CADTH Reimbursement Review
Cemiplimab (Libtayo)
Sponsor: Sanofi Genzyme, a division of Sanofi-Aventis Canada Inc.
Therapeutic area: Non–small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
CI
confidence interval
CrI
credible interval
DOR
duration of response
ECOG PS
Eastern Cooperative Oncology Group Performance Status
EORTC QLQ-C30
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
EORTC QLQ-LC13
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13
GHS
global health status
HR
hazard ratio
HRQoL
health-related quality of life
IRC
independent review committee
ITC
indirect treatment comparison
ITT
intention-to-treat
KPS
Karnofsky performance status
LCC
Lung Cancer Canada
MID
minimal important difference
mITT-1
modified intention-to-treat 1
NMA
network meta-analysis
NSCLC
non–small cell lung cancer
ORR
objective response rate
OR
odds ratio
OS
overall survival
PFS
progression-free survival
RECIST 1.1
Response Evaluation Criteria in Solid Tumors Version 1.1
RMST
restricted mean survival time
RPSFT
rank-preserving structural failure time
SAE
serious adverse event
SD
standard deviation
TEAE
treatment-emergent adverse event
TPS
Tumor Proportion Score
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Cemiplimab (Libtayo), sterile solution for IV infusion, 50 mg/mL |
Indication | First-line treatment of adult patients with NSCLC expressing PD-L1 (TPS ≥ 50%), as determined by a validated test, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC who are not candidates for surgical resection or definitive chemoradiation, or metastatic NSCLC |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | ACCESS (Australia-Canada-Singapore-Switzerland) work-sharing initiative |
NOC date | October 26, 2021 |
Sponsor | Sanofi Genzyme |
NOC = Notice of Compliance; NSCLC = non–small cell lung cancer; TPS = Tumor Proportion Score.
Introduction
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer-related deaths in Canada.1 An estimated 29,600 Canadians were diagnosed with lung cancer in 2021, representing approximately 13% of all new cancer cases, and an estimated 21,000 Canadians died from lung cancer, representing 25% of all cancer deaths in 2021.1 Tobacco smoking, including exposure to second-hand smoke, remains the main cause of lung cancer. Lung cancers are histologically divided into small cell lung cancer, which accounts for 15% of cases, and non–small cell lung cancer (NSCLC) — the most common type — which accounts for 85% of patients.2 Adenocarcinoma and squamous cell carcinoma are the 2 major histologic subtypes of NSCLC, accounting for approximately 40% to 60% and 30% of NSCLC cases, respectively.3 The overall survival (OS) for patients with NSCLC varies with disease stage. The estimated 5-year survival is 13% to 36% for patients with stage III disease, and only 10% for those with stage IV disease.1
A number of driver gene alterations have been identified in NSCLC. The most notable include BRAF mutation, EGFR exon 19 deletion and exon 21 L858R mutation, ALK translocation, and ROS1 rearrangement. EGFR mutations are identified in approximately 10% to 30% of patients with NSCLC, while ALK and ROS1 rearrangements occur in approximately 2% to 5% and 1% to 4% of NSCLC tumours, respectively.4-7 BRAF mutations are observed in 2% of patients with NSCLC.8 Treatment decisions are made based on tumour histologic subtype and the presence or absence of various oncogenic drivers, as well as other patient and disease-related characteristics. The expression of PD-L1 in malignant cells is also a biomarker for response to PD-1 and PD-L1 immune checkpoint inhibitors. Thus, PD-L1 immunohistochemistry is important in the selection of first-line therapy. At the time of diagnosis, the majority of patients with NSCLC are found to have advanced disease. For these patients, the goal of treatment is not curative and is focused on improving symptoms and quality of life, delaying disease progression, and extending OS.
Chemotherapy regimens alone have been widely replaced with PD-1 and PD-L1 checkpoint immunotherapy treatments (as monotherapy or in combination with chemotherapy) for the first-line treatment of patients with advanced or metastatic NSCLC without EGFR, ALK, or ROS1 aberrations and high tumour PD-L1 expression. Pembrolizumab monotherapy and pembrolizumab with chemotherapy are currently the only approved and publicly funded regimens in this setting. Nivolumab-ipilimumab plus 2 cycles of platinum-doublet has a positive CADTH recommendation but is not yet funded. Platinum-based doublet chemotherapy or single-agent chemotherapy can be used for patients with a contraindication to immunotherapy. Best supportive care is also an option.
Cemiplimab (Libtayo) is a human recombinant immunoglobulin G4 monoclonal antibody that binds to PD-1 and blocks its interaction with PD-L1 and PD-L2, antagonizing PD-L1–mediated T-cell inhibition and rescuing the antitumour response. Blocking the immune-inhibitor PD-1 and PD-L1 signalling pathway helps restore both helper and cytotoxic T-cell functioning, thereby increasing the number of effector T-cells able to recognize and attach to tumour cells. Cemiplimab has a Health Canada indication for the first-line treatment of adult patients with NSCLC expressing PD-L1 in 50% of tumour cells or more (Tumor Proportion Score [TPS] ≥ 50%), as determined by a validated test, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and are not candidates for surgical resection or definitive chemoradiation, or who have metastatic NSCLC. The sponsor’s reimbursement request is the same as the approved Health Canada indication.
The objective of this review was to evaluate the efficacy and safety of cemiplimab 50 mg/mL for IV infusion for the first-line treatment of adult patients with NSCLC expressing PD-L1 levels (TPS ≥ 50%), as determined by a validated test, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and are not candidates for surgical resection or definitive chemoradiation, or who have metastatic NSCLC.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, the Lung Health Foundation and Lung Cancer Canada (LCC), submitted patient input for this review. Most respondents from the Lung Health Foundation input stated that they experienced some symptoms as a result of their lung cancer, including shortness of breath (64%), fatigue (57%), depression (25%), cough (21%), difficulty fighting infection (21%), and chest tightness (14%). Some respondents indicated that the psychosocial effects of having a disease with a poor prognosis was more debilitating than the physical symptoms. Side effects of currently available treatments reported among participants included fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy. Respondents reported that they expected the following key outcomes to be improved from any new drug or treatment: stopping or delaying disease progression with minimal side effects, being able to access treatments that are effective for advanced disease, and being able to maintain some quality of life while on treatment. The LCC input evaluated respondents’ treatment preferences, with the assumption that patients will have the option to be treated closer to home at local community hospitals with cemiplimab due to its fixed dosing model. If given the choice between 2 equally efficacious treatment options, 91% of respondents would choose a therapy closer to home as it would provide benefits such as decreased travel time, savings on travel costs, and increased time with family and caregivers. A total of 97% of LCC respondents believed that having access to an additional treatment option closer to home would improve their health-related quality of life (HRQoL).
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH noted that pembrolizumab is currently the only approved and publicly funded standard of care monotherapy used for the first-line treatment of advanced or metastatic NSCLC in patients with no EGFR, ALK, or ROS1 aberrations and with PD-L1–positive tumours (TPS ≥ 50%). Cemiplimab monotherapy in PD-L1–positive (TPS ≥ 50%) NSCLC appears to be another treatment option with a similar mechanism of action in this setting. However, longer follow-up is needed to confirm efficacy is maintained and similar to other available options. The clinical experts consulted by CADTH indicated that the only predictive marker of response to PD-1 and PD-L1 inhibitors as monotherapy is PD-L1 testing, which is routinely done in all newly diagnosed patients with advanced or metastatic NSCLC. Clinical response (symptom assessment) and radiological surveillance are used to determine whether a patient is responding to treatment in clinical practice. Improvement in survival and quality of life (i.e., fewer symptoms, higher functional status, or stabilization of symptoms) would be considered a clinically meaningful response to treatment. Treatment response is evaluated clinically at each visit, and radiologically, approximately every 3 months to 4 months.
Clinician Group Input
Clinician input was received from the Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer Drug Advisory Committee, with 4 clinicians contributing to the submission. The clinician group noted that the most important goal of any treatment for NSCLC is to improve OS and improve progression-free survival (PFS). Monotherapy immunotherapy also has the additional benefit of avoiding chemotherapy. Patients most likely to benefit from cemiplimab are those with advanced or metastatic NSCLC and tumours having high levels of PD-L1 expression of 50% or more. Cemiplimab would be used as monotherapy in a first-line treatment for these patients and would be an alternative to pembrolizumab monotherapy, pembrolizumab plus platinum-doublet chemotherapy, or the combination of nivolumab-ipilimumab and 2 cycles of platinum-based chemotherapy. It would not be used as an additional therapy to currently available treatment options. In terms of response to treatment, the clinician group noted that the most meaningful response to treatment is the absence of disease progression, followed by improvement in disease-related symptoms that are assessed every 3 months in clinical practice. Disease progression or intolerable side effects were indicated as the primary reasons to discontinue therapy. The clinician group also noted that treatment continuation beyond progression should remain an option as some patients may benefit from continuing treatment beyond Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1)–defined progression.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for cemiplimab:
considerations for the initiation of therapy
considerations for the prescribing of therapy
the generalizability of trial populations to broader populations in the jurisdictions
care provision issues
system and economic issues
the potential need for a provisional funding algorithm.
The clinical experts consulted by CADTH provided advice on the potential implementation issues raised by the drug programs.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
The EMPOWER-Lung 1 study (also named R2810-ONC-1624) is an ongoing randomized, multi-centre, open-label, phase III study of cemiplimab monotherapy versus platinum-based doublet chemotherapy in patients with stage IIIB, stage IIIC, or stage IV NSCLC who were not candidates for treatment with definitive chemoradiotherapy, whose tumours expressed PD-L1 in at least 50% of tumour cells, with no EGFR, ALK, or ROS1 aberrations, and who had received no prior systemic treatment for their advanced disease. “Never-smokers” (people who had never smoked or who smoked fewer than 100 cigarettes in their lifetime) were ineligible for the study. The primary end points were OS and PFS, and the key secondary end point was objective response rate (ORR). Patient-reported outcomes included HRQoL. Overall, the mean age was 63 years (standard deviation [SD] = 8.4), and 85% of patients were men. A non-squamous histology was observed in 56% of patients, and the disease stage at screening was metastatic (stage IV) in 84% of patients. Patients had to have an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1 and approximately 73% of the patients in both treatment arms had an ECOG PS of 1. All patients were current or former smokers. Of 3,662 patients screened, 710 were randomized — 356 patients to the cemiplimab monotherapy arm and 354 patients to the chemotherapy arm. The mean duration of follow-up was 14.04 months overall (SD = 7.5), and in both treatment arms.
Efficacy Results
Due to issues with PD-L1 testing identified during the sponsor’s monitoring, samples from 235 patients tested before August 2018 had to be retested. Of these patients, 56 were found to have PD-L1 of less than 50% on retest. Consequently, a PD-L1 of 50% or more population was pre-specified to include only patients with PD-L1 of at least 50% on retest and those who were tested after August 2018 and were unaffected by testing irregularities. The PD-L1 of 50% or more population consisted of 563 patients (N = 283 for cemiplimab and N = 280 for chemotherapy). Efficacy end points were assessed in the intention-to-treat (ITT) population (N = 710) as well as in the PD-L1 of 50% or more population. Results for OS, ORR, PFS, and duration of response (DOR) in the PD-L1 of 50% or more population were consistent with those in the ITT population. Key efficacy results for the ITT population and the PD-L1 of 50% or more population in the trial are summarized in Table 2.
Overall Survival
As of the data cut-off date (March 1, 2020), the median OS was 22.1 months (95% confidence interval [CI] lower bound = 17.7 months) in the cemiplimab arm versus 14.3 months (95% CI, 11.7 months to 19.2 months) in the chemotherapy arm (P = 0.0022); the hazard ratio (HR) between groups was 0.676 (95% CI, 0.52 to 0.87).
Health-Related Quality of Life
Mean baseline scores for the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) global health status (GHS) scale were similar between patients in the cemiplimab and chemotherapy treatment arms. A mean change in score from baseline of greater than 5 points for the GHS was observed in the cemiplimab arm by cycle 2 (mean change = 5.16 [SD = 20.49]), above 9 points by cycle 6 (mean change = 9.38 [SD = 23.359]) and above 10 points by cycle 18 (mean change = 10.53 [SD = 25.71]). It stayed above 10 points, with wide variation, through to cycle 30. The mean change for GHS score in the chemotherapy arm was below 3 until cycle 12 and ranged from –8.33 (SD = 24.40) at cycle 18 to 5.56 (SD = 12.73) at cycle 21. The mean GHS scores numerically favoured cemiplimab post baseline up to cycle 6 and there were no consistent or notable differences between the arms at later time points.
Objective Response Rate
Complete response or partial response was observed in 36.5% of patients in the cemiplimab arm and 20.6% of patients in the chemotherapy arm. The odds ratio for comparison of cemiplimab to chemotherapy was 2.21 (95% CI, 1.58 to 3.10; P < 0.0001).
Progression-Free Survival
The median PFS was 6.2 months (95% CI, 4.5 months to 8.3 months) in the cemiplimab arm versus 5.6 months (95% CI, 4.5 months to 6.1 months) in the chemotherapy arm (P < 0.0001); the HR between groups was 0.593 (95% CI, 0.491 to 0.718).
Duration of Response
The Kaplan-Meier estimate of median DOR was 21.0 months (lower bound of 95% CI = 14.9 months) for cemiplimab, and 6.0 months (95% CI, 4.3 months to 6.4 months) for chemotherapy.
Table 2: Summary of Key Efficacy Results From the EMPOWER-Lung 1 Study
Outcome | ITT population | PD-L1 ≥ 50% populationa | ||
|---|---|---|---|---|
Cemiplimab (N = 356) | Chemotherapy (N = 354) | Cemiplimab (N = 283) | Chemotherapy (N = 280) | |
OS | ||||
Median (95% CI), monthsb | 22.1 (17.7 to NE) | 14.3 (11.7 to 19.2) | NR (17.9 to NE) | 14.2 (11.2 to 17.5) |
Stratified log-rank test, P valuec, d | 0.0022 | 0.0002 | ||
HR (95% CI)c, e | 0.676 (0.525 to 0.870) | 0.566 (0.418 to 0.767) | ||
Objective response | ||||
ORR (CR + PR), n (%) | 130 (36.5) | 73 (20.6) | 111 (39.2) | 57 (20.4) |
95% CI for ORRf | 31.5 to 41.8 | 16.5 to 25.2 | 33.5 to 45.2 | 15.8 to 25.6 |
Stratified CMH test, P valuef | < 0.0001 | < 0.0001 | ||
Odds ratio (95% CI)g | 2.214 (1.582 to 3.098) | 2.530 (1.736 to 3.687) | ||
PFS | ||||
Median (95% CI), monthsb | 6.2 (4.5 to 8.3) | 5.6 (4.5 to 6.1) | 8.2 (6.1 to 8.8) | 5.7 (4.5 to 6.2) |
Stratified log-rank test P valuec, d | < 0.0001 | < 0.0001 | ||
HR (95% CI) c, e | 0.593 (0.491 to 0.718) | 0.541 (0.433 to 0.675) | ||
DOR (CR + PR) | ||||
n | 130 | 73 | 111 | 57 |
Range, months | (1.9 to 23.3) | (1.3 to 16.5) | (1.9 to 23.3) | (1.3 to 14.5) |
≥ 6 months, n (%) | 90 (69.2) | 30 (41.1) | 73 (65.8) | 23 (40.4) |
≥ 12 months, n (%) | 36 (27.7) | 5 (6.8) | 22 (19.8) | 4 (7.0) |
≥ 18 months, n (%) | 15 (11.5) | 0 | 10 (9.0) | 0 |
Kaplan-Meier estimated DOR (CR + PR), months | ||||
Median (95% CI) | 21.0 (14.9 to NE) | 6.0 (4.3 to 6.4) | 16.7 (12.5 to 22.8) | 6.0 (4.3 to 6.5) |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CR = complete response; DOR = duration of response; HR = hazard ratio; ITT = intention-to-treat; NE = not estimable; NR = not reached; ORR = objective response rate; OS = overall survival; PD-L1 = programmed cell death-ligand 1; PFS = progression-free survival; PR = partial response.
aAnalyses were not adjusted for multiplicity.
bBased on the Kaplan-Meier method.
cStratified by histology (squamous, non-squamous).
dTwo-sided P value.
eBased on a stratified proportional hazards model (cemiplimab versus chemotherapy).
fClopper-Pearson exact CI.
gTwo-sided P value and odds ratio using stratified CMH test.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Harms Results
As of the data cut-off date, 88.2% of patients in the cemiplimab arm and 94.2% of patients in the chemotherapy arm had experienced at least 1 treatment-emergent adverse event (TEAE). In the cemiplimab arm, the most common TEAEs of any grade by preferred term experienced by 10% of patients or more were anemia (14.6%), decreased appetite (11.8%), and fatigue (10.1%). In the chemotherapy arm, the most common TEAEs of any grade by preferred term experienced by 10% of patients or more were anemia (50.0%), nausea (28.4%), alopecia (24.0%), decreased appetite (18.4%), neutropenia (18.4%), fatigue (17.0%), constipation (15.2%), thrombocytopenia (15.2%), vomiting, (14.3%), decreased neutrophil count (12.3%), peripheral neuropathy (10.8%), pneumonia (10.8%), and decreased platelet count (10.5%).
Grade 3 to grade 4 TEAEs occurred in 28% of patients in the cemiplimab arm and 39% of patients in the chemotherapy arm. The discontinuation of study treatment due to adverse events (AEs) was reported for 6.5% of patients in the cemiplimab arm and 4.1% of patients in the chemotherapy arm. Serious TEAEs were reported for 28.2% of patients in the cemiplimab arm and 27.5% of patients in the chemotherapy arm.
TEAEs that led to death occurred in 9.6% of patients treated with cemiplimab and 9.1% of patients treated with chemotherapy. In 9 (3%) patients treated with cemiplimab, the events leading to death were considered related to treatment, and included autoimmune myocarditis, cardiac failure, cardiopulmonary failure, respiratory failure, septic shock, cardiorespiratory arrest, nephritis, and tumour hyperprogression (n = 1 each).
In the cemiplimab arm, 17.5% of patients experienced at least 1 treatment-emergent immune-related AE, and in the chemotherapy arm, 2.3% of patients experienced at least 1 treatment-emergent immune-related AE. Most of these events were less than grade 3, with 3.7% of patients in the cemiplimab arm and 0.3% of patients in the chemotherapy arm experiencing an immune-related AE that was grade 3 or higher. Grade 4 and grade 5 immune-related AEs were only reported in the cemiplimab arm, occurring in 0.8% and 0.3% of patients, respectively.
Table 3: Summary of Key Harms Results From the EMPOWER-Lung 1 Study
Harms | Safety population | |
|---|---|---|
Cemiplimab (N = 355) | Chemotherapy (N = 342) | |
Patients with any TEAE, n (%) | 313 (88.2) | 322 (94.2) |
Patients with any serious TEAE, n (%) | 100 (28.2) | 94 (27.5) |
Patients with any TEAE resulting in treatment discontinuation, n (%) | 23 (6.5) | 14 (4.1) |
TEAEs leading to death, n (%) | 34 (9.6) | 31 (9.1) |
Notable harms | ||
Total number of immune-related AEs, n | 87 | 9 |
Patients with any immune-related AEs, n (%) | 62 (17.5) | 8 (2.3) |
Patients with infusion-related reactions | 20 (5.6) | 6 (1.8) |
AE = adverse event; TEAE = treatment-emergent adverse event.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Critical Appraisal
The EMPOWER-Lung 1 trial was centrally randomized and balanced baseline characteristics indicate that there is minimal concern for selection bias. This was an open-label trial, and although patient blinding would not have been possible given the differences in the 2 study treatment regimens, detection and performance bias that may result from lack of blinding of patients and investigators to assigned study treatments cannot be ruled out, especially for subjective patient-reported outcomes. Issues with PD-L1 testing were discovered when more than 50% of the planned population had been recruited, necessitating retesting of the 235 randomized patients at that point, but not all of them had remaining tissue samples (38%) and not all the retested samples proved PD-L1 of 50% or more (24%). As a result, analyses were also conducted in the population with confirmed PD-L1 TPS of 50% or more. The ITT population represents a truly randomized sample but includes some patients who did not in fact meet the inclusion criteria of the trial; the PD-L1 of 50% or more population is not strictly a randomized sample and serves as supportive data, as it may be more clinically relevant. The findings across these 2 populations were largely similar. Amendments to the protocol were made to allow patients who progressed on cemiplimab monotherapy to continue cemiplimab treatment with the addition of 4 cycles of histology-specific chemotherapy until further progression was observed. Similarly, patients in the chemotherapy arm were allowed to cross over to cemiplimab after initial disease progression on chemotherapy. This crossover was not accounted for in the main OS analysis and may have biased the findings in favour of chemotherapy (i.e., underestimated the effect of cemiplimab). Sensitivity analyses were conducted to account for the crossover effect, and these were consistent with the primary analyses.
The EMPOWER-Lung 1 trial included a heterogenous population of patients with NSCLC and a wide range of clinical presentations were well represented. However, a few patient groups were not included, notably non-smokers and patients who were immunocompromised or had a history of autoimmune diseases, and those with ECOG PS of 2 or more. Therefore, the generalizability of results to these patient groups may be limited. In addition, about 44% of patients presented with squamous histology, which is higher than what is expected in clinical practice (about 30%). The most important limitation of the evidence in terms of generalizability is the relevance of chemotherapy as a comparator in Canadian clinical practice where the standard of care for the treatment of patients with advanced or metastatic high PD-L1–expressing NSCLC and without oncogenic alterations includes immune checkpoint inhibitors. Pembrolizumab, with or without chemotherapy, is funded and is widely used for this indication. Therefore, the benefit of cemiplimab compared to chemotherapy in terms of improved survival in this patient population is limited in informing treatment choice in Canadian clinical practice.
Indirect Comparisons
Description of Studies
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Identified in the literature search were 2 additional published indirect treatment comparisons (ITCs), 1 of which included a comparison of cemiplimab against pembrolizumab plus chemotherapy. However, due to serious limitations in the published ITCs, conclusions could not be drawn based on the findings and the results are not included in this summary.
Efficacy Results
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Harms Results
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Critical Appraisal
Few inferences can be made from the results of the network meta-analysis (NMA) because of important limitations with the included studies and the methods and assumptions made in the NMA. The key limitation related to the choice of relevant comparators which did not include ||||||||||||| || ||||||||||| |||| ||||||||||||, a comparator considered to be relevant in the Canadian treatment landscape for patients with NSCLC expressing PD-L1 of 50% or more, though the most relevant comparator, ||||||||||||| |||||||||||, was captured. Therefore, the relevance of the systematic literature review and NMA to the Canadian context is unclear. The outcomes assessed were appropriate, though other important outcomes such as HRQoL were deemed not possible to analyze due to differences in study reporting. Moreover, several potential sources of heterogeneity exist across the trials that limit their comparison, including substantial heterogeneity across trial populations, such as tumour histology and smoking status. The available trials formed networks with no closed loops; therefore, it was not possible to validate the transitivity assumption of NMA and check for the consistency of results between direct and indirect comparisons. Random-effects models were attempted and determined not to be feasible to include in the base-case analysis due to the small number of included studies.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Conclusions
Based on clinical data from the EMPOWER-Lung 1 study, cemiplimab demonstrated a statistically significant and clinically meaningful survival benefit compared to chemotherapy in the treatment of patients with advanced NSCLC, no EGFR, ALK, or ROS1 aberrations, and PD-L1 expression of 50% or greater. ORR and duration of response were supportive of the primary efficacy results. Symptoms and HRQoL were assessed, but conclusions cannot be drawn for those outcomes. The toxicity (side effects) profile of cemiplimab was acceptable and compared favourably to chemotherapy. Chemotherapy is no longer the first-line standard of care in this patient population and the evidence from ITCs is insufficient for determining the comparative efficacy and safety of cemiplimab with other immune checkpoint inhibitors used in this setting. This includes ||||||||||||| |||||||||||, which is the most relevant comparator in the Canadian setting, given that cemiplimab monotherapy appears to provide an alternative immune checkpoint inhibitor monotherapy option to ||||||||||||| |||||||||||.
Introduction
Disease Background
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer-related deaths in Canada.1 In 2021, an estimated 29,600 Canadians were diagnosed with lung cancer, representing approximately 13% of all new cancer cases, and 21,000 Canadians died from lung cancer, representing 25% of all cancer deaths in 2021.1 It is estimated that 1 in 15 Canadian men will develop lung cancer during their lifetime and 1 in 18 will die from it.1 Similarly, 1 in 15 Canadian women will develop lung cancer during their lifetime and 1 in 20 will die from it.1 In Canada, the overall 5-year net survival for lung cancer from 2015 to 2017 was estimated to be 19% for men and 26% for women.1 The incidence of lung cancer remains low in patients younger than 40 years and it begins to rise and peaks between 65 and 84 years of age.10 Tobacco smoking, including exposure to second-hand smoke, remains the main cause of lung cancer, which is responsible for 72% of cases.1 Other known risk factors for lung cancer include exposure to asbestos, arsenic, radon, non–tobacco-related polycyclic aromatic hydrocarbons, and air pollution — these are suspected to contribute to the relatively high burden of non–smoking-related lung cancer, particularly in women. The most common symptoms of lung cancer are cough, dyspnea, hemoptysis, and chest pain, and systemic symptoms such as fatigue and weight loss.
Lung cancers are histologically divided into small cell lung cancer, which accounts for 15% of cases, and NSCLC, which is found in 85% of cases.2 Adenocarcinoma and squamous cell carcinoma are the 2 major histologic subtypes of NSCLC, accounting for approximately 40% to 60% and 30% of NSCLC cases, respectively.3 The OS for patients with NSCLC varies with disease stage. The estimated 5-year survival is 13% to 36% for patients with stage III disease, and only 10% for those with stage IV disease.1 The diagnostic evaluation entails imaging that can include chest X-ray, CT scan, PET, MRI and bone scans, and tissue biopsy for histologic confirmation and determination of the extent of the tumour to define the tumour-nodes-metastasis stage to guide treatment options.
A number of driver mutations have been identified in NSCLC. The most notable alterations are EGFR exon 19 deletion and exon 21 L858R mutation, ALK translocation, ROS1 rearrangement, and BRAF mutations. Tumours with these alterations are more common in never-smokers (never smoked or who smoked less than 100 cigarettes in their lifetime), long-time ex-smokers (longer than 10 years), or light smokers (less than 15 pack-years). Characterizing tumours according to histologic subtype and genetic composition has resulted in significant progress in the identification of response to certain drugs and personalized approaches in treating lung cancer.11 Targeted drugs that are active at these sites have led to a significant improvement in patient survival and quality of life compared to conventional cytotoxic therapies.12-14 Clinical practice guidelines recommend routine testing for oncogenic drivers in NSCLC tumours to guide treatment strategies; consequently, molecular testing has become an essential part of managing NSCLC in clinical practice.6,15 Driver alterations affect a small proportion of patients with NSCLC. EGFR mutations are identified in about 10% to 30% of patients with non-squamous NSCLC, while ALK and ROS1 rearrangements occur in about 2% to 5% and 1% to 4% of non-squamous NSCLC tumours, respectively.4-7 BRAF mutations are observed in 2% of patients with NSCLC.8
At the time of diagnosis, the majority of patients with NSCLC are found to have advanced disease that is inoperable, or that has spread to distant organs (i.e., metastatic disease). For these patients, the goal of treatment is not curative and is focused on improving symptoms and quality of life, delaying disease progression, and extending OS. Treatment decision is guided by patient-related and disease-related characteristics including age, ECOG PS, tumour stage, and histologic characteristics. Immune checkpoint inhibitors have dramatically changed the landscape of NSCLC treatment, particularly in the metastatic setting, by targeting the PD-1 and PD-L1 immune checkpoint. PD-1 plays a vital role in inhibiting immune responses; the inhibition of PD-1 promotes an effective immune response against cancer cells. The expression of PD-L1 in malignant cells is also a biomarker for response to PD-1 and PD-L1 immune checkpoint inhibitors. Thus, PD-L1 immunohistochemistry is important in the selection of first-line therapy.
Standards of Therapy
Until recently, patients with advanced or metastatic NSCLC without targetable driver mutations were treated with combination chemotherapy regimens — most commonly, platinum doublets based on tumour histology (e.g., cisplatin plus 1 of docetaxel, paclitaxel, pemetrexed, vinorelbine, or gemcitabine; carboplatin plus 1 of paclitaxel, pemetrexed, docetaxel, or gemcitabine). However, over the past decade, chemotherapy regimens alone have been widely replaced with PD-1 and PD-L1 checkpoint immunotherapy treatments (as monotherapy or in combination with chemotherapy) in this setting.
The most recent joint American Society of Clinical Oncology and Ontario Health (Cancer Care Ontario) clinical practice guideline update for the treatment of patients with stage IV NSCLC without driver alterations, with high PD-L1 expression (TPS ≥ 50%), squamous or non-squamous cell carcinoma, and performance status 0 or 1, recommends single-agent pembrolizumab as a first-line treatment.16 Based on input from clinicians consulted by CADTH for the purpose of this review, the most common first-line systemic treatment for advanced or metastatic PD-L1–strongly positive (≥ 50%) NSCLC without driver alterations used in current clinical practice in Canada is pembrolizumab monotherapy. Pembrolizumab plus platinum-doublet chemotherapy and combination nivolumab-ipilimumab with 2 cycles of platinum-based chemotherapy are therapies that can also be considered. Pembrolizumab monotherapy and pembrolizumab plus chemotherapy are funded for this patient population, while nivolumab-ipilimumab with chemotherapy has a positive CADTH recommendation but is not yet funded. Atezolizumab, with or without chemotherapy, is also approved in this setting but is not funded. Platinum-based doublet chemotherapy or single-agent chemotherapy can be used for patients with a contraindication to immunotherapy. Best supportive care is also an option.
Drug
Cemiplimab (Libtayo) is a human recombinant immunoglobulin G4 monoclonal antibody that binds to PD-1 and blocks its interaction with PD-L1 and programmed death ligand 2, antagonizing PD-L1–mediated T-cell inhibition and rescuing the antitumour response. Blocking the immune-inhibitor PD-1 and PD-L1 signalling pathway helps restore both helper and cytotoxic T-cell functioning, thereby increasing the number of effector T-cells able to recognize and attach tumour cells.
In the US, the FDA approved cemiplimab (with “rwlc” added as a suffix — cemiplimab-rwlc) for the first-line treatment of patients with advanced NSCLC whose tumours have high PD-L1 expression (TPS ≥ 50%) with no EGFR, ALK, or ROS1 aberrations. Cemiplimab was approved for the same indication by the European Medicines Agency’s Committee for Medicinal Products for Human Use.
Cemiplimab was approved in Canada for the first-line treatment of adult patients with NSCLC expressing PD-L1 in 50% of tumour cells or more (TPS ≥ 50%), as determined by a validated test, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and who are not candidates for surgical resection or definitive chemoradiation, or who have metastatic NSCLC. The sponsor’s reimbursement request is the same as the approved Health Canada indication.
Cemiplimab is supplied as a concentrate solution (50 mg/mL) for dilution as 250 mg/5 mL and 350 mg/7 mL. The recommended dose of cemiplimab is 350 mg administered as an IV infusion over 30 minutes every 3 weeks.17
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. Please refer to the Stakeholder Input document for the full patient group input submitted to CADTH.
Two patient groups, the Lung Health Foundation (previously known as the Ontario Lung Association) and LCC submitted patient input for this review. The input from the Lung Health Foundation was based on phone interviews with 3 lung cancer patients from Ontario, Manitoba, and Quebec, and an online survey completed by 13 patients and 1 care provider. LCC received patient input via an online survey, with responses from 33 lung cancer patients across 6 provinces. None of the respondents had experience with cemiplimab for the indication under review.
Most respondents from the Lung Health Foundation input stated that they experienced some symptoms as a result of their lung cancer, including shortness of breath (64%), fatigue (57%), depression (25%), cough (21%), difficulty fighting infection (21%), and chest tightness (14%). Some respondents indicated that the psychosocial effects of having a disease with a poor prognosis was more debilitating than the physical symptoms. Side effects of currently available treatments reported among participants included fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy. Respondents reported that they expected the following key outcomes to be improved from any new drug or treatment: stopping or delaying disease progression with minimal side effects, being able to access treatments that are effective for advanced disease, and being able to maintain some quality of life while on treatment.
The LCC input evaluated respondents’ treatment preferences, with the assumption that patients will have the option to be treated closer to home at local community hospitals with cemiplimab due to its fixed dosing model. If given the choice between 2 equally efficacious treatment options, 91% of respondents would choose a therapy closer to home as it would provide benefits such as decreased travel time, savings on travel costs, and increased time with family and caregivers. A total of 97% of LCC respondents believed that having access to an additional treatment option closer to home would improve their HRQoL.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of NSCLC.
Unmet Needs
The clinical experts consulted by CADTH noted that not all patients respond to or benefit from the current standard first-line systemic treatment options and eventually most patients will experience disease progression. Furthermore, some patients stop treatment early due to significant treatment toxicity. At the time of disease progression, options for second-line treatment have limited efficacy and carry increased toxicity. The goals of a new therapy in the first-line setting would be less toxicity and improved survival compared to current standard of care.
Place in Therapy
Cemiplimab is a PD-1 inhibitor with the same mechanism of action as pembrolizumab, currently in use in the first-line treatment of metastatic NSCLC as monotherapy or combined with chemotherapy. Atezolizumab is a PD-L1 inhibitor with a similar mechanism of action but is not in use as it is not publicly funded. No studies have directly compared cemiplimab to current available treatment options (pembrolizumab alone or with chemotherapy).
The clinical experts noted that cemiplimab monotherapy in PD-L1–strongly positive NSCLC appears to be another treatment option in the first-line setting of metastatic NSCLC. However, longer follow-up is needed to confirm that efficacy is maintained and is similar to other available options. The clinical experts believed that toxicity seemed to be slightly improved when doing cross-trial comparison, but this is likely the reflection of a better knowledge of the side-effect profile and more prompt management than a true difference in the safety profile among the current PD-1 and PD-L1 inhibitors. PD-1 and PD-L1 inhibitors are not interchangeable in patients with significant toxicity to immune therapy. The clinical experts indicated that cemiplimab may be used in the first-line setting in treatment-naive patients with metastatic NSCLC instead of an alternative PD-1 or PD-L1 inhibitor. They did not believe that it should be initiated after an alternative checkpoint inhibitor or chemotherapy alone as there is insufficient evidence on this sequence of therapies.
Patient Population
Patients with advanced or metastatic PD-L1–strongly positive NSCLC without EGFR, ALK, or ROS1 genomic alterations who are treatment-naive and have good performance status (i.e., ECOG PS of 0 or 1) would be best suited for first-line treatment with cemiplimab monotherapy. These patients would be identified during a medical oncology consultation at diagnosis of NSCLC. PD-L1 testing and molecular testing is widely available in all patients with newly diagnosed NSCLC. Patients least suitable for treatment with cemiplimab include those with poor performance status. However, the clinical experts consulted by CADTH also indicated that although the EMPOWER-Lung 1 trial excluded patients with an ECOG PS of 2 or greater, it would be reasonable to extend consideration for cemiplimab treatment to patients with ECOG PS of 2. Patients with pre-existing moderate-to-severe autoimmune conditions may be unsuitable for treatment with cemiplimab.
Assessing Response to Treatment
The clinical experts consulted by CADTH indicated that the only predictive marker of response to PD-1 and PD-L1 inhibitors as monotherapy is PD-L1 testing, which is routinely done in all newly diagnosed patients with advanced or metastatic NSCLC. Patients with PD-L1–strongly positive tumours (≥ 50%) are candidates for PD-1 or PD-L1 inhibitor monotherapy. Additionally, these patients are eligible for anti–PD-1 plus chemotherapy. Clinical response (symptom assessment) and radiological surveillance are used to determine whether a patient is responding to treatment in clinical practice. Improvement in survival and quality of life (i.e., fewer symptoms, higher functional status, or stabilization of symptoms) would be considered a clinically meaningful response to treatment. Treatment response is evaluated clinically at each visit and radiologically, approximately every 3 months to 4 months.
Discontinuing Treatment
The decision to discontinue treatment with cemiplimab is made by the treating oncologist on an individual basis. Treatment with cemiplimab may be discontinued in the case of unequivocal disease progression or severe life-threatening side effects (e.g., pneumonitis, immune myocarditis), or persistent lower-grade side effects.
Prescribing Conditions
Cemiplimab should be prescribed by medical oncologists and administered at a hospital outpatient clinic. PD-L1 testing to select patients for monotherapy is already in routine clinical use.
Additional Considerations
Cemiplimab could be an alternative first-line treatment option for patients with advanced or metastatic NSCLC. The clinical experts believed that based on current data, its efficacy and toxicity seemed to be similar to that of pembrolizumab monotherapy. Therefore, the cost of cemiplimab will be an important factor when considering its place in an already crowded space. In the PD-L1 of 50% or more patient population, there is clinical equipoise with regard to the treatment effect of immunotherapy monotherapy versus immunotherapy combined with chemotherapy. Given the favourable side-effect profile, single-agent pembrolizumab is often favoured over pembrolizumab plus chemotherapy. Cemiplimab will be competing for market share with single-agent pembrolizumab. Clinicians will view cemiplimab as having a similar side-effect profile to pembrolizumab with a shorter follow-up duration. Payers will like the option of having a competitive marketplace that will potentially improve costs.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. Please refer to the Stakeholder Input for the full clinical group input submitted to CADTH.
Clinician input was received from the Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer Drug Advisory Committee, which provides evidence-based clinical and health system guidance on drug-related issues in support of Cancer Care Ontario’s mandate, including the provincial drug reimbursement programs and the systemic treatment program. The clinician group noted that the most important goal of any treatment for NSCLC is to improve OS and improve PFS. Monotherapy immunotherapy also has the additional benefit of avoiding chemotherapy. Patients most likely to benefit from cemiplimab are those with advanced or metastatic NSCLC and tumours having high levels of PD-L1 expression (≥ 50%). Cemiplimab would be used as monotherapy in first-line treatment for these patients and would be an alternative to pembrolizumab monotherapy, or the combination of nivolumab-ipilimumab and 2 cycles of platinum-based chemotherapy. It would not be used as an additional therapy to currently available treatment options. In terms of response to treatment, the clinician group noted that the most meaningful response to treatment is the absence of disease progression followed by improvement in disease-related symptoms, which are assessed every 3 months in clinical practice. Disease progression or intolerable side effects were indicated as the primary reasons to discontinue therapy. The clinician group also noted that treatment continuation beyond progression should remain an option as some patients may benefit from continuing treatment beyond RECIST 1.1–defined progression.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for cemiplimab.
