CADTH Reimbursement Review
Tepotinib (Tepmetko)
Sponsor: EMD Serono, a division of EMD Inc., Canada
Therapeutic area: Locally advanced or metastatic non–small cell lung cancer
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ALP
alkaline phosphatase
ALT
alanine transaminase
ASCO
American Society of Clinical Oncology
AST
aspartate transaminase
CDM
Common Data Model
CI
confidence interval
CNS
central nervous system
CR
complete response
ctDNA
circulating tumour DNA
DOR
duration of response
ECOG
Eastern Cooperative Oncology Group
ECG
electrocardiogram
EMR
electronic medical records
EORTC
European Organisation for Research and Treatment of Cancer
EQ-5D-5L
5-Level EQ-5D
ESS
effective sample size
GGT
gamma-glutamyl transferase
HGF
hepatocyte growth factor
HR
hazard ratio
HRQoL
health-related quality of life
ITC
indirect treatment comparison
IRC
independent review committee
IV
intravenous
LCC
Lung Cancer Canada
LHF
Lung Health Foundation
MAIC
matching-adjusted indirect comparison
METex14
MET exon 14
MID
minimal important difference
MMRM
mixed model repeated measures
mITT
modified intention-to-treat population
MRI
magnetic resonance imaging
NGS
next-generation sequencing
NSCLC
non–small cell lung cancer
OH-CCO
Ontario Health – Cancer Care Ontario
ORR
objective response rate
OS
overall survival
PD-L1
programmed death-ligand 1
pERC
CADTH pan-Canadian Oncology Drug Review Expert Review Committee
PFS
progression-free survival
PR
partial response
PRO
patient-reported outcome
PS
performance status
QLQ-C30
Quality of Life Questionnaire Core 30
QLQ-LC13
Quality of Life Questionnaire Lung Cancer 13
RANO-BM
Response Assessment in Neuro-Oncology Brain Metastases
RECIST
Response Evaluation Criteria in Solid Tumours
RMST
restricted mean survival time
RWD
real-world data
RWE
real-world evidence
SAE
serious adverse event
SD
standard deviation
TTD
time to deterioration
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Tepotinib (Tepmetko), tablets, 225 mg tepotinib (as tepotinib hydrochloride), oral |
Indication | For the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring MET tyrosine kinase receptor exon 14 skipping alterations |
Reimbursement request | For the treatment of adult patients with advanced NSCLC harbouring MET exon 14 skipping alterations |
Health Canada approval status | NOC/c |
Health Canada review pathway | Expedited review (Project ORBIS) |
NOC/c date | May 27, 2021 |
Sponsor | EMD Serono, a division of EMD Inc., Canada |
MET = mesenchymal-epithelial transition; NOC/c = Notice of Compliance with Conditions; NSCLC = non–small cell lung cancer.
Introduction
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer deaths in Canada.1 Survival from lung cancer of all stages and histologic subtypes is poor, with an overall 5-year net survival of 19%.1 Lung cancer is classified into small cell lung cancer or non–small cell lung cancer (NSCLC), which accounts for approximately 88% of cases in Canada.1 The symptoms of advanced or metastatic NSCLC can be variable and often depend on the site of metastasis. At presentation, the most common signs and symptoms of NSCLC include persistent cough, shortness of breath, chest pain, wheezing, and hemoptysis.2 In patients with advanced NSCLC and distant metastasis, symptoms may include bone pain, headache, neurologic or psychiatric abnormalities, paraplegia, hepatomegaly, and pathological fractures.3
The mesenchymal-epithelial transition (MET) receptor tyrosine kinase is an oncogenic driver of NSCLC. Mutations that result in loss of exon 14 in the MET gene, called MET exon 14 (METex14) skipping alterations, lead to dysregulation and inappropriate signalling.4 METex14 skipping alterations occur in approximately 3% of NSCLC cases and are associated with poor prognosis, according to retrospective studies and the clinical experts consulted by CADTH.4-7 A retrospective study has also found that patients with NSCLC harbouring METex14 skipping mutations may be less responsive to immunotherapy.8 Currently, patients in Canada with advanced (i.e., stage IV and stage IIIB not amenable to curative treatment approaches) NSCLC harbouring METex14 skipping mutations are usually treated according to guidelines for advanced NSCLC without driver mutations.9,10 In the first-line setting, current treatments include immunotherapy, with or without chemotherapy. In the second- or later-line setting, single-agent chemotherapy, single-agent immunotherapy, or platinum-based chemotherapy doublets may be used.10
Tepotinib is an oral, ATP-competitive, and highly selective MET receptor tyrosine kinase inhibitor.11 Tepotinib is indicated for the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14skipping alterations. Documentation of METex14 skipping alteration status based on a validated METex14 assay is required before treatment with tepotinib. Tepotinib (225 mg tablets, as tepotinib hydrochloride) is taken orally, and the recommended dosage is 450 mg (2 tablets) once daily. Per the Health Canada product monograph, it is recommended that treatment be continued until disease progression or unacceptable toxicity.
The objective of this review is to perform a systematic review of the beneficial and harmful effects of tepotinib for the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping alterations.
After CADTH issued a draft CADTH pan-Canadian Oncology Drug Review Expert Review Committee (pERC) recommendation for tepotinib in March 2022, the following additional information was provided to CADTH.
The sponsor provided additional unpublished data from the pivotal VISION study with a data cut-off date of February 2021.
The sponsor provided an additional unanchored matching-adjusted indirect comparison (MAIC) comparing tepotinib to the combination of chemotherapy and immunotherapy. These data were not included in the submission to CADTH (the sponsor reported that the data became available only after the CADTH recommendation was issued). A comparison of tepotinib with a combination of chemotherapy and immunotherapy has been identified as an important gap in the evidence. The information has been summarized and critically appraised as an addendum to the CADTH report in Appendix 5.
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
CADTH received input from 2 patient advocacy groups: the Lung Health Foundation (LHF) and Lung Cancer Canada (LCC). The LHF collected information from an online survey with 13 patients and 1 caregiver, and from phone interviews with 2 patients. LCC collected data from phone and video interviews with 4 patients and 1 caregiver. Patients’ experiences with lung cancer varied from no symptoms to a negative impact on their ability to perform instrumental activities of daily living. The most frequently reported symptoms were shortness of breath, fatigue, and depression. Other notable concerns were anxiety and stigma related to their diagnosis that prevented them from fully engaging with their families and participating in social activities.
Patients indicated that they want treatments that stop or slow the progression of disease, prolong life, improve symptoms and quality of life, and have minimal side effects. The patients expressed that it is important to maintain independence and functioning. Patients reported that they struggled to navigate the health care system and access biomarker testing for MET mutations in Canada. The patients indicated that they want the equal access to biomarker testing across Canada at the time of diagnosis or early in treatment.
Input From the Clinical Experts Consulted by CADTH
CADTH received input from 2 clinical specialists with expertise in the diagnosis and management of NSCLC. The clinical experts reported that retrospective studies7,8 have indicated patients with METex14 alterations have a poor prognosis, and there appears to be less benefit from immunotherapy in these patients. Moreover, the clinical experts noted that patients with METex14 skipping mutations tend to be an elderly population that often experiences increased side effects from chemotherapy. Thus, better-tolerated treatments are needed. The clinical experts noted that there is currently no targeted treatment for NSCLC with METex14 mutations that is publicly funded. The clinical experts indicated that the goals of treatment in locally advanced (not amenable to curative treatment) or metastatic NSCLC are to improve overall survival (OS), progression-free survival (PFS), and response rate, as well as maintain quality of life. In addition, the clinical experts thought that new treatment options should minimize adverse events (AEs).
The clinical experts indicated that tepotinib would preferentially be used in the first-line setting for patients with METex14 skipping alterations because it is a targeted therapy. If patients received tepotinib or another MET receptor tyrosine kinase inhibitor as first-line treatment, later lines of therapy would consist of chemotherapy, immunotherapy, or chemotherapy plus immunotherapy, based on established provincial funding algorithms. If tepotinib was not used as first-line therapy, it would be used as second- or later-line therapy.
The clinical experts thought tepotinib should be used in patients with locally advanced or metastatic NSCLC harbouring a METex14 skipping mutation who meet the eligibility criteria used in the VISION trial. Patients with NSCLC with METex14 mutations would be identified by molecular biomarker testing using tumour tissue biopsy or liquid biopsy, followed by next-generation sequencing (NGS) panel testing. The clinical experts noted that it would be ideal for this testing to be done at the time of diagnosis of advanced NSCLC. The clinical experts would also consider using tepotinib in patients with an Eastern Cooperative Oncology Group performance status (ECOG PS) of 2 or more who otherwise met the VISION trial eligibility criteria.
The clinical experts reported that, in standard practice, response to treatment is assessed using CT or bone scans every 2 to 4 months, as clinically indicated. In addition, if patients present with disease-related symptoms before starting treatment, clinical assessments are conducted at regular intervals (e.g., every 3 months) to assess symptom control. The clinical experts reported that a clinically meaningful response to treatment would be improved survival and maintenance or improvement in quality of life. The clinical experts indicated that treatment with tepotinib would be discontinued if a patient experienced disease progression or intolerable treatment-related side effects.
CADTH received input from 3 clinician groups involving a total of 21 clinicians: Northeast Cancer Centre – Thoracic Cancer Clinicians, Ontario Health – Cancer Care Ontario’s (OH-CCO) Lung and Thoracic Cancers Drug Advisory Committee, and LCC’s Medical Advisory Committee. The clinician groups generally agreed with the input provided by the clinical experts consulted by CADTH. The clinician groups indicated that there is an unmet need for targeted therapy and improved outcomes in patients with advanced NSCLC harbouring METex14 skipping alterations. They thought that tepotinib would be preferentially offered as first-line therapy and as a subsequent therapy for those who had already received other treatments.
Drug Program Input
The drug programs noted that the VISION trial eligibility included patients with ECOG PS of 0 or 1 and asked whether patients with ECOG PS of 2 or more should be eligible for tepotinib. The clinical experts consulted by CADTH indicated that it would be reasonable to offer tepotinib to patients with an ECOG PS of 2 and 3.
The drug programs noted that patients currently receiving alternative first-line or subsequent lines of therapy would have a time-limited opportunity to switch to tepotinib at the time of public funding. The clinical experts indicated that, if a patient was responding to their current treatment, they would not switch the patient to tepotinib at the time of public funding. The clinical experts would keep the patient on their current treatment until they experienced progressive disease and then use tepotinib for the next line of therapy.
The drug programs noted that, in patients with advanced NSCLC with driver mutations (e.g., EGFR, ALK, ROS, BRAF) who receive targeted treatment in the first-line setting, chemotherapy is required before accessing immunotherapy, in line with previous pERC recommendations. The drug programs indicated that jurisdictions would use the same sequencing principles for therapies used after tepotinib, regardless of programmed death-ligand 1 (PD-L1) tumour proportion score. The drug programs noted that tepotinib may change the place in therapy of drugs reimbursed in subsequent lines. Last, the drug programs noted that testing for METex14 skipping alterations may not be routinely available in some jurisdictions and would need to be implemented.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
One ongoing, phase II, single-arm, open-label, multi-centre trial (VISION) was included in the CADTH systematic review.12-14 The primary objective of the VISION study was to assess the efficacy of tepotinib in patients with advanced (locally advanced or metastatic) NSCLC, as per objective response (confirmed complete response [CR] or partial response [PR]), determined according to Response Evaluation Criteria in Solid Tumours (RECIST) v1.1 criteria, based on independent review in patients that tested positive for METex14 skipping alterations or MET amplification. There were 3 cohorts in the VISION study (Cohorts A, B, and C). Patients were selected for each cohort based on defined MET alterations or MET amplification identified in tumour tissue and/or in circulating tumour DNA (ctDNA) derived from plasma (i.e., liquid biopsy). Patients with METex14 skipping alterations were enrolled in cohort A and cohort C under the same eligibility criteria and underwent the same study procedures. Cohort A was the pivotal cohort; cohort C was a confirmatory cohort added as a protocol amendment to extend and confirm the existing results for cohort A, and to expand the METex14 population in the study. After accrual for cohort A was complete, enrolment at sites was shifted from cohort A to cohort C (cohort A: N = 151; cohort A plus C: N = 254). Cohort B does not align with the Health Canada indication or reimbursement request; therefore, data for cohort B will not be presented in this review. All patients received 500 mg tepotinib hydrochloride hydrate, containing 450 mg tepotinib, orally once daily in 21-day cycles. Treatment was continued until disease progression, death, an AE leading to discontinuation, or withdrawal of consent. The primary outcome was objective response rate (ORR) by independent review committee (IRC) assessment. Secondary outcomes included OS; PFS by IRC; PFS by investigator; ORR by investigator; duration of response (DOR) by IRC; DOR by investigator; change from baseline and time to deterioration (TTD) by 10 points in 5-Level EQ-5D (EQ-5D-5L) visual analogue score (VAS), European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Core 30 (QLQ-C30) (global health status and quality of life score), and EORTC Quality of Life Questionnaire Lung Cancer 13 (QLQ-LC13) (coughing, dyspnea, and chest pain symptom scales); and safety. Data were analyzed descriptively; no statistical testing was performed.
The mean age of study patients in cohort A and cohort A plus C was 73 years. Most patients were White (70.9% in cohort A, 67.1% in cohort A plus C), had an ECOG PS of 1 (73.5% in cohort A, 72.2% in cohort A plus C), adenocarcinoma histology type (86.8% in cohort A, 81.2% in cohort A plus C), and stage IV disease at study entry (74.8% in cohort A, 64.3% in cohort A plus C). Approximately half of the patients in cohort A and cohort A plus C had received prior anticancer drug therapy for advanced or metastatic disease (54.3% and 51.0%, respectively). The most common types of prior anticancer therapies were cytotoxic therapy (49.7% in cohort A, 44.3% in cohort A plus C) and immunotherapy (25.8% in cohort A, 25.9% in cohort A plus C). Most patients had not had prior anticancer surgery (68.9% in cohort A, 67.5% in cohort A plus C). Per IRC assessment, 9.9% of patients in cohort A and 12.2% of patients in cohort A plus C had brain metastases at baseline.
Data from an interim analysis of VISION (data cut-off date of July 1, 2020) for the pivotal cohort A and pooled cohort A plus C (i.e., entire population with METex14 skipping alterations) are reported in this section. The results of key efficacy outcomes in the modified intention-to-treat (mITT) population are summarized in Table 2.
Overall Survival
Median OS was 17.6 (95% confidence interval [CI], 15.0 to 21.0) months in cohort A and 19.1 (95% CI, 15.3 to 22.1) months in cohort A plus C.
Progression-Free Survival
Median PFS by IRC was 8.9 (95% CI, 8.2 to 11.0) months in cohort A and 9.5 (95% CI, 8.2 to 11.2) months in cohort A plus C.
Objective Response Rate
The ORR by IRC was the primary end point in the VISION trial. The ORR by IRC was 45.0% (95% CI, 36.9 to 53.3) in cohort A and 46.4% (95% CI, 39.8 to 53.2) in cohort A plus C. All observed responses by IRC assessment were PR.
Duration of Response
Median DOR by IRC was 11.1 (95% CI, 8.4 to 18.5) months in cohort A and 11.1 (95% CI, 9.5 to 18.5) months in cohort A plus C.
Health-Related Quality of Life and Patient-Reported Outcomes
In cohort A, 40.4% of patients experienced a deterioration in EQ-5D-5L VAS score by 10 points, and median TTD was 8.3 (95% CI, 5.8 to 17.7) months. In cohort A plus C, 31.9% of patients experienced a deterioration in EQ-5D-5L VAS score by 10 points and median TTD was 8.3 (95% CI, 5.9 to 17.7) months.
In cohort A, 35.8% of patients experienced a deterioration in EORTC QLQ-C30 global health status and quality of life score by 10 points, and median TTD was 15.2 (95% CI, 6.0 to 33.2) months. In cohort A plus C, 29.1% of patients experienced a deterioration in global health status and quality of life score by 10 points, and median TTD was 15.2 (95% CI, 6.2 to 33.2) months.
In cohort A, 29.8% of patients experienced a deterioration by 10 points in the EORTC QLQ-LC13 cough symptom scale. and median TTD was 11.1 (95% CI, 11.1 to NE) months. For chest pain, 29.8% of patients experienced a deterioration by 10 points, and median TTD was 17.7 (95% CI, 11.1 to NE) months. For dyspnea, 50.3% of patients experienced a deterioration by 10 points, and median TTD was 5.5 (95% CI, 4.1 to 6.9) months.
In cohort A plus C, 20.5% of patients experienced a deterioration by 10 points in the EORTC QLQ-LC13 cough symptom scale, and median TTD was 13.8 (95% CI, 11.1 to NE) months. For chest pain, 22.0% of patients experienced a deterioration by 10 points, and median TTD was 17.7 (95% CI, 11.8 to NE) months. For dyspnea, 40.9% of patients experienced a deterioration by 10 points, and median TTD was 5.6 (95% CI, 4.1 to 6.9) months.
Harms Results
The results of key harms outcomes in the cohort A and cohort A plus C safety analysis sets as of the July 1, 2020, data cut-off date are reported here and summarized in Table 2.
Table 2: Summary of Key Results From VISION as of the July 1, 2020, Data Cut-Off Date
Outcome | Cohort A (mITT) N = 151 | Cohort A + C (mITT) N = 254 |
|---|---|---|
OS | ||
Patients with an event, n (%) | 75 (49.7) | 84 (33.1) |
Patients censored, n (%) | 76 (50.3) | 170 (66.9) |
Median duration of follow-up (95% CIa), months | 16.4 (13.6 to 18.5) | 9.9 (8.1 to 12.0) |
Median OSb (95% CIa), months | 17.6 (15.0 to 21.0) | 19.1 (15.3 to 22.1) |
PFS by IRC | ||
Patients with an event, n (%) | 87 (57.6) | 105 (41.3) |
PD | 56 (37.1) | 67 (26.4) |
Death | 31 (20.5) | 38 (15.0) |
Patients censored, n (%) | 64 (42.4) | 149 (58.7) |
Median duration of follow-up (95% CIa), months | 12.2 (11.0 to 14.0) | 7.0 (5.8 to 8.3) |
Median PFSb (95% CIa), months | 8.9 (8.2 to 11.0) | 9.5 (8.2 to 11.2) |
ORR by IRC (primary end point) | ||
Patients contributing to the analysis, n | 151 | 224 |
ORR, n (%) | 68 (45.0) | 104 (46.4) |
95% CIc | 36.9 to 53.3 | 39.8 to 53.2 |
DOR by IRC | ||
Patients with confirmed CR or PR, n | 68 | 104 |
Patients with an event (PD or death), n (%) | 31 (45.6) | 33 (31.7) |
Patients censored, n (%) | 37 (54.4) | 71 (68.3) |
Median duration of follow-up (95% CIa), months | 10.9 (9.7 to 16.7) | 7.0 (6.9 to 9.7) |
Median DORb (95% CIa), months | 11.1 (8.4 to 18.5) | 11.1 (9.5 to 18.5) |
EQ-5D-5L VAS | ||
Patients with a deterioration event, n (%) | 61 (40.4) | 81 (31.9) |
Median TTDb (95% CIa), months | 8.3 (5.8 to 17.7) | 8.3 (5.9 to 17.7) |
EORTC QLQ-C30 global health status and quality of life score | ||
Patients with a deterioration event, n (%) | 54 (35.8) | 74 (29.1) |
Median TTDb (95% CIa), months | 15.2 (6.0 to 33.2) | 15.2 (6.2 to 33.2) |
EORTC QLQ-LC13 cough symptom scale | ||
Patients with a deterioration event, n (%) | 45 (29.8) | 52 (20.5) |
Median TTDb (95% CIa), months | 11.1 (11.1 to NE) | 13.8 (11.1 to NE) |
EORTC QLQ-LC13 chest pain symptom scale | ||
Patients with a deterioration event, n (%) | 45 (29.8) | 56 (22.0) |
Median TTDb (95% CIa), months | 17.7 (11.1 to NE) | 17.7 (11.8 to NE) |
EORTC QLQ-LC13 dyspnea symptom scale | ||
Patients with a deterioration event, n (%) | 76 (50.3) | 104 (40.9) |
Median TTDb (95% CIa), months | 5.5 (4.1 to 6.9) | 5.6 (4.1 to 6.9) |
Harms, n (%) – Safety analysis set | N = 152 | N = 255 |
Aes | 151 (99.3) | 246 (96.5) |
SAEs | 85 (55.9) | 115 (45.1) |
WDAE (discontinuation of study treatment) | 42 (27.6) | 52 (20.4) |
Deaths | 76 (50.0) | 86 (33.7) |
Notable harms, n (%) — Safety analysis set | N = 152 | N = 255 |
Hepatotoxicity | ||
ALT increased | 18 (11.8) | 29 (11.4) |
AST increased | 13 (8.6) | 19 (7.5) |
Blood ALP increased | 11 (7.2) | 20 (7.8) |
GGT increased | 9 (5.9) | 14 (5.5) |
Renal toxicity | ||
Blood creatinine increased | 44 (28.9) | 64 (25.1) |
Acute kidney injury | 6 (3.9) | 9 (3.5) |
Renal failure | 6 (3.9) | 9 (3.5) |
Chronic kidney disease | 4 (2.6) | 9 (3.5) |
Interstitial lung disease | 2 (1.3) | 2 (0.8) |
Pneumonitis | 6 (3.9) | 6 (2.4) |
Peripheral edema | 114 (75.0) | 153 (60.0) |
AE = adverse event; ALP = alkaline phosphatase; ALT = alanine transaminase; AST = aspartate transaminase; CI = confidence interval; CR = complete response; DOR = duration of response; EORTC QLQ-C30 = European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; EQ-5D-5L VAS = 5-Level EQ-5D visual analogue scale; GGT = gamma-glutamyl transferase; IRC = independent review committee; mITT = modified intention-to-treat; NE = not estimable; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PR = partial response; SAE = serious adverse event; TTD = time to deterioration; WDAE = withdrawal due to adverse event.
a95% CI calculated using the Brookmeyer and Crowley method.
bMedian estimated using the Kaplan-Meier method.
c95% CI calculated using the Clopper-Pearson method.
Source: VISION Clinical Study Report.14
Adverse Events
In cohort A, 99.3% of patients experienced at least 1 treatment-emergent AE. The most frequently reported AE was peripheral edema (75.0%). Grade 3 or higher peripheral edema was reported in 18 (11.8%) patients in cohort A. Other frequently reported AEs in cohort A included nausea (35.5%), diarrhea (31.6%), hypoalbuminemia (29.6%), and increased blood creatinine (28.9%). In cohort A plus C, 96.5% of patients experienced at least 1 treatment-emergent AE. The most frequently reported AE was peripheral edema (60.0%). Grade 3 or higher peripheral edema was reported in 20 (7.8%) patients. Other frequently reported treatment-emergent AEs in cohort A plus C were nausea (26.3%), diarrhea (26.3%), increased blood creatinine (25.1%), and hypoalbuminemia (23.1%).
Serious Adverse Events
In cohort A, 55.9% of patients experienced at least 1 serious adverse event (SAE) as of the July 1, 2020, data cut-off date. The most frequently reported SAEs were pleural effusion (8.6%), disease progression (6.6%), and pneumonia (6.6%). In cohort A plus C, 45.1% of patients experienced at least 1 SAE. The most frequently reported SAEs were pleural effusion (6.7%), disease progression (4.7%), and pneumonia (4.7%).
Withdrawals Due to Adverse Events
In cohort A, a total of 42 (27.6%) of patients permanently discontinued study treatment due to an AE. The most frequently reported AEs leading to treatment discontinuation were peripheral edema (4.6%), pleural effusion (3.3%), and general physical health deterioration (2.6%). In cohort A plus C, 20.4% of patients permanently discontinued study treatment due to an AE. The most frequently reported AEs leading to treatment discontinuation were peripheral edema (3.5%) and pleural effusion (2.0%).
Mortality
In cohort A, 50.0% patients had died as of the July 1, 2020, data cut-off date. Causes of death were reported to be disease progression, in 39.5% of patients, and an AE, in 7.9% of patients. In cohort A plus C, 33.7% of patients had died. Causes of death were reported to be disease progression, in 25.9% of patients, and an AE, in 5.9% of patients.
Notable Harms
The most frequently reported hepatotoxicity-related AEs in cohort A and cohort A plus C were increased alanine transaminase (ALT) (11.8% and 11.4%, respectively), increased aspartate transaminase (AST) (8.6% and 7.5%, respectively), increased blood alkaline phosphatase (ALP) (7.2% and 7.8%, respectively), and increased gamma-glutamyl transferase (GGT) (5.9% and 5.5%, respectively). The most frequently reported renal toxicity-related AE in cohort A and cohort A plus C was increase in blood creatinine (28.9% and 25.1%, respectively). A total of 2 patients experienced interstitial lung disease, and 6 patients experienced pneumonitis in cohort A. As of the data cut-off date, 75.0% of patients in cohort A and 60.0% of patients in cohort A plus C experienced peripheral edema.
Critical Appraisal
For the primary end point and most secondary end points, an IRC was appropriately used. The primary limitations of the VISION trial were the absence of a comparator group and of statistical testing, and open-label administration of tepotinib. The time-to-event analyses were appropriate, although the data are difficult to interpret in a single-arm trial without a control group. Open-label administration of tepotinib may have affected subjectively measured outcomes, such as health-related quality of life (HRQoL) and patient-reported outcomes (PROs), response, and AEs, although the direction of the potential bias is unknown. In addition, there is uncertainty in the HRQoL and PRO data due to reduced sample sizes in later treatment cycles, which is likely to bias results in favour of tepotinib and overestimate treatment effects. Due to the absence of a comparator group and statistical testing, no definitive conclusions can be drawn on the efficacy of tepotinib based on the VISION trial.
The treatment regimen used in the VISION trial aligns with the Health Canada–recommended dosage. The VISION trial was an international, multi-centre study, but there were no sites in Canada. The clinical experts consulted by CADTH indicated that the eligibility criteria used in the VISION trial were appropriate and commonly used in NSCLC trials. The VISION trial restricted enrolment to patients with an ECOG PS of 0 or 1, and patients with NSCLC in Canada often have an ECOF PS of 2 or more. The VISION trial used liquid biopsy and/or tumour tissue biopsy to determine study patients’ METex14 skipping alteration status; the clinical experts indicated that both methods could be used in Canada.
Description of Studies
The sponsor submitted an indirect treatment comparison (ITC) consisting of 2 parts: an indirect comparison of individual patient data from the VISION trial to pooled real-world data (RWD) using propensity scoring, and an indirect comparison of individual patient data from the VISION trial to retrospective observational studies using an unanchored MAIC.15 In the indirect comparison using propensity scoring, tepotinib data from the VISION trial were compared to chemotherapy (without immunotherapy) and immunotherapy as monotherapy using a dataset derived from 4 real-world evidence (RWE) cohort studies and a database. For the indirect comparison using an unanchored MAIC, the VISION trial was compared to 3 retrospective observational studies including patients who were treated with chemotherapy or immunotherapy. The relative efficacy of tepotinib versus immunotherapy in combination with chemotherapy in NSCLC patients harbouring METex14 skipping alterations was not assessed. Efficacy was assessed in terms of PFS and OS. Safety outcomes were not assessed.
Efficacy Results
Overall, the authors of the ITC concluded that patients treated with tepotinib had a greater PFS time compared to patients treated with chemotherapy or immunotherapy. The authors also concluded that tepotinib conferred a benefit in OS compared to chemotherapy or immunotherapy, albeit of smaller magnitude than PFS. For the indirect comparisons of VISION to pooled RWD using propensity scoring, the Cox proportional hazard ratio (HR) for OS was 0.91 (95% CI, 0.62 to 1.35) in favour of tepotinib for the chemotherapy group and 0.91 (95% CI, 0.59 to 1.42) in favour of tepotinib for the immunotherapy group. For PFS, the Cox proportional HR was 0.49 (95% CI, 0.35 to 0.69) in favour of tepotinib for the chemotherapy group and 0.59 (95% CI, 0.39 to 0.90) in favour of tepotinib for the immunotherapy group. No HRs or other statistics were reported for the MAIC; the MAIC results were composed of Kaplan-Meier curves for visual comparison.
Harms Results
Harms outcomes were not assessed in the sponsor-submitted ITC.
Critical Appraisal
The description of the methods used in the ITC analyses lacked important details, which creates uncertainty in the data. The methods used to identify relevant studies and the criteria used for study selection were unclear. The sponsor submitted a systematic literature review16 that identified 2 of the 3 studies included in the MAIC; the third study included in the MAIC and the 4 RWD sources included in the indirect comparison using propensity scoring were not identified in this systematic literature review. Thus, it remains unclear how these studies were identified and selected for inclusion in the indirect comparisons. No a priori protocol for selecting studies for inclusion in the indirect comparisons specifically was reported. Furthermore, it is unclear why other studies that were identified in the sponsor’s systematic literature review were not included in the ITC. The quality of the RWD sources was not assessed by the authors of the ITC and therefore not considered in the ITC analysis. The quality of 2 studies included in the MAIC was determined to be low, and the quality of the third study was not assessed. Any potential risks of bias of the included data sources (i.e., methodological limitations) were not assessed and not reported. A limited assessment of heterogeneity was reported. Key gaps in the evidence provided by the ITC were that no safety outcomes were assessed and that chemotherapy in combination with immunotherapy was not included as a comparator.
Overall, substantial bias is expected in the results, given the inherent limitations of the study design. As a consequence, the results observed in the indirect treatment analyses are unlikely to be valid. The true effect may be substantially different than the results observed in the ITC. A number of key limitations related to the selection and assessment of studies and patients, as well as the methods used that could potentially bias the results, were identified. First, fundamental differences in study design between the VISION trial, RWD sources, and retrospective observational studies were noted, as were concerns over differences in the definition, assessment, and timing of the clinical end points. These differences could not be accounted for in the indirect comparisons. Second, there was limited assessment and reporting of clinically important heterogeneity, and the statistical analyses completed are unlikely to have accounted for all major differences. The generation of propensity scores and the unanchored MAIC did not include all important potential confounders or effect modifiers and prognostic factors. In the unanchored MAIC, a large reduction in effective sample size (ESS) was observed, which suggests there was likely significant heterogeneity between the VISION study and comparator studies. The results for comparisons with major reductions of ESS are not reliable. In addition, the indirect comparisons may have been biased by the differential distribution of invalid or missing data between the VISION clinical trial and retrospective datasets. Given these issues, there is substantial concern for the risk of bias in the sponsor-submitted ITCs, and no conclusions can be drawn from the data.
Regarding external validity, most patient data were from sites in the US. A number of important differences between the US and Canada likely exist with regard to the management of these patients, including differences in treatments available, health insurance coverage, and overall health care system structures, which would be expected to affect treatment eligibility and, thus, outcomes. In addition, the multiple studies included patients enrolled more than a decade ago, and these patients are unlikely to be representative of contemporary patients, as better therapies and supportive care are now available. This would be expected to bias the study results in favour of tepotinib.
No long-term extension studies or additional relevant studies were included in the sponsor’s submission to CADTH.
Conclusions
One ongoing phase II, single-arm trial (VISION) of tepotinib in patients with advanced (locally advanced or metastatic) NSCLC harbouring METex14 skipping alterations was identified in the systematic review conducted by CADTH. The VISION trial data were analyzed descriptively; no statistical hypotheses were tested. Per the clinical experts consulted by CADTH, the results suggested a potential beneficial effect of tepotinib on OS, PFS, ORR, and DOR, based on their clinical experience and expectations of the natural progression of the disease in patients with METex14 skipping alterations. The clinical experts indicated that there is significant unmet need for targeted treatment and improved outcomes in this patient population. However, due to the absence of a comparator arm and statistical testing, no definitive conclusions can be drawn regarding the efficacy of tepotinib based on the VISION trial. The HRQoL and PRO data from VISION suggested that quality of life may be maintained with tepotinib therapy per the clinical experts, but there is substantial uncertainty in these data due to decreased sample sizes in later treatment cycles, open-label administration of tepotinib, and absence of a comparator arm. As a result, the effect of tepotinib on HRQoL and PROs remains unknown. Almost all study patients reported treatment-emergent AEs, the most common of which was peripheral edema. The most frequently reported SAEs were pleural effusion, disease progression, and pneumonia. Few patients experienced interstitial lung disease or pneumonitis. The most common cause of death was disease progression. The clinical experts consulted by CADTH indicated that the safety of tepotinib was acceptable.
No direct evidence on the relative efficacy and safety of tepotinib versus standard of care therapies used to treat advanced NSCLC with METex14 skipping alterations in Canada (i.e., immunotherapy, chemotherapy, or immunotherapy with chemotherapy) was identified. Results from the indirect treatment analyses submitted by the sponsor suggested that tepotinib therapy may be associated with a benefit in PFS and OS compared to chemotherapy and immunotherapy. However, the ITCs are associated with substantial risk of bias and important limitations (i.e., methodological limitations, limited assessment of heterogeneity, reporting lacked important details, small sample sizes). In view of the substantial uncertainty in the ITC results, no conclusions can be drawn on the efficacy of tepotinib compared to chemotherapy or immunotherapy in patients with advanced NSCLC harbouring METex14 skipping alterations. Harms outcomes were not assessed in the ITC. The potential benefits and safety of tepotinib compared with other therapies remain unknown.
Introduction
Disease Background
Lung cancer is the most commonly diagnosed cancer and the leading cause of deaths from cancer in Canada.1 Survival from lung cancer of all stages and histologic subtypes is poor, with an overall 5-year net survival of 19%.1 In 2020, it was estimated that there would be 29,800 new cases of lung cancer diagnosed and 21,200 deaths from lung cancer that year.1 It is estimated that 1 in 17 Canadians will die from lung cancer.17
Lung cancer is classified into small cell lung cancer or NSCLC, which accounts for approximately 88% of cases in Canada.1 NSCLC is further classified into 3 main histologic subtypes: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma. The symptoms of metastatic NSCLC are often variable at presentation and depend on the site of metastasis. At presentation, the most common signs and symptoms of NSCLC include persistent cough, shortness of breath, chest pain, wheezing, and hemoptysis.2 In patients with advanced NSCLC and distant metastasis, signs and symptoms may include fatigue, loss of appetite, pain, bone pain, headache, neurologic or psychiatric abnormalities, paraplegia, hepatomegaly, and pathological fractures.3
The MET receptor tyrosine kinase and its ligand are oncogenic drivers of NSCLC. Mutations that result in loss of exon 14 in the MET gene, called METex14 skipping alterations, lead to dysregulation and inappropriate signalling.4 METex14 skipping alterations occur in approximately 3% of NSCLC cases and are associated with poor prognosis, according to retrospective studies and the clinical experts consulted by CADTH.4-7 A retrospective study has also found that patients with NSCLC harbouring METex14 skipping mutations may be less responsive to immunotherapy.8 Patients with NSCLC harbouring METex14 skipping mutations are more likely to be older, and the mutation occurs more frequently in adenocarcinoma.4,6 The clinical experts consulted by CADTH reported that METex14 skipping mutations can be seen in squamous cell and sarcomatoid lung cancer as well. There are several methods to detect METex14 skipping mutations in NSCLC using tumour tissue biopsy or liquid biopsy (e.g., NGS-based panel tests, immunohistochemistry, real-time polymerase chain reaction, Sanger sequencing).4,5
Standards of Therapy
The clinical experts consulted by CADTH indicated that, in current practice, patients in Canada with advanced NSCLC (i.e., stage IV and stage IIIB not amenable to curative treatment approaches) harbouring METex14 skipping mutations are usually treated according to guidelines for advanced NSCLC without oncogenic driver alterations. The clinical experts indicated that the goals of treatment in locally advanced (not amenable to curative treatment) or metastatic NSCLC are to improve OS, PFS, and response rate, as well as maintain HRQoL.
First-line treatment for patients with locally advanced or metastatic NSCLC without driver mutations is immunotherapy, with or without chemotherapy.9,10 According to the clinical experts consulted by CADTH, pembrolizumab, in combination with a platinum-doublet therapy, as well as nivolumab plus ipilimumab with 2 cycles of a platinum-doublet therapy, are treatment options, regardless of PD-L1 expression. For patients with PD-L1 tumour proportion score of 50% or more, single-agent pembrolizumab is an option.9 Patients ineligible for immunotherapy may receive chemotherapy in the first-line setting. A combination of 2 cytotoxic chemotherapies is recommended for these patients.10 Platinum-based doublet regimens are recommended over non-platinum therapy; non-platinum chemotherapy may be offered to patients who are ineligible for platinum therapy.10
In the second- or later-line setting, single-agent chemotherapy, single-agent immunotherapy, or platinum-based chemotherapy doublets may be used.10 According to the clinical experts consulted by CADTH, immunotherapy is generally used first-line for patients who are eligible; therefore, second-line immunotherapy is less common. In the post-chemotherapy setting, nivolumab, pembrolizumab, or atezolizumab may be used.10 In the third-line setting, docetaxel or pemetrexed may be recommended.10
In March 2021, the American Society of Clinical Oncology (ASCO) and the OH-CCO issued a joint guideline update, including guidance for patients with NSCLC with a METex14 skipping mutation.18 These guidelines recommend that, for patients with a METex14 skipping mutation, an ECOG PS of 0 to 2, and previously untreated NSCLC, clinicians may offer MET-targeted therapy with capmatinib or tepotinib, or standard first-line therapy based on nondriver mutation guidelines. For patients with a METex14 skipping mutation and an ECOG PS of 0 to 2, who have previously received or been ineligible for first-line chemotherapy with or without immunotherapy, clinicians may offer MET-targeted therapy with capmatinib or tepotinib. At the time of this review, capmatinib was being reviewed by Health Canada under the Notice of Compliance with Conditions (NOC/c) guidance (the Health Canada submission was accepted for review September 2021).19
Drug
Tepotinib is an oral, ATP-competitive, and highly selective MET receptor tyrosine kinase inhibitor.11 The MET receptor and its ligand hepatocyte growth factor (HGF) are involved in carcinogenesis and tumour progression. Oncogenic activation of MET has been shown to promote cancer cell proliferation, survival, migration and invasion, and tumour angiogenesis, as well as to mediate resistance to cancer therapies.11 Tepotinib targets MET receptor tyrosine kinase, including variants with exon 14 skipping alterations and inhibits HGF-dependent and -independent MET phosphorylation and MET-dependent downstream signalling pathways. Tepotinib is taken orally, and the recommended dosage is 450 mg as tepotinib hydrochloride once daily. Per the Health Canada product monograph, it is recommended that treatment be continued until disease progression or unacceptable toxicity.
Tepotinib underwent expedited review by Health Canada through Project ORBIS. On May 27, 2021, tepotinib was issued a Notice of Compliance with Conditions (NOC/c), pending the results of trials to verify its clinical benefit. Tepotinib is indicated for the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping alterations. Documentation of METex14 skipping alteration status, based on a validated METex14 assay, is required before treatment with tepotinib. The sponsor’s reimbursement request is for the treatment of adult patients with advanced NSCLC harbouring METex14 skipping alterations. Tepotinib has not been previously reviewed by CADTH.
Table 3: Key Characteristics of Tepotinib
Characteristic | Tepotinib |
|---|---|
Mechanism of action | Receptor tyrosine kinase inhibitor that targets MET and inhibits HGF-dependent and -independent MET phosphorylation and MET-dependent downstream signalling pathways |
Indicationa | For the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring MET tyrosine kinase receptor exon 14 skipping alterations |
Route of administration | Oral |
Recommended dosage | 450 mg (as tepotinib hydrochloride) once daily |
Serious adverse effects or safety issues | Hepatotoxicity Interstitial lung disease/pneumonitis Embryo-fetal toxicity |
Other | Tepotinib has been issued marketing authorization with conditions, pending the results of trials to verify its clinical benefit. |
HGF = hepatocyte growth factor; MET = mesenchymal-epithelial transition; NSCLC = non–small cell lung cancer.
aHealth Canada–approved indication.
Source: Product monograph.11
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
CADTH received input from 2 patient advocacy groups: the LHF and LCC.
The LHF (formerly the Ontario Lung Association) is a registered charity that advocates for patients with lung diseases and their caregivers. They collected information from an online survey with 13 patients and 1 caregiver (on or before August 31, 2021), and phone interviews with 2 patients (in September 2021). In addition, the LHF received input from a registered nurse and a certified respiratory educator. Patients’ experiences with lung cancer varied from no symptoms to negative impact on their instrumental activities of daily living. The most frequently reported symptoms were shortness of breath (64%), fatigue (57%), and depression (25%). Other notable concerns expressed by patients were anxiety associated with a poor prognosis and stigma attached to their diagnosis, which prevent them from fully engaging with their families and participating in social activities. Patients reported experiencing varying degrees of side effects from chemotherapy, ranging from hair loss and mild fatigue that are well-tolerated, to nausea, vomiting, and anemia that significantly affected their quality of life, even leading to hospitalization. Moreover, some patients experienced deconditioning and chronic fatigue after surgery, as well as tissue scarring, skin changes, and lung injury–related chronic obstructive pulmonary disease from radiation. Patients indicated that they want treatments that stop or slow the progression of disease, prolong life, and have minimal side effects. Given the poor prognosis of lung cancer, which is usually discovered in later stages, patients want treatments that are effective in advanced disease. Last, patients reported that they struggled to navigate the health care system and want equal access to biomarker testing at the time of diagnosis or early in treatment.
LCC is a national, registered organization that is a resource for lung cancer education, patient support, research, and advocacy. It collected data from phone and video interviews with 4 patients with NSCLC (in the US and Canada) and 1 caregiver (in Canada) in September 2021. All patients, including a patient for whom the caregiver answered the questionnaire, had confirmed METex14 skipping mutations. According to the survey, some patients found the diagnosis difficult to accept because the lung cancer was found at an advanced stage and the patient was otherwise healthy, and some were never exposed to smoking. In addition, patients indicated they became increasingly dependent on caregivers due to fatigue and functional deterioration, which increased mental distress and financial burden. The patients expressed that it is important to maintain independence and functioning so they can live in a manner similar to pre-diagnosis. Additionally, patients expressed that improving symptoms of NSCLC and quality of life, reducing side effects, delaying disease progression or achieving long-term remission, and living longer were important outcomes. There were 4 patients who had been treated with tepotinib, and all reported that their quality of life improved within a few months of starting therapy (e.g., reduced pain, returned to active lifestyle). One patient with a brain metastasis, who had previously tried another targeted therapy, became free of brain cancer after 8 months of tepotinib treatment. Patients also reported experiencing side effects from tepotinib treatment. One patient became hospitalized due to pulmonary edema and had to interrupt tepotinib therapy for 2 months. Two patients experienced edema at the extremities, which was managed with compression stockings. Patients who had previously tried other MET inhibitors and encountered tolerability and/or resistance issues appreciated having another treatment option. In terms of health care navigation, 1 patient did not have access to biomarker testing for MET mutations in Canada and had testing done in the US, where she paid $4,000 out of pocket after 50% reimbursement. This patient indicated that every Canadian with lung cancer should be able to access and afford the biomarker testing as well as the treatment itself so that they can benefit from targeted therapy.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of NSCLC.
The clinical experts consulted by CADTH indicated that the goals of treatment in locally advanced (not amenable to curative treatment) or metastatic NSCLC are to improve survival, delay progression, and increase clinical response rate, as well as maintain or improve HRQoL. In addition, the clinical experts thought that new treatment options should minimize AEs compared to current standard of care therapies.
The clinical experts noted that retrospective studies have indicated that patients with METex14 mutations have a poor prognosis, and there appears to be less benefit with the use of immunotherapy, even in patients with high PD-L1 expression.7,8 Moreover, the clinical experts noted that patients with METex14 skipping mutations tend to be an older population compared to other lung cancer patient populations with different targetable mutations. The clinical experts reported that elderly patients often experience increased side effects with chemotherapy; thus, treatments that are better tolerated are needed.
The clinical experts noted that there is currently no targeted treatment for NSCLC with METex14 mutations that is publicly funded in Canada. Patients with METex14 skipping alterations in Canada are currently treated per the funded treatment algorithm for locally advanced or metastatic NSCLC without an identified driver mutation.
Place in Therapy
The clinical experts indicated that tepotinib would preferentially be used in the first-line setting for patients with METex14 skipping alterations because targeted agents are usually preferred as initial therapy. If patients received tepotinib or another MET inhibitor as first-line treatment, later lines of therapy would consist of chemotherapy, immunotherapy, or chemotherapy plus immunotherapy, based on established provincial funding algorithms. If tepotinib was not used as first-line therapy, it would be used as second- or later-line therapy.
Patient Population
The clinical experts thought tepotinib should be used in patients with locally advanced or metastatic NSCLC harbouring a METex14 skipping mutation who meet the eligibility criteria used in the VISION trial. Patients with NSCLC with METex14 mutations would be identified by molecular biomarker testing using tumour tissue biopsy or liquid biopsy and NGS panel testing. Ideally, this testing would be done at the time of diagnosis of advanced NSCLC. The clinical experts noted that both tumour tissue biopsy and liquid biopsy were used in the VISION trial. The clinical experts would also consider using tepotinib in patients with an ECOG PS of 2 or more who otherwise met the VISION trial eligibility criteria.
Assessing Response to Treatment
The clinical experts indicated that response to treatment is assessed using CT or bone scans every 2 to 4 months, as clinically indicated. In addition, clinical assessments are conducted at regular intervals (e.g., every 3 months) to assess symptom control if patients present with disease-related symptoms before starting treatment. The clinical experts reported that a clinically meaningful response to treatment would be improved survival and maintenance or improvement in HRQoL.
Discontinuing Treatment
The clinical experts indicated that treatment with tepotinib would be discontinued if a patient experienced disease progression or intolerable treatment-related side effects.
Prescribing Conditions
The clinical experts indicated that tepotinib would be administered in the outpatient setting under the supervision of a medical oncologist.
This section was prepared by CADTH staff based on the input provided by clinician groups.
CADTH received input from 3 clinician groups comprising a total of 21 clinicians: Northeast Cancer Centre – Thoracic Cancer Clinicians, OH-CCO’s Lung and Thoracic Cancers Drug Advisory Committee, and LCC’s Medical Advisory Committee.
Northeast Cancer Centre – Thoracic Cancer Clinicians is a group of oncologists based in Sudbury, Ontario, who treat and advocate for older, rural patients, mostly francophone and Indigenous, throughout 12 satellite systemic therapy sites. They stated that patients with METex14 skipping mutations tend to be older, female, and non-smokers, and they have poorer prognosis. The clinicians reported that patients are currently treated without precision care and/or patient-directed therapy (i.e., systemic chemotherapy and immunotherapy) and are treated according to guidelines for NSCLC without mutations. These patients become refractory to the current treatments and experience frequent side effects. The clinician group indicated that there is a need for molecularly targeted and better-tolerated therapy that can delay disease progression, prolong OS, and improve quality of life. The oncologist group suggested that tepotinib could be used as any line of therapy for patients with good ECOG PS and METex14 alterations confirmed by either liquid or tissue biopsy. The clinician groups indicated that patients should be monitored for symptoms, disease progression, and survival every 3 months or at any change in ECOG PS. The group indicated that tepotinib should be discontinued if the patient’s PS deteriorates, intolerable AEs (e.g., edema) occur, or the disease progresses.
The OH-CCO’s Lung and Thoracic Cancers Drug Advisory Committee provides timely, evidence-based clinical and health system guidance. According to the OH-CCO, current first-line therapies for patients with METex14 mutation positive, stage IV NSCLC include pembrolizumab (if PD-L1 > 50%), platinum-based chemotherapy doublet plus pembrolizumab (with pemetrexed if PD-L1 is 50% or less and histologic subtype is non-squamous; with paclitaxel if PD-L1 is 50% or less and histologic subtype is squamous), or 2 cycles of platinum-based chemotherapy plus nivolumab and ipilimumab. Docetaxel or clinical trials could be considered as subsequent therapies. In addition, nondrug treatments, palliative or supportive care, and radiation could be used for symptomatic lesions. They stated that NSCLC is incurable, with a median OS of less than 2 years. Thus, there is an unmet need for a new therapy that can prolong survival, delay disease progression, reduce/improve symptoms, and shrink tumours. The clinician group reported that, unlike other targetable mutations, METex14 alterations occur in patients with squamous and sarcomatoid cancers, as well as certain pulmonary risk factors, such as smoking. Therefore, these patients fall into the unmet need group as well. According to the OH-CCO, because of tepotinib’s unique mechanism of action, tepotinib monotherapy would complement, not displace, other treatment options currently available. Based on OH-CCO and ASCO joint guidelines for stage IV NSCLC with targetable mutations, tepotinib would be preferentially offered in the community setting as first-line therapy and as a subsequent therapy to those who have already received other treatments. The OH-CCO indicated that re-treatment could be considered in rare cases, such as recurrence after completion of tepotinib therapy or interruption of tepotinib therapy for reasons other than disease progression. The group indicated that METex14 mutations should be identified in eligible patients by NGS platform testing. If MET mutation status is unknown, the patient should not be treated with tepotinib, the group thought. The clinician group indicated that, once tepotinib is initiated, patients should be monitored for improvement in symptoms, quality of life, tumour shrinkage, and disease progression every 4 to 8 weeks with imaging (e.g., radiography, CT scans), if imaging is needed. In cases of disease progression or intolerable side effects, the OH-CCO thought that tepotinib should be discontinued.
The LCC’s Medical Advisory Committee shared their recommendations on the management of NSCLC with MET mutations, which is in press at the journal Current Oncology as of this writing. The current therapies for treatment-naive patients are platinum-doublet, platinum-doublet with pembrolizumab (mostly for patients with PD-L1 of less than 50%; or patients with PD-L1 of 50% or more who are non-smokers, women, with a high burden of disease or symptoms), and pembrolizumab alone (if PD-L1 is 50% or more), unless this immunotherapy is contraindicated (e.g., due to auto-immune disease or active immunosuppressant therapy). For patients with metastatic NSCLC who had disease progression on prior systemic therapy, treatment options are platinum-doublet (if pembrolizumab was prior therapy); anti–PD-(L)1 therapies such as pembrolizumab, nivolumab, or atezolizumab (if platinum or pemetrexed, or platinum-doublet and pembrolizumab were previous therapy); and docetaxel (if disease progressed on platinum-doublet and pembrolizumab). The group reported that, since NSCLC with METex14 skipping mutations is aggressive, often resistant to anticancer therapies, and metastatic, the response rates to non-targeted therapies tend to be low, leading to poor prognosis. The group indicated that selective inhibition of MET, with few off-target effects, would be beneficial. The group reported that there is an unmet need for therapy that can improve OS, can rapidly improve symptoms, can delay disease progression, has intracranial activity for brain metastasis, has tolerable side effects, and minimizes health care resource utilization. Because METex14 skipping mutations often occur in older patients, smokers, those with sarcomatoid carcinoma, and a small percentage of those with squamous cell carcinoma, tepotinib could address unmet need in this population. The group suggested that tepotinib be offered as a single agent as early as possible, in the community setting, and in a line-agnostic manner to treatment-naive and pre-treated patients who have ECOG PS score 0 to 2 (and potentially 3) and a METex14 skipping mutation confirmed by NGS testing of either tumour or liquid biopsy. The group felt that NGS testing is unlikely to pose an obstacle to access to tepotinib because it is provincially funded in some circumstances and available through philanthropic or research settings in other cases. Furthermore, the group advocates that liquid biopsy testing should be considered across Canada to reduce the need for biopsy or repeat biopsy of tumour tissue. Last, the clinician group indicated that patients should have follow-up visits every 2 to 3 months to monitor for symptoms, with or without radiological evidence (e.g., CT imaging) of tumour shrinkage. The group stated that, if the patient wishes, safety issues arise, or the disease progresses, tepotinib should be discontinued.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Generalizability | |
The VISION trial eligibility included patients with ECOG PS of 0 or 1. Should patients with ECOG PS ≥ 2 be eligible? | The clinical experts indicated that it would be reasonable to offer tepotinib to patients with an ECOG PS of 2. One clinical expert indicated that they would not treat patients with tepotinib if they had an ECOG PS of 4 but would consider offering tepotinib to select patients with an ECOG PS of 3, because of the rapid responses seen with targeted therapies, in their experience. The second clinical expert indicated that they would consider offering tepotinib to patients with an ECOG PS > 2 because they thought the treatment could improve the patient’s ECOG PS. |
Patients currently receiving alternative first-line or subsequent lines of therapy would have a time-limited opportunity to switch to tepotinib. Should patients receiving these treatments be switched to tepotinib at the time of public funding, or would it be preferable to wait until disease progression on alternative therapies and use tepotinib as the next line of treatment? | The clinical experts indicated that, if patients were responding to their current treatment, they would not switch the patients to tepotinib at the time of public funding. The clinical experts would keep the patients on their current treatment until they experienced progressive disease and then use tepotinib for the next line of therapy. |
Funding algorithm | |
In patients with advanced NSCLC with driver mutations (e.g., EGFR, ALK, ROS, BRAF) who receive targeted treatment in the first-line setting, chemotherapy is required before accessing immunotherapy, in alignment with previous pERC recommendations. The drug programs would like to inform pERC that jurisdictions would use the same sequencing principles for subsequent therapies used after tepotinib, regardless of PD-L1 TPS. The drug programs noted that tepotinib may change place in therapy of drugs reimbursed in subsequent lines. | For consideration by pERC |
Care provision issues | |
Testing for METex14 skipping alterations may not be routinely available in some jurisdictions and would need to be implemented. | For consideration by pERC |
ECOG PS = Eastern Cooperative Oncology Group performance status; METex14 = MET exon 14; NSCLC = non–small cell lung cancer; PD-L1 = programmed death-ligand 1; pERC = CADTH pan-Canadian Oncology Drug Review Expert Review Committee; ROS = reactive oxygen species; TPS = tumour proportion score.
Clinical Evidence
The clinical evidence included in the review of tepotinib is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. No additional relevant studies were submitted by the sponsor or identified in the literature.
After CADTH issued a draft pERC recommendation for tepotinib in March 2022, the following additional information was provided to CADTH. The sponsor provided additional unpublished data from the pivotal VISION study with a data cut-off date of February 2021. The sponsor provided an additional unanchored MAIC comparing tepotinib to the combination of chemotherapy and immunotherapy. These data were not included in the submission to CADTH (the sponsor reported that the data became available only after the CADTH recommendation was issued). A comparison of tepotinib to a combination of chemotherapy and immunotherapy has been identified as an important gap in the evidence. The information has been summarized and critically appraised as an addendum to the CADTH report in Appendix 5.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of tepotinib (450 mg, as tepotinib hydrochloride) for the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping mutations.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping alterations. Subgroups:
|
Intervention | 450 mg tepotinib (equivalent to 500 mg tepotinib hydrochloride) orally once daily |
Comparator | Immunotherapy (e.g., pembrolizumab, nivolumab, nivolumab + ipilimumab, atezolizumab) Chemotherapy (e.g., platinum-doublet regimens, non–platinum-doublet regimens, pemetrexed, docetaxel, vinorelbine) Immunotherapy in combination with chemotherapy |
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, mortality, notable harms/harms of special interest (e.g., hepatotoxicity, renal toxicity, interstitial lung disease/pneumonitis, peripheral edema) |
Study Designs | Published and unpublished phase II, III, and IV RCTs |
AE = adverse event; CNS = central nervous system; DOR = duration of response; ECOG PS = Eastern Cooperative Oncology Group performance status; HRQoL = health-related quality of life; METex14 = MET exon 14; NSCLC = non–small cell lung cancer; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PRO = patient-reported outcome; RCT = randomized controlled trial; SAE = serious adverse events; vs. = versus; WDAE = withdrawal due to adverse events.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.20
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Tepmetko/tepotinib. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on September 23, 2021. Regular alerts updated the search until the meeting of the CADTH pERC on February 9, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature reference.21 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
One study was identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Table 6: Details of Included Studies
Study detail | VISION |
|---|---|
Designs and populations | |
Study design | OL, phase II, single-arm, multicohort study |
Locations | 130 sites in 11 countries in Europe, Asia, and the US |
Patient enrolment dates | September 6, 2016 — ongoing |
Enrolled as of July 1, 2020, DCO (N) |
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | 500 mg tepotinib hydrochloride hydrate (equivalent to 450 mg tepotinib) orally once daily |
Comparator(s) | None |
Duration | |
Phase | NA |
Pre-screening | Not specified (can be more than 28 days before the first dose of trial treatment) |
Screening | 28 days |
Treatment | Until disease progression, death, an AE leading to discontinuation, or withdrawal of consent |
Follow-up | 30 days |
Outcomes | |
Primary end point | ORR by IRC assessment (CR or PR as the best overall response) from first administration of study treatment to the first observation of PD |
Secondary and exploratory end points | Secondary:
Exploratory:
|
Notes | |
Publications | Paik et al. (2020)13 Sakai et al. (2021)22 |
AE = adverse event; CR = complete response; DCO = data cut-off; DOR = duration of response; ECG = electrocardiogram; ECOG PS = Eastern Cooperative Oncology Group performance status; EORTC = European Organization for the Research and Treatment of Cancer; EQ-5D-5L = 5-Level EQ-5D; HGF = hepatocyte growth factor; IRC = independent review committee; METex14 = MET exon 14; NA = not applicable; NSCLC = non–small cell lung cancer; OL = open-label; ORR = objective response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PK = pharmacokinetic; PR = partial response; QLQ-C30 = Quality of Life Questionnaire Core 30; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer 13; RECIST = Response Evaluation Criteria in Solid Tumours; TEAE = treatment-emergent adverse event.
Note: 4 additional reports were included from the sponsor’s submission to CADTH.12,14,23
aPatients with brain metastases whose condition was neurologically stable and whose glucocorticoid dose was being tapered were eligible to participate, as were patients with untreated asymptomatic brain metastases measuring 1 cm or less in diameter.
Source: VISION Clinical Study Reports,12,14 Paik et al. (2020).13
Description of Studies
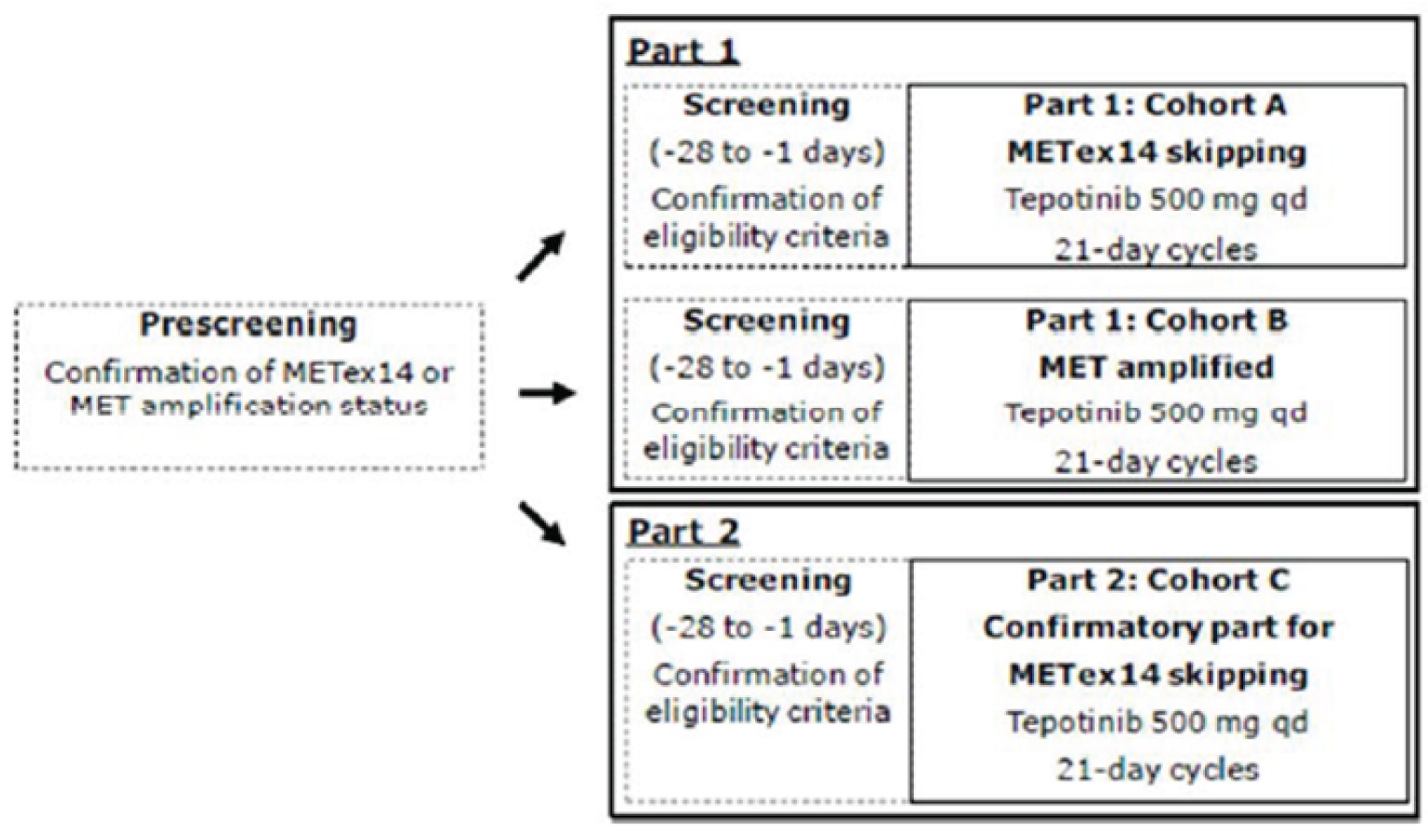
One ongoing, phase II, single-arm, open-label, multi-centre study (VISION) was included in this systematic review.12-14 The primary objective of the VISION study was to assess the efficacy of tepotinib in patients with advanced (locally advanced or metastatic) NSCLC, as per objective response (confirmed CR or PR), determined according to RECIST v1.1 criteria, based on independent review in patients that tested positive for METex14 skipping alterations or MET amplification. A total of 130 sites in 11 countries in Europe, Asia, and the US participated. There were 3 cohorts in the VISION study (Cohorts A, B, and C). Patients were selected for each cohort based on defined MET alterations or MET amplification identified in tumour tissue and/or in ctDNA derived from plasma. Patients with METex14 skipping alterations were enrolled in cohort A and cohort C under the same eligibility criteria and underwent the same study procedures. Cohort A was the pivotal cohort for the METex14 skipping alterations population; cohort C was a confirmatory cohort added as a protocol amendment to extend and confirm the existing results for cohort A, and to expand the METex14 skipping alteration population in the study. After accrual for cohort A was complete, enrolment at sites was shifted from cohort A to cohort C (cohort A: N = 151; cohort A plus C: N = 254). Cohort B included patients who tested positive for MET amplification only and were negative for METex14 skipping alterations. Cohort B does not align with the Health Canada indication or reimbursement request; therefore, data for cohort B will not be presented in this review.
The VISION study design is depicted in Figure 2. Determination of patients’ METex14 skipping alteration status or MET amplification was conducted during the pre-screening period (which could be more than 28 days before the first dose of study treatment) after patients had signed a pre-screening informed consent form. Tumour tissue for testing was obtained from archived samples or from freshly obtained biopsy tissue. ctDNA was isolated from freshly collected plasma samples (i.e., liquid biopsy). Parallel testing for METex14 skipping alterations in both tumour tissue and plasma was highly recommended, although not required. If, for any reasons, either tumour tissue biopsy or liquid biopsy material was not available, a positive test result from either specimen type was sufficient to enrol a patient. After confirmation of METex14 skipping alteration status or MET amplification, patients entered the 28-day screening period to confirm that they met the study’s eligibility criteria. During the treatment period, patients in all cohorts received tepotinib monotherapy as tepotinib hydrochloride hydrate at a dosage of 500 mg once daily in 21-day cycles. The 500 mg tepotinib hydrochloride hydrate contained 450 mg tepotinib (active moiety). Patients continued treatment until disease progression according to RECIST v1.1 criteria, death, an AE leading to discontinuation, or withdrawal of consent. An end-of-treatment visit was conducted within 14 days of the last dose of study treatment. A 30-day follow-up visit was performed at 30 days after the last dose of study treatment for all patients who discontinued study treatment permanently, including patients who completed an end-of-treatment visit.
The primary analysis for the US FDA was an interim analysis with a data cut-off date of January 1, 2020.12,13 The primary analysis population for this interim analysis was patients enrolled in cohort A who received their first dose of tepotinib before April 2, 2019 (N = 99).12,13 An updated interim analysis was conducted with a data cut-off date of July 1, 2020.14 At both interim analyses, enrolment in cohort A was complete and enrolment in cohort C was ongoing at the time of data cut-off.
Populations
Inclusion and Exclusion Criteria
Patients were included in cohort A or cohort C of the VISION trial if they were adults 18 years of age or older (20 years of age or older in Japan) with histologically or cytologically confirmed, locally advanced or metastatic NSCLC with METex14 skipping mutations. Prospective testing of METex14 skipping mutations was performed centrally on ctDNA obtained from plasma (liquid biopsy) or by evaluating RNA obtained from tumour tissue biopsy. Dual testing by the 2 biopsy methods was not a requirement for enrolment, although it was recommended. All patients had measurable disease according to RECIST v1.1 and an ECOG PS of 0 or 1. In addition, all patients had negative results on local testing for the presence of EGFR mutations or ALK rearrangements. Patients could have received up to 2 previous treatments for advanced or metastatic NSCLC. Patients with brain metastases whose condition was neurologically stable and whose glucocorticoid dose was being tapered were eligible to participate, as were patients with untreated asymptomatic brain metastases measuring 1 cm or less in diameter.
Baseline Characteristics
Baseline characteristics of patients enrolled in cohort A and pooled cohort A plus C of the VISION study are summarized in Table 7. The mean age of study patients was 73 years. Most patients were White (70.9% in cohort A, 67.1% in cohort A plus C), had an ECOG PS of 1 (73.5% in cohort A, 72.2% in cohort A plus C), adenocarcinoma histology type (86.8% in cohort A, 81.2% in cohort A plus C), and stage IV disease at study entry (74.8% in cohort A, 64.3% in cohort A plus C). Approximately half of the patients in cohort A and cohort A plus C had received prior anticancer drug therapy for advanced or metastatic disease (54.3% and 51.0%, respectively). The most common types of prior anticancer therapies were cytotoxic therapy (49.7% in cohort A, 44.3% in cohort A plus C) and immunotherapy (25.8% in cohort A, 25.9% in cohort A plus C). The most frequently administered prior drug therapies were carboplatin, pemetrexed, cisplatin, and pembrolizumab. Most patients had not had prior anticancer surgery (68.9% in cohort A, 67.5% in cohort A plus C). Per IRC assessment, 9.9% of patients in cohort A and 12.2% of patients in cohort A plus C had brain metastases at baseline.
Table 7: Summary of Baseline Characteristics — VISION Cohort A and Cohort A + C as of July 1, 2020, Data Cut-Off Date
Characteristic | Cohort A mITT (N = 151) | Cohort A + C SAS (N = 255) |
|---|---|---|
Age, mean (SD) | 73.0 (8.97) | 72.5 (9.04) |
Sex, n (%) | ||
Male | 79 (52.3) | 123 (48.2) |
Female | 72 (47.7) | 131 (51.8) |
Race, n (%) | ||
White | 107 (70.9) | 171 (67.1) |
Black or African American | 1 (0.7) | 3 (1.2) |
Asian | 38 (25.2) | 72 (28.2) |
Not collected at this site | 4 (2.6) | 7 (2.7) |
Other | 1 (0.7) | 1 (0.4) |
Missing | 0 | 1 (0.4) |
Geographic region, n (%) | ||
Europe | 77 (51.0) | 128 (50.2) |
North America | 39 (25.8) | 54 (21.2) |
Asia | 35 (23.2) | 73 (28.6) |
Smoking history, n (%) | ||
Never used nicotine | 65 (43.0) | 124 (48.6) |
Former nicotine users | 75 (49.7) | 114 (44.7) |
Regular nicotine users | 3 (2.0) | 7 (2.7) |
Missing | 8 (5.3) | 10 (3.9) |
ECOG PS | ||
0 | 40 (26.5) | 71 (27.8) |
1 | 111 (73.5) | 184 (72.2) |
Histologic subtype, n (%) | ||
Squamous | 14 (9.3) | 25 (9.8) |
Adenocarcinoma | 131 (86.8) | 207 (81.2) |
Adenosquamous | 2 (1.3) | 6 (2.4) |
Sarcomatoid | 3 (2.0) | 6 (2.4) |
Other | 1 (0.7) | 9 (3.5) |
Disease stage at study entry, n (%) | ||
IIIB | 3 (2.0) | 8 (3.1) |
IV | 113 (74.8) | 164 (64.3) |
IVA | 13 (8.6) | 27 (10.6) |
IVB | 22 (14.6) | 52 (20.4) |
Prior anticancer drug therapy for advanced NSCLC, n (%) | ||
Yes | 82 (54.3) | 130 (51.0) |
No | 69 (45.7) | 125 (49.0) |
Number of prior anticancer drug therapy lines, n (%) | ||
1 | 48 (31.8) | 81 (31.8) |
2 | 33 (21.9) | 47 (18.4) |
3 | 1 (0.7) | 2 (0.8) |
Type of prior anticancer drug therapies,a n (%) | ||
Cytotoxic therapy | 75 (49.7) | 113 (44.3) |
Monoclonal antibody therapy | 12 (7.9) | 17 (6.7) |
Small molecules | 1 (0.7) | 3 (1.2) |
Immunotherapy | 39 (25.8) | 66 (25.9) |
Most common prior anticancer drug therapies,a,b n (%) | ||
Carboplatin | 50 (33.1) | 73 (28.6) |
Pemetrexed | 35 (23.2) | 52 (20.4) |
Cisplatin | 26 (17.2) | 42 (16.5) |
Pembrolizumab | 25 (16.6) | 41 (16.1) |
Paclitaxel | 18 (11.9) | 22 (8.6) |
Pemetrexed disodium | 17 (11.3) | 27 (10.6) |
Carboplatin/paclitaxel | 12 (7.9) | 15 (5.9) |
Cisplatin/pemetrexed | 12 (7.9) | 17 (6.7) |
Bevacizumab | 11 (7.3) | 16 (6.3) |
Carboplatin/pemetrexed | 10 (6.6) | 14 (5.5) |
Nivolumab | 8 (5.3) | 11 (4.3) |
Prior anticancer radiotherapy, n (%) | ||
Yes | 74 (49.0) | 129 (50.6) |
No | 77 (51.0) | 126 (49.4) |
Prior anticancer surgery, n (%) | ||
Yes | 47 (31.1) | 83 (32.5) |
No | 104 (68.9) | 172 (67.5) |
Brain metastases at baseline by IRC, n (%) | ||
Present | 15 (9.9) | 31 (12.2) |
Absent | 136 (90.1) | 223 (87.5) |
Brain metastases at baseline by IRC or investigator, n (%) | ||
Present | 23 (15.2) | NR |
Absent | 128 (84.8) | NR |
ECOG PS = Eastern Cooperative Oncology Group performance status; IRC = independent review committee; mITT = modified intention-to-treat; NR = not reported; NSCLC = non–small cell lung cancer; SAS = safety analysis set; SD = standard deviation.
aAll prior therapy lines are considered, so patients may be included in more than 1 category.
bFrequency 5% or higher.
