evidence (RWE) patient cohort derived from the National Cancer Registration and Analysis Service (NCRAS) database in the UK, whereas PFS and TTD for all comparators were based on pseudo individual patient data from the same cohort digitized from the KM curves.1 Specifically, PFS was modelled on the proxy outcome of time to next treatment.1 The sponsor used 2 approaches to derive the comparative efficacy of dostarlimab against each comparator treatment, either by fitting parametric survival distributions to KM data, where possible, or by applying hazard ratios — derived from naive comparisons, matching-adjusted indirect comparisons (MAICs), or inverse probability treatment weighting (IPTW) analyses — to the estimated survival distributions of each corresponding outcome for dostarlimab (i.e., PFS, OS, and TTD).1
The parametric survival distribution chosen to extrapolate PFS for PLD was log-logistic, whereas the OS for PLD was estimated by deriving a hazard ratio from an MAIC via KM OS data in the UK RWE cohort and applying it to the parametric survival distribution for dostarlimab from the GARNET trial.1 The comparative efficacy (i.e., OS) of doxorubicin versus dostarlimab was assumed to be the same as for dostarlimab versus PLD. Parametric survival distributions were fitted to the KM curves for OS and PFS for carboplatin plus paclitaxel and for carboplatin plus PLD, respectively, with the log-logistic distribution as the chosen extrapolation. Similarly, the log-logistic distribution was the chosen parametric survival distribution fitted to the KM curves for PFS for doxorubicin, PLD, and CMT, whereas the Weibull distribution and the exponential distributions were chosen for carboplatin and for paclitaxel, respectively.1 OS rates for CMT, carboplatin, and paclitaxel were estimated by applying a hazard ratio for each treatment compared with dostarlimab to the projected OS distribution for dostarlimab, under the assumption of proportional hazards, based on the RWE cohort data.
Health state utility values were estimated with a regression model that adjusted for baseline utility and progression status using HRQoL data collected in the GARNET study with the EQ-5D-5-Levels (EQ-5D-5L) questionnaire and EQ-5D-5L values from the Canadian population. The sponsor incorporated utility values in the base case that differed by health state.1 The utility weight assigned to the PFS health state (0.784) was greater than the utility weight assigned to the progressed disease health state (0.740), and the same utility weights were applied for all treatments.1 Age- and sex-specific utility decrements were further applied to model the decline in HRQoL at each age and were based on EQ-5D utility scores for adults in the general population of England.4 To model the utility impacts of severe treatment-related adverse events (grade 3 or higher), the sponsor applied 1-off, treatment-specific disutilities, which were derived from several National Institute of Health Care Excellence gynecological cancer appraisals.5-7
The model included costs related to drug-acquisition costs, drug-administration and dispensing fees, dMMR and MSI-H EC screening costs, adverse events, subsequent therapy, surgery costs, resource use for each health state, and terminal care. Drug-acquisition costs (including subsequent therapy) for chemotherapy treatments, as well as exemestane, medroxyprogesterone, and anastrozole, were obtained from the IQVIA database, whereas those for tamoxifen and megestrol were obtained from the Ontario Drug Benefit Formulary.1,8 Drug-acquisition costs associated with CMT were weighted by the distribution of patients on these therapies in the UK RWE cohort from the NCRAS database for chemotherapy treatments and health care claims data from a Canadian study for hormone treatments within the mix.1 Drug-administration costs were estimated based on the mean time required by health care personnel (pharmacist, nurse, and physician) to administer chemotherapy per patient visit, or infusion chair time. Costs associated with screening for dMMR and MSI-H were obtained from a costing study of genomic profiling of patients with non–small cell lung cancer.9 Costs associated with adverse events were obtained from the Ontario Ministry of Health and Long-Term Care costing tool.10
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented below.
Base-Case Results
The sponsor’s base-case results were calculated as pairwise comparisons and as sequential analyses. In the sponsor’s sequential analyses, the CMT comparator was excluded. The sponsor submitted pairwise comparisons between dostarlimab and each individual comparator treatment. CADTH has presented the pairwise comparison for dostarlimab versus carboplatin plus paclitaxel, as it is the treatment with the highest number of observed events (i.e., least uncertainty) and is the treatment option that is next on the cost-effectiveness frontier (i.e., smallest number of incremental QALYs). All submitted pairwise comparisons are presented in Appendix 3.
In the sponsor’s base-case results of the pairwise comparisons between dostarlimab and individual comparators, incremental costs associated with dostarlimab ranged from $804,503 (compared with carboplatin plus paclitaxel) to $829,598 (compared with doxorubicin). Incremental QALYs gained ranged from 4.79 for dostarlimab versus carboplatin to 5.93 for dostarlimab versus doxorubicin or PLD. The incremental cost-effectiveness ratio (ICER) for dostarlimab ranged from $138,486 per QALY gained versus PLD to $171,989 per QALY gained versus carboplatin. Compared with CMT, incremental QALYs was 5.12 for dostarlimab and incremental costs were $816,233, with an ICER of $159,352 per QALY gained.
Results were driven by OS projections that predicted substantial differences in total LYs and increased drug-acquisition costs associated with dostarlimab. The sponsor’s submitted ICERs ranged from $138,000 to $172,000 (details in Appendix 3). Based on the sponsor’s base case, approximately 89% of the incremental benefit for dostarlimab was derived from the extrapolated period (beyond the single-arm trial period).
Table 3: Summary of the Sponsor’s Economic Evaluation Results (Dostarlimab Versus Carboplatin + Paclitaxel)
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. carboplatin + paclitaxel ($/QALY) |
|---|---|---|---|---|---|---|---|
Carboplatin + paclitaxel | 61,360 | Ref. | 2.12 | Ref. | 1.52 | Reference | Reference |
Dostarlimab | 865,863 | 804,503 | 8.89 | 6.77 | 6.42 | 4.90 | 164,193 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario and sensitivity analyses, which included various discount rates (0% and 3%); the adoption of alternative parametric curves for the extrapolation of OS (exponential and lognormal) and PFS (1-knot spline and 2-knot spline) for dostarlimab; the assumption that the OS efficacy of dostarlimab after treatment waning is equal to that of CMT starting 5 years after treatment initiation out to 10 years; the application of alternative data sources for OS hazard ratios of comparator therapies (i.e., Flatiron Health database for CMT1; the ZoptEC clinical trial for doxorubicin11; the McMeekin et al. [2015]12 study of paclitaxel and docetaxel; and the Mazgani et al. [2008]13 study of carboplatin plus paclitaxel); removal of the cap for the number of treatment cycles for chemotherapies; exclusion of age-related disutilities; and exclusion of IV drug administration costs.
Several scenarios resulted in notable increases in the ICER, including the adoption of alternative parametric curves for the extrapolation of OS and PFS. Notably, the sponsor’s model was highly sensitive to the use of the exponential parametric distributions for OS for dostarlimab (resulting in an ICER that ranged from $218,206 per QALY gained versus PLD to $450,209 per QALY gained versus carboplatin plus paclitaxel), and the use of the lognormal parametric distribution, which resulted in an ICER that ranged from $199,122 per QALY gained versus PLD to $279,935 per QALY gained versus carboplatin plus paclitaxel.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The comparative efficacy and safety of dostarlimab to relevant comparators is highly uncertain: A key limitation of the clinical efficacy (i.e., OS and PFS) data informing dostarlimab in the economic model is that it is based on the GARNET study, a nonrandomized, open-label, single-arm study without a comparator group. The primary objective of phase I (nonrandomized) studies is to document safety outcomes rather than efficacy. In the GARNET study, there were a limited number of patients in the efficacy dataset, and the interpretation of OS with dostarlimab is limited, owing to immature OS data and the short duration of follow-up. Additionally, the results for HRQoL and symptom-severity exploratory outcomes remained inconclusive, due to a number of important limitations, as identified by the CADTH clinical review. In the absence of comparative clinical evidence from the GARNET study to inform the efficacy (i.e., OS and PFS) and safety of dostarlimab, compared with comparator treatments, in the submitted model, and considering the lack of a common comparator, the sponsor undertook 6 indirect treatment comparisons (ITCs) — 3 MAIC reports and 3 IPTW reports — to assess the efficacy of dostarlimab compared with individual monotherapy and combination chemotherapy regimens. CADTH’s review of the clinical evidence identified a number of methodological limitations to the sponsor’s MAICs and IPTW analyses, which add considerable uncertainty to the cost-effectiveness analysis. Clinical experts consulted by CADTH indicated that in the absence of robust comparative data on PFS and OS, no firm conclusions could be drawn about how dostarlimab compares with other relevant treatment options.
Given the absence of direct evidence and the many limitations of the sponsor’s multiple ITCs, the cost-effectiveness of dostarlimab, compared with single-agent and compared with combination chemotherapies, is highly uncertain. The comparative effectiveness of dostarlimab versus relevant comparators remains highly uncertain.
The patient population in the comparator group was not matched for dMMR or MSI-H status and did not reflect the indicated population: The sponsor derived data on the comparator group from patient-level health data available from NCRAS, a synthetic real-world cohort of patients with advanced or recurrent EC in the UK. Although patients in cohort A1 in the GARNET trial included those with dMMR or MSI-H EC, per the indication, information on dMMR or MSI-H status was not available in the real-world cohort database. According to the clinical experts consulted by CADTH, dMMR or MSI-H status is a prognostic factor and strongly influences clinical management. This limitation in the sponsor’s submitted evidence means that the efficacy of dostarlimab, compared with all comparator treatments, is unknown for a dMMR and MSI-H population.
CADTH could not address this misalignment of the patient population through reanalysis. The cost-effectiveness of dostarlimab relative to all comparator treatments is unknown.
Model structure suggests a post-progression survival benefit: Results from the sponsor’s model suggest that dostarlimab is associated with longer survival after disease progression. Specifically, the sponsor’s results suggest a post-progression survival benefit for patients receiving dostarlimab, relative to carboplatin plus paclitaxel (the next-nearest comparator), such that roughly 42% of the incremental survival (2.84 LYs) would occur after patients have experienced disease progression and have discontinued dostarlimab (Table 13). The CADTH clinical review team noted that there is no evidence of a clear mechanism by which dostarlimab would continue to provide clinical benefit for patients with progressive disease, given that the GARNET study was a single-arm study and, therefore, a treatment effect between groups could not be assessed. The sponsor’s use of a PSM introduces structural assumptions about the relationship between PFS and OS that likely do not accurately reflect causal relationships within the disease pathway. These assumptions may produce a post-relapse survival bias that favours dostarlimab. Because of the structural independence between OS and PFS end points assumed in a PSM, extrapolations for each end point may reflect within-trial trends in the rates of progression and death. Last, the clinical experts consulted by CADTH observed that the results for carboplatin monotherapy yielded higher survival rates in this patient population than carboplatin plus docetaxel. This finding was considered implausible. Accordingly, CADTH omitted this finding from its analysis and chose the combination therapy as the comparator closest on the frontier to dostarlimab. For this reason, CADTH presented pairwise comparisons between dostarlimab and carboplatin plus paclitaxel.
CADTH asked the sponsor to provide additional evidence to support the implied post-progression benefit (2.84 incremental LYs and 1.98 incremental QALYs, compared with carboplatin plus paclitaxel); however, the sponsor did not provide any clinical evidence from the GARNET study to substantiate this claim. Rather, the sponsor asserted that RWE data suggested a post-progression benefit from dostarlimab would be possible because the proportion of patients likely to receive platinum-based therapy in the third-line would be higher than the proportion likely to receive currently available treatment options in the second-line; because a higher proportion of patients receiving dostarlimab in the model could receive active therapy after discontinuation; and because OS and PFS extrapolations applied in the sponsor’s base case represented the mechanism of action of a post-progression benefit for dostarlimab over time. The CADTH clinical review of the sponsor’s submitted evidence did not find evidence to support this explanation. The clinical experts consulted by CADTH noted that the sponsor’s chosen comparators did not match expected clinical practice and, therefore, did not support this explanation of the post-progression survival difference observed in the sponsor’s model. CADTH was unable to determine the extent to which the implied post-progression benefit was due to the effect of treatment with dostarlimab, structural bias in the PSM, or limitations in the comparator efficacy evidence.
Long-term extrapolations of the clinical efficacy (OS and PFS) of dostarlimab are likely overestimated: The sponsor fitted several parametric survival curves to extrapolate OS and PFS for patients who received dostarlimab over the lifetime time horizon (40 years), based on the observed period of the GARNET trial (median duration of follow-up = 16.3 months). According to the clinical experts consulted by CADTH, the estimates of OS and PFS beyond the observed period used in the GARNET study were unrealistically high and did not align with the anticipated prognosis for this patient population. The CADTH clinical review did not find evidence in the GARNET study to support the sponsor’s survival extrapolations.
The sponsor also fitted parametric survival curves for OS, PFS, and TTD for each individual comparator based on the UK RWE cohort data. The clinical experts consulted by CADTH indicated that the extrapolated OS and PFS estimates were overestimated and did not align with the natural history of Canadian patients seen in clinical practice. Based on the second-line therapies commonly used in Canadian clinical practice, the experts indicated that approximately 60% to 70% of patients would likely remain alive at 6 months, 30% at 12 months, and 10% or so at 24 months, followed by a rapid decline in OS and PFS.
Incremental QALYs for all comparators are likely overestimated as a result of this limitation. CADTH explored alternative assumptions in exploratory analyses (refer to Appendix 4).
The sponsor’s choice of comparators may not reflect current standard-of-care treatments offered in Canadian clinical practice: The sponsor excluded several chemotherapies (monotherapies and combined treatment regimens) that were identified to be relevant comparators for adults with recurrent or advanced dMMR or MSI-H EC who have progressed on or after prior treatment with a platinum-containing regimen (i.e., pembrolizumab, cisplatin, carboplatin plus docetaxel, carboplatin plus doxorubicin, or cisplatin plus doxorubicin), according to the clinical experts consulted by CADTH. Among the comparator treatments that were included, the clinical experts consulted by CADTH noted that several did not reflect Canadian clinical practice. Of note, the experts stated that hormonal therapy is rarely used in the indicated population and, importantly, there is no true standard of care for second-line therapies.
Additionally, the frequency of use of certain treatments as part of the standard of care in the second-line setting for the comparator group did not reflect Canadian clinical practice, particularly the distribution of patients who received a platinum doublet as a second-line therapy, such as carboplatin plus paclitaxel. The clinical experts consulted by CADTH noted that patients with dMMR or MSI-H disease are typically not sensitive to platinum-containing therapies and are therefore not eligible for re-treatment with platinum-containing regimens after progression. Accordingly, the frequency of use in the sponsor’s submission was not reflective of clinical practice for the indicated population. The clinical experts consulted by CADTH suggested that paclitaxel or liposomal doxorubicin would be the most frequently used comparators.
CADTH explored alternative assumptions in an exploratory analysis (refer to Appendix 4).
Patients who received dostarlimab were assumed to remain on treatment indefinitely over the lifetime time horizon, and the duration of treatment effect was assumed to last for a patient’s lifetime, without any treatment waning: In the sponsor’s base case, the sponsor did not apply a stopping rule or any treatment discontinuation criteria to patients who received dostarlimab. As such, patients were assumed to continue receiving dostarlimab indefinitely over the lifetime time horizon (40 years). Additionally, the sponsor assumed that the duration of treatment effect for dostarlimab would last for the patient’s lifetime (i.e., no treatment waning). Each of these assumptions were noted to be unlikely by the clinical experts consulted by CADTH. First, the clinical experts consulted by CADTH noted that 50% to 60% of patients were likely to discontinue treatment after 2 years, and the majority (approximately 90%) would likely be off treatment by 5 years. Second, CADTH’s clinical experts indicated that treatment-waning effects were likely to occur approximately 2 years after treatment initiation, based on their clinical experience with patients receiving immunotherapy in Canadian clinical practice.
CADTH explored alternative assumptions in an exploratory analysis (refer to Appendix 4).
Health state utility values for the progressed disease health state lacked face validity: In the economic model, health state utility values were estimated with EQ-5D-5L data collected from patients in the GARNET study. Based on the sponsor’s methodology, utility values for patients in the progressed disease health state (0.74) were similar to those for patients in the progression-free health state (0.78). The clinical experts consulted by CADTH indicated that patient quality of life typically worsens with disease progression. As such, health state utility values for the progressed disease health state lack face validity and likely overestimate patients’ post-progression quality of life in favour of dostarlimab. The magnitude of bias in favour of dostarlimab remains unknown, resulting in additional uncertainty about the impact of health state utility values on the ICER.
CADTH explored alternative assumptions in an exploratory analysis (refer to Appendix 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted As Limitations in the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Baseline characteristics of patient population. | Appropriate. The clinical experts consulted by CADTH agreed that the baseline patient characteristics of cohort A1 of the GARNET trial was reflective of patients in Canadian clinical practice for the present indication. The trial population was slightly younger and healthier (all ECOG ≤ 1), but the clinical experts consulted by CADTH noted that patients with ECOG 2 would be treated the same way in practice. Furthermore, no differential treatment effects would be expected from the different disease-management practices across countries. |
A lifetime time horizon of 40 years. | Appropriate. This time horizon is adequate to capture all lifetime associated costs and outcomes for the indicated population. |
The distribution of chemotherapies comprising the CMT basket was based on the distribution of therapies in the UK RWE cohort represented by NCRAS data.1 | Inappropriate. The UK RWE patient cohort did not reflect the indicated population. |
The distribution of hormone therapies comprising the CMT was derived from a health care claims study that examined treatment patterns and clinical outcomes in advanced and recurrent EC patients in Alberta, Canada.1 | Uncertain. |
For patients receiving dostarlimab, the estimated distribution of post-progression therapies was based on second-line treatments received by patients in the UK RWE cohort. For all other comparators, the distribution of post-progression therapies was based on third-line treatments received by patients in the UK RWE cohort. Additionally, 46% of patients receiving dostarlimab were assumed to have received a subsequent therapy after the discontinuation of dostarlimab. For comparator therapies, it was assumed that 30% of patients would receive subsequent treatment.1 | Inappropriate. Given that the comparator patient population did not reflect the indicated population (i.e., not appropriately matched for dMMR or MSI-H status), the proportion of patients who received subsequent therapies after discontinuation of dostarlimab, based on RWE data, is not generalizable to the indicated population. The generalizability of these estimates, as well as the estimated distributions of therapies received after progression, were also not validated by CADTH. However, the estimated proportion of patients expected to receive subsequent therapies after progression has a minimal impact on model results. |
The probabilities of grade 3 or higher AEs were derived from the GARNET study for dostarlimab, whereas probabilities for all other comparators were assumed to be equal to AEs seen with doxorubicin in the ZoptEC phase III randomized controlled trial.11 Grade 3 or higher treatment-related AEs included in the model reflected those with an incidence ≥ 5%.1 | Likely acceptable. |
General population mortality data obtained from Statistics Canada lifetables for 2017 to 201914 were used to cap OS estimates according to age-specific survival rates, such that the sponsor assumed that OS in the patient population would not exceed that of the general population at each age.1 | Appropriate. The clinical experts consulted by CADTH indicated that the prognosis for the indicated population is poor and that OS would rapidly decline after progression, suggesting that OS would not exceed that of the general population. |
Despite which treatment was received (dostarlimab or alternative therapy), all patients were assumed to be screened for dMMR or MSI-H status. As such, there are $0 incremental costs associated with IHC screening among patients who received dostarlimab. The cost for screening dMMR and MSI-H per patient was assumed to be $133.1 | Inappropriate. The clinical experts consulted by CADTH indicated that screening for dMMR or MSI-H status is not performed in all Canadian jurisdictions; it remains unknown what proportion of patients is unscreened. Even if all patients were assumed to be unscreened, the impact of IHC screening costs likely has a negligible impact on model results. |
AEs = adverse events; CMT = current mix of treatments; dMMR = deficient mismatch repair; EC = endometrial cancer; ECOG = Eastern Cooperative Oncology Group; IHC = immunohistochemistry; MSI-H = microsatellite instability-high; NCRAS = National Cancer Registration and Analysis Service; OS = overall survival; RWE = real-world evidence.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
As noted above, there are key limitations associated with the model structure, available clinical data for dostarlimab, and the comparative efficacy of relevant comparators. The use of the PSM in the current review is inappropriate, given that PSMs rely on mature OS data to produce reliable cost-effectiveness estimates. CADTH notes that the sponsor’s model predicts improbable estimates of incremental QALYs gained after disease progression, which was not supported by trial data. Importantly, the comparator population was based on data from a UK RWE patient cohort that was not matched for dMMR or MSI-H status and, thus, does not reflect the indicated population. Additionally, several included comparators were not reflective of clinical practice in Canada, and other relevant comparators were excluded. Although the clinical experts consulted by CADTH indicated that paclitaxel is the most commonly used second-line treatment, CADTH did not report results of this pairwise comparison due to sparse data. As noted in the summary of the sponsor’s economic evaluation, CADTH selected carboplatin plus paclitaxel as the main comparator; it is the treatment with the highest number of observed events (i.e., least uncertainty) and is next on the cost-effectiveness frontier. As such, the comparative effectiveness and safety of dostarlimab to relevant comparators is highly uncertain. The result of these limitations is that the costs and QALYs associated with the use of dostarlimab are highly uncertain. CADTH reanalyses cannot address this uncertainty in the clinical evidence. Consequently, CADTH was unable to conduct any base-case reanalysis of the sponsor’s model.