The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
The EMPOWER-Lung 1 trial comparator was platinum-doublet chemotherapy. This is likely not the most relevant comparator anymore. Should the relevant comparator be pembrolizumab alone or in combination with chemotherapy? How about nivolumab-ipilimumab in combination with chemotherapy (not yet funded, but has a positive CADTH recommendation)? | The most relevant comparator is pembrolizumab alone. On an individual level, the choice would be between cemiplimab monotherapy and pembrolizumab monotherapy. If a patient and/or clinician were to choose a treatment other than pembrolizumab monotherapy, there would be no compelling reason for them to consider cemiplimab monotherapy over that treatment. |
Considerations for initiation of therapy | |
There was about a 70% crossover rate from the chemotherapy-alone arm to receive cemiplimab. Should subsequent line treatment be made available on a time-limited basis for patients who would not have had the chance to receive cemiplimab in the first-line setting? And if so, would this only be extended to patients who received chemotherapy alone? | In clinical practice, most patients receive immunotherapy alone or in combination with chemotherapy in the first-line setting. Patients who have not received immunotherapy in the first-line setting should be considered to receive it in the second-line setting (unless contraindicated). Available second-line options are pembrolizumab, nivolumab, and atezolizumab. Second-line single-agent immunotherapy is readily available in all provinces for patients who progress on chemotherapy. The EMPOWER-Lung 1 data should not be extended to a second-line setting. |
In the EMPOWER-Lung 1 trial, at time of progression, patients were permitted to continue cemiplimab with the addition of histology-specific chemotherapy for up to 4 cycles. In clinical practice, should cemiplimab be continued beyond progressive disease as per the EMPOWER-Lung 1 trial? | In clinical practice, patients may continue immunotherapy for clinical benefit beyond disease progression. Adding chemotherapy to immune therapy at disease progression remains investigational. Based on the small number of patients from the EMPOWER-Lung 1 study who had chemotherapy added to immune therapy, no definitive conclusions can be derived. At present, the addition of chemotherapy to immunotherapy at progression is not funded. |
Patients who were never-smokers were not eligible for the EMPOWER-Lung 1 study. Should they be excluded if cemiplimab treatment is funded? | Non-smokers should not be excluded from treatment with cemiplimab. However, since they were excluded from the EMPOWER-Lung 1 trial, there is a lack of evidence on cemiplimab treatment outcomes for this population. |
Are patients who had previous adjuvant or neoadjuvant chemotherapy eligible to receive cemiplimab as was allowed in the EMPOWER-Lung 1 study? | Patients who had previous chemotherapy should be eligible to receive cemiplimab. In the EMPOWER-Lung 1 trial, patients had to be 6 months post-adjuvant/neoadjuvant chemotherapy to be eligible to participate. |
If a patient receives 108 weeks of treatment and subsequently relapses, should they be eligible for re-treatment and if so, would there be a maximum duration? | Patients should be eligible for re-treatment for 17 cycles (1 year) if extrapolating from the KEYNOTE-024 trial that allowed re-treatment for patients who stopped immune therapy early (before 2 years) because of complete response or for patients who completed 2 years of immune therapy and subsequently progressed. |
If a patient discontinues treatment before the completion of 108 weeks due to toxicity, but without relapse, could they restart and be treated to a maximum of 108 weeks? | Patients who discontinue treatment before completion due to toxicity can restart treatment as long as toxicity has resolved. |
Consider alignment with reimbursement criteria for pembrolizumab as well as nivolumab-ipilimumab in combination with chemotherapy. | For pERC consideration. |
Considerations for prescribing therapy | |
In the EMPOWER-Lung 1 study, the dose is 350 mg IV over 30 minutes every 3 weeks until progressive disease or 108 weeks (36 treatments over approximately 2 years). Is the 3 mg/kg IV over 30 minutes every 2 weeks dosing option for patients with low body weight currently available for advanced cutaneous squamous cell carcinoma also recommended for patients with NSCLC? | There is limited evidence to inform on alternative dosages other than that used in the EMPOWER-Lung 1 trial. Ideally, the fixed dose used in the clinical trial should be used. The weight-based dosing would be based on extrapolation from other disease settings. |
Consider alignment with prescribing criteria for pembrolizumab and nivolumab-ipilimumab. | For pERC consideration. |
Generalizability of trial populations to broader populations in the jurisdictions | |
In the EMPOWER-Lung 1 trial, patients were ECOG PS of 0 or 1. Can patients with ECOG PS > 1 be considered eligible | Patients with ECOG PS up to 2 may be considered for eligibility based on data showing efficacy of other treatments, such as chemotherapy, in these patients. |
Should treatment be funded for patients who have had pembrolizumab or nivolumab-ipilimumab in combination with chemotherapy and wish to switch to cemiplimab on a time-limited basis? | Switching is not generally necessary. If patients did not progress on pembrolizumab or nivolumab-ipilimumab, there is no benefit in switching to cemiplimab. |
Potential need for a provincial funding algorithm | |
PAG would like to note that other immune checkpoint inhibitors will not be funded in patients who experience disease progression while receiving cemiplimab. | For pERC consideration. |
Under what conditions would cemiplimab use be preferred over pembrolizumab, or nivolumab-ipilimumab in combination with chemotherapy? | With similar efficacy and toxicity profiles, the choice of first-line immunotherapy monotherapy will be based on access and physician choice. Similarly, the decision of first-line monotherapy vs. an immunotherapy-chemotherapy combination will be based on access, toxicity considerations, and volume of disease (ultimately physician choice) in the absence of evidence-based comparison data. Longer-term follow-up supports the use of pembrolizumab over cemiplimab. If funding allowed cemiplimab to continue with the addition of 4 cycles of chemotherapy at the time of progression, this would make it appealing to clinicians. |
Care provision issues | |
Pembrolizumab is currently reimbursed for this indication and nivolumab-ipilimumab in combination with chemotherapy is currently undergoing price negotiations. | For pERC consideration. |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; NSCLC = non–small cell lung cancer; PAG = Provincial Advisory Group; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee; vs. = versus.
Clinical Evidence
The clinical evidence included in the review of cemiplimab is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To evaluate the efficacy and safety of cemiplimab 50 mg/mL for IV infusion for the first-line treatment of adult patients with NSCLC expressing PD-L1 levels (TPS ≥ 50%), as determined by a validated test, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and who are not candidates for surgical resection or definitive chemoradiation, or who have metastatic NSCLC.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with NSCLC expressing PD-L1 levels (TPS ≥ 50%), with no EGFR, ALK, or ROS1 aberrations, who have previously untreated locally advanced NSCLC and are not candidates for surgical resection or definitive chemoradiation, or who have metastatic NSCLC Subgroups of interest
|
Intervention | Cemiplimab IV infusion, 350 mg Q3W |
Comparator |
|
Outcomes | Efficacy outcomes
Harms outcomes
Notable harms
|
Study designs | Published and unpublished phase III and phase IV randomized controlled trials |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; HRQoL = health-related quality of life; NSCLC = non–small cell lung cancer; OS = overall survival; PFS = progression-free survival; Q3W = every 3 weeks; TPS = Tumor Proportion Score; vs. = versus.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.18
Published literature was identified by searching the following bibliographic databases: Medline All (1946‒) via Ovid and Embase (1974‒) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy consisted of both controlled vocabulary, such as the US National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Libtayo (cemiplimab) and NSCLC. Clinical trials registries were searched: the US National Institutes of Health’s ClinicalTrials.gov, the WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on November 29, 2021. Regular alerts updated the search until the meeting of the CADTH pan-Canadian Oncology Drug Review Expert Committee on April 13, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the CADTH Grey Matters: A Practical Tool for Searching Health-Related Grey Literature checklist.19 Included in this search were the websites of regulatory agencies (the US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for NMAs dealing with NSCLC was run in MEDLINE All (1946‒) on November 29, 2021. No limits were applied to the search.
Findings From the Literature
A total of 249 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6.
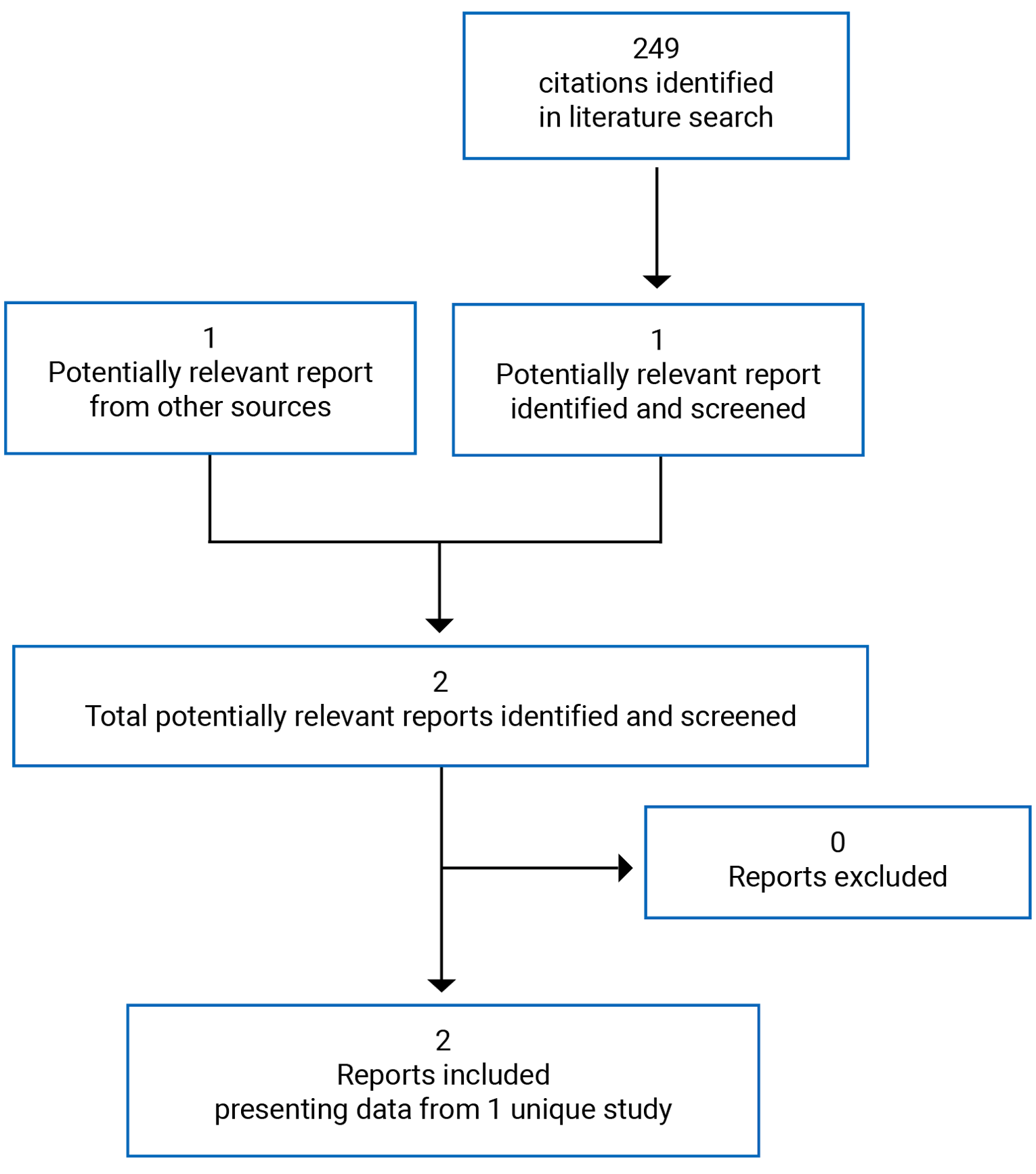
Table 6: Details of the EMPOWER-Lung 1 Study
Detail | Design and population |
|---|---|
Study design | Phase III, open-label, multi-centre RCT |
Locations | 138 centres in 24 countries (Australia, Belarus, Brazil, Bulgaria, Chile, China, Colombia, Czech Republic, Georgia, Greece, Hungary, Jordan, Lebanon, Malaysia, Mexico, Philippines, Poland, Romania, Russia, Spain, Taiwan, Thailand, Turkey, Ukraine) |
Study duration | May 29, 2017, to ongoing Patient enrolment: June 27, 2017, to February 27, 2020 |
Data cut-off date | March 1, 2020 (pre-specified interim/primary analysis) |
Number of patients randomized (randomization ratio) | 710 (1:1 = intervention:comparator) |
Main inclusion criteria |
|
Main exclusion criteria |
|
Drugs | |
Intervention | Cemiplimab monotherapy 350 mg IV Q3W for up to 108 weeks or until RECIST 1.1–defined progressive disease, unacceptable toxicity, death, or withdrawal of consent Patients who experienced progressive disease were given the option to continue treatment with cemiplimab 350 mg IV Q3W for up to 108 additional weeks, with the addition of histology-specific platinum-based doublet chemotherapy for 4 cycles. |
Comparator | 4 cycles to 6 cycles of investigator’s choice of standard of care platinum-based doublet chemotherapy Patients who experienced progressive disease were given the option to cross over to cemiplimab monotherapy 350 mg IV Q3W for up to 108 weeks. |
Duration | |
Phases | |
Screening (run-in) | Up to 28 days before randomization |
Open label |
|
Follow-up | Ongoing; follow-up visits every 6 weeks for approximately 6 months and then at 9 months and 12 months after the last dose of treatment. The last patient’s last visit is 48 months from enrolment of the last patient. |
Outcomes | |
Primary end points | OS and PFS by blinded IRC using RECIST 1.1 criteria |
Secondary end points |
|
Safety end points |
|
Notes | |
Publications | Sezer et al. (2021)20 |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CNS = central nervous system; CTLA-4 = anti-cytotoxic T-Lymphocyte-associated antigen 4; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; GFR = glomerular filtration rate; HCV = hepatitis C virus; HRQoL = health-related quality of life; IHC = immunohistochemistry; IRC = independent review committee; NSCLC = non–small cell lung cancer; PD-L1 = programmed death ligand 1; OS = overall survival; PIK3 = phosphatidyl inositol 3-kinase; PFS = progression-free survival; Q3W = every 3 weeks; RCT = randomized controlled trial; RECIST 1.1 = Response Evaluation Criteria in Solid Tumors Version 1.1; RNA = ribonucleic acid; ULN = upper limit of normal.
Note: One additional report was included (Health Canada reviewer’s report).3
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Description of the EMPOWER-Lung 1 Study
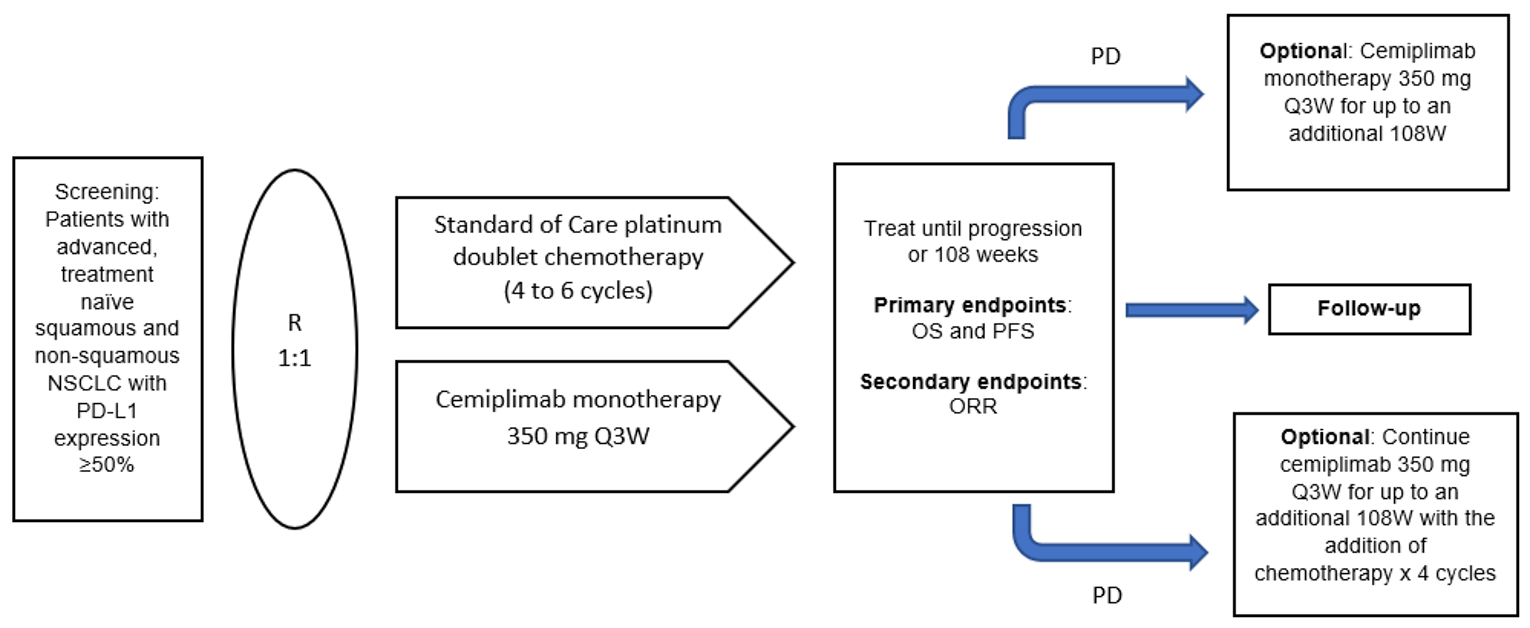
The EMPOWER-Lung 1 study (also named R2810-ONC-1624) is an ongoing phase III, open-label, multi-centre, randomized controlled trial with the objective of comparing the survival benefit of cemiplimab monotherapy to platinum-based doublet chemotherapy in the first-line treatment of patients with advanced or metastatic NSCLC whose tumours express PD-L1 in 50% of tumour cells or more, with no EGFR, ALK, or ROS1 aberrations, and who are not candidates for surgical resection or definitive chemotherapy. The trial was conducted in 138 sites across 24 countries (none in Canada). The clinical cut-off date for the primary analysis was March 1, 2020. The trial was funded by Regeneron Pharmaceuticals and Sanofi (Figure 2).
Randomization and treatment allocation: A total of 710 patients were randomized (1:1) to receive either cemiplimab monotherapy or the investigator’s choice of chemotherapy, which was decided for each patient before randomization. Randomization was performed according to a central randomization scheme provided by an interactive web response system stratified by the patient’s tumour histology (squamous versus non-squamous) and geographic region (Europe, Asia, or rest of the world). The stratification by geographical region was performed to balance treatment assignment within regions and was not included in the analysis of efficacy end points due to small numbers of some of the stratification categories.
Blinding: As EMPOWER-Lung 1 was an open-label study, there was no masking of either study investigators or patients to treatment allocation. To reduce bias in the assessment of disease progression, the assessment of end points was performed by an independent review committee (IRC) blinded to the patients’ study treatment assignment.
Study phases: The study consisted of a screening phase, a treatment phase, and a follow-up phase. Patients underwent a screening evaluation to determine their eligibility within 28 days before randomization. Baseline radiographic tumour assessments were also performed within 28 days before randomization. In the treatment phase, patients received either cemiplimab 350 mg administered intravenously every 3 weeks for up to 108 weeks or 4 cycles to 6 cycles of the investigator’s choice of platinum-doublet chemotherapy. Following disease progression, patients in the cemiplimab arm had the option to continue treatment with cemiplimab at the same dose and frequency for 108 additional weeks, with the addition of histology-specific platinum-based doublet chemotherapy for 4 cycles. Patients in the chemotherapy group whose disease had progressed were allowed to cross over to receive cemiplimab monotherapy for up to 108 weeks.
Protocol Amendments
There were several changes made to the study protocol throughout the duration of the study. Major amendments (from most to least recent) included: (1) adding 4 additional time points for interim analyses and updating the alpha spending function under the originally specified O’Brien-Fleming alpha spending framework, (2) changing OS to a primary objective and end point (from secondary) to provide more robust support for a registration application, (3) increasing the target enrolment from 300 patients to 700 patients to accommodate the anticipated weaker effect on PFS observed from other studies of anti–PD-1 in similar patient populations (the KEYNOTE-042 trial, of pembrolizumab),21 (4) adding the option to continue cemiplimab with the addition of 4 cycles of histology-specific chemotherapy after initial disease progression on cemiplimab monotherapy and removing the option for patients randomized to cemiplimab monotherapy to continue cemiplimab monotherapy until further progression, and (5) adding the option to cross over to cemiplimab after initial progression on chemotherapy.
NSCLC = non–small cell lung cancer; ORR = objective response rate; OS = overall survival; PD = progressive disease; PD-L1 = programmed cell death ligand 1; PFS = progression-free survival; Q3W = every 3 weeks; R = randomization; W = week.
Note: Study design was in effect during protocol amendment 8 when the primary end point was reached.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Populations
Inclusion and Exclusion Criteria
Eligible patients were 18 years or older, prior or current smokers, with histologically or cytologically confirmed stage IIIB or stage IIIC NSCLC who were not candidates for definitive chemoradiotherapy, or with untreated stage IV squamous or non-squamous NSCLC, with PD-L1 expressed in at least 50% of tumour cells (i.e., TPS ≥ 50%), and ECOG PS of 0 or 1. Patients were ineligible if they had never smoked (≤ 100 cigarettes in a lifetime), had active or untreated brain metastases, had tumours positive for EGFR mutations, ALK translocations, or ROS1 fusions, or had active, known, or suspected autoimmune disease that required systemic treatment during the previous 2 years. Patients with adequately treated clinically stable central nervous system metastases were eligible for the study.
Baseline Characteristics
Between June 27, 2017, and February 27, 2020, 710 patients who met the eligibility criteria were randomly assigned to receive cemiplimab (n = 356) or chemotherapy (n = 354). Due to issues with PD-L1 testing identified during the sponsor’s monitoring, samples from 235 patients tested before August 2018 had to be retested. Of these patients, 56 were found to have PD-L1 of less than 50% on retest. Consequently, a PD-L1 of 50% or more population was pre-specified to include only patients with PD-L1 of at least 50% on retest and those who were tested after August 2018 and were unaffected by testing irregularities. The PD-L1 of 50% or more population consisted of 563 patients (n = 283 for cemiplimab and n = 280 for chemotherapy).
In the ITT population, most patients were White (86.3%) and male (85.4%). More than half of the patients were younger than 65 years of age (54.9%), with a median (range) age of 63.0 (31 to 84) years. Almost 3-quarters of the patients had an ECOG PS of 1 (73.0%). All patients were either current (35.6%) cigarette smokers or past (64.4%) cigarette smokers. Overall, a higher percentage of patients presented with non-squamous histology compared to squamous histology (56.2% versus 43.8%, respectively), and the majority (83.8%) of patients had metastatic (stage IV) disease at screening versus locally advanced (stage IIIB or stage IIIC) disease (16.2%). The baseline demographic and disease characteristics were well balanced between the cemiplimab and chemotherapy arms in the ITT population and the PD-L1 of 50% or more population. The ITT population and the PD-L1 of 50% or more population also appeared similar on all noted characteristics (Table 7).
Table 7: Summary of Baseline Characteristics
Characteristic | ITT population | PD-L1 ≥ 50% population | ||
|---|---|---|---|---|
Cemiplimab (N = 356) | Chemotherapy (N = 354) | Cemiplimab (N = 283) | Chemotherapy (N = 280) | |
Age, years | ||||
Mean (SD) | 63.0 (8.17) | 63.3 (8.56) | 63.1 (8.17) | 63.9 (8.48) |
≥ 65, n (%) | 156 (43.8) | 164 (46.3) | 126 (44.5) | 133 (47.5) |
Sex, n (%) | ||||
Male | 312 (87.6) | 294 (83.1) | 248 (87.6) | 231 (82.5) |
Female | 44 (12.4) | 60 (16.9) | 35 (12.4) | 49 (17.5) |
Race, n (%) | ||||
White | 308 (86.5) | 305 (86.2) | 243 (85.9) | 240 (85.7) |
Black or African-American | 1 (0.3) | 3 (0.8) | 1 (0.4) | 3 (1.1) |
Asian | 39 (11.0) | 38 (10.7) | 31 (11.0) | 29 (10.4) |
American Indian or Alaska native | 6 (1.7) | 8 (2.3) | 6 (2.1) | 8 (2.9) |
Other/not reported | 2 (0.6) | 0 | 2 (0.7) | 0 |
Geographical region, n (%) | ||||
Europe | 275 (77.2) | 278 (78.5) | 215 (76.0) | 216 (77.1) |
Asia | 39 (11.0) | 38 (10.7) | 31 (11.0) | 29 (10.4) |
Rest of the world | 42 (11.8) | 38 (10.7) | 37 (13.1) | 35 (12.5) |
ECOG PS, n (%) | ||||
0 | 96 (27.0) | 96 (27.1) | 77 (27.2) | 75 (26.8) |
1 | 260 (73.0) | 258 (72.9) | 206 (72.8) | 205 (73.2) |
Smoking status, n (%) | ||||
Current smoker | 133 (37.4) | 120 (33.9) | 105 (37.1) | 92 (32.9) |
Past smoker | 223 (62.6) | 234 (66.1) | 178 (62.9) | 188 (67.1) |
Cancer stage at screening, n (%) | ||||
Locally advanced | 63 (18) | 52 (15) | 45 (16) | 42 (15) |
Metastatic | 293 (82) | 302 (85) | 238 (84) | 238 (85) |
Histology/cytology, n (%) | ||||
Squamous | 159 (45) | 152 (43) | 122 (43) | 121 (43) |
Non-squamous | 197 (55) | 202 (57) | 161 (57) | 159 (57) |
Brain metastases, n (%) | 44 (12) | 39 (11) | 34 (12) | 34 (12) |
ECOG PS = Eastern Cooperative Oncology Group Performance Status; ITT = intention-to-treat; PD-L1 = programmed cell death-ligand 1; SD = standard deviation.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017)9 and Sezer et al. (2021).20
Interventions
Cemiplimab
For patients randomly assigned to the cemiplimab arm, cemiplimab 350 mg was administered intravenously over a period of 30 minutes every 3 weeks for up to 108 weeks (i.e., 36 treatment cycles) or until RECIST 1.1–defined disease progression, unacceptable toxicity, death, or withdrawal of consent. Cemiplimab dose modification was not allowed.
Chemotherapy
For patients randomly assigned to the chemotherapy arm, platinum-based doublet chemotherapy (with and without maintenance therapy) was administered for 4 cycles to 6 cycles and according to the local prescribing information and practice guidelines. The investigators were allowed to choose from several alternatives, consistent with clinical treatment guidelines for chemotherapy in NSCLC at the time the trial was designed. Chemotherapy dose modification was allowed according to regional guidelines and standard of care. Treatments were continued for the specified duration of number of cycles or until RECIST 1.1–defined disease progression, unacceptable toxicity, death, or withdrawal of consent. Refer to Table 8 for the investigators’ choice of chemotherapy options.
Concomitant Treatments
Any treatment administered from the time of informed consent until 90 days after the last study treatment was considered concomitant medication. This included medications that were started before the study and were ongoing during the study as well as any therapies started in the follow-up period to treat a study drug–related AE.
While participating in the study, a patient could not receive any investigational agent for treatment of a tumour other than cemiplimab monotherapy or the study’s pre-specified chemotherapy regimen. Treatment with bevacizumab or necitumumab was not 1 of the protocol-defined treatment options. Patients for whom these 2 agents were deemed appropriate by the treating physician were asked not to enrol in the study. Other medications considered necessary for the patient’s welfare that were not expected to interfere with the evaluation of cemiplimab could be administered at the discretion of the investigator.
Crossover
An amendment was made to the protocol (amendment 6, August 22, 2018), allowing patients in the cemiplimab arm who progressed as per RECIST 1.1 criteria while on cemiplimab monotherapy to continue cemiplimab treatment with the addition of 4 cycles of histology-specific platinum-doublet chemotherapy until further progression was observed, provided the patient had not completed the 108-week treatment period. Patients in the chemotherapy group whose disease had progressed were allowed to cross over to receive cemiplimab monotherapy for up to 108 weeks.
Interim analysis 2, with a database lock date of April 14, 2020, was conducted when approximately 50% of expected OS events were observed; the primary end point of OS was reached. Following interim analysis 2, an amendment was made to the protocol (amendment 9) that allowed patients randomized to the chemotherapy arm to receive cemiplimab 350 mg every 3 weeks for up to 108 weeks.
Study Treatment Discontinuation
Patients who permanently discontinued from the study drug and did not withdraw from the study were asked to return to the clinic for all remaining study visits. Patients who permanently discontinued from the study drug and opted to withdraw from the study were asked to complete study assessments. The reasons for permanent discontinuation of cemiplimab included pregnancy, safety reasons or compliance issues at the discretion of the investigator or sponsor, and a patient with confirmed complete response who chose to stop treatment early after at least 6 months of treatment and was followed for the duration of study. Chemotherapy was permanently discontinued for safety reasons, compliance issues, intolerance due to toxicity, or other reasons as provided by regional guidelines and standard of care.
Table 8: Investigators’ Choice of Chemotherapy Regimens
Option | Chemotherapy regimen | Dosing frequency | Maintenance therapy |
|---|---|---|---|
1 | Pemetrexed 500 mg/m2 IV plus cisplatin 75 mg/m2 IV | Day 1 every 21 days for 4 to 6 cycles | Optional pemetrexed 500 mg/m2 IV day 1 every 21 days |
2 | Pemetrexed 500 mg/m2 IV plus carboplatin AUC of 5 mg/mL/minute IV or 6 mg/mL/minute IV | Day 1 every 21 days for 4 to 6 cycles | Optional pemetrexed 500 mg/m2 IV day 1 every 21 days |
3 | Paclitaxel 200 mg/m2 IV plus cisplatin 75 mg/m2 IV | Day 1 every 21 days for 4 to 6 cycles | No maintenance |
4 | Paclitaxel 200 mg/m2 IV plus carboplatin AUC of 5 mg/mL/minute IV or 6 mg/mL/minute IV | Day 1 every 21 days for 4 to 6 cycles | No maintenance |
5 | Gemcitabine 1,250 mg/m2 IV plus cisplatin 100 mg/m2 IV | Day 1 and day 8 (gemcitabine only) every 21 days for 4 to 6 cycles | No maintenance |
6 | Gemcitabine 1,250 mg/m2 IV plus carboplatin AUC of 5 mg/mL/minute IV or 6 mg/mL/minute IV | Day 1 and day 8 (gemcitabine only) every 21 days for 4 to 6 cycles | No maintenance |
AUC = area under the curve.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Outcomes
The efficacy end points identified in the CADTH review protocol that were assessed in the clinical trial included in this review are summarized as follows.
Efficacy
The primary end points were OS and PFS. OS was defined as the time from randomization to death or any cause. A patient who had not died was censored at the last known date of contact. PFS was defined as the time from randomization to the date of first documented disease progression as determined by the IRC using RECIST 1.1, or death from any cause. Patients were censored according to the following rules: patients without documented progression or death were censored at the last evaluable tumour assessment; patients without documented progression or death before initiation of new antitumour therapy were censored on the date of their last evaluable tumour assessment before, or on the date of the new antitumour therapy; patients who withdrew consent before taking any study treatment were censored at the date of randomization; and patients without any evaluable tumour assessments after randomization who did not die were censored on the date of randomization.
Secondary end points included ORR and DOR. ORR was defined as the proportion of patients with a best overall response of confirmed complete response or partial response. In performing this calculation, best overall response was determined between the date of randomization and the date of the first objectively documented progression or subsequent anticancer therapy, whichever came first. DOR was defined as the time between the date of first complete or partial response and the date of first documented disease progression or death from any cause (in patients with best overall response of complete or partial response).
Radiographic tumour assessments were carried out every 3 cycles (at week 9) and every 9 weeks thereafter until disease progression. Response was assessed according to RECIST 1.1 by IRC. Patients were followed up 2 weeks to 4 weeks after the last dose of study treatment, then 6 weeks after the last follow-up and every 9 weeks thereafter for the first year. Patients were then followed up for survival every 3 months.
HRQoL was measured by means of the EORTC QLQ-C30 and the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13 (EORTC QLQ-LC13). Patients completed questionnaires before any study procedures or visits, on day 1 of each cycle for the first 6 doses, and then on day 1 every 3 cycles (every 9 weeks).
The EORTC QLQ-C30 instrument is a generic questionnaire consisting of 30 items developed to assess symptoms and functioning of cancer patients. The instrument includes 5 functional scales, 4 symptom scales, 1 GHS scale, and 1 financial impact score. Most items are scored 1 (“not at all”) to 4 (“very much”) except for the items contributing to the GHS scale, which are scored 1 (“very poor”) to 7 (“excellent”). The recall period for each question is “during the past week.” An outcome variable consisting of a score from 0 to 100 is derived for each of the symptom scales or items according to the EORTC QLQ-C30 instructions. Higher scores on symptoms indicate a worse health state. Higher scores on the GHS and functioning scales indicate better health status or function.22
The EORTC QLQ-LC13 is a questionnaire measuring lung cancer symptoms and side effects from conventional chemotherapy and radiotherapy. It comprises 13 questions assessing lung cancer symptoms (cough, hemoptysis, dyspnea, and site-specific pain), treatment-related side effects (sore mouth, dysphagia, peripheral neuropathy, and alopecia), and pain medication. Except for a multi-item scale for dyspnea, all are single items. An outcome variable consisting of a score from 0 to 100 is derived for each of the symptom scales or items according to the EORTC QLQ-LC13 instructions. Higher scores on the symptom scales indicate greater symptom burden and therefore a worse health state.23
The validity and reliability of both instruments have been established in populations of patients with lung cancer. However, there is a dearth of information with respect to their responsiveness in this setting. A detailed discussion and critical appraisal of the EORTC QLQ-C30 and EORTC QLQ-LC13 is provided in Appendix 2.
Safety
Patient safety was assessed based on reported AEs, clinical laboratory data, vital signs, an electrocardiogram, and a physical examination. In general, these data were collected at baseline and on the first day of each cycle.
For safety variables, 3 observation periods were defined:
the pre-treatment period, defined as the time from signing the informed consent form to before the first dose of study drug
the on-treatment period, defined as the time from the day of the first dose of study drug to the day of the last dose of study drug plus 90 days or to 1 day before patients received their first dose of cemiplimab as crossover treatment or treatment of chemotherapy plus cemiplimab, or another anticancer systemic therapy, whichever was earlier
the post-treatment period, defined as the time starting 1 day after the end of the on-treatment period.
TEAEs were defined as AEs that developed or worsened during the on-treatment period and any treatment-related AEs that occurred during the post-treatment period but before the start of crossover treatment or treatment of chemotherapy plus cemiplimab. TEAEs included a list of immune-mediated preferred terms. All AEs with no alternative etiology associated with drug exposure were evaluated to determine possible immune etiology. Potential immune-related AEs requiring treatment with systemic corticosteroid or other immunosuppressants or events that were immune-mediated endocrinopathies were defined as immune-mediated AEs. These could occur any time from shortly after the first dose of treatment to several months after the last dose of treatment.
Infusion-related reactions included any AEs that occurred during an infusion or within 1 day after the infusion was completed.
All AEs reported were coded using the currently available version of the Medical Dictionary for Regulatory Activities. The verbatim text, the preferred term, and the primary System Organ Class were listed.
Statistical Analysis
Sample Size Calculations
The enrolment of 700 patients was planned and estimated to occur over 38 months (3 patients per month for the first 4 months, 12 patients per month from month 5 to month 8, 23 patients per month from month 9 to month 16, 30 patients per month from month 17 to month 20, and 20 patients per month thereafter). A dropout rate of 10% per year was assumed.
OS: Historically, in patients with stage IIIB, stage IIIC, or stage IV NSCLC treated with cisplatin or carboplatin plus paclitaxel every 3 weeks, the median OS has ranged from approximately 11.3 months to 14.2 months.24-26 For anti–PD-1 monotherapy, a delayed treatment effect in OS was observed in the KEYNOTE-042 and CheckMate 026 trials.21,24 In these studies, during the first 6 months of treatment, anti–PD-1 monotherapies had either no treatment effect or worse treatment effect in OS when compared to treatment with chemotherapy, especially in the patient population with PD-L1 expression of less than 50%. The sponsor assumed without crossover effect a median OS of 13 months for patients treated with chemotherapy alone, and a non-proportional HR between cemiplimab and chemotherapy, with an HR of 1.05 for the first 6 months and an HR of 0.58 after 6 months. With 5 interim analyses planned using the Lan-DeMets O’Brien-Fleming alpha spending function, and with 476 deaths expected at the final analysis of OS, the EMPOWER-Lung 1 study had approximately 86% power for detecting a significant OS effect at a 2-sided alpha of 0.04 and approximately 88% power for detecting a significant OS effect at a 2-sided alpha of 0.05.
PFS: Historically, in patients with stage IIIB, stage IIIC, or stage IV NSCLC treated with cisplatin or carboplatin plus paclitaxel every 3 weeks, the median PFS has ranged from approximately 4.8 months to 6.4 months.24-26 For PD-1 monotherapy, a delayed treatment effect in PFS was observed in KEYNOTE-042 and CheckMate 026 trials.21,24 In these studies, during the first 3 months of treatment, PD-1 monotherapies had either no treatment effect or worse treatment effect in PFS when compared to treatment with chemotherapy, especially in the patient population with PD-L1 expression of less than 50%. The sponsor assumed a median PFS of 6.4 months for patients treated with chemotherapy alone, and an HR between cemiplimab and chemotherapy, with an HR of 1.3 for the first 3 months and an HR of 0.5 after 3 months. With 525 PFS events, the EMPOWER-Lung 1 study had approximately 76% power for detecting a significant PFS effect at a 2-sided alpha of 0.01 and approximately 90% power for detecting a significant PFS effect at a 2-sided alpha of 0.05.
Analyses of Outcomes
The overall type I error rate for analyses of the primary end points of OS and PFS was controlled at a 2-sided alpha of 0.05 by the following alpha-reallocation strategy: the 2-sided alpha of 0.05 is initially split between the analyses of OS and PFS, with 0.04 for the OS analysis and 0.01 for the PFS analysis; the alpha allocated to PFS (0.01) was to be reallocated to OS if the PFS test was positive; and the alpha allocated to OS (0.04) was to be reallocated to PFS if the OS test was positive. The type I error rate for the interim and final analyses of OS was controlled using the O’Brien-Fleming spending function. The type I error rate for the analyses of primary and key secondary end points was controlled by a hierarchical testing procedure — namely, ORR was planned to only be tested if both analyses of OS and PFS were statistically significant. The main analyses did not account for the potential crossover or extended treatment that could occur after disease progression.
The primary end points (OS and PFS) were analyzed by stratified log-rank test using tumour histology (non-squamous versus squamous) as a stratification factor. HRs and corresponding 95% CIs were estimated by a stratified Cox regression model using the treatment as a covariate and tumour histology as a stratification factor. The distributions of OS and PFS were estimated using the Kaplan-Meier method.
The key secondary end point of ORR was tested only if both OS and PFS analyses were statistically significant, using the Cochran-Mantel-Haenszel test stratified by tumour histology (non-squamous versus squamous). The associated odds ratio and 95% CI were calculated. ORR and the corresponding 95% exact CI were calculated by the Clopper-Pearson method for each treatment arm.
The DOR determined for patients with an observed best overall response of complete or partial response was descriptive. Patients who never progressed while being followed were censored at the last valid tumour assessment.
For the scales of the EORTC QLQ-C30 and EORTC QLQ-LC13, the mean change from baseline score to each post-baseline visit was summarized descriptively, with a 10-point change considered to be clinically meaningful.
All safety end points were reported using descriptive statistics.
Summaries of TEAEs by treatment group included the following:
the number (n) and percentage (%) of patients with at least 1 TEAE by System Organ Class and preferred term
TEAEs by severity (National Cancer Institute’s Common Terminology Criteria for Adverse Events, version 4.03), presented by System Organ Class and preferred term
TEAEs by outcome
TEAEs leading to treatment discontinuation
TEAEs related to study drug
AEs of special interest
immune-related AEs
infusion-related reactions, defined as any AE occurring during the infusion or within 1 day after the infusion was completed).
Deaths and serious adverse events (SAEs) were listed and summarized by treatment group. Events of Common Terminology Criteria for Adverse Events grade 3 and grade 4 severity were summarized by treatment group. TEAEs leading to permanent treatment discontinuation were listed and summarized by treatment group.
Subgroup Analyses
HRs for OS and PFS were evaluated by pre-planned subgroups according to ECOG PS and tumour histology at baseline; these were subgroups of interest identified in the CADTH systematic review protocol. Subgroup analyses by the presence of brain metastasis and stage of cancer at baseline (locally advanced or metastatic) were added following study unblinding and database lock. Between-group treatment effect with a nominal 95% CI for these end points was estimated within each category. There was no multiplicity control. As such, all subgroup analyses are exploratory in nature.
Sensitivity Analyses
Sensitivity analyses for OS included using the rank-preserving structural failure time (RPSFT) model to account for the effect of the additional treatment after disease progression (crossover effect).27 The method relies on the assumption of common treatment effect, in which the treatment effect is the same for patients initially randomized to the experimental arm (cemiplimab in this case) and those who cross over from the control arm (chemotherapy in this case) to receive the experimental treatment.28 Re-censoring was applied to avoid potential bias from informative censoring introduced by the method. A second sensitivity analysis used the restricted mean survival time (RMST) method to account for the possible non-proportional hazards effect.