Source: VISION Clinical Study Report.14
Interventions
In the VISION study, patients received 500 mg tepotinib hydrochloride hydrate, which is equivalent to 450 mg tepotinib (the active moiety). Patients took tepotinib orally once daily, at approximately the same time each morning (± 2 hours), immediately after breakfast with a full glass of water (approximately 200 mL), during each 21-day cycle until disease progression per RECIST v1.1 criteria, withdrawal of consent, or an AE leading to discontinuation.
Any concomitant medications that were considered necessary for the patients’ welfare and would not interfere with tepotinib were permitted at the investigator’s discretion. Supportive treatment (e.g., bisphosphonates, agents for improving appetite), initiated before study entry, was allowed to continue. Changes in dose or schedule on study were discouraged. Initiation of prophylactic bisphosphonates during study treatment was avoided. Symptomatic treatment of brain metastasis with anticonvulsants known to have a reduced risk for drug interactions (e.g., lamotrigine, levetiracetam, pregabalin, or valproic acid) was allowed. Localized radiation therapy to alleviate symptoms such as bone pain was allowed, provided that the total dose delivered was in a palliative range and did not involve a target lesion used to determine response.
The following treatments were prohibited during the study: any other anticancer therapy, including chemotherapy, biologic therapy, hormonal therapy for anticancer purposes, targeted therapy, or investigational product other than tepotinib; drugs for which the product labelling included a contraindication for permeability glycoprotein, breast cancer resistance protein, organic cation transporter 1, organic cation transporter 2, multidrug and toxin extrusion protein 1, and multidrug and toxin extrusion protein 2 inhibiting drugs; and drugs that were known to induce permeability glycoprotein and thereby may decrease efficacy of tepotinib (e.g., avasimibe, carbamazepine, phenytoin, rifampin, Saint John’s Wort). Patients were withdrawn from the VISION study if they were using prohibited medicines for any reason.
Dose Reductions and Interruptions
If a patient experienced an AE in the VISION study, the investigator could either temporarily interrupt tepotinib treatment or continue tepotinib treatment at a lower dosage until the AE recovered to Grade 2 or less or to baseline values. Before Protocol Amendment 8 (January 17, 2020; see Table 9 for a summary of protocol amendments), the dosage was initially reduced to 300 mg tepotinib hydrochloride hydrate once daily. Further dosage reductions were made on a case-by-case basis in agreement with the sponsor. If the permitted lowest dosage was not tolerable, the patient was withdrawn from the study. After the implementation of Protocol Amendment 8, the standard dose reduction was changed to 250 mg tepotinib hydrochloride hydrate once daily. If the patient did not tolerate the 250 mg once daily dosage, or the AE did not resolve following treatment interruption, permanent treatment discontinuation was discussed with the sponsor. Following the dosage reductions, the daily dosage of tepotinib hydrochloride hydrate could be increased to 500 mg at the discretion of the investigator.
The maximum permitted period of continuous treatment interruption was 21 days. Before Protocol Amendment 8 (January 17, 2020), patients could be rechallenged at the initial dosage level of 500 mg tepotinib hydrochloride hydrate daily following a treatment interruption, or at a lower dosage level on a case-by-case basis, in agreement with the sponsor. Following the implementation of Protocol Amendment 8, re-exposure at the 250 mg tepotinib hydrochloride hydrate daily dosage level following a treatment interruption was permitted on a case-by-case basis.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the VISION trial and included in this review is provided in Table 8. These end points are further summarized in this section.
Efficacy Measurement for Primary and Secondary Outcomes
In VISION, a baseline tumour assessment was completed during the screening period. CT or MRI (MRI) with contrast enhancement was recommended for tumour assessment. Imaging studies, including CT or MRI of the chest, abdomen, and pelvis were performed at baseline to survey metastasis. Images from all patients at screening were independently reviewed by a single radiologist who checked the presence of measurable disease (measured in at least 1 dimension [longest diameter]) according to RECIST v1.1 before study treatment was started. Following the implementation of Protocol Amendment 8 (January 17, 2020; see Table 9 for a summary of protocol amendments), brain imaging by MRI with IV (IV) contrast enhancement was performed at baseline. MRI could be performed without contrast enhancement, if contrast was contraindicated. If MRI was not clinically feasible, CT of the brain with IV contrast enhancement could be used. For patients with brain metastases detected at baseline, subsequent brain imaging by MRI with IV contrast enhancement was performed for tumour assessment.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | VISION |
|---|---|
OS | Secondary |
PFS by IRC and by investigator assessment | Secondary |
ORR by IRC assessment | Primary |
ORR by investigator assessment | Secondary |
DOR by IRC and by investigator assessment | Secondary |
HRQoL and PROs (change from baseline and TTD of EQ-5D-5L VAS, EORTC QLQ-C30 global health status and quality of life, and EORTC QLQ-LC13 coughing, dyspnea, chest pain) | Secondary |
Intracranial CNS outcomes | Exploratory (post hoc) |
Safety | Secondary |
CNS = central nervous system; DOR = duration of response; EORTC = European Organization for the Research and Treatment of Cancer; EQ-5D-5L = 5-Level EQ-5D; HRQoL = health-related quality of life; IRC = independent review committee; ORR = objective response rate; OS = overall survival; PFS = progression-free survival; PRO = patient-reported outcome; QLQ-C30 = Quality of Life Questionnaire Core 30; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer 13; TTD = time to deterioration; VAS = visual analogue scale.
Source: VISION Clinical Study Reports,12,14 sponsor’s submission to CADTH.23
Table 9: Summary of Key Protocol Amendments in VISION
Amendment | Key changes |
|---|---|
Amendment 1 (September 21, 2016), Global | Removed reference to the mITT population in the CSP based on comments received from the Voluntary Harmonisation Procedure |
Amendment 2 (September 22, 2016), Global |
|
Amendment 3 (September 22, 2016), Local (France) | (French version) was updated according to Amendment 1 and Amendment 2 to synchronize the contents with the CSP used in other regions |
Amendment 4 (March 15, 2017), Global |
|
Amendment 5 (May 10, 2018), Global | Introduced an additional NSCLC MET amplification cohort (cohort B) into the study |
Amendment 6 (March 26, 2019), Global |
|
Amendment 7 (June 25, 2019), Global |
|
Amendment 8 (January 17, 2020), Global |
|
AE = adverse event; CSP = clinical study protocol; ctDNA = circulating tumour DNA; ILD = interstitial lung disease; METex14 = MET exon 14; mITT = modified intention-to-treat; NSCLC = non–small cell lung cancer; PD = progressive disease; PRO = patient-reported outcome.
Source: VISION Clinical Study Report,14 Paik et al. (2020).13
During the treatment period, patients had tumour assessments according to RECIST v1.1 every 6 weeks until 9 months and every 12 weeks thereafter, until disease progression, death, or withdrawal of consent. Patients who withdrew from treatment for reasons other than disease progression had tumour assessments performed every 6 weeks until 9 months and every 12 weeks thereafter, until radiologically documented disease progression, death, the end of study, or start of a new anticancer therapy, whichever occurred first. All tumour responses (PR and/or CR) were assessed every time with the same methods (CT or MRI) used at the first evaluation of the response.
For the determination of objective response and other RECIST-related outcomes, tumour evaluations were performed by the investigator and study centre radiologist as well as by independent review by the IRC. The decision to stop study treatment was primarily based on the investigator’s assessment.
Patients were followed-up every 3 months to collect information about survival and anticancer treatments.
Outcomes
In the VISION study, OS was a secondary outcome. OS time was measured as the time in months from first trial treatment administration to the date of death due to any cause. OS time was censored at the last date the patient was known to be alive, for patients not known to be deceased at time of analysis.
PFS by IRC and PFS by investigator assessment were secondary outcomes. PFS time was defined as the time in months from the first administration of trial treatment to the date of the first documentation of PD or death due to any cause within 84 days of the last evaluable tumour assessment, whichever occurred first. The PFS data were censored on the date of the last evaluable tumour assessment for patients who did not have an event (PD or death) or for patients with an event more than 84 days after the last evaluable tumour assessment. Patients who did not have an evaluable post-baseline tumour assessment were censored at the date of the start of trial treatment unless death occurred within 84 days of the first dose of trial treatment, in which case the death was considered an event.
The ORR confirmed by IRC was the primary outcome of the VISION study. Patients were identified as having an objective response if they achieved either a confirmed CR or PR from first administration of trial treatment to first observation of PD. Confirmation needed to take place by a tumour assessment at least 4 weeks (28 days) after the tumour assessments initially indicating CR or PR.
The ORR by investigator assessment was a secondary outcome. Objective response based on investigator assessment was derived and analyzed identically to the primary end point, apart from the use of the investigator’s evaluation instead of the IRC.
The DOR by IRC and DOR by investigator assessment were secondary outcomes. The DOR was evaluated only in patients who had an objective response. The DOR was defined as the time from when the CR or PR (whichever occurred first) criteria were first met until PD or death due to any cause within 84 days of the last evaluable tumour assessment, whichever occurred first. The DOR data were censored on the date of the last evaluable tumour assessment for patients who did not have an event (PD or death) or for patients with an event more than 84 days after the last evaluable tumour assessment.
In VISION, HRQoL and PROs were measured by 3 instruments: EQ-5D-5L, EORTC QLQ-C30, and EORTC QLQ-LC13. The questionnaires were completed every 6 weeks from cycle 1, day 1 until 9 months and every 12 weeks thereafter until disease progression, death, or withdrawal of consent. Change from baseline and TTD in EQ-5D-5L VAS score, EORTC QLQ-C30 global health status and quality of life score, and 3 EORTC QLQ-LC13 symptom scales (coughing, dyspnea, and chest pain) were assessed as secondary outcomes. A detailed discussion and critical appraisal of these outcomes is available in Appendix 4. In VISION, TTD was defined as the time between first dose and the first occurrence of a 10-point deterioration compared to the baseline score.23 For the TTD analysis, the PROs were censored at the date of last PRO assessment or date of start of treatment, whichever was later.
The EQ-5D-5L is a generic quality of life instrument that may be applied to a wide range of health conditions and treatments. The EQ-5D-5L consists of the EQ-5D Likert-type scale (index score) and the EQ VAS. The EQ-5D-5L VAS records the patient’s self-rated health on a vertical VAS on which the end points are labelled 0 (“the worst health you can imagine”) and 100 (“the best health you can imagine”). CADTH identified a minimal important difference (MID) of 7 to 11.5 points in patients with lung cancer.24 In VISION, the sponsor defined a clinically meaningful deterioration as a decrease of 10 points or more from baseline.23
The EORTC QLQ-C30 is a standardized questionnaire for evaluating the quality of life of patients with cancer participating in clinical trials. This questionnaire is complemented by the lung cancer–specific questionnaire, the QLQ-LC13. The EORTC QLQ-C30 consists of 5 functional scales, 3 symptom scales, 6 single items, and a global health status and quality of life scale. A higher score on a functional scale and on global health status and quality of life corresponds to higher function and better quality of life, while a higher score on a symptom scale corresponds to higher symptom burden. For the global health status and quality of life score, CADTH identified an MID of 4 to 9 points for improvement and 4 points for deterioration in patients with NSCLC.25 In VISION, the sponsor defined a clinically meaningful deterioration in global health status and quality of life score as a decrease of 10 points or more from baseline.23
The EORTC QLQ-LC13, which supplements the QLQ-C30, consists of lung cancer–related symptoms and treatment side effects. Higher scores represent increased symptom burden. No MID was identified in the literature for the EORTC-LC13 symptom scales. In VISION, the sponsor defined a clinically meaningful deterioration as an increase of 10 points or more from baseline.23
Intracranial central nervous system (CNS) outcomes were assessed in a post hoc exploratory analysis, which was reported in a poster and conference abstract included in the sponsor’s submission to CADTH.23 A retrospective analysis of brain lesions determined by CT or MRI was conducted by an IRC using Response Assessment in Neuro-Oncology Brain Metastases (RANO-BM) criteria. Responses were determined in patients with 1 or more evaluable post-baseline tumour assessment. Confirmation was not required. For patients with non-measurable lesions per RANO-BM, disease control in the brain was defined as no CR or PD.
To assess safety, the VISION study collected data on the number of patients with treatment-emergent AEs and deaths, laboratory parameters (hematology and coagulation, biochemistry, and urinalysis), vital signs, electrocardiograms, and physical examinations. Abnormal laboratory findings (Grade 1 to 3) and other abnormal investigational findings (e.g., on an electrocardiogram trace) were not reported as AEs unless they were associated with clinical signs and symptoms, led to treatment discontinuation or were considered otherwise medically important by the investigator. If a laboratory abnormality fulfilled these criteria, the identified medical condition (e.g., anemia, increased ALT) was reported as the AE rather than the abnormal value itself.
Statistical Analysis
In the VISION trial, no formal statistical hypotheses were tested. Data were analyzed descriptively. Analyses were performed by cohorts, and cohort A and cohort C (i.e., entire METex14 skipping alteration population) results were pooled and presented.
Determination of Sample Size
For all cohorts, the VISION study aimed to show an ORR assessed by IRC in the range of 40% to 50% and to demonstrate that the lower limit of the corresponding exact 2-sided 95% CI (according to Clopper-Pearson) for ORR exceeded 20% across lines of therapy. With a sample size of 60 patients per analysis set, a maximum width for the 95% CI of 26.4% would be achieved in the range for ORR of 40% to 60%.
The study planned to enrol patients in cohort A until at least 60 patients who had positive results of a liquid biopsy and at least 60 patients who had positive results of a tumour biopsy for METex14 skipping alterations were enrolled. Due to an anticipated overlap of patients who tested positive for METex14 skipping alterations in tumour tissue and in ctDNA derived from plasma, it was estimated that a total of approximately 100 patients would be enrolled in cohort A. Furthermore, the study planned to enrol at least 25 second- or later-line patients in cohort A.
Enrolment in cohort C was ongoing as of the most recent data cut-off date and was planned to continue until at least 60 patients who has positive results of a liquid biopsy and at least 60 patients who had positive results of a tumour biopsy for METex14 skipping alterations were enrolled. An overlap of patients who tested positive for METex14 skipping alterations in tumour tissue and in ctDNA derived from plasma was anticipated. Regardless of material used for inclusion in the study (i.e., liquid biopsy or tumour biopsy), the study planned to include at least 50 first-line, 30 second-line, and 20 third-line patients in cohort C.
Planned Analyses
Multiple analyses were planned in the VISION trial. Per the protocol, the primary efficacy analysis was conducted when the target enrolment population of at least 60 patients in both the subgroups of patients with positive results of a liquid biopsy and with positive results of a tissue biopsy had undergone at least 9 months of follow-up. For cohort A, the follow analyses were planned:
Interim futility analysis: Conducted when 12 patients who had positive results of a tumour tissue biopsy had completed 4 cycles of trial treatment (84 ± 3 days) or prematurely discontinued trial treatment for any reason. If 3 or fewer confirmed responders were observed, enrolment in cohort A of patients who tested positive for METex14 skipping alterations on tumour biopsy, but not on plasma ctDNA, would be discontinued. No stopping criteria were defined for any other interim analysis.
Interim analysis: Conducted when 12 patients who had positive results of a liquid biopsy had completed 4 cycles or prematurely discontinued trial treatment for any reason.
6-month follow-up analysis: Conducted when at least 60 patients who has positive results of a liquid biopsy and at least 60 patients who had positive results of a tumour biopsy had either been treated with tepotinib for 6 months or more, died or prematurely discontinued trial treatment for any reason.
9-month follow-up analysis: Conducted when at least 60 patients who had positive results of a liquid biopsy and at least 60 patients who had positive results of a tumour biopsy had either been treated with tepotinib for 9 months or more, died or prematurely discontinued trial treatment for any reason. This analysis was used as the primary analysis for the FDA. This analysis had a data cut-off date of January 1, 2020, and included 99 patients who received their first dose of tepotinib before April 2, 2019.12,13 Results for this analysis and the population of patients in cohort A who received their first dose of tepotinib before April 2, 2019, are provided in Appendix 3.
15-month follow-up analysis: Conducted once at least 60 patients who had positive results of a liquid biopsy and at least 60 patients who had positive results of a tumour biopsy had either been treated with tepotinib for at least 15 months, died or prematurely discontinued trial treatment for any reason, whichever came first. This analysis was introduced in accordance with feedback from the FDA. This corresponds to the analysis conducted with a data cut-off date of July 1, 2020.14
Periodic safety reviews
Final analysis: To be conducted when all patients in cohort A have discontinued trial drug and two-thirds of the patients have died.
The confirmatory cohort C was added to the VISION study as part of Protocol Amendment 6 (March 26, 2019; see Table 9 for a summary of protocol amendments). In addition to a separate analysis of cohort C, a pooled analysis of all subjects with METex14 skipping alterations was conducted (i.e., combining data from cohort A and cohort C). The following analyses were planned for the confirmatory part of the VISION study:
Primary (9-month follow-up) analysis: To be conducted once all patients have either been treated with tepotinib for at least 9 months, died, or prematurely discontinued trial treatment for any reason.
21-month follow-up analysis: To be conducted once all patients have either been treated with tepotinib for at least 21 months, died, or prematurely discontinued trial treatment for any reason.
Final analysis: To be conducted at the end of the confirmatory cohort C of the VISION study, defined as the time point at which all patients have discontinued trial drug and two-thirds of the patients have died.
Primary outcome analysis
The primary end point of VISION was ORR (CR or PR), determined according to RECIST v1.1 based on IRC assessment. The primary analysis of the primary end point was based on the mITT population. As the study is ongoing, analysis of ORR was conducted based on all patients enrolled, as well as all patients who had at least 2 post-baseline assessments or who discontinued treatment for any reason. No formal statistical hypotheses were tested. The number of patients achieving objective response and the ORR by IRC with the corresponding 2-sided exact Clopper-Pearson 95% CI were presented.
Sensitivity Analyses
Sensitivity analyses were performed using mITT populations of patients who had positive results of a tumour biopsy or liquid biopsy for METex14 skipping alterations. A further sensitivity analysis was conducted on the mITT analysis set, in which the start of any other anticancer treatment or procedure before study discontinuation was considered as PD, and any subsequent tumour assessments were not considered in the objective response assessment. If only partial dates were known for the start of other anticancer treatment or procedures, then the earliest possible date (based on partial date entered) up to the date of last dose of trial treatment was used.
Subgroup Analyses
Subgroup analyses were specified a priori. Subgroups of interest outlined in the protocol included ECOG PS (0 or 1), line of therapy (first-line, second-line, second- or later-line, third- or later-line), baseline brain metastases per IRC assessment (present or absent), and histological classification (adenocarcinoma, squamous, or other). Best objective response details and ORRs (based on IRC and investigator assessment) and corresponding 2-sided exact Clopper-Pearson 95% CIs were presented for each of the identified subgroups. If there were fewer than 10 patients in any subgroup, then the results were presented in listings rather than calculating ORRs and 95% CIs.
Secondary Outcome Analysis
Secondary end point analyses were performed on the mITT population. The ORR based on investigator assessment was derived and analyzed identically to the primary end point, apart from the use of the investigator’s evaluation rather than that of the IRC. For DOR and PFS, the end points were assessed using both the IRC results and the investigator assessment.
For OS, PFS, and DOR, Kaplan-Meier estimates were presented with a summary of associated statistics (median and 3-, 6-, 9-, 12-, 15-, 18-month rate estimates and estimates for every 3 months thereafter, as applicable) including the corresponding 2-sided 95% CIs. The CIs for the median were calculated according to Brookmeyer and Crowley (1982) and CIs for the survival function estimates at the defined time points were derived using the log-log transformation according to Kalbfleisch and Prentice (1980). The estimate of the standard error was computed using Greenwood’s formula. Kaplan-Meier plots were also presented.
For the HRQoL and PROs, data were presented for the EQ-5D-5L VAS, EORTC QLQ-C30 global health status and quality of life score, and 3 symptom scales of the EORTC QLQ-LC13 (coughing, dyspnea, and chest pain). A TTD analysis was planned for these outcomes. Kaplan-Meier estimates were presented, including the corresponding 2-sided 95% CIs. The CIs for the median were calculated according to Brookmeyer and Crowley (1982). Kaplan-Meier plots were also presented.
For the EORTC QLQ-LC13 symptom scales, a mixed-effect model repeated measures (MMRM) analysis was performed to evaluate longitudinal change from baseline. The MMRM including change from baseline as the dependent variable; patient, analysis visit, and baseline score as a covariate; and baseline score by analysis visit interaction to account for nonconstant baseline effect across visits.
For all secondary end points except for HRQoL and PROs, subgroup analyses were performed for the same subgroups as the primary end point, depending on whether there was a sufficient number of patients (at least 5) in each subgroup. No subgroup analyses were performed on the HRQoL and PROs.
Handling of Missing Data
To impute missing tumour assessment dates, a missing day was imputed as the 15th of the month, if month and year were documented. If the imputation was earlier than the date of start of treatment, the day of start of treatment was taken. In all other cases, missing or incomplete dates were not imputed. To impute missing death dates, if month and year were available, the day was imputed as the 15th of the month, unless this resulted in a date earlier than the last date the patient was known to be alive. In that case, the date of death was imputed as the day after the last date the patient was known to be alive. No imputation was done for missing HRQoL and PRO data.
Analysis Populations
In the VISION trial, the mITT population included all patients who were administered at least 1 dose of tepotinib and had METex14 skipping alterations confirmed by a validated central laboratory assay.
The safety analysis set included all patients enrolled in the VISION trial who were administered at least 1 dose of tepotinib.
Protocol Amendments and Deviations
A total of 8 protocol amendments were implemented during the VISION trial, and the key changes made with these amendments are summarized in Table 9. In Protocol Amendment 4, the eligible study population was expanded to include all subtypes of NSCLC and patients who were treatment-naive and would receive tepotinib as first-line treatment. Before this amendment, only patients with adenocarcinoma were eligible, and patients were required to have failed 1 to 2 lines of systemic therapy, including a platinum-doublet-containing regimen. Cohort B and cohort C were added as part of Protocol Amendments 5 and 6, respectively. Following Protocol Amendment 6, the VISION trial consisted of 2 parts: part 1 included pivotal cohort A (METex14 skipping alterations) and cohort B (MET amplification); part 2 included cohort C (confirmatory part for METex14 skipping alterations).
In cohort A, 104 (68.4%) patients had at least 1 important protocol deviation. Important protocol deviations reported in 10% or more of patients were related to laboratory assessment criteria (40.1%), SAE criteria (14.5%), informed consent (13.2%), study procedures criteria (13.2%), and efficacy criteria (11.8%). Nine (5.9%) of patients had at least 1 clinically important protocol deviation: 3 patients had missed tumour imaging scans due to the COVID-19 pandemic, 5 patients deviated from key entry criteria, and 1 patient did not have METex14 skipping alterations confirmed by a validated central laboratory assay.
In cohort A plus C, 149 (58.4%) patients had at least 1 important protocol deviation. Protocol deviations reported in 10% or more of patients were related to laboratory assessment criteria (25.9%), study procedures criteria (15.3%), informed consent (11.8%), and SAE criteria (10.2%). Twelve (4.7%) patients in cohort A plus C had at least 1 clinically important protocol deviation. In addition to the deviations described earlier for cohort A, a tumour evaluation or staging imaging scan (CT or MRI) was not performed at a required time point for 3 patients.
Results
Patient Disposition
The disposition of patients in cohort A and cohort A plus C of the VISION study as of July 1, 2020, data cut-off date is summarized in Table 10. A total of 7,658 patients were pre-screened to determine MET alteration status in tissue and blood samples. Following identification of a METex14 skipping alteration in the pre-screening period, 168 patients were screened for enrolment in the pivotal cohort A. In total, 15 patients failed screening and were discontinued from the study because they did not meet the eligibility criteria (n = 9), they withdrew consent (n = 1), they died (n = 4), or “other” reasons (n = 1). A total of 152 patients were enrolled in cohort A and treated with at least 1 dose of tepotinib. One patient enrolled and treated in cohort A was excluded from the mITT population because the patient tested negative for METex14 skipping alterations in the liquid biopsy and had tumour biopsy data that were not evaluable.
To maintain recruitment for the confirmatory cohort C, cohort A recruitment was not interrupted in the study, and enrolment at sites was gradually shifted from cohort A to cohort C. As of the July 1, 2020, data cut-off, 282 patients were screened for enrolment in the pooled cohort A plus C. In total, 21 patients failed screening and discontinued the study because they did not meet the eligibility criteria (n = 13), they withdrew consent (n = 1), they had AEs (n = 1), they died (n = 5), or “other” reasons (n = 1). Following screening, 255 patients were enrolled in the study in the pooled cohort A plus C and treated with at least a single dose of tepotinib.
Exposure to Study Treatments
Patient exposure to tepotinib in the VISION study is summarized in Table 11. In cohort A, the mean duration of exposure to tepotinib was 9.38 (standard deviation [SD] = 7.63) months. Most patients (64.5%) were exposed to a relative dose intensity of 90% to 100%. Overall, 37.5% of patients had at least 1 dose reduction. Most patients (59.9%) experienced a therapy delay, which was defined as at least 2 days between 2 administrations of tepotinib. Therapy delays were attributed to AEs (48.0%), missed doses (29.6%), and other reasons (19.1%).
In cohort A plus C, the mean duration of exposure to tepotinib was 7.05 (SD = 6.71) months. Most patients (69.4%) were exposed to a relative dose intensity of 90% to 100%. Overall, 29.8% of patients had at least 1 dose reduction, and 50.2% of patients experienced therapy delays. Therapy delays were attributed to AEs (42.0%), missed doses (22.4%), and other reasons (15.7%). Data were missing for 7.5% of patients.
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in this section. Data from the updated interim analysis (data cut-off date of July 1, 2020) for the overall pivotal cohort A (N = 151) and pooled cohort A plus C (i.e., entire METex14 skipping population; N = 254) are reported here.14
See Appendix 3 for detailed efficacy data, including results from the primary analysis for the FDA (data cut-off date of January 1, 2020; N = 151 in cohort A and N = 180 in cohort A plus C) and the primary analysis population for the FDA submission, which consisted of patients in cohort A who received the first dose of tepotinib before April 2, 2019 (N = 99).12,13
Overall Survival
In the VISION trial, OS was a secondary end point. Results for OS in cohort A and cohort A plus C as of the July 1, 2020, data cut-off are summarized in Table 12. The Kaplan-Meier curve for cohort A is depicted in Figure 3; the Kaplan-Meier curve for cohort A plus C is depicted in Figure 4. In cohort A, the median duration of follow-up for OS was 16.4 (95% CI, 13.6 to 18.5) months, and the median OS was 17.6 (95% CI, 15.0 to 21.0) months. In cohort A plus C, the median duration of follow-up for OS was 9.9 (95% CI, 8.1 to 12.0) months, and the median OS was 19.1 (95% CI, 15.3 to 22.1) months.
Results of the subgroup analyses of OS conducted in cohort A and cohort A plus C are summarized in Table 13. The median OS was broadly consistent across subgroups. In the pivotal cohort A, the median OS was 17.6 (95% CI, 9.7 to 29.7) months in the first-line therapy subgroup and 19.7 (95% CI, 15.0 to 21.0) months in the second- or later-line therapy subgroup.
Table 10: Patient Disposition — VISION as of the July 1, 2020, Data Cut-Off Date
Disposition | Cohort A | Cohort A + C |
|---|---|---|
Pre-screened for MET alteration status, N | 7,658 | |
Screened, N | 168 | 282 |
Active in screening, N | 1 | 6 |
Discontinued during screening, N | 15 | 21 |
Eligibility criteria not met | 9 | 13 |
Withdrew consent | 1 | 1 |
AE | 0 | 1 |
Death | 4 | 5 |
Other | 1 | 1 |
Enrolled and treated, N | 152 | 255 |
Discontinued treatment, N (%) | 124 (81.6) | 154 (60.4) |
AE | 26 | 31 |
Protocol noncompliance | 1 | 1 |
Death | 12 | 16 |
PD | 77 | 93 |
Withdrew consent | 5 | 7 |
Other | 3 | 6 |
Treatment ongoing, N (%) | 28 (18.4) | 101 (39.6) |
mITT, N | 151 | 254 |
Safety, N | 152 | 255 |
AE = adverse event; mITT = modified intention-to-treat; PD = progressive disease.
Source: VISION Clinical Study Report.14
Table 11: Exposure to Tepotinib in VISION Cohort A and Cohort A + C (Safety Analysis Set) as of July 1, 2020, Data Cut-Off Date
Tepotinib exposure | Cohort A (N = 152) | Cohort A + C (N = 255) |
|---|---|---|
Mean duration of therapy (SD), months | 9.38 (7.63) | 7.05 (6.71) |
Mean dose intensity (SD), mg per 3-week cycle | 9,195.76 (1,755.06) | 9,420.99 (1,689.85) |
Relative dose intensity, n (%) | ||
< 60% | 12 (7.9) | 15 (5.9) |
60% to 80% | 32 (21.1) | 44 (17.3) |
80% to 90% | 10 (6.6) | 19 (7.5) |
90% to 100% | 98 (64.5) | 177 (69.4) |
Patients who had ≥ 1 dose reduction, n (%) | 57 (37.5) | 76 (29.8) |
Patients who had therapy delays, n (%) | 91 (59.9) | 128 (50.2) |
SD = standard deviation.
Source: VISION Clinical Study Report.14
Table 12: OS Results in VISION, Cohort A and Cohort A + C (mITT Population) as of the July 1, 2020, Data Cut-Off Date
End point | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
Patients with an event, n (%) | 75 (49.7) | 84 (33.1) |
Patients censored, n (%) | 76 (50.3) | 170 (66.9) |
Median duration of follow-up (95% CIa), months | 16.4 (13.6 to 18.5) | 9.9 (8.1 to 12.0) |
Median OSb (95% CIa), months | 17.6 (15.0 to 21.0) | 19.1 (15.3 to 22.1) |
CI = confidence interval; mITT = modified intention-to-treat; OS = overall survival.
aCalculated using the Brookmeyer and Crowley method.
bEstimated using the Kaplan-Meier method.
Source: VISION Clinical Study Report.14
Table 13: Subgroup Analyses of OS in Cohort A and Cohort A + C (mITT Population) as of the July 1, 2020, Data Cut-Off Date
Subgroup | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
Line of treatment | ||
First-line, n | 69 | 125 |
Patients who died, n (%) | 35 (50.7) | 42 (33.6) |
Median OSa (95% CIb), months | 17.6 (9.7 to 29.7) | 17.6 (9.9 to 29.7) |
Second- or later-line, n | 82 | 129 |
Patients who died, n (%) | 40 (48.8) | 42 (32.6) |
Median OSa (95% CIb), months | 19.7 (15.0 to 21.0) | 19.8 (15.2 to 22.3) |
Presence of brain metastases at baseline by IRC | ||
Present, n | 15 | 31 |
Patients who died, n (%) | 7 (46.7) | 8 (25.8) |
Median OSa (95% CIb), months | 22.1 (8.0 to NE) | 22.1 (9.5 to NE) |
Absent, n | 136 | 223 |
Patients who died, n (%) | 68 (50.0) | 76 (34.1) |
Median OSa (95% CIb), months | 17.6 (15.2 to 21.0) | 19.1 (15.3 to 22.3) |
Histological subtype | ||
Adenocarcinoma, n | 131 | 207 |
Patients who died, n (%) | 63 (48.1) | 70 (33.8) |
Median OSa (95% CIb), months | 19.7 (15.3 to 23.6) | 19.7 (15.8 to 23.6) |
Squamous, n | 14 | 24 |
Patients who died, n (%) | 8 (57.1) | 10 (41.7) |
Median OSa (95% CIb), months | 13.5 (3.5 to 21.0) | 13.5 (4.3 to 21.0) |
Other, n | 6 | 23 |
Patients who died, n (%) | 4 (66.7) | 4 (17.4) |
Median OSa (95% CIb), months | 5.8 (0.3 to NE) | NE (8.5 to NE) |
ECOG PS | ||
0, n | 40 | 70 |
Patients who died, n (%) | 14 (35.0) | 15 (21.4) |
Median OSa (95% CIb), months | 24.9 (19.1 to NE) | 24.9 (19.1 to NE) |
1, n | 111 | 184 |
Patients who died, n (%) | 61 (55.0) | 69 (37.5) |
Median OSa (95% CIb), months | 15.8 (12.1 to 19.8) | 15.8 (12.3 to 19.8) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group performance status; IRC = independent review committee; mITT = modified intention-to-treat; NE = not estimable; OS = overall survival.
aEstimated using the Kaplan-Meier method.
bCalculated using the Brookmeyer and Crowley method.
Source: VISION Clinical Study Report.14
Table 14: PFS Results in VISION, Cohort A and Cohort A + C (mITT Population) as of July 1, 2020, Data Cut-Off Date
End point | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
PFS by IRC (secondary end point) | ||
Patients with an event, n (%) | 87 (57.6) | 105 (41.3) |
PD | 56 (37.1) | 67 (26.4) |
Death | 31 (20.5) | 38 (15.0) |
Patients censored, n (%) | 64 (42.4) | 149 (58.7) |
Median duration of follow-up (95% CIa), months | 12.2 (11.0 to 14.0) | 7.0 (5.8 to 8.3) |
Median PFSb (95% CIa), months | 8.9 (8.2 to 11.0) | 9.5 (8.2 to 11.2) |
PFS by investigator (secondary end point) | ||
Patients with an event, n (%) | 98 (64.9) | 121 (47.6) |
PD | 78 (51.7) | 95 (37.4) |
Death | 20 (13.2) | 26 (10.2) |
Patients censored, n (%) | 53 (35.1) | 133 (52.4) |
Median duration of follow-up (95% CIa), months | 16.5 (13.8 to 20.2) | 8.3 (8.1 to 11.1) |
Median PFSb (95% CIa), months | 8.5 (6.9 to 11.0) | 8.3 (6.9 to 10.6) |
CI = confidence interval; IRC = independent review committee; mITT = modified intention-to-treat; PD = progressive disease; PFS = progression-free survival.
aCalculated using the Brookmeyer and Crowley method.
bEstimated using the Kaplan-Meier method.
Source: VISION Clinical Study Report.14
Figure 3: Kaplan-Meier Curve Showing OS in Cohort A (mITT Population, N = 151) as of the July 1, 2020, Data Cut-Off Date
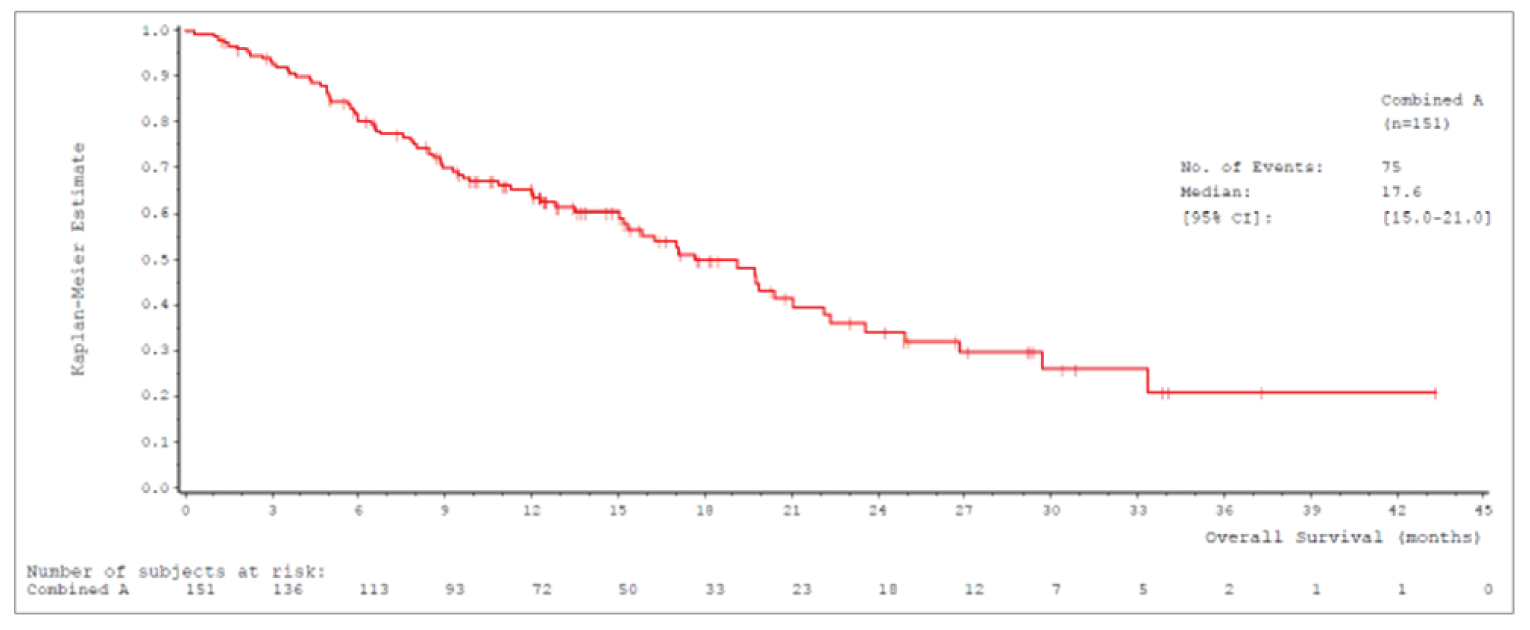
CI = confidence interval; mITT = modified intention-to-treat; OS = overall survival.
Source: VISION Clinical Study Report.14
Figure 4: Kaplan-Meier Curve Showing OS in Cohort A + C (mITT Population, N = 254) as of the July 1, 2020, Data Cut-Off Date
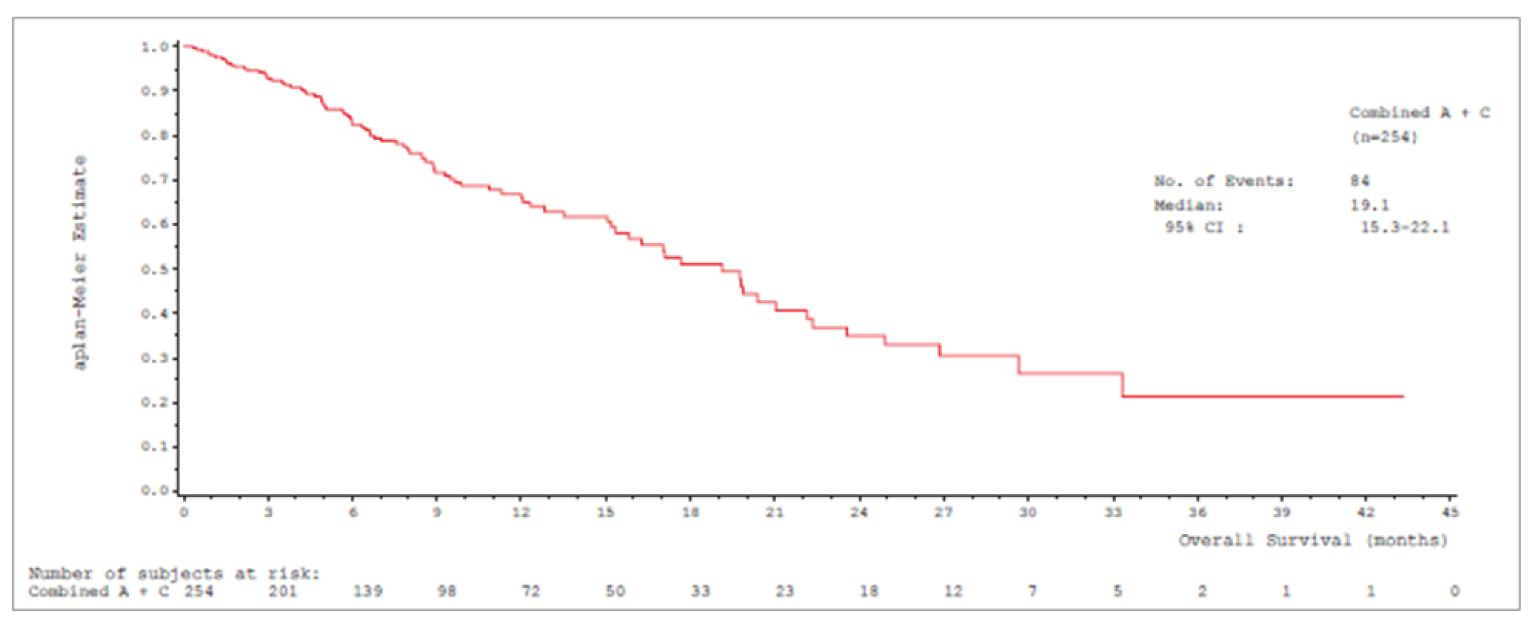
CI = confidence interval; mITT = modified intention-to-treat; OS = overall survival.
Source: VISION Clinical Study Report.14
Progression-Free Survival
In the VISION trial, PFS by IRC and PFS by investigator assessment were secondary end points. Results for PFS in the cohort A and cohort A plus C as of the July 1, 2020, data cut-off are summarized in Table 14. The Kaplan-Meier curve of PFS by IRC in cohort A is depicted in Figure 5; the Kaplan-Meier curve of PFS by IRC in cohort A plus C is depicted in Figure 6. In cohort A, 57.6% of patients had an event (i.e., progressive disease or death) by IRC assessment, whereas 64.9% of patients had an event per investigator assessment. Median PFS by IRC was 8.9 (95% CI, 8.2 to 11.0) months, and median PFS by investigator was 8.5 (95% CI, 6.9 to 11.0) months in cohort A. In cohort A plus C, 41.3% of patients had an event per IRC assessment, whereas 47.6% had an event per investigator assessment. Median PFS by IRC was 9.5 (95% CI, 8.2 to 11.2) months, and median PFS by investigator was 8.3 (95% CI, 6.9 to 10.6) months in cohort A plus C.
Results of the subgroup analyses of PFS by IRC conducted in cohort A and cohort A plus C are summarized in Table 15. The median PFS by IRC was broadly consistent across all subgroups. In the pivotal cohort A, median PFS was 8.5 (95% CI, 6.8 to 11.3) months in the first-line therapy subgroup and 10.9 (95% CI, 8.2 to 12.7) months in the second- or later-line therapy subgroup.
Results of the subgroup analyses of ORR by IRC for cohort A are summarized in Table 17. The ORRs based on IRC assessment were similar across subgroups and consistent with the ORR in overall cohort A.
Figure 5: Kaplan-Meier Curve Showing PFS by IRC in Cohort A mITT Population (N = 151), as of July 1, 2020, Data Cut-Off Date
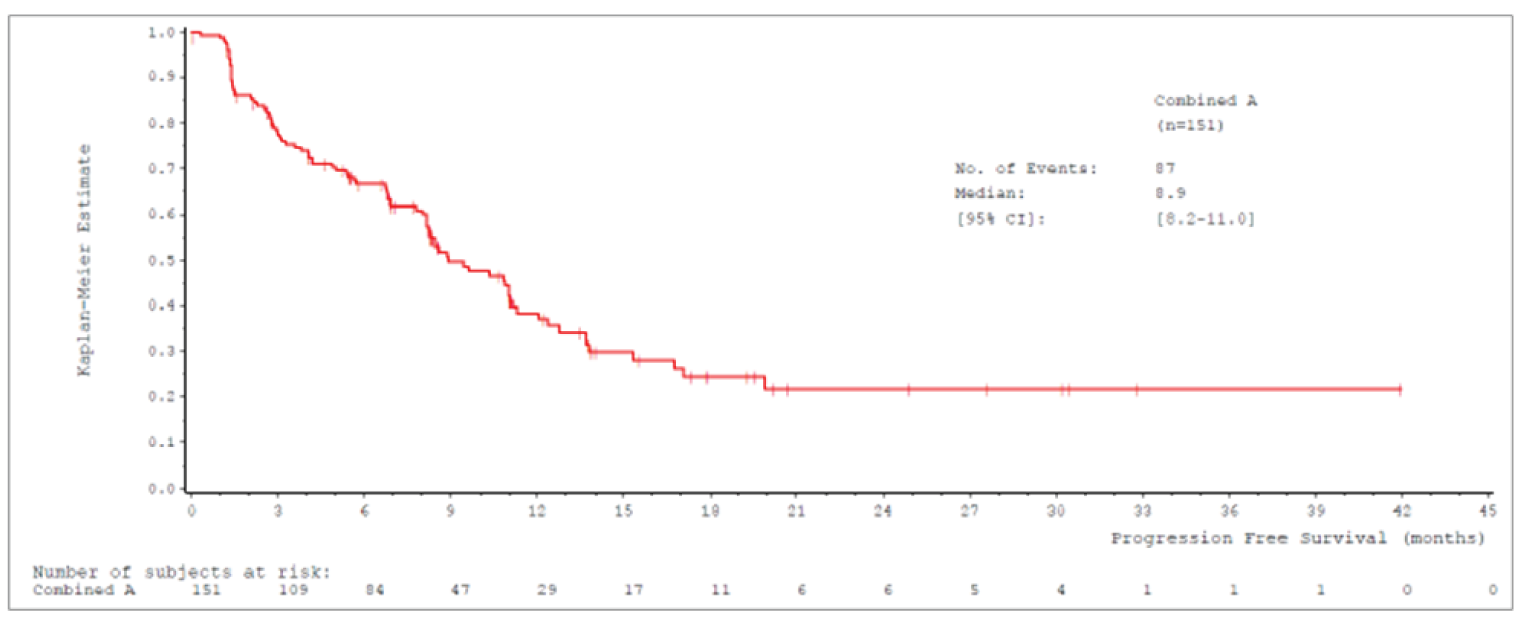
CI = confidence interval; IRC = independent review committee; mITT = modified intention-to-treat; PFS = progression-free survival.
Source: VISION Clinical Study Report.14
Figure 6: Kaplan-Meier Curve Showing PFS by IRC in Cohort A + C mITT Population (N = 254), as of July 1, 2020, Data Cut-Off Date
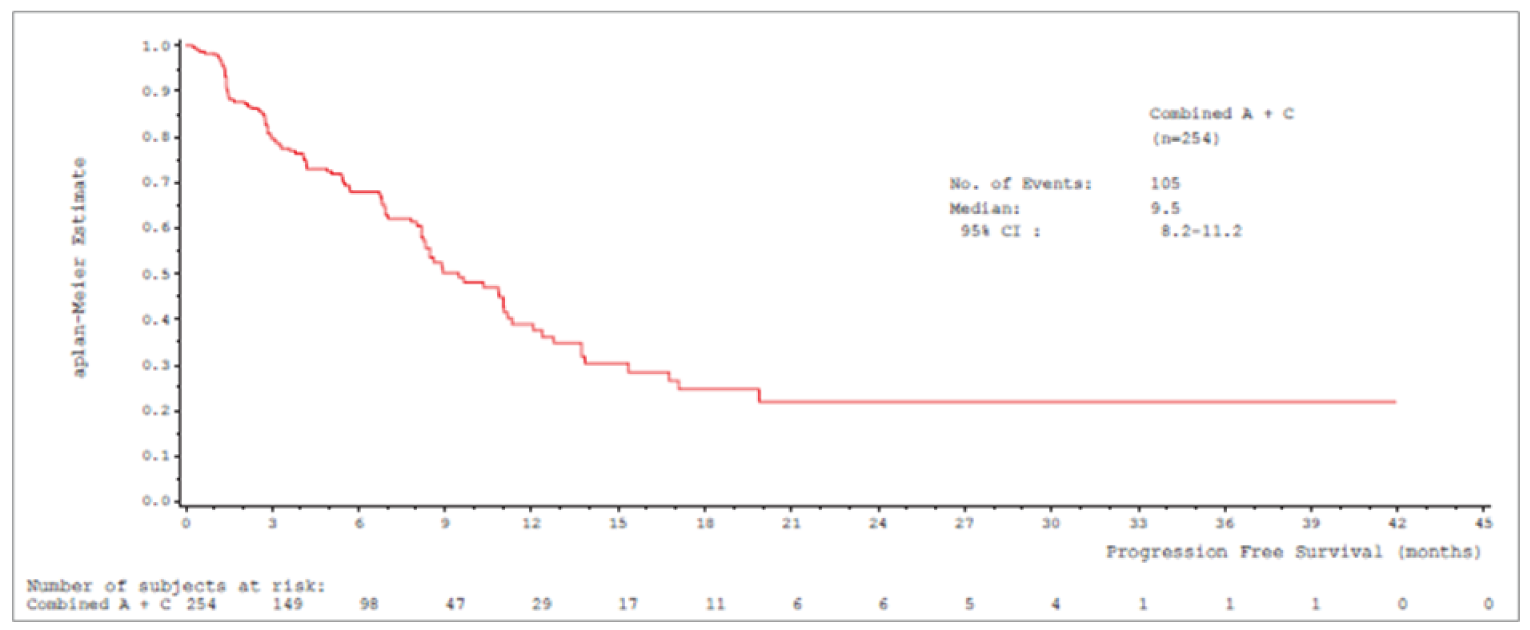
CI = confidence interval; IRC = independent review committee; mITT = modified intention-to-treat; PFS = progression-free survival.
Source: VISION Clinical Study Report.14
Table 15: Subgroup Analyses of PFS by IRC in Cohort A and Cohort A + C (mITT Population) as of July 1, 2020, Data Cut-Off Date
Subgroup | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
Line of treatment | ||
First-line, n | 69 | 125 |
Patients with events, n (%) | 39 (56.5) | 49 (39.2) |
Median PFSa (95% CIb), months | 8.5 (6.8 to 11.3) | 8.5 (7.8 to 11.3) |
Second- or later-line, n | 82 | 129 |
Patients with events, n (%) | 48 (58.5) | 56 (43.4) |
Median PFSa (95% CIb), months | 10.9 (8.2 to 12.7) | 10.9 (8.2 to 12.7) |
Presence of brain metastases at baseline by IRC | ||
Present, n | 15 | 31 |
Patients with events, n (%) | 7 (46.7) | 10 (32.3) |
Median PFSa (95% CIb), months | 10.9 (6.8 to NE) | 9.5 (6.8 to NE) |
Absent, n | 136 | 223 |
Patients with events, n (%) | 80 (58.8) | 95 (42.6) |
Median PFSa (95% CIb), months | 8.9 (8.2 to 11.3) | 8.9 (8.2 to 11.3) |
Histological subtype | ||
Adenocarcinoma, n | 131 | 207 |
Patients with events, n (%) | 74 (56.5) | 88 (42.5) |
Median PFSa (95% CIb), months | 9.7 8.2 to 12.1) | 9.7 (8.2 to 12.1) |
Squamous, n | 14 | 24 |
Patients with events, n (%) | 9 (64.3) | 11 (45.8) |
Median PFSa (95% CIb), months | 3.3 (1.4 to NE) | 5.5 (2.1 to NE) |
Other, n | 6 | 23 |
Patients with events, n (%) | 4 (66.7) | 6 (26.1) |
Median PFSa (95% CIb), months | 5.8 (0.3 to 8.5) | 8.5 (3.1 to 8.5) |
ECOG PS | ||
0, n | 40 | 70 |
Patients with events, n (%) | 15 (37.5) | 19 (27.1) |
Median PFSa (95% CIb), months | NE (8.5 to NE) | NE (8.5 to NE) |
1, n | 111 | 184 |
Patients with events, n (%) | 72 (64.9) | 86 (46.7) |
Median PFSa (95% CIb), months | 8.3 (6.8 to 10.9) | 8.3 (6.9 to 10.3) |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group performance status; IRC = independent review committee; mITT = modified intention-to-treat; NE = not estimable; PFS = progression-free survival.
aEstimated using the Kaplan-Meier method.
bCalculated using the Brookmeyer and Crowley method.
Source: VISION Clinical Study Report.14
Objective Response Rate
Results for the ORR by IRC and ORR by investigator are summarized in Table 16. The ORR by IRC was the primary end point in the VISION trial. The ORR by IRC was 45.0% (95% CI, 36.9 to 53.3) in cohort A and 46.4% (95% CI, 39.8 to 53.2) in cohort A plus C. All observed responses by IRC assessment were PR. The ORR by investigator assessment was a secondary end point. For cohort A, the ORR by investigator assessment was 53.0% (95% CI, 44.7 to 61.1). For cohort A plus C, the ORR by investigator was 43.7% (95% CI, 37.5 to 50.0). Most observed responses by investigator assessment were PR.
Table 16: ORR Results in VISION, Cohort A and Cohort A + C (mITT Population) as of July 1, 2020, Data Cut-Off Date
End point | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
ORR by IRC (primary end point) | ||
Patients contributing to the analysis,a n | 151 | 224 |
ORR, n (%) | 68 (45.0) | 104 (46.4) |
95% CIb | 36.9 to 53.3 | 39.8 to 53.2 |
Best overall responsec | ||
CR | 0 | 0 |
PR | 68 (45.0) | 104 (46.4) |
SD | 38 (25.2) | 53 (23.7) |
PD | 26 (17.2) | 34 (15.2) |
Not evaluable | 19 (12.6) | 33 (14.7) |
ORR by investigator (secondary end point) | ||
Patients contributing to the analysis,a n | 151 | 254 |
ORR, n (%) | 80 (53.0) | 111 (43.7) |
95% CI | 44.7 to 61.1 | 37.5 to 50.0 |
Best overall response | ||
CR | 3 (2.0) | 3 (1.2) |
PR | 77 (51.0) | 108 (42.5) |
SD | 33 (21.9) | 54 (21.3) |
PD | 23 (15.2) | 34 (13.4) |
Not evaluable | 15 (9.9) | 55 (21.7) |
CI = confidence interval; CR = complete response; IRC = independent review committee; mITT = modified intention-to-treat; ORR = objective response rate; PD = progressive disease; PR = partial response; SD = stable disease.
aOnly patients with 2 ore more post-baseline assessments or who discontinued study treatment for any reason are included in the analysis.
b95% CI calculated using the Clopper-Pearson method.
cCR and PR had to be confirmed. SD had to last at least 12 weeks.
Source: VISION Clinical Study Report.14
Table 17: Subgroup Analyses of ORR by IRC in Cohort A and Cohort A + C (mITT Population) as of July 1, 2020, Data Cut-Off Date
Subgroup | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
Line of treatment | ||
First-line, n | 69 | 125 |
ORR, n (%) | 31 (44.9) | 52 (41.6) |
95% CIa | 32.9 to 57.4 | 32.9 to 50.8 |
Second- or later-line, n | 82 | 129 |
ORR, n (%) | 37 (45.1) | 52 (40.3) |
95% CIa | 34.1 to 56.5 | 31.8 to 49.3 |
Presence of brain metastases at baseline by IRC | ||
Present, n | 15 | 31 |
ORR, n (%) | 8 (53.3) | 14 (45.2) |
95% CIa | 26.6 to 78.7 | 27.3 to 64.0 |
Absent, n | 136 | 223 |
ORR, n (%) | 60 (44.1) | 90 (40.4) |
95% CIa | 35.6 to 52.9 | 33.9 to 47.1 |
Histological subtype | ||
Adenocarcinoma, n | 131 | 207 |
ORR, n (%) | 63 (48.1) | 87 (42.0) |
95% CIa | 39.3 to 57.0 | 35.2 to 49.1 |
Squamous, n | 14 | 24 |
ORR, n (%) | 3 (21.4) | 7 (29.2) |
95% CIa | 4.7 to 50.8 | 12.6 to 51.1 |
Other, n | 6 | 23 |
ORR, n (%) | 2 (33.3) | 10 (43.5) |
95% CIa | 4.3 to 77.7 | 23.2 to 65.5 |
ECOG PS | ||
0, n | 40 | 70 |
ORR, n (%) | 23 (57.5) | 35 (50.0) |
95% CIa | 40.9 to 73.0 | 37.8 to 62.2 |
1, n | 111 | 184 |
ORR, n (%) | 45 (40.5) | 69 (37.5) |
95% CIa | 31.3 to 50.3 | 30.5 to 44.9 |
CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group performance status; IRC = independent review committee; mITT = modified intention-to-treat; ORR = objective response rate.
a95% CI calculated using the Clopper-Pearson method.
Source: VISION Clinical Study Report.14
Duration of Response
The DOR by IRC assessment and DOR by investigator assessment were secondary end points in the VISION trial. Results for DOR in cohort A and cohort A plus C as of the July 1, 2020, data cut-off date are summarized in Table 18.
In cohort A, a total of 68 patients had an objective response (confirmed CR or PR) by IRC assessment, and 31 (45.6%) of those patients experienced PD or death as of the July 1, 2020, data cut-off date. Median DOR by IRC was 11.1 (95% CI, 8.4 to 18.5) months. A total of 80 patients had an objective response by investigator assessment, and 39 (48.8%) of those patients experienced PD or death. Median DOR by investigator assessment was 12.7 (95% CI, 9.7 to 18.3) months.
In cohort A plus C, a total of 104 patients had an objective response by IRC assessment, and 33 (31.7%) of those patients experienced PD or death as of the July 1, 2020, data cut-off date. Median DOR by IRC was 11.1 (95% CI, 9.5 to 18.5) months. A total of 111 patients had an objective response by investigator assessment, and 43 (38.7%) of those patients experienced PD or death. Median DOR by investigator assessment was 12.5 (95% CI, 9.7 to 18.3) months.
Results of the subgroup analyses of DOR by IRC are summarized in Table 19. The median DOR by IRC assessment across subgroups was broadly consistent with the main analysis.
Health-Related Quality of Life and Patient-Reported Outcomes
The VISION trial assessed change from baseline and TTD by 10 points in the EQ-5D-5L VAS score; EORTC QLQ-C30 global health status and quality of life score; and EORTC QLQ-LC13 cough, chest pain, and dyspnea symptom scale scores as secondary end points. Results of the TTD analysis of each HRQoL and PRO outcome are summarized in Table 20. No subgroup analyses were performed on the HRQoL and PROs.
Table 18: DOR Results in VISION, Cohort A and Cohort A + C (mITT Population) as of July 1, 2020, Data Cut-Off Date
End point | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
DOR by IRC | ||
Patients with confirmed CR or PR, n | 68 | 104 |
Patients with an event (PD or death), n (%) | 31 (45.6) | 33 (31.7) |
Patients censored, n (%) | 37 (54.4) | 71 (68.3) |
Median duration of follow-up (95% CI),a months | 10.9 (9.7 to 16.7) | 7.0 (6.9 to 9.7) |
Median DORb (95% CIa), months | 11.1 (8.4 to 18.5) | 11.1 (9.5 to 18.5) |
DOR by Investigator | ||
Patients with confirmed CR or PR, n | 80 | 111 |
Patients with an event (PD or death), n (%) | 39 (48.8) | 43 (38.7) |
Patients censored, n (%) | 41 (51.3) | 68 (61.3) |
Median duration of follow-up (95% CI),a months | 15.2 (12.5 to 19.4) | 9.7 (7.0 to 15.1) |
Median DORb (95% CIa), months | 12.7 (9.7 to 18.3) | 12.5 (9.7 to 18.3) |
CI = confidence interval; CR = complete response; DOR = duration of response; IRC = independent review committee; mITT = modified intention-to-treat; PD = progressive disease; PR = partial response.
aCalculated using the Brookmeyer and Crowley method.
bEstimated using the Kaplan-Meier method.
Source: VISION Clinical Study Report.14
Table 19: Subgroup Analyses of DOR by IRC in Cohort A and Cohort A + C (mITT Population) as of July 1, 2020, Data Cut-Off Date
Subgroup | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
Line of treatment | ||
First-line, n | 69 | 125 |
Patients with confirmed CR or PR, n | 31 | 52 |
Patients with an event (PD or death), n (%) | 13 (41.9) | 13 (25.0) |
Median DORa (95% CIb), months | 10.8 (6.9 to NE) | 10.8 (7.2 to NE) |
Second- or later-line, n | 82 | 129 |
Patients with confirmed CR or PR, n | 37 | 52 |
Patients with an event (PD or death), n (%) | 18 (48.6) | 20 (38.5) |
Median DORa (95% CIb), months | 11.1 (9.5 to 18.5) | 11.1 (9.5 to 18.5) |
Presence of brain metastases at baseline by IRC | ||
Present, n | 15 | 31 |
Patients with confirmed CR or PR, n | 8 | 14 |
Patients with an event (PD or death), n (%) | 4 (50.0) | 5 (35.7) |
Median DORa (95% CIb), months | 9.5 (5.5 to NE) | 9.5 (5.5 to NE) |
Absent, n | 136 | 223 |
90 | 60 | |
Patients with an event (PD or death), n (%) | 27 (45.0) | 28 (31.1) |
Median DORa (95% CIb), months | 11.1 (9.7 to 18.5) | 12.4 (9.7 to NE) |
Histological subtype | ||
Adenocarcinoma, n | 131 | 207 |
Patients with confirmed CR or PR, n | 63 | 87 |
Patients with an event (PD or death), n (%) | 29 (46.0) | 30 (34.5) |
Median DORa (95% CIb), months | 11.1 (9.5 to 18.5) | 11.1 (9.5 to 18.5) |
Squamous, n | 14 | 24 |
Patients with confirmed CR or PR, n | 3 | 7 |
Patients with an event (PD or death), n (%) | 1 (33.3) | 2 (28.6) |
Median DORa (95% CIb), months | NE (8.3 to NE) | 8.3 (2.9 to NE) |
Other, n | 6 | 23 |
Patients with confirmed CR or PR, n | 2 | 10 |
Patients with an event (PD or death), n (%) | 1 (50.0) | 1 (10.0) |
Median DORa (95% CIb), months | NE (5.8 to NE) | NE (5.8 to NE) |
ECOG PS | ||
0, n | 40 | 70 |
Patients with confirmed CR or PR, n | 23 | 35 |
Patients with an event (PD or death), n (%) | 6 (26.1) | 7 (20.0) |
Median DORa (95% CIb), months | NE (9.7 to NE) | NE (9.7 to NE) |
1, n | 111 | 184 |
Patients with confirmed CR or PR, n | 45 | 69 |
Patients with an event (PD or death), n (%) | 25 (55.6) | 26 (37.7) |
Median DORa (95% CIb), months | 10.1 (7.2 to 15.7) | 10.1 (7.2 to 15.7) |
CI = confidence interval; CR = complete response; DOR = duration of response; IRC = independent review committee; mITT = modified intention-to-treat; NE = not estimable; PD = progressive disease; PR = partial response.
aEstimated using the Kaplan-Meier method.
bCalculated using the Brookmeyer and Crowley method.
Source: VISION Clinical Study Report.14
Table 20: TTD by 10 Points in HRQoL and PROs in VISION, Cohort A and Cohort A + C (mITT Population) as of July 1, 2020, Data Cut-Off Date
End point | Cohort A (N = 151) | Cohort A + C (N = 254) |
|---|---|---|
EQ-5D-5L VAS | ||
Patients with a deterioration event, n (%) | 61 (40.4) | 81 (31.9) |
Median TTDa (95% CIb), months | 8.3 (5.8 to 17.7) | 8.3 (5.9 to 17.7) |
EORTC QLQ-C30 Global Health Status and Quality of Life | ||
Patients with a deterioration event, n (%) | 54 (35.8) | 74 (29.1) |
Median TTDa (95% CIb), months | 15.2 (6.0 to 33.2) | 15.2 (6.2 to 33.2) |
EORTC QLQ-LC13 Coughing | ||
Patients with a deterioration event, n (%) | 45 (29.8) | 52 (20.5) |
Median TTDa (95% CIb), months | 11.1 (11.1 to NE) | 13.8 (11.1 to NE) |
EORTC QLQ-LC13 Chest Pain | ||
Patients with a deterioration event, n (%) | 45 (29.8) | 56 (22.0) |
Median TTDa (95% CIb), months | 17.7 (11.1 to NE) | 17.7 (11.8 to NE) |
EORTC QLQ-LC13 Dyspnea | ||
Patients with a deterioration event, n (%) | 76 (50.3) | 104 (40.9) |
Median TTDa (95% CIb), months | 5.5 (4.1 to 6.9) | 5.6 (4.1 to 6.9) |
CI = confidence interval; EORTC = European Organization for the Research and Treatment of Cancer; EQ-5D-5L = 5-Level EQ-5D; HRQoL = health-related quality of life; NE = not estimable; mITT = modified intention-to-treat; PRO = patient-reported outcome; QLQ-C30 = Quality of Life Questionnaire; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer 13; TTD = time to deterioration; VAS = visual analogue scale.
aEstimated using the Kaplan-Meier method.
bCalculated using the Brookmeyer and Crowley method.
Source: VISION Clinical Study Report.14
EQ-5D-5L VAS
At baseline, 89.4% (135/151) of patients in cohort A completed the EQ-5D-5L questionnaire and had a resulting VAS score. The mean baseline VAS score was 62 (SD = 20.4). A boxplot of change from baseline in EQ-5D-5L VAS score in cohort A is depicted in Figure 7. Mean values were generally stable over time. The mean change from baseline from cycle 3 up to cycle 21 ranged from 0 to 7 across cycles in cohort A. The number of patients contributing to the analysis deceased at later cycles: 115 at cycle 3, 108 at cycle 5, 98 at cycle 7, 82 at cycle 9, 66 at cycle 11, 59 at cycle 13, 43 at cycle 17, 28 at cycle 21, 14 at cycle 25, 13 at cycle 29, and 75 at end-of-treatment or 30-day safety follow-up.
A boxplot of change from baseline in VAS score in cohort A plus C is depicted in Figure 8. Trends observed in cohort A plus C are similar to those observed in cohort A. At baseline, 84.6% (215 of 254) of patients in cohort A plus C completed the EQ-5D-5L questionnaire and had a resulting VAS score. The number of patients contributing to the analysis deceased at later cycles: 180 at cycle 3, 157 at cycle 5, 133 at cycle 7, 99 at cycle 9, 73 at cycle 11, 62 at cycle 13, 43 at cycle 17, 28 at cycle 21, 14 at cycle 25, 13 at cycle 29, and 91 at end-of-treatment or 30-day safety follow-up.
Figure 7: Boxplot of Change From Baseline in EQ-5D-5L VAS by Time Point, Cohort A mITT Population (N = 151); July 1, 2020, Data Cut-Off Date
C = cycle; D = day; EOT/FU = end-of-treatment/30-day safety follow-up; EQ-5D-5L = 5-Level EQ-5D; EQ VAS = EQ-5D visual analogue scale; mITT = modified intention-to-treat; VAS = visual analogue scale.
Note: Visits with 10 patients or fewer are not summarized and presented. ||
Source: VISION Clinical Study Report.14
Figure 8: Boxplot of Change From Baseline in EQ-5D-5L VAS by Time Point, Cohort A + C mITT Population (N = 254); July 1, 2020, Data Cut-Off Date
C = cycle; D = day; EOT/FU = end-of-treatment or 30-day safety follow-up; EQ-5D-5L = 5-Level EQ-5D; EQ VAS = EQ-5D visual analogue scale; mITT = modified intention-to-treat; VAS = visual analogue scale.
Note: Visits with 10 patients or fewer are not summarized and presented.
Source: VISION Clinical Study Report.14
In cohort A, 40.4% (n = 61) of patients experienced a deterioration in EQ-5D-5L VAS score by 10 points as of the July 1, 2020, data cut-off date. Median TTD in EQ-5D-5L VAS score was 8.3 (95% CI, 5.8 to 17.7) months. In cohort A plus C, 31.9% (n = 81) of patients experienced a deterioration in EQ-5D-5L VAS score by 10 points, and median TTD was 8.3 (95% CI, 5.9 to 17.7) months.
EORTC QLQ-C30 Global Health Status and Quality of Life Score
In cohort A, 89.4% (135 of 152) of patients completed the EORTC QLQ-C30 questionnaire at baseline. The mean global health status and quality of life score at baseline was 54.3 (SD = 24.20). A boxplot of change from baseline in global health status and quality of life score by time point for cohort A is depicted in Figure 9. Mean values were generally stable over time, with mean changes from baseline from cycle 3 up to cycle 21 ranging from −0.3 to 10.2 across cycles. The number of patients contributing to the analysis deceased at later cycles: 115 at cycle 3, 108 at cycle 5, 99 at cycle 7, 82 at cycle 9, 66 at cycle 11, 59 at cycle 13, 43 at cycle 17, 28 at cycle 21, 14 at cycle 25, 13 at cycle 29, and 76 at end-of-treatment or 30-day safety follow-up.
