Drugs, Health Technologies, Health Systems
Health Technology Review
Newborn Screening for Metachromatic Leukodystrophy
This multi-part report includes:
Executive Summary
Recommendation
Evidence Review
Executive Summary
Executive Summary
What Is Newborn Screening?
Newborn screening is a population-level public health intervention that involves testing done shortly after birth to check for serious and rare conditions during an early or asymptomatic stage. The goal of newborn screening is to enable earlier detection of serious and rare conditions to facilitate early treatment that may lead to improved health outcomes in newborns and young children. In Canada, newborn screening activities are within the scope of the provincial and territorial governments, as they provide health service delivery for their residents.
What Is the Purpose of This Recommendation and Evidence Review?
In support of the Government of Canada’s National Strategy for Drugs for Rare Diseases, Canada’s Drug Agency (CDA-AMC) convened the Newborn Screening Advisory Panel to develop pan-Canadian guidance and recommendations, which were published in March 2025. As part of their guidance, the Newborn Screening Advisory Panel recommended that a pilot evidence review be undertaken in the short term (i.e., 1 to 2 years) to inform recommendations concerning whether to add a condition to the Recommended Pan-Canadian Newborn Screening List.
The Newborn Screening Advisory Panel recommends 6 guiding principles as a core set of values to guide work related to newborn screening in Canada: the health rights of the newborn; equity; effectiveness, safety, and quality; transparency; collaboration; and sustainability. The Newborn Screening Advisory Panel considered the health rights of the newborn to be an overarching principle. Ethical justification for newborn screening rests on the expectation that all newborns who are screened can reasonably access timely, effective, and publicly funded diagnosis, treatment, and follow-up interventions and services, when appropriate, within the health system in which screening is offered.
What Is Metachromatic Leukodystrophy?
Metachromatic leukodystrophy (MLD) is a rare inherited condition that causes nerve damage. Depending on an individual’s age at the time of symptom development, metachromatic leukodystrophy is broadly classified as either early-onset or late-onset.
In its early-onset forms (i.e., late infantile and early juvenile), MLD progresses rapidly, causes substantial disability, and results in premature mortality. Children with early-onset MLD are rarely identified before symptom development. This can result in a long journey to receive a diagnosis and can limit timely access to supportive care or, where available, effective treatment. Identifying affected individuals before symptoms develop may provide benefit by offering timely access to available treatment.
Late-onset MLD (i.e., late juvenile and adult) often involves a slower progression that occurs over the course of years and can result in significant morbidity and premature mortality.
MLD was identified as an emerging condition of relevance for newborn screening in Canada in consultation with key experts from the newborn screening community in Canada and the Newborn Screening Advisory Panel. The recent development of a potential disease-modifying gene therapy for early-onset MLD has led jurisdictions to consider the addition of MLD to newborn screening programs internationally. This gene therapy has not yet been submitted for regulatory approval in Canada. MLD is not currently screened for in any jurisdiction in Canada as part of newborn screening programs.
How Did CDA-AMC Evaluate Newborn Screening for MLD?
CDA-AMC conducted an evidence review on the benefits and harms of newborn screening for MLD by considering the Newborn Screening Advisory Panel’s guidance and recommendations, reviewing the literature, and considering evidence from a wide range of sources, including input from experts, patient organizations, and interested parties. The evidence review informed deliberations by the Health Technology Expert Review Panel (HTERP) to make recommendations as to whether MLD should be added to the Recommended Pan-Canadian Newborn Screening List.
What Is HTERP?
The mandate of HTERP is advisory in nature and is to participate in the development of guidance or recommendations for CDA-AMC projects on medical devices, diagnostic tests, and clinical interventions, inclusive of models and programs of care.
HTERP comprises 7 core members (1 chair, 1 ethicist, 1 health economist, 1 patient member, 2 health care practitioners, and 1 health technology assessment specialist) who serve for all topics under consideration during their term of office. In addition to these core members, HTERP also includes up to 5 expert members appointed to provide their expertise on a specific topic. To develop the recommendations on newborn screening for MLD, HTERP appointed 2 clinicians with expertise in neurogenetics and leukodystrophies, 2 members with expertise in newborn screening programs, and 1 member who was able to speak to the perspectives of caregiving for people with MLD.
Recommendation
Health Technology Expert Review Panel Recommendation
the treatment is reviewed and approved for use by Health Canada
the treatment receives a recommendation in favour of reimbursement from a health technology assessment body in Canada
the treatment is publicly funded.
To make this recommendation, HTERP considered the following information:
the Evidence Review by CDA-AMC
input received from an open call that included responses from a family member, a patient group, a clinician, and a pharmaceutical company
the Newborn Screening Advisory Panel’s “Toward a Future Pan-Canadian Coordinated Approach for Newborn Screening: A Report From the Advisory Panel” guidance document.1
HTERP acknowledges the following:
Disease-modifying gene therapy: One of the Newborn Screening Advisory Panel’s recommendations indicates that newborn screening (NBS) should be for those conditions for which there are effective treatments available to individuals who receive screening. While treatment for early-onset MLD is not available in Canada, HTERP recognized that it is important to prepare for the potential addition of conditions to the Recommended Pan-Canadian Newborn Screening List in support of the National Strategy for Drugs for Rare Diseases.
Costing information: As part of the CDA-AMC Evidence Review, a budget impact analysis was conducted to support HTERP’s deliberations. However, the unique complexities of NBS contexts within each jurisdiction in Canada, the time frame, and the available evidence meant that the cost estimates of the budget impact analysis had limitations and uncertainties. The cost of implementation and potential opportunity costs were therefore not factored into the recommendation made by HTERP.
Capacity and infrastructure needs: The addition of MLD to the Recommended Pan-Canadian Newborn Screening List may pose disproportionate challenges related to screening capacity and infrastructure needs in different jurisdictions, and there remains uncertainty about the relative affordability for NBS programs across the country.
Rationale for the Recommendation
Table 1 provides an overview of HTERP’s rationale for their recommendation following CDA-AMC’s multicriteria deliberative framework,2 in accordance with the Newborn Screening Advisory Panel’s recommended criteria for adding a condition.
Table 1: HTERP’s Rationale for the Recommendation
Advisory panel’s recommended criteria for adding a condition | CDA-AMC deliberative framework domains |
|---|---|
The condition:
| Unmet clinical need:
|
Test:
Treatment:
| Clinical value:
|
Other considerations:
| Economic considerations, distinct social and ethical considerations, and impact on health systems:
|
CDA-AMC = Canada’s Drug Agency; HTERP = Health Technology Expert Review Panel; MLD = metachromatic leukodystrophy; NBS = newborn screening.
Additional Guidance
HTERP recognizes that there may be implementation challenges with the addition of MLD to provincial and territorial NBS panels; therefore, HTERP has outlined the following considerations to support health systems' readiness.
The Condition (MLD)
Case definitions should be developed and refined to establish consensus regarding subtypes of the condition. MLD is typically classified into 4 subtypes: late infantile, early juvenile, late juvenile, and adult. Consensus is required to better differentiate early-onset and late-onset forms.
Screening is expected to identify late-onset and uncertain-onset cases, in addition to early-onset cases. Additional screening data will help clarify these phenotypes and inform appropriate management pathways.
The Test
Three-tier screening algorithms (i.e., sulfatide levels, followed by ARSA enzyme activity, followed by molecular testing) should be implemented and used to achieve high specificity and reduce false-positive results.
There should be ongoing adjustment of screening test thresholds to determine an optimal balance (i.e., maximizing positive predictive value while maintaining perfect sensitivity) of sensitivity and specificity in real-world conditions.
The Treatment
Clinical pathways and protocols should be developed for monitoring and managing patients with late-onset disease who are identified by NBS, in addition to those with early-onset disease.
Other Considerations
Additional capacity and investments in infrastructure (e.g., equipment and new processes) may be required, including the development and implementation of procedures to manage second-tier ARSA enzyme activity.
Equity and accessibility should be considered in the design and implementation of screening programs for MLD.
Real-world data should be collected to assess the impact of adding MLD to NBS programs by comparing outcomes (e.g., new MLD cases, earlier age of diagnosis, health care utilization) between prescreening and postscreening contexts.
Continue to follow best practice standards for consent for NBS in Canada.
With uncertainty in costing information, system preparedness should include assessment of readiness with intentional, advance preparation that includes engagement and collaboration across NBS programs, clinicians, and patient groups.
HTERP Core Members
Aude Motulsky, Health Technology Assessment Specialist, Quebec
Brian Chan, Health Economist, Ontario
Duncan Steele, Ethicist, Alberta
Leslie Anne Campbell, Chair, Nova Scotia
Prachi Khanna, Patient Member, British Columbia
Sandor Demeter, Health Care Practitioner, Manitoba
Tasleem Nimjee, Health Care Practitioner, Ontario
No conflicts of interest were identified or reported by the core or expert members.
References
1.CDA-AMC. Toward a Future Pan-Canadian Coordinated Approach for Newborn Screening: A Report From the Advisory Panel. 2025: https://www.cda-amc.ca/sites/default/files/DRD/HC0079-NBS_Recommendations_Report.pdf. Accessed 23 May/25.
2.CDA-AMC. Expert Committee Deliberation at Canada’s Drug Agency. 2025: https://www.cda-amc.ca/sites/default/files/MG%20Methods/expert_committee_deliberation.pdf. Accessed 16 January 2026.
Evidence Review
Abbreviations
AE
adverse event
allo-HSCT
allogeneic hematopoietic stem cell transplant
AO
adult onset
arsa-cel
atidarsagene autotemcel
BIA
budget impact analysis
CDA-AMC
Canada’s Drug Agency
CI
confidence interval
DB
double blind
DBS
dried bloodspot
EJ
early juvenile
ES
early symptomatic
HTERP
Health Technology Expert Review Panel
LC-MS/MS
liquid chromatography tandem mass spectrometry
LI
late infantile
LJ
late juvenile
LSD
lysosomal storage disorder
MLD
metachromatic leukodystrophy
MoH
Ministry of Health
NBS
newborn screening
NH
natural history
NPV
negative predictive value
PP
per protocol
PPV
positive predictive value
PS
presymptomatic
RCT
randomized controlled trial
RR
relative risk
SAE
serious adverse event
SD
standard deviation
UPLC-MS/MS
ultra-performance liquid chromatography tandem mass spectrometry
Evidence Review
This evidence review, led by Canada’s Drug Agency (CDA-AMC), was a foundational piece of information used to inform the Health Technology Expert Review Panel (HTERP)’s deliberations and recommendation.
Summary
What Did We Do?
We sought to action the Newborn Screening Advisory Panel’s recommended criteria and evidence review process for adding a condition to the Recommended Pan-Canadian Newborn Screening List and considered evidence from a wide range of sources, including input from experts and interested parties, to assess the:
potential benefits and harms associated with newborn screening (NBS) for metachromatic leukodystrophy (MLD)
accuracy of proposed screening algorithms for MLD
comparative impact of initiating treatment in asymptomatic, rather than symptomatic, individuals
availability and acceptability of NBS for MLD
implementation considerations associated with adding MLD to NBS
budget impact of implementing NBS for MLD up to the point of diagnosis
social and ethical considerations associated with the potential addition of MLD to NBS.
What Did We Find?
Early-onset (i.e., late infantile [LI] and early juvenile [EJ]) MLD is the most severe form of the disease, characterized by rapid neurodegeneration and greatly reduced life expectancy, and accounts for an estimated 75% to 90% of cases. The birth prevalence of MLD (including all subtypes) is estimated at less than 1 per 100,000 live births worldwide. No central registry has been identified to track incidence and subtype distribution in Canada.
We were unable to identify direct, comparative clinical evidence on the benefits and harms of universal NBS for MLD versus no screening.
Existing evidence suggests that both 2-tier and 3-tier screening algorithms can detect early-onset MLD with high specificity. A 3-tier approach (i.e., sulfatide levels, followed by ARSA enzyme activity, followed by molecular testing) appears to be the most effective, with a demonstrated positive predictive value (PPV) of 100% and a false-positive rate of 0% in included studies.
Across input gathered for this review from clinical experts, NBS laboratory professionals, and the MLD community, there was general support for expanding NBS to include MLD. However, most emphasized that implementation should not proceed without effective, disease-modifying treatment for early-onset disease in Canada.
Currently there are no disease-modifying treatment options approved in Canada for LI MLD, the most severe form of early-onset disease. While allogeneic hematopoietic stem cell transplant (allo-HSCT) appears to have modest benefits in presymptomatic (PS) EJ MLD, clinical experts indicated that this is not typically considered an appropriate treatment option for early-onset disease. There is a potential disease-modifying gene therapy for early-onset MLD (atidarsagene autotemcel [arsa-cel]). However, it has not yet been submitted for regulatory review in Canada; therefore, evidence of its effectiveness was not assessed as part of this review.
Adding MLD to NBS would require new laboratory and specialist capacity, adding operational and capital costs estimated to range between $4.5 million and $30.1 million for screening and diagnosis across Canada (excluding Quebec) over 10 years (ranging from $84,686 to $832,443 per additional potentially treatable patient). These costs are unlikely to be uniform across jurisdictions and are highly sensitive to assumptions regarding ability to share laboratory and specialist capacity across jurisdictions, as well as the incidence rate of MLD.
Individuals affected with late-onset and indeterminate MLD will be identified during screening for early-onset MLD. Existing prevalence data suggest that early-onset disease will account for the majority of newly identified individuals; however, true prevalence is uncertain and estimates may shift if universal NBS for MLD is implemented. As a result, the potential addition of MLD to the Recommended Pan-Canadian Newborn Screening List raises questions about the appropriate scope of benefits (i.e., whether identification of late-onset or indeterminate MLD should be considered a benefit or a harm). Relatedly, it raises questions of whether potential burdens of prolonged monitoring are justified, given that late-onset and indeterminate findings are not immediately actionable.
Approach
Context
NBS enables earlier detection of serious conditions and rare diseases to offer early treatment, with the aim of improving health outcomes in infants and children. NBS involves testing newborns, often by collecting a small amount of blood from the baby’s heel, to identify treatable conditions at an early or asymptomatic stage. As part of public health prevention, it aims to screen all newborns in the general population to identify affected individuals to initiate treatment before the onset of potentially irreversible symptoms.1
NBS activities in Canada are within the scope of the provincial and territorial governments, as they provide health service delivery for their residents. Because each province and territory governs its own mandate, NBS policies, practices, and processes are not uniform across the provincial and territorial programs.2 As a part of the Government of Canada’s National Strategy for Drugs for Rare Diseases, CDA-AMC convened a Newborn Screening Advisory Panel to provide guidance to enhance the pan-Canadian coordination of NBS, including the consistency of conditions screened for in newborns across Canada.
In March 2025, the Newborn Screening Advisory Panel made a set of nonbinding short-term and long-term recommendations across 7 areas,3 including a Recommended Pan-Canadian Newborn Screening List of 25 conditions for NBS and recommendations to pilot the recommended criteria and processes for adding a condition to the pan-Canadian list. The Newborn Screening Advisory Panel, along with interested parties who provided input, also proposed a set of 29 emerging conditions of relevance for NBS for monitoring and potential assessment. MLD was 1 of the identified emerging conditions.
CDA-AMC actioned the Newborn Screening Advisory Panel’s recommendation and conducted a pilot evidence review to support HTERP make a recommendation as to whether MLD should be added to the Recommended Pan-Canadian Newborn Screening List. It was selected for this pilot because it is a rare, debilitating condition that emerges in infancy or early childhood and has recently been assessed for inclusion in NBS programs internationally due to a recently developed gene therapy.4 This gene therapy, arsa-cel, has not yet been submitted for regulatory or reimbursement approval in Canada. As a result, the current treatment landscape for MLD does not include arsa-cel and so evidence on its effectiveness was not assessed as part of this review.
This evidence review helped to inform HTERP’s deliberations and recommendations on whether MLD should be added to the Recommended Pan-Canadian Newborn Screening List.
Objectives
We drew on the Newborn Screening Advisory Panel’s recommended criteria for adding a condition to develop our research questions and approach to identify and summarize evidence on NBS for MLD. A description of how research questions were mapped onto the recommended criteria can be found in Appendix 4 of the Supplemental Material. In this evidence review, we:
describe the natural history (NH), severity, and epidemiology of early-onset MLD
identify, appraise, and synthesize the evidence on the benefits and harms of NBS for early-onset MLD, the accuracy of screening strategies for identifying early-onset MLD, and the effectiveness of treatment for early-onset MLD
describe the availability and acceptability of the MLD screening test
identify available treatments for MLD, and describe their acceptability to health professionals and to the public
identify social, ethical, implementation, resource, and cost considerations of NBS for early-onset MLD.
Evidence Assessment Methods
To describe the NH, severity, and epidemiology of early-onset MLD, we conducted a literature review (including web searches, a review of grey literature and other sources, and a review of scoping materials) and used a narrative summary to report findings. Information from patients, clinicians, caregivers, and other people with lived and living experience was also drawn upon to describe the severity and impacts of living with the condition.
To identify the clinical benefits and harms of universal newborn bloodspot screening for early-onset MLD compared to no universal NBS, we conducted a rapid review, informed by the updated Cochrane Rapid Review Methods Guidance for Rapid Reviews of Effectiveness.5
To assess the accuracy of 2-tier and 3-tier dried bloodspot (DBS) NBS strategies for early-onset MLD and to assess whether early initiation of treatment leads to improved outcomes compared to initiation of treatment following clinical presentation, we summarized and updated the evidence presented in a rapid review commissioned by the UK National Screening Committee, published in 2025.6
We conducted an implementation review using an environmental scan of information from grey and published literature sources (as available), key informant interviews with experts across Canada, input from clinical experts contributing to the pilot evidence review, and input from submissions to an open call for feedback. A narrative summary of the findings describes the availability and acceptability of NBS for MLD in Canada, as well as key health system impacts anticipated from implementing NBS for MLD.
We conducted a budget impact analysis (BIA) of implementing universal newborn DBS screening for early-onset MLD compared to no screening in Canada, specifically from the perspective of a publicly funded health care system and considering only the costs associated with screening and diagnostic testing.
To identify social and ethical considerations associated with newborn DBS screening for early-onset MLD, we conducted an ethics review guided by 4 key domains: lived experiences of those affected by MLD, evidence for including MLD in NBS, clinical implementation of MLD screening, and broader health system impacts.
Details on the methodology for each component of the evidence assessment, including the literature searches and inclusion criteria, can be found in the Supplemental Material. To help inform the research questions and conduct of this evidence review, we engaged with NBS and metabolic disease experts across Canada. The evidence review was also peer-reviewed.
Call for Community Input
Between May 29, 2025, and July 19, 2025, we invited patient, caregiver, clinician, and industry input (referred to as “community input”) for our review of NBS for early-onset MLD through an open call for input on our website. This input, along with the evidence review, was provided to HTERP to support their deliberations as they developed a recommendation on whether to add MLD to the Recommended Pan-Canadian Newborn Screening list. The input we received will be published in full on our website.
The Condition
MLD is typically classified into 4 subtypes based on the age of symptom onset: LI, EJ, LJ, and adult onset (AO).
Early-onset (i.e., LI and EJ) MLD is the most severe form of the disease, characterized by rapid neurodegeneration and greatly reduced life expectancy, and accounts for an estimated 75% to 90% of cases. It is the primary focus of this pilot evidence review.
The birth prevalence of MLD (including all subtypes) is estimated at less than 1 per 100,000 live births worldwide. No central registry has been identified to track incidence and subtype distribution in Canada.
This section addresses the following research question.
What Is the Natural History, Severity, and Epidemiology of Early-Onset MLD?
Overview of MLD
MLD is a rare, inherited, autosomal recessive lysosomal storage disorder (LSD) caused by mutations in the ARSA gene, leading to a deficiency of the ARSA enzyme.7-9 This deficiency results in the accumulation of sulfatides in the central and peripheral nervous systems, causing progressive demyelination and neurodegeneration.7,8 Approximately 350 pathogenic variants of the ARSA gene have been described.10,11
MLD is typically classified into 4 subtypes based on the age of symptom onset (Table 1).9,12 Because NBS is intended to identify serious diseases of infancy and early childhood for which an effective treatment exists,3 the primary focus of this pilot evidence review is NBS for the early-onset phenotype of MLD. It is important to note, however, that the screening test for MLD will inevitably also identify late-onset and uncertain-onset phenotypes, necessitating consideration of how these patients should be managed. While the clinical evidence summarized here pertains only to early-onset disease, discussion of the treatment of late-onset phenotypes is also included when relevant.