Scenario Analysis Results
Because a CADTH base-case reanalysis was not performed, price-reduction analyses were conducted using only the sponsor’s base case. As such, deterministic price-reduction analyses are subject to the key limitations of the sponsor’s model, noted above, and are based on publicly available prices of the comparator treatments. Based on the sponsor’s submitted base case, a reduction of 74% in the price of dostarlimab would be required for dostarlimab to be cost-effective at a conventional threshold of $50,000 per QALY, compared with carboplatin plus paclitaxel (Appendix 4). It is important to note that this price-reduction estimate is based on estimates of incremental OS that are likely not representative of the true incremental benefit of dostarlimab, including single-arm trial results, unmatched comparator populations, and overestimated and highly uncertain survival estimates. The directionality of bias in the sponsor’s submission suggests that the price reduction would need to be higher than the estimated 74%. Further details of this exploratory analysis are provided in Appendix 4.
Although CADTH did not conduct any formal reanalyses of the sponsor’s model, an exploratory analysis was undertaken to explore the impact that changes to model assumptions would have on the ICER. The key limitations of the sponsor’s base-case analysis noted in the CADTH appraisal of the sponsor’s economic evaluation apply to this exploratory analysis, including the fundamental limitation that there is no direct evidence to support the comparative efficacy of dostarlimab to multiple relevant comparator treatments. As such, this exploratory analysis should not be interpretated as a CADTH base case, as there remains uncertainty regarding the true effect of dostarlimab. The key insight from this exploratory analysis is that the cost-effectiveness estimate of dostarlimab is highly influenced by assumptions related to OS. Details of this exploratory analysis are provided in Appendix 4.
Issues for Consideration
Pembrolizumab is indicated for this patient population; however, it is currently not funded in Canada though it has received regulatory approval. As pembrolizumab is not currently funded, its use is relatively uncommon at present, as noted by drug plans; however, its use is expected to increase quickly over the next few years among patients with private insurance. The cost-effectiveness of dostarlimab compared with pembrolizumab is unknown.
The current submission is for the use of dostarlimab monotherapy for the treatment of adults with recurrent or advanced dMMR or MSI-H EC that has progressed on or after prior treatment with a platinum-containing regimen. Based on clinical experts’ responses to drug plan feedback, there is the potential for indication creep among patients who received platinum-based therapy only in early-stage disease, those who received more than 2 lines of therapy for advanced or recurrent disease, and those who are currently receiving systemic therapy for recurrent dMMR or MSI-H (with prior platinum-based therapy). Off-label use of dostarlimab among patients with advanced or recurrent EC is likely to result in an increased budget impact.
Drug plan feedback noted that dMMR and MSI-H testing is not a standard or reflexive test across jurisdictions, The clinical experts consulted by CADTH indicated that all patients with EC of any stage should receive dMMR and MSI-H testing at the time of diagnosis, and that it is not costly and is easily performed.
The submitted economic model did not include an option to assess a weight-based dosing regimen for dostarlimab; rather, 2 fixed-dosing regimens (i.e., 500 mg every 3 weeks and 1,000 mg every 6 weeks) were considered on the basis of the Health Canada indication and the GARNET study protocol. The clinical experts consulted by CADTH indicated that dostarlimab may be administered as either a flat-rate dose or a weight-based dose, based on the maximum capped dose derived from several pharmacokinetic studies in support of weight-based dosing of immunotherapies. As dostarlimab is an immunotherapy with monoclonal antibodies similar to pembrolizumab, weight-based dosing for dostarlimab is likely to be similarly implemented in Canadian practice, despite the absence of weight-based dosing information in the product monograph and will likely have an impact on associated drug costs. The cost-effectiveness of dostarlimab’s weight-based dosing regimen, however, is unknown.
Overall Conclusions
The CADTH review of the clinical evidence found no direct comparative evidence between dostarlimab and any relevant treatment comparator in the treatment of adults with recurrent or advanced dMMR or MSI-H EC. There is indirect evidence to suggest that dostarlimab may improve PFS, compared with standard of care (chemotherapeutic regimens); however, no strong conclusions can be drawn because of the many limitations in the indirect comparisons. The sponsor’s submitted ITCs were based on data from patients whose dMMR or MSI-H status was unknown, meaning that they are not aligned with the indication for dostarlimab. Thus, the effectiveness of dostarlimab remains highly uncertain relative to any currently reimbursed treatment.
The sponsor submitted a model comparing the cost-effectiveness of dostarlimab with second-line treatments (PLD, doxorubicin, paclitaxel, CMT, carboplatin plus PLD, carboplatin plus paclitaxel, and carboplatin). According to the comparative evidence in the submitted model, carboplatin plus paclitaxel was the next most effective treatment on the efficiency frontier, with the least uncertainty around OS and PFS parameters. The sponsor’s estimate of the pairwise ICER for dostarlimab versus carboplatin plus paclitaxel was $164,193 per QALY, and the probability that dostarlimab is cost-effective was 0% at a willingness-to-pay threshold of $50,000 per QALY.
CADTH was not able to conduct a reanalysis because of foundational limitations with the sponsor’s model and submitted clinical evidence. Notably, the efficacy of comparator treatments in dMMR or MSI-H EC patients was unknown and, thus, the cost-effectiveness of dostarlimab compared with second-line treatments in the indicated population is unknown. The sponsor’s choice of a PSM produced QALY estimates that were likely overestimated. The model was highly sensitive to long-term extrapolation assumptions about OS and PFS, with a sizable majority of incremental QALYs observed in the extrapolated period (i.e., beyond the period of the trial). The sponsor’s model predicts a post-progression survival benefit for dostarlimab that is not supported by the trial evidence. CADTH was unable to address the critical limitation regarding the apparent post-progression survival benefit because of constraints introduced by the submitted model structure. Exploratory analysis found that the model is highly sensitive to several model assumptions, including long-term survival extrapolation, treatment discontinuation, and treatment-effectiveness waning after 24 months. Based on the CADTH exploratory analysis, a price reduction of 83% would be needed for dostarlimab to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY; however, this estimate is subject to limitations in the sponsor’s submission, most crucially the inappropriateness of the estimated efficacy of comparator treatments in the dMMR and MSI-H population. Additional price reduction may be warranted.
Treatment of dMMR or MSI-H EC with dostarlimab increases costs compared with other currently available treatments. CADTH could not estimate the amount of health improvement that may result from treatment with dostarlimab because of highly uncertain clinical efficacy data and a lack of information about the efficacy of comparator treatments. Because of the methodological limitations identified in the model and clinical evidence, the cost-effectiveness of dostarlimab is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dostarlimab, 500 mg/10 mL vial (50 mg/mL) intravenous infusion. Mississauga (ON): GlaxoKlineSmith; 2021 Sept.
2.Julius JM, Tanyi JL, Nogueras-Gonzalez GM, et al. Evaluation of pegylated liposomal doxorubicin dose on the adverse drug event profile and outcomes in treatment of recurrent endometrial cancer. Int J Gynecol Cancer. 2013;23(2):348-354. PubMed
3.Oaknin A, Tinker AV, Gilbert L, et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020;6(11):1766-1772. PubMed
4.Ara R, Brazier JE. Using health state utility values from the general population to approximate baselines in decision analytic models when condition-specific data are not available. Value Health. 2011;14(4):539-545. PubMed
5.National Institute for Health and Care Excellence. Niraparib for maintenance treatment of relapsed, platinum-sensitive ovarian, fallopian tube and peritoneal cancer. (Technology appraisal guidance TA528) 2018: https://www.nice.org.uk/guidance/ta528. Accessed 2022 Jan 6.
6.National Institute for Health and Care Excellence. Olaparib for maintenance treatment of BRCA mutation-positive advanced ovarian, fallopian tube or peritoneal cancer after response to first-line platinum-based chemotherapy. (Technology appraisal guidance TA598) 2019: https://www.nice.org.uk/guidance/ta598. Accessed 2022 Jan 6.
7.National Institute for Health and Care Excellence. Rucaparib for maintenance treatment of relapsed platinum-sensitive ovarian, fallopian tube or peritoneal cancer. (Technology appraisal guidance TA611) 2019: https://www.nice.org.uk/guidance/ta611. Accessed 2022 Jan 6.
8.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Dec 23.
9.Johnston KM, Sheffield BS, Yip S, Lakzadeh P, Qian C, Nam J. Costs of in-house genomic profiling and implications for economic evaluation: a case example of non-small cell lung cancer (NSCLC). J Med Econ. 2020;23(10):1123-1129. PubMed
10.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2021 Dec 23.
11.Miller DS, Scambia G, Bondarenko I, et al. ZoptEC: Phase III randomized controlled study comparing zoptarelin with doxorubicin as second line therapy for locally advanced, recurrent, or metastatic endometrial cancer (NCT01767155). J Clin Oncol. 2018;36(15_suppl):5503-5503.
12.McMeekin S, Dizon D, Barter J, et al. Phase III randomized trial of second-line ixabepilone versus paclitaxel or doxorubicin in women with advanced endometrial cancer. Gynecol Oncol. 2015;138(1):18-23. PubMed
13.Mazgani M, Le N, Hoskins PJ. Reuse of carboplatin and paclitaxel in patients with relapsed endometrial cancer--the British Columbia Cancer Agency experience. Gynecol Oncol. 2008;111(3):474-477. PubMed
14.Life tables, Canada, provinces and territories 1980/1982 to 2017/2019. Ottawa (ON): Statistics Canada; 2021: https://www150.statcan.gc.ca/n1/pub/84-537-x/84-537-x2020001-eng.htm. Accessed 2022 Jan 6.
15.Cancer Care Ontario. CRBP regimen: CARBOplatin. [no date][no date]: . Accessed 2021 Dec 23.
16.CADTH Reimbursement Review: pembrolizumab (Keytruda). Can J Health Technol 2021;1(9). https://www.cadth.ca/sites/default/files/DRR/2021/PC0235-Keytruda-Combined.pdf. Accessed 2022 Jan 6.
17.Cancer Care Ontario. CISplatin. [no date][no date]: . Accessed 2021 Dec 23.
18.Cancer Care Ontario. PACLitaxel. [no date][no date]: . Accessed 2021 Dec 23.
19.Cancer Care Ontario. DOXOrubicin. [no date][no date]: . Accessed 2021 Dec 23.
20.Cancer Care Ontario. CRBPPACL regimen: PACLitaxel-CARBOplatin. [no date][no date]: . Accessed 2021 Dec 23.
21.Cancer Care Ontario. CRBPDOCE regimen: DOCEtaxel-CARBOplatin. [no date][no date]: . Accessed 2021 Dec 23.
22.Cancer Care Ontario. CISPDOXO regimen: CISplatin-DOXOrubicin. [no date][no date]: . Accessed 2021 Dec 23.
23.DeltaPA. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Dec 23.
24.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dostarlimab, 500 mg/10 mL vial (50 mg/mL) intravenous infusion. Mississauga (ON): GlaxoKlineSmith; 2021 Sept.
25.Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics 2021. Toronto (ON): Canadian Cancer Society, Statistics Canada, Public Health Agency of Canada; 2021: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-.pdf?rev=2b9d2be7a2d34c1dab6a01c6b0a6a32d&hash=01DE85401DBF0217F8B64F2B7DF43986REF. Accessed 2022 Jan 6.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CADTH Cost Comparison Table for dMMR or MSI-H Recurrent or Advanced Endometrial Cancer That Has Progressed on or Following Prior Treatment With a Platinum-Containing Regimen
Treatment | Strength / concentration | Form | Price | Recommended dosage | Average daily cost ($) | 28-day cost ($) |
|---|---|---|---|---|---|---|
Dostarlimab | 500 mg/10 mL vial (50 mg/mL) | Vial | 10,270.0000a | Initial: 500 mg every 3 weeks (doses 1 to 4); Subsequent: 1,000 mg every 6 weeks beginning 3 weeks after dose 4 (dose 5 onwards) | 487.71 | 13,655 |
Pembrolizumab | 100 mg | Vial | 4,400.0000b | 200 mg every 3 weeks | 419.05 | 11,733 |
Single-agent chemotherapies | ||||||
Carboplatin | 50 mg | Vial | 70.0000 | AUC 5 to 6 (750 to 900 mg), day 1, every 21 days, total of 6 to 8 cycles c | 46.90 to 56.90 | 1,313 to 1,593 |
150 mg | 210.0000 | |||||
450 mg | 599.9985 | |||||
600 mg | 775.00 | |||||
Cisplatin | 1 mg/mL (50 mL) | Vial | 323.0000 (6.46 per mL) | Day 1: 50 to 75 mg/m2 (or days 1 to 5: 15 to 20 mg/m2) Every 3 to 4 weeksd | 38.57 | 1,080 |
1 mg/mL (100 mL) | 270.0000 (2.70 per mL) | |||||
Paclitaxel | 6 mg/mL (5 mL in 5 mL) [30 mg] | Vial | 300.0000 (60.0 per mL) | 175 mg/m2 over 3 hours every 3 weekse | 157.14 | 4,400 |
6 mg/mL (50 mL in 50 mL) [300 mg] | 3,740.0000 (74.8 per mL) | |||||
Doxorubicin | 2 mg/mL (10 mL) [20 mg] | Vial | 665.4700 782.9100 | Q1W: 10 to 20 mg/m2 bolus Q3W: 60 to 75 mg/m2 bolus (40 to 60 mg/m2 when used in combination) Q4W: 20 to 30 mg/m2/day bolus for 3 consecutive daysf | 3.33 to 10.00 | 93 to 280 |
2 mg/mL (25 mL) [50 mg] | 252.2500 255.0000 285.0000 360.37000 | |||||
10 mg/mL (5 mL) [50 mg] | 70.0000 106.1200 | |||||
100 mg | 1,304.2000 | |||||
150 mg | 1,081.000 | |||||
200 mg | 770.0000 973.0000 | |||||
Platinum-based chemotherapies | ||||||
Carboplatin + Paclitaxelg | ||||||
Carboplatin | 50 mg | Vial | 70.0000 | AUC 4 to 6 (600 to 900 mg) | 36.90 to 56.90 | 1,033 to 1,593 |
150 mg | 210.0000 | |||||
450 mg | 599.9985 | |||||
600 mg | 840.0000 | |||||
Paclitaxel | 6 mg/mL (5 mL in 5 mL) [30 mg] | Vial | 300.0000 (60.0 per mL) | 175 mg/m2 on day 1 | 157.14 | 4,400 |
6 mg/mL (50 mL in 50 mL) [300 mg] | 3,740.0000 (74.8 per mL) | |||||
Carboplatin + Paclitaxel | Every 21 days for 6 cycles max | 194.04 to 214.04 | 5,433 to 5,993 | |||
Carboplatin + Docetaxelh | ||||||
Carboplatin | 50 mg | Vial | 70.0000 | AUC 5 to 6 (750 to 900 mg) | 46.90 to 56.90 | 1,313 to 1,593 |
150 mg | 210.0000 | |||||
450 mg | 599.9985 | |||||
600 mg | 840.0000 | |||||
Docetaxel | 20 mg | Vial | 249.0000 | 75 mg/m2 | 59.24 | 1,659 |
80 mg | 497.0000 925.000 | |||||
160 mg | 990.0000 1940.4000 1,850.0000 | |||||
Carboplatin + Docetaxel | Every 21 days, for 6 cycles | 106.14 to 116.14 | 2,972 to 3,252 | |||
Cisplatin + Doxorubicin i | ||||||
Cisplatin | 1 mg/mL (50 mL) [50 mg] | Vial | 323.0000 (6.4600 per mL) | 50 mg/m2 on day 1 | 12.86 | 360 |
1 mg/mL (100 mL) [100 mg] | 270.0000 (2.7000 per mL) | |||||
Doxorubicin | 2 mg/mL (10 mL) [20 mg] | Vial | 665.4700 782.9100 | 50 mg/m2 on day 1 | 6.67 | 187 |
2 mg/mL (25 mL) [50 mg] | 252.2500 255.0000 285.0000 360.37000 | |||||
10 mg/mL (5 mL) [50 mg] | 70.0000 106.1200 | |||||
100 mg | 1,304.2000 | |||||
150 mg | 1,081.000 | |||||
200 mg | 770.0000 973.0000 | |||||
Cisplatin + Doxorubicin | Every 21 days, for a total of 6 cycles | 19.53 | 547 | |||
Hormone therapies | ||||||
Medroxyprogesterone | 2.5 mg | Tablet | 0.1183 | 10 mg daily, for 12 to 14 days | 0.17 | 5 |
5 mg | 0.2365 0.4730 | |||||
10 mg | 0.1670 | |||||
100 mg | 1.2057j | |||||
150 mg | 31.6900 | |||||
Megestrol | 40 mg | Tablet | 1.3340 | 80 to 320 mg or 125 mg/m2 daily in divided doses | 2.67 to 5.34 | 75 to 150 |
160 mg | 5.8151 | |||||
Tamoxifen | 10 mg | Tablet | 0.1750 | 20 to 40 mg daily in single or divided doses | 0.35 to 0.70 | 10 to 20 |
20 mg | 0.3500 | |||||
Letrozole (Generic) | 2.5 mg | Tablet | 1.3780 | 2.5 mg daily | 1.38 | 39 |
Exemestane (Generic) | 25 mg | Tablet | 1.3263 5.7533 | 25 mg daily | 1.33 | 37 |
Note: Prices of chemotherapies were obtained from the DeltaPA IQVIA database (accessed November 3, 2021), and the prices of hormone therapies were obtained from the Ontario Drug Benefit Formulary, unless otherwise indicated, and do not include dispensing fees. Cost calculations assume a body surface area of 1.80 m2 where applicable. For all target AUC calculations, dose calculations followed guidance from the Cancer Care Ontario product monograph for carboplatin: Target AUC is 4 to 6. Carboplatin is dosed according to the following formula: Maximum carboplatin dose (mg) = target AUC (mg/mL per min) × (125 + 25); maximum dose is based on a capped GFR estimate at 125 mL/min for patients with normal renal function.15
aSponsor’s submitted price for dostarlimab.1
bPembrolizumab pricing obtained from CADTH CDR pharmacoeconomic report.16
cCarboplatin product monograph.15
dCisplatin product monograph.17
ePaclitaxel product monograph.18
fDoxorubicin.19
gCarboplatin + paclitaxel product monograph.20
hCarboplatin + docetaxel product monograph.21
ICisplatin + doxorubicin product monograph.22
jPricing for medroxyprogesterone obtained from the DeltaPA database.23
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to limitations for the sponsor’s inappropriate comparator population. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | No | Refer to key limitations for the sponsor’s inappropriate use of partitioned survival modelling approach. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Sponsor’s Estimates of Long-Term Progression-Free Survival for Dostarlimab
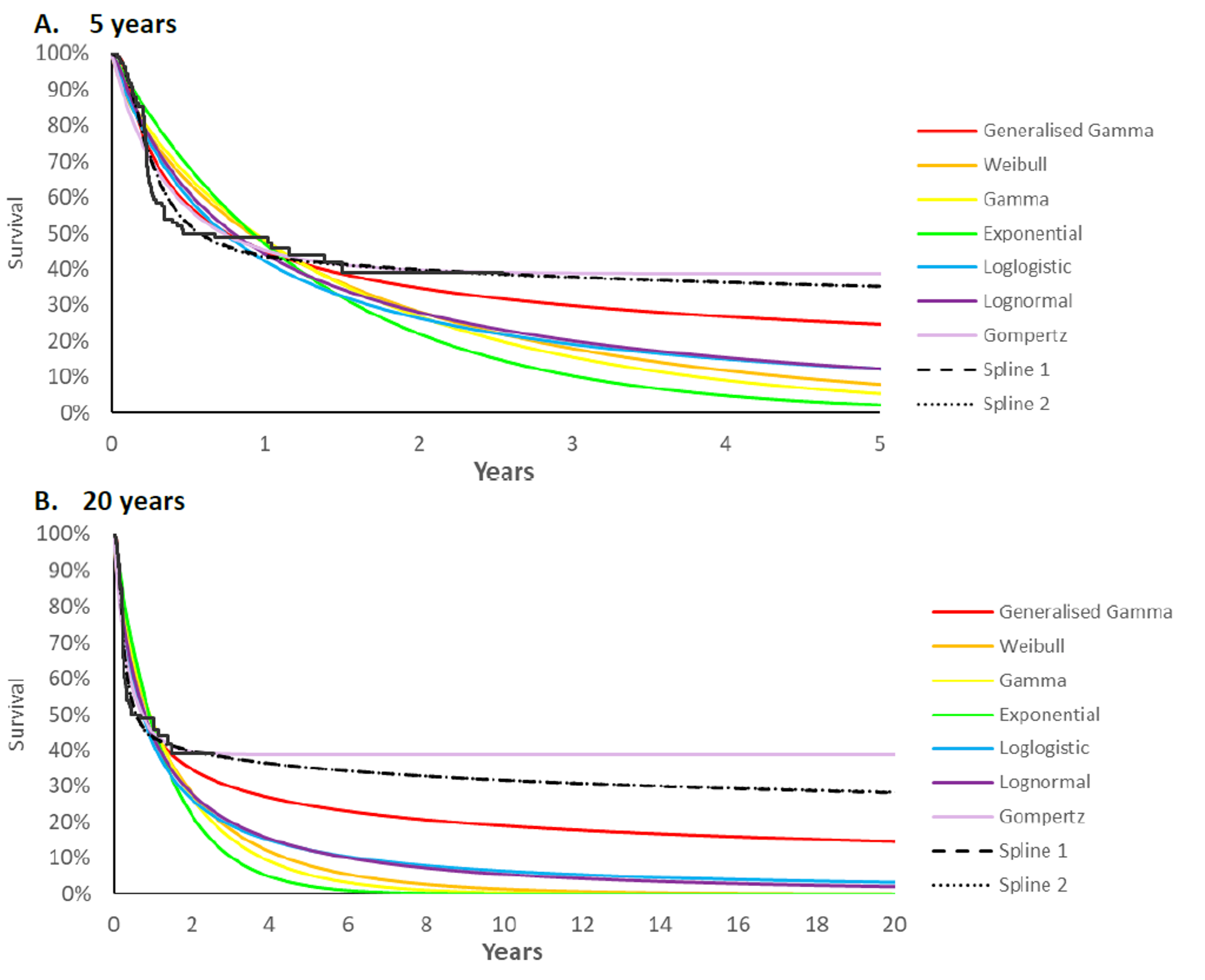
Note: the generalized gamma distribution was chosen in the sponsor’s base case.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 2: Sponsor’s Estimates of Long-Term Overall Survival for Dostarlimab
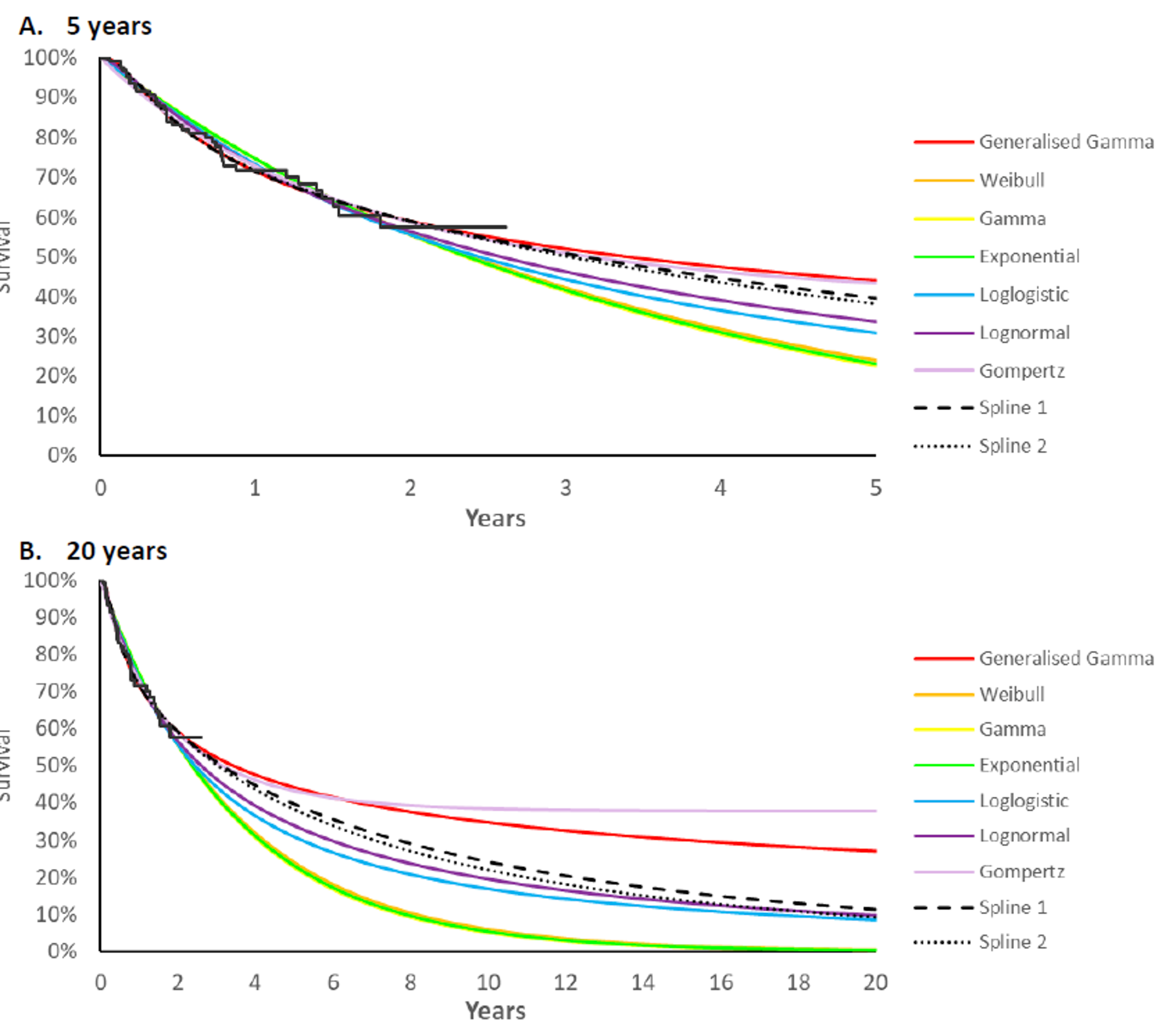
Note: the generalized gamma distribution was chosen in the sponsor’s base case.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
While the sponsor included multiple comparator treatments to assess the cost-effectiveness of dostarlimab, only the ICER for dostarlimab versus carboplatin + paclitaxel is presented below. The carboplatin + paclitaxel subgroup had the largest number of observations in the RWE survival cohort evidence used within the model. Accordingly, there is the least amount of parametric uncertainty for this comparator. Carboplatin + paclitaxel also had the highest comparative efficacy to dostarlimab (i.e., next-highest QALYs) relative to other subgroups. The true efficacy of carboplatin + paclitaxel within the indicated population remains unknown.