In cohort A plus C, 84.6% (215 of 254) of patients completed the EORTC QLQ-C30 questionnaire at baseline. A boxplot of change from baseline in global health status and quality of life score in cohort A plus C is depicted in Figure 10. The number of patients contributing to the analysis deceased at later cycles: 180 at cycle 3, 157 at cycle 5, 134 at cycle 7, 99 at cycle 9, 73 at cycle 11, 62 at cycle 13, 43 at cycle 17, 28 at cycle 21, 14 at cycle 25, 13 at cycle 29, and 92 at end-of-treatment or 30-day safety follow-up.
Figure 9: Boxplot of Change From Baseline in EORTC QLQ-C30 Global Health Status or Quality of Life Score by Time Point, Cohort A mITT Population (N = 151); July 1, 2020, Data Cut-Off Date
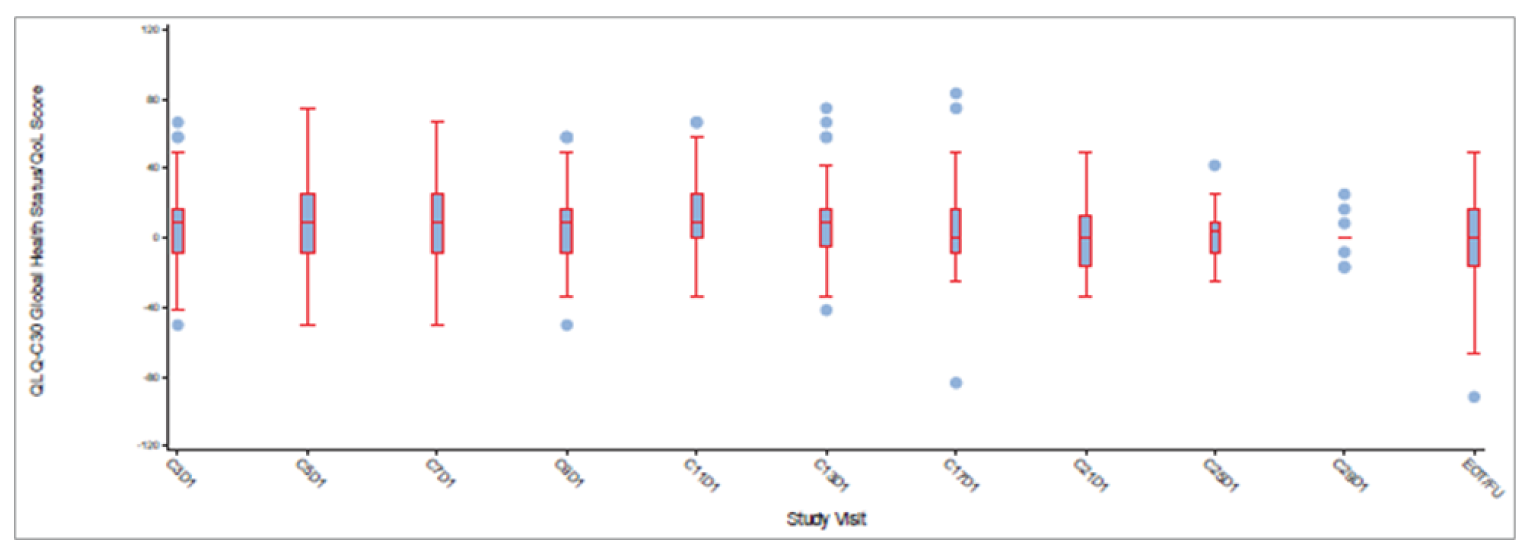
C = cycle; D = day; EORTC = European Organisation for Research and Treatment of Cancer; EOT/FU = end-of-treatment or 30-day safety follow-up; mITT = modified intention-to-treat; QLQ-C30 = Quality of Life Questionnaire Core 30; QoL = quality of life.
Note: Visits with 10 patients or fewer are not summarized and presented.
Source: VISION Clinical Study Report.14
Figure 10: Boxplot of Change From Baseline in EORTC QLQ-C30 Global Health Status or Quality of Life Score by Time Point, Cohort A + C mITT Population (N = 254); July 1, 2020, Data Cut-Off Date
C = cycle; D = day; EORTC = European Organisation for Research and Treatment of Cancer; EOT/FU = end-of-treatment or 30-day safety follow-up; mITT = modified intention-to-treat; QLQ-C30 = Quality of Life Questionnaire Core 30; QoL = quality of life.
Note: Visits with 10 patients or fewer are not summarized and presented.
Source: VISION Clinical Study Report.14
As of the July 1, 2020, data cut-off date, 35.8% (n = 54) of patients in cohort A experienced a deterioration in QLQ-C30 global health status and quality of life score by 10 points, and median TTD was 15.2 (95% CI, 6.0 to 33.2) months. In cohort A plus C, 29.1% (n = 74) of patients experienced a deterioration in global health status and quality of life score by 10 points, and median TTD was 15.2 (95% CI, 6.2 to 33.2) months.
EORTC QLQ-L13 Symptom Scales (Coughing, Dyspnea, and Chest Pain)
In cohort A, 89.4% (135 of 152) of patients completed the EORTC QLQ-LC13 questionnaire at baseline. The number of patients with an evaluable questionnaire decreased over time: 108 at cycle 5, 82 at cycle 9, 59 at cycle 13, 43 at cycle 17, 28 at cycle 21, 14 at cycle 25, 13 at cycle 29, 8 at cycle 33, 6 at cycle 37, 3 at cycle 41, and 2 at cycle 45 as of the July 1, 2020, data cut-off date.
In cohort A plus C, 84.6% (215 of 254) of patients completed the EORTC QLQ-LC13 questionnaire at baseline. Similarly, the number of patients an evaluable questionnaire decreased over time: 180 at cycle 3, 157 at cycle 5, 99 at cycle 9, 62 at cycle 13, 43 at cycle 17, 28 at cycle 21, 14 at cycle 25, 13 at cycle 29, 8 at cycle 33, 6 at cycle 37, 3 at cycle 41, and 2 at cycle 45.
An MMRM was performed on the QLQ-LC13 symptom scales assessed in the VISION trial (coughing, dyspnea, and chest pain). The best variance-covariance pattern was chosen based on model fit using Akaike information criterion among unstructured, Toeplitz, first-order autoregressive, and variance components. The QLQ-LC13 symptom scales range in score from 0 to 100, and a decrease in score represents an improvement in symptoms. The least squares mean changes from baseline in the EORTC QLQ-LC13 symptom scales of coughing, dyspnea, and chest pain are depicted in Figure 11, Figure 12, and Figure 13, respectively. A decrease in QLQ-LC13 symptom scale score represents an improvement; an increase in QLQ-LC13 symptom scale score represents increased symptom burden.
Figure 11: Line Plot of LS Mean Change From Baseline by Visit in EORTC QLQ-LC13 Coughing Symptom Score, Cohort A (N = 151; Left) and Cohort A + C (N = 254; Right) mITT Population as of July 1, 2020, Data Cut-Off Date
BSL = baseline; C = cycle; D = day; EORTC = European Organisation for Research and Treatment of Cancer; LS = least squares; mITT = modified intention-to-treat; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer-13; SEM = standard error of mean.
Source: VISION Clinical Study Report.14
Figure 12: Line Plot of LS Mean Change From Baseline by Visit in EORTC QLQ-LC13 Dyspnea Symptom Score, Cohort A (N = 151; Left) and Cohort A + C (N = 254; Right) mITT Population as of July 1, 2020, Data Cut-Off Date
BSL = baseline; C = cycle; D = day; EORTC = European Organisation for Research and Treatment of Cancer; LS = least squares; mITT = modified intention-to-treat; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer-13; SEM = standard error of mean.
Source: VISION Clinical Study Report.14
Figure 13: Line Plot of LS Mean Change From Baseline by Visit in EORTC QLQ-LC13 Chest Pain Symptom Score, Cohort A (N = 151; Left) and Cohort A + C (N = 254; Right) mITT Population as of July 1, 2020, Data Cut-Off Date
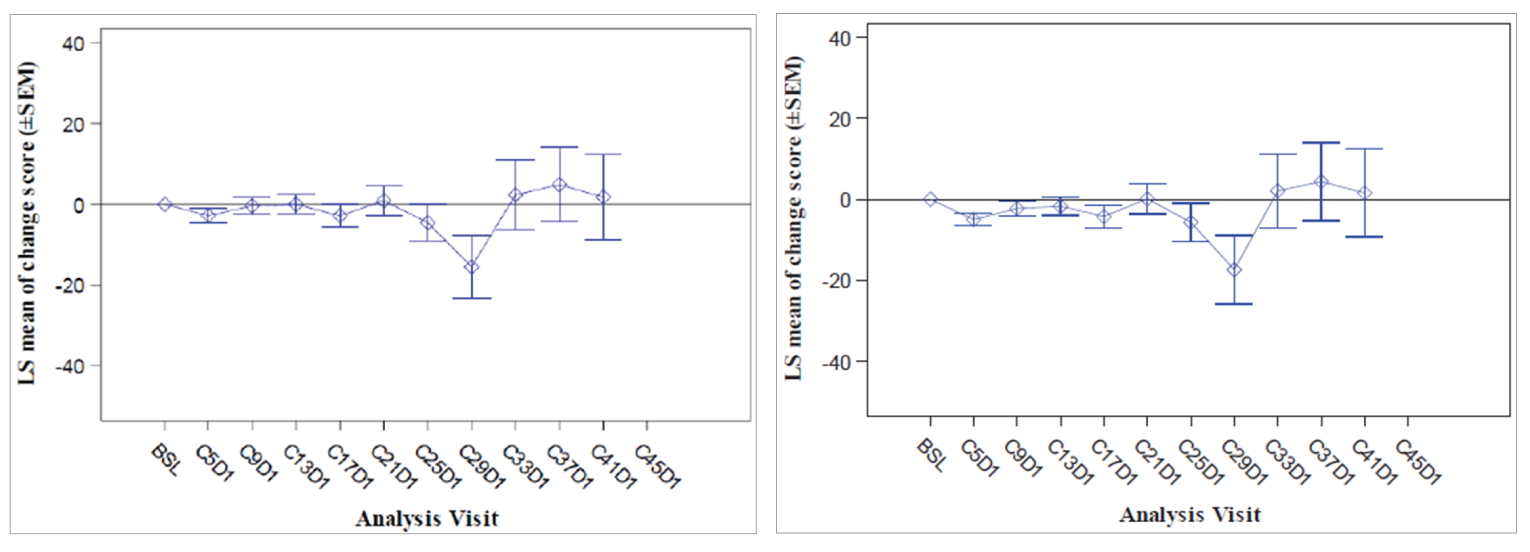
BSL = baseline; C = cycle; D = day; EORTC = European Organisation for Research and Treatment of Cancer; LS = least squares; mITT = modified intention-to-treat; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer-13; SEM = standard error of mean.
Source: VISION Clinical Study Report.14
In cohort A, 29.8% (n = 45) of patients experienced a deterioration by 10 points in coughing symptom score, and median TTD was 11.1 (95% CI, 11.1 to NE) months. For chest pain, 29.8% (n = 45) of patients experienced a deterioration event, and median TTD was 17.7 (95% CI, 11.1 to NE) months. For dyspnea, 50.3% (n = 76) of patients experienced a deterioration event, and median TTD was 5.5 (95% CI, 4.1 to 6.9) months.
In cohort A plus C, 20.5% (n = 52) of patients experienced a deterioration by 10 points in coughing symptom score, and median TTD was 13.8 (95% CI, 11.1 to NE) months. For chest pain, 22.0% (n = 56) of patients experienced a deterioration event, and median TTD was 17.7 (95% CI, 11.8 to NE) months. For dyspnea, 40.9% (n = 104) of patients experienced a deterioration event, and median TTD was 5.6 (95% CI, 4.1 to 6.9) months.
Intracranial CNS Outcomes
Intracranial CNS outcomes in cohort A were reported in an abstract and poster included in the sponsor’s submission to CADTH.23 According to these reports, 23 (15%) of the 152 patients enrolled in cohort A had brain metastases by RECIST v1.1 criteria at baseline, determined by IRC or investigator. New lesions in the brain were identified by the investigator in 6 of 152 patients. Four of the patients who developed new brain lesions also had brain metastases at baseline. A post hoc retrospective analysis of brain lesions determined by CT or MRI was conducted by an IRC using RANO-BM criteria, with a data cut-off date of July 1, 2020. A total of 15 patients had brain metastases at baseline that were evaluable according to RANO-BM criteria. Of 7 patients with measurable CNS disease per RANO-BM, all of whom had received prior radiotherapy, intracranial best-observed responses were PR (n = 5), SD (n = 1), and PD (n = 1). Of 8 patients with non-measurable brain lesions only, 7 achieved intracranial disease control and 1 had PD. Of the 7 patients with disease control, 3 had CR. No data on intracranial CNS outcomes were reported for cohort A plus C.
Harms
Only those harms identified in the review protocol are reported in this section. See Table 21 for detailed harms data on cohort A and cohort A plus C (safety analysis set) as of the July 1, 2020, data cut-off date.
Table 21: Summary of Harms in VISION, Cohort A and Cohort A + C (Safety Analysis Set) as of July 1, 2020, Data Cut-Off Date
Harms | Cohort A (N = 152) | Cohort A + C (N = 255) |
|---|---|---|
Patients with ≥ 1 treatment-emergent AE | ||
n (%) | 151 (99.3) | 246 (96.5) |
Most common events,a n (%) | ||
Peripheral edema | 114 (75.0) | 153 (60.0) |
Nausea | 54 (35.5) | 67 (26.3) |
Diarrhea | 48 (31.6) | 67 (26.3) |
Hypoalbuminemia | 45 (29.6) | 59 (23.1) |
Blood creatinine increased | 44 (28.9) | 64 (25.1) |
Dyspnea | 38 (25.0) | 46 (18.0) |
Fatigue | 30 (19.7) | 38 (14.9) |
Decreased appetite | 28 (18.4) | 40 (15.7) |
Constipation | 26 (17.1) | 40 (15.7) |
Pleural effusion | 25 (16.4) | 34 (13.3) |
Vomiting | 25 (16.4) | 33 (12.9) |
Asthenia | 24 (15.8) | 31 (12.2) |
Back pain | 23 (15.1) | 28 (11.0) |
Cough | 22 (14.5) | 31 (12.2) |
Pneumonia | 21 (13.8) | 26 (10.2) |
Amylase increased | 20 (13.2) | 21 (8.2) |
ALT increased | 18 (11.8) | 29 (11.4) |
Alopecia | 17 (11.2) | 24 (9.4) |
Upper abdominal pain | 16 (10.5) | 22 (8.6) |
Dry skin | 16 (10.5) | 21 (8.2) |
Patients with ≥ 1 SAE | ||
n (%) | 85 (55.9) | 115 (45.1) |
Most common events,b n (%) | ||
Pleural effusion | 13 (8.6) | 17 (6.7) |
Disease progression | 10 (6.6) | 12 (4.7) |
Pneumonia | 10 (6.6) | 12 (4.7) |
General physical health deterioration | 9 (5.9) | 9 (3.5) |
Dyspnea | 8 (5.3) | 10 (3.9) |
Patients who permanently discontinued study treatment due to AEs | ||
n (%) | 42 (27.6) | 52 (20.4) |
Most common events,c n (%) | ||
Peripheral edema | 7 (4.6) | 9 (3.5) |
Pleural effusion | 5 (3.3) | 5 (2.0) |
General physical health deterioration | 4 (2.6) | 4 (1.6) |
Dyspnea | 3 (2.0) | 4 (1.6) |
Pneumonitis | 3 (2.0) | 3 (1.2) |
Disease progression | 3 (2.0) | 4 (1.6) |
Genital edema | 3 (2.0) | 3 (1.2) |
Deaths | ||
n (%) | 76 (50.0) | 86 (33.7) |
Primary reason for death, n (%) | ||
Disease progression | 60 (39.5) | 66 (25.9) |
AE | 12 (7.9) | 15 (5.9) |
Unknown | 4 (2.6) | 4 (1.6) |
Missing | 0 | 1 (0.4) |
Notable harms | ||
Hepatotoxicity, n (%) | ||
ALT increased | 18 (11.8) | 29 (11.4) |
AST increased | 13 (8.6) | 19 (7.5) |
Blood ALP increased | 11 (7.2) | 20 (7.8) |
GGT increased | 9 (5.9) | 14 (5.5) |
Blood albumin decreased | 4 (2.6) | 4 (1.6) |
Hepatic function abnormal | 2 (1.3) | 1 (0.4) |
Hepatic steatosis | 1 (0.7) | 1 (0.4) |
Hepatocellular injury | 1 (0.7) | 1 (0.4) |
Hypertransaminasemia | 1 (0.7) | 1 (0.4) |
Liver disorder | 1 (0.7) | 1 (0.4) |
Liver function test increased | 1 (0.7) | 3 (1.2) |
Blood bilirubin increased | 1 (0.7) | 1 (0.4) |
Hepatic enzyme increased | 1 (0.7) | 1 (0.4) |
Renal toxicity, n (%) | ||
Blood creatinine increased | 44 (28.9) | 64 (25.1) |
Acute kidney injury | 6 (3.9) | 9 (3.5) |
Renal failure | 6 (3.9) | 9 (3.5) |
Chronic kidney disease | 4 (2.6) | 9 (3.5) |
Creatinine renal clearance decreased | 2 (1.3) | 4 (1.6) |
Renal impairment | 2 (1.3) | 8 (3.1) |
Renal injury | 1 (0.7) | 1 (0.4) |
Interstitial lung disease, n (%) | 2 (1.3) | 2 (0.8) |
Pneumonitis, n (%) | 6 (3.9) | 6 (2.4) |
Peripheral edema, n (%) | 114 (75.0) | 153 (60.0) |
AE = adverse event; ALP = alkaline phosphatase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; GGT = gamma-glutamyl transferase; SAE = serious adverse event.
aFrequency > 10% in cohort A or cohort A + C.
bFrequency > 4% in cohort A or cohort A + C.
cFrequency > 2% in cohort A or cohort A + C.
Source: VISION Clinical Study Report.14
Adverse Events
In cohort A, 99.3% of patients experienced at least 1 treatment-emergent AE as of the July 1, 2020, data cut-off date. The most frequently reported AE was peripheral edema (75.0%). Grade 3 or higher peripheral edema was reported in 18 (11.8%) patients. Other frequently reported AEs included nausea (35.5%), diarrhea (31.6%), hypoalbuminemia (29.6%), and increased blood creatinine (28.9%).
In cohort A plus C, 96.5% of patients experienced at least 1 treatment-emergent AE. The most frequently reported AE was peripheral edema (60.0%). Grade 3 or higher peripheral edema was reported in 20 (7.8%) patients. Other frequently reported treatment-emergent AEs were nausea (26.3%), diarrhea (26.3%), increased blood creatinine (25.1%), and hypoalbuminemia (23.1%).
In cohort A, 55.9% of patients experienced at least 1 SAE as of the July 1, 2020, data cut-off date. The most frequently reported SAEs were pleural effusion (8.6%), disease progression (6.6%), and pneumonia (6.6%).
In cohort A plus C, 45.1% of patients experienced at least 1 SAE. The most frequently reported SAEs were pleural effusion (6.7%), disease progression (4.7%), and pneumonia (4.7%).
Withdrawals Due to Adverse Events
In cohort A, 27.6% of patients permanently discontinued study treatment due to an AE. The most frequently reported AEs leading to treatment discontinuation were peripheral edema (4.6%), pleural effusion (3.3%), and general physical health deterioration (2.6%).
In cohort A plus C, 20.4% of patients permanently discontinued study treatment due to an AE. The most frequently reported AEs leading to treatment discontinuation were peripheral edema (3.5%) and pleural effusion (2.0%).
Mortality
In cohort A, 50.0% patients had died as of the July 1, 2020, data cut-off date. Causes of death were reported to be disease progression in 39.5% of patients and an AE in 7.9% of patients.
In cohort A plus C, 33.7% of patient had died. Causes of death were reported to be disease progression in 25.9% of patients and an AE in 5.9% of patients.
Notable Harms
The most frequently reported hepatotoxicity-related AEs in cohort A and cohort A plus C were increased ALT (11.8% and 11.4%, respectively), increased AST (8.6% and 7.5%, respectively), increased blood ALP (7.2% and 7.8%, respectively), and increased GGT (5.9% and 5.5%, respectively). The most frequently reported renal toxicity-related AE in cohort A and cohort A plus C was increased blood creatinine (28.9% and 25.1%, respectively). A total of 2 patients experienced interstitial lung disease and 6 patients experienced pneumonitis, all in cohort A. As of the data cut-off date, 75.0% of patients in cohort A and 60.0% of patients in cohort A plus C had experienced peripheral edema.
Critical Appraisal
Internal Validity
For the primary end point and most secondary end points, an IRC was appropriately used. However, in some secondary assessments, only investigator judgment was used to assess occurrence of the end point, which could lead to observer bias due to the open-label nature of the study. In general, the investigator assessments were in line with the IRC when both were completed on the same end point, although, for ORR, there were marginally fewer patients with a CR (noted by the IRC) during follow-up.
VISION is an open-label, single-arm study. There is no direct evidence comparing tepotinib to a control arm. Furthermore, no statistical testing was performed; data were analyzed descriptively. Due to these limitations of the study design, no definitive conclusions can be drawn from the VISION study regarding the efficacy of tepotinib relative to a comparator, which increases the risk of bias in the estimation of treatment effects due to the potential for confounding related to fluctuations in health status and other unidentified prognostic factors that could affect subjectively assessed outcomes. The open-label, single-arm design can increase the risk of bias in reporting outcomes that are subjective in measurement and interpretation (e.g., response, HRQoL, and AEs) and likely results in overestimating the treatment effect. Outcomes such as OS time and mortality are less likely to be affected. The risk of bias due to the open-label design may be unavoidable, given the unmet need in this population. The potential for this bias was also reduced by using IRC assessment for key study outcomes, such as ORR, DOR, and PFS.
In the original VISION study protocol, only patients with adenocarcinoma were eligible, and patients were required to have failed at 1 or 2 lines of systemic therapy, including a platinum-doublet-containing regimen. In a protocol amendment that was implemented after enrolment in the study had begun, the eligible study population was expanded to include all subtypes of NSCLC and patients who were treatment-naive (i.e., would receive tepotinib as first-line treatment). Significant changes to the study eligibility criteria after participant enrolment had begun may have introduced bias. The direction of this potential bias is unknown. However, the sponsor did conduct subgroup analyses of treatment-naive patients versus patients that were previously treated, and the results were broadly consistent with the overall analyses.
Most patients enrolled in VISION had at least 1 important protocol deviation. This adds to the uncertainty of the results observed.
The analysis populations used in the VISION trial were appropriate. The efficacy outcomes were analyzed descriptively in a mITT population, which excluded 1 patient who was enrolled and treated in cohort A, because the patient tested negative for METex14 skipping alterations in the liquid biopsy and had tumour biopsy data that were not evaluable. Exclusion of 1 patient is unlikely to affect outcomes. Safety outcomes were assessed in all patients who were treated with tepotinib. The clinical experts indicated that exposure to tepotinib in VISION was adequate to assess the efficacy and safety outcomes.
The time-to-event analyses were appropriate, but the data are difficult to interpret in a single-arm trial without a comparator. The median duration of follow-up at the time of the analysis was sufficient. Survival times (median OS and median PFS) were estimated from the Kaplan-Meier models, and patients that did not have an event were censored, which is appropriate.
Subgroup analyses were performed on the outcomes OS, PFS, ORR, and DOR. These subgroup analyses were specified a priori. However, many of the subgroups had limited sample size, resulting in imprecise results for many of the subgroups and uncertainty in the data. There were no tests for statistical differences between subgroups, as the sponsor was only interested in evaluating the overall robustness and consistency of treatment effects.
Intracranial CNS outcomes were analyzed post hoc as an exploratory analysis, which was reported in a conference abstract and poster included in the sponsor’s submission to CADTH only. Furthermore, intracranial CNS outcomes were assessed in patients in cohort A that presented with brain metastases at baseline, as opposed to the overall cohort A patient population. This limits interpretation of the intracranial CNS outcome data because data were not reported for all enrolled study patients (e.g., the proportion of all study patients who experienced disease progression in the CNS).
The VISION study assessed HRQoL and PROs using the EQ-5D-5L, EORTC QLQ-C30, and EORTC QLQ-LC13 questionnaires. However, data were not reported in the Clinical Study Reports for the overall cohort A or cohort A plus C for all components of these instruments. Analyses of the PROs were focused on the EQ-5D-5L VAS, global health status and quality of life from the QLQ-C30, and 3 symptoms scales from the QLQ-LC13. The TTD analysis for this subset of scores was pre-specified in the study protocol. Exclusion of the other components of the instruments may have created bias. The sponsor defined a clinically significant deterioration as a change in 10 points for each of the PROs assessed in the VISION trial. CADTH identified an MID of 7 to 11.5 points for the EQ-5D-5L VAS and 4 points for deterioration in EORTC QLQ-C30 global health status and quality of life score. No MID was identified in the literature for the EORTC QLQ-LC13 symptom scales.
HRQoL outcomes had significant missing data at later time points, affecting the interpretability of trends over time and creating potential for bias in those that remain; the outcomes may therefore not reflect the overall population (i.e., patients who did not continue to complete assessments or who died may have had poorer HRQoL). Therefore, it cannot be concluded that HRQoL is maintained or improves over time. Regarding the EORTC QLQ-LC13 data, the sponsor noted in the Clinical Study Report that data late in the study (i.e., after cycle 25) may not be valid or reliable, due to small sample sizes. The number of patients contributing to the analysis of the EORTC QLQ-C30 global health status and quality of life score and EQ-5D-5L VAS similarly decreased to relatively small sample sizes at later cycles as well. The decreased number of patients contributing to the analysis of the HRQoL and PROs over time creates uncertainty in the data. All HRQoL and PRO data are likely biased in favour of tepotinib and overestimate effects due to the missing data at baseline and follow-up because patients who respond to treatment or have no AEs are more likely to continue in the study and complete the questionnaires. Regarding the MMRM analysis of the EORTC QLQ-LC13 data, the assumption of data missing at random is unlikely to hold true. As a result, the MMRM is unlikely to be a valid method to evaluate the changes in QLQ-LC13 over the follow-up period. The investigators have assumed missing data were missing at random, which is not supported by the losses to follow-up and reasons for discontinuations noted. Moreover, the MMRM approach further assumes that patients missing data would continue to behave or change in a fashion similar to those with ongoing data points. This assumption is strong and unverifiable, and may increase the bias in the observed results, particularly when patients discontinue therapy due to AEs or lack of efficacy, as observed in this study. Last, no index scores for the EQ-5D-5L were provided, and, as a result, the net treatment effect across domains could not be captured.
External Validity
The VISION trial was an international, multi-centre study that included sites in Europe, the US, and Asia. There were no study sites in Canada. The treatment regimen used in the VISION trial aligns with Health Canada’s recommended dose, which is 450 mg tepotinib as tepotinib hydrochloride.
The clinical experts consulted by CADTH indicated that the eligibility criteria used in the VISION trial were appropriate and commonly used in NSCLC trials. It was noted that the VISION study included patients with intracranial CNS metastases at baseline who were neurologically stable and whose glucocorticoid dose was being tapered and patients with untreated CNS metastases who were asymptomatic and had lesions 1 cm or less in diameter. The clinical experts indicated that this reflects the advanced NSCLC patient population in Canada, as a proportion of patients present with brain metastases at baseline and/or develop brain metastases as their disease progresses. However, the clinical experts also noted that the VISION trial restricted enrolment to patients with an ECOG PS of 0 or 1, and patients with NSCLC in Canada often have an ECOG PS of 2 or more.
The clinical experts noted that the study patient population was older than patients typically seen in NSCLC trials, which reflects the population of NSCLC patients with METex14 skipping mutations in Canada. Most patients enrolled in the VISION trial were also White, which the clinical experts indicated was expected because METex14 skipping mutations are typically found in elderly White patients. Although there were some differences in the baseline characteristics of the patients in the study versus the Canadian patient population, the clinical experts indicated that these differences were minor and unlikely to affect generalizability of the study results.
The clinical experts noted that METex14 skipping alterations are rare, which was reflected in the number of patients pre-screened for MET alteration status in the VISION trial. Furthermore, the clinical experts noted that the VISION trial used liquid and/or tumour tissue biopsy to determine the patients’ METex14 skipping alteration status. The clinical experts indicated that both methods could be used in Canada to identify patients with METex14 skipping alterations.
The clinical experts consulted by CADTH indicated that they would preferentially use tepotinib in the first-line setting. In VISION, approximately half of the study patients had received prior anticancer drug therapy and therefore received tepotinib as second- or later-line therapy. The clinical experts indicated that a greater proportion of patients received tepotinib as second- or later-line therapy than they would expect to observe in Canadian practice, and they thought that this is likely due to the eligibility criteria in the original trial protocol (i.e., patients were required to have failed at 1 or 2 lines of systemic therapy), which was later amended to include patients who would receive tepotinib as first-line treatment. The clinical experts noted some differences in previous anticancer drug treatments received by the study patients from what they would expect with standard practice in Canada. More patients appeared to be treated with single-agent platinum therapy than the clinical experts expected, and the most frequently used therapies (≥ 5%) did not include a triplet combination with pembrolizumab. The clinical experts noted that this may have been due to the elderly population, as well as the timing of the trial and approval or availability of other treatments in the countries that trial was conducted in.
The patient groups that provided input on this review indicated that they want treatments that improve symptoms, improve or maintain HRQoL, increase survival, delay disease progression, and help patients achieve long-term remission. This aligns with the following outcomes assessed in the VISION trial: HRQoL and PROs (EORTC QLQ-C30 global health status and quality of life; QLQ-LC13 coughing, dyspnea, chest pain; and EQ-5D-5L VAS), OS, PFS, ORR, and DOR. Similarly, OS was identified as the most important outcome by the clinical experts consulted by CADTH and was assessed as a secondary outcome in VISION. The clinical experts also identified PFS, response rate (i.e., ORR), and maintenance of quality of life as important outcomes.
Patients in the VISION trial had access to study physicians more frequently than patients would in standard practice. In the VISION trial, imaging for tumour assessment was conducted every 6 weeks until 9 months and every 12 weeks thereafter, until disease progression, death, or withdrawal of consent. The clinical experts indicated that, in standard practice, imaging is done every 2 to 4 months. In standard practice, the follow-up visits are done every 4 to 8 weeks in Canada, per the clinical experts. The difference in access to imaging and follow-up visits could affect generalizability of results. A higher level of care may have biased outcomes and resulted in an overestimation of the treatment effect.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
As there was no direct evidence comparing tepotinib to other therapies for the treatment of advanced NSCLC harbouring METex14 skipping alterations, CADTH reviewed the indirect evidence. In addition to reviewing the sponsor’s submission, CADTH conducted a literature search to identify potentially relevant ITCs in patients with advanced NSCLC. A focused literature search for ITCs dealing with non–small cell lung cancer was run in MEDLINE All (1946–) on September 23, 2021. No language or date limits were applied to the search. No potentially relevant ITCs were identified in the literature search. One sponsor-submitted ITC15 is included in this review. The objective of this section of the Clinical Review Report is to summarize and critically appraise the indirect evidence.
Description of Indirect Comparisons
The sponsor-submitted ITCs15 consisted of 2 parts: indirect comparisons of individual patient data from the VISION trial to pooled RWD using propensity scoring, and indirect comparisons of individual patient data from the VISION trial to retrospective observational studies using unanchored MAIC methods.15 In the indirect comparison using propensity scoring, tepotinib data from cohort A of the VISION trial was compared to chemotherapy (without immunotherapy), immunotherapy alone, and crizotinib using a pooled dataset derived from 4 RWE data sources. Crizotinib was not identified as a relevant comparator in the CADTH systematic review protocol outlined in Table 5. Thus, results for the indirect comparison of tepotinib to crizotinib are not included in CADTH’s summary of the ITC results. For the indirect comparisons using unanchored MAICs, cohort A of the VISION trial was compared to 3 retrospective observational studies of patients with NSCLC harbouring METex14 skipping alterations who were treated with chemotherapy or immunotherapy.
Methods of the Sponsor-Submitted ITC
Objectives
The objective of the sponsor-submitted ITC was to estimate the comparative effectiveness of tepotinib relative to commonly used therapies (i.e., chemotherapy and immunotherapy).
Study Selection Methods
The sponsor-submitted ITC analyses included studies and RWE that provided information on patients with NSCLC harbouring METex14 skipping alterations. The methods used to select studies for inclusion were not clearly described in the technical report15 or in CADTH’s requests for additional information.15,26,27 No a priori protocol for selecting studies for inclusion in the indirect comparisons was reported. The eligibility criteria for study inclusion and exclusion (i.e., population, intervention, comparators, outcomes) were not clearly described in the technical report beyond studies that provided information on patients with NSCLC harbouring METex14 skipping alterations.
The sponsor provided a systematic literature review on METex14 skipping alterations in NSCLC.16 For their review, the following databases were searched: PubMed, Embase, and conference proceedings from the ASCO, the World Conference on Lung Cancer, the Society for Health Economics and Outcomes Research (ISPOR), and the European Society of Medical Oncology. The electronic database searches were performed on January 20, 2020, and updated on August 3, 2020; the conference website searches were conducted on September 4, 2020. Publications included in the sponsor’s systematic literature review were required to meeting the following eligibility criteria:
indication: NSCLC with METex14 mutation skipping
interventions: any pharmacologic treatment
types of publication: any study design
patient population: adult patients with NSCLC who harbour METex14 mutation skipping
outcomes:
clinical outcomes
humanistic outcomes
economic outcomes
prognostic outcomes
epidemiological outcomes
publications published in English until January 22, 2020, for the first report and publications published until September 24, 2020, for the updated literature review.
Overall, the sponsor’s systematic literature review identified 17 unique studies assessing clinical burden in patients with NSCLC harbouring METex14 skipping alterations. All identified studies were non-randomized interventional studies or observational studies. This list of studies identified 2 of the 3 studies included in the unanchored MAIC only. It is unclear how the third study in the MAIC was identified and selected for inclusion. In addition, the sponsor’s systematic literature review did not identify the 4 real-world cohort studies included in the indirect comparison using propensity scoring. The sponsor’s systematic literature review concluded that, given the lack of head-to-head comparison, unanchored ITCs may be needed with updated data on novel MET tyrosine kinase inhibitors to determine relative effectiveness. The feasibility of conducting unanchored comparisons was not further discussed in the sponsor’s systematic literature review report.
For the indirect comparison using propensity scoring, the authors of the ITC reported that they gained access to patient-level data from 4 noninterventional, real-world cohort studies. Data from the studies were imported into a Common Data Model (CDM). For the unanchored MAIC, the authors included studies published in the literature that reported outcomes in patients with NSCLC harbouring METex14 skipping alterations. The ITC technical report indicated that a MAIC was conducted to compare the retrospective studies to VISION because patient-level data were not available to the sponsor. The methods used to extract data from the 3 studies included in the unanchored MAIC were not reported. Study quality was not assessed by the authors of the ITC. A limited assessment of heterogeneity was provided as a narrative.15,27
The sponsor-submitted ITC assessed 2 efficacy outcomes: OS and PFS. It is unclear whether these outcomes were pre-specified. No safety outcomes were included in the ITC.
ITC Analysis Methods
Indirect Comparison to VISION Study Using Propensity Scoring
Data from 4 real-world cohort studies were imported into a CDM. The patient-level data were aligned to the VISION study using inclusion and exclusion criteria, which were implemented in the following order:
include age 18 years or older
exclude stages I-IIIA
exclude missing both disease stage and advanced or metastatic disease status
exclude ECOG 2 or more
exclude missing both PFS or time to next treatment or death and OS
include METex14 skipping population
exclude ALK positive
exclude EGFR positive.
Before the eligibility criteria were applied, there were 360 patients in the overall dataset. After the eligibility criteria were applied, 140 patients with data for 273 treatment lines remained in the CDM.
Two mutually exclusive analysis groups were derived based on therapy type: patients who received chemotherapy (potentially in combination with other medicines, but not immunotherapy or MET inhibitors), and patients who received immunotherapy alone. As there were more treatment lines than patients, a maximum of 1 treatment line was included per patient in each analysis. When there was more than 1 line available for a given analysis, a random line was selected. For example, for a patient with first-line immunotherapy and then 2 lines of chemotherapy, the immunotherapy line would be included in the immunotherapy comparison, with 1 of the 2 chemotherapy lines selected randomly for the chemotherapy comparison. Similarly, a patient who received a MET inhibitor and then chemotherapy would be included as a previously treated chemotherapy patient.
Propensity scoring was then conducted on the immunotherapy and chemotherapy groups, with propensity scores calculated on variables selected by clinical input. A standardized mortality ratio weighting approach was used to reweight the RWD to match the tepotinib data from the VISION study. The authors indicated that reweighting was used instead of matching to use all available data. The authors of the ITC chose variables included in the calculation of the propensity score based on input from 2 clinical experts (lung cancer oncologists) they consulted. These clinicians reviewed a list of possible covariates and deemed the following variables to be relevant:
prior treatment experience
mean age
metastatic or stage 4 disease (versus non-metastatic)
sex
adenocarcinoma histology
smoking history.
The authors of the ITC noted that, per their clinical experts, ECOG PS would ideally have been included in the models, but this was not possible due to the amount of missing data.
The authors presented baseline demographic characteristics for the chemotherapy and immunotherapy analysis sets before and after weighting. ESS measures were reported.
The ITC reported data for the outcomes OS and PFS. In the ITC technical report, the authors indicate that they considered response rate but did not evaluate this outcome due to a large volume of data that were missing or not reported. Kaplan-Meier curves were presented for OS and PFS for visual comparison. It was unclear whether PFS by investigator assessment or IRC assessment from the VISION trial was used. In addition, the authors reported Cox proportional HRs and restricted mean survival time (RMST) for the propensity score results. Statistical tests were performed on unweighted and weighted data, for both PFS and OS, in all comparisons.
When PFS data were unavailable, time to next treatment or death, or time on treatment, was used as a proxy. Data-cleaning rules were used so that censoring was consistent across end points. When patients continued treatment without an OS event, they were assumed to be censored for OS at their last contact point for treatment. When a patient’s death was confirmed, any data after the death were discarded because the authors thought they were likely either estimated or predicted data. If a patient’s treatment times added up to more than their survival time, the final line of treatment time was shortened so that the treatment time data matched the OS time.
Subgroup analyses were conducted by line of treatment (previously untreated versus previously treated). It is unclear whether the subgroup analyses were pre-specified. The same approach to propensity score weighting was taken as with the overall groups, with standardized mortality ratio weights used to reweight the comparator group to match the characteristics of the VISION study patients from the same line of treatment. Kaplan-Meier curves were presented for visual comparison.
A sensitivity analysis was conducted, using only patients with ECOG PS data available. It is unclear whether this sensitivity analysis was pre-specified.
Indirect Comparison to VISION Study Using MAIC
The authors selected 3 retrospective studies for inclusion in the unanchored MAIC. The technical report did not include a justification for selection of the model or choice of the comparator studies. The methods used to extract data from the studies were not reported. An unanchored MAIC was performed because the index trial (VISION) is a single-arm study.
Patient characteristics available from each of the studies were presented to clinical experts to select variables for matching. The following characteristics were selected and adjusted for:
prior treatment experience
mean age
sex
smoking status
adenocarcinoma histology
metastatic disease.
As noted earlier, the authors of the ITC reported that that ECOG PS ideally would have been included in the models, but this was not possible due to missing data.
The MAIC package in R was used to reweight the patient-level tepotinib data from the VISION study to match the 3 selected retrospective studies. ESS measures were reported, and the authors presented demographic data before and after weighting. A limited assessment of heterogeneity was provided as a narrative. The authors did not report any steps taken to address potential heterogeneity.
OS and PFS were analyzed. It was unclear whether PFS by investigator assessment or IRC assessment from the VISION trial was used. Kaplan-Meier curves were presented for visual comparison only. No subgroup or sensitivity analyses were performed.
Results of Indirect Comparison to VISION Using Propensity Scoring
Summary of Included Studies
The authors provided a limited description of the RWE studies and database that were pooled for the indirect comparison using propensity scoring, which are summarized in this section and in Table 22.
Table 22: Summary of Studies — Indirect Comparison Using Propensity Scoring
Study | Study design | Study sites | N | Outcomes captured |
|---|---|---|---|---|
0015 | Noninterventional real-world retrospective cohort study | Community oncology practices in the US | 39 | PFS OS Response rate |
0035 | Noninterventional real-world retrospective cohort study | Israel, Netherlands, Taiwan, and US | 86 | TTNTD |
COTA | RWE database | US and Canada | 202 | PFS OS TTNTD |
Wong et al. (2021) | Retrospective review | Canada (British Columbia only) | 41 | Time on treatment OS |
OS = overall survival; PFS = progression-free survival; RWE = real-world evidence; TTNTD = time to next treatment or death.
Source: Sponsor’s technical report.15
The noninterventional 0015 study consisted of data collected from the Concerto HealthAI US real-world database taken from electronic medical records (EMRs) used in a real-world retrospective cohort study. Most sites were community oncology practices in the US. The study period used was January 1, 2004, until September 30, 2019. The dataset included 39 patients with MET alterations, with 76 treatment lines. Outcomes captured included PFS, OS, and response rate. As the data were taken from clinical practice, all assessments were done by an investigator.
The noninterventional 0035 study consisted of EMR data collected via chart abstraction at 6 sites in 4 countries (Israel, the Netherlands, Taiwan, the US). The inclusion period was January 1, 2010, to September 30, 2018. Patients were followed from an index treatment exposure date (first recorded exposure to systemic therapy) until death, loss-to follow-up, or the end of available data. The dataset included 86 patients harbouring a MET alteration, with details of 165 treatment lines. The data captured did not include response. PFS was not directly captured and was estimated from time to next treatment or death.
The COTA RWE database is a de-identified data source drawn from EMRs of mainly academic, for-profit, and community oncologist provider sites and hospital systems in the US. In total, 202 patient records were available for a total of 680 lines of therapy. These data were collected from August 15, 2008, to February 10, 2020.
The Wong et al. (2021) study was a retrospective review of treatments and outcomes for patients with metastatic NSCLC harbouring METex14 skipping alterations in British Colombia, Canada, from January 2016 to September 2019. The objective of the study was to identify patients treated in British Columbia with METex14 skipping to understand prevalence, biology, and response to treatment, and to identify molecular signatures that may predict for response or resistance to targeted MET therapy in the setting of advanced disease. Data were available for 41 patients with METex14 skipping alterations, although not all received treatment. Treatments used are not identified beyond class for platinum-doublet chemotherapy and immunotherapy.
Baseline Patient Characteristics
Baseline characteristics of patients in the immunotherapy and chemotherapy analysis sets from the CDM compared to patients treated with tepotinib in the VISION study are summarized in Table 23.
Table 23: Baseline Patient Characteristics of the Analysis Sets Used in the Indirect Comparison With Propensity Scoring
Characteristic | Tepotinib (VISION) | Chemotherapy (CDM) | Immunotherapy (CDM) |
|---|---|---|---|
Sample size, n | 151 | 66 | 51 |
0015 | NA | 10 | 9 |
0035 | NA | 23 | 10 |
COTA | NA | 22 | 18 |
Wong et al. (2021) | NA | 11 | 14 |
Age, mean (SD) | 73.0 (9.0) | 69.9 (8.5) | 71.7 (10.0) |
Prior treatment, n (%) | |||
Untreated | 82 (54.3) | 29 (43.9) | 31 (60.8) |
Experienced | 69 (45.7) | 37 (56.1) | 20 (39.2) |
Sex, n (%) | |||
Male | 79 (52.3) | 38 (57.6) | 24 (47.1) |
Female | 72 (47.7) | 28 (42.4) | 27 (52.9) |
Race, n (%) | |||
White | 107 (70.9) | 34 (51.5) | 29 (56.9) |
Asian | 38 (25.2) | 16 (24.2) | 2 (3.9) |
Black or African American | 0 | 1 (1.5) | 1 (2.0) |
Other | 0 | 1 (1.5) | 3 (5.9) |
Not available | 4 (2.6) | 14 (21.2) | 16 (31.4) |
Smoking history, n (%) | |||
Yes | 78 (51.7) | 37 (56.1) | 31 (60.8) |
No | 73 (48.3) | 29 (43.9) | 20 (39.2) |
Disease stage, n (%) | |||
IIIB/IIIC | 3 (2.0) | 6 (9.1) | 2 (3.9) |
IV/IVB | 148 (98.0) | 49 (74.2) | 34 (66.7) |
Not available | 0 | 11 (16.7) | 15 (29.4) |
Metastatic disease, n (%) | |||
Yes | 148 (98.0) | 64 (97.0) | 51 (100.0) |
No | 3 (2.0) | 2 (3.0) | 0 |
Histology, n (%) | |||
Adenocarcinoma | 131 (86.8) | 49 (74.2) | 40 (78.4) |
Squamous | 14 (9.3) | 7 (10.6) | 6 (11.8) |
Others | 6 (4.0) | 5 (7.6) | 3 (5.9) |
CDM = common data model; NA = not applicable; SD = standard deviation.
Source: Sponsor’s technical report.15
In cohort A (N = 151) of the VISION study, the mean age was 73.0 years. Most patients were White (70.9%), had an ECOG PS of 1 (73.5%), adenocarcinoma histology type (86.8%), and stage IV disease at study entry (74.8%). Overall, 54.3% of patients had received prior anticancer drug therapy for advanced or metastatic disease, and 51.7% had a smoking history. The most frequently administered prior drug therapies were carboplatin, pemetrexed, cisplatin, and pembrolizumab.
The chemotherapy analysis set (N = 66) had a mean age of 69.9 years. Most patients were treatment-experienced (56.1%), had a history of smoking (56.1%), had metastatic disease (97.0%), and had adenocarcinoma on histology (74.2%). The proportion of patients in the chemotherapy analysis set that had adenocarcinoma histology was lower than VISION, as was the mean age. Data on ECOG PS were not reported in the ITC technical report. Most chemotherapy regimens contained pemetrexed (37 of 66) or platinum-based chemotherapy (51 of 66). The most frequently reported treatment regimen was carboplatin and pemetrexed (30.3%).
The immunotherapy analysis set (N = 51) had a mean age of 71.7 years. The majority of patients were previously untreated (60.8%), had a history of smoking (60.8%), and had adenocarcinoma on histology (78.4%). The proportion of patients who were previously untreated was higher, and the proportion of patients who had adenocarcinoma on histology was lower in the immunotherapy analysis set compared to VISION. All patients (100%) had metastatic disease. Data on ECOG PS were not reported in the ITC technical report. Of the immunotherapies that were specified, the most frequently received were pembrolizumab (43.1%) and nivolumab (21.6%).
Results
The unweighted and weighted baseline patient characteristics for the chemotherapy and immunotherapy analysis sets compared to patients treated with tepotinib in VISION are summarized in Table 24. The authors of the ITC reported that the matching worked as intended in the chemotherapy dataset. However, the authors noted that there remained some imbalances in the immunotherapy dataset, likely due to low patient numbers. Metastatic disease remained more than a standardized mean difference of 0.1, which is an often-used metric in the literature to indicate imbalance in the covariate.
Table 24: Baseline Patient Characteristics of the Chemotherapy and Immunotherapy Analysis Sets in the CDM Before and After Standardized Mortality Ratio Weighting, Compared to VISION
Characteristic | Tepotinib (VISION) | Chemotherapy (CDM) | Immunotherapy (CDM) | ||
|---|---|---|---|---|---|
Unweighted | Weighted | Unweighted | Weighted | ||
Sample size or ESS, n | 151 | 66 | 152.0 | 51 | 149.8 |
0015 | NA | 10 | 20.3 | 9 | 22.1 |
0035 | NA | 23 | 54.7 | 10 | 36.8 |
COTA | NA | 22 | 50.8 | 18 | 43.4 |
Wong et al. (2021) | NA | 11 | 26.1 | 14 | 47.5 |
Age, mean (SD) | 73.0 (9.0) | 69.9 (8.5) | 72.7 (7.0) | 71.7 (10.0) | 72.7 (9.5) |
Prior treatment, n (%) | |||||
Untreated | 82 (54.3) | 29 (43.9) | 86.1 (56.7) | 31 (60.8) | 79.8 (53.2) |
Experienced | 69 (45.7) | 37 (56.1) | 65.8 (43.3) | 20 (39.2) | 70.0 (46.8) |
Sex, n (%) | |||||
Male | 79 (52.3) | 38 (57.6) | 81.7 (53.7) | 24 (47.1) | 83.8 (56.0) |
Female | 72 (47.7) | 28 (42.4) | 70.3 (46.3) | 27 (52.9) | 66.0 (44.0) |
Race, n (%) | |||||
White | 107 (70.9) | 34 (51.5) | 77.1 (50.7) | 29 (56.9) | 79.7 (53.2) |
Asian | 38 (25.2) | 16 (24.2) | 39.2 (25.8) | 2 (3.9) | 9.4 (6.3) |
Black or African American | 0 | 1 (1.5) | 1.1 (0.7) | 1 (2.0) | 1.7 (1.1) |
Other | 0 | 1 (1.5) | 2.0 (1.3) | 3 (5.9) | 7.2 (4.8) |
Not available | 4 (2.6) | 14 (21.2) | 32.6 (21.4) | 16 (31.4) | 51.8 (34.6) |
Smoking history, n (%) | |||||
Yes | 78 (51.7) | 37 (56.1) | 78.4 (51.6) | 31 (60.8) | 74.7 (49.9) |
No | 73 (48.3) | 29 (43.9) | 73.6 (48.4) | 20 (39.2) | 75.1 (50.1) |
Disease stage, n (%) | |||||
IIIB/IIIC | 3 (2.0) | 6 (9.1) | 13.2 (8.7) | 2 (3.9) | 5.1 (3.4) |
IV/IVB | 148 (98.0) | 49 (74.2) | 112.6 (74.1) | 34 (66.7) | 91.6 (61.1) |
Not available | 0 | 11 (16.7) | 26.1 (17.2) | 15 (29.4) | 53.1 (35.4) |
Metastatic disease, n (%) | |||||
Yes | 148 (98.0) | 64 (97.0) | 149.4 (98.3) | 51 (100.0) | 149.8 (100.0) |
No | 3 (2.0) | 2 (3.0) | 2.5 (1.7) | 0 | 0 |
Histology n (%) | |||||
Adenocarcinoma | 131 (86.8) | 49 (74.2) | 133.6 (87.9) | 40 (78.4) | 130.5 (87.1) |
Squamous | 14 (9.3) | 7 (10.6) | 7.5 (4.9) | 6 (11.8) | 11.8 (7.9) |
Others | 6 (4.0) | 5 (7.6) | 4.9 (3.2) | 3 (5.9) | 3.5 (2.3) |
CDM = common data model; ESS = effective sample size; NA = not applicable; SD = standard deviation.
Source: Sponsor’s technical report.15
The Kaplan-Meier curves of OS and PFS for the chemotherapy analysis set compared to tepotinib in VISION are depicted in Figure 14 and Figure 15, respectively. The Kaplan-Meier curves of OS and PFS for the immunotherapy analysis set compared to tepotinib in VISION are depicted in Figure 16 and Figure 17, respectively.
Figure 14: Kaplan-Meier Plot of OS for the Unweighted and Weighted Chemotherapy Analysis Set From RWD Compared to VISION
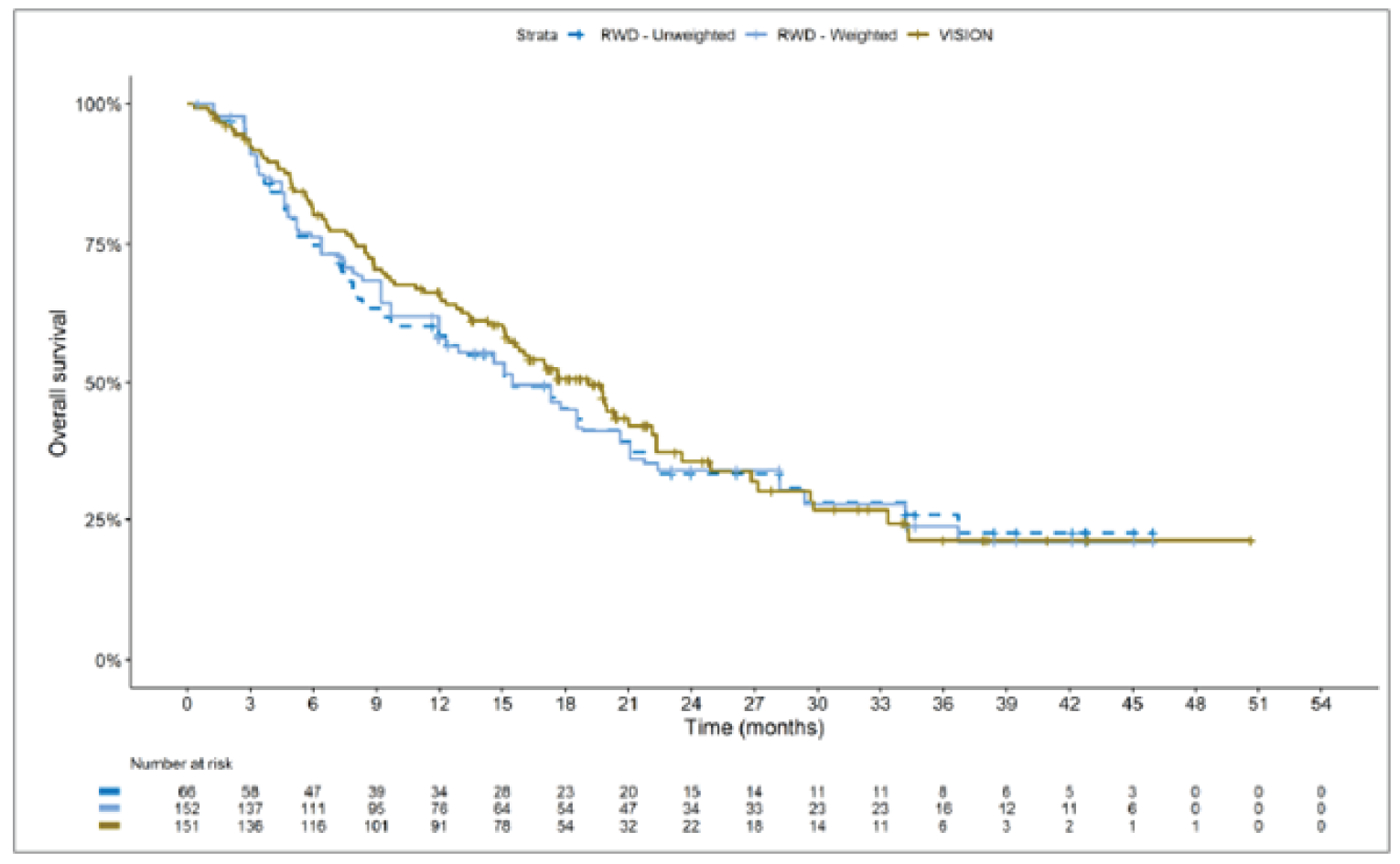
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 15: Kaplan-Meier Plot of PFS for the Unweighted and Weighted Chemotherapy Analysis Set From RWD Compared to VISION
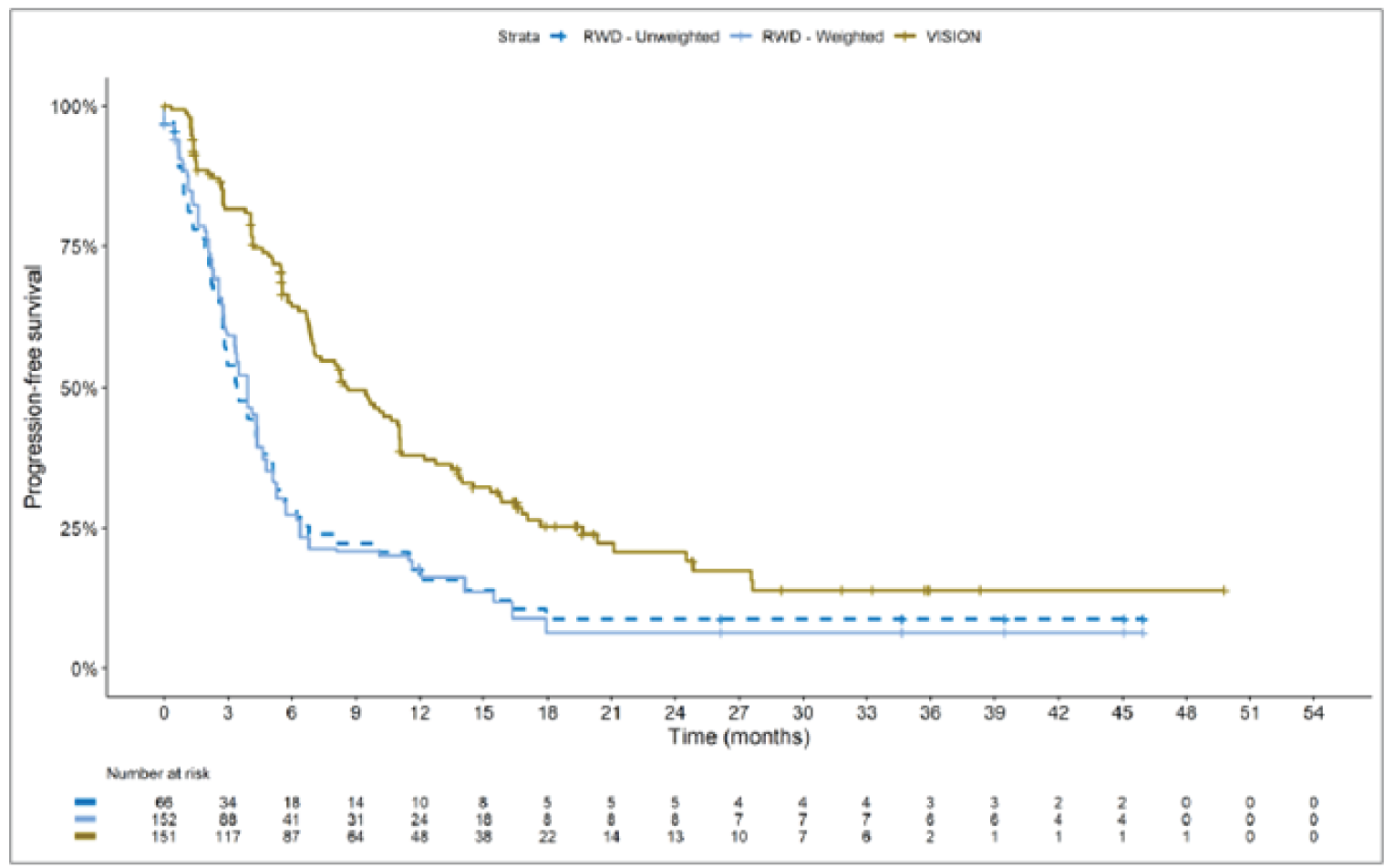
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 16: Kaplan-Meier Plot of OS for the Unweighted and Weighted Immunotherapy Analysis Set From RWD Compared to VISION
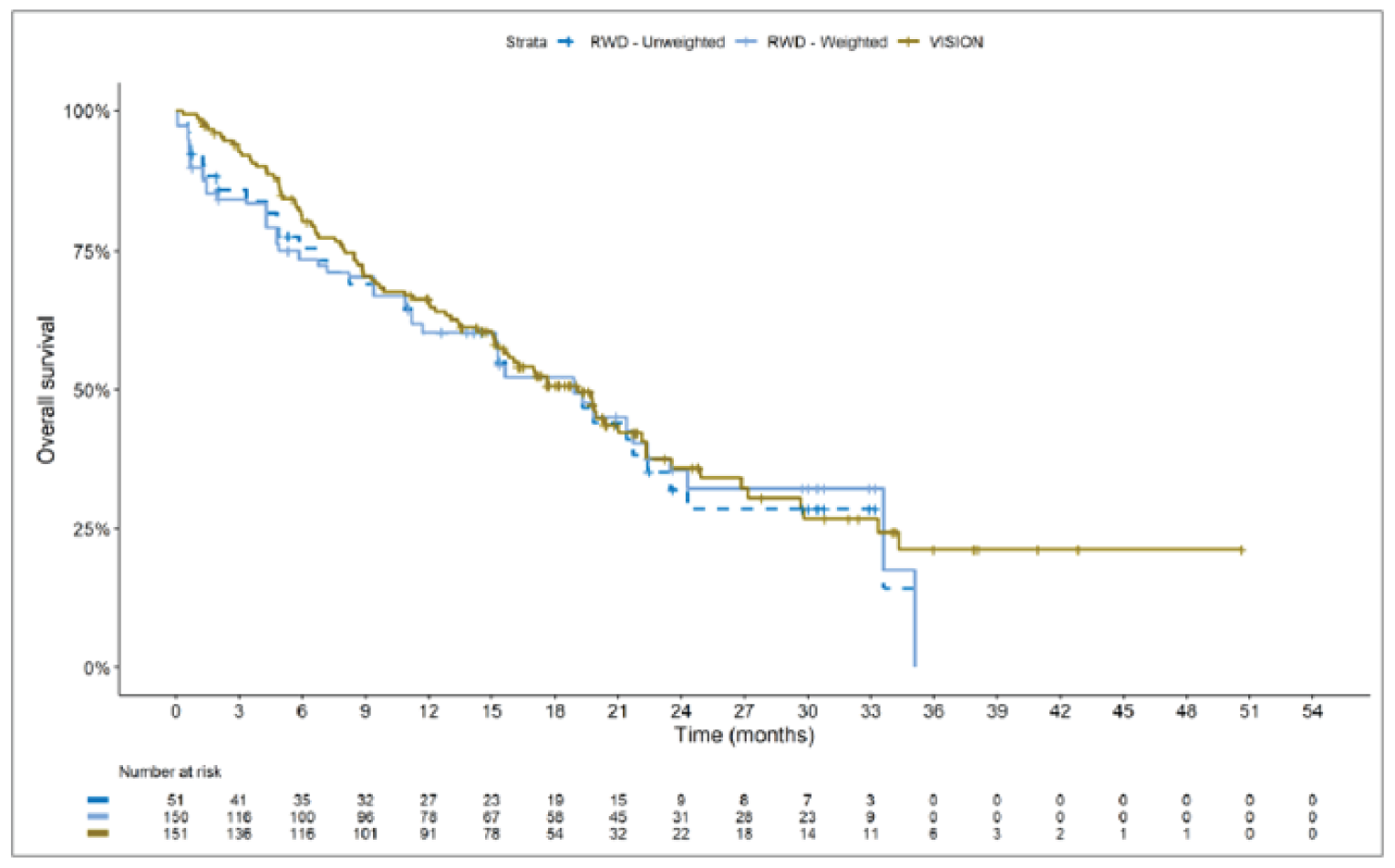
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 17: Kaplan-Meier Plot of PFS for the Unweighted and Weighted Immunotherapy Analysis Set From RWD Compared to VISION
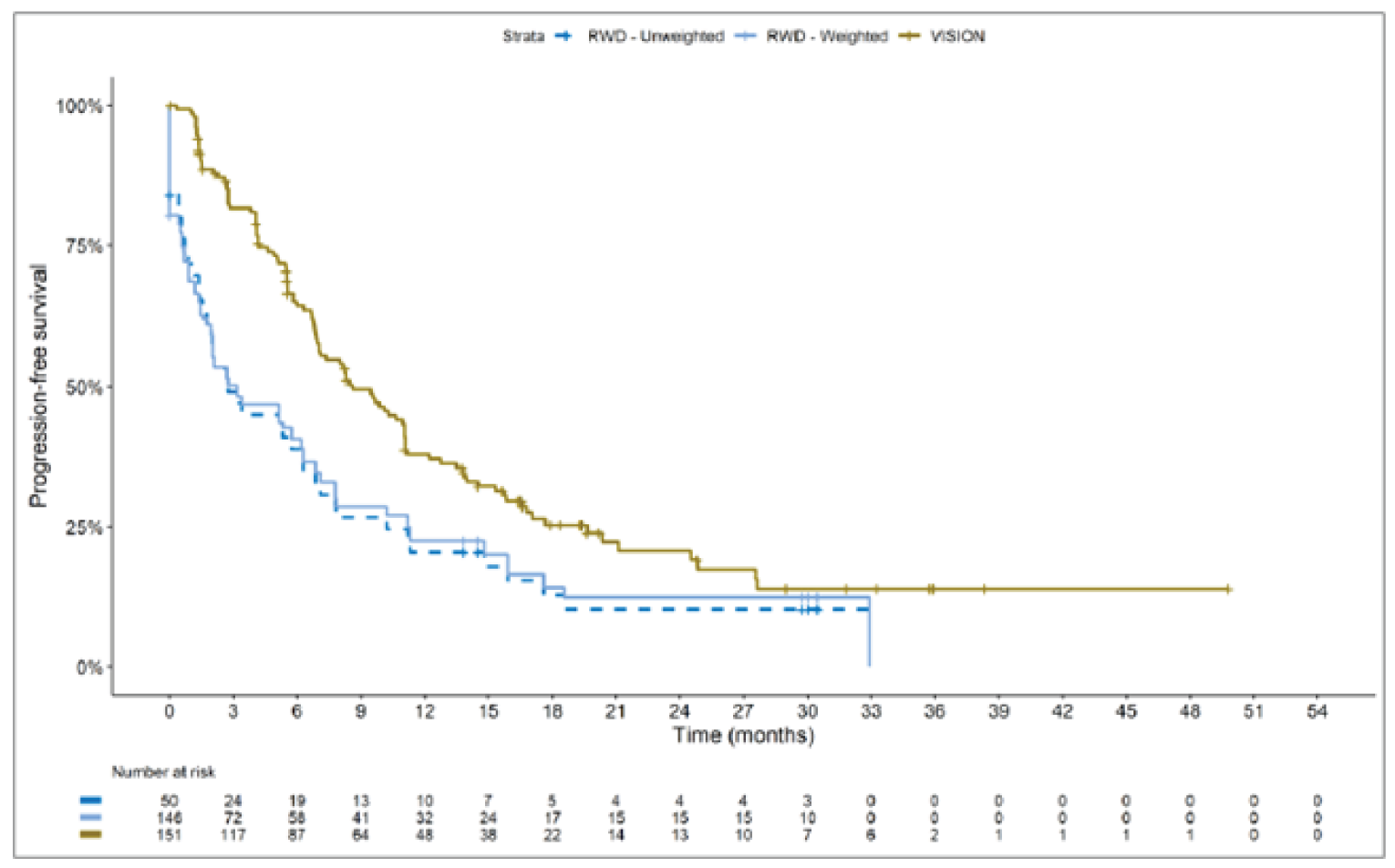
RWD = real-world data.
Source: Sponsor’s technical report.15
Propensity score results for OS and PFS for the immunotherapy and chemotherapy analysis sets compared to tepotinib in the VISION study are summarized in Table 25. An HR of less than 1 favoured tepotinib. For OS, the Cox proportional HR was 0.91 (95% CI, 0.62 to 1.35) for chemotherapy and 0.91 (95% CI, 0.59 to 1.42) for immunotherapy. The RMST for OS was 18.30 months and 18.60 months in the chemotherapy and immunotherapy groups, respectively, compared to 19.25 months in the tepotinib group. For PFS, the Cox proportional HR was 0.49 (95% CI, 0.35 to 0.69) for chemotherapy and 0.59 (95% CI, 0.39 to 0.90) for immunotherapy. The RMST for PFS was 7.52 months and 8.15 months in the chemotherapy and immunotherapy groups, respectively, compared to 12.71 months in the tepotinib group. The authors of the ITC indicated that the Cox proportional hazards assumption may not have been met.
Table 25: Summary of Propensity Score Results of Immunotherapy and Chemotherapy vs. Tepotinib
Outcome | Tepotinib (N = 151) | Chemotherapy (N = 66, ESS = 152) | Immunotherapy (N = 51, ESS = 150) |
|---|---|---|---|
OS | |||
Median OS (95% CI), months | 19.1 (15.3 to 22.3) | 15.5 (9.7 to 28.2) | 18.9 (11.2 to NE) |
RMST, months | 19.25 | 18.30 | 18.60 |
Cox HRa (95% CI) | NA | 0.91 (0.62 to 1.35) | 0.91 (0.59 to 1.42) |
P value | NA | 0.6495 | 0.6820 |
PFS | |||
Median PFS (95% CI), months | 8.6 (7.0 to 11.1) | 3.9 (2.8 to 5.1) | 3.2 (1.4 to 7.8) |
RMST, months | 12.71 | 7.52 | 8.15 |
Cox HRa (95% CI) | NA | 0.49 (0.35 to 0.69) | 0.59 (0.39 to 0.90) |
P value | NA | < 0.0001 | 0.0131 |
CI = confidence interval; Cox = Cox proportional hazards; ESS = effective sample size; HR = hazard ratio; NA = not applicable; NE = not estimable; OS = overall survival; PFS = progression-free survival; RMST = restricted mean survival time; vs. = versus.
aHR less than 1.0 favours tepotinib.