Table 1: Metachromatic Leukodystrophy Subtypes and Distributions
Phenotype | Subtype | Symptom onset | Percentage of cases6 |
|---|---|---|---|
Early-onset | Late infantile | < 30 months | 50% to 60% |
Early juvenile | 30 months to < 7 years | 20% to 40% | |
Late-onset | Late juvenile | 7 years to < 17 years | |
Adult | ≥ 17 years | 10% to 20% |
Source: Data adapted from Gomez-Ospina.6
Early-onset MLD, which comprises the LI and EJ subtypes, is the most severe form of the disease.9,13 LI MLD is characterized by rapid neurocognitive decline, motor regression, hypotonia, seizures, and eventual loss of all voluntary functions.7,8,14,15 Most children with this form of MLD have a life expectancy of around 5 years.16 EJ MLD follows a slower but still progressive trajectory.7,16 Initial symptoms may include behavioural changes, cognitive regression, gait disturbances, and incontinence. Over time, patients lose motor and language skills and require full-time care.8,14,15 Across both early-onset subtypes of MLD, life expectancy is greatly reduced, with most affected children not reaching adulthood.7,16
We did not identify any recent reports describing the epidemiology of MLD in Canada. The birth prevalence of MLD (including all subtypes) is estimated at less than 1 per 100,000 live births worldwide.7,9 There are data from outside of Canada estimating a higher prevalence in some Indigenous populations (i.e., those from the western Navajo Nation and those with Yup’ik ancestry of southern Alaska).9,12,13 However, underdiagnosis and delayed diagnosis17 — particularly in early-onset forms — could contribute to underreported prevalence.18 As of 2025, no centralized registry to track MLD incidence or subtype distribution has been identified in Canada.
Current Screening, Diagnosis, and Treatment Paradigm in Canada
Using published literature sources and in consultation with NBS and metabolic disease experts across Canada, we proposed an algorithm that is likely representative of the current testing, diagnosis, and treatment pathway for MLD in Canada (Figure 1). None of the provincial or territorial NBS programs in Canada currently include MLD in their panels.3 As a result, most patients in Canada are identified after symptom onset, with the diagnostic journey differing depending on whether there is a known family history. In families with a prior diagnosis, at-risk siblings are typically monitored closely and may be tested before symptoms emerge. For children without this history, the path to diagnosis is often prolonged and emotionally exhausting. Families have described months or years of uncertainty, during which early symptoms are attributed to more common pediatric conditions.
Once MLD is suspected, diagnosis typically involves a multimodal assessment. Brain MRI often reveals a characteristic leukodystrophy pattern, while peripheral nerve studies can show demyelinating neuropathy. Biochemical testing, including the measurement of urinary sulfatide levels and ARSA enzyme activity in leukocytes (similar to the 2-tier screening strategies), can confirm deficiency and distinguish MLD from ARSA pseudodeficiency. Definitive confirmation of diagnosis comes from molecular genetic testing of the ARSA gene (and/or PSAP or SUMF1 genes, if SapB deficiency or multiple sulfatase deficiency disorders are suspected). This combination of sulfatide level measurements, enzyme activity, and molecular testing is similar to the 3-tier screening strategy discussed later in this report. Other evaluations that may support diagnosis include gallbladder imaging to inspect for polyps, hydrops, or wall thickening (unusually common in MLD), and physiotherapy or occupational therapy assessments to help quantify functional impairment and inform supportive care.19,20
Following biochemical and molecular confirmation, specialists (i.e., physicians specializing in metabolic disease, neurologists) may undertake an assessment to predict the likely disease onset category (early-onset, late-onset, or uncertain-onset). This determination integrates genotype-phenotype correlations, age at symptom onset, and any emerging clinical signs.21,22 For complex or borderline cases, particularly when the pathogenicity of detected variants is unclear, the case may be referred to a multidisciplinary panel of experts for discussion.23 Such panels may draw on external expertise or published case data to support classification.23 Accurate onset prediction is important for counselling families, planning surveillance, and determining eligibility for time-sensitive treatments.
There is currently no disease-modifying treatment for LI MLD available in Canada; supportive and palliative care remain standard once rapid neurodegeneration has begun. However, a potential disease-modifying treatment (arsa-cel, which is an ex vivo autologous hematopoietic stem cell gene therapy) has received regulatory approval by the European Medicines Agency in the European Union (December 2020) and by the FDA in the US (March 2024) in patients with PS LI and EJ MLD, as well as early symptomatic (ES) EJ MLD. At the time of writing this report (September 2025), this treatment had not yet been submitted in Canada for consideration for regulatory or reimbursement approval. The safety and efficacy of this treatment for improving survival, motor and cognitive function, and maintaining brain myelin and language skills has been assessed in 2 studies (NCT0150182 and NCT03392987). The outcomes of these studies are summarized and appraised in the Treatment section of this report.
Treatment for individuals diagnosed with juvenile onset or AO MLD in Canada may include allo-HSCT. This treatment is only available for some carefully selected, PS or minimally symptomatic patients and involves the transplant of healthy donor cells, capable of producing the functional ARSA enzyme, into the patient’s bone marrow. This is typically a one-time procedure, with follow-up to ensure that engraftment was successful and that the recipient has begun to produce their own healthy cells. The outcomes of 1 cohort study assessing allo-HSCT in patients with juvenile MLD are summarized and appraised in the Treatment section of this report. While safety outcomes were not reported in that study, the known challenges and risks of allo-HSCT for other conditions include the need for suitable donors, narrow treatment windows, and the risk of engraftment failure and graft-versus-host disease.12,24
Figure 1: Current MLD Diagnosis and Treatment Paradigm (Reference Scenario)
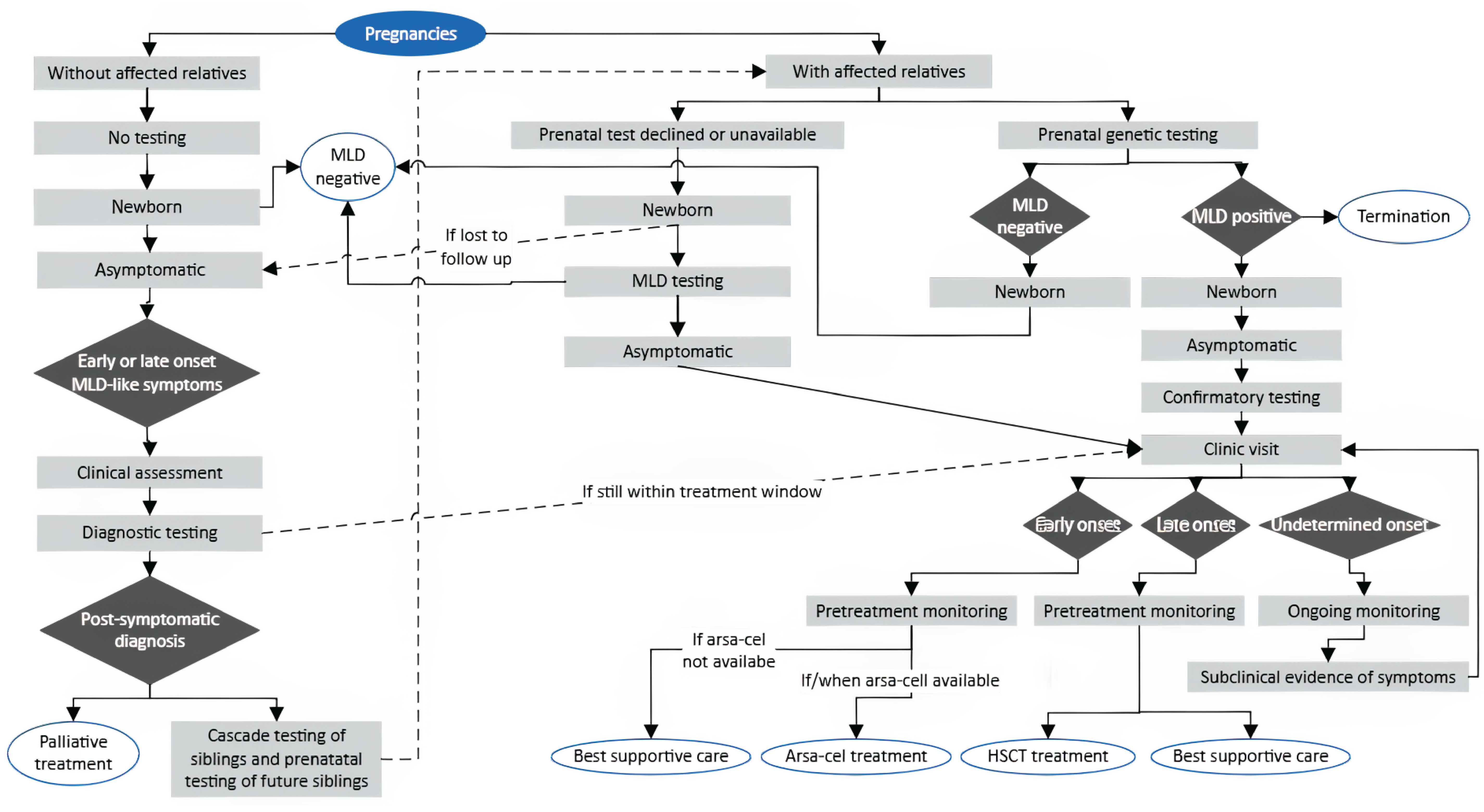
Arsa-cel = atidarsagene autotemcel; HSCT = hematopoietic stem cell transplant; MLD = metachromatic leukodystrophy; NBS = newborn screening.
Note: This figure was designed using published literature sources15,20,21,23 and in consultation with NBS and metabolic disease experts across Canada.
Benefits and Harms of Universal NBS for MLD
This section addresses the following research question.
What Are the Benefits and Harms of Universal Newborn Bloodspot Screening for Early-Onset MLD as Compared to No Universal NBS?
In 2011, the Centers for Disease Control (CDC) recognized improvements in, and expansion of, universal NBS as 1 of the top 10 public health achievements in the first decade of the 21st century.25 While universal NBS for a growing number of conditions has been credited with important benefits to newborns affected by rare and other conditions, universal NBS can also introduce potential harms.26 Consequently, an assessment of the available evidence describing the direct benefits and harms of universal NBS as compared to no universal NBS for any new condition, including MLD, is essential.
Details describing the methods used to identify and synthesize evidence are reported in Appendix 1 of the Supplemental Material.
Quantity of Research Available
There were 322 records identified in the literature searches following the removal of duplicates. Following screening of titles and abstracts, 313 records were excluded and 9 potentially relevant reports from the electronic search were retrieved for full-text review. No potentially eligible studies addressing a direct comparison between universal NBS for MLD and no NBS for MLD were identified from the grey literature search.
Of the 9 potentially relevant sources, 5 publications were excluded because they described a universal NBS program for MLD without a direct comparison to no NBS for MLD, and 4 publications were excluded because they were not full reports (i.e., they were conference abstracts) and did not have sufficient information or data to assess eligibility. A diagram presenting the results of the screening and selection is presented in Appendix 3 of the Supplemental Material.
Interpretation
No eligible studies were identified in answer to our research question concerning the benefits and harms of universal NBS for MLD as compared directly to no universal NBS for MLD, which limits our ability to describe the direct benefits and harms of universal NBS for MLD. One reason for the limited evidence to inform this research question may be that NBS for MLD remains a relatively new endeavour, with some pilot programs under way and only very recent efforts to implement NBS for MLD in some jurisdictions.27 The rare nature of MLD and the severity of early-onset forms make the generation of high-quality evidence (e.g., randomized trials of universal screening versus no screening) likely to be infeasible. The best available evidence might come from observational studies that compare settings with and without universal NBS for MLD. In the absence of this direct evidence, reviews of diagnostic test accuracy and treatment benefits and harms can provide indirect evidence to inform an understanding of the potential benefits and harms of a universal NBS program for MLD.
Guidelines have been published in the UK and the US, which recommend universal NBS for MLD given the recent availability of arsa-cel in those jurisdictions.19,20 Data generated from jurisdictions within which universal NBS for MLD has been implemented may provide data to increase understanding concerning the benefits and harms of universal NBS for early-onset MLD.
The Test
There are few studies assessing the accuracy of 2-tier and 3-tier screening algorithms for MLD, which are affected by small numbers and risk of bias.
Estimates of PPV were variable; however, sensitivity, specificity, and negative predictive value (NPV) approached 100%. The number of false-positives in 2-tier screening strategies was small and was reduced to zero with the addition of a third tier.
NBS for early-onset MLD is not currently available in any province or territory in Canada.
Most key informants and literature sources consulted as part of the environmental scan informing the implementation review agreed that the acceptability of NBS for early-onset MLD is dependent upon the availability of an effective treatment.
A critical consideration around implementing NBS for early-onset MLD is the performance of the screening algorithm, including its ability to reliably differentiate early-onset from late-onset disease, as well as mitigate false-positive, incidental, and uncertain findings.
This section addresses the following research questions.
What Is the Accuracy of 2-Tier or 3-Tier DBS NBS Strategies for Early-Onset MLD? What Is the Availability and Acceptability of DBS Testing for Early-Onset MLD?
Accuracy of 2-Tier or 3-Tier DBS NBS Strategies for Early-Onset MLD
To address this research question, we summarized and updated the evidence presented in a rapid review commissioned by the UK National Screening Committee, which was published in 2025.6 The aim of this report, henceforth referred to as the “UK review,” was to assess the published evidence relevant to the potential inclusion of MLD in the UK’s recommended NBS panel. Using a mapping exercise and an assessment of its methodological quality using A MeaSurement Tool to Assess systematic Reviews 2 (AMSTAR 2)28 (refer to Appendix 3 of the Supplemental Material), and given the similarities across the publicly funded health systems in the UK and Canada, it was determined that there was sufficient alignment in scope, criteria, and research questions between the UK review and this pilot evidence review to warrant this evidence assessment strategy. It is important to note that while the UK review’s research question on screening test accuracy focused on single-tier and 2-tier NBS strategies, after consultation with clinical experts in Canada, we chose to remove single-tier strategies and add 3-tier strategies to our research question, as it was suggested that a 3-tier strategy is the most relevant to the Canadian context.
The UK review included 3 studies relevant to this question: a feasibility study conducted in the US (N = 27,335; Hong et al. [2021]),29 a large pilot study from Germany (N = 109,259; Laugwitz et al. [2024]),23 and a smaller prepilot study from the UK (N = 3,687; Wu et al. [2024]).30 A literature search update conducted on June 13, 2025, using the same methodology as the UK review identified 1 additional publication for inclusion: a prospective pilot study from Italy (N = 42,262; Malvagia et al. [2025]).31
Two studies included in the UK review (Hong et al. [2021]29 and Wu et al. [2024]30), along with the study by Malvagia et al. (2025),31 evaluated a 2-tier screening algorithm (originally developed by Hong et al. [2021]) that involved the quantification of sulfatides in DBS samples using ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) as the first tier and the measurement of ARSA enzyme activity using the same platform as the second tier. Hong et al. (2021)29 also evaluated a second algorithm that measured ARSA enzyme activity in DBS samples as the first tier and sulfatides as the second tier. There were some variations in sulfatide targets and cut-off values across studies. Laugwitz et al. (2024)23 evaluated a 3-tier screening algorithm that included the same first 2 tiers, and genetic sequencing of the ARSA, PSAP, and SUMF1 genes in DBS samples as the third tier. Given that their research question focused only on 2-tier strategies, and to facilitate comparison between studies, the authors of the UK review calculated and reported accuracy estimates for this study as if it were a 2-tier algorithm, treating genetic sequencing as part of the confirmatory diagnostics. As we are interested in both 2-tier and 3-tier strategies, we have presented the accuracy outcomes for both (Table 2).
Risk of Bias and Applicability of Studies
A detailed critical appraisal of the included studies can be found in Appendix 3 of the Supplemental Material. Although the 3 studies included in the UK review were primarily designed to assess the feasibility of implementing NBS programs rather than diagnostic test accuracy, the authors suggested that it was important to assess their limitations with respect to their ability to answer the research question, regardless of their design and primary aim. Therefore, the risk of bias and applicability was assessed using the Quality Assessment of Diagnostic Accuracy Studies 2 (QUADAS-2) tool32 for diagnostic test accuracy studies. All 4 included studies were considered at unclear or high risk of bias in the Index Test and Flow and Timing domains due to midstudy adjustments in screening cut-off values; the application of the reference standard only to screen-positive cases; and, in some cases, the exclusion of samples for which second-tier screening was not undertaken from analyses. These issues hindered the ability of the studies to accurately estimate the number of true and false negatives, and risk overestimating sensitivity by assuming all screen-negative infants are unaffected (i.e., the NPV is assumed to be 100%). The specificity may also be overestimated, but the degree of overestimation is likely small. Our independent assessment of the study by Malvagia et al. (2025)31 highlighted similar concerns. All studies also presented potential conflicts of interest, as several authors were involved in the clinical trials funded by Orchard Therapeutics, the manufacturer of arsa-cel.
Despite the large sample sizes of several of the included studies, the rarity of MLD further limited the available evidence, as the sample size was still inadequate to identify any MLD-positive cases in 2 of the 4 studies. This restricted the ability to estimate the true incidence, true sensitivity, and PPV. Although not an objective of the screening tests, this also restricted the evaluation of methods to differentiate between early-onset and late-onset phenotypes.
Three of the included studies were conducted in Europe and 1 was conducted in the US. While any differences in MLD incidence and ARSA variants across groups with various ethnic origins is not well characterized, this may limit the generalizability of findings to more ethnically diverse populations, such as those found in Canada.
Summary of Outcomes
A summary of relevant outcomes is provided in Table 2, with more detailed outcomes provided in Appendix 3 of the Supplemental Material.
MLD incidence and differentiation between early-onset and late-onset disease: Patients who were MLD-positive were found in 2 of 4 included studies (Laugwitz et al. [n = 3] and Hong et al. [n = 1]), with estimated incidences of 2.75 in 100,000 and 3.66 in 100,000, respectively. Subtype predictions were determined based on clinical data, genotype, and residual ARSA activity in leukocytes.23 Two of the patients who were MLD-positive were diagnosed as having early-onset disease (both EJ) and 1 as having late-onset disease (LJ or adult), and for 1 patient, the predicted age of onset was not reported.29 Hong et al. (2021) also noted that while the other newborn who was screen-positive but MLD-negative was MLD heterozygous — and it was considered highly likely that they would be unaffected — it was possible, based on their genotype, that they would display mild symptoms later in life.
Sensitivity and specificity: In the 2 studies with confirmed MLD-positive cases,23,29 both sensitivity and specificity approached 100%. While the 2 publications that did not identify any MLD-positive cases could not estimate real-world sensitivity or PPV, both studies performed retrospective validation of the algorithm with patients who had been genetically confirmed to be MLD-positive. Malvagia et al. (2025)31 reported correctly identifying 15 nonneonatal samples using their 2-tier algorithm. Wu et al. (2024)30 found 1 case of LI MLD during the validation phase, in an individual who had C16:0 sulfatide levels lower (0.15 µmol/L) than the cut-off proposed in the study by Hong et al. (2021)29 (≥ 0.17 µmol/L). If the C16:0 cut-off were lowered to 0.15 μmol/L, the first-tier positive rate would increase from 0.3% to 0.76%.
Table 2: Summary of Outcomes for Screening Algorithm Accuracy
Study | Screening algorithm | Screen-detected MLD, n | Sensitivity, % | Specificity,a % | PPV, % | NPV,a % | False-positive fraction,b % |
|---|---|---|---|---|---|---|---|
UK review6 | |||||||
Hong et al. (2021)29 N = 27,335 | Algorithm A: 2-tier First tier: C16:0 sulfatides Second tier: ARSA enzyme activity | 1 (incidence: 3.66 in 100,000)c | 100 | 99.99 | 50 (95% CI, 12.35 to 87.65)a | 100 | 0.0037 |
Hong et al. (2021)29 N = 2,287 | Algorithm B: 2-tier First tier: ARSA enzyme activity Second tier: C16:0 sulfatides | 0 | NE | NE | NE | NE | NR |
Wu et al. (2024)30 N = 3,687 | 2-tier First tier: C16:0 sulfatides Second tier: ARSA enzyme activity | 0 | NE | 100 | NE | 100 | 0 |
Laugwitz et al. (2024)23 N = 109,259 | 2-tierd First tier: C16:0 and C16:1-OH sulfatides Second tier: ARSA enzyme activity | 3 (incidence: 2.75 in 100,000)c | 100 | 99.98 | 15 (95% CI, 9.89 to 22.11)a | 100 | 0.016 |
Laugwitz et al. (2024)23 N = 109,259 | 3-tier First tier: C16:0 and C16:1-OH sulfatides Second tier: ARSA enzyme activity Third tier: Genetic sequencing of ARSA, PSAP, and SUMF1 | 3 | 100 | 100 | 100 | 100 | 0 |
Literature search update | |||||||
Malvagia et al. (2025)31 N = 42,262 | 2-tier First tier: C16:0, C16:1-OH, C16:0-OH, C16:1 sulfatides Second tier: ARSA enzyme activity Recall step (if second tier positive): Repeat sulfatides and ARSA enzyme activity on new DBS sample | 0 | NE | 100 | NE | 100 | 0 |
DBS = dried bloodspot; MLD = metachromatic leukodystrophy; NE = not estimable; NPV = negative predictive value; NR = not reported; PPV = positive predictive value.
aNumbers estimated with the assumption that no MLD cases were missed and participants whose DBS did not receive second-tier testing were excluded from the analysis.
bAfter the full screening algorithm.
cCalculated.
dLaugwitz et al. evaluated a 3-tier screening algorithm. However, to compare across studies, the UK review reported accuracy estimates as if it were a 2-tier strategy. We have included both the 2-tier and 3-tier accuracy estimates here.