Three sensitivity analyses were performed for PFS using different censoring rules and progressive disease definitions. The first was the same as the primary analysis except that it considered the initiation of new antitumour treatment as a progressive disease event for patients without documented progressive disease or death before initiation of new antitumour treatment. In the second sensitivity analysis, a patient who had progressive disease or death after missing 2 or more tumour assessments was censored at the last evaluable tumour assessment before missing 2 or more tumour assessments. The third sensitivity analysis was performed based on investigator-determined progressive disease events.
Interim Analyses
The study protocol specified 5 interim analyses before the final analysis, to be done by an independent statistics group and reviewed by the independent data-monitoring committee. The first interim analysis of OS was planned after approximately 1-third of OS events expected according to the sample size calculations (159 deaths) were observed, and the second interim analysis was planned after approximately 50% of expected events (238 deaths) were observed. The first interim analysis was conducted after 164 deaths had occurred and the OS result did not meet the significance threshold. The second interim analysis was conducted after 249 deaths had occurred and was based on the data cut-off date of March 1, 2020. The nominal alpha for the second interim analysis was adjusted to 0.00255 by the Lan-DeMets approach with the O’Brien-Fleming spending function based on actual number of events observed.
Efficacy results based on the second interim analysis were reviewed by the independent data-monitoring committee, which recommended that the randomized portion of the study be stopped given that cemiplimab met the pre-specified threshold for demonstration of superior OS benefit compared to chemotherapy. Subsequently, all eligible patients randomly assigned to chemotherapy were given the option to cross over to cemiplimab.
Analysis Populations
There were 3 pre-specified (a priori–defined) analysis populations.
The full analysis population (ITT), which included all randomized patients (N = 710). The ITT population was based on the treatment allocated (as randomized). Within this report, conclusions are based on the ITT population.
The PD-L1 of 50% or more population (modified intention-to-treat 1 [mITT-1]; N = 563), which included all randomized patients who were enrolled based on tests performed after August 2018 whose tumours expressed PD-L1 in at least 50% of tumour cells based on the 22C3 pharmDx PD-L1 assay at entry performed in accordance with approved labelling, including assay instructions for use (n = 475). Also included were patients who were tested before August 2018, but whose PD-L1 samples required retesting due to PD-L1 quality testing issues and, upon retest, were confirmed as having PD-L1 tumour expression in at least 50% of tumour cells based on a PD-L1 assay performed in accordance with approved labelling, including assay instructions for use (n = 88).
The safety analysis population, which included all randomized patients who received at least 1 dose of any study drug (N = 697). This population was based on the treatment received (as treated).
Efficacy end points were assessed in the ITT population as well as in the PD-L1 of 50% or more population. Safety was assessed in all patients who received at least 1 dose of the assigned study treatment.
Figure 3 shows patient disposition by PD-L1 testing status and retest results.
Figure 3: Analysis Populations by PD-L1 Testing Status and Retest
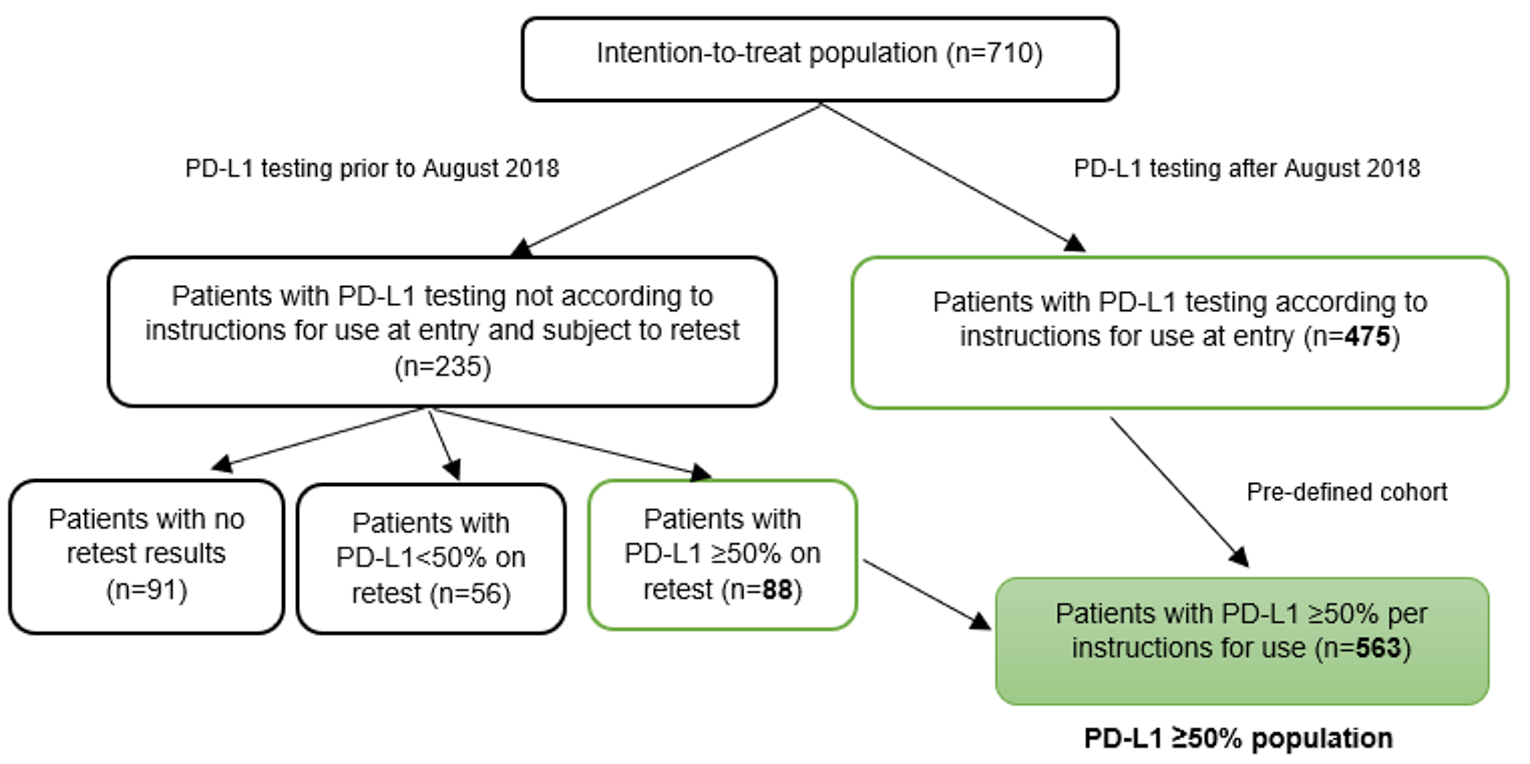
PD-L1 = programmed cell death-ligand 1.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Results
Patient Disposition
Details on patient disposition are provided in Table 9. Of 3,662 patients screened for eligibility, 710 (19.4%) were enrolled and randomized. At the time of data cut-off (March 1, 2020), of the 710 patients (i.e., the ITT population), treatment was ongoing for 184 patients (25.9%) and 513 patients (72.3%) were off treatment. Thirteen patients (1 in the cemiplimab arm and 12 in the chemotherapy arm) were randomized but not treated. Study treatment was completed in 155 (21.8%) patients: 6 (1.7%) patients in the cemiplimab arm and 149 (42.1%) patients in the chemotherapy arm. The difference in treatment completion between the 2 treatment arms may have been at least partially due to the different duration of study treatment in the cemiplimab arm (administered every 3 weeks up to 108 weeks) compared with the chemotherapy arm (administered for 4 cycles to 6 cycles every 3 weeks). The most common reason for treatment discontinuation was disease progression (37.4% in the cemiplimab arm and 23.7% in the chemotherapy arm).
At the time of the data cut-off, 163 (45.8%) patients in the cemiplimab arm and 208 (58.8%) patients in the chemotherapy arm had discontinued the study. Withdrawal of consent was more common in the chemotherapy group (12.7%) than the cemiplimab group (7.9%). The mean study follow-up duration in the ITT population was 14.04 (SD = 7.6) months overall in both treatment arms. The overall median duration of follow-up was 13.09 months in the cemiplimab arm and 13.08 months in the chemotherapy arm.
Table 9: Patient Disposition (Intention-to-Treat Population in the EMPOWER-Lung 1 Study)
Patient disposition | Cemiplimab | Chemotherapy | Total |
|---|---|---|---|
Screened, N | 3,662 | ||
Randomized, N (%)a | 356 (9.7) | 354 (9.7) | 710 (19.4) |
Randomized and not treated (any study drug), n (%) | 1 (0.3) | 12 (3.4) | 13 (1.8) |
Treatment ongoing, n (%) | 139 (39.0) | 45 (12.7) | 184 (25.9) |
Off treatment, n (%) | 216 (60.7) | 297 (83.9) | 513 (72.3) |
Treatment completed | 6 (1.7) | 149 (42.1) | 155 (21.8) |
Treatment discontinued | 210 (59.0) | 148 (41.8) | 358 (50.4) |
Primary reason for treatment discontinuation | |||
Adverse event | 23 (6.5) | 14 (4.0) | 37 (5.2) |
Death | 29 (8.1) | 25 (7.1) | 54 (7.6) |
Loss to follow-up | 3 (0.8) | 4 (1.1) | 7 (1.0) |
Patient decision | 9 (2.5) | 7 (2.0) | 16 (2.3) |
Physician decision | 5 (1.4) | 5 (1.4) | 10 (1.4) |
Disease progression | 133 (37.4) | 84 (23.7) | 217 (30.6) |
Withdrawal of consent | 8 (2.2) | 9 (2.5) | 17 (2.4) |
Study ongoing, n (%) | 193 (54.2) | 146 (41.2) | 339 (47.7) |
Off study, n (%) | 163 (45.8) | 208 (58.8) | 371 (52.3) |
Study completed | 3 (0.8) | 1 (0.3) | 4 (0.6) |
Primary reason for study discontinuation | |||
Death | 96 (27.0) | 111 (31.4) | 207 (29.2) |
Loss to follow-up | 5 (1.4) | 6 (1.7) | 11 (1.5) |
Patient decision | 17 (4.8) | 16 (4.5) | 33 (4.6) |
Sponsor decision | 1 (0.3) | 1 (0.3) | 2 (0.3) |
Physician decision | 1 (0.3) | 3 (0.8) | 4 (0.6) |
Disease progression | 12 (3.4) | 21 (5.9) | 33 (4.6) |
Withdrawal of consent | 28 (7.9) | 45 (12.7) | 73 (10.3) |
Other | 0 | 4 (1.1) | 4 (0.6) |
aThe percentage refers to the proportion of the screened population. All other percentages below the “Randomized, N (%)” fields refer to the proportion of the randomized population.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Protocol Deviations
During the course of the study, 177 important protocol deviations were reported in 112 patients, but these were evenly distributed between the 2 treatment arms and were considered unlikely to jeopardize the integrity of the data.
Exposure to Study Treatments
As of the data cut-off date of March 1, 2020, in the safety population (N = 697), 355 patients received at least 1 dose of cemiplimab and 342 patients received at least 1 dose of chemotherapy. The overall median duration of exposure was 27.3 weeks (range = 0.3 weeks to 115.0 weeks) for cemiplimab and 17.7 weeks (range = 0.6 weeks to 86.7 weeks) for chemotherapy. A total of 197 (55.5%) patients were treated with cemiplimab for 24 weeks or more and 84 (23.7%) patients were treated for 48 weeks or more. In the chemotherapy arm, 54 (15.8%) patients were treated with cemiplimab for 24 weeks or more and 10 (2.9%) patients were treated for 48 weeks or more. The shorter duration of exposure for the chemotherapy arm was mostly due to the majority of patients completing study treatment after 4 cycles to 6 cycles (equal to 12 weeks to 18 weeks) in the chemotherapy arm, compared to those in the cemiplimab arm (108 weeks). As per protocol, only those patients in the chemotherapy arm who were receiving maintenance pemetrexed (a subgroup of those with adenocarcinoma histology) stayed on treatment longer than 4 cycles to 6 cycles (Table 10).
In the cemiplimab arm, 28.2% of patients had at least 1 infusion interruption or dose delay compared with 45.3% of patients in the chemotherapy arm. At least 1 infusion interruption was reported in 4% of patients treated with cemiplimab and 2% of patients treated with chemotherapy.
Table 10: Summary of Treatment Exposure (Safety Population)
Factors | Cemiplimab (n = 355) | Chemotherapy (n = 342) | ||||
|---|---|---|---|---|---|---|
Cisplatin (n = 74) | Carboplatin (n = 271) | Pemetrexed (n = 137) | Paclitaxel (n = 140) | Gemcitabine (n = 68) | ||
Median (IQR) duration of exposure, weeks | 27.3 (12.0 to 46.4) | 13.7 (9.1 to 18.0) | 16.3 (10.6 to 18.3) | 17.9 (10.6 to 32.9) | 17.7 (11.0 to 18.3) | 16.4 (8.7 to 18.8) |
Median number of doses administered | 9 | 5 | 5 | 5 | 6 | 10 |
Number of doses administered, n (%) | ||||||
< 6 | 125 (35) | 43 (58) | 143 (53) | 69 (50) | 66 (47) | 18 (27) |
6 to 12 | 91 (26) | 31 (42) | 128 (47) | 40 (29) | 74 (53) | 27 (40) |
12 to 24 | 100 (28) | 0 | 0 | 26 (19) | 0 | 23 (34) |
24 to 36 | 33 (9) | 0 | 0 | 2 (2) | 0 | 0 |
≥ 36 | 6 (2) | 0 | 0 | 0 | 0 | 0 |
IQR = interquartile range.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017)9 and Sezer et al. (2021).20
Prior and Concomitant Therapy
Overall, 164 (23.1%) patients in the ITT population received prior cancer-related therapy — most frequently, radiotherapy (n = 122 [17.2%] patients). Patients could have received adjuvant or neoadjuvant platinum-doublet chemotherapy, if they had developed recurrent or metastatic disease more than 6 months after completing therapy. This included 83 (23.3%) patients in the cemiplimab arm and 81 (22.9%) patients in the chemotherapy arm. Of these, 33 (4.6%) patients had prior cancer-related systemic therapy, with a mean time from the end of that therapy to randomization of 28.7 (SD = 39.2) months. Prior cancer-related surgery was reported for 33 (9.3%) patients in the cemiplimab arm and 36 (10.2%) patients in the chemotherapy arm. Prior cancer-related radiotherapy was reported for 63 (17.7%) patients in the cemiplimab arm and 59 (16.7%) patients in the chemotherapy arm (Table 11). The proportions of patients with prior and concomitant therapies in the PD-L1 of 50% or more population were similar to those in the ITT population.
|| ||| |||||| ||||||||||| |||| |||||||| |||||| || ||| |||||||||| ||| ||| ||||| || ||| |||||||||||| |||| |||| || ||||| | ||||||||||| |||||||||| |||||| ||| |||||| ||| |||| |||||| ||||||| || ||||||||||| ||||||||||| || ||| |||||||||| ||| |||| |||||||||| ||||||| ||| || ||| |||||||||||| ||| |||| ||||||||||||||| ||| |||||||| ||| |||||||| ||||||||||| ||| |||||||||||||| |||||||| ||||| ||| |||||||||||| ||||||||| |||||||| |||||||||||||| ||| |||||||| ||| |||||||| ||| |||||||||| |||||||| |||||||| ||||||||||||||||| ||||| || |||||||| ||||||||||||||| |||| ||| ||||||| |||||| ||| |||||| ||| |||||||||||||| || |||||||| ||||||||||||||| ||| ||||||||| || |||| || ||||||||||| ||| ||||||||||||| ||||||||||| || |||||||| ||||||||| |||||||||||||
Table 11: Prior Therapy (Intention-to-Treat Population)
Therapy | Cemiplimab (N = 356) | Chemotherapy (N = 354) | Total (N = 710) |
|---|---|---|---|
Number of patients with any prior cancer-related therapy,a n (%) | 83 (23.3) | 81 (22.9) | 164 (23.1) |
Number of patients with any prior cancer-related systemic therapy, n (%) | 13 (3.7) | 20 (5.6) | 33 (4.6) |
Therapy setting, n (%) | |||
Adjuvant | 9 (2.5) | 15 (4.2) | 24 (3.4) |
Neoadjuvant | 4 (1.1) | 7 (2.0) | 11 (1.5) |
Time from end of last prior regimen to randomization (months) | |||
n | 12 | 20 | 32 |
Mean (SD) | 18.7 (19.5) | 34.8 (46.7) | 28.7 (39.2) |
Number of patients with any prior cancer-related surgery,b n (%) | 33 (9.3) | 36 (10.2) | 69 (9.7) |
Number of patients with any prior cancer-related radiotherapy, n (%) | 63 (17.7) | 59 (16.7) | 122 (17.2) |
SD = standard deviation.
aAny prior cancer-related therapy includes patients who have had systemic therapy, surgery (excluding diagnostic procedures), or radiotherapy.
bPrior cancer-related surgery excludes diagnostic procedures.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Subsequent Anticancer Therapy
In the ITT population, about 20% of patients in the cemiplimab arm and 44% of patients in the chemotherapy arm received at least 1 subsequent anticancer therapy. Of note,14.3% in the cemiplimab arm received cemiplimab plus chemotherapy as extended treatment, as allowed by amendment 6 of the protocol (Table 12). In the chemotherapy arm, 156 out of 203 (77%) progressors by data cut-off went on to receive subsequent systemic therapy (1 received surgery) and 150 (42.4%) of 354 randomized patients crossed over to cemiplimab.
Table 12: Subsequent Anticancer Therapies (Intention-to-Treat Population)
Therapy | Cemiplimab (N = 356) | Chemotherapy (N = 354) |
|---|---|---|
Patients with any subsequent anticancer therapies, n (%) | 71 (19.9) | 157 (44.4) |
Radiotherapy a | 0 | 0 |
Surgery | 0 | 1 (0.3) |
Systemic therapy | 71 (19.9) | 156 (44.1) |
Cemiplimab as crossover treatment | 0 | 150 (42.4) |
Cemiplimab plus chemotherapy as extended treatment | 51 (14.3) | 0 |
Other systemic therapies | 23 (6.5) | 18 (5.1) |
Carboplatin | 12 (3.4) | 10 (2.8) |
Paclitaxel | 9 (2.5) | 5 (1.4) |
Cisplatin | 6 (1.7) | 1 (0.3) |
Pemetrexed | 5 (1.4) | 1 (0.3) |
Gemcitabine | 3 (0.8) | 4 (1.1) |
Vinorelbine | 3 (0.8) | 2 (0.6) |
Docetaxel | 2 (0.6) | 4 (1.1) |
Afatinib | 1 (0.3) | 0 |
Bevacizumab | 1 (0.3) | 0 |
Etoposide | 1 (0.3) | 3 (0.8) |
Nintedanib esilate | 1 (0.3) | 1 (0.3) |
Pembrolizumab | 0 | 2 (0.6) |
aExcludes radiotherapies with palliative intent.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported as follows.
Overall Survival
In the ITT population, 249 deaths had occurred by the data cut-off date (35% of the ITT population); the study met its OS primary end point by showing a statistically significant increase in survival with cemiplimab versus chemotherapy in the targeted population. Median OS was 22.1 months (lower bound of 95% CI = 17.7; upper bound = not estimable) in the cemiplimab arm versus 14.3 months (95% CI, 11.7 months to 19.2 months) in the chemotherapy arm (P = 0.0022). The HR for OS comparing cemiplimab to chemotherapy was 0.676 (95% CI, 0.52 to 0.87). The estimated probability of surviving from baseline through 12 months and 24 months was 70.3% (95% CI, 64.4% to 75.4%) and 48.6% (95% CI, 39.2% to 57.3%), respectively, for patients in the cemiplimab arm compared with 55.7% (95% CI, 49.2% to 61.7%) and 29.7% (95% CI, 18.8% to 41.4%), respectively, for patients in the chemotherapy arm (Table 13 and Figure 4).
In the PD-L1 of 50% or more population, OS results were similar to those in the ITT population (Table 13 and Figure 8 in Appendix 3).
In the subgroup analyses in both the ITT population and the PD-L1 of 50% or more population, point estimates for HR were supportive of OS benefit with cemiplimab (Table 14 and Table 36 in Appendix 3).
Factor | ITT population | PD-L1 ≥ 50% population | ||
|---|---|---|---|---|
Cemiplimab (N = 356) | Chemotherapy (N = 354) | Cemiplimab (N = 283) | Chemotherapy (N = 280) | |
Number of deaths, n (%) | 108 (30.3) | 141 (39.8) | 70 (24.7) | 105 (37.5) |
Number of censored patients, n (%) | 248 (69.7) | 213 (60.2) | 213 (75.3) | 175 (62.5) |
Median (95% CI), monthsa | 22.1 (17.7 to NE) | 14.3 (11.7 to 19.2) | NR (17.9 to NE) | 14.2 (11.2 to 17.5) |
Stratified log-rank test, P valueb,c | 0.0022 | 0.0002d | ||
HR (95% CI)b, e | 0.676 (0.525 to 0.870) | 0.566 (0.418 to 0.767) | ||
Estimated survival probability, % (95% CI)a | ||||
6 months | 81.2 (76.4 to 85.1) | 76.2 (71.0 to 80.6) | 83.7 (78.5 to 87.7) | 74.4 (68.3 to 79.5) |
12 months | 70.3 (64.4 to 75.4) | 55.7 (49.2 to 61.7) | 72.4 (65.6 to 78.1) | 53.9 (46.2 to 61.1) |
18 months | 56.1 (48.1 to 63.3) | 43.3 (35.8 to 50.4) | 60.5 (49.6 to 69.8) | 39.6 (29.4 to 49.5) |
24 months | 48.6 (39.2 to 57.3) | 29.7 (18.8 to 41.4) | 50.4 (36.4 to 62.9) | 27.1 (13.7 to 42.5) |
CI = confidence interval; HR = hazard ratio; ITT = intention-to-treat; NE = not estimable; NR = not reached; PD-L1 = programmed cell death-ligand 1.
aBased on the Kaplan-Meier method.
bStratified by histology (squamous, non-squamous).
cTwo-sided P value.
dP value was not controlled for multiplicity.
eBased on stratified proportional hazards model (cemiplimab versus chemotherapy).
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Figure 4: Kaplan-Meier Curve of Overall Survival (Intention-to-Treat Population)
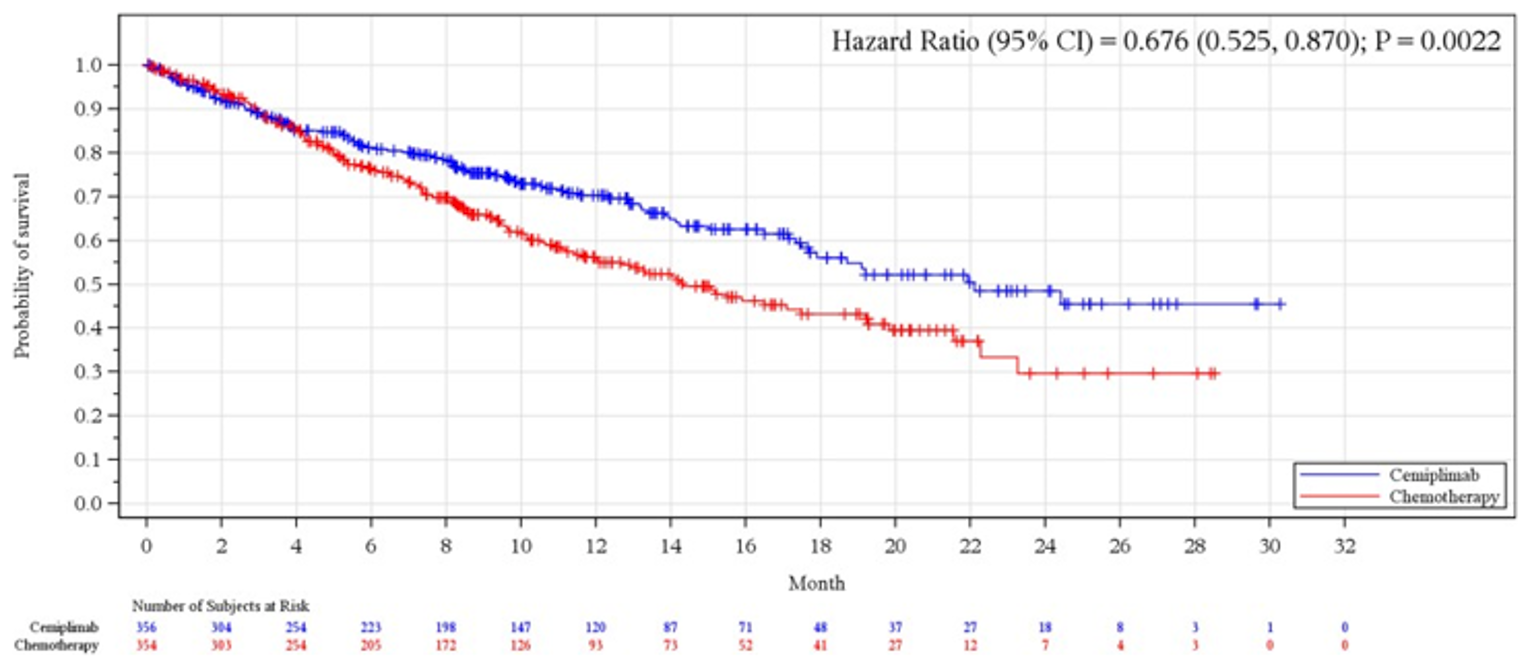
CI = confidence interval.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Table 14: Overall Survival by Subgroup (Intention-to-Treat Population)
Factors | Cemiplimab (N = 356) | Chemotherapy (N = 354) | HR (95% CI)a | ||
|---|---|---|---|---|---|
Event (%) | Median time (95% CI)b | Event (%) | Median time (95% CI)b | ||
ECOG PS | |||||
0 | 25/96 (26.0) | ||||||||||||||| | 32/96 (33.3) | ||||||||||||||| | 0.777 (0.459 to 1.315) |
1 | 83/260 (31.9) | ||||||||||||||| | 109/258 (42.2) | ||||||||||||||||| | 0.658 (0.493 to 0.877) |
Histologyc | |||||
Squamous | 45/159 (28.3) | ||||||||||||||| | 65/152 (42.8) | ||||||||||||||||| | 0.526 (0.358 to 0.773)d |
Non-squamous | 63/197 (32.0) | ||||||||||||||| | 76/202 (37.6) | ||||||||||||||| | 0.827 (0.592 to 1.156)d |
Brain metastasis at baseline | |||||
Yes | 9/44 (20.5) | ||||||||||||| | 15/39 (38.5) | |||||||||||||| | 0.444 (0.185 to 1.065) |
No | 99/312 (31.7) | ||||||||||||||| | 126/315 (40.0) | ||||||||||||||||| | 0.707 (0.542 to 0.922) |
Cancer stage at screening | |||||
Locally advanced | 17/63 (27.0) | ||||||||||||| | 16/52 (30.8) | ||||||||||||| | 0.847 (0.426 to 1.682) |
Metastatic | 91/293 (31.1) | ||||||||||||||| | 125/302 (41.4) | ||||||||||||||||| | 0.677 (0.517 to 0.888) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; NE = not estimable; NR = not reached.
aBased on stratified proportional hazards model (cemiplimab versus chemotherapy).
bBased on the Kaplan-Meier method.
cAccording to the clinical database at baseline.
dBased on unstratified proportional hazards model (cemiplimab versus chemotherapy).
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Sensitivity Analyses of Overall Survival
In the sensitivity analyses using the RMST method conducted for OS in the ITT population, mean survival times were 18.9 (95% CI, 17.5 to 20.3) months in the cemiplimab arm and 15.8 (95% CI, 14.4 to 17.3) months in the chemotherapy arm.
In the sensitivity analyses using the RPSFT method that were conducted for OS in the ITT population to account for crossover effects, the HR was 0.580 (95% CI, 0.438 to 0.770); the median OS was 22.1 (95% CI, 17.7 to not estimable) months for cemiplimab versus 12.0 (95% CI, 9.7 to 14.2) months for chemotherapy.
Improvement in Cancer-Related Symptoms and Health-Related Quality of Life
|||||||| |||||||||| ||||| ||| ||| ||||| ||||||| ||| |||||||| |||| ||||| ||||| ||| || ||| ||||||| ||||||||||||| |||||||||| ||||| ||||||||| ||| ||||| |||||||||| |||| |||| |||| ||| ||||||| ||||| || ||| ||||| |||||||| ||| ||||||||| ||| |||||||| |||||||||| ||| |||||||| || |||||||| ||| ||| ||||||||||| |||||||| ||| |||||| |||| ||| |||||||| |||||||| ||||||||||||| |||| ||||| || ||||||| ||||||||| ||||| |||| |||| | |||||||| |||| ||||||||||| || ||| |||||||||| ||| ||| | || ||| |||||||||||| ||||
Mean baseline scores for the EORTC QLQ-C30 GHS and functioning scales were similar between patients in the cemiplimab and chemotherapy treatment arms. The functioning scale scores at baseline were in line with EORTC QLQ-C30 reference values corresponding to NSCLC and indicating moderate to high levels of functioning (mean baseline functioning scale scores ranged between 74.04 and 89.32 of a possible score of 100). The mean GHS score at baseline was 59.0 (SD = 21.53) and 59.7 (SD = 20.80) in the cemiplimab and chemotherapy arms, respectively, indicating impaired quality of life (Figure 5).
A mean change in score from baseline of greater than 5 points for the GHS was observed in the cemiplimab arm by cycle 2 (mean change = 5.16 [SD = 20.49]), above 9 points by cycle 6 (9.38 [SD = 23.359]), and above 10 points by cycle 18 (10.53 [SD = 25.71]). It stayed above 10 points, with wide variation, through to cycle 30. The mean change for GHS score in the chemotherapy arm was below 3 until cycle 12 ||| |||||| |||| ||||| ||||||| || ||||| || || |||| ||||||| || ||||| ||| ||| |||| ||| |||||| ||||||||||| |||||||| |||||||||| ||||||||||||| || || ||||| | ||| ||||| |||| || |||||||||| || ||||||| ||||||||||| ||||||| ||| |||| || ||||| |||| |||||||
Mean baseline scores for the 9 symptom scales or items were similar between patients in the cemiplimab and chemotherapy arms, indicating moderate to low symptom burden at baseline, and in line with EORTC QLQ-C30 reference values corresponding to NSCLC. By cycle 9, there was a mean decrease from baseline in symptom scale score greater than 10 points in the cemiplimab arm for the fatigue, pain, dyspnea, insomnia, and appetite loss scales, |||| |||| |||| |||||| |||| |||||||| || | ||||| |||||||| |||||| |||||||| |||||| |||||||| |||||| |||||||| ||| |||||| ||||||||| ||||||||||||| ||||||||||||||| |||||| |||||||| |||||| |||| |||| || |||| ||||||||| |||||
For the EORTC QLQ-LC13’s lung cancer symptoms, a mean decrease in score from baseline of greater than 10 points was observed for pain (chest), pain (arm/shoulders), and cough in both treatment arms. For treatment-related side effects such as sore mouth, dysphagia, peripheral neuropathy, and alopecia, mean scores remained consistent as compared to baseline among patients in the cemiplimab arm. Similar results were observed in the chemotherapy arm, except for notably higher mean scores in the chemotherapy arm for alopecia from cycle 2 to cycle 6 and peripheral neuropathy at cycle 5 and cycle 6.
Figure 5: Longitudinal Plots of EORTC QLQ-C30 Global Health Status

This figure has been redacted at the sponsor’s request.
Objective Response Rate
An IRC-assessed objective response (complete response or partial response) was observed in 36.5% (96% CI, 31.5% to 41.8%) of patients in the cemiplimab arm, and 20.6% (95% CI, 16.5% to 25.2%) of patients in the chemotherapy arm. The odds ratio for comparison of cemiplimab to chemotherapy was 2.21 (95% CI, 1.58 to 3.10; P < 0.0001). The results for ORR in the PD-L1 of 50% or more population were similar to the ITT results (Table 15).
Table 15: Best Overall Objective Tumour Response Rate
Response | ITT population | PD-L1 ≥ 50% population | ||
|---|---|---|---|---|
Cemiplimab (N = 356) | Chemotherapy (N = 354) | Cemiplimab (N = 283) | Chemotherapy (N = 280) | |
Best overall tumour response, n (%) | ||||
CR | 11 (3.1) | 3 (0.8) | 6 (2.1) | 3 (1.1) |
PR | 119 (33.4) | 70 (19.8) | 105 (37.1) | 54 (19.3) |
Stable disease | 101 (28.4) | 168 (47.5) | 76 (26.9) | 135 (48.2) |
Non-CR/non-PD | 2 (0.6) | 4 (1.1) | 2 (0.7) | 1 (0.4) |
PD | 68 (19.1) | 52 (14.7) | 52 (18.4) | 41 (14.6) |
Not evaluable | 55 (15.4) | 57 (16.1) | 42 (14.8) | 46 (16.4) |
Response | ||||
ORR: CR + PR | 130 (36.5) | 73 (20.6) | 111 (39.2) | 57 (20.4) |
95% CI for ORRa | 31.5 to 41.8 | 16.5 to 25.2 | 33.5 to 45.2 | 15.8 to 25.6 |
Stratified CMH test, P valueb | < 0.0001 | < 0.0001c | ||
Odds ratio (95% CI)b | 2.214 (1.582 to 3.098) | 2.530 (1.736 to 3.687) | ||
Kaplan-Meier estimated duration of response | ||||
n | 130 | 73 | 111 | 57 |
Median (95% CI), months | 21.0 (14.9 to NE) | 6.0 (4.3 to 6.4) | 16.7 (12.5 to 22.8) | 6.0 (4.3 to 6.5) |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CR = complete response; ITT = intention-to-treat; NE = not estimable; ORR = objective response rate; PD = progressive disease; PD-L1 = programmed cell death-ligand 1; PR = partial response.
aClopper-Pearson exact CI.
bTwo-sided P value and odds ratio using stratified Cochran-Mantel-Haenszel test.
cP value was not adjusted for multiplicity.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Progression-Free Survival
The median PFS in the cemiplimab arm versus chemotherapy arm was 6.2 (95% CI, 4.5 to 8.3) months versus 5.6 (95% CI, 4.5 to 6.1) months, respectively (P < 0.0001). The HR between treatment arms was 0.593 (95% CI, 0.491 to 0.718). The estimated probability of PFS from baseline through 12 months and 24 months was 37.8% (95% CI, 31.9% to 43.6%) and 21.7% (95% CI, 14.7% to 29.7%), respectively, for patients in the cemiplimab arm compared with 7.2% (95% CI, 4.3% to 11.2%) and not estimable, for patients in the chemotherapy arm (Table 16 and Figure 6).
In the PD-L1 of 50% or more population, PFS results were similar to the ITT population (Table 16 and Figure 9 in Appendix 3).
In both the ITT population and the PD-L1 of 50% or more population, the overall PFS benefit with cemiplimab was observed in all subgroups of interest identified for this review (Table 17 and Table 37 in Appendix 3).
Table 16: Progression-Free Survival
Outcome | ITT population | PD-L1 ≥ 50% population | ||
|---|---|---|---|---|
Cemiplimab (N = 356) | Chemotherapy (N = 354) | Cemiplimab (N = 283) | Chemotherapy (N = 280) | |
Number of events, n (%) | 201 (56.5) | 262 (74.0) | 147 (51.9) | 197 (70.4) |
Progressive disease, n (%) | 158 (44.4) | 203 (57.3) | 119 (42.0) | 150 (53.6) |
Death, n (%) | 43 (12.1) | 59 (16.7) | 28 (9.9) | 47 (16.8) |
Number of censored patients, n (%) | 155 (43.5) | 92 (26.0) | 136 (48.1) | 83 (29.6) |
Median (95% CI), monthsa | 6.2 (4.5 to 8.3) | 5.6 (4.5 to 6.1) | 8.2 (6.1 to 8.8) | 5.7 (4.5 to 6.2) |
Stratified log-rank test, P valueb, c | < 0.0001 | < 0.0001d | ||
HR (95% CI)b, e | 0.593 (0.491 to 0.718) | 0.541 (0.433 to 0.675) | ||
Estimated event-free probability, % (95% CI)a | ||||
6 months | 53.1 (47.4 to 58.5) | 48.0 (42.2 to 53.6) | 57.2 (50.7 to 63.2) | 48.4 (41.8 to 54.7) |
12 months | 37.8 (31.9 to 43.6) | 7.2 (4.3 to 11.2) | 40.7 (33.7 to 47.5) | 7.1 (3.6 to 12.1) |
18 months | 28.0 (21.7 to 34.7) | 3.9 (1.8 to 7.5) | 27.8 (19.4 to 36.7) | NE |
24 months | 21.7 (14.7 to 29.7) | NE | 20.4 (11.7 to 30.8) | NE |
CI = confidence interval; HR = hazard ratio; ITT = intention-to-treat; NE = not estimable; PD-L1 = programmed cell death-ligand 1.
aBased on the Kaplan-Meier method.
bStratified by histology (squamous, non-squamous).
cTwo-sided P value.
dP value was not adjusted for multiplicity.
eBased on stratified proportional hazards model (cemiplimab versus chemotherapy).
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Figure 6: Kaplan-Meier Curve of Progression-Free Survival (Intention-to-Treat Population)
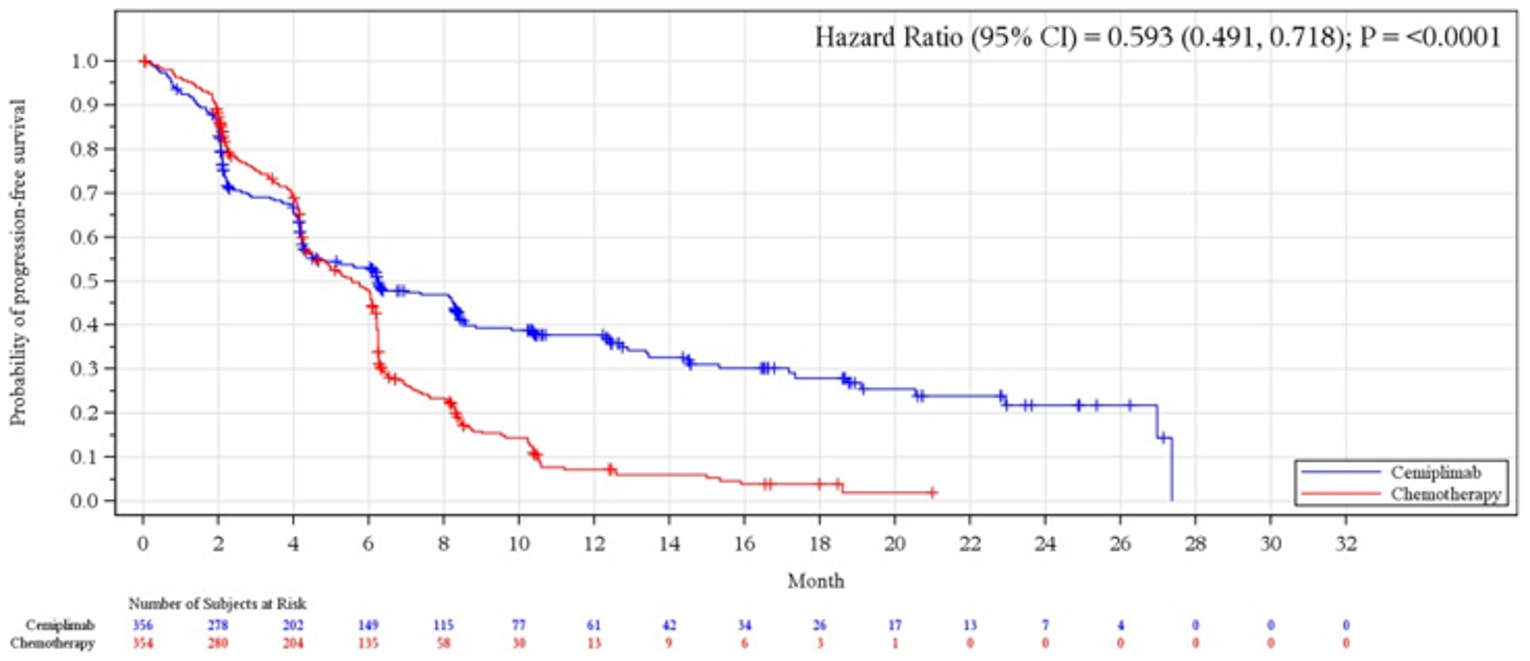
CI = confidence interval.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Table 17: Progression-Free Survival by Subgroup (Intention-to-Treat Population)
Factors | Cemiplimab (N = 356) | Chemotherapy (N = 354) | HR (95% CI)a | ||
|---|---|---|---|---|---|
Event (%) | Median time (95% CI)b | Event (%) | Median time (95% CI)b | ||
ECOG PS | |||||
0 | 52/96 (54.2) | ||| ||||| ||||| | 65/96 (67.7) | ||| ||||| |||| | 0.681 (0.470 to 0.987) |
1 | 149/260 (57.3) | ||| ||||| |||| | 197/258 (76.4) | ||| ||||| |||| | 0.573 (0.459 to 0.715) |
Histologyc | |||||
Squamous | 93/159 (58.5) | ||| ||||| |||| | 117/152 (77.0) | ||| ||||| |||| | 0.530 (0.398 to 0.704)d |
Non-squamous | 108/197 (54.8) | ||| ||||| |||| | 145/202 (71.8) | ||| ||||| |||| | 0.652 (0.505 to 0.841)d |
Brain metastasis at baseline | |||||
Yes | 19/44 (43.2) | |||| ||||| ||| | 31/39 (79.5) | ||| ||||| |||| | 0.491 (0.268 to 0.899) |
No | 182/312 (58.3) | ||| ||||| |||| | 231/315 (73.3) | ||| ||||| |||| | 0.620 (0.507 to 0.758) |
Cancer stage at screening | |||||
Locally advanced | 40/63 (63.5) | ||| ||||| |||| | 37/52 (71.2) | ||| ||||| |||| | 0.588 (0.362 to 0.956) |
Metastatic | 161/293 (54.9) | ||| ||||| |||| | 225/302 (74.5) | ||| ||||| |||| | 0.603 (0.490 to 0.742) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio.
aBased on stratified proportional hazards model (cemiplimab versus chemotherapy).
bBased on the Kaplan-Meier method.
cAccording to the clinical database at baseline.
dBased on unstratified proportional hazards model (cemiplimab versus chemotherapy).