Source: Sponsor’s technical report.15
Based on visual comparison of the Kaplan-Meier curves and the data presented in Table 25, the authors of the ITC concluded that, when the patient populations were balanced, patients treated with tepotinib had a greater PFS compared to patients treated with chemotherapy or immunotherapy. The authors also concluded that tepotinib conferred a benefit of smaller magnitude in OS compared to chemotherapy and immunotherapy.
Subgroup analyses were performed by line of treatment (untreated or treatment-naive versus previously treated). Results of the subgroup analyses are summarized in Appendix 3 (Table 44). The Kaplan-Meier plots for visual comparison are also presented in Appendix 3 (Figure 28 to Figure 35). In general, the subgroup analyses of PFS were consistent with the overall cohort analysis. The subgroup analysis results for OS varied by comparison and subgroup.
The results of the sensitivity analysis using only patients with an ECOG PS available were generally consistent with the overall analysis of PFS and OS.
Critical Appraisal of Indirect Comparison Using Propensity Scoring
The methods used in the sponsor-submitted ITC lacked important details, which creates uncertainty in the data. The methods used to identify and select the RWE cohort studies that contributed RWD to the CDM were not reported. The sponsor’s systematic literature review16 did not identify any of the 4 RWE cohort studies. The criteria used to select these studies were unclear. No a priori protocol was reported for selecting the studies used in the ITC specifically. Furthermore, it is unclear why other studies that were identified in the sponsor’s systematic literature review were not included in this ITC. As a result, there is significant risk of bias in study selection, and there is a lack of certainty that all relevant studies were identified. The authors of the ITC did not assess the quality of the RWE cohort studies and therefore did not consider their quality in the ITC analysis. Any potential risks of bias of the included data sources (i.e., methodological limitations) were not assessed or reported. A limited assessment of heterogeneity was reported. In addition to reporting issues, a number of methodological limitations were noted.
First, the authors of the ITC used propensity scoring to indirectly compare an interventional, prospective trial (VISION) to RWD from retrospective, noninterventional cohort studies and databases. Differences in the study designs could not be accounted for in the indirect comparison. This may be problematic, as there are notable differences in treatments and procedures received by patients, data collection methods, and assessments. In addition, the indirect comparisons may have been biased by the differential distribution of invalid or missing data between the VISION clinical trial and retrospective datasets.
The authors of the ITC applied inclusion and exclusion criteria to the patient-level data imported into the CDM based on the eligibility criteria used in VISION trial. Although the data were restricted to better align with VISION, the authors of the ITC noted that the remaining patients in the immunotherapy and chemotherapy analysis sets of RWD cannot be confirmed as eligible for the VISION study (i.e., they may have been excluded for other reasons if they had been prospectively screened). In addition, details regarding key baseline characteristics (e.g., ECOG PS, presence of brain metastases at baseline) of the patients included in the RWD were not provided. As a consequence, there may be clinical differences between the RWD groups and the VISION study patients that could bias the results of the indirect comparison.
The comparison with immunotherapy may have been biased by the fact that this therapy is restricted to tumours with high PD-L1 expression in the first-line setting, which was not accounted for in the VISION trial.
A key assumption of propensity score analyses to ensure unbiased estimates is that all confounders that predict either receipt of therapy or the outcome itself are included in the development of the propensity score. However, the variables included for matching in the indirect comparison do not reflect all confounders. Per the author’s clinical experts consulted on the sponsor-submitted ITC, ECOG PS would ideally have been included in the models but was not included due to the large number of patients missing these data. The clinical experts consulted by CADTH also agreed that ECOG PS is an important confounder. The clinical experts indicated that, in clinical practice, many patients with an ECOG PS of 2 or higher are treated, whereas they were excluded from the VISION trial. Therefore, ECOG PS is likely unbalanced between the groups. In terms of histology, patient numbers allowed only the proportion of adenocarcinoma to be matched. Last, the clinical experts consulted by CADTH noted that the presence of brain metastases at baseline is an important confounder that was not accounted for in this analysis. Since not all confounders were included, there is a substantial risk of bias in the results. Moreover, to ensure unbiased estimates with the use of propensity scores, balance among all major confounders must be achieved, both those included in the propensity score development itself and other potential confounders. Balance among the limited list of confounders included in the propensity score development was achieved in the chemotherapy analysis set. However, there remained some imbalances in the immunotherapy dataset despite weighting, likely due to low patient numbers. The imbalances further contribute to the risk of bias and uncertainty in the results.
In addition, in propensity score analyses, after estimating the propensity score, the overlap (region of common support) of the distributions of the estimated propensity score for the treatment and comparison groups should be examined graphically or through formal goodness-of-fit tests before moving on to the propensity score application step. However, no information on the overlap or presence of extreme propensity score values between the groups was presented.
Clinically relevant outcomes, OS and PFS, were assessed in the sponsor-submitted ITC. However, the definitions of OS and PFS used in the RWD were not clear. The methods of assessing OS and PFS used in the studies that the RWD was extracted from were not reported. When PFS data were unavailable, the authors used time to next treatment or death or time on treatment as a proxy. The clinical experts consulted by CADTH indicated that this was a reasonable approach, and data-cleaning rules were used to try to ensure censoring was consistent across end points for patients in the immunotherapy and chemotherapy analysis sets. However, differences in how these outcomes were captured in VISION compared to the RWD sources (e.g., when assessments occurred, whether they were investigator-assessed versus IRC-assessed, use of other end points as proxy for PFS) contributed to uncertainty in the data.
In addition, the authors of the ITC noted limitations in their analysis of OS and PFS. First, the authors of the ITC indicated that the Cox proportional hazards assumption may not have been met, which is expected to introduce bias in the Cox proportional HRs, although the direction of bias is unclear. Second, the authors reported that there is a substantial amount of post-progression treatment in the immunotherapy and chemotherapy groups in the CDM, which is likely to confound the OS results. These limitations further contribute to the uncertainty in the PFS and OS results. The ITC did not provide data on other important efficacy outcomes (e.g., ORR, HRQoL). Safety outcomes (e.g., overall AEs, SAEs, discontinuation rates) were also not reported.
Overall, results of the indirect comparison of RWD to VISION using propensity scoring are expected to be biased, given the inherent limitations of the study design and conduct, and the reporting of the methods and results lacked important details. As a result, no conclusions can be made from the data. A number of key limitations related to the selection and assessment of patients, as well as the propensity score methods, were identified that could potentially bias the results. First, fundamental differences between the VISION trials and retrospective RWE databases and cohort studies for inclusion in the CDM were noted, as were concerns over differences in the assessment and timing of the clinical end points. Second, and most important, clinically important heterogeneity was incompletely assessed and reported, and the statistical analyses completed are unlikely to have accounted for all major differences. The generation of propensity scores did not include all important potential confounders and may have been biased by the differential distribution of invalid or missing data between the VISION clinical trial and retrospective datasets. Propensity score methods can control only for measured confounders, and systematic differences may remain between the VISION patients and those selected from the RWD sources.
With regard to external validity, 1 of the 4 sources of RWD included sites from Canada (British Columbia only). The majority of patient data were from sites in the US. A number of important differences likely exist between the US and Canada with regard to the management of these patients, differences in treatments available, health insurance coverage, and overall health care system structures, which would be expected to affect outcomes. In addition, the databases used included patients enrolled early in the studies, more than a decade ago, and these patients are unlikely to be representative of contemporary patients, because better therapies and supportive care are now available, which would be expected to bias the study results in favour of tepotinib. Immunotherapy with or without chemotherapy is the standard of care for first-line treatment of NSCLC in Canada. The indirect comparison using propensity scoring included chemotherapy without immunotherapy and immunotherapy alone as comparators, which were appropriate. Tepotinib was not compared to chemotherapy in combination with immunotherapy, which represents a significant gap in the evidence.
Results of the Unanchored MAIC
Summary of Included Studies
The authors of the sponsor-submitted ITC15 provided a limited description of the 3 retrospective studies included in the unanchored MAIC, and additional information was provided by the sponsor.27 Details of the included studies and their designs are summarized in Table 26. Baseline characteristics of the patient populations in the retrospective studies and VISION are summarized in Table 27. Some sources of heterogeneity were identified by the authors of the ITC and the sponsor, which are summarized in this section.
The Awad et al. (2019) study28 was a multi-centre retrospective study of 148 patients with METex14 mutations from 12 institutions. Most patients were from the US. Patients were included in the OS analysis if they were diagnosed on or after January 2010 and data were available until December 2016.28 The study describes the patient characteristics and outcomes seen in a real-world METex14 population treated with different classes of therapy, focusing primarily on whether a MET inhibitor improves outcomes. The MET population in the Awad et al. (2019) study consisted of patients treated with crizotinib, glesatinib, capmatinib, savolitinib, tepotinib, cabozantinib, or merestinib. The authors noted that this patient population likely included patients who were enrolled in the VISION trial. The authors used the group of patients who did not receive a MET inhibitor in the MAIC only. This non-MET population consisted of 34 patients with a median age of 70, and median OS of 8.1 months. Ten (37%) patients had brain metastases at diagnosis. In the non-MET population, most patients received platinum- or pemetrexed-based regimens. The authors of the ITC noted that the lines of therapy differed from those in the VISION study, with a smaller proportion of patients receiving tepotinib first-line. In addition, the authors of the ITC highlighted that there were differences from the VISION study in cancer histology (76% adenocarcinoma), and MET alteration type, as a variety of MET alterations were included (i.e., not limited to METex14 skipping mutations). According to the author’s assessment, the Awad et al. (2019) study can provide historical control data, primarily for previously treated patients, although there are limitations to its interpretation, given the lack of reporting subsequent treatments. The Awad et al. (2019) study assessed OS and PFS, but it did not report PFS data for the group of patients that were not treated with a MET inhibitor in the study publication. OS was defined as the date of diagnosis of stage IV disease until death due to any cause.28
The Sabari et al. (2018) study8 was a multi-centre retrospective study of 147 patients investigating the efficacy of immunotherapy in patients with METex14 skipping alterations. Patients identified between January 2014 and May 2017 were eligible.8 The authors of the ITC indicated that this study had authors and centres similar to those in the Awad et al. (2019) study. OS and PFS were assessed, but definitions of these outcomes were not reported.8 Immunotherapies received by the patients included pembrolizumab, nivolumab, atezolizumab, durvalumab, and ipilimumab plus nivolumab.15 The authors of the ITC indicated that the patient population was older than a typical NSCLC population (median age 73 years), which was similar to the VISION study population. The authors also reported that the study population was broader than VISION, with 46 patients not having metastatic disease and fewer having adenocarcinoma. The Sabari et al. (2018) study reported PFS and OS data for 24 patients treated with immunotherapy. The line of treatment that immunotherapy was received in was not reported. The authors of the ITC noted that other limitations of the study were a small sample size and heterogeneity of the immunotherapy received.
The Guisier et al. (2020) study29 investigated the effectiveness of immunotherapy in NSCLC harbouring multiple alterations (BRAF, MET, HER2, and RET). The time period of the patient data collected was not reported. Of the 107 patients enrolled in the study, 30 had MET mutations and were the population of interest for the sponsor’s unanchored MAIC. The sponsor-submitted technical report indicated that these patients had METex14 skipping alterations. However, the Guisier et al. (2020) study publication29 did not specify the types of MET mutations that were eligible. It is unclear whether the population with MET mutations from this study included only patients with METex14 skipping alterations. The authors of the ITC indicated that the baseline characteristics were similar to those in the VISION study population, based on metastatic sites and adenocarcinoma histology (93%). However, the authors of the ITC also indicated that the patient population did not align with the VISION study because 23% of patients had an ECOG PS of 2 and most patients were second- or later-line (4 of 30 patients were first-line). OS and PFS were assessed in the Guisier et al. (2020) study. OS was defined as the time from the introduction of immunotherapy to death.29 PFS was defined as the time from initiation of immunotherapy to progression on immunotherapy (radiological progression per RECIST v1.1 criteria or clinical progression) or death.29
A quality assessment of the Awad et al. (2019) and Sabari et al. (2018) studies was provided in the sponsor’s systematic literature review.16 Quality of the studies was assessed by the percentage of positive responses to the questions in the Downs and Black Checklist. The quality score was 57% for the Awad et al. (2019) study and 43% for the Sabari et al. (2018) study. The Guisier et al. (2020) study was not included in the sponsor’s systematic literature review, and a quality assessment of this study was not provided in the ITC technical report.
Table 26: Summary of Studies — Unanchored MAIC
Study | Awad et al. (2019) | Sabari et al. (2018) | Guisier et al. (2020) |
|---|---|---|---|
Study design | Retrospective analysis | Retrospective analysis | Retrospective analysis |
Study sites | 12 sites in the US | 2 sites in the US | 21 sites in France |
Primary study objective | To determine whether MET TKIs impact clinical outcomes of METex14 mutant NSCLC | To conduct an analysis of patients with METex14 skipping alterations, evaluating PD-L1 expression, tumour mutational burden, and response to immunotherapy | To assess ICI efficacy (ORR, DOR, PFS, and OS) for NSCLC harbouring BRAF, HER2, or MET mutations, or RET translocations |
Patient population | Patients with METex14 NSCLC | Patients with METex14-altered lung cancers of any stage | Adult patients with metastatic NSCLC with BRAF-, HER2-, or MET-activating mutations, or RET translocations and treatment with single-agent anti–PD-1 or PD-L1 ICI |
Enrolled, N | 148 | 147 | 107 |
Cohorts |
|
|
|
Treatments | Patients who received a MET inhibitor: crizotinib, glesatinib, capmatinib, savolitinib, tepotinib, cabozantinib, or merestinib Patients who never received a MET inhibitor: most received platinum or pemetrexed-based chemotherapy regimens | Immunotherapy | Immunotherapy |
Outcomes assessed | OS PFS (data reported for patients treated with MET TKIs only) | Response per RECIST Duration of therapy DOR PFS OS | Best response Response rate DCR DOR PFS OS |
OS definition used | Date of diagnosis of stage IV disease until death due to any cause; patients who were alive at the time of analysis were censored on the last date of contact | NR | Time from the introduction of ICI to death |
PFS definition used | Start date of TKI treatment until the date of clinical or radiographic progression or death, as assessed by the investigator | NR | Time from initiation of ICI to progression on ICI; progression was defined RECIST v1.1 radiological or clinical progression (deteriorated clinical status preventing systemic treatment) or death |
DCR = disease control rate; DOR = duration of response; ICI = immune-checkpoint inhibitors; MAIC = matching-adjusted indirect comparison; METex14 = MET exon 14; NR = not reported; NSCLC = non–small cell lung cancer; ORR = objective response rate; OS = overall survival; PD-L1 = programmed death-ligand 1; PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumours; TKI = tyrosine kinase inhibitor.
Source: Sponsor’s technical report,15 Awad et al. (2019),28 Sabari et al. (2018),8 Guisier et al. (2020),29 and sponsor’s response to CADTH’s request for information.27
Table 27: Baseline Characteristics for the VISION Study and Comparator Groups Used in the MAIC
Characteristic | Awad et al. (2019) | Sabari et al. (2018) | Guisier et al. (2020) | VISION |
|---|---|---|---|---|
Patients included in the MAIC, n | 34 | 147 | 30 | 151 |
Untreated, n (%) | 9 (26) | NR (46) | 4 (13) | 69 (46) |
Age, mean (SD) | 70 (NR) | 73 (NR) | 64.3 (11.8) | 73.0 (8.97) |
Male, n (%) | 17 (50) | 58 (39) | 19 (63) | 79 (52) |
Never smoker, n (%) | 13 (38) | 52 (35) | 11 (37) | 73 (48) |
Adenocarcinoma histology, n (%) | 26 (76) | 109 (74) | 28 (93) | 109 (72) |
Metastatic disease, n (%) | NR | 101 (69) | NR | 148 (98) |
MAIC = matching-adjusted indirect comparison; NR = not reported; SD = standard deviation.
Source: Sponsor’s technical report,15 Awad et al. (2019),28 Sabari et al. (2018),8 Guisier et al. (2020),29 and VISION Clinical Study Report.14
The tepotinib data from the VISION study was reweighted to match the 3 retrospective studies. A visual comparison of Kaplan-Meier curves was conducted for each comparison. No HRs or other statistics were reported.
Results of the reweighting of the VISION data to match the Awad et al. (2019) study are provided in Table 28. Patients were not matched using the characteristic of metastatic disease because these data were not reported in the Awad et al. (2019) study. After weighting, the sample size of the tepotinib group decreased from |||||| patients to an ESS of |||||| patients. The Kaplan-Meier plot of OS for the Awad et al. (2019) study data compared to the unweighted and MAIC-weighted VISION data is depicted in Figure 18. Since the Awad et al. (2019) study did not report PFS data for the group of patients that were not treated with MET inhibitors, no comparison was made for PFS.
Results of the reweighting of the VISION data to match the Sabari et al. (2018) study are provided in Table 29. After weighting, the sample size of the tepotinib group decreased from 151 patients to an ESS of 51.1 patients. The authors indicated that the large reduction in sample size was due to the lower proportion of patients with metastatic disease in the Sabari et al. (2018) study. The Kaplan-Meier plots of OS and PFS for the Sabari et al. (2018) study data compared to the unweighted and weighted VISION data are depicted in Figure 19 and Figure 20, respectively.
Results of the reweighting of the VISION data to match the Guisier et al. (2020) study are reported in Table 30. After weighting, the sample size of the tepotinib group decreased from 151 patients to an ESS of 79.5. The authors of the ITC reported that this notable loss in sample size was largely due to the Guisier et al. (2020) study population containing mostly treatment-experienced patients. The Kaplan-Meier plots of OS and PFS for the Guisier et al. (2020) study data compared to the unweighted and weighted VISION data are depicted in Figure 21 and Figure 22 respectively.
Table 28: VISION Patient Characteristics, Before and After Application of MAIC to Match Awad et al. (2019)
Characteristic | Tepotinib — Unweighted | Tepotinib — Weighted | Awad et al. |
|---|---|---|---|
N/ESS | |||||||||||| | |||||||||||| | |||||||||||| |
Untreated, % | |||||||||||| | |||||||||||| | |||||||||||| |
Age, mean (SD) | |||||||||||| | |||||||||||| | |||||||||||| |
Male, % | |||||||||||| | |||||||||||| | |||||||||||| |
No smoking history, % | |||||||||||| | |||||||||||| | |||||||||||| |
Adenocarcinoma histology, % | |||||||||||| | |||||||||||| | |||||||||||| |
ESS = effective sample size; MAIC = matching-adjusted indirect comparison; NR = not reported; SD = standard deviation.
Source: Sponsor’s technical report.15
Figure 18: Kaplan-Meier Plot of OS of Awad et al. (2019) Compared to the Unweighted and Weighted VISION Data
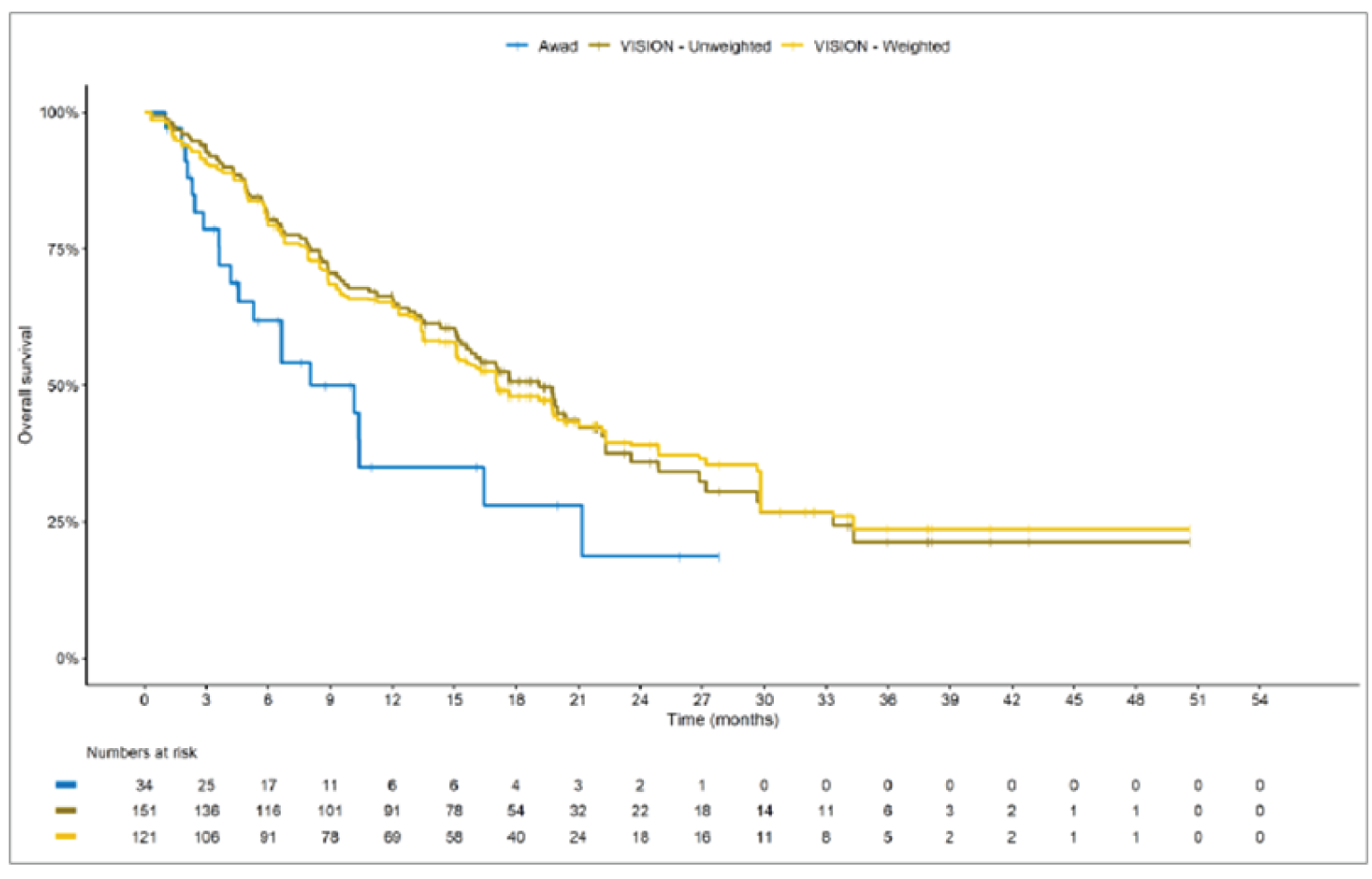
Source: Sponsor’s technical report.15
Table 29: VISION Patient Characteristics, Before and After Application of MAIC to Match Sabari et al. (2018)
Characteristic | Tepotinib — Unweighted | Tepotinib — Weighted | Sabari et al. |
|---|---|---|---|
N/ESS | 151.0 | 51.1 | 147 |
Untreated, % | 46 | 46 | 46 |
Age, mean (SD) | 73.0 (8.97) | 73.0 (NR) | 73 (NR) |
Male, % | 52 | 39 | 39 |
No smoking history, % | 48 | 35 | 35 |
Adenocarcinoma histology, % | 87 | 74 | 74 |
Metastatic disease, % | 98 | 69 | 69 |
ESS = effective sample size; MAIC = matching-adjusted indirect comparison; NR = not reported; SD = standard deviation.
Source: Sponsor’s technical report.15
Figure 19: Kaplan-Meier Plot of OS of Sabari et al. (2018) Compared to the Unweighted and Weighted VISION Data
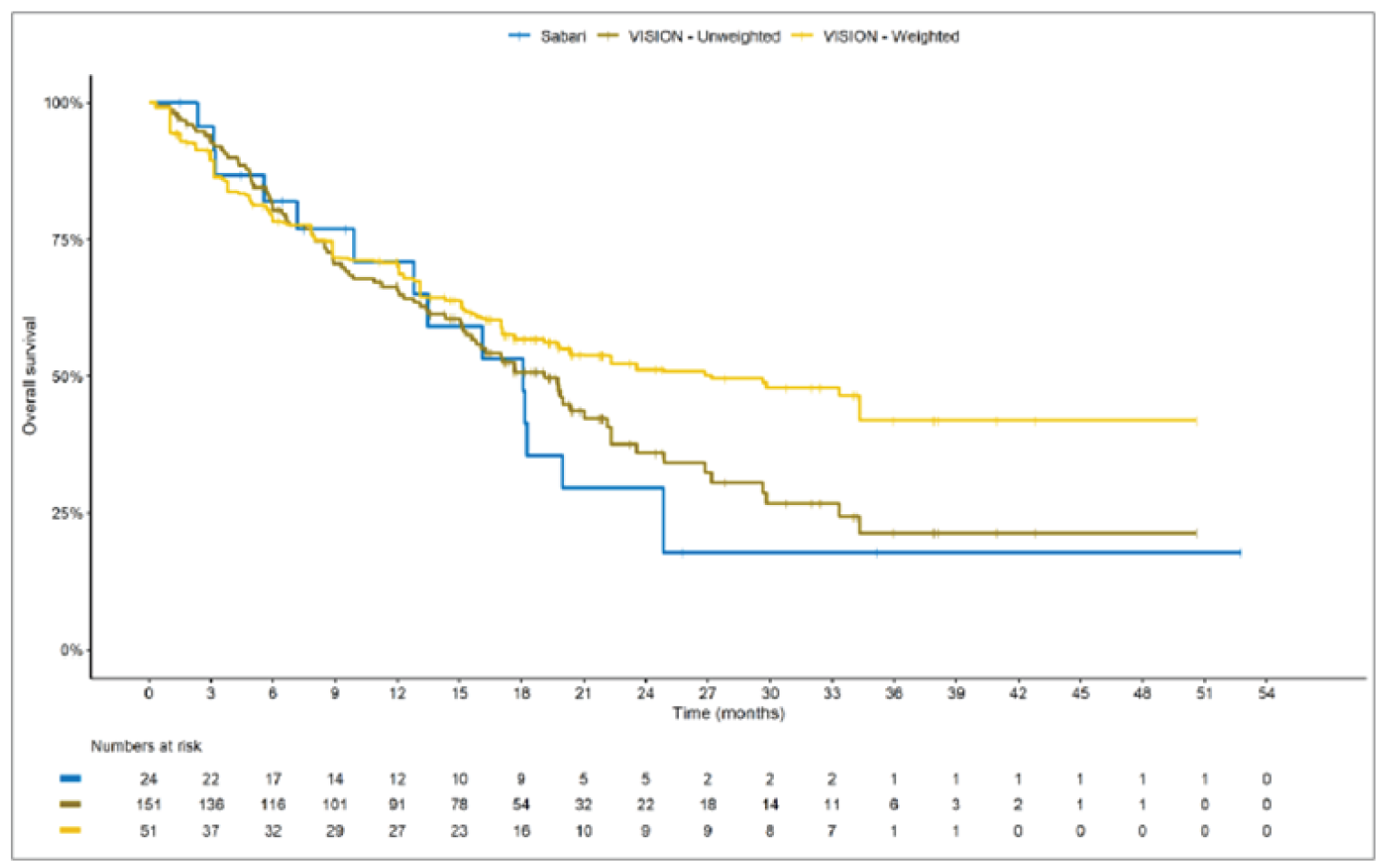
Source: Sponsor’s technical report.15
Figure 20: Kaplan-Meier Plot of PFS of Sabari et al. (2018) Compared to the Unweighted and Weighted VISION Data
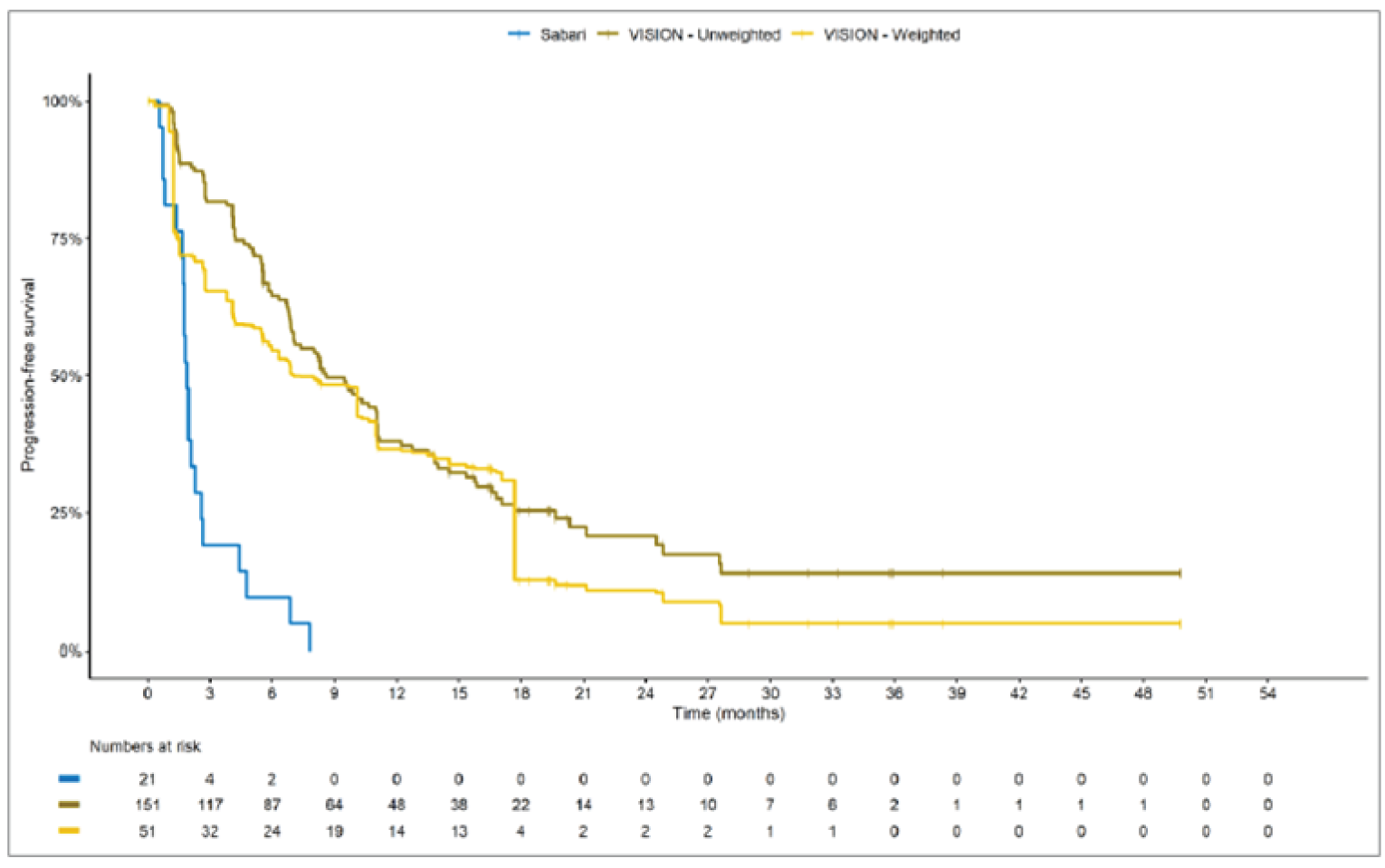
Source: Sponsor’s technical report.15
Table 30: VISION Patient Characteristics, Before and After Application of MAIC to Match Guisier et al. (2020)
Characteristic | Tepotinib — Unweighted | Tepotinib — Weighted | Guisier et al. |
|---|---|---|---|
N/ESS | 151.0 | 79.5 | 30 |
Untreated, % | 46 | 13 | 13 |
Age, mean (SD) | 73.0 (8.97) | 64.3 (NR) | 64.3 (NR) |
Male, % | 52 | 63 | 63 |
No smoking history, % | 48 | 37 | 37 |
Adenocarcinoma histology, % | 87 | 93 | 93 |
Metastatic disease, % | 98 | NR | NR |
ESS = effective sample size; MAIC = matching-adjusted indirect comparison; NR = not reported; SD = standard deviation.
Source: Sponsor’s technical report.15
Figure 21: Kaplan-Meier Plot of OS of Guisier et al. (2020) Compared to the Unweighted and Weighted VISION Data
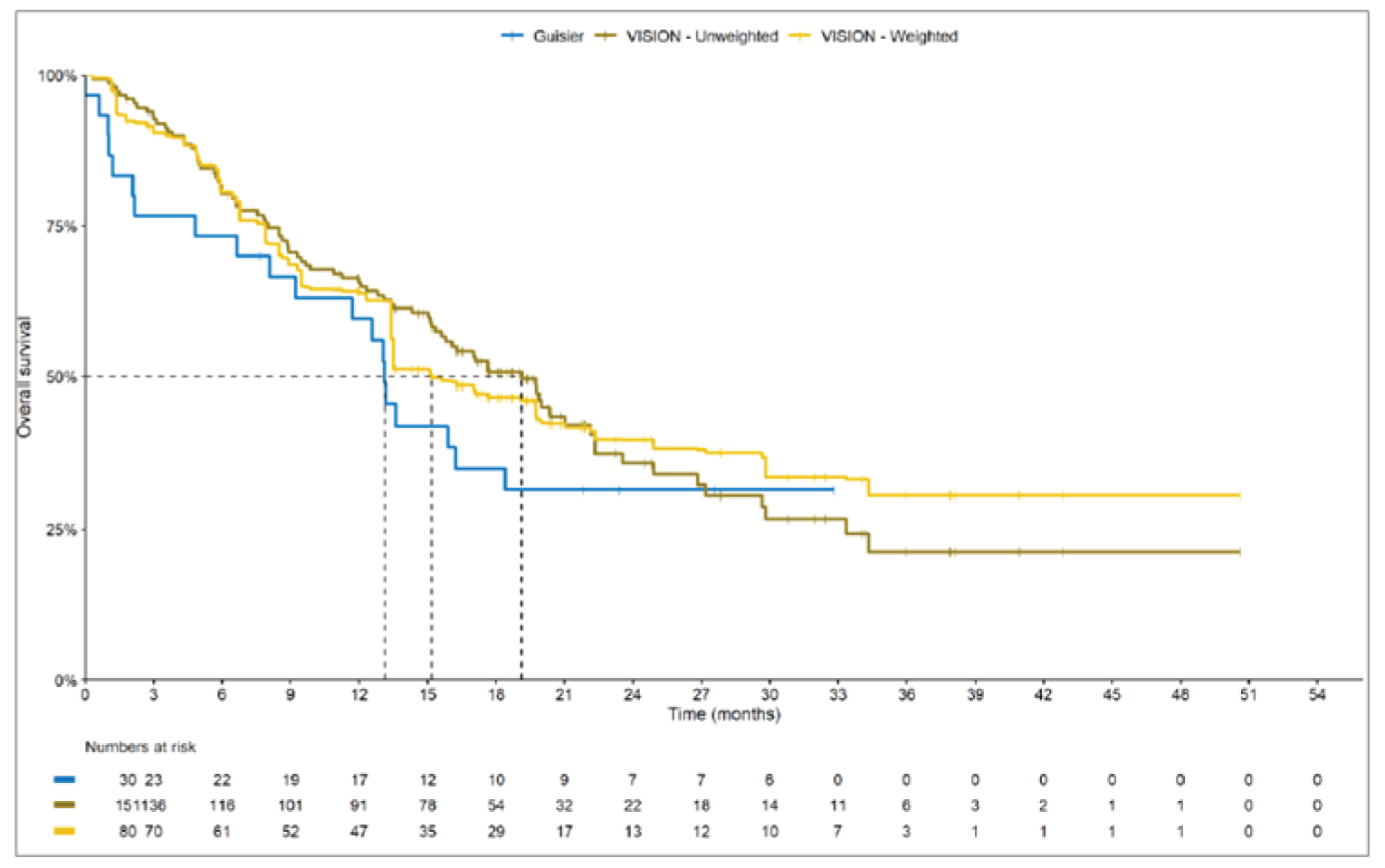
Source: Sponsor’s technical report.15
Figure 22: Kaplan-Meier Plot of PFS of Guisier et al. (2020) Compared to the Unweighted and MAIC-Weighted VISION Data
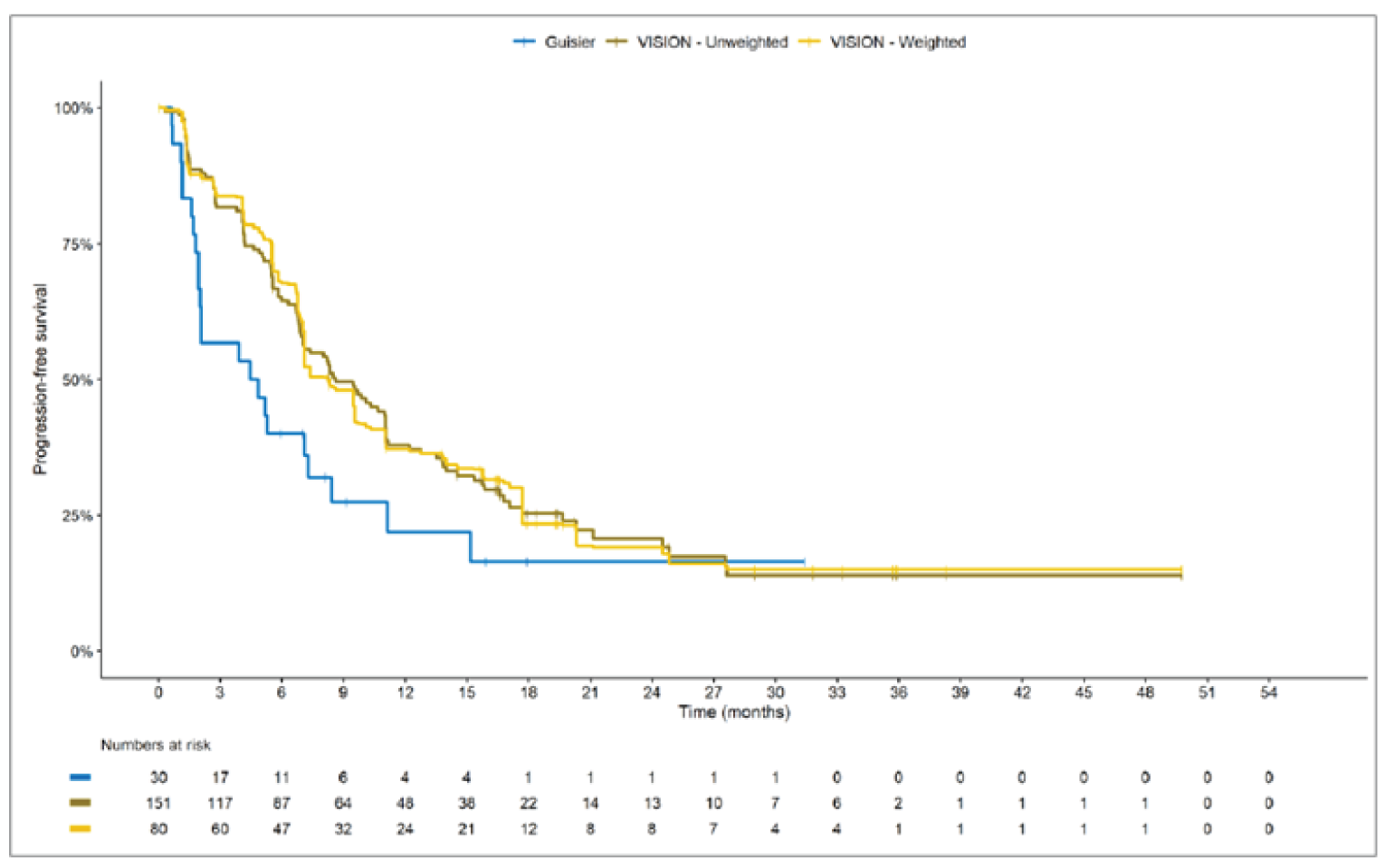
Source: Sponsor’s technical report.15
Critical Appraisal of the Unanchored MAIC
There were substantial methodological limitations of the unanchored MAIC. First, a MAIC can adjust only for heterogeneity that is directly related to differences in baseline patient characteristics. Other sources of heterogeneity, such as those related to differences in study design, definitions of study outcomes, or changes in the management of support of patients over time, cannot be adjusted for. The prospective, interventional VISION trial was compared to retrospective observational studies. This may be problematic, as there are notable differences in treatments and procedures received by patients, data collection methods, and assessments, and these differences in study design could not be accounted for in the MAIC. In addition, differences (or potential differences) in the studies’ outcome definitions could not be assessed due to lack of reporting. In the VISION study, OS was defined as the time from first trial treatment administration to the date of death, and a similar definition was used in the Guisier et al. (2020) study. However, the Awad et al. (2019) study defined OS as the date of diagnosis of stage IV disease until death due to any cause. The Sabari et al. (2018) study publication did not report definitions of PFS and OS; thus, it is unknown whether the study’s definition of outcomes differed from that in VISION. The definition of PFS used in the Guisier et al. (2020) study was similar to the definition used in the VISION study. Differences in study outcome definitions could not be accounted for in the MAIC; thus, these differences create uncertainty in the results.
Furthermore, an unanchored indirect comparison using MAIC methods provides an unbiased comparison only if all prognostic and effect-modifying factors are included in the weighting process. The variables included in the unanchored MAIC do not reflect all important effect modifiers and prognostic factors. The technical report for the sponsor-submitted ITC indicated that ECOG PS would ideally have been included in the model, but it was not possible due to the amount of missing data. Patients included in the retrospective studies were likely sicker (i.e., included patients with ECOG PS of 2 or more) than patients in VISION, which restricted enrolment to ECOG 0 or 1. The clinical experts consulted by CADTH indicated that ECOG PS and the presence of brain metastases at baseline are important factors and thus should have been included the model to provide an unbiased comparison. In addition, patient numbers allowed the proportion of adenocarcinoma to be matched only in terms of histology, and metastatic disease was not matched in the comparisons to the Awad et al. (2019) and Guisier et al. (2020) studies because data for this characteristic were not reported. Exclusion of important confounders in the matching introduces bias and creates substantial uncertainty in the results. Unanchored forms of population-adjusted indirect comparisons make the much stronger assumption of “conditional constancy of absolute effects.” This means that the absolute treatment effects are assumed to be constant at any given level of the effect modifiers and prognostic variables, and all effect modifiers and prognostic variables must be known. This assumption is unlikely to have been met in this unanchored MAIC; therefore, no conclusions can be made from the data.
In the comparisons, a large reduction in ESS was observed, which suggests there was likely significant heterogeneity between the VISION study and comparator studies. The results for indirect comparisons with major reductions of ESS indicate that the weights are highly variable due to a lack of population overlap and that the resulting estimate may not be reliable.
In addition, the methods and findings of the MAIC lacked important details, which creates further uncertainty in the results. The sponsor submitted a systematic literature review, which identified the Sabari et al. (2018) and Awad et al. (2019) studies included in the unanchored MAIC. However, the third study (Guisier et al. [2020]) was not identified in the sponsor’s systematic literature review, so it is unclear how this study was identified and selected for inclusion in the MAIC. Furthermore, it is unclear why other studies identified in the sponsor’s systematic literature review were not included in the ITC. The methods used to identify studies specifically for the submitted unanchored MAIC and the selection criteria were unclear. As a result, there is significant risk of bias in study selection and a lack of certainty that all relevant studies were identified. The data extraction methods were not reported. The quality of the Awad et al. (2019) and Sabari et al. (2018) studies was low; the quality of the Guisier et al. (2020) study was not assessed by the authors of the ITC and therefore not considered in the ITC analysis. Potential risks of bias in the included studies were not assessed and not reported. A limited assessment of heterogeneity was reported. Kaplan-Meier curves were presented for visual comparison only.
The MAIC assessed OS and PFS, which were identified as important outcomes by the clinical experts, clinician groups, and patient groups that provided input on this review. The OS results were likely confounded by subsequent therapies (i.e., post-progression treatments), which were not accounted for in the MAIC. In addition, the differences in definitions of OS time between studies may have also introduced bias in the results. The ITC did not include data on ORR, DOR, or HRQoL, which were also identified as important outcomes. In addition, safety outcomes were not reported in the ITC, which represents an important gap in the evidence.
Some of the patients included in the retrospective studies were from earlier time periods, and they would not be representative of current patients receiving contemporary treatments and would be expected to have worse outcomes. This may cause overestimation of the effects of tepotinib. Since the Awad et al. (2019) and Sabari et al. (2018) studies were from similar authors and centres, patients may have been included more than once. This would be expected to bias the study results because the same patients may have been included more than once in the estimates.
With regard to external validity, none of the studies included sites from Canada. Most patients in the retrospective studies (2 of 3 studies) were from sites in the US. A number of important differences likely exist between the US and Canada with regard to the management of these patients, most notably insurance coverage, which would be expected to affect outcomes.
In Canada, the standard of care for patients with advanced NSCLC is chemotherapy with or with immunotherapy, or immunotherapy alone. In the population from the Awad et al. (2019) study used for comparison, most patients were treated with chemotherapy. In the Sabari et al. (2018) and Guisier et al. (2020) studies, patients were treated with immunotherapy. Tepotinib was not compared to a combination of chemotherapy and immunotherapy in the MAIC, which represents a gap in the evidence.
Other Relevant Evidence
No long-term extension studies or additional relevant studies were included in the sponsor’s submission to CADTH.
Discussion
Summary of Available Evidence
One ongoing, phase II, single-arm, open-label trial (VISION) was included in the systematic review. The objective of the VISION study was to assess the efficacy of tepotinib in patients with advanced (locally advanced or metastatic) NSCLC, as per objective response (confirmed CR or PR) determined according to RECIST v1.1, based on independent review in patients who tested positive for METex14 skipping alterations or MET amplification. Patients with METex14 skipping alterations were enrolled in cohort A and cohort C under the same eligibility criteria and underwent the same study procedures. cohort A was the pivotal cohort (N = 151); cohort C was a confirmatory cohort added as a protocol amendment, and enrolment at sites was shifted from cohort A to cohort C once accrual for cohort A was completed (cohort A plus C: N = 254). All patients received 500 mg tepotinib hydrochloride hydrate, containing 450 mg tepotinib, orally once daily in 21-day cycles. Treatment was continued until disease progression per RECIST v1.1 criteria, death, an AE leading to discontinuation, or withdrawal of consent. The primary outcome was ORR by IRC assessment. Secondary outcomes included OS; PFS by IRC; PFS by investigator; ORR by investigator; DOR by IRC; DOR by investigator; change from baseline and TTD in EQ-5D-5L VAS, EORTC QLQ-C30 (global health status and quality of life score), EORTC QLQ-LC13 (coughing, dyspnea, and chest pain symptom scales); and safety. Intracranial CNS outcomes were assessed in a post hoc exploratory analysis. Data were analyzed descriptively; no statistical testing was performed.
In VISION, the mean age of study patients was 73 years in cohort A and cohort A plus C. Most patients were White, had an ECOG PS of 1, adenocarcinoma histology type, and stage IV disease at study entry. Approximately half of the study patients had received prior anticancer drug therapy for advanced or metastatic disease. The most common types of prior anticancer therapies were cytotoxic therapy and immunotherapy. The most frequently administered prior drug therapies were carboplatin, pemetrexed, cisplatin, and pembrolizumab. Most patients had not had prior anticancer surgery. Per IRC assessment, 9.9% of patients in cohort A and 12.2% of patients in cohort A plus C had brain metastases at baseline.
Since the primary clinical review of tepotinib consisted of a single-arm trial, a review of the available indirect evidence was conducted. The ITCs submitted by the sponsor were summarized and critically appraised. The objective of the ITC analyses was to estimate the comparative effectiveness of tepotinib relative to commonly used therapies (i.e., chemotherapy and immunotherapy). The ITCs consisted of an indirect comparison of VISION to pooled RWD using propensity scoring and unanchored MAICs comparing VISION to 3 retrospective observational studies. The authors reported results for clinical efficacy in terms of OS and PFS, comparing tepotinib to chemotherapy without immunotherapy and immunotherapy alone. Harms outcomes were not assessed.
No long-term extension studies or additional relevant studies were included in the sponsor’s submission to CADTH.
Interpretation of Results
Efficacy
The clinical experts consulted by CADTH indicated that the goals of treatment in locally advanced (not amenable to curative treatment) or metastatic NSCLC are to improve survival, delay progression, and improve response rate, as well as improve or maintain HRQoL. They noted that METex14 skipping alterations are a rare mutation, and no targeted treatments are publicly funded in Canada. Patients with advanced NSCLC and METex14 mutations are currently treated per guidelines for advanced NSCLC without driver mutations, and therapy under these guidelines is immunotherapy with or without chemotherapy. The clinical experts reported that retrospective studies7,8 have indicated that patients with NSCLC harbouring METex14 skipping mutations have a poor prognosis and there may be less benefit from the use of immunotherapy. The clinical experts indicated that there is unmet need in this patient population. Similar views were expressed by the clinician groups that provided input on this review.
The patient groups indicated that they want treatments that improve symptoms, improve or maintain HRQoL, increase survival, delay disease progression, and help patients achieve long-term remission. The patients also expressed that it is important to maintain independence and functioning. This aligns with the following outcomes assessed in the VISION trial: HRQoL and PROs, OS, PFS, ORR, and DOR.
OS, PFS, ORR, DOR, and HRQoL and PROs were identified as important outcomes in the CADTH review protocol, and these outcomes were assessed as secondary end points in the VISION study. Intracranial CNS outcomes were also identified as important in the CADTH review protocol.
For the primary end point, the VISION study aimed to show an ORR assessed by IRC of 40% to 50%, and to demonstrate that the lower limit of the corresponding exact 2-sided 95% CI exceeded 20%. The VISION trial achieved its primary end point. As of the July 1, 2020, data cut-off, ORR by IRC assessment was 45.0% (95% CI, 36.9% to 53.3%) in the pivotal cohort A. All responses were PR. The clinical experts consulted by CADTH indicated that these results were clinically meaningful. Results were similar in the pooled confirmatory cohort A plus C (i.e., entire METex14 skipping population). Furthermore, pre-specified subgroup analyses by line of treatment, presence of brain metastases at baseline, histological subtype, and ECOG PS were broadly numerically consistent with the overall analysis. No statistical testing was done on the differences between subgroups.
Time-to-event outcomes such as PFS and OS are difficult to interpret in single-arm trials. The clinical experts consulted by CADTH indicated that the Kaplan-Meier curves and median OS and PFS in the pivotal cohort A were clinically meaningful and suggested a benefit with tepotinib, although there is uncertainty due to the absence of a comparator arm. Similarly, the clinical experts indicated that the ORR and DOR results suggested a clinically meaningful benefit and that response to tepotinib was durable, although these data are difficult to interpret without a comparator arm as well. PFS, ORR, and DOR were assessed by both the IRC and the investigator, and results by investigator assessment were generally consistent with the results by IRC assessment. Pre-specified subgroup analyses of each secondary end point were broadly consistent with the overall analysis, although median OS and median PFS were numerically greater in the second- or later-line of therapy group compared to the first-line group. The reason for this numerical difference between subgroups in uncertain.
The HRQoL and PROs were assessed by change from baseline and TTD by 10 points for each PRO score. CADTH identified an MID of 7 to 11.5 points for the EQ-5D-5L VAS and 4 points for deterioration in EORTC QLQ-C30 global health status and quality of life score.24,25 No MID was identified in the literature for the EORTC QLQ-LC13 symptom scales. Regarding change from baseline, the EQ-5D-5L and EORTC QLQ-C30 global health status and quality of life scores remained generally stable over time in cohort A and cohort A plus C, although there is uncertainty in the data due to the relatively small number of patients contributing to the analysis at later cycles. The missing data would be expected to overestimate the true effect of tepotinib on HRQoL outcomes. The sponsor indicated that results of the MMRM analysis of the QLQ-LC13 symptom scales were uncertain after cycle 25, due to small sample sizes. For the earlier cycles, the MMRM analysis of the QLQ-LC13 symptom scales indicated that there may have been some improvement in coughing, whereas dyspnea and chest pain remained relatively stable. However, the MRMM analysis would be expected to overestimate the treatment effects, as it is unlikely the assumptions of the MRMM have been met in this study. Overall, the clinical experts consulted by CADTH indicated that the HRQoL and PRO results suggested that HRQoL may be maintained with tepotinib therapy. However, there is substantial uncertainty in the data due to the decreasing number of patients that completed the questionnaires over time, which is likely to lead to an overestimation of treatment effects, and due to the absence of a control group. Furthermore, the VISION trial was open-label, which may have introduced bias in the reporting of subjective outcomes such as HRQoL and PROs.
In the original VISION study protocol, only patients with adenocarcinoma were eligible, and patients were required to have failed 1 to 2 lines of systemic therapy, including a platinum-doublet-containing regimen. In a protocol amendment that was implemented after enrolment in the study had begun, the eligible study population was expanded to include all subtypes of NSCLC and patients that were treatment-naive (i.e., would receive tepotinib as first-line treatment). These significant changes to the eligibility criteria implemented after participant enrolment had begun may have introduced bias. The direction of this bias is unknown. However, the sponsor did conduct subgroup analyses of treatment-naive patients versus patients who had been previously treated, and the results of these subgroup analyses were broadly consistent with the overall analyses.
The primary limitations of the VISION trial that impact the interpretation of results were the absence of a comparator group and statistical testing. Due to these limitations of the study design, no definitive conclusions can be drawn concerning the comparative effectiveness of tepotinib based on the VISION trial data alone.
There was no direct evidence available to assess the relative efficacy of tepotinib versus current standard of care therapies for patients with advanced NSCLC with METex14 skipping alterations. The sponsor submitted an ITC that included an indirect comparison of VISION to pooled RWD using propensity scoring and an unanchored MAIC comparing VISION to 3 retrospective observational studies. Relative efficacy was assessed in terms of PFS and OS. The comparators were chemotherapy without immunotherapy and immunotherapy alone. Key gaps in the evidence provided by the ITC were that no safety outcomes were assessed and that chemotherapy in combination with immunotherapy was not included as a comparator. Although the ITC reported a benefit in PFS and OS with tepotinib treatment compared to chemotherapy and immunotherapy, there is potential for significant bias in the ITC results. The methods and findings of the ITC were not adequately detailed, and several methodological limitations were noted. Differences in the study designs could not be accounted for in the indirect comparisons. This may be problematic, as there are notable differences in treatments and procedures received by patients, data collection methods, and assessments. There are concerns that not all effect modifiers and prognostic factors have been identified and adjusted for in the analyses, and that there are limitations regarding availability of data to allow for, including key variables in the weighting process. Most important, the authors of the ITC conducted a limited assessment of heterogeneity, and there is likely considerable heterogeneity among the patient populations of the pooled RWD, retrospective observational studies, and VISION trial. The small ESS in the studies used in the MAICs suggests that substantial differences exist between the patient population in the VISION trial and in the retrospective observational studies. These differences were not fully accounted for with the statistical analyses employed. Due to the methodological limitations, limited reporting of methods and results, and small sample sizes in the indirect comparisons, there is substantial risk of bias and uncertainty in the results. As a result, no conclusion can be made based on the ITC results.
Harms
The patient groups that provided input for this review indicated that they want treatments with minimal side effects. Similarly, the clinical experts consulted by CADTH indicated that new treatments would ideally minimize AEs compared to current standard of care therapies. Moreover, the clinical experts noted that patients with NSCLC harbouring METex14 skipping mutations tend to be an elderly population, and elderly patients often experience increased side effects with chemotherapy. Thus, treatments that are better tolerated are needed.
The evidence regarding the safety of tepotinib was derived from the VISION trial. Almost all study patients experienced at least 1 treatment-emergent AE. The most frequently reported AE was peripheral edema, which was a notable harm identified in the CADTH systematic review protocol. The clinical experts noted that peripheral edema is relatively manageable in most patients. Other common AEs included nausea, diarrhea, hypoalbuminemia, increased blood creatinine, and dyspnea. The clinical experts consulted by CADTH indicated that the toxicity profile of tepotinib may be preferable to that of standard of care therapies for some patients. The VISION trial was open-label, which may have affected the reporting of AEs. The direction of potential bias due to open-label administration of tepotinib is unknown.
As of the July 1, 2020, data cut-off date, 27.6% of patients in cohort A and 20.4% in cohort A plus C permanently discontinued study treatment due to an AE. The most common AEs leading to treatment discontinuation were peripheral edema, pleural effusion, and general physical health deterioration. The incidence of SAEs was 55.9% in cohort A and 45.1% in cohort A plus C. The most frequently reported SAEs were pleural effusion, disease progression, and pneumonia. Overall, 50% of patients in cohort A and 33.7% in cohort A plus C had died. The most common cause of death was disease progression.
Tepotinib has serious warnings and precautions in the Health Canada product monograph11 for hepatotoxicity and interstitial lung disease or pneumonitis, which were included as notable harms in the CADTH systematic review protocol. The most frequently reported hepatotoxicity-related AEs were increased ALT, AST, blood ALP, and GGT. A total of 2 patients experienced interstitial lung disease, and 6 patients experienced pneumonitis. Another notable harm specified in the review protocol was renal toxicity, and the most common treatment-emergent AE related to renal toxicity was increased blood creatinine.
Harms outcomes were not assessed in the sponsor-submitted ITC; therefore, the relative safety of tepotinib compared to standard of care therapies remains unknown.
Conclusions
One ongoing phase II, single-arm trial (VISION) of tepotinib in patients with advanced (locally advanced or metastatic) NSCLC harbouring METex14 skipping alterations was identified in the systematic review conducted by CADTH. The VISION trial data were analyzed descriptively; no statistical hypotheses were tested. According to the clinical experts consulted by CADTH, the results suggested there may be a potential beneficial effect of tepotinib on OS, PFS, ORR, and DOR based on their clinical experience and expectations of the natural progression of the disease in patients with METex14 skipping alterations. The clinical experts indicated that there is significant unmet need for targeted treatment and improved outcomes in this patient population. However, due to the absence of a comparator arm and statistical testing, no definitive conclusions can be drawn regarding the efficacy of tepotinib based on the VISION trial. The HRQoL and PRO data from VISION suggested that quality of life may be maintained with tepotinib therapy per the clinical experts, but there is substantial uncertainty in these data due to decreased sample sizes at later treatment cycles, open-label administration of tepotinib, and absence of a comparator arm. As a result, the effect of tepotinib on HRQoL and PROs remains unknown. Almost all study patients reported treatment-emergent AEs, the most common of which was peripheral edema. The most frequently reported SAEs were pleural effusion, disease progression, and pneumonia. Few patients experienced interstitial lung disease or pneumonitis. The most common cause of death was disease progression. The clinical experts consulted by CADTH indicated that the safety of tepotinib was acceptable.
No direct evidence was identified concerning the relative efficacy and safety of tepotinib versus standard of care therapies used to treat advanced NSCLC with METex14 skipping alterations in Canada (i.e., immunotherapy, chemotherapy, or immunotherapy with chemotherapy). Results from the indirect treatment analyses submitted by the sponsor suggested that tepotinib therapy may be associated with a benefit in PFS and OS compared to chemotherapy and immunotherapy. However, the ITCs are associated with substantial risk of bias and important limitations were identified (i.e., methodological limitations, limited assessment of heterogeneity, reporting lacked important details, small sample sizes). In view of the substantial uncertainty in the ITC results, no conclusions can be drawn concerning the efficacy of tepotinib compared to chemotherapy or immunotherapy in patients with advanced NSCLC harbouring METex14 skipping alterations. Harms outcomes were not assessed in the ITC. The potential benefits and safety of tepotinib compared with other therapies remain unknown.
References
1.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics: a 2020 special report on lung cancer. Toronto (ON): Canadian Cancer Society; 2020: Accessed 2021 Nov 10.
2.Goebel C, Louden CL, McKenna Jr R, Onugha O, Wachtel A, Long T. Diagnosis of non-small cell lung cancer for early stage asymptomatic patients. Cancer Genomics Proteomics. 2019;16(4):229-244. PubMed
3.Hammerschmidt S, Wirtz H. Lung cancer: current diagnosis and treatment. Dtsch Arztebl Int. 2009;106(49):809-818; quiz 819-820. PubMed
4.Drusbosky LM, Dawar R, Rodriguez E, Ikpeazu CV. Therapeutic strategies in METex14 skipping mutated non-small cell lung cancer. J Hematol Oncol. 2021;14(1):129. PubMed
5.Fujino T, Suda K, Mitsudomi T. Lung cancer with MET exon 14 skipping mutation: genetic feature, current treatments, and future challenges. Lung Cancer (Auckl). 2021;12:35-50. PubMed
6.Vuong HG, Ho ATN, Altibi AMA, Nakazawa T, Katoh R, Kondo T. Clinicopathological implications of MET exon 14 mutations in non-small cell lung cancer - a systematic review and meta-analysis. Lung Cancer. 2018;123:76-82. PubMed
7.Tong JH, Yeung SF, Chan AW, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22(12):3048-3056. PubMed
8.Sabari JK, Leonardi GC, Shu CA, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018;29(10):2085-2091. PubMed
9.Hanna NH, Schneider BJ, Temin S, et al. Therapy for stage IV non–small-cell lung cancer without driver alterations: ASCO and OH (CCO) joint guideline update. Toronto (ON): Cancer Care Ontario; 2020: Accessed 2021 Nov 3.
10.Ellis PM, Vella ET, Ung YC, Lung Cancer Disease Site Group. Systemic treatment for patients with advanced non-small cell lung cancer. Toronto (ON): Cancer Care Ontario; 2016: Accessed 2021 Nov 3.
11.PrTepmetkoTM (tepotinib): 225 mg tablets [product monograph]. Mississauga (ON): EMD Serono Canada; 2021 May 27.
12.Clinical Study Report: MS200095-0022. A Phase II single-arm trial to investigate tepotinib in advanced (locally advanced or metastatic) non-small cell lung cancer with MET exon 14 (METex14) skipping alterations or MET amplification (VISION) [internal sponsor's report]. Darmstadt (Germany): Merck KGaA; 2020 May 14.
13.Paik PK, Felip E, Veillon R, et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med. 2020;383(10):931-943. PubMed
14.Clinical Study Report: MS200095-0022. A Phase II single-arm trial to investigate tepotinib in advanced (locally advanced or metastatic) non-small cell lung cancer with MET exon 14 (METex14) skipping alterations or MET amplification (VISION) [internal sponsor's report]. Darmstadt (Germany): Merck KGaA; 2020 Oct 10.
15.Conduct of an unbiased comparison in NSCLC with MET skipping [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: PrTepmetkoTM (tepotinib) 225 mg tablets. Mississauga (ON): EMD Serono Canada; 2021 Aug 27.
16.Garcia A, el Alili H, Gezin A, et al. Systematic literature review (SUR) update of METex 14 mutation skipping in non-small cell lung cancer (NSCLC) [sponsor's internal report]. In: Drug Reimbursement Review sponsor submission: PrTepmetkoTM (tepotinib) 225 mg tablets. Mississauga (ON): EMD Serono Canada; 2021 Aug 27. 2020.
17.Canadian Cancer Statistics Advisory Committee. Canadian cancer statistics 2019. Toronto (ON): Canadian Cancer Society; 2019: Accessed 2021 Nov 10.
18.Hanna NH, Robinson AG, Temin S, et al. Therapy for stage IV non-small-cell lung cancer with driver alterations: ASCO and OH (CCO) joint guideline update. J Clin Oncol. 2021;39(9):1040-1091. PubMed
19.Drug and health product submissions under review (SUR): new drug submissions under review. Ottawa (ON): Health Canada; 2021: Accessed 2021 Nov 3.
20.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
21.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: Accessed 2021 Sep 22.
22.Sakai H, Morise M, Kato T, et al. Tepotinib in patients with NSCLC harbouring MET exon 14 skipping: Japanese subset analysis from the Phase II VISION study. Jpn J Clin Oncol. 2021;51(8):1261-1268. PubMed
23.Drug Reimbursement Review sponsor submission: PrTepmetkoTM (tepotinib) 225 mg tablets [internal sponsor's package]. Mississauga (ON): EMD Serono Canada; 2021 Aug 27.
24.Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70-70. PubMed
25.Maringwa JT, Quinten C, King M, et al. Minimal important differences for interpreting health-related quality of life scores from the EORTC QLQ-C30 in lung cancer patients participating in randomized controlled trials. Support Care Cancer. 2011;19(11):1753-1760. PubMed
26.EMD Serono response to October 7, 2021 DRR request for additional information regarding tepotinib DRR review [internal additional sponsor's information]. Mississauga (ON): EMD Serono Canada; 2021.
27.EMD Serono response to October 27, 2021 DRR request for additional information regarding tepotinib DRR review [internal additional sponsor's information]. Mississauga (ON): EMD Serono Canada; 2021.
28.Awad MM, Leonardi GC, Kravets S, et al. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: a retrospective analysis. Lung Cancer. 2019;133:96-102. PubMed
29.Guisier F, Dubos-Arvis C, Viñas F, et al. Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET translocation: GFPC 01-2018. J Thorac Oncol. 2020;15(4):628-636. PubMed
30.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
31.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
32.Nicklasson M, Bergman B. Validity, reliability and clinical relevance of EORTC QLQ-C30 and LC13 in patients with chest malignancies in a palliative setting. Qual Life Res. 2007;16(6):1019-1028. PubMed
33.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376. PubMed
34.Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Eur J Cancer. 1994;30a(5):635-642. PubMed
35.EQ-5D-5L user guide: basic information on how to use the EQ-5D-5L instrument Rotterdam (NL): EuroQol Research Foundation; 2019: Accessed 2021 Nov 2.
36.2014 Alberta population norms for EQ-5D-5L. Calgary (AB): Health Quality Council of Alberta; 2014: Accessed 2021 Nov 2.
37.Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727-1736. PubMed
38.Giesinger JM, Kieffer JM, Fayers PM, et al. Replication and validation of higher order models demonstrated that a summary score for the EORTC QLQ-C30 is robust. J Clin Epidemiol. 2016;69:79-88. PubMed
39.Quality of Life FAQ. Brussels (BE): EORTC; 2021: Accessed 2021 Nov 2.
40.EORTC QLQ-C30 scoring manual. Belgium (BE): EORTC; 2001: Accessed 2021 Nov 2.
41.Revicki D, Hays RD, Cella D, Sloan J. Recommended methods for determining responsiveness and minimally important differences for patient-reported outcomes. J Clin Epidemiol. 2008;61(2):102-109. PubMed
42.Lung Ccncer (update of QLQ-LC13). Brussels (BE): EORTC: Accessed 2021 Nov 2.
43.Koller M, Hjermstad MJ, Tomaszewski KA, et al. An international study to revise the EORTC questionnaire for assessing quality of life in lung cancer patients. Ann Oncol. 2017;28(11):2874-2881. PubMed
44.Koller M, Shamieh O, Hjermstad MJ, et al. Psychometric properties of the updated EORTC module for assessing quality of life in patients with lung cancer (QLQ-LC29): an international, observational field study. Lancet Oncol. 2020;21(5):723-732. PubMed
45.EMD Serono additional information regarding tepotinib DRR review provided April 6, 2022 [internal additional sponsor's information]. Missisauga (ON): EMD Serono Canada.
46.Hatswell A, Vioix H, Provost M, Liu F. Supplementary analyses for tepotinib vs. chemotherapy plus immunotherapy in advanced NSCLC [sponsor's internal report]. Nottingham, UK: Delta Hat Limited; 2022. Accessed April 6, 2022.
47.Rodríguez-Abreu D, Powell SF, Hochmair MJ, et al. Pemetrexed plus platinum with or without pembrolizumab in patients with previously untreated metastatic nonsquamous NSCLC: protocol-specified final analysis from KEYNOTE-189. Ann Oncol. 2021;32(7):881-895. PubMed
48.Bronserud MM, Iachina M, Green A, Groenvold M, Jakobsen E. Patient reported outcome data as performance indicators in surgically treated lung cancer patients. Lung Cancer. 2019;130:143-148. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: September 23, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit retrieval by study type.
Limits:
Publication date limit: none
Humans
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(tepotinib* or Tepmetko* or EMD-1214063 or EMD1214063 or MSC-2156119* or MSC2156119* or WHO 9934 or WHO9934 or 1IJV77EI07 or VY5YX2TQ1F).ti,ab,ot,kf,hw,nm,rn.
1 use medall
*tepotinib/ or (tepotinib* or Tepmetko* or EMD-1214063 or EMD1214063 or MSC-2156119* or MSC2156119* or WHO 9934 or WHO9934).ti,ab,kw,dq.
3 use oemezd
4 not (conference abstract or conference review).pt.
2 or 5
remove duplicates from 6
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search -- tepotinib AND carcinoma, non–small cell lung
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search terms -- tepotinib (intervention) AND non–small cell lung cancer (condition)
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms -- tepotinib
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms – tepotinib AND lung cancer
Grey Literature
Search dates: September 13-22, 202
Keywords: Tepmetko, tepotinib, non–small cell lung cancer
Limits: none
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Ryoo BY, Cheng AL, Ren Z, et al. Randomised Phase 1b/2 trial of tepotinib vs sorafenib in Asian patients with advanced hepatocellular carcinoma with MET overexpression. Br J Cancer. 2021 Jul;125(2):200-208. PubMed: PM33972742 | Study population |
Takamori S, Matsubara T, Fujishita T, et al. Dramatic intracranial response to tepotinib in a patient with lung adenocarcinoma harbouring MET exon 14 skipping mutation. Thorac Cancer. 2021 03;12(6):978-980. PubMed: PM33533182 | Study design |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 33: Patient Disposition — VISION, as of the January 1, 2020, Data Cut-Off Date
Patient disposition | VISION | |
|---|---|---|
Cohort A | Cohort A + C | |
Pre-screened for MET alteration status, N | 6,708 | |
Screened, N | 169 | 206 |
Active in screening, N | 2 | 5 |
Discontinued during screening, N | 15 | 20 |
Eligibility criteria not met | 8 | 11 |
Withdrew consent | 1 | 1 |
AE | 0 | 1 |
Death | 4 | 4 |
Other | 2 | 3 |
Enrolled and treated, N | 152 | 181 |
Discontinued treatment, N (%) | 92 (60.5) | 95 (52.5) |
AE | 23 | 25 |
Protocol noncompliance | 1 | 1 |
Death | 12 | 13 |
PD | 51 | 51 |
Withdrew consent | 3 | 3 |
Other | 2 | 2 |
Treatment ongoing, N (%) | 60 (39.5) | 86 (47.5) |
Cohort A mITT April 2, 2019, N | 99 | NA |
Overall mITT, N | 151 | 181 |
Safety, N | 152 | 181 |
AE = adverse event; MET = mesenchymal-epithelial transition; mITT = modified intention-to-treat; mITT April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; PD = progressive disease.