Table 7: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Dostarlimab | Carboplatin + paclitaxela | Incremental (vs. carboplatin + paclitaxel) |
|---|---|---|---|
Discounted LYs | |||
Total | 8.89 | 2.12 | 6.77 |
Progression-free survival | 5.18 | 1.25 | 3.93 |
Progressed disease | 3.71 | 0.87 | 2.84 |
Discounted QALYs | |||
Total | 6.42 | 1.52 | 4.62 |
Progression-free survival | 3.82 | 0.98 | 2.90 |
Progressed disease | 2.61 | 0.63 | 1.64 |
Adverse events | –0.01 | –0.08 | 0.08 |
Discounted costs ($) | |||
Total | 865,863 | 61,360 | 804,503 |
Study treatment and administration costs | 802,538 | 24,090 | 778,448 |
Routine monitoring costs | 36,018 | 7,371 | 28,647 |
Subsequent treatment costs | 5,740 | 3,936 | 1,804 |
End of life costs | 21,484 | 24,046 | –2,562 |
Adverse event costs | 81 | 1,916 | –1,835 |
ICER ($/QALY) | 164,193 | ||
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; vs. = versus.
aReference product is the next most efficacious comparator based on the results of sponsor’s submitted indirect treatment comparison, and the best survival evidence from the RWE data.
Detailed Results of the Sponsor’s Economic Evaluation
Table 8: Summary of the Sponsor’s Economic Evaluation Results (Dostarlimab Versus Pairwise Doxorubicin)
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. Doxorubicin ($/QALY) |
|---|---|---|---|---|---|---|---|
Doxorubicin | 36,264 | Reference | 0.76 | Ref. | 0.50 | Reference | Reference |
Dostarlimab | 865,863 | 829,598 | 8.89 | 8.12 | 6.42 | 5.93 | 139,936 |
CMT = current mix of treatments; ICER = incremental cost-effectiveness ratio; LY = life-year; PLD = pegylated liposomal doxorubicin; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Table 9: Summary of the Sponsor’s Economic Evaluation Results (Dostarlimab Versus PLD)
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. PLD |
|---|---|---|---|---|---|---|---|
PLD | 44,178 | Reference | 0.75 | Reference | 0.49 | Reference | Reference |
Dostarlimab | 865,863 | 821,685 | 8.89 | 8.13 | 6.42 | 5.93 | 138,486 |
ICER = incremental cost-effectiveness ratio; LY = life-year; PLD = pegylated liposomal doxorubicin; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Table 10: Summary of the Sponsor’s Economic Evaluation Results (Dostarlimab Versus CMT)
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. CMT |
|---|---|---|---|---|---|---|---|
CMT | 49,629 | Reference | 1.86 | Ref. | 1.30 | Reference | Reference |
Dostarlimab | 865,863 | 816,233 | 8.89 | 8.13 | 6.42 | 5.93 | 159,352 |
CMT = Current mix of treatments; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus
Source: Sponsor’s pharmacoeconomic submission.1
Table 11: Summary of the Sponsor’s Economic Evaluation Results (Dostarlimab Versus Carboplatin)
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. carboplatin |
|---|---|---|---|---|---|---|---|
Carboplatin | 41,856 | Reference | 2.35 | Ref. | 1.63 | Reference | Reference |
Dostarlimab | 865,863 | 824,006 | 8.89 | 6.54 | 6.42 | 4.79 | 171,989 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus
Source: Sponsor’s pharmacoeconomic submission.1
Table 12: Summary of the Sponsor’s Economic Evaluation Results (Dostarlimab Versus Paclitaxel)
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. paclitaxel |
|---|---|---|---|---|---|---|---|
Paclitaxel | 53,796 | Reference | 1.34 | Ref. | 0.92 | Reference | Reference |
Dostarlimab | 865,863 | 812,067 | 8.89 | 7.54 | 6.42 | 5.51 | 147,467 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus
Source: Sponsor’s pharmacoeconomic submission.1
Table 13: Summary of the Sponsor’s Economic Evaluation Results (Dostarlimab Versus Carboplatin + PLD)
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. carboplatin + PLD ($/QALY) |
|---|---|---|---|---|---|---|---|
Carboplatin + PLD | 54,484 | Reference | 1.37 | Ref. | 1.91 | Reference | Reference |
Dostarlimab | 865,863 | 811,379 | 8.89 | 6.98 | 6.42 | 5.05 | 160,664 |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Scenario Analyses
While CADTH did not conduct any formal reanalyses of the sponsor’s model, the economic review team performed an exploratory analysis to explore the impact of several key limitations on the ICER which included applying an alternate parametric survival distribution for OS; applying an alternate parametric survival distribution for PFS; applying a treatment stopping rule at 2 years with 60% of patients stopping treatment; applying a treatment-waning effect starting at 24 months; and revising the pricing of carboplatin.
Of note, the fundamental limitations in the sponsor’s model persist within this exploratory analysis. There is no direct evidence to support the comparative efficacy of dostarlimab to multiple relevant comparator treatments. Therefore, this exploratory analysis should not be interpreted as a formal CADTH reanalysis to which credence should be given to the results; in particular, the incremental QALY benefit estimated as part of this exploratory analysis remains unlikely to be representative of the true effect of dostarlimab, such that the corresponding ICER is unlikely to be reflective of the true cost-effectiveness of dostarlimab. Instead, the key insight from this exploratory analysis is that the cost-effectiveness estimate of dostarlimab is highly influenced by the uncertainty in the OS and PFS data, and treatment-waning effects applied at 24 months.
Despite the many limitations associated with the sponsor’s submitted ITCs, and that carboplatin + paclitaxel is unlikely to be used in the second-line setting for the indicated population according to the clinical experts consulted by CADTH, carboplatin + paclitaxel was selected as the comparator of choice to explore the cost-effectiveness against dostarlimab in the CADTH exploratory analysis as the number of data points available within the RWE cohort for OS and PFS were greater than other treatments and it demonstrated better efficacy relative to other treatments. However, the cost-effectiveness of dostarlimab compared with all single-agent and combination chemotherapies, and hormone therapy, is highly uncertain. Due to several limitations of the KM data for OS and PFS informed by the available clinical evidence and feedback from the clinical experts consulted by CADTH pertaining to the clinical plausibility of the long-term extrapolations for OS and PFS in the sponsor’s base case, CADTH selected alternate survival distributions for OS and PFS based on clinical plausibility as part of the exploratory analysis. Additionally, CADTH applied a stopping rule at 2 years following treatment initiation with 60% of patients discontinuing treatment. CADTH further applied a treatment-waning effect beginning at year 2, such that patients who initially received dostarlimab would experience similar treatment effects as carboplatin + paclitaxel. When an alternative parametric survival distribution (Exponential) was chosen for the long-term extrapolation of the OS data, the predicted incremental gain in life-years was 82% lower than in the sponsor’s base case, resulting in lower incremental QALYs (0.98) and a higher estimated ICER ($460,362 per QALY). When an alternative parametric survival distribution (Lognormal) was chosen for the long-term extrapolation of the PFS data, the predicted incremental gain in life-years was 82% lower than in the sponsor’s base case, resulting in lower incremental QALYs (0.96) and a higher estimated ICER ($470,639 per QALY).
Table 14: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH exploratory value or assumption |
|---|---|---|
1. Extrapolation of overall survival curves for dostarlimab | OS: Generalized gamma | OS: Exponential |
2. Extrapolation of progression-free survival curves for dostarlimab | PFS: Generalized gamma | PFS: Lognormal |
3. Stopping rule | None | Assume that 60% of patients stop treatment at 2 years |
4. Treatment waning | None | Applied treatment waning, starting at 24 months |
CADTH exploratory analysis | Reanalyses 1 + 2 + 3 + 4 | |
OS = overall survival; PFS = progression-free survival.
Table 15: Summary of the Stepped Analysis of the CADTH Exploratory Analysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base casea,b | Carboplatin + paclitaxelc | 61,360 | 1.52 | Reference |
Dostarlimab | 865,863 | 6.42 | 164,193 | |
CADTH reanalysis 1d | Carboplatin + paclitaxelc | 61,388 | 1.51 | Reference |
Dostarlimab | 513,050 | 2.49 | 460,362 | |
CADTH reanalysis 2 | Carboplatin + paclitaxelc | 61,388 | 1.51 | Reference |
Dostarlimab | 867,913 | 2.12 | 179,306 | |
CADTH reanalysis 3 | Carboplatin + paclitaxelc | 61,388 | 1.51 | Reference |
Dostarlimab | 505,243 | 6.12 | 96,172 | |
CADTH reanalysis 4 | Carboplatin + paclitaxelc | 61,388 | 1.51 | Reference |
Dostarlimab | 671,529 | 3.33 | 333,955 | |
CADTH exploratory analysisa,d | Carboplatin + paclitaxelc | 61,698 | 1.54 | Reference |
Dostarlimab | 436,486 | 2.65 | $336,627 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aThe sponsor’s and CADTH base case results are probabilistic while remaining analyses are deterministically presented.
bReference product is the next most efficacious comparator based on the results of sponsor’s submitted indirect treatment comparison, and the best survival evidence from the RWE data.
cSponsor’s submitted base case: dostarlimab vs. carboplatin + paclitaxel only.
dProbabilistic results are presented for the CADTH exploratory reanalysis, which includes key limitations identified by CADTH that were modifiable in the sponsor’s economic model. All other analyses are presented deterministically.
CADTH undertook several scenario analyses on the CADTH exploratory analysis to determine the impact of alternative assumptions on the exploratory analysis of cost-effectiveness for dostarlimab compared with carboplatin + paclitaxel. This included:
Comparison against the most relevant comparators in Canadian clinical practice according to the clinical experts consulted by CADTH: paclitaxel.
No treatment waning.
Lower health state utility value (0.625) based on the Progressed Disease health state utility value for the ZoptEC trial.1
The CADTH exploratory analysis was most sensitive to the cost-effectiveness comparison against paclitaxel, the most commonly used treatment in Canadian clinical practice according to CADTH’s clinical experts, which was informed by efficacy data from the sponsor’s indirect treatment comparisons.
Table 16: Summary of CADTH Exploratory Scenario Analyses
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. Carboplatin + paclitaxel ($/QALY) |
|---|---|---|---|---|---|---|---|
Sponsor’s submitted base case | |||||||
Carboplatin + paclitaxel | 61,360 | Reference | 2.12 | Reference | 1.52 | Reference | Reference |
Dostarlimab | 865,863 | 804,503 | 8.89 | 6.77 | 6.42 | 4.90 | 164,193 |
CADTH’s exploratory analysis | |||||||
Carboplatin + paclitaxel | 61,389 | Reference | 2.12 | Reference | 1.53 | Reference | Reference |
Dostarlimab | 436,918 | 375,528 | 3.53 | 1.40 | 2.67 | 1.13 | 330,962 |
CADTH scenario: dostarlimab vs. paclitaxel | |||||||
Paclitaxel | 52,370 | Reference | 0.82 | Reference | 0.54 | Reference | Reference |
Dostarlimab | 439,656 | 387,285 | 3.52 | 2.71 | 2.63 | 2.09 | 185,452 |
CADTH scenario: No treatment waning | |||||||
Carboplatin + paclitaxel | 61,388 | Reference | 2.12 | Reference | 1.51 | Reference | Reference |
Dostarlimab | 386,542 | 324,154 | 3.28 | 1.16 | 2.47 | 0.96 | 338,781 |
CADTH scenario: Lower health state utility value | |||||||
Carboplatin + paclitaxel | 61,388 | Reference | 2.12 | Reference | 1.43 | Reference | Reference |
Dostarlimab | 439,656 | 378,267 | 3.52 | 1.41 | 2.52 | 1.09 | 345,699 |
CADTH scenario: No stopping rule | |||||||
Carboplatin + paclitaxel | 61,388 | Reference | 2.12 | Reference | 1.51 | Reference | Reference |
Dostarlimab | 564,528 | 503,139 | 3.52 | 1.41 | 2.63 | 1.13 | 446,759 |
ICER = incremental cost-effectiveness ratio; PLD = pegylated liposomal doxorubicin; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Note: The sponsor’s base case results and CADTH’s exploratory analysis results are presented probabilistically while all scenarios are deterministic.
Price-Reduction Analysis
As no formal CADTH reanalysis was performed, price-reduction analyses were conducted using only the sponsor’s base case assumptions. This deterministic analysis was subject to the key limitations of the sponsor’s model as noted in the CADTH Appraisal of the Sponsor’s Economic Evaluation section. Based on the CADTH exploratory analysis, a reduction in the price of dostarlimab by 83% would be required for dostarlimab to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY compared to carboplatin + paclitaxel. It is important to note that this price-reduction estimate is based on estimates of incremental life-years (and hence QALYs) that are highly uncertain and may not be representative of the true incremental effect of dostarlimab. Consequently, the price reduction required for dostarlimab to be cost-effective remains unknown.
Table 17: Price-Reduction Analyses of Sponsor’s Base-Case and CADTH Exploratory Analysis
Price reduction | ICERs for dostarlimab vs. carboplatin + paclitaxel ($) | |
|---|---|---|
Sponsor’s base case | CADTH exploratory analysis | |
No price reduction | $174,693 | $335,880 |
74% | $46,594 | $78,389 |
83% | $31,014 | $47,072 |
ICER = incremental cost-effectiveness ratio; NA = not applicable; vs. = versus.
Notes: All analyses in this table are deterministic and are subject to limitations within the sponsor’s economic model.
Text in bold signifies the approximate point at which the value hits a threshold of $50,000 per QALY.
Appendix 5: Submitted BIA and CADTH Appraisal
Table 18: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA) estimating the incremental budget impact of reimbursing dostarlimab for patients with mismatch repair deficient (dMMR) or microsatellite instability-high (MSI-H) recurrent or advanced endometrial cancer (EC) that has progressed on or following prior treatment with a platinum-containing regimen. The BIA was undertaken from the perspective of the Canadian public drug plans over a 3-year time horizon, and the sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec). In the reference scenario, patients were assumed to receive doxorubicin, PLD, carboplatin, carboplatin + paclitaxel, carboplatin + PLD, paclitaxel, and hormonal therapies, in the second-line setting following failure on first-line platinum-based chemotherapy. In the new drug scenario, dostarlimab was assumed to be reimbursed and prescribed as second-line therapy.24
The sponsor estimated the eligible population using an epidemiologic approach was derived via several assumptions and inputs to first estimate the incident population (i.e., newly eligible patients advanced or recurrent EC patients who meet the eligibility criteria for dostarlimab upon its introduction) and the prevalent population (i.e., previously eligible patients who would have met eligibility criteria for treatment with dostarlimab in the years before its introduction).24 The sponsor assumed that a proportion of patients eligible for treatment at the start of each year would initiate treatment in each quarter of the year, to calculate quarterly costs per year.24 This was accomplished by further providing breakdown of the proportion of patients on dostarlimab or other comparator treatment by progression status at each quarter year using PFS and OS curves for each treatment, respectively. The estimated numbers of patients on each treatment in each quarter were then multiplied by corresponding estimates of the quarterly costs of each treatment to ascertain quarterly treatment-specific costs per year.24
In the sponsor’s base case, drug-acquisition costs, dispensing fees, mark-up costs, costs of subsequent therapy, costs of screening for dMMR/MSI-H, and health care resource utilization costs (i.e., pharmacist, nurse, physician, and infusion chair time) were captured. Drug wastage was considered in calculations, with dosing based on Cancer Care Ontario’s regimen monographs. Drug-administration costs were not included.24
Key inputs to the BIA are documented in Table 19.