PPV and NPV: When comparing the 2-tier algorithms in those studies that found MLD cases, the PPV was highly variable between studies, with Hong et al. (2021)29 reporting 50% (95% confidence interval [CI], 12.35% to 87.65%) and Laugwitz et al. (2024)23 reporting 15% (95% CI, 9.89% to 22.11%) PPV. When genetic testing was included as the third tier, the PPV in the Laugwitz et al. (2024)23 study increased to 100%. The NPV was assumed to be 100% in all included studies, given that none of the studies systematically applied the reference standard to newborns who were screen-negative, conducted record reviews, or performed surveillance of this group to identify any false-negatives. As a result, the estimate relied on the assumption that no MLD cases were missed. Of note, Laugwitz et al. (2024)23 did conduct genetic testing on 210 samples that were first-tier–positive and second-tier–negative, and on 151 samples that were first-tier–positive but had no second-tier testing. None of these samples were found to have clinically relevant variants.
False-positive fraction: In algorithm A of Hong et al. (2021),29 195 of 27,335 individuals screened positive on the first tier and 2 of 195 individuals screened positive after the second tier. After confirmatory genetic sequencing, 1 of these individuals was confirmed to be MLD-positive (while the other was interpreted as a heterozygous carrier [MLD-unaffected]). The false-positive fractions after 2 tiers of testing in the included studies were 0.0037%,23,29 0.016%,23,31 and 0.01%.31 Of note, the Malvagia et al. (2025) study included a “recall step” following the second tier that involved retesting both sulfatide and ARSA enzyme activity on new DBS samples. Four of 10 individuals were recalled due to inconclusive results (insufficient material on the DBS to perform the ARSA enzyme test) rather than being second-tier–positive, and were excluded from the false-positive fraction (FPF) calculation. None of the recalled individuals tested positive after the repeat analyses. No patients tested positive on the second tier in the study by Wu et al. (2024).30 When calculated after 3 tiers, the study by Laugwitz et al. (2024) had an FPF of 0%.
Interpretation
The reported estimates of MLD incidence are higher than the estimated worldwide range of 0.16 to 1.85 per 100,000.13 Given the rarity of the disease and the sample size limitations, this is to be expected. Further, it is a known phenomenon that systematic screening, as conducted in these studies, often reveals higher disease prevalence than previously estimated from clinical diagnosis alone.33 There were very limited data to evaluate the ability of the screening tests to predict age of onset or to support the estimated percentages of early-onset versus late-onset disease.
The sensitivity and specificity of all NBS tests are dependent on decisions regarding cut-off values and a balance between the risk of false-negatives and false-positives. Adjusting cut-off values to reduce the risk of false-negatives often has the effect of increasing the false-positive rate.34 False-negatives are usually more harmful than false-positives because an affected newborn may be missed, whereas false-positives lead to unnecessary recalls and additional testing, which can cause a nonoptimal allocation of health care resources and parental anxiety, with long-term consequences on the child-parent relationship.34 As with any condition screened for by NBS, if NBS for MLD is implemented, it will likely involve ongoing adjustment of the cut-off values for biomarkers to find an ideal balance. Both the sensitivity and specificity presented in the included studies were calculated with assumptions. Namely, the number of true negatives and false-negatives (and, therefore, sensitivity, specificity, PPV, and NPV estimates) assumed that no MLD cases were missed. While this approach is more realistic given that performing genetic sequencing on all newborns is not considered practicable, it reduces the quality of test accuracy estimates.
Conclusions
Overall, the included studies had concerns for bias common in research on rare diseases, where small sample sizes and the lack of concurrent control groups are unavoidable. No screen positives were identified in some studies, and it was not likely to be feasible to apply the reference standard to all screen-negative infants. There is, therefore, a lack of data to inform true estimates of accuracy (PPV in some studies, NPV, sensitivity, specificity). That said, the reported sensitivity, specificity, and NPV all approached 100%. There was high variability in estimates of PPV calculated from 2 studies for the same 2-tier screening strategy, given the relatively small number of screen-positive infants contributing to the estimates; however, the addition of a third tier in 1 study increased the PPV to 100%. The false-positive fraction in 2-tier screening strategies was small (0.0037% to 0.02%) and was improved to 0% with the addition of genetic sequencing as the third tier. The prevalence of MLD in the tested population remains subject to uncertainty.6
A Proposed NBS Algorithm Model
Using input from clinical and laboratory experts across Canada, we proposed an algorithm that represents a likely testing and care pathway for NBS for MLD, should it be implemented (Figure 2). This algorithm is intended as a guiding example, not a prescriptive approach, and serves as the reference case for the BIA presented later on in this report.
The algorithm includes 3 stages. Stage 1 is a 3-tiered DBS screening strategy adopted from Laugwitz et al. (2024).23 In this stage, tier 1 analyzes C16:0 and C16:1-OH sulfatide levels via UPLC-MS/MS, with samples showing C16:0 levels greater than or equal to 17.0 µmol/L or C16:1-OH levels greater than or equal to 0.05 µmol/L considered first-tier–positive. Tier 2 measures ARSA activity using the same platform, with activity less than or equal to 0.015 µmol/L/hr considered second-tier-positive. Tier 3 involves genetic sequencing of ARSA, SUMF1, and PSAP genes, with clinically relevant ARSA variants considered screen-positive.
Stage 2 involves confirmatory diagnostics and disease onset prediction, informed by published literature12,19,21 and expert opinion. Once a newborn is found to be screen-positive, they would be called in for a clinic visit with a physician specializing in metabolic disorders (or most responsible physician) for leukocyte ARSA enzyme activity testing, urine sulfatide testing, and parental genetic sequencing to clarify inheritance. With these results, the physician would confirm the MLD diagnosis and determine the patient’s likely age of onset (early, late, or uncertain). The diagnosis would be delivered at a follow-up visit.
Stage 3 covers pretreatment monitoring based on predicted onset, and follows published guidelines on the clinical management of MLD19,20 and expert opinion. For LI or EJ (early-onset) MLD, monitoring involves brief assessments every 3 months and 1 comprehensive assessment before treatment (if available). Late-onset cases receive brief assessments every 6 to 12 months and comprehensive assessments every 12 to 24 months, on an alternating schedule. Monitoring frequency for undetermined-onset cases is age-dependent, as shown in Figure 2, and treatment timing depends on the emergence of subclinical symptoms.
Accurately screening for MLD presents several challenges, all of which are effectively addressed through this 3-tier strategy. First, ARSA pseudodeficiency alleles can reduce ARSA enzyme activity in laboratory testing without causing disease, leading to false-positives in screening strategies that rely on enzyme-based testing only. However, newborns with these pseudodeficiency alleles typically do not have elevated sulfatide levels, so a screening strategy (such as this one) that measures sulfatide levels in the first tier can effectively filter them out.
Second, 2 potential incidental findings are relevant to MLD screening: SapB deficiency and multiple sulfatase deficiency (MSD). SapB deficiency generally preserves normal ARSA enzyme activity in vitro, so affected individuals would likely test negative at the second tier, reducing the chance of detection. MSD, while rare, may still be identified; for this reason, a care pathway is included for such cases. Incorporating SUMF1 and PSAP gene analysis at the third tier helps differentiate these conditions from MLD, ensuring they do not contribute to false-positive results.
Finally, although high sulfatide levels and low enzyme activity are indicative of MLD, genetic testing may reveal ARSA variants of unknown significance, with which the age of onset is uncertain. Individuals with these findings are classified in an “uncertain onset” category and placed under routine monitoring. This allows for early recognition of emerging symptoms and timely intervention if disease progression occurs.
Availability and Acceptability of DBS Testing for Early-Onset MLD
DBS testing for MLD is not currently available in any of the NBS programs across Canada. Acceptability to health professionals and to the public was therefore the focus of this question.
Acceptability in the context of health care interventions has been defined as: “…a multi-faceted construct that reflects the extent to which people delivering or receiving a healthcare intervention consider it to be appropriate, based on anticipated or experienced cognitive and emotional responses to the intervention” (p. 1).35 As it concerns NBS in Canada, acceptability can also be conceptualized using the criteria for adding or reassessing a condition as recommended in recent pan-Canadian guidance.3 Incorporating these concepts and criteria, our assessment of NBS for MLD focused on the acceptability of the proposed algorithm, as well as broader considerations concerning the acceptability of screening for MLD in Canada.
Figure 2: Proposed NBS Algorithm for MLD
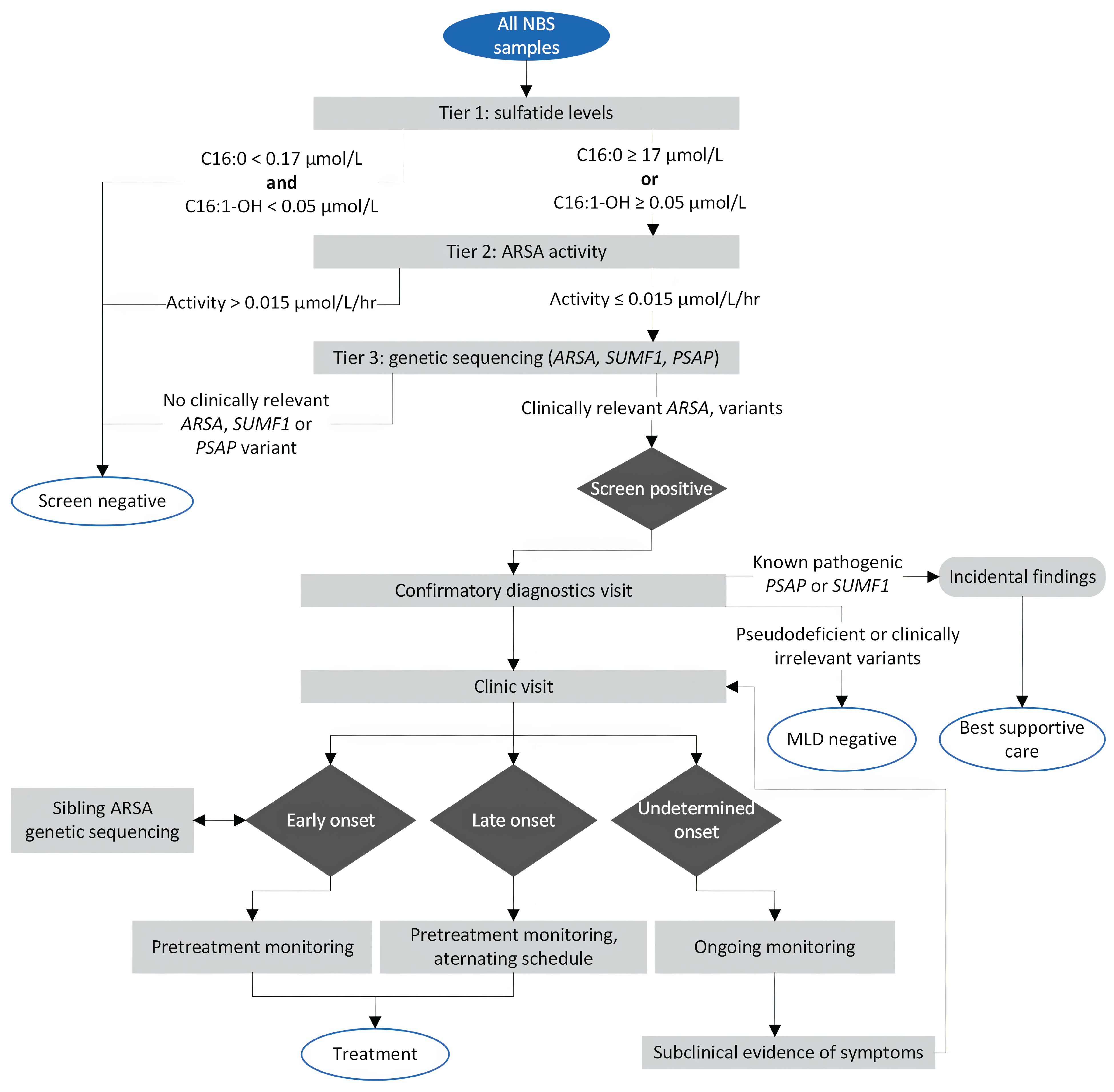
MLD = metachromatic leukodystrophy; NBS = newborn screening.
Acceptability of NBS for MLD
As part of the implementation review, we sought key informants with knowledge and expertise of NBS and/or inherited metabolic disease from all of the provincial and territorial jurisdictions. We conducted interviews with expert key informants from 6 jurisdictions in Canada (British Columbia, Alberta, Manitoba, Ontario, Nova Scotia, and Newfoundland) in July 2025. Most of the key informants we interviewed were clear that implementing NBS for MLD is not currently acceptable in the absence of an available, effective treatment for early-onset disease in Canada. Nonetheless, some key informants pointed to potential benefits of NBS for MLD in addition to providing an opportunity for treatment (if 1 becomes available). One such potential benefit suggested was avoidance of the multiple health care visits associated with clinical ascertainment of MLD in the absence of NBS (often referred to as the “diagnostic odyssey”).30,36 With MLD, clinical ascertainment can be lengthy and filled with uncertainty, anxiety, and distress for families, as described in a submission responding to the CDA-AMC open call for input on this pilot evidence review. Despite these potential benefits of NBS for MLD, criteria that inform the addition of a condition to NBS panels require that an effective treatment be available for affected infants.3
As described previously, the acceptability of NBS for MLD is further complicated by the fact that the algorithm will identify both early-onset and late-onset forms of the disease, whereas the emerging treatment, arsa-cel, is indicated for early-onset disease only, for which it is available in other countries.37 One key informant questioned the appropriateness of NBS for MLD altogether, given the natural course of the disease, where timing of symptom onset for all subtypes occurs after the neonatal and early infantile periods, and therefore falls outside of the intended scope of NBS.3 This concern was also highlighted by another key informant, who reported their opinion that, while all leukodystrophies (including MLD) may not align with the conventional aims of NBS due to the timing of symptom onset, screening for MLD will be necessary if arsa-cel treatment becomes available. It was suggested that NBS may be the only currently available and feasible means for identifying patients with early-onset MLD presymptomatically, to ensure they can have timely access to and benefit from treatment, if available. While most key informants agreed that it is not ideal to identify late-onset forms of MLD with NBS, it was acknowledged that the implementation of NBS for MLD will identify these infants with late-onset disease, which would require NBS programs and associated clinical communities of practice to develop protocols and monitoring that can also manage these patients effectively.
Nonetheless, support in principle for NBS for MLD was expressed by many key informants and those providing community input, some of whom indicated that preparing for the implementation of NBS for MLD is warranted, given their confidence and assumptions that arsa-cel for early-onset MLD may become available in Canada in the foreseeable future. Key informants felt that preparing to implement NBS for MLD in the jurisdictions across Canada must begin with an assessment of readiness, which is a key concept when assessing acceptability and considerations around the implementation of NBS for a new condition.38,39 As 1 key informant described, readiness among both NBS programs and the relevant communities of clinical practice (as well as the health systems and social context within which they are situated) is key to ensuring the success of NBS for a new condition in bringing benefits to affected patients. As this key informant described, past successful experiences in Canada when implementing NBS for a new condition have benefited from intentional, advance preparation, including engagement and collaboration across NBS programs and communities of clinical practice that would manage referrals for the new condition. It was also noted that patient advocacy groups can play an important role in establishing readiness, by advancing awareness and understanding of the condition and advocating for effective health interventions.
Finally, several key informants with clinical expertise emphasized the sense of urgency among NBS programs and associated communities of clinical practice in Canada to ensure that arsa-cel for early-onset MLD is made available in Canada as soon as possible. Their support, in principle, echoes much of the widespread support expressed for NBS for MLD in the literature,23,40 with recent guidelines in the US and Europe supporting NBS for MLD where treatment is available.19,20
Acceptability of the Proposed NBS Algorithm for MLD
All key informants and clinical experts contributing to this pilot evidence review agreed that the proposed NBS algorithm for MLD appeared to be a reasonable approach and would likely be used in the NBS programs across Canada, if they were mandated by their Ministries of Health (MoHs) to implement NBS for MLD. Concerns and considerations around implementing the proposed algorithm included capital investment required in jurisdictions where laboratory infrastructure does not have current capacity to process the tests for the algorithm; the extent to which MLD disease subtypes could accurately be identified; and the need for validation and refinement of the algorithm to optimize accuracy, including mitigation of false-positive findings and management of incidental and uncertain findings.
For some jurisdictions, it was not clear from the key informant interviews whether the NBS algorithm for MLD could be implemented using current laboratory infrastructure and resources, and for most jurisdictions, it was clear that there is not current capacity and that capital and other investments would be required. First-tier sulfatide testing for the proposed MLD NBS algorithm can be multiplexed with other analytes processed on high-sensitivity liquid chromatography tandem mass spectrometry (LC-MS/MS);41 however, it was confirmed by NBS laboratory experts in Canada that there is currently no NBS program in Canada that uses LC-MS/MS for first-tier screening, which would necessitate upfront investment in all jurisdictions. Nonetheless, 1 jurisdiction was identified in which other conditions that are currently being added to the NBS panel would also allow for MLD testing to be multiplexed in the near future. Notably, it was also clarified by NBS laboratory experts that there are inhibitory substances in DBS that interfere with the measurement of ARSA activity in the second tier of the proposed NBS algorithm for MLD. Techniques for resolving this problem have been developed31,42,43 and it has been addressed in jurisdictions outside of Canada where NBS for MLD has already been implemented. Canadian NBS laboratories may also consider this issue before implementing NBS for MLD, and NBS laboratory experts in Canada indicated that the anticipated associated costs would likely not be considerable. The overall implications of these considerations for acceptability include cost, time, and other resource requirements for most jurisdictions in Canada, if NBS for MLD were to be recommended and implemented.
As to the second of the considerations discussed around the proposed algorithm, literature sources corroborated the concern, highlighting that while the algorithm has demonstrated accuracy in distinguishing MLD by disease subtype,22,44 there remains a risk that not all cases of MLD could definitively be differentiated by disease subtype.9 Key informants acknowledged that, similar to the implementation of NBS for other diseases in the past, NBS laboratories across Canada would have to invest time and resources into validating and refining the proposed algorithm for MLD as part of its implementation. Downstream from the implementation of the algorithm is the interpretation of the information generated by the algorithm, including decisions concerning referral, diagnosis, and treatment of patients with MLD (which is discussed in more detail in the Health System Considerations section).
False-Positive, Incidental, and Uncertain Findings
In addition to concerns around the interpretation of NBS findings for MLD disease subtypes, the unintended identification of findings other than MLD by the proposed algorithm (including false-positive results, incidental, and uncertain findings) was discussed by all key informants, clinical experts contributing to the pilot evidence review, and clinicians responding to the CDA-AMC open call for input. Notably, most key informants did not express significant concern with the risk of false-positive or incidental findings if NBS for MLD were to be implemented, generally citing the proposed algorithm and the data generated to date around its performance and accuracy. As described previously, pilot studies assessing 2-tier and 3-tier screening strategies have demonstrated low false-positive rates;23,29-31 however, and as corroborated by 1 clinical expert contributing to the pilot evidence review, the evidence remains limited, and none has yet been published from within the Canadian context.
Specific to considerations concerning implementation and acceptability, 1 key informant suggested that the performance and accuracy of the algorithm as demonstrated by empirical studies may differ from the real-world context of NBS programs, including those in Canada. The same key informant emphasized that a clear and operationalizable definition of the primary target of NBS for MLD, as well as classification of nonprimary targets (with some examples presented in Appendix 3 of the Supplemental Material), would have to be established to ensure the algorithm could be optimized.
Detailed findings are presented in Appendix 3 of the Supplemental Material. The potential health systems impacts and implications of these considerations around the proposed NBS algorithm for MLD are further described in the Health System Considerations section.
The Treatment
Disclaimer: CDA-AMC summarized and updated the evidence presented in a rapid review conducted by the UK National Screening Committee, published in 2025. The following summary by CDA-AMC was used to fulfill the Newborn Screening Advisory Panel’s recommendations related to treatment as 1 of the factors for consideration to support the evidence review, notably that NBS programs should require that an effective treatment be available to individuals who receive the test.
CDA-AMC acknowledges that no targeted treatment for early-onset MLD has yet been submitted for consideration for regulatory or reimbursement approval in Canada; therefore, any decisions to include MLD as part of jurisdictional NBS testing would need to be conditional upon a targeted treatment for early-onset MLD that meets all of the following criteria:
the treatment is reviewed and approved for use by Health Canada
the treatment receives a recommendation in favour of reimbursement from a health technology assessment body in Canada
the treatment is publicly funded.
There are few studies assessing the effectiveness of treatment for early-onset metachromatic leukodystrophy, which are affected by small numbers and risk of bias.
No disease-modifying treatment for LI MLD (e.g., arsa-cel) has been submitted for consideration for regulatory or reimbursement review in Canada at the time of this review (September 2025).
This section addresses the following research question.
Does Early Initiation of Treatment Lead to Improved Outcomes for Early-Onset MLD Compared to Initiation of Treatment Following Clinical Presentation?