Source: VISION Clinical Study Reports12,14
Table 34: Summary of Exposure to Tepotinib in VISION
Exposure | January 1, 2020 DCO | July 1, 2020 DCO | ||||
|---|---|---|---|---|---|---|
Cohort A SAS April 2, 2019 | Cohort A SAS Overall | Cohort A + C SAS | Cohort A mITT April 2, 2019 | Cohort A SAS Overall | Cohort A + C SAS | |
n | 100 | 152 | 181 | 99 | 152 | 255 |
Mean duration of therapy (SD), months | 9.32 (7.48) | 7.74 (6.57) | 6.78 (6.42) | 10.40 (8.89) | 9.38 (7.63) | 7.05 (6.71) |
Mean dose intensity, (SD), mg per 3-week cycle | 9,110.06 (1,765.93) | 9,340.40 (1,619.54) | 9,485.34 (1,540.31) | 9,052.13 (1,848.46) | 9,195.76 (1,755.06) | 9,420.99 (1,689.85) |
Relative dose intensity, n (%) | ||||||
< 60% | 9 (9.0) | 10 (6.6) | 10 (5.5) | 9 (9.1) | 12 (7.9) | 15 (5.9) |
60% to 80% | 23 (23.0) | 29 (19.1) | 31 (17.1) | 25 (25.3) | 32 (21.1) | 44 (17.3) |
80% to 90% | 7 (7.0) | 12 (7.9) | 13 (7.2) | 5 (5.1) | 10 (6.6) | 19 (7.5) |
90% to 100% | 61 (61.0) | 101 (66.4) | 127 (70.2) | 60 (60.6) | 98 (64.5) | 177 (69.4) |
Patients that had ≥ 1 dose reduction | 38 (38.0) | 53 (34.9) | 54 (29.8) | 39 (39.4) | 57 (37.5) | 76 (29.8) |
Patients that had therapy delays | 61 (61.0) | 84 (55.3) | 91 (50.3) | 61 (61.6) | 91 (59.9) | 128 (50.2) |
Cohort A April 2, 2019 = Cohort A with first dose of tepotinib before April 2, 2019; DCO = data cut-off; mITT = modified intention-to-treat; SAS = safety analysis set; SD = standard deviation.
Source: VISION Clinical Study Reports12,14
Table 35: Summary of OS Results in VISION, mITT Population
End point | January 1, 2020, DCO | July 1, 2020, DCO | ||||
|---|---|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 180) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 254) | |
Patients with an event, n (%) | 41 (41.4) | 57 (37.7) | 59 (32.8) | 63 (63.6) | 75 (49.7) | 84 (33.1) |
Patients censored, n (%) | 49 (49.5) | 94 (62.3) | 121 (67.2) | 36 (36.4) | 76 (50.3) | 170 (66.9) |
Median duration of follow-up (95% CI), months | 12.5 (9.7, 17.2) | 11.8 (9.1, 14.6) | 9.1 (6.8, 11.8) | 25.1 (20.3, 29.3) | 16.4 (13.6, 18.5) | 9.9 (8.1, 12.0) |
Median OS (95% CI), months | 17.1 (12.0, 26.8) | 19.1 (12.3, 26.8) | 19.1 (12.3, 26.8) | 17 (12.0, 20.4) | 17.6 (15.0, 21.0) | 19.1 (15.3, 22.1) |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; DCO = data cut-off; mITT = modified intention-to-treat; OS = overall survival.
Source: VISION Clinical Study Reports12,14
Figure 23: Kaplan-Meier Curve Showing OS in Cohort A Patients Who Received the First Dose of Tepotinib Before April 2, 2019, mITT Population (N = 99); January 1, 2020, Data Cut-Off Date
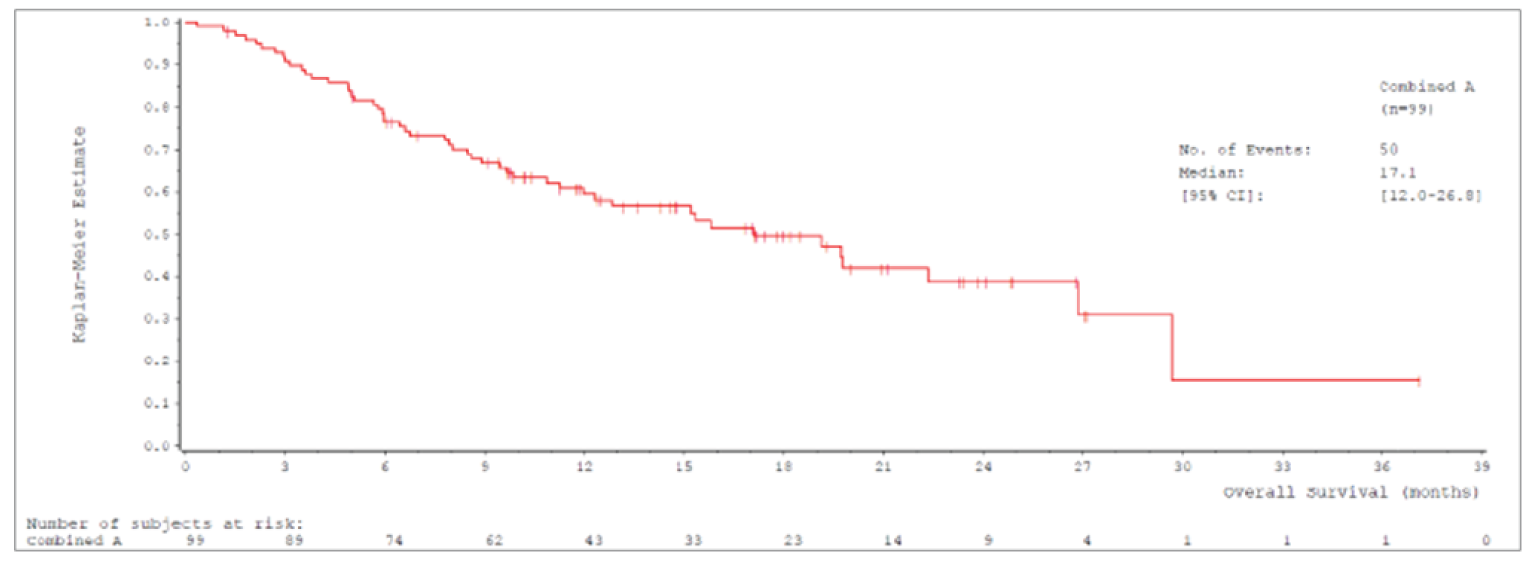
CI = confidence interval; mITT = modified intention-to-treat; OS = overall survival.
Source: VISION Clinical Study Report.12
Figure 24: Kaplan-Meier Curve Showing OS in Cohort A Overall mITT Population (N = 151); January 1, 2020, Data Cut-Off Date
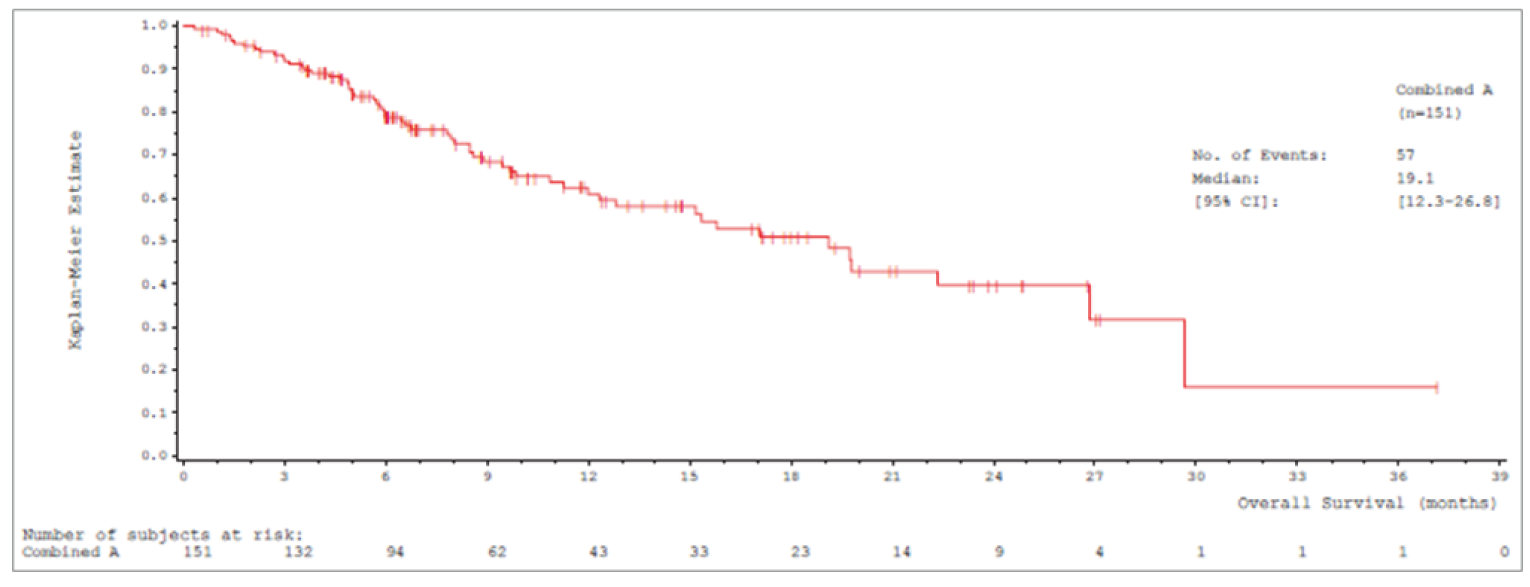
CI = confidence interval; mITT = modified intention-to-treat; OS = overall survival.
Source: VISION Clinical Study Report.12
Table 36: Subgroup Analyses of OS in Cohort A, mITT Population
Subgroup | January 1, 2020 DCO | July 1, 2020 DCO | ||
|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | |
Line of treatment | ||||
First-line, n | 43 | 69 | 43 | 69 |
Patients who died, n (%) | 21 (48.8) | 25 (36.2) | 28 (65.1) | 35 (50.7) |
Median OS (95% CI), months | 19.1 (8.5, 29.7) | 19.1 (8.5, 29.7) | 15.3 (8.5, 23.6) | 17.6 (9.7, 29.7) |
Second- or later-line, n | 56 | 82 | 53 | 82 |
Patients who died, n (%) | 29 (51.8) | 32 (39.0) | 32 (60.4) | 40 (48.8) |
Median OS (95% CI), months | 17.1 (11.3, 26.8) | 17.1 (12.0, 26.8) | 19.7 (12.8, 21.0) | 19.7 (15.0, 21.0) |
Presence of brain metastases at baseline by IRC | ||||
Present, n | 11 | 15 | 11 | 15 |
Patients who died, n (%) | 5 (45.5) | 5 (33.3) | 6 (54.5) | 7 (46.7) |
Median OS (95% CI), months | 12 (5.9, NE) | 12.0 (8.0, NE) | 17.1 (5.9, NE) | 22.1 (8.0, NE) |
Absent, n | 88 | 136 | 88 | 136 |
Patients who died, n (%) | 45 (51.1) | 52 (38.2) | 57 (64.8) | 68 (50.0) |
Median OS (95% CI), months | 19.1 (11.3, 26.8) | 19.1 (12.8, 26.8) | 17.0 (12.1, 20.4) | 17.6 (15.2, 21.0) |
Histological subtype | ||||
Adenocarcinoma, n | 89 | 131 | 89 | 131 |
Patients who died, n (%) | 44 (49.4) | 49 (37.4) | 55 (61.8) | 63 (48.1) |
Median OS (95% CI), months | 19.1 (12.3, 26.8) | 19.1 (12.8, 29.7) | 17.1 (12.3, 22.1) | 19.7 (15.3, 23.6) |
Squamous, n | 7 | 13 | 7 | 14 |
Patients who died, n (%) | 3 (42.9) | 4 (30.8) | 5 (71.4) | 8 (57.1) |
Median OS (95% CI), months | NE (2.1, NE) | NE (3.0, NE) | 13.5 (2.1, 21.0) | 13.5 (3.5, 21.0) |
Other, n | 3 | 7 | 3 | 6 |
Patients who died, n (%) | 3 (100.0) | 4 (57.1) | 3 (100.0) | 4 (66.7) |
Median OS (95% CI), months | 3.1 (0.3, 8.5) | 8.5 (0.3, 8.5) | 3.1 (0.3, 8.5) | 5.8 (0.3, NE) |
ECOG PS | ||||
0, n | 22 | 40 | 22 | 40 |
Patients who died, n (%) | 8 (36.4) | 10 (25.0) | 11 (50.0) | 14 (35.0) |
Median OS (95% CI), months | 29.7 (11.3, 29.7) | 29.7 (11.3, 29.7) | 24.9 (11.3, NE) | 24.9 (19.1, NE) |
1, n | 77 | 111 | 77 | 111 |
Patients who died, n (%) | 42 (54.5) | 47 (42.3) | 52 (67.5) | 61 (55.0) |
Median OS (95% CI), months | 15.2 (9.7, 19.8) | 15.3 (9.9, 22.3) | 15.2 (9.7, 19.7) | 15.8 (12.1, 19.8) |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group performance status; IRC = independent review committee; mITT = modified intention-to-treat; NE = not estimable; OS = overall survival.
Source: VISION Clinical Study Reports12,14
Table 37: Summary of PFS Results in VISION, mITT Population
End point | January 1, 2020, DCO | July 1, 2020, DCO | ||||
|---|---|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 180) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 254) | |
PFS by IRC (Secondary End point) | ||||||
Patients with an event, n (%) | 53 (53.5) | 72 (47.7) | 75 (41.7) | 67 (67.7) | 87 (57.6) | 105 (41.3) |
PD | 29 (29.3) | 43 (28.5) | 44 (24.4) | 41 (41.4) | 56 (37.1) | 67 (26.4) |
Death | 24 (24.2) | 29 (19.2) | 31 (17.2) | 26 (26.3) | 31 (20.5) | 38 (15.0) |
Patients censored, n (%) | 46 (46.5) | 79 (52.3) | 105 (58.3) | 32 (32.3) | 64 (42.4) | 149 (58.7) |
Median duration of follow-up (95% CI), months | 11.3 (6.8, 16.5) | 8.3 (6.8, 11.0) | 6.8 (5.5, 8.3) | 17.9 (13.9, 20.7) | 12.2 (11.0, 14.0) | 7.0 (5.8, 8.3) |
Median PFS (95% CI), months | 8.5 (6.7, 10.9) | 8.6 (8.0, 11.0) | 8.6 (7.8, 11.0) | 8.5 (6.8, 11.0) | 8.9 (8.2, 11.0) | 9.5 (8.2, 11.2) |
PFS by Investigator (Secondary End point) | ||||||
Patients with an event, n (%) | 57 (57.6) | 74 (49.0) | 77 (42.8) | 68 (68.7) | 98 (64.9) | 121 (47.6) |
PD | 41 (41.4) | 53 (35.1) | 54 (30.0) | 50 (50.5) | 78 (51.7) | 95 (37.4) |
Death | 16 (16.2) | 21 (13.9) | 23 (12.8) | 18 (18.2) | 20 (13.2) | 26 (10.2) |
Patients censored, n (%) | 42 (42.4) | 77 (51.0) | 103 (57.2) | 31 (31.3) | 53 (35.1) | 133 (52.4) |
Median duration of follow-up (95% CI), months | 13.8 (8.2, 18.4) | 9.6 (8.2, 13.8) | 8.2 (5.6, 10.9) | 20.2 (17.3, 24.9) | 16.5 (13.8, 20.2)) | 8.3 (8.1, 11.1) |
Median PFS (95% CI), months | 8.5 (5.8, 11.0) | 9.7 (7.0, 12.2) | 9.7 (7.0, 12.2) | 8.6 [6.7, 12.2] | 8.5 (6.9, 11.0) | 8.3 (6.9, 10.6) |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; DCO = data cut-off; IRC = independent review committee; mITT = modified intention-to-treat; PD = progressive disease; PFS = progression-free survival.
Source: VISION Clinical Study Reports12,14
Figure 25: Kaplan-Meier Curve Showing PFS by IRC in Cohort A Patients Who Received the First Dose of Tepotinib Before April 2, 2019, mITT Population (N = 99); January 1, 2020, Data Cut-Off Date
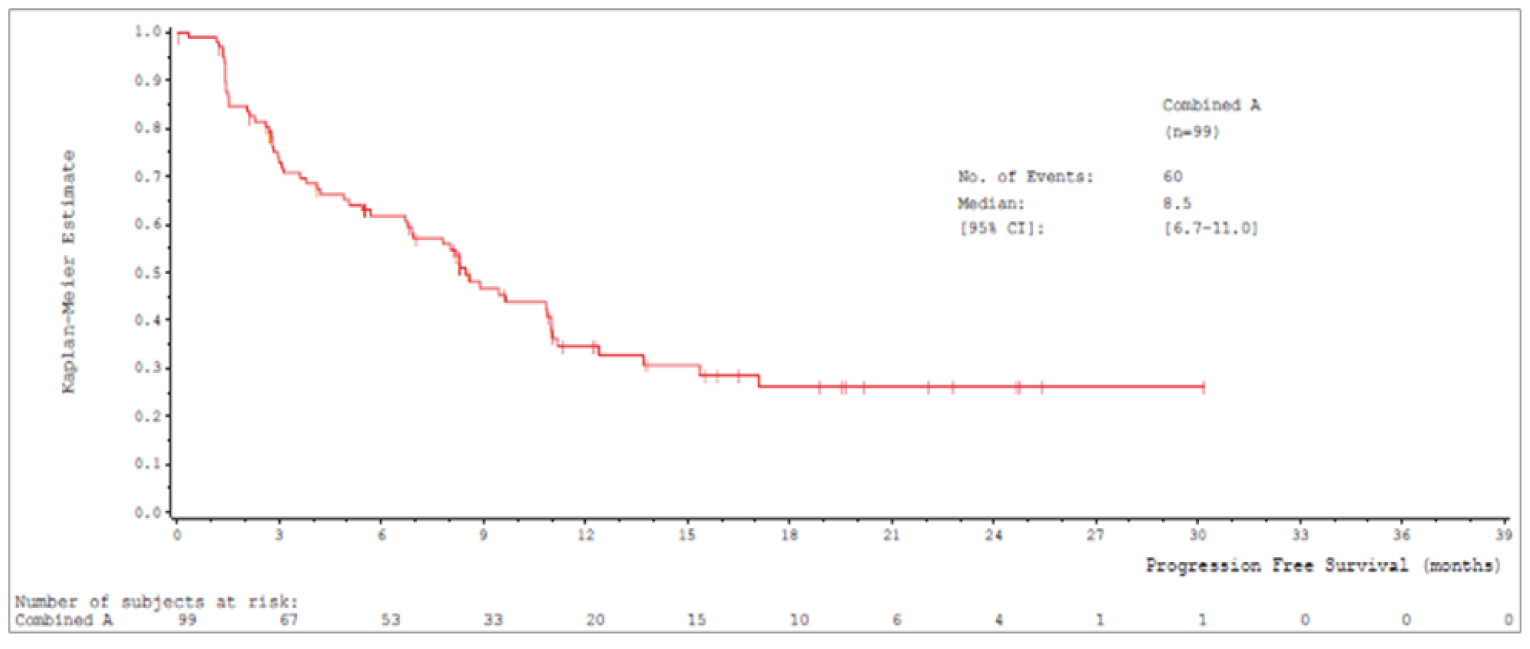
CI = confidence interval; IRC = independent review committee; mITT = modified intention-to-treat; PFS = progression-free survival.
Source: VISION Clinical Study Report.12
Figure 26: Kaplan-Meier Curve Showing PFS by IRC in Cohort A Overall mITT Population (N = 151); January 1, 2020, Data Cut-Off Date
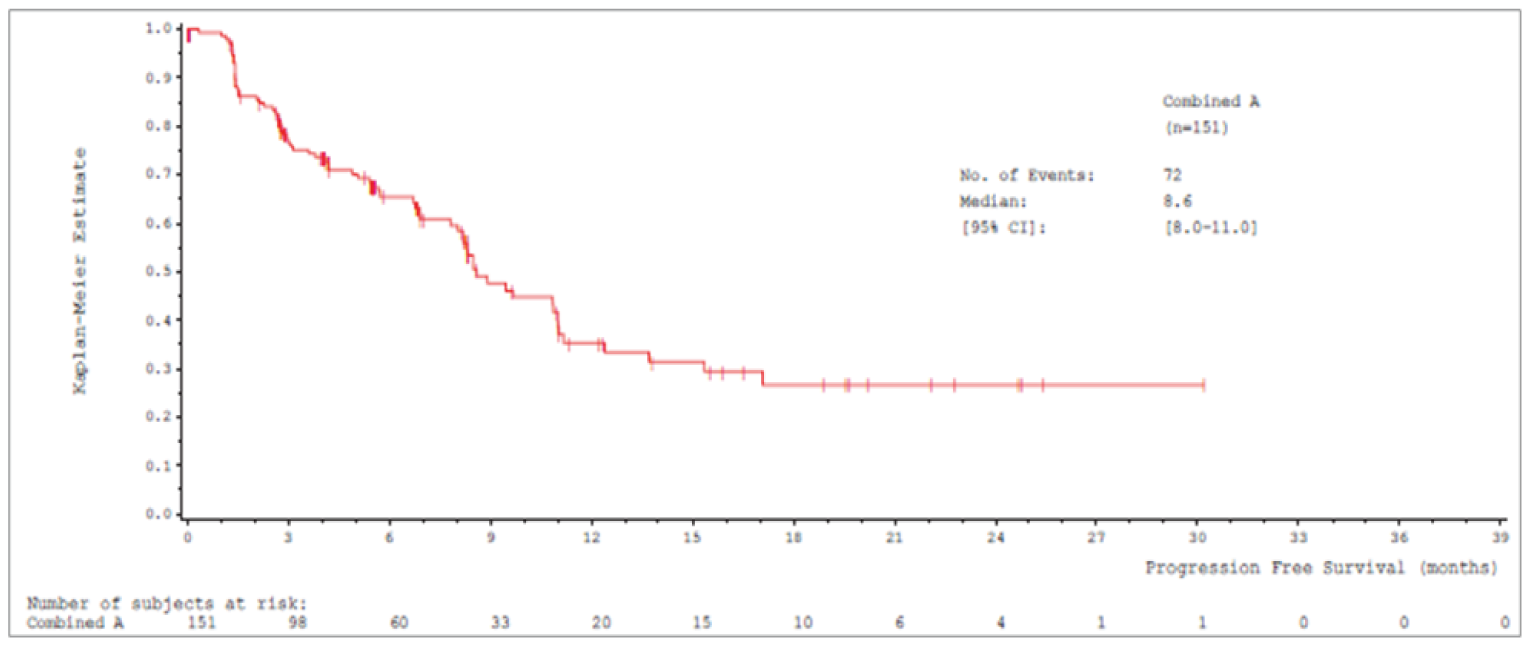
CI = confidence interval; IRC = independent review committee; mITT = modified intention-to-treat; PFS = progression-free survival.
Source: VISION Clinical Study Report.12
Figure 27: Kaplan-Meier Curve Showing PFS by IRC in Cohort A Patients Who Received the First Dose of Tepotinib Before April 2, 2019, mITT Population (N = 99); July 1, 2020, DATA CUT-OFF DATE
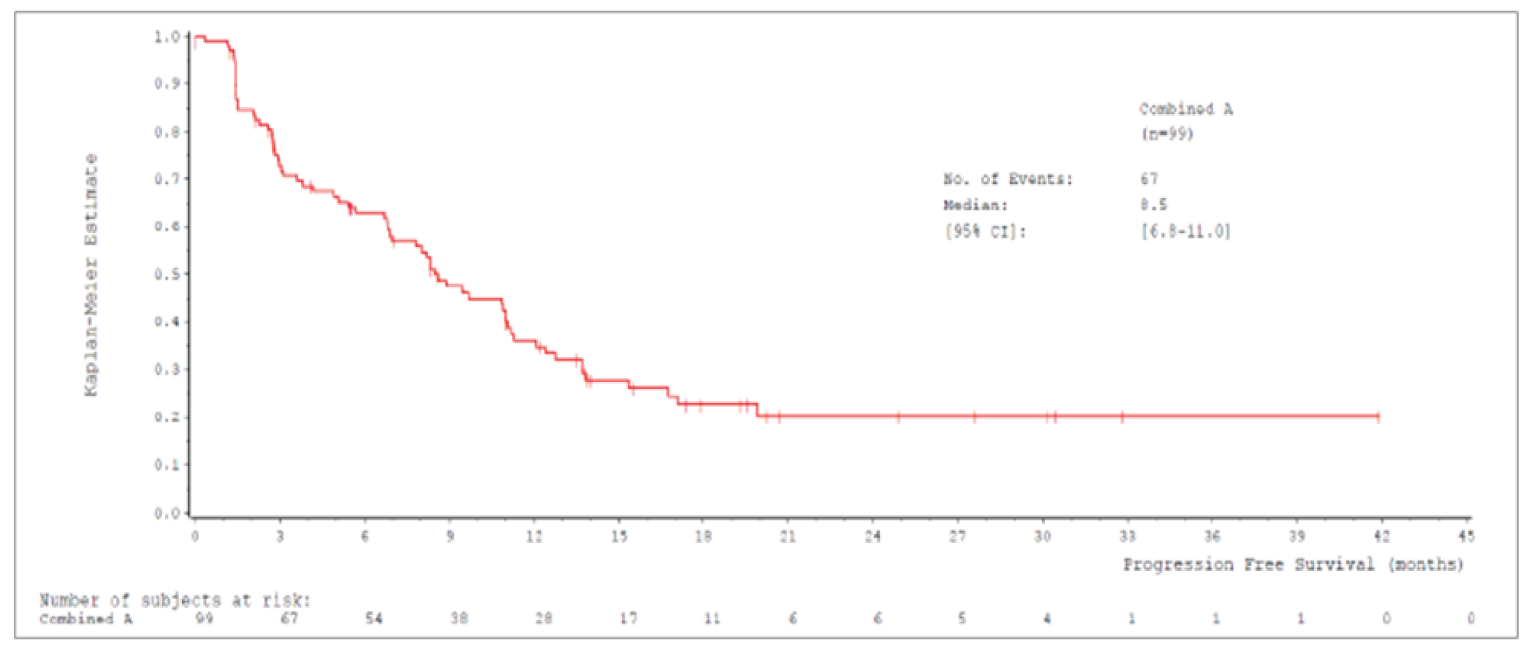
CI = confidence interval; IRC = independent review committee; mITT = modified intention-to-treat; PFS = progression-free survival.
Source: VISION Clinical Study Report.12
Table 38: Subgroup Analyses of PFS by IRC in Cohort A, mITT Population
Subgroup | January 1, 2020 DCO | July 1, 2020 DCO | ||
|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | |
Line of treatment | ||||
First-line, n | 43 | 69 | 43 | 69 |
Patients with events, n (%) | 27 (62.8) | 33 (47.8) | 29 (67.4) | 39 (56.5) |
Median PFS (95% CI), months | 8.0 (3.1, 10.8) | 8.5 (5.4, 15.3) | 8.0 (3.8, 11.3) | 8.5 (6.8, 11.3) |
Second- or later-line, n | 56 | 82 | 56 | 82 |
Patients with events, n (%) | 33 (58.9) | 39 (47.6) | 38 (67.9) | 48 (58.5) |
Median PFS (95% CI), months | 9.5 (6.7, 11.2) | 9.5 (6.9, 11.2) | 10.9 (6.7, 12.7) | 10.9 (8.2, 12.7) |
Presence of brain metastases at baseline by IRC | ||||
Present, n | 11 | 15 | 11 | 15 |
Patients with events, n (%) | 5 (45.5) | 6 (40.0) | 6 (54.5) | 7 (46.7) |
Median PFS (95% CI), months | 10.9 (8.0, NE) | 10.9 (8.0, NE) | 10.9 (6.8, NE) | 10.9 (6.8, NE) |
Absent, n | 88 | 136 | 88 | 136 |
Patients with events, n (%) | 55 (62.5) | 66 (48.5) | 61 (69.3) | 80 (58.8) |
Median PFS (95% CI), months | 8.3 (5.1, 11.0) | 8.5 (6.9, 11.0) | 8.3 (5.4, 11.0) | 8.9 (8.2, 11.3) |
Histological subtype | ||||
Adenocarcinoma, n | 89 | 131 | 89 | 131 |
Patients with events, n (%) | 52 (58.4) | 62 (47.3) | 59 (66.3) | 74 (56.5) |
Median PFS (95% CI), months | 8.9 (6.9, 11.2) | 8.9 (8.0, 11.2) | 9.5 (6.9, 11.3) | 9.7 (8.2, 12.1) |
Squamous, n | 7 | 13 | 7 | 14 |
Patients with events, n (%) | 5 (71.4) | 6 (46.2) | 5 (71.4) | 9 (64.3) |
Median PFS (95% CI), months | 3.0 (1.4, NE) | 11.0 (1.4, NE) | 3 (1.4) | 3.3 (1.4, NE) |
Other, n | 3 | 7 | 3 | 6 |
Patients with events, n (%) | 3 (100.0) | 4 (57.1) | 3 (100.0) | 4 (66.7) |
Median PFS (95% CI), months | 3.1 (0.3, 8.5) | 8.5 (0.3, 8.5) | 3.1 (0.3, 8.5) | 5.8 (0.3, 8.5) |
ECOG PS | ||||
0, n | 22 | 40 | 22 | 40 |
Patients with events, n (%) | 10 (45.5) | 12 (30.0) | 10 (45.5) | 15 (37.5) |
Median PFS (95% CI), months | 11.0 (6.8, NE) | 11.0 (8.5, NE) | NE (6.8, NE) | NE (8.5, NE) |
1, n | 77 | 111 | 77 | 111 |
Patients with events, n (%) | 50 (64.9) | 60 (54.1) | 57 (74.0) | 72 (64.9) |
Median PFS (95% CI), months | 8.0 (4.2, 9.7) | 8.2 (5.7, 9.7) | 8 (4.9, 9.7) | 8.3 (6.8, 10.9) |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group performance status; IRC = independent review committee; mITT = modified intention-to-treat; NA = not applicable; NE = not estimable; PFS = progression-free survival.
Source: VISION Clinical Study Reports12,14
Table 39: Summary of ORR Results in VISION, mITT Population
End point | January 1, 2020, DCO | July 1, 2020, DCO | ||||
|---|---|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 180) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 254) | |
ORR by IRC (Primary End point) | ||||||
Patients contributing to the analysisa, n | 99 | 146 | 154 | 99 | 151 | 224 |
ORR, n (%) | 46 (46.5) | 65 (44.5) | 67 (43.5) | 45 (45.5) | 68 (45.0) | 104 (46.4) |
95% CIb | 36.4, 56.8 | 36.3, 53.0 | 35.5, 51.7 | 35.4, 55.8 | 36.9, 53.3 | 39.8, 53.2 |
Best overall responsec | ||||||
CR | 0 | 0 | 0 | 0 | 0 | 0 |
PR | 46 (46.5) | 65 (44.5) | 67 (43.5) | 45 (45.5) | 68 (45.0) | 104 (46.4) |
SD | 19 (19.2) | 37 (25.3) | 39 (25.3) | 20 (20.2) | 38 (25.2) | 53 (23.7) |
PD | 19 (19.2) | 23 (15.8) | 24 (15.6) | 19 (19.2) | 26 (17.2) | 34 (15.2) |
Not evaluable | 15 (15.2) | 21 (14.4) | 24 (15.6) | 15 (15.2) | 19 (12.6) | 33 (14.7) |
ORR by Investigator (Secondary End point) | ||||||
Patients contributing to the analysisa, n | 99 | 146 | 154 | 99 | 151 | 254 |
ORR, n (%) | 55 (55.6) | 80 (54.8) | 83 (53.9) | 55 (55.6) | 80 (53.0) | 111 (43.7) |
95% CI | 45.2, 65.5 | 46.4, 63.0 | 45.7, 61.9 | 45.2, 65.5 | 44.7, 61.1 | 37.5, 50.0 |
Best overall response | ||||||
CR | 2 (2.0) | 3 (2.1) | 3 (1.9) | 2 (2.0) | 3 (2.0) | 3 (1.2) |
PR | 53 (53.5) | 77 (52.7) | 80 (51.9) | 53 (53.5) | 77 (51.0) | 108 (42.5) |
SD | 17 (17.2) | 31 (21.2) | 32 (20.8) | 17 (17.2) | 33 (21.9) | 54 (21.3) |
PD | 18 (18.2) | 19 (13.0) | 19 (12.3) | 18 (18.2) | 23 (15.2) | 34 (13.4) |
Not evaluable | 9 (9.1) | 16 (11.0) | 20 (13.0) | 9 (9.1) | 15 (9.9) | 55 (21.7) |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; CR = complete response; DCO = data cut-off; IRC = independent review committee; mITT = modified intention-to-treat; NA = not applicable; PD = progressive disease; ORR = objective response rate; PD = progressive disease; PR = partial response; SD = stable disease.
aOnly patients with 2 or more post-baseline assessments or who discontinued study treatment for any reason are included in the analysis.
b95% exact CI using the Clopper-Pearson method.
cCR and PR had to be confirmed. SD had to last at least 12 weeks.
Source: VISION Clinical Study Reports12,14
Table 40: Subgroup Analyses of ORR by IRC (Primary End Point) in Cohort A, mITT Population With 2 Post-Baseline Assessments
Subgroup | January 1, 2020 DCO | July 1, 2020 DCO | ||
|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 146) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | |
Line of treatment | ||||
First-line, n | 43 | 65 | 43 | 69 |
ORR, n (%) | 19 (44.2) | 29 (44.6) | 19 (44.2) | 31 (44.9) |
95% CI | 29.1, 60.1 | 32.3, 57.5 | 29.1, 60.1 | 32.9, 57.4 |
Second- or later-line, n | 56 | 81 | 56 | 82 |
ORR, n (%) | 27 (48.2) | 36 (44.4) | 26 (46.4) | 37 (45.1) |
95% CI | 34.7, 62.0 | 33.4, 55.9 | 33.0, 60.3 | 34.1, 56.5 |
Presence of brain metastases at baseline by IRC | ||||
Present, n | 11 | 14 | 11 | 15 |
ORR, n (%) | 6 (54.5) | 8 (57.1) | 6 (54.5) | 8 (53.3) |
95% CI | 23.4, 83.3 | 28.9, 82.3 | 23.4, 83.3 | 26.6, 78.7 |
Absent, n | 88 | 132 | 88 | 136 |
ORR, n (%) | 40 (45.5) | 57 (43.2) | 39 (44.3) | 60 (44.1) |
95% CI | 34.8, 56.4 | 34.6, 52.1 | 33.7, 55.3 | 35.6, 52.9 |
Histological subtype | ||||
Adenocarcinoma, n | 89 | 127 | 89 | 131 |
ORR, n (%) | 43 (48.3) | 61 (48.0) | 42 (47.2) | 63 (48.1) |
95% CI | 37.6, 59.2 | 39.1, 57.1 | 36.5, 58.1 | 39.3, 57.0 |
Squamous, n | 7 | 12 | 7 | 14 |
ORR, n (%) | 2 (28.6) | 2 (16.7) | 2 (28.6) | 3 (21.4) |
95% CI | 3.7, 71.0 | 2.1, 48.4 | 3.7, 71.0 | 4.7, 50.8 |
Other, n | 3 | 7 | 3 | 6 |
ORR, n (%) | 1 (33.3) | 2 (28.6) | 1 (33.3) | 2 (33.3) |
95% CI | 0.8, 90.6 | 3.7, 71.0 | 0.8, 90.6 | 4.3, 77.7 |
ECOG PS | ||||
0, n | 22 | 37 | 22 | 40 |
ORR, n (%) | 13 (59.1) | 22 (59.5) | 13 (59.1) | 23 (57.5) |
95% CI | 36.4, 79.3 | 42.1, 75.2 | 36.4, 79.3 | 40.9, 73.0 |
1, n | 77 | 109 | 77 | 111 |
ORR, n (%) | 33 (42.9) | 43 (39.4) | 32 (41.6) | 45 (40.5) |
95% CI | 31.6, 54.6 | 30.2, 49.3 | 30.4, 53.4 | 31.3, 50.3 |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group performance status; IRC = independent review committee; mITT = modified intention-to-treat; ORR = objective response rate.
Source: VISION Clinical Study Reports12,14
Table 41: Summary of DOR Results in VISION, mITT Population
End point | January 1, 2020, DCO | July 1, 2020, DCO | ||||
|---|---|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 180) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 254) | |
DOR by IRC | ||||||
Patients with confirmed CR or PR, n | 46 | 65 | 67 | 45 | 68 | 104 |
Patients with an event (PD or death), n (%) | 21 (45.7) | 24 (36.9) | 24 (35.8) | 25 (55.6) | 31 (45.6) | 33 (31.7) |
Patients censored, n (%) | 25 (54.3) | 41 (63.1) | 43 (64.2) | 20 (44.4) | 37 (54.4) | 71 (68.3) |
Median duration of follow-up (95% CI), months | 14.6 (9.7, 18.3) | 9.7 (4.2, 14.6) | 9.7 (5.6, 14.6) | 16.7 (11.2, 28.8) | 10.9 (9.7, 16.7) | 7.0 (6.9, 9.7) |
Median DOR (95% CI), months | 11.1 (7.2, NE) | 9.9 (7.2, NE) | 9.9 (7.2, NE) | 11.1 (8.4, 18.5) | 11.1 (8.4, 18.5) | 11.1 (9.5, 18.5) |
DOR by Investigator | ||||||
Patients with confirmed CR or PR, n | 55 | 80 | 83 | 55 | 80 | 111 |
Patients with an event (PD or death), n (%) | 27 (49.1) | 30 (37.5) | 30 (36.1) | 29 (52.7) | 39 (48.8) | 43 (38.7) |
Patients censored, n (%) | 28 (50.9) | 50 (62.5) | 53 (63.9) | 26 (47.3) | 41 (51.3) | 68 (61.3) |
Median duration of follow-up (95% CI), months | 15.1 (12.5, 18.3) | 10.1 (6.9, 13.4) | 9.7 (5.6, 13.2) | 18.2 (15.2, 22.7) | 15.2 (12.5, 19.4) | 9.7 (7.0, 15.1) |
Median DOR (95% CI), months | 14.0 (9.7, 18.3) | 14.0 (9.7, 18.3) | 14.0 (9.7, 18.3) | 14.0 (9.7, NE) | 12.7 (9.7, 18.3) | 12.5 (9.7, 18.3) |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; CR = complete response; DCO = data cut-off; DOR = duration of response; IRC = independent review committee; mITT = modified intention-to-treat; NE = not estimable; PD = progressive disease; PR = partial response.
Source: VISION Clinical Study Reports.12,14
Table 42: Subgroup Analyses of DOR by IRC in Cohort A, mITT Population
Subgroup | January 1, 2020 DCO | July 1, 2020 DCO | ||
|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | |
Line of treatment | ||||
First-line, n | 43 | 69 | 43 | 69 |
Patients with confirmed CR or PR, n | 19 | 29 | 19 | 31 |
Patients with an event (PD or death), n (%) | 7 (36.8) | 9 (31.0) | 9 (47.4) | 13 (41.9) |
Median DOR (95% CI), months | NE (5.8, NE) | 7.2 (5.8, NE) | NE (5.8, NE) | 10.8 (6.9, NE) |
Second- or later-line, n | 56 | 82 | 56 | 82 |
Patients with confirmed CR or PR, n | 27 | 36 | 26 | 37 |
Patients with an event (PD or death), n (%) | 14 (51.9) | 15 (41.7) | 16 (61.5) | 18 (48.6) |
Median DOR (95% CI), months | 9.9 (8.3, NE) | 9.9 (8.3, NE) | 11.1 (8.4, 18.5) | 11.1 (9.5, 18.5) |
Presence of brain metastases at baseline by IRC | ||||
Present, n | 11 | 15 | 11 | 15 |
Patients with confirmed CR or PR, n | 6 | 8 | 6 | 8 |
Patients with an event (PD or death), n (%) | 3 (50.0) | 3 (37.5) | 4 (66.7) | 4 (50.0) |
Median DOR (95% CI), months | 9.5 (6.6, NE) | 9.5 (6.6, NE) | 9.0 (5.5, NE) | 9.5 (5.5, NE) |
Absent, n | 88 | 136 | 88 | 136 |
Patients with confirmed CR or PR, n | 40 | 57 | 39 | 60 |
Patients with an event (PD or death), n (%) | 18 (45.0) | 21 (36.8) | 21 (53.8) | 27 (45.0) |
Median DOR (95% CI), months | 11.1 (7.0, NE) | 11.1 (7.2, NE) | 12.4 (8.3, NE) | 11.1 (9.7, 18.5) |
Histological subtype | ||||
Adenocarcinoma, n | 89 | 131 | 89 | 131 |
Patients with confirmed CR or PR, n | 43 | 61 | 42 | 63 |
Patients with an event (PD or death), n (%) | 19 (44.2) | 22 (36.1) | 23 (54.8) | 29 (46.0) |
Median DOR (95% CI), months | 11.1 (7.2, NE) | 11.1 (7.2, NE) | 11.1 (9.5, NE) | 11.1 (9.5, 18.5) |
Squamous, n | 7 | 13 | 7 | 14 |
Patients with confirmed CR or PR, n | 2 | 2 | 2 | 3 |
Patients with an event (PD or death), n (%) | 1 (50.0) | 1 (50.0) | 1 (50.0) | 1 (33.3) |
Median DOR (95% CI), months | NE (8.3, NE) | NE (8.3, NE) | NE (8.3, NE) | NE (8.3, NE) |
Other, n | 3 | 7 | 3 | 6 |
Patients with confirmed CR or PR, n | 1 | 2 | 1 | 2 |
Patients with an event (PD or death), n (%) | 1 (100) | 1 (50.0) | 1 (100) | 1 (50.0) |
Median DOR (95% CI), months | 5.8 (NE, NE) | 5.8 (NE, NE) | 5.8 (NE, NE) | NE (5.8, NE) |
ECOG PS | ||||
0, n | 22 | 40 | 22 | 40 |
Patients with confirmed CR or PR, n | 13 | 22 | 13 | 23 |
Patients with an event (PD or death), n (%) | 3 (23.1) | 4 (18.2) | 3 (23.1) | 6 (26.1) |
Median DOR (95% CI), months | NE (9.7, NE) | NE (7.2, NE) | NE (9.7, NE) | NE (9.7, NE) |
1, n | 77 | 111 | 77 | 111 |
Patients with confirmed CR or PR, n | 33 | 43 | 32 | 45 |
Patients with an event (PD or death), n (%) | 18 (54.5) | 20 (46.5) | 22 (68.8) | 25 (55.6) |
Median DOR (95% CI), months | 8.4 (6.6, 15.7) | 8.4 (6.6, 15.7) | 9.5 (6.9, 15.4) | 10.1 (7.2, 15.7) |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; CR = complete response; DCO = data cut-off; DOR = duration of response; IRC = independent review committee; mITT = modified intention-to-treat; NE = not estimable; PD = progressive disease; PR = partial response.
Source: VISION Clinical Study Reports12,14
Table 43: Summary of TTD by 10 Points in HRQoL and PROs in VISION, mITT Population
End point | January 1, 2020 DCO | July 1, 2020 DCO | ||||
|---|---|---|---|---|---|---|
Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 180) | Cohort A April 2, 2019 (N = 99) | Cohort A Overall (N = 151) | Cohort A + C (N = 254) | |
EQ-5D-5L VAS | ||||||
Patients with a deterioration event, n (%) | 36 (36.4) | 55 (36.4) | 58 (32.2) | 37 (37.4) | 61 (40.4) | 81 (31.9) |
Median time to deterioration (95% CI), months | 11.1 (5.8, 19.4) | 6.9 (5.6, 17.7) | 6.9 (5.6, 17.7) | 11.1 (5.8, 19.4) | 8.3 (5.8, 17.7) | 8.3 (5.9, 17.7) |
EORTC QLQ-C30 Global Health Status Score | ||||||
Patients with a deterioration event, n (%) | 31 (31.3) | 48 (31.8) | 49 (27.2) | 31 (31.3) | 54 (35.8) | 74 (29.1) |
Median time to deterioration (95% CI), months | 15.2 (5.6, 33.2) | 15.2 (5.6, 33.2) | 15.2 (5.8, 33.2) | 15.2 (5.6, 33.2) | 15.2 (6.0, 33.2) | 15.2 (6.2, 33.2) |
EORTC QLQ-LC13 Cough | ||||||
Patients with a deterioration event, n (%) | 24 (24.2) | 38 (25.2) | 40 (22.2) | 26 (26.3) | 45 (29.8) | 52 (20.5) |
Median time to deterioration (95% CI), months | 19.3 (11.1, NE) | 13.8 (11.1, NE) | 13.8 (11.1, NE) | 13.8 (11.1, NE) | 11.1 (11.1, NE) | 13.8 (11.1, NE) |
EORTC QLQ-LC13 Chest Pain | ||||||
Patients with a deterioration event, n (%) | 24 (24.2) | 34 (22.5) | 35 (19.4) | 24 (24.2) | 45 (29.8) | 56 (22.0) |
Median time to deterioration (95% CI), months | 24.9 (11.1, NE) | 24.9 (11.1, NE) | 24.9 (17.7, NE) | 24.9 (17.7, NE) | 17.7 (11.1, NE) | 17.7 (11.8, NE) |
EORTC QLQ-LC13 Dyspnea | ||||||
Patients with a deterioration event, n (%) | 45 (45.5) | 70 (46.4) | 74 (41.1) | 46 (46.5) | 76 (50.3) | 104 (40.9) |
Median time to deterioration (95% CI), months | 5.6 (3.3, 11.1) | 4.5 (4.1, 6.9) | 4.5 (3.7, 5.6) | 5.6 (3.3, 11.1) | 5.5 (4.1, 6.9) | 5.6 (4.1, 6.9) |
CI = confidence interval; cohort A April 2, 2019 = cohort A with first dose of tepotinib before April 2, 2019; DCO = data cut-off; EORTC QLQ-C30 = European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire; EORTC QLQ-LC13 = European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Lung Cancer 13; EQ-5D-5L = 5-Level EQ-5D; NE = not estimable; mITT = modified intention-to-treat; VAS = visual analogue scale.
Source: VISION Clinical Study Reports12,14
Table 44: Subgroup Analyses of PFS and OS by Line of Treatment, Indirect Comparison of Immunotherapy and Chemotherapy Groups to Tepotinib (VISION) Using Propensity Scoring
Subgroup | Data | Tepotinib | Immunotherapy (Weighted) | Chemotherapy (Weighted) |
|---|---|---|---|---|
PFS | ||||
Untreated | N | 69 | 20 | 49 |
ESS | NA | 69 | 68 | |
Median (95% CI), months | 9.7 (6.9-15.3) | 6.2 (1.4-NA) | 3.4 (2.8-4.6) | |
RMST, months | 13.74 | 11.56 | 5.45 | |
Cox HR | NA | 0.81 (0.41-1.62) | 0.35 (0.22-0.57) | |
P value | NA | 0.5598 | < 0.0001 | |
Previously treated | N | 82 | 32 | 34 |
ESS | NA | 80 | 80 | |
Median (95% CI), months | 8.3 (6.7-11.1) | 2.7 (0.7-7.8) | 3.5 (2.6-6.4) | |
RMST, months | 10.50 | 5.21 | 6.44 | |
Cox HR | NA | 0.48 (0.29-0.77) | 0.58 (0.35-0.96) | |
P value | NA | 0.0025 | 0.0344 | |
OS | ||||
Untreated | N | 69 | 20 | 49 |
ESS | NA | 69 | 68 | |
Median (95% CI), months | 17.6 (13.4-29.8) | 22.4 (9.4-NE) | 18.8 (14.6-38.3) | |
RMST, months | 18.26 | 19.15 | 18.33 | |
Cox HR | NA | 1.21 (0.56-2.61) | 1.04 (0.62-1.76) | |
P value | NA | 0.6198 | 0.8660 | |
Previously treated | N | 82 | 32 | 34 |
ESS | NA | 80 | 80 | |
Median (95% CI), months | 19.8 (15.6-22.3) | 15.3 (10.9-NE) | 17.3 (9.2-NE) | |
RMST, months | 19.07 | 16.89 | 18.45 | |
Cox HR | NA | 0.76 (0.44-1.30) | 0.95 (0.54-1.68) | |
P value | NA | 0.3158 | 0.8581 | |
CI = confidence interval; ESS = effective sample size; HR = hazard ratio; NA = not applicable; NE = not estimable; RMST = restricted mean survival time.
Source: Sponsor’s technical report15
Figure 28: Subgroup Analysis — Kaplan-Meier Plot of OS for RWD Indirect Comparison to VISION Using Propensity Scoring; Weighted and Unweighted Chemotherapy Analysis Set, Untreated Patients
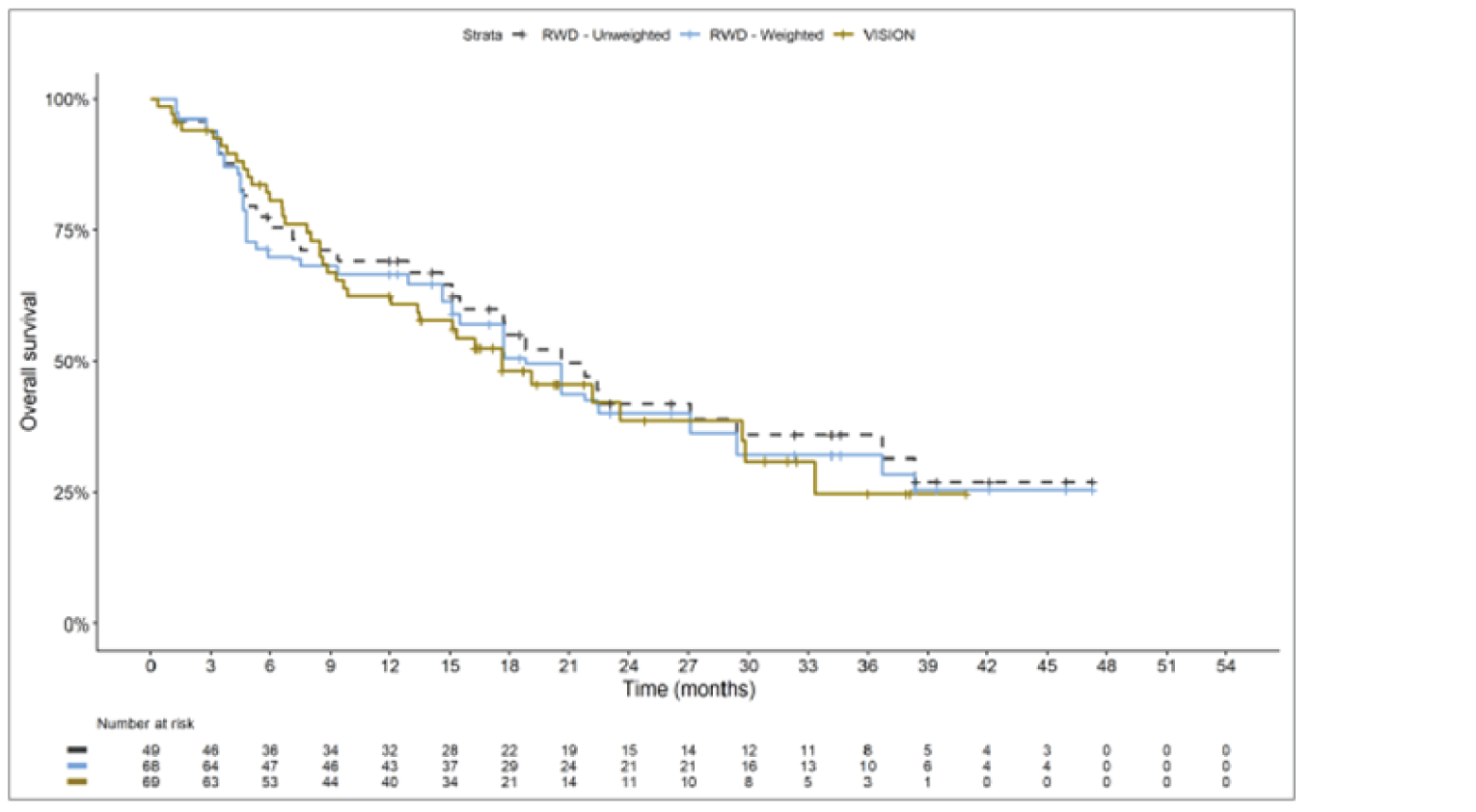
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 29: Subgroup Analysis — Kaplan-Meier Plot of OS for RWD Indirect Comparison to VISION Using Propensity Scoring; Weighted and Unweighted Chemotherapy Analysis Set, Previously Treated Patients
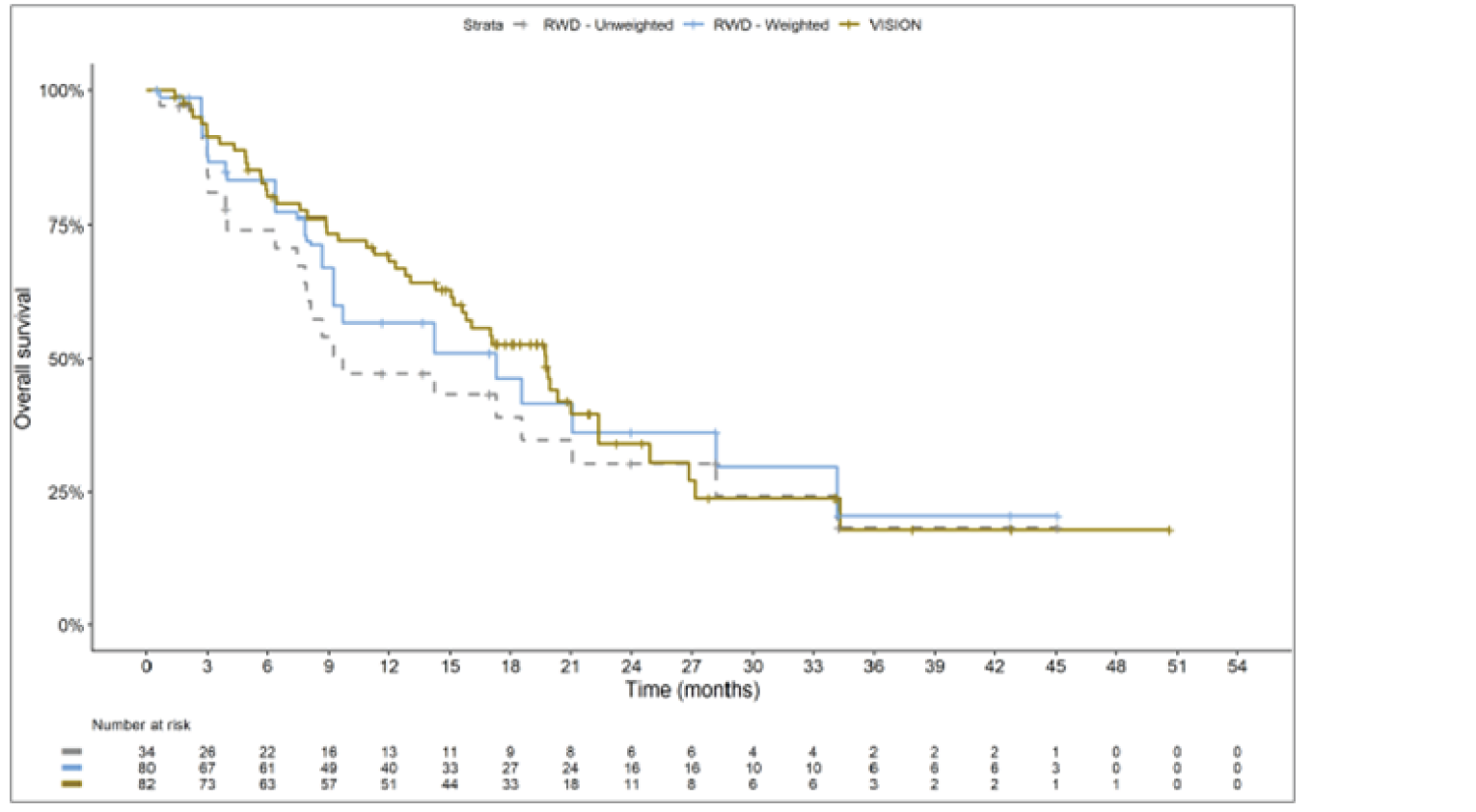
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 30: Subgroup Analysis — Kaplan-Meier Plot of PFS for RWD Indirect Comparison to VISION Using Propensity Scoring; Weighted and Unweighted Chemotherapy Analysis Set, Untreated Patients
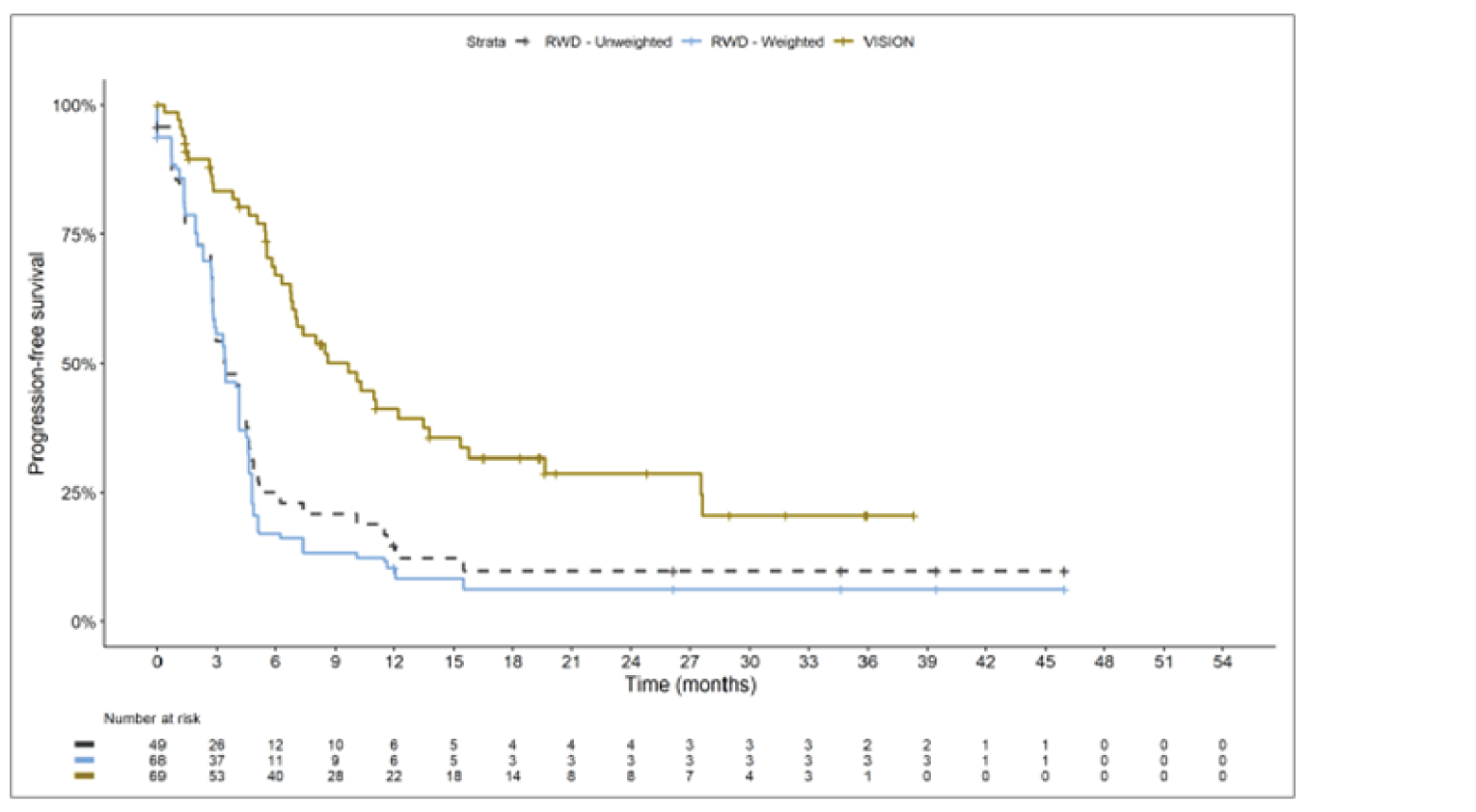
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 31: Subgroup Analysis — Kaplan-Meier Plot of PFS for RWD Indirect Comparison to VISION Using Propensity Scoring; Weighted and Unweighted Chemotherapy Analysis Set, Previously Treated Patients
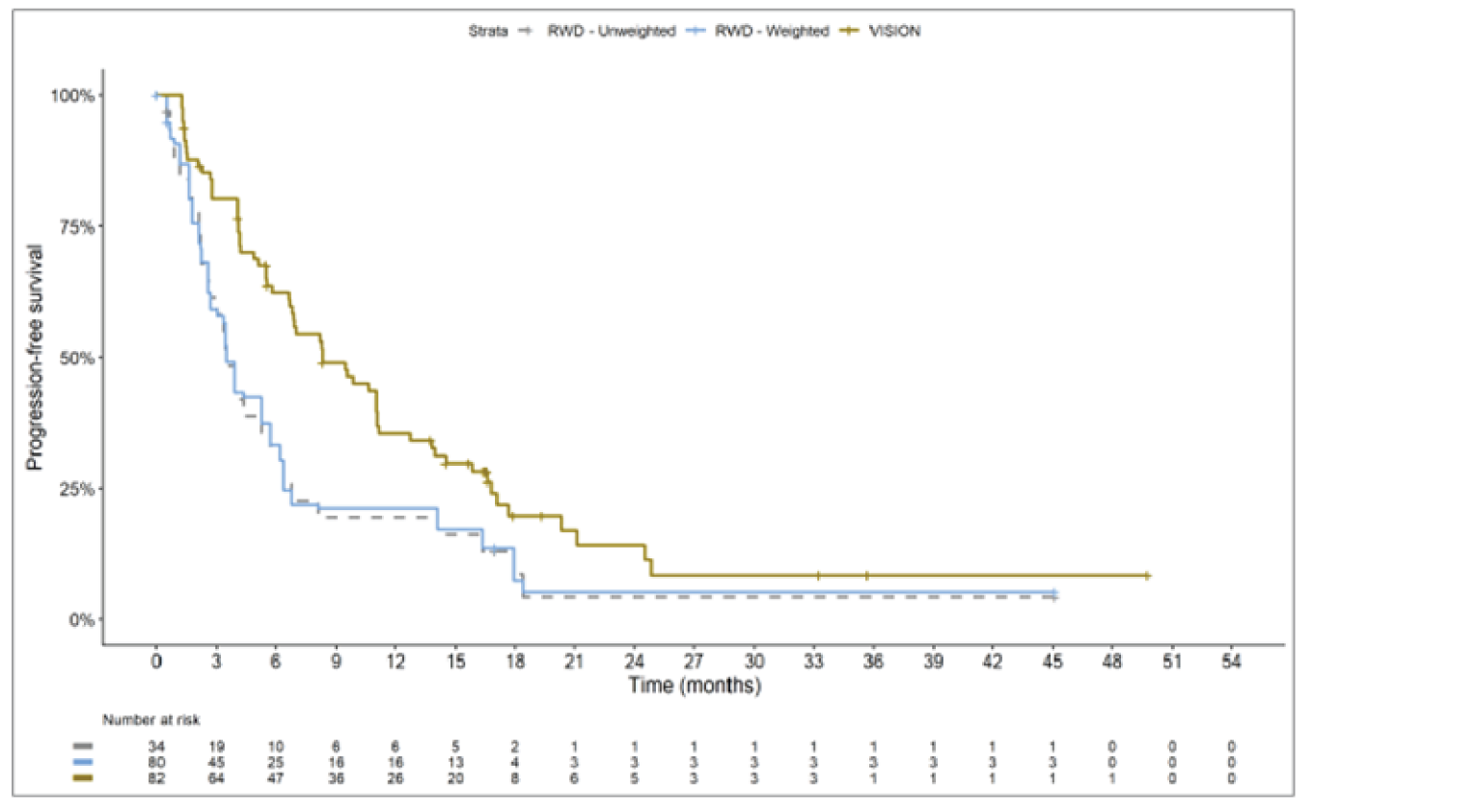
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 32: Subgroup Analysis — Kaplan-Meier Plot of OS for RWD Indirect Comparison to VISION Using Propensity Scoring; Weighted and Unweighted Immunotherapy Analysis Set, Untreated Patients
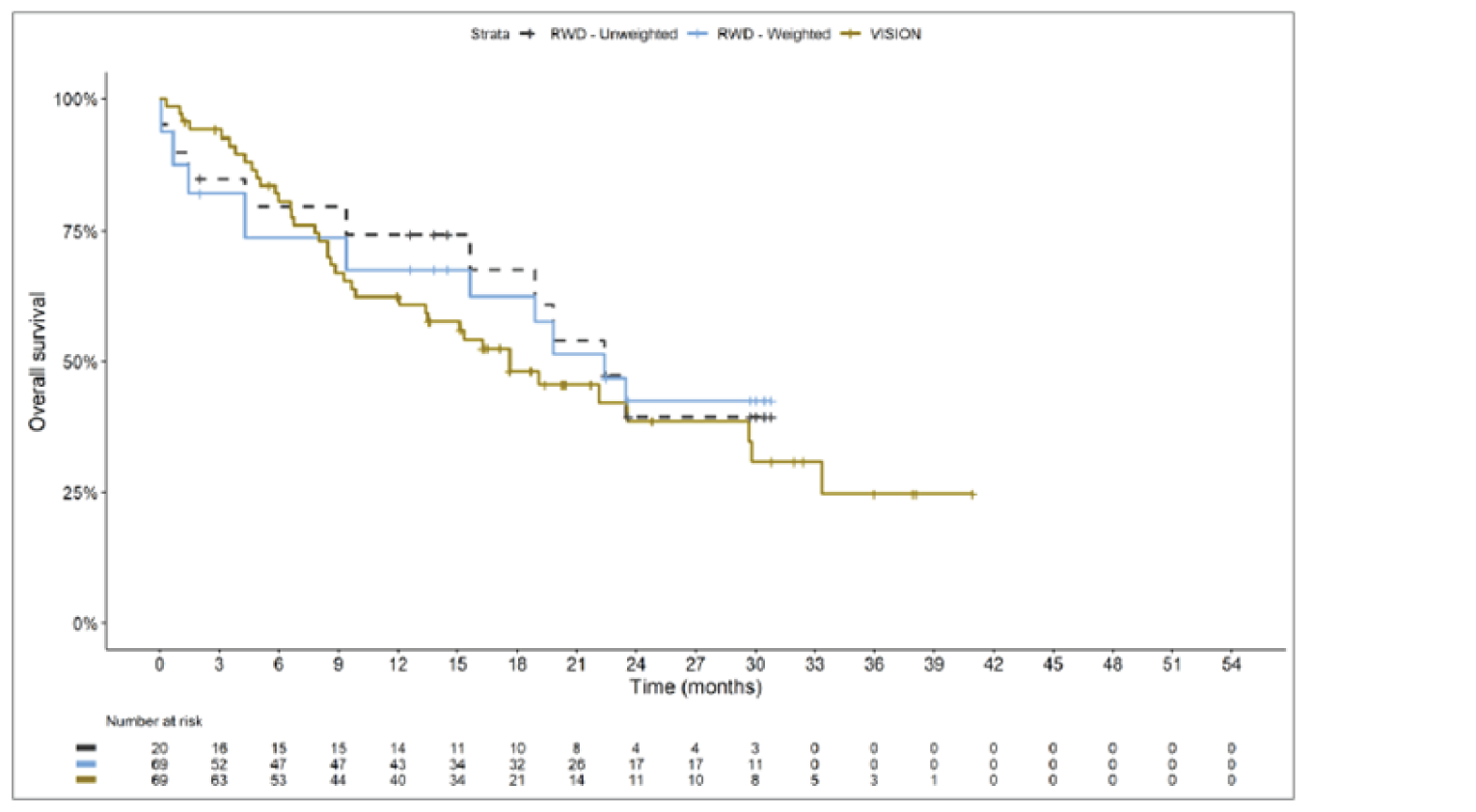
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 33: Subgroup Analysis — Kaplan-Meier Plot of OS for RWD Indirect Comparison to VISION Using Propensity Scoring; Weighted and Unweighted Immunotherapy Analysis Set, Previously Treated Patients
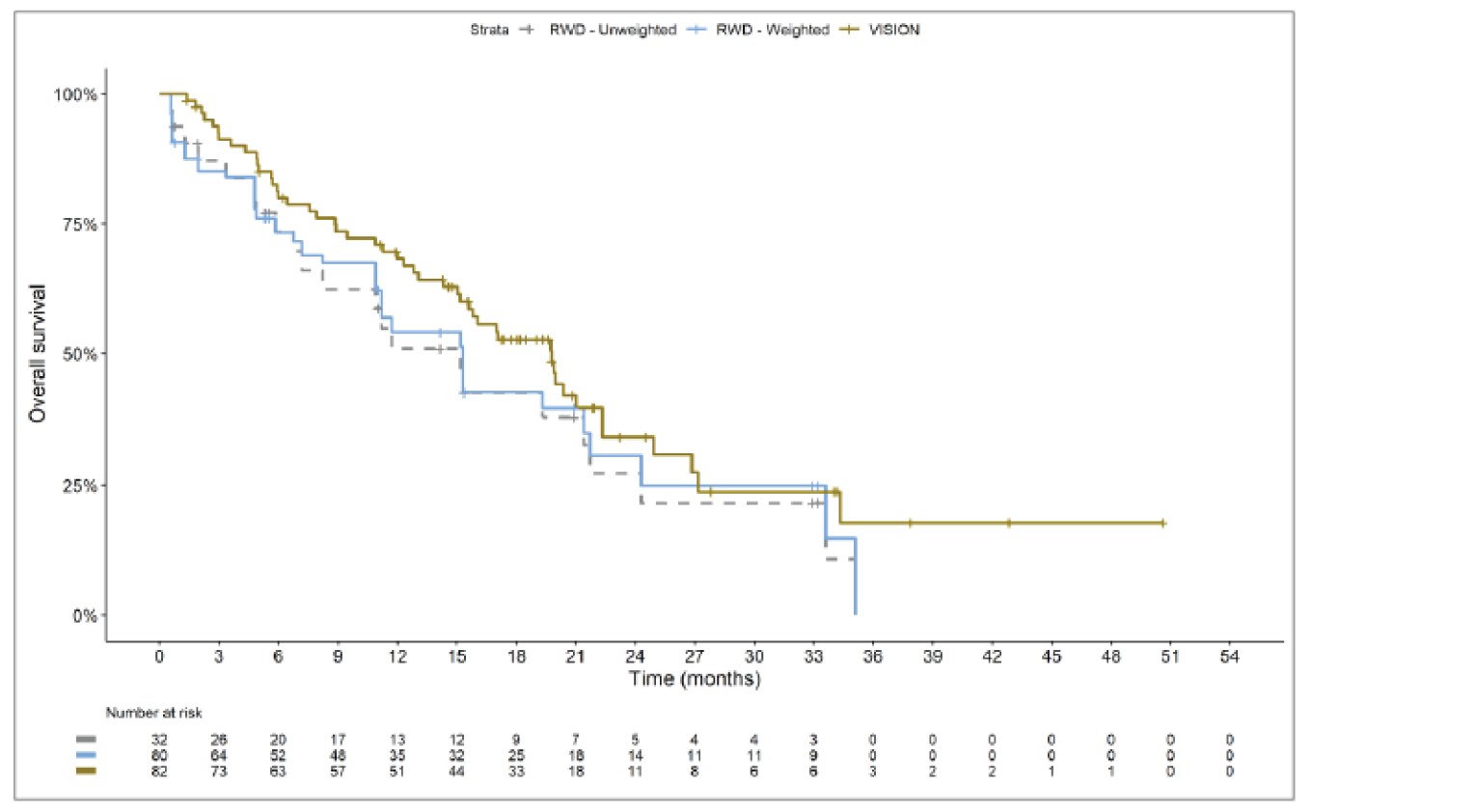
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 34: Subgroup Analysis — Kaplan-Meier Plot of PFS for RWD Indirect Comparison to VISION Using Propensity Scoring; Weighted and Unweighted Immunotherapy Analysis Set, Untreated Patients
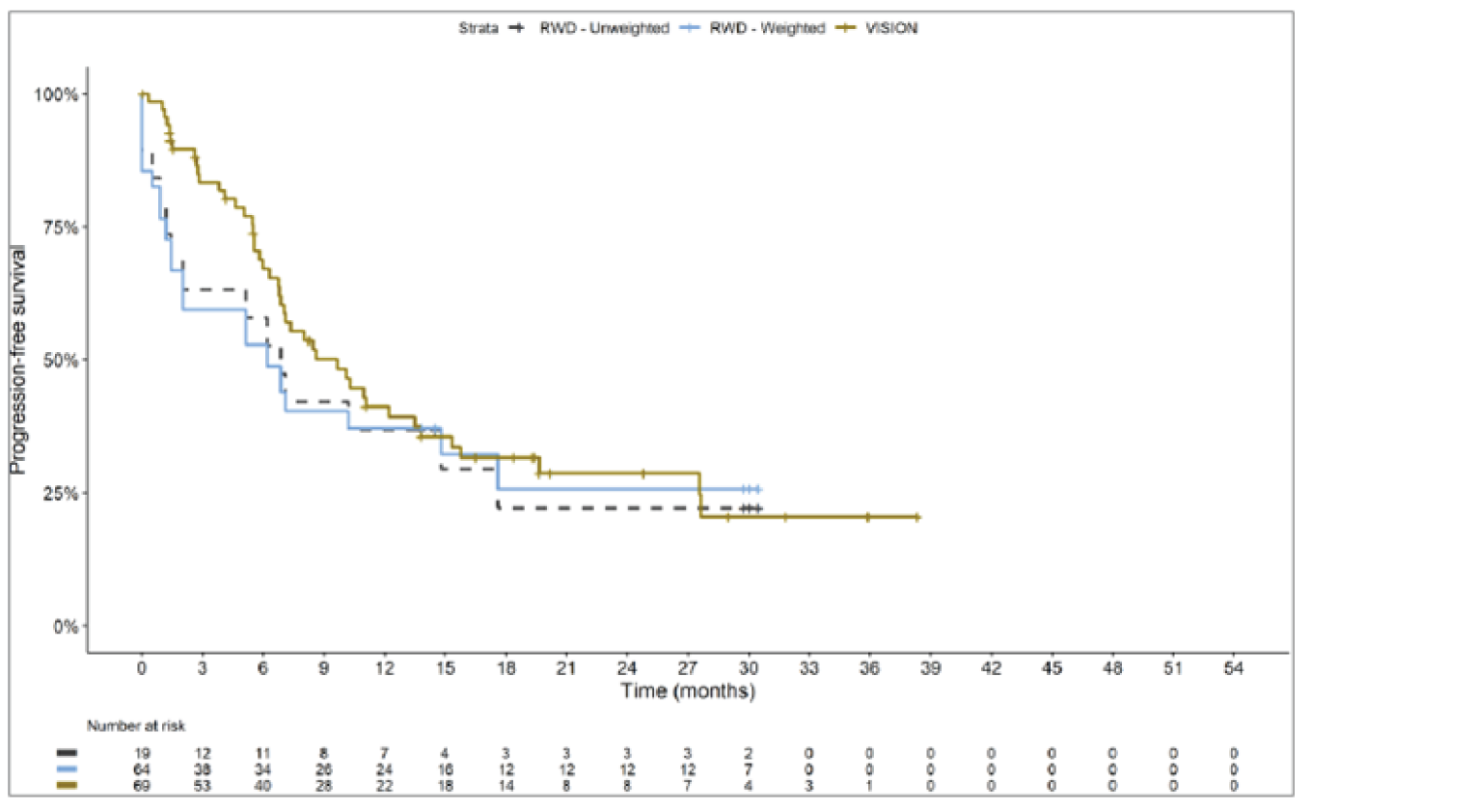
RWD = real-world data.