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Sensitivity Analyses of PFS
In the sensitivity analyses using the RMST method that were conducted for PFS in the ITT population, the mean PFS times were 9.6 (95% CI, 8.7 to 10.6) months in the cemiplimab arm and 6.0 (95% CI, 5.5 to 6.5) months in the chemotherapy arm. These findings were compatible with the main analysis.
Duration of Response
In the ITT population, the range of the observed DOR was 1.9 months to 23.3 months in the cemiplimab arm and 1.3 months to 16.5 months in the chemotherapy arm, with 69.2% of responding patients in the cemiplimab arm and 41.1% of responding patients in the chemotherapy arm having an observed DOR of 6 months or more (Table 18).
||||| || |||||||| ||||||||| ||| |||||||| ||| || ||| ||||| | ||| |||||||||| |||||| |||| ||| || |||| |||||| || ||| |||||||||| ||| ||| ||| || |||| |||||| || ||| |||||||||||| |||| || ||| |||||||||| ||| |||||||||||| ||||| ||||| ||| ||||| || |||||||||| ||||||||| ||||||||||||| |||| |||||||| ||| | | |||||| |||||||||||
Table 18: Observed Duration of Response
DOR | ITT population | PD-L1 ≥ 50% population | ||
|---|---|---|---|---|
Cemiplimab (N = 130) | Chemotherapy (N = 73) | Cemiplimab (N = 111) | Chemotherapy (N = 57) | |
Observed DOR (CR or PR), monthsa | ||||
Range | 1.9 to 23.3 | 1.3 to 16.5 | |||| |||| | |||| |||| |
Observed DOR (CR and PR), n (%)a | ||||
< 6 months | 40 (30.8) | 43 (58.9) | || |||||| | || |||||| |
≥ 6 months | 90 (69.2) | 30 (41.1) | || |||||| | || |||||| |
≥ 12 months | 36 (27.7) | 5 (6.8) | || |||||| | | ||||| |
≥ 18 months | 15 (11.5) | 0 | || ||||| | |||||||| |
≥ 24 months | 0 | 0 | |||||||| | |||||||| |
Kaplan-Meier estimated DOR (CR or PR), months | ||||
Number of events, n (%) | 37 (28.5) | 49 (67.1) | || |||||| | || |||||| |
Median (95% CI) | 21.0 (14.9 to NE) | 6.0 (4.3 to 6.4) | |||| |||||| ||||| | ||| ||||| |||| |
CI = confidence interval; CR = complete response; DOR = duration of response; IRC = independent review committee; ITT = intention-to-treat; NE = not estimable; PD-L1 = programmed cell death-ligand 1; PR = partial response.
aBased on patients with confirmed CR or PR confirmed by IRC.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Harms
Adverse Events
As of the data cut-off date, 88.2% of patients in the cemiplimab arm and 94.2% of patients in the chemotherapy arm experienced at least 1 TEAE (Table 19). In the cemiplimab arm, the most common TEAEs of any grade by preferred term experienced by 10% of patients or more were anemia (14.6%), decreased appetite (11.8%), and fatigue (10.1%). In the chemotherapy arm, the most common TEAEs of any grade by preferred term experienced by 10% of patients or more were anemia (50.0%), nausea (28.4%), alopecia (24.0%), decreased appetite (18.4%), neutropenia (18.4%), fatigue (17.0%), constipation (15.2%), thrombocytopenia (15.2%), vomiting (14.3%), decreased neutrophil count (12.3%), peripheral neuropathy (10.8%), pneumonia (10.8%), and decreased platelet count (10.5%).
Grade 3 to grade 4 TEAEs, regardless of attribution, occurred in 28% of patients in the cemiplimab arm and 39% of patients in the chemotherapy arm.
Table 19: Summary of Treatment-Emergent Adverse Events (Safety Population)
Characteristic | Cemiplimab (N = 355) | Chemotherapy (N = 342) |
|---|---|---|
Total number of TEAEs, n | 1,976 | 2,610 |
NCI grade 3/4/5 TEAEs, n | 255 | 458 |
Total number of serious TEAEs, n | 165 | 177 |
Patients with any TEAE, n (%) | 313 (88.2) | 322 (94.2) |
Patients with any NCI grade 3/4/5 TEAE, n (%) | 132 (37.2) | 166 (48.5) |
Patients with any TEAE resulting in death, n (%) | 34 (9.6) | 31 (9.1) |
Total number of serious TEAEs, n | 165 | 177 |
Patients with any serious TEAE, n (%) | 100 (28.2) | 94 (27.5) |
Patients who discontinued study treatment due to serious TEAEs, n (%) | 14 (3.9) | 6 (1.8) |
Patients with any serious TEAE leading to a dose delay/infusion interruption, n (%) | 34 (9.6) | 28 (8.2) |
Patients with any serious TEAE leading to a dose reduction, n (%) | 0 | 7 (2.0) |
Patients with any serious TEAE resulting in death, n (%) | 34 (9.6) | 31 (9.1) |
CTCAE = Common Terminology Criteria for Adverse Events; NCI = National Cancer Institute; TEAE = treatment-emergent adverse event.
Note: TEAEs that resulted in death comprise all TEAEs with a fatal outcome, including those related to disease progression. NCI grades were coded using CTCAE, version 4.03. A patient is counted only once for multiple occurrences within a category.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Serious Adverse Events
As of the data cut-off date, 28.2% of patients in the cemiplimab arm and 27.5% of patients in the chemotherapy arm had experienced at least 1 serious TEAE (Table 19). In the cemiplimab arm, the only serious TEAE by preferred term experienced by 2% of patients or more was pneumonia (4.8%) (Table 20). In the chemotherapy arm, the most common serious TEAEs by preferred term experienced by 2% of patients or more were pneumonia (5.0%), anemia (3.8%), and febrile neutropenia (2.3%) (Table 20).
Table 20: Serious Treatment-Emergent Adverse Events Experienced by Patients (in Greater Than 1% in Any Group) by System Organ Class and Preferred Term (Safety Population)
System Organ Class, n (%) Preferred term, n (%) | Cemiplimab (N = 355) | Chemotherapy (N = 342) |
|---|---|---|
Number of patients with any serious TEAE, n (%) | 100 (28.2) | 94 (27.5) |
Infections and infestations | 30 (8.5) | 32 (9.4) |
Pneumonia | 17 (4.8) | 17 (5.0) |
Septic shock | 4 (1.1) | 2 (0.6) |
Respiratory, thoracic, and mediastinal disorders | 28 (7.9) | 18 (5.3) |
Pneumonitis | 6 (1.7) | 0 |
Pulmonary embolism | 6 (1.7) | 2 (0.6) |
Dyspnea | 4 (1.1) | 4 (1.2) |
Pleural effusion | 4 (1.1) | 3 (0.9) |
Respiratory failure | 4 (1.1) | 2 (0.6) |
General disorders and administration site conditions | 12 (3.4) | 8 (2.3) |
Deatha | 5 (1.4) | 1 (0.3) |
Gastrointestinal disorders | 8 (2.3) | 10 (2.9) |
Vomiting | 0 | 4 (1.2) |
Blood and lymphatic system disorders | 6 (1.7) | 24 (7.0) |
Anemia | 3 (0.8) | 13 (3.8) |
Febrile neutropenia | 1 (0.3) | 8 (2.3) |
Neutropenia | 1 (0.3) | 4 (1.2) |
Thrombocytopenia | 0 | 6 (1.8) |
TEAE = treatment-emergent adverse event.
Note: A patient is counted only once for multiple occurrences within a category.
aCause unknown.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Treatment Discontinuation Due to Adverse Events
As of the data cut-off date, 6.5% of patients in the cemiplimab arm and 4.1% of patients in the chemotherapy arm had experienced at least 1 TEAE resulting in discontinuation of study treatment. In the cemiplimab arm, the most common TEAEs leading to treatment discontinuation (experienced by > 1 patient) were pneumonitis (1.1%), ischemic stroke (0.6%), and increased aspartate aminotransferase (0.6%). In the chemotherapy arm, the most common TEAEs leading to treatment discontinuation (experienced by > 1 patient) were thrombocytopenia (0.9%) and anemia (0.6%) (Table 21).
In the cemiplimab arm, 28.2% of patients had TEAEs resulting in delay or interruption of treatment. In the chemotherapy arm, 31.0% of patients experienced a delay or interruption to their treatment due to TEAEs. The most frequently reported AEs resulting in dose delay or interruption of study treatment were pneumonia in the cemiplimab arm and myelotoxicity in the chemotherapy arm.
Table 21: Treatment Discontinuation Due to Adverse Events (Safety Population)
System Organ Class, n (%) Preferred term, n (%) | Cemiplimab (N = 355) | Chemotherapy (N = 342) |
|---|---|---|
Number of patients with any TEAE resulting in treatment discontinuation, n (%) | 23 (6.5) | 14 (4.1) |
Respiratory, thoracic, and mediastinal disorders | 5 (1.4) | 0 |
Pneumonitis | 4 (1.1) | 0 |
Nervous system disorders | 4 (1.1) | 2 (0.6) |
Ischemic stroke | 2 (0.6) | 1 (0.3) |
Investigations | 3 (0.8) | 2 (0.6) |
Aspartate aminotransferase, increased | 2 (0.6) | 0 |
Blood and lymphatic system disorders | 0 | 6 (1.8) |
Anemia | 0 | 2 (0.6) |
Thrombocytopenia | 0 | 3 (0.9) |
Patients with any TEAE leading to a dose delay/infusion interruption, n (%) | 100 (28.2) | 106 (31.0) |
Patients with any TEAE leading to a dose reduction, n (%) | 0 | 51 (14.9) |
AE = adverse event; TEAE = treatment-emergent adverse event.
Note: All AEs were coded using the Medical Dictionary for Regulatory Activities Version 20.0. A patient is counted only once for multiple occurrences within a System Organ Class and/or preferred term.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Death
TEAEs that led to death occurred in 9.6% of patients (34 of 355 patients) treated with cemiplimab and 9.1% of patients (31 of 342 patients) treated with chemotherapy. In 9 (3%) patients treated with cemiplimab, the events leading to death were considered related to treatment, and included autoimmune myocarditis, cardiac failure, cardiopulmonary failure, respiratory failure, septic shock, cardiorespiratory arrest, nephritis, and tumour hyperprogression (n = 1 each). The events leading to death were considered related to treatment in 7 (2%) patients treated with chemotherapy and were pneumonia (n = 2), pulmonary embolism (n = 2), cardiac arrest (n = 1), lung abscess (n = 1), and myocardial infarction (n = 1).
Notable Harms
Immune-Related Adverse Events
In the cemiplimab arm, 17.5% of patients experienced at least 1 treatment-emergent immune-related AE, and in the chemotherapy arm, 2.3% of patients experienced at least 1 treatment-emergent immune-related AE (Table 22). Most of these events were categorized as less than grade 3, with 3.7% of patients in the cemiplimab arm and 0.3% of patients in the chemotherapy arm experiencing an immune-related AE that was grade 3 or higher. Grade 4 and grade 5 immune-related AEs were only reported in the cemiplimab arm, occurring in 0.8% and 0.3% of patients, respectively. In the cemiplimab arm, 1 patient each reported a grade 4 immune-related AE of pneumonitis, immune-mediated pneumonitis, and immune-mediated hepatitis, and a grade 5 immune-related AE of nephritis. One patient in the cemiplimab arm died from immune-related nephritis.
Treatment-emergent immune-related AEs led to treatment discontinuation in 2.5% of patients in the cemiplimab arm and none of the patients in the chemotherapy arm. Serious immune-related AEs were reported for 3.7% of patients in the cemiplimab arm and none of the patients in the chemotherapy arm.
Table 22: Summary of Treatment-Emergent Immune-Related Adverse Events (Safety Population)
Characteristic | Cemiplimab (N = 355) | Chemotherapy (N = 342) |
|---|---|---|
Total number of immune-related AEs, n | 87 | 9 |
Total number of serious immune-related AEs, n | 15 | 0 |
Patients with any immune-related AEs, n (%) | 62 (17.5) | 8 (2.3) |
Patients with NCI grade 3/4/5 immune-related AEs, n (%) | 13 (3.7) | 1 (0.3) |
Patients with any serious immune-related AEs, n (%) | 13 (3.7) | 0 |
Patients who discontinued treatment due to an immune-related AE, n (%) | 9 (2.5) | 0 |
Patients with any immune-related AE leading to a dose delay/infusion interruption, n (%) | 24 (6.8) | 0 |
Patients with any immune-related AE leading to a dose reduction, n (%) | 0 | 1 (0.3) |
Patients with any immune-related AE resulting in death, n (%) | 1 (0.3) | 0 |
AE = adverse event; CTCAE = Common Terminology Criteria for Adverse Events; NCI = National Cancer Institute.
Note: NCI grades were coded using CTCAE, version 4.03. A patient was counted only once for multiple occurrences within a category.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Infusion-Related Reactions
As of the data cut-off date, 5.6% of patients in the cemiplimab arm and 1.8% of patients in the chemotherapy arm had experienced at least 1 infusion-related reaction. No patients in the cemiplimab arm and 0.6% of patients in the chemotherapy arm experienced grade 3 or higher infusion-related reactions. One patient each in both the cemiplimab and chemotherapy arms experienced serious infusion reactions. One patient each in both the cemiplimab arm and the chemotherapy arm required permanent discontinuation from study treatments due to an infusion-related reaction.
Critical Appraisal
Internal Validity
Treatment assignment in the trial was based on a central randomization scheme, which would ensure concealment of the randomized arms until allocation. Balanced baseline characteristics across treatment arms suggest that randomization was successful, and that the risk of selection bias was low. Following allocation, the trial was open label, although patient blinding would have been impractical and challenging given the differences in the 2 study treatment regimens. Performance and detection bias that may result from the lack of blinding of patients and investigators to assigned study treatments cannot be ruled out. For example, a patient’s knowledge of their assigned treatment could result in over-estimation or underestimation of safety end points and patient-reported outcomes like symptoms and HRQoL. Investigator knowledge of the treatment arm could have resulted in different concomitant supportive care being offered to patients in the 2 treatment arms. However, since OS is an objective outcome and an IRC that was blinded to treatment assignment was used to adjudicate tumour responses, the primary efficacy end points are unaffected by the lack of blinding of patients and investigators.
Statistical power calculations were reported, and target sample size (n = 710) was achieved. However, the sample size was based on the results observed in clinical studies with other anti–PD-1 treatments, assuming non-proportional hazards, but not the crossover effect. Lack of adjustment for crossover and extended treatment would have likely biased the findings in favour of chemotherapy (i.e., underestimated the effect of cemiplimab). However, the results for the sensitivity analyses for OS accounting for the effect of crossover (RPSFT) and non-proportional hazards (RMST) were compatible with the main analysis, suggesting that this was not a concern from a sample size perspective.
Issues with PD-L1 testing were discovered when more than 50% of the planned population had been recruited, necessitating retesting of the 235 randomized patients at that point. However, not all of them had remaining tissue samples (38%) and not all the retested samples proved PD-L1 of 50% or more (24%). The ITT population represents a truly randomized sample but includes some patients who did not in fact meet the inclusion criteria of the trial; the mITT-1 population (i.e., PD-L1 ≥ 50% population) is not strictly a randomized sample but has been included in this report as supportive data, as it may be more clinically relevant. Though selection bias in the mITT-1 population is possible, concern for this is low as those in the mITT-1 population appeared largely similar to the ITT population in their baseline characteristics. The results based on this subpopulation were consistent with the primary ITT analyses.
The primary and secondary efficacy end points of OS and IRC-assessed PFS, ORR, and DOR are considered appropriate for the disease setting. Some uncertainties are noted in the analyses of the primary efficacy end points. Since OS and PFS were tested as co-primary end points (OS was promoted in protocol amendment 7), an alpha-reallocation strategy was implemented. This may not be the ideal approach as the results could be regarded as positive if either 1 of the 2 end points reaches statistical significance. However, analyses of both OS and PFS reached statistical significance, so this is not a major concern. Furthermore, while 5 interim analyses were planned for OS, none was planned for PFS, with 3 of the OS interim analyses conducted before the planned primary PFS analysis — even though PFS is expected to mature faster than OS. The study was stopped at interim analysis for OS before the primary PFS analysis, which is unusual. In addition, of 461 censored patients, 11% of patients in the cemiplimab arm and 19% of patients in the chemotherapy arm withdrew consent to be followed for survival. As the findings are the result of an interim analysis, there is some increased risk that the benefits of cemiplimab were overestimated.
Although results from the secondary end points of ORR and DOR support the benefit of cemiplimab over chemotherapy, a relatively large proportion of patients in both treatment arms are indicated as being “not evaluable” for overall tumour response. The major reasons for non-evaluability were “first time point not yet reached” and “death.” However, overall frequencies are comparable between the 2 treatment arms, which reduces the potential for attrition bias. Analyses of symptoms and HRQoL were limited by large losses to follow-up over time and a lack of between-group statistical testing.
As of protocol amendment 6 (August 22, 2018), patients randomized to receive cemiplimab who experienced RECIST 1.1–defined progression during cemiplimab monotherapy were allowed to continue cemiplimab treatment with the addition of 4 cycles of histology-specific chemotherapy until further progression was observed, provided the patient had not completed the 108-week treatment period and protocol-specified criteria were met. Of note, before amendment 6, a considerable number of progressors from the cemiplimab arm continued with cemiplimab — as allowed by the protocol — without adding chemotherapy (17 out of 24 progressors). The same protocol amendment also allowed patients to cross over to cemiplimab after initial disease progression on chemotherapy. However, given that the primary analyses based on the ITT population were statistically significant and that sensitivity analyses using the RPSFT method that accounts for the crossover effect were consistent with the primary analyses, the effect of crossover on OS is likely minimal and unlikely to change the conclusions based on the primary analyses of OS. Of note, after the data cut-off date of March 1, 2020, patients in the chemotherapy arm were given the option to cross over to the cemiplimab arm even if they had not progressed on chemotherapy. While the amendment does not affect the OS results presented to date, it impacts any future analyses of OS. Likewise, future updates of PFS analyses are unlikely to provide an unbiased estimate due to the crossover of patients in the chemotherapy arm to the cemiplimab arm even before progression.
External Validity
The EMPOWER-Lung 1 trial included a heterogenous population of patients with NSCLC and a wide range of clinical presentations were well represented. A few patient groups were not included — notably, non-smokers and patients who were immunocompromised or had a history of autoimmune diseases, and those with ECOG PS of 2 of higher. The rationale for excluding never-smokers was based on findings from some studies suggesting that this group of patients does not benefit from PD-L1 monotherapy.20,24,26 Previous trials in the same setting, notably the KEYNOTE-042 study with pembrolizumab, included non-smokers.21 The clinical experts consulted by CADTH for this review also indicated that non-smokers should not be excluded from treatment with immune checkpoint inhibitors including cemiplimab. Of note, the Health Canada indication for cemiplimab does not specify smoking status. Nonetheless, the exclusion of this group of patients from the EMPOWER-Lung 1 study limits the generalizability of evidence with respect to the efficacy of cemiplimab in non-smokers, among whom the clinical experts consulted by CADTH mentioned that the response to cemiplimab might be expected to be less robust. With regard to patients with autoimmune diseases, most previous trials of PD-1 checkpoint inhibitors excluded these patients, and the clinical experts noted that in clinical practice, the suitability of immunotherapy for the treatment of patients with autoimmune conditions requires careful considerations of benefits and risks to the patient. In addition, about 44% of patients presented with squamous histology, which is higher than what is expected in clinical practice (about 30%). However, the inclusion of a notable proportion of patients with locally advanced NSCLC who were not candidates for definitive chemoradiation (16.2%), as well as patients with brain metastases (11.7%), compared to other studies of immune checkpoint inhibitors in the same setting, makes this study’s patient population somewhat more reflective of real-world clinical practice.
The study recruited patients mainly from Asia and Europe; no patients from the US or Canada were recruited. This was due to the approval of pembrolizumab monotherapy for first-line treatment of metastatic NSCLC with high tumour PD-L1 expression. In countries where pembrolizumab was considered as standard of care, a trial design comparing cemiplimab with chemotherapy would have been considered unethical (though at the start of enrolment in June 2017, pembrolizumab was not funded in some provinces in Canada). Therefore, the study was conducted in countries where anti–PD-1 agents were not approved, not reimbursed, or not readily available. The clinical experts consulted by CADTH noted that the lack of representation of Canadian patients does not reduce the generalizability of results with respect to the efficacy of cemiplimab compared to chemotherapy in advanced or metastatic NSCLC in the Canadian clinical practice, although chemotherapy is rarely used as first-line treatment in this setting except for patients with a contraindication to immunotherapy.
The most important limitation of the evidence in terms of generalizability is the relevance of chemotherapy as a comparator in Canadian clinical practice. While chemotherapy regimens are still the standard of care in countries where therapies based on immune checkpoint inhibitors are not approved or available, the current standard of care in Canada for the treatment of patients with advanced or metastatic high PD-L1 expressing NSCLC and without oncogenic alterations includes immune checkpoint inhibitors. Several regimens are approved, among which pembrolizumab with or without chemotherapy is funded and is widely used for this indication. Therefore, the benefit of cemiplimab compared to chemotherapy in terms of improved survival in this patient population is limited in informing treatment choice in Canadian clinical practice.
Indirect Evidence
Sections have been redacted at the sponsor’s request.
Objectives and Methods for the Summary of Indirect Evidence
|||||||||| ||| |||| |||||||| || |||||||||||| || |||||||| |||| |||||||| || |||||||||| ||||| |||||||||| ||||| | |||| |||||||| || |||||||||||| |||||||| || |||||||||| |||||||| ||||||| ||||| |||||||| ||||||||||| |||||| ||||| |||||| |||||||||| ||||||||||| ||| ||||||||| ||| |||| ||||||| || ||||| ||| || |||||| |||||||| ||||||||| |||||||||| || ||||| ||||||||| ||| |||||||| || |||||||||| ||||| |||||||||| ||||| | |||| | |||||| ||||||||||||||| |||||||| |||||||| ||| ||||||||||| ||||| ||||||||| | |||||||||| |||||| || |||||||| ||||||||||| |||||||| |||||||| ||||||||| ||||||||||| |||||| || |||||||| |||| |||||||| || |||||||||| ||||| |||||||||| ||||| | |||| || |||||||| || ||||||||| ||| ||||||||| |||||||||| || |||||| | ||||||| |||||||||| |||||| ||| |||| ||||||| |||| ||||| ||| ||| || ||||||| ||| |||||| | || |||||||| ||| ||||| || |||||| |||| ||||||| || ||| ||||||| ||||||| |||||||||| ||| |||| |||| |||||||| |||| |||||||| ||| ||||||||| || ||| |||||||| ||||| || ||| |||||||| |||||| |||| ||||| ||||||| |||||||| || |||||| | ||||| || ||| ||||||||| |||||||||| || | |||||||||| ||||||| ||| ||||||||||||||||| ||| ||| | ||||||||| ||||| | || || || ||| |||||| ||| | || ||||| || ||| |||||| |||| ||||||||| ||||||||||| ||| |||||||||| |||||||||| ||| ||||||||||||||||| ||| ||| |||| || |||||| ||| |||||||||||||||| ||||||
Description of Indirect Comparisons
||| ||||||||||||||||| ||| ||||||| ||||||||||||| ||||| || |||||||| ||| |||||||| |||||||| |||||||| ||| |||||| || |||||||||| ||||||||||| ||||||| |||||||| ||||||||||| || |||||||| |||| |||||||| || |||||||||| ||||| |||||||||| ||||| | ||| ||| |||| ||| |||||||| ||||| |||||||| |||||||| ||| ||||||| ||||||||| | |||||||||| |||||| || ||||| || |||||||| |||||||| ||||||| ||| ||||||||| || ||| |||| ||| |||||||| |||||||| |||| ||| |||| |||| ||||| | || | ||||||||| |||| ||||| | || | ||||||||||||||| |||| ||| |||||||||||||||| ||| || ||||||||| ||||
||| |||||||| || || || ||| ||||||32 ||| ||||| || |||||||||31 |||||||| |||||||| |||| || |||||||| ||| ||| || |||||||||| ||||||||| ||||||| ||| |||||||| |||| ||||| |||||||||| ||||| ||||||||| ||||||||||| ||||||||||||| |||||||||||| ||||||||| ||| |||||| || ||||||| |||||||| || ||| ||||||||| ||| ||| ||||| |||||||||| || ||| |||||||||| ||| |||||||| ||||| || Table 23|
Table 23: Study Selection Criteria and Methods for Indirect Treatment Comparisons
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Methods of Sponsor-Submitted Indirect Treatment Comparison
Objectives
|| ||| ||||||| || |||||||||||| |||||||| |||||| ||||||| ||| |||||||| ||||||||| ||| |||||||| |||| |||| |||||||| || ||||| |||||||| ||||||| ||| |||||||| || |||||||||| ||||| |||||||||| ||||| | |||| ||| ||||||||| || |||| ||| ||| || |||||||| ||||||||||| |||||||| ||| |||||| |||| ||| |||||||||| |||||||| || ||||||||| |||||||| ||||||||| ||| ||| ||||||||| || |||||||| || |||||||||| ||||| |||||||| |||||||||| ||||| |||||
Study Selection Methods
||| |||||||| ||||||| |||||||| || |||||||||| |||||| |||||||||| ||||||| |||||||||| |||||||| ||| ||||||||| |||||| |||||| |||||||||| ||| |||||||| |||||| |||||||| |||||||| |||| |||||||| || |||||||| ||| ||||| ||| || |||||||| |||||||||| || |||||| ||| |||||||| ||||| |||||||| |||||||||||||||||||| ||| |||||||| ||||| |||| |||||||||| ||||||||||| ||| ||| ||||| |||| || ||||| ||| |||||||| |||||||||| ||||||||||| || |||||||| |||| |||||||| ||||||| || |||||||| |||||||| ||||||| |||||||| ||||||| ||| ||||||| |||||||| ||||||| ||||| |||||||||| || |||| |||||| ||||||| |||||||| |||| |||||| |||||||||| ||||||| ||| ||||||| ||| ||||||||||||| || |||||| ||||||| |||||| ||||||||| || |||||||||| || ||||||||| |||||||||| ||||||| ||| ||||||||||||| ||| |||| ||||||||| || |||||||| ||||||||| |||||||||||| |||| ||| ||| |||| |||| |||||||||| |||| ||| |||||||||| ||||||||||||||||| |||| |||||||| || |||| |||| ||||| || || ||| |||| || ||||| |||||||| ||||||| ||||| |||||||| ||||||||| ||| |||||||| || |||||||||| ||||| |||||||||| ||||| | |||| ||||||||| || ||||| | ||||||||| || ||||||||| ||||||||| | || |||| || ||| |||||||| |||||||| ||| |||||| |||||||||| ||| ||||||||| || ||||||| ||||||||| ||||||| |||| |||||||| || |||| ||| ||| |||||| |||||||| || |||||||| || |||| ||||||||||||||| ||||||||||| || ||||||||| |||||| |||||||||||| ||| ||||||||| |||| |||||||| ||| ||||||||| ||||| |||||||||| ||| ||||||||| ||||||||||||| |||||||| ||||||||| ||| |||| |||| |||||| ||| ||||| || ||||| | |||||| ||| |||||||| |||||||||| ||| ||||||||||||| ||||||| ||||||||| |||| |||||||| || ||||||||| |||||| || ||||||||||| |||| | ||||| ||||||||| ||| ||||||||| |||||||| ||| |||||||| ||||||||| |||||||| ||||||||| || ||| ||||||||| ||| ||||||||| |||||||| ||||||||| ||| ||||| ||||| |||||| ||| |||| ||||||||||| ||| ||||| ||||||||| ||||||||||| ||| | ||||| ||||| ||| |||||||||| || || ||| ||||||| ||||||||||| ||| ||| |||||| |||| |||||||||| |||||||||| ||||||||||||||| |||| |||||||||| |||| ||| |||||||||| ||| |||||||||| ||| ||||||||| || || ||||||||||| ||||||||| ||| |||||||| ||| |||||||||||||| ||| |||| || |||||||| ||| ||||||||| ||| ||||||| |||||||||||| ||||| |||| |||||||||| ||||||||||| ||| ||||||||| ||| |||||||||| ||||||||||||| ||| |||||||||| ||||||||| ||| |||||| |||| |||||||| ||| ||||||||| || ||| |||| |||| || |||||| |||| |||||||| ||| ||||||||| ||| ||||| || ||| ||| || ||| ||||||||| ||| ||||||||| ||||| |||| ||||||| |||| ||||||| || ||| |||| |||| |||| |||| ||||||||| |||||||| || |||||||||| |||| ||||||||| ||||||||| || ||| ||||||||| || |||||||| |||||| |||||||| |||||||||| |||||||||| |||||||||| ||||||||| ||||||||||||| ||||||| ||| ||||||| |||||||||| |||||||| |||| ||||||||| ||||||||| || ||| ||||||||| || ||| ||||||||||| |||| |||||||| ||| |||||| ||||| ||| ||| |||||||| |||| ||||||||||| ||||| || |||| ||| |||||||| ||||| ||| |||||||| ||||||||||||||| |||| || |||| ||||| ||||||| || |||| |||| |||||||| | |||||||| |||||||| ||||||||||| |||||||||| |||||||||||| |||||||| || ||||||||||||| |||||||||| ||| ||||||| |||||||||| |||||||||| ||||||| ||||| ||||||||| ||||||| |||||||||| ||| ||||| ||||||| || ||||||||| |||||||| |||||||| |||||||| |||| ||| |||| |||| || ||||||||||| |||||| ||| |||| || ||||||||| |||||| |||||||| || |||||||| |||| |||| |||||||| ||| |||||||| |||| ||||||| ||||||| ||||| |||||| |||||||| |||||||||| |||| || |||||||| |||||| |||| ||| |||||||| || ||||||| |||||||| |||||| |||||||| |||||||| |||||||| ||| ||||||||| || ||| |||||||||| || |||||||||| ||||||||||||||| ||| |||| ||||| ||| ||||| | || ||| ||||||||| ||| |||| ||||| ||| ||||| ||||| ||||||||||||||||| ||| |||| ||||| ||| ||||| ||||| ||||||| ||||||| |||||| |||||| ||||||||| ||| ||||||||||||||||||| ||||||||| |||||||||| ||||||||||||||||| ||||||| |||||||| |||| ||||| |||||| |||||||| |||| |||||||| |||||| |||||||| |||||||||| ||||||| |||||||| ||||||||| ||||||||||||||| ||| || ||| |||||||||| ||| ||||||||||||||||||| ||| |||||||| |||| || |||||| ||| ||| ||||| |||||||| |||||| |||||| ||||||||||| |||||||| ||| ||||||| |||||| |||||||||
Indirect Treatment Comparison Analysis Methods
||| ||||||| || ||| ||||||||| ||| |||| | |||||||| ||| ||||||||| ||||| |||||| |||| ||| |||||| ||||||| |||| |||||| |||| |||||||||| ||| ||||||||| ||| |||||||||| || ||| |||||||| ||||||||||| ||||||||| ||||| || |||||||| || || ||||||| |||||||||| || |||||||||| ||||| ||| |||||| |||||||| |||||| ||| || ||| ||||| |||| |||||||||||| |||||| |||||||||| |||| ||||||| || ||||||||||||| |||||||| || |||| |||||||||| ||||| ||| |||||||| ||||| ||| |||||||| ||| |||||||| ||||| ||| ||| |||| |||||||| ||| ||| || ||| ||| ||||| ||| |||||||||| || |||||||||||| |||||||| ||| ||||||||| ||||| | |||||||||| ||||| |||| | |||||||||||||| |||||| |||||||||| ||| ||| ||| || |||| ||||||||||||| |||||||| |||||| || |||| |||||||||| || ||| ||||||| ||||||||| || ||| ||||||||| |||||||| || |||| || ||| |||||| ||||||||||||||| ||||| ||||||||||||| |||| | |||| || | ||| | |||||||| || |||||| |||| |||||||| |||| ||| |||||||||||| ||||||| |||||||||| ||| ||||||||| |||||| ||||| |||||||||| ||||||||||| |||||||||| || ||| | || | ||| || | ||| |||| || |||| || || |||| || || |||| || ||||| |||||||||||| ||| |||||||| ||||||||| ||||||| |||||| |||| |||||||| ||||| ||| ||||||||| |||||||||||| || ||||||||| |||| ||| || |||||| || | || ||| | |||||||||| |||||||||| ||| |||||| |||||||| |||| |||| ||||| | ||||| ||| | ||||| ||||||||||| ||| || ||||||||| ||| || |||||| || ||| | |||||||||| |||||||||| ||| |||||| |||||||| |||| |||| ||||| | ||||| ||| | ||||| |||||||||||| ||||||||||||||| |||||| |||| |||| ||| |||| |||| ||||||| ||| ||||||||| |||||||| ||||| ||| |||||||||||| ||||||| |||| |||| ||||||| |||||||| || || ||| |||||||||| |||||||| |||| ||||||||| || ||||||| ||| ||| |||||||| |||||||| || || ||||| ||| |||||| |||||||| |||| ||| ||||||| |||| ||||||||| ||||| || ||| |||||||||| || |||||||| |||||||||||| ||| ||||| ||||| | |||||||| |||||||||| ||||| |||| | |||||||| |||||||||| ||| ||||| ||||| |||||| ||||||||||||||| ||||| ||||||||||||| ||| ||| |||||||||| |||| |||| |||| | |||| || | ||| | |||||||| || ||||||| |||||||| ||||||||| ||||||| |||| ||||||||| || |||||||| ||||||||| ||||||||||||| || ||||||||| |||||||| ||||||||| ||||||| ||||||| ||| |||||||| ||||||||||||| |||||||| |||| ||| |||||||| |||||||| ||| |||||||||| || ||| |||||| ||| ||| |||||||| ||||||||| ||||||| ||||| |||| ||||||||||| |||| ||| ||||| ||| |||||| ||||||||||| || ||| ||||||||| |||||||||||||| ||| ||||||| || ||| ||| ||||| |||||||| ||| |||| ||||||||| || ||||| || |||||||||||| |||| |||||||| ||||||||| |||||| ||||||||| ||||||| ||| ||||||||||||| || |||||||| ||||| |||| ||| |||| ||| ||| ||||||||| ||||| ||||||||||||| |||||||| ||||| || |||||| |||| ||||||||| |||| |||||||| ||||||||| ||||||| ||||||||| || |||||||||||| |||| ||| ||||||||||||| |||||||| ||||| || |||||||| ||| ||||| || ||| ||||| ||| ||||||| |||| |||| ||||||||| || |||| ||| ||||||||||||| |||||||| |||||||| |||||||||||| ||||||||| |||||||| ||||||| |||| ||||||||| || ||||| || ||| || ||||| ||| ||||||| ||||||||| || ||| |||||||| ||||||| ||| |||||| ||||||||| ||||| ||| ||| |||||| |||||||||| ||||||| ||| ||||||||| || ||||| || ||| || |||||||||||||
Table 24: Sponsor-Submitted Indirect Treatment Comparison Analysis Methods
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Results of Sponsor-Submitted Indirect Treatment Comparison
Summary of Included Studies
|| | ||||| || ||||| |||||||| |||||||||| |||| ||| |||||||||| |||||||||| ||||||| || ||||||| ||||||||||||| | ||||| |||| |||||||| || ||| |||||||||| |||||| ||| ||||| ||||| |||||| |||| |||||||||||| ||| |||||||||||||| |||||||||||||| ||| ||||||||||| ||||||||| |||||||| ||||||||||||| ||| ||||||||| ||| ||||||||||| ||||||||| ||||| |||||||| || ||||||||||| ||||||| |||||||||| |||| ||||||||||| ||| ||| ||||||||| ||||||||||| || ||||| ||||||||| ||| |||| |||||| ||| ||||||||||| ||||||||| ||||| ||| |||||||| |||| ||| |||| |||| ||||||||| |||||||||| ||| |||||| |||| |||||| ||||||||||||| |||||||||||| ||| ||||||||||| ||||||||||||| ||||| |||||| |||| |||||||| |||| ||| |||| |||| |||||||| ||| || ||||||||||| || ||| ||||||||||||| || ||| |||||| |||| || ||||||||| ||||| |||||||||| ||||||| ||| |||||||| |||||||||| ||||||||| | |||| |||| |||||||| ||||||||| ||| |||||||||||| || |||||||||||| ||| ||||||||||| |||||||
||| |||||| |||||||| |||| ||| ||||||||| |||| |||| |||||| |||||||| || |||||||||| ||||||| |||||||||| |||| || ||| ||||||||||||| ||| |||| ||||| ||||||||||||| ||||||| || ||| |||| || ||||||||||| |||||||||| || |||||||| ||||||||||||| |||| ||| ||| |||||||| ||| |||| ||||| ||||||||||||| ||||||| || ||| |||| || |||||||||| ||| ||||||| |||||||||| |||| |||||| |||| |||| ||||||| ||||||||| ||||| || ||| |||||||| ||||||||| || ||| |||| |||| |||||||| |||||||
Table 25 |||||||||| ||| ||| ||||||||||||||| || ||| |||||| |||||||| || ||| |||| |||| |||||||| || ||| ||||||||||||||||| |||| ||| | ||||||| |||| ||||| ||| |||| ||||||||| || |||||||| |||||||||| |||||||||||| | ||| ||||||||||| ||||| || ||||||| |||||||| |||| ||||| |||||||||| | |||| |||||| || |||||||||||| | ||||| ||| || ||||| |||| || ||| |||||||| || ||| ||||| ||||| || ||||| ||||| ||||||||| || | |||||||| ||| |||||||||| || ||| |||||||| |||| |||| ||||| ||||| ||| ||||||||||| ||||| |||||||| |||||||| |||| ||||| |||||||||| ||| ||| ||||||||| ||| |||||||||| |||||||| || ||| ||||||||||||||||| ||| ||| ||| |||||||| || |||||||| |||| ||||| |||||||||| ||||| ||||| ||||||||| |||||||| |||| ||||||| ||||||| |||| ||||||||||| ||||||||| |||| ||||| || |||||||| ||||| ||||||||||| ||| |||||||||||| | |||||||| ||||| ||| || || ||| |||| || ||| ||||||||||||| ||||||||| ||| ||||||||| || |||||||||||| | ||| |||||||| ||||||||||| || |||||||||| || |||||| || |||||||||| |||| ||| |||||||| || || ||||||||||||| ||||| |||||||| ||||||||||| || |||||||||||||| |||||| |||| |||||||||||| ||||| ||||||| |||||||||| |||||||||||| || |||||||||||| ||| |||| ||||||||| ||||||||| ||| |||| |||||||| ||||||||||| || || |||||||||||| ||||| ||||||| ||||||||||||| |||||||||||| ||||| ||| || |||||||||| ||||||||| || |||||||||||||||||||||| |||||||| |||| ||||||||| ||||||| |||||| |||||||| ||||| |||| |||| ||||||||||| ||||||| |||||||||||| | ||| ||||||||||| |||| ||||||| || ||||| |||||||| ||||||||||| |||||||||||||||| ||||||||| || ||||||||| ||| ||||||| |||||| ||| ||||||| ||||||||| | ||||||||||| |||||||| ||||||||| |||| |||||||||||| | |||| ||| |||||||| |||| ||||| |||| ||| ||| ||||||||||| ||||||||| ||||||||| ||| ||||||| || |||| ||||||||||| |||||||| ||| ||||||||| || | ||||| ||||||| || |||| |||||||||||||| ||||||||| || |||||||||||| | ||| |||| |||||| || ||| ||||||||| ||||||| ||||||||| ||||||| |||| ||| ||||||||||| ||| ||||||||||| |||||| |||| ||| ||||| |||||||| ||||||||| || |||| ||| |||| ||||||| |||||||||||||
Table 25: Characteristics of Trials Included in the Indirect Treatment Comparison
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
||||||| |||||||| ||||||||||||||| |||||| ||| | |||| |||| |||||| ||| ||||||||| || Table 26| |||| |||| |||| ||||||||||| |||||| |||| ||||||| ||||||| |||||| ||| | ||||||| ||||||||||||| ||| ||||||| ||||||| ||| |||||| |||| ||||||| || |||||||||||| ||||||| ||||||| ||||||| |||||||||| ||| ||||||| |||||| || |||||||||||| |||| ||| || ||| ||||||| |||||||||| ||| |||| ||| |||| ||||| |||||| |||||||| || ||||| || ||||| || ||||||||||| ||| |||||||||||| || ||||||||||| |||| |||||||| |||| ||||| ||||||| |||| || |||| |||||||| || ||||||||||| |||| |||||||| |||| || ||||| ||||| ||||||| ||| |||||||||||| | |||| ||| ||| ||||||| ||| ||||| |||||||| ||| ||||||||||| ||||||| |||||||||| ||| | ||||||| |||||||||| || |||||||||||| |||||||||| ||||| || |||||| |||||||| || ||||||||||| ||| |||||||||||| | |||| ||| ||||| || ||| |||||||||||| |||||||||| ||||||||||| |||| |||| |||||||| |||||||| |||| ||||| || |||||||| ||||||| ||| ||||||| |||||||||| || ||||||||||| ||| | || ||| ||||| ||| ||||||| ||| ||| ||||||| |||||||||| || |||||||||||| | ||| ||| || ||||| ||||| ||| |||||||| |||||||| |||||||||||| ||| |||||||||| |||||| ||| | ||||||| ||||||||||| ||| ||||||||||| |||||||||| |||| ||||||||| ||||||||||| |||||| |||| ||| |||| || ||| |||||| || ||| |||||| |||||||| ||||||||| ||| |||||| || ||| |||| |||| |||||||| ||| |||||| ||||||||||| ||||||||| || || ||||||||||| |||||| |||||||||| ||| |||||| |||| ||||| |||| || |||||||| |||| ||||||||||| ||| ||| |||||| ||||| | || | ||||||||| ||| ||| ||||||||| ||| |||| |||||||| ||| |||| |||| |||||||| ||||||| ||||||||| || |||| ||||||||||| ||| |||||||||||| ||||||| || |||| ||| |||||||| ||||||||| || ||| |||||||| ||||||||||||||| |||| || |||| ||||| ||| |||||| |||| |||||||||| || |||| ||| |||| || ||||| |||| ||| ||||||||| || ||| |||| || |||||||| || ||| ||||| |||||| |||| ||| |||| |||||||||| ||||||||||| |||||
Table 26: Summary of Patient Characteristics of Trials Included in the Indirect Treatment Comparison
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Table 27: Assessment of Homogeneity for Sponsor-Submitted Indirect Treatment Comparison
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Results
||| |||| |||| ||||||| |||||||| ||||| |||||||| |||||||||||| || ||||||||||| ||| ||||||||||| ||| ||| |||||||| |||| ||| ||||||||| || ||||||||| ||||||| |||||| ||||| | || || ||||| |||||||| |||||||||||| | ||| |||||||||||| ||||| |||||||| ||| ||||| || ||||||||| ||||| |||| || |||||| ||||| || ||| |||| |||| ||||||||
Figure 7: Network of Studies Included in the Base-Case Analysis

Note: This figure has been redacted at the sponsor’s request.