Table 19: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Total female population in Canada | 15,214,105 |
Annual growth rate | 1.1% |
Annual incidence of uterine cancer (per 100,000) | 39.2% |
Annual % change in incidence | 0.5% |
Proportion with endometrial cancer | 95.0% |
Proportion with Stage I | 74.6% |
Proportion with Stage II | 6.2% |
Proportion with Stage III | 12.0% |
Proportion with Stage IV | 7.2% |
Stage III and IV with progression on 1L PBCT | |
Stage III receiving 1L PBCT | 72.3% |
Stage IV receiving 1L PBCT | 72.3% |
Stage III with progression and receive active therapy | 70.0% |
Stage IV with progression and receive active therapy | 70.0% |
Stage I and II with recurrence and progression on 1L PBCT | |
Lifetime recurrence with Stage I | 6.5% |
Stage I receiving PBCT in the neoadjuvant/adjuvant | 6.1% |
Stage I receiving PBCT in the recurrent setting | 25.9% |
Stage I with progression and receive active therapy | 70.0% |
Lifetime recurrence with Stage II | 20.0% |
Stage II receiving PBCT in the neoadjuvant/adjuvant setting | 19.8% |
Stage II receiving PBCT in the recurrent setting | 25.9% |
Stage II with progression and receive active therapy | 70.0% |
Newly eligible cases with prior PBCT | 658 / 669 / 680 |
Previously eligible cases | |
Life expectancy (years) for dMMR/MSI-H EC with prior PBCT | 2 |
Proportion of patients electing to start treatment in each year | 70% / 100% / 0% |
Total newly and previously eligible patients | 1,581 / 870 / 680 |
Proportion tested for dMMR/MSI-H | 100% |
Proportion with dMMR/MSI-H | 26% |
Total eligible patients at the start of each year | |
% aged < 65 years | 50% |
% aged ≥ 65 years | 50% |
% covered aged < 65 years | 54% |
% covered aged ≥ 65 years | 100% |
Number of patients eligible for drug under review | 316 / 174 / 136 |
Market uptake (3 years) | |
Uptake (reference scenario) | |
Doxorubicin | 4.3% / 4.3% / 4.3% |
Pegylated liposomal doxorubicin | 20.9% / 20.9% / 20.9% |
Carboplatin | 8.6% / 8.6% / 8.6% |
Carboplatin + paclitaxel | 31.5% / 31.5% / 31.5% |
Carboplatin + liposomal doxorubicin | 15.4% / 15.4% / 15.4% |
Paclitaxel | 4.3% / 4.3% / 4.3% |
Hormonal therapies | 15% / 15% / 15% |
Uptake (new drug scenario) | |
Dostarlimab | 50% / 70% / 70% |
Doxorubicin | 2.2% / 1.3% / 1.3% |
Pegylated liposomal doxorubicin | 10.5 / 6.3% / 6.3% |
Carboplatin | 4.3% / 2.6% / 2.6% |
Carboplatin + paclitaxel | 15.7% / 9.4% / 9.4% |
Carboplatin + liposomal doxorubicin | 7.7% / 4.6% / 4.6% |
Paclitaxel | 2.2% / 1.3% / 1.3% |
Hormonal therapies | 7.5% / 4.5% / 4.5% |
Cost of treatment (per patient) | |
Cost of treatment per year | |
Dostarlimab | $178,624.64 |
Doxorubicin | $4,620.00 |
Pegylated liposomal doxorubicin | $19,964.10 |
Carboplatin | $9,300.02 |
Carboplatin + paclitaxel | $29,100.02 |
Carboplatin + liposomal doxorubicin | $29,264.12 |
Paclitaxel | $19,800.00 |
Hormonal therapies | |
Tamoxifen | $12.91 |
Anastrozole | $35.13 |
Exemestane | $48.93 |
Megestrol | $1,122.13 |
Medroxyprogesterone | $106.76 |
1L = first-line; dMMR = mismatch repair deficient; MSI-H = high microsatellite instability; PBCT = platinum-based chemotherapy.
Summary of the Sponsor’s BIA Results
The sponsor estimated that the budget impact of reimbursing dostarlimab as second-line treatment for patients with dMMR/MSI-H recurrent or advanced EC would be $17,209,586 in year 1, $24,690,413 in year 2, and $28,464,128 in year 3 for a 3-year total budget impact of $70,364,128.24
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty in the estimated population size eligible for dostarlimab: The sponsor undertook an epidemiological approach to estimate the size of the population eligible for dostarlimab. This required deriving inputs from the published literature and applying several assumptions to derive estimates for the incident and prevalent populations in a multi-step approach. The clinical experts consulted by CADTH indicated that although the final estimated population appeared to be largely reasonable, several estimates of the target population derived from the sponsor’s assumptions and inputs may be associated with some uncertainty. First, the clinical expert consulted by CADTH noted that the sponsor’s assumed annual incidence appeared to be slightly higher than expected. CADTH’s clinical expert indicated that according to 2021 statistics reported by a Canadian Cancer report,25 there will be approximately 8,000 new cases with an age-standardized incidence rate of 37.2 per 100,000 women. Additionally, 1 of the experts noted that the sponsor’s assumed proportion of patients with Stage IV progression who would receive active therapy (70%) appeared to be reasonable while the other noted that this proportion may be lower (50%). Additionally, the clinical experts consulted by CADTH indicated that the assumed proportion of patients with dMMR/MSI-H could be as high as 30%.
CADTH undertook several scenario analyses to explore the uncertainty in the estimated population size. In scenario analyses, CADTH explored alternate assumptions by (a) decreasing the annual incidence of uterine cancer to 37.2%; (b) decreasing the proportion of patients with Stage IV disease progression requiring active therapy to 50%; and (c) applied the alternate assumption that 30% of patients will have a dMMR or MSH-H status.
Exclusion of relevant comparators: As per the Health Canada indication and the sponsor’s submitted reimbursement request, the submitted pharmacoeconomic model for dostarlimab is indicated for the treatment of monotherapy for the treatment of adult patients with dMMR/MSI-H recurrent or advanced EC that has progressed on or following prior treatment with a platinum-containing regimen. Feedback from the clinical experts consulted by CADTH for this review indicates that the sponsor excluded several chemotherapy agents from the BIA (monotherapies and combined treatment regimens) that were identified to be relevant comparators for the indicated population (i.e., pembrolizumab, cisplatin, carboplatin plus docetaxel, carboplatin plus doxorubicin, and cisplatin plus doxorubicin). Additionally, the sponsor included hormonal therapies (i.e., medroxyprogesterone, megestrol, tamoxifen, anastrozole, and exemestane), which is not a relevant comparator according to the clinical experts consulted by CADTH and is commonly offered to patients with a more limited recurrence risk (e.g., low volume disease burden with an estrogen/progesterone receptor positive). As such, the incremental budget impact of dostarlimab is associated with uncertainty.
CADTH was unable to address this limitation.
The market share distribution of comparators in the reference scenario is uncertain: In the sponsor’s submitted BIA, the baseline market share distribution for the comparator treatments in the reference scenario did not reflect clinical expectations of treatments offered in the second-line setting, as indicated by CADTH’s clinical expert and drug plan feedback. CADTH’s clinical expert noted that commonly used second-line therapies in the Canadian clinical practice include paclitaxel or liposomal doxorubicin, however, in the sponsor’s base case, carboplatin + paclitaxel was assigned the largest market share while doxorubicin and paclitaxel were assigned the smallest market share. CADTH’s clinical expert further indicated that the purpose for first-line chemotherapy (either for metastatic disease, or for recurrence management) is suggestive of the expected market share distribution of second-line chemotherapy. Specifically, the clinical experts consulted by CADTH noted that patients who progress after first-line therapy do so quickly, in which case a platinum doublet such as carboplatin + paclitaxel would not be used unless patients were platinum-sensitive. Given that the indicated population is dMMR/MSI-H, these patients have aggressive disease and are platinum-refractory rather than platinum-sensitive, thus, it is unlikely for such a high proportion of patients to be retreated with carboplatin + paclitaxel.
CADTH did not address this limitation. Due to the uncertainty in the baseline market share distribution in the reference scenario, CADTH revised the baseline market share distribution to reflect the clinical expert opinion in a scenario analysis.
The anticipated uptake of dostarlimab in the new drug scenario is uncertain: The sponsor assumed that dostarlimab would have an anticipated market uptake rate of 50% in year 1, 70% in year 2, and 70% in year 3 while displacing comparator therapies proportionally to their estimated market shares in the reference scenario (i.e., market share for all other comparators would be halved in year 1, further reduced by 20% in year 2, and remain steady after that). The clinical expert consulted by CADTH indicated that while the sponsor’s anticipated uptake rate of dostarlimab in the new drug scenario appeared to be reasonable, there remains uncertainty to how dostarlimab will displace other comparators, as dostarlimab is expected to likely displace hormonal therapies and doxorubicin more than others given the low response rates of other treatments. Additionally, CADTH’s clinical expert noted that the introduction of dostarlimab would fully displace carboplatin + paclitaxel and
CADTH addressed this limitation by revising the market share distribution in the new drug scenario to reflect the expert’s feedback.
The sponsor assumed continued treatment over the 3-year time horizon, which likely overestimated drug costs: In the base case, the sponsor assumed that all patients would continue to remain on dostarlimab over the 3-year time horizon. The clinical experts consulted by CADTH indicated that approximately 50 to 60% of patients would likely stop treatment after 2 years of treatment. Based on expert feedback, CADTH revised the treatment stopping rule to 24 months, and revised the proportion of patients expected to stop treatment at 2 years to 60%.
CADTH addressed this limitation by permitting 60% of patients to stop treatment at 2 years.
The sponsor’s submitted BIA is based on the same clinical evidence (i.e., OS and PFS) as its pharmacoeconomic analysis, and may overestimate drug costs: The sponsor incorporated extrapolated OS and PFS curves up to 48 months (reflecting baseline and years 1, 2 and 3) to estimate the proportion of patients who remained alive, and those who were on treatment, respectively, in each year over the 3-year time horizon in their submitted BIA. The sponsor applied the same treatment-specific OS and PFS curves as in their pharmacoeconomic base case to calculate quarterly drug costs over the 3-year time horizon in their submitted BIA. As several limitations were identified with the efficacy data in the GARNET study, the long-term extrapolations of this clinical evidence are highly uncertain. As such, CADTH opted to use trial evidence only (i.e., Kaplan-Meier OS and PFS data from the GARNET study), as it represents OS and PFS estimates without any statistical adjustments. CADTH selected the relevant time points from this data to align with the forecasted period in the BIA. OS and PFS estimates from year 2 or 24 months onwards were assumed to remain the same for the remainder of the time horizon in the BIA.
CADTH addressed this limitation by applying the unadjusted Kaplan-Meier data from the GARNET study for OS and PFS in the BIA case. In a scenario analysis, CADTH explored the impact of arbitrarily assuming less efficacious estimates for OS by 20% at each time point.
CADTH Reanalyses of the BIA
Table 20: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Market share in the reference scenario years 1 to 3 | Doxorubicin = 4.3% Pegylated Liposomal Doxorubicin = 20.9% Carboplatin = 8.6% Carboplatin + Paclitaxel = 31.5% Carboplatin + Liposomal Doxorubicin = 15.4% Paclitaxel = 4.3% Hormonal therapies = 15% | Doxorubicin = 30% Pegylated Liposomal Doxorubicin = 20% Carboplatin = 10% Carboplatin + Paclitaxel = 30% Carboplatin + Liposomal Doxorubicin = 5% Paclitaxel = 5% Hormonal therapies = 0% |
2. Market share in the new drug scenario years 1 to 3 | Dostarlimab = 50% / 70% / 70% Doxorubicin = 2.2% / 1.3% / 1.3% Pegylated Liposomal Doxorubicin = 10.5% / 6.3% / 6.3% Carboplatin = 4.3% / 2.6% / 2.6% Carboplatin + Paclitaxel = 15.7% / 9.4% / 9.4% Carboplatin + Liposomal Doxorubicin = 7.7% / 4.6% / 4.6% Paclitaxel = 2.2% / 1.3% / 1.3% Hormonal therapies = 7.5% / 4.5% / 4.5% | Dostarlimab = 50% / 70% / 80% Doxorubicin = 15% / 10% / 6.5% Pegylated Liposomal Doxorubicin = 10% / 5% / 1.5% Carboplatin = 5% / 2.5% / 1.0% Carboplatin + Paclitaxel = 15% / 10% / 10% Carboplatin + Liposomal Doxorubicin = 2.5% / 1.3% / 0.6% Paclitaxel = 2.5% / 1.3% / 0.6% Hormonal therapies = 0% (less than 1%) |
3. Stopping rule | None |
|
4. Overall survival and progression-free survival of dostarlimab | OS and PFS curves based on the extrapolations as per sponsor’s submitted economic model, extrapolated up to 48 months: OS: Generalized Gamma PFS: Generalized Gamma | OS and PFS curves based on Kaplan-Meier curves from the GARNET study, extrapolated up to 48 months: OS: Kaplan-Meier PFS: Kaplan-Meier |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
Applying the changes in Table 20 resulted in a minor increase in the estimated budget impact under the drug plan perspective to $ over 3 years. The results of the CADTH stepwise reanalyses are presented in summary format in Table 21 and a more detailed breakdown is presented in Table 22.
Table 21: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $70,364,128 |
CADTH reanalysis 1 | $71,769,719 |
CADTH reanalysis 2 | $71,361,803 |
CADTH reanalysis 3 | $64,792,567 |
CADTH reanalysis 4 | $71,026,852 |
CADTH base case | $70,366,188 |
BIA = budget impact analysis.
Table 22: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $2,727,684 | $24,015,775 | $28,756,447 | $31,631,431 | $84,403,653 |
New drug | $2,727,684 | $6,806,188 | $4,066,034 | $3,167,303 | $14,039,526 | |
Budget impact | $0 | $17,209,586 | $24,690,413 | $28,464,128 | $70,364,128 | |
CADTH base case | Reference | $2,727,136 | $24,014,929 | $28,755,935 | $31,631,053 | $84,401,917 |
New drug | $2,727,136 | $6,804,674 | $4,064,791 | $3,166,263 | $14,035,729 | |
Budget impact | $0 | $17,210,255 | $24,691,144 | $28,464,789 | $70,366,188 |
BIA = budget impact analysis.
CADTH conducted the following additional scenario analyses from the drug plan perspective (Scenarios 1 to 5, Table 23):
Decreasing the annual incidence of uterine cancer to 37.2% based on 2021 Canadian Cancer statistics.25
Applying the alternate assumption that approximately 50% of patients will have Stage IV disease with progression and require active therapy.
Applied the alternate assumption that 30% of patients will have a dMMR or MSH-H status.
Applied the arbitrary assumption that the overall survival is 20% less efficacious at each time point on the Kaplan-Meier OS curve.
Applied an 83% reduction in the price of dostarlimab to align with the point at which the ICER is within the willingness-to-pay threshold of $50,000 per QALY in the CADTH exploratory reanalysis.
The model results were most sensitive to changes in the proportion of patients with a dMMR/MSI-H status.
Table 23: CADTH Scenario Analyses
Stepped analysis | Budget impact | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
CADTH scenario analysis 1: annual incidence of 37.2% | Reference | $2,588,964 | $6,459,913 | $3,858,847 | $3,005,844 | $13,324,604 |
New drug | $2,588,964 | $22,798,205 | $27,299,006 | $30,028,455 | $80,125,667 | |
Budget impact | $0 | $16,338,292 | $23,440,159 | $27,022,612 | $66,801,063 | |
CADTH scenario analysis 2: 50% of patients will have Stage IV disease with progression and require active therapy | Reference | $2,475,010 | $5,597,675 | $3,304,500 | $2,562,167 | $11,464,342 |
New drug | $2,475,010 | $21,555,153 | $25,855,396 | $25,158,485 | $72,569,034 | |
Budget impact | $0 | $15,957,478 | $22,550,896 | $22,596,318 | $61,104,691 | |
CADTH scenario analysis 3: 30% of patients will have a dMMR or MSH-H status | Reference | $3,146,693 | $7,116,806 | $4,201,295 | $3,257,504 | $14,575,605 |
New drug | $3,146,693 | $27,404,921 | $32,872,190 | $31,986,148 | $92,263,259 | |
Budget impact | $0 | $20,288,115 | $28,670,895 | $28,728,644 | $77,687,653 | |
CADTH scenario analysis 4: overall survival is 20% less efficacious at each time point | Reference | $2,727,134 | $6,167,898 | $3,641,122 | $2,823,170 | $12,632,191 |
New drug | $2,727,134 | $23,348,266 | $28,179,037 | $27,444,379 | $78,971,681 | |
Budget impact | $0 | $17,180,367 | $24,537,915 | $24,621,209 | $66,339,490 | |
CADTH scenario analysis 5: 83% price reduction | Reference | $2,727,136 | $6,804,674 | $4,064,791 | $3,166,263 | $14,035,729 |
New drug | $2,727,136 | $7,374,769 | $6,403,330 | $6,562,156 | $20,340,254 | |
Budget impact | $0 | $570,094 | $2,338,539 | $3,395,893 | $6,304,526 |
Stakeholder Input
Patient Input
Canadian Cancer Society
Information Gathering
The Canadian Cancer Society gathered perspectives through distributing a survey as well as accepting testimonials from patients and caregivers. We received a total of six testimonials and 22 survey responses.Of the 22 survey respondents, there were six that had taken and two that had cared for someone who has taken Dostarlimab (eight in total). The data was gathered within the time frame of October 22 – November
All patients and caregivers who contributed a testimonial had experience with Dostarlimab.
Please note that all survey questions were directed toward the patient’s experience. When a caregiver wasasked a question, the survey indicated the questions referred to the patient.
Demographic Information for Survey Respondents
Demographic information collected from the survey is displayed below. Please note that not all surveyoptions that were offered are shown within the charts as they are limited to the options respondents actually selected.
Which of the following best describes you?
There were 20 survey respondents who were patients who currently have or previously had endometrial cancer, and two respondents who indicated they are currently or were previously a caregiver for someonewith endometrial cancer.
What Province or Territory do you currently reside in?
The majority of respondents resided in Quebec (45%) and British Columbia (27%). The other 28% resided in Ontario, Alberta, Saskatchewan and Manitoba.
Figure 1: Geographic Location of Respondents
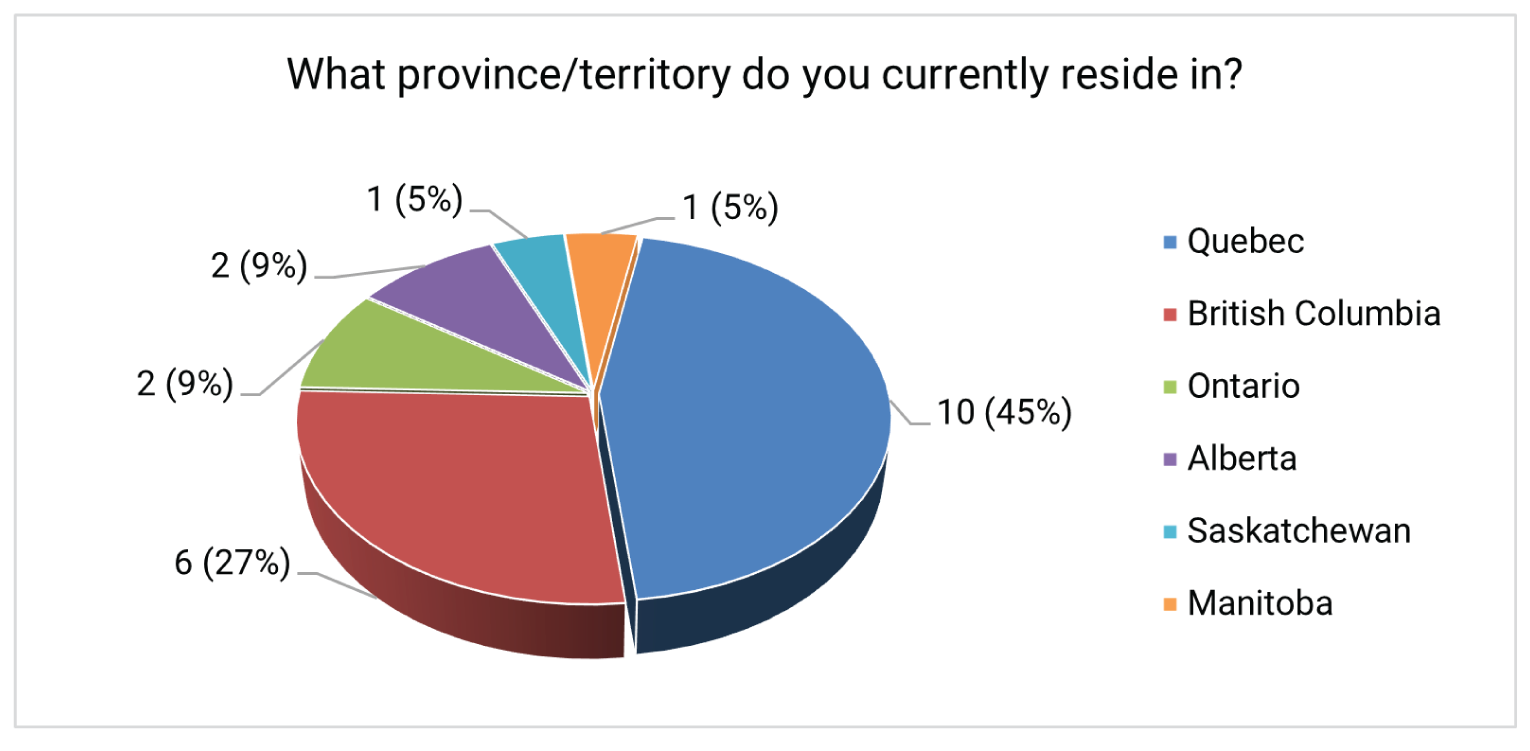
Note: Both caregiver respondents resided in Quebec. Of the 8 with experience with Dostarlimab, 6 resided in QC and 2 resided in BC.
How old are you?
The majority of survey respondents were between 50 and 59 and 70 and 79 years old (68%). Please refer to Figure 2 for other age brackets surveyed.