Summary of Included Studies
To address this research question, we summarized and updated the evidence presented in the UK review.6 For that research question, the authors of the review searched for studies assessing the effectiveness and safety of arsa-cel or any other intervention for MLD, where the condition was identified presymptomatically (through population screening or other means) and published between January 2012 and October 2024. Three relevant publications were identified in the UK review. Two publications (Fumagalli et al. [2022] and Sessa et al. [2016])37,45 presented evidence from the same phase I/II clinical trial of the gene therapy arsa-cel, infused as a fresh cell product, for the treatment of patients with PS or ES LI or EJ MLD (NCT01560182), and from patients who accessed treatment through early access programs (EAPs). The third publication (Groeschel et al. [2016]) was a retrospective cohort study that compared the long-term outcomes of patients with juvenile MLD who received allo-HSCT to an untreated NH cohort.46 None of the studies compared the effectiveness of treatments for MLD in early detection (screening or cascade testing) versus late detection (symptomatic presentation), as outlined by the research question. The 2 studies compared outcomes in patients who received treatment to NH cohorts of individuals who did not undergo treatment (secondary eligibility criteria).
A literature search update conducted on June 13, 2025, using the same methodology as the UK review identified 2 additional publications for inclusion. One publication (Fumagalli et al. [2025]) provided an integrated analysis of clinical outcomes on the same study and EAPs as the publication by Fumagalli et al. (2022), combined with evidence from another prospective, open-label, single-arm, phase II interventional trial (NCT03392987) evaluating the safety and efficacy of arsa-cel infused as a cryopreserved cell product.47 We did not locate a distinct publication of the NCT03392987 study. The second publication was a secondary, post hoc analysis of a subset of the patients in the study by Fumagalli et al. (2025), with PS LI MLD, and reported solely on nerve conduction velocity, a surrogate end point to peripheral neuropathy and a diagnostic pillar for MLD.48 The results and appraisal of the study by Fumagalli et al. (2022), as reported in the UK review, are presented in Appendix 2 of the Supplemental Material for information purposes, as it contributed to the earlier conclusions about arsa-cel reported in the UK review.
Methodological Quality of Studies
The study by Groeschel et al. (2016) was assessed by the authors of the UK review using the Risk Of Bias In Non-randomised Studies - of Interventions (ROBINS-I) tool for assessing risk of bias in nonrandomized studies of interventions.49 It was rated as being at critical risk of bias, with the key areas of concern being the absent consideration of potential confounding, missing data, and issues related to the selection of participants. Other methodological weaknesses included a small sample size, which reduces the certainty in the results, and a retrospective design with no attempt to match the historical cohort by age and disease.46 A summary of the results of the ROBINS-I assessment is provided in Appendix 3 of the Supplemental Material. Our independent assessment of the publication by Fumagalli et al. (2025) judged the report (containing studies NCT0150182 and NCT03392987) to be at serious risk of bias due to inadequate consideration of potential confounding, lack of clarify about missing data, and potential issues in the collection of outcome data. The 2 studies assessing arsa-cel were funded by the manufacturer (Orchard Therapeutics).
Evidence on the safety and effectiveness of allo-HSCT was limited to 1 study.46 Evidence on arsa-cel included 4 publications, based on 2 trials.
All of the included publications related to assessment of arsa-cel37,45,47,50 were based on 2 studies conducted in Italy. While the authors noted that patients from 22 different countries travelled to the clinical site, the majority were “white of Caucasian European heritage” [wording from original source] (72%).47 This may limit the generalizability of findings to more ethnically diverse populations, such as that of Canada. In studies that reported on sex, how sex was defined was not reported. No other factors that might contribute to an assessment of health inequities (using the PROGRESS-Plus framework51,52) — including, for example, socioeconomic status or place of residence — were reported. All allo-HSCT transplants reported by Groeschel et al. (2016) occurred between 1991 and 2012, so the outcomes reported may not be representative of those achievable in current practice.46
Summary of Outcomes
Atidarsagene Autotemcel
The UK review authors summarized the outcomes of the prospective, nonrandomized, phase I/II clinical trial (NCT01560182) and EAPs as presented in 2 publications.37,45 This study reported on 16 patients with LI MLD (15 PS and 1 ES), and 13 patients with juvenile MLD (5 PS and 8 ES). The median follow-up was 3.16 years (range, 0.64 to 7.51 years). The NH cohort comprised 31 patients (19 LI and 12 EJ).37,45 Fumagalli et al. (2025) included all of these patients plus an additional 10 patients from the NCT03392987 study, for a total of 39 patients who received treatment (28 with LI MLD, 8 with PS EJ, and 11 with ES EJ), and 49 in the NH cohort.47 The median follow-up was 6.76 years (range, not reported to 12.19 years), with a data cut-off of November 1, 2022.47 As previously mentioned, the reported results focused on the study by Fumagalli et al. (2025),47 given its longer follow-up and expanded cohort.
Severe motor impairment (Gross Motor Function Classification in Metachromatic Leukodystrophy [GMFC-MLD] level ≥ 5)–free survival: Survival free from severe motor impairment was defined as surviving and maintaining a level less than or equal to 4 on the GMFC-MLD scale (ability to sit, crawl or roll, or better). Consistent with the study by Fumagalli et al. (2022),37 in the integrated analysis in Fumagalli et al. (2025), a longer time to severe motor impairment was found in the LI MLD group. At 6 years of age, 100% (95% CI, 100% to 100%) of patients with LI MLD who had received treatment were alive and free from severe motor impairment compared to 0% (95% CI not evaluable) in untreated patients with LI MLD (P < 0.001). Of note, severe motor impairment developed at 7.2 years of age in 1 patient who had received treatment. At 2 years after treatment, estimates of severe motor impairment–free survival favoured the PS LI group of patients who had received treatment compared to the NH cohort, with a between-group difference of 60% (95% CI, 26% to 79%). For comparisons with the PS EJ and ES EJ groups, point estimates favoured the NH cohort, but the CIs were wide and crossing the null, suggesting uncertainty about which group may be favoured. The between-group differences were −13% (95% CI, −53% to 12%) for the PS EJ group, and −5% (95% CI, −39% to 26%) for the ES EJ group. At 5 years after treatment, estimates favoured the PS LI group of patients who had received treatment compared to the NH cohort, with a between-group difference of 85% (95% CI, 55% to 98%). Comparisons for PS EJ and ES EJ disease both favoured the treated groups, but with less certainty. Between-group differences were 44% (95% CI, −18% to 82%) for the PS EJ group, and 69% (95% CI, 25% to 93%) for the ES EJ group. At 10 years of age, 87.5% (95% CI, 38.7% to 98.1%; P = 0.04) of patients with PS EJ MLD who had received treatment and 80.0% (95% CI, 40.9% to 94.6%; P < 0.001) of patients with ES EJ MLD who had received treatment were surviving without severe motor impairment, compared to 11.2% (95% CI, 0.9% to 36.4%) of patients with EJ MLD who had not received treatment.
Gross Motor Function Measure 88 (GMFM-88): The GMFM-88 is scored as an average percentage over 5 dimensions (range, 0% to 100%), with higher scores indicating better gross motor function. Between-group differences refer to the difference in adjusted means, calculated as Treated – NH (i.e., a between-group difference of 70% indicates that the treated group outperformed the NH cohort by 70%; a negative difference indicates that NH outperformed the treated group). Consistent with Fumagalli et al. (2022), the integrated analysis of Fumagalli et al. (2025) reported that, at 2 years after treatment, arsa-cel was associated with better GMFM-88 scores. Between-group differences were 70.44 (95% CI, 62.28 to 78.60) for the LI MLD group, 52.35 (95% CI, 25.52 to 79.17) for the PS EJ group, and 47.24 (95% CI, 22.86 to 71.62) for the ES EJ group.47 At 5 years after treatment, estimates also favoured the LI MLD group who had received treatment compared to the NH cohort, with a between-group difference of 78.68 (95% CI, 55.96 to 100.00). The point estimates for the PS EJ and ES EJ groups at 5 years favoured arsa-cel, but CIs were wide and crossing the null, suggesting uncertainty about which group may be favoured. The between-group differences were 75.97 (95% CI, −100.00 to 100.00) for the PS EJ group and 39.86 (95% CI, −14.45 to 94.18) for the ES EJ group.47
Severe cognitive impairment–free survival: Consistent with the study by Fumagalli et al. (2022),37 in the integrated analysis in Fumagalli et al. (2025), a longer time to severe cognitive impairment was found in the LI MLD group. At 6 years of age, 100% (95% CI, 100% to 100%) of patients with PS LI MLD who had received treatment were alive and free from severe cognitive impairment, compared to 8.8% (95% CI, 1.5% to 24.3%) of patients with LI MLD who had not received treatment. Of note, 1 patient with PS LI MLD who had received treatment had confirmed severe cognitive impairment at 6.4 years of age. At 10 years of age, the estimated percentages of patients with PS EJ and ES EJ disease who had received treatment and were surviving without severe cognitive impairment were 87.5% (95% CI, 38.7% to 98.1%) and 64.8% (95% CI, 25.3% to 87.2%), respectively, compared to 7.5% (95% CI, 0.5% to 28.4%) of patients with EJ MLD who had not received treatment.
Brain MRI severity scores: Brain MRI severity scores range from 0 to 31.5, where a score of 0 is considered normal and a score of 31.5 is markedly abnormal.45,53 Consistent with the study by Fumagalli et al. (2022), Fumagalli et al. (2025) reported that, at 2 and 5 years after treatment, the brain MRI severity scores were lower in patients treated with arsa-cel compared to patients who had not received treatment. The between-group differences at 5 years after treatment were −20.73 (95% CI, −23.71 to −17.75) for the PS LI group, −21.16 (95% CI, −27.05 to −15.28) for the PS EJ group, and −14.63 (95% CI, −26.01 to −3.26) for the ES EJ group.
Nerve conduction velocity (NCV): NCV was reported as an NCV score (m/s) in the publication by Zambon (2025) (updated analysis with patients with LI MLD from Fumagalli et al. [2025]).50 This was not directly comparable to the NCV index reported in the study by Fumagalli et al. (2022).37 In this context, higher (faster) NCV scores can be assumed to indicate a healthier nerve while lower scores may indicate MLD-associated demyelination. According to Zambon et al. (2025), patients with LI MLD who had received treatment showed higher NCV values in 3 different motor and sensory nerves (the deep peroneal nerve, median nerve, and ulnar nerve) compared to patients in the NH group at 3 years after treatment, suggesting decreased neuropathy. A younger age at treatment corresponded to higher ulnar nerve and median nerve NCV scores (P = 0.0208; P = 0.0223) at 2 years after treatment.50
Language loss: Consistent with the study by Fumagalli et al. (2022), Fumagalli et al. (2025) reported that, at 6 years of age, 100% ((95% CI, 100% to 100%) of patients with LI MLD who had received treatment had no loss of language skills, compared to 4.2% (95% CI, 0.3% to 17.7%) of patients with LI MLD who had not received treatment. At 10 years of age, 100% (95% CI, 100% to 100%) of patients with PS EJ MLD who had received treatment and 66.7% (95% CI, 28.2% to 87.8%) of patients with ES EJ MLD who had received treatment had no loss of language skills, compared to 26.5% (95% CI, 4.6% to 56.3%) of patients with EJ MLD who had not received treatment.
Harms
All-cause mortality: All-cause mortality was defined as the percentage of deaths by any cause during a certain period. At the time of publication of the study by Fumagalli et al. (2025), a total of 3 deaths had occurred in patients who had received treatment across the 2 interventional trials and EAPs, with a median follow-up of 7.4 years (range, 3.2 to 13.4 years).47 All deaths occurred in patients with ES EJ MLD: 2 due to disease progression and 1 due to ischemic stroke following an infectious event approximately 1 year after treatment. At 6 years of age, 100% (95% CI, 100% to 100%) of patients with LI MLD who had received treatment were alive, compared to 59% (95% CI, 37.2% to 75.5%) of patients with LI MLD who had not received treatment. Overall survival (time-to-event) was similar among patients with PS EJ and ES EJ MLD who had received treatment compared to patients with EJ MLD who had not received treatment.
Adverse events (AEs): In the study by Fumagalli et al. (2025) assessing arsa-cel, AEs were reported only for the treatment group.47 The most frequent grade 3 or higher AEs (occurring in at least 20% of patients) were febrile neutropenia (82%), stomatitis (74%), gait disturbance (46%), muscle spasticity (36%), cognitive disorder (31%), motor dysfunction (31%), and neutropenia (21%). Serious AEs (SAEs) included hepatic veno-occlusive disease and atypical hemolytic uremic syndrome in 1 patient with PS LI MLD, grade 4 muscle spasticity in 2 patients with ES EJ MLD, 4 SAEs of dysphagia, and 2 grade 3 seizure events.
Anti-ARSA antibodies were detected in 6 of 39 patients (15%). Five of the 6 events resolved after a short course of rituximab; 1 event was ongoing at the data cut-off date. There was an absence of clonal abnormalities or oncogenic events. No evidence of neutrophil engraftment failure was found.
Allogeneic Hematopoietic Stem Cell Transplant
Evidence assessing the effectiveness of allo-HSCT for juvenile MLD was limited to 1 retrospective study,46 summarized in the UK review.6 This study found that allo-HSCT was associated with reductions in both the proportion of patients with progression to severe motor impairment (40% in the treated group versus 68% in the untreated group; relative risk [RR] = 0.59; 95% CI, 0.33 to 1.04; P = 0.04) and the proportion of patients with language loss (40% in the treated group versus 68% in the untreated group; RR = 0.59; 95% CI, 0.33 to 1.04; P = 0.07) at 10 years after disease onset. HSCT also appeared to be associated with improved brain MRI severity score compared to patients in the control group, but the difference was not statistically significant and it was unclear whether assessments occurred at the same time points in both groups. The mean brain MRI scores were reported to be 18.6 (standard deviation [SD] = 10.2) in the allo-HSCT group compared to 22.6 (SD = 5.5) in the untreated group (P = 0.06).
All-cause mortality in patients who received treatment (6 of 24 [25%]), at a median follow-up of 7.5 years (range, 3 to 19.7 years), was similar to that in patients who did not receive treatment (11 of 41 [27%]). Follow-up duration was not reported in the untreated group. Four of the deaths in the treated group were transplant-related rather than MLD progression–related.
Harms
Harms data were not reported in the study assessing allo-HSCT.
International Approvals and Reimbursement Status of Arsa-Cel
Although arsa-cel has not been submitted for regulatory or reimbursement review in Canada at the time of this review, it has been approved for use in several other countries and is publicly reimbursed in a number of jurisdictions (Table 3).
Table 3: Arsa-Cel Regulatory Approvals and Reimbursement
Regulatory approval and reimbursement | Location or country |
|---|---|
Regulatory approval of arsa-cel | Lenmeldy: US Libmeldy: European Union, Iceland, Liechtenstein, Norway, Switzerland, UK |
Public reimbursement of arsa-cel | Belgium, Denmark, Finland, France,a Germany, Iceland, Ireland, Italy, the Netherlands, Norway, Spain, Sweden, UK, US |
arsa-cel = atidarsagene autotemcel.
aAvailable only under special or early access.
Conclusions
Overall, evidence for the effectiveness of treatment for early-onset MLD is limited to 2 small, single-arm, manufacturer-sponsored trials for arsa-cel (n = 39), and 1 retrospective study for allo-HSCT (n = 31), with concerns for risk of bias. As is common in the study of rare diseases, the studies lacked randomized comparison groups; it is not clear whether these would have been feasible to enrol. However, this introduces risk of bias due to the limited (for studies of arsa-cel) or no (for the study of allo-HSCT) consideration of the potential for confounding.
Health System Considerations for Implementation
Implementing NBS for MLD will have important administrative, cost, and workload impacts for NBS programs, laboratories, and associated communities of clinical practice across Canada.
A key factor of success when considering any new condition for NBS panels is effective communication between the provincial and territorial NBS programs and the MoHs.
NBS laboratories that implement screening for MLD will bear upfront costs and effort that will vary across provincial and territorial jurisdictions based on existing infrastructure and resources. There may be opportunity to consider centralization of NBS for MLD to increase efficiency.
Impacts to clinical resources from implementing NBS for MLD will depend on the accuracy of the screening algorithm and its performance in identifying early-onset disease as a primary target. Secondary, incidental, false-positive and indeterminate findings from NBS for MLD will have to be defined and could impact clinical services, depending on the volume of these referrals. There may be opportunity to consider convening expert groups to support the management of complex referrals and/or cases.
This section addresses the following research question.
What Are the Key Health Systems Considerations for Implementing Universal Newborn DBS Screening for Early-Onset MLD in the Provincial and Territorial Programs Across Canada?
Implementation of NBS for MLD would have important health systems implications for all provincial and territorial jurisdictions in Canada. In general, these implications would be broadly relevant to 3 features or areas of the health system: health administration and decision-making (including approval processes and budgets); NBS programs and laboratories; and clinical health services, where infants identified with MLD by NBS would be referred, diagnosed, monitored, and treated.
Administrative and Decision-Making Considerations
To understand some of the key health systems considerations at the administrative and decision-making levels, conversations with key informants focused on the processes for considering and deciding on the addition of conditions to NBS panels and how this has been undertaken with other conditions in the past. While processes and procedures vary across the jurisdictions (with some being more formalized and others being less structured), a common theme among key informants was the importance of gaining support and funding from within the provincial or territorial MoH. Several key informants highlighted the complexity of NBS54 and the challenges in effectively translating that complexity to policy-makers and decision-makers. Key factors mentioned for the success of working with the MoHs included effective administrative structures (i.e., the structures within which NBS programs are situated) that promote dialogue, relationships, trust, and clear communication. Of these identified factors, clear and effective communication emerged as a recurring theme (i.e., when and where communication between the MoH and NBS program has been clear and effective, the addition of conditions to the NBS panel in the past has been perceived as a success). In jurisdictions where there are less established or less effective channels of communication, some key informants described instances of perceived misalignment between the NBS program and MoH concerning decisions about NBS. Another factor that was apparent in the effectiveness of relationships between the MoHs and NBS programs was the way and extent to which an NBS program is integrated within MoH administrative structures, and how well this facilitates clear and effective communication and collaboration.
Some key informants pointed out that an important anticipated challenge before implementing NBS for MLD would be gaining approval and funding for arsa-cel therapy for early-onset MLD across the provincial and territorial jurisdictions, particularly given its known high cost in other countries.55 Nonetheless, most key informants emphasized that it is essential that NBS programs anticipate implementation of NBS for MLD, with the expectation that arsa-cel is likely to become available for early-onset MLD in Canada. However, there was 1 key informant who highlighted opportunity cost considerations around implementing NBS for MLD, the importance of undertaking decision-making within the broader context of more common diseases of infancy and childhood, and the importance of considering the overall health care budget and needs of the population. Another key informant suggested that consideration of NBS for any condition for which a novel therapy is emerging (including MLD) should be undertaken in parallel with any associated reimbursement decisions for the emerging therapy, as both decisions impact one another. While 1 study from the UK56 concluded that NBS for MLD is cost-effective in their context, no cost-effectiveness data on NBS for MLD were identified in Canada, and reimbursement recommendations have not been made in Canada as the drug has not yet been submitted for approval and has not been through the reimbursement review process to inform decisions around implementation.
Given the level of acceptability assessed around NBS for MLD (as described in The Test section), most key informants felt that NBS programs should prepare for its implementation early, as the process could take months to more than a year if it were to be approved. The duration of implementation is typically dependent on what is needed in each jurisdiction and NBS program, including whether and how much funding from the MoH may be necessary. Preparation and readiness to implement NBS for MLD would include clearly defining primary and secondary targets as differentiated from incidental findings and undertaking engagement work within relevant communities of practice. Several key informants pointed out that gathering data and insights from other jurisdictions within which NBS for MLD has already been implemented would also be important and useful when preparing to implement it. Another key informant highlighted that ongoing monitoring and evaluation of the performance, impacts, and outcomes of NBS for MLD among the provincial and territorial NBS programs would be essential, and that the establishment and oversight of an expert review panel would be ideal. An example of this kind of approach has been described in the US, where the MLD RUSP Approval and Newborn Screening Implementation Program (RANSIP) was convened to gather experts who collaborate and build consensus around the implementation of NBS for MLD.57
Laboratory Considerations and Impacts
Most key informants agreed that the NBS laboratories would be substantially impacted by the addition of MLD to provincial and territorial NBS programs. Laboratory impacts would involve ensuring laboratory instrumentation, resources, and space are sufficient; developing and validating the tests that comprise the algorithm; providing training for staff; and managing changes to workflow, documentation, and information technology (IT) systems. While some key informants anticipated that these impacts could be substantial, others did not foresee impacts to laboratories being burdensome, which may at least partially reflect the differences in current infrastructure and resources across the jurisdictions.