Overall Survival
||| ||||||| ||| || || ||| |||| || |||| ||||||||| ||| ||| |||| |||| || ||| |||| ||||| ||||||||||||||||| || ||| |||| |||| |||||||| ||||||||| || |||||||||||| |||||||||||| |||||||||| |||||||||| ||| |||| || | | ||| || | |||| |||| | ||||||| ||| ||||||||| | |||||||||| ||||||||||| |||||||||||||| ||| || ||||||||||||| ||| |||||||| ||||||| || ||| |||| |||| ||||| ||||||| |||||||||| |||||||||| ||| ||| ||||| |||||||||||| ||||||||||| |||||||| ||| ||||||||| |||||||||||||| |||||||| || ||| ||||||| |||| |||| |||| ||||||| || ||| |||||||||| ||| |||||||| |||| || |||||||||||| || ||| |||| |||||| ||| || ||| |||||||| ||||||||||| ||||||||| |||| ||| ||||||||| || ||| |||||| ||||||| ||||||||| ||| ||||||| |||||| || |||||||||| ||||||| |||||||||| ||| ||||||||||||| |||| ||||||| || || ||| || ||| |||| |||| |||||||| || |||| || ||| |||||||| ||||||||||| |||||||||
Table 28: Hazard Ratios Estimated From Fractional Polynomial Network Meta-Analysis Overall Survival; Base Case
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
|||||||||||| ||| |||| |||| || ||| |||| |||| |||||||| || ||| || |||||| || ||| |||||||| |||| || |||||||| |||||||| |||||||||| | ||||||||| || ||| |||||||||||| ||||||| |||||||||||||||||||||||||| || ||||||| ||| ||||| ||| ||| ||||| ||||||| ||| ||||||||| ||||| |||||||| ||| ||| |||||| ||||||| |||| |||| || |||| |||| |||| || ||| |||| ||||| |||| ||||| ||||||||||||||| ||| ||||| ||||||| |||| ||||||||| || ||| ||||||||| |||||||| |||||||| ||| |||||||| |||| |||||||| ||| |||||||||||| |||||| |||| ||||||| ||| ||||| ||| || |||| || || |||||| |||| ||| ||||||||| |||||||||| || |||||||||| ||||||||| || ||||||| ||| ||| |||||||| |||||||| |||| |||||||||| |||| ||| |||| |||| |||||||| ||| ||| ||| |||||||| | |||||||||| ||||||| |||||||||| ||| ||||||||||||| || |||||||| |||| |||||||| || |||||||||||| ||||||
Table 29: Hazard Ratio Estimated From Constant Hazard Ratio; Overall Survival Sensitivity Analyses
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Progression-Free Survival
||| ||||||| ||| ||| || ||| |||| || |||| ||||||||| ||| ||| |||| |||| || ||| |||| ||||| || Figure 7 ||||| || ||| |||| |||| |||||||| ||||||||| || |||||||||||| |||||||||||| |||||||||| |||||||||| ||| |||| || | | ||| || | || |||| | ||||||| ||| ||||||||| | |||||||||| ||||||||||| |||||||||||||| ||| || ||||||||||||| ||| |||||||| ||||||| || ||| |||| |||| ||||| ||||||| |||||||||| |||||||||| ||| ||| ||| ||| ||||| || Table 30| ||||||||||| |||||||| ||| ||||| || |||||||| | Table 39| ||| ||||||| |||| |||| |||| ||||||| || |||| |||||||||| ||| |||||||| |||| || |||||||||||| || ||| |||| |||||| ||| || ||| |||||||| ||||||||||| ||||||||| |||| ||| ||||||||| || ||| |||||| ||||||| ||||||||| ||| ||||||| |||||| |||| |||||||||| ||| |||||||| |||| ||||||||||||| || ||| |||| |||| |||||||| ||||||||| || ||| ||||||| |||| |||||| |||||||| ||||||||||| |||||||| ||||||||| |||||| |||| |||||| || ||| || |||||| || ||||||| |||||||||||| |||||||||||| |||| ||| ||||||||| || ||||||||||| |||| ||| |||||||| ||| |||| ||| ||||||||||| || |||||||||||| | ||||||||| |||||||| || ||||| |||| |||| || |||||||||||| |||| ||||||||||| |||||||| |||||| |||||||||||| |||||||||| ||||||| |||||||||| ||| ||||||||||||| |||| ||||||| || ||||
Table 30: Hazard Ratios Estimated From Fractional Polynomial Network Meta-Analysis Progression-Free Survival (Base Case)
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
|||||||||||| ||| |||| |||| || ||| |||| |||| |||||||| || ||| || |||||| || ||| |||||||| |||| || |||||||| |||||||| |||||||||| | ||||||||| || ||| |||||||||||| ||||||| ||||||||||||| || || ||| ||||||| ||| ||||| ||| ||| ||||| |||||| ||| ||||||||| ||||| |||||||| ||| ||| |||||| |||||||||| || || |||||||| |||| || |||||||||||| |||| || |||||||||| ||||||| |||||||||| ||| |||||||||||||| |||| ||||| || ||||||||| ||| ||| ||||| ||||||| |||| ||||||||| || ||| ||||||||| |||||||| |||||||| |||||||| |||| |||||||| ||| |||||||||||| |||||| |||||||||||| ||| ||||| ||| || |||| || || |||||| |||| ||| ||||||||| |||||||||| || |||||||||| ||||||||| ||| ||||||| ||| ||| |||||||| |||||||||| || |||||||||| ||| || ||||||||||||| ||| |||||||||| ||| ||||||||||||| ||| ||| |||||||| |||| |||||| ||||||||| |||||||| ||| ||||||||
Table 31: Hazard Ratio Estimated From Constant Hazard Ratio (Progression-Free Survival Sensitivity Analyses)
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Objective Response
||| ||| ||| |||| ||| |||| ||||||||| ||||||| ||| ||||| || ||| |||| ||| || ||| |||| ||||| || |||||||| ||||| || ||| ||||||| || ||| ||||||||||||| |||| |||| ||||||||| ||||| |||| ||||||||||| |||||||| ||| |||||||||| || ||||||||| || ||| |||| ||||| |||||||||| ||| |||||||| |||| |||| || |||||||||||| ||| | ||||| ||| |||| |||| || ||||| ||| ||||||||||||| ||| | ||||| ||| |||| |||| || ||||||
Table 32: Odds Ratios Estimated From Fixed-Effects and Random-Effects Network Meta-Analysis for Objective Response Rate (Base-Case and Sensitivity Analyses)
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Safety
||| ||| |||||| ||||| ||| |||||||| ||||||| ||| ||| |||| || |||| |||| ||| ||| |||||||| |||||||| ||||||||| ||||| ||| |||| ||| ||||||||| || ||||| | || | ||||||||| ||||||| |||||| ||||||||| ||||| ||| ||| ||||||| || ||| ||||||||||||| ||| ||| ||||| | || | ||||||||| ||||||| ||||||| |||||| | || | ||||||||||||||| ||||||| ||||||| ||| |||||||||||||||| ||| || ||||||||| ||||||| |||||| ||| |||||||||| || |||||||| | |||||||||| ||| |||||||| |||| || |||||||||||| || ||| |||||| ||||||||| |||| ||| ||||||||| || |||||||||||||||| ||| || ||||||||| ||||||| ||||||| ||||| ||| |||||||||||| |||||||||| || ||| | |||||| ||||||||| ||||||| |||||||||| ||| ||||||||||||||
Table 33: Odds Ratios Estimated From Fixed-Effects Network Meta-Analysis of Safety End Points
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Critical Appraisal of Sponsor-Submitted Indirect Treatment Comparison
The key limitations of the NMA are related to several major clinical assumptions: there are several notable differences in the characteristics of patients across the studies, which impact the interpretation of the NMA results; the network is small in size and structure, which prevented the use of a random-effects model as the base-case analysis; and there is a lack of closed loops, which precluded the assessment of consistency between direct and indirect comparisons.
The NMA was based on a systematic review that identified studies according to pre-specified inclusion criteria. The sponsor provided sufficient information on the search criteria, study selection, data extraction, and risk of bias assessment to ascertain that these were rigorously conducted. The key limitation related to selection criteria which did not include ||||||||||||| || ||||||||||| |||| ||||||||||||, a comparator considered to be relevant in the Canadian treatment landscape for patients with NSCLC expressing PD-L1 of 50% or more. It should be noted that while ||||||||||||| || ||||||||||| |||| |||||||||||| is used in Canada, the most relevant comparator for cemiplimab, ||||||||||||| |||||||||||, was captured in the sponsor’s submission. A full list of excluded publications and the reasons for their exclusion was not provided; therefore, it was not possible to assess whether potentially relevant studies may have been excluded.
Overall, the outcomes assessed were appropriate; however, the sponsor deemed HRQoL, an outcome identified as being important to patients, not possible to analyze based on differences in study reporting of HRQoL data.
Based on the NMA report, the outcomes assessed in the included trials appeared similar with respect to the definitions used. All trials used the RECIST 1.1 tumour response criterion, and trials relevant to this review were assessed according to an IRC.
There were several differences in study characteristics across the trials that are noted as potential sources of heterogeneity. Of the 3 studies that are included in the base-case analysis | |||| ||||||||||| ||| ||||||||| || || ||| |||||||||| || |||||||| |||||||||| ||||| | |||| ||| |||||||| |||| ||| ||||| |||||| |||| |||| |||||||| |||||||| |||||||| |||| ||| |||| |||||||||| ||||||| ||||||||| || ||||| ||||||| ||||||||||| ||||||| || |||||||||||| ||||||||| ||||||||||||| ||| |||||| |||||||| || ||||||| |||||||||| ||| |||||||||| ||||||||||| ||| ||| ||||| ||| ||||||||||| ||||||||||| ||||||| ||| || |||||||||||| |||||||| || ||||| |||| || ||||||||||||| |||||||||||, while EMPOWER-Lung 1 allowed crossover in both arms, patients receiving investigator’s choice chemotherapy could transition to cemiplimab monotherapy and patients receiving cemiplimab monotherapy could transition to receiving cemiplimab in combination with chemotherapy. The ITC authors noted that this may bias the results; therefore, multiple statistical methods were implemented to account for crossover in both trials and presented as sensitivity analyses. ||| ||||||| |||| ||| ||||||||| |||||||| |||||||| || ||| ||||||| |||||||| ||| ||| |||| | ||||||||||| |||||| || ||| ||||||| || ||| || |||| |||||||| |||||| || |||||||||| ||||||| |||||||||| ||| ||||||||||||| |||| ||||||| || ||| || ||||| |||||||| || || ||||||| ||||||| ||| ||||||| |||||||||| |||||| ||||| |||| ||||| ||||||||| ||| ||| ||||||||| ||||||| || |||| ||||||||||| ||| |||||||||||| ||
The ITC authors noted that there was significant heterogeneity in the baseline characteristics across the included trials, specifically with regard to tumour histology, which the ITC authors noted is expected to influence treatment effects. The authors also highlighted that while the preferred method of accounting for this heterogeneity is to incorporate these characteristics as covariates via meta-regression analysis, this was infeasible due to an insufficient number of studies. Instead, a subgroup analysis was conducted for squamous versus non-squamous patients using constant HRs. Constant HRs were used despite the violation of the proportional hazard assumption in the primary analysis, due to lack of Kaplan-Meier curves available in the histologic subgroup data. While these results did not show a significant impact on results due to tumour histology, the trials from which the data originated were not powered to detect differences in treatment effect stratified by tumour histology. Therefore, the impact that heterogeneity in tumour histology across trials had on the base-case analysis is unknown. Further heterogeneity in patient characteristics were found in the proportion of never-smokers across trials. The EMPOWER-Lung 1 study did not enrol any never-smokers, |||||||||| |||||||| |||| ||| ||||| ||||| ||||||| || ||| ||||||||||||| ||| || |||||||||||| ||||| ||||||||||||| ||| ||||||||||| |||||||| ||| ||| ||| || ||| ||||||||||||| ||| || |||||||||||| ||||| ||||||||||||| The authors noted that the benefit of a PD-1 blockade has not been shown in never-smokers, indicating that excluding these patients while the pembrolizumab trials include them would potentially bias the results in favour of cemiplimab.
The available trials formed sparse networks with no closed loops; therefore, it was not possible to validate the transitivity assumption of NMA and check for consistency of results between direct and indirect comparisons. The results are therefore based on indirect comparisons only, which reduces certainty in the findings. Random-effects models were attempted and determined not to be feasible to include in the base-case analysis due to the small number of included studies. Consequently, as connections were only described by a single trial for cemiplimab, base-case results are based on a fixed-effects model, as random-effects analyses were not meaningful.
Safety results show that cemiplimab was not different from ||||||||||||| with respect to grade 3 and higher all-cause AEs, grade 3 and higher immune-mediated AEs, and discontinuations due to all-cause AEs. The results of these ITCs and NMAs focusing on overall AEs reported in trials should be interpreted with consideration of the differences in study design, definitions, and reporting of AEs, which increase the uncertainty of the results. Given the variable length in follow-up across trials, there is potential for biased results in the frequency of AEs as some may occur rarely or may only become apparent long after the beginning of treatment.
Methods of He et al. (2021)32
Objectives
The objective of the NMA by He et al. (2021)32 was to evaluate the efficacy of first-line, single-agent immune checkpoint inhibitors versus combination immune checkpoint inhibitors in patients with advanced NSCLC and PD-L1 of 50% or more. For the purpose of this review, only those interventions and comparators that aligned with the CADTH protocol will be discussed.
Study Selection Methods
A search was conducted of PubMed, Embase, Cochrane Library, and the ClinicalTrials.gov website from inception until December 2020. In addition, abstract listings from relevant meetings were searched. Reference lists from review articles, commentaries, included studies, and conference publications were also scanned. Studies were included if they were phase III randomized controlled trials that evaluated the efficacy of first-line immune checkpoint inhibitors or chemotherapy for the treatment of patients with advanced NSCLC, reported 1 or more efficacy or safety end points, and were published in English. Selection was conducted by 2 independent reviewers who assessed the eligibility of records according to the inclusion criteria. The Cochrane Risk of Bias tool (version 1) was used to assess the risk of bias of included trials. Data extraction was performed by 2 reviewers. Efficacy outcomes were evaluated by IRCs to reduce potential assessment bias from the investigator. The treatment-related AEs (TRAEs) were assessed in the “as treated” population, which included all patients who underwent randomization and received at least 1 dose of the assigned combination treatment. The primary efficacy outcomes assessed were OS, PFS, and ORR. The primary safety outcome was TRAEs grade 3 to grade 5.
Indirect Treatment Comparison Analysis Methods
The authors performed a Bayesian NMA; the priors used were not specified. The HR or OR and their 95% credible intervals (CrIs) were used to summarize comparisons about efficacy outcomes and safety. For each outcome measure, a fixed-effects or random-effects model was used depending on the amount of heterogeneity observed, and analyses were performed using Markov chain Monte Carlo methods. The number of chains, burn-ins, and iterations were not reported. There was no justification provided for the chosen statistical model. There was no mention of convergence assessment or measures of model fit.
A pairwise meta-analysis (methods not described) on indirect comparisons and subgroup analysis based on histologic type were conducted. The authors assumed that characteristics that might affect outcomes were similar across the studies, but it is not clear what characteristics were assessed or how this was accomplished. Consistency could not be assessed as there were no closed loops. Statistical heterogeneity was assessed using Cochran’s Q test and the statistical inconsistency index (I2). Sensitivity analyses to explore sources of heterogeneity were planned but were not conducted as no substantial heterogeneity was observed.
Egger’s regression test with a funnel plot was used to assess the publication bias, and a P value of less than 0.10 was considered to present potential publication bias.
Results of He et al. (2021)32
Summary of Included Studies
Out of a total of 1,695 articles identified from the systematic literature search, 14 trials (N = 3,448 patients) were included in the NMA. The trials included patients with advanced NSCLC with PD-L1 of 50% or more. The NMA included 13 studies assessing OS, 12 studies assessing PFS, and 11 studies assessing ORR. There was 1 trial comparing cemiplimab to chemotherapy (the EMPOWER-Lung 1 study). Only those indirect comparisons that included interventions or comparators specified in the CADTH protocol were included in this report.
There were some differences in study patient characteristics across the included trials. Of note, the EMPOWER-Lung 1 trial only included patients with PD-L1 expression of 50% or more, while others included a varied proportion of patients with PD-L1 of less than 50% (often less than half the patients). In addition, the trial only included current or former smokers, unlike the other studies that included some non-smokers. The EMPOWER-Lung 1 trial also included a greater proportion of males compared to the other included trials. Characteristics such as cancer stage were not reported; hence, it is not possible to determine whether this was similar across trials. The length of follow-up was not provided. All trials of interest to the CADTH review were open label, including the EMPOWER-Lung 1 trial. Egger’s regression test was carried out and publication bias was not detected for any analysis.
Results
The evidence formed a star-shaped network with chemotherapy as the central node. In direct comparisons, cemiplimab demonstrated improvement in OS (HR = 0.57; 95% CrI, 0.43 to 0.77) and PFS (HR = 0.54; 95% CrI, 0.43 to 0.68) compared to chemotherapy. Findings were similar in subgroup analysis by histologic subtype. ORR was better for cemiplimab than for chemotherapy (odds ratio [OR] = 2.53; 95% CrI, 1.74 to 3.72). Cemiplimab had lower odds of TRAEs of grade 3 to grade 5 compared to chemotherapy (OR = 0.63; 95% CrI, 0.46 to 0.85).
Based on indirect comparisons, there was little to no difference in OS between cemiplimab, pembrolizumab, and pembrolizumab plus chemotherapy. The CrIs for these analyses were wide. For the analysis of PFS, the performance of pembrolizumab plus chemotherapy was better than cemiplimab alone (HR = 0.68; 95% CrI, 0.47 to 0.98), but there was little to no difference between cemiplimab and pembrolizumab (HR = 1.38; 95% CrI, 0.50 to 3.79). For ORR, there was little to no difference between cemiplimab and pembrolizumab plus chemotherapy, but cemiplimab performed better than pembrolizumab (OR = 1.59; 95% CrI, 1.00 to 2.54). The risk of TRAEs of grade 3 to grade 5 was higher with pembrolizumab plus chemotherapy compared to cemiplimab (OR = 2.01; 95% CrI, 1.35 to 2.98) and was higher with cemiplimab than pembrolizumab (OR = 2.01; 95% CrI, 1.38 to 2.95).
Critical Appraisal of He et al. (2021)32
There were many limitations of the NMA conducted by He et al. (2021),32 primarily due to the inadequate reporting of the methodology used. Criteria such as convergence and model fit appear to have not been assessed. The studies were compared in NMA, assuming that the transitivity assumption was met; however, examination of the baseline characteristics across studies reveals notable differences in potential prognostic factors that violate this assumption. There is a lack of clarity on whether the assessment of outcomes was comparable across the included trials. The choice of fixed effects versus random effects was not specified a priori and instead was based on the heterogeneity of the findings, potentially introducing investigator bias. The model was sparse with no closed loops; hence, conclusions were reliant on indirect findings that were subject to uncertainty. Other limitations included the influence of factors such as differences in immune checkpoint inhibitors and chemotherapy regimens, which could have introduced further intransitivity. Additionally, the PD-L1 assay methods and sensitivity were not consistent across all studies.
The EMPOWER-Lung 1 study (the only trial that evaluated cemiplimab) did not include non-smoking patients, who tend to have diminished treatment effects with immune checkpoint inhibitors. Hence, the treatment effect of cemiplimab may be overestimated compared to studies that did include this patient population. Due to small sample sizes and a sparse network, many of the effect estimates were largely imprecise. The subgroup analysis based on histology was performed only for direct comparisons, and was not very applicable to some trials that primarily included 1 histology over the other (e.g., non-squamous NSCLC). In indirect comparisons, the impact on efficacy outcomes of individual patient characteristics, such as age, smoking status, or the presence of liver or brain metastases, was not assessed. Given that there were no closed loops, all findings were based only on indirect evidence, which further increases uncertainty.
Given the level of uncertainty presented by these limitations, credible conclusions cannot be drawn regarding the efficacy of cemiplimab in patients with NSCLC compared to pembrolizumab or the combination of pembrolizumab plus chemotherapy. HRQoL was not assessed, though this was identified as an important outcome to patients.
Methods of Majem et al. (2021)31
Objectives
The objective of the ITC by Majem et al. (2021)31 was to evaluate the efficacy of available PD-L1 and PD-1 inhibitor monotherapies for the first-line treatment of patients with high PD-L1 expression (≥ 50%) and locally advanced or metastatic NSCLC compared to chemotherapy.
Study Selection Methods
A systematic search was conducted in PubMed to identify all eligible trials from inception until November 1, 2020. Additional searches were undertaken of selected conference proceedings. Only phase III trials conducted in patients with locally advanced or advanced NSCLC and PD-L1 of 50% or more, not previously treated for their metastatic disease, and receiving first-line PD-L1 or PD-1 inhibitor monotherapy were eligible for inclusion. Outcomes included were PFS and OS. Details of the process for study selection and data extraction were not reported in the publication. Risk of bias was assessed using the Cochrane Risk of Bias tool (version 1).
ITC Analysis Methods
The ITCs were conducted using the Bucher method, with chemotherapy as the common comparator. Only the subsets of patients with PD-L1 of 50% or more were included. There was no justification of the analysis method chosen. Cox proportional HRs along with their corresponding 95% CIs were used as the summary estimates of relative treatment effects. For dichotomous data, ORs were estimated. The study authors stated that the results were not altered by sensitivity analyses. However, sensitivity analyses were not reported within the study. Analyses were not controlled for multiplicity. When assessing OS, results from both KEYNOTE studies were grouped for pembrolizumab comparisons since there was no significant heterogeneity between the studies (I2 = 0.0%; P = 0.6978). When assessing PFS, results were considered separately for pembrolizumab comparisons since significant heterogeneity (I2 = 80.65%; P = 0.0064) was observed between KEYNOTE studies.
Results of Majem et al. (2021)31
Summary of Included Studies
Out of the 79 records identified, 6 clinical trials (N = 2,111 patients) were included in the ITCs. Of these 6 trials, only 3 included interventions relevant to the pre-specified CADTH protocol, including cemiplimab (n = 1) and pembrolizumab (n = 2). Two trials (KEYNOTE-024 and EMPOWER-Lung 1) included only patients with PD-L1 expression levels of 50% or more. The trials were similar in terms of age and ECOG PS. However, the EMPOWER-Lung 1 trial had a higher proportion of males than the other trials and fewer non-squamous tumours, and was the only trial that did not include non-smokers. In addition, participants in the EMPOWER-Lung 1 trial who responded to cemiplimab treatment were permitted to continue receiving treatment in combination with chemotherapy in the event of progressive disease. The length of follow-up was not described. All trials with comparators of interest to the CADTH review were open label, including EMPOWER-Lung 1.
Results
In direct comparisons, OS was longer with cemiplimab compared to chemotherapy (HR = 0.57; 95% CI, 0.42 to 0.77), as was PFS (HR = 0.54; 95% CI, 0.43 to 0.68; P < 0.0001). In indirect comparisons, there was little to no difference in OS for cemiplimab compared to pembrolizumab (HR = 0.84; 95% CI, 0.59 to 1.19; P = 0.319). PFS was longer with cemiplimab compared to pembrolizumab when using the results of KEYNOTE-042 (HR = 0.67; 95% CI, 0.49 to 0.90; P = 0.008) but not when using the results of KEYNOTE-024 (HR = 1.08; 95% CI, 0.74 to 1.59; P = 0.621).
Critical Appraisal of Majem et al. (2021)31
A number of limitations were present within the ITCs that primarily stem from inadequate reporting of the methods used and limitations based on available study data. A review of the baseline characteristics indicates that there was some variability across studies in potential prognostic factors (e.g., smoking status). Other differences that were not discussed in the study included the length of follow-up and variable PD-L1 assay methods. The conclusions were reliant on indirect findings that were imprecise and subject to considerable uncertainty. Other limitations included the influence of factors such as differences in immune checkpoint inhibitors and chemotherapy regimens, which could have introduced further heterogeneity across direct comparisons.
The treatment effects of cemiplimab in the EMPOWER-Lung 1 trial may be overestimated given the lack of non-smokers, in whom the treatment effects of immune checkpoint inhibitors tend to be diminished, as compared to the pembrolizumab trials. For PFS, the effect of cemiplimab compared to pembrolizumab differed based on the trial of pembrolizumab used in the analysis (KEYNOTE-024 versus KEYNOTE-042). Interim findings of the EMPOWER-Lung 1 study were used for OS, whereas the final analysis was used for the KEYNOTE studies.
Given the level of uncertainty presented by these limitations, credible conclusions cannot be drawn regarding the efficacy of cemiplimab in patients with NSCLC compared to pembrolizumab. Safety and HRQoL were not assessed, despite these being important outcomes to patients and clinicians.
Summary
The sponsor-submitted NMA, while confirming benefit over chemotherapy, did not clearly demonstrate that cemiplimab was favoured, not favoured, or similar to ||||||||||||| in the analyses. The significant limitations associated with the NMA preclude drawing definitive conclusions on the comparative efficacy and safety of cemiplimab. These included the small and sparsely populated networks, reliance on only indirect comparisons, and the considerable clinical and methodological heterogeneity that was not adequately addressed. In addition to being hindered by many of the same limitations of the sponsor-submitted NMA, the published ITCs were poorly reported, which precluded full appraisal of their quality and the development of credible conclusions. Though 1 of these ITCs included a comparator of interest to the CADTH review not found in the other ITCs (pembrolizumab plus chemotherapy), considerable weaknesses in methodology of the published ITC mean that the findings of analyses comparing cemiplimab to pembrolizumab plus chemotherapy are subject to considerable uncertainty.
Other Relevant Evidence
No other relevant evidence was identified for this review.
Discussion
Summary of Available Evidence
The evidence base for this review consists of 1 randomized controlled trial, 1 ITC submitted by the sponsor, and 2 published ITCs. The EMPOWER-Lung 1 study is an ongoing randomized, multi-centre, open-label, phase III study of cemiplimab monotherapy versus platinum-based doublet chemotherapy in patients with stage IIIB, stage IIIC, or stage IV NSCLC who were not candidates for treatment with definitive chemoradiotherapy, whose tumours expressed PD-L1 in at least 50% of tumour cells, with no EGFR, ALK, or ROS1 aberrations, and who had received no prior systemic treatment for their advanced disease. The primary end points were OS and PFS and the key secondary end point was ORR. In total, 710 patients were randomized — 356 patients to the cemiplimab monotherapy arm and 354 patients to the chemotherapy arm. The median age of patients in the trial was 63 years, and 85% of patients were men. A non-squamous histology was observed in 56% of patients, and the disease stage at screening was metastatic (stage IV) in 84% of patients. About 23% of patients had received some prior therapy, most commonly radiotherapy, for their disease. The main limitations of the clinical evidence are the exclusion of never-smokers and crossover between treatment arms. In addition, chemotherapy, the comparator in the trial, is not reflective of standard of care in Canada.
The sponsor-submitted NMA evaluated the comparative efficacy and safety of cemiplimab versus competing interventions for the treatment of patients with NSCLC expressing PD-L1 of 50% or more. In the base-case analysis, cemiplimab was compared with chemotherapy, and with pembrolizumab for OS, PFS, ORR, and safety outcomes. Bayesian NMA methods were used for the comparisons. However, few inferences can be made from the results of the NMA due to important limitations with the included studies and the methods and assumptions made in the NMA. Importantly, the choice of relevant comparators did not include pembrolizumab in combination with chemotherapy, a comparator considered to be relevant in the Canadian setting, though the most relevant comparator, pembrolizumab monotherapy, was captured. The outcomes assessed were appropriate; however, other important outcomes such as HRQoL were deemed not possible to analyze. The published ITCs included a comparison with pembrolizumab plus chemotherapy, but these, too, had important methodological limitations and conclusions could not be drawn based on the findings.
Interpretation of Results
Efficacy
In the EMPOWER-Lung 1 study, cemiplimab resulted in statistically significantly longer OS and PFS compared to chemotherapy in patients with advanced NSCLC with PD-L1 of 50% or more and no EGFR, ALK, or ROS1 aberrations. With a median follow-up of 13 months and after 249 OS events up to the data cut-off date, representing 35% of the ITT population, the primary OS end point was met. OS showed a statistically significant difference between the 2 arms in favour of cemiplimab (HR = 0.68; 95% CI, 0.53 to 0.87), with median OS of 22.1 months in the cemiplimab arm and 14.3 months in the chemotherapy arm (P = 0.0022), an improvement of 7.8 months. Subgroup analysis of OS suggested a benefit of cemiplimab over chemotherapy across most subgroups, although the degree of benefit was highly variable. PFS results were also in favour of cemiplimab, with a median IRC-assessed PFS of 6.2 months versus 5.6 months in the chemotherapy arm, although the difference of 0.6 months may not be clinically important. The ORR (36% versus 21% in the cemiplimab arm and chemotherapy arm, respectively) and median DOR results (21 months versus 6 months in the cemiplimab and chemotherapy arms, respectively) further support the benefit observed with OS and PFS. However, despite these positive results, the chosen comparator of the EMPOWER-Lung 1 study is no longer the standard of care in Canada for the high PD-L1–expressing NSCLC population. Therefore, the demonstrated superiority of cemiplimab to chemotherapy for improving survival outcomes may be of low clinical relevance and is a major limitation of the evidence. A comparison of the efficacy of cemiplimab to relevant comparators (as noted in the CADTH protocol), pembrolizumab monotherapy and pembrolizumab plus platinum-based chemotherapy, was available only in indirect comparisons. These were subject to uncertainty due to the sparsity of the networks and evidence of violations of the underlying assumptions of the NMA methodology, which precluded definitive conclusions regarding the efficacy of cemiplimab compared to these relevant comparators.
An important aspect of treatment desired by both the clinicians and patient groups consulted by CADTH was improvement in cancer-related symptoms and patients’ HRQoL. The overall HRQoL (i.e., GHS) improved in both treatment arms and the mean score results appeared to favour cemiplimab for the early time points (up to the start of cycle 6). The results of HRQoL measures were impacted by decreasing sample sizes and, consequently, very large SDs at the later time points, and between-group statistical comparisons were not conducted. In addition, the trial was open label and it is possible that these subjective measures were impacted by detection and performance biases. As a result, definitive conclusions about the impact of treatment with cemiplimab versus chemotherapy on symptoms and HRQoL cannot be drawn. These outcomes were not included within the indirect comparisons to other relevant comparators (pembrolizumab monotherapy, and pembrolizumab + platinum-based chemotherapy).
Locally advanced or metastatic NSCLC is incurable, and therefore is considered a serious and life-threatening disease. The treatment goals in patients with locally advanced or metastatic NSCLC are to reduce tumour size, slow or delay progression and metastasis, reduce treatment-related toxicity, optimize quality of life, and prolong OS. The findings of the EMPOWER-Lung 1 study provide support to existing evidence that anti–PD-1 therapies provide greater survival benefits in this population of patients, particularly those whose tumours have higher levels of PD-L1 expression, compared with chemotherapy regimens.21 Two anti–PD-1 therapies, pembrolizumab and cemiplimab, have demonstrated survival benefit as monotherapy in the first-line NSCLC setting, but currently only 1 of these therapies (pembrolizumab) is publicly funded. As the prognosis for patients with locally advanced or metastatic NSCLC remains poor, there is still an unmet need for novel therapies with improved treatment benefit and/or safety profiles. It may be advantageous to have additional agents that convincingly demonstrate a survival benefit as monotherapy in the first-line NSCLC setting. As the clinical experts consulted by CADTH noted, cemiplimab offers an additional treatment option rather than filling an unmet treatment need.
Pembrolizumab monotherapy is the standard of care first-line treatment in this patient population, recommended by clinical practice guidelines, and is widely funded and used in clinical practice. Therefore, clinicians may not readily take up another similar drug in the same class if no clear advantage compared to the established standard of care is demonstrated. No head-to-head evidence is available regarding the comparative efficacy of cemiplimab with other anti–PD-1 monotherapies used in the same setting (e.g., pembrolizumab). Evidence from the indirect comparisons is inconclusive and did not clearly demonstrate a benefit of cemiplimab compared to pembrolizumab.
Harms
Cemiplimab exhibited an acceptable toxicity profile, as expected from an anti–PD-1 checkpoint inhibitor. The incidence and severity of immune-related AEs were also reasonable, considering cemiplimab’s anti–PD-1 mechanism of action, and most events were clinically manageable. It is important to note that at the data cut-off date, 39.2% of patients in the cemiplimab arm were still on cemiplimab treatment and only 1.7% of patients had completed the planned 108 weeks of treatment. Therefore, no firm conclusions can be drawn regarding the safety of cemiplimab over the proposed 2-year treatment period in this setting. It is possible that with longer follow-up, a greater frequency of AEs would be observed.
Most of the patients from both arms experienced AEs. A higher proportion of patients in the chemotherapy arm experienced grade 3 or higher AEs compared to the cemiplimab arm (48% versus 37%). The incidence of SAEs was similar in the 2 treatment arms (approximately 28%), although the type of SAE varied by treatment arm. While pneumonia was the most common SAE and equally prevalent in both arms (n = 17), other respiratory SAEs were more frequent in the cemiplimab arm; pneumonitis and pulmonary embolism occurred in 12 patients, compared to 2 in the chemotherapy arm. In the chemotherapy arm, myelotoxic effects such as anemia and febrile neutropenia were more frequent.
The proportion of patients who discontinued treatment permanently because of AEs was slightly higher in the cemiplimab arm compared to the chemotherapy arm (6.5% versus 4.1%). However, the AE profile was notably different in the 2 treatment arms. Patients in the chemotherapy arm presented a higher incidence and severity of myelotoxicity, alopecia, nausea, constipation, decreased appetite, neuropathy, and fatigue, whereas immune-related AEs and associated endocrine disorders were more frequent in the cemiplimab arm. In the cemiplimab arm, 5 patients discontinued because of respiratory AEs, most of them pneumonitis, and in the chemotherapy arm, 6 patients discontinued because of hematological toxicity events. Although similar proportions of AEs, SAEs, and AEs leading to treatment discontinuation were observed in the 2 treatment arms, it appears that cemiplimab is generally well tolerated and compares favourably in relation to potentially more serious chemotherapy-related AEs.
The incidence of immune-related AEs in patients in the cemiplimab arm (17.5%) is comparable to that of previous studies of cemiplimab and to that seen with other anti–PD-1 and anti–PD-L1 agents used in clinical practice. The most frequent immune-related AEs were hypothyroidism (5.6%), hyperthyroidism (4.2%), pneumonitis (2.3%), hepatitis (1.7%), rash (1.7%), and colitis (1.1%). Approximately 80% of all immune-related AEs were grade 1 or grade 2 events and were clinically manageable.