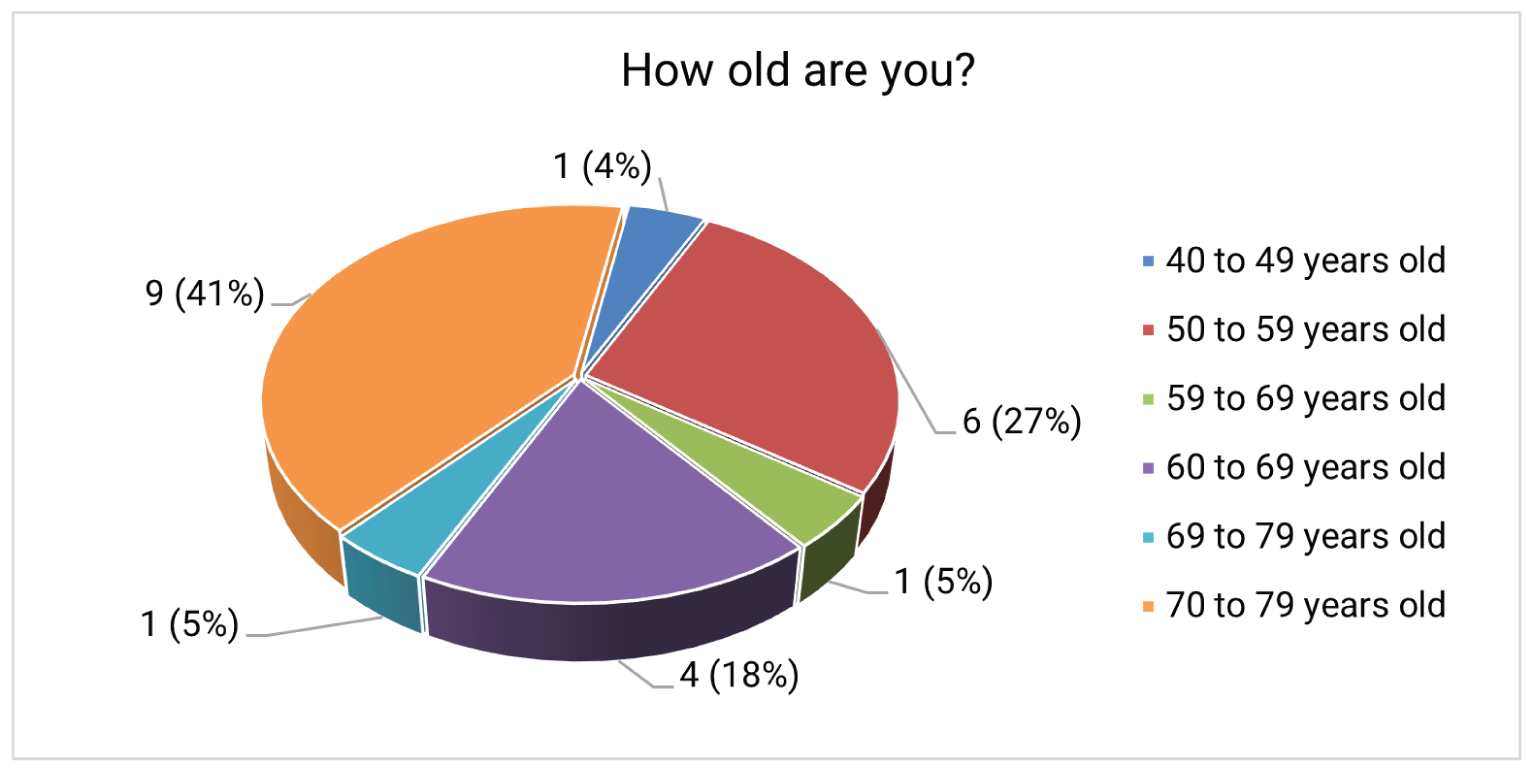
Note: Of the 8 patients with experience with Dostarlimab, two were aged 50 – 59, two were aged 60 – 69and four were aged 70 – 79.
What is your gender?
All respondents indicated they identify as their biological gender at birth (female), except one who self identified as male who also had experience with Dostarlimab.
Disease Experience
How much of an impact do symptoms associated with endometrial cancer have on your day-to-day activities and quality of life? (select all that apply).
The ability to conduct household chores scored highest as a day-to-day activity where patients experienceddifficulty, with ten responses in the moderate to significant impact range. The ability to travel and exercise was not far behind, with nine moderate to significant impact responses. The ability to work had the next most substantial impact, with eight responses in the moderate to significant range. There were 56 moderate to significant impact options selected across the 22 participants (34%).
Table 1: Impact of Symptoms on Day-to-Day Life and Quality of Life
Task | No impact | Small impact | Moderate impact | Significant impact | I’m not sure | N/A |
|---|---|---|---|---|---|---|
Ability to work | 8 (36%) | 0 (0%) | 4 (18%) | 4 (18%) | 0 (0%) | 6 (27%) |
Ability to travel | 8 (36%) | 3 (14%) | 4 (18%) | 5 (23%) | 1 (4.5%) | 1 (4.5%) |
Ability to exercise | 8 (36%) | 4 (18%) | 5 (23%) | 4 (18%) | 0 (0%) | 1 (4.5%) |
Ability to conduct household chores | 10 (45%) | 1 (4.5%) | 9 (41%) | 1 (4.5%) | 0 (0%) | 1 (4.5%) |
Ability to fulfill family obligations | 9 (41%) | 5 (23%) | 5 (23%) | 2 (9%) | 0 (0%) | 1 (4.5%) |
Ability to spend time with family and friends | 13 (59%) | 2 (9%) | 6 (27%) | 0 (0%) | 0 (0%) | 1 (4.5%) |
Ability to concentrate | 12 (54.5%) | 4 (18%) | 2 (9%) | 3 (14%) | 0 (0%) | 1 (4.5%) |
Ability to fulfill practical needs (dressing, bathing, preparing meals) | 14 (64%) | 5 (23%) | 0 (0%) | 2 (9%) | 0 (0%) | 1 (4.5%) |
Note: “ Not applicable” was also an option if the task was not relevant to them ( for example, i f they didn’t have a job and therefore would not answer the question on their ability to work).
Note: Percentages have been rounded
Specify any other areas of your life that have been impacted and how significant the impact is:
“Emotional stability - significant impact”
“I have a dance school and was unable to uphold all my duties to keep it going. Needed lots of support from family and friends. Chemo was so debilitating and this immunotherapy treatment haschanged my life. Able to live again. I hope other women can benefit from this immunotherapy instead of the huge side effects of chemo.”
“My social life was greatly impacted. I could not go out as I wished.”
“[The] ability to get travel insurance.”
“Very tired lots of naps.
Experiences With Currently Available Treatments
What is the greatest financial barrier related to your treatment(s)?
The most significant financial barriers identified included travel costs and loss of income due to absencefrom work. Overall, 59% of all responders reported a financial barrier related to their treatment.
Of the four respondents who indicated travel costs were their greatest financial barrier, three had experience with Dostarlimab. Both respondents who indicated drug costs were a major barrier were also users of Dostarlimab. Additionally, three of the four respondents who selected “loss of income due to absence from work” had experience with Dostarlimab. A total of 61% of those who indicated they have financial barriers (8 people) were those who have used or cared for someone who used the drug under evaluation. These results may reflect to the increased costs affiliated with accessing this drug via a clinicaltrial (see question 1, section 5).
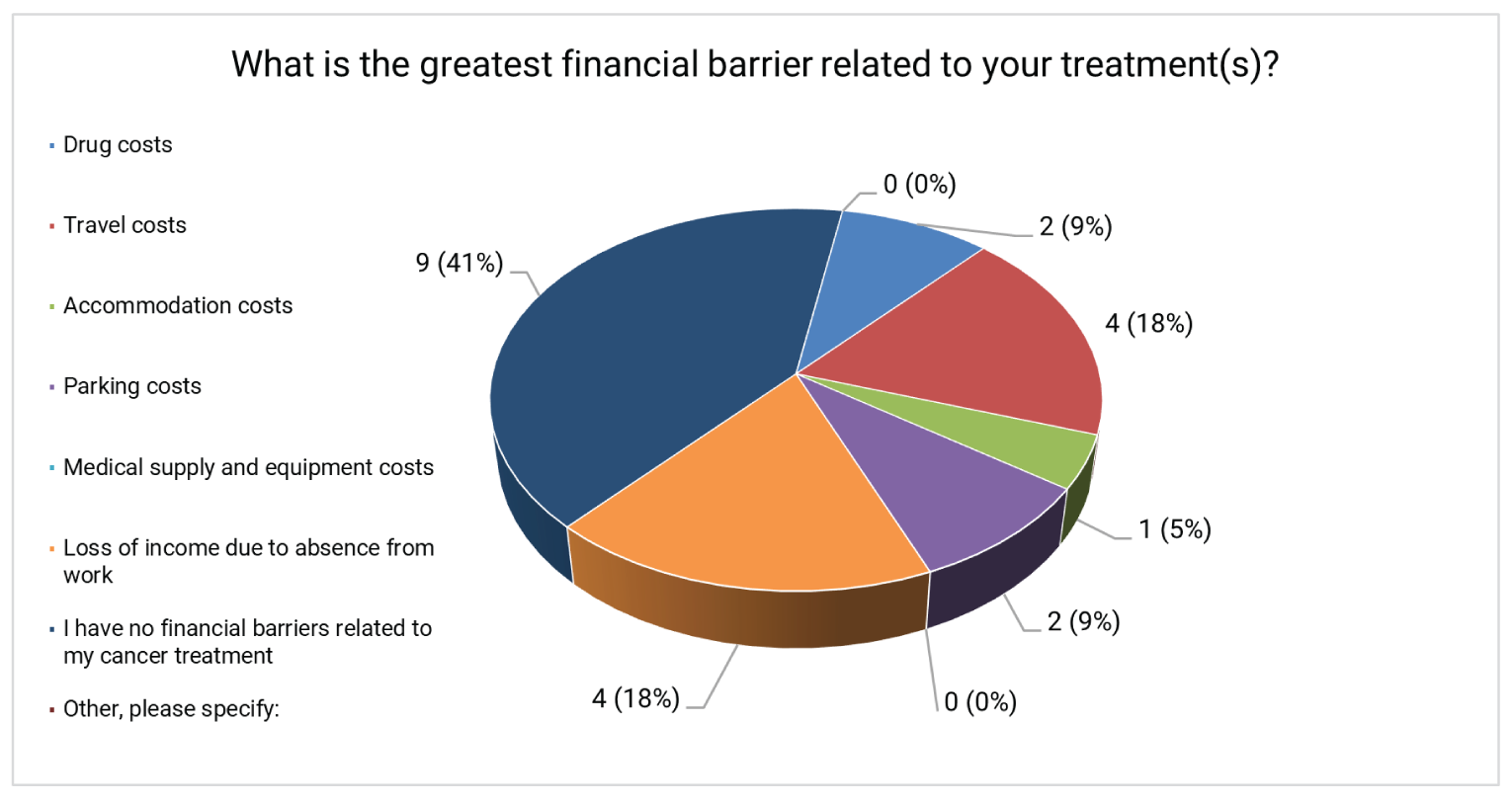
How is your cancer currently being treated? (select all that apply)
The majority of responders indicated they were not receiving treatment at this time (42%) or that they were undergoing immunotherapy (38%). Eight of nine responders who indicated they were undergoingimmunotherapy also stated they had experience with Dostarlimab.
One responder was doing a combination of chemotherapy, surgery, and hormone therapy while taking Anastrozole and Caelyx. The other person undergoing chemotherapy was taking Carboplatin and Paclitaxel.The responder that selected “Other” previously had surgery and declined chemotherapy and radiation.
How much of an impact do the following cancer treatment side effects have on your daily life?
Table 2 below depicts how impactful prevalent cancer treatment side effects were to this group. Please note that respondents could indicate “not applicable” if they did not have a specific side effect so “no
impact” means the side effect was present at some point, but to such a small degree it did not impact the person’s life. It is also pertinent to note that even if the individual indicated they are currently not receiving treatment (see Figure 4), they still experienced side effects from prior cancer treatments.
The most significant side effect impacts included changes in libido and sexual function with ten responses (45% of people) in the moderate to significant impact range and fatigue with nine responses (41% of people) in the moderate to significant impact range. There were a total of 62 responses in the moderate to significant range, leaving 22% of all responses in this range.
Table 2: Impact of Treatment Side Effects Experienced By Respondents
Side Effects | No impact | Small impact | Moderate impact | Significant impact | I’m not sure | N/A |
|---|---|---|---|---|---|---|
Fatigue | 4 (18%) | 5 (23%) | 6 (27%) | 3 (14%) | 0 (0%) | 4 (18%) |
Hair loss | 11 (50%) | 2 (9%) | 2 (9%) | 2 (9%) | 0 (0%) | 5 (23%) |
Easy bruising and bleeding | 13 (59%) | 3 (14%) | 1 (4.5%) | 0 (0%) | 0 (0%) | 5 (23%) |
Frequent infections | 14 (64%) | 2 (9%) | 1 (4.5%) | 0 (0%) | 0 (0%) | 5 (23%) |
Anemia (low red blood cell counts) | 12 (54.5%) | 2 (9%) | 2 (9%) | 0 (0%) | 1 (4.5%) | 5 (23%) |
Nausea and vomiting | 12 (54.5%) | 5 (23%) | 0 (0%) | 0 (0%) | 0 (0%) | 5 (23%) |
Appetite changes | 8 (36%) | 5 (23%) | 4 (18%) | 0 (0%) | 1 (4.5%) | 4 (18%) |
Constipation | 10 (45%) | 1 (4.5%) | 5 (23%) | 1 (4.5%) | 2 (9%) | 3 (14%) |
Diarrhea | 13 (59%) | 1 (4.5%) | 2 (9%) | 1 (4.5%) | 1 (4.5%) | 4 (18%) |
Peripheral neuropathy (numbness, tingling and pain in the nerves) | 4 (18%) | 6 (27%) | 1 (4.5%) | 4 (4.5%) | 0 (0%) | 7 (32%) |
Kidney problems | 14 (64%) | 0 (%) | 2 (9%) | 0 (0%) | 1 (4.5%) | 5 (23%) |
Weight changes | 8 (36%) | 4 (18%) | 3 (14%) | 2 (9%) | 1 (4.5%) | 4 (18%) |
Concentration and focus | 7 (32%) | 7 (32%) | 1 (4.5%) | 2 (9%) | 0 (%) | 5 (23%) |
Changes in libido or sexual function | 6 (27%) | 1 (4.5%) | 4 (18%) | 6 (27%) | 1 (4.5%) | 4 (18%) |
Fertility problems | 5 (23%) | 0 (0%) | 0 (%) | 1 (4.5%) | 0 (0%) | 16 (73%) |
Pain | 8 (36%) | 5 (23%) | 4(18%) | 1 (4.5%) | 0 (0%) | 4 (18%) |
Mouth, tongue, and throat problems such as sores and pain with swallowing | 12 (54.5%) | 3 (14%) | 1 (4.5%) | 0 (0%) | 0 (0%) | 6 (27%) |
Note: “Not applicable” was an option if the side effect was not relevant to them (for example, if have the side effect at all).
Note: Percentages have been rounded.
If there are any other side effects caused by your current cancer treatment(s), please specify what they are and how significant their impact is on your life:
“Skin toxicity. Significant impact”
“Stamina - difficulty standing for more than 30 minutes or standing & Chopping food. Sleep more than normal.”
Bladder incontinence – significant, Hot flashes - small impact
“sécheresse de la peau (dry skin)”
“Vision is blurry for distance like watching TV, computer screen, can't read street signs, can't make out facial features at 10 feet away. Vision goes back to normal after chemo ends.”
“Arthritis with moderate impact.”
“Bikini line incision for total hysterectomy and/ omentectomy seems to have caused tummy to droop a bit like a floppy breast.”
“Arthritis”
What improvements would you like to see in new treatments that are not achieved in currently available treatments? For example: effectiveness for relieving certain symptoms or side effects, affordability, ease of use etc.
Survey respondents indicated they would like fewer side effects such as skin issues, fatigue, bladder control,stamina, vaginal bleeding after intercourse, vaginal dryness, hair loss, pain, concentration problems (chemotherapy “fog”) and arthritis. They also indicated they would like to see more drug affordability as cancer can become expensive during treatment as usual activities are interrupted.
Treatment access was also an issue for one respondent as they indicated the drugs Carboplatin and Paclitaxel were the only pharmaceutical options in their province (Manitoba). They went on to note that other provinces and parts of the world (US and Europe) have more options.
Quotes from Testimonials Outside the Survey
Many testimonials described the harsh debilitating side effects they or their family member experienced with traditional treatments as well as an overall lack of efficacy. All patients who submitted testimonials underwent several lines of conventional therapy with poor results that led to significant reductions in quality of life, multiple side effects, stress, financial distress, and lost time. Patients and caregivers described some of these conventional treatments to have a cascading effect which led to further complications.
“The first line of chemotherapy gave me very bad side effects, pain and complications. They gave me first and second line chemotherapy sessions, both had very bad side effects and were not effective.….At that point, they gave me radiation to shrink the tumour, and yet that was only for one small tumour that I had….My abdominal pain become worse and worse. The pain medications were not effective.”
“Prior to my diagnosis I was a dance teacher for 30 years and working over 30 h a week at my dance school. Dancing and teaching children was my passion and a dream come true….The treatment suggested by my medical team was frightening and I wanted to seek alternative treatments. They explained it was not a viable option and surgery was the best decision at the time. I had my uterus and ovaries removed…it was stage 1A endometrial cancer…It wasn’t long after that pain became an issue…I was told my cancer came back and I will need to do chemotherapy. I was devastated. I had to go to the hospital every single week for treatment during my 2015 summer. Every treatment felt like a truck ran over me. I was unwell and very weak. I saw my muscles deteriorating and I couldn’t teach and dance anymore. I lost my hair, my identity, my person…finance became an issue because I had to hire other dance teachers to save my business; my income was greatly reduced. Once my chemotherapy was completed, towards the end of 2015, I started to feel better…Only to be told that my cancer has come back once again. I was on another type of chemotherapy (Caelys) for the whole year of 2016…I also had multiple appointments at the hospital, because my ureters were blocked, multiple double J changes and constant pain and discomfort caused by the double J. I couldn’t sit or lie down. I slept standing up…Toward the end of 2016, the chemotherapy that I was on didn’t work anymore. My pain was excruciating, and I couldn’t bear it anymore….[they] removed the double J to alleviate my pain…I had my bladder and rectum removed and had extensive surgery…Cancer treatment didn’t seem to be efficient and I was considered a palliative candidate…As a dancer, it meant the end of my dream and my passion but I wasn’t ready to give up.”
“I had major surgery August 20, 2014, entailing a total removal of my reproductive system, 6 hours and 4 hours in recovery. Then a full course of very aggressive chemotherapy and radiation. To say that I was at my lowest is an understatement, I have never been so ill…after a 10 day stay in the hospital I had lost 40 pounds. Very weak needing constant hep with the basic necessitates of life”.
“August 26 2016 it was the beginning of 6 cycles of Taxol/Carboplatin every 3 weeks. Her last treatment was on December 2, 2016. The chemotherapy sessions were very hard on my mom. She had a lot of adverse reactions. She was hospitalized a few times. She needed a lot of blood transfusions, neutrophil injections and her chemotherapy doses were constantly adjusted. My sister remembers so vividly the moment that our mother started losing her hair. As she was brushing her hair at the hospital to make her feel better, she was slowly brushing off chunks of hair off her head. She looked frail, different, almost ghost like as time passed. Although this was expected, the emotions that this was real, and it was actually happening flooded us like a nightmare…Doctor Jardon told us that the first line of chemotherapy sessions did not work”.
“Prior to my mother’s diagnosis, she was a janitor in an office building and she loved her work…she was very involved in the community and the children loved her. She is involved with ceremonies, traditions and she teaches [Cree] customs to children…My mother would help during hunting season. She would mostly clean, cut and prepare the meat for the conservation…After my mother received chemo treatment, we had to stay at a hotel (far from the greatest) for 2 weeks before we could go back home….I remembered my mother losing her hair and having nausea. She wasn’t involved with the community like she used to and couldn’t help her family with the trapping seasons. She also had to quit her job”.
Improved Outcomes & Experience With Drug Under Review
The following data is from the eight responders that have experience using Dostarlimab or care for someone who has used it.
How did you access TBC (Dostarlimab)?
All survey responders (and patients described in testimonials) received Dostarlimab during a clinical trial.
What were the benefits and disadvantages you experienced while taking TBC (Dostarlimab) and how did they impact your life? For example, side effects, ease of use, effectiveness, cost etc.
“ No side effects”.
“Highly effective”.
“The best treatment ever”.
“This drug saved my life. I would rather deal with the side effects than not take the drug. I see no disadvantages in taking this drug”.
Benefits...very few side effects. I have skin problems [and] thyroid issues, but it is being remedied by medications. It seems quite effective, it has not cost me anything”.
“Effectiveness”
“This drug saved my mother's life and every day I am grateful that she is able to receive treatment. She constantly tells me that she would rather take this drug and deal with the side effects than not take the drug at all.”
“My Mother did not have any side effects. [The] treatment [was] easy.”
Quotes from Testimonials
“It did save my life. I told my family that I am feeling much better and here I am today after 5 years well without major side effects except my arthritis. I survived to see my two daughters getting married and the birth of my two grandchildren. I am thankful for immunotherapy, it extended my life. Even with all the side effects I have now, I would like to continue taking the treatment and I do recommend it to other patients”.
“As of today, I have received over 30 treatments. Although I have a colostomy and urostomy, I don’t feel many side effects from this current treatment [Dostarlimab]. If I have to compare this medication to the previous chemotherapies (Paclitaxel, Carboplatin, Caelyx), radiation, and the second surgery, it’s easier thank a walk in the park! When I think of the past, I wish this medication was available from the start; I would probably not have gone through this whole nightmare and surgeries…I was able to resume my life. Teaching is still an issue because of my stomas but I am much more implicated as a director of my dance studio. I have many dancing groups and we were able to win over 100 competitions”.