Cost impacts of adding a new condition to the NBS panel are dependent on the tests required to perform the screening algorithm, the testing volume of the laboratory, as well as existing infrastructure and resources already available in the laboratory. Two clinical experts explained that the first-tier sulfatide test cannot be multiplexed on routine acylcarnitine LC-MS/MS platforms, which would require most NBS labs in Canada to develop a standalone test for MLD. For labs within which NBS for MLD would be implemented as a standalone test, costs would be greater, as they would require a sensitive, dedicated LC-MS/MS instrument and time for staff to prepare and run the equipment each day. For MLD, 1 key informant and a clinical expert contributing to the pilot evidence review suggested that the addition of both the sulfatide and ARSA enzymology testing could require 1 to 2 additional LC-MS/MS instruments for a laboratory, depending on whether there is currently sufficient backup capacity. One clinical expert explained that these instruments have an approximate 10-year usable lifespan and an annual service cost that is generally about 10% of the purchase cost. Any staffing costs to the lab associated with the addition of NBS for MLD would also be dependent on existing laboratory resources, testing volumes, and staffing levels, with 1 clinical expert estimating that the addition of a 0.5 to 1.0 full-time equivalent (FTE) technologist may be needed for an average Canadian NBS lab.
To manage the anticipated impacts and costs of adding MLD to NBS programs efficiently, some key informants suggested that NBS for MLD could be centralized to 1 or more labs in Canada that have current infrastructure and capacity to implement it, with other jurisdictions sending samples out for MLD screening. Notably, this has been discussed as a measure to support and enable NBS for MLD in the US, as well.41 There was variable interest in and support for this idea across those participating in the key informant interviews, with some acknowledging that it could mitigate the capital investment requirements for some jurisdictions and others pointing out that there would still be significant cost and logistical implications for labs sending out samples, as they would have to pay for coordination and processing of samples, shipping, documentation, and other associated costs. One key informant highlighted additional considerations with this kind of approach, including the availability of sufficient bloodspot samples for each infant to accommodate multiple tiers of screening for multiple conditions across more than 1 lab, as well as the administrative burden, which may not reduce the overall impact of implementing NBS for MLD.
Clinical Considerations and Impacts
Implications of Implementing NBS for MLD
Most key informants agreed that clinical resources would be impacted substantially following implementation of NBS for MLD, including the management of increased referrals from NBS in metabolic clinics, which would add to the workload for physicians and other health care workers. For MLD, there would likely be biochemical genetic and/or neurology clinical assessments, which could include brain MRI, nerve conduction studies and gall bladder imaging, as well as clinical assessments by occupational therapists and physiotherapists. While 1 key informant pointed out that the numbers of referrals for MLD from NBS may not create unmanageable impacts, the growing wave of genetic and genomic technologies is likely to continue increased demand for expanded NBS,58,59 which would increase impacts to clinical care across time, as the need for expertise and resources necessary to interpret NBS findings and manage referrals would increase accordingly.
Nonetheless, 2 experts pointed out that NBS for MLD may reduce overall impacts on clinical care services, as the potential for a diagnostic odyssey could be reduced or eliminated. As these experts pointed out, all symptomatic patients with MLD currently undergo clinical assessment with the same imaging and specialist services, with NBS referrals for MLD resulting in these assessments occurring earlier and with the benefit of information as to which assessments need to be conducted. Conversely, another key informant questioned whether the concept of NBS for MLD may inappropriately constitute a case-finding effort and could become a driver of substantial impacts on clinical care services, where unmanageable numbers of infants with uncertain diagnoses and prognoses could be unnecessarily medicalized. Most key informants did not express these concerns, however, generally pointing to data that have been generated to date around the performance and accuracy of the screening algorithm.23,29-31 Our assessment of those data is in The Test section.
The most common concern raised among key informants as well as clinical experts contributing to the pilot review about impacts on clinical services after implementing NBS for MLD was ensuring that early-onset disease could be effectively and reliably differentiated from late-onset disease. While all key informants agreed that early-onset MLD should be the primary target of NBS if treatment becomes available, they acknowledged that late-onset disease would also be identified by NBS and that predicting timing of symptom onset could be challenging.20 While some key informants suggested that late-onset MLD may constitute a secondary finding of interest, others characterized it as an incidental or false-positive finding and an inappropriate target of NBS, underscoring the importance of establishing consensus around case definitions and classification. Similarly, perspectives on how late-onset MLD identified by NBS may best be managed varied across key informants, with some suggesting that it is an important target that would necessitate careful follow-up monitoring and potentially future treatment with HSCT, whereas others simply acknowledged that it would be an important and difficult challenge that would have to be addressed. Notably, 1 key informant suggested that the identification of late-onset MLD may be 1 of several factors that render MLD an inappropriate target of NBS.
Likewise, the management of uncertain findings and complex cases of MLD (e.g., attenuated forms of the disease or variants of unknown significance, for which the diagnosis and treatment plan may be unclear) referred from NBS could have a substantial impact on clinics. Research to date indicates that a small proportion of patients with MLD could have poor prognostic certainty (largely based on rare gene variants), and while there are efforts to ameliorate this uncertainty,21 as 1 clinical expert contributing to this pilot evidence review pointed out, they are not yet in routine clinical use. Consequently, predicting the timing of symptom onset and deciding when to initiate treatment could be a challenge for clinical care providers, particularly following the implementation of NBS for MLD when patients would be identified before symptom onset.
To address this, guidelines within the US and UK contexts for the management of MLD identified by NBS have been published, including recommendations for the monitoring and management of cases where disease subtype is uncertain.19,20 To further support clinicians in Canada if NBS for MLD were to be implemented, several key informants suggested that regularly convening a group of experts to discuss and support clinical practice and decision-making for uncertain or complex referrals and/or cases of MLD could help to address this challenge, describing other examples of this approach (both within Canada and in other jurisdictions) that currently exist for other conditions identified through NBS. Notably, there are examples of this kind of approach to managing MLD in the US and in Europe.9,57 Several key informants agreed that this could alleviate the workload and impact on individual clinicians and ensure optimal care for patients with MLD that incorporates the insights and expertise of a broader set of clinical experts.
Additional concerns included the potential for NBS for MLD to generate false-positive findings, with 1 clinical expert explaining that every false-positive finding from NBS is costly, as it involves a referral, a specialist physician visit, and follow-up genetic testing. In addition, this expert explained that there are negative impacts of false-positive findings from NBS for families, which are considerable for a disease as serious as MLD. Other key informants expressed similar concerns about the potential for false-positive findings from NBS for MLD and the impacts that this would have on metabolic clinics as well as families, including the potential for medicalizing unaffected infants and children.26 Some key informants did not express concern around the prospect of false-positive and incidental findings from NBS for MLD, mostly due to the data generated to date around the performance of the proposed algorithm.60
Incidental findings were also discussed, with most key informants acknowledging the risk of identifying patients affected with MSD or SapB deficiency but also pointing to the ultrarare nature of these conditions and the low likelihood that these incidental findings would have substantial impact on health systems. Notably, several key informants explained that they have never seen these conditions across the duration of their careers, many of which spanned decades across multiple jurisdictions within and outside of Canada. Most key informants also acknowledged that, due the ultrarare nature of these conditions,61 any cases identified would have to be managed individually, as there is unlikely to be a standard of care or available treatments.27
Implications of Implementing Treatment for Early-Onset MLD
Currently, no targeted disease-modifying therapy has been submitted for regulatory or reimbursement approval in Canada for early-onset MLD. The establishment of NBS for MLD would necessarily involve careful consideration around preparing to deliver novel treatments for early-onset MLD, if they become available, are approved for use, and are reimbursed in Canadian jurisdictions.
As an ex vivo, autologous hematopoietic stem cell gene therapy, the manufacturer of arsa-cel has indicated that it can only be administered in qualified treatment centres (QTCs) that have undergone an extensive assessment using prespecified criteria and an established process (Orchard Therapeutics, London, UK: personal communication, Jul 18, 2025). Consequently, 1 or more treatment centres in Canada would necessarily have to undergo this qualification assessment and process, which would have administrative, logistical, and training implications for the facilities, physicians, and other health care staff who would have to administer the new treatment.
In Europe, there are currently a limited number of QTCs, all of which accept referrals from other EU countries.9 Similarly, in the US, there are 5 QTCs that can accept MLD patients from other states.62 It is currently not clear as to the number or location of treatment centres in Canada that may qualify if arsa-cel were to be approved for use and reimbursement, but it is likely that there would be few and they would be concentrated in urban areas. A treatment eligibility board has been established in Europe to allow for expert collaboration concerning treatment and management of newly diagnosed patients with MLD.9 This resonates with the ideas expressed by key informants informing the implementation review concerning the management of MLD identified by NBS, and demonstrating the feasibility and utility of this concept, which could be applied within Canada as well.
Detailed findings are presented in Appendix 3 of the Supplemental Material.
Budget Impact of Implementation
If potential disease-modifying treatments for MLD are made available in Canada, the implementation of a 3-tier NBS program for MLD is estimated to increase the identification of potentially treatable patients (incremental: 22 to 42 with early-onset MLD, 6 to 12 with late-onset MLD, over 10 years Canada-wide, excluding Quebec).
This represents a total incremental cost to the health care systems in Canada that can range from $4.5 million to $30.1 million within a 10-year period ($84,686 to $832,443 per additional potentially treatable patient with MLD identified) in screening and diagnostic costs. The largest proportion (47% to 86%) of these incremental costs is estimated to be operational costs (including reagents, technician time, consumables). Capital costs (acquisition and maintenance of additional mass spectrometer capacity) are still important components of the incremental budget impact (14% to 53%), much of which may be incurred up front (if amortization agreements are not possible).
These estimates are uncertain and are most sensitive to implementation assumptions concerning the current capacity of mass spectrometers and the ability to optimize the capacity of equipment within and across jurisdictions (e.g., multiplex testing, capacity-sharing or centralization to high-volume jurisdictions, and so on.)
This section addresses the following research question.
What Is the 10-Year Budget Impact of Implementing Universal Newborn DBS Screening for Early-Onset MLD Compared to No Universal Screening in Canada, Specifically From the Perspective of a Publicly Funded Health Care System and Considering Only the Costs Associated With Screening and Diagnostic Testing?
Study Design and Methods
A BIA was conducted using an Excel-based tool developed for this project. This tool has the flexibility to conduct various scenario analyses and to report estimates of budget impact by cost category and year. While the current tool can adjust the newborn population by province or territory, due to a lack of specific epidemiological and costing data, more meaningful results by jurisdiction were not feasible. The BIA is limited to only consider screening and diagnostic costs, due to a lack of information on the availability and costs associated with potentially disease-modifying treatments (such as arsa-cel for LI and EJ MLD and allo-HSCT for LJ and AO MLD), pretreatment monitoring resource needs associated with such therapies, the potential outcomes of these treatments, and the resource needs of palliative and other care for patients with MLD in the absence of disease-modifying treatment, once the MLD diagnosis is made.
Patient Population
The patient population consisted of all newborns in Canada born within the time horizon of the analysis (2026 to 2035), along with those born earlier who were projected to develop MLD within the time horizon. As universal NBS for early-onset MLD will inevitably also identify patients affected by late-onset MLD, the BIA included patients who would develop any onset phenotype of MLD. To inform the size of the population, for 2023 and earlier years, we used Statistics Canada data on the number of live births per year by jurisdiction of residence of the mother. To project 2024 and later years, we used the Statistics Canada M1 medium growth projected population of persons younger than 1 year of age as a proxy for the number of live births per year.63,64
Strategies Assessed
The reference scenario was considered equivalent to the current strategy without NBS for MLD (no NBS, Figure 1). We assumed that pregnancies where there is a family history of MLD would be offered prenatal testing and/or testing at birth to identify newborns with MLD, with the majority of such patients identified within the potential treatment window for potential disease-modifying treatments. The treatment window was assumed to include PS patients for LI MLD, and PS or ES patients for EJ, LJ, and AO MLD, as defined in other jurisdictions’ regulatory approvals for arsa-cel65-67 and guidelines for allo-HSCT.68 Smaller proportions of patients with EJ, LJ, and AO MLD without a family history were also assumed to be identified within their respective treatment windows through diagnostic assessment and testing (full details are provided in Appendix 1 of the Supplemental Material). All other patients were assumed to develop symptoms and receive diagnostic assessment but be diagnosed beyond their treatment windows.
The NBS scenario assumed that all newborns in Canada would receive 3-tier NBS for MLD, as described in the Newborn Screening Algorithm section of this report (proposed model in Figure 2) and in further detail by Laugwitz et al. (2024),23 starting in year 1 of the analysis (2026). For the base case, we assumed that MLD screening could not be multiplexed with other NBS programs that are currently or soon will be implemented, and that existing mass spectrometers are already operating at full capacity. We assumed newborns screening positive on all 3 tests would receive genetic or metabolic specialist consultation, confirmatory testing in the form of urine sulfatide levels and white blood cell enzymology for ARSA activity, and genetic sequencing of the parents’ ARSA variants (if their ARSA status was previously unknown). In this NBS scenario, patients who were born before NBS for MLD initiation but projected to develop MLD within the time horizon were assumed to have similar diagnostic pathways and outcomes to the reference scenario (i.e., retroactive screening did not occur). We assumed that NBS would not alter the incidence of MLD in the population but would alter when MLD would be identified.
Of note, due to a lack of data, we assumed that NBS for MLD would not impact family planning, and thus we assumed that earlier identification of MLD due to NBS would not alter the number of children born with MLD within the time horizon. Nonetheless, NBS may ultimately lead to altered reproductive choices (refer to Ethical Considerations in the Use of NBS for EO MLD), rendering this assumption uncertain. We assumed prenatal testing of infants with a known family history of MLD would continue as in current clinical practice in addition to NBS. Thus, costs associated with prenatal testing were not included in the analysis, as they were assumed to be the same in the no NBS and NBS scenarios.
Time Horizon, Discounting, and Inflation
The base case and all sensitivity analyses were conducted using a 10-year time horizon (2026 to 2035), with results presented in 1-year increments. We adopted this time horizon to capture the development of symptoms for most of those born before NBS and reflect the usual lifespan of mass spectrometers. In line with the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) guidelines for conducting BIAs, no discounting was applied.69 We adopted an inflation rate of 2% per annum, in line with the Bank of Canada’s target rate, which was applied to all costs.70
Perspective
The BIA was conducted from the perspective of Canada’s provincial and territorial health care systems (excluding Quebec). As such, only costs covered by the health care payer were captured, which may include screening costs (reagents, consumable supplies, compensation), confirmatory testing costs, medical appointments, diagnostic radiology and diagnostic testing, and capital costs associated with the increased need for mass spectrometers for testing. Indirect costs (such as productivity losses for parents or guardians, out-of-pocket costs, and time lost) were not considered in the analysis. To calculate the number of additional mass spectrometers required for the implementation of NBS for MLD, we assumed 7 NBS programs across Canada, excluding Quebec (some jurisdictions provide NBS services to others, as described in Appendix 1 of the Supplemental Material), and that these programs do not have any existing mass spectrometer capacity to absorb the volume of new tests (i.e., the worst case scenario that existing mass spectrometers are operating at full capacity, and the analysis rounds up the number of new machines required).
Inputs and Data Sources
The input parameters used in the base-case analysis are summarized in Appendix 1 of the Supplemental Material. All inputs were deterministic for the purposes of this analysis.
Epidemiological inputs were informed by the probabilities of positive results on each NBS tier, informed by the Laugwitz et al. (2024)23 finding of 3 patients with MLD among 109,259 newborns screened. These were adjusted for our estimated PPVs and NPVs for the 3 tiers together, which were informed by clinical expert opinion on the proportion of identified ARSA variants that were of unknown significance. As a result, the base-case analysis assumed an MLD incidence of 2.8 per 100,000 live births, or 1 in 35,191. An alternate rate was explored in the scenario analysis.
NH inputs for MLD were derived from the literature and validated with clinical experts, including the distribution of MLD phenotypes,12,56,71 the probability of patients having no known family history of MLD,56 the probability of patients being diagnosed within the treatment window in current practice based on their family history,56 and the typical age of symptom onset or PS identification by phenotype, if not identified at birth.72
Costs associated with NBS for MLD included reagent and consumable costs, medical laboratory technician costs, and an associated productivity rate (the proportion of tests that were newborn screens, rather than calibration and quality control). Additionally, costs associated with the purchase and maintenance of LC-MS/MS were included. The majority of costs associated with NBS were derived from personal communications with a laboratory expert (Dr. Graham Sinclair, Newborn Screening Laboratory Head, BC Children's Hospital, Vancouver: email communications, Aug 5, 2025, and Aug 28, 2025), as well as from a Newborn Screening Ontario Molecular Diagnostic Testing price list73 and the Government of Canada’s Job Bank.74 Costs associated with confirmatory testing for patients with positive NBS results for MLD or positive prenatal tests, as well as those associated with diagnosing older patients presymptomatically or postsymptomatically, were estimated based on clinical expert input, personal communications with the same laboratory expert, the Ontario Schedule of Benefits for Physician Services,75 Discovery DNA,76 and GenomeQuébec.77
Scenario Analyses
The incidence of MLD used in the base case was derived from Laugwitz et al. (2024),23 which is based on small numbers and is at the highest end of those reported in the literature (0.63 to 3.7 per 100,000).13,56 As MLD is a rare disease, even small variations in the number of cases identified within a study can have a large impact on calculated incidence. While it is a known phenomenon that systemic screening often reveals higher disease prevalence than previously estimated through clinical diagnosis alone,33 we conducted a scenario analysis assuming half the base-case incidence (1.4 per 100,000 live births) to explore the impact of this uncertainty. Additionally, while the base case accounts for a 50% redundancy in mass spectrometry machine time required to allow for short-term maintenance, and rounds capacity needs up to the next whole machine, it does not include the costs of spare whole machines (i.e., the need for an additional whole mass spectrometer in case of failure or large backlog). We therefore conducted a scenario analysis where each screening program purchased and maintained 1 additional mass spectrometer beyond that already estimated (British Columbia plus Yukon, Alberta plus Northwest Territories, Saskatchewan, Manitoba, Ontario plus Nunavut, and the Maritimes, while the excess capacity needs of Newfoundland and Labrador were assumed to be fulfilled by Ontario). We also conducted a scenario analysis that did not round up the additional required mass spectrometry capacity for each NBS program; instead, the total estimated increased capacity requirement of mass spectrometers specifically for MLD was reported, allowing programs to estimate the actual number of machines required given the potential for current mass spectrometry resources to have additional capacity and/or for other NBS testing to use the remaining time on newly purchased equipment (and thus have those costs attributed to NBS for other conditions) and/or for more efficient sharing of resources within and between screening programs. Finally, feedback from some provincial jurisdictions indicated that multiplexing may become possible in some jurisdictions in the near future (refer to section titled The Test: Acceptability of the Proposed NBS Algorithm for MLD), and thus a scenario analysis was performed assuming multiplexing of the first-tier sulfatide screening test (i.e., best case scenario), which also reported actual estimates of mass spectrometry equipment required, rather than the number of additional machines required rounded up. Parameter changes for these scenarios are available in Appendix 1 of the Supplemental Material.
Results
The incremental resource use, cost, and diagnostic outcomes of implementing NBS for MLD under a Canadian health care payer perspective, excluding Quebec, up to the point of diagnosis of MLD, are provided in Table 4. These include disaggregated costs; the estimated number of screening tests that would be conducted at each tier; the incremental number of patients with MLD who would be identified per year (within and after the treatment window); and the incremental resource use of medical laboratory technicians, mass spectrometers, and physician appointments that would take place as a result of NBS for MLD implementation. Annual results and 10-year results for the no NBS scenario (reference) and NBS scenario can be found in Appendix 3 of the Supplemental Material.
In the base-case analysis, within a 10-year time horizon, in the absence of NBS for MLD (reference scenario), we estimated that approximately 79 patients would be diagnosed with MLD, of which 59 would have early-onset MLD (LI or EJ) and 20 would have late-onset (LJ or AO) MLD. Of these, 18 with early-onset MLD and 16 with late-onset MLD would be diagnosed within the treatment window, while 46 would be diagnosed after the treatment window. If NBS for MLD were implemented (NBS scenario), we estimated that 98 patients would be diagnosed with MLD within the time horizon (due to NBS leading to earlier diagnoses, as screened patients would be diagnosed in their year of birth rather than later). Of these, 60 patients with early-onset MLD and 27 patients with late-onset MLD would be diagnosed within the treatment window, with 11 diagnosed past the window, almost all of whom were born before the implementation of NBS. As such, the incremental number of patients diagnosed with MLD within the time horizon (NBS versus no NBS) would be 19 people, with 42 additional patients with early-onset MLD and 12 additional patients with late-onset MLD diagnosed within the treatment window. Thirty-five fewer patients would be diagnosed after the treatment window had passed.