Compared to other drugs in its class (notably pembrolizumab, which is the current standard of care) in the sponsor-submitted ITC, there was no evidence for a difference in the safety profile of cemiplimab compared to pembrolizumab with regard to grade 3 to grade 5 all-cause AEs, grade 3 to grade 5 immune-mediated AEs, and discontinuation due to all-cause AEs. The significant limitations associated with the ITC preclude drawing definitive conclusions on the comparative safety of cemiplimab with other anti–PD-1 agents.
Conclusions
Based on clinical data from the EMPOWER-Lung 1 study, cemiplimab demonstrated a statistically significant and clinically meaningful survival benefit compared to chemotherapy in the treatment of patients with advanced NSCLC, no EGFR, ALK, or ROS1 aberrations, and PD-L1 expression of 50% or greater. ORR and duration of survival were supportive of the primary efficacy results. Symptoms and HRQoL were assessed, but conclusions cannot be drawn for those outcomes. The toxicity profile of cemiplimab was acceptable and compared favourably to chemotherapy. Chemotherapy is no longer the first-line standard of care in this patient population and the evidence from ITCs is insufficient for determining comparative efficacy and safety of cemiplimab with other immune checkpoint inhibitors used in this setting. This includes ||||||||||||| |||||||||||, which is the most relevant comparator in the Canadian setting given that cemiplimab monotherapy appears to provide an alternative immune checkpoint inhibitor monotherapy option to ||||||||||||| |||||||||||.
References
1.Canadian Cancer Society. Lung cancer statistics. 2021: https://cancer.ca/en/cancer-information/cancer-types/lung/statistics. Accessed 2022 Mar 11.
2.Peters S, Adjei AA, Gridelli C, et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23 Suppl 7:vii56-64. PubMed
3.Health Canada reviewer's report: Libtayo (cemiplimab) [internal sponsor's report]. City (PROV): Sanofi-aventis Canada; 2021.
4.Chen YM. Update of epidermal growth factor receptor-tyrosine kinase inhibitors in non-small-cell lung cancer. J Chin Med Assoc. 2013;76(5):249-257. PubMed
5.Kawaguchi T, Koh Y, Ando M, et al. Prospective analysis of oncogenic driver mutations and environmental factors: Japan molecular epidemiology for lung cancer study. J Clin Oncol. 2016;34(19):2247-2257. PubMed
6.Planchard D, Popat S, Kerr K, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv192-iv237.
7.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693-1703. PubMed
8.Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol. 2014;25(1):138-142. PubMed
9.Clinical Study Report: R2810-ONC-1624. A global, randomized, phase 3, open-label study of REGN2810 (ANTI-PD-1 antibody) versus platinum-based chemotherapy in first-line treatment of patients with advanced or metastatic PD-L1 + non-small cell lung cancer [internal sponsor's report]. Tarrytown (NY): Regeneron Pharmaceuticals, Inc.; 2017 May 29.
10.Duma N, Santana-Davila R, Molina JR. Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc. 2019;94(8):1623-1640. PubMed
11.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175-180. PubMed
12.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394. PubMed
13.Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963-1971. PubMed
14.Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113-125. PubMed
15.National Comprehensive Cancer Network. Non-small cell lung cancer. NCCN clinical practice guidelines in oncology (NCCN guidelines)2022.
16.Hanna NH, Robinson AG, Temin S, et al. Therapy for stage IV non-small-cell lung cancer with driver alterations: ASCO and OH (CCO) joint guideline update. J Clin Oncol. 2021;39(9):1040-1091. PubMed
17.Libtayo (cemiplimab for injection): solution for infusion, 50mg/mL; Antineoplastic Agent, Monoclonal Antibody[product monograph]. Laval (QC): Sanofi-Aventis Canada Inc; 2021 Oct 8.
18.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
19.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Nov 30.
20.Sezer A, Kilickap S, Gumus M, et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet. 2021;397(10274):592-604. PubMed
21.Mok TSK, Wu YL, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819-1830. PubMed
22.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
23.Fayers PM. Interpreting quality of life data: population-based reference data for the EORTC QLQ-C30. Eur J Cancer. 2001;37(11):1331-1334. PubMed
24.Carbone DP, Reck M, Paz-Ares L, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376(25):2415-2426. PubMed
25.Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379(21):2040-2051. PubMed
26.Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823-1833. PubMed
27.Robins M, Tsiatis A. Correcting for non-compliance in randomized trials using rank preserving structural failure time models. Communications in Statistics -Theory and Methods. 1991;20(8).
28.Latimer NR, Abrams KR. NICE DSU technical support document 16: adjusting survival time estimates in the presence of treatment switching. London (UK): National Institute for Health and Care Excellence (NICE); 2014: https://www.ncbi.nlm.nih.gov/books/NBK310374/pdf/Bookshelf_NBK310374.pdf. Accessed 2022 Mar 11.
29.Network meta-analysis of clinical trials supporting cemiplimab monotherapy in advanced or metastatic non-small cell lung cancer with PD-L1 ≥50%: Technical report for the R2810-ONC-1624 true PD-L1 ≥50% ITT population (mITT-1) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cemiplimab (Libtayo), 350 mg IV. Mississauga (ON): Sanofi Genzyme; 2021 May 31.
30.Papadopoulos KP, Autio K, Golan T, et al. Phase I study of MK-4166, an anti-human glucocorticoid-induced tnf receptor antibody, alone or with pembrolizumab in advanced solid tumors. Clin Cancer Res. 2021;27(7):1904-1911. PubMed
31.Majem M, Cobo M, Isla D, et al. PD-(L)1 inhibitors as monotherapy for the first-line treatment of non-small-cell lung cancer patients with high PD-L1 expression: a network meta-analysis. J Clin Med. 2021;10(7):26. PubMed
32.He M, Zheng T, Zhang X, et al. First-line treatment options for advanced non-small cell lung cancer patients with PD-L1 >= 50%: a systematic review and network meta-analysis. Cancer Immunol Immunother. 2021;16:16. PubMed
33.Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika. 1994;81(3):515-526.
34.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU technical support document 2: a generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. London: National Institute for Health and Care Excellence (NICE); 2011 (updated 2014): https://www.ncbi.nlm.nih.gov/books/NBK310366/pdf/Bookshelf_NBK310366.pdf. Accessed 2022 Mar 11.
35.Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med. 2016;375(19):1823-1833. PubMed
36.Wu YL, Zhang L, Fan Y, et al. Randomized clinical trial of pembrolizumab vs chemotherapy for previously untreated Chinese patients with PD-L1-positive locally advanced or metastatic non-small-cell lung cancer: KEYNOTE-042 China Study. Int J Cancer. 2021;148(9):2313-2320. PubMed
37.Herbst RS, Giaccone G, de Marinis F, et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N Engl J Med. 2020;383(14):1328-1339. PubMed
38.Rizvi NA, Cho BC, Reinmuth N, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: the MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020;6(5):661-674. PubMed
39.Teckle P, Peacock S, McTaggart-Cowan H, et al. The ability of cancer-specific and generic preference-based instruments to discriminate across clinical and self-reported measures of cancer severities. Health Qual Life Outcomes. 2011;9:106. PubMed
40.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-1028. PubMed
41.Ozturk A, Sarihan S, Ercan I, Karadag M. Evaluating quality of life and pulmonary function of long-term survivors of non-small cell lung cancer treated with radical or postoperative radiotherapy. Am J Clin Oncol. 2009;32(1):65-72. PubMed
42.Maringwa JT, Quinten C, King M, et al. Minimal important differences for interpreting health-related quality of life scores from the EORTC QLQ-C30 in lung cancer patients participating in randomized controlled trials. Support Care Cancer. 2011;19(11):1753-1760. PubMed
43.Drug Reimbursement Review sponsor submission: LIBTAYO (cemiplimab for injection) 350 mg every three (3) weeks as an intravenous (IV) infusion [internal sponsor's package]. Mississauga (ON): Sanofi Genzyme; 2021 Oct 13.
44.Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144. PubMed
45.Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Eur J Cancer. 1994;30A(5):635-642. PubMed
46.Scott N, Fayers P, Aaronson N, et al. EORTC QLQ-C30. Reference values. Brussels (Belgium): European Organization for Research and Treatment of Cancer (EORTC); 2008: https://www.eortc.org/app/uploads/sites/2/2018/02/reference_values_manual2008.pdf. Accessed 2022 Mar 11.
47.Aggarwal J, Chakraborty S, Ghosh Laskar S, et al. Reference data for standardized quality of life questionnaires in Indian patients with brain metastases from non-small cell lung cancer: results from a prospective study. Cureus. 2017;9(4):e1149. PubMed
48.Maringwa JT, Quinten C, King M, et al. Minimal important differences for interpreting health-related quality of life scores from the EORTC QLQ-C30 in lung cancer patients participating in randomized controlled trials. Support Care Cancer. 2011;19(11):1753-1760. PubMed
49.Koller M, Hjermstad M, Tomaszewski K, et al. An international study to revise the EORTC questionnaire for assessing quality of life in lung cancer patients. Ann Oncol. 2017;28(11):2874-2881. PubMed
50.Montazeri A, Gillis CR, McEwen J. Quality of life in patients with lung cancer: a review of literature from 1970 to 1995. Chest. 1998;113(2):467-481. PubMed
51.Brabo EP, Paschoal ME, Biasoli I, et al. Brazilian version of the QLQ-LC13 lung cancer module of the European Organization for Research and Treatment of Cancer: preliminary reliability and validity report. Qual Life Res. 2006;15(9):1519-1524. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: November 29, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for 1 character |
? | Truncation symbol for 1 or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq = # | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Cemiplimab* or Libtayo* or regn2810 or regn 2810 or sar 439684 or sar439684 or 6QVL057INT).ti,ab,ot,kf,kw,hw,nm,rn.
1 use medall
*cemiplimab/
(Cemiplimab* or Libtayo* or regn2810 or regn 2810 or sar 439684 or sar439684).ti,ab,kf,dq.
3 or 4
5 use oemezd
(conference abstract or conference review).pt.
6 not 7
2 or 8
remove duplicates from 9
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search terms: Libtayo (cemiplimab) and non–small cell lung cancer (NSCLC)
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
Search terms: Libtayo (cemiplimab) and non–small cell lung cancer (NSCLC)
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms: Libtayo (cemiplimab) and non–small cell lung cancer (NSCLC)
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms: Libtayo (cemiplimab) and non–small cell lung cancer (NSCLC)
Grey Literature
Search dates: November 28 - November 29, 2021
Keywords: Libtayo, cemiplimab, non–small cell lung cancer, NSCLC
Limits: No publication limits
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
Appendix 2: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and minimal important difference [MID]):
EORTC QLQ-C30
EORTC QLQ-LC13
Findings
The findings about the validity, reliability, responsiveness, and MID of each outcome measure are summarized in Table 35.
Table 35: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EORTC QLQ-C30 | Cancer-specific self-reported measure of HRQoL. 30-item questionnaire, consisting of 5 functional scales (physical, role, emotional, social, and cognitive), 9 symptom scales (fatigue, nausea/vomiting, pain, dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties), and a GHS scale. A higher score for functional scales and for GHS represents better functioning ability or HRQoL. A higher score for symptom scales represents a worsening of symptoms.22 | In studies with lung cancer patients: Validity: Moderate to strong correlations between the 5QLQ-C30 functioning scales (r = 0.41 to 0.77); FACT-G and QLQ-C30 scales (r = 0.64 to 0.76)39; HADS with all QLQ-C30 functioning scales (r = 0.28 to 0.75); BPI scales with all QLQ-C30 scales except for nausea/vomiting (r = 0.20 to 0.72),40 supporting convergent validity. Known-groups approach: Able to differentiate across different measures of cancer severity: cancer stages (d = 0.49); ECOG PS (d = 0.65); and self-reported health status (d = 1.36).39 Reliability: Cronbach’s alpha ranging from 0.56 to 0.93 with 7 scales having acceptable internal consistency (Alpha > 0.70).41 Responsiveness: No relevant studies identified. | In a study with NSCLC patients: MID estimates for improvement (deterioration) using the ECOG PS and weight change as anchors: physical functioning: 9 and 5 (4 and 6) role functioning: 14 and 7 (5 and 5) social functioning: 5 and 7 (7 and 9) GHS: 9 and 4 (4 and 4) fatigue: 14 and 5 (6 and 11) pain: 16 and 2 (3 and 7).42 The EMPOWER-Lung 1 trial defined a clinically meaningful improvement in the GHS score as a ≥ 10-point change.43 In a study with lung cancer patients: an anchor-based approach in which patients who reported “a little” change on the SSQ had subsequent changes on a scale of the EORTC QLQ-C30 of 5 to 10 points.44 |
EORTC QLQ-LC13 | The QLQ-LC13 is a tumour-specific questionnaire used to supplement the EORTC QLQ-C30 and contains 13 items related to lung cancer symptoms and treatment side effects including: a 3-item scale assessing dyspnea and 9 single items: pain in chest, pain in arm or shoulder, pain in other parts, coughing, hemoptysis, sore mouth or tongue, dysphagia, peripheral neuropathy, and alopecia.22 Higher scores on the symptom scales indicate worse symptoms.22 | Validity: Good ability to differentiate between subgroups of patients supporting construct validity: significantly higher scores for all pain items among patients with metastatic vs. local disease before treatment (P < 0.01). Changes in symptom measures over time were significantly associated with either chemotherapy (tingling in arms and legs and hair loss) or radiotherapy (difficulty swallowing) (P < 0.001).45 Reliability: Good internal consistency reliability for the dyspnea multi-item scale (Alpha = 0.81).45 Responsiveness: No relevant studies identified. | No relevant studies identified. |
HADS = hospital anxiety and depression scale; HRQoL = health-related quality of life; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; GHS = global health status; LC13 = Lung Cancer Supplement; MID = minimal important difference; SSQ = subjective significance questionnaire; vs. = versus.
A search of the literature was conducted to examine the psychometric properties of the instruments among patients with lung cancer.
European Organisation for Research and Treatment of Cancer, 30 Item Core Quality of Life Questionnaire
Description and Scoring
The EORTC QLQ-C30 is 1 of the most used patient-reported outcome measures in oncology clinical trials.22 It is a multidimensional, cancer-specific, self-administered, measure of HRQoL. This questionnaire is intended to be complemented by tumour-specific questionnaire modules or supplements, such as the EORTC QLQ-LC13 for patients with lung cancer.45 Version 3.0 of the instrument, as used in the EMPOWER-Lung 1 trial, is the current standard version of the instrument and differs from version 2.0 as it now has a 4-point scale (“not at all,” “a little,” “quite a bit” and “very much”) for the first 5 items, an increase from a 2-point scale (“yes” and “no”).22 The EORTC QLQ-C30 uses a 1-week recall period in assessing function and symptoms and consists of 5 functional scales (physical, role, emotional, cognitive, and social functioning), 3 symptom scales (fatigue, nausea/vomiting, and pain), and 6 single items (dyspnea, insomnia, appetite loss, constipation, diarrhea, and financial difficulties). These items are rated on a scale from 1 (not at all) to 4 (very much). This instrument also includes a GHS section with questions rated on a 1 to 7 scale (very poor to excellent, respectively).22
Raw scores for each scale are computed as the average of the items that contribute to a particular scale.22 This scaling approach is based upon the assumption that it is appropriate to provide equal weighting to each item that comprises a scale. There is also an assumption that, for each item, the interval between response options is equal (for example, the difference in score between “not at all” and “a little” is the same as “a little” and “quite a bit”). Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, more severe symptoms on the symptom scales, and better HRQoL on the global quality of life scale. Thus, a decline in scores on symptom scales would reflect an improvement, whereas an increase in scores on function and quality of life scales would reflect an improvement.
||| ||||||| |||||| ||||| |||||||| |||||||| ||||||| ||||| ||||||| |||||| || ||||||||| |||||| |||||||| || ||| ||||| ||||||| ||||||||| |||||| |||||||||| This manual contains pre-treatment reference data from more than 23,000 patients obtained from EORTC studies, clinical trials, and other epidemiological studies.47 Data are presented for all cancer patients and specific disease sites (e.g., lung cancer) as well as for gender, age, and cancer stage subgroups for certain disease sites.46
Psychometric Properties
Validity
Teckle et al. (2011)39 examined the validity of the English version of the instrument among 184 patients with breast (36%), lung (33%), or colorectal (31%) cancer in Canada. Convergent validity was assessed by comparing the instrument to the Functional Assessment of Cancer Therapy General (FACT-G). The FACT-G physical well-being scale was highly correlated with role function (r = 0.64) and the FACT-G emotional well-being scale was highly correlated with the EORTC QLQ-C30 emotional functioning scale (r = 0.76). The social domains of EORTC QLQ-C30 and FACT-G were poorly correlated (r = 0.13) potentially because each scale measures different aspects of social problems faced by cancer patients, with the EORTC QLQ-C30 emphasizing social problems caused by physical hardships, indicating that the 2 scales are not interchangeable. Teckle et al. assessed the discriminant validity of the instrument by examining its ability to differentiate between disease severities. Cohen’s D was used to categorize effect sizes as small (d = 0.2 to 0.5), medium (d = 0.5 to 0.8), or large (d > 0.8). The instrument was able to differentiate between: cancer stages 1 to 2 versus cancer stages 3 to 4 with a small effect size (d = 0.49); patients with ECOG PS 0 versus ECOG PS 1 to 3 with a medium effect size (d = 0.65); and between those with self-reported health status excellent to good versus self-reported health status fair to very poor with a large effect size (d = 1.36).
Recent studies have assessed the validity of different language versions of the instrument among patients with lung cancer. A study by Ozturk et al. (2009)41 examined a Turkish version of the instrument among 28 patients with NSCLC. The study used a known-groups approach to examine correlations between the instrument and the Karnofsky performance status (KPS) scale. Those with higher KPS scores (90 to 100) had better physical functioning (r = 0.41) and an inverse relationship was found between lower KPS scores (70 to 80) and constipation (r = 0.44), indicating the ability to differentiate between illness severities.
Reliability
Teckle et al. reported good internal consistency reliability for the GHS scale and all functioning scales ranging from 0.76 (role functioning) to 0.82 (cognitive functioning).39 In the study by Ozturk et al., the Turkish version of the EORTC QLQ-C30 showed good internal consistency reliability, with Cronbach’s alpha values ranging from 0.56 (social functioning) to 0.93 (role functioning), with 7 of the 9 multi-item scales having an alpha value of greater than 0.70.41
Responsiveness
No studies evaluating the responsiveness of the instrument in lung cancer patients were identified.
Minimal Important Difference
In the EMPOWER-Lung 1 study, a clinically meaningful improvement in the ||| ||||| of the EORTC QLQ-C30 was defined as an increase of 10 points or more, but no justification was provided.9 One study conducted in breast cancer (n = 246) and small cell lung cancer patients (n = 111) in 1998 used an anchor-based approach to estimate the MID. Patients who reported “a little” change (for better or worse) on the subjective significance questionnaire had corresponding changes on a function or symptom scale of the EORTC QLQ-C30 of approximately 5 to 10 points.44
Maringwa et al. (2011)48 used the WHO/ECOG PS and weight change as anchors to estimate the MID in each scale using a pooled dataset of 812 NSCLC patients undergoing treatment. The ECOG PS consists of values from 0 (no cancer symptoms) to 4 (bedbound). MID estimates were based on a 1-category change in ECOG PS and a 5% to less than 20% change in weight. The respective MID for improvement for each scale using the ECOG PS and weight change as anchors were: 9 and 5 for physical functioning; 14 and 7 for role functioning; 5 and 7 for social functioning; 9 and 4 for GHS; 14 and 5 for fatigue; and 16 and 2 for pain. MID estimates for deterioration for the ECOG PS and weight change anchors were: 4 and 6 for physical functioning; 5 and 5 for role functioning; 7 and 9 for social functioning; 4 and 4 for GHS; 6 and 11 for fatigue; and 3 and 7 for pain, respectively.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire, 13-Item Lung Cancer Module
Description and Scoring
The EORTC QLQ-LC13 is a tumour-specific questionnaire developed in 1994 and is used to supplement the EORTC QLQ-C30. The instrument contains 13 items about lung cancer-related symptoms and treatment side effects including: a 3-item scale assessing dyspnea (difficulty breathing) and 9 single items: pain in chest, pain in arm or shoulder, pain in other parts, coughing, coughing up blood, sore mouth or tongue, trouble swallowing, tingling in hands and feet, and hair loss.22 All items are on a 4-point scale from 1 (“not at all”) to 4 (“very much”). As in the EORTC QLQ-C30, higher scores on functioning scales indicate better functioning while higher scores on the symptom scales indicate worse symptoms. The instrument is intended for use among patients receiving chemotherapy and/or radiotherapy and therefore may not capture all the symptoms and side effects of the immunotherapy under review in the EMPOWER-Lung 1 trial. The EORTC has since updated this instrument with the EORTC QLQ-LC29, which contains 12 of the 13 original items and is supplemented with an additional 17 items to account for the effects older and newer cancer therapies including surgery and targeted therapy, although this instrument was not used in the EMPOWER-Lung 1 trial.49
Psychometric Properties
Validity
Bergman et al. (1994)45 found the EORTC QLQ-LC13 to be a valid instrument for measuring treatment-specific symptoms in patients with non-respectable lung cancer receiving chemotherapy or radiotherapy, particularly when combined with the QLQ-C30 questionnaire.45,50 The study assessed 2 field studies across 17 countries among 883 and 735 lung cancer patients in each study. Construct validity of the instrument was assessed using a known-groups approach which demonstrated a good ability to discriminate between subgroups of patients with lung cancer.45 When examining combined data from both studies, patients with metastatic disease had significantly higher levels of pain before treatment than those with local disease for all 3 pain items (P < 0.01). However, disease stage was not significantly related to self-reported coughing, hemoptysis, or dyspnea. Except for a sore mouth, the changes in toxicity measures over time were significantly associated with either chemotherapy (tingling in arms and legs and hair loss) or radiotherapy (difficulty swallowing). Significantly higher pre-treatment pain and dyspnea scores were noted for those with poorer ECOG PS (2 to 4) than those with a better ECOG PS (0 to 1) (P < 0.001). Similar results were found in a study by Brabo et al. (2006),51 which examined the Brazilian Portuguese version of the instrument among 60 lung cancer patients. Statistically significant differences were noted for shoulder/arm pain (P < 0.024) and dyspnea (P < 0.004) when comparing pre-treatment scores between patients with an ECOG PS of 0 to 1 versus those with an ECOG PS of 2 to 4.
Reliability
Bergman et al. found that the 3-item dyspnea scale had an alpha coefficient of 0.81, illustrating good internal consistency reliability.45 Both Bergman et al. and Brabo et al. found that the 3 pain items in the LC13 did not produce a scale with acceptable reliability estimates for group comparisons (Alphaalpha = 0.54 and 0.44).45,51 However, in both studies, the 3 pain items performed reliably when combined with the 2 pain items of the QLQ-C30 (alpha = 0.80 and 0.73), although this was not done in the EMPOWER-Lung 1 trial.
Responsiveness
No studies evaluating the responsiveness of the instrument in lung cancer patients were identified.
Minimal Important Difference
No studies evaluating the MID of the instrument in lung cancer patients were identified.
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Figure 8: Kaplan-Meier Curve of Overall Survival in the EMPOWER-Lung 1 Trial (PD-L1 of 50% or More Population)
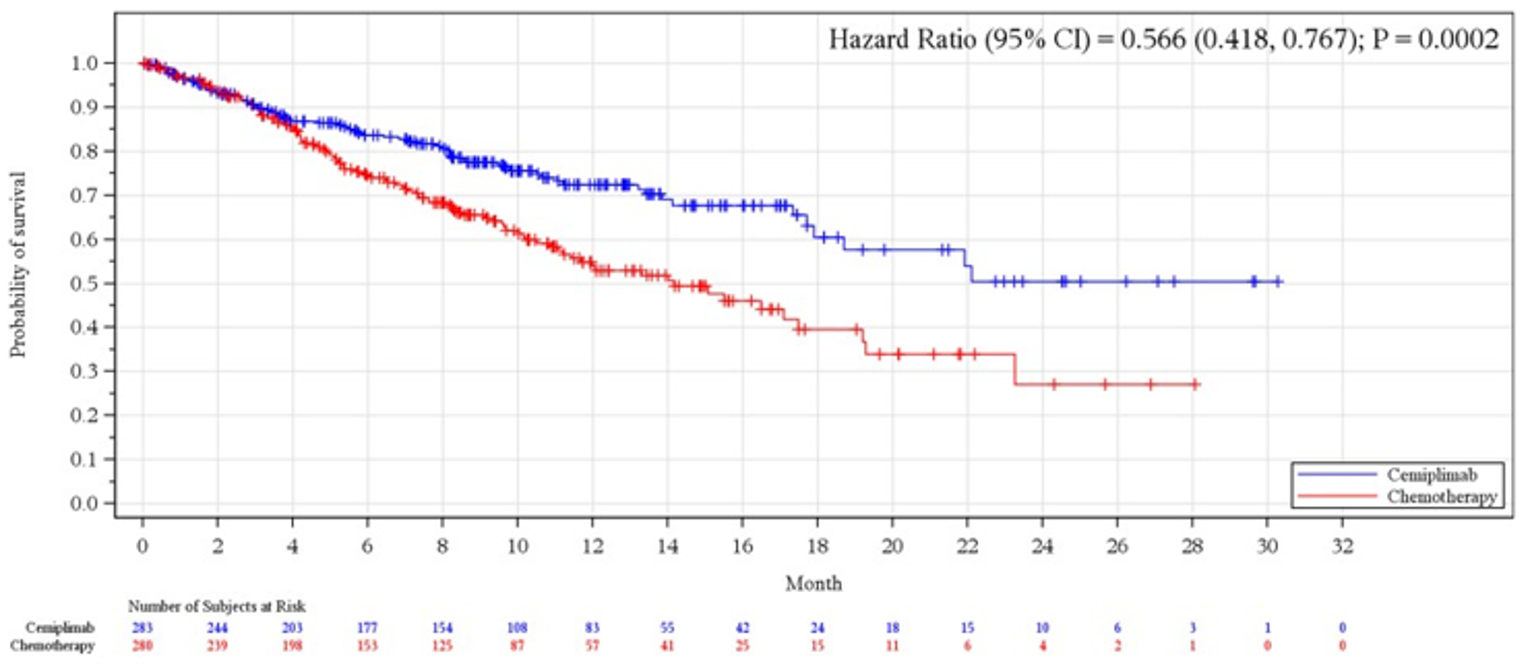
CI = confidence interval; PD-L1 = programmed cell death-ligand 1.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Table 36: Overall Survival by Subgroup in the EMPOWER-Lung 1 Trial (PD-L1 of 50% or More Population)
Factors | Cemiplimab (N = 283) | Chemotherapy (N = 280) | HR (95% CI)a | ||
|---|---|---|---|---|---|
Event (%) | Median time (95% CI)b | Event (%) | Median time (95% CI)b | ||
ECOG PS | |||||
0 | 18/77 (23.4) | 17.9 (17.3 to NE) | 23/75 (30.7) | 16.5 (11.4 to NE) | 0.766 (0.409 to 1.436) |
1 | 52/206 (25.2) | NR (18.7 to NE) | 82/205 (40.0) | 13.3 (10.2 to 19.2) | 0.536 (0.378 to 0.762) |
Histologyc | |||||
Squamous | 30/122 (24.6) | 21.9 (17.3 to NE) | 48/121 (39.7) | 15.5 (10.9 to 19.3) | 0.483 (0.304 to 0.767)d |
Non-squamous | 40/161 (24.8) | NR (17.9 to NE) | 57/159 (35.8) | 14.2 (9.7 to NE) | 0.639 (0.426 to 0.958)d |
Brain metastasis at baseline | |||||
Yes | 4/34 (11.8) | 18.7 (17.3 to NE) | 12/34 (35.3) | 11.7 (7.0 to NE) | 0.168 (0.037 to 0.762) |
No | 66/249 (26.5) | NR (17.9 to NE) | 93/246 (37.8) | 14.2 (11.1 to 17.5) | 0.603 (0.439 to 0.829) |
Cancer stage at screening | |||||
Locally advanced | 9/45 (20.0) | NR (17.7 to NE) | 15/42 (35.7) | 15.5 (9.6 to NE) | 0.479 (0.202 to 1.135) |
Metastatic | 61/238 (25.6) | 22.1 (18.7 to NE) | 90/238 (37.8) | 14.2 (10.9 to 17.5) | 0.589 (0.425 to 0.816) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; NE = not estimable; NR = not reached; PD-L1 = programmed cell death-ligand 1.
aBased on stratified proportional hazards model (cemiplimab versus chemotherapy).
bBased on the Kaplan-Meier method.
cAccording to clinical database at baseline.
dBased on unstratified proportional hazards model (cemiplimab versus chemotherapy).
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Figure 9: Kaplan-Meier Curve of Progression-Free Survival in the EMPOWER-Lung 1 Trial (PD-L1 of 50% or More Population)
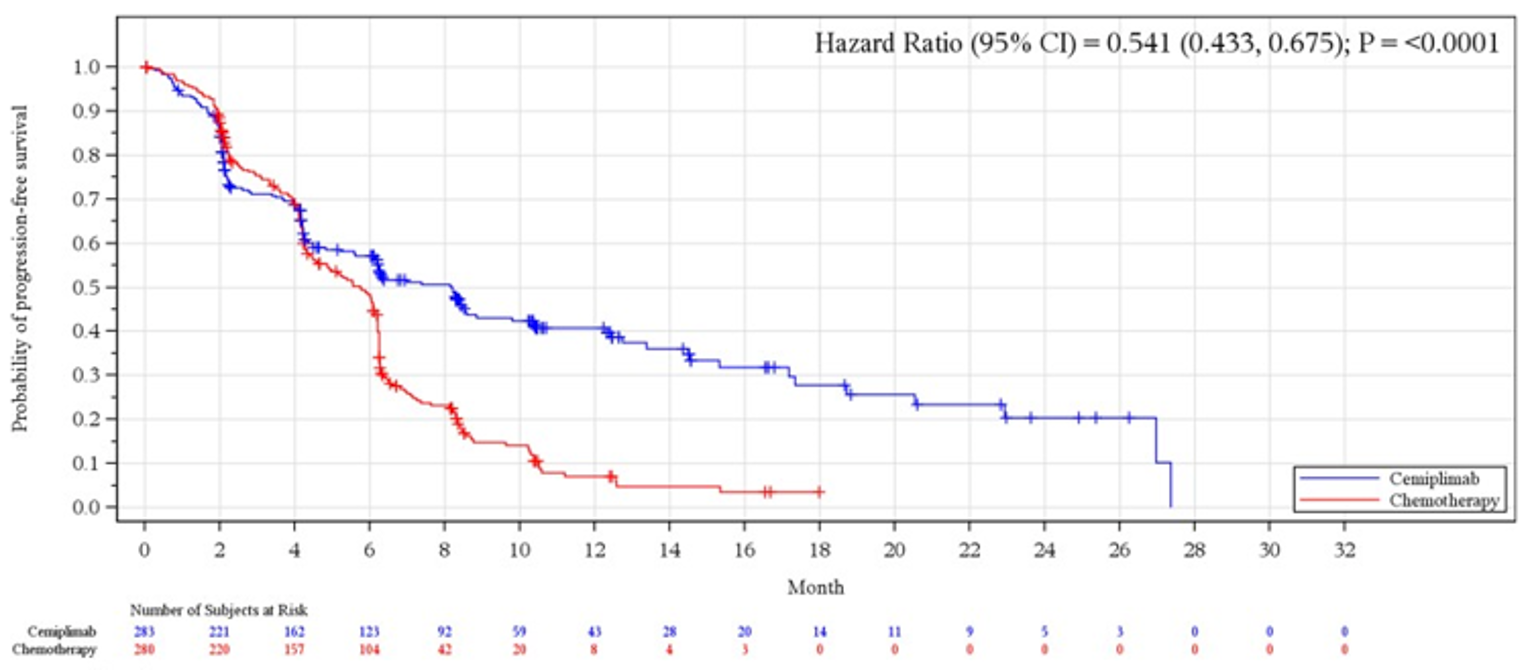
CI = confidence interval; PD-L1 = programmed cell death-ligand 1.
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Table 37: Progression-Free Survival by Subgroup in the EMPOWER-Lung 1 Trial (PD-L1 of 50% or More Population)
Factors | Cemiplimab (N = 283) | Chemotherapy (N = 280) | HR (95% CI)a | ||
|---|---|---|---|---|---|
Event (%) | Median time (95% CI)b | Event (%) | Median time (95% CI)b | ||
ECOG PS | |||||
0 | 39/77 (50.6) | 8.2 (4.3 to 12.7) | 46/75 (61.3) | 6.1 (4.4 to 6.8) | 0.591 (0.378 to 0.924) |
1 | 108/206 (52.4) | 7.4 (5.2 to 10.4) | 151/205 (73.7) | 5.6 (4.3 to 6.2) | 0.524 (0.405 to 0.679) |
Histologyc | |||||
Squamous | 67/122 (54.9) | 8.3 (4.8 to 10.4) | 90/121 (74.4) | 6.0 (4.5 to 6.2) | 0.475 (0.339 to 0.665)d |
Non-squamous | 80/161 (49.7) | 8.1 (4.3 to 12.7) | 107/159 (67.3) | 5.4 (4.2 to 6.2) | 0.598 (0.444 to 0.805)d |
Brain metastasis at baseline | |||||
Yes | 13/34 (38.2) | 10.4 (4.2 to NE) | 26/34 (76.5) | 5.3 (2.2 to 6.5) | 0.451 (0.222 to 0.916) |
No | 134/249 (53.8) | 7.4 (5.6 to 8.6) | 171/246 (69.5) | 5.8 (4.5 to 6.2) | 0.561 (0.443 to 0.710) |
Cancer stage at screening | |||||
Locally advanced | 27/45 (60.0) | 8.4 (4.5 to 15.3) | 28/42 (66.7) | 6.2 (4.6 to 6.6) | 0.489 (0.272 to 0.879) |
Metastatic | 120/238 (50.4) | 7.0 (5.2 to 10.4) | 169/238 (71.0) | 5.4 (4.3 to 6.1) | 0.554 (0.435 to 0.705) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; PD-L1 = programmed cell death-ligand 1.
aBased on stratified proportional hazards model (cemiplimab versus chemotherapy).
bBased on the Kaplan-Meier method.
cAccording to clinical database at baseline.
dBased on unstratified proportional hazards model (cemiplimab versus chemotherapy).
Source: Clinical Study Report for the EMPOWER-Lung 1 study (2017).9
Table 38: Hazard Ratios Estimated From Fractional Polynomial Network Meta-Analysis Overall Survival (Sensitivity Analysis)
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Table 39: Hazard Ratios Estimated From Fractional Polynomial Network Meta-Analysis Progression-Free Survival (Sensitivity Analyses)
|||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
Note: This table has been redacted at the sponsor’s request.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
HR
hazard ratio
NMA
network meta-analysis
NSCLC
non–small cell lung cancer
OS
overall survival
PFS
progression-free survival
PSM
partitioned survival model
QALY
quality-adjusted life-year
TTD
time to treatment discontinuation
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Cemiplimab (Libtayo), sterile solution for IV infusion, 50 mg/mL |
Submitted price | $8,200 per 350 mg vial |
Indication | First-line treatment of adult patients with NSCLC expressing PD-L1 (TPS ≥ 50%), as determined by a validated test, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC who are not candidates for surgical resection or definitive chemoradiation, or metastatic NSCLC |
Health Canada approval status | NOC |
Health Canada review pathway | ACCESS (Australia-Canada-Singapore-Switzerland) work-sharing initiative |
NOC date | October 26, 2021 |
Reimbursement request | As per indication |
Sponsor | Sanofi Genzyme |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; NSCLC = non–small cell lung cancer; TPS = Tumor Proportion Score.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis PSM |
Target populations | Adult patients with NSCLC expressing PD-L1 (TPS ≥ 50%), as determined by a validated test, with no EGFR, ALK, or ROS1 aberrations, who have:
|
Treatment | Cemiplimab |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (30 years) |
Key data sources | Phase III R2810-ONC-1624 (EMPOWER-1) trial and sponsor’s conducted NMA (KEYNOTE-024 trial) |
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
ICER = incremental cost-effectiveness ratio; LY = life-year; NMA = network meta-analysis; NSCLC = non–small cell lung cancer; OS = overall survival; PFS = progression-free survival; PSM = partitioned survival model; QALY = quality-adjusted life-year; TPS = Tumor Proportion Score; vs. = versus; WTP = willingness-to-pay.
Conclusions
The sponsor derived efficacy outcomes for cemiplimab and chemotherapy from the sponsor’s pivotal EMPOWER-1 trial. Given the lack of direct head-to-head studies for cemiplimab compared to pembrolizumab, the sponsor conducted a network meta-analysis (NMA) to derive hazard ratios (HRs) for a relative treatment benefit for pembrolizumab and cemiplimab compared to chemotherapy. However, there was uncertainty in the results from the NMA due to sparse networks and heterogeneity in baseline characteristics across included trials, and thus the comparative clinical efficacy between cemiplimab and pembrolizumab in terms of OS and PFS is uncertain.
CADTH identified several limitations in the economic analyses submitted by the sponsor. The predicted PFS and OS for cemiplimab and pembrolizumab in the sponsor’s submission lacked clinical validity. Clinical experts consulted by CADTH also indicated that chemotherapy was an inappropriate comparator for cemiplimab because patients typically only receive chemotherapy if they are ineligible for immunotherapy in the first-line setting. Clinical experts also noted that the utility values used for pre-progression and post-progression health states did not align with clinical expectations and that the treatment dosage for pembrolizumab and subsequent treatment regimens did not reflect standard of care in Canada. Clinical experts indicated that despite the uncertain indirect comparative evidence, it may be expected that cemiplimab and pembrolizumab have similar clinical efficacy and safety profiles.
CADTH performed reanalysis by excluding chemotherapy as a comparator; assuming equal OS, PFS, and AE rates between cemiplimab and pembrolizumab; and using an alternative model to extrapolate PFS. In addition, CADTH applied weight-based dosing with vial sharing for pembrolizumab, revised the subsequent treatments, and used alternate utility values based on clinical feedback. Results from the CADTH base case showed that cemiplimab resulted in higher costs by $125,981 per patient ($266,281 versus $140,300) and equal quality-adjusted life-years (QALYs) compared to pembrolizumab; a minimum price reduction of 61% is required for cemiplimab to provide cost savings when compared with pembrolizumab. Results from a scenario analysis indicated that when a 2-year treatment stopping rule was applied to cemiplimab, aligning with the EMPOWER-1 trial and pembrolizumab funding criteria, cemiplimab costs decreased but were still higher than that of pembrolizumab by $27,090 ($167,390 versus $140,300). In this scenario, a minimum price reduction of 25% is required for cemiplimab to provide cost savings when compared with pembrolizumab.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission).
Patient input was received from Lung Cancer Canada and the Lung Health Foundation (previously known as the Ontario Lung Association), collected via an online survey and telephone interviews across Canada. Patients and caregivers described how living with lung cancer affects their quality of life, mental health, ability to work, social lives, and daily routines. Symptoms that affected quality of life included shortness of breath, fatigue, depression, cough, compromised immune system, and chest tightness. Some respondents reported weight loss, diminished appetite, and challenges with physical and emotional intimacy. Both patients and caregivers reported experiencing severe physiologic burdens and emotional exhaustion from coping with the poor prognosis of advanced disease. Patients also reported anxiety that was further heightened by delayed accessibility to tests and treatments due to systemic and geographical barriers, especially during the COVID-19 pandemic. Current treatments included surgery, radiation, chemotherapy, targeted therapy, and immunotherapy. The benefits experienced with current treatments included prolonged life, delayed disease progression, and reduced severity of symptoms. However, these treatments also had severe side effects including, but not limited to, fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy, which impacted quality of life, the ability to work, and daily routine. Generally, patients showed a preference for earlier biomarker testing and the availability of multiple treatment options, and a willingness to accept some side effects of treatment. Patients expressed a desire for treatments that reduced or stopped disease progression with minimal side effects, improved quality of life for both the patients and caregivers, were effective in advanced cases, and easier to administer (i.e., a fixed dose regimen and reduced travel time).