“I am now approaching my third year of this trial…if it were to stop, will the cancer return and will I be able to have the same treatment or will I be shoved to the side and maybe given something else that will not work for me? I hope that this drug will be approved and that many women in my situation will have the outcome I have had. I have given three years of my life for the future of a medical miracle, I only hope I will be able to enjoy my life with my grandchildren which were born in this time frame. I have also regained 40 pounds and with adjustments I am living a wonderful life”.
“She started looking better and better. Everyone who saw her told her she looked amazing. My sister got married in May 2018 and I rushed to get married in November of the same year since my mom was doing well. We went wedding dress shopping and she was able to attend all of our events. She looked amazing. We were filled with so much joy and hope. But the best news came a month before my wedding. Tyanno called my mom and told her they do not see any tumours on her scan…I hope that everyone realized how beneficial this treatment is; how many lives it can save, but most of all. How many families could have a second chance at life.”
“Having her treatment every 6 weeks allowed my mother to be present for the family and community…Since 2018,my mom resumed her regular life. From the moose to the geese-trapping season, my mother is present to help the family. She cleans, cuts and prepares the meat for conservation. She takes part in cree ceremonies and she is much more involved in our small town community. …I could never have imagined her so well while receiving cancer treatment. I am hoping this medication could be given in a center closer to our home and not in a clinical research department so far away. It would reduce the amount of resources needed for my mother to receive treatment. She continues her Dostarlimab every 6 weeks and her cancer is very well controlled.”
“This treatment gave me the opportunity to live my life relatively normal, continue to work, support my family, and gave me at least 2 years (and still counting) of time. It was less invasive than some of the chemo options. And mild side effects. I could work, live life normally. With my family. This trial changed my life. It gave me time. And, people that are living with cancer, that is what they want. TIME.”
Was TBC (Dostarlimab) easier to use than previous therapies?
All survey responders indicated that Dostarlimab was easier to use than previous therapies. Six responders explained it was easier to use because there were no side effects to minimal side effects compared to other treatments. One responder said it was easier because the treatment was every six weeks. One responder indicated they felt the short time of infusion made it easier than other therapies.
The responses above from the survey mirror the messaging received in the testimonials. Patients and caregivers expressed that side effects were reduced significantly in number and severity, which reduced suffering and improved their quality of life significantly. Some also indicated they felt they may have been able to avoid other medical interventions if this treatment had been available sooner. They also expressed Dostarlimab was more convenient, economical and accessible with the six-week infusion schedule, particularly for those living in rural areas such as a native reservation. This schedule allowed them more time to do the things that positively impactedtheir quality of life, rather than frequently being away from home and having so much precious time allocated to cancer treatment.
Anything Else?
Please provide any additional information that you feel may be useful for the evaluation of TBC (Dostarlimab).
“[I] hope it becomes available to more women so they don’t suffer like I did for so long with extensivechemo- radiation-surgery. All bad experiences. This immunotherapy is the best. Thanks to my Drs for giving this to me.”
“I was a good candidate”.
“I strongly recommend this drug. It gave my family a second chance. My mother meant the world tous and the thought of losing her was unbearable.”
Across survey responses and testimonials, patient and caregiver responses frequently echoed similar sentiments.
Patients are asking for treatments that are more effective, but with non-debilitating side effects so theycan continue to live an acceptable quality of life with less suffrage.
Patients would like to have more life outside of their treatment rather than occupying their time with numerous visits to the clinic.
Patients want to achieve the longest remission possible for themselves and have more time with theirfamily.
Patients would like more treatment options, and they want them available in all provinces for moreequitable healthcare.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drugreview processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict ofInterest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail thehelp and who provided it.
Patients within the network of the Division of Gynecologic Oncology at McGill University Health Center participated inthe survey along with patients within the CCS network.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes,please detail the help and who provided it.
The network of the Division of Gynecologic Oncology at McGill University Health Center provided the testimonials which were written by patients. One nurse assisted a caregiver by writing down his verbal testimonial as indicated in the testimonial itself. They also shared our survey directly with patients.
No one assisted CCS with the analysis of the survey or testimonials. CCS was the sole author of this submission.
List any companies or organizations that have provided your group with financial payment over the pasttwo years AND who may have direct or indirect interest in the drug under review.
Table 3: Conflict of Interest Declaration for Canadian Cancer Society
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
N/A | — | — | — | — |
GSK has not provided funds to CCS. Please let us know if you need information related to funding from other pharma companies that provide funds to CCS. To the best of our knowledge there are no existing conflicts of interest.
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Canadian Cancer Society
Date: November 5, 2021
Clinician Input
British Columbia Cancer Provincial Gynecological Oncology Tumour Group
About the British Columbia Cancer Provincial Gynecological Oncology Tumour Group
The BC Cancer Provincial Gynecological Oncology Tumour Group is comprised of clinicians, pathologists and researchers all involved in the treatment of patients with gynecological malignancies in the province of British Columbia. Clinicians include Medical Oncologists, Radiation Oncologists, Gynecological Oncologists and General Practitioners in Oncology. The tumour group is responsible for encouraging best standards of practice, updating treatment guidelines and advocating for access to novel therapies shown to have clinical efficacy.
Information Gathering
Please describe how you gathered the information included in the submission.
The information was gathered primarily from the GARNET trial, an open label single arm study with multiple cohorts, investigating the safety/efficacy of dostarlimab. There were two parts of the study, with part 2b enrolling patients who were diagnosed with advanced endometrial cancer and who had a mismatch repair deficient (MMRd)/microsatellite instability high (MSI- H) profile. Patients received dostarlimab IV, 500 mg q3weeks for 4 doses, then 1000 mg q6 weeks until disease progression/treatment discontinuation/withdrawal.
Current Treatments
Describe the current treatment paradigm for the disease.
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: There is a large unmet need for women diagnosed with advanced endometrial cancer.
Approximately 7400 women were diagnosed with endometrial cancer in 2020 in Canada, making it the most common gynecological cancer afflicting Canadian women. With 1300 people dying from the disease in 2020, it is the second most lethal gynecological cancer. Women diagnosed with early stage disease have a good prognosis, however, in the setting of having recurrent or metastatic disease, median overall survival is poor, with a 10-20% predicted 5 year survival.
In the first line setting, the standard treatment for advanced/metastatic disease would be platinum based chemotherapy, typically with carboplatin and paclitaxel, response rates in the range of 50% and median progression free survival of 13 months. In certain situations, such as hormone sensitive slow growing cancers or frail patients, endocrine therapy may be used instead of chemotherapy. Radiation therapy is an option for the palliation of symptoms.
Upon progression following first line therapy, there are no effective second line treatments available to Canadian women. The usual approach is to attempt single agent cyto-toxic therapy, however response rates are low, in the range of 15-20%, and a median progression free survival of 3 months.
Recently, pembrolizumab (an immunotherapy agent) was granted Health Canada NOC/c approval for patients diagnosed with MMRd/MSI-H endometrial cancer who have progressed on prior therapy with no other reasonable options available. Although efficacy data for pembrolizumab from the Keynote 158 trial in this setting has demonstrated a response rate of 57.1%, including a complete response rate of 16.1% and long duration of response, there is no public reimbursement for this agent for endometrial cancer.
Given the above, there is an urgent need for access to novel therapeutic agents that are well tolerated and efficacious, for the treatment of patients with advanced endometrial cancer in Canada.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: The goal of treatment would be to delay progression, prevent symptoms from disease, maintain/improve quality of life, and potentially prolong life. This would all be done while trying to prevent/minimize toxicities. Click here to enter response.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience.
Response: Unfortunately, once patients progress beyond first line treatment in the advanced/metastatic disease setting, the treatment goals are not met with the currently available therapies, which are single agent chemotherapy agents with poor response rates. Treatments that have demonstrated efficacy with minimal toxicity are not publicly re-imbursed.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Prior to recent data and regulatory approvals, patients with metastatic endometrial cancer had only one effective line of treatment available to them. Approximately 30% of patients have a MMRd/MSI-H profile (which includes Lynch Syndrome patients, who account for approximately 5% of all endometrial cancer cases). Studies have demonstrated that this patient population has the greatest potential to benefit from the use of immunotherapy/immune checkpoint inhibitors.
Place in therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: For patients with metastatic endometrial cancer and who are MMRd/MSI-H, upon progression of first line platinum-based chemotherapy, dostarlimab would be used as monotherapy. The GARNET trial has demonstrated that dostarlimab causes disease regression, and therefore is not only providing symptomatic management. An objective response rate of 42.3%, including 12.7% with complete response and 29.6% with partial response. The median duration of response was not reached, after a median follow-up of 11.2 months. The estimated maintenance of response was 96.4% at 6 months and 76.8% at 12 months. In addition, the most common Grade 3 treatment-related adverse events were anemia (2.9%), colitis (1.9%) and diarrhea (1.9%). Therefore, dostarlimab provides clinically meaningful anti-tumour activity with a very favourable safety profile, and not just symptomatic management. It is anticipated that dostarlimab and other immunotherapy agents will change the treatment pathway for patients with MMRd/MSI-H endometrial cancer.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: In the first line setting, patients diagnosed with metastatic endometrial cancer should be offered platinum based chemotherapy. Following progression of disease, in the setting of having a MMRd/MSI-H profile, it is recommended to proceed with immune checkpoint inhibition, such as dostarlimab, given that other treatment options (i.e. single agent non-platinum chemotherapy) offer poor rates of response. If there is rapid relapse (< 12 mo) of disease upon completion of adjuvant platinum-taxane-based chemotherapy (e.g. for stage I, II, or completely resected stage III disease) re- challenge with chemotherapy is not likely to be beneficial and is associated with significant toxicity (e.g. neuropathy, alopecia, fatigue, low blood counts). For those with dMMR or MSI-H disease, it would be recommended to treat with an immune checkpoint inhibitor in this setting.
Finally, there may be patients for whom either refuse chemotherapy or for whom chemotherapy may be contra-indicated or is already known to be very poorly tolerated. Treating with an immune checkpoint inhibitor as the first line of therapy may be appropriate in such selected cases, as long as the dMMR or MSI-H status of the cancer is confirmed.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: Dostarlimab would be offered to patients in the second line setting, who have progressed on platinum-based chemotherapy and who have a MMRd/MSI-H profile. This would shift the non-platinum single agent chemotherapy agents to the third line setting, examples being doxorubicin and gemcitabine.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Patients who are diagnosed with advanced endometrial cancer and who have a dMMR or MSI-H profile are most likely to respond given data from the GARNET trial investigating dostarlimab as well as other studies involving immune checkpoint inhibitors (e.g. pembrolizumab). There are no special disease characteristics that would refine this population further.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify). Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Patients would be identified after being diagnosed with endometrial cancer and undergoing biomarker testing looking for MMRd/MSI-H. This is routinely done on all patients diagnosed with endometrial cancer, and would identify eligible patients.
Which patients would be least suitable for treatment with the drug under review?
Response: At this time, patients who do not have a MMRd/MSI-H profile would not be suitable for this particular treatment.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review? If so, how would these patients be identified?
Response: As stated previously, patients diagnosed with uterine cancer are routinely tested for the presence or absence of MMRd/MSI-H. Patients with this profile have been shown to have the greatest chance of response to immune checkpoint inhibitors such as dostarlimab.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Patients would be monitored through regular clinical visits, physical exam, tumour markers if relevant, review of symptoms and periodic imaging with CT scan.
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: A meaningful response would entail symptom improvement along with evidence of disease stability/response via imaging. It would be important to also see patients have an improved functional status and be able accomplish their activities of daily living.
How often should treatment response be assessed?
Response: Typically, patients will be seen/examined every 3 weeks once treatment has started, to assess tolerance and for the presence of any toxicities. This may be extended to 6 weeks after cycle 5 of treatment, when the dostarlimab treatment intervals is lengthened. Imaging is typically done every 12 weeks, or to investigate new symptoms or physical findings.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify)
Response: Treatment discontinuation will happen in the setting of disease progression or toxicities that indicate that it is not safe to continue on with the current therapy.
What settings are appropriate for treatment with the drug under review?
Response: This treatment can be delivered in the community setting, outpatient clinics and speciality clinics/comprehensive cancer centres. There should be appropriate staff available (physician/nursing, etc) for monitoring and evaluating any potential adverse reactions.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: N/A
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: As highlighted above, given that endometrial cancer is the most common gynecological cancer affecting Canadian women, there is a large unmet need for women diagnosed with advanced disease. Data for immune checkpoint inhibitors such as dostarlimab have consistently demonstrated efficacy and should become part of standard of care. In addition, the goal of researchers has been to identify biomarkers to identify patients who are most likely to benefit from therapy, and in this case the dMMR or MSI-H status allows treatment to be used in the population most likely to benefit from therapy, reducing exposure to large populations for whom the agents will have little to no benefit, thus resulting in cost savings to the system.
Canadian patients have partnered in this research, as many participated in the GARNET study. As a result, many clinicians in Canada have patients who have responded well to immune checkpoint inhibition and continue to benefit from treatment with both complete responses and unusually long periods of disease control. Among those who respond, the benefit and treatment tolerance far exceed any results observed with either standard chemotherapy or hormone based treatments.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No. This submission was compiled using published clinical data and local experts only.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Aalok Kumar
Position: Medical Oncologist, BC Cancer Surrey; Provincial Systemic Gynecological Cancer Chair, BC Cancer
Date: 25-10-2021
Table 4: Conflict of Interest Declaration for British Columbia Cancer Provincial Gynecological Oncology Tumour Group Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSC | X | — | — | — |
AZ | X | — | — | — |
Merck | X | — | — | — |
Declaration for Clinician 2
Name: Anna Tinker
Position: Medical Oncologist, BC Cancer Vancouver. BC Cancer Gyne Oncology Provincial Tumour Group Chair
Date: 25/10/2021
Table 5: Conflict of Interest Declaration for British Columbia Cancer Provincial Gynecological Oncology Tumour Group Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AstraZeneca (honoraria and research grants) | — | — | — | X |
GSK | X | — | — | — |
Eisai | X | — | — | — |
Viatris | X | — | — | — |
Declaration for Clinician 3
Name: Susan Ellard
Position: Medical Oncologist and Department Leader, Medical Oncology, BC Cancer — Kelowna
Date: 26-10-2021
Table 6: Conflict of Interest Declaration for British Columbia Cancer Provincial Gynecological Oncology Tumour Group Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK | X | — | — | — |
AstraZeneca | — | X | — | — |
Merck | — | X | — | — |
Bristol Myers Squibb | X | — | — | — |
Declaration for Clinician 4
Name: Janice Kwon
Position: Gynecologic Oncologist; BC Gynecology Tumour Group Surgical Chair
Date: 26-10-2021
Table 7: Conflict of Interest Declaration for British Columbia Cancer Provincial Gynecological Oncology Tumour Group Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Astra Zeneca (hononaria and research funding) | — | — | — | X |
Declaration for Clinician 5
Name: Alannah Smrke
Position: Medical Oncologist BC Cancer Vancouver
Date: 26-10-2021
Table 8: Conflict of Interest Declaration for for British Columbia Cancer Provincial Gynecological Oncology Tumour Group Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Medison | X | — | — | — |
GSK | X | — | — | — |
McGill University Health Centre
About the McGill University Health Centre
This submission is from the doctors and nurses of Division of Gynecologic Oncology, McGill University Health Centre (MUHC). At the MUHC, patients with endometrial cancer are treated by a team consisting of three gynecologic oncologists, one medical oncologist, 4 fellows (in a Royal College of Physicians and Surgeons of Canada accredited training program), one clinical nurse specialist, one pivot nurse and eight oncology trained research nurses. We have 16 in-patient beds. We were the first team in Quebec to be awarded level IV (supra-regional) status by the Provincial Government (Programme Québécois de Cancérologie- Ministère de la Santé et des Services Sociaux) and have maintained this position since. Our research unit - Women’s Health Research Unit (WHRU) is active in investigator initiated basic and translation research supported by peer reviewed grants as well as in cooperative group (NRG) and industry sponsored clinical trials. Our websites are:
https://www.mcgill.ca/obgyn/divisions/gynecologic-oncology
https://rimuhc.ca/women-s-health
https://rimuhc.ca/whru-our-team
Information Gathering
The information in this submission is from our experience with own patients, our research findings (we have attached 2 papers) and our knowledge of the literature. With respect to dostarlimab, we started using it in the context of the GARNET trial in June 2017 and have more than 4 years of experience with it.
Current Treatments
Describe the current treatment paradigm for the disease.
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: Endometrial cancer is the 4th most common cancer in Canadian women. Approximately 7,400 women are diagnosed each year and 1,300 die from the disease. Most Canadian women are diagnosed with localized disease and are cured by surgery +/- adjuvant radiotherapy/chemotherapy. If diagnosed after the disease has disseminated (stage IV), it is difficult to cure. The disturbing fact about endometrial cancer is that it is one of the few cancers in Canada and other high resource countries inwhich the age-adjusted death rates have been rising steadily by 1.9% each year from 2009. Importantly, there is an alarming doubling of incidence in women 30-49 years. Thus, it is very important that we prevent recurrences, and if recurrences do occur, use the most appropriate treatment in terms of efficacy and toxicity.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: The most important goal is to prolong good quality life. Prolonging poor quality life with interventions that are associated with significant toxicity serves very little purpose.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience.
Response: For patients who recur with disseminated disease after the gold standard treatment of carboplatin and taxol, there is currently NO good treatment. To-date, >20 trials have investigated various chemotherapies and biologic therapies in this subset of patients; response rates achieved with chemotherapy is below 10%, median duration of response is <4 months and median overall survival less than a year. The grade ≥3 toxicity associated with these 2nds, 3rd line chemotherapies are high, exceeding 70%.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: The drug under review, dostarlimab is an anti-PD1, which is highly effective in patients with mismatch repair deficient endometrial cancer. Only about 20% endometrial cancers - about 1800/year irecurr in Canada. The sizable proportion of these are isolated recurrences and can be treated with surgery +/- radiation +/- chemotherapy. The 5-yr survival of newly diagnosed stage IV disease is 21%, and for recurrent chemo-naïve disease it is 21%. However, recurrence after the gold standard treatment of platinum and taxol, has a dismal prognosis with a 5-year survival of 8-9%. Fortunately about 30% of these patients have tumours that have a deficient mismatch repair pathway (dMMR). These tumours are exquisitely sensitive to dostarlimab. So dostarlimab is indicated for this subpopulation – a niche group
Place In Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: For patients with dMMR tumours, which recur after platinum-based chemotherapy, dostarlimab should be offered. It is highly effective, works quickly and has minimal toxicity. We have tried pembrolizumab in these patients as we have access to pembrolizumab from our hospital pharmacy, if we made a special case for it. Pembrolizumab works in dMMR patients but much more slowly than dostarlimab and has more side effects. However, as it available to us outside of a clinical trial but dostarlimab is not, we have quite a lot of experience with pembrolizumab for dMMR recurrence. If the patient has a high burden of disease, because pembrolizumab works much more slowly, we combine it with chemotherapy to get a handle on the disease. This increases toxicity. However, even in the face of a high burden of disease, dostarlimab works alone and patients experience relief with a couple of cycles. Grade ≥3 toxicity with dostarlimab, is very low, experienced by <3% of patient. So, in our experience, dostarlimab is the most humane, fiscally prudent option for best option for treating patients with dMMR tumours, which has recured after platinum-based chemotherapy.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: It is false economy to delay starting a highly effective, treatment with minimal toxicity in this niche population. Trying 2nd line chemotherapy alone in these patients is not advisable. It does not work and just causes toxicity. Patients spent most of their time in the ER and inpatient unit with symptoms from the increasing burden of cancer or side effects of the chemotherapy.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: After dostarlimab has failed in patients who had failed platinum-based chemotherapy, in our hands nothing else has worked. We counsel such patients and offer palliative/supportive care. However, if patients have responded well, but the drug was interrupted for any other reason and the disease has recurred, it would be appropriate to restart, and in our experience it works again.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: The patients most likely to respond are dMMR patients. However, some patients who are MMR proficient have also responded and the responses have been durable. We are not quite certain which is the best biomarker to identify the subgroup of MMR proficient patients who respond, but we think it may be evaluation of tumour mutational burden. However, this needs more study.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify). Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: The gratifying aspect of this treatment is that the biomarker test for identifying dMMR patients is readily available in any tertiary care pathology laboratory. The test is inexpensive, and interpretation is standardized and within the skill set of any gynecologic pathologist.
Which patients would be least suitable for treatment with the drug under review?
Response: Only a subset of MMR proficient endometrial cancer patients who recur after platinum-based chemotherapy are likely to respond to dostarlimab. So, for such patients, we need better biomarkers.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review? If so, how would these patients be identified?
Response: The response rate of dMMR endometrial cancer patients to dostarlimab is high, between 42-70%. In patients who respond, the responses are durable with 77% of patients continuing to respond for >12 months.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Patients are best monitored by CT scan and symptoms. If there is disease progression in 3 cycles, we follow up in 6 weeks to ensure it is not pseudo-progression and if progression is confirmed, we discontinue the drug.
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: Clinical symptom relief and CT scans are the best way to monitor patients. Stable disease, partial response and complete response on CT, with minimal or manageable toxicity is a good reason to continue the treatment.
How often should treatment response be assessed?
Response: Initially, the dostarlimab is best given in 500mg doses every 3 weeks for 4 cycles and then as 1000mg every 6 weeks. After the first 3 cycles, it is best to repeat the CT to assess response. If there is no progression, and the patient is doing well the CT monitoring can be spaced out to 12 weeks.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify)
Response: The treatment should be discontinued if there is disease progression and pseudo- progression is ruled out by a repeat CT in 6 weeks.
What settings are appropriate for treatment with the drug under review?