In terms of cost, again within a 10-year time horizon, we estimated that the no NBS case would cost $670,000 up to the point of diagnosis, consisting of confirmatory and diagnostic testing costs. Implementation of NBS for MLD would result in an additional cost of $23.7 million, up to the point of diagnosis. This consists of $14.1 million (60%) in direct operational screening costs, $9.6 million (40%) in mass spectrometer costs (capital and maintenance costs), and $197,000 (< 1%) in confirmatory MLD testing costs (after a positive screening or prenatal result), with savings of $214,000 (< −1%) in diagnostic testing costs (for patients who were not identified at birth). The Canadian health care systems would be required to provide, on average, approximately 279,340 sulfatide DBS tests (in the first tier), 974 ARSA activity DBS tests (in the second tier), and 85 genetic sequencing DBS tests (in the third tier) per year. These would require, on average, 7.2 extra FTEs of lab technicians per year, spread across the various NBS programs, and an additional 9 mass spectrometers (rounding up each NBS program’s needs), with a negligible impact over time on the number of doctor appointments up to the point of diagnosis. Over 10 years, assuming an additional 54 patients are identified within the treatment window, the total average incremental cost per additional potentially treatable patient (with early-onset or late-onset MLD) as a result of NBS for MLD would be $443,078, with an average 3-tier cost of $8.50 per newborn screened.
Results for the scenario analyses can be found in Appendix 3 of the Supplemental Material. When the probability of a positive first-tier screening test, and thus the incidence rate of MLD, is assumed to be half that of the base case, the incremental cost of implementing NBS for MLD was estimated to be $23.1 million, with 22 additional patients with early-onset MLD and 6 additional patients with late-onset MLD identified within the treatment window, compared to no NBS. Assuming this halved incidence rate, the incremental cost per additional potentially treatable patient would be $832,443, with an average cost of $8.26 per infant screened. In a scenario where every independent screening program is assumed to require an additional mass spectrometer, the number of additional potentially treatable patients remains unchanged (42 with early-onset MLD, 12 with late-onset MLD). However, 15 additional mass spectrometers would be required rather than 9, increasing the total incremental cost to $30.1 million, resulting in an incremental cost of $562,774 per additional potentially treatable patient, and an average cost of $10.79 per newborn screened. In a less budget-intense scenario in terms of mass spectrometer needs, where the estimated additional machine capacity required for MLD screening was reported precisely rather than rounding up each NBS program’s requirements, the total incremental cost decreased to $18.8 million. This was due to reductions in acquisition and maintenance costs to reflect only the additional yearly capacity of 4.3 mass spectrometers required (rather than 9 in the base case or 15 in the extra capacity scenario) compared to no NBS, and would result in an incremental cost of $350,129 per additional potentially treatable patient, and an average cost of $6.71 per newborn screened. If multiplexing of the tier 1 sulfatide test was implemented across the country, the number of additional potentially treatable patients identified remain unchanged (42 with early-onset MLD, 12 with late-onset MLD); however, the total incremental cost further decreased to $4.5 million due to reductions in the use of consumables, as well as lab technician and mass spectrometer time (requiring only an additional 0.6 device capacity and 1 FTE lab technician per year compared to no NBS). This would be the least budget-intense implementation scenario for a 3-tier NBS program for MLD, with an average total incremental cost to the health system of approximately $84,686 per additional potentially treatable patient, up to the point of diagnosis, with an estimated average 3-tier cost of $1.62 per newborn screened.
Limitations and Key Uncertainties
The BIA may overestimate the incidence of MLD in Canada, potentially inflating the numbers of patients diagnosed within the treatment window through NBS. It may also overstate certain costs, which could be reduced if existing system capacity allows for multiplexing NBS for MLD with other screening tests, if there is already some surplus mass spectrometer availability, or if system capacity can be coordinated across programs. Conversely, the BIA may underestimate other costs. It excludes the potential time and resources needed to refine the screening algorithm, train staff, adjust laboratory workflows, and manage logistics (e.g., documentation, shipping, IT systems, and data coordination). While it accounts for the purchase of LC-MS/MS machines and maintenance costs over 10 years, it omits infrastructure needs, such as laboratory space and equipment replacement. Postdiagnosis monitoring, treatment costs, and other subsequent health care costs were also excluded, and it was not possible to estimate these at the current time, nor to estimate the health care cost of caring for patients who have not been treated. To do so would require a disease simulation model to account for the costs and effects of treatments compared to no treatment.
Clinical expert input noted that the dedication of part of a genetic counsellor workload may be useful in researching and reporting on ARSA variants of unknown significance as they are found through NBS. This has not been included within the BIA, although an additional genetic or metabolic specialist appointment has been assigned for each patient estimated to have a variant of unknown significance.
Table 4: Incremental Budget, Resource Use, and Diagnosis Results for NBS vs. No NBS, Canada Excluding Quebec
Results, incremental | 2026 | 2027 | 2028 | 2029 | 2030 | 2031 | 2032 | 2033 | 2034 | 2035 | 10-year total |
|---|---|---|---|---|---|---|---|---|---|---|---|
Number of NBS screens conducted | |||||||||||
First-tier sulfatide DBS | 279,000 | 276,100 | 275,200 | 275,600 | 276,700 | 277,800 | 279,500 | 281,800 | 284,300 | 287,400 | 2,793,400 |
Second-tier ARSA activity DBS | 973 | 963 | 960 | 961 | 965 | 969 | 975 | 983 | 991 | 1,002 | 9741 |
Third-tier genetic sequencing DBS | 85 | 84 | 83 | 84 | 84 | 84 | 85 | 85 | 86 | 87 | 847 |
Incremental costs for screening and diagnosis | |||||||||||
Direct NBS costs (3-tier DBS tests) | $1.3 million | $1.3 million | $1.3 million | $1.4 million | $1.4 million | $1.4 million | $1.5 million | $1.5 million | $1.5 million | $1.6 million | $14.1 million |
NBS mass spectrometer costs (purchase and maintenance) | $5.0 million | $468,180 | $477,544 | $487,094 | $496,836 | $506,773 | $516,909 | $527,247 | $537,792 | $548,547 | $9.6 million |
Confirmatory testing costs (after positive NBS) | $17,991 | $18,160 | $18,463 | $18,859 | $19,313 | $19,778 | $20,297 | $20,873 | $21,480 | $22,148 | $197,362 |
Diagnostic testing costs (for patients not identified through NBS or prenatal tests) | $0 | –$18,227 | –$18,399 | –$18,705 | –$23,943 | –$24,448 | –$25,001 | –$25,633 | –$29,386 | –$30,153 | –$213,895 |
Total incremental costs for screening and diagnosis | $6.4 million | $1.8 million | $1.8 million | $1.8 million | $1.9 million | $1.9 million | $2.0 million | $2.0 million | $2.1 million | $2.1 million | $23.7 million |
Incremental number of patients diagnosed per year | |||||||||||
Number of patients with early-onset MLD (LI or EJ) diagnosed within treatment windowa | 4.29 | 4.21 | 4.20 | 4.21 | 4.09 | 4.10 | 4.13 | 4.17 | 4.20 | 4.25 | 41.85 |
Number of patients with late-onset MLD (LJ or AO) diagnosed within treatment windowa | 1.27 | 1.26 | 1.25 | 1.25 | 1.26 | 1.26 | 1.27 | 1.28 | 0.79 | 0.81 | 11.71 |
Number of patients with MLD diagnosed after treatment windowa | 0.00 | −3.40 | −3.36 | −3.35 | −4.08 | −4.09 | −4.10 | −4.12 | −4.15 | −4.18 | −34.83 |
Total incremental number of patients diagnosed with MLD per yeara | 5.56 | 2.07 | 2.09 | 2.11 | 1.26 | 1.28 | 1.30 | 1.33 | 0.85 | 0.88 | 18.73 |
Incremental resource use | |||||||||||
Lab technicians required for tier 1 and 2 screening (FTEs) | 7.22 | 7.14 | 7.12 | 7.13 | 7.16 | 7.18 | 7.23 | 7.29 | 7.35 | 7.43 | 72.25 |
Additional mass spectrometer capacity required for tier 1 and 2 screening (machine-years) | 9.00 | 9.00 | 9.00 | 9.00 | 9.00 | 9.00 | 9.00 | 9.00 | 9.00 | 9.00 | 90.00 |
Incremental physician appointments up to point of diagnosis | 11.95 | 1.53 | 1.60 | 1.65 | −0.89 | −0.86 | −0.82 | −0.79 | −2.27 | −2.23 | 8.87 |
AO = adult onset; DBS = dried bloodspot; EJ = early juvenile; FTE = full-time equivalent; LI = late infantile; LJ = late juvenile; MLD = metachromatic leukodystrophy; NBS = newborn screening; vs. = versus.
aThe number of patients identified is highest in 2026 (year 1), as the model assumes that all patients with MLD born in 2026 are identified by NBS, and patients born in previous years are still being identified as in the reference case. By 2027 (year 2), all modelled patients with LI MLD born before NBS implementation have previously been identified and will in the future be identified by NBS in their year of birth. Similarly, by 2030 (year 5) and 2034 (year 9), all modelled patients with EJ MLD and LJ MLD, respectively, born before NBS implementation have also previously been identified. Those with AO MLD will continue to be identified as in the reference case for longer than the time horizon of the analysis.
Social and Ethical Considerations
Early-onset MLD leads to a rapid and devastating decline in children, placing substantial emotional, psychosocial, and financial impacts on families, who must grapple with their child’s swift deterioration and the absence of effective treatment options.
MLD illustrates an underlying tension in NBS more broadly, where there is debate on what should constitute the appropriate scope of benefits. As NBS programs expand to include more conditions that test the boundaries of traditional screening criteria, there is some interest in extending the scope of benefits to include elements outside of direct clinical benefit to the newborn (e.g., family planning, limiting diagnostic odysseys). While NBS for MLD offers the promise of earlier diagnosis, its implementation risks identifying late-onset or indeterminate cases that may never require treatment, raising complex ethical, emotional, and systemic questions about the balance between expanding knowledge and imposing long-term uncertainty on families.
NBS is typically conducted under an opt-out model of consent to protect and privilege the rights of the child and their health. However, when NBS for a condition identifies other late-onset subtypes and where such early identification has uncertain benefits and risks, as in the case of MLD, ethical concerns are raised. Addressing these may call for considering a more explicit informed consent and relationally supportive practices to respect parental autonomy and caregiving roles.
While adding MLD to NBS may offer clinical benefits, its defensibility also depends on addressing potential structural inequities in follow-up care. Without this, families in rural or remote areas, newcomers to Canada, and some racialized groups may face disproportionate impacts (e.g., travel and associated out-of-pocket costs, language barriers, or unnecessary prolonged surveillance) while receiving fewer potential benefits.
This section addresses the following research question.
What Are the Social and Ethical Considerations Associated With Newborn DBS Screening for Early-Onset MLD?
Ethical Considerations in the Diagnosis, Treatment, and Experiences of MLD
The defensibility of adding MLD to NBS panels depends, in part, on the experiences of living with MLD and the extent to which current treatments can alter disease trajectories. As detailed in this section, children face a rapid and devastating decline, families experience serious caregiver impacts, and diagnoses often come only after long delays. At the same time, current treatment options remain limited, with only narrow windows of opportunity for intervention and limited understanding of benefit.
Disease Burden
MLD is a rare, progressive LSD caused by pathogenic variants in the ARSA and PSAP genes.14 As noted in the The Condition section, MLD has a broad phenotype that is composed of 2 early-onset and 2 late-onset subtypes. While existing data suggest that early-onset subtypes appear at a higher rate, true prevalence estimates across subtypes remain uncertain, as case identification has historically relied on clinical presentation, which may favour recognition of severe early-onset disease.14,21 NH is also incompletely understood, particularly for juvenile and adult forms, where diagnostic challenges and uncertainty about the pathogenic significance of some gene variants complicate efforts to track progression and prognosis.14 Clinical experts noted that implementation of NBS (if recommended) would likely shift current prevalence estimates by identifying more late-onset cases or variants of unknown significance.
Children with early-onset MLD experience a rapid and devastating disease trajectory, with loss of motor and cognitive function typically occurring within months of diagnosis and accompanied by severe neurologic pain.12 For families, this translates into profound psychosocial, emotional, and financial impacts. Many caregivers reported some leaving employment to provide full-time care and described feelings of exhaustion, isolation, and the collapse of normal family routines.78 Caregiver input echoed this, highlighting the distress of watching a previously healthy child become fully dependent within months. These demands can strain family relationships and leave lasting impacts on parents and siblings, including grief, guilt, and ongoing anxiety about the child’s prognosis.78,79 Knowing the diagnosis in the absence of effective treatment options may compound these impacts, as caregivers must navigate the certainty of decline with few avenues for intervention or hope for reprieve.
Diagnostic Challenges and Disparities
As described previously, most cases of MLD in Canada are currently identified after symptom onset, with early diagnosis occurring almost exclusively when there is a known family history (e.g., asymptomatic sibling of an affected child). Caregiver input and published literature describe lengthy and distressing diagnostic odysseys, often involving repeated consultations and misdiagnoses before MLD is confirmed.78 Clinical experts indicated that this may be exacerbated for families in rural or remote areas, where specialist services are limited. They added that newcomers whose first language is neither English nor French may face additional communication and navigation challenges in accessing follow-up testing and specialist care. Diagnostic delays are especially harmful in early-onset forms, where the disease progresses rapidly and opportunities for intervention, when available, are lost.
Evidence from the US highlights additional potential inequities in diagnosis: children from racialized populations and those with public or no insurance were significantly less likely to receive timely diagnoses or specialty care, even after controlling for geography.80 There are also uncertainties about phenotypic variation and the distribution of genetic variants across populations, which complicate accurate classification of disease-causing alleles in racialized populations.20 Studies suggest that racialized populations may face an elevated risk of pseudodeficiencies (reduced enzyme activity that is not disease-causing), leading to inaccurate identification of LSDs.81 While direct parallels to the Canadian context are uncertain, similar structural determinants of health could perpetuate inequities in diagnosis, even if NBS were in place.
Current Treatments
At present, treatment options remain limited. In Canada, outside of supportive care, there is no approved disease-modifying therapy for EI MLD, even when identified presymptomatically. Allo-HSCT may benefit select PS patients with EJ, LJ, and AO disease, but its use is constrained by the need for suitable donors (particularly challenging for diverse populations), narrow treatment windows, and the risk of serious adverse effects (e.g., graft-versus-host disease).12,24 Clinical experts pointed to 1 autologous gene therapy product (arsa-cel) as a promising intervention for PS individuals with LI and EJ disease (and potentially patients with ES, EJ disease), with favourable outcomes reported in other jurisdictions.12,24 However, arsa-cel has not been submitted for reimbursement or regulatory review in Canada at the time of this review.
Ethics of Evidence and Evaluation of NBS for MLD
As detailed in the The Test section, the CDA-AMC review team assessed evidence from 4 studies examining the accuracy of 3 screening algorithms for MLD. Two involved 2 tiers of testing and the third involved 3 tiers. In this section, we briefly identify ethical implications associated with study outcomes, the identification of traditionally off-target findings, and the generalizability of findings to diverse populations.
Accuracy of Algorithms
Evidence suggests that both 2-tier and 3-tier screening algorithms can detect MLD with high specificity. A 3-tier approach (sulfatide levels, followed by ARSA enzyme, followed by molecular testing) appears particularly strong, with a PPV approaching 100% and 0 false-positives. By contrast, 2-tier strategies have shown more variability, with PPVs ranging from 15% to 50%. While these results are encouraging, the small number of cases identified and risk of bias across studies limit confidence in how well these findings would generalize to practice. The ethical consideration is not that accuracy of screening algorithms is poor, but rather that premature confidence in limited evidence could influence policy decisions toward adoption without fully accounting for this uncertainty. If implemented, a program that fails to meet expectations may not only have long-term consequences for (missed) patients and their families, but it could also undermine public confidence in NBS.
Off-Target and Uncertain Findings
Beyond the question of accuracy, even well-performing algorithms raise challenges when they identify newborns outside the intended early-onset target population. Detection of uncertain genotype-phenotype and late-onset cases (traditionally considered out of scope in NBS) appears unavoidable. Although existing evidence indicates that early-onset disease comprises the majority of cases (75% to 90%), clinical experts indicated that prevalence estimates would likely shift if MLD were added to NBS. Reporting late-onset and uncertain genotype-phenotype findings (despite limited prognostic value) places families and clinicians in the position of making long-term monitoring and life-planning decisions under substantial uncertainty. For patients and their families, the key question is whether anxiety, long-term surveillance, and potential impacts on life-planning are warranted in the context of findings that are not immediately actionable or, in the case of indeterminate findings, may never become actionable. At the policy level, it raises the question of whether directing public resources toward such cases (with uncertain long-term implications) represents a fair and sustainable use of NBS capacity.
Generalizability
None of the included studies reported participant characteristics such as sex, ethnicity, or other PROGRESS-Plus51 factors. This limits our ability to assess whether screening algorithms perform consistently across diverse population groups or whether certain population groups face elevated risks of false-positive or false-negative results. This is especially concerning given broader evidence out of the US that suggests racialized populations may be more likely to be identified with LSD pseudodeficiencies.81
Ethical Considerations in the Use of NBS for Early-Onset MLD
Clinical experts and laboratory professionals engaged by CDA-AMC, as well as caregiver input provided in response to a CDA-AMC open call, were generally supportive of adding MLD (ideally using a 3-tier screening algorithm) to the recommended NBS list. They suggested its addition was of “critical importance” given the rapid progression of early-onset (particularly LI) forms, the need for early (PS) intervention, and the high risk of premature mortality if left untreated.20,21,24 While acknowledging the evidentiary limitations of proposed algorithms, most clinical experts and laboratory professionals considered these acceptable in light of the devastating nature of the disease. At the same time, they stressed that MLD would not meet established screening criteria3 in the absence of an effective treatment option for early-onset disease and plan on how to manage identification of late-onset cases. However, given their anticipation of future treatment options becoming available in Canada, some experts suggested it would be prudent to begin preparing for inclusion of MLD to the recommended NBS list now.
These perspectives not only highlight the urgency of early intervention but also expose the tension between existing screening criteria and emerging expectations of benefit. Since its inception, NBS has been guided by Wilson and Junger’s principles for screening (reflected in the Newborn Screening Advisory Panel’s criteria guiding this review), designed to protect against potential harm to newborns and their families; however, advances in both screening technology and treatment options increasingly test those boundaries.82 In what follows, we examine 3 interrelated domains of ethical concern (and their relevance to MLD) around the scope of benefits considered acceptable in NBS, the implications of off-target and uncertain findings, and the importance of consent and equity in ensuring that burdens and benefits are fairly distributed.
Scope of Benefits of NBS
As screening technologies and treatment options have advanced, interest has grown in expanding NBS to include conditions outside the boundaries of Wilson and Junger’s criteria.82-84 This shift has heightened debate about the role of NBS and, in particular, what should constitute the appropriate scope of benefits.83,85,86 Two overarching approaches have been described, 1 targeted and the other broad. The targeted-scope model (taking a public health emergency orientation82,86) limits screening to severe, early-onset conditions for which effective treatment is available at the time of detection.85,86 Benefits are narrowly (and explicitly) defined as improved clinical outcomes for the newborn, while indirect gains (such as earlier diagnosis and family planning) are considered secondary.3,85-87 By contrast, the broad-scope model (taking a public health service orientation82,86) treats benefits more expansively, including earlier diagnosis, reproductive planning, and enabling moderate interventions to reduce morbidity.83,86 Here, informational and preparatory gains are seen as direct benefits (even if no disease-modifying treatment is available) and greater weight is placed on parental values and autonomy insofar as earlier knowledge may enable families to make more informed choices about care and future planning.85-87
In a broad-scope framing, reproductive choice is often highlighted as a direct benefit. However, some authors caution that grounding NBS in reproductive decision-making risks shifting the focus away from child health and toward judgments about which lives are valued.88 This, they argue, reflects historical eugenics logics, where screening could become justified less by benefit to the individual newborn than by its potential to shape population-level reproductive choices about what constitutes a life worth living.88 Others highlight public support for expanded screening, although concerns remain about uncertainty, psychosocial impact, and the balance of risks and benefits.87,89 Indeed, Canadian publications suggest support for expanded screening, although preferences shift toward voluntary, choice-based approaches when treatment is limited.87,89 Parents have also identified potential consequences of expanded screening by suggesting that NBS can disrupt early bonding, create lasting guilt, and produce “patients-in-waiting” whose futures are clouded by uncertainty.90 While preventive care pathways following abnormal or late-onset results are potentially life-saving, these can be intensive and shift responsibility onto families, potentially exacerbating burdens for those with fewer resources.90
Rather than neatly fitting within either a targeted or broad-scope model, NBS for MLD has the potential to straddle these boundaries and raises questions about how “benefit” should be defined in practice. Screening for MLD would clearly align with a targeted model in its aim to identify severe, early-onset disease if an effective early intervention were available. However, there is currently no effective treatment option for early-onset disease in Canada. Should an effective treatment option become available, the identification of newborns with no immediate need for intervention (i.e., late-onset and indeterminate cases) remains unavoidable. While identifying at-risk infants may also be considered a benefit, given the potential impact on diagnostic odysseys, family planning, and early access to support services (among other things), these potential benefits are more clearly located within a broad-scope model of benefit.
For this reason, in addition to considering the value of adding MLD should a disease-altering intervention like arsa-cel become available in Canada, decision-makers are inevitably faced with the need to weigh the relative value of expanding the Pan-Canadian Newborn Screening List to include conditions with broader-scope benefits against the potential harms of such an expansion. As described subsequently, this can be challenging to do given the uncertainty surrounding true prevalence of early-onset and late-onset MLD.