Source: Sponsor’s technical report.15
Figure 35: Subgroup Analysis — Kaplan-Meier Plot of PFS for RWD Indirect Comparison to VISION Using Propensity Scoring; Weighted and Unweighted Immunotherapy Analysis Set, Previously Treated Patients
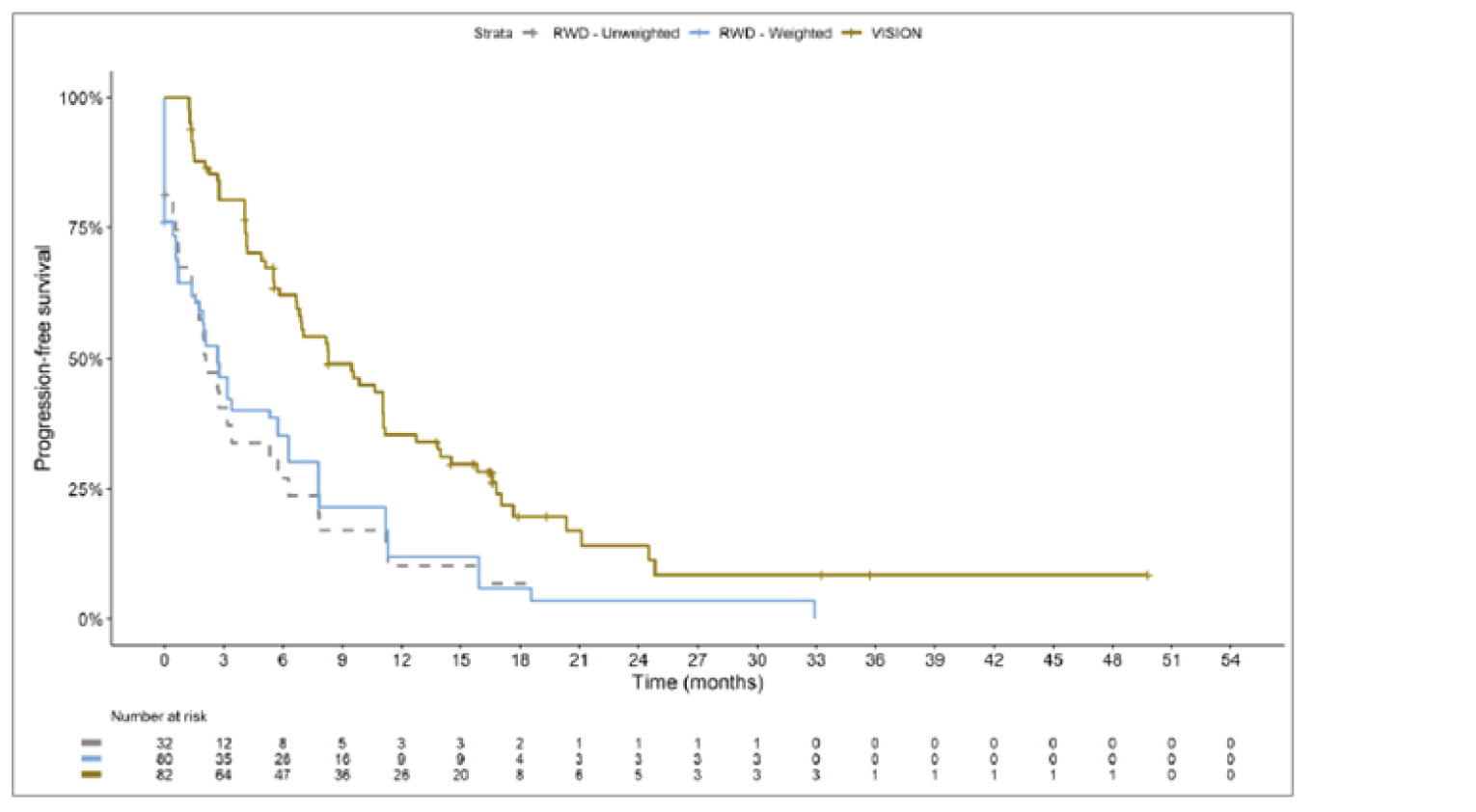
RWD = real-world data.
Source: Sponsor’s technical report.15
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures that were reported in the VISION trial and review their measurement properties (validity, reliability, responsiveness to change, and MID):
European Quality of Life Five Dimension Five Level (EQ-5D-5L) instrument: Visual analogue scale (VAS)
European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30): Global Health Status and Quality of Life (QoL) score
European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13 Module (EORTC QLQ-LC13): coughing, dyspnea, pain symptom scales
Findings
A focused literature search was conducted to identify the psychometric properties and MID of each of the stated outcome measures. The findings about validity, reliability, responsiveness, and MID of each outcome measure are summarized in Table 45.
Interpretation of the reliability and validity metrics were based on the following criteria:
Internal consistency (Cronbach alpha) and test–retest reliability: ≥ 0.7 is considered acceptable.30
Validity – Correlations based on Cohen classification31:
≤ 0.3 = weak
0.3 to ≤ 0.5 = moderate
> 0.5 = strong
Table 45: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about Measurement Properties | MID |
|---|---|---|---|
EQ-5D-5L | EQ-5D-5L is a generic, preference-based HRQoL questionnaire consisting of an index score and VAS score. The index score is based on 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Score ranges from 0 (“dead”) to 1 (“perfect health”) where negative scores represent “worse than dead.” The EQ VAS ranges from 0 (worst health imaginable) to 100 (best health imaginable). | Measurement properties have not been assessed in patients with NSCLC | VAS: MID estimates for patients with lung cancer are 7.5 – 11.5 based on ECOG PS (0.5 SD = 8), 7 - 10 based on FACT-G (0.5 SD = 9).24 |
EORTC QLQ-C30 | The EORTC QLQ-C30 is a standardized, patient self-administered questionnaire for evaluating the quality of life of patients with cancer. Consists of 4 types of scales:
Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores ranging from 1 to 4. For the 2 items that form the global health status and QoL scale, the response format is a 7-point Likert-type scale, with anchors between 1 (very poor) and 7 (excellent). Raw scores from each scale are converted to a 0-100 scale using a linear transformation, with a higher score reflecting better function on the function scale, higher symptom burden on the symptom scales, and better quality of life on global health status and QoL scale. | Validity: Construct validity for the global health status and QoL score has been demonstrated with WHO PS (P < 0.0001) and a standard 6-minute walk test,32 as well as ECOG PS and weight loss pre- and on-treatment (P < 0.001 to P < 0.05).33 Moderate correlation with walking distance > 200 m (r > 0.4) and a correlation (r value not reported) with spirometry were reported.32 Reliability: Global health status and QoL showed acceptable reliability coefficient (alpha > 0.7).32 Reliability coefficient (alpha > 0.7) was acceptable both pre- and during treatment periods.33 Responsiveness: Group differences (improved vs. deteriorated based on ECOG PS) over 28 days between pre- and on-treatment periods showed a statistically difference in global quality of life (P < 0.01) scale. No such difference was identified in patients whose ECOG PS remained unchanged.33 | For patients with NSCLC, the MID for improvement and deterioration for global health status/ QoL are 4-9 and 4, respectively.25 |
EORTC QLQ-LC13 | The EORTC QLQ-LC13 module, which supplements the QLQ-C30, is a self-reported, lung cancer–specific questionnaire with 13 items addressing symptoms associated with lung cancer and its standard treatment:
All items are scored on a 4-point categorical scale ranging from 1 (not at all) to 4 (very much), except for the 1 item on pain medication, which has dichotomous response categories (no or yes). All scale and item scores are linearly transformed to a 0-100 scale, with higher scores representing increased symptom burden. | Validity: Construct validity has been established between pain score and disease type (P < 0.001). Also, based on ECOG PS, construct validity was confirmed in dyspnea, coughing, and pain (P < 0.001) scores.34 Correlation between spirometry result and dyspnea score was found to be weak (r = 0.24). BPI intensity score and QLQ-LC13 pain score were found to be modestly correlated (r > 0.4).32 Reliability: Reliability coefficient (Cronbach alpha) range for dyspnea scores was 0.81-0.83. However, internal consistency was found to be unacceptable for pain scores (alpha = 0.53 – 0.54) when QLQ-LC13 was used alone without QLQ-C30 questionnaire pain items.34 Reliability estimate for dyspnea scale has been confirmed to be acceptable, i.e., alpha = 0.76 in another study.32 Responsiveness: Dyspnea, coughing, and pain scores improved significantly over time between pre-treatment and on-treatment period (P < 0.001 for all except for extra thoracic pain which showed P < 0.05). Responsiveness of chest pain (P < 0.01), dyspnea (P < 0.001) and coughing (P < 0.001) to change in ECOG PS was also noted.34 | Unknown in patients with NSCLC |
BPI = Brief Pain Inventory; ECOG PS = Eastern Cooperative Oncology Group Performance Status; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30; EORTC QLQ-LC13 = European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Lung Cancer 13; EQ-5D-5L = 5-Level EQ-5D; FACT-G = Functional Assessment of Cancer Therapy - General; HRQoL = health-related quality of life; MID = minimal important difference; NSCLC = non–small cell lung cancer; QoL = quality of life; SD = standard deviation; VAS = visual analogue scale; WHO PS = World Health Organization performance status.
5-Level EQ-5D Instrument
The EQ-5D-5L is a generic quality of life instrument developed by the EuroQol Group. It may be applied to a wide range of health conditions and treatments.35 The EQ-5D-5L was developed by the EuroQol Group as an improvement to the EQ-5D 3-level (EQ-5D-3L) instrument, to measure small and medium health changes and reduce ceiling effects. As a generic measure of HRQoL that can capture the net effect of treatment benefits and harms, the EQ-5D-5L provides valuable information from a patient perspective. In addition, the EQ-5D-5L is used in clinical trials to obtain utility weights for economic models.36 The EQ-5D-5L consists of the EQ-5D descriptive system and the EQ VAS. The EQ VAS records the respondent’s self-rated health on a vertical VAS37 where the end points are labelled 0 (“the worst health you can imagine”) and 100 (“the best health you can imagine”). The respondents are asked to mark an X on the point of the VAS that best represents their health on that day. The VAS scores can be summarized and analyzed as continuous data.35,36
The EQ-5D-5L has been extensively validated across countries around the world and in various conditions. However, the psychometric properties of the EQ-5D-5L have not been assessed in patients with NSCLC, therefore its validity, reliability, and responsiveness to change have not been discussed further in this report.
Minimal Important Difference
Pickard et al. (2007)24 conducted a retrospective analysis on 534 cancer patients, including 50 patients with lung cancer, to estimate an MID for the EQ-5D-5L VAS based on anchor-based and distribution-based methods. Based on ECOG PS grade as an anchor, the estimated MID in patients with lung cancer ranged from 7.5 to 11.5 (0.5 SD = 8). The Functional Assessment of Cancer Therapy - General (FACT-G) quintile-based MID for patients with lung cancer ranged from 7 to 10 (0.5 SD = 9).
In the VISION study, the sponsor defined a clinically meaningful deterioration in EQ-5D-5L VAS as a ≥ 10-point decrease from baseline.14,23
Other Considerations and Limitations
Since the EQ-5D-5L is intended to measure HRQoL in the general population, there may be a mismatch between its domains or dimensions and HRQoL in patients with NSCLC that are impacted by treatments and/or the disease. Also, Pickard et al. (2007) used an older version of EQ-5D-3L VAS in their study.24 In discussion with the CADTH review team, however, it was determined to be only a slight difference between the old EQ-5D-3L VAS, which has the numerical scale overlapped on the VAS, and the most recent version of EQ-5D-3L or 5L VAS, where the VAS has been harmonized between the 3L and 5L and the scale has been placed to the right side of the VAS. Therefore, the MID estimate using the old version of EQ-5D-3L VAS was reported as no major discrepancies in interpretation are expected.
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30
Description
The EORTC QLQ-C30 is one of the most commonly used PRO measures in oncology clinical trials.38 It is a multi-dimensional, cancer-specific, evaluative measure of HRQoL. It was designed specifically for the purpose of assessing changes in participants’ HRQoL in clinical trials, in response to treatment.39 The questionnaire consists of 30 questions that are scored to create 5 multi-item functional scales (physical, role, cognitive, emotional, social), 3 multi-item symptom scales (fatigue, pain, nausea and vomiting), 6 single-item symptom scales (dyspnea, insomnia, appetite loss, constipation, diarrhea, financial impact), and a 2-item global health status and quality of life scale. In the VISION trial, data were presented for the global health status and quality of life scale only. It is available in 90 different languages and is intended for use in adult populations only. Version 3.0 of the questionnaire is the most current version and has been in use since December of 1997.39 The sponsors’ study used the most recent version (i.e., version 3).
Scoring
The EORTC QLQ-C30 uses a 1-week recall period in assessing function and symptoms. Most questions have 4 response options (“not at all,” “a little,” “quite a bit,” “very much”), with scores on these items ranging from 1 to 4. For the 2 items from the global health status and quality of life scale, however, the response format is a 7-point Likert-type scale, with anchors between 1 (very poor) and 7 (excellent).
Raw scores for each scale are computed as the average of the items that contribute to a particular scale. This scaling approach is based upon the assumption that it is appropriate to provide equal weighting to each item that comprises a scale. There is also an assumption that, for each item, the interval between response options is equal (for example, the difference in score between “not at all” and “a little” is the same as “a little” and “quite a bit,” at a value of 1 unit). All scales and single-item measures range in score from 0 to 100. Higher scores for the functioning scales and global health status and quality of life denote a better level of functioning/QoL (i.e., a better state of the patient), while higher scores on the symptom scales indicate a higher level of symptom burden (i.e., a worse state of the patient). Each raw scale score is converted to a standardized score that ranges from 0 to 100 using a linear transformation, with a higher score reflecting better function on the function scales, better QoL on the global health status and quality of life scale, and higher symptom burden on the symptom scales (i.e., higher scores simply reflect higher levels of response on that scale). According to the EORTC QLQ-C30’s scoring algorithm, if there are missing items for a scale, the score for the scale can still be computed if there are responses for at least half of the items. In calculating the scale score, the missing items are simply ignored — an approach that assumes that the missing items have values equal to the average of those items for what the respondent completed.40
Validity
Nicklasson et al. (2007)32 conducted a construct validity test with 112 Swedish patients diagnosed with lung cancer or pleural mesothelioma, including 85 (76%) patients with NSCLC, not amenable to curative or life-prolonging treatment. The results were based on a known groups approach with WHO PS and a standard 6-minute walk test, and significant interaction effects were observed with global health status and quality of life (P < 0.0001). In a correlation analysis employing walking distance (> 200m, n = 58) as a continuous variable, a moderate correlation (r > 0.4) with global health status and quality of life was observed. With spirometry, a correlation (r = not reported) with global health status and quality of life was observed such that patients with an FEV1 predicted value <50% (n = 27) scored worse than did patients with an FEV1 predicted value ≥50% (n = 61).
Another group, Aaronson et al. (1993),33 tested construct validity in 305 patients with nonresectable lung cancer (of 287 patients with reported histologic types, 63.1% had NSCLC) undergoing either radiotherapy or chemotherapy from 13 countries including Canada. Based on a known groups approach, patients with better ECOG PS scores at the pre-treatment stage reported significantly higher global health status and quality of life scores (ANOVA: n = 295, P < 0.001 to P < 0.05) and statistically significant group differences as expected according to their ECOG PS while on treatment (ANOVA: n = 265, P < 0.001 to P < 0.05). Similarly, statistically significant group differences were observed in pre-treatment when patients having less weight loss reported better global health status and quality of life scores as expected (ANOVA: n = 295, P < 0.001 to P < 0.05).
Reliability
Nicklasson et al. (2007)32 performed reliability testing in the same population as described in the Validity section. Reliability of the global health status and quality of life scale showed an internal consistency of 0.70 or higher, which is an accepted threshold for group comparisons.
Aaronson et al. (1993)33 tested reliability in the same population as described in the Validity section. The reliability coefficients (Cronbach alpha) for the global health status and quality of life were 0.86 before treatment and 0.89 during treatment.
Responsiveness to Change
According to the Aaronson et al. (1993)33 ANOVA with divided patient samples based on ECOG PS (improved / decrease in score of at least 1 = 13%, unchanged = 57%, deteriorated / increase in score of at least 1 = 30% of patients), between-group differences over time (averaged 28 days, SD = 19 days) were statistically significant in global health status and quality of life scale (P < 0.01). For example, a group of patients (n = 34) whose ECOG PS score improved over time had a mean (SD) pre-treatment global health status score of 53.3 (21.8) and on-treatment global health status score of 62.9 (19.4). Those patients (n = 79) whose ECOG PS score deteriorated over time had a mean (SD) pre-treatment score of 56.2 (25.5) and on-treatment global health status score of 50.5 (25.0). No changes were noted in QLQ-C30 scores among those patients whose PS had remained unchanged.
Minimal Important Difference
Maringwa et al. (2011)25 estimated MIDs based on anchor-based method by pooling data from 2 RCTs on EORTC. Total 812 patients with palliative, locally advanced, and/or metastatic NSCLC that are undergoing treatment were enrolled. As for anchors chosen, physician-rated WHO PS and weight change were used based on their relevance to patients with NSCLC. Effect size of 0.2 SD, 0.5 SD, and threshold of 1 standard error of mean (SEM) of HRQoL scores have been reported as distribution-based MIDs to compare with the anchor-based MIDs.
MID estimates for improvement (i.e., 1 category change in PS, 5% to less than 20% weight gain) in global health status were 9 and 4. The respective MID estimate for deterioration (i.e., 1 category change in PS, 5 - <20% weight loss) was 4. MID estimates based on distribution method were 4 (0.25 SD), 11 (0.5 SD), and 9 (SEM).
In the VISION study, the sponsor defined a clinically meaningful deterioration in global health status and quality of life as a ≥10-point decrease from baseline.14,23
Other Considerations and Limitations
The limitation of MID estimation performed by Maringwa et al. (2011)25 is poor correlations between changes in either anchor (WHO PS or weight) and QLQ-C30. For example, for changes in global health status scores and changes in both anchors, the correlations coefficients range from 0.10 to 0.14 in absolute values. The Spearman rank correlation of at least 0.30 is suggested to be acceptable association.41
European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Lung
Cancer 13
Description
The EORTC QLQ-LC13 is a self-reported, lung cancer–specific questionnaire with 13 items addressing symptoms associated with lung cancer and its standard treatment. This module supplements the QLQ-C30 questionnaire. While the QLQ-C30 questionnaire covers basic components of HRQoL relevant to a wide range of patient populations, the QLQ-LC13 measures specific symptoms and side effects experienced by lung cancer patients receiving non-surgical treatment. When administered together with the QLQ-C30 questionnaire, the QLQ-LC13 assesses disease- and treatment-specific symptoms for lung cancer patients participating in clinical trials.42
The QLQ-LC13 supplementary module comprises both multi-item and single-item measures of lung cancer-associated symptoms (i.e., coughing, hemoptysis, dyspnea, and pain) and side effects from conventional chemotherapy and radiotherapy (i.e., hair loss, neuropathy, sore mouth, and dysphagia). Only dyspnea and pain domains are comprised of multi-items and the rest are single items. All items employ a 1-week time frame. With the exception of the 1 item on pain medication, which has dichotomous response categories (no or yes), all items are scored on a 4-point categorical scale ranging from 1 (not at all) to 4 (very much). For ease of presentation and interpretation, all scale and item scores are linearly transformed to a 0 to 100 scale, with higher scores representing increased symptom levels. Missing items are ignored if at least half of the items are filled per dimension.42
In the VISION study, data were reported for coughing, dyspnea, and chest pain.14,23
Validity
Bergman et al. (1994)34 tested construct validity with 883 patients with non-resectable lung cancer (NSCLC: 62%) receiving either chemotherapy or radiotherapy from 17 countries (Canadian patients: n = 115). Two known groups, namely disease stage (local, locoregional, metastatic) and ECOG PS, were selected. Patients with metastatic disease reported higher levels of pain and more frequent use of pain medication (P < 0.001) compared to patients with local disease. However, stage of disease was not significantly related to coughing or dyspnea. In contrast, ECOG PS divided into 2 levels (0-1 versus 2-4), was related significantly to pre-treatment dyspnea and coughing with elevated symptom score levels found primarily in patients with a poorer PS (P < 0.001). Statistically significant differences in pain scores were also observed as a function of ECOG PS (P < 0.001).
Nicklasson et al.32 conducted another validity study with 112 Swedish patients (NSCLC: n = 85, 76%) diagnosed with lung cancer or pleural mesothelioma not amenable to curative or life-prolonging treatment. The finding showed that correlation between spirometry results and QLQ-LC13 3-item dyspnea scale was weak, although statistically significant (n = 96; r = 0.24; P < 0.05). Furthermore, the convergent validity testing indicated that the Brief Pain Inventory (BPI) intensity subscale correlated modestly (r > 0.4) with the QLQ-LC13 pain items.
Reliability
Based on the study results from Bergman et al. (1994),34 the QLQ-LC13 dyspnea items formed a 3-item scale with internal consistency estimates exceeding the minimum levels (alpha > 0.7) required for group comparisons. When combined with the QLQ-C30 questionnaire item on dyspnea, the reliability of the 3-item scale was improved further, with Cronbach alpha coefficients averaging 0.85 in both studies.
In contrast, the QLQ-LC13 pain items did not form a reliable scale. When combined with the QLQ-C30 questionnaire item(s) on pain, the alpha coefficient improved considerably, indicating that the QLQ-LC13 pain items correlated better with the general pain item(s) in the QLQ-C30 questionnaire than with each other. However, in study 2, the reliability of the combined (QLQ-LC13 and QLQ-C30) 5-item pain scale was inferior to that of the 2-item QLQ-C30 scale alone.
Table 46: Reliability (Cronbach Alpha) of Multi-Item Scales in the Pre-Treatment QLQ-LC13
Content area, Source | No. of items | Scale properties | |||||||
|---|---|---|---|---|---|---|---|---|---|
Study 1 | Study 2 | ||||||||
Mean | SD | alpha | Mean | SD | alpha | ||||
Dyspnea | |||||||||
QLQ-LC13 | 3 | 33.9 | 27.2 | 0.83 | 34.0 | 25.6 | 0.81 | ||
QLQ-LC13 + C36/30 | 4 | 36.0 | 26.6 | 0.86 | 36.2 | 25.2 | 0.85 | ||
Pain | |||||||||
QLQ-LC13 | 3* | 23.0 | 22.0 | 0.53 | 21.7 | 21.7 | 0.54 | ||
QLQ-LC13 + C36 | 4 | 25.6 | 23.0 | 0.71 | -- | ||||
QLQ-LC13 + C30 | 5 | — | 25.0 | 23.7 | 0.80 | ||||
QLQ-C30† | 2 | — | 29.9 | 31.3 | 0.83 | ||||
No. = number; QLQ-C30 = Quality of Life Questionnaire Core 30; QLQ-C36 = Quality of Life Questionnaire Core 36; QLQ-LC13 = Quality of Life Questionnaire Lung Cancer 13; SD = standard deviation
Study 1 (n = 537) is from time period 1987-1989 and study 2 (n = 346) is from time period 1990-1991.
*The item on pain medication was excluded due to its response format.
†Two-item pain scale from the core questionnaire, for comparison.
Source: Bergman et al., The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Multicenter Study Eur J Cancer, 30(5):635-42. Copyright © 1994, with permission from Elsevier.34s
The finding for dyspnea scale has also been confirmed by Nicklasson et al.32 study (n = 112), which showed reliability estimate for dyspnea of alpha = 0.76.
Responsiveness to Change
In the Bergman et al. (1994)34 study, patients completed the QLQ-LC13 questionnaire once prior to the start of treatment (‘pre-treatment’) and following the first course of radiotherapy or second course of chemotherapy (‘on-treatment’). The results showed that dyspnea, coughing, and pain scores changed significantly over time in the expected direction (i.e., declined or improved; P < 0.001 for all 3 domains except for extra thoracic pain item which showed P < 0.05).
When compared between groups divided by ECOG PS (‘improved at least 1 level,’ ‘unchanged,’ ‘deteriorated at least 1 level’), ANOVA revealed significant interaction effects (group differences over time) for dyspnea (P < 0.001), chest pain (P < 0.01) and coughing (P < 0.01).
Minimal Important Difference
No information on an MID for the QLQ-LC13 was found in the literature. In the VISION study, the sponsor defined a clinically meaningful deterioration in the QLQ-LC13 symptom scales as a ≥ 10-point increase from baseline.14,23
Other Considerations and Limitations
The study conducted by Bergman et al. (1994)33 did not specify the time interval between ‘pre-treatment’ or ‘on-treatment’ when testing responsiveness to change. Furthermore, 1 item on perceived medication effectiveness in QLQ-LC13 questionnaire was excluded for further analyses because it caused confusion among patients, potentially due to its positive wording in contrast to the rest of questions. This exclusion reduces the reliability estimate of the multi-item pain scales of the QLQ-LC13. As for responsiveness to change, the results would have been more robust had Bergman et al. (1994) administered the questionnaire either multiple times throughout the study or after few cycles of therapies to capture accumulated effects of treatments. Lastly, an updated lung cancer module, QLQ-LC29, was published in 2017.43 The QLQ-LC29 better reflects the QoL impact by major treatment advances compared to QLQ-LC13, which was published in 1994 and its psychometric properties have been assessed.44
Appendix 5: Additional Information Provided to CADTH for the Reconsideration Process
Note that this appendix has not been copy-edited.
Background
Following the issuance of the draft CADTH pERC recommendation for tepotinib in March 2022, the following additional information was provided to CADTH.
The sponsor provided additional unpublished data from the pivotal VISION study with a data cut-off date of February 1, 2021.
The sponsor provided an additional unanchored MAIC comparing tepotinib to the combination of chemotherapy and immunotherapy. A comparison of tepotinib to a combination of chemotherapy and immunotherapy has been identified as an important gap in the evidence.
These data were not included in the submission to CADTH. After the CADTH recommendation was issued, the sponsor reported that the data only became available after their submission to CADTH.
Updated VISION Study Data
Summary of Results
The sponsor submitted data for the pre-specified cohort A and the confirmatory cohort C with a data cut-off date of February 1, 2021.45
Regarding efficacy outcomes, data were reported for the end points ORR, DOR, PFS, and OS. Results were reported for cohort A, cohort C, and the combined cohort A plus C. Results are presented in Table 47. Median duration of follow-up time at the time of this analysis were not reported. Results were generally consistent with the previous analyses with earlier data cut-off dates.
Regarding harms outcomes, data on treatment-emergent AEs were reported for the combined VISION cohort A plus C (N = 291) as of the February 1, 2021 data cut-off. The data are summarized in Table 48. Updated data on SAEs, withdrawals due to adverse events, and deaths were not reported. The AE data were generally consistent with the previous analyses with earlier data cut-off dates.
Table 47: Summary of Updated Efficacy Data From VISION (February 2021 Data Cut-Off)
Efficacy Outcome | Overall | First Line | Second or Later Line |
|---|---|---|---|
Cohort A (all treated patients), N | 152 | 69 | 83 |
ORR, n (%) [95% CIa] | 71 (46.7) [38.6, 55.0] | 35 (50.7) [38.4, 63.0] | 36 (43.4) [32.5, 54.7] |
Median DORb (95% CIc), months | 15.4 (9.7, 32.7) | 32.7 (7.2, NE) | 12.4 (9.5, 18.5) |
Median PFSb (95% CIc), months | 10.8 (8.3, 12.4) | 10.3 (8.0, 15.3) | 11.0 (8.2, 12.7) |
Patients with an event, n (%) | 89 (58.6) | 38 (55.1) | 51 (61.4) |
Median OSb (95% CIc), months | 19.1 (15.2, 22.1) | 17.6 (9.9, 29.7) | 19.7 (15.0, 22.3) |
Patients with an event, n (%) | 88 (57.9) | 39 (56.5) | 49 (59.0) |
Cohort C (patients with first dose before November 1, 2020), N | 123 | 68 | 55 |
ORR, n (%) [95% CIa, %] | 64 (52.0) [42.8, 61.1] | 39 (57.4) [44.8, 69.3] | 25 (45.5) [32.0, 59.4] |
Median DORb (95% CIc), months | 10.8 (8.3, NE) | NE (8.3, NE) | 10.8 (4.2, NE) |
Median PFSb (95% CIc), months | 10.4 (7.0, NE) | 10.4 (7.0, NE) | 12.1 (6.4, NE) |
Patients with an event, n (%) | 42 (34.1) | 22 (32.4) | 20 (36.4) |
Median OSb (95% CIc), months | NE (14.4, NE) | 14.4 (10.4, NE) | NE (NE, NE) |
Patients with an event, n (%) | 31 (25.2) | 21 (30.9) | 10 (18.2) |
Cohort A + C (all treated patients in Cohort A + Patients in Cohort C with first dose before November 1, 2020), N | 275 | 137 | 138 |
ORR, n (%) [95% CIa] | 135 (49.1) [43.0, 55.2] | 74 (54.0) [45.3, 62.6] | 61 (44.2) [35.8, 52.9] |
Median DORb (95% CIc), months | 13.8 (9.9, 19.4) | 32.7 (9.0, NE) | 11.1 (8.4, 18.5) |
Median PFSb (95% CIc), months | 10.8 (8.5, 12.4) | 10.4 (8.4, 15.3) | 11.0 (8.2, 12.4) |
Patients with an event, n (%) | 131 (47.6) | 60 (43.8) | 71 (51.4) |
Median OSb (95% CIc), months | 19.7 (15.6, 22.1) | 17.6 (13.4, 29.7) | 19.9 (15.8, 22.3) |
Patients with an event, n (%) | 119 (43.3) | 60 (43.8) | 59 (42.8) |
CI = confidence interval; DOR = duration of response; n = number; NE = not estimable; ORR = objective response rate; OS = overall survival; PFS = progression-free survival.
a95% CI calculated using the Clopper-Pearson method.
bKaplan-Meier estimates.
c95% CI calculated using the Brookmeyer and Crowley method.
Source: Additional information provided on April 6, 2022.45
Table 48: Summary of Updated Harms Data From VISION (February 2021 Data Cut-Off) — Safety Analysis Set
Harm | Cohort A + C (N = 291) |
|---|---|
Patients with ≥1 AE, n (%) | 287 (98.6) |
Most common events,a n (%) | |
Peripheral edema | 191 (65.6) |
Nausea | 88 (30.2) |
Notable harms | |
Hepatotoxicity, n (%) | |
ALT increased | 37 (12.7) |
AST increased | 24 (8.2) |
Renal toxicity, n (%) | |
Blood creatinine increased | 76 (26.1) |
Chronic kidney disease | 13 (4.5) |
Renal impairment | 9 (3.1) |
Interstitial lung disease, n (%) | NR |
Pneumonitis, n (%) | NR |
Peripheral edema, n (%) | 191 (65.6) |
AE = adverse event; ALT = alanine aminotransaminase; AST = aspartate aminotransferase; n = number; NR = not reported.
aFrequency ≥ 30%.
Source: Additional information provided on April 6, 2022.45
Critical Appraisal
Reporting of the efficacy and harms data from the analysis with the February 2021 data cut-off date was incomplete. Data were not reported for all efficacy outcomes. Furthermore, median duration of follow-up for the outcomes was not reported.
As previously discussed in this report, the primary limitations of the VISION study are the open-label, single-arm design and descriptive analysis (i.e., no statistical testing). Due to these limitations of the study design, no definitive conclusions can be drawn from the VISION study regarding the efficacy of tepotinib relative to a comparator. The data from the updated analysis with a more recent data cut-off date do not change the conclusions drawn by the CADTH review team.
Indirect Evidence
Description of the ITC
The sponsor provided supplementary ITC analyses for tepotinib versus chemotherapy plus immunotherapy in advanced NSCLC.46 In this ITC, tepotinib used in patients with advanced NSCLC harbouring METex14 skipping mutations (VISION study, subpopulation of patients that were previously untreated in cohort A plus C) was compared to the combination of pembrolizumab + pemetrexed + platinum chemotherapy in patients with wildtype NSCLC (KEYNOTE-189 study) in terms of PFS and OS using an unanchored MAIC.
Per the sponsor, this MAIC was initially undertaken as part of the company response to the National Institute for Health and Care Excellence (NICE) Appraisal Consultation Document for tepotinib after the first committee meeting on January 12, 2022 and provided to NICE on February 22, 2022.
Methods
Study Selection Methods
The authors chose to include the KEYNOTE-189 study, based on the pivotal clinical trial of pembrolizumab with pemetrexed and platinum chemotherapy for untreated, metastatic, non-squamous non–small cell lung cancer.
The authors of the ITC reported that this study was identified by conducting the previous literature review presented in the original submission to CADTH and confirmed by 3 of the sponsor’s clinical experts in the UK in the review process of NICE for tepotinib. However, the systematic literature review16 provided by the sponsor in response to CADTH’s request for additional information did not identify the KEYNOTE-189 study; the sponsor’s systematic literature review was focused on NSCLC harbouring METex14 skipping alterations. Therefore, the methods by which the KEYNOTE-189 study was selected remain unclear.
Analysis Methods
The methods used to extract data from the KEYNOTE-189 study were not reported. An unanchored MAIC was performed because the index trial (VISION) is a single-arm study. The unanchored MAIC compared patients from VISION cohort A plus C the received tepotinib in the first-line setting (previously untreated subpopulation) to the KEYNOTE-189 study.
The MAICs were implemented using the ‘maic’ R package. The following characteristics were selected and adjusted for:
Percentage of patients previously untreated
ECOG (where available i.e., clinical trials)
Age
Sex
Adenocarcinoma
Smoking
Metastatic vs advanced
OS and PFS were analyzed. It was not clear whether PFS by investigator assessment or IRC assessment from the VISION trial was used.
ESS measures were reported, and the authors presented demographics before and after weighting. A limited assessment of heterogeneity was provided as a narrative. The authors did not report any steps taken to address potential heterogeneity.
Results
Summary of Included Studies
The authors of the sponsor-submitted ITC provided a limited description of the comparator study included in the unanchored MAIC.46 KEYNOTE-18947 was a phase III RCT comparing the first-line treatment of pembrolizumab in combination with pemetrexed and platinum-based chemotherapy versus pemetrexed with platinum in patients with advanced NSCLC. The publication selected for data extraction for the MAIC was Rodriguez-Abreu et al. (2021),47 which reported the protocol-specified final analysis from KEYNOTE-189. Eligible patients were randomized 2:1 to receive pembrolizumab 200 mg (n = 410) or placebo (n = 206) every 3 weeks (for up to 35 cycles, ∼2 years) plus 4 cycles of pemetrexed (500 mg/m2) and investigators’ choice of cisplatin (75 mg/m2) or carboplatin (area under the curve 5 mg/min/mL) every 3 weeks, followed by pemetrexed until progression. Patients assigned to placebo plus pemetrexed–platinum could cross over to pembrolizumab upon progression if eligibility criteria were met. The primary end points were OS and PFS. The authors of the ITC did not report a quality assessment of the KEYNOTE-189 study.
Baseline characteristics and the results of the reweighting of the VISION data to match the KEYNOTE-189 study are provided in Table 49. After weighting, the sample size of the tepotinib group decreased from 148 patients to an ESS of 38.7 patients. The authors of the ITC noted that there were large differences between the patient populations of VISION and KEYNOTE-189 regarding the characteristics that were analyzed. In addition, there are also differences in MET status, which was not measured in KEYNOTE-189.
Table 49: Patient Characteristics – VISION Cohort A + C (Previously Untreated Subpopulation) Before and After MAIC to KEYNOTE-189
Characteristic | VISION Cohort A + C (Previously Untreated) Unweighted | VISION Cohort A + C (Previously Untreated) Weighted | KEYNOTE-189 |
|---|---|---|---|
n/ESS | 148 | 38.7 | 410 |
Previously treated, % | 0.0 | 0.0 | 0.0 |
Age | |||
Mean (SD) age, years | 73.7 (NR) | 68.0 (NR) | NR (NR) |
Median (IQR) age, years | 73.8 (NR) | 65.8 (NR) | 65.0 (NR) |
Age >65 years, % | 81.8 | 50.0 | NR |
Male, % | 50.0 | 62.0 | 62.0 |
ECOG 0, % | 27.7 | 45.1 | 45.1 |
Smoking, % | 54.1 | 88.3 | 88.3 |
Adenocarcinoma, % | 79.1 | 96.1 | 96.1 |
Metastatic/stage 4 disease, % | 94.6 | 99.5 | 99.5 |
ESS = effective sample size; ECOG = Eastern Cooperative Oncology Group; n = number; NR = not reported.
Source: Supplementary MAIC Report48
Results of the MAIC are presented in Table 50. The authors also provided a figure depicting the Kaplan-Meier curves for both OS and PFS (Figure 36). The 95% CIs for the Cox proportional HRs included the null and there were no statistically significant differences between groups for both PFS and OS.
Table 50: PFS and OS Results From MAIC Comparing VISION Cohort A + C (Previously Untreated Subpopulation) to KEYNOTE-189
Outcome | VISION Cohort A + C (Previously Untreated) Unweighted | VISION Cohort A + C (Previously Untreated) Weighted | KEYNOTE-189 |
|---|---|---|---|
n/ESS | 148 | 38.7 | 410 |
PFS | |||
Median PFS (95% CI), months | 8.6 (7.1, 12.4) | 13.5 (10.1, NE) | 9.2 (8.4, 10.9) |
24-month RMST | 11.9 | 14.6 | 11.9 |
Cox PH (95% CI) | 0.99 (0.77, 1.28) | 0.67 (0.42, 1.07) | — |
P value | 0.94 | 0.091 | — |
OS | |||
Median OS (95% CI), months | 17.6 (13.5, 29.8) | 23.6 (22.1, NE) | 22.3 (19.9, 25.1) |
24-month RMST | 15.8 | 19.4 | 17.3 |
Cox PH (95% CI) | 1.26 (0.94, 1.68) | 0.71 (0.39, 1.3) | — |
P value | 0.118 | 0.269 | — |
CI = confidence interval; ESS = effective sample size; n = number; NE = not estimable; NR = not reported; OS = overall survival; PH = proportional hazard; PFS = progression-free survival; RMST = restricted mean survival time.
Source: Supplementary MAIC Report.48
Figure 36: Kaplan-Meier Plots of PFS and OS for the MAIC-Weighted VISION Cohort A + C (Previously Untreated Subpopulation) Compared to KEYNOTE-189
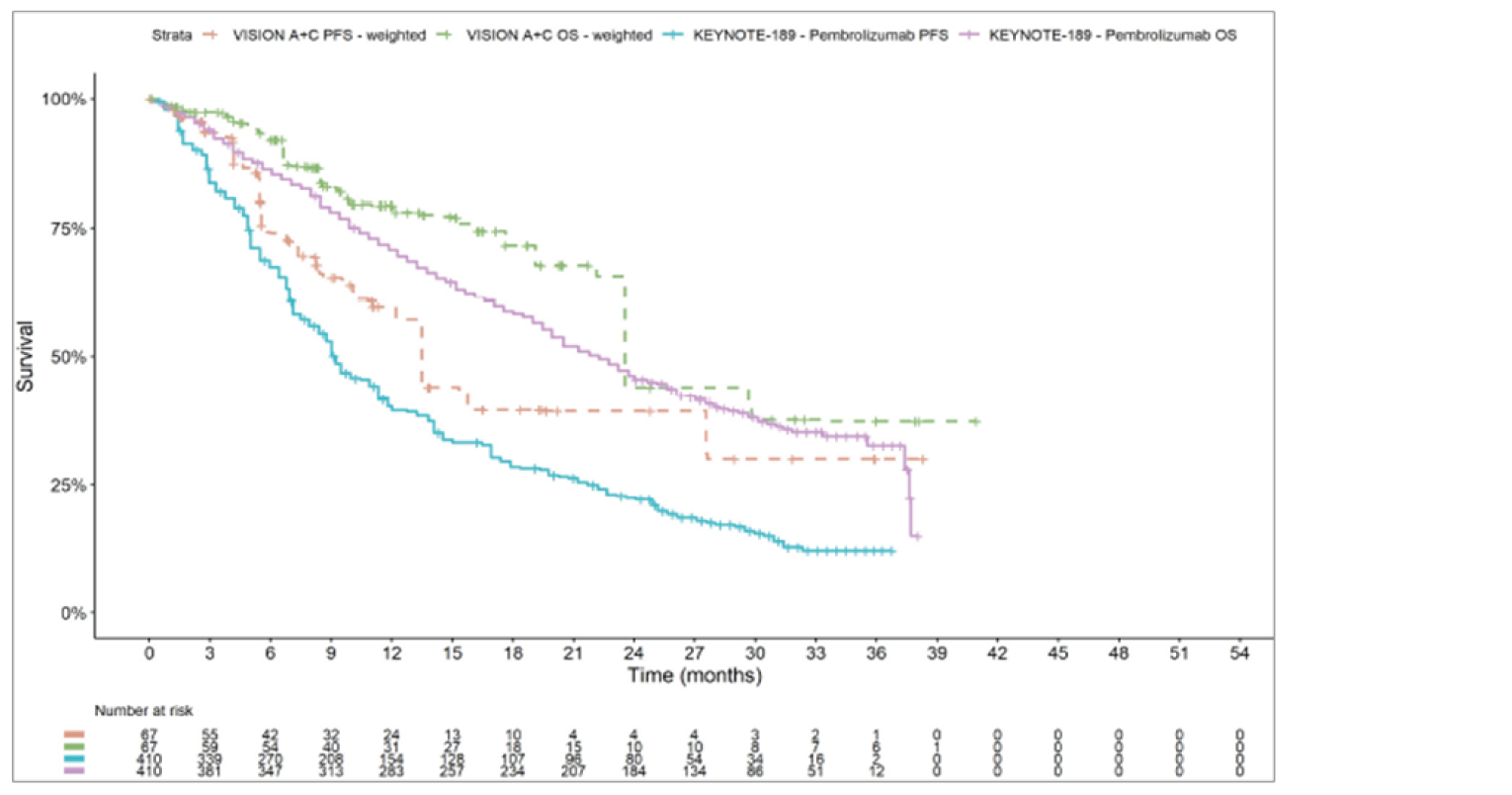
OS = overall survival; PFS = progression-free survival.
Source: Supplementary MAIC Report.46
Critical Appraisal
The MAIC assessed OS and PFS, which were identified as important outcomes by the clinical experts, clinician groups, and patient groups that provided input on this review. The MAIC was conducted to address an important evidence gap identified by the CADTH pERC in the draft recommendation. However, there are methodological limitations, heterogeneity between studies, and thus significant uncertainty in the results.
An important limitation of this supplemental ITC is that the comparison is between patients with wild type NSCLC (i.e., did not have a known driver mutation) and patients with advanced NSCLC harbouring METex14 skipping alterations. The METex14 skipping alteration status of patients enrolled in the KEYNOTE-189 study is unknown, and the authors of the MAIC noted that it is likely the vast majority of patients enrolled in KEYNOTE-189 did not have the METex14 skipping alteration because it is a rare mutation in advanced NSCLC. In addition, the MAIC used only previously untreated patients from VISION cohort A plus C (i.e., tepotinib used in the first-line setting).
In the comparisons, a large reduction in ESS was observed, which suggests there was likely significant heterogeneity between the VISION study and comparator study. Moreover, only a limited number of characteristics were included in the matching process and residual confounding between the groups is expected. The results for comparisons with major reductions of ESS indicates that the weights are highly variable due to a lack of population overlap, and that the resulting estimate may not be reliable.
There were substantial methodological limitations of the unanchored MAIC. First, a MAIC can only adjust for heterogeneity that is directly related to differences in baseline patient characteristics. Other sources of heterogeneity, such as those related to differences in study design, definitions of study outcomes, or changes in the management or support of patients over time, cannot be adjusted for. The VISION trial was a single-arm phase II trial whereas the KEYNOTE-189 study was a phase III, placebo-controlled RCT. In the VISION study, OS was defined as the time from first trial treatment administration to the date of death and PFS was defined as the PFS time was defined as the time from the first administration to the date of the first documentation of PD or death due to any cause. It is unclear whether PFS by IRC assessment or PFS by investigator assessment data from the VISION study were used in this MAIC. In the KEYNOTE-189 study, OS was defined as the time from randomization to death from any cause and PFS was defined as time from randomization to disease progression, as assessed by blinded, independent central radiologic review, or death from any cause, whichever occurred first.49 Patients in the placebo-combination group in whom disease progression was verified by blinded, independent central radiologic review were eligible to cross over to receive pembrolizumab monotherapy. Differences in study design and outcome definitions could not be accounted for in the MAIC, thus these differences create uncertainty in the results.
Furthermore, an unanchored indirect comparison using MAIC methods will only provide an unbiased comparison if all prognostic and effect-modifying factors are included in the weighting process. The variables included in the unanchored MAIC do not reflect all important effect modifiers and prognostic factors. Exclusion of important confounders in the matching introduces bias and creates substantial uncertainty in the results. Unanchored forms of population-adjusted indirect comparisons make the much stronger assumption of “conditional constancy of absolute effects.” This means that the absolute treatment effects are assumed constant at any given level of the effect modifiers and prognostic variables, and all effect modifiers and prognostic variables are required to be known. This assumption is unlikely to have been met in this unanchored MAIC therefore no conclusions can be made from the data.
In addition, the methods and findings of the MAIC lacked important details, which creates further uncertainty in the results. The sponsor submitted a systematic literature review that did not identify the KEYNOTE-189 study so it is unclear how this study was identified and selected for inclusion in the MAIC. There may be bias in the study selection methods.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
EQ-5D-5L
5-Level EQ-5D
ICER
incremental cost-effectiveness ratio
ITC
indirect treatment comparison
LY
life-year
METex14
MET exon 14
NGS
next-generation sequencing
NSCLC
non–small cell lung cancer
OS
overall survival
PD
progressed disease
PDC
platinum doublet chemotherapy
PFS
progression-free survival
QALY
quality-adjusted life-year
RWE
real-world evidence
TTNTD
time to next treatment or death
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Tepotinib (Tepmetko), oral tablets, 225 mg |
Submitted price | Tepotinib, 225 mg, tablet: $153.96 |
Indication | For the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring MET tyrosine kinase receptor exon 14 skipping alterations |
Health Canada approval status | NOC/c |
Health Canada review pathway | Expedited review (Project ORBIS) |
NOC date | May 27, 2021 |
Reimbursement request | For the treatment of adult patients with advanced NSCLC harbouring MET exon 14 skipping alterations |
Sponsor | EMD Serono, a division of EMD Inc., Canada |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; NOC/c = Notice of Compliance with Conditions; NSCLC = non–small cell lung cancer.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | CUA PSM |
Target population(s) | Adult patients with locally advanced unresectable or metastatic NSCLC harbouring MET tyrosine kinase receptor exon 14 skipping alterations |
Treatment | Tepotinib |
Comparators | Line-agnostic population: Immunotherapy (pembrolizumab, nivolumab, atezolizumab) First-line (1L) population: Immunotherapy + PDC (pembrolizumab + pemetrexed + carboplatin/cisplatin) 2L+ population: Chemotherapy (pemetrexed + carboplatin/cisplatin, docetaxel) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs; LYs |
Time horizon | Lifetime (10 years) |
Key data source |
|
Submitted results | Three sponsor base cases: Line-agnostic population: Tepotinib produced more QALYs and was less costly (i.e., dominant) than immunotherapy 1L population: Tepotinib produced fewer QALYs and was less costly than immunotherapy + PDC. The ICER for immunotherapy + PDC vs. tepotinib was $297,422 per QALY (incremental costs: $199,719; incremental QALYs: 0.6715) 2L+ population: ICER for tepotinib vs. chemotherapy was $370,130 per QALY (incremental costs: $67,476; incremental QALYs: 0.1823) |
Key limitations |
|
CADTH reanalysis results |
|
1L = first line; 2L+ = second or later line; CUA = cost-utility analysis; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; METex14 = MET exon 14; NSCLC = non–small cell lung cancer; PDC = platinum doublet chemotherapy; PSM = partitioned survival model; QALY = quality-adjusted life-year; vs. = versus.
Conclusions
Due to the absence of direct or reliable indirect evidence on the relative benefit of tepotinib in comparison to currently funded alternatives with regard to progression-free survival (PFS) and overall survival (OS), the cost-effectiveness of tepotinib is associated with substantial uncertainty. No definitive conclusions could be drawn on the efficacy of tepotinib compared to chemotherapy or immunotherapy with or without platinum doublet chemotherapy (PDC) in patients with advanced non–small cell lung cancer (NSCLC) harbouring MET exon 14 (METex14) skipping alterations.
The considerable uncertainty pertaining to the clinical effectiveness evidence prevents a reliable estimation of the cost-effectiveness of tepotinib relative to current treatment alternatives. As a result, CADTH could not determine a base-case estimate. CADTH undertook an exploratory reanalysis that focused on cost-effectiveness in the first-line and second- and later line settings separately as well as using consistent methodology to estimate treatment duration; assuming pembrolizumab weight-based dosing with vial sharing; revising subsequent treatment assumptions, including and correcting METex14 testing costs; revising chemotherapy adverse event (AE) management costs; and deriving an estimate versus immunotherapy in the first-line population. The sponsor’s clinical efficacy assumptions were retained in exploratory analyses, and a scenario analysis was performed assuming equal efficacy, with regard to PFS and OS, between tepotinib and comparators in the first-line setting.
In the first-line CADTH exploratory reanalysis, tepotinib was dominated (i.e., more costly and less effective) by immunotherapy monotherapy, regardless of whether METex14 testing costs were included. In a further exploratory reanalysis versus immunotherapy plus PDC, tepotinib was less costly and less effective. This is based on uncertain clinical evidence provided by the sponsor which shows tepotinib producing worse outcomes than immunotherapy. In the second- or later-line CADTH exploratory reanalysis, tepotinib was more costly and produced more quality-adjusted life-years (QALYs) versus chemotherapy, with an incremental cost-effectiveness ratio (ICER) of $836,523 per QALY. When excluding METex14 testing costs ($46,630 to identify each eligible patient), the ICER was $551,240 per QALY. Since the true testing costs are uncertain, the ICER is expected to be between this upper and lower limit.
Overall, testing costs, relative clinical benefit, and place in therapy are key considerations when determining the cost-effectiveness of tepotinib. Due to the rarity of METex14, many individuals need to be tested to identify a single patient eligible for treatment with tepotinib. In the first-line setting, based on public list prices, tepotinib is a lower-cost alternative to pembrolizumab plus PDC, but more expensive than pembrolizumab monotherapy. If tepotinib is considered equally effective as pembrolizumab; is priced no more than pembrolizumab monotherapy; and if plans incur no additional testing costs due to implementing METex14 testing, then it may represent a cost-effective alternative at a threshold of $50,000 per QALY. If incremental testing costs are incurred (i.e., if testing for METex14 is not routine and jurisdictions need to implement it), then price reductions up to 38% will be required to compensate for these additional testing costs. In the second- or later-line setting, if no testing costs are incurred, price reductions of more than 70% are needed; if testing costs are incurred, then cost-effectiveness may not be achieved even with 100% price reductions. Since the true testing costs are uncertain, the required discount is expected to be between these upper and lower limits.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission).
CADTH received input from 2 patient advocacy groups: the Lung Health Foundation and Lung Cancer Canada. The Lung Health Foundation collected information from an online survey with 13 patients and 1 caregiver, and phone interviews with 2 patients. Lung Cancer Canada collected data from phone and video interviews with 4 patients and 1 caregiver. Patients’ experiences with lung cancer varied from no symptoms to having the disease negatively impact their ability to perform activities of daily living. Patients indicated that they want treatments that stop or slow the progression of disease, prolong life, improve symptoms and quality of life, and have minimal side effects. Patients reported that they struggled to navigate the health care system and to gain access to biomarker testing for MET mutations in Canada.
CADTH received input from 2 clinical specialists with expertise in the diagnosis and management of NSCLC. The clinical experts reported that retrospective studies have indicated patients with METex14 alterations have a poor prognosis, and there appears to be less benefit with immunotherapy in these patients. The clinical experts indicated that the goals of treatment in locally advanced (not amenable to curative treatment) or metastatic NSCLC are to improve OS, PFS, and response rate, as well as maintain quality of life. In addition, the clinical experts thought that new treatment options should minimize AEs. The clinical experts indicated that tepotinib would preferentially be used in the first-line setting for patients with METex14 skipping alterations because it is a targeted therapy. If patients received tepotinib or another MET receptor tyrosine kinase inhibitor as first-line treatment, later lines of therapy would consist of chemotherapy, immunotherapy, or chemotherapy plus immunotherapy, based on established provincial funding algorithms. If tepotinib was not used as first-line therapy, it would be used as second- or later-line therapy. Patients with METex14 mutated NSCLC would most commonly be identified by next-generation sequencing (NGS) or panel testing. The clinical experts indicated that treatment with tepotinib would be discontinued if a patient experienced disease progression or intolerable treatment-related side effects. A clinical expert noted that, although the VISION trial is non-randomized, the efficacy end points seem to compare favourably to historical cohorts.
CADTH received input from 3 clinician groups consisting of a total of 21 clinicians: Northeast Cancer Centre – Thoracic Cancer Clinicians, Ontario Health – Cancer Care Ontario’s Lung and Thoracic Cancers Drug Advisory Committee, and Lung Cancer Canada’s Medical Advisory Committee. The clinician groups generally agreed with the input provided by the clinical experts consulted by CADTH. The clinician groups indicated that there is an unmet need for targeted therapy and improved outcomes in patients with advanced NSCLC harbouring METex14 skipping alterations. They thought that tepotinib would be preferentially offered as first-line therapy and offered as a subsequent therapy to those who had already received other treatments.
The drug programs noted that, in patients with advanced NSCLC with driver mutations (e.g., EGFR, ALK, ROS, BRAF) who receive targeted treatment in the first-line setting, chemotherapy is required before accessing immunotherapy, in line with previous CADTH pan-Canadian Oncology Drug Review Expert Review Committee (pERC) recommendations. The drug programs indicated that jurisdictions would use the same sequencing principles for subsequent therapies used after tepotinib, regardless of programmed death-ligand 1 tumour proportion score. The drug programs noted that tepotinib may change place in therapy of drugs reimbursed in subsequent lines. Last, the drug programs noted that METex14 skipping alteration testing may not be routinely available in some jurisdictions and would need to be implemented.
Several of these concerns were addressed in the sponsor’s model:
OS, PFS, and quality of life (including disutilities due to AEs) were incorporated into the model.
Costs and disutilities related to AEs were included.
In addition, CADTH addressed some of these concerns as follows:
The costs of implementing METex14 testing costs were considered in CADTH’s reanalysis.
Potential changes to the funding algorithm were considered in the budget impact analysis.
A scenario analysis was performed, assuming equal efficacy between tepotinib and immunotherapy with or without PDC in the first-line population, rather than the sponsor’s assumption that tepotinib is clinically inferior.
Economic Review
The current review is for tepotinib (Tepmetko) for adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping alterations.1
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of tepotinib versus 3 treatment comparators in 3 populations stratified by line of therapy (line agnostic, first line, second or later line).1 The model population consisted of adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping alterations. This population was aligned with the Health Canada indication and differs slightly from the reimbursement request (Table 1). Immunotherapy was modelled as the line-agnostic comparator; this treatment group included a mix of pembrolizumab, nivolumab, and atezolizumab. Immunotherapy plus PDC was assumed to be pembrolizumab plus pemetrexed plus carboplatin or cisplatin and was modelled as the first-line comparator. Finally, chemotherapy was modelled as the second- or later-line comparator, which included a mix of pemetrexed plus carboplatin or cisplatin and docetaxel.
The sponsor’s assumed dosage regimen for tepotinib is 450 mg administered orally once daily.1 Treatment with tepotinib should continue until disease progression or unacceptable toxicity. At the sponsor’s submitted price of $9,237.60 per package of 60 tepotinib tablets ($153.96 per tablet), the total drug acquisition cost of each monthly treatment cycle in the model was $9,340.24 for 60.6 tablets. Patients were assumed to be treated at a relative dose intensity of 100%, with full drug wastage and no vial sharing. The weighted average costs per monthly treatment cycle of immunotherapy, immunotherapy plus PDC, and chemotherapy were $11,609, $19,861, and $3,805, respectively. Details for the modelled treatment costs of all comparators are summarized in Table 10, Appendix 3.
The clinical outcomes of interest were QALYs and life-years (LYs). The economic analysis was undertaken over a lifetime time horizon (10 years), from the perspective of the publicly funded health care payer. Discounting of 1.5% per annum was applied to both costs and outcomes.
Model Structure
A partitioned survival model was developed in Microsoft Excel.1 The model comprised 3 health states, characterized by PFS, progressed disease (PD), and death (Figure 1, Appendix 3). The modelled cycle length was 1 month. The proportion of patients who were progression-free, experienced PD, or were dead at any time over the model horizon was derived from extrapolations of PFS and OS.
All patients entered the model in the PFS health state. Patients could then move to the PD state and then to the death state, or directly from the PFS state to the death state. The proportion of patients with PD was estimated as the difference between the OS and PFS curves. Treatment duration for tepotinib was modelled using extrapolated time-to-next-treatment-or-death (TTNTD) curves from VISION, while treatment duration for comparators was assumed to be equal to extrapolated PFS curves, as TTNTD data were not available.
Model Inputs
The mean population characteristics modelled for patient age (75 years for first line; 72 years for second or later line), weight (65.91 kg), and body surface area (1.73 m2) were aligned with the VISION trial.2
Clinical parameters were obtained primarily from data sources assessing treatments for adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping alterations. For comparisons with immunotherapy (line-agnostic population) and chemotherapy (second- or later-line population), clinical parameters were obtained from VISION and a real-world evidence (RWE) cohort derived from 4 observational studies across North America and Europe. Propensity score weighting was used to balance key patient characteristics.3 To account for missing PFS data (only 31% recorded) in the pooled RWE cohort, TTNTD data were used as a proxy. The relative efficacy of immunotherapy plus PDC (first-line population) was estimated using a naïve indirect treatment comparison (ITC), in which OS and PFS hazard ratios of pembrolizumab plus chemotherapy versus chemotherapy from the subgroup of patients 65 years and older of the KEYNOTE-189 trial were applied to the chemotherapy arm of the pooled RWE cohort.4
Extrapolations for OS, PFS (tepotinib), PFS and TTNTD (immunotherapy and chemotherapy) were selected based on goodness of fit statistics (Akaike information criterion or Bayesian information criterion), expert opinion, and visual fit. Models were independently fitted to data from each treatment group. For the tepotinib arm, PFS and OS were each modelled using the exponential function. For the immunotherapy arm, PFS and OS were modelled using the gamma function. For the chemotherapy arm, PFS and OS were modelled using the log-normal and gamma functions, respectively. Background mortality adjustments were applied such that the hazard of death for patients was not lower than the hazard of death of the general population.
Five-level EQ-5D (EQ-5D-5L) data were collected in the VISION trial.5 The sponsor calculated utility values for the intention-to-treat population as the EQ-5D time-trade-off index score using Canadian EQ-5D-5L tariffs.6 In the base case, utility values were based on health state (i.e., PFS versus PD), independent of treatment. AE disutility values were primarily drawn from published literature on advanced NSCLC.7,8
The model included costs related to drug acquisition, subsequent therapy, drug administration, AEs, and health state resource use. Drug acquisition costs for the comparator treatments were drawn from CADTH Economic Guidance Reports.9-12 The second-line subsequent treatment distribution was obtained from VISION2 for tepotinib, and from treatment guidelines and clinical expert opinion for comparators. The third-line subsequent treatment distribution was based on clinical expert opinion. Drug administration costs and health state resource use costs were drawn from Ontario’s Schedule of Benefits and published sources.13-15 The sponsor included treatment-related AEs (grades 3 or higher) experienced by at least 5% of patients, with incidence rates were based on the VISION trial and other published sources.16-18 AE management costs were derived from the Canadian Institute for Health Information Patient Cost Estimator.19 Health state resource use estimates for disease management were based on a prior UK National Institute for Health and Care Excellence technology appraisal in NSCLC.20 The cost of METex14 testing was not included in the reference case analysis.
Evaluation of the first-line and second- or later-line populations used efficacy data and cost and resource use estimates specific to the relevant line of therapy. Evaluation of the line-agnostic population used line-agnostic efficacy data for each comparator, while costs were calculated as a weighted average of the first-line and second- or later-line analysis costs, based on the distribution of first-line patients (44.5%) and second- or later-line patients (55.5%) patients randomized in the VISION trial.2
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base case and 3,000 iterations for scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented in this section.
Base-Case Results
The sponsor submitted 3 base-case analyses for populations stratified by line of therapy (line agnostic, first line, second or later line).
In the sponsor’s base-case analysis for the line-agnostic population, tepotinib was associated with an expected cost of $140,499 and 1.6033 QALYs over a 10-year time horizon. Treatment with tepotinib was dominant (i.e., less costly and produced more QALYs) than immunotherapy (Table 3). At a willingness-to-pay of $50,000 per QALY, the probability of tepotinib being cost-effective was 88%.
In the sponsor’s base-case analysis for the first-line population, tepotinib was associated with an expected cost of $161,516 and 1.6278 QALYs over a 10-year time horizon. Treatment with tepotinib was less costly and produced fewer QALYs than immunotherapy plus PDC. Thus, the ICER for immunotherapy plus PDC versus tepotinib was $297,422 (Table 3).
In the sponsor’s base-case analysis for the second- or later-line population, tepotinib was associated with an expected cost of $125,839 and 1.6046 QALYs over a 10-year time horizon. Treatment with tepotinib was more costly and produced more QALYs than chemotherapy (Table 3). The probabilistic ICER for treatment with tepotinib versus chemotherapy was $370,130/QALY. At a willingness-to-pay of $50,000 per QALY, the probability of tepotinib being cost-effective was 0%.
For all analyses, the majority of QALYs were accrued in the PFS health state for tepotinib, and in the PD health state for comparators. Furthermore, the majority of QALYs were accrued within the period for which follow-up data were available. Additionally, at the end of the 10-year time horizon, less than 5% of patients were estimated to remain alive in the tepotinib and comparator arms in all analyses. Further detailed results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. comparator ($/QALY) |
|---|---|---|---|---|---|
Line-agnostic population | |||||
Tepotinib | $140,499 | Reference | 1.6033 | Reference | Reference |
Immunotherapy | $174,107 | $33,608 | 1.5584 | NA | Dominated by tepotinib |
1L population | |||||
Tepotinib | $161,516 | Reference | 1.6278 | Reference | Reference |
Immunotherapy + PDC | $361,235 | $199,719 | 2.2993 | 0.6715 | $297,422 vs. tepotinib |
2L+ population | |||||
Chemotherapy | $58,364 | Reference | 1.4223 | Reference | Reference |
Tepotinib | $125,839 | $67,476 | 1.6046 | 0.1823 | $370,130 vs. chemotherapy |
ICER = incremental cost-effectiveness ratio; NA = not applicable; PDC = platinum doublet chemotherapy; QALY = quality-adjusted life-year; vs. = versus.