Response: Dostarlimab has minimal toxicity, and this is its strength. So, it can be given in the community setting, outpatient clinic etc. Patients should be educated about potential toxicity and should be monitored for AEs. However, we treat pts from the cree nation which is two plane rides away from Montreal. Patients are assessed, have blood tests, and if all is well, they are given the infusion, and leave. From our experience it would be appropriate to work closely with local physicians and share responsibility and care of patients with local doctors.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: NA.
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: We believe that the humane, sensible and cost effective treatment of recurrent dMMR tumour which has failed platinum based chemotherapy is to use dostarlimab as opposed is repeating chemotherapy that does not work. We have previously reported on total direct healthcare costs associated with repeated regimens of cytotoxic chemotherapy. We reported that the proportion of patients in whom total direct health care costs exceeded $79,000/patient was only 19% when the first recurrence was treated with chemotherapy. But total direct healthcare rose steeply with every subsequent recurrence and cytotoxic regimen, so that by the time we used 2 cytotoxic regimens, the direct health care costs exceeded $79,000/patient in 47% of patients. The steep rise in healthcare costs is associated with frequent inpatient admissions that these patients needed to tide them over acute symptoms. Although the population studied for the above study were ovarian cancer patients, the cytotoxic chemotherapies we use for recurrent and advanced endometrial cancer are the same as those we use for ovarian cancer, except that they are even less effective for recurrent endometrial cancer (paper attached).
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Lucy Gilbert, MD MSc, FRCOG
Position: Director, Division of Gynecologic Oncology, McGill University Health Centre
Date: 29/10/2021
Table 9: Conflict of Interest Declaration for McGill University Health Centre Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK for participation in Advisory Boards and Steering Committee | — | X | — | — |
Institutional Grants received from GSK (RUBY, FIRST and GARNET clinical trials) | — | — | — | X |
Declaration for Clinician 2
Name: Xing Zeng MDCM, FRCSC
Position: Gynecologic Oncologist
Date: 29/10/2021
Table 10: Conflict of Interest Declaration for McGill University Health Centre Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Institutional Grants received from GSK (RUBY, FIRST and GARNET clinical trials) | — | — | — | X |
Declaration for Clinician 3
Name: Victoria Mandilaras MDCM, FRCSC
Position: Medical Oncologist
Date: 29-10-2021
Table 11: Conflict of Interest Declaration for McGill University Health Centre Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK | X | — | — | — |
Ontario Health Cancer Care Ontario’s Drug Advisory Committee
About Ontario Health Cancer Care Ontario’s Drug Advisory Committee
Please describe the purpose of your organization. Include a link to your website (if applicable).
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug- related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
This input was jointly discussed with the listed DAC members.
Current Treatments
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines?Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: For patients who failed platinum treatment, currently there’s no standard of care treatment and minimal options. Patients receiving treatment beyond 1L are likely to have chemo-resistant disease. There is a lack of data for treatment for these patients.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: Prolong life, delay disease progression, reduce severity of symptoms, improve QoL, reduce burden on caregivers, maintain independence, minimize toxicities
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience
Response: Less than 15% treatment response and rapid progression and death. Median PFS is around 3 to 3.5 months in this population. There is no standard second-line options and no meaningful and effective treatment for these patients. Second line agent (doxorubicin) is toxic and challenging in older population.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Endometrial cancer population is rising in Canada – with rising incidence and mortality (Ref: Canadian Cancer Society); 30% patients will have dMMR. Historically, these patients are insufficiently studied by clinical trials and resulting in inequity in care compared to other disease sites. Additionally, these patients are not represented by advocacy groups which may result in inequitable access to treatment. Endometrial cancer is a heterogeneous disease. It’s only recently that biomarkers are identified in this disease. There is a class effect to immunotherapy in dMMR tumours that is especially important in endometrial cancer.
Place In Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: If recommended by the Expert Committee, dostarlimab will be the first targeted agent funded for endometrial cancer. It addresses a significant gap and provide the patients with an effective and meaningful treatment beyond first-line.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective. If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: There are no other funded options available
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: This will be given after carbo-Taxol. No effect on sequencing.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: As per the indication - treatment of adult patients with recurrent or advanced mismatch repair deficient (dMMR) or microsatellite instability-high (MSI-H) endometrial cancer (EC) that has progressed on or following prior treatment with a platinum containing regimen
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify). Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Reflex IHC for MMR protein is standard of care for all EC and is funded in Ontario. There’s now improved understanding of the different classification of EC and predictive value of dMMR and immunotherapy response. These patients will now need effective treatment as a result of the companion diagnostics.
Which patients would be least suitable for treatment with the drug under review?
Response: Patients with contraindications to immune checkpoint inhibitor; patients with intact MMR.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review? If so, how would these patients be identified?
Response: mismatch repair deficient (dMMR) by IHC or MSI-H by PCR
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Standard of care – physical exam and imaging; symptom surveillance
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: Partial, complete response or stable disease. Improvement of disease symptoms. GARNET trial demonstrated some patients have durable response.
How often should treatment response be assessed?
Response: As per standard of care
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify)
Response: disease progression, adverse events, additional treatment needed; patient’s choice
What settings are appropriate for treatment with the drug under review?
Response: Outpatient clinic
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: N/A
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: There is a significant unmet need for these patients. Although there are other Health Canada approved drug (pembrolizumab) for this indication, but it is not currently funded.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Sarah Ferguson
Position: Ontario Cancer Lead, gynecologic oncologist
Date: 25 Oct 2021
Table 12: Conflict of Interest Declaration for OH-CCO’s Drug Advisory Committee Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK — No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Helen MacKay
Position: Head, Division of Medical Oncology & Hematology – Sunnybrook
Date: 17 Sep 2021
Table 13: Conflict of Interest Declaration for OH-CCO’s Drug Advisory Committee Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK — No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Stephen Welch
Position: Medical oncologist
Date: 17 Sep 2021
Table 14: Conflict of Interest Declaration for OH-CCO’s Drug Advisory Committee Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK — No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Orit Freedman
Position: Gynecologic oncologist
Date: 17 Sep 2021
Table 15: Conflict of Interest Declaration for OH-CCO’s Drug Advisory Committee Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK — No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr. Taymaa May
Position: Surgical oncologist
Date: 17 Sep 2021
Table 16: Conflict of Interest Declaration for OH-CCO’s Drug Advisory Committee Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK — No COI | — | — | — | — |
Declaration for Clinician 6
Name: Dr. Julie Francis
Position: Gynecologic oncologist
Date: 17 Sep 2021
Table 17: Conflict of Interest Declaration for OH-CCO’s Drug Advisory Committee Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK — No COI | — | — | — | — |
Declaration for Clinician 7
Name: Dr. Leah Jutzi
Position: Gynecologic oncologist
Date: 17 Sep 2021
Table 18: Conflict of Interest Declaration for OH-CCO’s Drug Advisory Committee Clinician 7
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK — No COI | — | — | — | — |
Declaration for Clinician 8
Name: Dr. Josee-Lyne Ethier
Position: Medical Oncologist
Date: 23 Sep 2021
Table 19: Conflict of Interest Declaration for OH-CCO’s Drug Advisory Committee Clinician 8
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK | X | — | — | — |
Princess Margaret Cancer Centre
About Princess Margaret Cancer Centre
The Gynecologic Cancers Disease Site Group at the Princess Margaret is a multi- and inter-disciplinary team of oncologists and allied health professionals, with a clinical focus in the management of patients diagnosed and living with gynecologic cancers. The physician team consists of medical, radiation and gynecologic oncologists, with support from radiologists and pathologists with expertise in gynecologic cancers.
Information Gathering
Peer-reviewed manuscripts of completed clinical trials and conference proceedings were reviewed to provide the information included in this submission.
Current Treatments
Describe the current treatment paradigm for the disease.
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: Endometrial cancer is a clinically heterogenous disease. Although the majority of patients present with early-stage, potentially curable disease, 20% of patients who present with more aggressive histology cancers have a high risk of recurrent disease that is non-curative. For these patients, systemic therapy is the mainstay of treatment, but options are limited, and responses short-lived. Standard treatment options for patients with recurrent/metastatic endometrial cancer include endocrine therapy with aromatase inhibitors and progestins, and cytotoxic chemotherapy. Unfortunately, responses are not very durable. Once patients experience disease progression on the standard 1st line platinum/taxane regimens, options are extremely limited and 2nd-line chemotherapy regimens have response rates of less than 20% and provide benefit for 3 to 4 months.
An improved understanding of the molecular background of endometrial cancer including the ability to characterize specific molecular subgroups, has defined new treatment options for patients with recurrent disease. These include molecularly targeted agents, including anti-angiogenic agents, and immunomodulatory approaches such as the immune checkpoint inhibitors. Endometrial cancer is also recognized as being a more highly immunogenic tumour, making immunotherapy strategies particularly attractive options. Defects in mismatch repair (MMR) leading to microsatellite instability (MSI) defines a patient subgroup with high mutational tumour burden (TMB) that is likely to benefit from immune checkpoint inhibitor treatments.
Across different tumour types, endometrial cancer appears to have one of the highest frequencies of MSI or MMRd, with up to 30% patients with recurrent endometrial cancer in this subgroup. A smaller percentage of patients harbour oncogenic variants in POLE, which also leads to high tumour mutational burden.
At the current time, patients with endometrial cancer can only access molecularly targeted agents or immunotherapy approaches through a clinical trial. However, with the growing body of literature supporting the use of immune checkpoint inhibitors in patients with tumours with high tumour mutational burden related to MMR-defects (MMR-d) or POLE mutation, the inability to access these agents uniformly represents a significant gap in care for Canadian patients with recurrent/advanced endometrial cancer. There is currently no patient-support or compassionate access program available, and none of the immune checkpoint inhibitors are funded through provincial programs.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: Standard 2nd-line chemotherapy options include dose-dense paclitaxel and doxorubicin. Median progression-free-survival (PFS) on these regimens is 3 to 4mths, and toxicity is not insignificant. An ideal treatment would provide more durable disease control (PFS is an acceptable surrogate of disease control), with an acceptable tolerability profile leading to no adverse effects on patients’ quality of life. Ideally, an agent should also lead to improved overall survival in comparison with an accepted standard of care.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience.
Response: The current 2nd line options of chemotherapy provide very limited benefit for patients and are associated with significant toxicity including fatigue, neurotoxicity and myelosuppression. The benefit of chemotherapy is even more limited in patients with low grade endometrial cancers. In the subgroup of endometrial cancer patients with high mutational tumour burden due to MMR-d (MSI-hi) or POLE-mutation, immune checkpoint inhibitors, and specifically PD-1 and PD-L1 inhibitors have been demonstrated to have response rates of 40 to 50% with durable disease control. Specifically, pembrolizumab monotherapy had an overall response rate (ORR) of 57% in 49 patients with MMR-d or MSI-hi endometrial cancer. More importantly, responses were durable, with > 90% lasting longer than 9-months, which is remarkable given approximately 50% patients had already experienced disease progression on > 2 lines of systemic therapy. This study included a total of 151 patients with a range of tumour types; 15% experienced > Grade 3 treatment-related adverse events and approximately 10% had to discontinue treatment due to treatment-related toxicity (Marabelle A et al, JCO 2019). The more recent experience with dostarlimab monotherapy in 103 patients with MMR-d endometrial cancer, 40% of whom had received 2 or more prior lines of therapy, was equally impressive, with an ORR of 44%. Notably, in a larger cohort of 316 patients evaluable for safety, treatment-related adverse events > Grade 3 occurred in < 15% and lead to discontinuation of therapy in < 5% patients (Berton D, ASCO 2021).
This data supports efficacy and tolerability of PD-1 inhibitors in patients with MMR-d or MSI-high endometrial cancer. However, at this time there is no path to access these agents apart from clinical trial participation or self-funding, leading to inequities in care that can be provided.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Endometrial cancer is considered an immunogenic tumour and there are multiple on-going studies evaluating different combination therapies which include immunomodulatory drugs. At this time however, it is quite clear that the subgroup of patients with tumours with high TMB related to MMR-d/MSI or POLE mutations, are a group of patients who can be predicted to have a good response to monotherapy with a PD-1 inhibitor such as dostarlimab.
Place In Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Recent studies of immune checkpoint inhibitors in patients with MMRd endometrial cancer with disease progression after platinum/taxane based therapy has demonstrated improvements in both PFS and QoL. KN-158 evaluated efficacy of pembro in patients with MSI-hi/MMR-d tumours and included 49 patients with endometrial cancer, demonstrating a response rate (RR) of 57% which included 8 pts (16%) with complete response (CR) and 20 pts (41%) with partial response. 22 pts (9%) had to d/c treatment due to adverse events. PD-L1 inhibitors avelumab and durvalumab have had ORR of 27% and 43% respectively in patients with MMR-d tumours.
Response rates in patients with MMR-proficient tumours (MMR-p) are lower. Based on the current data, monotherapy with a PD-1 inhibitor like dostarlimab should be considered for patients with MMR-d tumours who have had disease progression after 1st line platinum-based therapy.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Dostarlimab has demonstrated efficacy in patients who have had disease progression on at least one line of systemic therapy, with approximately 40% patients having received 2 or more prior lines of therapy. Based on this data, the recommendation at this time would be for patients to be considered for standard 1st line therapy with a platinum/taxane combination. At the time of disease progression, provided patients maintain good performance status (ECOG 0/1) and preserved end-organ function, the current body of evidence supports the use of PD-1 monotherapy such as dostarlimab.
Studies are on-going evaluating the benefits of dostarlimab and other immune checkpoint inhibitors in the first-line setting (i.e. in combination with chemotherapy), and therefore this strategy would only be recommended within the confines of a clinical trial.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: After progression on dostarlimab patients can be considered for standard of care chemotherapy or clinical trials. At this time, there is limited data to support the use of dostarlimab-combination therapy after progression on dostarlimab. This would not be recommended outside of the confines of a clinical trial.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: N/A
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify). Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.) Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: The most relevant selection strategy would be to identify patients with MMR-d/MSI-high tumours, and with tumours harbouring POLE mutation. MMR IHC is a common and fairly standard histological test that can be easily done on request for patients with recurrent endometrial cancer. Otherwise, patients may also be identified if they have had had somatic molecular profiling through a translational research program.
Which patients would be least suitable for treatment with the drug under review?
Response: The current body of data suggests that patients with MMR-proficient endometrial cancer (i.e. without defects in MMR pathway) are much less likely to benefit from monotherapy with an iCPI. Recently completed and on-going studies suggest that certain subsets of these patients may benefit from approaches that combine an iCPI with another molecularly targeted agent (eg anti- angiogenic or PARP inhibitor) and therefore, this group of patients should be considered for other therapeutic strategies including clinical trials.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review? If so, how would these patients be identified?
Response: Patients can be identified by tumour MMR immunohistochemistry, MSI PCR or mutation testing for POLE (if available). PD-L1 status does not appear to predict for tumour response and would not be required
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Response to immune checkpoint inhibitor therapy would be based on tumour assessment by CT or MR completed every 2 to 3 cycles of therapy (i.e. every 6 to 9 wks)
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms.
Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: A clinically meaningful response would be maintaining radiographic disease control (i.e. tumour response or stabilization on CT/MR) with good tolerance of treatment (i.e. </= grade 2 treatment- related adverse effects) and stable or improving symptoms of disease. Assessment of radiographic response is objective, however determination of clinical benefit will have an element of subjectivity.
How often should treatment response be assessed?
Response: Treatment response should be assessed radiographically with computed tomography (CT) or magnetic resonance imaging (MR) every 2 to 3 cycles of therapy (ie every 6 to 9 weeks).
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify).
Disease Response: Patients would continue on treatment as long as there was a confirmed favourable response to therapy which includes a complete/partial tumour response or disease stabilization, with stable or improved symptoms and good tolerability. There are patients who may have improved or stable symptoms but evidence of tumour progression, with increase of existing or development of new lesions. This may be consistent with the phenomena of pseudo- progression. If patients are clinically stable then they would continue on treatment, If next set of imaging demonstrates progression patients should discontinue therapy.
Adverse Events: Treatment should be held for moderate-severe immune-related toxicity and managed as per standard guidelines (Haanen J, Carbonnel F, Robert C et al. Annals of Oncology 2017; 28 (4)). Re-challenge would be per discretion of treating physician in conversation with patient, and usually can be considered if severity of immune-related event was < Grade 3. (Dolladille C et al; JAMA Oncology 2020)
What settings are appropriate for treatment with the drug under review?
Response: Patients being managed at a cancer centre by oncologists with expertise in (1) systemic therapy for gynecologic cancers and (2) managing immune-related adverse events.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: N/A
References
Green A, Feinberg J and Makker V. A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. ASCO Educational Book 40, 2020
Haanen J, Carbonnel F, Robert C et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology 2017; 28(4): 119-42
Dolladile C, Ederhy S, Sassier M et al. Immune Checkpoint Inhibitor Rechallenge after Immune-related Adverse Events in Patients with Cancer. JAMA Oncology 2020; 6(6): 865-871
Marabelle A, Le D, Ascierto P et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. JCO 2019; 38: 1-10
Berton D, Banerjee S, Curigliano G et al. Antitumour Activity of Dostarlimab in Patients with Mismatch Repair-Deficient (dMMR) Tumours: A Combined Analysis of 2 Cohorts in the GARNET study. 2021 ASCO Jun 4-8; Abst #2564
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
GSK team provided slides from the GARNET study presented at ASCO 2021.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Neesha Dhani
Position: Staff Medical Oncologist, Gynecologic Cancers Disease Site Group
Date: Oct 26, 2021
Table 20: Conflict of Interest Declaration for Princess Margaret Cancer Centre Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Amit Oza
Position: Head, Division of Medical Oncology & Hematology, Staff Medical Oncologist, Gynecologic Cancers Disease Site Group
Date: 28-Oct-2021
Table 21: Conflict of Interest Declaration for Princess Margaret Cancer Centre Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Stephanie Lheureux
Position: Staff Medical Oncologist, Gynecologic Cancers Disease Site Group
Date: Oct 28-2021
Table 22: Conflict of Interest Declaration for Princess Margaret Cancer Centre Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Robert Grant
Position: Staff Medical Oncologist, Gastrointestinal & Gynecologic Cancers Disease Site Groups
Date: Oct 28-2021
Table 23: Conflict of Interest Declaration for Princess Margaret Cancer Centre Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Saskatchewan Cancer Agency Gynecological Oncologists
About Saskatchewan Cancer Agency Gynecological Oncologists
There are a total of six Gynecological Oncologists (GO’s) in the province covering 2 sites which is the Saskatchewan Cancer Agency (SCA). Regina and Saskatoon each have 3 practitioners.
Information Gathering
We, as a group have a deep interest in having treatment options for recurrent endometrial cancer as it is so common and minimal treatment options. There are PDL-1 available in USA and a collegue used to work in USA, and so we know the pembro literature well. Subsequently the GARNET study became available. We did reach out to GSK to know more and had meeting with them.
Current Treatments
Response: After the GARNET study was reported this become clear that a level 3 study will not be performed. The FDA has approved similar agents and states and the efficacy and safety profile are very similar. My colleague who worked in the states for 4 years has experience with a similar PD 1 immune therapy with excellent result and minimal toxicity. This study supports that Dostarlimab has very similar outcomes and should be considered as having more competition means lowered costs for the HC system.
Candida needs access to this drug as well. Especially based the data, this year volume of these patient and the fact that is been used in the United States again my patient has been accessing a similar drug through an outside source thing age cost and it will impact on lifestyle probably forever.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Response: Quality of life is always most important indicator in research studies for cancer especially in recurrence disease. I certainly a.m. Of the mind that if you have a palliative patient a treatment that is improving there symptoms I rely last on the imaging and her tumour markers if relevant.
Prolonging life for me as an oncologist is not the main goal although for some patients it is. In my recent at anecdotal N=1 I have a patient with the biggest oligometastasis in her chest that I have ever seen. This is refractory to hormone therapy and chemotherapy and her level of pain was almost ready to be admitted to a palliative care unit for thoracic tap block. As we do not have access to a PD-L targeted agent she is paying out of pocket and within 2 weeks of her 1st treatment she was out at the beach. This is so incredibly meaningful as a care provider but all of the examples above certainly commented the discussion. Just for some page the goals and expectations are different but this has separate control of targeting many of these areas as it is quite well tolerated
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Response: I think all of these apply. Many endometrial cancer is chemo refractory because there slow growing. If they recur the will eventually become refractory to current treatment and some patients cannot tolerate doublet chemotherapy which is certainly the standard of care Sami progress sooner.
Immunotherapy also with better target agent that intuitively makes sense to use especially we have the knowledge of the MMR status and standard care. As such the targeted component of history is why should be considered for implantation into level 3 evidence even without level 3 evidence specific to this particular drug
Which patients have the greatest unmet need for an intervention such as the drug under review?
Response: Endometrial cancer is the common gynecological malignancy and rate going up steadily over the last 30 years. This trend will continue the increase of obese continues to rise. Currently we have no robust treatments when these patients progress/recur. There are new novel therapies for much rarer cancers and is a continue frustration of myself my colleagues we have no meaningful treatment options for such a large component of her patient population.
Place In Therapy
How would the drug under review fit into the current treatment paradigm?
Response: Similar drugs have been approved. There response as already chief to the paradigm in the United States. Patient's her paying out of pocket her seeing dramatic FX and make since is now weighs mainstay of cancer treatment to look at immunotherapy and more targeted agents which this is looking at MMR deficient cancers. So no this is not the 1st treatment that would be approved to address the underlying disease process however it study shows similar responses and should be an option for practitioners.