Off-Target and Uncertain Findings
Although biochemical markers for LSDs are typically reliable and easily detectable (e.g., sulfatide accumulation and ARSA enzyme activity in MLD),91 the marked phenotypic heterogeneity of these disorders — particularly the coexistence of early-onset and late-onset forms — complicates judgments about whether population-level screening is justified.91,92 The identification of late-onset cases is unavoidable in LSD screening, raising questions about uncertain benefits, indeterminate prognoses, and the risks of generating long-term clinical and psychosocial burdens for families.91,93
MLD pilot programs assessed in the The Test section identified newborns across a wide phenotypic spectrum, ranging from EJ (i.e., early-onset) to late-onset cases, as well as indeterminate and even “likely unaffected” individuals. These outcomes highlight the risks and burdens of identifying newborns with MLD for whom no immediate intervention is needed, including heightened parental anxiety, prolonged monitoring, and additional strain on health system resources resulting from additional monitoring. While shifts in the understanding of epidemiology and NH of MLD are expected, clinical experts stressed that the scale and consequences of these shifts cannot be reliably predicted in advance. While some noted that such findings can expand epidemiological knowledge and inform therapeutic development, others question whether these benefits outweigh the personal and systemic costs borne by families flagged through screening. At the same time, caregiver input emphasized that earlier diagnosis can also bring relief, alleviating uncertainty, enabling life planning, and improving access to supportive services. This is supported by some evidence suggesting that individuals with a broad range of late-onset LSDs (no participants had MLD) would have appreciated knowing sooner (e.g., via NBS), as they thought it could have influenced their reproductive decision-making, health behaviours, and career planning, as well as potentially foster a sense of validation and understanding from others, even if these benefits coexist with potential harms.94 Nonetheless, others have suggested that in the case of both late-onset and uncertain prognosis, the lifelong potential that one may become affected with the condition in question can echo the psychological and emotional distress of the uncertainty of a prolonged diagnostic odyssey.95
The experience of screening for Krabbe disease in New York State underscores these trade-offs. In its first 5 years, screening more than 1 million newborns for Krabbe disease identified 11 infants projected to develop early-onset disease and 19 projected to develop late-onset disease.96,97 While it was anticipated that nearly 90% of new cases would be early-onset, based on existing prevalence data at the time,97 what emerged instead was that more than 60% of identified infants fell outside the traditional target population of NBS (i.e., early-onset). While these findings expanded knowledge of the epidemiology of Krabbe disease, they also extended psychosocial distress and prolonged uncertainty of late-onset disease or indeterminate findings to far more families than expected.96,97 This mismatch between expectations and outcomes illustrates how NBS can reshape assumptions about disease prevalence, and why such shifts cannot be treated as value-neutral.
As an LSD (like Krabbe) with uncertain true prevalence, clinical experts acknowledged that our understanding of MLD is likely to shift if NBS were introduced. While current expectations suggest that 75% to 90% of identified cases would be early-onset, screening could identify an increasing number of late-onset and indeterminate findings, raising questions about the impacts of these findings on families, care providers, and health systems.
Informed Consent and Relationality
NBS has historically been justified as a mandatory or near-mandatory public health intervention on the grounds of protecting children’s health and preventing serious harm.98 Many programs, including those in Canada, therefore function under an “opt-out” model, in which consent for screening is implied rather than explicitly sought.82 This model can be defended when applied to well-established conditions where the benefits of early detection are clear and the harms of missing a case are severe.82 However, it becomes more ethically challenging in the context of conditions like MLD, given the uncertain benefits and risks of screening, including predictive accuracy concerns and the possibility of late-onset and indeterminate findings. In such cases, explicit informed consent from parents before screening may facilitate respecting parental autonomy, which would then mitigate potential psychological harms to parents and give them the opportunity to decide what kind of information they wish to receive.96 There is some concern that requiring informed consent could lead to lower rates of uptake and limit the utility of NBS programs.99 This has not borne out in practice.99
In addition, relational perspectives on NBS emphasize that screening practices should not only protect individual choice through informed consent practices, but also actively support parents in their caregiving role.99 Empirical studies suggest that while many parents consent readily to NBS because of the trust they place in the health care system, others feel poorly informed or pressured by circumstances.100 From a relational perspective, the timing, quality, and follow-up of screening are ethically significant because NBS is not only an individual choice but also impacts how parents care for, and relate to, their child.
Equity
Equity considerations highlight that even if MLD were added to NBS programs nationwide, the distribution of benefits (e.g., earlier detection, potential access to therapies) and burdens — particularly structural and logistical demands, such as travel, out-of-pocket costs, and long-term monitoring — would likely remain uneven across families or communities. While the process of initial screening would likely be delivered equitably, the addition of MLD could raise several structural concerns related to follow-up visits and care for newborns who are positive for MLD and their families. Access to metabolic specialists, neurologists, geneticists, and advanced therapies tends to be concentrated in major urban centres, meaning families in rural, remote, and underserved regions could face disproportionate burdens, including extensive travel, lost income from time off work, and out-of-pocket expenses. Newcomers to Canada whose first language is neither English nor French may also face linguistic and navigation barriers in accessing follow-up testing and specialist care. Evidence from the US further shows that racialized groups living with leukodystrophies, including MLD, may experience reduced access to specialty care, even when geography is accounted for.80 Such disparities suggest that structural barriers are compounded by social and systemic inequities, raising the risk that some families will bear disproportionate burdens with fewer corresponding benefits.
Addressing inequities cannot be solved by technical fixes alone; it will require attention to the broader social conditions that shape families’ well-being, including economic security, access to transportation, and culturally safe care. From this perspective, the ethical defensibility of adding MLD to NBS rests not only on its clinical benefits but also on whether it contributes to reducing, rather than compounding, systemic disadvantages.
Health Systems Considerations
Decisions about cost, capacity, and coordination shape whether screening programs are experienced as fair, proportionate, and trustworthy. In this sense, evaluating health system readiness is inseparable from considering how resources are allocated, how consistently families are supported across jurisdictions, and how program governance maintains public confidence.
Introducing NBS for MLD would entail direct costs (laboratory reagents, additional staff) and subsequent indirect costs (family travel, out-of-pocket expenses, psychosocial supports) that must be weighed against the proportion of infants expected to benefit from early detection, particularly where a proportion of screen-positive cases will be late-onset or indeterminate and require prolonged monitoring. Excluding upfront capital expenditures (i.e., the purchase cost of new mass spectrometers), our BIA base case estimated that the additional operating costs to jurisdictions will hover somewhere between $1.8 million and $2.1 million annually over the next 10 years. Given the rarity of MLD, some laboratory professionals expressed concern that adding MLD could stretch limited program capacity and divert resources from other early childhood public health interventions with more immediate returns.
Infrastructure and Workforce Capacity
Implementing NBS for MLD would require laboratory capacity for multitier testing, access to timely confirmatory diagnostics (e.g., enzyme activity assays, genetic sequencing), and specialist consultation in metabolic or leukodystrophy clinics. These demands are likely to increase pressure unevenly across jurisdictions and NBS programs, depending on existing infrastructure and workforce capacity. These demands may result in some jurisdictions deciding not to implement NBS for MLD, leading to inequitable access across Canada.3 Even if adopted, variability in existing infrastructure may lead to inconsistencies across jurisdictions in the choice of screening algorithms (including cut-off thresholds across tiers), follow-up protocols, and classification of screen-positive cases. Such variation may create uncertainty for families moving between jurisdictions, challenge the portability of benefits, and complicate the evaluation of program effectiveness. While such inconsistency would not be unique to MLD, the pan-Canadian Recommended Newborn Screening List is intended to foster greater consistency,3 making these disparities particularly salient.
Expanding NBS to include MLD would also place new demand on metabolic specialists, genetic counsellors, neurologists, allied health providers, and family care providers. Increased follow-up for early-onset cases, along with ongoing monitoring and counselling for late-onset or indeterminate cases, may stretch specialist capacity and heighten burdens on family care providers who already play a central role in newborn care.101 Given the concentration of specialist services in urban centres, small jurisdictions with limited specialist availability may face longer turnaround times for diagnosis and treatment initiation, or rely more heavily on family care providers,101 potentially widening geographic inequities.
Governance
The governance of NBS programs is ultimately a question of trust: parents and the public need to be confident that decisions about expanding NBS are transparent, accountable, and grounded in strong oversight. Some have criticized NBS programs for their opaque decision-making, weak oversight, and limited quality control.102 Protecting families and maintaining trust in NBS programs therefore requires strong governance structures that ensure accountability and transparency at a policy level.102 Established frameworks for fair decision-making in health policy underline the need for transparent processes, relevant rationales, and opportunities for review when decisions about adding conditions are made. Clinical experts and laboratory professionals also highlighted the potential role of a national registry to track outcomes, refine protocols, and support evidence-informed adjustments over time, which are important elements for fairness, sustainability, and public trust.
Implications for Decision-Makers
The Condition
Current evidence underscores the severity and substantial burden of early-onset forms of MLD, which are estimated to account for most cases of MLD (75% to 90%). These estimates are shaped by clinical ascertainment and family history, which identifies severe, rapidly progressing cases and may underestimate the prevalence of late-onset disease. Adding MLD to NBS would generate more data on the incidence of MLD and its subtypes, as screening captures a broader range of affected individuals.
If implemented, data generated from NBS for MLD could shift our understanding of the proportions of early-onset and late-onset MLD, with potential implications for both program goals and resource planning, underscoring the importance of evaluation and considering the need for reassessment over time. There may be opportunities to learn from and share data and information from NBS programs internationally that have already implemented NBS for MLD, to better elucidate the incidence of subtypes as well as the benefits, harms, and implementation considerations for NBS for MLD.
The Test and Accuracy of Screening Algorithms
Evidence and input from experts suggest that if NBS for MLD were adopted in Canada, a 3-tier strategy (sulfatide levels, followed by ARSA enzyme activity, followed by molecular testing) is likely to be the most accurate and preferred approach. Nonetheless, current algorithms may not always reliably distinguish early-onset from late-onset disease and will generate incidental or uncertain findings. Decision-makers may consider whether the current evidence base and expert input provide sufficient confidence with which to proceed.
The Acceptability, Benefits, and Harms of NBS
Most experts consulted for this review agreed that early-onset MLD is an acceptable addition to NBS in Canada, as long as an effective treatment option becomes available. In general, members of the NBS community consulted for this review emphasized the importance of making NBS for MLD available if an effective treatment option becomes available and expressed confidence in the experience and ability of Canadian NBS programs and associated clinical communities of practice to effectively manage NBS referrals for both early-onset and late-onset MLD.
The risk of identifying late-onset MLD and incidental and uncertain findings remains an important consideration with implications for newborns, their families, and health systems. The identification of late-onset MLD has the potential to be considered as a benefit and a harm of NBS for MLD depending on how the scope of benefits is defined. The development of processes for the consent for screening and communication of results, as well as ensuring appropriate follow-up care for affected individuals, may help shape public and professional trust and the acceptability of NBS for MLD.
The Treatment and Its Effectiveness
The Newborn Screening Advisory Panel’s criteria emphasize that NBS is most appropriate when effective treatments are available. For MLD in Canada, there are currently no disease-modifying treatments available for early-onset disease. While a gene therapy (i.e., arsa-cel) for early-onset MLD is available in some international jurisdictions, it has not been submitted for consideration for regulatory or reimbursement approval in Canada. This means decisions about adding MLD are closely tied to the regulatory and reimbursement landscape of disease-modifying therapies for early-onset MLD.
Implementation and Health System Considerations
Experts engaged by CDA-AMC generally agreed that it would be possible to implement NBS for MLD across Canada. However, implementing NBS for MLD would have an impact on provincial and territorial programs, particularly in smaller jurisdictions where relatively more extensive resources and infrastructure may be required to accommodate NBS for MLD.
Long-term sustainability may be supported through coordinated pan-Canadian approaches (e.g., supportive advisory bodies and structures) to mitigate jurisdictional inequities and benefit from economies of scale. Additionally, there may be opportunities to consider centralized capacity for screening for MLD.
Evidence Reviews and the Recommended Pan-Canadian Newborn Screening List
This pilot evidence review sought to action relevant short-term recommendations from the Newborn Screening Advisory Panel as a learning opportunity, which included the following:
Adopt the proposed criteria and processes for adding and reassessing a condition for the Recommended Pan-Canadian Newborn Screening List. This would include developing transparent deliberative processes to support recommendations for adding or reassessing a condition.
Pilot the proposed processes for adding a condition by conducting an evidence review on a candidate condition and developing recommendations. This would provide learnings for coordination among interested parties and explore the development of a secretariat support function (p. 9).3
This first pilot evidence review focused on integrating the recommended criteria; trialling the evidence review process by leveraging existing infrastructure from a pan-Canadian health organization; and implementing features of the Newborn Screening Advisory Panel’s recommended deliberation, recommendation, engagement, and communication processes. This will provide learnings and inputs that will be used to further develop and refine processes in alignment with those recommended by the Newborn Screening Advisory Panel.
Key Findings and Uncertainties
Table 5 provides a summary of the key findings and uncertainties from the evidence review, in relation to the Newborn Screening Advisory Panel Criteria.
Table 5: Summary of Key Findings and Uncertainties
Domain | Newborn Screening Advisory Panel criteria | Key findings from the evidence review | Key uncertainties |
|---|---|---|---|
The condition | The condition should be serious and one that arises in children and/or leads to morbidity and mortality in childhood. | Early-onset MLD (i.e., late infantile and early juvenile) is a serious condition that arises in infancy or childhood and has severe, deleterious impacts and outcomes for patients and families. | Late-onset MLD (i.e., late juvenile and adult onset) arises in late childhood, adolescence, or adulthood, and cannot always be differentiated from early-onset disease by NBS. |
The epidemiology (including incidence and variation across regions and jurisdictions) and NH of the condition should be adequately understood. | The birth prevalence of MLD (all subtypes) is estimated at less than 1 per 100,000 live births worldwide. MLD is classified as either early-onset or late-onset. These 2 broad classifications are typically broken down further into 4 subtypes based on the age of symptom onset: late infantile (≤ 30 months), early juvenile (30 months to < 7 years), late juvenile (7 years to < 17 years), and adult (≥ 17 years). Early-onset (i.e., late infantile and early juvenile) MLD is the most severe form of the disease, characterized by rapid neurodegeneration and greatly reduced life expectancy, and accounts for an estimated 75% to 90% of cases. | Data describing the epidemiology and NH remain limited due to the rarity of the condition. Experts consulted on the pilot evidence review agreed that NBS will increase knowledge and understanding of the epidemiology and NH of MLD. The estimated prevalence of subtypes may change should NBS for MLD be adopted. | |
Differences in the incidence and variation in test performance in subpopulations, particularly in those who are underserved or underrepresented, should be characterized and adequately understood. | Data describing the incidence and variation in test performance across subpopulations were not identified. | Differences in the incidence and variation in test performance in subpopulations are not known. | |
The test | There should be a robust, scalable, safe, and validated screening test. | Evidence from a small number of studies across several jurisdictions suggests that both 2-tier and 3-tier screening algorithms can detect early-onset MLD with the reported sensitivity and specificity approaching 100%. PPV estimates were 15% and 50% in 2 studies assessing the same 2-tier strategy. The addition of a third tier increased the PPV to 100%. Estimates of NPV were high (100%). The number of false-positives in 2-tier screening strategies was small (0.0037% to 0.02%) and was improved to 0% with the addition of genetic sequencing as the third tier. | Data describing the performance and accuracy of the screening test are limited in quantity and quality. There is no information on true negatives or false-negatives because the application of the reference standard to all screen-negative newborns was not considered practical. Thus, estimates of NPV were based on the assumption that no MLD cases were missed without methods to identify false-negatives. The NBS algorithm for MLD may not always be able to distinguish early-onset disease from late-onset disease. |
The benefits of screening should outweigh the physical and psychological harms caused by the screening test. | The benefits of screening for early-onset MLD are dependent on the availability of effective treatment; there is currently no effective, disease-modifying treatment for late infantile MLD available in Canada. Where effective treatment is available for early-onset MLD, screening allows for identification of early-onset disease and treatment in the presymptomatic period. There are no physical harms caused by the screening test. | Adding MLD to NBS could result in increased identification of late-onset cases. Late-onset forms of disease are not primary targets of NBS for MLD. It is unclear if the identification of late-onset and indeterminate cases should be considered a harm or a benefit of NBS for MLD. The identification of late-onset and indeterminate cases of MLD through NBS may cause harm (e.g., psychological, inappropriate monitoring). However, it may be considered a benefit (e.g., enable earlier detection and access to treatment.) | |
The screening test, diagnosis, and treatment should be socially and ethically acceptable to health professionals and the public. | Most experts consulted agreed that the Newborn Screening Algorithm and diagnostic process for early-onset MLD were acceptable. There is currently no disease-modifying treatment option available in Canada for early-onset MLD. Because NBS for early-onset MLD may also identify late-onset forms of the disease (which are not an appropriate target of NBS), ethical and social implications may include consideration of explicit consent models. The risk of identifying late-onset cases, and false-positive, incidental, and uncertain findings may have considerable consequences for marginalized families, particularly those living in rural and/or remote locations. | The acceptability of NBS for MLD to the public is not known. | |
The treatment | There should be an effective treatment or intervention for newborns identified through early detection, with evidence of early treatment leading to better health outcomes and reduced morbidity and/or mortality than late treatment. | While an emerging treatment for late infantile MLD has been developed, it has not been submitted for regulatory or reimbursement review in Canada. | It is unknown whether the emerging treatment will become available in Canada. Data describing the effectiveness of the emerging treatment in other jurisdictions are limited in quantity and quality. |
There is an agreed policy on the further diagnostic investigation of newborns with a positive screening test. There should be agreed evidence-based policies covering appropriate treatments and the cases in which they can be offered. | There are recent guidelines informing the management of MLD identified through NBS but they are not specific to the Canadian context. No policies in Canada were identified informing the diagnostic investigation or treatment of MLD. However, clinical experts in Canada are trained and manage current cases of MLD that are ascertained clinically. | There is no published consensus informing the diagnosis and treatment of MLD in Canada. | |
Other considerations | The budgetary impact of case-finding (including screening, diagnosis, and treatment) should be considered in relation to not screening. | A 3-tier NBS program for MLD is estimated to have an incremental budget impact on Canada’s provincial and territorial health systems that could range from $4.5 million to $30.1 million, which includes screening and diagnostic costs up to the point of diagnosis over a 10-year period. The variation depends on key implementation assumptions. A 3-tier NBS program for MLD is estimated to represent an average incremental cost per new potentially treatable patient (early-onset or late-onset MLD) that could range from $84,686 to $832,443. | Current estimates of the incidence of MLD in Canada remain uncertain and could affect the costs per new potentially treatable patient. Estimates of the costs associated with implementation of NBS for MLD, including capital and operational costs for NBS laboratories, as well as postdiagnosis clinical monitoring and follow-up, remain uncertain and could affect budget impact estimates. |
Services and facilities for diagnosis and treatment should be available to newborns who are screened. | Services and facilities for diagnosis and supportive care of early-onset MLD in Canada currently exist for those whose disease is ascertained clinically. Services and facilities for diagnosis and treatment of early-onset MLD identified by NBS would have to be developed in Canada alongside implementation of NBS. Services and facilities to accommodate the increased demand for monitoring and treatment of late-onset MLD would have to be developed in Canada alongside implementation of NBS. | Impacts to NBS programs, laboratories and associated clinical communities of practice could be substantial and may vary across provincial and territorial jurisdictions. |
MLD = metachromatic leukodystrophy; NBS = newborn screening; NH = natural history; NPV = negative predictive value; PPV = positive predictive value.
References
1.Wilson K KS, Potter BK, Geraghty MT, Chakraborty P. Developing a National Newborn Screening Strategy for Canada. 2010: https://ruor.uottawa.ca/server/api/core/bitstreams/dfba8fe8-d355-46c0-9ae0-7f07fe72176f/content. Accessed 8 January 2025.
2.Groulx-Boivin E, Osman H, Chakraborty P, et al. Variability in Newborn Screening Across Canada: Spinal Muscular Atrophy and Beyond. Can J Neurol Sci. 2024;51(2):203-209. PubMed
3.CDA-AMC. Toward a Future Pan-Canadian Coordinated Approach for Newborn Screening: A Report From the Advisory Panel. 2025: https://www.cda-amc.ca/sites/default/files/DRD/HC0079-NBS_Recommendations_Report.pdf. Accessed 23 May/25.
4.Fahim SM, Lin G, Suh K, et al. Atidarsagene autotemcel for metachromatic leukodystrophy. J Manag Care Spec Pharm. 2024;30(2):201-205. PubMed
5.Garritty C, Hamel C, Trivella M, et al. Updated recommendations for the Cochrane rapid review methods guidance for rapid reviews of effectiveness. BMJ. 2024;384:e076335. PubMed
6.Westwood ME RMZ, Chen J, Armstrong N, Noake C. Newborn screening for metachromatic leukodystrophy (MLD): A rapid evidence review. 2025: https://view-health-screening-recommendations.service.gov.uk/metachromatic-leukodystrophy/. Accessed 6 May 2025.