Clinician input was received from the Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer Drug Advisory Committee. Current treatments were described as pembrolizumab monotherapy (most common), pembrolizumab in combination with platinum-based chemotherapy, nivolumab-ipilimumab plus 2 cycles of platinum-based chemotherapy, and atezolizumab monotherapy (not approved). Clinician input noted that the goal of treatment is to improve overall survival (OS) and progression-free survival (PFS). More treatments that meet these goals are needed. Clinicians noted that patients with advanced and metastatic non–small cell lung cancer (NSCLC) with PD-L1 expression of 50% or more are more likely to benefit from cemiplimab and PD-L1 testing is common for newly diagnosed NSCLC patients. Cemiplimab, if approved, should be administered at cancer centres by a medical oncologist trained in the delivery of immunotherapy. In clinical practice, patient response is measured by response rates (assessments by repeat imaging) and patient-reported improvement in symptoms every 3 months on therapy. Clinicians noted that patients would discontinue therapy in the case of disease progression or intolerable side effects. Further, clinician input noted that patients benefiting from therapy should be allowed to continue treatment beyond progression defined by Response Evaluation Criteria in Solid Tumors (RECIST) criteria.
Drug plan input received for this review noted that platinum-based chemotherapy is not a relevant comparator and nivolumab-ipilimumab in combination with chemotherapy was not publicly funded at the time the review was conducted. Feedback also noted to consider aligning reimbursement and prescribing criteria with that of pembrolizumab and nivolumab-ipilimumab in combination with chemotherapy.
Several of these concerns were addressed in the sponsor’s model:
The sponsor considered pembrolizumab monotherapy as a comparator.
OS, PFS, and quality of life (including disutilities due to adverse events [AEs]) were incorporated into the model.
In addition, CADTH addressed some of these concerns as follows:
As suggested by drug plans, CADTH excluded chemotherapy as a comparator for the revised base case in the pharmacoeconomic report.
CADTH performed a scenario analysis by applying a 2-year treatment stopping rule for cemiplimab to align with the funding criteria for pembrolizumab.
Economic Review
The current review is for cemiplimab (Libtayo) for the first-line treatment of patients with NSCLC and 50% or more PD-L1 expression, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and who are not candidates for surgical resection or definitive chemoradiation, or who have metastatic NSCLC.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis evaluating the cost-effectiveness of cemiplimab versus pembrolizumab monotherapy and chemotherapy for patients with NSCLC expressing PD-L1 (in ≥ 50% of tumour cells), with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and who are not candidates for surgical resection or definitive chemoradiation, or who have metastatic NSCLC. The target population aligns with the Health Canada–indicated population and reimbursement request.1
Cemiplimab 350 mg is administered intravenously every 3 weeks, until progression. At the submitted price of $8,200 per 350 mg dose, the monthly cycle cost is $11,955.56. The sponsor-modelled per-cycle costs for the comparator treatments were $12,824.61 for pembrolizumab monotherapy and $5,732.02 for chemotherapy. A 2-year stopping rule was applied for pembrolizumab, aligning with the funding criteria.1
The analysis was performed from the perspective of Canada’s publicly funded health care system. Costs and QALYs for each treatment regimen were simulated over a time horizon of 30 years. Future costs and QALYs were discounted at an annual rate of 1.5% per annum.
Model Structure
The sponsor submitted a partitioned survival model (PSM) consisting of 3 health states: 1) pre-progression, 2) post-progression, and 3) death (Figure 1). Patients begin in the pre-progression health state and transition either directly to the death state, or to the post-progression health state before moving to the death state. The proportion of patients in the pre-progression state was determined using treatment-specific PFS curves, and the proportion of patients in the death state was modelled based on treatment-specific OS curves. The proportion of patients in the post-progression health state was estimated as the difference between the proportion of patients alive and the proportion of patients in the pre-progression health state.1
Model Inputs
The baseline characteristics of the modelled patient cohort were based on the patient population in the EMPOWER-1 trial for cemiplimab versus chemotherapy, a phase III trial that focused on patients with NSCLC with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and who are not candidates for surgical resection or definitive chemoradiation, or have progressed after treatment with definitive chemoradiation, or metastatic NSCLC. The mean age of the patient cohort was 63 years. The sponsor’s base case was based on the true PD-L1 of 50% or more population (i.e., the modified intention-to-treat 1 population) of the EMPOWER-1 trial.
The submitted model considered chemotherapy (pemetrexed and cisplatin, gemcitabine and cisplatin, and paclitaxel and cisplatin) and pembrolizumab as comparators. The sponsor derived PFS and OS curves for chemotherapy and HRs for PFS and OS for cemiplimab compared to chemotherapy from the EMPOWER-1 trial. The comparative efficacy of pembrolizumab compared to chemotherapy was obtained from the KEYNOTE-024 trial. An NMA was performed to derive relative treatment effects for cemiplimab and pembrolizumab. Outcomes for all treatments were adjusted for (direct and indirect) crossover using the 2-stage correction method. PFS and OS outcomes for chemotherapy were extrapolated beyond the trial duration by fitting parametric survival models to the trial data. Model selection was based on statistical fit (Akaike information criterion [AIC], Bayesian information criterion [BIC, deviance information criterion [DIC]), clinical plausibility, and external validation against real-world data. Time-varying HRs for relative treatment effect for cemiplimab and pembrolizumab were modelled using second-order fractional polynomial models. The sponsor assumed that that HRs for relative treatment effect for cemiplimab and pembrolizumab were constant beyond 36 months. The extrapolated survival outcomes were corrected for general population mortality. The sponsor used an exponential distribution to extrapolate OS and a Weibull distribution to model PFS for the chemotherapy in combination with the best-fitting fractional polynomial model for the time-varying HRs. The sponsor used PFS curves to represent treatment duration for all treatments. A treatment stopping rule of 2 years was applied for patients receiving pembrolizumab.
The analysis included all grade 3 to grade 4 AEs that occurred in either grade in 5% or more of patients for cemiplimab and chemotherapy, and grade 3 to grade 4 AEs that occurred in either grade in 10% or more of patients for pembrolizumab. AE rates for cemiplimab and chemotherapy were based on the EMPOWER-1 trial, and those for pembrolizumab were obtained from the KEYNOTE-024 trial. AE costs were applied to the first cycle of the model for each treatment arm; AE unit costs were obtained from the Ontario Case Costing Initiative.2
Pre-progression and post-progression health utility for patients with advanced NSCLC and with PD-L1 of 50% or more was derived using data from the EMPOWER-1 trial. In the trial, data on health-related quality of life was measured using the EORTC QLQ-C30 instrument. The sponsor used a mapping algorithm by Noel et al. (2020) to map scores from EORTC QLQ-C30 to EQ-5D 5-Level utility values.3 The model also incorporated utility decrements for AEs, which were obtained from a study by Nafees et al. (2008).4
Costs were stratified in terms of pre-progression and post-progression costs. Costs in the pre-progression health state included drug costs (acquisition and administration costs), monitoring costs associated with routine care, and AE costs. Costs related to post-progression included monitoring costs associated with routine care and the cost of terminal care when patients transition to the death state.
The sponsor assumed a flat dose for cemiplimab and pembrolizumab, and as a result the model did not incorporate drug wastage. For cemiplimab, drug dosage and frequency of administration were based on the phase III EMPOWER-1 trial. The cost of drug administration was sourced from the Ontario Schedule of Benefits,5 where the cost of administration varied between interventions depending on the expected duration of administration. Resource use estimates for disease management for pre-progression and post-progression health states were obtained from a previous Health Technology Assessment submission for pembrolizumab, and unit costs were obtained from the Ontario Schedule of Benefits.6-9 The 1-time cost of subsequent treatment was applied to progressed patients in either treatment arm. The model assumed that patients who progressed on single immuno-oncology agents receive histology-specific chemotherapy, including carboplatin-pemetrexed for non-squamous patients and platinum-doublet chemotherapy for squamous patients. A 1-time cost of terminal care was applied for patients who transitioned to the death health state.10
Summary of Sponsor’s Economic Evaluation Results
The sponsor presented probabilistic base-case results based on 5,000 iterations (Table 3). The probabilistic findings are presented as follows. The submitted analyses were based on publicly available prices.
Base-Case Results
Results from the sponsor’s sequential analysis showed that pembrolizumab was extendedly dominated through chemotherapy and cemiplimab, and the incremental cost-effectiveness ratio for cemiplimab was $26,521 per QALY compared to chemotherapy (Table 3). Disaggregated results are reported in Table 10 in Appendix 3. The majority of QALYs for cemiplimab were accrued in the post-progression period and beyond the trial follow-up. The deterministic and probabilistic results were similar.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Chemotherapy | 138,952 | 1.23 | Reference |
Cemiplimab | 195,360 | 3.36 | 26,521 |
Pembrolizumab | 185,995 | 2.47 | Extendedly dominateda |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aThe ICER for pembrolizumab vs. chemotherapy is higher than the ICER for cemiplimab vs. chemotherapy.
Source: Sponsor’s Pharmacoeconomic Submission.1
Sensitivity and Scenario Analysis Results
The sponsor performed 1-way sensitivity analyses by varying parameters for the survival models used to extrapolate OS and PFS curves for all treatments, utility values, and disease management costs. Results from 1-way analysis showed that changes to the OS curves and PFS curves were key drivers of cost-effectiveness results.
The sponsor performed a series of scenario analyses by varying the following model parameters: crossover adjustment, the use of conservative or optimistic curve selections for OS and PFS, time horizon, and the application of a 2-year treatment cap for cemiplimab. The costs of cemiplimab were reduced by 14% when the 2-year stopping rule was applied. The sponsor’s results were robust to changes in methods used to adjust for crossover and time horizon.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
A high degree of uncertainty in the comparative efficacy of cemiplimab and pembrolizumab: Due to the lack of head-to-head clinical evidence for cemiplimab and pembrolizumab, the relative treatment benefit for cemiplimab and pembrolizumab was derived using an NMA. The evidence network for the NMA consisted of ||||||||||||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||. However, there exists uncertainty in the results from the NMA due to sparse networks and heterogeneity in baseline characteristics across included trials (tumour histology, proportion of non-smokers). As a result, the presence and magnitude of benefit for cemiplimab compared to pembrolizumab is highly uncertain. Clinical experts consulted by CADTH advised that cemiplimab and pembrolizumab are expected to have comparable OS and PFS, which should align with the results from the KEYNOTE-024 trial, as well as similar safety profiles.
CADTH performed reanalysis by assuming equal PFS, OS, and AE incidence for cemiplimab and pembrolizumab.
Sponsor included treatments not used in Canadian practice: The sponsor compared cemiplimab to pembrolizumab and chemotherapy. Clinical experts consulted by CADTH indicated that chemotherapy should not be considered as a relevant comparator for cemiplimab because patients are only expected to receive chemotherapy if they are ineligible for immunotherapy in the first-line setting.
CADTH performed reanalysis by excluding chemotherapy from the list of comparators.
A lack of clinical validity in survival outcomes: The predicted OS and PFS for pembrolizumab and cemiplimab in the sponsor’s model lacked clinical validity. Based on the sponsor’s extrapolation of OS, cemiplimab had an OS benefit relative to pembrolizumab even after the trial duration, despite having worse modelled mean PFS than pembrolizumab. Moreover, the 5-year PFS and OS for pembrolizumab predicted from the sponsor’s model were substantially lower than the 5-year results reported in the KEYNOTE-024 trial.
CADTH performed reanalysis using an alternative model to extrapolate PFS to better align with results reported in the KEYNOTE-024 trial.
Structural uncertainty due to the use of a PSM: The sponsor used a PSM to calculate long-term costs and outcomes for each treatment. Although the PSM approach is commonly used in health technology assessments of oncology treatments, the model does not explicitly model progression and subsequent treatments and could inaccurately reflect the long-term treatment effect of the modelled intervention. The impact of subsequent treatments on survival and health utility could not be captured in the sponsor’s analyses due to constraints of the modelling approach used.
CADTH was unable to assess the concerns identified in this limitation.
Estimation of pembrolizumab costs not reflective of current standard of care: The sponsor calculated the costs of pembrolizumab based on a flat dose assumption (200 mg every 3 weeks). However, public drug plan and clinical expert feedback suggested that weight-based dosing (2 mg/kg up to a cap of 200 mg every 3 weeks) with vial sharing is commonly implemented across jurisdictions. Thus, the costs of pembrolizumab are overestimated.
CADTH performed reanalysis by using weight-based dosing (2 mg/kg up to a cap of 200 mg every 3 weeks) with vial sharing to estimate drug costs for pembrolizumab.
Subsequent treatment regimen not reflective of current standard of care: The sponsor’s model assumed that patients who progressed on single immuno-oncology agents received histology-specific chemotherapy, including carboplatin-pemetrexed for non-squamous patients (|% of progressed patients) and platinum-doublet chemotherapy for squamous patients (|% of progressed patients). Clinical experts consulted by CADTH suggested that the subsequent treatment regimen modelled by the sponsor was not reflective of standard of care in Canada.
CADTH performed reanalysis using revised estimates for subsequent treatment regimens for second-line chemotherapy based on clinical expert feedback to reflect current standard of care in Canada.
A lack of face validity in health utility values: The sponsor applied a pre-progression and post-progression utility value of 0.86 and 0.83, respectively. These health utility values were derived by applying a mapping algorithm to EORTC QLQ-C30 data collected in the EMPOWER-1 trial. The mapping algorithm used to derive utility values was developed based on a cohort of patients with head and neck cancer; these utility values may not reflect patients with NSCLC. Moreover, the health utility values for pre-progression derived by the sponsor were higher than the age-specific health utility norm for the general population in Canada (0.84). As well, the decrement in utility as a result of progression modelled by the sponsor did not align with clinical expectations. Clinical experts consulted by CADTH indicated that utility values for patients in the pre-progression health state are expected to be marginally lower than those for the general population and that cancer progression is expected to substantially reduce health-related quality of life.
CADTH performed reanalysis by using utility values derived from the KEYNOTE-024 trial, which were more closely aligned with clinical expectations.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Treatment duration for all treatments was based on their respective PFS curves. | Inappropriate. Clinical experts consulted by CADTH suggested that, if available, TTD data should be used to derive treatment duration for all treatment arms due to differences in the definition of PFS across the trials. CADTH explored the impact of deriving treatment duration for cemiplimab based on TTD curves as a scenario analysis. However, CADTH was unable to apply this assumption for pembrolizumab as TTD curves for pembrolizumab were not provided in the sponsor’s model. |
The derived time-varying HRs for relative treatment benefit for cemiplimab and pembrolizumab compared to chemotherapy improved over time. The sponsor applied a conservative assumption that the HRs for cemiplimab and pembrolizumab remained constant after 36 months. The sponsor’s model assumed that treatment effect for cemiplimab and pembrolizumab remained constant after trial duration; however, no waning in treatment effect was assumed. | Clinical experts consulted by CADTH indicated that treatment waning is expected in clinical practice; however, no long-term data exists on the extent of treatment waning. The impact of this assumption on the cost-effectiveness results was uncertain but was partially addressed in the CADTH reanalysis through alternate clinical efficacy assumptions. |
For the sponsor’s base case, the sponsor applied a treatment stopping rule of 24 months for pembrolizumab and until progression for cemiplimab, based on their respective trial protocols. | Appropriate. |
The sponsor’s model assumed that 100% of progressed patients received subsequent therapy. | Inappropriate. Clinical experts consulted by CADTH indicated that only 40% to 50% of patient who received the first-line treatment went on to receive subsequent therapy. The submitted model was not flexible to test alternate assumptions. CADTH was unable to address this limitation; however, it is not expected to have a large impact on results. |
HR = hazard ratio; PFS = progression-free survival; TTD = time to treatment discontinuation.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH reanalyses addressed several limitations within the economic model. The CADTH base case was derived by applying the following changes: excluding chemotherapy as a comparator; assuming equal OS, PFS, and AE rates for cemiplimab and pembrolizumab; using an alternate parametric survival model to extrapolate PFS; applying weight-based dosing with vial sharing for pembrolizumab; revising the subsequent treatments; and using alternate health-state utility values.
Table 5 details each change made to derive the CADTH revised base case, which was conducted in a stepwise approach to highlight the impact of each change. The summary of results from the reanalyses are presented in Table 6.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
CADTH reanalysis 1: Comparators |
| Pembrolizumab |
CADTH reanalysis 2: OS for cemiplimab and pembrolizumab | OS for cemiplimab and pembrolizumab was derived using an NMA and long-term extrapolations. Predicted 5-year OS:
| OS was assumed to be equal between cemiplimab and pembrolizumab, using the cemiplimab OS model that more closely aligned with the KEYNOTE-024 trial.a Predicted 5-year OS:
|
CADTH reanalysis 3: PFS for cemiplimab and pembrolizumab | PFS for cemiplimab and pembrolizumab was derived using an NMA and long-term extrapolations. Predicted 3-year PFS:
Predicted 5-year PFS:
| PFS was assumed to be equal between cemiplimab and pembrolizumab, using a more optimistic modelb to extrapolate PFS for cemiplimab that more closely aligned with the KEYNOTE-024 trial.c Predicted 3-year PFS:
Predicted 5-year PFS:
|
CADTH reanalysis 4: Treatment dosage for pembrolizumab | Flat dose of 200 mg, every 3 weeks | Weight-based dosage (2 mg/kg up to a maximum dose of 200 mg, every 3 weeks) with vial sharing |
CADTH reanalysis 5: Subsequent treatment regimen | For patients who receive chemotherapy as a subsequent treatment upon progression, subsequent treatments were assumed to be:
| Of those patients receiving chemotherapy in a second-line setting, distribution of subsequent treatments was revised to be:
For patients who receive cisplatin + gemcitabine or cisplatin + paclitaxel as a subsequent therapy, treatment dosage was revised to 6 cycles. |
CADTH reanalysis 6: Health utility value |
|
|
CADTH reanalysis 7: Adverse events |
| AE rates assumed to be equal between treatments |
CADTH base case | Combined revisions 1 + 2 + 3 + 4 + 5 + 6 + 7 | |
AD = adverse event; NMA = network meta-analysis; OS = overall survival; PFS = progression-free survival.
aFive-year OS reported in the KEYNOTE-024 trial was 31.90%.
bModel used to extrapolate PFS in CADTH base case: fractional polynomial model, with P1 being 0 and P2 being 0.
cFive-year PFS reported in the KEYNOTE-024 trial was 12.8%. It should be noted that the predicted PFS using the CADTH reanalysis was still slightly lower than estimations from KEYNOTE-024; however, the sponsor’s model was not flexible to assess more appropriate scenarios.
dSourced from the National Institute for Health and Care Excellence submission TA531 (2018).11
Results from the CADTH base case showed that cemiplimab resulted in higher costs ($266,281 versus $140,300) and equal QALYs (2.81 versus 2.81) compared to pembrolizumab. The probability of cemiplimab being cost-effective at a willingness-to-pay threshold of $50,000 was 0%. Disaggregated results from the CADTH base case are presented in Table 11. Results from the stepped analysis showed that the cost-effectiveness of cemiplimab was highly sensitive to assumptions regarding relative OS and PFS benefits for cemiplimab compared to pembrolizumab and the parametric survival model used to extrapolate PFS. Additionally, applying weight-based dosing with vial sharing for pembrolizumab increased the incremental cost-effectiveness ratio. Cost-effectiveness findings were not significantly impacted by changes to the AE rates, subsequent treatment regimen, and health utility values.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis (Deterministic)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | Chemotherapy | 138,980 | 1.23 | Reference |
Pembrolizumab | 187,207 | 2.42 | Extendedly dominated | |
Cemiplimab | 191,981 | 3.29 | 25,643 | |
CADTH reanalysis 1: Exclusion of chemotherapy as a comparator | Pembrolizumab | 187,207 | 2.42 | Reference |
Cemiplimab | 191,981 | 3.29 | 5,458 | |
CADTH reanalysis 2: Equal OS for cemiplimab and pembrolizumab | Pembrolizumab | 187,666 | 3.30 | Reference |
Cemiplimab | 191,981 | 3.29 | Dominated | |
CADTH reanalysis 3: Equal PFS for cemiplimab and pembrolizumab | Pembrolizumab | 171,986 | 2.43 | Reference |
Cemiplimab | 253,077 | 3.31 | 92,194 | |
CADTH reanalysis 4: Weight-based dosage for pembrolizumab | Pembrolizumab | 147,225 | 2.42 | Reference |
Cemiplimab | 191,981 | 3.29 | 51,165 | |
CADTH reanalysis 5: Changing the subsequent treatment distribution and dosage | Pembrolizumab | 191,647 | 2.42 | Reference |
Cemiplimab | 196,304 | 3.29 | 5,323 | |
CADTH reanalysis 6: Using alternative health state–specific health utility values | Pembrolizumab | 187,207 | 2.04 | Reference |
Cemiplimab | 191,981 | 2.74 | 6,871 | |
CADTH reanalysis 7: Assuming equal AE rates | Pembrolizumab | 186,833 | 2.42 | Reference |
Cemiplimab | 191,981 | 3.29 | 5,887 |
AE = adverse event; ICER = incremental cost-effectiveness ratio; OS = overall survival; PFS = progression-free survival; QALY = quality-adjusted life-year.
Table 7: Summary of the CADTH Reanalysis Results (Probabilistic)
Drug | Total costs | Total QALYs | ICER |
|---|---|---|---|
Pembrolizumab | 140,300 | 2.81 | Reference |
Cemiplimab | 266,281 | 2.81 | Cemiplimab more costly and equally effective |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
CADTH performed a series of scenario analyses to test the impact of the following model parameters and assumptions: applying a 2-year treatment stopping rule for cemiplimab to align with the EMPOWER-1 trial and funding criteria for pembrolizumab, applying time to treatment discontinuation (TTD) data to derive the treatment duration for cemiplimab, and incorporating AE rates for cemiplimab and pembrolizumab based on their respective trial results. Details regarding the scenario analyses performed have been presented in Appendix 4.
Results from the scenario analyses indicated that the cost-effectiveness results were sensitive to the treatment stopping rule assumptions, but robust to other changes in the model’s assumptions and input parameters. Applying a 2-year stopping rule to cemiplimab (aligning with pembrolizumab) lowered cemiplimab costs; however, cemiplimab still remained costlier compared to pembrolizumab ($167,390 versus $140,300). QALYs were not impacted (2.81 versus 2.81). Results from all scenario analyses are presented in Table 12 in Appendix 4.
Results from a price reduction analysis on the CADTH reanalysis showed that a minimum price reduction of 61% is required for cemiplimab to provide cost savings when compared with pembrolizumab. Additionally, a price reduction analysis was performed for the CADTH scenario analysis where a treatment stopping rule of 2 years was applied for cemiplimab to align with the EMPOWER-1 trial and funding criteria for pembrolizumab. In this scenario, a minimum price reduction of 25% was required for cemiplimab to provide cost savings when compared with pembrolizumab (Table 13 in Appendix 4).
Issues for Consideration
Treatment duration for cemiplimab: The sponsor and CADTH base case assumed that cemiplimab is treated to progression, aligning with the product monograph, which states that cemiplimab may be continued until symptomatic disease progression or unacceptable toxicity. In the EMPOWER-1 trial, cemiplimab was administered until progressive disease or 108 weeks (i.e., 36 treatments; approximately 2 years); this duration is approximately aligned with the funding criteria for pembrolizumab in Canada, which set the maximum of duration at 2 years (35 administrations). If the funding criteria for cemiplimab were to be aligned with pembrolizumab, cemiplimab costs would be expected to be lower, aligning with the relevant scenario analysis.
Comparator pricing based on publicly available prices: The modelled price of pembrolizumab is based on publicly accessible list prices and does not reflect existing confidential pricing that has been negotiated by public plans. When existing confidential discounts on pembrolizumab are considered, greater price reductions than those referenced in this report would be required to achieve cost-effectiveness.
Relevant comparators: Nivolumab-ipilimumab plus 2 cycles of platinum-doublet chemotherapy was not included as a comparator in the economic model, based on clinical expert feedback that this regimen is not commonly used and that cemiplimab is not likely to displace nivolumab-ipilimumab plus platinum-doublet chemotherapy. However, this assumption is subject to uncertainty, and clinical practice may differ once nivolumab-ipilimumab plus platinum-doublet chemotherapy is publicly funded. Based on publicly available list prices, pembrolizumab is the lowest cost relevant comparator for cemiplimab, and thus the inclusion of nivolumab-ipilimumab plus platinum-doublet chemotherapy in the economic analysis would not impact the results. However, once nivolumab-ipilimumab plus platinum-doublet chemotherapy is funded, drug plans may consider ensuring that cemiplimab costs no more than either pembrolizumab or nivolumab-ipilimumab plus platinum-doublet chemotherapy while considering confidential discounts.
Overall Conclusions
The sponsor derived efficacy outcomes for cemiplimab and chemotherapy from the EMPOWER-1 trial. Given the lack of direct head-to-head studies for cemiplimab compared to pembrolizumab, the sponsor conducted an NMA to derive HRs for a relative treatment benefit for pembrolizumab and cemiplimab compared to chemotherapy. However, there was uncertainty in the results from the NMA due to sparse networks and heterogeneity in baseline characteristics across included trials, and thus the comparative clinical efficacy between cemiplimab and pembrolizumab in terms of OS and PFS is uncertain.
CADTH identified several limitations in the economic analyses submitted by the sponsor. The predicted PFS and OS for cemiplimab and pembrolizumab in the sponsor’s submission lacked clinical validity. Clinical experts consulted by CADTH also indicated that chemotherapy was an inappropriate comparator for cemiplimab because patients typically only receive chemotherapy if they are ineligible for immunotherapy in the first-line setting. Clinical experts also noted that the utility values used for pre-progression and post-progression health states did not align with clinical expectations and that the treatment dosage for pembrolizumab and subsequent treatment regimens did not reflect standard of care in Canada. Clinical experts indicated that despite the uncertain indirect comparative evidence, it may be expected that cemiplimab and pembrolizumab have similar clinical efficacy and safety profiles.
CADTH performed reanalysis by excluding chemotherapy as a comparator; assuming equal OS, PFS, and AE rates between cemiplimab and pembrolizumab; and using an alternative model to extrapolate PFS. In addition, CADTH applied weight-based dosing with vial sharing for pembrolizumab, revised the subsequent treatments, and used alternate utility values based on clinical feedback. Results from the CADTH base case showed that cemiplimab resulted in higher costs by $125,981 per patient ($266,281 versus $140,300) and equal QALYs compared to pembrolizumab; a minimum price reduction of 61% is required for cemiplimab to provide cost savings when compared with pembrolizumab. Results from a scenario analysis indicated that when a 2-year treatment stopping rule was applied to cemiplimab, aligning with the EMPOWER-1 trial and pembrolizumab funding criteria, cemiplimab costs decreased but were still higher than those of pembrolizumab by $27,090 ($167,390 versus $140,300). In this scenario, a minimum price reduction of 25% is required for cemiplimab to provide cost savings when compared with pembrolizumab.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: LIBTAYO (cemiplimab for injection) 350 mg every three (3) weeks as an intravenous (IV) infusion [internal sponsor's package]. Mississauga (ON): Sanofi Genzyme; 2021 Oct 13.
2.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 Mar 21.
3.Noel CW SR, Su J, et al. . Mapping the EORTC QLQ-C30 and QLQ-H&N35, onto EQ-5D-5L and HUI-3 indices in patients with head and neck cancer. Head Neck Oncol. 2020. PubMed
4.Nafees B, Stafford M, Gavriel S, Bhalla S, Watkins J. Health state utilities for non small cell lung cancer. Health Qual Life Outcomes. 2008;6:84. PubMed
5.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf. Accessed 2022 Mar 21.
6.Maslove L, Gower N, Spiro S, Rudd R, Stephens R, West P. Estimation of the additional costs of chemotherapy for patients with advanced non-small cell lung cancer. Thorax. 2005;60(7):564-569. PubMed
7.National Institute for Health and Care Excellence. Advanced breast cancer: diagnosis and treatment. (Clinical guideline CG81) 2009; https://www.nice.org.uk/guidance/cg81. Accessed 2022 Mar 21.
8.Drug Reimbursement Review sponsor submission: LIBTAYO (cemiplimab for injection) 350 mg every three (3) weeks as an intravenous (IV) infusion [internal sponsor's package]. Mississauga (ON): Sanofi Genzyme; 2021 Oct 13.
9.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Mar 21.
10.Bremner KE, Krahn MD, Warren JL, et al. An international comparison of costs of end-of-life care for advanced lung cancer patients using health administrative data. Palliat Med. 2015;29(10):918-928. PubMed
11.National Institute for Health and Care Excellence. Pembrolizumab for untreated PD-L1-positive metastatic non-small-cell lung cancer. (Technology appraisal guidance TA531) 2018; https://www.nice.org.uk/guidance/ta531. Accessed 2022 Mar 21.
12.Libtayo (cemiplimab): concentrate for solution for infusion (50mg/mL). Antineoplastic agent, monoclonal antibody [product monograph]. Laval (QC): sanofi-aventis Canada Inc.; 2019 Apr 10.
13.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: LIBTAYO (cemiplimab for injection) 350 mg every three (3) weeks as an intravenous (IV) infusion [internal sponsor's package]. Mississauga (ON): Sanofi Genzyme; 2021 Oct 13.
14.CADTH reimbursement review: pembrolizumab (keytruda). Therapeutic area: classical Hodgkin lymphoma. (Canadian Journal of Health Technologies volume 1 issue 12)2021: https://www.cadth.ca/sites/default/files/DRR/2021/PC0236%20Keytruda-combined-report.pdf. Accessed 2022 Mar 21.
15.Keytruda (pembrolizumab): solution for infusion 100 mg/4 mL vial. Antineoplastic agent, monoclonal antibody [product monograph] Kirkland (QC): Merck Canada Inc.; 2021 Nov 24.
16.Canadian Partnership Against Cancer. Distribution of cases by stage at diagnosis for non-small cell lung cancer – 2013 diagnosis year. 2013.
17.Dietel M, Savelov N, Salanova R, et al. Real-world prevalence of programmed death ligand 1 expression in locally advanced or metastatic non-small-cell lung cancer: The global, multicenter EXPRESS study. Lung Cancer. 2019;134:174-179. PubMed
18.Uramoto H, Tanaka F. Recurrence after surgery in patients with NSCLC. Transl Lung Cancer Res. 2014;3(4):242-249. PubMed
19.Boyd JA, Hubbs JL, Kim DW, Hollis D, Marks LB, Kelsey CR. Timing of local and distant failure in resected lung cancer: implications for reported rates of local failure. J Thorac Oncol. 2010;5(2):211-214. PubMed
20.Canadian Cancer Society. Canadian cancer statistics: a 2020 special report on lung cancer. 2020; https://cdn.cancer.ca/-/media/files/cancer-information/resources/publications/2020-canadian-cancer-statistics-special-report/2020-canadian-cancer-statistics-special-report-en.pdf. Accessed 2022 Mar 22.
21.Statistics Canada. Table 17-10-0005-01 Population estimates on July 1st, by age and sex. 2021; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2022 Mar 22.
22.Brenner DR, Weir HK, Demers AA, et al. Projected estimates of cancer in Canada in 2020. CMAJ. 2020;192(9):E199-E205. PubMed
23.Sezer A, Kilickap S, Gümüş M, et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. The Lancet. 2021;397(10274):592-604. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Non–Small Cell Lung Cancer Expressing PD-L1 in 50% or More of Tumour Cells (Tumor Proportion Score of 50% or More)
Treatment | Concentration | Form | Price ($) | Recommended dosage | Average daily cost ($) | Average 21-day course ($) | Average 28-day cost ($) |
|---|---|---|---|---|---|---|---|
Cemiplimab (Libtayo) | 50 mg/mL | 5 mL 7 mL Vial IV infusion | 8,200.0000a | 350 mg every 3‑week cycle | 390.48 | 8,200 | 10,933 |
Immunotherapy | |||||||
Pembrolizumab (Keytruda) | 100 mg/4mL | 4 mL Vial IV infusion | 4,400.0000b | 2 mg/kg every 3 weeksc | 293.33 | 6,160 | 8,213 |
Note: Prices calculated by CADTH do not include dispensing fees. Cemiplimab is noted as a single-use product in product monograph.12 Dosing is based on Health Canada product monographs except for pembrolizumab, for which weight-based dosing and vial sharing was used. For treatments using weight-based dosing (i.e., pembrolizumab), CADTH assumed 70 kg or 1.8 m2.
aSponsor’s submitted price.13
bPrice obtained from CADTH’s review of pembrolizumab.14
cDosage is capped at 200 mg every 3 weeks.15
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/no | Commentsa |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CADTH appraisal section. |
Model has been adequately programmed and has sufficient face validity | No | Refer to CADTH appraisal section. |
Model structure is adequate for decision problem | No | Refer to CADTH appraisal section. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | Acceptable. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | Acceptable. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | Acceptable. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Results for Cemiplimab, Pembrolizumab, and Chemotherapy (Discounted, Probabilistic)
Outcome | Cemiplimab | Pembrolizumab | Chemotherapy |
|---|---|---|---|
Progression-free survival time (in months) | 11.84 | 13.77 | 5.40 |
Post-progression survival time (in months) | 40.28 | 23.27 | 12.66 |
Life-years | 4.34 | 3.09 | 1.51 |
Discounted total life-years | 4.03 | 2.94 | 1.47 |
Discounted QALYs; pre-progression | 0.83 | 0.97 | 0.39 |
Discounted QALYs; progressive disease | 2.53 | 1.50 | 0.85 |
Discounted QALYs lost due to adverse events | 0.00 | 0.00 | 0.00 |
Discounted total QALYs | 3.36 | 2.47 | 1.23 |
Discounted drug acquisition and admin cost; pre-progression | $144,500 | $135,269 | $26,518 |
Discounted drug acquisition and admin cost; progressive disease | $15,206 | $15,633 | $74,730 |
Discounted pre-progression routine care costs | $1,103 | $1,285 | $512 |
Discounted post-progression routine care costs | $4,230 | $2,518 | $1,417 |
Discounted adverse event costs | $465 | $839 | $4,620 |
Discounted total cost | $195,360 | $185,995 | $138,952 |
QALY = quality-adjusted life-year.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 11: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Cemiplimab | Pembrolizumab | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 3.95 | 3.95 | 0 |
Pre-progression | 1.37 | 1.37 | 0 |
Progressive disease | 2.58 | 2.58 | 0 |
Discounted QALYs | |||
Total | 2.81 | 2.81 | 0 |
Pre-progression | 1.10 | 1.10 | 0 |
Progressive disease | 1.72 | 1.72 | 0 |
Lost due to adverse events | 0.00 | 0.00 | 0 |
Costs ($) | |||
Total cost | 266,281 | 140,300 | 125,981 |
Drug acquisition and admin cost; pre-progression | 211,069 | 85,088 | 125,982 |
Disease management cost; pre-progression | 1,610 | 1,610 | 0 |
Drug acquisition and admin cost; progressive disease | 19,671 | 19,671 | 0 |
Disease management cost; progressive disease | 3,595 | 3,595 | 0 |
Adverse event costs | 464 | 464 | 0 |
Societal costs | 0 | 0 | 0 |
ICER ($/QALY) | Cemiplimab dominated | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
CADTH performed the following scenario analyses:
Applying a 2-year treatment stopping rule for cemiplimab: CADTH performed a scenario analysis by applying a treatment stopping rule of 2 years to cemiplimab to align with the EMPOWER-1 trial and funding criteria for pembrolizumab.
Using TTD data to derive treatment duration for cemiplimab: Clinical experts consulted by CADTH indicated that if available TTD data should be used to determine treatment duration for all treatments. CADTH performed a scenario analysis by using modelled TTD data from the EMPOWER-1 trial to derive treatment duration for cemiplimab. CADTH was unable to apply this assumption for pembrolizumab as TTD curves for pembrolizumab were not included in the sponsor’s model.
Applying AE rates for cemiplimab and pembrolizumab that were obtained from their respective trials: CADTH performed a scenario analysis by incorporating AE rates for cemiplimab and pembrolizumab obtained from their respective trials. For this scenario, CADTH applied the AE rates for cemiplimab and pembrolizumab that were modelled in the sponsor’s base case.
Table 12: CADTH Scenario Analysis
Scenario | Drug | Total costs | Total QALYs | ICER |
|---|---|---|---|---|
CADTH base case | Pembrolizumab | 140,300 | 2.81 | Reference |
Cemiplimab | 266,281 | 2.81 | Cemiplimab more costly and equally effective | |
Applying a 2-year treatment stopping rule for cemiplimab | Pembrolizumab | 140,300 | 2.81 | Reference |
Cemiplimab | 167,390 | 2.81 | Cemiplimab more costly and equally effective | |
Using TTD data to derive treatment duration for cemiplimab | Pembrolizumab | 140,300 | 2.81 | Reference |
Cemiplimab | 290,124 | 2.81 | Cemiplimab more costly and equally effective | |
Applying AE rates for cemiplimab and pembrolizumab reported in their respective trials | Pembrolizumab | 140,674 | 2.814 | Reference |
Cemiplimab | 266,281 | 2.815 | 694,658,090 |
AE = adverse event; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TTD = time to treatment discontinuation.
Table 13: CADTH Price Reduction Analyses
Price reduction | ICERs for cemiplimab vs. comparators | ||
|---|---|---|---|
Sponsor base case | CADTH reanalysis: cemiplimab vs. pembrolizumaba | CADTH scenario analysis – cemiplimab vs. pembrolizumab (2-year treatment stopping rule applied to cemiplimab)a | |
No price reduction | No price reduction was required as the ICER of cemiplimab was lower than $50,000/QALY at the submitted price. ICER for cemiplimab vs. chemotherapy = 26,521b | Cemiplimab has equal QALYs and is more costly than pembrolizumab | Cemiplimab has equal QALYs and is more costly than pembrolizumab |
10% | NA | ||
20% | NA | ||
30% | NA | Cemiplimab has equal QALYs and is less costly than pembrolizumabc | |
40% | NA | NA | |
50% | NA | NA | |
60% | NA | NA | |
70% | NA | Cemiplimab has equal QALYs and is less costly than pembrolizumabd | NA |
ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
aCemiplimab and pembrolizumab are assumed to produce the same number of QALYs; therefore, the intervention that is deemed cost-effective at any willingness-to-pay threshold is the 1 with lower costs.
bPembrolizumab was extendedly dominated through chemotherapy and cemiplimab.
cAt a 25% discount for cemiplimab, total costs for cemiplimab and pembrolizumab are equal.
dAt a 61% discount for cemiplimab, total costs for cemiplimab and pembrolizumab are equal.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
BIA = budget impact analysis; CUA = cost-utility analysis.
Summary of Sponsor’s Budget Impact Analysis
The sponsor’s submitted budget impact analysis (BIA)13 assessed the expected budgetary impact of reimbursing cemiplimab for the first-line treatment of adult patients with NSCLC expressing PD-L1 levels in 50% or more of tumour cells as determined by a validated test, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and who are not candidates for surgical resection or definitive chemoradiation, or metastatic NSCLC. The BIA was conducted from the public drug program perspective over a 3-year time horizon (2023 to 2025). Key inputs to the BIA are documented in Table 15.
The sponsor estimated population size using an epidemiology-based approach, with data obtained from published literature16-19 and Canadian Cancer Society statistics20 to estimate the number of new (incident) patients eligible for treatment with cemiplimab. Standard of care included pembrolizumab monotherapy and chemotherapies used in first-line treatment (cisplatin + pemetrexed; cisplatin + paclitaxel; and cisplatin + gemcitabine). Age-standardized incidence rates of lung cancer for each jurisdiction were based on published literature.21,22 It was assumed that 89% of lung cancer cases are NSCLC based on published data from 2012 to 2016 in Canada.20 The proportion of NSCLC cases classified as stage IIIB to stage IV was 53.2% and the remaining 46.8% were stage I to stage IIIA.16 The sponsor assumed 35.3% of NSCLC cases stage I to stage IIIA have distant recurrence of disease which were included in the estimated population.18,19 Furthermore, it was assumed that 55% of patients are negative for EGFR or ALK mutations and 25% of EGFR or ALK negative patients have expression of PD-L1 in 50% or more of tumour cells.17 The sponsor assumed 90% of eligible patients receive treatment. Subsequent therapy costs were not considered.