I do not see that in the near future to be considered first-line treatment based on the study profile but certainly there is some people that believe should be first-line treatment. There is some evidence that adding another PDL inhibitor to Lenvatinib has activity and another drug is being studied currently with carboplatin and paclitaxel. The treatment X of the stroke are high enough that it would give some prolonged disease-free progression however there is certainly a possibility that for some patients would not be there last line of treatment
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Yes, this treatment would be provided to patients after first line, platinum containing drugs at the time of recurrence. There are more studies on another PDL-1 inhibitor and dostarlimab
How would this drug affect the sequencing of therapies for the target condition?
Response: At the current time this would be after first-line treatment. This drug would not be used again further treatment regimen many evidence that I am aware of.
Which patients would be best suited for treatment with the drug under review?
Response: Those that are DNA mismatch repair deficient/and high levels of MSI-H instability. This testing is being done on all colorectal and endometrial cancers and, as such, if we have the test already being we should be able to offer targeted agents based on this testing. It is being done anyway as it will make treatment even more cost effective because they will be more targeted and appropriate although it is not standard of care, tumour mutational burden (TMB) may also help target who is going to respond better.
Some of these patients did respond to hormone therapy and sometimes this can cause a very sustained response to treatment this to no avail standard chemotherapy often has minimal benefit.
Patients with recurrence disease all want treatment options, especially if the performance status allows an for such a common cancer does not ongoing for straight the that we have such minimal options for these patients.
I do not foresee that recommendations for treatment would be different based on disease characteristics. As there is some risk of pneumonitis with her people would be hesitant to give a patient with a significant disease burden in the chest this drug, I am not sure, but we do need options.
How would patients best suited for treatment with the drug under review be identified?
Response: Recurrent disease (measurable). Platinum given and considered resistant and no more than 2 treatments prior. MLH deficient. PDL-1 naïve. No contraindications.
Which patients would be least suitable for treatment with the drug under review?
Response: Anything poor performance status would be considered. MMR proficient be change with the use it with another drug over time, and some would be hesitant within exist pulmonary condition.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: This would not actually change at all from current practice because all of her endometrial cancers are determined to be MMR deficient her proficient at the time of their biopsy or hysterectomy if not done prior.
Is there some other tests such is mutation burden that may be of help but these are not currently in practice routinely. What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Response: Although not sometimes completely documented in imaging outside of a study protocol RECIST criteria would be the standard but again in recurrence setting with palliative intent treatment this should be equally weighed with symptoms improvement if the patient is initiated on treatment with symptomatic progression. There are no tumour markers to follow this particular cancer so symptoms, radiographic imaging plus or minus exam which is often less help this particular cancer be utilized.
Certainly when disease is in the chest & symptomatic lymphadenopathy in the retroperitoneal can be easy to follow-up
What would be considered a clinically meaningful response to treatment?
Response: I think there is variability b/t all physicians but I certainly hope and believe that, as stated above physicians will look at the whole patient including imaged and patient symptom profile. ECOG this assessed at every visit and quality of life should be part of any drug implementation and I believe this drug has demonstrated this as well as its other drugs in its class for tolerability and maintain patient's quality of life How often should treatment response be assessed?
How often should treatment response be assessed?
Response: Symptom based for some but at onset of treatment likely at 3 -6 month intervals imaging would be utilized and then likely less often thereafter. Laboratory values as well as respiratory/ pulmonary status will also be assessed to ensure that treatment response is occurring without toxicity.
Sometimes a challenge with this particular drug classes that disease is often in the lung and there can be symptomatic pneumonitis with this drug which can sometimes be challenging to delineate the cause.
What factors should be considered when deciding to discontinue treatment?
Response: Measurable or clinical progression.
Toxicity: anemia that is profound transfusion, patient intolerability for non-safety related reasons such as fatigue, nausea, diarrhea, pruritus. Pneumonitis, renal failure, hyponatremia
What settings are appropriate for treatment with the drug under review?
Response: Immediate reactions are rare and so the cancer agency may allow in Community outreach centers
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: N/A
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: The lack of level 3 evidence should not be a penalty for this drug as the class of drug has proven its efficacy in this disease site and its parameters for use. Endometrial cancer is COMMON and lacks robust treatment options in recurrent setting and it should be a priority for Health Canada to approve appropriate options as it affect thousands of families annual. Its efficacy, tolerability, and lack of severe toxicity should be of tremendous excitement for moving through approval process easily and in EXPEDITIOUSLY. Other drugs for rarer tumours and less robust/significant benefit have approval and it is time for this common cancer treatment to be of equal importance.
We implore the thorough assessment and for approval of Dostarlimab.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Having a new colleague who worked in the State we are very keen to have immunotherapy access in Canada. She is constantly searching the literature and supplies with information. In gaining information we did have an academic presentation from GSK with respect to this drug at our request.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No, we did not.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Vickie J martin
Position: Gynecological Oncologist
Date: 20/10/2021
Table 24: Conflict of Interest Declaration for Saskatchewan Cancer Agency Gynecological Oncologists Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr Laura Hopkins
Position: Gynecological Oncologists
Table 25: Conflict of Interest Declaration for Saskatchewan Cancer Agency Gynecological Oncologists Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
The Society of Gynecologic Oncology of Canada
About the Society of Gynecologic Oncology of Canada
The Society of Gynecologic Oncology of Canada (GOC) is a non-profit multidisciplinary organization. It is the national society representing health care professionals including physicians, nurses, and scientists Involved in the treatment and prevention of gynecologic cancer. GOC strives to improve the care of women with, or who are at risk of, gynecologic cancer by raising standards of practice, encouraging ongoing research, promoting innovation in prevention, care and discovery and advancing awareness.
Information Gathering
The data for this submission is from the GARNET trial. This multi-center, open label, single-arm, multi-cohort study evaluated the safety and efficacy of dostarlimab monotherapy in 2 parts, dose escalation and expansion. Part 2B enrolled patients into 4 expansion cohorts, including a cohort with mismatch repair deficient (MMRd)/Microsatellite- instability high (MSI-H) endometrial cancers (cohort A1). The trial assessed the antitumour activity and safety of dostarlimab. Patients received dostarlimab intravenously at 500 mg every 3 weeks for 4 doses, then 1000 mg every 6 weeks until disease progression, treatment discontinuation, or withdrawal.
Current Treatments
Describe the current treatment paradigm for the disease.
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: Treatment options for women diagnosed with endometrial cancer represent a critical unmet need. Endometrial cancer is the most common cancer gynecologic cancer affecting women in Canada with an estimated 7400 women diagnosed in 2020. It’s the second most lethal gynecologic cancer, approximately 1300 will die of the disease in Canada in 2020. Whilst prognosis remains good for those diagnosed with early-stage disease, for those with recurrent or metastatic endometrial cancer median overall survival is short. The standard of care first-line treatment for advanced or metastatic disease is platinum-based chemotherapy, or less commonly, endocrine therapy for hormone sensitive slow growing cancers.
Radiation therapy can be used but is only applied to the palliation of cancer related symptoms and in a focal manner.
There are no effective second-line treatments accessible for Canadian women. Response rates to second-line cytotoxic agents are low (< 20%) with median Progression Free Survival (mPFS) of 3-4 months. Health Canada recently issued a NOC/c approval for immunotherapy agent pembrolizumab for dMMR or MSI-H endometrial cancers that have progressed following prior therapy and that have no satisfactory alternative treatment options. Pembrolizumab in dMMR endometrial cancer has a high response rate (57.1% ORR) a notable complete response rate (16.1% CR) and long durations of response (DOR not reached: at 12 months, 89% of patients were still in response) (phase 2 data, Keynote 158 trial). However, there is no funded access (e.g., no public funding, no Special Access Programs and no Compassionate Access) to pembrolizumab for dMMR or MSI-H endometrial cancer in Canada. Only those with insurance coverage or the capacity to self-pay have access to pembrolizumab, creating a major disparity in access to treatment. Broadly, Canadian women with dMMR or MSI-H endometrial cancer do not have access to immunotherapy.
Hormone therapy is accessible for a subset of endometrial cancers, typically those with slower growing disease that is low grade (FIGO grade 1 or 2) and ER/PR positive. Such treatments are available across Canada as they are inexpensive and funded by most jurisdictions, however, they are not highly effective (RR <40%, PFS ~3 mo) thus most women experience disease progression and require additional therapy.
Access to new, well-tolerated, therapeutic options for Canadian women diagnosed with endometrial cancer is urgently needed. GOC is advocating on behalf of its membership and Canadian women diagnosed with endometrial cancer who do not have a national advocacy organization to represent them.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: As with most oncology treatment regimens in the recurrent setting, goals are to delay progression, reduce disease burden and symptoms from the cancer, improve quality of life, and if, possible prolong life. The recurrent endometrial cancer setting, primary goals would be to improve progression free survival while minimizing toxicities and side effects from treatments.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments. Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience.
Response: Presently, none of the treatment goals in Section 4 are being met in patients who have already received standard first-line therapy. All women with recurrent endometrial cancer become refractory to standard therapy and there is both a lack of novel treatment and limited/no access to any proven treatments.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Until recently, there has only been one-line of treatment for metastatic endometrial cancer. Therefore, there is a need in the entire population of those with recurrent disease. However, up to 30% of recurrent endometrial cancers are MSI-H or dMMR. This includes ~5% with endometrial cancers related to Lynch Syndrome, as hereditary cancer syndrome which increases the risk of endometrial cancers (as well as other cancers such as colorectal carcinoma). This biomarker defined population has the greatest potential to benefit from immune checkpoint inhibitor therapy.
Place In Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Dostarlimab would be used as monotherapy in the advanced or metastatic endometrial cancers that are dMMR or MSI-H who progressed on or after prior platinum treatment. The drug would be used in biomarker defined population that is most likely to benefit from treatment. The drug causes disease regression and therefore does directly affect the underlying disease process. This agent, and others in this class, are expected to shift the paradigm in the treatment of dMMR disease, including dMMR endometrial cancer.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Patients with recurrent metastatic endometrial cancer should receive platinum-based therapy first. When those with dMMR tumours have progression following first-line platinum-based, we would recommend proceeding with treatment using a with an immune checkpoint inhibitor such as dostarlimab. Other treatment options have poor response rates and short durations of benefit, (RR <30%, mPFS < 3 mo).
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: Dostarlimab would be offered after progression on platinum-based therapy (in the dMMR cohort only). After progression on dostarlimab, 3rd line agents such as adriamycin, paclitaxel, gemcitabine, could be offered.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Patients with recurrent dMMR or MSI-H endometrial cancer having progressed on platinum-based therapy. As previously mentioned, this is a niche population of patients who stand to benefit the most from dostarlimab treatment, as current second line therapies have poor response rates a lower duration of responses.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify). Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.). Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Patients are identified by biomarker testing (MMR or MSI testing) which is performed on all uterine cancers in Canada. This would identify eligible patients for this therapy once they progressed on platinum-based therapy. Which patients would be least suitable for treatment with the drug under review?
Which patients would be least suitable for treatment with the drug under review?
Response: Patients with pMMR endometrial cancer would not be as suitable for this monotherapy treatment.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review? If so, how would these patients be identified?
Response: Yes, current standard of care in Canadian centres includes universal testing of all endometrial carcinomas for mismatch repair deficiency (MMR). This is a highly targeted and small population with a known biomarker (with established, validated testing) that identifies those who are most likely to respond.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Standard clinical monitoring of therapy including physical exam and symptom review and intermittent imaging by CT scan.
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: Clinically meaningful response would be improvement of symptoms and physical findings related to the recurrent endometrial cancer (pain, bleeding, shortness of breath, etc), while maintaining or improving performance status and ability to perform activities of daily living. Evidence of disease regression on imaging studies would be used to support the clinical findings. In many cases, disease stabilization is also a very meaningful endpoint, especially if the patient had a good baseline performance status and few disease-related symptoms.
How often should treatment response be assessed?
Response: Initial monitoring every 3 weeks following treatment initiation to assess for treatment tolerance and assessment of toxicities (diarrhea, hypertension, hypothyroidism, fatigue, anemia). Imaging would generally be ordered every 8-12 weeks by local standards.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify, e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify).
Response: Treatment should be discontinued if patients experience disease progression or serious toxicities that preclude re-challenge with the same agent.
What settings are appropriate for treatment with the drug under review?
Response: This therapy is suitable to be delivered in community, outpatient, and specialty clinics. Patients should be under the care of a treating physician with experience in the monitoring and evaluation of possible toxicities caused by immune checkpoint inhibitor therapy. This group of agents is now routinely used by most oncologists.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: N/A
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: N/A
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict-of-interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
N/A
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
N/A
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Jennifer Brown Broderick
Position: Gynecologic Oncologist
Date: 24-10-2021
Table 26: Conflict of Interest Declaration for the Society of Gynecologic Oncology of Canada Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Alon Altman
Position: Gynecologic Oncologist
Date: 27-10-2021
Table 27: Conflict of Interest Declaration for the Society of Gynecologic Oncology of Canada Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK | X | — | — | — |
Merck, AstraZeneca | X | — | — | — |
Pfizer, Clovis, Novasure, Array | — | — | — | — |
Declaration for Clinician 3
Name: Michael Fung-Kee-Fung
Position: Gynecologic Oncologist
Date: 29-10-2021
Table 28: Conflict of Interest Declaration for the Society of Gynecologic Oncology of Canada Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK | — | X | — | — |
AstraZeneca | X | — | — | — |
Sunnybrook Health Sciences Centre, Division of Gynecologic Oncology
About Sunnybrook Health Sciences Centre, Division of Gynecologic Oncology
The division of gynecologic oncology at the Odette Cancer Centre, Sunnybrook Health Sciences includes five gynecologic oncologists and two medical oncologists. The goal of our division is to treat women with female genital tract cancer. All members of the division have academic appointments at the University of Toronto.
https://sunnybrook.ca/content/?page=occ-gynae-about
Information Gathering
The information for this submission was gathered by performing a literature search. This included searching previously published articles through PubMed and gathering information from abstract presentations. The information was gathered by the members of our division.
Further, our division has participated in multiple clinical trials in which the investigational drug was an immunotherapy, similar to Dostarlimab, and we therefore have experience in assessing the drug and the specific side effect profile of these agents.
Current Treatments
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: Treatment of patients with recurrent endometrial cancer who have previously received one line of platinum-based chemotherapy include: second line chemotherapy, radiation therapy or surgery. The preferred modality is based on extent and location of disease at time of recurrence. In most cases of localized pelvic recurrence in patients that have not previously received radiation treatment, pelvic radiation is the preferred modality. In most cases, when disease recurrence extends beyond the pelvis systemic therapy is the preferred option. Currently, the main options for systemic therapy include endocrine therapy (progesterone) for low grade endometrial cancer and chemotherapy for recurrent high- grade cancer. Multiple single agent chemotherapy regimens including; liposomal doxorubicin, topotecan, oxaliplatin, docetaxel and bevacizumab have been assessed in clinical trials with an overall response rate estimated at 7 to 20% and a duration of response less than six months. Due to the low response rates with a limited duration of response, new treatments are needed. The combination of Pembrolizumab and Lenvatinib has demonstrated promising results in patients with recurrent endometrial cancer. In a study including 108 patients an overall response rate of 38% was demonstrated; among subgroups the response rate was 63.6% in MSI-H tumours and 36.2% in MSI-S tumours. The median duration of response was 21.2 months. However, the side effect profile was significant with 66.9% of patients experiencing a grade 3 or 4 toxicity. It is currently not funded in Canada.
Treatment Goals
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: The goal of treatment in recurrent endometrial cancer is to prolong progression free survival while maintaining a good quality of life. This can be challenging to achieve when using standard chemotherapy regimens, especially when multi-regimen chemotherapy is used where significant toxicity is seen.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience.
Response: The current available treatments for recurrent endometrial cancer lack both efficacy and a durable response. Those that have had previous chemotherapy treatment have a low response rate of around 10- 15% and the duration of response is limited.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: Endometrial cancer has been traditionally categorized using histologic subtype. Recently, molecular characterization has provided a better understanding of endometrial cancer from a prognosis perspective and potentially can better predict response to treatment. Hence, the option to treat patients with targeted therapy using molecular characterization is an exciting opportunity that has the potential to identify treatment options that will produce a high response rate.
Patients included in this submission are those with a MMR deficiency (dMMR). dMMR is found in approximately 35% of patients with endometrial cancer and therefore, although it represents a subgroup it includes a high percentage of patients.
Further, the patient population included patients with recurrent endometrial cancer that were previously treated with a platinum based doublet chemotherapy and that had ≤2 lines of chemotherapy.
Place In Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: The role of Dostarlimab would be for treatment of patients with recurrent endometrial cancer that are MMR deficient and have previously been treated with one line of platinum based chemotherapy. These patients currently have very limited options and therefore Dostarlimab offers an excellent treatment option with a limited and tolerable side effect profile making it a better option for our patients.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Of the 71 evaluable patients with dMMR endometrial cancer that were enrolled in the GARNET study the overall response rate was 42.3% with 12.7% of patients achieving a complete response. They not only demonstrated a high response rate but also a durable response with 93.3% of patients demonstrating a duration of response longer than six months. The average duration of response for second line chemotherapy is around 6 months. Dostarlimab was well tolerated with only 5.6% of patients discontinuing drug due to side effects. Hence, Dostarlimab provides an excellent second line option (after platinum-based chemotherapy) for patients with dMMR and should be considered the preferred treatment regimen. Further, given the poor response rates demonstrated in previous studies and few treatment options available for these patients we find this data compelling and hence, strongly agree that Dostarlimab should be made accessible to our patients.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: Dostarlimab would replace the current standard of care for patients with recurrent endometrial cancer that are dMMR that have been treated with platinum-based chemotherapy. Patients that progress on Dostarlimab would then be eligible for second line chemotherapy.
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Patients that are best suited for treatment with Dostarlimab are those with recurrent endometrial cancer that have received a prior line of platinum-based chemotherapy but no more than 2 prior lines of treatment and have a documented dMMR as assessed by IHC. These patients have a 42.3% chance of response and a 93.3% chance of a duration of response six months or longer.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify). Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.). Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: Patients with recurrent endometrial cancer are treated by gynecologic oncologists or medical oncologists at cancer centres. These patients will be identified by their treating oncologist. Progression of disease will be determined by a combination of clinical symptoms and radiologic evidence on CT scan. A biopsy can be obtained in cases in which the diagnosis of recurrent endometrial cancer is uncertain.
Which patients would be least suitable for treatment with the drug under review?
Response: Patients with endometrial cancer that would be less suitable for treatment with the Dostarlimab include those that have not yet been treated with platinum based chemotherapy and patients without mismatch repair deficiency as seen on IHC. Further, patients with a poor performance status (ECOG 3 or above), inadequate organ function, those with a poor medical risk due to a serious uncontrolled medical disorder and those with a known immunodeficiency or currently on systemic steroids or other immunosuppressant medications.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: In the GARNET study responses to Dostarlimab were seen in patients with type I and II endometrial cancer 40% and 47.6%, respectively), in those with 1 or 2 lines of previous chemotherapy and in 50% of those with a response to their most recent platinum based chemotherapy. Hence, all patients with dMMR recurrent endometrial cancer that have had at least 1 but no more than 2 lines of prior chemotherapy should be offered treatment.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: The outcome that was used in the GARNET trial is response to treatment as per RECIST v1.1 criteria. This is an acceptable means of assessing response in clinical practice and should be used in this case.
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: A clinically meaningful response in this patient population would include improvement in symptoms e.g. a reduction in abdominal or pelvic pain, a reduction in urinary or bowel symptoms, increased appetite and energy levels. A progression free survival of 6 months or more would be clinically meaningful in this patient population.
How often should treatment response be assessed?
Response: Response should be assessed on imaging (CT scan) every 12 weeks. Due to the nature of immunotherapies and the possibility of pseudoprogression, patients with progression of disease on the first CT scan after initiation of treatment, but no other symptoms, should continue treatment until further imaging demonstrates progression of disease.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify)
Response: Dostarlimab should be discontinued if radiologic evidence of unequivocal progression of disease is demonstrated or if patients develop intolerable side effects. The main side effects that warrant consideration of holding or discontinuing Dostarlimab are: grade 3 anemia, pneumonitis, grade 3 colitis, grade 3 asthenia, grade 3 myalgia, pemphigoid or grade 3 increase in transaminases.
What settings are appropriate for treatment with the drug under review?
Response: Dostarlimab should be administered in a chemotherapy suite with the appropriate supervision of an oncologist familiar with immunotherapy side effects.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: N/A
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: No
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No outside help was provided.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No help was provided in analyzing or interpreting the data.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Danielle Vicus
Position: Staff Gynecologic Oncologist
Date: 26-10-2021
Table 29: Conflict of Interest Declaration for Sunnybrook Health Sciences Centre, Division of Gynecologic Oncology Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK | — | X | — | — |
Merck | X | — | — | — |
Declaration for Clinician 2
Name: Al Covens
Position: Department Head, Gynecologic Oncology
Date: 26-10-2021
Table 30: Conflict of Interest Declaration for Sunnybrook Health Sciences Centre, Division of Gynecologic Oncology Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
GSK | X | — | — | — |
Declaration for Clinician 3
Name: Lilian Gien
Position: Gynecologic Oncologist, Gynecology Site Group Lead Odette Cancer Centre
Date: 29/10/2021
Table 31: Conflict of Interest Declaration for Sunnybrook Health Sciences Centre, Division of Gynecologic Oncology Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Merck | X | — | — | — |
GSK | X | — | — | — |
Declaration for Clinician 4
Name: Raymond Osborne
Position: Staff Gynecologic Oncologist
Date: 29-10-2021
Table 32: Conflict of Interest Declaration for Sunnybrook Health Sciences Centre, Division of Gynecologic Oncology Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Rachel Kupets
Position: Staff Gynecologic Oncologist
Date: 29-10-2021
Table 33: Conflict of Interest Declaration for Sunnybrook Health Sciences Centre, Division of Gynecologic Oncology Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.