7.Groeschel S, Kehrer C, Krageloh-Mann I. Standardized description of rare diseases-the natural course and treatment of metachromatic leukodystrophy. Rev Neurol. 2014;58(CONGRESO1):C84-C90.
8.Harrington M, Whalley D, Twiss J, et al. Insights into the natural history of metachromatic leukodystrophy from interviews with caregivers. Orphanet J Rare Dis. 2019;14(1):89. PubMed
9.Asbreuk M, Schoenmakers DH, Adang LA, et al. Metachromatic Leukodystrophy: New Therapy Advancements and Emerging Research Directions. Neurology. 2025;105(2):e213817. PubMed
10.Cesani M, Lorioli L, Grossi S, et al. Mutation Update of ARSA and PSAP Genes Causing Metachromatic Leukodystrophy. Hum Mutat. 2016;37(1):16-27. PubMed
11.National Library of Medicine. ClinVar. n.d.: https://www.ncbi.nlm.nih.gov/clinvar/. Accessed 9 September 2025.
12.Gomez-Ospina N. Arylsulfatase A Deficiency. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews. 2024 April 25 ed. Seattle (WA): University of Washington, Seattle.
13.Chang S-C, Bergamasco A, Bonnin M, Bisonó TA, Moride Y. A systematic review on the birth prevalence of metachromatic leukodystrophy. Orphanet J Rare Dis. 2024;19(1):80. PubMed
14.Chang SC, Eichinger CS, Field P. The natural history and burden of illness of metachromatic leukodystrophy: a systematic literature review. Eur J Med Res. 2024;29(1):181. PubMed
15.van Rappard DF, Boelens JJ, Wolf NI. Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab. 2015;29(2):261-273. PubMed
16.Zamani G. Metachromatic leukodystrophy: Overveiw. Iran. 2014;8(4):5-6.
17.Thomas S, Morrison A, Morton G, Roberts P, Clark V, Imrie J. The burden of disease in metachromatic leukodystrophy: results of a caregiver survey in the UK and Republic of Ireland. Orphanet J Rare Dis. 2024;19(1):87. PubMed
18.Ashrafi MR, Amanat M, Garshasbi M, et al. An update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies. Expert Rev Neurother. 2020;20(1):65-84. PubMed
19.Laugwitz L, Schoenmakers DH, Adang LA, et al. Newborn screening in metachromatic leukodystrophy - European consensus-based recommendations on clinical management. Eur J Paediatr Neurol. 2024;49:141-154. PubMed
20.Adang LA, Bonkowsky JL, Boelens JJ, et al. Consensus guidelines for the monitoring and management of metachromatic leukodystrophy in the United States. Cytotherapy. 2024;26(7):739-748. PubMed
21.Trinidad M, Hong X, Froelich S, et al. Predicting disease severity in metachromatic leukodystrophy using protein activity and a patient phenotype matrix. Genome Biol. 2023;24(1):172. PubMed
22.Santhanakumaran V, Groeschel S, Harzer K, et al. Predicting clinical phenotypes of metachromatic leukodystrophy based on the arylsulfatase A activity and the ARSA genotype? - Chances and challenges. Mol Genet Metab. 2022;137(3):273-282. PubMed
23.Laugwitz L, Mechtler TP, Janzen N, et al. Newborn Screening and Presymptomatic Treatment of Metachromatic Leukodystrophy. N Engl J Med. 2024;391(13):1256-1258. PubMed
24.Jonckheere AI, Kingma SDK, Eyskens F, Bordon V, Jansen AC. Metachromatic leukodystrophy: To screen or not to screen? Eur J Paediatr Neurol. 2023;46:1-7. PubMed
25.Koppaka R; Centers for Disease Control and Prevention (CDC). Ten Great Public Health Achievements—United States, 2001--2010. MMWR Morb Mortal Wkly Rep.; 2011: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6019a5.htm. Accessed 25 August 2025.
26.Chakraborty P, Potter BK, Hayeems RZ. Maximizing Benefits and Minimizing Harms: Diagnostic Uncertainty Arising From Newborn Screening. Pediatrics. 2021;148(6). PubMed
27.Lam WKK, Ream MA, Grosse SD, et al. Evidence Regarding Metachromatic Leukodystrophy Newborn Screening. Pediatrics. 2025;12:12. PubMed
28.Shea BJ, Reeves BC, Wells G, et al. AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both. BMJ. 2017;358:j4008. PubMed
29.Hong X, Daiker J, Sadilek M, et al. Toward newborn screening of metachromatic leukodystrophy: results from analysis of over 27,000 newborn dried blood spots. Genet Med. 2021;23(3):555-561. PubMed
30.Wu THY, Brown HA, Church HJ, et al. Improving newborn screening test performance for metachromatic leukodystrophy: Recommendation from a pre-pilot study that identified a late-infantile case for treatment. Mol Genet Metab. 2024;142(1):108349. PubMed
31.Malvagia S, Bettiol A, Porcaro M, et al. Newborn Screening for Metachromatic Leukodystrophy in Tuscany: The Paradigm of a Successful Preventive Medicine Program. Int J Neonatal Screen. 2025;11(2):24. PubMed
32.Whiting PF, Rutjes AW, Westwood ME, et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann Intern Med. 2011;155(8):529-536. PubMed
33.Wilson JMG, Jungner G, World Health Organization. Principles and practice of screening for disease. Geneva (CH): World Health Organization; 1968: https://iris.who.int/handle/10665/37650. Accessed 2024 Jun 27.
34.Malvagia S, Forni G, Ombrone D, la Marca G. Development of Strategies to Decrease False Positive Results in Newborn Screening. Int J Neonatal Screen. 2020;6(4). PubMed
35.Sekhon M, Cartwright M, Francis JJ. Acceptability of healthcare interventions: an overview of reviews and development of a theoretical framework. BMC Health Serv Res. 2017;17(1):88. PubMed
36.Azzopardi PJ, Upshur REG, Luca S, et al. Health-care providers' perspectives on uncertainty generated by variant forms of newborn screening targets. Genet Med. 2020;22(3):566-573. PubMed
37.Fumagalli F, Calbi V, Natali Sora MG, et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet. 2022;399(10322):372-383. PubMed
38.Health Resources & Services Administration. Committee Approach to Evaluating the Condition Review Report (Decision Matrix). 2024: https://www.hrsa.gov/advisory-committees/heritable-disorders/decision-matrix. Accessed 17 January 2025.
39.Scyne Advisory and the Department of Health and Aged Care. Newborn Bloodspot Screening Expansion READINESS ASSESSMENT EXECUTIVE SUMMARY. 2024: https://www.health.gov.au/sites/default/files/2024-06/newborn-bloodspot-screening-expansion-readiness-assessment-executive-summary_0.pdf. Accessed 12 August 2025.
40.Sivananthan S, Davison J, Chakrapani A, Gissen P. Metachromatic leukodystrophy: A single centre review of features at diagnosis and barriers to accessing treatment. Mol Genet Metab. 2023;Conference: WORLDSymposiumTM 2023. Orlando United States. 138(2) (no pagination).
41.ACHDNC. THE ADVISORY COMMITTEE ON HERITABLE DISORDERS IN NEWBORNS AND CHILDREN IN-PERSON/WEBINAR. 2024: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/meetings/achdnc-aug-2024-day2-transcript.pdf. Accessed 19 August 2025.
42.Hong X, Kumar AB, Daiker J, et al. Leukocyte and Dried Blood Spot Arylsulfatase A Assay by Tandem Mass Spectrometry. Anal Chem. 2020;92(9):6341-6348. PubMed
43.Shaff A, Basheeruddin K, Bekri S, et al. Newborn screening for metachromatic leukodystrophy: Preparation of reagents and methodology for measurement of sulfatides and arylsulfatase A enzymatic activity in dried blood spots. Mol Genet Metab. 2025;145(3):109138. PubMed
44.Calbi V, Fumagalli F, Fratini ES, et al. Blood sulfatides as disease biomarker for metachromatic leukodystrophy: Disease characterization, early diagnosis, and response to treatment. Mol Genet Metab. 2023;Conference: WORLDSymposiumTM 2023. Orlando United States. 138(2) (no pagination).
45.Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016;388(10043):476-487. PubMed
46.Groeschel S, Kühl J-S, Bley AE, et al. Long-term Outcome of Allogeneic Hematopoietic Stem Cell Transplantation in Patients With Juvenile Metachromatic Leukodystrophy Compared With Nontransplanted Control Patients. JAMA Neurology. 2016;73(9):1133-1140. PubMed
47.Fumagalli F, Calbi V, Gallo V, et al. Long-Term Effects of Atidarsagene Autotemcel for Metachromatic Leukodystrophy. N Engl J Med. 2025;392(16):1609-1620. PubMed
48.Cameron CL, Kang PB, Burns TM, Darras BT, Jones HR, Jr. Multifocal slowing of nerve conduction in metachromatic leukodystrophy. Muscle Nerve. 2004;29(4):531-536. PubMed
49.Sterne JA, Hernán MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ. 2016;355:i4919. PubMed
50.Zambon AA, Rancoita PMV, Quattrini A, et al. Effects of atidarsagene autotemcel gene therapy on peripheral nerves in late-infantile metachromatic leukodystrophy. Brain. 2025;26:26. PubMed
51.O'Neill J, Tabish H, Welch V, et al. Applying an equity lens to interventions: using PROGRESS ensures consideration of socially stratifying factors to illuminate inequities in health. J Clin Epidemiol. 2014;67(1):56-64. PubMed
52.Oliver S KJ, Caird J, Lorenc T, Oliver K, Harden A, Thomas J, Greaves A, Oakley A. Health Promotion, Inequalities and Young People's Health: A Systematic Review of Research. EPPI-Centre, Social Science Research Unit, Institute of Education, University of London; 2008: https://eppi.ioe.ac.uk/cms/Portals/0/PDF%20reviews%20and%20summaries/Inequalities%20Young%20People%20R2008Oliver.pdf. Accessed July 31, 2025.
53.Biffi A, Cesani M, Fumagalli F, et al. Metachromatic leukodystrophy - mutation analysis provides further evidence of genotype-phenotype correlation. Clin Genet. 2008;74(4):349-357. PubMed
54.Suhr D, Suhr TR, Gelb MH. Scientific, clinical, social, and policy status and considerations for implementing newborn screening of metachromatic leukodystrophy. Mol Genet Metab. 2020;129(2):S149-S150.
55.Ozturk Y. Superdrugs & Supertherapies: High prices to pay for miracles. Acta Pharmaceutica Sciencia. 2025;63(1):XXV-XXIX.
56.Bean K, Jones SA, Chakrapani A, et al. Exploring the Cost-Effectiveness of Newborn Screening for Metachromatic Leukodystrophy (MLD) in the UK. Int. 2024;10(3):26. PubMed
57.Suhr D, Suhr TR, Mathiesen T, Therrell BL. RANSIP newborn screening program: Working to bring early diagnostics, inclusion, and therapeutic access to metachromatic leukodystrophy patients. Mol Genet Metab. 2021;132(2):S104-S105.
58.Bhatia S, Le Cam Y, Carrion J, et al. Strengthening health systems for access to gene therapy in rare genetic disorders. Mol Ther Methods Clin Dev. 2024;32(2):101220. PubMed
59.Watson MS, Lloyd-Puryear MA, Howell RR. The Progress and Future of US Newborn Screening. Int J Neonatal Screen. 2022;8(3). PubMed
60.Gelb MH, Shaff A, Basheeruddin K, et al. Consensus Assay Methodology for Analysis of Sulfatides and Arylsulfatase a Enzymatic Activity in Dried Blood Spots for Newborn Screening of Metachromatic Leukodystrophy. Ssrn. 2025;04.
61.Cappuccio G, Alagia M, Brunetti-Pierri N. A systematic cross-sectional survey of multiple sulfatase deficiency. Mol Genet Metab. 2020;130(4):283-288. PubMed
62.Orchard Therapeutics North America. TREATMENT CENTERS. 2025: https://lenmeldy.com/treatment-centers/. Accessed 20 August 2025.
63.Table 17-10-0057-01. Projected population, by projection scenario, age and gender, as of July 1 (x 1,000). 2025. https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1710005701. Accessed 2025 July 15.
64.Table 13-10-0414-01. Live births, by place of residence of mother. 2024. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310041401. Accessed 2025 July 15.
65.NICE. Atidarsagene autotemcel for treating metachromatic leukodystrophy. HST18. 2022; https://www.nice.org.uk/guidance/hst18. Accessed 2025 Jul 16.
66.US Food & Drug Administration. LENMELDY Summary Basis for Regulartory Action. 2024; https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/lenmeldy. Accessed 2025 September 08.
67.European Medicines Agency. Libmeldy: autologous CD34+ cells encoding ARSA gene. 2020; https://www.ema.europa.eu/en/medicines/human/EPAR/libmeldy. Accessed 2025 September 08.
68.Schoenmakers DH, Asbreuk M, Martin T, et al. Key lessons from the first international treatment eligibility committee: the case of metachromatic leukodystrophy. Eur J Paediatr Neurol. 2025;57:72-81. PubMed
69.Sullivan SD, Mauskopf JA, Augustovski F, et al. Budget impact analysis-principles of good practice: report of the ISPOR 2012 Budget Impact Analysis Good Practice II Task Force. Value Health. 2014;17(1):5-14. PubMed
70.Bank of Canada. Inflation-control target. 2021; https://www.bankofcanada.ca/rates/indicators/key-variables/inflation-control-target/. Accessed 2025 Aug 01.
71.Wang RY, Bodamer OA, Watson MS, Wilcox WR, Diseases AWGoDCoLS. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13(5):457-484. PubMed
72.Morton G, Thomas S, Roberts P, Clark V, Imrie J, Morrison A. The importance of early diagnosis and views on newborn screening in metachromatic leukodystrophy: results of a Caregiver Survey in the UK and Republic of Ireland. Orphanet J Rare Dis. 2022;17(1):403. PubMed
73.Newborn Screening Ontario Molecular Diagnostics. Molecular Diagnostic Testing. 2025; https://www.newbornscreening.on.ca/media/t1mhhpni/molecular-price-list-jan-2025_v3.pdf. Accessed 2025 September 02.
74.Job Bank: Medical laboratory Technologist in Canada. 2024; https://www.jobbank.gc.ca/marketreport/wages-occupation/4187/ca. Accessed 2025 Aug 26.
75.Ontario Ministry of Health. Schedule of benefits for physician services under the Health Insurance Act: (February 14, 2025 (effective Mar 3, 2025)). 2025: https://www.ontario.ca/files/2025-03/moh-schedule-benefit-2025-03-19.pdf. Accessed 2025 Jul 15.
76.Discovery DNA. Whole Genome Sequencing. 2025; https://www.discoverydna.ca/product-page/clinically-accredited-whole-genome-sequencing. Accessed 2025 August 22.
77.GenomeQuebec. Promotions: Next-generation sequencing services from the Centre d’expertise et de services Génome Québec. 2025; https://genomequebec.com/en/technological-services/promotions/. Accessed 2025 Aug 22.
78.Ammann-Schnell L, Groeschel S, Kehrer C, Frölich S, Krägeloh-Mann I. The impact of severe rare chronic neurological disease in childhood on the quality of life of families-a study on MLD and PCH2. Orphanet J Rare Dis. 2021;16(1):1-16. PubMed
79.Koto Y, Ueki S, Yamakawa M, Sakai N. Experiences of patients and their family members with metachromatic leukodystrophy, adrenoleukodystrophy, and Krabbe disease: a qualitative systematic review protocol. JBI Evidence Synthesis. 2023;21(5):1027-1033. PubMed
80.Bonkowsky JL, Wilkes J, Bardsley T, Urbik VM, Stoddard G. Association of Diagnosis of Leukodystrophy With Race and Ethnicity Among Pediatric and Adolescent Patients. JAMA Network Open. 2018;1(7):e185031. PubMed
81.Strenk ME, Berrios C, Garrett JR. Addressing the Burdens That Newborn Screening Imposes on Underserved Communities. The American Journal of Bioethics. 2023;23(7):79-82. PubMed
82.Ross LF. Mandatory versus voluntary consent for newborn screening? Kennedy Inst Ethics J. 2010;20(4):299-328. PubMed
83.Forman J, Coyle F, Levy-Fisch J, Roberts P, Terry S, Legge M. Screening criteria: the need to deal with new developments and ethical issues in newborn metabolic screening. J Community Genet. 2013;4(1):59-67. PubMed
84.Wasserstein MP, Orsini JJ, Goldenberg A, et al. The future of newborn screening for lysosomal disorders. Neurosci Lett. 2021;760(no pagination). PubMed
85.van Dijk T, Kater A, Jansen M, et al. Expanding Neonatal Bloodspot Screening: A Multi-Stakeholder Perspective. Front Pediatr. 2021;9:706394. PubMed
86.Nicholls SG, Wilson BJ, Etchegary H, et al. Benefits and burdens of newborn screening: public understanding and decision-making. Per Med. 2014;11(6):593-607. PubMed
87.Hayeems RZ, Miller FA, Bombard Y, et al. Expectations and values about expanded newborn screening: a public engagement study. Health Expect. 2015;18(3):419-429. PubMed
88.Paul DB. Eugenics Redux: “Reproductive Benefit” as a Rationale for Newborn Screening. Hastings Cent Rep. 2018;48 Suppl 2:S12-S13. PubMed
89.Liang NSY, Watts-Dickens A, Chitayat D, Babul-Hirji R, Chakraborty P, Hayeems RZ. Parental Preferences for Expanded Newborn Screening: What Are the Limits? Children (Basel). 2023;10(8). PubMed
90.Grob R. Qualitative Research on Expanded Prenatal and Newborn Screening: Robust but Marginalized. Hastings Cent Rep. 2019;49 Suppl 1(Suppl 1):S72-S81.
91.Wasserstein MP, Caggana M, Bailey SM, et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet Med. 2019;21(3):631-640. PubMed
92.Wasserstein MP, Orsini JJ, Goldenberg A, et al. The future of newborn screening for lysosomal disorders. Neurosci Lett. 2021;760:136080. PubMed
93.Davids L, Sun Y, Moore RH, et al. Health care practitioners' experience-based opinions on providing care after a positive newborn screen for Pompe disease. Mol Genet Metab. 2021;134(1-2):20-28. PubMed
94.Lisi EC, Ali N. Opinions of adults affected with later-onset lysosomal storage diseases regarding newborn screening: A qualitative study. J Genet Couns. 2021;30(6):1544-1558. PubMed
95.Timmermans S, Buchbinder M. Potentializing Newborn Screening. Current Anthropology. 2013;54(S7):S26-S35.
96.Dees RH, Kwon JM. The Ethics of Krabbe Newborn Screening. Public Health Ethics. 2013;6(1):114-119.
97.Grob R, Roberts S, Timmermans S. Families' Experiences with Newborn Screening: A Critical Source of Evidence. Hastings Cent Rep. 2018;48 Suppl 2:S29-S31. PubMed
98.Anderson R, Rothwell E, Botkin JR. Newborn screening: ethical, legal, and social implications. Annu Rev Nurs Res. 2011;29:113-132. PubMed
99.Van der Burg S, Oerlemans A. Fostering Caring Relationships: Suggestions to Rethink Liberal Perspectives on the Ethics of Newborn Screening. Bioethics. 2018;32(3):171-183. PubMed
100.Moody L, Choudhry K. Parental views on informed consent for expanded newborn screening. Health Expect. 2013;16(3):239-250. PubMed
101.Hayeems RZ, Miller FA, Carroll JC, et al. Primary care role in expanded newborn screening: After the heel prick test. Can Fam Physician. 2013;59(8):861-868. PubMed
102.Miller FA. The Sad Story of Newborn Screening for Krabbe: The Need for Good Governance. Public Health Ethics. 2013;6(1):123-126.
ISSN: 2563-6596
Canada’s Drug Agency (CDA-AMC) is a pan-Canadian health organization. Created and funded by Canada’s federal, provincial, and territorial governments, we’re responsible for driving better coordination, alignment, and public value within Canada’s drug and health technology landscape. We provide Canada’s health system leaders with independent evidence and advice so they can make informed drug, health technology, and health system decisions, and we collaborate with national and international partners to enhance our collective impact.
Disclaimer: CDA-AMC has taken care to ensure that the information in this document was accurate, complete, and up to date when it was published, but does not make any guarantee to that effect. Your use of this information is subject to this disclaimer and the Terms of Use at cda-amc.ca.
The information in this document is made available for informational and educational purposes only and should not be used as a substitute for professional medical advice, the application of clinical judgment in respect of the care of a particular patient, or other professional judgments in any decision-making process. You assume full responsibility for the use of the information and rely on it at your own risk.
CDA-AMC does not endorse any information, drugs, therapies, treatments, products, processes, or services. The views and opinions of third parties published in this document do not necessarily reflect those of CDA-AMC. The copyright and other intellectual property rights in this document are owned by the Canadian Agency for Drugs and Technologies in Health (operating as CDA-AMC) and its licensors.
Questions or requests for information about this report can be directed to Requests@CDA-AMC.ca.