Note: The submitted analysis is based on publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor assessed a limited set of model parameters in probabilistic scenario analyses. The scenarios consist of adjusting the discount rate, including biomarker testing for tepotinib, and using time-to-death utilities. All scenarios resulted in a change in the ICER of less than 15%. The greatest change occurred when time-to-death utilities were applied in the first-line population, which resulted in the ICER decreasing by 12%. In an additional scenario in which crizotinib was included as a line-agnostic comparator, tepotinib had incremental costs and QALYs of $30,048 and 0.447, resulting in a mean probabilistic ICER of $67,159 per QALY.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations of the sponsor’s analysis that have notable implications for the economic analysis:
Inclusion of line-agnostic efficacy data in a base-case analysis: The sponsor’s base-case analyses included 3 independently modelled subpopulations stratified by line of therapy (line agnostic, first line, second or later line). As per the CADTH Guidelines, researchers should examine any potential sources of heterogeneity that may lead to differences in parameter-input values across distinct subgroups, and a stratified analysis will allow decision-makers to identify any differential results across subgroups.21 Therefore, it is more appropriate to focus on the first-line and second- or later-line analyses presented.
Based on data from the VISION trial, the pooled RWE cohort, and feedback from clinical experts, it is understood that treatment practices and clinical outcomes differ by line of therapy. Therefore, line-agnostic efficacy data should not be used to estimate cost-effectiveness. Furthermore, immunotherapy was the line-agnostic comparator, and efficacy estimates were derived from a pool of patients receiving various regimens (e.g., pembrolizumab, ipilimumab, nivolumab, durvalumab). When multiple comparators are relevant to the funding decision, each treatment should be considered on its own, and then individually assessed in a sequential analysis. These analyses lacked regimen-specific comparative efficacy and safety parameters for the individual treatment regimens. Due to the limitations associated with using line-agnostic efficacy data and a pooled basket of immunotherapy regimens, the line-agnostic analysis should not be used to estimate cost-effectiveness.
Since the sponsor’s set of base-case analyses included subgroups stratified by line of therapy (i.e., first line and second- or later-line), the CADTH reanalysis focused on those populations.
Uncertainty associated with the ITC-derived estimates: For comparisons with immunotherapy (line-agnostic population) and chemotherapy (second- or later-line population), clinical parameters were estimated from a propensity score–weighted analysis using data from VISION and an RWE cohort. According to the CADTH Clinical Review report, the sponsor’s ITC is associated with substantial risk of bias, and important limitations were identified (i.e., methodological limitations, limited assessment of heterogeneity, poor reporting, small sample sizes). Therefore, no conclusions can be drawn on the efficacy of tepotinib compared to chemotherapy or immunotherapy in patients with advanced NSCLC harbouring METex14 skipping alterations. Furthermore, the relative efficacy of immunotherapy plus PDC (first-line population) was estimated using a naive ITC in which the OS and PFS hazard ratios of pembrolizumab plus chemotherapy versus chemotherapy from the subgroup of patients 65 years and older in the KEYNOTE-189 trial was applied to the chemotherapy arm of the pooled RWE cohort. Naive ITCs do not adjust for the different distributions of prognostic factors and treatment effect modifiers between populations, which are expected to differ between patients in the KEYNOTE-189 trial and the real-world setting. Further, the sponsor did not provide an assessment outlining the feasibility of this method. As a result, the naive ITC is associated with an unknown amount of bias, and no conclusions can be drawn from the analysis.
Since the VISION trial is a single-arm study, the ITCs conducted by the sponsor are the only evidence available to attempt to estimate the comparative clinical efficacy of tepotinib. CADTH noted that the sponsor’s submission modelled tepotinib to be clinically inferior (i.e., deliver fewer QALYs) when compared with immunotherapy plus PDC in the first-line population, and to provide only marginal QALY gains when compared with chemotherapy in the second- or later-line population. CADTH retained the sponsor’s comparative efficacy estimates when conducting exploratory analyses, as the sponsor provided no alternative estimates that were deemed more plausible. A scenario analysis was also performed to assess the impact of setting PFS and OS to be equal between treatment arms, rather than the sponsor’s assumption that tepotinib was clinically inferior, in the first-line population to reflect clinical expert opinion.
Lack of face validity in survival outcomes: The sponsor’s long-term extrapolations across analyses lacked face validity, as they assumed little to no relationship between PFS and OS. For example, tepotinib was modelled to have a PFS benefit but worse OS when compared with immunotherapy plus PDC in the first-line population. Modelled curves for each treatment arm also crossed in most cases. Feedback from clinical experts noted that there is expected to be a relationship between PFS and OS in patients with NSCLC, notwithstanding uncertainty in how METex14 skipping alterations would affect this.
Like the ITC limitation noted above, CADTH was unable to address the uncertainty associated with the ITCs, and, thus, the uncertainty associated with long-term survival outcomes similarly could not be addressed.
Inconsistent approach to model treatment duration: Treatment duration for tepotinib was modelled using extrapolated TTNTD curves from VISION. However, treatment duration for comparators was assumed to be equal to extrapolated combined TTNTD and PFS curves, as TTNTD data were incomplete. The use of inconsistent methodology to model treatment duration between arms likely leads to bias in estimating treatment costs between the intervention and comparator arms. This bias, due to inconsistent methodology, may favour tepotinib, since the use of TTNTD data led to lower treatment costs than assuming treatment duration is equal to PFS in the tepotinib arm. Therefore, using TTNTD in one arm and PFS and TTNTD in the other may introduce a bias: treatment costs may be overestimated in the arm that uses TTNTD and PFS. CADTH also notes that additional uncertainty is introduced in the comparator arm because of missing data and differences in data collection between clinical trial and real-world settings.
Since complete treatment duration data were not available for comparators, CADTH set treatment duration to be equal to PFS in the tepotinib arm to limit the risk of bias in favour of tepotinib resulting from inconsistent methodologies used to derive treatment duration.
Omission of relevant comparators in the base-case analyses: The 3 base-case analyses (i.e., line agnostic, first line, second or later line) included only 1 unique comparator each. Specifically, immunotherapy was modelled as the only line-agnostic comparator; immunotherapy plus PDC was modelled as the only first-line comparator; and chemotherapy was modelled as the only second- or later-line comparator.
Based on feedback from clinical experts, some patients likely receive either chemotherapy or immunotherapy alone as a first-line treatment; therefore, both treatments should also be included in the first-line analysis. It was further noted that vinorelbine as well as ipilimumab plus nivolumab, with or without PDC, are likely relevant comparators, although they are not frequently used. In the CADTH exploratory analysis, PDC costs were removed in the first-line analysis to derive a cost-effectiveness estimate versus immunotherapy in the first-line setting. CADTH was unable to address the other limitations with respect to exclusion of other comparators.
Limited PFS data and use of TTNTD as a proxy for comparators: To account for missing PFS data (only 31% recorded) in the pooled RWE cohort, TTNTD data were used as a proxy for PFS. Since the majority of the PFS data are missing, the PFS estimates for tepotinib are highly uncertain.
Clinical expert feedback noted that the use of TTNTD as a proxy is overall reasonable but may result in an overestimation of PFS, as some patients may remain on treatment for a period after they progress, due to less frequent imaging assessments in clinical practice than in clinical trials. CADTH was unable to address the lack of reliable PFS data.
Inappropriate estimation of METex14 testing costs and exclusion in the base-case analysis: The sponsor excluded METex14 testing in the reference case analysis on the basis that no incremental cost would be expected, since most NGS testing centres in Canada will soon be reporting METex14 results. Based on clinical expert and drug plan feedback, it was noted that the METex14 biomarker will be available as a component of some NGS platforms. However, it is uncertain to what extent it will be available across jurisdictions, and testing may need to be implemented in regions where it is currently unavailable. The sponsor performed a scenario analysis in which METex14 testing costs were included; however, the analysis applied the cost of a test only to the tepotinib arm and did not consider the number of tests needed to identify and treat each eligible patient. Since METex14 skipping alterations are rare, many tests would need to be administered to identify each patient. Thus, the testing costs in the sponsor’s scenario analysis are significantly underestimated. Furthermore, the sponsor calculated the cost per test based on a weighted average of immunohistochemistry or NGS. Based on feedback from clinical experts, immunohistochemistry is not expected to be used to test for METex14 skipping alterations.
The cost of METex14 testing was included in the CADTH exploratory analyses and was revised to reflect the number of tests needed to identify and treat each eligible patient, based on the prevalence of METex14 skipping alterations, assuming all tests are based on NGS.
Overestimation of AE costs for chemotherapy: AE incidence rates for comparators were derived from published sources, since they were not captured in the RWE. The sponsor assumed that chemotherapy was associated with AE management costs approximately 7 times higher than tepotinib, primarily driven by higher AE rates for docetaxel when compared with other chemotherapy components in the basket (i.e., PDC). Feedback from clinical experts noted that, although chemotherapy is expected to have higher AE rates, the sponsor’s AE management costs for docetaxel are likely overestimated, as docetaxel is expected to have toxicity similar to that of PDC.
In the CADTH exploratory reanalysis, CADTH revised the AE management costs for the docetaxel component of the chemotherapy basket to be equal to that of PDC.
Estimation of pembrolizumab costs: The sponsor calculated pembrolizumab drug costs using fixed dosage (200 mg every 3 weeks), as specified in the product monograph. However, public drug plan and clinician feedback noted that capped weight-based dosage (2 mg/kg up to a cap of 200 mg) with vial sharing is common across jurisdictions. Thus, the costs of pembrolizumab are overestimated.
CADTH recalculated pembrolizumab drug costs using capped weight-based dosage (2 mg/kg up to a cap of 200 mg) with vial sharing.
Inaccurate estimation of subsequent treatments: The sponsor assumed that 55% of patients would receive a second-line subsequent therapy. Furthermore, crizotinib was included as a second-line subsequent treatment. Feedback from clinical experts consulted by CADTH noted that 55% of patients receiving a second-line subsequent therapy is an overestimation. Furthermore, the clinical experts noted that crizotinib is unlikely to be used if tepotinib is available and that crizotinib is unavailable through public funding sources.
CADTH revised the proportion of patients receiving a second-line subsequent therapy to align with what would be expected in clinical practice. Crizotinib was also removed from the subsequent therapy distribution.
Price of certain chemotherapy and immunotherapy drugs do not align with data from DeltaPA: The sponsor derived drug cost data from prior CADTH submissions. The costs used for docetaxel, carboplatin, cisplatin, pemetrexed, and atezolizumab were deemed inappropriate, as they differed from list prices currently available in Canada.
The price of docetaxel, carboplatin, cisplatin, pemetrexed, and atezolizumab were revised to the current Canadian list prices, as reflected in DeltaPA.
Additionally, the sponsor made the following key assumptions, which have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Patient characteristics in the model were informed by the VISION trial | Acceptable |
Exclusion of crizotinib as a relevant comparator from the base-case analysis | Acceptable: Clinical experts consulted by CADTH agreed that crizotinib is not likely to be used if tepotinib is available, and crizotinib is not available through public funding sources |
Subsequent treatment after 2L was assumed, based on Canadian clinical expert feedback, to be docetaxel monotherapy | Acceptable |
The lifetime time horizon is 10 years | Uncertain: Clinical experts noted that this time horizon may be long, since only a small proportion (approximately 10%) of patients are expected to be alive after 5 years; however, the sponsor’s long-term extrapolations for tepotinib were consistent with this assumption |
Utility values were derived from EQ-5D-5L data collected in the VISION trial | Acceptable: The sponsor’s calculated utility values for the PFS and PD health states (0.78 and 0.71) were relatively consistent with prior submissions in NSCLC; use of alternative utility values derived from literature would have a limited impact on the results |
Patients are treated at 100% dose intensity | Acceptable: It is unclear how dose intensity would differ between tepotinib and comparator regimens; however, this is not expected to have a large impact on the results and is considered acceptable by clinical experts |
Use of a basket of regimens to estimate costs in the chemotherapy arm; chemotherapy costs in the 2L+ analysis were assumed to be a weighted average of carboplatin + pemetrexed (10%), cisplatin + pemetrexed (10%), and docetaxel (80%) | Uncertain but conservative: Clinical experts consulted by CADTH noted that the proportion of patients receiving docetaxel may be overestimated; however, docetaxel is associated with lower costs than PDC |
Carboplatin dosage of 4 to 6 cycles of 325 mg every 3 weeks | Uncertain but conservative: The carboplatin product monograph states that the recommended dosage of carboplatin is 400 mg/m2 every 4 weeks; thus, the sponsor’s assumptions are conservative |
2L = second line; 2L+ = second and later line; EQ-5D-5L = 5-Level EQ-5D; NSCLC = non–small cell lung cancer; PD = progressed disease; PDC = platinum doublet chemotherapy; PFS = progression-free survival.
CADTH Reanalyses of the Economic Evaluation
CADTH could not address several key limitations associated with the sponsor’s economic evaluation, primarily the lack of robust comparative clinical effectiveness data. Due to these limitations, all reanalyses subsequently undertaken by CADTH are considered exploratory.
CADTH’s exploratory reanalysis (Table 5) evaluated the impact of making alternative cost and resource use assumptions; making alternative treatment duration assumptions; using pembrolizumab capped weight-based dosage with vial sharing; removing PDC costs from immunotherapy plus PDC; revising AE management costs for chemotherapy; and correcting and including METex14 testing costs.
As noted previously, CADTH was unable to address the uncertainty associated with the ITCs and was therefore unable to estimate the comparative clinical efficacy of tepotinib. CADTH retained the sponsor’s comparative efficacy estimates when conducting exploratory analyses, since the sponsor provided no alternative estimates that were deemed more plausible. A scenario analysis was also performed to assess the impact of setting PFS and OS to be equal between treatment arms in the first-line setting, rather than the sponsor’s assumption that tepotinib was clinically inferior.
Table 5: Summary of CADTH Exploratory Analyses
Exploratory analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Populations considered in the base-case analyses | Line-agnostic, 1L, 2L+ | 1L, 2L+ (i.e., excluding line-agnostic) |
2. Incorrect chemotherapy and immunotherapy drug costs | Drug prices for docetaxel, carboplatin, cisplatin, pemetrexed, and atezolizumab drawn from previous CADTH submissions | Drug costs for docetaxel, carboplatin, cisplatin, pemetrexed, and atezolizumab revised based on current data from the DeltaPA database |
Changes to derive the CADTH exploratory reanalysis | ||
1. Treatment duration | Based on extrapolated TTNTD curves from VISION for tepotinib, and assumed to be equal to PFS for comparators | Treatment duration set to be equal to PFS for tepotinib and comparators |
2. Pembrolizumab dosage | Fixed, 200 mg q.3.w. | Weight-based, 2 mg/kg q.3.w. |
3. Subsequent treatments | 55% of patients receive a 2L subsequent therapy Crizotinib included in subsequent therapy distribution | 45% of patients receive a 2L subsequent therapy Crizotinib excluded from subsequent therapy distribution |
4. Interventions included in comparator regimen | Pembrolizumab + PDC | Pembrolizumab (i.e., removed PDC from regimen cost) |
5. Adverse event management costs for docetaxel in the chemotherapy basket | Based on published sources | Assumed to be equal to that of PDC in the chemotherapy basket |
6. a) METex14 testing | Excluded | Included |
6. b) Cost of METex14 testing | Cost per test based on weighted average cost of immunohistochemistry (30%; $133/test) and hybrid capture based on NGS (70%; $1,400/test) Average cost per test applied to tepotinib arm $1,019 per treated patient | Cost per test recalculated to assume 100% of tests are based on NGS ($1,400/test) Cost per test recalculated to consider the number of tests needed to identify and treat each eligible patient (i.e., 33.3) based on the estimated prevalence of METex14 skipping alterations (3%) $46,667b per treated patient |
CADTH exploratory reanalysis – 1L (vs. immunotherapy) | — | 1 + 2 + 3 + 4 + 6a + 6b |
CADTH exploratory reanalysis – 1L (vs. immunotherapy + PDC) | — | 1 + 2 + 3 + 6a + 6b |
CADTH exploratory reanalysis – 2L | — | 1 + 5 + 6a + 6b |
1L = first line; 2L = second line; 2L+ = second and later line; METex14 = MET exon 14; PDC = platinum doublet chemotherapy; NGS = next-generation sequencing; QALY = quality-adjusted life-year; TTNTD = time to next treatment or death; vs. = versus.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or standard errors in probabilistic analyses) that are not identified as limitations.
bAssuming an average of 33.3 tests each costing $1,400 need to be administered to identify 1 patient based on an assumed 3% prevalence of METex14 skipping alterations.
Exploratory Analysis Results
The cost-effectiveness findings were sensitive to multiple cost and resource use assumptions, primarily the use of pembrolizumab weight-based dosage with vial sharing, and the correction and inclusion of METex14 testing costs.
The first-line CADTH exploratory reanalysis was conducted with revised treatment duration estimates; pembrolizumab weight-based dosage; revised subsequent treatment estimates; exclusion of PDC costs; and inclusion and correction of METex14 testing costs. Tepotinib was dominated (i.e., more costly and less effective) by immunotherapy (Table 6). In an exploratory reanalysis in which the costs of PDC were included, tepotinib was less costly and less effective than immunotherapy plus PDC (Appendix 4).
The second- or later-line CADTH exploratory reanalysis was conducted using revised treatment duration estimates; revised AE management costs; and inclusion and correction of METex14 testing costs. Tepotinib was more costly and produced more QALYs than chemotherapy, with an ICER of $836,523 per QALY (Table 7). When excluding METex14 testing costs, the ICER was $551,240 per QALY (Appendix 4). Since the true testing costs are uncertain, the ICER is expected to be between this upper and lower limit.
Given the limitations previously identified, these results should be viewed as an exploration of the inherent uncertainty in the clinical data that underpins the economic evaluation.
Table 6: Summary of the CADTH Exploratory Reanalysis Results for Tepotinib vs. Immunotherapy — 1L
Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Immunotherapy | $169,166 | 3.1031 | 2.2994 | Reference |
Tepotinib | $232,922 | 2.1397 | 1.6218 | Dominated by immunotherapy |
1L = first line; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Table 7: Summary of the CADTH Exploratory Reanalysis Results for Tepotinib vs. Chemotherapy — 2L+
Drug | Total costs ($) | Total LYs | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Chemotherapy | $40,984 | 1.9391 | 1.4264 | Reference |
Tepotinib | $185,536 | 2.1376 | 1.5992 | $836,523 |
2L+ = second or later line; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; vs. = versus.
Scenario Analysis Results
CADTH undertook several price-reduction analyses.
In the first-line population, tepotinib was more costly and less effective than immunotherapy monotherapy (pembrolizumab). Price-reduction analyses therefore identify the price at which tepotinib is sufficiently less costly than pembrolizumab, such that, despite the greater clinical benefits with pembrolizumab, pembrolizumab would no longer be cost-effective. In the first-line population, if the decision-maker is willing to pay $50,000 per QALY, a price reduction of 65% for tepotinib would be needed. In a scenario in which METex14 testing costs were excluded, a 35% price reduction for tepotinib would be required.
In a scenario in which OS and PFS were set to be equal between treatments in the first-line setting, tepotinib remained more expensive than immunotherapy monotherapy (pembrolizumab). A price reduction of 38% would be required for tepotinib to have costs equal to pembrolizumab in this scenario. CADTH noted that, when METex14 testing costs were excluded, tepotinib remained a more costly alternative to pembrolizumab monotherapy.
In the second-line setting, a price reduction of more than 99% is required for tepotinib to be considered cost-effective versus chemotherapy at a willingness-to-pay threshold of $50,000 per QALY. When METex14 testing costs were excluded, the price reduction required for tepotinib to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY was 71%.
Details on the exploratory analyses, scenario analyses, and price-reduction analyses are provided in Appendix 4.
Issues for Consideration
Drug may change place in therapy of comparator drugs: In alignment with previous CADTH pan-Canadian Oncology Drug Review Expert Review Committee (pERC) recommendations, patients with advanced NSCLC with driver mutations who receive targeted treatment in the first-line setting must receive chemotherapy before accessing immunotherapy. Feedback from public drug plans noted that jurisdictions would use the same sequencing principles for therapies used after tepotinib, regardless of programmed death-ligand 1 tumour proportion score.
Implementation of METex14 testing: Feedback from CADTH participating drug plans noted that METex14 skipping alteration testing may not be routinely available in some jurisdictions and would need to be implemented.
Overall Conclusions
Due to the absence of direct or reliable indirect evidence on the relative benefit of tepotinib in comparison to currently funded alternatives regarding PFS and OS, the cost-effectiveness of tepotinib is associated with substantial uncertainty. No definitive conclusions could be drawn on the efficacy of tepotinib compared to chemotherapy or immunotherapy, with or without PDC, in patients with advanced NSCLC harbouring METex14 skipping alterations.
The considerable uncertainty of the clinical effectiveness evidence prevents a reliable estimation of the cost-effectiveness of tepotinib relative to current treatment alternatives. As a result, CADTH could not determine a base-case estimate. CADTH undertook an exploratory reanalysis that focused on cost-effectiveness in the first-line and second-line settings separately, as well as using consistent methodology to estimate treatment duration; assuming pembrolizumab weight-based dosage with vial sharing; revising subsequent treatment assumptions, including and correcting METex14 testing costs; revising chemotherapy AE management costs; and deriving an estimate versus immunotherapy in the first-line population. The sponsor’s clinical efficacy assumptions were retained in exploratory analyses, and a scenario analysis was performed assuming equal efficacy, regarding PFS and OS, between tepotinib and comparators in the first-line setting.
The sponsor’s line-agnostic base-case analysis was excluded since there is considerable heterogeneity across different lines of therapy in terms of comparators and prognosis. As a result, the CADTH exploratory reanalysis focused on the first-line and second- or later-line populations. In the first-line CADTH exploratory reanalysis, tepotinib was dominated (i.e., more costly and less effective) by immunotherapy, regardless of whether METex14 testing costs were included. In a further exploratory reanalysis versus immunotherapy plus PDC, tepotinib was less costly and less effective. This is based on uncertain clinical evidence provided by the sponsor, which shows tepotinib producing worse outcomes than immunotherapy. In the second- or later-line CADTH exploratory reanalysis, tepotinib was more costly and produced more QALYs than chemotherapy, with an ICER of $836,523 per QALY. When excluding METex14 testing costs ($46,667 to identify each eligible patient), the ICER was $551,240 per QALY. Since the true testing costs are uncertain, the ICER is expected to be between this upper and lower limit.
Overall, testing costs and place in therapy are key considerations when determining the cost-effectiveness of tepotinib. Due to the rarity of METex14, many individuals need to be tested to identify a single patient eligible for treatment with tepotinib. In the first-line setting, based on public list prices, tepotinib is a lower-cost alternative to pembrolizumab plus PDC but more expensive than pembrolizumab monotherapy. If tepotinib is considered equally effective as pembrolizumab; if it is priced no more than pembrolizumab monotherapy; and if plans incur no additional testing costs due to implementing METex14 testing, then tepotinib may represent a cost-effective alternative at a threshold of $50,000 per QALY. If incremental testing costs are incurred (i.e., if testing for METex14 is not routine and jurisdictions need to implement it), then price reductions up to 38% will be required to compensate for these additional testing costs. In the second- or later-line setting, if no testing costs are incurred, price reductions of more than 70% are needed; if testing costs are incurred, then cost-effectiveness may not be achieved even with 100% price reductions.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: PrTepmetkoTM (tepotinib) 225 mg tablets. Mississauga (ON): EMD Serono Canada; 2021 Aug 27.
2.Clinical Study Report: MS200095-0022. A phase II single-arm trial to investigate tepotinib in advanced (locally advanced or metastatic) non-small cell lung cancer with MET exon 14 (METex14) skipping alterations or MET amplification (VISION) [internal sponsor's report]. Darmstadt (Germany): Merck KGaA; 2020 May 14.
3.Conduct of an unbiased comparison in NSCLC with MET skipping [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: PrTepmetkoTM (tepotinib) 225 mg tablets [internal sponsor's package]. Mississauga (ON): EMD Serono Canada; 2021 Aug 27.
4.Rodriguez-Abreu D, Powell SF, Hochmair MJ, et al. Pemetrexed plus platinum with or without pembrolizumab in patients with previously untreated metastatic nonsquamous NSCLC: protocol-specified final analysis from KEYNOTE-189. Ann Oncol. 2021;32(7):881-895. PubMed
5.Paik PK, Felip E, Veillon R, et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med. 2020;383(10):931-943. PubMed
6.Quality-of-life analysis of the VISION trial data for tepotinib in non-small-cell lung cancer [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: PrTepmetkoTM (tepotinib) 225 mg tablets [internal sponsor's package]. Mississauga (ON): EMD Serono Canada; 2021 Aug 27. 2021.
7.Doyle S, Lloyd A, Walker M. Health state utility scores in advanced non-small cell lung cancer. Lung Cancer. 2008;62(3):374-380. PubMed
8.Nafees B, Stafford M, Gavriel S, Bhalla S, Watkins J. Health state utilities for non small cell lung cancer. Health Qual Life Outcomes. 2008;6:84. PubMed
9.Atezolizumab (Tecentriq) for non-small cell lung cancer. pan-Canadian Oncology Drug Review final economic guidance report. Ottawa (ON): CADTH; 2018: https://www.cadth.ca/sites/default/files/pcodr/pcodr_atezolizumab_tecentriq_nsclc_fn_egr.pdf. Accessed 2021 Oct 13.
10.Pembrolizumab (Keytruda) for nonsquamous non-small cell lung cancer. pan-Canadian Oncology Drug Review final economic guidance report. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/pcodr/Reviews2019/10153PembroNSQ-NSCLC_fnEGR_NOREDACT-ABBREV_Post_31May2019_final.pdf. Accessed 2021 Oct 13.
11.Pembrolizumab (Keytruda) for non-small cell lung cancer. pan-Canadian Oncology Drug Review final economic guidance report. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/pcodr/pcodr_pembrolizumab_keytruda_nsclc_1stln_fn_egr.pdf. Accessed 2021 Oct 13.
12.Nivolumab (Opdivo) for non-small cell lung cancer. pan-Canadian Oncology Drug Review final economic guidance report. Ottawa (ON): CADTH; 2016: https://www.cadth.ca/sites/default/files/pcodr/nivolumab_opdivo_nsclc_fn_egr.pdf. Accessed 2021 Oct 13.
13.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2021 Sep 29.
14.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
15.Schedule of benefits for physician services under the Health Insurance Act: effective Oct 1, 2021. Toronto (ON): Ontario Ministry of Health; 2021: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2021 Sep 29.
16.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123-135. PubMed
17.PrKeytruda® (pembrolizumab): 50 mg powder for solution for infusion; solution for infusion 100 mg/4 mL vial [product monograph]. Kirkland (QC): Merck Canada; 2021 Jul 15.
18.PrTecentriq® (atezolizumab): 60 mg/mL concentate for solution for infusion; 1200 mg/20 mL and 840 mg/14 mL single use vials [product monograph]. Mississauga (ON): Hoffmann-La Roche; 2021 Jun 11.
19.Patient cost estimator. 2021; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2021 Oct 9.
20.Erlotinib and gefitinib for treating nonsmall-cell lung cancer that has progressed after prior chemotherapy. NICE Technology appraisal guidance TA374. London (UK): National Institute for Health and Care Excellence; 2015: https://www.nice.org.uk/guidance/ta374/resources/erlotinib-and-gefitinib-for-treating-nonsmallcell-lung-cancer-that-has-progressed-after-prior-chemotherapy-pdf-82602789240517. Accessed 2021 Oct 25.
21.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/dv/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2021 Sep 29.
22.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: PrTepmetkoTM (tepotinib) 225 mg tablets. Mississauga (ON): EMD Serono Canada; 2021 Aug 27.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and CADTH participating drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Non–Small Cell Lung Cancer
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | 21-day course ($) | Average |
|---|---|---|---|---|---|---|---|
Tepotinib | |||||||
Tepotinib | 225.0 | Tablet | 153.96 | 450 mg once daily | 307.92 | 6,466 | 8,622 |
Chemotherapy | |||||||
Docetaxel | 20 mg/1 mL 80 mg/4 mL 160 mg/8 mL | Vial for IV infusion | 249.0000 497.0000 990.0000 | 100 mg/m2 every 3 weeks | $71.00 | $1,491.00 | $1,988 |
PDCa | — | — | — | — | $203.14 to $207.19 | $4,266 to $4,351 | $5,688 to $5,801 |
Carboplatin | 50 mg/5 mL 150 mg/15mL 450 mg/45mL 600 mg/60mL | Vial for IV infusion | 70.0000 210.0000 599.9985 775.0200 | 325 mg every 3 weeksb | $23.33 | $490 | $653 |
Cisplatin | 50 mg/50 mL 100 mg/100 mL | Vial for IV infusion | 135.0000 270.0000 | 75 mg/m2 every 3 weeksc | $19.29 | $405 | $540 |
Pemetrexed | 100 mg 1,000mg | Powder for solution for infusion | 429.0000 4,290.0000 | 500 mg/m2 every 3 weeks | $183.86 | $3,861 | $5,148 |
Pemetrexed | 100 mg 1,000mg | Powder for solution for infusion | 429.0000 4,290.0000 | 500 mg/m2 every 3 weeks | $183.86 | $3,861 | $5,148 |
Immunotherapy | |||||||
Atezolizumab | 1,200 mg/20 mL 840 mg/14 mL | Vial for IV infusion | 6,453.0000 4,517.10 | 1,200 mg every 3 weeks | $307.29 | $6,453 | $8,604 |
Nivolumab | 40 mg 100 mg | Vial for IV infusion | 782.2200 1,955.5600 | 3 mg/kg every 2 weeks | $335.24 | $7,040 | $9,387 |
Pembrolizumab | 100 mg/4 mL vial | Solution for infusion | 4,400.0000 | 2 mg/kg every 3 weeks | $284.88 | $5,982 | $7,977 |
Immunotherapy + PDC | |||||||
Pembrolizumab + PDCa | — | — | — | — | $488.02 to $492.07 | $10,248 to $10,333 | $13,665 to $13,778 |
Pembrolizumab | 100 mg/4 mL vial | Solution for infusion | 4,400.0000 | 2 mg/kg every 3 weeks | $284.88 | $5,982 | $7,977 |
PDC | — | — | — | — | $203.14 to $207.19 | $4,266 to $4,351 | $5,688 to $5,801 |
PDC = platinum doublet chemotherapy.
Note: All prices are from IQVIA DeltaPA, and do not include dispensing fees. Dosing is based on Health Canada product monographs except for pembrolizumab. For treatments using weight-based or BSA-based dosing, CADTH assumed 68 kg or 1.76m2. Cost calculations assume no vial sharing except for pembrolizumab.
aPDC is comprised of pemetrexed + carboplatin or cisplatin.
bCarboplatin dose calculated according to an area under the curve concentration using the Calvert Formula, and assuming patient characteristics of the VISION trial and creatinine clearance of 60 mL/min; includes a fix duration of 4 cycles for immunotherapy + PDC and 6 cycles for PDC.
cFixed duration of 4 cycles for immunotherapy + PDC and 6 cycles for PDC.
dCalculated using weight-based dosing with vial sharing rather than monograph flat dosing to better align with jurisdictional practices.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The sponsor did not allow flexibility to assess all relevant comparators in each base-case analysis stratified by line of therapy. |
Model has been adequately programmed and has sufficient face validity | No | The model was adequately programmed, with no obvious programming errors, but had limited validity. For example, the parametric extrapolations in the 1L and 2L+ analyses assumed implausible relationships between OS and PFS. |
Model structure is adequate for decision problem | No | The use of a partitioned survival model did not appear to adequately capture the decision problem. Modelled parametric curves for each treatment arm crossed in most cases, assumed implausible relationships between OS and PFS, and had little flexibility to assess alternate efficacy assumptions. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | The sponsor varied model parameters in the probabilistic analysis. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Parameter uncertainty was adequately incorporated within the model. Structural uncertainty was partially but not fully explored through scenario analyses. For example, no scenario analyses were performed to explore how the use of different parametric extrapolations for OS and PFS impact the outcomes. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | The model was easy to navigate and transparent. The report was generally well organized. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Table 10: Summary of Treatment Comparator Costs in Sponsor’s Economic Evaluation
Drug regimen | Cost per treatment dose | Cost per monthly treatment cycle |
|---|---|---|
Drug under review | ||
Tepotinib | $307.92 | $9,340.24 |
Modelled comparators | ||
Immunotherapy monotherapy (Line-agnostic)
| $6,776.00 $3,911.10 $8,800.00 | $11,609 |
Immunotherapy + PDC (1L)
| $8,800.00 $5,006.40 $4,978.50 | $19,861 |
Chemotherapy (2L+)
| $5,006.40 $4,978.50 $2,055.60 | $3,805 |
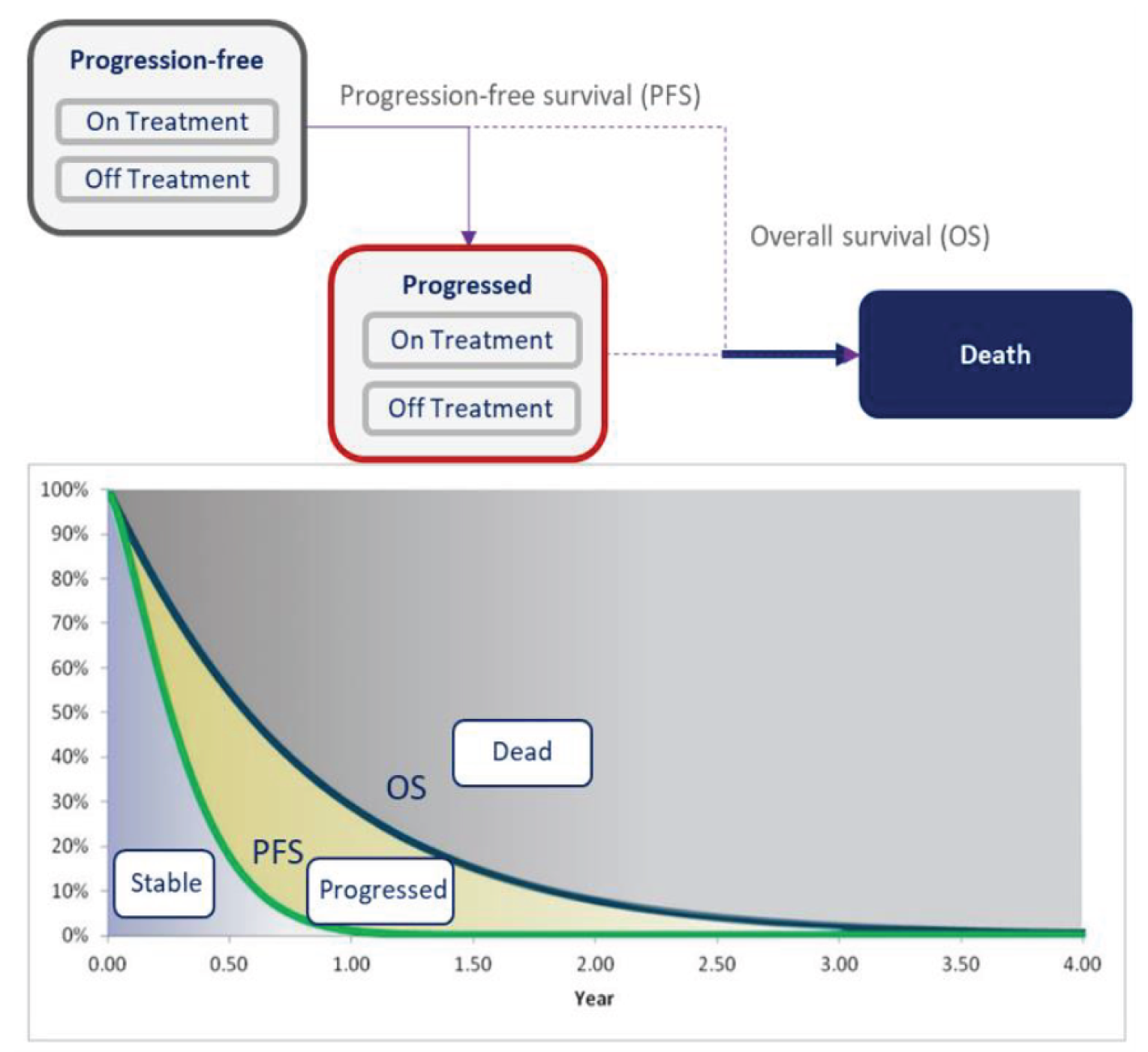
OS = overall survival; PFS = progression-free survival.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of Sponsor’s Economic Evaluation Results — 1L
Outcome | Tepotinib | Immunotherapy + PDC |
|---|---|---|
TOTAL LYs | 2.1474 | 3.1027 |
Progression-free LYs | 1.3723 | 1.2407 |
Post-progression LYs | 0.7751 | 1.8620 |
On-treatment LYs | 1.0551 | 1.2803 |
Off-treatment LYs | 1.0922 | 1.8223 |
TOTAL QALYs | 1.6278 | 2.2993 |
Progression-free QALYs | 1.0763 | 1.0041 |
Post-progression QALYs | 0.5527 | 1.2993 |
Decrement due to AE | -0.0012 | -0.0041 |
TOTAL costs | $161,516 | $361,235 |
Drug acquisition | $122,756 | $315,616 |
Administration | $0 | $7,765 |
Treatment monitoring | $190 | $598 |
AE management | $1,523 | $3,651 |
Disease management | $5,520 | $8,544 |
Subsequent treatment | $31,527 | $25,061 |
Testing costs | $0 | $0 |
Incremental results for tepotinib vs. immunotherapy | ||
Incremental costs | –$199,719 (tepotinib less costly versus immunotherapy + PDC) | |
Incremental LYs | –0.9553 | |
Incremental QALYs | –0.6715 (tepotinib less effective versus immunotherapy + PDC) | |
ICER per QALY | Tepotinib less costly and less effective (ICER vs. tepotinib: $297,422) | |
AE = adverse event; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; PDC = platinum doublet chemotherapy; QALY = quality-adjusted life-year; vs. = versus.
Note: The submitted results were based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Table 12: Disaggregated Summary of Sponsor’s Economic Evaluation Results — 2L+
Outcome | Tepotinib | Chemotherapy |
|---|---|---|
TOTAL LYs | 2.1454 | 1.9436 |
Progression-free LYs | 1.0741 | 0.6578 |
Post-progression LYs | 1.0713 | 1.2858 |
On-treatment LYs | 0.9409 | 0.6645 |
Off-treatment LYs | 1.2045 | 1.2791 |
TOTAL QALYs | 1.6046 | 1.4223 |
Progression-free QALYs | 0.8419 | 0.5208 |
Post-progression QALYs | 0.7638 | 0.9120 |
Decrement due to AE | –0.0012 | –0.0105 |
TOTAL costs | $125,839 | $58,364 |
Drug acquisition | $110,229 | $32,316 |
Administration | $0 | $1,859 |
Treatment monitoring | $170 | $156 |
AE management | $1,522 | $10,236 |
Disease management | $5,766 | $5,475 |
Subsequent treatment | $8,153 | $8,322 |
Testing costs | $0 | $0 |
Incremental results for tepotinib vs. chemotherapy | ||
Incremental costs | $67,476 | |
Incremental LYs | 0.2018 | |
Incremental QALYs | 0.1823 | |
ICER per QALY | $370,130 | |
AE = adverse event; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
Note: The submitted results were based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Exploratory Analysis
Table 13: Summary of CADTH Reanalysis Results — 1L
Scenario | Treatment | Total costs | Total QALYs | ICER vs. Tepotinib |
|---|---|---|---|---|
Sponsor submitted base case | ||||
Sponsor’s base case | Tepotinib | $161,516 | 1.6278 | Ref. |
Immunotherapy + PDC | $361,235 | 2.2993 | $297,422 vs. tepotinib | |
Corrected base case | ||||
Sponsor’s corrected base case | Tepotinib | $159,964 | 1.6272 | Reference |
Immunotherapy + PDC | $334,100 | 2.2987 | $259,332 | |
CADTH exploratory analyses | ||||
CADTH reanalysis 1: treatment duration | Tepotinib | $195,975 | 1.6246 | Reference |
Immunotherapy + PDC | $332,502 | 2.3032 | $201,197 | |
CADTH reanalysis 2: pembrolizumab dosage | Tepotinib | $158,164 | 1.6334 | Reference |
Immunotherapy + PDC | $269,177 | 2.2992 | $166,759 | |
CADTH reanalysis 3: subsequent treatments | Tepotinib | $152,466 | 1.6263 | Reference |
Immunotherapy + PDC | $327,793 | 2.3017 | $259,585 | |
CADTH reanalysis 4: interventions included in the comparator regimen | Tepotinib | $160,185 | 1.6245 | Reference |
Immunotherapy | $241,277 | 2.2990 | $120,222 | |
CADTH reanalysis 6a + 6b: METex14 testing | Tepotinib | $206,920 | 1.6339 | Reference |
Immunotherapy + PDC | $333,583 | 2.2935 | $192,016 | |
CADTH exploratory reanalysis versus immunotherapy (1 + 2 + 3 + 4 + 6a + 6b) | Immunotherapy | $169,166 | 2.2994 | Reference |
Tepotinib | $232,922 | 1.6218 | Dominated by immunotherapy | |
CADTH exploratory reanalysis versus immunotherapy + PDC (1 + 2 + 3 + 6a + 6b) | Tepotinib | $233,073 | 1.6239 | Reference |
Immunotherapy + PDC | $263,554 | 2.2940 | $45,487 | |
1L = first line; ICER = incremental cost-effectiveness ratio; METex14 = MET exon 14; PDC = platinum doublet chemotherapy; QALY = quality-adjusted life-year; vs. = versus.
Table 14: Summary of CADTH Reanalysis Results — 2L+
Scenario | Treatment | Total costs | Total QALYs | ICER vs. Chemotherapy |
|---|---|---|---|---|
Sponsor submitted base case | ||||
Sponsor’s base case | Chemotherapy | $58,364 | 1.4223 | Ref. |
Tepotinib | $125,839 | 1.6046 | $370,130 vs. chemotherapy | |
Corrected base case | ||||
Sponsor’s corrected base case | Chemotherapy | $48,362 | 1.4214 | Reference |
Tepotinib | $123,555 | 1.6034 | $413,211 | |
CADTH exploratory analyses | ||||
CADTH reanalysis 1: treatment duration | Chemotherapy | $48,438 | 1.4206 | Reference |
Tepotinib | $137,951 | 1.6041 | $487,801 | |
CADTH reanalysis 5: adverse event management costs for docetaxel in the chemotherapy basket | Chemotherapy | $41,112 | 1.4288 | Reference |
Tepotinib | $123,623 | 1.5971 | $490,030 | |
CADTH reanalysis 6a + 6b: METex14 testing | Chemotherapy | $48,477 | 1.4218 | Reference |
Tepotinib | $169,992 | 1.6080 | $652,443 | |
CADTH exploratory reanalysis: (1 + 5 + 6a + 6b) | Chemotherapy | $40,984 | 1.4264 | Reference |
Tepotinib | $185,536 | 1.5992 | $836,523 | |
2L = second line; ICER = incremental cost-effectiveness ratio; METex14 = MET exon 14; QALY = quality-adjusted life-year; vs. = versus.
Detailed Results of CADTH’s Exploratory Analyses
Table 15: Disaggregated Summary of CADTH’s Economic Evaluation Results — 1L
(vs. Immunotherapy)
Outcome | Tepotinib | Immunotherapy (mono) |
|---|---|---|
TOTAL LYs | 2.1397 | 3.1031 |
Progression-free LYs | 1.3742 | 1.2810 |
Post-progression LYs | 0.7656 | 1.8222 |
On-treatment LYs | 1.3742 | 1.2810 |
Off-treatment LYs | 0.7656 | 1.8222 |
TOTAL QALYs | 1.6218 | 2.2994 |
Progression-free QALYs | 1.0771 | 1.0041 |
Post-progression QALYs | 0.5459 | 1.2994 |
Decrement due to AE | –0.0012 | –0.0041 |
TOTAL costs | $232,922 | $169,166 |
Drug acquisition | $158,497 | $137,111 |
Administration | $0 | $4,063 |
Treatment monitoring | $248 | $599 |
AE management | $1,521 | $3,656 |
Disease management | $5,498 | $8,550 |
Subsequent treatment | $20,380 | $15,187 |
Testing costs | $46,778 | $0 |
Incremental results for tepotinib vs. immunotherapy | ||
Incremental costs | $63,756 | |
Incremental LYs | –0.9634 | |
Incremental QALYs | –0.6776 | |
ICER per QALY | Dominated by immunotherapy | |
AE = adverse event; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
Note: The submitted results were based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Table 16: Disaggregated Summary of CADTH’s Economic Evaluation Results — 1L
(vs. Immunotherapy + PDC)
Outcome | Tepotinib | Immunotherapy + PDC |
|---|---|---|
TOTAL LYs | 2.1423 | 3.0955 |
Progression-free LYs | 1.3763 | 1.2836 |
Post-progression LYs | 0.7659 | 1.8119 |
On-treatment LYs | 1.3763 | 1.2836 |
Off-treatment LYs | 0.7659 | 1.8119 |
TOTAL QALYs | 1.6239 | 2.2940 |
Progression-free QALYs | 1.0791 | 1.0064 |
Post-progression QALYs | 0.5459 | 1.2917 |
Decrement due to AE | –0.0012 | –0.0041 |
TOTAL costs | $233,073 | $263,554 |
Drug acquisition | $158,659 | $227,863 |
Administration | $0 | $7,778 |
Treatment monitoring | $248 | $599 |
AE management | $1,522 | $3,657 |
Disease management | $5,501 | $8,528 |
Subsequent treatment | $20,413 | $15,129 |
Testing costs | $46,730 | $0 |
Incremental results for immunotherapy + PDC vs. tepotinib | ||
Incremental costs | $30,482 (immunotherapy + PDC more costly versus tepotinib) | |
Incremental LYs | 0.9533 (immunotherapy + PDC more effective versus tepotinib) | |
Incremental QALYs | 0.6701 | |
ICER per QALY | $45,487 | |
AE = adverse event; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; PDC = platinum doublet chemotherapy; QALY = quality-adjusted life-year; vs. = versus.
Note: The submitted results were based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Table 17: Disaggregated Summary of Sponsor’s Economic Evaluation Results — 2L+
Outcome | Tepotinib | Chemotherapy |
|---|---|---|
TOTAL LYs | 2.1376 | 1.9391 |
Progression-free LYs | 1.0761 | 0.6622 |
Post-progression LYs | 1.0615 | 1.2768 |
On-treatment LYs | 1.0761 | 0.6622 |
Off-treatment LYs | 1.0615 | 1.2768 |
TOTAL QALYs | 1.5992 | 1.4264 |
Progression-free QALYs | 0.8434 | 0.5191 |
Post-progression QALYs | 0.7569 | 0.9105 |
Decrement due to AE | –0.0012 | –0.0032 |
TOTAL costs | $185,536 | $40,984 |
Drug acquisition | $125,420 | $24,506 |
Administration | $0 | $1,857 |
Treatment monitoring | $193 | $155 |
AE management | $1,520 | $2,832 |
Disease management | $5,719 | $5,443 |
Subsequent treatment | $6,054 | $6,190 |
Testing costs | $46,630 | $0 |
Incremental results for tepotinib vs. chemotherapy | NA | |
Incremental costs | $144,552 | |
Incremental LYs | 0.1985 | |
Incremental QALYs | 0.1728 | |
ICER per QALY | $836,523 | |
AE = adverse event; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
Note: The submitted results were based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Scenario Analyses
Table 18: CADTH Scenario Analyses
Scenario | CADTH Base Case | CADTH Scenario |
|---|---|---|
Scenario Analyses | ||
1. METex14 testing costs | Included | Excluded |
2. Overall survival and progression-free survival | Based on extrapolated data from the VISION trial for tepotinib, and from a naive ITC for immunotherapy + PDC | OS and PFS set to be equal between treatments arms |
Table 19: CADTH Scenario Analysis Results — 1L
Drug | Total Costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
CADTH exploratory reanalysis – 1L (tepotinib versus immunotherapy) | |||
Immunotherapy | $169,166 | 2.2994 | Reference |
Tepotinib | $232,922 | 1.6218 | Dominated by immunotherapy |
Scenario 1: Exclude METex14 testing costs | |||
Immunotherapy | $167,768 | 2.2951 | Reference |
Tepotinib | $187,154 | 1.6242 | Dominated by immunotherapy |
Scenario 2: Overall survival and progression-free survival equal between immunotherapy and tepotinib | |||
Immunotherapy | $175,679 | 1.6206a | Reference |
Tepotinib | $233,193 | 1.6235a | $19,763,012a |
1L = first line; ICER = incremental cost-effectiveness ratio; METex14 = MET exon 14; QALY = quality-adjusted life-year
aSmall differences in QALY’s are caused by differences in disutility’s due to adverse events, the large ICER is due to small QALY differences approaching zero.
Table 20: CADTH Price-Reduction Analyses — 1L
Scenario | ICERs for immunotherapy +/- PDC vs. tepotinib ($ / QALY) | |||
|---|---|---|---|---|
Price reduction | Sponsor base case (immunotherapy + PDC vs. Tepotinib) | CADTH exploratory reanalysis (immunotherapy vs. tepotinib) | CADTH scenario 1 – exclude METex14 testing costs (immunotherapy vs. tepotinib) | CADTH scenario 2 – equal OS and PFS (tepotinib vs. immunotherapy) |
No price reduction | $297,422 | Immunotherapy dominants tepotinib | Immunotherapy dominants tepotinib | $19,763,012 |
10% | $314,294 | Immunotherapy dominants tepotinib | Immunotherapy dominants tepotinib | $14,622,982 |
20% | $331,165 | Immunotherapy dominants tepotinib | $16,429 | $9,482,952 |
30% | $348,037 | Immunotherapy dominants tepotinib | $39,090 | $4,342,923 |
40% | $364,908 | Immunotherapy dominants tepotinib | $61,751 | Tepotinib dominates immunotherapy |
50% | $381,780 | $16,421 | $84,412 | Tepotinib dominates immunotherapy |
60% | $398,652 | $38,523 | $107,073 | Tepotinib dominates immunotherapy |
70% | $415,523 | $60,625 | $129,734 | Tepotinib dominates immunotherapy |
80% | $432,395 | $82,727 | $152,395 | Tepotinib dominates immunotherapy |
90% | $449,266 | $104,829 | $175,056 | Tepotinib dominates immunotherapy |
99% | $464,451 | $124,721 | $195,451 | Tepotinib dominates immunotherapy |
ICER = incremental cost-effectiveness ratio; METex14 = MET exon 14; QALY = quality-adjusted life-years; vs. = versus.
Table 21: CADTH Scenario Analysis Results — 2L+
Drug | Total Costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
CADTH exploratory reanalysis – 2L (tepotinib vs. chemotherapy) | |||
Chemotherapy | $40,984 | 1.4264 | Reference |
Tepotinib | $185,536 | 1.5992 | $836,523 |
Scenario 1: Exclude METex14 testing costs | |||
Chemotherapy | $41,031 | 1.4271 | Reference |
Tepotinib | $138,597 | 1.6041 | $551,240 |
2L = second line; ICER = incremental cost-effectiveness ratio; METex14 = MET exon 14; QALY = quality-adjusted life-year; vs. = versus.
Table 22: CADTH Price-Reduction Analyses — 2L+
Scenario | ICERs for tepotinib vs. chemotherapy ($ / QALY) | ||
|---|---|---|---|
Price reduction | Sponsor base case | CADTH exploratory reanalysis | CADTH scenario 1 – exclude METex14 testing costs |
No price reduction | $370,132 | $836,523 | $551,240 |
10% | $309,692 | $763,980 | $480,586 |
20% | $249,252 | $691,436 | $409,931 |
30% | $188,812 | $618,893 | $339,277 |
40% | $128,372 | $546,349 | $268,622 |
50% | $67,932 | $473,806 | $197,968 |
60% | $7,492 | $401,262 | $127,313 |
70% | Dominant | $328,719 | $56,659 |
80% | Dominant | $256,175 | Dominant |
90% | Dominant | $183,632 | Dominant |
99% | Dominant | $118,342 | Dominant |
2L = second line; ICER = incremental cost-effectiveness ratio; METex14 = MET exon 14; QALY = quality-adjusted life-years; vs. = versus.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 23: CADTH Summary Findings From the Sponsor’s BIA
Key Take-Aways of the BIA |
|---|
|
BIA = budget impact analysis.
Summary of Sponsor’s BIA
The sponsor’s submitted budget impact analysis investigated the adoption of tepotinib for the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping alterations. Current treatment options for patients in the sponsor’s reference scenario included immunotherapy plus PDC, immunotherapy, and chemotherapy. The BIA was undertaken from the public payer perspective for the Canadian setting using an epidemiology approach over a 3-year projected time horizon (2023 to 2025) as well as a baseline year (2022). The BIA investigates the Canadian population, excluding Quebec. Key inputs to the BIA are documented in Table 24. The sponsor assumed that the relevant first-line comparators were immunotherapy and immunotherapy plus PDC. In the second- or later-line setting, the sponsor assumed the relevant comparators were immunotherapy and chemotherapy. Outcomes of the line-agnostic (first-line and second- or later-line combined) population were calculated as the sum of the independent first-line and second- or later-line populations, considering the relevant comparators for each line of therapy. To derive the market size the sponsor assumed 100% of the target population was eligible for public coverage and that tepotinib uptake would reach a market size of |||||||% of the first-line population and |||||||% of the second- or later-line population over 3 years. It was assumed that tepotinib would take a higher proportion of market share away from immunotherapy plus PDC and chemotherapy than it takes from immunotherapy in the first-line and second- or later-line settings. Three-year mean treatment duration for tepotinib was modelled using extrapolated TTNTD curves from VISION, while treatment duration of comparators was modelled using extrapolated combined PFS/TTNTD curves. The costs of drug administration, subsequent treatment, and biomarker testing were not included in the sponsor’s base case. Patients were assumed to weigh 65.91 kg to derive weight-based costing estimates.
The BIA was found to be sensitive to the tepotinib market share, treatment duration assumptions, and correction/inclusion of METex14 testing. Key inputs to the BIA are documented in Table 28.
Figure 2: Sponsor’s Estimation of the Size of the Eligible Population
1L = first line; 2L = second line; m = metastatic; METex14 = MET exon 14; NSCLC = non–small cell lung cancer; pCODR = CADTH pan-Canadian Oncology Drug Review.
Source: Sponsor’s budget impact analysis.22
Table 24: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / |
|---|---|
Target Population | |
Annual population growth rate | 1.1% |
METex14 testing rate | ||||||||||||||||||||||||||||||||||| |
Number of patients eligible for tepotinib | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| |
Market Uptake (3 years) | |
Uptake (reference scenario) | See Table 25 |
Annual Drug Costs (per patient) | |
Tepotinib Immunotherapy Immunotherapy + PDC c Chemotherapy | $112,083 $128,687a - $152,533b $238,503 $45,715 |
1L = first line; 2L = second line; m = metastatic; METex14 = MET exon 14; PDC = platinum doublet chemotherapy.
aImmunotherapy based on 2L treatment mix (10% atezolizumab; 40% nivolumab; 50% pembrolizumab).
bImmunotherapy based on 1L treatment mix (100% pembrolizumab).
cComprised of pembrolizumab + pemetrexed + cisplatin or carboplatin.
Table 25: Sponsor’s Estimation of Market Update in the New Drug Scenario
Drug | Reference Scenario | New Drug Scenario Year 1 | New Drug Scenario Year 2 | New Drug Scenario Year 3 |
|---|---|---|---|---|
1L Population | ||||
Tepotinib | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Immunotherapy | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Immunotherapy + PDC | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
2L Population | ||||
Tepotinib | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Immunotherapy | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Chemotherapy | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||||||||||||| |
1L = first line; 2L = second line; NSCLC = non-small cell lung cancer.
Source: Sponsor’s Budget Impact Analysis22
Summary of the Sponsor’s BIA Results
Based on the sponsor’s analysis, the 3-year budget impact of tepotinib for the treatment of adult patients with locally advanced unresectable or metastatic NSCLC harbouring METex14 skipping alterations is a saving of $16,965,241. The annual budget impact was –$2,407,700 in Year 1, –$6,301,130 in Year 2, and –$8,256,410 Year 3 (Table 26). Cost savings were primarily generated through reduced first-line drug acquisition costs (Table 21).
Stepped analysis | Annual Cost Year 1 | Annual Cost Year 2 | Annual Cost Year 3 | Three-year total |
|---|---|---|---|---|
Reference scenario | $44,291,205 | $66,422,016 | $67,634,305 | $178,347,527 |
New drug scenario | $41,883,505 | $60,120,886 | $59,377,895 | $161,382,286 |
Incremental costs | –$2,407,700 | –$6,301,130 | –$8,256,410 | –$16,965,241 |
Source: Sponsor’s Budget Impact Analysis.22
Table 27: Three-Year Cost Breakdown
Stepped analysis | Reference scenario | New drug scenario | Incremental costs |
|---|---|---|---|
1L Drug acquisition | $155,384,343 | $128,241,397 | –$27,142,946 |
2L+ Drug acquisition | $13,121,351 | $24,050,659 | $10,929,308 |
Drug mark-up | $9,841,833 | $9,082,654 | –$759,179 |
Dispensing fees | $0 | $7,576 | $7,576 |
Totals | $178,347,527 | $161,382,286 | –$16,965,241 |
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Inconsistent approach to model treatment duration: 3-year mean treatment duration was based on the CUA where tepotinib was modelled using extrapolated TTNTD curves from VISION and treatment duration for comparators was assumed to be equal to extrapolated combined TTNTD/PFS curves, as TTNTD data were not complete. Furthermore, the sponsor’s calculated 3-year mean did not align with the CUA model resulting in erroneous values for some comparators.
Similar to the CUA, CADTH set treatment duration to be equal to PFS in the tepotinib arm to limit the potential risk of bias in favour of tepotinib that may have been introduced by the sponsor as a result of inconsistent methodologies used to derive treatment duration; 3-year mean treatment durations were then recalculated based on these changes.
Inaccurate estimation of pembrolizumab costs: The sponsor calculated pembrolizumab drug costs using fixed dosing (200 mg every 3 weeks) as specified in the product monograph. However, public drug plan and clinician feedback noted that capped weight-based dosing (2 mg/kg up to a cap of 200 mg) with vial sharing is commonly implemented across jurisdictions. Thus, the costs of pembrolizumab are overestimated.
CADTH recalculated pembrolizumab drug cost using weight-based dosing (2 mg/kg up to a cap of 200 mg) with vial sharing.
Comparator market shares: Chemotherapy was not included as a first-line comparator. However, input from clinical experts noted that patients who are not eligible for immunotherapy will receive chemotherapy for all lines of treatment. This is further supported by the RWE collected by the sponsor where a proportion of patients received chemotherapy in the first-line setting.
CADTH revised the market shares to include chemotherapy in the first-line setting to better align with expert input and the sponsor’s RWE. Tepotinib was then assumed to capture a proportional market share from all comparators in the first-line setting, including chemotherapy.
Tepotinib market capture: Tepotinib was assumed to capture market share from both immunotherapy and chemotherapy in the second- or later-line setting. Based on clinical expert input, if tepotinib is given in the second- or later-line setting, it would only displace chemotherapy since immunotherapy would have already been given in the first-line setting if possible.
CADTH revised the second- or later-line market capture estimates so that tepotinib captures market share from chemotherapy only in the second- or later-line setting.
Inappropriate estimation of METex14 testing costs: The sponsor performed a scenario analysis where METex14 testing was included. The sponsor calculated the cost per test based on a weighted average of immunohistochemistry or NGS. Based on feedback from clinical experts, immunohistochemistry is not expected to be used to test for METex14 skipping alterations. Furthermore, the cost of METex14 testing was calculated such that only patients receiving tepotinib would be tested for METex14 skipping alterations. This is not appropriate as it underestimates the total patients who receive a test.
The cost of METex14 testing was revised to reflect the number of tests need to identify and treat each eligible patient based on the prevalence of METex14 skipping alterations, assuming all tests are based on NGS.
Price of certain chemotherapy and immunotherapy drugs do not align with data from DeltaPA: The sponsor sourced drug cost data from prior CADTH submissions. The costs used for docetaxel, carboplatin, cisplatin, pemetrexed, and atezolizumab were deemed inappropriate as they differed from list prices currently available in Canada.
The price of docetaxel, carboplatin, cisplatin, pemetrexed, and atezolizumab were revised to the current Canadian list prices as reflected in DeltaPA.
CADTH Reanalyses of the BIA
CADTH conducted multiple revisions as part of the reanalyses. A scenario analysis was also included to assess the impact of incremental costs associated with METex14 testing, assuming no testing is currently being performed and jurisdictions would need to implement.
Table 28: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Incorrect chemotherapy and immunotherapy drug costs | Drug prices for docetaxel, carboplatin, cisplatin, pemetrexed, and atezolizumab sourced from previous CADTH submissions | Drug costs for docetaxel, carboplatin, cisplatin, pemetrexed, and atezolizumab revised based on current data from the Delta PA database |
Changes to derive the CADTH base case | ||
1. Treatment duration | Based on extrapolated TTNTD curves from VISION for tepotinib, and assumed to be equal to PFS for comparators Sponsor’s calculated 3-year mean did not align with CUA model resulting in erroneous values for some comparators. | Treatment duration set to be equal to PFS for both tepotinib and comparators. 3-year mean re-calculated based on this assumption. |
2. Pembrolizumab dosage | Fixed 200 mg Q3W | Weight-based 2 mg/kg Q3W |
3. a) Comparator market shares | Chemotherapy not included as a 1L treatment. | Chemotherapy included as a 1L treatment; tepotinib assumed to capture market share proportionally from all comparators in the 1L setting. |
3. b) Tepotinib market capture | Tepotinib captures market share from both immunotherapy and chemotherapy in the 2L setting. | Tepotinib captures market share exclusively from chemotherapy in the 2L setting. |
4. a) METex14 testing | Excluded | Included |
4. b) Cost of METex14 testing | Cost per test based on weighted average cost of immunohistochemistry (30%; $133/test) and hybrid capture based on next generation sequencing (70%; $1,400/test) Average cost per test applied to patients receiving tepotinib $1,019 per tested patient that receives tepotinib | Cost per test recalculated to assume 100% of tests are based on NGS ($1,400/test) Cost per test applied to all tested NSCLC patients using the sponsor’s assumed testing rates (57.5% / 85.0% / 85.0% in Year 1 / Year 2 / Year 3). $1400 per tested patient, regardless of whether they receive tepotinib |
CADTH base case | Reanalysis 1 + 2 + 3 | |
CADTH scenario analysis - Include METex14 testing costs | Reanalysis 1 + 2 + 3 + 4 | |
BIA = budget impact analysis; METex14 = MET exon 14; NGS = next generation sequencing; NSCLC = non–small cell lung cancer; Q3W = every 3 weeks; TTNTD = time to next treatment or death.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses, etc.) that are not identified as limitations.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 29 and a more detailed breakdown is presented in Table 30.
CADTH reanalyses estimated that the introducing tepotinib for the modelled indication will be associated with an incremental cost of $13,498,247 in the first three years in the scenario without METex14 testing costs. If METex14 testing is not available on existing NGS platforms and needs to be implemented, the incremental cost would increase to $69,931,737.
Table 29: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | –$16,965,241 |
Sponsor’s corrected base case | –$11,873,622 |
CADTH reanalysis 1: treatment duration | –$13,950,361 |
CADTH reanalysis 2: pembrolizumab dosage | $3,973,087 |
CADTH reanalysis 3: comparator market shares | $647,871 |
CADTH reanalysis 4: METex14 testing | $44,559,868 |
CADTH Base Case (1 + 2 + 3) | $13,498,247 |
CADTH Scenario Analysis (1 + 2 + 3 + 4) - include incremental METex14 testing costs | $69,931,737 |
BIA = budget impact analysis; METex14 = MET exon 14.
Table 30: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $22,591,740 | $44,291,205 | $66,422,016 | $67,634,305 | $178,347,527 |
New drug | $22,591,740 | $41,883,505 | $60,120,886 | $59,377,895 | $161,382,286 | |
Budget impact | $0 | –$2,407,700 | –$6,301,130 | –$8,256,410 | –$16,965,241 | |
CADTH Base Case | Reference | $14,264,957 | $28,084,986 | $42,170,001 | $43,048,998 | $113,303,985 |
New drug | $14,264,957 | $29,158,974 | $46,472,037 | $51,171,222 | $126,802,232 | |
Budget impact | $0 | $1,073,988 | $4,302,036 | $8,122,224 | $13,498,247 | |
CADTH Scenario Analysis – include incremental METex14 testing costs | Reference | $14,264,957 | $28,084,986 | $42,170,001 | $43,048,998 | $113,303,985 |
New drug | $14,264,957 | $43,248,033 | $67,528,442 | $72,459,247 | $183,235,722 | |
Budget impact | $0 | $15,163,047 | $25,358,441 | $29,410,249 | $69,931,737 |
BIA = budget impact analysis; METex14 = MET exon 14.
Stakeholder Input
Patient Input
Lung Cancer Canada
About Lung Cancer Canada
Lung Cancer Canada is a registered national charitable organization that serves as Canada’s leading resource for lung cancer education, patient support, research and advocacy. Lung Cancer Canada is a member of the Global Lung Cancer Coalition and is the only organization in Canada focused exclusively on lung cancer.
https://www.lungcancercanada.ca/
Lung Cancer Canada is registered with CADTH.
Information Gathering
Data Collection
The information and data discussed throughout this document was collected through phone and video interviews with patients and caregivers, who very kindly gave us their time to share their thoughts and experiences with the disease and treatment in question. All interviews were conducted in September 2021.
Demographic Data
NSCLC due to MET exon-14 skipping mutations is rare, as the estimated prevalence rates of this mutation make up about 1.7% of all NSCLCs. All of the patients discussed have the MET exon 14 skipping mutation, and 4 out of 5 patients interviewed have experience with tepotinib. The patients that Lung Cancer Canada had interviewed were in the older demographic, with 3 out of 5 patients being retired by the time of their diagnosis. Specific treatment experience can be found in section 6.
Name | Gender | Age at diagnosis | Patient or Caregiver | Source | MET exon-14 skipping alteration? | Experience with tepotinib? | Line of treatment with tepotinib | Location |
|---|---|---|---|---|---|---|---|---|
LS | Female | 51 | Patient | Interview | Yes | Yes | 3rd-line | Canada |
LW | Male | 72 | Patient | Interview | Yes | Yes | 1st-line | Canada |
DR | Female | 72 | Patient | Interview | Yes | Yes | 2nd-line | USA |
VM | Female | 40 | Caregiver | Interview | Yes | Yes | 3rd-line | Canada |
CR | Female | 70 | Patient | Interview | Yes | No | N/A | Canada |
Disease Experience
DR has been living a very active lifestyle ever since she was young, and even today at 72 years old, she is still thriving, exercising, and enjoying spending time with her family thanks to the discoveries and research that has been done in the field of lung cancer. Being diagnosed with lung cancer was a real kick in the butt for her as it was almost the last thing she expected to face when she went into the ER for what her doctors thought was bronchitis. She had never smoked a day in her life, was never exposed to any second-hand smoke, and considers herself very healthy and athletic. The reality of many lung cancer patients starts out like DR’s, and unfortunately, most tumours are only caught at advanced stages, and thus, leaves patients like DR with little time to grasp and reflect on their diagnoses before starting treatment.
The MET proto-oncogene encodes for the receptor tyrosine kinase, and the consequences of MET alteration leads to decreased turnover of the MET protein and increased MET signalling, which ultimately leads to oncogenesis (Paik et al., 2020). MET exon 14 skipping is an incredibly rare mutation that is the driver of only 3-4% of non–small-cell lung cancer (NSCLC) patients, most frequently affecting those who are never-smokers (Heist et al., 2016). NSCLCs are the most common type of lung cancer, occurring in 80-85% of all lung cancer cases. A fellow Canadian MET patient, CL, would not have had the chance to be treated in the APL-101 targeted therapy clinical trial if she had not been able to access biomarker testing that was only available to her from the United States. Having the ability to access biomarker testing ensures that patients do not get left behind with treatment options that are not specific to their needs, and so physicians are able to know what they are actually treating rather than going in blind. Targeted therapies have recently emerged as an important (and sometimes, their only) mean of disease management for many NSCLC patients who have targetable mutations found through biomarker testing, such as tepotinib.
Tepotinib is a highly selective MET inhibitor and has shown promising results in advanced NSCLC patients, including possible intracranial activity in inhibiting MET-dependant tumour cells, which has slowed the progression of disease and resulting metastases, particularly in the brain (Stenger, 2021). In the phase two VISION trial, the overall response rate recorded for tepotinib in 152 patients with advanced-stage NSCLC was 46%, and this rate stayed consistent when used across multiple lines of treatment (particularly second and third lines) due to the targeted nature of the medication (Paik et al., 2020). Median progression-free survival was seen to be 8.5 months and a response duration of 11.1 months (Roth et al., 2020). These are incredibly promising results, as this time allows for patients who previously had unsuccessful treatment regimens to experience improved quality of life and be able to manage their disease through an oral targeted therapy.