The sponsor assumed pembrolizumab takes up 99% of market share under standard care, and that cemiplimab only displaces market share from pembrolizumab (i.e., no change in market share of chemotherapies as under the reimbursement of cemiplimab). The sponsor adopted a treatment duration of 6.3 months for cemiplimab versus 7 months on pembrolizumab.13,23
The sponsor included only drug acquisition costs. The recommended dose of cemiplimab was 350 mg every 3 weeks. Pembrolizumab was dosed at 200 mg every 3 weeks. Dosing of chemotherapies was based on published literature assuming 100% dose intensity.23
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population
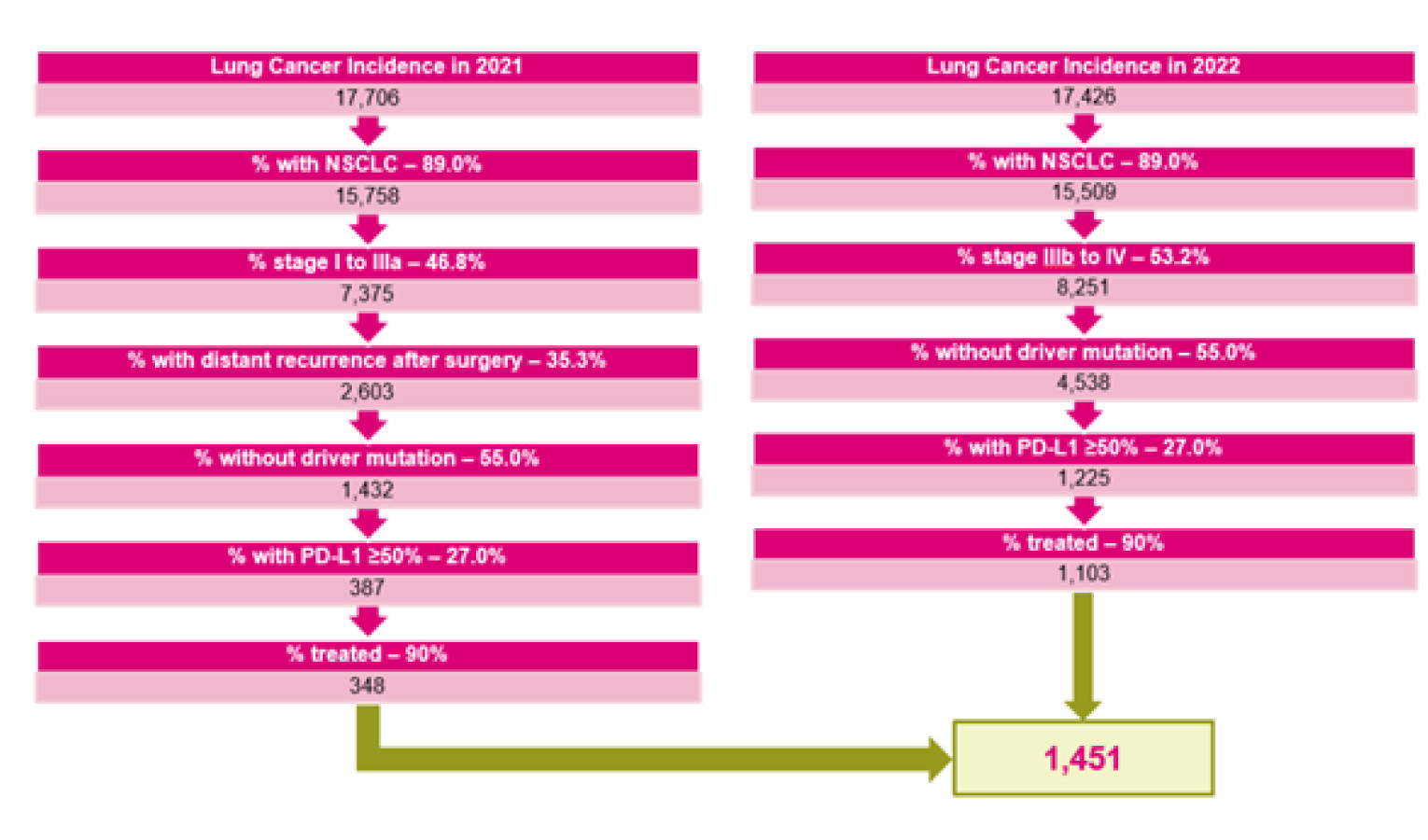
NSCLC = non–small cell lung cancer.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (year 1/year 2/year 3) |
|---|---|
Target population | |
Lung cancer incidence | 0.0560% / 0.0544% / 0.0528% |
Percentage with NSCLC | Refer to Figure 2 |
Percentage of stage IIIB to stage IV | |
Percentage of stage IIIB to stage IIIA | |
Percentage with distant recurrence | |
Percentage without driver mutation | |
Percentage with PD-L1 ≥ 50% | |
Percentage treated | |
Percentage plan-eligible | 100.0% |
Number of patients eligible for drug under review | 1,428/1,406/1,386 |
Market uptake (3 years) | |
Uptake (reference scenario) Pembrolizumab Chemotherapy | 99.0%/99.0%/99.0% 1.0%/1.0%/1.0% |
Uptake (new drug scenario) Cemiplimab Pembrolizumab Chemotherapy | 6.0%/14.5%/15.9% 93.0%/84.5%/83.1% 1.0%/1.0%/1.0% |
Cost of treatment (per patient) | |
Cost of treatment over course Cemiplimab Pembrolizumab Chemotherapy | $74,825 $89,222 $21,922a |
NSCLC = non–small cell lung cancer.
aAverage cost of cisplatin plus pemetrexed ($28,170), cisplatin plus paclitaxel ($25,830), and cisplatin plus gemcitabine ($11,765).
Summary of the Sponsor’s Budget Impact Analysis Results
The sponsor estimated the net 3-year budget impact of introducing cemiplimab for the first-line treatment of adult patients with NSCLC expressing PD-L1 levels in 50% or more of tumour cells, with no EGFR, ALK, or ROS1 aberrations, who have locally advanced NSCLC and who are not candidates for surgical resection or definitive chemoradiation, or metastatic NSCLC to be cost savings of $7,343,746 (year 1 = $1,235,533; year 2 = $2,935,505; year 3 = $3,172,708).
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
Estimation of pembrolizumab costs not reflective of current standard of care: The sponsor calculated the costs of pembrolizumab based on a flat dose (200 mg every 3 weeks) assumption. However, public drug plan and clinical expert feedback suggested that weight-based dosing (2 mg/kg up to a cap of 200 mg) with vial sharing is commonly implemented across jurisdictions. Thus, the costs of pembrolizumab are overestimated.
CADTH performed reanalysis by using weight-based dosing (2 mg/kg up to a cap of 200 mg) with vial sharing to estimate drug costs for pembrolizumab.
Inconsistent approach to model treatment duration: In the BIA, the sponsor assumed patients are treated with cemiplimab for 6.3 months and with pembrolizumab for 7 months, respectively, which was misaligned with the pharmacoeconomic model. Treatment duration was updated to reflect the mean treatment duration in the pharmacoeconomic model.
CADTH recalculated the mean treatment duration of pembrolizumab and cemiplimab to align with the CADTH base case for the cost-utility analysis.
There is uncertainty in the number of eligible patients: The sponsor estimated the number of patients eligible for cemiplimab treatment using an epidemiologic approach, with inputs based on assumptions and data from published literature, which are associated with uncertainty. CADTH was unable to validate the sponsor’s adopted estimate of the proportion of patients without a driver mutation (55%) in the original source.17 Further, the original source does not include information on all the genetic mutations (EGFR, ALK, and ROS1) and thus, may not be appropriate to derive an estimate of the proportion of patients without any relevant driver mutations. According to the clinical expert consulted for this review by CADTH, the proportion of patients without a driver mutation (EGFR, ALK, or ROS1) was overestimated by the sponsor, and is expected to be in the range of 12% to 18%. Clinical experts further noted that the sponsor’s assumption that 90% eligible patients are treated is also likely overestimated. The clinical experts noted the patient population with the indication of interest is predominantly elderly and individuals with poor performance status, such as those with a smoking history, would not be treated. According to the clinical experts, the proportion of patients treated is expected to be in the range of 50% to 70%.
In CADTH reanalysis, 18% of patients were assumed to be without a driver mutation and 60% of patients were assumed to receive treatment based on feedback from clinical experts.
There is uncertainty in the projected market share of cemiplimab: The sponsor assumed cemiplimab has a market share of 15.9% by year 3. The clinical experts noted that uptake may be lower since pembrolizumab may be preferred as it has longer-term follow-up data.
In scenario analysis, CADTH explored the impact of uncertainty in market share assumptions by assuming an arbitrary relative 25% reduction in market share of cemiplimab in each year.
CADTH Reanalyses of the Budget Impact Analysis
CADTH revised the sponsor’s base case by correcting estimation of pembrolizumab acquisition costs, aligning treatment duration of pembrolizumab and cemiplimab with the cost-utility analysis, assuming 18% of patients are without a driver mutation and 60% of patients receive treatment.
Table 16: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Treatment duration | Misaligned with the pharmacoeconomic model | Aligned with the pharmacoeconomic model |
2. Percentage of patients without driver mutation | 55% | 18% |
3. Percentage of patients treated | 90% | 60% |
4. Treatment dosage for pembrolizumab | Flat dose of 200 mg, every 3 weeks | Weight-based dosage (2 mg/kg up to a maximum dosage of 200 mg, every 3 weeks) with vial sharing |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
The results of the CADTH stepwise reanalysis are presented in summary format in Table 17 and a more detailed breakdown is presented in Table 18. Based on the CADTH base case, the budget impact of the reimbursing cemiplimab in the target population is expected to be $2,341,491 in year 1, $5,563,150 in year 2, and $6,012,679 in year 3, with a 3-year total of $13,917,320.
Table 17: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total |
|---|---|
Submitted base case | –$7,343,746 |
CADTH reanalysis 1 | $45,182,360 |
CADTH reanalysis 2 | –$2,403,408 |
CADTH reanalysis 3 | –$4,895,831 |
CADTH reanalysis 4 | $6,309,416 |
CADTH base case | $13,917,320 |
CADTH also conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 18.
Assuming a 25% reduction in market share of cemiplimab.
Adopting 2-year stopping rule for cemiplimab, align with the EMPOWER-1 trial and funding criteria for pembrolizumab.
Table 18: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $128,451,942 | $126,455,489 | $124,536,412 | $122,692,265 | $373,684,167 |
New drug | $128,451,942 | $125,219,956 | $121,600,908 | $119,519,557 | $366,340,421 | |
Budget impact | $0 | –$1,235,533 | –$2,935,505 | –$3,172,708 | –$7,343,746 | |
CADTH base case | Reference | $26,737,084 | $26,321,525 | $25,922,072 | $25,538,215 | $77,781,813 |
New drug | $26,737,084 | $28,663,016 | $31,485,222 | $31,550,895 | $91,699,133 | |
Budget impact | $0 | $2,341,491 | $5,563,150 | $6,012,679 | $13,917,320 | |
CADTH scenario analysis: 25% reduction in market share of cemiplimab | Reference | $26,737,084 | $26,321,525 | $25,922,072 | $25,538,215 | $77,781,813 |
New drug | $26,737,084 | $28,077,643 | $30,094,435 | $30,047,725 | $88,219,803 | |
Budget impact | $0 | $1,756,118 | $4,172,363 | $4,509,509 | $10,437,990 | |
CADTH scenario analysis: 2-year stopping rule for cemiplimaba | Reference | $26,737,084 | $26,321,525 | $25,922,072 | $25,538,215 | $77,781,813 |
New drug | $26,737,084 | $26,849,265 | $27,175,929 | $26,893,390 | $80,918,584 | |
Budget impact | $0 | $527,740 | $1,253,857 | $1,355,175 | $3,136,771 |
aAligning with the relevant CADTH scenario analysis for the cost-utility analysis.
Stakeholder Input
Patient Group Input
Lung Cancer Canada
About Lung Cancer Canada
Lung Cancer Canada is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research and advocacy. Lung Cancer Canada is a member of the Global Lung Cancer Coalition and is the only organization in Canada focused exclusively on lung cancer. https://www.lungcancercanada.ca/
Lung Cancer Canada is registered with CADTH.
Information Gathering
This treatment was recently approved in the US and there were no trial sites in Canada. As such Lung Cancer Canada was unable to interview any patients who have experience with cemiplimab for this submission. The current standard of care for these patients is pembrolizumab which is an effective treatment. The question relevant to this submission is the value of an additional treatment choice. Cemiplimab is clinically efficacious and clinicians agree it is as efficacious as the current standard of care. However cemiplimab may allow patients to be treated closer to home as it is a fixed dose regimen. LCC conducted a survey open to all lung cancer patients to probe the value of being treated closer to home and the illustrate the patient cost of travel. We also present information gathered from previous submissions.
The survey was conducted on SurveyMonkey and promoted on LCC’s social media and through LCC patient committees based in Canada. Responses were collected between October 22 and October 26, 2021.
The survey received 33 responses.
42% Ontario
15% Quebec, BC and Alberta
9% Manitoba
3% New Brunswick
Participants’ identities remained anonymous, and no identifying information was asked within the survey.
Disease Experience
Non-small cell lung cancer (NSCLC) is the most common type of lung cancer and occurs in 80-85% of all lung cancer cases. There have been many recent advancements in lung cancer research that have allowed for the continued development and release of treatment options that extend beyond chemotherapy. However inequities in treatment accessibility have long been a major barrier for many cancer patients, especially for those in rural areas and small communities. For these patients, traveling long distances to a major city may be their only option, which also often comes with a hefty price tag that adds up due to travel costs. The reality is that these systemic and geographical barriers impede timely access to diagnosis, treatment, and care that makes all the difference for lung cancer patients. According to the Canadian Partnership Against Cancer, rural residents have very limited access to healthcare services and lead to several negative impacts that only widen the gap between health inequities. The three-year lung cancer survival rate for those in rural communities was less than that of patients in urban areas, at 26% and 29% respectively (Canadian Partnership Against Cancer, 2020). Thus, the need for additional treatment options that can be delivered at community hospitals away from city centers is extremely prominent.
For immunotherapy, the current standard of care is a weight-based dose treatment. Though this allows for the fine-tuning of dosages that cater specifically to each individual patient, it also comes with drawbacks that tie in with accessibility of treatment for patients. This component makes it more complicated for community centres to administer as small patient numbers can lead to a higher potential for wastage or for administrators to schedule as they try to group patients to avoid wastage. However, the need to travel long distances to cancer centres for some individuals is difficult as there are many financial, emotional, and mental barriers that do not allow for ease of access to care. Those with lower socioeconomic status are also more likely to be diagnosed with advanced disease (stage III or IV) and therefore have more limited treatment options that may help their case (Canadian Partnership Against Cancer, 2020). Those that have lower socioeconomic status also have poorer outcomes. Fixed based dosing can benefit these patients who are impacted by such challenges, as they can be treated in smaller infusion centers at community hospitals, with numerous additional benefits, such as cost-effectiveness, better patient compliance, and easier access to care. Cemiplimab has the ability to address these and add to the treatment paradigm for such patients in Canada.
Cemiplimab is an immunotherapy treatment for advanced NSCLC patients who have a PDL-1 level of over 50% without ALK, ROS1, or EGFR mutations. It is indicated for first line treatment, and results from the EMPOWER-Lung1 phase 3 trial have showed very promising results in its efficacy and potential side effects. It saw a 43% reduction of risk of death compared to standard platinum chemotherapy, a median overall survival of 22 months compared to 14 months respectively, and 6.2 months of median progression-free survival compared to 5.6 months with chemotherapy (Sanofi Pharmaceuticals, 2021). Overall, cemiplimab’s potential to bring another treatment alternative to the current standard of care is needed to allow for the diversification of options available to patients. Advanced NSCLC has a very low five-year survival rate at only 19% in Canada, and so the need for additional therapeutic options for such difficult-to-treat patients is clear.
Experiences With Currently Available Treatments
Please refer to Lung Cancer Canada’s previous submission for pembrolizumab Keytruda) for first-line treatment of NSCLC.
Existing treatments that target solely on the PD-1 and PDL-1 protein that contribute to advanced non-small cell lung cancer in patients that do not have any oncogenic driver mutations include other types of immune checkpoint inhibitors that inhibit oncogenesis, such as pembrolizumab. For immunotherapy treatments, pembrolizumab is the current treatment paradigm for NSCLC patients who have PDL-1 expression of over 50% and whose tumours do not have a known EGFR, ALK, or ROS 1 mutation. It has seen a median progression free survival of 10.3 months, objective response rate of 44.8%, and overall survival at 6 months was significantly longer with pembrolizumab than chemotherapy, at 80.2% of patients alive versus 72.4% respectively (Reck et al., 2016). The main adverse events recorded form pembrolizumab included diarrhea, fatigue, nausea, and fever (Reck et al., 2016).
In Lung Cancer Canada’s previous CADTH submissions for pembrolizumab, it has been presented that patients on treatment are able to maintain a high quality of life level. Pembrolizumab has been seen to be effective at reducing tumour size and controlling symptoms, Patients are able to be independent as side effects are highly manageable and patients are able to engage in life, perform tasks and even work without caregiver assistance. This means that caregivers do not have to take time off work to care for their loved one or take them to treatments, thus minimizing the financial impact of the treatment. Patients are able to maintain a high level of functionality. It is a treatment that positively impacts the mental health of patients and their families as it is a very effective treatment.
Overall, cemiplimab will act as an alternative to the current standard of care for the first-line treatment of the aforementioned population, pembrolizumab. Based on the scientific research, both pembrolizumab and cemiplimab’s efficacy rates, tolerability, and list of common adverse events are similar, and thus, comparable to each other. Cemiplimab trials also took in more difficult-to-treat patients in comparison to pembrolizumab, which is therefore more representative of the real population of lung cancer patients, and still had positive and comparable results to pembrolizumab. Thus, CADTH can be additionally sure that cemiplimab’s potential impact on patients will be comparable in a real-world scenario.
Improved Outcomes
There have been many incredible advancements in lung cancer research in recent years that have changed the treatment paradigm for patients in Canada. With immunotherapy, the treatment options for patients become more limited in comparison to targeted therapies, as one’s specific oncogenic mutation is not specifically targeted, and thus, leaves a more widespread impact on them with notable side effects. In this case, pembrolizumab is the only option that is available to this population, and adding a second treatment options will allow for an alternative in the market. In a new therapy, patients most value:
Having manageable side effects and improvements in managing their NSCLC symptoms
Being able to have a full and worthwhile quality of life
Being able to maintain their independence and functionality to minimize the burden on their caregivers and loved ones
Delaying disease progression and settling patients into long-term remission for improved survivorship
In this case, as there is a comparable funded immunotherapy, patients expect cemiplimab to be as equally effective as the current standard of care
Experience With Drug Under Review
As mentioned previously, the reimbursement of cemiplimab will offer an alternative option for Canadian patients with advanced PDL-1 >50% non-small cell lung cancer. Pembrolizumab is already indicated, approved and funded in this population, and thus, will bring additional benefits for patients to diversify the market and offer treatments closer to home with the fixed dosing model cemiplimab provides. As the efficacy, safety, mechanism, and targeted patient populations between pembrolizumab and cemiplimab are almost the same, the main variable that cemiplimab will bring to the current treatment paradigm is offering an additional treatment value to patients as it a fixed-dose treatment. The current standard of care uses weight-based dosing. This may benefit those who live further from major hospitals in city centers and allow them to be treated closer to home. As mentioned above, this component makes it more complicated for community centres to administer as small patient numbers can lead to a higher potential for wastage or for administrators to schedule as they try to group patients to avoid wastage. Travel and accessibility of treatment has been a barrier to care for many patients, and this is where the benefits of cemiplimab as an alternative will come into play. Lung Cancer Canada was unable to gather patients with experience with cemiplimab, so a short survey addressed to all lung cancer patients was conducted to evaluate the impact of travel on treatment choices, in the context of having cemiplimab more readily available. This section will summarize the survey results.
Most patients would rather choose a treatment option closer to home.
91% of patients said if they were given the option between two equally as efficacious treatment options, with one near their home and the other at a large cancer center at least one hour away, they would choose the one close to home. This sets the case with cemiplimab and pembrolizumab, as both treatments are virtually equally as efficacious and are comparable to each other. The fixed dose model that cemiplimab offers will allow for infusions to be done at local community hospitals across the country and will fill the gap in accessibility and variety of treatment options for patients across Canada. There needs to be equity in healthcare that can allow patients to access the care they need, without having to sacrifice or compromise on important variables such as efficacy of treatment, financial implications, and quality of life. Only 9% of respondents would rather travel to the larger cancer center further away, which is the current case for not only pembrolizumab, but many others who cannot access chemotherapy near them, or in smaller provinces where most services are located in a central city. Having cemiplimab accessible in smaller community hospitals scattered around the country will bring ease of access and equity to cancer care.
The top benefits of being treated close to home were travel-related.
The two top-ranked benefits of being treated close to home were “decreased travel time to/from hospitals” (voted by 94% of respondents); travel cost savings (i.e. gas, parking costs), which was voted by 76%, and increased time with family and caregivers (64%). The implications that traveling long distances for doctors’ appointments, cancer treatment, scans, and diagnostic tests has on patients is substantial, and having the alternative options that cuts down on these costs is a significant factor that patients value. In addition, some patients need to relocate or move away from home for extended periods of time during treatment in a different city, which is the case for many chemotherapy and immunotherapy patients. Needing to find a place to stay overnight, or being away from family for weeks at a time carries many additional mental and emotional burdens, let alone the expensive costs and financial implications this has.
Patients believed that staying close to home for treatment could allow for more independence and better quality of life.
Other benefits that patients valued in the survey included increased recovery time at home, quicker return to life activities, ability to stay in a familiar environment, improvements in mental health, and increased flexibility in being able to drive themselves rather than depending on caregivers. Many of these benefits have implications on patients’ independence and quality of life, and being able to stay at home during recovery, be close to family, and remain in a familiar environment has numerous benefits for lung cancer patients’ mental health. Shorter recovery times and decreased dependence on caregivers to drive patients to appointments, in turn, brings the patient’s independence back and allows them the flexibility and freedom to stick to their own schedule rather than relying on others. Cemiplimab has shown to have minimal and manageable side effects, and thus, in most cases would not impede a patient’s independence, but rather allow them to return to life quicker.
Cancer team expertise was the top concern patients had in accessing treatment close to home.
Though there are many benefits to having cemiplimab available close to home for patients, some acknowledge the drawbacks this may have on their healthcare. The top concern respondents ranked in the survey was the expertise of local cancer teams, in which 70% voted for. However, patients will still nonetheless be connected to their primary care physicians and oncologists, and will be attending infusion treatments at local hospitals, in which cemiplimab follows a fixed dosing model that is relatively simpler than weight based. Other drawbacks that respondents mentioned include privacy concerns (i.e., ability to remain anonymous in a small community), and side effect management. 30% of respondents said they do not have any concerns about being treated at local hospitals.
Most patients agreed that adding an additional treatment option close to home will make a huge difference in quality of life.
97% of participants agreed that having an additional treatment option, such as cemiplimab, that can be administered at smaller community hospitals closer would make a positive difference in lung cancer patients’ quality of life. As mentioned above, patients saw the numerous benefits that this could have in the treatment paradigm as currently, there is only one option available for the PDL-1 > 50% advanced NSCLC population in Canada. As cemiplimab is intended to be a first-line treatment, the sequencing and standard of care for second and third-line therapy remains the same; thus, there is no departure from the current standard of care. Being able to grasp their independence, improve their quality of life, reduce dependence on caregivers, and shorter recovery times are all potential benefits cemiplimab will have on the target population, and the patients surveyed agree as well. 70% of patients within the aforementioned 97% agreed it will make a very significant difference in quality of life, while the remaining 27% agreed it will have noticeable positive impacts. One respondent said it will not make a difference, and nobody thought it would have negative impacts. Therefore, adding an alternative treatment option will allow the treatment paradigm in Canada to move forward and bring equity to healthcare.
Companion Diagnostic Test
There is no additional companion diagnostic test associated with cemiplimab, as PDL-1 is currently tested in all provinces.
Anything Else?
CADTH will consider this submission as a treatment that is an alternative to pembrolizumab which is an effective treatment for NSCLC, and will deliberative the necessity of an additional treatment in this space. Clinically, the two treatments perform similarly. Practically, fixed dosing (cemiplimab) vs weight-based dosing (pembrolizumab) helps facilitate the delivery of care to be brought closer to the patient. The challenges of travel have been well documented and the benefits of treatment closer to home have been illustrated with the survey results. Access and equity are tied to delivery of care and the dosing in cemiplimab can help administrators and care teams design strategy to overcome this barrier with NSCLC patients. Additionally, the introduction of a second molecule in this space allows for marketplace competition. The sustainability of our healthcare system is a social responsibility. Options mean patients are given choice and can make informed decisions based on their individual situations. For these reasons, Lung Cancer Canada urges CADTH to make a positive funding recommendation for this submission.
In closing, Lung Cancer Canada would like to thank CADTH for the opportunity to submit. We applaud CADTH’s commitment to listen to patient voice. However, we note a few developments with disappointment. In the past, the complete draft economic and clinical guidance reports were released at the same time as the initial recommendation. Currently we only receive a summary. It is extremely challenging for us to provide input without the full summary. This situation is akin to asking to evaluate a paper for publication while only being provided with an abstract. It cannot be done responsibly. CADTH has also changed the threshold QALY to $50,000, in line with non-cancer treatments. However we believe that the cost of cancer treatments cannot be compared to non-cancer treatments and that the values differ. This was one of the premises behind the original decision to establish JODR and then PCODR. We encourage re-examination of these practices.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
N/A
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
N/A
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Conflict of Interest Declaration for Lung Cancer Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
N/A | — | — | — | — |
Lung Health Foundation / The Ontario Lung Association
About Lung Health Foundation
The Ontario Lung Association (now named Lung Health Foundation) is registered with the CADTH and pCODR (www.lunghealth.ca). The Lung Health Foundation (Ontario Lung
Association) is a registered charity that assists and empowers people living with or caring for others with lung disease. It is a recognized leader, voice and primary resource in the prevention and control of respiratory illness, tobacco cessation and prevention, and its effects on lung health. The Foundation provides programs and services to patients and health-care providers, invests in lung research and advocates for improved policies in lung health. It is run by a board of directors and has approximately 46 employees, supported by thousands of dedicated volunteers.
Information Gathering
The information provided from the Lung Health Foundation in this submission was obtained from an online survey completed by 13 lung cancer patients and one caregiver, as well as three phone interviews with people currently living with lung cancer. Information on age, gender and geographical location was not collected from any of the 14 online respondents. All of the online respondents completed the survey on or before August 31, 2021. The phone interviews were conducted from September - October 2021 with two female patients and one male patient. The patients are located in Ontario, Manitoba and Quebec. Input from a Registered Nurse as well as a Certified Respiratory Educator was obtained for this submission. Those individuals reviewed sections related to disease experience, experiences with available treatments and outcomes based on information received from monthly patient support groups and phone consultations.
One research study was consulted to inform the answer to question 4: Janse, S., Janssen, E., Huwig, T., Basu Roy, U., Ferris, A., Presley, C. J., & Bridges, J. F. (2021). Line of therapy and patient preferences regarding lung cancer treatment: a discrete-choice experiment. Current Medical Research and Opinion, 37(4), 643-653.
Disease Experience
The respondents had varying experiences with their lung cancer diagnosis. One reported they experienced no symptoms from the actual disease. Most patients report only feeling some pain in their side or chest tightness. The psychosocial effects of having an illness with a poor prognosis are more debilitating, according to some respondents. One patient describes fearing that they had only 6-18 months left to live and struggled to cope because they have young children. Another respondent shared the difficulties of being diagnosed with lung cancer during the COVID-19 pandemic. Tests and treatments were delayed and this was a great source of anxiety for the patient. The patient worried about the cancer metastasizing and having a poorer prognosis as a result of the delays.
Other symptoms and challenges these patients experienced as a result of their lung cancer were shortness of breath (64%), fatigue (57%), depression (25%), cough (21%), difficulty fighting infection (21%) and chest tightness (14%). Weight loss, diminished appetite and challenges with physical and emotional intimacy were also noted by a few respondents.
When asked whether this condition affected their day-to-day life, 60% of respondents indicated that it greatly impacted their ability to complete instrumental activities of daily living, 38% indicated it negatively impacted their work, and 28% their leisure activities and hobbies.
Patients described having a challenging time maintaining relationships with families and friends. They felt short tempered and impatient and this made them feel isolated. Patients also described withdrawing from social activities because of the stigma attached to a lung cancer diagnosis. To quote one of the respondents, “I did not want anyone to know I had lung cancer, I wanted people to still have empathy for my children.”
Family members and caregivers of those living with lung cancer share the same psychosocial burdens as the patients and have the added responsibility of providing care. Being a caregiver affects their ability work, relationships with family and friends and their emotional well-being.
As well, their independence and ability to travel and socialize are often impacted. Having to take time off work to drive those they are caring for to get groceries, run errands or attend medical appointments can be problematic for caregivers and feelings of fatigue and emotional exhaustion are not uncommon
Experiences With Currently Available Treatments
The treatments tried by the respondents include surgery, radiation, chemotherapy, targeted therapy and immunotherapy. The medications tried include Cisplatin, Docetaxel, Gefitinib, Entrectnib, Alectinib, Brigatinib, Tagrisso, Nivolumab and Ipilimumab.
The benefits experienced with the treatments were: prolonged life, delayed disease progression and a reduction in the severity of disease-related symptoms. Although these benefits were noted, most patients struggled with lingering side effects. Respondents who received surgery reported deconditioning and chronic fatigue. Some of the side effects reported from radiation were fatigue, skin changes, hair loss and tissue scarring. One patient reported that they now have COPD related to lung tissue scarring from radiation.
With medications, the side effects reported included fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy. Side effects from chemotherapy severely impacted the patients’ quality of life, ability to work and in some cases, the ability to perform activities of daily living. One of the respondents reports that while on chemotherapy, because of the hair loss and side effects, he was visibly ill which severely impacted his self-esteem.
When asked about challenges with access to treatment, the respondents reported that they struggled to navigate the healthcare system. In some cases, they were not clear where to go for information and support.
Respondents would not only like to see biomarker testing done earlier, but also done for all biomarkers. This will allow patients to receive targeted therapy. Some patients felt that taking treatments before biomarker testing led them to suffer unnecessarily with side effects from medications that provided no therapeutic benefit. In addition, patients value having treatment options when they are deciding on their best course of action. Research has shown that lung cancer patient preferences for treatments can vary widely dependent on clinical and demographic attributes and that patients active role in decision-making can work to improve health outcomes (Janse et al., 2021).
Improved Outcomes
Key treatment outcomes for this group of lung cancer patients include stopping or slowing the progression of the disease with minimal side effects. Patients would also like to see medications that are effective for advanced disease. Due to the poor outcomes associated with advanced disease, patients describe feeling very anxious about any sign or prospect of disease progression.
Patients state that if treatments were more effective in treating lung cancer at any stage, then a diagnosis would not feel like a “death sentence”. One of the respondents reported that after she was given a prognosis of 6-18months, she became withdrawn and struggled to cope. She stated, “I did not want to go anywhere or do anything, I just wanted to spend every last second with my children". This isolation negatively impacted her quality of life and mental well- being.
Side effects are also a great source of distress for patients. Some reported that they had no symptoms from the actual cancer but struggled with the side effects from treatment more.
Patients would like treatments with minimal side effects so that they can carry on with regular activities while on treatment. The importance of maintaining some quality of life cannot be overstated.
Caregivers report having to make decisions about treatment options. Seeing patients suffer through side effects is particularly challenging and they report often feeling conflicted as they want the patient to receive treatment but do not want them to suffer through side effects.
When choosing therapy, patients are also interested in the efficacy of the medication. One respondent commented that they would be more receptive to side effects if there was a guarantee that the medication would stop or slow down the progression of lung cancer.
Experience With Drug Under Review
No patients within this evidence group submission had experience with the medication under review.
Companion Diagnostic Test
Although patients in this submission group do not have experience with the drug under review, they did receive biomarker tests for other treatments. The majority of the respondents who went through the testing indicated they wished it had been done sooner. Depending on the stage of the cancer diagnosis, biomarker testing was not always an option at diagnosis.
One of the respondents reported that they would have been preferred to be tested for all the biomarkers in one test. They felt testing for a few at a time lengthened the process which caused additional stress and worry about disease progression.
Anything Else?
Not applicable.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation.
Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 2: Conflict of Interest Declaration for Lung Health Foundation / The Ontario Lung Association
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme | — | — | X | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Lung Health Foundation/Ontario Lung Association
Date: October 28, 2021
Clinician Group Input
Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer Drug Advisory Committee (Lung DAC)
About Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer Drug Advisory Committee (Lung DAC)
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
This input was jointly discussed via email.
Current Treatments
Describe the current treatment paradigm for the disease.
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: The population of interest is patients with advanced and metastatic NSCLC not amenable to curative treatment approaches, that have tumours expressing PD-L1 at levels of 50% or higher. This represents approximately one third of patients with advanced and metastatic NSCLC. There are several treatment options for this group of patients including pembrolizumab monotherapy, pembrolizumab in combination with platinum based chemotherapy and nivolumab, ipilimumab plus two cycles of platinum based chemotherapy. There are also published data supporting atezolizumab monotherapy but this is not currently approved. These agents all have data demonstrating improved PFS, overall survival and objective response rates. The most commonly recommended therapy for this group of patients is pembrolizumab monotherapy.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Examples: Prolong life, delay disease progression, improve lung function, prevent the need for organ transplant, prevent infection or transmission of disease, reduce loss of cognition, reduce the severity of symptoms, minimize adverse effects, improve health-related quality of life, increase the ability to maintain employment, maintain independence, reduce burden on caregivers.
Response: The most important goals are improved overall survival and improved progression free survival. Monotherapy immunotherapy also has the additional information of avoidance of chemotherapy
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 4, please describe goals (needs) that are not being met by currently available treatments.
Examples: Not all patients respond to available treatments. Patients become refractory to current treatment options. No treatments are available to reverse the course of disease. No treatments are available to address key outcomes. Treatments are needed that are better tolerated. Treatment are needed to improve compliance. Formulations are needed to improve convenience
Response: The implementation of immunotherapy in the first line therapy of metastatic NSCLC has resulted in significant improvements for patients. Most trials demonstrate improvements in median survival of 6 to 12 months and meaningful improvements in survival at 3 and 5 years. Nevertheless most patients relapse and die of their disease. Therefore there is a need for improved therapies. Cemiplimab would represent an additional treatment option for this group of patients
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: The patients most likely to benefit from cemiplimab are those patients with advanced and metastatic NSCLC and tumours having high levels of PD-L1 expression (=> 50%).
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Is there a mechanism of action that would complement other available treatments, and would it be added to other treatments? Is the drug under review the first treatment approved that will address the underlying disease process rather than being a symptomatic management therapy? Would the drug under review be used as a first-line treatment, in combination with other treatments, or as a later (or last) line of treatment? Is the drug under review expected to cause a shift in the current treatment paradigm?
Response: Cemiplimab would be used as monotherapy for the first line treatment of advanced and metastatic NSCLC and tumours with high level of PD-L1 expression (=> 50%). Cemiplimab would be an alternative treatment to pembrolizumab monotherapy, or the combination of nivolumab, ipilimumab and 2 cycles of platinum based chemotherapy. It would not be used as an additional therapy to currently available treatment options
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
If so, please describe which treatments should be tried, in what order, and include a brief rationale.
Response: Cemiplimab would be used as first-line therapy for advanced and metastatic NSCLC in patients with high PD-L1 expresison. It would not be appropriate to use other therapies first.
How would this drug affect the sequencing of therapies for the target condition?
If appropriate for this condition, please indicate which treatments would be given after the therapy has failed and specify whether this is a significant departure from the sequence employed in current practice. Would there be opportunity to treat patients with this same drug in a subsequent line of therapy? If so, according to what parameters?
Response: Cemiplimab would replace existing first-line treatment options of pembrolizumab monotherapy, pembrolizumab plus chemotherapy, and nivolumab, ipilimumab and 2 cycles of chemotherapy
Which patients would be best suited for treatment with the drug under review?
Which patients are most likely to respond to treatment with the drug under review? Which patients are most in need of an intervention? Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Patients with advanced and metastatic NSCLC and tumours with high PD-L1 expression (=> 50%) and no specific contraindications to an immune checkpoint inhibitor.
How would patients best suited for treatment with the drug under review be identified?
Examples: Clinician examination or judgement, laboratory tests (specify), diagnostic tools (specify). Is the condition challenging to diagnose in routine clinical practice? Are there any issues related to diagnosis? (e.g., tests may not be widely available, tests may be available at a cost, uncertainty in testing, unclear whether a scale is accurate or the scale may be subjective, variability in expert opinion.). Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)? Should patients who are pre-symptomatic be treated considering the mechanism of action of the drug under review?
Response: PD-L1 is reflexively tested on newly diagnosed patients with NSCLC. Therefore patients would be readily identified.
Which patients would be least suitable for treatment with the drug under review?
Response: Patients with advanced and metastatic NSCLC and tumours with negative or low PD-L1 expression.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
If so, how would these patients be identified?
Response: Subgroup analyses from the Empower-1 trial do not identify patients more or less likely to benefit from cemiplimab other than PD-L1 expression => 50%.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: Outcomes to determine if a patient is benefiting from therapy are response rates assessed on repeat imaging, as well as improvement in patient symptoms assessed by patient assessment
What would be considered a clinically meaningful response to treatment?
Examples: Reduction in the frequency or severity of symptoms (provide specifics regarding changes in frequency, severity, and so forth). Attainment of major motor milestones. Ability to perform activities of daily living. Improvement in symptoms. Stabilization (no deterioration) of symptoms. Consider the magnitude of the response to treatment. Is this likely to vary across physicians?
Response: The most meaningful response to treatment is the absence of disease progression. Additional measures of response are improvement in disease related symptoms
How often should treatment response be assessed?
Response: In routine clinical practice, response should be assessed every three months on therapy.
What factors should be considered when deciding to discontinue treatment?
Examples: Disease progression (specify; e.g., loss of lower limb mobility). Certain adverse events occur (specify type, frequency, and severity). Additional treatment becomes necessary (specify)
Response: Disease progression or intolerable side effects would be the primary reasons to discontinue therapy. It is important to recognise that patients benefiting from therapy may benefit from continuing treatment beyond RECIST defined progression. This should remain an option.
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: Cemiplimab should be administered in an established centre providing cancer systemic therapy and under the supervision of a medical oncologist trained in the administration of immunotherapy.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: NA
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: Cemiplimab appears to offer similar benefit to other first line immunotherapy agents and should be considered as an alternative agent for funding.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Gail Darling
Position: Ontario Cancer Lead/Thoracic Surgeon
Date: 28/Oct/2021
Table 3: Declaration for Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer DAC Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi-Genzyme — No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Peter Ellis
Position: Division Head of Medical Oncology Juravinski Cancer Centre, Member of Lung DAC
Date: 7-Oct-2021
Table 4: Declaration for Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer DAC Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi | X | — | — | — |
Declaration for Clinician 3
Name: Dr. Stacey Hubay
Position: Medical oncologist; Member of Lung DAC
Date: 27-Oct-2021
Table 5: Declaration for Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer DAC Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi-Genzyme — No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr. Mohammad Rassouli
Position: Medical oncologist; Member of Lung DAC
Date: 27-Oct-2021
Table 6: Declaration for Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancer DAC Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi-Genzyme — No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.