Quality of life, patient, and caregiver experience are also other critical aspects when managing non-small cell lung cancer, especially with the approval of a new oral therapy treatment like tepotinib. With the relatively older population of patients that lung cancer affects, this also ties in with unique patient and caregiver burdens that contribute to overall disease experience. With non-small-cell lung cancer, most patients are diagnosed at late stages, and approximately 70% of new diagnoses being locally advanced or metastatic disease, specifically brain or central nervous system metastases (Nadler et al., 2020). This has many implications on patient and caregiver burden in multiple dimensions, as progression of disease can lead to decreased functionality, inability to perform activities of daily living, increasing requirement on caregivers to care for patients, financial burdens, and overall, decreased quality of life (Polanski et al., 2016). Tepotinib has shown to be successful in targeting MET alterations and providing patients with the luxury of delaying disease progression and allowing them to live a quality life while being able to manage their disease effectively. We hope that with CADTH’s consideration of reimbursement of tepotinib, all MET patients can have the opportunity to be able to return to doing the activities they love and return to a state of independence and functionality similar to their pre-diagnosis selves, just like DR.
Experiences With Currently Available Treatments
Chemotherapy and immunotherapy are the current standards of care in Canada for first line treatment for patients with this MET mutation and NSCLC. Multitargeted tyrosine kinase inhibitors are also occasionally used to target tumour cells, though have been met with variable results and limited success due to its non-specificity to the MET oncogene (Wong et al., 2021). However, targeted therapy has since emerged as an important mean of disease management for NSCLC patients with a targetable mutation, including MET. It has seen much more promising results with significantly less symptom burdens on the patient compared to chemotherapy.
Chemotherapy left an incredibly harsh and heavy side effect burden.
VM had an incredibly difficult cancer journey since she was diagnosed in 2017 with stage 3B NSCLC . She started on Cisplastin (chemotherapy) as her first treatment, which initially showed good results, but one month into treatment, it left her with many of the negative side effects traditionally associated with chemotherapy, leaving her very sick and hospitalized due to low blood count. She required a lot of help from caregivers and family, which left her devastated because she wanted to be great mother and wife but was unable to do much. VM ultimately relapsed with metastases to her liver and bones, and had to stop chemotherapy as it was not working. It was only 7 months after diagnosis that she was able to get biomarker testing done to confirm she had the rare MET exon-14 alteration.
LS also started off her cancer journey in December 2020 with chemotherapy as her first line of treatment. She witnessed how her mother reacted very poorly to chemotherapy about 20 years prior being in lots of pain and fatigue, which did not end up working in her case. As a result, LS expected the same reaction in herself, but surprisingly, she did not have many side effects apart from hair loss and some general fatigue. She was able to still complete her daily tasks on most days, though others felt very tiring and was on and off in her energy levels. She had 3 rounds of chemotherapy treatment over the course of a month and was completed by February 2021.
Other therapies were not effective in managing their disease.
Once VM was able to get the biomarker testing to confirm that she had the MET mutation, she started on crizotinib, which worked very well for about 9 months until she had another major relapse in May 2018. Her health declined significantly due to the many metastases that were in her brain, and unfortunately, left her unable to walk, communicate, and care for herself at all. She had to be fed, washed, and constantly cared for as she was bedridden, it was like “she was a totally different person”. She constantly had nightmares that woke her up every 2 hours overnight. Her caregivers were under the impression that VM was not suffering, though they did not know for sure because VM was in such a terrible condition that she was unable to communicate or respond to questions. She had an extremely poor quality of life, to the point that her caregivers were considering end-of-life care for VM, until their second miracle came when she started tepotinib.
LS also started on a different targeted therapy after completing chemotherapy that worked very well for the first 1.5 months, but suddenly started to have a bad reaction to it that landed her in the hospital twice while she was on the drug. As she is located in a relatively rural area in New Brunswick, she was limited in her access to better healthcare and other treatments, particularly since her local doctors were not as knowledgeable on any of the potential side effects of the drug. She ended up hospitalized for two weeks with severe vomiting in which she had to be on a feeding tube to regain her strength. She was left in a weak state with effects on her memory, that is until she started tepotinib in April 2021.
DR had her fair share of ups and downs since she started her cancer journey in February 2021 as a 72 year-old retired female. Her physicians refused to start her on any treatments until they knew the exact mutation that was driving her tumours, which turned out the be the MET exon 14 alteration, and thus, was started on capmatinib a month later. It initially worked very well and was extremely successful in shrinking her tumour, resolving her cough, and reducing the swelling in her lymph nodes that was initially impeding her ability to eat and swallow. However, about 6 weeks into treatment, she developed a rare but documented side effect of the drug that left her with a full-body itch that was extremely debilitating. She was placed on anti-histamines to attempt to manage the itch which was not very effective, so she was taken off the treatment for a while, during which the itch went away and she felt better. She then tried dose reduction to ¼ dose, but the itch came back, and so did signs of disease spread to her bones and skin. This side effect was incredibly atrocious and almost unbearable for DR, until she was switched over to tepotinib in June.
Improved Outcomes
There have been many incredible advancements in lung cancer research in recent years that have changed the treatment paradigm for patients in Canada. With MET exon 14 skipping being a rare mutation that results in NSCLC tumours while still being a new discovery in lung cancer research, there has not been many previous opportunities for the development and refinement of new targeted therapy treatments for MET patients, until now. It has been seen that MET-targeted therapies, including tepotinib, have been met with incredible success that gives patients their livelihoods back and allows them to plan further down the line for a possible future that previous therapies could not give them. When choosing a therapy, some of the most crucial outcomes that patients want to have include:
Improved management of their symptoms of non-small cell lung cancer
Allowing patients to have a full and worthwhile quality of life
Having manageable side effects
Allowing patients to live longer and maintain their independence and functionality so minimize the burden on their caregivers and loved ones
Delaying disease progression and settling patients into long-term remission for improved survivorship
Experience With Drug Under Review
Prior to tepotinib, patients carried a very high symptom burden.
Prior to tepotinib, VM’s caregivers and family were feeling that she was in end of life care. While she was on another targeted therapy, it initially worked but soon had to terminate treatment once scans had shown metastases in her liver, bones, and eventually, her brain. Her health declined very rapidly and left her with severe symptoms that impeded her functionality. She was unable to talk, communicate, walk, and was bed-ridden with significant impairments in her cognitive function in that she was almost “a totally different person” to her caregivers.
While on a different treatment, LS had some significant bouts of symptom flare-ups that landed her in the hospital for two weeks for severe nausea, vomiting, and lack of appetite in that she was on a feeding tube to regain her strength and energy. Her previous treatment left her with some memory issues, severe nausea, and a possible infection before she switched to tepotinib.
LW’s cancer journey started in June 2020 and started tepotinib as his first-line of treatment. His initial symptoms that alluded to his diagnosis of NSCLC included upper back pain and numbness on one side of his chest that eventually impeded his ability to sleep comfortably. His back and chest pain was initially very severe in that he was unable to sleep through the night without taking acetaminophen every few hours to relieve the pain, but then became a more dull and persistent pain afterwards. It started to interfere with his daily life and ability to use the bathroom, and eventually, the active lifestyle he was used to leading.
DR also had significant bouts of symptom flare-ups while on other previous targeted therapies that led to her being on and off of treatments a few times during her cancer journey. She went to the ER in February 2021 for what was thought to be bronchitis, but ultimately found lung cancer and pneumonia instead. She had a chronic cough that was debilitating, shortness of breath that was addressed with cough medicine, her lymph nodes were so swollen that it impeded her ability to eat and swallow, so she was only on soft foods. Her health quickly deteriorated after diagnosis, and a week after leaving the hospital, she had a stroke that resulted from a clotting factor from the cancer. She then had a severe side effect from her previous targeted therapy that left her in a very atrocious full-body itch that continued even after dose reduction, which eventually had to be terminated. Ever since DR started tepotinib, the symptoms and side effects from the previous treatment have not returned, and is managing the treatment reasonably well. She is hopeful that her physical health will allow her to return to some level of normalcy as she continues with the treatment.
Tepotinib has seen to relieve symptoms in a relatively quick time frame.
The patients Lung Cancer Canada had interviewed for this submission had much faster disease progression than previous patients interviewed. When LW started tepotinib in October, he would need to take medication a few times through the night to relieve the pain when in bed; however, within 1-2 months of treatment, all of his initial symptoms of the cancer had resolved, with very minimal hip, chest, and back pain left, and no more numbness on one side of his body. He continues to be able to lead the active lifestyle he used to have prior to diagnosis, and has no issues going to sleep at night due to the pain.
Once she stared tepotinib, LS noticed she felt significantly better within a month or so, and hasn’t had any nausea, vomiting, or shortness of breath since she started the treatment. She is able to go for walks, care for her garden, and hopefully in the future, return to work.
Tepotinib is seen to work on brain metastases and yielded drastic improvements in functionality in patients who were previously very weak.
As seen in VM’s case, she was essentially bedridden and extremely weak with a poor quality of life before she started tepotinib. VM had a number of brain metastases prior to starting tepotinib, which left her with incredibly significant cognitive impairments in which she was unable to communicate, respond to any questions, and care for herself at all. When VM started tepotinib in July 2018, the astonishing difference it made in her was like a miracle to her family; “she was a totally different person before she started tepotinib, but once she did, she slowly came back as herself again. It was a miracle!”. Within about 6 weeks after starting treatment, she slowly made her way back to her old self and was able to regain the independence and lifestyle she once had before diagnosis. She slowly regained her energy as her brain metastasis were shrinking, and she was able to talk, communicate, walk to the bathroom, had no shortness of breath, and even called her friends on the phone for the first time in months. This paved the way for her family and friends to make up for the time they lost while her health was poor.
Within a few months, she was able to dance, socialize with friends at events, cook a Christmas meal, and walk to the bus stop with her kids before school. By the 8 month-mark, she was free of brain metastases. The extra year that VM was on tepotinib gave her back her life that she had been deprived of prior to treatment, and allowed her to enjoy the time she had missed out on with her kids, husband, family, and friends. Overtime with tepotinib, VM regained her independence and ability to perform activities of daily living and get back to caring for her family. This also relieved the burden on her caregivers, and she was able to return to being a mother and wife again, thanks to the success of tepotinib.
Tepotinib has minimal and manageable side effects.
The most common adverse events that have been documented and attributed to tepotinib include peripheral edema, nausea, diarrhea, increased blood creatinine, decreased appetite, and hypoalbuminemia, amongst other less common side effects (Paik et al., 2020). In comparison to adverse events seen with other therapies, such as chemotherapy or radiation, these are all relatively minor side effects that carry a lessened burden on the patient and the resulting impact it may have on caregivers as well.
The patients that Lung Cancer Canada had interviewed for this submission have been on tepotinib for 3 months at the minimum, and the side effects that they experienced were relatively similar to the ones listed above. Two patients noted they had edema in their extremities (wrist, legs, feet), but was manageable with compression socks and did not impede their physical activity too much. Other effects that patients have noted include constipation, fatigue, decreased appetite, minor nausea, and skeletal discomfort. One patient did note they had a potential allergic reaction to the drug; their skin became very itchy and scratchy, and easily bruised. However, it is manageable with over-the-counter Claritin and does not impact her ability to perform daily activities too often.
In DR’s case, she had developed a rare side effect of full body itches while on a different targeted therapy prior to tepotinib, which persisted even with dose reduction, and eventually had to switch to tepotinib after scans had showed disease progression. Once she started tepotinib in June 2021, the itches have subsided and never returned, which was a relief for her. However, she did have to take a 3 week hiatus from tepotinib about 2 months into treatment when she was hospitalized for severe vomiting, difficulty breathing, and low blood pressure. A chest X-ray showed her lungs were 80% full of fluid, so she had a pleurodesis procedure completed that resulted in a chest tube being placed. As of early September 2021, she is back on tepotinib and has had some skeletal discomfort that is manageable, as well as a minor cough and pleural effusion. The adverse events she experienced on tepotinib though, were much more bearable than the previous therapy’s adverse events to the point of termination.
Tepotinib helped patients get back to many of the activities they could do before diagnosis, including returning to work.
Overall, one of the most important outcomes and goals that lung cancer patients long for is to be able to return to a life that can resemble what they had pre-diagnosis. Long-term stability in their disease and potential remission is what the ultimate clinical goals are for oncologists, researchers, and clinical teams that aid these patients in getting back to a quality of life that is fulfilling. The patients that Lung Cancer Canada had interviewed were all working towards this goal of return to life, where some have already achieved this quality of life that very closely resembles what they had before diagnosis, all thanks to tepotinib.
Though she has had many ups and downs in her cancer journey since her diagnosis 7 months ago, DR has realized that tepotinib is her best chance at being able to manage her disease because the MET exon 14 skipping oncogenic mutation is so rare, that she knows she is lucky to even have a treatment like tepotinib that has been shown to be effective at targeting such a rare mutation that is driving her cancer. DR considers herself to be very athletic and has been extremely active and in good health all her life, so being diagnosed with lung cancer as a never-smoker was a real shock and “kick in the face” to her. She has continued to do physical activity as much as she can while on tepotinib, such as walking and jogging occasionally, and her ultimate goal is to be able to cycle and swim again once her health improves. Tepotinib is her ticket to normalcy, and she is hopeful for the future she has with the drug.
After starting tepotinib in April 2021, LS has been able to stay active and independent while being able to go further than just simply performing daily activities, such as socialize with friends, eat out, and spend time outdoors. Though not as often as she used to, LS still goes for walks with her dogs most days, and her energy levels are significantly better than when she had chemotherapy as her first line of treatment that she is hopeful she can return to caring for the barn animals she used to have prior to diagnosis by next summer. Returning to work as a teacher’s aid is also one of the goals LS believes she would be ready to get back to within a couple of months when her health improves and is more stable. Tepotinib has given patients like LS the ability to make plans for the future further down the line, revived hopes and dreams, and set meaningful goals for their future.
With tepotinib being his first line of treatment, LW had been able to maintain a similar level of functionality and independence that he had prior to his diagnosis with lung cancer 15 months ago. He had always been a very active individual and often spent a lot of time outdoors with his wife and family. However, one of the symptoms he experienced that led to his diagnosis of lung cancer was numbness and pain in his chest and back, and eventually interfered with his bathroom activities, in addition to not being able to physically do as much as he used to. However, he never shied away from physical activity, he was still able to walk, jog, and hike to a certain extent throughout his cancer journey because he never had any debilitating adverse events from tepotinib that interfered with his physicality.
Tepotinib has allowed patients to regain their independence and relieve the burden on caregivers.
Having regained her functionality and independence has allowed LS to rely much less on caregivers. This has allowed her husband the flexibility and relief to focus on providing for the family financially by being able to return to work out-of-province for the next few months to save up. She still has family staying close by to her so she is not completely alone if she does need extra help, but this also allows for some flexibility in their caregivers’ life to have some of the heavy burdens of caring for a patient to be taken off their shoulders.
Tepotinib helped LW maintain his level of functionality and independence in that the disease never had any major impacts on his physical abilities, since this was his very first line of treatment. This is one of the most important outcomes that patients like LW want in a treatment. He does not require much help from caregivers as much as he used to prior to starting treatment, and he even continues to be able to hike, fish, kayak, and bike often, and even went on a 50km round-trip bike ride recently. He is no longer impacted physically, and this has left him optimistic about the future with his cancer journey.
Tepotinib gave children time to accept their parent’s diagnosis.
The success that patients have seen with tepotinib also allows for families time to fully grasp and accept their loved ones’ diagnosis. While she was sick and bedridden prior to tepotinib, VM’s oldest daughter, who was 16 at the time, had to take on the extremely difficult role of being the caregiver for her two younger siblings while their father was at work. This also left incredibly significant mental and emotional effects on not only her, but also her other siblings, occasionally to the unfortunate point where VM’s youngest daughter would rather not see or speak to her mother because the thought of seeing her mother so sick was too much for her. Once VM started recovering and regaining her energy, functionality, and independence back, her eldest daughter was able to relieve a lot of the caregiver burden and weight that had been on her shoulders trying to care for her family and siblings while her mom was sick. When VM was slowly starting to recover and becoming more like herself again with the help of tepotinib, her youngest daughter started visiting and speaking to her mother again. The impact that a parents’ diagnosis can have on their children is insurmountable, and the success of tepotinib has allowed time for families to accept their loved one’s diagnosis.
The success of tepotinib positively impacted patients and caregivers’ mental health.
The worries and helpless feelings of seeing a loved one so weak from their disease are unfortunately so common in patient’s caregivers, families, and friends, and negatively impacts their mental health. For LS, being diagnosed during the holidays at the end of 2020 was a real test for her family, but she had a lot of support from friends and loved ones to help her get through her cancer journey. While on chemotherapy, she was also battling depression and had many more bad days than good where she would want to sleep all day. When she was first diagnosed, she was still trying to prove to people around her that she was still healthy, feeling well, and thus, tried to continue to do all the activities that she used to before she got sick. She pushed herself to continue to cook, exercise, care for her barn animals, and garden, but soon realized she needed to set her boundaries and had to stop. However, once she started tepotinib, the success of the drug in her case gave her the ability and energy to perform all these activities again and made her much happier once she was able to set boundaries and know when she needs to stop.
Being a retired physician, LW was used to seeing cancer patients from the other side of the desk, but never fully grasped the feelings his patients had to go through. When his diagnosis came, his only goal was to celebrate the next Christmas as he did not think he would make it. He prepared all his affairs and accepted the diagnosis right away. Now with tepotinib showing very promising results as his first treatment, he is much more optimistic and positive about his prognosis and shared how incredibly amazing it is just be able to simply take a few tablets a day and be on with his day. He used to have no appreciation for the inconveniences a cancer diagnosis and all that entails, has on his patients’ lives, but now he understands and is cautiously looking forward to the future. Tepotinib has enabled families to accept the diagnosis and given people something to look forward to in their futures and share real-life moments that had previously seemed impossible.
Tepotinib can help extend patients’ lives and expands the market for more treatment options in the case of tolerability issues.
Currently, there are two other MET-based targeted therapy drugs that have showed promising results, crizotinib and capmatinib. Crizotinib is a PAN inhibitor and does have some MET activity, but tepotinib and capmatinib have much higher affinities and specificities for MET, and thus, are the ideal therapies for MET patients. Understanding there is another MET inhibitor on the market, expanding the number of therapeutic options that are available for Canadian lung cancer patients with the reimbursement of tepotinib is critical, as the issues of tolerability and ultimately, drug resistance are ones that must be considered. Just like in both VM, LS, and DR’s cases, they initially started on other MET inhibitors prior to tepotinib, however, they were left with extremely heavy symptom burdens and did not end up working in their case. Thus, having the ability and choice to switch to another MET inhibitor, tepotinib, was critical for them as it essentially restored their functionality, independence, energy levels, and significantly improved their symptoms.
When VM’s health rapidly declined while on another MET inhibitor, it was very hard to bear for her family and they felt like all hope was lost. Her family made the extremely tough decision to refuse a clinical trial in the United States because it was too expensive, and this left them with virtually no other choice but to consider palliative care. However, the opportunity came to join the trial for tepotinib, which changed her life. Once they had started on tepotinib, VM was able to spend time with her family and friends again, communicate, exercise, and continue doing the activities she loved and missed. As seen with VM’s case, having the flexibility and freedom to be able to have another targeted therapy for MET was critical and life-changing as it meant that once she build tolerance and drug resistance to the first treatment, she still had another to fall back on. Expanding the market and paradigm of therapeutic options for lung cancer patients is critical to move forward in the field and allow patients to have a chance at curing their disease.
Companion Diagnostic Test
CL was diagnosed in September 2020 with NSCLC with the MET exon 14 skipping mutation, though her journey of getting the biomarker test to narrow down her specific treatments to a targeted therapy was one that many lung cancer patients in Canada face. 3 weeks after she had an annual check-up with her primary care physician who did not find anything through the tests she ran, CL had some back pain that led to her shocking diagnosis of lung cancer. Spots in her lungs and lymph nodes were found on her chest x-ray, which was a drastic change, as only 4 months ago in April, they could not find anything. After going through immunotherapy and chemotherapy as her first line of treatment, there was some improvement in the lung tumours, but had no effect on the ones in her lymph nodes. As a result, her physicians were suspicious of MET, but Canada did not have the biomarker testing at the time that was needed to confirm the mutation. As a result, her pathology report had to be sent to the United States to be able to test for the MET mutation, which in fact, came back positive for the rare exon 14 skipping mutation that drove her cancer. The hefty price tag that this came with was a tough choice CL had to make, but luckily she was reimbursed 50% of the cost, though still leaving her with a nearly $4000 bill. Only then were her targeted treatment options opening up as her physicians knew what they were specifically treating instead of going in blind. CL’s story is one that is so common in Canadian patients that shows the story of how comprehensive biomarker testing in Canada is urgently needed. Being able to access diagnostic testing for her specific mutation opened up the APL-101 study for her, and this targeted therapy clinical trial has since been working very successfully in her case, scans have shown shrinkage in her tumours, which has allowed her to maintain stable disease and maintain her functionality and quality of life with very minor side effects.
As CL said to Lung Cancer Canada, “we really need the facilities in Canada to be able to test for the biomarkers that drive these cancers and personalize treatment for each patient to their specific needs, so that we all just don’t go in blindly. The more testing and targeted therapies we have, the better it is for everyone. Patients get left behind when physicians don’t know what their treating”. MET testing is included in some of the provincial panels. The importance of accessible and affordable biomarker testing for all lung cancer patients in Canada cannot be overstated and is one that should be available for all.
Anything Else?
Therapies targeting the specific signatures in the cancer cell have been a paradigm shift for lung cnacer patients. Targeted therapies have helped patients beat the 19% 5-year survival rate. Tepotinib is a targeted therapy that acts on MET. Currently, capmatinib is another treatment that may soon apply for reimbursement. There is a place for both capmatinib and tepotinib in treatment as the issues of tolerability and ultimately, drug resistance are ones that must be considered.
When VM’s health rapidly declined while on another MET inhibitor, it was very hard to bear for her family and they felt like all hope was lost. Her family made the extremely tough decision to refuse a clinical trial in the United States because it was too expensive, and this left them with virtually no other choice but to consider palliative care. However, the opportunity came to join the trial for tepotinib, which changed her life. Once they had started on tepotinib, VM was able to spend time with her family and friends again, communicate, exercise, and continue doing the activities she loved and missed. As seen with VM’s case, having the flexibility and freedom to be able to have another targeted therapy for MET was critical and life-changing as it meant that once she build tolerance and drug resistance to the first treatment, she still had another to fall back on.
We also understand that CADTH will perform an economic assessment of this treatment. We ask CADTH to continue to recognise that the life-threatening situation in cancer differs from non-cancer conditions. The threshold QALY for cancer cannot be the same as non-cancer treatments. We ask CADTH to re-evaluate the threshold that is used to evaluate cancer treatments.
Finally, we continue to ask CADTH to release the draft economic and clinical reports when the initial recommendation is released. The summary is able to act as a quick reference. However, it is extremely challenging to contribute meaningful feedback unless we are allowed to see the full summary.
References
Heist, R. S., Shim, H. S., Gingipally, S., Mino-Kenudson, M., Le, L., Gainor, J. F., ... & Iafrate, A. J. (2016). MET exon 14 skipping in non-small cell lung cancer. The Oncologist, 21(4), 481-486. DOI: 10.1634/theoncologist.2015-0510
Nadler, E., Espirito, J. L., Pavilack, M., Baidoo, B., & Fernandes, A. (2020). Real-world disease burden and outcomes of brain metastases in EGFR mutation-positive non-small-cell lung cancer. Future Oncology, 16(22), 1575-1584. DOI: 10.2217/fon-2020-0280
Paik, P. K., Felip, E., Veillon, R., Sakai, H., Cortot, A. B., Garassino, M. C., ... & Le, X. (2020). Tepotinib in non–small-cell lung cancer with MET exon 14 skipping mutations. New England Journal of Medicine, 383(10), 931-943. DOI: 10.1056/NEJMoa2004407
Polanski, J., Jankowska-Polanska, B., Rosinczuk, J., Chabowski, M., & Szymanska-Chabowska, A. (2016). Quality of life of patients with lung cancer. OncoTargets and Therapy, 9, 1023–1028. DOI: 10.2147/OTT.S100685
Roth, K. G., Mambetsariev, I., & Salgia, R. (2020). Prolonged survival and response to tepotinib in a non-small-cell lung cancer patient with brain metastases harboring MET exon 14 mutation: a research report. Cold Spring Harbor Molecular Case Studies, 6(6), a005785. DOI: 10.1101/mcs.a005785
Stenger, M. (2021, March 10). Tepotinib for Metastatic NSCLC WITH MET Exon 14–skipping alterations. The ASCO Post. https://ascopost.com/issues/march-10-2021/tepotinib-for-metastatic-nsclc-with-met-exon-14-skipping-alterations/.
Wong, S. K., Alex, D., Bosdet, I., Hughesman, C., Karsan, A., Yip, S., & Ho, C. (2021). MET exon 14 skipping mutation positive non-small cell lung cancer: Response to systemic therapy. Lung Cancer, 154, 142-145. DOI: 10.1016/j.lungcan.2021.02.030
Patient Group Conflict of Interest Declaration — Lung Cancer Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures — Lung Cancer Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
EMD Serono | — | — | X | — |
Lung Health Foundation/The Ontario Lung Association
About Lung Health Foundation/The Ontario Lung Association
The Ontario Lung Association (now named Lung Health Foundation) is registered with the CADTH and pCODR (www.lunghealth.ca). The Lung Health Foundation (Ontario Lung Association) is a registered charity that assists and empowers people living with or caring for others with lung disease. It is a recognized leader, voice and primary resource in the prevention and control of respiratory illness, tobacco cessation and prevention, and its effects on lung health. The Foundation provides programs and services to patients and health-care providers, invests in lung research and advocates for improved policies in lung health. It is run by a board of directors and has approximately 46 employees, supported by thousands of dedicated volunteers.
Information Gathering
The information provided from the Lung Health Foundation in this submission was obtained from an online survey completed by 13 lung cancer patients and one caregiver, as well as two phone interviews with people currently living with lung cancer. Information on age, gender and geographical location was not collected from any of the 14 online respondents. All of the online respondents completed the survey on or before August 31, 2021. The phone interviews were conducted in September 2021 with two female lung cancer patients, one living in Ontario and the second living in Manitoba. Input from a Registered Nurse as well as a certified respiratory educator was obtained for this submission. Those individuals reviewed sections related to disease experience, experiences with available treatments and outcomes.
Statistics in response #6 were pulled from: Hong, L., Zhang, J., Heymach, J. V., & Le, X. (2021). Current and future treatment options for MET exon 14 skipping alterations in non-small cell lung cancer. Therapeutic Advances in Medical Oncology, 13, 1758835921992976.
Disease Experience
CADTH involves clinical experts in every review to explain disease progression and treatment goals. Here we are interested in understanding the illness from a patient’s perspective. Describe how the disease impacts patients’ and caregivers’ day-to-day life and quality of life. Are there any aspects of the illness that are more important to control than others?
The respondents had varying experiences with their lung cancer diagnosis. One reported they experienced no symptoms from the actual disease but the psychosocial effects of having an illness with a poor prognosis were quite debilitating. This patient described fearing that they had only 6-18 months left to live and struggled to cope because they have young children. Another respondent shared the difficulties of being diagnosed with lung cancer during the COVID-19 pandemic. Tests and treatments were delayed and this was a great source of anxiety for the patient. The patient worried about the cancer metastasizing and having a poorer prognosis as a result of the delays.
Other symptoms and challenges these patients experienced as a result of their lung cancer were shortness of breath (64%), fatigue (57%), depression (25%), cough (21%), difficulty fighting infection (21%) and chest tightness (14%). Weight loss, diminished appetite and challenges with physical and emotional intimacy were also noted by a few respondents.
When asked whether this condition affected their day-to-day life, 60% of respondents indicated that it greatly impacted their ability to complete instrumental activities of daily living, 38% indicated it negatively impacted their work, and 28% their leisure activities and hobbies.
Patients described having a challenging time maintaining relationships with families and friends. They felt short tempered and impatient and this made them feel isolated. Patients also described withdrawing from social activities because of the stigma attached to a lung cancer diagnosis. To quote one of the respondents, “I did not want anyone to know I had lung cancer, I wanted people to still have empathy for my children.”
Experiences With Currently Available Treatments
CADTH examines the clinical benefit and cost-effectiveness of new drugs compared with currently available treatments. We can use this information to evaluate how well the drug under review might address gaps if current therapies fall short for patients and caregivers. Describe how well patients and caregivers are managing their illnesses with currently available treatments (please specify treatments). Consider benefits seen, and side effects experienced and their management. Also consider any difficulties accessing treatment (cost, travel to clinic, time off work) and receiving treatment (swallowing pills, infusion lines).
The treatments tried by the respondents include surgery, radiation, chemotherapy, targeted therapy and immunotherapy. The medications tried include Cisplatin, Docetaxel, Gefitinib, Entrectnib, Alectinib, Brigatinib and Tagrisso.
The benefits experienced with the treatments were: prolonged life, delayed disease progression and a reduction in the severity of disease-related symptoms. Although these benefits were noted, most patients struggled with lingering side effects. Respondents who received surgery reported deconditioning and chronic fatigue. Some of the side effects reported from radiation were fatigue, skin changes, hair loss and tissue scarring. One patient reported that they now have COPD related to lung tissue scarring from radiation.
With oral and subcutaneous medications, the side effects reported included fatigue, nausea, vomiting, mood changes, diminished appetite, weight loss, hair loss, anemia, and neuropathy. Side effects from chemotherapy severely impacted the patients’ quality of life, ability to work and in some cases, the ability to perform activities of daily living.
When asked about challenges with access to treatment, the respondents reported that they struggled to navigate the healthcare system. In some cases, they were not clear where to go for information and support.
Respondents would not only like to see biomarker testing done earlier, but also done for all biomarkers. This will allow patients to receive targeted therapy. Some patients felt that taking treatments before biomarker testing led them to suffer unnecessarily with side effects from medications that provided no therapeutic benefit.
Improved Outcomes
Key treatment outcomes for this group of lung cancer patients include stopping or slowing the progression of the disease with minimal side effects. Patients would also like to see medications that are effective for advanced disease. Due to the poor outcomes associated with advanced disease, patients describe feeling very anxious about any sign or prospect of disease progression.
Patients state that if treatments were more effective in treating lung cancer at any stage, then a diagnosis would not feel like a “death sentence”. One of the respondents reported that after she was given a prognosis of 6-18months, she was withdrawn and struggled to cope. She stated, “I did not want to go anywhere or do anything, I just wanted to spend every last second with my children". This isolation negatively impacted her quality of life and mental well-being.
Side effects are also a great source of distress for patients. Some reported that they had no symptoms from the actual cancer but struggled with the side effects from treatment more.
Patients would like treatments with minimal side effects so that they can carry on with regular activities while on treatment. The importance of maintaining some quality of life cannot be overstated.
When choosing therapy, patients are also interested in the efficacy of the medication. One respondent commented that they would be more receptive to side effects if there was a guarantee that the medication would stop or slow down the progression of lung cancer.
Experience With Drug Under Review
No patients within this evidence group submission had experience with the medication under review.
3-4% of patients with NSCLC have METex14 mutations (Hong, L., Zhang, J., Heymach, J. V., & Le, X. (2021). Current and future treatment options for MET exon 14 skipping alterations in non-small cell lung cancer. Therapeutic Advances in Medical Oncology, 13, 1758835921992976). This mutation is associated with poorer overall survival compared to patients without the mutation, and with metastatic disease. Treatment options for this small cohort of lung cancer patients are limited and access to treatments that improve progression-free survival and maintain patient quality of life are desperately needed. Given the high rate of brain metastases in those with this mutation, patients desire treatment options that improve survival and PFS. Having variation in options is an important factor brought forth by patients.
Companion Diagnostic Test
Although patients in this submission group do not have experience with the drug under review, they did receive biomarker tests for other treatments. The majority of the respondents who went through the testing indicated they wished it had been done sooner. Depending on the stage of the cancer diagnosis, biomarker testing was not always an option at diagnosis.
One of the respondents reported that they would have been preferred to be tested for all the biomarkers in one test. They felt testing for a few at a time lengthened the process which caused additional stress and worry about disease progression.
Anything Else?
Is there anything else specifically related to this drug review that CADTH reviewers or the expert committee should know?
Not applicable
Patient Group Conflict of Interest Declaration — Lung Health Foundation / The Ontario Lung Association
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation.
Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
N/A
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 3: Conflict of Interest Declaration for Lung Health Foundation/The Ontario Lung Association
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Clinician Input
Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancers Drug Advisory Committee
About Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancers Drug Advisory Committee
OH-CCO’s Drug Advisory Committees provide timely evidence-based clinical and health system guidance on drug-related issues in support of CCO’s mandate, including the Provincial Drug Reimbursement Programs (PDRP) and the Systemic Treatment Program.
Information Gathering
This input was jointly discussed via email.
Current Treatments
Describe the current treatment paradigm for the disease.
Focus on the Canadian context. Please include drug and non-drug treatments. Drugs without Health Canada approval for use in the management of the indication of interest may be relevant if they are routinely used in Canadian clinical practice. Are such treatments supported by clinical practice guidelines? Treatments available through special access programs are relevant. Do current treatments modify the underlying disease mechanism? Target symptoms?
Response: Patients with MET positive stage IV NSCLC would received immunotherapy first-line pembrolizumab (PD-L1 > 50%), or platinum-based chemotherapy plus pembrolizumab as initial treatment – chemotherapy with either carboplatin/pemetrexed/pembrolizumab for PD-L1 less than 50% and non-squamous, or carboplatin/paclitaxel/pembrolizumab if squamous and PD-L1 less than 50%. An alternative first line therapy would be 2 cycles of platinum based chemotherapy plus nivolumab and ipilimumab. Docetaxel or clinical trials would be considered as subsequent therapy. Current treatments delay symptoms and progression, and prolong life a modest to moderate amount. For non-drug treatments, treatment consists of palliative care/supportive care, and radiation therapy for some symptomatic lesions.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Response: Prolong life, delay disease progression, delay progression of symptoms, tumor shrinkage, improved symptoms, improved PFS and improved OS
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Response: This is incurable disease with a median OS of less than 2 years. Not everyone responds to therapy and better treatments are needed. Despite advances, most patients will still progress and need additional therapies to delay death and symptoms further. The option of an oral therapy has many advantages including better efficacy.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Would these patients be considered a subpopulation or niche population? Describe characteristics of this patient population. Would the drug under review address the unmet need in this patient population?
Response: These patients are a subpopulation of advanced and metastatic NSCLC defined by the presence of a MET exon 14 skipping molecular abnormality in squamous or non-squamous patients. This is a different "targeted therapy" population than other targeted agents in lung cancer such as EGFR or ALK patients, as Met ex 14 may occur in squamous patients, and in patients with other pulmonary risk factors like smoking. The drug addresses partly an unmet need in this population.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: It is a mechanism of action unique from other available treatments, and would complement them. It would not be added simultaneously with other treatments, but would be used in addition (i.e. after or before).
Based on OH CCO-ASCO guidelines for the management of stage IV NSCLC with targetable mutations, tepotinib would be used preferentially as first line therapy – prior to standard chemo/immuno in some patients. There would be a prevalent population of patients who received prior therapy already who should be offered the drug as subsequent therapy. The drug may be used in subsequent line of treatment in others. It will likely be used prior to docetaxel therapy in most patients.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Based on high efficacy (high ORR and long PFS), tepotinib would be used as initial therapy.
In some patients it would be appropriate to recommend patients try other treatments first, but this is a clinical decision with the physician and patient. Current ASCO/CCO guidelines list this as a "may use" option in first or second line, and others as "may use" as well.
How would this drug affect the sequencing of therapies for the target condition?
Response: If used first, then chemotherapy or immunotherapy or the combination would be used subsequently. If used second, then docetaxel may be used subsequently. Retreatment would be under extremely unusual parameters. The only situation this could be considered is if a patient were to develop an independent Met ex 14 positive metastatic lung cancer while off therapy, or if a patient required to come off tepotinib for a non-progressive reason.
Which patients would be best suited for treatment with the drug under review?
Response: Patients with stage IV NSCLC with met ex 14 skipping mutation.
How would patients best suited for treatment with the drug under review be identified?
Response: Most non-squamous patients undergo next generation sequencing on their tumour, which identifies Met ex 14 with modern assays. This is usually reflex at time of diagnosis. Met ex 14, given the incidence in squamous cell patients and sarcomatoid, would ideally be tested for in squamous patients as well.
Which patients would be least suitable for treatment with the drug under review?
Response: Patients without a MET mutation abnormality or if the results are unknown
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: Yes, with current molecular testing using NGS platforms.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Response: Response rate and time until disease progression. Also improvement in patient symptoms, QoL
What would be considered a clinically meaningful response to treatment?
Response: Tumor shrinkage and improvement in disease related symptoms
How often should treatment response be assessed?
Response: Every visit with history/physical (every ~ 4-8 wks). Imaging (Xrays, CT scans) as necessary or up to physician discretion
What factors should be considered when deciding to discontinue treatment?
Response: Disease progression or intolerable side effects
What settings are appropriate for treatment with the drug under review?
Response: Community setting (oral take home cancer drug)
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
If so, which specialties would be relevant?
Response: N/A
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: This is yet another molecularly targeted therapy with high response rates and better PFS than chemotherapy
Conflict of Interest Declarations — About Ontario Health (Cancer Care Ontario) Lung and Thoracic Cancers Drug Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support to the DAC in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Gail Darling
Position: Cardiothoracic Surgeon/Ontario Cancer Lead
Date: 20-Sep-2021
Table 4: Conflict of Interest Declaration for Lung Health Foundation/The Ontario Lung Association — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
EMD Serono Canada – no COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Andrew Robinson
Position: Medical oncologist
Date: 10-Sep-2021
Table 5: Conflict of Interest Declaration for Lung Health Foundation/The Ontario Lung Association — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
EMD Serono Canada – no COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Peter Ellis
Position: Medical oncologist JCC Hamilton
Date: 13 Sep 2021
Table 6: Conflict of Interest Declaration for Lung Health Foundation/The Ontario Lung Association — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
EMD Serono Canada – no COI | — | — | — | — |
Northeast Cancer Centre – Thoracic Cancer Clinicians
About Northeast Cancer Centre – Thoracic Cancer Clinicians
We are comprised of the oncologists who treat thoracic cancers at the Northeast Cancer Centre in Sudbury ON. Our team has a focus on providing distributed treatments across twelve satellite systemic therapy sites. Our patients are predominantly older, live in more rural areas and are Francophone or Indigenous.
Our aim is to advocate for the diverse group of thoracic cancer patients in our region.
Information Gathering
Please describe how you gathered the information included in the submission.
https://www.nejm.org/doi/full/10.1056/NEJMoa2004407
Current Treatments
Response: 3-4% of all patients with NSCLC (adenocarcinoma) and 2% of patients with squamous cell carcinoma have a METex14 skipping alteration. At this time, they are treated according to non-mutated NSCLC guidelines which includes traditional systemic chemotherapy and immunotherapy.
Treatment Goals
Response: Prolong overall survival. Delay progression. Improve quality of life.
Treatment Gaps (Unmet Needs)
Response: Available treatments are not precision-care or patient-directed therapy. Patients become refractory to current treatments and the number of side effects is quite high. Treatments are needed that are molecularly-targeted and better tolerated.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Response: Patients with METex14 alterations with NSCLC. They tend to be older, female, and non-smokers.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: It could be used in 1st line, 2nd or subsequent line for metastatic NSCLC
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: At this time – if a patient had a METex14 alteration – using tepotinib at any time during their treatment would be ideal.
How would this drug affect the sequencing of therapies for the target condition?
Response: It would not. It would simply add another subsequent line of therapy
Which patients would be best suited for treatment with the drug under review?
Response: Any patient with a METex14 alteration in their NSCLC
How would patients best suited for treatment with the drug under review be identified?
Response: Using either liquid or tissue biopsy
Which patients would be least suitable for treatment with the drug under review?
Response: Patients without a METex14 alteration or patients with poor ECOG status.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: Yes – if they harbour a METex14 alteraction
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Response: PFS, OS, quality of life, response rate
What would be considered a clinically meaningful response to treatment?
Response: Reduction in symptoms, delay of progression, increased overall survival
How often should treatment response be assessed?
Response: Every 3 months or with a change in performance status
What factors should be considered when deciding to discontinue treatment?
Response: Patient performance status, adverse events (edema), disease progression
What settings are appropriate for treatment with the drug under review?
Examples: Community setting, hospital (outpatient clinic), specialty clinic
Response: Patients should be treated with this oral agent at home
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: N/A
Additional Information
Response: Tepotinib is an extremely important advance for patients who have METex14 skipping alterations. These tend to be found in patients who are older, women, and never smokers. Patients with METex14 skipping alterations may experience a poorer prognosis and they have a worse progression free survival with chemotherapy and respond poorly to immunotherapy. It is imperative that targeted treatments are developed and approved for these patients as soon as possible. Tepotinib is easy to take (once daily) and has an onset within 2-3 months of treatment. Median duration of response was 11.1 months in the VISION study. Tepotinib is generally quite safe and improved quality of life with low rates of treatment discontinuation due to AEs.
Conflict of Interest Declarations — Northeast Cancer Centre – Thoracic Cancer Clinicians
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Lacey Pitre
Position: Medical Oncologist, Lead Cancer Clinical Trials, CCO Lead Northeast region
Date: Sept. 15, 2021
Table 7: Conflict of Interest Declaration for Northeast Cancer Centre – Thoracic Cancer Clinicians — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Astra Zeneca – April 2021 | — | — | — | — |
Declaration for Clinician 2
Name: Geordie Linford
Position: Medical Oncologist
Date: September 15, 2021
Table 8: Conflict of Interest Declaration for Northeast Cancer Centre – Thoracic Cancer Clinicians — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen 2021 | — | — | — | — |
Declaration for Clinician 3
Name: Luisa Bonilla
Position: Medical Oncologist
Date: September 15, 2021
Table 9: Conflict of Interest Declaration for Northeast Cancer Centre – Thoracic Cancer Clinicians — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Lung Cancer Canada Medical Advisory Committee
Information Gathering
Information gathered for inclusion in this submission came from several sources. First, a Canadian consensus series of meetings has been held and our recommendations on the management of MET-altered NSCLC is in the process of being published at Current Oncology. In addition, a pubmed search was conducted on MET skipping and the relevant literature was reviewed. We also reviewed the abstracts from recent meeting proceedings including the ASCO and ESMO annual meetings and the World Congress on Lung Cancer from 2019-2021.
Current Treatments
Response: The current standard of care for treatment-naïve advanced or metastatic non-small cell lung cancer (NSCLC) based on reimbursement in all the provinces whose tumours harbour MET skipping alterations includes:
platinum doublet chemotherapy based on histology;
platinum doublet chemotherapy plus pembrolizumab for those with PDL-1 expression <50%, and possibly those with PDL-1 expression > 50% who are non-smokers, female, high disease or symptom burden; and
pembrolizumab alone for those with PDL-1 expression > 50%.
Options 2 and 3 are contraindicated in those who have active autoimmune disease or who have organ or bone marrow transplantation on active immunosuppressants.
For mNSCLC patients who progressed on prior systemic therapy, the options include:
Platinum doublet for those who had received pembrolizumab as first-line therapy,
Anti-PD(L)1 therapy, including pembrolizumab, nivolumab and atezolizumab, for those who had received platinum/pemetrexed as first-line therapy (but with the adoption of platinum doublet and pembrolizumab as first-line therapy, this represents a very small number of patients), and
Docetaxel for those who have progressed on platinum doublet and pembrolizumab.
MET exon 14 skipping mutant NSCLC is generally associated with aggressive disease, resistance to anti-cancer therapies, and poor prognosis when not treated with MET inhibitors (Salgia,R, et al. Cancer Treat Rev 2020, 87, 102022; Tong, J et al. Clin Cancer Res 2016, 22, 3048–3056; Lee, G. et al. J Thorac Oncol 2017, 12, 1233–1246). Several retrospective analyses have been published looking at the historical benefit of use of these standard systemic therapies specifically in patients with MET skipping aberrations. As MET skipping is uncommon, the number of patients in each study is small. In a multinational cohort of 70 patients (36 with response data), the objective response rate for platinum doublet chemotherapy was 23% (Bittoni, M et al. Lung Cancer. 2021: Vol 159 pp 96-106). No responses to immunotherapy were reported either single agent or in combination.
A broader publication looking at the use of immune checkpoint inhibitors in patients with driver mutations was published from the IMMUNOTARGET registry. This study included 36 patients with MET skipping mutations and concluded that the overall response rates (ORR) to ICIs in this group with MET skipping mutations is low, approximately 16%, with a median PFS of approximately 2–5 months although some long-term beneficiaries were seen (Mazieres, J et al. Ann Oncol 2019, 30, 1321–1328).
Colleagues from British Columbia also published their case series of 41 patients (33 with response data) identified with MET skipping aberrations (Wong, S. et al. Lung Cancer. 2021: Vol 154 pp 142-145). In patients treated with platinum doublet, objective response rate was low at 9% with stable disease achieved in 64%. In the BC cohort, the objective response rate to IO was 7% with stable disease achieved in 43%. Time to immunotherapy treatment discontinuation was on average only 2.4 months. Response rates to combination chemotherapy and IO were not reported as these therapies have only recently been made available in Canada.
Beyond the standard therapies that are described above, there are agents that are approved by Health Canada for other indications that have MET inhibiting activity. The agent with the most data is crizotinib – currently approved for use in mNSCLC for patients with ALK and ROS1 translocations. The antitumor activity and safety of crizotinib (250 mg twice daily) was assessed in the PROFILE-1001 trial that included 69 patients with advanced NSCLCs harbouring MET skipping mutations (Drilon, A. et al. Nat Med 2020, 26, 47–51). Objective response rate was 32% among 65 response-evaluable patients. The median duration of response was 9.1 months. Median PFS was 7.3 months and 54% of participants were progression-free at 6 months. Median OS was estimated at 20.5 months. The publication on the BC cohort described above, also reported on the benefits of crizotinib use in a real-world cohort. Physician-assessed response was partial response (PR) 21%, stable disease (SD) 33%, progressive disease (PD) 25% and not evaluable in 21%. The majority of these patients were treated in the 1st line setting (88% of 24 patients reported). While the data on crizotinib appears numerically superior to that of non-targeted therapies such as chemotherapy or immunotherapy, more selective inhibitors of MET with fewer off target effects have become available such as tepotinib. Crizotinib is currently only available through self-pay or private insurance in most provinces. Compassionate access was previously available but not currently.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Response: In the advanced or metastatic NSCLC setting, the goals of therapy are, in the order of priority.
Improvement in mOS: the most conclusive endpoint for all anti-cancer systemic therapy. The challenge in patients with driver mutant lung cancer has been that, with excellent response rates even in later lines of therapy, identifying OS benefits is challenging. Conduct of randomized phase 3 trials in many circumstances has not been attempted, especially when the fraction of patients with a particular molecular abnormality is low (rate for MET skipping is ~2% of mNSCLC in the BC published data). Precedence has been set with positive CADTH recommendations in ROS1, BRAF and NTRK in similar situations without randomized OS data.
Rapidity of and prolonged improvement in lung cancer related symptoms measured by median time-to-response, ORR, or progressive disease rate and mPFS: As the majority of advanced or metastatic NSCLC are symptomatic at the time of initial diagnosis and at the time of progression from prior therapy, early and prolonged symptoms improvement without disease progression radiologically will provide clinically relevant improvement in health-related quality-of-life.
Toxicity: Incidences of Grade 2 toxicity experienced daily, and Grade 3 or higher clinically important toxicity and dose reduction or dose discontinuation are especially important to consider for any systemic therapy. For one, constant grade 2 toxicity, such as nausea, vomiting, diarrhea, and so on, can negatively impact on the quality-of-life (QoL) of patients and oral medication adherence. The latter can further adversely affect the real-life efficacy or effectiveness of an oral therapy. Second, as mentioned above, advanced, or metastatic NSCLC patients have high symptom burden, which can further impair patient well-being in the setting of frequent and clinically significant toxicity.
Prevention or treatment of brain metastases: Up to 40% of advanced or metastatic NSCLC can present with brain metastases during their treatment journey. As reported by Peters et al. [Cancer Treat Rev. 2016;45(2):139-162], brain metastases have a negative impact on QoL and carry a poor prognosis. Only a small number of mNSCLC patients will be candidates for surgical resection and/or stereotactic brain radiation. The majority will be treated with whole brain radiation (WBRT), which carries significant short-term and long-term toxicity, such as immediate memory loss, loss of higher cortical function and fatigue, can negatively impair the functional status, independence and QoL of patients. Therefore, brain penetrating systemic therapy, not only treat but also prevent/delay brain metastases, improve QoL and preserve functional status of mNSCLC patients.
Resource utilization: Intravenous systemic therapy is given every 3-6 weeks, requiring resources for clinical assessment, laboratory investigation and drug administration for 1-3 hours, depending on the regimen used. But oral therapy can potentially reduce resources used, especially if there is a low incidence of grade 2 toxicity requiring clinical intervention and grade 3 or 4 toxicity. This is especially important in the Canadian setting due to clinic and chemotherapy administration space constraints.
Impact of COVID on safety on systemic therapy: With ongoing issue with COVID, oral therapy reduces the patient footprint in cancer centres, which can reduce the chance of outbreak and the exposure to potential COVID infection. Oral therapy can minimize disruption of therapy. Currently, chemotherapy, radiation and immunotherapy are considered to have increased risk for serious outcome from COVID due to their effect on the immune system, as compared to targeted agents.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Response:
Improvement in mOS: At this time, there are no randomized data of tepotinib versus standard therapy nor are there plans for this data to be generated. In the phase 2 VISION study, the median duration of overall survival was 17.1 months ((95% CI, 12.0 to 26.8) according to data that were not mature (Paik, P. et al. N Engl J Med 2020;383:931-43). In the BC cohort, median overall survival for metastatic patients treated with any systemic therapy was 15.4 months (95% confidence interval 9.3–21.6) – the majority of whom received the 1st generation MET inhibitor, crizotinib. In contrast, the median overall survival from a cohort of patients with MET skipping alterations with good performance status who were treated with standard of care that did not include MET inhibitors was 8.1 months (Awad, M, et al. J Clin Oncol 2017, 35:15_suppl, 8511).
Rapidity of and prolonged improvement in lung cancer related symptoms measured by median time-to-response, ORR, or progressive disease rate and mPFS: In the phase 2 VISION study, in treatment-naïve patients (n = 65), ORR was 44.6%, median DOR was 10.8 months (Table 2), and PFS was 8.5 months. In previously-treated patients (n = 81), ORR was 45.7%, median DOR was 11.1 months, and median PFS was 10.9 months. Responses were rapid, with onset usually within 6 weeks after the initiation of treatment. As discussed above, there is little published on the combination of chemotherapy and immunotherapy in patients with MET skipping alterations. Response rates to platinum doublet have ranged between 9-23% in the retrospective series discussed above which is lower than what would be expected from patients with wild type NSCLC. The response rate to immunotherapy is also lower than what would be expected from patients with wild type NSCLC.
Toxicity: Side effects of tepotinib are similar to other agents with activity against MET. The majority of side effects are grade 1 and 2. The data from a presentation of the VISION data at the European Society of Medical Oncology (ESMO) Annual meeting in 2020 by Mazieres, J, et al – detailing 255 patients treated with tepotinib. Peripheral edema is identified in the majority of patients. GI side effects such as nausea and diarrhea are reported in 20% of patients similar to many other TKIs. Laboratory toxicities include elevated creatinine, low albumin and asymptomatic elevations in amylase / lipase. Treatment related adverse events led to dose reductions in 27.8% of patients and to permanent discontinuations in 10.6% of patients. Treatment discontinuation for chemotherapy in NSCLC is typically between 10-11% as reported in recent first line trials comparing immunotherapy and chemotherapy or the combination of chemotherapy and immunotherapy versus chemotherapy (Keynote 24 and 189, respectively). The health related quality of life data has also been presented by Garrasino, M. et al at ESMO 2020. Improvements in cough, dyspnea and chest pain that persisted for over 24 weeks were identified.
Prevention or treatment of brain metastases: Clinical data on the efficacy of tepotinib in patients with brain metastases is limited but has been seen in preclinical models. Clinical data was presented at the ESMO 2020 Annual Meeting by Vitari, S, et al as well as by Patel, J et al at the American Society of Clinical Oncology Annual meeting in 2021. The majority of the patients included in these presentations had prior treatment with radiation for their brain metastases. The limited data suggest similar intracranial activity as extracranial activity. Further data on the efficacy of tepotinib in patients with brain metastases is anticipated from the VISION C cohort.
Resource utilization: Tepotinib is an orally administered agent that will utilize no chemotherapy administration services. Although clinical assessments for toxicity and response are needed, these assessments can occur with virtual or in-person clinic visits with supplemental laboratory evaluations. The majority of patients who receive TKIs are assessed twice per month during the initial 2 months of treatment followed by monthly or bi-monthly evaluations. This is significantly less than what is experienced by patients treated with chemotherapy or immunotherapy or the combination.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Response: The population with the greatest unmet need is that with mNSCLC with MET skipping alterations. METex14 skipping mutations are detected in 2–4% of lung adenocarcinoma cases (Liang, H, et al. Onco Targets Ther 2020, 13, 2491–2510). These mutations usually occur in older patients (median age of 72 years) with a higher percentage of ever-smokers compared to patients with tumours harbouring other oncogenic alterations such as EGFR/ALK/ROS1 (Schrock, A, et al. J Thorac Oncol 2016, 11, 1493–1502). A high frequency of MET skipping mutations has been reported in the NSCLC non- squamous subtype of pulmonary sarcomatoid carcinoma ranging from 5%–32% of patients (Liu, X. et al. J Clin Oncol 2016, 34, 794–802; Saffroy, R. et al. Oncotarget 2017, 8, 42428–42437). METex14 skipping mutations have also been found in a very small percentage (1%) of patients with squamous cell carcinoma (Lam, V et al. Clin Lung Cancer 2019, 20, 30–36). Patients with MET skipping mutations have a high frequency of multifocal and extrathoracic metastases – mainly to the bone, brain, and adrenal glands (Digumarthy, R. et al. Cancers (Basel) 2019, 11, 2033). mNSCLC patients with MET skipping mutations would be considered a niche population. Tepotinib would address the unmet need in this population.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: Tepotinib would be the first drug specifically approved for patients with MET skipping driver mutations. Standardly we use targeted therapies for driver mutations as single agents as is the case with tepotinib. We would not recommend adding it to other treatments. This would be a new line of treatment and our clinical preference would be to use it in a line-agnostic fashion allowing both treatment naïve and pre-treated patients access. This would be a shift in treatment paradigm as right now the minority of Canadian patients who are found to have MET skipping mutations can access therapy given lack of approved agents or consistent provincial funding.
Newly diagnosed MET skipping mutated NSCLC:
1st line: Tepotinib
2nd line: It is unclear whether the most optimal second-line therapy should be platinum doublet or platinum doublet plus pembrolizumab as minimal data is available
3rd line: Docetaxel, or anti-PD(L)1 therapy for those who have not received such agents in prior lines of therapy can be considered
Previously treated MET skipping mutated NSCLC: Based on the efficacy (ORR, mPFS, DOR), patients who were identified to have MET skipping mutations after receiving prior therapy should receive tepotinib given very low response rates to agents such as docetaxel or single agent IO.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Based on the treatment paradigm for mNSCLC of offering our best therapy as early as possible, those with a driver mutation should be treated with the corresponding tyrosine kinase inhibitor based on higher ORR, longer mPFS and intracranial activity, MET skipping mutated mNSCLC should be treated with a MET targeted agent, such as tepotinib, once the driver mutation is documented. To date, all available systemic therapy for mNSCLC, including chemotherapy, anti-PD(L)1 therapeutics and their combinations have not demonstrated better outcome or toxicity profile.
How would this drug affect the sequencing of therapies for the target condition?
Response:
Newly diagnosed mNSCLC with MET skipping: Based on the ORR and mPFS of tepotinib relative to chemotherapy, immunotherapy, or their combinations, tepotinib should be offered as first-line therapy in all newly diagnosed mNSCLC patients with MET skipping alterations with a score of ECOG 0-3.
Previously treated mNSCLC with MET skipping: The efficacy of tepotinib is similar in patients who are treatment naïve and those who have had prior chemotherapy treatment. Patients with MET skipping alterations who have previously been treated with systemic treatment should also be offered treatment with tepotinib given the superior response rate in comparison to that expected from single agent treatment with immunotherapy or docetaxel.
Which patients would be best suited for treatment with the drug under review?
Response: The most appropriate mNSCLC patients to be treated with tepotinib are those whose tumour (histology or cytology) or circulating tumour DNA in blood have documented MET skipping alterations detected by a validated molecular diagnostic preferably next generation sequencing (NGS) ideally with RNA capability. MET skipping alterations can be detected with amplicon based DNA panel NGS as well as single analyte strategies such as qRT-PCR or RNA in situ hybridization. DNA based NGS panels can detect MET skipping alterations but often do not have adequate coverage to capture all clinically relevant mutations – thus they are not preferred. Single analyte testing would be prohibitively costly given the frequency of this aberration and would only be recommended for confirmation in a case by case scenario. MET immunohistochemistry has poor correlation with MET skipping mutations detected through other means and is not recommended.
How would patients best suited for treatment with the drug under review be identified?
Response: Ideally, all advanced and metastatic NSCLC, regardless of histological subtype, should have either tumour or blood tested for MET skipping mutations. MET skipping alterations are most commonly identified in patients with adenocarcinomas as well as sarcomatoid lung cancers. MET skipping mutations are also identified in squamous mNSCLC patients but less commonly so than in adenocarcinomas – many of these patients with MET skipping squamous lung cancers will be light or never smokers who are already recommended for molecular testing in the guidelines. Every province either has implemented or will be implementing NGS including MET skipping due to cost effectiveness over gene-by-gene molecular diagnostics including EGFR, ALK, ROS1, BRAF V600E, HER-2, KRAS, NTRK 1-3 and RET. Thus, the molecular diagnostic for MET mutation is not likely be an obstacle for access to tepotinib. In some circumstances this testing is provincially funded and in others it is through philanthropic or research dollars. Consideration should be given to appropriate ongoing funding for pathology and molecular genetics to support testing across all these indications listed above.
We do recommend repeat NGS testing in those patients with preserved performance status after initial treatment who have had panels that did not test for the breadth of genes above. Most of the genes that would have been missed are those that require RNA based NGS testing which is the current technology being validated and implemented nation-wide.
Blood testing for MET skipping was utilized in the VISION clinical trials. Patients who were identified through testing of circulating tumour DNA in the blood had nearly identical response rates to those tested on tumour tissue (48 vs 50%). In patients where archival tissue has been exhausted, NGS testing of circulating tumour DNA would be a less morbid alternative to re-biopsy with subsequent tissue testing and should be strongly considered for nationwide validation and funding. Multiple agents are currently either under review or have been recently reviewed that will require re-testing of patient’s archival samples. Strategies should be put into place to facilitate testing protocols such as NGS testing of “liquid” biopsies to reduce the need for re-biopsy. Access to biopsies is already a limited resource across the country.
Which patients would be least suitable for treatment with the drug under review?
Response: The benefit of tepotinib is demonstrated in NSCLC patients with MET skipping alterations both in the treatment-naïve and previously treated setting. It is debatable whether ECOG 3-4 patients with MET skipping would benefit. Given tumour shrinkage was reported in 89% of patients in the VISION trial with rapid onset of that response (majority within 6 weeks), one can argue that those with ECOG 3 can be offered tepotinib. Only MET skipping mutation negative NSCLC patients and MET skipping mutant NSCLC with ECOG 4 will not be candidate for tepotinib.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review? If so, how would these patients be identified?
Response: The only known predictor for response is the presence of MET skipping alterations identified in either tissue or blood. Other further refinement of those most likely to exhibit a response beyond that subgroup are unknown.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? Are the outcomes used in clinical practice aligned with the outcomes typically used in clinical trials?
Response: CT imaging is the standard with response determined by the treating physician. This assessment modality would be similar to investigator assessed treatment response in clinical trials.
What would be considered a clinically meaningful response to treatment?
Response: In clinical practice, the definition of a clinically meaningful response to anti-cancer therapy such as tepotinib is defined as:
documentation of lung cancer-related symptoms stabilization or improvement by frequency and severity with or without radiological evidence of tumour shrinkage, or
documentation of radiographic reduction of documented sites of known disease at baseline.
How often should treatment response be assessed?
Response: Response to other oral tyrosine kinase inhibitors in Canada is typically assessed every 2-3 months. Given the initial response to tepotinib is usually seen by 6 weeks, this standard would be appropriate.
What factors should be considered when deciding to discontinue treatment?
Response: In clinical practice, tepotinib will continue until one or more of the following conditions is/are fulfilled:
Toxicity despite multiple dose reductions
Patient wishes
Concurrent medical condition(s) that will jeopardize patient safety while receiving tepotinib
Disease progression except:
those who have oligoprogression that are amendable to local therapy such as radiation or surgery. Based on study by Gomez et al. from MD Anderson Cancer Centre, patients who experienced oligoprogression had an improvement in both mPFS (14.2 months versus 4.4 months. P=0.022) and mOS (37.6 months versus 9.4 months, p=0.034) with aggressive local therapy over observation or continuation of systemic therapy. See Canadian consensus statement on this (Laurie, S. et al. Curr Oncol. 2019 Feb;26(1):e81-e93)
those who have newly diagnosed or progression of brain metastases who should continue with tepotinib after receiving brain radiation or surgery if appropriate.
those who have asymptomatic disease. Our Canadian (and global practice) for patients with driver mutations is to continue treatment until there is no longer clinical benefit as represented by overt progression on imaging associated with increased symptom burden.
What settings are appropriate for treatment with the drug under review?
Response: Tepotinib would typically be given in a community setting in a patient’s home as it is an oral anti-cancer therapy.
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: N/A
Conflict of Interest Declarations — Lung Cancer Canada Medical Advisory Committee
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
None.
Declaration for Clinician 1
Name: Dr Rosalyn Juergens
Date: November 12, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Honoraria
Table 10: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 1
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Bristol-Myers Squibb | Advisory role and honoraria | X | — | — | — |
Astra Zeneca | Advisory role and honoraria | — | X | — | — |
Merck Sharp and Dohme | Advisory role and honoraria | X | — | — | — |
Roche | Advisory role and honoraria | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 2
Name: Geoffrey Liu
Date: November 11, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Research/educational grants
Table 11: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 2
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Takeda Canada | Advisory Board, Health Technology Assessment Submission Advice, Speaker’s Bureau, past 10 years | — | — | X | — |
Takeda Canada | (To institution, not individual) Observational Study funding, past 10 years | — | — | — | X |
Hoffman La Roche | Advisory Board, Health Technology Assessment Submission Advice, past 10 years | — | — | X | — |
Pfizer | Advisory Board, Health Technology Assessment Submission Advice, part 10 years | — | — | X | — |
AstraZeneca | Advisory Board, Health Technology Assessment Submission Advice, Speaker’s Bureau, past 10 years, | — | — | X | — |
AstraZeneca | (To institution, not individual) Observational Study funding, past 10 years | — | — | — | X |
Bristol Myers Squibb | Advisory Board | X | — | — | — |
Boehringer Ingerheim | (To institution, not individual) Observational Study funding, past 10 years | — | — | X | — |
Abbvie | Advisory Board, past 10 years | — | X | — | — |
Merck | Advisory Board, Health Technology Assessment Submission Advice, past 10 years | — | X | — | — |
EMD Serono | Speaker’s Bureau, past 10 years | X | — | — | — |
Novartis | Advisory Board,past 10 years | — | — | X | — |
Glaxo Smith Kline | Advisory Board, past 10 years | — | X | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review? If yes, please list them in the following box.
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 3
Name: Barbara Melosky
Date: November 10, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Table 12: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 3
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Novartis | Advisory Board | X | — | — | — |
Roche | Advisory Board | X | — | — | — |
Merck | Advisory Board | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No, I do not have holdings or other interests in organizations that may have a direct or indirect interest in the drug under review.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups
No, I do not have personal or commercial relationships either with a drug or health technology manufacturer or other interest groups.
Declaration for Clinician 4
Name: Dr Paul Wheatley-Price
Date: November 11, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Table 13: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 4
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Astra Zeneca | Advisory Role | — | X | — | — |
Boehringer Ingeiheim | Advisory Role | X | — | — | — |
Bristol-Myers Squibb | Advisory Role | X | — | — | — |
Merck | Advisory Role | — | X | — | — |
Novartis | Advisory Role | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 5
Name: Dr Jeffrey Rothenstein
Date: November 12, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Honoraria
Table 14: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 5
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Roche | Advisory Role and Honoraria | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 6
Name: Dr Nicole Bouchard
Date: November 12, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Conference attendance
Research/educational grants
Table 15: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 6
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Astra Zeneca | Advisory Role/Conference | X | — | — | — |
Bristol-Myers Squibb | Advisory Role/Research | X | — | — | — |
Merck | Advisory Role /Research/Conference | X | — | — | — |
Bayer | Advisory Role | X | — | — | — |
Pfizer | Conference/Research | X | — | — | — |
Roche | Advisory Role | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
Yes. Expert for INESSS (diagnosis and treatment for Lung Cancer in Quebec)
Declaration for Clinician 7
Name: Dr Normand Blais
Date: November 11, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Table 16: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 7
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Novartis | Medical advisor | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
N/A
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
N/A
Declaration for Clinician 8
Name: Dr David Dawe
Date: November 13, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Honoraria
Research/educational grants
Table 17: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 8
Name of Organization | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
AstraZeneca | Advisory boards | X | — | — | — |
Merck | Advisory Boards | X | — | — | — |
AstraZeneca | Research Grant | — | — | X | — |
Boehringer-Ingelheim | Honoraria | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 9
Name: Dr Mahmoud Abdelsalam
Date: October 16, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Honoraria
Travel grants
Table 18: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 9
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
BMS | Advisory role, Honoraria and travel grants | — | X | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review? If yes, please list them in the following box.
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 10
Name: Dr Stephanie Snow
Date: November 12, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Research/educational grants
Table 19: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 10
Bristol-Myers Squibb | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Amgen | Advisory Role | X | — | — | — |
Astra Zeneca | Advisory Role | — | — | X | — |
Astra Zeneca | Research Grant | X | — | — | — |
Bayer | Advisory Role | — | X | — | — |
Bristol-Myers Squibb | Advisory Role | — | — | X | — |
Eisai | Advisory Role | X | — | — | — |
Merck | Advisory Role | — | — | X | — |
Novartis | Advisory Role | X | — | — | — |
Pfizer | Advisory Role | X | — | — | — |
Purdue | Advisory Role | X | — | — | — |
Roche | Advisory Role | — | — | X | — |
Taiho | Advisory Role | — | X | — | — |
Takeda | Advisory Role | — | X | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 11
Name: Parneet Cheema
Date: November 21, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Honoraria
Table 20: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 11
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Bristol Myers Squibb | Advisory board/Honoraria | X | — | — | — |
Merck | Advisory board/Honoraria | X | — | — | — |
Astrazeneca | Advisory board/Honoraria | X | — | — | — |
Roche | Advisory board/Honoraria | X | — | — | — |
Novartis | Advisory board/Honoraria | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 12
Name: Dr. Donna Maziak
Date: September 12, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
No.
What form of payment did you receive?
N/A
Table 21: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 12
Bristol-Myers Squibb | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
No COI | — | — | — | — | |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 13
Name: Dr. Sunil Yadav
Date: September 12, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Honoraria
Table 22: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 13
Bristol-Myers Squibb | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Bristol-Myers Squibb | Advisory Board | X | — | — | — |
Astra Zeneca | Advisory Board and Speaking | X | — | — | — |
Merck | Advisory Board and Speaking | — | — | X | — |
Roche | Advisory Board and Speaking | — | X | — | — |
Takeda | Advisory Board and Speaking | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review?
No.
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups?
No.
Declaration for Clinician 14
Name: Dr. Callista Phillips
Date: November 25, 2020
Payment Received
Have you received any payments over the previous two years from any company or organization that may have a direct or indirect interest in the drug under review?
Yes.
What form of payment did you receive?
Advisory role (e.g., advisory boards, health technology assessment submission advice)
Honoraria
Table 23: Conflict of Interest Declaration for Lung Cancer Canada Medical Advisory Committee — Clinician 14
Company | Nature or description of activities or interests | Check Appropriate Dollar Range | |||
|---|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | ||
Astra Zeneca | Advisory Board Stage 3 NSCLC | X | — | — | — |
Bayer | National Consultancy meeting and Train the Trainer- Larotrectenib in NTRK fusion positive cancers | X | — | — | — |
Roche | Lung regional Consultancy meeting | X | — | — | — |
Holdings or Other Interests
Have you received or are in possession of stocks or options of more than $10,000 (excluding mutual funds) for organizations that may have a direct or indirect interest in the drug under review? If yes, please list them in the following box.
N/A
Affiliations, Personal or Commercial Relationships
Do you have personal or commercial relationships either with a drug or health technology manufacturer (including the manufacturer’s parent corporation, subsidiaries, affiliates, and associated corporations) or other interest groups? If yes, please provide the names of the companies and organizations, and outline the nature of these relationships, in the following box.
N/A
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.